
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvancing predictive toxicology: overcoming hurdles and shaping the future
Sara
Masarone†
 a,
Katie V.
Beckwith†
b,
Matthew R.
Wilkinson†
a,
Shreshth
Tuli†
a,
Amy
Lane
a,
Sam
Windsor†
a,
Jordan
Lane†
*a and
Layla
Hosseini-Gerami†
*a
a,
Katie V.
Beckwith†
b,
Matthew R.
Wilkinson†
a,
Shreshth
Tuli†
a,
Amy
Lane
a,
Sam
Windsor†
a,
Jordan
Lane†
*a and
Layla
Hosseini-Gerami†
*a
aIgnota Labs Ltd, Cambridge, UK. E-mail: jordan.lane@ignotalabs.ai
bYusuf Hamied Department of Chemistry, University of Cambridge, UK
First published on 6th January 2025
Abstract
Modern drug discovery projects are plagued with high failure rates, many of which have safety as the underlying cause. The drug discovery process involves selecting the right compounds from a pool of possible candidates to satisfy some pre-set requirements. As this process is costly and time consuming, finding toxicities at later stages can result in project failure. In this context, the use of existing data from previous projects can help develop computational models (e.g. QSARs) and algorithms to speed up the identification of compound toxicity. While clinical and in vivo data continues to be fundamental, data originating from organ-on-a-chip models, cell lines and previous studies can accelerate the drug discovery process allowing for faster identification of toxicities and thus saving time and resources.
Introduction
Modern drug discovery adopts a survival-of-the-fittest discovery approach to finding candidate molecules (Fig. 1). This process begins with vast compound libraries, which are progressively refined through a combination of in silico, in vitro, and in vivo experiments. As the pipeline proceeds, testing becomes increasingly expensive whilst the number of viable candidate molecules decreases, resulting in poor odds that even a single structure will make it to the clinic. Evidence for the diminishing effectiveness of this approach is clear, with as many as 90% of drug discovery projects failing.1–4 Furthermore, the number of patents accepted for novel compounds has shown a significant decline as it becomes ever harder to find new chemical entities (NCEs) that meet the approval requirements for widespread use.5 Failed projects incur both significant financial losses—ranging from approximately $1 million in early-stage research to over $2.6 billion by the final stages of clinical development—and a negative impact on the industry-wide drive toward improved sustainability, as the time and resource investment often yields no positive return. Fig. 1 illustrates the drug discovery funnel, where candidate compounds are progressively narrowed from more than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecules in the basic research phase to just one approved drug. The integration of in silico, in vitro, and in vivo approaches across the funnel highlights how these methodologies interact to filter compounds effectively at different stages. For example, early-stage techniques such as virtual screening and database mining (in silico) are complemented by target identification and lead optimization, which combine computational models with in vitro assays. Preclinical research involves safety pharmacology, toxicokinetics, and animal studies (in vivo), with each step informing the next through iterative feedback loops. This integration emphasizes the importance of predictive toxicology across all stages, reducing attrition rates by identifying potential safety risks earlier in the pipeline.
000 molecules in the basic research phase to just one approved drug. The integration of in silico, in vitro, and in vivo approaches across the funnel highlights how these methodologies interact to filter compounds effectively at different stages. For example, early-stage techniques such as virtual screening and database mining (in silico) are complemented by target identification and lead optimization, which combine computational models with in vitro assays. Preclinical research involves safety pharmacology, toxicokinetics, and animal studies (in vivo), with each step informing the next through iterative feedback loops. This integration emphasizes the importance of predictive toxicology across all stages, reducing attrition rates by identifying potential safety risks earlier in the pipeline.
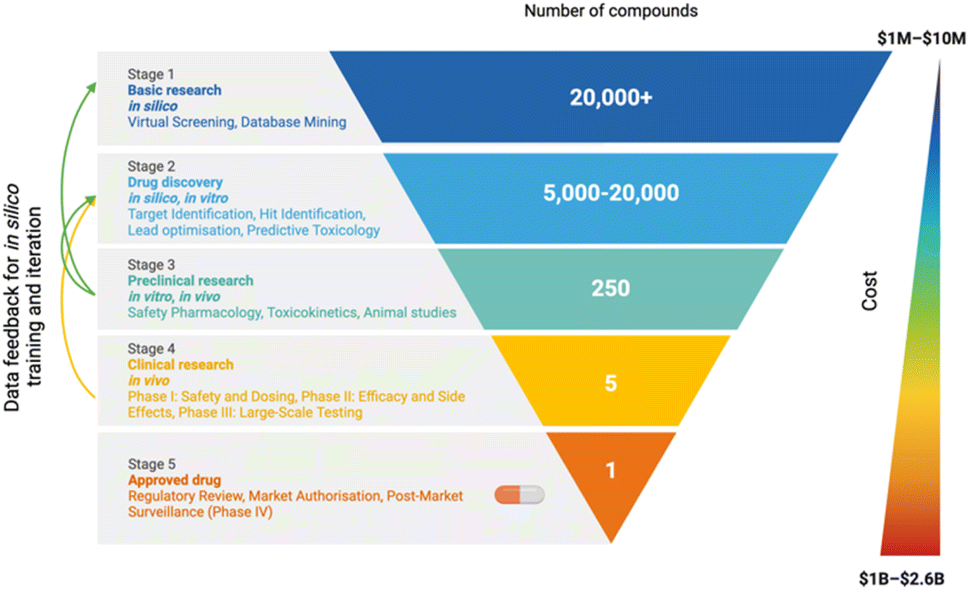 | ||
| Fig. 1 Drug discovery funnel from Stage 1 (basic research) to Stage 5 (approved drug). This process involves a combination of in silico, in vitro and in vivo experiments, increasing in complexity and cost as the drug progresses. Data from in vitro and in vivo experiments can be used to train in silico models, and models can inform on which experiments to perform. On the right hand side the cost of each phase, from $1m up to $2.6b. | ||
Safety concerns halt 56% of projects which, after efficacy, makes it the largest contributor to project failure.6 Despite safety being the single most important factor in determining a drug's chances of approval, safety assessment is often neglected until the late stages of the discovery timeline.7 There are significant barriers preventing safety from becoming an early priority. For example, although safety assessments can be carried out using in vitro systems, the cost and time burden associated with these experiments as well as the sheer number of potential toxicity endpoints to screen against, makes conventional large-scale testing impossible. This is especially true for small or medium BioTech companies with limited resources. Instead, strategic decisions must be made, selecting limited numbers of compounds and endpoints for testing. This narrow approach increases the risk of overlooking toxic effects, that will ultimately halt the project further down the development timeline. Furthermore, in vitro tests do not fully capture the interactions a drug makes in living organisms (in vivo).8In vivo models offer better translation to clinical observations compared to in vitro, but translation from pre-clinical species to human findings is still far from perfect.9 In addition, in vivo studies' inherent reliance on animal testing is expensive and raises significant ethical concerns. In a bid to create a solution for large-scale yet clinically relevant toxicity screening, in silico approaches offer a promising solution to address the limitations of wet lab and animal testing. To fully realise their potential, in silico solutions require careful implementation to foster widespread adoption and trust.
Artificial Intelligence (AI) has seen a surge in popularity with data-driven models delivering state-of-the-art performance across tasks previously thought to be only possible with manual involvement. In drug discovery, this unlocks a vast wave of potential across the entire lifecycle of pharmaceuticals.10 By definition, AI learns from prior experience to make informed predictions on a given task. In contrast to traditional wet lab experimentation, where negative data from failed projects is archived and ignored, integrating this data with AI can inform future research. Instead, there is value in the failed project data, as the experience and relationships it uncovered can be carried forward to provide informed decisions on where to target practical efforts in the future. Crucially, to be useful in practice, these models must be robust enough to accurately generalise to novel chemical structures.
This perspective article highlights the recent advancements in predictive toxicology and their potential impact on safety assessments in drug research and development. The article explores the concept of in silico toxicology and the benefits it brings compared to traditional approaches. To present a comprehensive perspective on the field, the utilisation of AI and Machine Learning (ML) is examined specifically focusing on its integration with systems biology, ‘omics’ data, and cell painting techniques for advancing predictive toxicology. In addition, the challenges that limit the applicability of these methods in practice are discussed. This includes limited data availability, representative chemical space coverage, and difficulties in predicting in vivo responses. This article also provides perspectives on how the challenges can be best addressed to advance the field.
Big drivers
The integration of AI into drug discovery is driven by several key factors, including regulatory initiatives, economic incentives, and the need to reduce both time and costs associated with drug development. It is well known that traditional methods of drug discovery are time consuming as clinical trials can take years to complete and can require billions of dollars to bring a drug to market.11 This prolonged timeline is due to the sequential phases of drug development, including initial research, preclinical testing, multiple phases of clinical trials, and regulatory approval. At each stage drugs can fail due to poor efficacy, safety, or drug selectivity/design, contributing to the overall inefficiency of the process.1The financial burden is also an important driver, with costs escalating due to the need for extensive laboratory testing, large-scale clinical trials, and the deployment of specialised personnel and resources. This results in the cost of failure being substantial; for every successful drug, numerous candidates fail at various stages, leading to sunk costs that must be absorbed by pharmaceutical companies. These failures often occur late in the development process, particularly during clinical trials, where safety and efficacy issues frequently emerge, leading to the termination of projects after significant investment.
Incentives and decisions from governments and policy makers are also driving the adoption of data driven technology. One such example is the FDA's forward-looking initiative – FDA 2.0.12 This encourages the adoption of advanced technologies to streamline drug approval processes. The initiative aims to modernize regulatory frameworks, making them more adaptable to innovative methodologies like AI, ultimately facilitating faster and more efficient drug development cycles. One key focus of this is to not only embrace new technologies, but to eliminate the moral issues surrounding drugs discovery, with a particular focus on animal testing in this case.
A paper recently released by the FDA discussed the use of ML to screen and design compounds to accelerate de novo drug design and to elucidate drug target interactions. The Center for Drug Evaluation and Research (CDER) AI Steering committee was also established to facilitate and coordinate the use of AI in the pharmacology industry. This aims to facilitate the creation of frameworks in collaboration with other partners or companies and ultimately guide the use of ML in this field. The FDA discussion paper also touches upon important aspects to consider when developing ML models, such as data bias, the ethics around the use of AI in the clinic, transparency and explainability.13
From a legislation perspective, another crucial motivator for the use of AI in drug discovery is the Inflation Reduction Act, which imposes cost containment measures on pharmaceutical companies. This legislation enforces a controlled price inflation having a profound impact on the pharmaceutical landscape. Although a welcome relief for patients, control of drug pricing has a knock-on effect for research and development efforts within pharma, an area with $83 billion spend in 2019.14 Pricing controls will impact R&D spending as well as stake holder decisions around the market strategy and intellectual property controls. This is especially impactful on early-stage assets, where the level or risk is much larger for achieving a significant return on investment. With more constrained budgets, the opportunity for both risk and cost reduction from AI methods is an ever more critical lifeline for pharmaceutical development. AI technologies offer a tangible solution by accelerating the drug discovery process, reducing costs, as compounds can be screened using in silico technologies and predictive modelling and making the entire process more efficient.10
Status quo of toxicity assessment
Toxicity evaluation is a critical component in drug development to ensure that potential therapeutic candidates are both efficacious and safe. Before testing in the clinic, two different model systems are used to generate data about the risk of toxicity: namely in vitro (outside the living organism) and in vivo (inside the living organism). Both methods provide unique insights into a compound's toxicological profile, but each differs in complexity and effectiveness in aligning with clinical toxicity observations (Fig. 2). | ||
| Fig. 2 Toxicity studies are carried out in increasingly complex systems, from simple 2D assays through to human studies. In silico models, powered by AI, provide a high-throughput, mechanistically enabled approach to predictive toxicology, complementing in vitro and in vivo systems. These models offer rapid insights and allow iterative hypothesis testing before resource-intensive experiments. Each step in complexity offers higher clinical relevance but comes with greater costs and lower throughput. | ||
In vitro data
In vitro toxicity data are collected from a range of biochemical or cellular assays designed to replicate a specific aspect of more complex biology. For example, to assess cardiotoxicity risk, a proxy in vitro assay determines whether a compound inhibits the ion channel encoded by the human ether-a-go-go related gene (hERG) – a known mechanism of drug-induced long QT syndrome which causes cardiac death. Assays are optimised for speed, reproducibility and reliability by exercising significant control over confounding variables. When using heavily controlled model systems, there is a balance between elucidating mechanistic understanding and in vivo correlation.Recent advances in in vitro toxicity assessment aim to improve physiological relevance and include the use of spheroid and organ-on-a-chip technologies. Growing cells in 3D environments (rather than as a 2D layer on a plate) allow the cells to develop better intercellular and cell-matrix communication, which strongly influences the physiological attributes of individual cells in vivo.15 For example, a study compared the response of 2D HepG2 (an immortal human hepatocyte cell line) and 3D cultured spheroids to a range of liver toxicants finding that the 3D system was more representative of the in vivo liver response.16
Despite their improved in vivo relevance over 2D cultures, 3D cultures lack the microenvironmental complexity and precise control over physiological conditions that organ-on-a-chip systems offer. These microfluidic devices replicate the structural and functional units of human organs, allowing for accurate simulation of human responses to drugs and chemicals under physiologically relevant conditions.17 This technology also enables real-time analysis of cellular responses which is important as toxicity responses are often time dependent.18
Although ideally in vitro assays aim to represent the underlying biology, there are frequently complexities and dynamics that they cannot capture, despite the advances in spheroids and microfluidics technology. Complexities around the cell line background, species, immortalisation of cancer cell lines and lab-to-lab variability can all affect the quality and reproducibility of these in vitro results.19 Despite the limitations, without the reductionist approach of in vitro assays, understanding the mechanism of action would be impossible to determine. Mitochondrial toxicity is a prominent example of this. A range of clinical toxicity issues are caused by mitochondrial toxicity,20 but without probing the underlying cellular processes, attributing mitochondrial toxicity as the underlying cause is impossible.
In vivo data
In vivo toxicity refers to the study of the effects of a substance within a living organism (usually in species such as rat or pig), representing a significant step up in both complexity and clinical relevance compared to in vitro methods. In vivo analysis tests a compound's effect within a living system, encompassing the full spectrum of biological interactions such as absorption, metabolism, distribution, and excretion (ADME). This approach evaluates multiple parameters, including behavioural changes, histopathological alterations, and biochemical responses.In vivo studies are crucial because they provide a more comprehensive and realistic view of how a compound behaves in a complex biological system. Unlike in vitro assays, which are optimised for speed and reproducibility but may lack certain biological complexities, in vivo models offer data that is more predictive of human responses. This makes them an essential step in the drug development process, as they can uncover potential issues that might not be evident in simpler models.21
Despite their advantages, in vivo studies come with significant ethical and practical limitations. There is a strong consensus that animal testing should be minimised, and all efforts should be made to find alternatives – termed the “3Rs” of reduction, replacement and refinement.22 Additionally, while in vivo models provide valuable insights, they often face criticism for not perfectly mimicking human diseases or toxicological responses due to differences in species homology.23 An analysis of pre-clinical and phase I trial data on 108 oncology drugs showed a poor correlation between animal and human outcomes (positive predictive value = 0.65).24 The FDA's mandate for testing in two non-human species underscores the uncertainty regarding the relevance of animal models to human biology. Furthermore, the complexity of whole-organism studies makes it challenging to pinpoint specific mechanisms of toxicity, necessitating more extensive studies with increased animal and compound numbers, thus increasing time and costs. These limitations highlight the need for clinical data, which provide the highest level of relevance and accuracy in assessing human responses to new drugs.
Clinical data
Clinical data is obtained from human studies, where the drug is administered to human volunteers, providing the most direct assessment of clinical toxicity and efficacy. Clinical trials test a drug's safety, efficacy, and toxicity in humans. These trials are typically conducted in phases, with Phase II/III trials focusing on specific disease populations. The data collected includes information on adverse effects, therapeutic benefits, pharmacokinetics (PK), and pharmacodynamics (PD).Human clinical trials are indispensable because they offer the most accurate and direct assessment of how a drug will perform in the target population. While in vitro and in vivo models are essential for preliminary testing and risk reduction, they cannot fully replicate the complexity of human biology. Clinical trials provide comprehensive data on human-specific factors such as co-morbidities, interactions with other medications, and individual variations in metabolism, sex, ethnicity, and lifestyle. This level of detail is crucial for determining the real-world safety and efficacy of a new drug.
Despite being the gold standard, the complexity of clinical trials,25 with varied patient-specific factors and population-level differences, makes it challenging to identify root causes of observed effects without the support of in vitro and in vivo experiments. The interplay of these factors underscores the necessity of preceding preclinical studies to support and interpret clinical data accurately.
Towards improved understanding with data-centric approaches
Traditional in vitro, in vivo, and clinical studies often struggle with the vast complexity and volume of data generated, leading to slower progress and sometimes incomplete mechanistic insights. AI addresses the inherent limitations of experimental data by rapidly analysing and interpret large datasets, identifying patterns and making predictions with greater precision and speed. This capability allows researchers to integrate data from various sources, providing a more comprehensive understanding of toxicological responses.AI models created from in vitro data can create a platform that facilitates mechanistic understanding via in silico analysis. This approach, despite limited by the lack of extensive data, allows for a rapid, iterative assessment of potential toxicophores across a broad range of specific biological endpoints. By using this broad assessment sweep, areas of interest can be identified for further focus. For compounds with notable effects that are predicted in silico, predictions can be supplemented by in vivo data, allowing for a more comprehensive picture of a drug's toxicity profile. This step-by-step progression ensures judicious use of resources whilst upholding the rigour of analysis that traditional lab-based testing offers.
More recent works have improved the throughput of in vivo systems offering an opportunity to build AI models on data that was previously too limited in size. Literature has also shown an increased capacity to harness data from novel platforms such as organ-on-a-chip and 3D cell culture systems. Leveraging these approaches allows users to merge the richness of in vivo system data with the scalability of in vitro studies.26 By validating the outputs of more controlled models on in vivo translatable biology, more robust, translatable AI models can be developed for efficient screening with high-quality validation.
Owing to the swift insights from in vitro data and the comprehensive perspective from in vivo data, establishing a dynamic in silico feedback system allows researchers to iterate and refine their hypotheses and experiments in near real-time. This will diminish redundancy and enhance the pace of safety evaluation and consequently, the wider drug discovery process.
In silico toxicology and the power of AI and ML
In silico toxicology is a broad field encompassing methods beyond just AI and ML. Examples of such methods include:(1) Read across – uses toxicity data from well-characterised compounds to directly infer the effects of structurally related, untested compounds, based on the principles of chemical similarity.
(2) Structural alerts – identifies specific chemical structures/substructures (called ‘alerts’) that are known to be associated with toxicological outcomes. If a compound contains one of these alerts, it may exhibit the associated toxicity.
(3) Quantitative structure activity relationships (QSAR) – establish a relationship between chemical features and observed biological activity to construct a mathematical model to predict compound effects.
Although it is important to consider that in silico toxicology includes all these methods, this article will focus specifically on the use of AI for predicting toxicity. ML exists as a subsection of AI. In this article specifically, the term ML will refer to any algorithm uses data to learn a specific task. Although the term ML also encompasses Deep Learning (DL), this article will not discuss the use of neural networks in this field – an area of ML that specifically uses neural networks. When AI is discussed, it will encompass both ML and DL methods.
Both ML and DL have seen an explosion of interest across the drug discovery process. Successful applications include molecular property prediction,27 synthesis design,28 protein structure elucidation29 and smart manufacturing of pharmaceutical products.30 Although the applications beyond toxicity are out of scope for this article, it is important to consider how significant adopting AI methods will be for the drug discovery industry as a whole.31 The primary value added when utilising AI models for predicting toxicity comes from unlocking safety evaluation data from the moment the structure of a drug is chosen. Unlike in vitro and in vivo tests, in silico models do not require compound synthesis. While these models require high-quality, curated data for both training and validation, they can still be particularly advantageous in early drug development where sample quantities are at a premium due to synthesis being challenging and expensive. In silico screening from the beginning of the drug discovery timeline also allows for significant cost savings as molecules likely to exhibit toxic behaviour are not progressed through the necessary development stages to reach the point of wet lab pre-clinical toxicity assessments.32 Thus, there is a clear financial advantage to avoiding sunk costs due to failed projects at this stage.
Aside from financial incentives, AI methods offer far superior throughput compared with laboratory experimentations. In vitro assays for a single compound can take several days to collect results from, compared to inference times in the seconds scale for in silico tools. By assessing a greater number of compounds, trends can better be explored which helps inform structural changes to compounds during development. Interpretability methods offered by ML and DL models further assist this as discussed in the Interpretability and trust section.
Despite the advantages, the use of AI for toxicity prediction is still an emerging technology. Clear barriers that limit adoption persist making the use of AI models challenging. Research efforts targeting these areas are critical in enabling the industry can unlock the vast potential on offer. Toxicity is a complex, multivariate problem which makes training accurate AI models highly challenging. Asking a model to predict the overall clinical toxicity of a compound would require a significantly more information-rich input than what is available from just the molecular structure. It is well established that clinical toxicity results from a wide variety of factors. Where model systems are used to make assessments, AI practitioners must also begin with a simplified system inspired by the same wet lab testing done practically. This is important as capturing complete information regarding an in vitro assay requires significantly less information than accurately representing in vivo systems. In fact, in vitro assays are a useful starting point for modelling, as by design they aim to control variables such that only differences in the chemical structure give rise to the observed effect. Factors such as bioavailability, delivery routes and patient level differences are not accounted for and hence do not affect in vitro assays. These assays are routinely used to gather toxicity data during drug discovery, meaning training data is available in quantities suitable for applying AI methods. It must be noted that although common practice in drug discovery settings, there is limited translation of in vitro outcomes to clinical toxicity.23 As such, extending the predictive power of AI models beyond digitalised in vitro twins remains an open research challenge.
Applying AI methods in practice
As is true with assays, discrete biological endpoints are chosen for modelling, examples of which include but are not limited to cardiac ion channels, mitochondrial agents and neurotransmitter receptors. From the perspective of toxicity screening, data-driven models are applied to predict the toxicity of a candidate against specific biological endpoints. This form of modelling can be described as structure-to-property, meaning that the model accepts chemical structures as inputs, and returns a prediction regarding a specific property.33,34 Other important decisions about the context under which the model is trained must also be made. These include using suitable concentration thresholds, selecting appropriate assays, choosing cell types/species and determining how and when data points from different assays can be combined into the training dataset.Once a model is trained, it can be called upon to make inferences, and its ability to do so must be assessed. Properly assessing the performance of chemical models is difficult and requires careful consideration to ensure misleading indicators of success are avoided. To further the evaluation process, exploring interpretability methods is vital to foster trust and draw useful conclusions about how the model navigates high-dimensional data.
Limitations and challenges
Chemical space generalisation
ML models trained on chemical structure data often face challenges in generalisation. The robustness of a model—namely its ability to accurately predict properties of novel compounds—largely depends on the diversity of its training data.The concept of “chemical space” encompasses all potential chemical structures, with estimates suggesting the number of “drug-like” molecules is in the order of 1060.35 Given its immense size, it's not feasible to gather toxicity data for every compound. As a result, in silico models are typically limited to specific sections of this space. This limitation restricts a model's applicability domain (AD), making predictions for unfamiliar chemicals potentially unreliable. This constraint is of particular concern when models inform critical decisions. Additionally, the presence of activity cliffs, where structurally similar compounds have vastly different toxicity levels and activity profiles (Fig. 3), poses a modelling challenge as models relying on structural similarities can be misled by these nuances.36 In the example in Fig. 3, the addition of a hydroxy group increased the inhibitory activity of a compound by almost three orders of magnitude.37 Expanding the training dataset to include a broader range of chemical structures enhances a model's predictive capabilities. In certain scenarios, data augmentation can also be performed, being careful not to introduce additional bias, especially with very small datasets. Transfer learning, which relies on transferring knowledge from a pre-trained model can also be considered, although pretrained large models can be hard to come across in cheminformatics.38
 | ||
| Fig. 3 Activity cliffs refer to a scenario where a small structural change leads to a large change in activity. Here, the compound on the left has a pKi of 4.37 against blood coagulation factor Xa, while the compound on the right (with a hydroxy group added) increases the pKi to 7.11, almost three orders of magnitude greater. | ||
Incorporating domain-specific knowledge, such as mechanistic/pathway-based information, or higher order data (non-chemical structure-based, e.g., omics or cell painting data39) can refine predictions, especially when navigating the challenges posed by activity cliffs.40
Data availability and sharing
Although, as discussed, increasing the size and diversity of a training set can lead to great improvements in model robustness, in practice this is often difficult to achieve due to the lack of access to such data. Many datasets containing valuable chemical and toxicological information are locked behind company walls due to confidentiality concerns and the intellectual property (IP) surrounding chemical structures.41 This is even true of data relating to failed projects or series which no longer hold value to the company that created the data. This restriction on data sharing hinders the progress of building models which can better generalise by covering a broader chemical space.The push towards open access and the Findable, Accessible, Interoperable and Reusable (FAIR) principles aims to address these challenges.42 Open access advocates for free and unrestricted access to research outputs, ensuring that this data can be readily accessed and utilised by researchers, ultimately benefitting all. The FAIR principles further strengthen this approach by ensuring that data is not only accessible, but presented in such a way that it can be easily found, integrated with other datasets, and reused for various research purposes.
However, while these principles are promising, their implementation is not without challenges. Concerns regarding IP, competitive advantage and data misuse are significant barriers to broader data sharing. To harness the benefits of open access and FAIR data, there is a need for collaborative efforts between academia, industry and regulatory bodies. By creating frameworks that protect proprietary interests while promoting data sharing (by having some incentives, for example), the scientific community can work towards more robust and reliable ML models.
Two positive examples of the implementation of FAIR practices come from Roche and AstraZeneca that have implemented these principles to enhance the use and sharing of clinical trial data for scientific insights. Roche focuses on a “learn-by-doing” approach and prospective “FAIRification”, while AstraZeneca uses scientific use cases and iterative data modelling to drive translational medicine research and foster data stewardship. Both initiatives highlight the importance of cultural shifts and structured processes to achieve scalable, reusable data systems.43
Evaluation
Regardless of the modelling approach adopted, evaluation of performance must be carried out by selecting appropriate criteria and metrics. It is widely accepted that splitting data into training, validation and testing sets is best practice for evaluating AI models, however, in chemical applications of AI, the choice of the splitting approach requires careful consideration. Although users can assign data points to the different subsets at random, literature in this field has shown that this often provides an overly optimistic performance evaluation.44 This is because random splitting does not ensure that compounds presented to the model during inference are chemically dissimilar. The alternative is to adopt a scaffold splitting approach, where compounds with similar substructures are bucketed and assigned to subsets such that the training, validation and testing subsets are chemically dissimilar. In contrast to random splitting, scaffold splits offer an overly harsh performance criteria as in reality it is unlikely a model will be required to predict on entirely unseen molecules. In practise it is often useful to evaluate models using both approaches to properly understand the circumstances by which an algorithm will perform well or poorly. In addition, an independent validation set (coming from a different data set) provides fundamental insights into the model's behaviour in a real-world scenario.Choosing appropriate metrics to evaluate a model is vital. In the field of toxicity, metric choice must reflect the target application and the distribution of samples across the dataset used to train the model. Class imbalance is incredibly common in toxicity prediction tasks. Biologically it is much more likely that a compound will be inactive with respect to a particular target and so datasets combining active and inactive compounds regularly have many more inactive compounds. Although not inherently limiting, users must select evaluation metrics that are not misrepresented when working with imbalanced data. To illustrate this, consider a dataset of 90:10 inactive to active compounds. The model can be 90% accurate by assigning inactive labels all the time. In this case, the classifier has no skill but accuracy of 90% is a seemingly impressive performance statistic. The same is true for AUROC, which is regularly reported in the literature for model evaluation.45 In addition to these, F1 score can be effective for evaluating imbalanced datasets as it balances precision and recall, but it can obscure class-specific performance and is less informative when class distributions are highly skewed. Matthews Correlation Coefficient (MCC) is also a robust metric for imbalanced data, it provides a single value that considers all confusion matrix components, allowing for a more balanced view of model performance across classes.
In general, all these metrics must be interpreted with proper consideration to the datasets they represent if they are to be meaningful.
In addition to model validation “per se” (as described in this section), new models can also be evaluated against pre-existent models by benchmarking them. This can help gain an understanding of whether a new technique has improved upon commonly used methods and what are its strengths and weaknesses compared to other models.
Technical challenges
To mitigate technical challenges in cheminformatics, such as data scarcity and the difficulty of modelling specific cases like activity cliffs (where structurally similar molecules exhibit vastly different activities), transfer learning and data augmentation can be highly effective. Transfer learning leverages pre-trained models on large, diverse chemical datasets, allowing for the adaptation of these models to smaller, task-specific datasets, thereby improving predictions without needing extensive new data. Data augmentation techniques, such as generating synthetic molecular structures or adding noise to existing data, can be used, although caution should be used to avoid generating biased synthetic data.Benchmarking
Developing datasets that can be used as benchmarks is paramount for any AI application. Across cheminformatics, benchmarking is particularly challenging as data is scarce and generating accurate, representative samples is expensive and time-consuming. Given the project-specific details of individual drug discovery programs, having specific benchmark datasets like those seen in other AI applications is less appropriate for cheminformatics tasks. Wet lab experiments are known to have a significant degree of variability between users, research groups and institutions and so capturing a single set of results that will represent all of these poses a significant challenge to the research community.46Initiatives have emerged over recent years to try and tackle the benchmarking challenge. The most well-adopted example of this is the Therapeutic Data Commons (TDC)47 and the Tox21 dataset from.48 TDC offers a variety of cheminformatics benchmarks including toxicity, however the datasets included are limited in size and are not accompanied by relevant scientific context regarding how they were generated. Despite being a promising initiative, Tox21 has been widely criticised for its data and metadata quality, and literature has documented its ineffectiveness for modelling.49,50 It must also be considered that both tools only offer in vitro data. This means that the challenge of comparing performance on in vivo data is one that continues to remain unsolved at the time of writing.
Initiatives have emerged over recent years to try and tackle the benchmarking challenge. A platform, Polaris51 was launched to implement, host and run benchmarks in computational drug discovery. The aims of this platform are to address the performance gap seen between test set metrics and applications to real-life drug discovery projects, and to close the gap between modellers and downstream users. This provides a valuable resource to the community towards the development of toxicity models which are practically useful and relevant in drug discovery.
Without suitable benchmarks, assessing the performance of different modelling techniques is not possible. During the AI lifecycle, performance changes can be attributed to data quality and size as well as ML or DL model design and hyperparameter tuning. To truly assess and compare model performance, the effects of the training data must be minimised by keeping consistent cross-validation splits, labels and the number of data points. Only by doing so can meaningful performance conclusions be drawn.
Interpretability and trust
Beyond the technical and scientific limitations of in silico toxicity modelling approaches, there is a significant psychological barrier to the wider adoption of these methods. A contributing factor is the divergence in expertise and expectations between the developers and users of these models. The primary users, medicinal chemists and toxicologists, have a distinct set of priorities and concerns compared to ML specialists, the model developers.For ML researchers, the goal is to develop or refine algorithms to achieve the best possible performance metrics. This can lead to the use of complex “black-box” DL methods. While such approaches might squeeze out an additional fraction of accuracy compared to simpler, inherently interpretable methods such as tree-based algorithms, they can be obscure in their operation, making it challenging for non-experts to understand or trust. The goal of the medicinal chemist or toxicologist is to obtain clarity and reliability. An incremental increase in accuracy is of secondary importance if they cannot discern why a prediction was made, the nature of the data on which the model was trained, or its relevance and reliability for their specific chemical series.
Addressing this challenge requires a shift in focus. While technical advancements are essential, equal weight should be given to the communication and presentation of model results. Efforts should be channelled towards creating interfaces and explanations that translate the complexities of ML into insights that are meaningful to toxicologists and chemists, as only then will there be a genuine alignment between computational advancements and practical toxicological applications, fostering greater confidence and integration of in silico methodologies within the field.
Methods to increase interpretability and trust in predictive toxicology models include the use of permutation feature importance and SHapley Additive exPlanations (SHAP).52 These provide importance scores to individual features, enabling an explanation of predicted outcomes. Structural features of high importance for predicting a toxicity outcome can be mapped to the original compound structure to produce ‘toxicophores’ of relevant chemical moieties in causing the unwanted interaction.53 Other examples of SHAP for drug discovery include its application to compound potency and multitarget activity prediction and its use for metabolic stability analyses.54,55 For example, Rodríguez-Pérez identified crucial groups for B-cell lymphoma 2 protein (Bcl-2) inhibition such as 2-amino-3-chloro-pyridine, and Wojtuch presented a case study showing that an aromatic ring with the chlorine atom attached increases metabolic stability.
Once these explanations have been generated, they should be reviewed and assessed by an expert human, such as a medicinal chemist. For example, a tertiary amine moiety is a known driver of hERG inhibition,56 and hence a predictive hERG model which gives a high importance to this chemical feature indicates that the model has learned the underlying causes of the molecular interaction, increasing trust and confidence in its predictions.
Towards understanding in vivo toxicity responses
The task of bridging the gap between controlled in vitro environments and the complex realities of in vivo systems has been a persistent challenge in computational toxicology.57 Ensuring that predictions are relevant to in vivo responses is paramount for both drug development and safety assessment, so this presents a significant gap in the field that must be addressed. Here we describe some potential approaches and how they can shape the future of in silico toxicity assessment.In vivo relevance
Due to the relative abundance of in vitro toxicology data vs. in vivo, many computational models for toxicity are built on data derived from biochemical or cell-based assays, on endpoints which are themselves merely models or proxies for in vivo adverse events. Due to the ADME properties of compounds, effects seen in isolated purified proteins or cellular systems may not be recapitulated in a living organism.58 A compound, despite displaying toxicity-related activity in an in vitro setting, may be poorly absorbed when administered to an organism, potentially attenuating its effect. Post absorption, its distribution might not be systemic; it could predominantly localise to specific tissues or organs, influencing its PD properties. For example, compounds active in in vitro neurotoxicity assays but which are not able to be distributed past the blood–brain barrier (BBB), may not show in vivo neurotoxicity. Metabolism introduces another layer of complexity. The organism's enzymatic machinery can transform compounds, potentially producing toxic metabolites from a non-toxic parent compound or vice versa. Furthermore, the rate and pathway of compound excretion can affect toxicity, e.g., if compounds are slow to be excreted and accumulate in the kidney, renal toxicity may occur after repeated dosing, which is not detectable in single-dose in vitro assays.Beyond these PK considerations, physiological interactions intrinsic to living organisms further complicate the extrapolation. The interplay between organs, systemic responses, and immune-mediated reactions can markedly modulate a compound's toxicological profile.59 This biological complexity is why it is a challenge to predict direct in vivo or clinical endpoints based on chemical structure alone; a chemical representation is not sufficient information to predict idiosyncratic responses such as DILI. This is exemplified in a review of computational models for DILI prediction by Vall et al., who remark that higher order data types such as genomics, gene expression or imaging data may improve predictability of in vivo responses.60 The task is to bridge the observational gap between controlled in vitro environments and the dynamic realities of in vivo systems.
Integrating predictions and data modalities
In toxicology assessment, it is important to distinguish between hazard (a compound's inherent potential to cause harm) and risk (the likelihood of that harm manifesting under specific conditions). Models built on discrete in vitro endpoints, though individually informative of toxicity hazard, provide a portion of the broader toxicity risk. To transition from these isolated insights to a comprehensive understanding of in vivo risk, the integration of data modalities and predictions derived from in vitro assays could serve as a suitable approach (Fig. 4).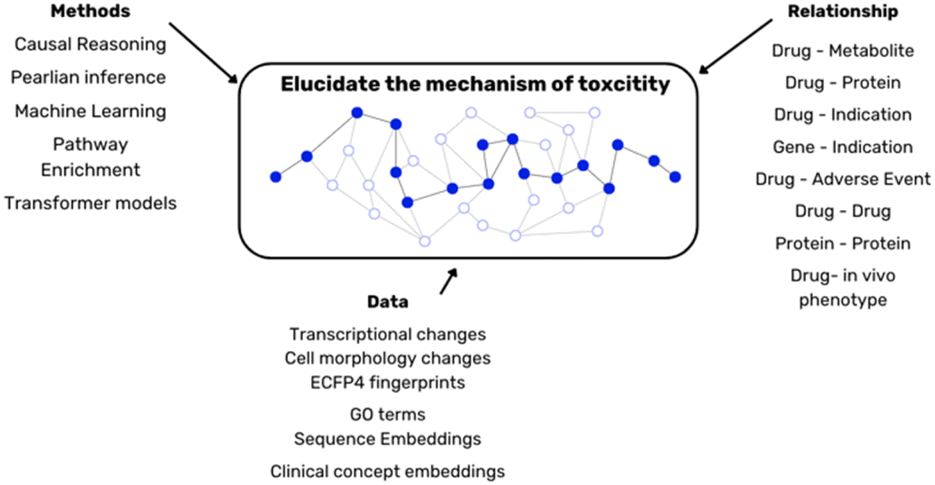 | ||
| Fig. 4 Different methods, data and heterogeneous relationships are required to elucidate the biological complexity of toxicity. | ||
One strategy for predicting in vivo organ-level toxicity is to integrate results from in silico predictions across multiple in vitro endpoints. For example, the prediction of DILI can benefit from combining predictions from models focused on bile salt export pump inhibition, mitochondrial toxicity, and liver (e.g., HepG2 cell) cytotoxicity. As each of these models addresses specific mechanisms that may contribute to DILI, their combined predictions can offer a holistic understanding of liver injury risk. This has been exemplified in work by Seal et al.,61 where this combined predictive approach outperformed direct predictions of DILI based on chemical structure alone. However, a prevailing challenge is mapping these discrete endpoints to organ-level responses. Recent efforts have aimed at statistically assessing the likelihood of adverse events arising from off-target or secondary pharmacology effects.62,63 As research progresses in this domain, the aim is to better understand the relationships and synergies between different endpoints to discern which combinations offer the most informative insights into risk.
Due to advancements in high-throughput technologies, the integration of ‘omics’ data has gained traction as a method for modelling compound responses. By considering the interplay between different biological processes, this approach captures a closer approximation of the system's response, offering a depth that complements traditional compound structure-based assessments.64,65 For example, genomics information can be used to understand the genetic predispositions that may influence a compound's effects, informing toxicity risk on a personalised level (pharmacogenomics).66 Transcriptomics data provides a snapshot of cellular response to compound perturbation and can be used to understand the mechanisms of toxicity of a compound (toxicogenomics).65 Such data is available on a large scale in the public domain through platforms such as LINCS L1000,67 and the datasets Open TG-GATEs68 and DrugMatrix69 link compound-induced gene expression data to in vivo findings (in rat) such as clinical chemistry, histopathology and toxic effects. Other ‘omics’ modalities, such as metabolomics and phosphoproteomics, offer views on compound metabolic pathways and protein signalling activity, respectively. The strength of ‘omics’ lies not in these individual datasets but in their integration. By combining these modalities, researchers can attain a layered, comprehensive view of compound-induced changes, from the genetic level to the functional metabolic outcomes. This allows for a more granular prediction and understanding of toxicities, facilitating a holistic approach to risk assessment. However, this integration is not without challenges. ‘Omics’ data often come with high biological variability and noise, making the extraction of meaningful signals a complex task – and the signal-to-noise ratio varies greatly across different modalities.70
The integration of ADME and PK/PD data, and predictions built on such data, can also aid in assessing in vivo risk.71 Such insights are critical for bridging the gap between in vitro findings and in vivo implications. When combined with toxicological predictions based on in vitro data, ADME and PK/PD data provide a clearer picture of the real-world exposure scenarios. For instance, while an in vitro assay might indicate hepatotoxicity, PK data might reveal that the compound doesn't reach the liver in significant concentrations, adjusting the perceived risk.
Methods for elucidating systems-level toxicology
Toxicology's progression into the era of big data has necessitated the development and application of advanced computational methods to accommodate the data influx. Drawing meaningful conclusions from integrated datasets is complex, requiring data storage frameworks and methodological approaches that can handle the intricacy of biological systems. Systems biology stands at the forefront of these efforts, emphasising the interconnected nature of biological systems and the emergent properties that result from the interactions within an organism.Knowledge graphs and network-based data structures have aided in this data integration challenge. These structured data representations capture intricate relationships between various biological entities, from genes and proteins to metabolic pathways. One example of a pre-made knowledge graph tailored for computational toxicity is ComptoxAI,72 providing links between chemical exposures, pathways and systems nodes that explain toxic outcomes (780038 distinct chemicals included as of July 2022). Beyond mere data storage, knowledge graphs facilitate efficient data retrieval and serve as robust platforms for advanced computational analyses. One of their significant strengths is the ability to integrate the results of machine ML models, such as predictions based on in vitro endpoints, into a coherent and interconnected framework. By doing so, they provide an enriched environment where predictions from different models can be combined with high-order data modalities, offering a holistic understanding of potential toxicological risks. This has been exemplified by Hao et al., who used data from ComptoxAI to predict causal chains of compound-gene, gene-pathway and pathway-toxicity interactions with a graph-based DL approach called AIDTox.73 An example of a prediction output can be seen in ref. 73. Fig. 5 shows the predicted important biological processes leading to dasatinib causing HEK293 cell death such as interaction with CYP1A2 leading to metabolism of lipids. This allows for a more granular understanding of, and potential elucidation of new mechanisms of drug-induced toxicity.
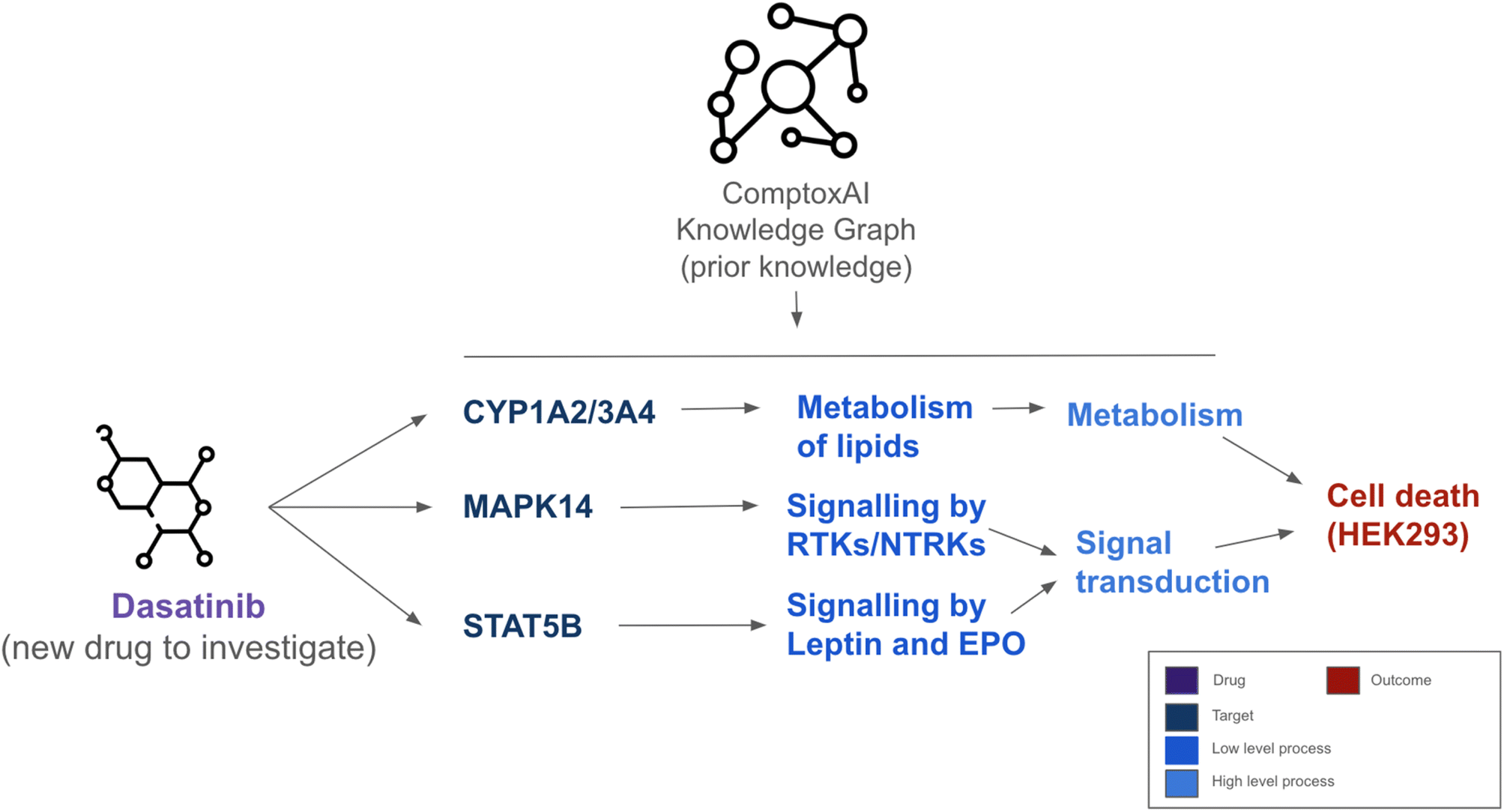 | ||
| Fig. 5 Explained prediction of Dasatinib causing HEK293 cell death. The figure illustrates the targets hit by Dasatinib and the low and high level pathways involved in the HEK393 cell death. On top, a diagram illustrating the role of ComptoxAI, a knowledge graph that provides additional knowledge to map these processes. Figure adapted with permission from (https://github.com/yhao-compbio/AIDTox). | ||
A particularly promising area is the application of causal reasoning techniques on knowledge graphs across multiple ‘omics’ layers.74 This approach can uncover the sequence of molecular events leading to a toxic outcome, providing insights that are more nuanced and closer to the real-world biological intricacies. For example, Trairatphisan et al.75 used a causal reasoning approach, CARNIVAL,76 to uncover aberrant cell signalling in DILI, leveraging Open TG-GATEs repeat-dosing transcriptional and in vivo histopathological data to identify a regulatory pathway among liver fibrosis-inducing compounds. By deciphering the intricacies of molecular pathways, causal reasoning can identify potential intervention points for mitigating adverse effects, or even reveal previously unknown off-target effects of compounds.
Conclusions
The integration of AI and ML into drug discovery and toxicology represents a transformative shift in the pharmaceutical development approach. Even in this early adoption stage, there is clearly significant potential for improving the efficiency and success rates of pharmaceutical development. By leveraging in silico models, researchers can harness vast amounts of data from previous projects, including failed ones, to inform future efforts. This approach not only reduces the time and cost associated with traditional wet lab and in vivo testing but also enhances the ability to predict and mitigate toxicity early in the drug development pipeline. Digital tools showcase a proactive discovery approach, allowing key decision makers to target resources towards the most promising projects. In doing so, projects are de-risked especially when tackling novel areas of chemistry or biology to development of NCEs and first in class products. This promises development landscape with a reduced financial burden incurred by late-stage clinical failures. It must also be noted that AI toxicology is not just about predicting in vivo outcomes, but also provides a wealth of resources for hypothesis generation and troubleshooting when safety issues inevitably arise. The ability to integrate diverse, multimodal data sources and condense high dimensional relationships into actionable insights is a powerful tool for drug hunters to use.Despite the evident advantages, several challenges must be addressed to fully realize the potential of AI in predictive toxicology. These include data availability, the need for comprehensive and representative datasets, and the difficulties in translating in vitro and in silico findings to in vivo contexts. Overcoming these hurdles requires collaborative efforts across academia, industry, and regulatory bodies to promote data sharing and develop robust, interpretable models that can be trusted by practitioners. By nature, in silico toxicology is a highly interdisciplinary field and collaboration between wet lab scientists and AI developers is critical in building useful tools with maximum impact.
The future of drug discovery and toxicology will be increasingly data-driven, with AI and ML playing a central role in navigating the complexities of biological systems and predicting pharmaceutical outcomes. By integrating diverse data modalities and refining computational methods, we can move towards more accurate and efficient toxicity assessments, ultimately improving the safety and efficacy of new therapeutic candidates.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
All authors marked with equal contribution were collectively responsible for conceptualization, content curation, and writing this manuscript. Final review and editing was done by SM and MRW with critical feedback from all authors. Figures were made by JL and AL.Conflicts of interest
All authors are current or past employees of Ignota Labs Ltd (http://www.ignotalabs.ai/).Acknowledgements
All authors extend their gratitude to the members of the Ignota Labs scientific advisory board for their invaluable feedback and guidance. The authors also thank James Bradshaw for contributions to scoping the content for this article. KVB acknowledges support by the Engineering and Physical Sciences Research Council (EPSRC) [EP/SO24220/1].Notes and references
- D. Sun, W. Gao, H. Hu and S. Zhou, Why 90% of clinical drug development fails and how to improve it?, Acta Pharm. Sin. B, 2022, 12(7), 3049–3062 CrossRef CAS PubMed.
- I. Kola and J. Landis, Can the pharmaceutical industry reduce attrition rates?, Nat. Rev. Drug Discovery, 2004, 3(8), 711–716 CrossRef CAS PubMed.
- M. J. Waring, J. Arrowsmith, A. R. Leach, P. D. Leeson, S. Mandrell and R. M. Owen, et al., An analysis of the attrition of drug candidates from four major pharmaceutical companies, Nat. Rev. Drug Discovery, 2015, 14(7), 475–486 CrossRef CAS PubMed.
- A. D. Hingorani, V. Kuan, C. Finan, F. A. Kruger, A. Gaulton and S. Chopade, et al., Improving the odds of drug development success through human genomics: modelling study, Sci. Rep., 2019, 9(1), 18911 CrossRef CAS PubMed.
- E. Işık and Ö. Orhangazi, Profitability and drug discovery, Ind. Corp. Change, 2022, 31(4), 891–904 CrossRef.
- D. Cook, D. Brown, R. Alexander, R. March, P. Morgan and G. Satterthwaite, et al., Lessons learned from the fate of AstraZeneca's drug pipeline: a five-dimensional framework, Nat. Rev. Drug Discovery, 2014, 13(6), 419–431 CrossRef CAS PubMed.
- J. M. McKim, Building a Tiered Approach to In Vitro Predictive Toxicity Screening: A Focus on Assays with In Vivo Relevance, Comb. Chem. High Throughput Screen., 2010, 13(2), 188–206 CrossRef CAS PubMed.
- E. Madorran, A. Stožer, S. Bevc and U. Maver, In vitro toxicity model: Upgrades to bridge the gap between preclinical and clinical research, Bosn. J. Basic Med. Sci., 2020, 20(2), 157–168 CAS.
- C. H. C. Leenaars, C. Kouwenaar, F. R. Stafleu, A. Bleich, M. Ritskes-Hoitinga and R. B. M. De Vries, et al., Animal to human translation: a systematic scoping review of reported concordance rates, J. Transl. Med., 2019, 17(1), 223 CrossRef PubMed.
- D. Paul, G. Sanap, S. Shenoy, D. Kalyane, K. Kalia and R. K. Tekade, Artificial intelligence in drug discovery and development, Drug Discov. Today, 2021, 26(1), 80–93 CrossRef CAS PubMed.
- O. J. Wouters, M. McKee and J. Luyten, Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018, JAMA, 2020, 323(9), 844–853 CrossRef PubMed.
- J. J. Han, FDA Modernization Act 2.0 allows for alternatives to animal testing, Artif. Organs, 2023, 47(3), 449–450 CrossRef PubMed.
- Using Artificial Intelligence and Machine Learning in the Development of Drugs and Biological Products[Internet]. [cited 2024 Nov 29]. Available from: https://www.fda.gov/media/167973/download.
- S. D. Sullivan, Medicare Drug Price Negotiation in the United States: Implications and Unanswered Questions, Value Health, 2023, 26(3), 394–399 CrossRef PubMed.
- V. Zingales, M. R. Esposito, N. Torriero, M. Taroncher, E. Cimetta and M. J. Ruiz, The Growing Importance of Three-Dimensional Models and Microphysiological Systems in the Assessment of Mycotoxin Toxicity, Toxins, 2023, 15(7), 422 CrossRef CAS PubMed.
- S. J. Fey and K. Wrzesinski, Determination of Drug Toxicity Using 3D Spheroids Constructed From an Immortal Human Hepatocyte Cell Line, Toxicol. Sci., 2012, 127(2), 403–411 CrossRef CAS PubMed.
- Y. Cong, X. Han, Y. Wang, Z. Chen, Y. Lu and T. Liu, et al., Drug Toxicity Evaluation Based on Organ-on-a-Chip Technology: A Review, Micromachines, 2020, 11(4), 381 CrossRef PubMed.
- K. K. Rozman and J. Doull, Dose and time as variables of toxicity, Toxicology, 2000, 144(1), 169–178 CrossRef CAS PubMed.
- C. Hirsch and S. Schildknecht, In Vitro Research Reproducibility: Keeping Up High Standards, Front. Pharmacol, 2019, 10, 1484 CrossRef PubMed.
- Y. T. Lin, K. H. Lin, C. J. Huang and A. C. Wei, MitoTox: a comprehensive mitochondrial toxicity database, BMC Bioinf., 2021, 22(10), 369 CrossRef PubMed.
- M. R. Fielden and K. L. Kolaja, The role of early in vivo toxicity testing in drug discovery toxicology, Expet Opin. Drug Saf., 2008, 7(2), 107–110 CrossRef CAS PubMed.
- P. Flecknell, Replacement, reduction and refinement, ALTEX, 2002, 19(2), 73–78 Search PubMed.
- G. A. Van Norman, Limitations of Animal Studies for Predicting Toxicity in Clinical Trials: Is it Time to Rethink Our Current Approach?, JACC, 2019, 4(7), 845–854 Search PubMed.
- J. T. Atkins, G. C. George, K. Hess, K. L. Marcelo-Lewis, Y. Yuan and G. Borthakur, et al., Pre-clinical animal models are poor predictors of human toxicities in phase 1 oncology clinical trials, Br. J. Cancer, 2020, 123(10), 1496–1501 CrossRef PubMed.
- N. Markey, B. Howitt, I. El-Mansouri, C. Schwartzenberg, O. Kotova and C. Meier, Clinical trials are becoming more complex: a machine learning analysis of data from over 16,000 trials, Sci. Rep., 2024, 14(1), 3514 CrossRef CAS PubMed.
- S. Deng, C. Li, J. Cao, Z. Cui, J. Du and Z. Fu, et al., Organ-on-a-chip meets artificial intelligence in drug evaluation, Theranostics, 2023, 13(13), 4526–4558 CrossRef CAS PubMed.
- W. P. Walters and R. Barzilay, Applications of Deep Learning in Molecule Generation and Molecular Property Prediction, Acc. Chem. Res., 2021, 54(2), 263–270 CrossRef CAS PubMed.
- S. Q. Zhang, L. C. Xu, S. W. Li, J. C. A. Oliveira, X. Li and L. Ackermann, et al., Bridging Chemical Knowledge and Machine Learning for Performance Prediction of Organic Synthesis, Chem.–Eur. J., 2023, 29(6), e202202834 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov and O. Ronneberger, et al., Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596(7873), 583–589 CrossRef CAS PubMed.
- N. S. Arden, A. C. Fisher, K. Tyner, L. X. Yu, S. L. Lee and M. Kopcha, Industry 4.0 for pharmaceutical manufacturing: Preparing for the smart factories of the future, Int. J. Pharm., 2021, 602, 120554 CrossRef CAS PubMed.
- M. Mock, S. Edavettal, C. Langmead and A. Russell, AI can help to speed up drug discovery — but only if we give it the right data, Nature, 2023, 621(7979), 467–470 CrossRef CAS PubMed.
- H. Norlen, A. P. Worth and S. Gabbert, A Tutorial for Analysing the Cost-effectiveness of Alternative Methods for Assessing Chemical Toxicity: The Case of Acute Oral Toxicity Prediction, Altern. Lab. Anim., 2014, 42(2), 115–127 CrossRef CAS PubMed.
- J. Chang and J. C. Ye, Bidirectional generation of structure and properties through a single molecular foundation model, Nat. Commun., 2024, 15(1), 2323 CrossRef CAS PubMed.
- S. Shermukhamedov, D. Mamurjonova, T. Maihom and M. Probst, Structure to Property: Chemical Element Embeddings for Predicting Electronic Properties of Crystals, J. Chem. Inf. Model., 2024, 64(15), 5762–5770 Search PubMed.
- J. L. Reymond, The Chemical Space Project, Acc. Chem. Res., 2015, 48(3), 722–730 CrossRef CAS PubMed.
- D. van Tilborg, A. Alenicheva and F. Grisoni, Exposing the Limitations of Molecular Machine Learning with Activity Cliffs, J. Chem. Inf. Model., 2022, 62(23), 5938–5951 CrossRef CAS PubMed.
- M. Dablander, T. Hanser, R. Lambiotte and G. M. Morris, Exploring QSAR models for activity-cliff prediction, J. Cheminf., 2023, 15(1), 47 Search PubMed.
- A. Mumuni and F. Mumuni, Data augmentation: A comprehensive survey of modern approaches, Array, 2022, 16, 100258 CrossRef.
- S. Seal, O. Spjuth, L. Hosseini-Gerami, M. García-Ortegón, S. Singh and A. Bender, et al., Insights into Drug Cardiotoxicity from Biological and Chemical Data: The First Public Classifiers for FDA Drug-Induced Cardiotoxicity Rank, J. Chem. Inf. Model., 2024, 64(4), 1172–1186 CrossRef CAS PubMed.
- B. Baillif, J. Wichard, O. Méndez-Lucio and D. Rouquié, Exploring the Use of Compound-Induced Transcriptomic Data Generated From Cell Lines to Predict Compound Activity Toward Molecular Targets, Front. Chem., 2020, 8 Search PubMed . Available from: https://www.frontiersin.org/articles/10.3389/fchem.2020.00296.
- A. Thakkar, T. Kogej, J. L. Reymond, O. Engkvist and E. Jannik Bjerrum, Datasets and their influence on the development of computer assisted synthesis planning tools in the pharmaceutical domain, Chem. Sci., 2020, 11(1), 154–168 RSC.
- M. D. Wilkinson, M. Dumontier, IjJ. Aalbersberg, G. Appleton, M. Axton and A. Baak, et al., The FAIR Guiding Principles for scientific data management and stewardship, Sci. Data, 2016, 3(1), 160018 CrossRef PubMed.
- I. Harrow, R. Balakrishnan, H. Küçük McGinty, T. Plasterer and M. Romacker, Maximizing data value for biopharma through FAIR and quality implementation: FAIR plus Q, Drug Discovery Today, 2022, 27(5), 1441–1447 CrossRef CAS PubMed.
- G. W. Bemis and M. A. Murcko, The properties of known drugs. 1. Molecular frameworks, J. Med. Chem., 1996, 39(15), 2887–2893 CrossRef CAS PubMed.
- Z. Wu, B. Ramsundar, E. N. Feinberg, J. Gomes, C. Geniesse and A. S. Pappu, et al., MoleculeNet: a benchmark for molecular machine learning, Chem. Sci., 2018, 9(2), 513–530 RSC.
- G. A. Landrum and S. Riniker, Combining IC50 or Ki Values from Different Sources Is a Source of Significant Noise, J. Chem. Inf. Model., 2024, 64(5), 1560–1567 CrossRef CAS PubMed.
- K. Huang, T. Fu, W. Gao, Y. Zhao, Y. Roohani and J. Leskovec, et al., Artificial intelligence foundation for therapeutic science, Nat. Chem. Biol., 2022, 18(10), 1033–1036 CrossRef CAS PubMed.
- A. M. Richard, R. Huang, S. Waidyanatha, P. Shinn, B. J. Collins and I. Thillainadarajah, et al., The Tox21 10K Compound Library: Collaborative Chemistry Advancing Toxicology, Chem. Res. Toxicol., 2021, 34(2), 189–216 Search PubMed.
- J. H. Hsieh, A. Sedykh, R. Huang, M. Xia and R. R. Tice, A Data Analysis Pipeline Accounting for Artifacts in Tox21 Quantitative High-Throughput Screening Assays, J. Biomol. Screen, 2015, 20(7), 887–897 CrossRef CAS PubMed.
- D. J. Cooper and S. Schürer, Improving the Utility of the Tox21 Dataset by Deep Metadata Annotations and Constructing Reusable Benchmarked Chemical Reference Signatures, Molecules, 2019, 24(8), 1604 CrossRef CAS PubMed.
- Polaris[Internet]. [cited 2024 Aug 1]. Available from: https://polarishub.io.
- S. J. Belfield, M. T. D. Cronin, S. J. Enoch and J. W. Firman, Guidance for good practice in the application of machine learning in development of toxicological quantitative structure-activity relationships (QSARs), PLoS One, 2023, 18(5), e0282924 CrossRef CAS PubMed.
- R. Rodríguez-Pérez and J. Bajorath, Interpretation of Compound Activity Predictions from Complex Machine Learning Models Using Local Approximations and Shapley Values, J. Med. Chem., 2020, 63(16), 8761–8777 CrossRef PubMed.
- R. Rodríguez-Pérez and J. Bajorath, Interpretation of machine learning models using shapley values: application to compound potency and multi-target activity predictions, J. Comput. Aided Mol. Des., 2020, 34(10), 1013–1026 CrossRef PubMed.
- A. Wojtuch, R. Jankowski and S. Podlewska, How can SHAP values help to shape metabolic stability of chemical compounds?, J. Cheminf., 2021, 13(1), 74 Search PubMed.
- A. Garrido, A. Lepailleur, S. M. Mignani, P. Dallemagne and C. Rochais, hERG toxicity assessment: Useful guidelines for drug design, Eur. J. Med. Chem., 2020, 195, 112290 CrossRef CAS PubMed.
- Q. Zhang, J. Li, A. Middleton, S. Bhattacharya and R. B. Conolly, Bridging the Data Gap From in vitro Toxicity Testing to Chemical Safety Assessment Through Computational Modeling, Front. Public Health, 2018, 6 Search PubMed . Available from: https://www.frontiersin.org/articles/10.3389/fpubh.2018.00261.
- A. Bender and I. Cortés-Ciriano, Artificial intelligence in drug discovery: what is realistic, what are illusions? Part 1: Ways to make an impact, and why we are not there yet, Drug Discovery Today, 2021, 26(2), 511–524 CrossRef CAS PubMed.
- M. Mosedale and P. B. Watkins, Understanding Idiosyncratic Toxicity: Lessons Learned from Drug-Induced Liver Injury, J. Med. Chem., 2020, 63(12), 6436–6461 CrossRef CAS PubMed.
- A. Vall, Y. Sabnis, J. Shi, R. Class, S. Hochreiter and G. Klambauer, The Promise of AI for DILI Prediction, Front. Artif. Intell., 2021, 4, 638410 CrossRef PubMed.
- S. Seal, D. P. Williams, L. Hosseini-Gerami, O. Spjuth and A. Bender, Improved Early Detection of Drug-Induced Liver Injury by Integrating Predicted in vivo and in vitro Data, bioRxiv, 2024, 575128 Search PubMed . Available from: https://www.biorxiv.org/content/10.1101/2024.01.10.575128v1.
- J. J. Sutherland, D. Yonchev, A. Fekete and L. Urban, A preclinical secondary pharmacology resource illuminates target-adverse drug reaction associations of marketed drugs, Nat. Commun., 2023, 14(1), 4323 CrossRef CAS PubMed.
- I. A. Smit, A. M. Afzal, C. H. G. Allen, F. Svensson, T. Hanser and A. Bender, Systematic Analysis of Protein Targets Associated with Adverse Events of Drugs from Clinical Trials and Postmarketing Reports, Chem. Res. Toxicol., 2021, 34(2), 365–384 Search PubMed.
- M. A. Trapotsi, L. Hosseini-Gerami and A. Bender, Computational analyses of mechanism of action (MoA): data, methods and integration, RSC Chem. Biol., 2022, 3(2), 170–200 RSC.
- B. Alexander-Dann, L. L. Pruteanu, E. Oerton, N. Sharma, I. Berindan-Neagoe and D. Módos, et al., Developments in toxicogenomics: understanding and predicting compound-induced toxicity from gene expression data, Mol. Omics, 2018, 14(4), 218–236 RSC.
- G. Zhang and D. W. Nebert, Personalized medicine: Genetic risk prediction of drug response, Pharmacol. Ther., 2017, 175, 75–90 CrossRef CAS PubMed.
- A. Subramanian, R. Narayan, S. M. Corsello, D. D. Peck, T. E. Natoli and X. Lu, et al., A Next Generation Connectivity Map: L1000 platform and the first 1,000,000 profiles, Cell, 2017, 171(6), 1437–1452 CrossRef CAS PubMed.
- Y. Igarashi, N. Nakatsu, T. Yamashita, A. Ono, Y. Ohno and T. Urushidani, et al., Open TG-GATEs: a large-scale toxicogenomics database, Nucleic Acids Res., 2015, 43(Database issue), D921–D927 CrossRef CAS PubMed.
- D. L. Svoboda, T. Saddler and S. S. Auerbach, An Overview of National Toxicology Program's Toxicogenomic Applications: DrugMatrix and ToxFX, in Advances in Computational Toxicology: Methodologies and Applications in Regulatory Science, ed. H. Hong, Challenges and Advances in Computational Chemistry and Physics, Springer International Publishing, Cham, 2019. pp. 141–157, DOI:10.1007/978-3-030-16443-0_8.
- R. Yamada, D. Okada, J. Wang, T. Basak and S. Koyama, Interpretation of omics data analyses, J. Hum. Genet., 2021, 66(1), 93–102 CrossRef PubMed.
- O. Obrezanova, A. Martinsson, T. Whitehead, S. Mahmoud, A. Bender and F. Miljković, et al., Prediction of In Vivo Pharmacokinetic Parameters and Time–Exposure Curves in Rats Using Machine Learning from the Chemical Structure, Mol. Pharmaceutics, 2022, 19(5), 1488–1504 CrossRef CAS PubMed.
- J. D. Romano, Y. Hao, J. H. Moore and T. M. Penning, Automating Predictive Toxicology Using ComptoxAI, Chem. Res. Toxicol., 2022, 35(8), 1370 Search PubMed.
- Y. Hao, J. D. Romano and J. H. Moore, Knowledge graph aids comprehensive explanation of drug and chemical toxicity, CPT Pharmacometrics Syst. Pharmacol., 2023, 12(8), 1072–1079 Search PubMed.
- A. Dugourd, C. Kuppe, M. Sciacovelli, E. Gjerga, A. Gabor and K. B. Emdal, et al., Causal integration of multi-omics data with prior knowledge to generate mechanistic hypotheses, Mol. Syst. Biol., 2021, 17(1), e9730 CrossRef PubMed.
- P. Trairatphisan, T. M. de Souza, J. Kleinjans, D. Jennen and J. Saez-Rodriguez, Contextualization of causal regulatory networks from toxicogenomics data applied to drug-induced liver injury, Toxicol. Lett., 2021, 350, 40–51 Search PubMed.
- A. Liu, P. Trairatphisan, E. Gjerga, A. Didangelos, J. Barratt and J. Saez-Rodriguez, From expression footprints to causal pathways: contextualizing large signaling networks with CARNIVAL, npj Syst. Biol. Appl., 2019, 5(1), 1–10 CAS.
Footnote |
| † Indicates equal contribution. |
| This journal is © The Royal Society of Chemistry 2025 |