
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N,N,N-Tridentate ligand promoted cobalt-catalyzed direct carbonylation of chloroacetonitrile to 2-cyano substituted acetates and amides†
Chao
Xu
ab,
Zhi-Peng
Bao
ac,
Le-Cheng
Wang
ac and
Xiao-Feng
Wu
 *abc
*abc
aDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 116023, Dalian, Liaoning, China. E-mail: xwu2020@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, China
cLeibniz-Institut für Katalyse e. V., Albert-Einstein-Straβe 29a, 18059 Rostock, Germany
First published on 10th March 2025
Abstract
2-Cyano-N-acetamides and 2-cyanoacetates are of great importance in the pharmaceutical industry, fueling the search for novel synthesis approaches. Transition-metal-catalyzed carbonylation, especially with cobalt, has potential but also suffers limitations, especially in reactions involving alkyl halides. Herein, a mild cobalt-catalyzed direct aminocarbonylation and alkoxycarbonylation of chloroacetonitrile promoted by an N,N,N-tridentate ligand was established. The targeted 2-cyano substituted acetates and amides are obtained in good to excellent yields. The preparation process is scalable and the compounds can be further transformed into bioactive molecules.
Introduction
2-Cyano-N-acetamides and 2-cyanoacetates have long occupied a pivotal position within the pharmaceutical industry.1 Take teriflunomide as an example, it serves as a pyrimidine synthesis inhibitor, boasting anti-inflammatory and immunomodulatory properties. This compound plays a crucial role in treating patients suffering from relapsing–remitting multiple sclerosis.1a Tyrphostin AG 494, on the other hand, represents a potent and selective EGFR tyrosine kinase inhibitor.1b Among 2-cyanoacetates, enbucrilate has found its application as a surgical tissue adhesive.1c Additionally, octocrylene functions as a sunscreen agent in sun protection products, effectively absorbing UV rays (Fig. 1a).1d Considering the significance of these compounds, the continuous exploration of novel synthetic methods has always been the pursuit of chemists. | ||
| Fig. 1 Background and synopsis of current work. | ||
Transition-metal-catalyzed carbonylation reactions occupy a forefront position in the field of the synthesis of carbonyl-containing compounds in both industry and academia.2 Alkoxycarbonylation and aminocarbonylation of organic halogens have been well-studied with the participation of noble metals, especially palladium-catalyzed reactions (Fig. 1b-1).3 In addition, although palladium-catalyzed systems are usually efficient, the requirement for expensive palladium catalysts and phosphine ligands is suboptimal. In 1938, the cobalt-catalyzed Fischer–Tropsch reaction has attracted extensive attention and is still widely used in industry to produce aldehydes from olefins.4 In 1960, the cobalt-catalyzed carbonylation of methanol was first commercialized by BASF.5 Undoubtedly, cobalt-catalyzed carbonylation has great potential, and its more cost-effective nature endows it with a greater possibility of industrialization. Although the direct coupling of cobalt-catalyzed aryl halides and alkenyl halides with nucleophiles has been extensively reported (Fig. 1b-2),6 even for palladium-catalyzed carbonylation, the relevant reactions involving alkyl halides or pseudohalides are still less investigated,7 especially aminocarbonylation (Fig. 1b-3).8 Based on the achievements reported up to now, two factors might be impeding the development of cobalt-catalyzed carbonylation chemistry. Firstly, carbon monoxide has a strong inclination to coordinate closely with cobalt metal, thereby forming stable cobalt carbonyl complexes. Secondly, the catalytic activity of cobalt shows little responsiveness to the added ligands.9
Recently, the work on directing-group-assisted cobalt-catalyzed intramolecular aminocarbonylation has been reported (Fig. 1b-4).10 Douglas and co-workers pioneered the use of 8-aminoquinoline11 as a directing group and conducted a detailed study on the reaction mechanism involving this directing group.12 In recent years, the Shi's research group reported the directing-group-assisted cobalt-catalyzed enantioselective intramolecular aminocarbonylation.13 These studies inspired us to believe that the coordination mode of the directing group can effectively enhance the sensitivity of cobalt to ligands. We envisioned installing the directing group onto the ligand, and by means of multidentate chelation, preventing the formation of overly stable cobalt carbonyl and enhancing the activity of carbonylation. Herein, we report a cobalt-catalyzed direct aminocarbonylation and alkoxycarbonylation of chloroacetonitrile promoted by an N,N,N-tridentate ligand, enabling the direct synthesis of a wide variety of 2-cyano-N-acetamides and 2-cyanoacetate compounds (Fig. 1c).
Results and discussion
Screening conditions
Our experiment was initiated using chloroacetonitrile and aniline as the model substrates. Initially, we employed commercially available phosphine ligands and nitrogen ligands (for details, see ESI†), but the expected product was not detected (Table 1, entry 1). Only the nucleophilic substitution product was isolated. This indicates that the activation of alkyl halides by the metal needs to be fast enough to distinguish from the highly prone nucleophilic substitution reaction. Subsequently, a series of N,N,N-tridentate ligands of 8-aminoquinoline were prepared (Fig. 2). The electronic effect of the ligands significantly influenced the reaction yield (Table 1, entries 2–6). The ligand with electron-withdrawing group at the 5-position of pyridine gave yields of 78% and 79%, independently. Subsequently, considering atom economy, ligand L17 was synthesized, and it achieved a yield of 80% (Table 1, entry 8). The screening of cobalt salts determined that CoCl2·6H2O was a more suitable metal salt (Table 1, entry 9). Under standard conditions, the use of 1 mol% palladium acetate did not yield the target product (Table 1, entry 10). The use of Ni(acac)2 and Cu(acac)2 did not result in the formation of the target product either. Without the participation of manganese powder, no target product was obtained (Table 1, entry 11). When Zn or silane were used as reducing agents, product 1 could be obtained with a slightly lower yield (Table 1, entries 12 and 13). Reducing the reaction temperature to 50 °C and the reaction pressure to 5 bar could achieve a yield of 98% (Table 1, entries 14 and 15). It is worth noting that when the substrate chloroacetonitrile is replaced with bromoacetonitrile, the target compound can be obtained in excellent yield (95%) at room temperature (Table 1, entry 16).|
|
|||
|---|---|---|---|
| Entry | Catal | Ligand | 3a [%] |
| a Reaction conditions: S1 (0.45 mmol), S2 (0.3 mmol), Co(acac)2 (10 mol%), ligand (10 mol%), Mn (20 mol%), CO (30 bar), Na2CO3 (1.5 eq.), MeCN (1.5 mL) stirred at 60 °C for 16 h. b Yields were determined by GC with dodecane as an internal standard and calculated based on aniline. c Isolated yield. d Other reducing agents. e 50 °C. f 5 bar. g Bromoacetonitrile, room temperature. | |||
| 1 | Co(acac)2 | L1–L10 | 0 |
| 2 | Co(acac)2 | L11 | 0 |
| 3 | Co(acac)2 | L12 | 19 |
| 4 | Co(acac)2 | L13 | 25 |
| 5 | Co(acac)2 | L14 | 32 |
| 6 | Co(acac)2 | L15 | 79 |
| 7 | Co(acac)2 | L16 | 78 |
| 8 | Co(acac)2 | L17 | 80 |
| 9 | CoCl 2 ·6H 2 O | L17 | 99(92) |
| 10 | Pd(OAc)2 | L17 | 0 |
| 11 | w/o Mn | L17 | 0 |
| 12 | Znd | L17 | 90 |
| 13 | MeSiH(EtO)2d | L17 | 92 |
| 14 | CoCl2·6H2Oe | L17 | 98 |
| 15 | CoCl2·6H2Of | L17 | 98 |
| 16 | CoCl2·6H2Og | L17 | 95 |

 | ||
| Fig. 2 N,N,N-Tridentate ligands used in this work. | ||
With the optimized reaction conditions in place, we tested the practicality and limitations of this carbonylation transformation for a series of aromatic amines and alcohols (Scheme 1). This transformation demonstrated a broad substrate scope and functional group tolerance. In the first stage, we tested a variety of aromatic amines (1–14) bearing electron-donating or electron-withdrawing groups. The corresponding carbonylation products could be obtained in good to excellent yields. For substrates with both a phenolic hydroxyl group and an amino group, a single target compound 8 could be obtained with high selectivity (87%). It is worth mentioning that nitro compounds have always been difficult to be compatible in palladium-catalyzed carbonylation reactions. In this system, the product 14 with the nitro group retained could be obtained in a 77% yield. Subsequently, by examining the electronic effect at the meta-position of the aromatic ring, both electron-withdrawing groups and electron-donating groups could give good to excellent yields (15–17). The steric effect was then investigated. The steric hindrance at the ortho-position significantly affected the reaction yield (18–19). When p-phenylenediamine was used as the substrate, in the presence of 4 equivalents of chloroacetonitrile, the mono-aminocarbonylation product 20 was obtained in a yield of 80%. Naphthalene ring could give an 89% yield. Heterocycle was also investigated and the target product 22 was isolated in a 60% yield.
 | ||
| Scheme 1 Scope of amines and alcohols. a Reaction conditions: S1 (0.45 mmol), amines or alcohols (0.3 mmol), CoCl2·6H2O (10 mol%), L17 (10 mol%), Mn (20 mol%), CO (5 bar), Na2CO3 (1.5 eq.), MeCN (1.5 mL) stirred at 50 °C for 16 h. isolated yield. | ||
Subsequently, research on alcohol substrates was carried out. At first, benzyl alcohols were explored, and the corresponding esters were produced in satisfactory yields. The Bpin group showed excellent compatibility with the reaction system, and the corresponding product 27 had a yield of 68%. Furfuryl alcohol and 2-thiophenemethanol were also included in the scope of the study, and the yields of the corresponding target compounds were 70% (29) and 86% (30) respectively. In addition, the functional group tolerance of functionalized substrate alcohols was investigated. Phenethyl alcohol (28), cyclic alcohols (31, 32), ethers (33), halogens (34, 35), alkenes (36), and alkynes (37) all exhibited good tolerance. Several crucial 2-cyanoacetate compounds (38–41) were synthesized in good to excellent yields. It is worth noting that these compounds are important chemical intermediates (Scheme 2). Remarkably, biologically active amines and alcohols such as benzocaine, fenchol, nerol, cholesterol, lenalidomide, D/L-menthol, testosterone, pregnenolone were all successfully converted into the corresponding carbonylated products (42–49). Thiophenol as nucleophile was also tested under our standard conditions and thioether from the direct nucleophilic substitution was obtained. For chloroacetonitrile, no desired product was detected if the CN group was replaced with NO2 or alkyl.
 | ||
| Scheme 2 Scope of amines and alcohols. | ||
To further demonstrate the application value of the current strategy, we conducted transformation experiments to prepare useful drugs starting from simple anilines and alcohols (Scheme 3). The triazole ring 50 can be obtained by the cyclization of compound 1 and benzyl azide.14 It has the effect of inhibiting vascular endothelial growth factor (VEGF), blocking the signal through VEGF receptors, and thus inhibiting malignant angiogenesis. In addition, the presence of the triazole ring demonstrates the great potential of structural modification and target labeling. Furazan compounds are an important class of heterocyclic compounds with a wide range of applications in the field of organic chemistry, covering distinct areas such as the preparation of bioactive molecules and energetic materials.15 The furazan compound 51 was obtained in a yield of 81% through a simple three-step synthesis.16 Compound 52 can be used as a novel AKR1C3 inhibitor and serves as a potential new treatment for castration-resistant prostate cancer.17 Leflunomide is marketed as a drug for the treatment of rheumatoid arthritis, and teriflunomide 53 is the active metabolite of leflunomide.18 Moreover, compound 38 can be employed in the preparation of enbucrilate,19 a surgical tissue adhesive, while compound 39 can be utilized for formulating sunscreen agents.20,21
 | ||
| Scheme 3 Later-stage of pharmaceutical derivatives. | ||
A scale-up reaction was performed successfully, and the desired product 2 can be obtained in 95% yield by expanding this reaction at 10 times with even 5 mol% catalyst (Scheme 4a). To delve deeper into the reaction mechanism, we carried out several control experiments. Initially, under standard conditions, when the radical inhibitors BHT (2,4-di-tert-butyl-4- methylphenol) or TEMPO were introduced into our model reaction, only a trace amount of the target product 1 was detected (Scheme 4b). Furthermore, compound (4,4-diphenylbut-3-enenitrile) was formed through the trapping of the cyanomethyl radical by the radical inhibitor 1,1-DPE (1,1-diphenylethylene) (Scheme 4c). Zero-valent cobalt is not the active valence state for the reaction. When Co2(CO)8 was used as the catalyst, no target product was detected (Scheme 4d).
 | ||
| Scheme 4 Scale-up reaction and control experiments. | ||
Based on our experiments and relevant literature,22 we propose a possible reaction mechanism (Scheme 5). First, the reactive species B is formed from the divalent cobalt precursor A under the reaction conditions with the participation of a reducing agent. Subsequently, the CoILn B reacts with chloroacetonitrile to yield complex C through a rapid single-electron radical process. The key acyl-metal intermediate D is formed by the migratory insertion of CO into the carbon-metal bond of intermediate C. Then, with the assistance of a base, the nucleophile completes the nucleophilic attack to generate intermediate E. Finally, the product is obtained via reductive elimination, and the monovalent cobalt B is regenerated to enter the next cycle.
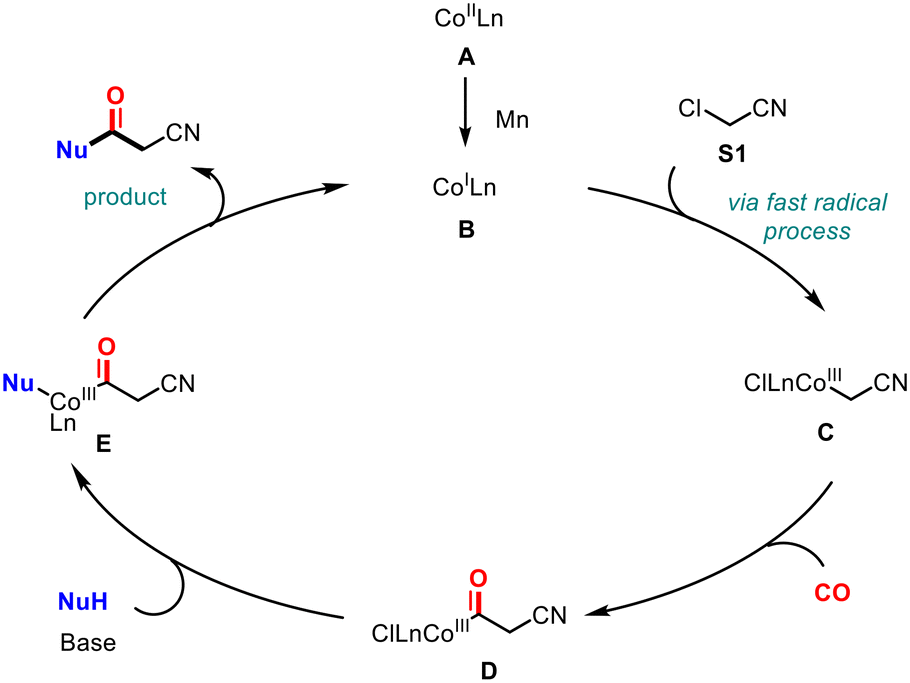 | ||
| Scheme 5 Proposed mechanism. | ||
In conclusion, a cobalt-catalyst system promoted by an N,N,N-tridentate ligands has been developed. Chloroacetonitrile can be efficiently activated by this system to achieve intermolecular direct aminocarbonylation and alkoxycarbonylation reactions. Mechanistic experiments indicate that this catalytic process undergoes a single-electron radical pathway. A variety of 2-cyano-N-acetamides and 2-cyanoacetates were synthesized from simple and inexpensive amines and alcohols. Moreover, good functional-group compatibility is exhibited by this reaction. The potential for further applications of this method is demonstrated by the subsequent strategies.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the financial support from the National Key R&D Pro-gram of China (2023YFA1507500).References
- (a) P. Gottle, A. Manousi, D. Kremer, L. Reiche, H.-P. Hartung and P. Kury, J. Neuroinflammation, 2018, 15, 76–87 CrossRef PubMed; (b) N. Osherov and A. Levitzki, FEBS Lett., 1997, 410, 187–190 CrossRef CAS PubMed; (c) S. H. Caldwell, E. E. Hespenheide, B. D. Greenwald, P. G. Northup and J. T. Patrie, Aliment. Pharmacol. Ther., 2007, 26, 49–59 CrossRef CAS PubMed; (d) L. A. Baker, M. D. Horbury and V. G. Stavros, Opt. Express, 2016, 24, 10700–10709 CrossRef CAS PubMed.
- (a) L. Kollar, Modern Carbonylation Methods, Wiley-VCH, Weinheim, 2008 CrossRef; (b) M. Beller, Catalytic Carbonylation Reactions, Springer, Berlin, 2006 CrossRef; (c) X.-F. Wu, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1–35 CrossRef CAS PubMed; (d) I. Ryu and N. Sonoda, Angew. Chem., Int. Ed. Engl., 1996, 35, 1050–1066 CrossRef; (e) J.-B. Peng, F.-P. Wu and X.-F. Wu, Chem. Rev., 2019, 119, 2090–2127 CrossRef CAS PubMed.
- For selected reviews on carbonylation of aryl halides and alkyl halides, see: (a) M. Beller and X.-F. Wu, Carbonylation Activation of C-X Bonds, Transition Metal Catalyzed Carbonylation Reactions, Springer, Berlin, 2013 CrossRef; (b) L.-J. Cheng and N. P. Mankad, Acc. Chem. Res., 2021, 54, 2261–2274 CrossRef CAS PubMed; (c) S. Sumino, A. Fusano, T. Fukuyama and I. Ryu, Acc. Chem. Res., 2014, 47, 1563–1574 CrossRef CAS PubMed; (d) X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986–5009 RSC; (e) L.-P. Wu, X.-J. Fang, Q. Liu, R. Jackstell, M. Beller and X.-F. Wu, ACS Catal., 2014, 4, 2977–2989 CrossRef CAS; (f) I. Ryu, Chem. Soc. Rev., 2001, 30, 16–25 RSC.
- R. Franke, D. Selent and A. Borner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- J.-B. Peng, H.-Q. Geng and X.-F. Wu, Chem, 2019, 5, 526–552 CAS.
- (a) M.-L. Yang, Y.-K. Liu, X. Qi, Y.-H. Zhao and X.-F. Wu, Green Synth. Catal., 2024, 5, 211–269 CrossRef CAS; (b) A. M. Veatch, S.-B. Liu and E. J. Alexanian, Angew. Chem., Int. Ed., 2022, 61, e202210772 CrossRef CAS PubMed; (c) B. Loubinoux, B. Fixari, J. J. Brunet and P. Caubere, J. Organomet. Chem., 1976, 105, C22–C24 CrossRef CAS; (d) J. J. Brunet, C. Sidot, B. Loubinoux and P. Caubere, J. Org. Chem., 1979, 44, 2199–2202 CrossRef CAS.
- (a) B. T. Sargent and E. J. Alexanian, Angew. Chem., Int. Ed., 2019, 58, 9533–9536 CrossRef CAS PubMed; (b) Y.-X. Wang, P. Wang, H. Neumann and M. Beller, ACS Catal., 2023, 13, 6744–6753 CrossRef CAS.
- A. M. Veatch and E. J. Alexanian, Chem. Sci., 2020, 11, 7210–7213 RSC.
- L.-C. Wang, B. Chen and X.-F. Wu, Angew. Chem., Int. Ed., 2022, 61, e202203797 CrossRef CAS PubMed.
- (a) L. Lukasevics and L. Grigorjeva, Org. Biomol. Chem., 2020, 18, 7460–7466 RSC; (b) L. Grigorjeva and O. Daugulis, Org. Lett., 2014, 16, 4688–4690 CrossRef CAS PubMed; (c) T. T. Nguyen, L. Grigorjeva and O. Daugulis, Chem. Commun., 2017, 53, 5136–5138 RSC; (d) F. Ling, C.-R. Ai, Y.-P. Lv and W.-H. Zhong, Adv. Synth. Catal., 2017, 359, 3707–3712 CrossRef CAS; (e) L. Lukasevics, A. Cizikovs and L. Grigorjeva, Org. Lett., 2021, 23, 2748–2753 CrossRef CAS PubMed; (f) L. Zeng, H. Li, S. Tang, X. Gao, Y. Deng, G. Zhang, C.-W. Pao, J.-L. Chen, J.-F. Lee and A.-W. Lei, ACS Catal., 2018, 8, 5448–5453 CrossRef CAS.
- (a) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef CAS PubMed; (b) L. Grigorjeva and O. Daugulis, Angew. Chem., Int. Ed., 2014, 53, 10209–10212 CrossRef CAS PubMed; (c) T. T. Nguyen, L. Grigorjeva and O. Daugulis, ACS Catal., 2016, 6, 551–554 CrossRef CAS PubMed; (d) L. Grigorjeva and O. Daugulis, Org. Lett., 2014, 16, 4684–4687 CrossRef CAS PubMed.
- L. Lukasevics, N. George, X.-Q. Wang, L. Grigorjeva and O. Daugulis, J. Am. Chem. Soc., 2025, 147, 2476–2490 CrossRef CAS PubMed.
- M.-Y. Teng, Y.-J. Wu, J.-H. Chen, F.-R. Huang, D.-Y. Liu, Q.-J. Yao and B.-F. Shi, Angew. Chem., Int. Ed., 2024, 63, e202318803 CrossRef CAS PubMed.
- A. S. Kiselyova, M. Semenovab and V. V. Semenov, ACS Med. Chem. Lett., 2009, 19, 1344–1348 CrossRef PubMed.
- (a) A. B. Sheremetev, N. N. Makhova and W. Friedrichsen, Adv. Heterocycl. Chem., 2001, 78, 65–188 CrossRef CAS; (b) S. Guo, Y. Song, Q. Huang, H. Yuan, B. Wan, Y. Wang, R. He, M. G. Beconi, S. G. Franzblau and A. P. Kozikowski, J. Med. Chem., 2010, 53, 649–659 CrossRef CAS PubMed.
- A. J. Neel and R. Zhao, Org. Lett., 2018, 20, 2024–2027 CrossRef CAS PubMed.
- S. Endo, H. Oguri, J. Segawa, M. Kawai, D.-W. Hu, S. Xia, T. Okada, K. Irie, S. Fujii, H. Gouda, K. Iguchi, T. Matsukawa, N. Fujimoto, T. Nakayama, N. Toyooka, T. Matsunaga and A. Ikari, J. Med. Chem., 2020, 63, 10396–10411 CrossRef CAS PubMed.
- S. Gerndt, C. C. Chen, Y. K. Chao, Y. Yuan, S. Burgstaller, S. A. Rosato, E. Krogsaeter, N. Urban, K. Jacob, O. N. P. Nguyen, M. T. Miller, M. Keller, A. M. Vollmar, T. Gudermann, S. Zierler, J. Schredelseker, M. Schaefer, M. Biel, R. Malli, C. Wahl-Schott, F. Bracher, S. Patel and C. Grimm, eLife, 2020, 9, e54712 CrossRef CAS PubMed.
- Y. R. Carriles, R. Á. Brito, R. M. Sánchez, E. S. Acevedo, P. R. Domínguez and W. D. Mueller, Molecules, 2014, 19, 6220–6227 CrossRef PubMed.
- J. Huang, et al., (DSM Nutritional Products Ltd) PCT/EP2008/000372, 2008.
- A. Kikuchi, Y. Hata, R. Kumasaka, Y. Nanbu and M. Yagi, Photochem. Photobiol., 2013, 89, 523–528 CrossRef CAS PubMed.
- (a) P. Yang and W. Shu, Angew. Chem., Int. Ed., 2022, 61, e202208018 CrossRef CAS PubMed; (b) Y. Li, D. Liu, L. Wan, J. Zhang, X. Lu and Y. Fu, J. Am. Chem. Soc., 2022, 144, 13961–13972 CrossRef CAS PubMed; (c) Y. Wang, D.-R. Wang, S.-L. Wang, Q.-L. Chong, Z.-H. Zhang and F.-K. Meng, Angew. Chem., Int. Ed., 2025, 64, e202413313 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00129c |
| This journal is © The Royal Society of Chemistry 2025 |