
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The CuB site in particulate methane monooxygenase may be used to produce hydrogen peroxide†
Kristoffer J. M.
Lundgren
 a,
Lili
Cao
ab,
Magne
Torbjörnsson
a,
Erik D.
Hedegård
ac and
Ulf
Ryde
*a
a,
Lili
Cao
ab,
Magne
Torbjörnsson
a,
Erik D.
Hedegård
ac and
Ulf
Ryde
*a
aDepartment of Computational Chemistry, Lund University, Chemical Centre, P. O. Box 124, SE-221 00 Lund, Sweden. E-mail: Ulf.Ryde@compchem.lu.se; Tel: +46-46 2224502
bMolecular AI, Discovery Sciences R&D AstraZeneca, Gothenburg, Pepparedsleden 1, SE-431 83 Mölndal, Sweden
cDepartment of Physics, Chemistry and Pharmacy, University of Southern Denmark, Campusvej 55, 5230 Odense, Denmark
First published on 16th January 2025
Abstract
Particulate methane monooxygenase (pMMO) is the most efficient of the two groups of enzymes that can hydroxylate methane. The enzyme is membrane bound and therefore hard to study experimentally. For that reason, there is still no consensus regarding the location and nature of the active site. We have used combined quantum mechanical and molecular mechanical (QM/MM) methods to study the reactivity of the CuB site with a histidine brace and two additional histidine ligands. We compare it with the similar active site of lytic polysaccharide monooxygenases. We show that the CuB site can form a reactive [CuO]+ state by the addition of three electrons and two protons, starting from a resting Cu(II) state, with a maximum barrier of 72 kJ mol−1. The [CuO]+ state can abstract a proton from methane, forming a Cu-bound OH− group, which may then recombine with the CH3 group, forming the methanol product. The two steps have barriers of 59 and 52 kJ mol−1, respectively. However, in many of the steps, formation and dissociation of H2O2 or HO2− compete with the formation of the [CuO]+ state and the former steps are typically more favourable. Thus, our calculations indicate that the CuB site is not employed for methane oxidation, but may rather be used for the formation of hydrogen peroxide. This conclusion concurs with recent experimental investigations that excludes the CuB site as the site for methane oxidation.
Introduction
Methane is one of the hardest organic substrates to hydroxylate, with a C–H bond dissociation energy of 435 kJ mol−1.1 In biology, there are only two families of enzymes that can perform this reaction. The first is the soluble methane monooxygenase,2,3 which contains a highly conserved diiron active site.4,5 This is a well-characterised group of enzymes, whose reaction mechanism is rather clear.6–10 The second group is the particulate methane monooxygenase (pMMO), which normally is the predominant enzyme in methanotrophic bacteria.1,11 However, this group has been much harder to study and characterise because the purification of this large membrane-bound enzyme is difficult.The first X-ray structure of a pMMO was published in 2005 (at 2.8 Å resolution), showing a heterotrimeric pmoA3pmoB3pmoC3 structure with one mononuclear and one dinuclear Cu site in the soluble periplasmic parts of the pmoB subunit, and a monomeric Zn site in the pmoC subunit, inside the membrane (these three metal sites will be called A, B and C in the following; cf.Fig. 1).12 The CuA site involves two His ligands and distant a Glu ligand. The CuB site has three His ligands, whereas the C site includes an Asp and two His ligands. In 2008, another X-ray structure of pMMO at 3.8 Å resolution confirmed the presence of the dinuclear CuB site, but not the mononuclear CuA site. Site C was occupied by Cu.13 Three years later, a crystal structure at 2.68 Å resolution also proposed Cu ions in site B, but it was mononuclear in one of the three protomers and dinuclear in the other two.14 Three additional crystal structures of pMMO were published in a study of the Zn inhibition of the enzyme.15 They all showed a single Cu ion in site B, Cu or Zn binding to site A, depending on the Zn concentration, and sometimes another Zn site.
 | ||
| Fig. 1 pMMO with the three metal sites, CuA in red, CuB in yellow and CuC in magenta (from the 3RGB structure). (a) shows the entire trimer of trimers, whereas (b) shows only one subunit with pmoA in cyan, pmoB in blue and pmoC in green. The CuD is close to the CuC site but the two sites are never occupied simultaneously in the same crystal or cryo-EM structures. | ||
The dinuclear nature of site B was originally suggested by X-ray absorption near-edge (EXAFS) measurements on pmoB.14,16–18 In 2018, we re-investigated the crystal structures,19 employing quantum refinement, i.e. X-ray crystallography with the CuB site supported by quantum mechanical (QM) calculations. The results showed that there is actually no support for a dinuclear copper site B from the crystallographic data. Instead, it is best described by a single Cu ion, coordinated by four protein ligands and possibly an axial water molecule. The same year, another crystal structure of pMMO in bicelles also showed a mononuclear CuB site and the accompanying EXAFS data were consistent with a mononuclear site.20 Two studies in 2019 with EPR and mass spectrometry, respectively, confirmed the mononuclear nature of all Cu sites in pMMO21,22 and this was later confirmed also for other types of methanotrophs.23 On the other hand, an EXAFS study from another group still suggested a dinuclear CuB site,24 but this conclusion was later attributed to background contamination and photodamage.25
Due to the rather poor resolution of the crystal structures, the metal content, as well as the location of the active site in pMMO have been highly controversial. Mössbauer spectroscopy data led to the proposal that the active site is site C occupied by a diiron cluster instead of zinc.26 The argument was mainly based on the residues around site C, with two conserved His and carboxylate groups, i.e. similar to that in the soluble methane monooxygenases. However, experiments conducted by Rosenzweig and co-workers demonstrated that addition of Cu could recover over 90% of the methane oxidation activity from the metal-free protein, while adding iron alone did not restore any activity.27 Moreover, some methane oxidation activity was detected for a construct involving the soluble regions of pmoB that contain CuB, whereas no activity was detected in the absence of this site.27 Likewise, mutation experiments indicated that the activity is localised in site B instead of site A.27 Further biochemical and spectroscopic studies from the same group suggested that CuB is the active site and that the reaction mechanism involves a peroxide intermediate.28,29 However, in 2019 they reinterpreted the experiments and argued that instead the mononuclear CuC site is the active site.21,22 In particular, they showed that only the ligands of CuC are strictly conserved in all pMMOs and that destruction of the CuC site abolish methane oxidation activity.21
In contrast, Chan and coworkers have long argued that pMMO contains 12–15 Cu ions per protomer, most of which are lost when preparing the protein for crystallisation, and that the active site involves a trinuclear Cu3 cluster, located in a negatively charged cavity at the interface between pmoA and pmoC (called site E in the following).7,10,30–32 In fact, they have constructed a 12-peptide fragment of pmoA from this site that binds three Cu ions and can slowly oxidise methane in presence of O2 or H2O2.33 The have also made biomimetic Cu3 models that can oxidise methane.33
In 2021, two cryogenic electron microscopy (cryo-EM) studies of pMMO were published. One presented eight cryo-EM structures of pMMO from three organisms and in different types of nanodiscs.34 All showed 2–3 mononuclear Cu sites, coinciding with the crystallographic CuA, CuB and CuC sites, but in three of the structures, the CuC ion had instead moved to a nearby site, with one Asn and two His ligands (called site D in the following). The other study reported one cryo-EM structure which showed a mononuclear CuA site and a dinuclear CuB site, but no metal ion in sites C or D.30 On the other hand, two Cu ions were found in site E, but with strange geometries (only one weak ligand to each Cu ion at distances of 2.6–3.0 Å). Finally, a mononuclear site with a Glu ligand and a dinuclear site with Thr, Asn and Asp ligands were found in the water-exposed part of pmoB. They belong to a region that has been termed the CuI sponge and have been suggested to provide a buffer of reducing equivalents.24,35
In addition to the experimental investigations, several computational studies of pMMO have been performed. Yoshizawa and coworkers have shown that both a mononuclear CuIII-oxo species and dinuclear bis(μ-oxo)CuIICuIII are reactive enough to oxidise CH4, although the latter species is more easily formed.36–38 The calculations of the mononuclear Cu ion were based on site A, whereas those on the dinuclear site were based on site B. Solomon and coworkers have studied the active species in the Cu-ZSM-5 zeolite that also oxidises methane and suggested that the active species is a CuII–O–CuII structure11 and Gupta and coworkers have studied a model complex with a suggested reactive CuIII–O–CuII core.39 We have studied methanol oxidation by a mononuclear CuB site using QM-cluster calculations.19 Starting from a [CuO]+ complex, a three-step reaction was found, with an initial hydrogen abstraction from CH4 (22 kJ mol−1 barrier), binding of the resulting methyl radical to Cu and rebound of the OH group to the methyl radical (53 kJ mol−1 barrier).
In 2021, Peng et al. presented a combined QM and molecular mechanics (QM/MM) study of pMMO.40 They showed that duroquinol cannot reduce a mononuclear CuB site, but it can reduce the CuC site. Once reduced, CuC can bind O2 and it can be reduced to a [CuO]+ state (with another duroquinol molecule), which can oxidise CH4. The rate-limiting step is the cleavage of the O–O bond with a barrier of 72 kJ mol−1. Recently, they used QM/MM calculations to compare the reactivity of the CuC and CuD sites, based on the cryo-EM structures from Rosenzweig et al., suggesting that CuD is the more reactive site.41 This agrees with ENDOR studies of the binding of trifluoro-ethanol.42 Recently, Siegbahn studied the reactivity of the CuD site with QM-cluster calculations and showed that it may oxidise both CH4 and NH3, using a CuII(OH)2 reactive intermediate.43
It is notable that in the CuB site, the amino-terminal His residue of pmoB coordinates to Cu with both the sidechain and the amino-terminal –NH2 group. Interestingly, this motif is also observed for the active-site Cu ion in the lytic polysaccharide monooxygenases (LPMOs), where it was coined a histidine brace.44–47 The LPMOs is a group of enzymes that cleave polysaccharide chains by hydroxylating one of the C–H bonds, similar to the pMMO reactivity.44–47 The LPMOs have been thoroughly studied by computational methods. It has been shown that the reaction mechanism involves a [CuO]+ species that abstracts a hydrogen atom from the substrate, after which the Cu-bound OH group rebinds to the substrate.48–53 Moreover, the active site of LPMOs is known to activate O2 and form H2O2 in absence of substrate.54,55
Clearly, the reaction mechanism of methane hydroxylation by pMMO is not fully elucidated and experimental structures seem to spur more questions than answers. In this paper, we use QM/MM calculations to study the reactivity of the mononuclear CuB site. Even if it is no longer considered to be the active site of pMMO, its similarity to the active site of LPMO is intriguing and we therefore compare its reactivity with that of the LPMO active site. In particular, we also study alternative reactions and the results indicate that the CuB site actually may be poised towards the formation of hydrogen peroxide, rather than oxidation of methane.
Methods
The proteins
We have studied the CuB site in pMMO. The calculations were based on the 2.8 Å crystal structure of pMMO from Methylococcus capsulatus (PDB code 3RGB)14 and included only one pmoA–pmoB–pmoC monomer of the trimeric structure. No attempts were made to model the missing residues in the structures. All crystallographic water molecules were kept in the calculations.The protonation states of all residues were determined from a detailed study of the hydrogen-bond pattern and the solvent accessibility. It was checked by the PROPKA56 and Maestro software.57 The latter software was allowed to rotate the amide sidechain of the Asn and Gln residues. All Arg, Lys, Asp, and Glu residues were assumed to be charged, except Asp-47B, Asp-132B, Asp-166C, Glu-100B, Glu-96C, Glu-100C, Glu-154C, Glu-176C, and Lys-55B (the last letter in the residue numbers indicates the subunit according to the PDB files; note that subunits A, B and C are pmoB, pmoA and pmoC, respectively). Residues His-139A, 192A, 40B, 168B, 232B, 160C and 173C were assumed to be protonated on the ND1 atom, His-307A and 11B were presumed to be protonated on both the ND1 and NE2 atoms (and therefore positively charged), whereas the remaining His residues were modelled with a proton on the NE2 atom.
MD equilibration
All MM calculations were performed with the Amber software.58 For the protein, we used the Amber ff14SB force field59 and water molecules were described by the TIP3P model.60 For the metal sites, we employed restrained electrostatic potential charges,61 obtained from electrostatic potentials calculated at the TPSS/def2-SV(P) level of theory62,63 and sampled with the Mertz–Kollman scheme, although at a higher-than-default point density (∼2000 per atoms64).65 Metal sites outside the QM systems were kept at the crystal-structure geometry.The protein was solvated in a sphere with a radius of 62 Å around the geometrical centre of the protein, giving 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 788 atoms in total. The added protons and water molecules were optimised by a 10 ns simulated annealing calculation (up to 370 K), followed by a minimisation, keeping the other atoms fixed at the crystal-structure positions. Bond lengths involving H atoms were constrained with the SHAKE algorithm,66 allowing a time step of 1 fs.
788 atoms in total. The added protons and water molecules were optimised by a 10 ns simulated annealing calculation (up to 370 K), followed by a minimisation, keeping the other atoms fixed at the crystal-structure positions. Bond lengths involving H atoms were constrained with the SHAKE algorithm,66 allowing a time step of 1 fs.
QM/MM calculations
The QM/MM calculations were performed with the ComQum software.67,68 In this approach, the protein and solvent are split into two subsystems: system 1 (the QM region) was relaxed by QM methods, whereas system 2 contained the remaining part of the protein and the solvent. It was kept fixed at the original coordinates (equilibrated crystal structure).In the QM calculations, system 1 was represented by a wavefunction, whereas all the other atoms were represented by an array of partial point charges, one for each atom, taken from the MM setup. Thereby, the polarisation of the QM system by the surroundings is included in a self-consistent manner (electrostatic embedding). When there is a bond between systems 1 and 2 (a junction), the hydrogen link-atom approach was employed: the QM system was capped with hydrogen atoms (hydrogen link atoms, HL), the positions of which are linearly related to the corresponding carbon atoms (carbon link atoms, CL) in the full system.67,69 All atoms were included in the point-charge model, except the CL atoms.70
The total QM/MM energy in ComQum was calculated as67,68
 | (1) |
 is the MM energy of the QM system, still truncated by HL atoms, but without any electrostatic interactions. Finally,
is the MM energy of the QM system, still truncated by HL atoms, but without any electrostatic interactions. Finally,  is the classical energy of all atoms (in both the QM and MM regions) with CL atoms and with the charges of the QM region set to zero (to avoid double-counting of the electrostatic interactions). Thus, ComQum employs a subtractive scheme with electrostatic embedding and van der Waals link-atom corrections.71 No cutoff is used for any of the interactions in the three energy terms in eqn (1).
is the classical energy of all atoms (in both the QM and MM regions) with CL atoms and with the charges of the QM region set to zero (to avoid double-counting of the electrostatic interactions). Thus, ComQum employs a subtractive scheme with electrostatic embedding and van der Waals link-atom corrections.71 No cutoff is used for any of the interactions in the three energy terms in eqn (1).
The geometry optimisations were continued until the energy change between two iterations was less than 10−6 a.u. (2.6 J mol−1) and the maximum norm of the Cartesian gradients was below 10−3 a.u.
QM calculations
The QM system consisted of the Cu ion,19 two methyl-imidazole groups modelling the sidechains of His-137A and 139A, H2N–CH(CH2-imidazole)–CONH–CH2–CONH–(CH2)3–COO−, as a model of the amino-terminal residue His-33A (sidechain and backbone) and the connecting Gly-34A and Glu-35A residues, as well as four water molecules. The glutamate sidechain was included because it is the only residue that may provide protons needed for the reaction and the water molecules because they may facilitate the transfer of the protons. The QM system is shown in Fig. 2. | ||
| Fig. 2 The quantum systems used for the CuB site with the coordinating and surrounding residues marked. | ||
All QM calculations were performed with the Turbomole software (versions 7.1, 7.2, 7.6.1 and 7.8).72 We employed two DFT methods, TPSS62 and B3LYP,73–75 and two basis sets of increasing size, def2-SV(P)63 and def2-TZVPD.76 The calculations were sped up by expanding the Coulomb interactions in an auxiliary basis set, the resolution-of-identity (RI) approximation.77,78 Empirical dispersion corrections were included with the DFT-D3 approach79 and Becke–Johnson damping,80 as implemented in Turbomole.
Geometries were optimised by TPSS-D3/def2-SV(P) calculations (used in our previous studies of pMMO19 and LPMO49,52,55) and then more accurate energies were obtained with B3LYP-D3/def2-TZVPD calculations. It is common practice to use a cheaper method to obtain geometries and a more expensive and accurate method for energies,81 and it is important to use both a pure and a hybrid DFT functional to see whether the energies are stable. Reported energies and spin populations (obtained by a Mulliken population analysis) are from the B3LYP/def2-TZVPD calculations.
Results and discussion
We have studied the formation of a reactive [CuO]+ state, the oxidation of CH4 and putative side reactions for the CuB sites in pMMO. An overview over all studied reactions is shown in Fig. 3. As is evident from this figure, the net reaction involves addition of two electrons and two protons (after a priming reduction of the CuII resting state), i.e. CuI + O2 + 2e− + 2H+ → [CuO]+ + H2O. The [CuO]+ state may then hydroxylate the substrate: [CuO]+ + CH4 → [CuOH·CH3]+ → CuI + CH3OH. In cases where the order of the reduction and protonation is unclear (or could occur in a concerted manner), we consider the two processes separately. The details of the O2 activation and methane hydroxylation are described below in separate sections. | ||
| Fig. 3 Reactions in the activation of O2 hydroxylation of methane by the Cu sites in pMMO. | ||
The CuB site is shown in Fig. 2. The Cu ion is bound to three His residues, one of which binds with a His brace consisting of both the imidazole sidechain and the amino-terminal backbone –NH2 group. This site shows a conspicuous similarity to the active Cu site in the LPMOs and we therefore also perform a detailed study of the proton-transfer reactions to compare with similar calculations for LPMO.52
The resting state and the initial reduction
First, we discuss the structural changes accompanying the first reduction of the resting Cu2+ state (reaction 1 → 2 in Fig. 3). In the resting (oxidised) state 1 (shown in Fig. 4), the Cu ion binds to four N atoms, viz. the ND1 atoms of His-33A and His-137A (called NA and NB in the following), the NE2 atom of His-139A (called NC) atoms and the His-33A backbone (amino-terminal) –NH2 group (called NN). The four Cu–N distances show a smaller variation (1.94–2.08 Å) than in the quantum-refined structure of the same enzyme (1.85–2.22 Å).19 The four N atoms form an approximate square plane, as is expected for CuII. The Mulliken spin population on the Cu ion is 0.57e. A water molecule (Wat1) also coordinates to Cu at a distance of 2.30 Å. It also forms a hydrogen bond to Glu-35A with a H–O distance of 1.66 Å and to another water molecule (Wat2) with a H–O distance of 1.89 Å. On the opposite side of Cu, there is another water molecule (Wat4), but at a non-coordinating distance of 3.13 Å. This distance is longer than in a recent molecular dynamics and QM/MM study (2.32–2.39 Å)82 but in reasonable agreement with the recent cryo-EM structures (3.4–3.6 Å).34 | ||
| Fig. 4 Oxidised resting state 1 (left) and reduced state 2 (right). Distances are in Å. | ||
In the reduced state 2, the Cu–NA bond is broken, 2.94 Å, and NA instead receives a hydrogen bond from Wat4 (cf.Fig. 4). Likewise, Wat1 has dissociated from Cu (3.21 Å), but it retains its hydrogen bonds to Glu-35A and Wat2. This is expected because the d10 CuI ion prefers a lower coordination number than the d9 CuII ion. In both structures, a fourth water molecule (Wat3) forms a hydrogen bond to the carboxyl group of Glu-35A. This shows that once the CuB site is reduced, the water molecule automatically dissociates, opening for the binding of O2, contrary to the suggestion that the high coordination number would inhibit O2 binding.82 This has been taken as an advantage of the CuC or CuD sites,9,23,82 but most likely only reflect the oxidation states of the Cu ion in the available crystal and cryo-EM structures. Interestingly, in the reduced state of the LPMOs, there is a vacant coordination site trans to the NN ligand,83–85 but in the pMMOs this site is occupied by NB. The site that is opened by dissociation of NA from Cu in the reduced state is too small to house O2 and the same applies to the site opened by the dissociation of Wat1. Instead, the most natural binding site is trans to the former binding site of Wat1 (cf.Fig. 3).
Formation of the O2-bound state
We next consider the [CuO2]+ state (3 in Fig. 3), i.e. the state formed by binding of O2 to the reduced active site. We studied it in the triplet, open-shell and closed-shell singlet states. The triplet state is 17 and 99 kJ mol−1 more stable than the open- and closed-shell singlet states, respectively. The spin populations on Cu and O2 in the triplet state are 0.37 and 1.46e, indicating that it is mainly composed of CuI and triplet 3O2. The optimised structure is shown in Fig. 5. O2 binds end-on to Cu with a Cu–O–O angle of 121°. The triplet and open-shell spin states have nearly identical structures besides that 33 has a somewhat longer Cu–O bond (2.23 Å vs. 2.18 Å). | ||
| Fig. 5 QM/MM optimised [CuO2]+ (3) in triplet state (left) and [CuO2]0 (4) in the doublet state (right). Distances are in Å. | ||
It is notable that in the CuB site of pMMO, O2 binds perpendicular to the histidine-brace plane, in contrast to the LPMOs, where O2 and H2O2 bind within the histidine-brace plane.52,86 The reason is that the fourth coordination position in the histidine-brace plane is occupied by the third His ligand in pMMO. For LPMOs, it has been argued that the reactivity with H2O2 is caused by in-plane binding of H2O2, allowing an interaction between Cu 3dx2−y2 and the empty H2O2 σ* orbitals; the histidine brace was argued to increase the ligand-field splitting of the Cu 3d orbitals, lowering the gap between the Cu 3dx2−y2 and H2O2 σ* orbitals, which in turn lowers the barrier for the homolytic O–O cleavage.87 However, a later paper showed that the presence of a substrate (not considered in ref. 87) also lowers the H2O2 σ* orbital.88 Thus, arguments based on the ligand-field imposed by the histidine brace in the LPMOs cannot simply be transferred to the pMMO CuB reactivity. Moreover, as can be seen in Fig. 5, the structures of 3 is quite distorted and the O–Cu–NB angle (145°) is actually larger than the NN–Cu–NB angle (133°; making it unclear which atoms are coplanar). NA accepts a hydrogen bond from Wat4, which brings the Cu ion out of the imidazole ring plane of His-33A.
Formation of the [CuOOH]+ state
Initial attempts to optimise an intermediate with a protonated O2 unit (5 in Fig. 3) led to release of the O2H group from the metal site. We note that for the LPMOs, such a release of O2H was not seen and this protonation could occur before reduction, yielding a [CuOOH]2+ moiety (5), but the associated energies were quite uphill.89 Consequently, we instead focus on the pathway in which 3 ([CuO2]+) is first reduced to 4. This reduction leads to a doublet [CuO2] structure, also shown in Fig. 5. The spin populations on Cu (0.00e) and O2 (0.97e) indicate that it consists of CuI and 2O2˙−. Compared to the geometry of 3, the sidechain of His-33A (NA) has dissociated from the copper ion, consistent with the CuI oxidation state.The next step is the protonation of 4 to a [CuOOH]+ species, 6. While the related LPMO enzymes have amino-acid residues optimally located for facile proton transfer to the [CuO2]+ unit,55,89 no such residues exist in pMMO. With inspiration from an ongoing debate regarding involvement of the terminal NH2 group in LPMOs,11,90 we decided to investigate if the amino-terminal –NH2 group could be involved in protonation of the superoxide. We investigated a reaction chain where a proton is added to O2 from the nearby Glu-35A residue via the amino-terminal –NH2 group. Therefore, a proton was added to the OE2 atom of Glu-35A (denoted 6a, cf.Fig. 6). NA is still dissociated from Cu and the spin densities on Cu and O2 are still 0.00 and 0.97e.
 | ||
| Fig. 6 Reactant 6a, first transition state (TS6a/b) and intermediate 6b, as well as the second transition state (TS6b/c) and product 6c for the protonation of [CuO2]. Distances are in Å. | ||
From this structure, the proton on Glu-35A can be transferred to NN with a barrier of 72 kJ mol−1. In fact, the proton transfers via a Wat1 molecule and the transition state essentially involves a H3O+ ion (TS6a/b in Fig. 6). The product (6b in Fig. 6) has a triply protonated NN atom, which has dissociated from Cu. The structure is 92 kJ mol−1 more stable than the 6a structure. The spin populations on Cu and O2 are 0.02 and 0.99e.
One of the protons on NN forms a strong hydrogen bond to the non-coordinating O atom of the [CuO2]0 moiety with a H–O distance of 1.52 Å. Therefore, it is not surprising that this proton can easily be transferred to [CuO2]0 with a barrier of only 4 kJ mol−1, obtained at a H–O distance of 1.35 Å. The product (6c) contains a [CuOOH]+ unit. It is 105 kJ mol−1 more stable than the 6b structure and 197 kJ mol−1 stable than the starting 6a structure. The optimised structures for the transition state and the product 6c are shown in Fig. 6. The spin populations of 6c are 0.50e on Cu and 0.30e on the O2H group, indicating something in between the CuI–HO2˙ and CuII–HO2− states, with a dominance of the latter state, because the spin population of Cu is only slightly less than for the resting oxidised state 1 (0.57e). Consequently, all four N atoms coordinate to the Cu ion again with distances similar to those of 6a. However, the Cu–O bond length has decreased to 2.06 Å and the O–O bond length has increased to 1.44 Å.
Thus, we can conclude that the formation of 6c is possible with a net activation energy of 72 kJ mol−1 and a strongly exothermic reaction energy. Thus, 6a can be converted to 6c, provided that Glu-35A can be protonated. The energy profile is shown in Fig. 7.
 | ||
| Fig. 7 Energy profile (kJ mol−1) for the reactions involving 6a, 6b and 6c. | ||
Formation of the Cu-oxyl species
For the generation of the oxyl intermediate, [CuO]+ (9c), we considered two putative reaction paths from 6c, viz. by first adding a proton and then an electron (6 → 8 → 9) or vice versa (6 → 7 → 9; cf.Fig. 3). We again involved Glu-35A as proton donor, so that the proton was first added to OE2.If we first protonate 6c without any reduction, giving 8, the proton directly transfers from OE2 to the terminal amino group without any barrier (8b; the reaction is down-hill by ∼51 kJ mol−1). Further transfer of the proton from the amino group to the inner (Cu-coordinating) O1 atom of HO2 does not lead to any cleavage of the O–O bond or any product (the energy increases monotonously and the proton goes spontaneously back if the restraint is released). However, if it is instead transferred to the outer O2 atom, HOOH is formed, which dissociates from the CuII ion (8d). It has no spin and the spin population on Cu is 0.57e. This reaction has a barrier of only 36 kJ mol−1 and it is exothermic by 125 kJ mol−1. Both structures are in the doublet spin state and they are shown in Fig. 8. The energy profile is shown in Fig. 9.
 | ||
| Fig. 8 Intermediates 8b and 8d. Distances in Å. | ||
 | ||
| Fig. 9 Energy profile (kJ mol−1) for the reactions involving 8a, 8b and 8d. | ||
Reducing 6c before protonation leads to 7c, shown in Fig. 10. It is most stable in the closed-shell singlet state (188 kJ mol−1 below the triplet), indicating a CuI–HO2− state. As expected, NA dissociates from Cu. The Cu–O distance is 2.05 Å and the O–O distance is 1.52 Å. Interestingly, H2O2 can easily dissociate from this state to the second coordination sphere, abstracting a proton from Wat1, which then coordinates to Cu as OH− (7d in Fig. 10). The reaction has a barrier of only 10 kJ mol−1 and is downhill by 45 kJ mol−1. The energy profile is shown in Fig. 11. A structure with instead HO2− dissociated has the same energy within 1 kJ mol−1.
 | ||
| Fig. 10 Intermediates 7c and 7d. Distances in Å. | ||
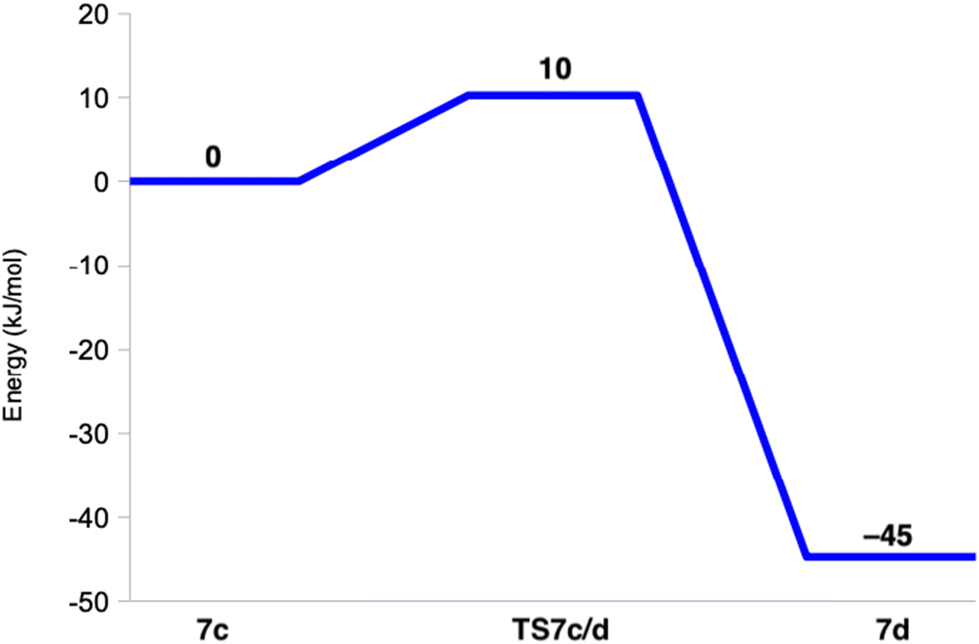 | ||
| Fig. 11 Energy profile (kJ mol−1) for the reaction from 7c to 7d. | ||
Next, we added a proton to OE2 of Glu-35A (9a), but it is automatically transferred to the amino-terminal NH2 group (9b in Fig. 12), a down-hill reaction by ∼99 kJ mol−1, without any barrier. Both states are most stable in the closed-shell singlet states, with the triplet state ∼35 kJ mol−1 higher in energy (no open-shell singlet could be found). Interestingly, for the triplet state, 9a is stable, but not 9b.
 | ||
| Fig. 12 Intermediates 9b, 9c and 9d (distances in Å). | ||
From 9b, a proton can be transferred from the amino terminal to the non-coordinated O2 atom of the substrate, leading to a cleavage of the O–O bond. The product is 9c, the [CuO]+ + H2O reactive intermediate, shown in Fig. 12. The new water molecule forms a strong hydrogen bond to the oxo group, as does Wat4, with H–O distances of 1.75–1.77 Å. The Cu ion is coordinated to all four N atoms, but the distance to NN is quite long, 2.35 Å. The Cu–O distance is 1.93 Å. The product is most stable in the triplet state, with the open-shell singlet 10 kJ mol−1 higher in energy. The spin populations on Cu and O are 0.57 and 1.14e, indicating that it consists of CuII and a O˙− ion (−0.57 and 0.86e on the open-shell singlet). Since the reactant is a closed-shell singlet and the product a triplet, the transition state is found at the crossing of these two spin surfaces, at a H–O distance of 1.37 Å and with an activation energy of only 13 kJ mol−1. The 9c product is 165 kJ mol−1 more stable than the 9b state. The energy profile is shown in Fig. 13.
 | ||
| Fig. 13 Energy profile (kJ mol−1) for the reaction from 9a and 9b to 9c or 9d. | ||
An alternative reaction transfers the proton to the Cu-coordinated O1 atom, forming instead HOOH (9d in Fig. 12). This reaction proceeds in the closed-shell singlet state (i.e. with Cu remaining in the +I state and HO2− transforming to HOOH). The proton on the amino terminal transfers to the O1 atom with a barrier of only 13 kJ mol−1. The formed HOOH dissociates from Cu and forms hydrogen bonds to Wat4 (1.66 Å) and the backbone CO group of His-33A. The sidechain of the latter has also dissociated from Cu. The 9d product is 199 kJ mol−1 more stable than the 9b structure with the proton on the amino terminal and it is 34 kJ mol−1 more stable than 9c.
CH4 hydroxylation
Finally, we investigated the abstraction of a hydrogen atom from the methane substrate. The calculations were started from the 10 structure, in which the newly formed water molecule in 9c was replaced by a methane molecule. We investigate both triplet and open-shell singlet spin states (the closed-shell singlet state is 58 kJ mol−1 higher in energy for 10 and ∼145 kJ mol−1 higher for 11; the latter could only be optimised with restraints). Optimised structures for this reaction are depicted in Fig. 14. The reactant is most stable in the triplet state, with the open-shell singlet 5 kJ mol−1 higher in energy. In 310, CH4 forms a weak hydrogen bond to [CuO]1+ (2.58 Å CH–O distance). The Cu ion coordinates to all four N atoms, but the Cu–NN distance is rather long (2.34 Å). The Cu–O distance is also quite long (1.93 Å) and the O atom bears a high spin density (1.16e; 0.56e on Cu), still suggesting a CuII–O˙ state.51 | ||
| Fig. 14 Optimised geometries of 310, 3TS10/11, 311 and product 12 for the methane hydroxylation reaction. Distances are in Å. | ||
The intermediate formed after the reaction (11) is also most stable in the triplet spin state, with the open-shell singlet 5 kJ mol−1 higher in energy. In this structure, a hydrogen atom is extracted from methane to form [CuOH]+ (with an O–H bond length of 0.98 Å) and a CH3 radical. Thereby, the Cu–O distance is elongated to 2.00 Å. The spin populations on Cu, OH and CH3 are 0.61, 0.14 and 0.98e, indicating mainly a CuII–OH−–CH3˙ state. The intermediate is 11 kJ mol−1 more stable than the reactant state.
The transition state (TS10/11) on the triplet surface is found at a H–O distance of 1.20 Å (the C–H distance is 1.32 Å). It has spin populations of 0.59, 0.72 and 0.48 and −0.06e on the Cu, O, C and the transferred H atoms. The activation energy is 59 kJ mol−1. This is similar to what we found with a simple QM-cluster model.19 The transition state on the open-shell surface has an activation energy of 70 kJ mol−1. The energy profile is shown in Fig. 15.
 | ||
| Fig. 15 Energy profile (kJ mol−1) for the reaction from 10 to 12. | ||
Next, we continued the reaction by an attack of the Cu-bound OH group on the methyl radical of structure 11. The product (12 in Fig. 14) contains CuI and CH3OH and is therefore most stable in the closed-shell singlet state. The reaction is strongly downhill and the product is 222 kJ mol−1 more stable than the reactant 10 and 210 kJ mol−1 more stable than the intermediate 11. Both methanol and His-33A have dissociated from the CuI ion. Thus, the reaction involves a transition from the triplet spin state of 11 to a closed-shell singlet state of the product (12), so the transition state is the triplet–singlet crossing point, which is found at a C–O distance of 2.13 Å. The activation energy is 52 kJ mol−1.
The observation that 10, TS10/11, and 11 all are most stable in the triplet state and the product is a singlet is in agreement with previous small-model studies.19,36 Overall, the QM/MM calculations predict a barrier of 59 kJ mol−1 for the reaction, with the C–H bond activation being rate-determining.
Conclusions
We have studied the reactivity of the His-brace CuB site in pMMO. We considered several possible reaction mechanisms summarised in Fig. 3. The resting oxidised CuII state (1) is first reduced to CuI (2), which binds O2 (3) in a slightly bent (121° Cu–O–O angle) end-on fashion, forming a complex that is mainly CuI–3O2. This complex seems to be first reduced to CuI–2O2˙− (4), before it can be protonated to a [CuOOH]+ complex (6), which is a mixture of the CuI–2HO2˙ and the CuII–HO2− states. Interestingly, we have recently seen the same for the LPMO active site, where the intermediate corresponding to 3 also was significantly more favourable to protonate after it was reduced.89 The proton can be transferred from the nearby Glu-35A residue to O2via the amino-terminal –NH2 group with a barrier of 72 kJ mol−1. This is significantly higher than 6–10 kJ mol−1 obtained from LPMO,89 which may be ascribed to both differences within the first coordination sphere (the LPMOs are only coordinated by two histidine residues) and the second-coordination sphere where amino-acid residues can facilitate facile proton transfer. A second reduction to the CuI–HO2− state (7) seems to be needed before it can bind a second proton. Again the proton can be transferred from Glu-35A to HO2 with a barrier of only 13 kJ mol−1. Transferring the proton to the non-coordinating O atom leads to cleavage of the O–O bond to form [CuO]+ + H2O (9c). The [CuO]+ structure is most stable in the triplet state and is best described as CuII–O˙−. The latter structure can abstract a proton from CH4 with a barrier of 59 kJ mol−1. The transition state is late, with a H–O distance of 1.2 Å. The hydrogen abstraction gives a CuII–OH−–CH3˙ intermediate. Finally, the OH− group can recombine with the methyl radical, forming a methanol product. This reaction involves a change from a triplet to a closed-shell singlet state. The barrier is slightly lower than for the hydrogen abstraction, 52 kJ mol−1.However, in many steps, there are alternative reactions leading towards formation of peroxide. 6c can be protonated with a low barrier (36 kJ mol−1), forming a dissociated H2O2 product in a strongly exothermic reaction (125 kJ mol−1). Likewise, dissociation of H2O2 or HO2− from 7 is favourable by 45 kJ mol−1 and proceeds with a minimal barrier of 10 kJ mol−1. Finally, protonation of the coordinating O1 atom in 9b is a competing reaction to the protonation of the non-coordinating O2 atom and it proceeds with the same barrier (13 kJ mol−1) and leads to a product with H2O2 dissociated from the CuI ion, that is 34 kJ mol−1 more stable than the reactive 9c structure. These reactions will be further favoured by the gain in entropy by the dissociation of H2O2 or HO2−. Therefore, it seems to be an inevitable conclusion that the CuB site is poised for the production of H2O2 rather than for the oxidation of methane. Such a conclusion agrees with the recent suggestions that CuD, rather than CuB, is the actual catalytic site of pMMO.9,34,42 The results may suggest that H2O2 is produced in the CuB for use in the catalytic cycle in the CuD site, as has also been suggested by Peng et al.,40 although they suggested that the peroxide is formed directly at the CuD site. H2O2 production has also been experimentally observed for a truncated construct of the PmoB subunit containing the CuB site.24
We have also observed that the CuB site has a labile ligand that can be used to satisfy the differing coordination preferences of CuI (a low coordination number) and CuII (square planar/pyramidal or octahedral). It is mostly NA of His-33A, which may dissociate and form a hydrogen bond to a second-sphere water molecule for CuI.
It should be noted that there are some further families of proteins that also contain a Cu ion bound to a histidine-brace motif, but do not perform oxidative reactions e.g. the Cu sites in CopC, X325 and Bim1. CopC is a Cu chaperon that is involved in bacterial homeostasis.91 It binds Cu(II) with a bidentate amino-terminal His ligand, another His residue, an Asp ligand and sometimes a water molecule.92 The two His sidechains are at cis positions. A conserved second-sphere Glu residue is believed to bring stability to the active site and enhance the Cu binding by forming a hydrogen bond to the amino terminal.93 H2O2 production has been experimentally observed for some mutants of this Glu group of CopC.92 The active sites of X325 and Bim1 have the same set of ligands, but the two His sidechains bind at trans positions.93,94 Further QM studies are needed to understand the role of the Asp ligand and why such sites cannot perform oxidative reactions. Studies of Cu complexes of simple peptides have shown that the reactivity is tuned by small details in the coordination sphere.95,96
This study nicely illustrates how computational studies can be used to describe and predict reactivity of biological sites. However, more studies are still needed to understand the reactivity of the pMMOs. In particular, accurate estimates of the reduction potential and proton affinity of the various intermediates would be crucial to judge what states can really be reached by addition of electrons from biochemical donors and protons from the solvent.
Author contributions
K. J. M. L.: investigation, writing – review & editing, visualization; L. C.: investigation, data curation, writing – original draft, writing – review & editing, visualization; M. T.: investigation; E. D. H.: conceptualization, writing – review & editing, supervision; U. R.: conceptualization, methodology, software, formal analysis, investigation, resources, data curation, writing – review & editing, visualization, supervision, project administration, funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This investigation has been supported by grants from the Swedish Research Council (projects 2018-05003 and 2022-04978), from COST through Action CM1305 (ECOSTBio), from China Scholarship Council, from eSSENCE: the e-science collaboration and from the Royal Physiographic Society in Lund. EDH thanks The Villum Foundation, Young Investigator Program (grant no. 29412), the Swedish Research Council (grants no. 2019-04205 and 2022-04978), and Independent Research Fund Denmark (grant no. 0252-00002B and 2064-00002B) for support. The computations were enabled by resources provided by LUNARC, the Centre for Scientific and Technical Computing at Lund University, and by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) at HPC2N at Umeå University, NSC at Linköping University and PDC centre for high performance computing at KTH Royal Institute of Technology, partially funded by the Swedish Research Council through grant agreements no. 2022-06725 and 2018-05973.References
- M. A. Culpepper and A. C. Rosenzweig, Crit. Rev. Biochem. Mol. Biol., 2012, 47, 483–492 CrossRef CAS PubMed.
- M. Merkx, D. A. Kopp, M. H. Sazinsky, J. L. Blazyk, J. Müller and S. J. Lippard, Angew. Chem., Int. Ed., 2001, 40, 2782–2807 CrossRef CAS PubMed.
- A. Messerschmidt, Handbook Metalloprot, Wiley, 40 edn, 2001 Search PubMed.
- N. Elango, R. Radhakrishnan, W. A. Froland, B. J. Wallar, C. A. Earhart, J. D. Lipscomb and D. H. Ohlendorf, Protein Sci., 1997, 6, 556–568 CrossRef CAS PubMed.
- A. C. Rosenzweig, C. A. Frederick, S. J. Lippard and P. Nordlund, Nature, 1993, 366, 537–543 CrossRef CAS PubMed.
- R. Banerjee, J. C. Jones and J. D. Lipscomb, Annu. Rev. Biochem., 2019, 88, 409–431 CrossRef CAS PubMed.
- V. C. C. Wang, S. Maji, P. P. Y. Chen, H. K. Lee, S. S. F. Yu and S. I. Chan, Chem. Rev., 2017, 117, 8574–8621 CrossRef CAS PubMed.
- M. O. Ross and A. C. Rosenzweig, J. Biol. Inorg. Chem., 2017, 22, 307–319 CrossRef CAS PubMed.
- F. J. Tucci and A. C. Rosenzweig, Chem. Rev., 2024, 124, 1288–1320 CrossRef CAS PubMed.
- S. I. Chan, V. C. C. Wang, P. P. Y. Chen and S. S. F. Yu, J. Chin. Chem. Soc., 2022, 69, 1147–1158 CrossRef CAS.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- R. L. Lieberman and A. C. Rosenzweig, Nature, 2005, 434, 177–182 CrossRef CAS PubMed.
- A. S. Hakemian, K. C. Kondapalli, J. Telser, B. M. Hoffman, T. L. Stemmler and A. C. Rosenzweig, Biochemistry, 2008, 47, 6793–6801 CrossRef CAS PubMed.
- S. M. Smith, S. Rawat, J. Telser, B. M. Hoffman, T. L. Stemmler and A. C. Rosenzweig, Biochemistry, 2011, 269, 26344–26348 Search PubMed.
- S. Sirajuddin, D. Barupala, S. Helling, K. Marcus, T. L. Stemmler and A. C. Rosenzweig, J. Biol. Chem., 2014, 289, 21782–21794 CrossRef PubMed.
- R. L. Lieberman, D. B. Shrestha, P. E. Doan, B. M. Hoffman, T. L. Stemmler and A. C. Rosenzweig, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3820–3825 CrossRef CAS PubMed.
- A. C. Rosenzweig, Biochem. Soc. Trans., 2008, 36, 1134–1137 CrossRef CAS PubMed.
- R. L. Lieberman, K. C. Kondapalli, D. B. Shrestha, A. S. Hakemian, S. M. Smith, J. Telser, J. Kuzelka, R. Gupta, A. S. Borovik, S. J. Lippard, B. M. Hoffman, A. C. Rosenzweig and T. L. Stemmler, Inorg. Chem., 2006, 45, 8372–8381 CrossRef CAS PubMed.
- L. Cao, O. Caldararu, A. C. Rosenzweig and U. Ryde, Angew. Chem., Int. Ed., 2018, 57, 162–166 CrossRef CAS PubMed.
- S. Y. Ro, M. O. Ross, Y. W. Deng, S. Batelu, T. J. Lawton, J. D. Hurley, T. L. Stemmler, B. M. Hoffman and A. C. Rosenzweig, J. Biol. Chem., 2018, 293, 10457–10465 CrossRef PubMed.
- M. O. Ross, F. MacMillan, J. Wang, A. Nisthal, T. J. Lawton, B. D. Olafson, S. L. Mayo, A. C. Rosenzweig and B. M. Hoffman, Science, 2019, 364, 566–570 CrossRef CAS PubMed.
- S. Y. Ro, L. F. Schachner, C. W. Koo, R. Purohit, J. P. Remis, G. E. Kenney, B. W. Liauw, P. M. Thomas, S. M. Patrie, N. L. Kelleher and A. C. Rosenzweig, Nat. Commun., 2019, 10, 2675 CrossRef PubMed.
- R. J. Jodts, M. O. Ross, C. W. Koo, P. E. Doan, A. C. Rosenzweig and B. M. Hoffman, J. Am. Chem. Soc., 2021, 143, 15358–15368 CrossRef CAS PubMed.
- Y. J. Lu, M. C. Hung, B. T. Chang, T. L. Lee, Z. H. Lin, I. K. Tsai, Y. S. Chen, C. S. Chang, Y. F. Tsai, K. H. Chen, S. I. Chan and S. S. Yu, J. Inorg. Biochem., 2019, 196, 110691 CrossRef CAS PubMed.
- G. E. Cutsail 3rd, M. O. Ross, A. C. Rosenzweig and S. DeBeer, Chem. Sci., 2021, 12, 6194–6209 RSC.
- M. Martinho, D. W. Choi, A. A. DiSpirito, W. E. Antholine, J. D. Semrau and E. Münck, J. Am. Chem. Soc., 2007, 129, 15783–15785 CrossRef CAS PubMed.
- R. Balasubramanian, S. M. Smith, S. Rawat, L. A. Yatsunyk, T. L. Stemmler and A. C. Rosenzweig, Nature, 2010, 465, 115–119 CrossRef CAS PubMed.
- M. A. Culpepper, G. E. Cutsail, 3rd, W. A. Gunderson, B. M. Hoffman and A. C. Rosenzweig, J. Am. Chem. Soc., 2014, 136, 11767–11775 CrossRef CAS PubMed.
- M. A. Culpepper, G. E. Cutsail 3rd, B. M. Hoffman and A. C. Rosenzweig, J. Am. Chem. Soc., 2012, 134, 7640–7643 Search PubMed.
- W. H. Chang, H. H. Lin, I. K. Tsai, S. H. Huang, S. C. Chung, I. P. Tu, S. S. F. Yu and S. I. Chan, J. Am. Chem. Soc., 2021, 143, 9922–9932 Search PubMed.
- S. I. Chan, V. C. Wang, J. C. Lai, S. S. Yu, P. P. Chen, K. H. Chen, C. L. Chen and M. K. Chan, Angew. Chem., Int. Ed., 2007, 46, 1992–1994 CrossRef CAS PubMed.
- S. I. Chan, W.-H. Chang, S.-H. Huang, H.-H. Lin and S. S. F. Yu, J. Inorg. Biochem., 2021, 225, 111602 CrossRef CAS PubMed.
- S. I. Chan, Y. J. Lu, P. Nagababu, S. Maji, M. C. Hung, M. M. Lee, I. J. Hsu, P. D. Minh, J. C. Lai, K. Y. Ng, S. Ramalingam, S. S. Yu and M. K. Chan, Angew. Chem., Int. Ed., 2013, 52, 3731–3735 CrossRef CAS PubMed.
- C. W. Koo, F. J. Tucci, Y. He and A. C. Rosenzweig, Science, 2022, 375, 1287–1291 CrossRef CAS PubMed.
- S. S. Yu, C. Z. Ji, Y. P. Wu, T. L. Lee, C. H. Lai, S. C. Lin, Z. L. Yang, V. C. Wang, K. H. Chen and S. I. Chan, Biochemistry, 2007, 46, 13762–13774 Search PubMed.
- K. Yoshizawa and Y. Shiota, J. Am. Chem. Soc., 2006, 128, 9873–9881 CrossRef CAS PubMed.
- Y. Shiota and K. Yoshizawa, Inorg. Chem., 2009, 48, 838–845 CrossRef CAS PubMed.
- S. Itoyama, K. Doitomi, T. Kamachi, Y. Shiota and K. Yoshizawa, Inorg. Chem., 2016, 55, 2771–2775 CrossRef CAS PubMed.
- S. Arora, P. Rawal and P. Gupta, Chem. Commun., 2024, 30, e202303722 CAS.
- W. Peng, X. Qu, S. Shaik and B. Wang, Nat. Catal., 2021, 4, 266–273 CrossRef CAS.
- W. Peng, Z. Wang, Q. Zhang, S. Yan and B. Wang, J. Am. Chem. Soc., 2023, 145, 25304–25317 CrossRef CAS PubMed.
- F. J. Tucci, R. J. Jodts, B. M. Hoffman and A. C. Rosenzweig, Nat. Catal., 2023, 6, 1194–1204 CrossRef CAS PubMed.
- P. E. M. Siegbahn, J. Phys. Chem. B, 2024, 128, 5840–5845 CrossRef CAS PubMed.
- P. V. Harris, D. Welner, K. C. McFarland, E. Re, J.-C. Navarro Poulsen, K. Brown, R. Salbo, H. Ding, E. Vlasenko, S. Merino, F. Xu, J. Cherry, S. Larsen and L. Lo Leggio, Biochemistry, 2010, 49, 3305–3316 CrossRef CAS PubMed.
- R. J. Quinlan, M. D. Sweeney, L. Lo Leggio, H. Otten, J.-C. N. Poulsen, K. S. Johansen, K. B. R. M. Krogh, C. I. Jørgensen, M. Tovborg, A. Anthonsen, T. Tryfona, C. P. Walter, P. Dupree, F. Xu, G. J. Davies and P. H. Walton, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15079–15084 CrossRef CAS PubMed.
- G. Vaaje-Kolstad, B. Westereng, S. J. Horn, Z. Liu, H. Zhai, M. Sørlie and V. G. H. Eijsink, Science, 2010, 330, 219–222 CrossRef CAS PubMed.
- P. H. Walton and G. J. Davies, Curr. Opin. Chem. Biol., 2016, 31, 195–207 CrossRef CAS PubMed.
- S. Kim, J. Ståhlberg, M. Sandgren, R. S. Paton and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 149–154 CrossRef CAS PubMed.
- E. D. Hedegård and U. Ryde, J. Biol. Inorg. Chem., 2017, 22, 1029–1037 CrossRef PubMed.
- L. Bertini, R. Breglia, M. Lambrughi, P. Fantucci, L. De Gioia, M. Borsari, M. Sola, C. A. Bortolotti and M. Bruschi, Inorg. Chem., 2018, 57, 86–97 CrossRef CAS PubMed.
- B. Wang, E. M. Johnston, P. Li, S. Shaik, G. J. Davies, P. H. Walton and C. Rovira, ACS Catal., 2018, 8, 1346–1351 CrossRef CAS.
- E. D. Hedegård and U. Ryde, Chem. Sci., 2018, 9, 3866–3880 RSC.
- B. Bissaro, B. Streit, I. Isaksen, V. G. H. Eijsink, G. T. Beckham, J. L. DuBois and Å. K. Røhr, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1504–1513 CrossRef CAS PubMed.
- R. Kittl, D. Kracher, D. Burgstaller, D. Haltrich and R. Ludwig, Biotechnol. Biofuels, 2012, 5, 79 CrossRef CAS PubMed.
- O. Caldararu, E. Oksanen, U. Ryde and E. D. Hedegård, Chem. Sci., 2019, 10, 576–586 RSC.
- M. H. M. Olsson, C. R. Søndergaard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- L. L. C. Schrödinger, Maestro; Schrödinger release 2019-2, 2019 Search PubMed.
- D. A. Case, J. T. Berryman, R. M. Betz, D. S. Cerutti, T. E. Cheatham, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, T. Luchko, R. Luo, B. Madej, K. M. Merz, G. Monard, P. Needham, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. E. Roitberg, R. Salomon-Ferrer, C. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, D. M. York and P. A. Kollman, AMBER 14, University of California, San Francisco, 2014 Search PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401–146401 CrossRef PubMed.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- E. Sigfridsson and U. Ryde, J. Comput. Chem., 1998, 19, 377–395 CrossRef CAS.
- B. H. Besler, K. M. Merz and P. A. Kollman, J. Comput. Chem., 1990, 11, 431–439 CrossRef CAS.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- U. Ryde, J. Comput.-Aided Mol. Des., 1996, 10, 153–164 CrossRef CAS PubMed.
- U. Ryde and M. H. M. Olsson, Int. J. Quantum Chem., 2001, 81, 335–347 CrossRef CAS.
- N. Reuter, A. Dejaegere, B. Maigret and M. Karplus, J. Phys. Chem. A, 2000, 104, 1720–1735 CrossRef CAS.
- L. Hu, P. Söderhjelm and U. Ryde, J. Chem. Theory Comput., 2011, 7, 761–777 CrossRef CAS PubMed.
- L. Cao and U. Ryde, Front. Chem., 2018, 6, 89–89 CrossRef PubMed.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2014, 4, 91–100 CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1372 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–289 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154119 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- M. R. A. Blomberg, T. Borowski, F. Himo, R.-Z. Liao and P. E. M. Siegbahn, Chem. Rev., 2014, 114, 3601–3658 CrossRef CAS PubMed.
- W. D. B. Da Silva, R. P. Dias and J. C. S. Da Silva, Phys. Chem. Chem. Phys., 2022, 24, 16611–16621 RSC.
- K. E. Frandsen, T. J. Simmons, P. Dupree, J. C. Poulsen, G. R. Hemsworth, L. Ciano, E. M. Johnston, M. Tovborg, K. S. Johansen, P. von Freiesleben, L. Marmuse, S. Fort, S. Cottaz, H. Driguez, B. Henrissat, N. Lenfant, F. Tuna, A. Baldansuren, G. J. Davies, L. Lo Leggio and P. H. Walton, Nat. Chem. Biol., 2016, 12, 298–303 CrossRef CAS PubMed.
- T. Tandrup, S. J. Muderspach, S. Banerjee, G. Santoni, J. O. Ipsen, C. Hernandez-Rollan, M. H. H. Norholm, K. S. Johansen, F. Meilleur and L. Lo Leggio, IUCrJ, 2022, 9, 666–681 CrossRef CAS PubMed.
- W. B. O'Dell, P. K. Agarwal and F. Meilleur, Angew. Chem., Int. Ed., 2017, 56, 767–770 CrossRef PubMed.
- L. Ciano, G. J. Davies, W. B. Tolman and P. H. Walton, Nat. Catal., 2018, 1, 571–577 CrossRef CAS.
- H. Lim, M. T. Brueggemeyer, W. J. Transue, K. K. Meier, S. M. Jones, T. Kroll, D. Sokaras, B. Kelemen, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2023, 145, 16015–16025 CrossRef CAS PubMed.
- E. K. Wieduwilt, L. Lo Leggio and E. D. Hedegard, Dalton Trans., 2024, 53, 5796–5807 RSC.
- M. M. Hagemann, E. K. Wieduwilt, U. Ryde and E. D. Hedegard, Inorg. Chem., 2024, 63, 21929–21940 CrossRef CAS PubMed.
- J. Haak, O. Golten, M. Sørlie, V. Eijsink and G. Cutsail III, Chem. Sci., 2025, 16, 233–254 RSC.
- T. J. Lawton, G. E. Kenney, J. D. Hurley and A. C. Rosenzweig, Biochemistry, 2016, 55, 2278–2290 CrossRef CAS PubMed.
- J. O. Ipsen, C. Hernandez-Rollan, S. J. Muderspach, S. Brander, A. B. Bertelsen, P. E. Jensen, M. H. H. Norholm, L. Lo Leggio and K. S. Johansen, FEBS Lett., 2021, 595, 1708–1720 CrossRef CAS PubMed.
- S. Brander, I. Horvath, J. O. Ipsen, A. Peciulyte, L. Olsson, C. Hernandez-Rollan, M. H. H. Norholm, S. Mossin, L. L. Leggio, C. Probst, D. J. Thiele and K. S. Johansen, Sci. Rep., 2020, 10, 16369 CrossRef CAS PubMed.
- A. Labourel, K. E. H. Frandsen, F. Zhang, N. Brouilly, S. Grisel, M. Haon, L. Ciano, D. Ropartz, M. Fanuel, F. Martin, D. Navarro, M. N. Rosso, T. Tandrup, B. Bissaro, K. S. Johansen, A. Zerva, P. H. Walton, B. Henrissat, L. L. Leggio and J. G. Berrin, Nat. Chem. Biol., 2020, 16, 345–350 CrossRef CAS PubMed.
- G. Y. Park, J. Y. Lee, R. A. Himes, G. S. Thomas, N. J. Blackburn and K. D. Karlin, J. Am. Chem. Soc., 2014, 136, 12532–12535 CrossRef CAS PubMed.
- S. Schwab, J. Shearer, S. E. Conklin, B. Alies and K. L. Haas, J. Inorg. Biochem., 2016, 158, 70–76 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt03301a |
| This journal is © The Royal Society of Chemistry 2025 |