
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The selective oxidation of methanol to formaldehyde using novel iron molybdate catalysts prepared by supercritical antisolvent precipitation†
Jack R.
Pitchers
*a,
James
Carter
a,
Aziz
Genç
a,
Thomas J. A.
Slater
 a,
David J.
Morgan
a,
Alice
Oakley
b,
Bart D.
Vandegehuchte
c,
Stuart H.
Taylor
a and
Graham J.
Hutchings
*a
a,
David J.
Morgan
a,
Alice
Oakley
b,
Bart D.
Vandegehuchte
c,
Stuart H.
Taylor
a and
Graham J.
Hutchings
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Translational Research Hub, Maindy Road, Cardiff, UK. E-mail: pitchersjr@cardiff.ac.uk; hutch@cardiff.ac.uk
bSchool of Chemistry, Southampton University, Highfield Campus, Southampton, Hampshire, UK
cTotalEnergies OneTech Belgium, Zone Industrielle C, 7181 Feluy, Belgium
First published on 31st March 2025
Abstract
Iron molybdate catalysts have been extensively explored for the oxidation of methanol to formaldehyde. However, low surface area catalysts are typically formed, and iron-rich phases still exist from common preparation methods, leading to lower selectivity. The use of supercritical antisolvent precipitation to form novel precursors led to catalysts with improved productivity compared to alternative precipitation techniques. Using isoconversion studies, new structure–performance relationships have been uncovered. The novel iron molybdate catalysts provided an improved formaldehyde production of 42.5 mmolCH2O gcat−1 h−1 for the best performing catalyst, whilst specific productivity was used as a descriptor to probe intrinsic properties of the catalysts. Improved performance was achieved by increased agglomerate size and by phase purity, both controlled by the precursor structure. Both properties improve the supply of oxygen to the amorphous MoOx surface phase from the reducible crystalline phase.
Introduction
The synthesis of formaldehyde (CH2O) is an important process, with formaldehyde predominantly used for the production of resins in building materials.1 Among the best catalysts for formaldehyde production are iron molybdate catalysts, which usually exhibit higher yield at lower temperatures compared to silver-based catalysts.1 First developed in the 1930's by Adkins and Peterson, Mo-based catalysts were found to be selective to CH2O, whilst Fe-based catalysts were active in the conversion of MeOH.2 When Mo and Fe were combined, the catalyst was both active and selective. Since the initial discovery, much research has been oriented to understanding the influence of the different phases that are present.3–6 Fe2O3 catalysts combust MeOH, hypothesized to be due to the formation of a formate intermediate,3 in contrast to the methoxy intermediate formed on Mo oxide catalysts. Therefore, catalysts with excess Mo (MoO3–Fe2(MoO4)3) are typically pursued to ensure the absence of Fe-rich regions, as well as the stability against deactivation via sublimation of Mo under reaction conditions.7 The role of the various phases present in high-performance FeMoOx catalysts have previously been explored: an amorphous MoOx layer forms at the surface, covering the crystalline Fe2(MoO4)3 core.5 Fe2(MoO4)3 is understood to improve catalytic activity due to its high oxygen mobility, replenishing the surface with oxygen that has previously been removed during the redox cycle of MoOxvia the Mars–van Krevelen mechanism.8The performance of iron molybdate catalysts is still limited regarding low surface area and phase purity. Coprecipitation, a commonly used method, forms catalysts with low specific surface area,5 limiting activity as well as forming Fe-rich regions due to the precipitation mechanism occurring over a wide range of pH.9 Yeo and coworkers prepared FeMoOx catalysts via mechanochemistry,10 which achieved a higher specific surface area compared to analogous coprecipitated catalysts. However, Fe-rich regions formed in the pores due to the decomposition of oxalic acid in the preparation. Pudge and coworkers found that CH2O selectivity was improved by increased phase homogeneity, which in turn was influenced by varying the acid used in sol–gel synthesis,11 isolating Mo and Fe more effectively to form fewer Fe-rich areas.
One method to improve surface area and phase purity of catalyst precursors is supercritical antisolvent (SAS) precipitation. SAS precipitation exploits the properties of supercritical CO2 (scCO2) to reduce upon contact the solvation strength of a solvent.12 In this case, Fe(OAc)2 and MoO2(AcAc)2 dissolved in the solvent form binary metal precipitates as catalyst precursors when introduced to a vessel containing scCO2.13 The density of scCO2 can be carefully controlled by varying pressure, molar content of CO2 and temperature,12,14–16 therefore influencing the antisolvent properties and, consequently, of the precipitate. The solvent itself also determines the precipitate properties, via its miscibility with scCO2,16 whilst precursor concentration and solution flow rate have been shown to influence particle size,17,18 and therefore catalytic performance. Miscibility is important due to precipitation occurring through mass transfer of scCO2 in the solvent droplet which eventually reduces surface tension between the scCO2 and solvent to form a single phase,19 although water as a cosolvent presents low miscibility and can achieve access to specific phases.20 SAS precipitation typically forms amorphous, homogeneous precipitates21 due to the rapid precipitation in the process. The control in phase purity from the homogeneity is of particular interest with FeMoOx catalysts to theoretically reduce the presence of Fe rich areas. Aerogels are typically formed too, as supercritical drying extracts the solvent from the precipitate while preserving the morphology, increasing the surface area.
Whilst the influence of Mo/Fe ratio on methanol oxidation is well established, more in-depth structure–performance relationships are of limited understanding. Herein, we explore the formation of novel iron molybdate catalysts using precursors formed via SAS precipitation, exhibiting how the technique can influence Mo/Fe ratio, particle size, carbonate content and phase purity. The novel preparation technique was undertaken to not only improve CH2O productivity, but uncover new structure–performance relationships. By varying multiple SAS precipitation conditions – pressure, metal precursor concentration, water cosolvent content and solution flow rate – the specific surface area of iron molybdate catalysts formed was enhanced in comparison to other methods in the literature, whilst the phase purity is hypothesised to be improved which is demonstrated for some catalysts in this work.
Experimental
Preparation of SAS precipitated iron molybdate catalysts
To prepare novel iron molybdate catalysts, MoO2(AcAc)2 and Fe(OAc)2 were dissolved in a methanol/water solvent mixture, with a molar ratio of 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, with the appropriate concentration of metal precursors according to Table 3. The solution was stirred for 16 hours.
:1, with the appropriate concentration of metal precursors according to Table 3. The solution was stirred for 16 hours.
To undertake SAS precipitation, apparatus for supercritical antisolvent precipitation was used (parts manufactured by Waters and assembled by Sci-Med); the configuration is shown in Fig. S1.a.† Clean solvent was pumped through the system via the HPLC pump to reach a steady state between the solvent and the supercritical CO2 (scCO2). Once steady state was reached, the metal solution was flowed into the precipitation vessel, via a coaxial nozzle (Fig. S1.b†). All SAS parameters were set according to Table 3. Once the metal solution had been pumped through the system, the samples were dried under supercritical conditions to remove any remaining solvent.
SAS precipitation parameters were selected to maximise the range of the conditions, whilst considering scientific principles. Water cosolvent content was selected over the range 1–20% to ensure precipitation occurred and to probe change in precipitate properties. Water content of 20% upper limit was selected to investigate if sub-supercritical conditions would form a precipitate. Metal precursor concentration was limited in the upper limit by solubility limits of MoO2(AcAc)2 and Fe(OAc)2 in MeOH/H2O and limited in the lower limit to ensure formation a substantial mass of catalyst. Solution flow rate was limited by the HPLC pump capacity, but a 5 mL min−1 to 10 mL min−1 range was relatively large compared to the absolute values used. Pressure was varied from 80 bar to 140 bar with the lower limit being close to the supercritical pressure of CO2, whilst the upper limit was used for safe operation.
Characterisation techniques
Microwave plasma atomic emission spectroscopy (MP-AES) was undertaken using an Agilent Technologies 4100 MP-AES spectrometer with Agilent MP expert software to measure the elemental composition. A calibration plot was obtained by diluting standard metal solutions with deionised water. Samples (20 mg) were dissolved in dilute aqua regia solution (100 mL).Scanning electron microscopy (SEM) imaging was undertaken to determine the SAS precipitates and catalysts morphology using a Tescan Maia3 field emission gun scanning electron microscope (FEG-SEM).
Aberration-corrected scanning transmission electron microscopy (AC-STEM) was performed using a probe-corrected Thermo Fisher Scientific Spectra 200 Cold-FEG operating at 200 kV. The instrument was equipped with a HAADF detector, and the imaging was done at a probe current of 120 pA and convergence angle of 30 mrad. Samples were dry dispersed onto 300 mesh copper grids coated with a holey carbon film. Energy-dispersive X-ray (EDX) mapping was performed using a Super-X G2 detector.
Fourier transformed infrared (FT-IR) spectroscopy data was collected using a Bruker FT-IR spectrometer, over the range 500–4000 cm−1.
Thermogravimetric analysis (TGA) was undertaken to measure the changes in mass loss on calcination. The SAS precipitates were loaded into the apparatus and air was flowed at 20 mL min−1 with the temperature ramped from ambient to 700 °C at a ramp rate of 10 °C min−1.
Surface area analysis was undertaken using Brunauer–Emmett–Teller (BET) theory using a Quantachrome Quadrasorb SI instrument. Degassing under vacuum for calcined samples was undertaken at 150 °C for 3 hours, whilst uncalcined samples were degassed at 80 °C for 4 hours. A 5-point N2 isotherm was measured with relative pressure, P/P0, in the range of 0.05 to 0.30.
X-ray photoelectron spectroscopy (XPS) was used to determine the surface Mo/Fe ratio. Spectra were collected using a Kratos Axis Ultra DLD system. A monochromatic Al Kα X-ray source was used, operating at 120 W (10 mA × 12 kV). A pass energy of 40 eV was used for high resolution scans with step sizes of 0.1 eV, whilst survey spectra were collected using a pass energy of 160 eV, with a step size of 1 eV. Spectra were acquired in the hybrid mode and slot aperture yielding an analysis area of approximately 300 × 700 μm2. Charging of the sample was minimised by a magnetically confined charge compensation system. Data was analysed using CasaXPS v2.3.26.22 Calibration was made to the of the lowest C 1s peak of adventitious carbon taken to be 284.8 eV, with secondary checks made on the suitability of the carbon.23 Quantification was performed using a Shirley background to account for electron scattering and modified Wagner sensitivity factors were used as supplied by the manufacturer.
Powder X-ray diffraction (XRD) analysis was undertaken to confirm crystalline phases present. Patterns were collected with the use of a Bruker AXS D2 phaser with Cu Kα X-ray source, with a wavelength of 1.54 Å. Patterns were collected over a 2θ range of 10–80°, a slit size of 0.2 mm, step size of 0.0126° and time per step of 0.5 seconds. International Centre for Diffraction Data (ICCD) standard Powder Diffraction files were used to identify catalyst phases.
Evaluation of catalytic performance
Catalytic performance tests for methanol oxidation to formaldehyde were undertaken in a fixed bed reactor. Isoconversion studies were conducted at 250 °C. The MeOH:O2:N2 ratio was 5:10:85 for all testing, with a total flow rate of 30 mL min−1. Catalyst mass was varied to achieve a conversion between 13.9 and 17%.
Calculation of conversion (Xrct) of methanol was as follows:
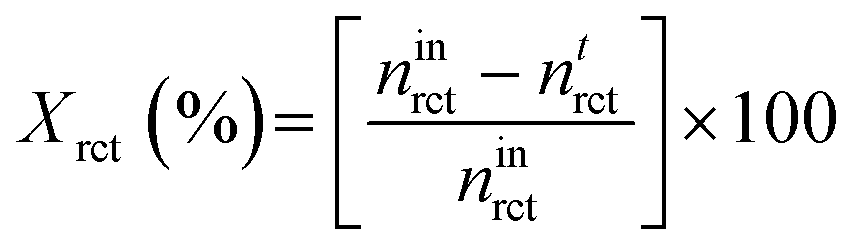
Selectivity (S) towards product i was as follows:
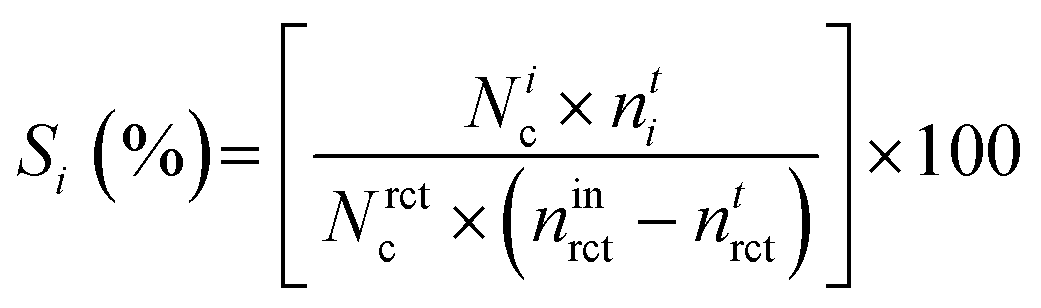
Carbon balance was calculated as follows:
| Carbon balance (%) = ∑Si |
Carbon balance was within error for all tests.
Conversion was standardised by calculating the mass-normalised activity and specific activity. Mass-normalised activity was calculated as follows:
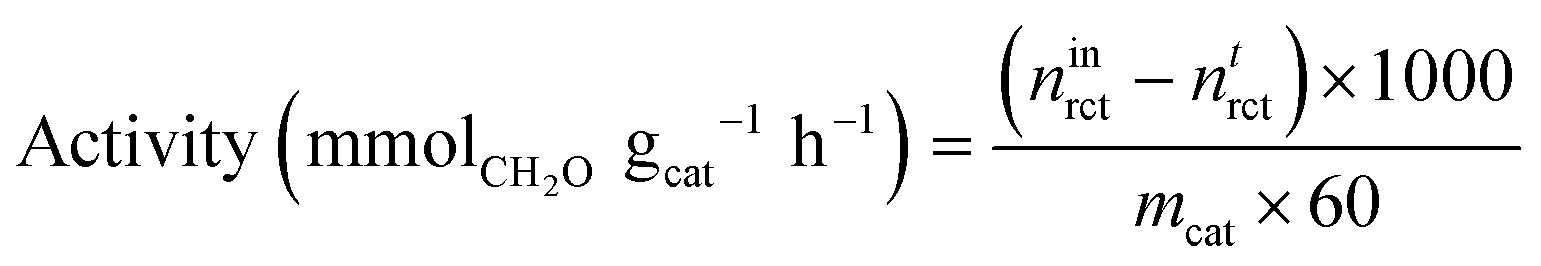
Specific activity was calculated as follows:

Mass-normalised productivity and specific productivity were calculated to standardise the yield. Mass-normalised productivity was calculated as follows:
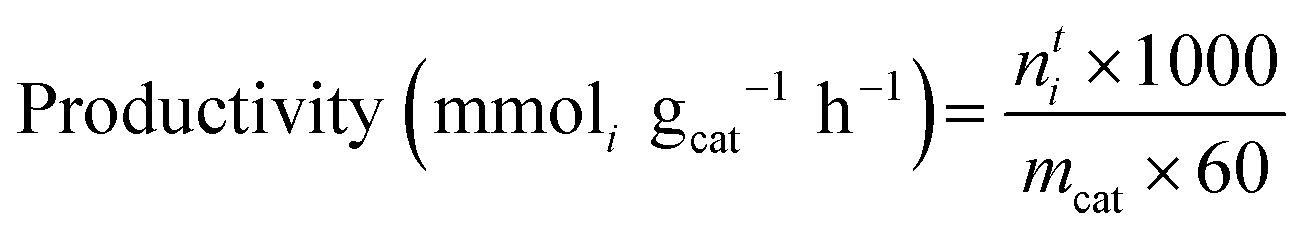
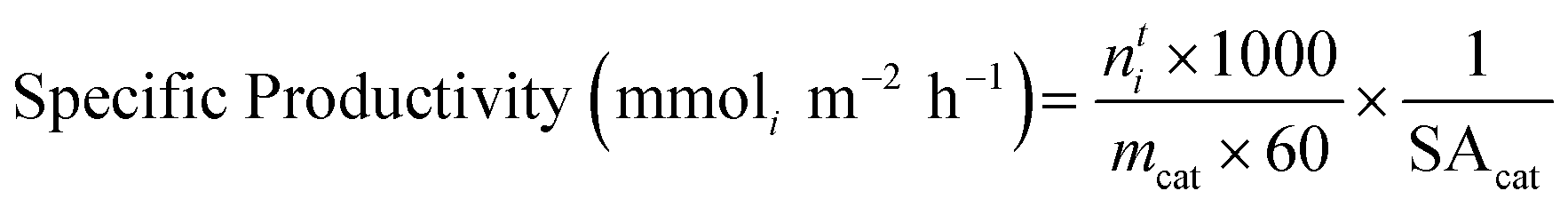
Results and discussion
Catalyst precursor structure
Catalyst precursors formed via SAS precipitation were amorphous (shown by Fig. S2†), typical of the precipitation mechanism. The range in pressure (80–160 bar), precursor concentration (5–10 g L−1), water cosolvent content (1–20%) and solution flow rate (5–10 mL min−1) were all determined by ensuring the system was sufficiently supercritical to undertake precipitation combined with the apparatus limitations. Fourier transform infrared (FT-IR) spectra of the catalyst precursors highlighted differences in chemical composition (Fig. 1). All precursors feature a large broad peak from 700–900 cm−1, typically assigned to the Mo–O vibration of MoO4 in Fe2(MoO4)3.24 Bands for FeMo_80P-10C-1W-5F (in which P, C, W and F represent pressure (bar), precursor concentration (g L−1), water cosolvent content (%) and solution flow rate (mL min−1) in the SAS precipitation of each sample, respectively – with further details in Table 3), FeMo_140P-5C-20W-10F and FeMo_110P-7.5C-10.5W-7.5F at 1280, 1350, 1527 and 1561 cm−1, were assigned to two separate carbonate environments, similar to rosasite minerals, representing the ν3 asymmetric CO32− stretch.25 The SAS precipitation mechanism in this study is therefore hypothesised to produce precipitates with a general structure of FeyMoz(CO3)a·xH2O. Carbonate is hypothesised to form when using H2O as a cosolvent for SAS precipitation due to the reaction between scCO2 and H2O under reaction conditions, forming carbonic acid which then decomposes to carbonate.21 The formation of carbonate in the precipitates can be controlled by varying the conditions. For example, samples FeMo_80P-10C-1W-5F, FeMo_140P-5C-20W-10F and FeMo_110P-7.5C-10.5W-7.5F all exhibit a carbonate content in the precipitates. FeMo_80P-10C-1W-5F has the lowest water cosolvent content of all samples and the largest carbonate content, leading to the understanding that, whilst water is required to form carbonates, the conditions required are to be near supercritical for the solvent–CO2 mixture. Higher water contents are known to shift the mixture critical point to higher pressure and temperature, which consequently may lead to less carbonate formation.26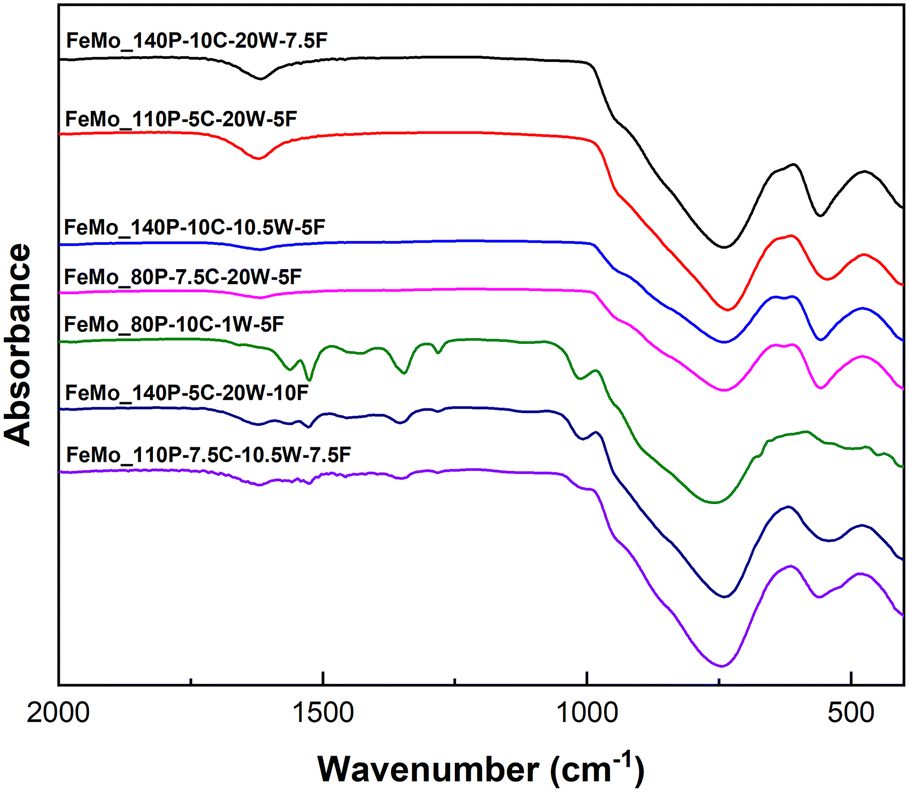 | ||
| Fig. 1 FT-IR spectra for SAS precipitated iron molybdate precursors. | ||
Particle size varied across all samples, as indicated in Table 1 and by SEM imaging of catalyst precursors in Fig. S3–S9† (with Fig. S10† showing particle size distribution). SEM imaging highlighted significantly different particle diameters, depending on the conditions used. This is a well-known phenomenon with SAS precipitation, where both the miscibility between the solvent and scCO2 and the scCO2 density and molar fraction exhibit a strong impact. Higher pressures and lower water contents will decrease particle size due to the shorter growth time during precipitation. This correlates with the trends seen in particle size in the current precipitates formed, with FeMo_110P-5C-20W-5F and FeMo_140P-10C-10.5W-5F having larger average particle sizes (Fig. S10†) of 0.172 and 0.226 μm, respectively, as well as FeMo_140P-5C-20W-10F exhibiting the smallest average particle size of 0.093 μm, due to a high pressure of 140 bar, and a low precursor concentration of 5 g L−1.This allows particles to grow more before precipitation occurs at supersaturation, as the concentration is further from the solubility limits.27
| Sample | Mass loss on calcination (%) | Average uncalcined particle size (μm) | Mo/Fe ratio of FeMoOx catalysts | Semi-quantitative MoO3 content of FeMoOx catalysts (%) | Specific surface area (m2 g−1) | ||
|---|---|---|---|---|---|---|---|
| Bulk (MP-AES) | Surface (XPS) | Uncalcined | Calcined | ||||
| FeMo_140P-10C-20W-7.5F | 0.68 | 0.143 | 1.58 | 2.6 | 8 | 11.2 | 7.1 |
| FeMo_110P-5C-20W-5F | 0.34 | 0.162 | 1.37 | 2.3 | 0 | 15.1 | 9.9 |
| FeMo_140P-10C-10.5W-5F | 1.30 | 0.216 | 1.66 | 2.6 | 7 | 13.5 | 7.9 |
| FeMo_80P-7.5C-20W-5F | 8.18 | 0.170 | 1.55 | 2.3 | 9 | 5.6 | 8.4 |
| FeMo_80P-10C-1W-5F | 11.15 | 0.104 | 1.61 | 2.6 | 14 | 14.9 | 9.9 |
| FeMo_140P-5C-20W-10F | 7.00 | 0.093 | 1.50 | 2.4 | 5 | 14.8 | 10.4 |
| FeMo_110P-7.5C-10.5W-7.5F | 7.82 | 0.104 | 1.60 | 2.3 | 7 | 20.5 | 11.2 |
Thermogravimetric analysis (TGA) for all samples (Fig. S11†) showed an initial mass loss at ∼100 °C, which was attributed to the removal of solvent from the precipitates. A difference in mass loss was then measured across the catalysts. Precipitates with carbonate present, as observed in FT-IR (Fig. 1), exhibited a mass loss at ∼370 °C. The samples containing carbonates also exhibited a mass gain at ∼320 °C, suggesting oxidation, likely to be the conversion of Fe2+ to Fe3+, as Mo6+ is present in both the metal precursor and the final catalyst phases. Samples that did not present any carbonate content showed no mass loss or gain after the initial removal of solvent, and therefore during the SAS precipitation for FeMo_140P-10C-20W-7.5F, FeMo_110P-5C-20W-5F and FeMo_140P-10C-10.5W-5F are hypothesised to undergo oxidation of Fe2+, whilst potentially forming amorphous iron molybdate precursors (as no mass change was observed). The precipitates with no carbonate content may contain amorphous FeMoOx, as no defining bands for carbonate were identified, no mass gain and limited mass loss on calcination, and had similar FT-IR spectra when compared to the calcined catalysts.28 However, crystallisation must occur during calcination as the catalyst develops a crystalline structure (Fig. 2).
 | ||
| Fig. 2 XRD patterns for iron molybdate catalysts formed from SAS precursors. | ||
Catalyst structure
Catalyst precursors were calcined in air at 500 °C for 2 hours with a ramp rate of 5 °C min−1, based on the TGA data for the SAS precipitates. The following characterisation is focused on catalyst structure after calcination.The bulk Mo/Fe ratio (Table 1) show that the nominal value of 1.5 was almost achieved for all samples, with the lowest bulk Fe/Mo ratio of 1.37 achieved for FeMo_110P-5C-20W-5F and the highest Fe/Mo ratio of 1.66 for FeMo_140P-10C-10.5W-5F. The slight variation in ratio is due to the difference in relative solubility limits in the solvent between the Mo and Fe precursors and how the SAS conditions influence the precipitation mechanism.
XRD patterns (Fig. 2) showed the presence of Fe2(MoO4)3 (2θ = 15.1° and 23.2° – ISCD 80449) and MoO3 (2θ = 27.3° – ISCD 152313) for all samples. The presence of crystalline MoO3 suggests also the presence of Fe-rich regions due to the intended Mo/Fe ratio of 1.5 being the stoichiometric ratio of Fe2(MoO4)3. However, Fe-rich crystalline phases, Fe2O3, were not detected (2θ = 33.1° and 35.6° – ICSD 15840), suggesting either amorphous or nanocrystalline Fe-rich regions.
The MoO3 content was measured semi-quantitatively using relative diffraction intensity ratios and was highest for FeMo_80P-10C-1W-5F despite having an Mo/Fe ratio of 1.61. FeMo_80P-10C-1W-5F exhibits a much higher MoO3 content than FeMo_140P-10C-10.5W-5F, despite the similar Mo/Fe ratio, which suggests the preparation method can influence homogeneity. FeMo_80P-10C-1W-5F led to a larger mass loss than FeMo_140P-10C-10.5W-5F, with FT-IR showing FeMo_80P-10C-1W-5F to have a high carbonate content; a high carbonate content is hypothesized to decrease phase purity due to the two distinct metal environments which are present.
AC-STEM (Fig. 3) of FeMo_80P-10C-1W-5F and FeMo_110P-5C-20W-7.5F provided clear images showing the layered crystalline structure of the samples. STEM-EDX data (Fig. S12 and S13†) reveal the presence of Mo-rich surface layers with a thickness of ∼1.5 and 2 nm, respectively, corroborating well with the XPS measurements (Table 2), when compared to the bulk ratio (Table 2) of the catalyst, measured by MP-AES. It is observed that the amorphous layers in both samples increase in thickness during imaging, generated by e-beam induced amorphization during the AC-STEM analysis (Fig. S14†). Mo surface enrichment has been previously established and is important for improved selectivity,29 by limiting the presence of Fe at the surface, known to combust methanol to CO and CO2. The XPS spectra of catalysts (Fig. S15†) showed the Mo6+ oxidation state present at the surface, with binding energies for Mo3d/2 and Mo5d/2 analogous to MoO3.
 | ||
| Fig. 3 a) HAADF-STEM image of FeMo_80P-10C-1W-5F, showing crystalline structure. b) FFT obtained from imaging of FeMo_80P-10C-1W-5F, with insert showing the nanoparticle structure of orthorhombic Fe2(MoO3)4 phase. c) HAADF-STEM image of FeMo_110P-5C-20W-7.5F, showing crystalline structure. d) FFT obtained from imaging of FeMo_110P-5C-20W-7.5F, with insert showing the nanoparticle structure of orthorhombic Fe2(MoO3)4 phase. | ||
| Sample | WHSV (gMeOH gcat−1 h−1) | Conversion (%) | Selectivity (%) | Productivity (mmolCH2O gcat−1 h−1) | Specific productivity (mmolCH2O m−2 h−1) | ||
|---|---|---|---|---|---|---|---|
| CH2O | COx | DMM | |||||
| FeMo_140P-10C-20W-7.5F | 7.4 | 14.8 | 74.6 | 2.3 | 4.1 | 25.4 | 3.6 |
| FeMo_110P-5C-20W-5F | 10.7 | 15.8 | 80.5 | 4.8 | 4.0 | 42.5 | 4.3 |
| FeMo_140P-10C-10.5W-5F | 7.9 | 17.0 | 80.7 | 1.7 | 3.9 | 33.7 | 4.3 |
| FeMo_80P-7.5C-20W-5F | 8.4 | 14.0 | 72.3 | 2.5 | 4.9 | 26.6 | 3.2 |
| FeMo_80P-10C-1W-5F | 6.7 | 15.4 | 79.1 | 5.9 | 4.4 | 25.6 | 2.6 |
| FeMo_140P-5C-20W-10F | 9.8 | 13.9 | 78.0 | 3.7 | 5.5 | 33.3 | 3.2 |
| FeMo_110P-7.5C-10.5W-7.5F | 9.4 | 15.7 | 79.3 | 5.2 | 4.3 | 36.6 | 3.3 |
| Sample name | Pressure (bar) | Precursor concentration (g L−1) | Water content (%) | Solution flow rate (mL min−1) |
|---|---|---|---|---|
| FeMo_140P-10C-20W-7.5F | 140 | 10 | 20 | 7.5 |
| FeMo_110P-5C-20W-5F | 110 | 5 | 20 | 5 |
| FeMo_140P-10C-10.5W-5F | 140 | 10 | 10.5 | 5 |
| FeMo_80P-7.5C-20W-5F | 80 | 7.5 | 20 | 5 |
| FeMo_80P-10C-1W-5F | 80 | 10 | 1 | 5 |
| FeMo_140P-5C-20W-10F | 140 | 5 | 20 | 10 |
| FeMo_110P-7.5C-10.5W-7.5F | 110 | 7.5 | 10.5 | 7.5 |
Fig. S16–S22† show SEM images which compare precursor morphology with the catalyst morphology. The catalysts retain a relatively similar structure after calcination, which was confirmed by the correlation between uncalcined and calcined specific surface area (Fig. S23†). Some change in the spherical morphology is shown, likely due to the change in crystallinity on calcination. However, it can be concluded that the precursor morphology was retained, with similar agglomerate sizes before and after calcination.
Isoconversion studies and structure–performance relationships
Isoconversion methanol oxidation tests were undertaken to compare performance between the different iron molybdate catalysts (Table 2). A conversion range of 13.9–17.0% was achieved by varying the weight hourly space velocity (WHSV). Productivity was improved for FeMo_110P-5C-20W-5F and FeMo_140P-10C-10.5W-5F when compared to other precipitation methods (Table S1†). An increase in productivity correlated with an increase in specific surface area, as shown in Fig. 4. However, productivity does not just rely on surface area. For example, FeMo_110P-5C-20W-5F has a lower specific surface area than FeMo_140P-5C-20W-10F and FeMo_110P-7.5C-10.5W-7.5F, but a higher productivity, suggesting that more than just the surface area is important for the productivity. | ||
| Fig. 4 The relationship between CH2O productivity and specific surface area. | ||
Structure–performance relationships were constructed by considering the specific productivity as the main performance indicator. As shown in Table 2, there is a significant difference in specific productivity between all catalysts despite the comparable conversion.
A common thread in research on FeMoOx catalysts is that the Mo/Fe ratio is considered as the most impactful parameter, with clear differences in performance from an Mo/Fe ratio from 1.5 to 3, with 2.2 often explored5 as this is the ratio commonly found for industrial catalysts.30,31 In the present range of catalysts, the Mo/Fe ratio was seemingly not the most determinant factor in this study (Fig. 5). The Mo/Fe ratio is therefore not the main factor in defining performance within this set of catalysts.
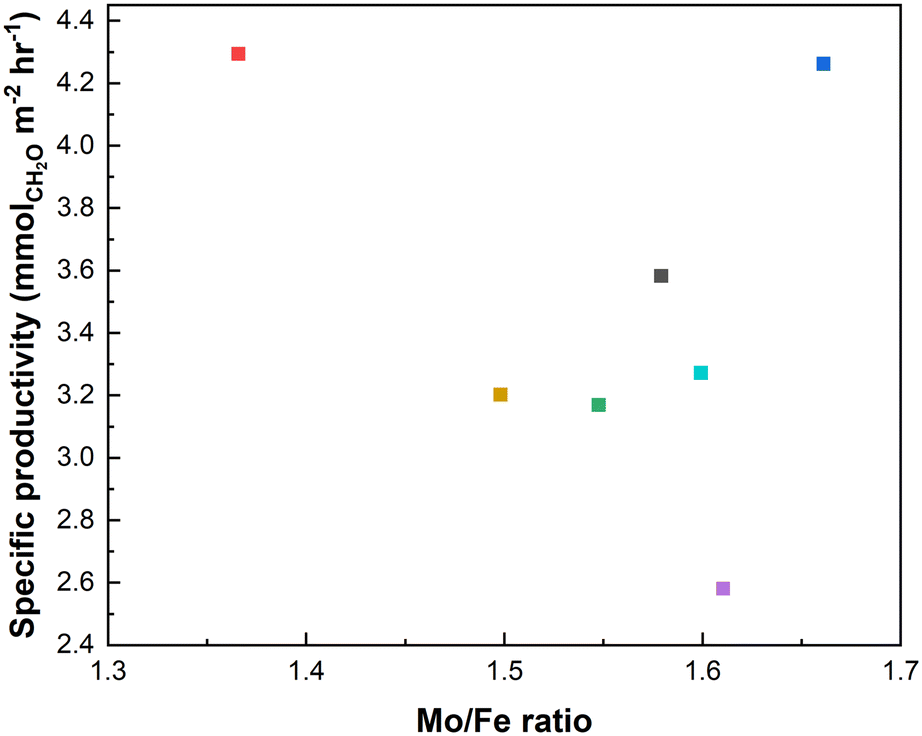 | ||
| Fig. 5 Specific productivity of CH2O in relation to Mo/Fe ratio. | ||
An increase in particle size loosely correlates to an increase in specific productivity (Fig. 6). The Mo layer thickness of FeMo_80P-10C-1W-5F and FeMo_110P-5C-20W-7.5F were compared, due to exhibiting the lowest and the highest CH2O productivity, respectively. HAADF-STEM images in Fig. 3 confirmed the presence of an amorphous overlayer, typically seen for FeMoOx catalysts,5 although further consideration of beam induced amorphization is required. Fig. S12 and S13† show the EDX analyses of FeMo_80P-10C-1W-5F and FeMo_110P-5C-20W-7.5F, with a Mo-rich layer thickness of ∼2 and ∼1.5 nm, respectively.
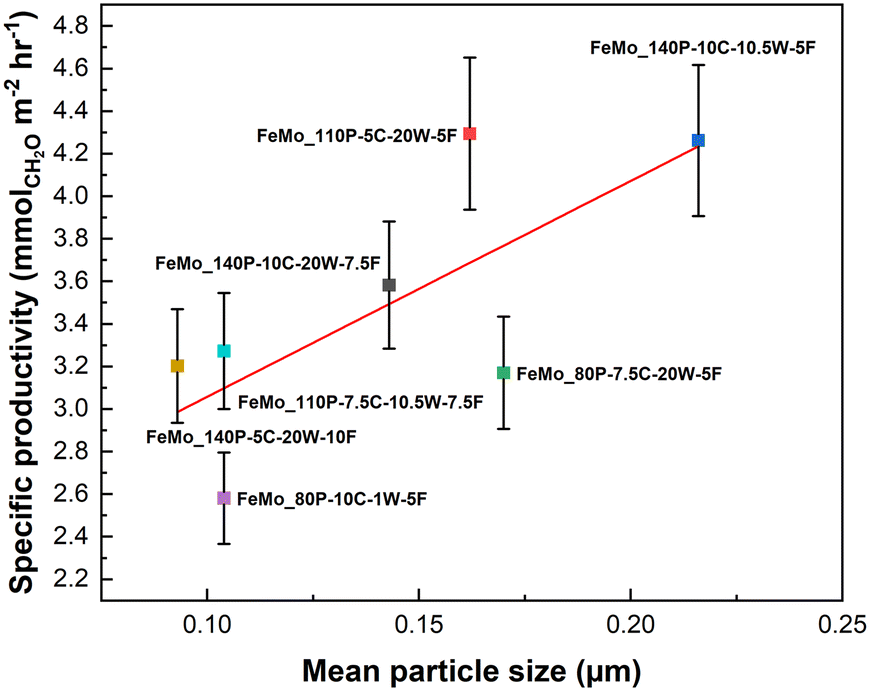 | ||
| Fig. 6 The relationship between mean particle size of the precipitates and specific CH2O productivity. Errors based on repeat testing of FeMo_140P-5C-20W-10F. | ||
The increase in particle size is hypothesised to improve diffusion from the crystalline Fe2(MoO4)3, which has a high oxygen mobility, to the MoOx surface layer, through the increase of contact area between MoOx and Fe2(MoO4)3. The hypothesis agrees with operando and transient spectroscopy of FeMoOx catalysts, with Fe2(MoO4)3 being reduced during the reaction.32 Oxygen diffusion leads to more rapid reoxidisation of the reduced surface of Mo4+ to Mo6+, the latter being the selective cation for the Mars–van Krevelen mechanism within FeMoOx catalysts.33 The increase in specific productivity with precipitate particle size also compliments Bowker's hypothesis in requiring a large Fe2(MoO4)3 content to ensure a larger oxygen reservoir.33 However, the full use of this oxygen reservoir is unlikely under the isoconversion conditions due to the excess of oxygen in the feed and the low conversion limiting the consumption of lattice oxygen. Further studies to understand the interface between Fe2(MoO4)3 and amorphous MoOx surface layer is key to understanding the importance of each phase.
Whilst the scatter of the data for the catalysts specific productivity is evident for the relationship with mean particle size, this is due to the variation of other influencing properties across the range of catalysts. For example, FeMo_80P-10C-1W-5F has a similar mean particle size to FeMo_140P-5C-20W-10F, but a much higher mass loss on calcination, and therefore the performance is much lower. Mass loss was an influencing property on performance and is discussed in the following section.
To control mean particle size, the time until dissolution must be controlled through selection of SAS precipitation conditions. Dissolution of precipitates occurs through the expansion of the liquid solvent using supercritical CO2, lowering the solubility limit.34 SAS conditions influence the ability of the supercritical CO2 to dissolute the precipitates. A larger particle size is a result of a slower dissolution rate and droplet size, as the solvent droplet grows in the vessel until dissolution occurs.
From the experimental approach, a lower solution flow rate (5 mL min−1) leads to a larger mean particle size. This is likely related to the breakup into smaller droplets for higher flow rates due to a larger capillary number, a measure between inertia forces and surface tension.35 A smaller droplet will form a smaller particle, and this is in agreement with trends observed.
The outlier of the trend for low solution flow rate resulting in a large mean particle size is FeMo_80P-10C-1W-5F. This may be due to the significant difference in water content, compared to the other catalysts. Water has a low miscibility with supercritical CO2 and leads to the formation of a two state system, with surface tension of the liquid solvent being maintained,36 and therefore the mixture critical point (MCP) is significantly shifted to elevated values when a higher water content is used. When there is a low water content, such as with FeMo_80P-10C-1W-5F, the MCP is relatively close to the synthesis parameters compared to higher water contents, and droplet diameter is limited in growth before precipitation, as shown in Fig. S25.† Therefore, a balance between all parameters must be considered to control the dissolution rate to achieve a specific mean particle size.
An increase in specific productivity also correlated surprisingly well with a decrease in mass loss on calcination (Fig. 7). This could be due to multiple reasons relating to the precipitate composition and the change in properties on calcination. Firstly, the lower mass loss in precipitates related to the carbonate content (a higher carbonate content led to a higher mass loss). The materials with high carbonate content (FeMo_80P-10C-1W-5F, FeMo_140P-5C-20W-10F and FeMo_110P-7.5C-10.5W-7.5F) may show more segregation between Mo and Fe due to the distinct carbonate regions (Fig. 1). The distinct regions would reduce phase purity, which has been shown to decrease performance by decreased oxygen diffusion to the surface from the crystalline core as this would increase the MoO3 present. MoO3 has been shown to exhibit a lower oxygen mobility in comparison to Fe2(MoO4)3 (ref. 37) and therefore would hinder the oxygen diffusion if present when compared to a pure Fe2(MoO4)3 phase. A hindered oxygen diffusion would lead to a lower presence of Mo6+, formed through reoxidation, and hence a lower productivity.
 | ||
| Fig. 7 The relationship between mass loss of the SAS precipitates and specific CH2O productivity. Errors based on repeat testing of FeMo_140P-5C-20W-10F. | ||
To control mass loss through controlling carbonate content of SAS precipitates, the conditions of the synthesis must be supercritical or close to supercritical. This is due to the need for the conditions to form a reaction between H2O and supercritical CO2. In general, precipitates formed with high pressures (140 bar) have a lower mass loss, although water content is relatively high when compared to the largest mass loss of FeMo_80P-10C-1W-5F. This is important, as water does need to be present to form carbonates, but if the water content is too high it may lead to conditions which are not sufficiently supercritical to allow the reaction between H2O and CO2, despite the elevated pressures. Le Chatelier's principle may also lead to effects limiting carbonate formation. With the reaction between H2O and CO2 being H2O + CO2 ⇌ H2CO3 ⇌ 2H+ + CO32−,21 the dissociation of carbonic acid (H2CO3) may be limited at higher pressures. However, further exploration regarding the effects of water content and carbonate content are limited in the literature and require further examination.
As shown in Fig. 6 and 7, FeMo_110P-5C-20W-5F and FeMO_140P-10C-10.5W-5F exhibit both a larger mean precipitate particle size and lower extent of mass loss, both of which have been found to be beneficial properties when forming FeMoOx catalysts from SAS precipitates. The lower productivity of FeMo_80P-10C-1W-5F can be assigned to the smaller precipitate particle size and higher extent of mass loss. FeMo_140P-10C-20W-7.5F highlights the requirement of both properties to achieve a high CH2O productivity, as the sample exhibits a similar extent of mass loss to FeMo_110P-5C-20W-5F and FeMo_140P-10C-10.5W-5F, yet has a lower CH2O productivity, due to the small particle size of the precursors.
The extent of mass loss and particle size relate to the importance of Fe2(MoO4)3 and its role with supplying oxygen to the surface MoOx layer. However, mass loss, which relates to phase purity determined by carbonate content, has a stronger correlation to specific productivity. The presence of Fe in the mixed metal phase provides increased oxygen mobility,38 which leads to a greater supply of oxygen to the surface, and therefore maximising phase purity of Fe2(MoO4)3, whilst ensuring Mo coverage at the surface to control selectivity,29 is of upmost importance. Controlling crystalline Mo rich phases, which have a lower oxygen mobility than the mixed metal phase,37 to provide high oxygen mobility but sufficient surface coverage should be explored in future studies.
Conclusions
Novel iron molybdate catalyst precursors have been formed using SAS precipitation. Once calcined, the precursors formed FeMoOx catalysts which exhibited varying physico-chemical properties, including Mo/Fe ratio, particle size and phase purity. Isoconversion studies have unveiled phase purity and particle size to be of importance and both relate to the role of Fe2(MoO4)3 to form active catalysts. The spherical morphology of SAS precipitates allowed probing the dependence of performance on particle size, with an increase in precipitate particle size leading to increased productivity, whilst the change in SAS precipitate chemical composition influenced the variation of phase purity.An increase in specific productivity with particle size contrasts with a decrease in mass-normalised productivity due to the inverse relationship between particle size and specific surface area. Therefore, future work should focus on improving porosity to increase both the specific surface area of the catalysts as well as the surface area of the crystalline Fe2(MoO4)3 core.
A decrease in segregation of Fe and Mo is hypothesised to increase specific CH2O productivity. The use of TGA to measure mass loss on calcination showed that the extent of mass loss is proportional to the carbonate content, and correlated to a lower CH2O productivity. An increased carbonate content is hypothesised to stimulate phase segregation due to the presence of two distinct carbonate environments. The SAS method has been proven to be versatile in reducing carbonate content in catalyst precursors to a minimum and therefore maximising phase purity. The technique is therefore useful in producing high performance methanol oxidation catalysts.
Data availability
All data is available in the paper and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was carried out as part of the Consortium on Metal Nanocatalysis funded by TotalEnergies. We would like to thank the European Regional Development Fund, The Welsh European Funding Office and The Wolfson Foundation for part-funding the CCI Electron Microscopy Facility.References
- H. I. Mahdi, N. N. Ramlee, D. H. da S. Santos, D. A. Giannakoudakis, L. H. de Oliveira, R. Selvasembian, N. I. W. Azelee, A. Bazargan and L. Meili, Mol. Catal., 2023, 537, 112944 CrossRef CAS.
- H. Adkins and W. R. Peterson, J. Am. Chem. Soc., 1931, 53, 1512–1520 CrossRef CAS.
- M. P. House, A. F. Carley, R. Echeverria-Valda and M. Bowker, J. Phys. Chem. C, 2008, 112, 4333–4341 CrossRef CAS.
- M. R. Sun-Kou, S. Mendioroz, J. L. G. Fierro, J. M. Palacios and A. Guerrero-Ruiz, J. Mater. Sci., 1995, 30, 496–503 CrossRef CAS.
- E. Söderhjelm, M. P. House, N. Cruise, J. Holmberg, M. Bowker, J. O. Bovin and A. Andersson, Top. Catal., 2008, 50, 145–155 CrossRef.
- A. M. Beale, S. D. M. Jacques, E. Sacaliuc-Parvalescu, M. G. O'Brien, P. Barnes and B. M. Weckhuysen, Appl. Catal., A, 2009, 363, 143–152 CrossRef CAS.
- K. V. Raun, L. F. Lundegaard, J. Chevallier, P. Beato, C. C. Appel, K. Nielsen, M. Thorhauge, A. D. Jensen and M. Høj, Catal. Sci. Technol., 2018, 8, 4626–4637 RSC.
- K. Routray, W. Zhou, C. J. Kiely, W. Grünert and I. E. Wachs, J. Catal., 2010, 275, 84–98 CrossRef CAS.
- A. P. V. Soares, M. F. Portela and A. Kiennemann, Catal. Rev.: Sci. Eng., 2004, 47, 125–174 CrossRef.
- B. R. Yeo, G. J. F. Pudge, K. G. Bugler, A. V. Rushby, S. Kondrat, J. Bartley, S. Golunski, S. H. Taylor, E. Gibson, P. P. Wells, C. Brookes, M. Bowker and G. J. Hutchings, Surf. Sci., 2016, 648, 163–169 CrossRef CAS.
- G. J. F. Pudge, G. J. Hutchings, S. A. Kondrat, K. Morrison, E. F. Perkins, A. V. Rushby and J. K. Bartley, Catal. Sci. Technol., 2022, 12, 4552–4560 RSC.
- E. Reverchon, E. Torino, S. Dowy, A. Braeuer and A. Leipertz, Chem. Eng. J., 2010, 156, 446–458 CrossRef CAS.
- P. J. Smith, S. A. Kondrat, J. H. Carter, P. A. Chater, J. K. Bartley, S. H. Taylor, M. S. Spencer and G. J. Hutchings, ChemCatChem, 2017, 9, 1621–1631 CrossRef CAS PubMed.
- E. Reverchon, I. De Marco and E. Torino, J. Supercrit. Fluids, 2007, 43, 126–138 CrossRef CAS.
- E. Alonso, I. Montequi, S. Lucas and M. J. Cocero, J. Supercrit. Fluids, 2007, 39, 453–461 CrossRef CAS.
- V. Prosapio, E. Reverchon and I. De Marco, Chem. Eng. Trans., 2015, 763–768 Search PubMed.
- P. Franco, M. Martino, V. Palma, A. Scarpellini and I. De Marco, Int. J. Hydrogen Energy, 2018, 43, 19965–19975 CrossRef CAS.
- M. C. Iannaco, S. Mottola, V. Vaiano, G. Iervolino and I. De Marco, J. CO2 Util., 2024, 85, 102878 CrossRef CAS.
- E. Reverchon and I. De Marco, Chem. Eng. J., 2011, 169, 358–370 CrossRef CAS.
- R. P. Marin, S. Ishikawa, H. Bahruji, G. Shaw, S. A. Kondrat, P. J. Miedziak, D. J. Morgan, S. H. Taylor, J. K. Bartley, J. K. Edwards, M. Bowker, W. Ueda and G. J. Hutchings, Appl. Catal., A, 2015, 504, 62–73 CrossRef CAS.
- Z.-R. Tang, S. A. Kondrat, C. Dickinson, J. K. Bartley, A. F. Carley, S. H. Taylor, T. E. Davies, M. Allix, M. J. Rosseinsky, J. B. Claridge, Z. Xu, S. Romani, M. J. Crudace and G. J. Hutchings, Catal. Sci. Technol., 2011, 1, 740–746 RSC.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- D. J. Morgan, Surf. Interface Anal., 2024, 1–8 Search PubMed.
- T. Hwan Kim, B. Ramachandra, J. Sik Choi, M. Saidutta, K. Yeon Choo, S.-D. Song and Y.-W. Rhee, Catal. Lett., 2004, 98, 161–165 Search PubMed.
- R. L. Frost, D. L. Wain, W. N. Martens and B. J. Reddy, Spectrochim. Acta, Part A, 2007, 66, 1068–1074 CrossRef PubMed.
- R. P. Marin, S. A. Kondrat, R. K. Pinnell, T. E. Davies, S. Golunski, J. K. Bartley, G. J. Hutchings and S. H. Taylor, Appl. Catal., B, 2013, 140–141, 671–679 CAS.
- M. Rossmann, A. Braeuer, S. Dowy, T. G. Gallinger, A. Leipertz and E. Schluecker, J. Supercrit. Fluids, 2012, 66, 350–358 CAS.
- Y. V. Kalashnykov, J. Saudi Chem. Soc., 2018, 22, 372–379 Search PubMed.
- M. P. House, A. E. Mervyn, S. Ae and M. Bowker, Catal. Lett., 2008, 122, 210–213 CAS.
- M. Bowker, R. Holroyd, A. Elliott, P. Morrall, A. Alouche, C. Entwistle and A. Toerncrona, Catal. Lett., 2002, 83, 165–176 CAS.
- A. Andersson, M. Hernelind and O. Augustsson, Catal. Today, 2006, 112, 40–44 CAS.
- L. Schumacher, M. Radtke, J. Welzenbach and C. Hess, Commun. Chem., 2023, 6, 1–9 Search PubMed.
- M. Bowker, Top. Catal., 2015, 58, 606–612 CAS.
- A. Martín and M. J. Cocero, Adv. Drug Delivery Rev., 2008, 60, 339–350 Search PubMed.
- H. Kröber and U. Teipel, J. Supercrit. Fluids, 2002, 22, 229–235 Search PubMed.
- N. S. Nesterov, V. P. Pakharukova and O. N. Martyanov, J. Supercrit. Fluids, 2017, 130, 133–139 CrossRef CAS.
- W. Kuang, Y. Fan and Y. Chen, Langmuir, 2000, 16, 5205–5208 CrossRef CAS.
- M. Bowker, A. E. Richard, H. Ae, M. House, A. R. Bracey, A. E. Chanut Bamroongwongdee, A. E. Mervyn, S. Ae and A. Carley, Top. Catal., 2008, 48, 158–165 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00211g |
| This journal is © The Royal Society of Chemistry 2025 |