Catalytic static mixers enable the continuous hydrogenation of cannabidiol and tetrahydrocannabinol†
Received
30th January 2025
, Accepted 16th March 2025
First published on 17th March 2025
Abstract
In this work, we investigated the catalytic hydrogenation of cannabidiol (CBD), delta-8-tetrahydrocannabinol (Δ8-THC) and delta-9-tetrahydrocannabinol (Δ9-THC) by using catalytic static mixer (CSM) technology within a shell-and-tube reactor. Hydrogenation of these compounds is typically reported in batch at milligram quantities and affords a mixture of products. We were interested in developing a robust preparative-scale synthesis of 8,9-dihydrocannabidiol (H2CBD) and tetrahydrocannabidiol (H4CBD) from CBD, and hexahydrocannabinol (HHC) from Δ8-THC and Δ9-THC. We examined the influence of different noble metal-based CSMs (Pt/alumina, Pd/alumina, Pd-electroplated and Ru/alumina) and different operating conditions on the reaction performance. Pd/alumina CSMs were found to be unsuitable due to the formation of impurities, which partly arose due to double bond isomerization. Pd-electroplated CSMs displayed very low activity. Ru/alumina CSMs were observed to undergo rapid catalyst deactivation. Pt/alumina CSMs displayed high activity and good selectivity, even though signs of deactivation were still present at temperatures higher than 80 °C. We linked this deactivation to a combined influence of internal mass transfer limitation and accumulation of adsorbed molecules on the metal surface. After a careful fine-tuning of the operating conditions over Pt/alumina CSMs, we could obtain H2CBD, H4CBD and HHC in high yield from the corresponding cannabinoid derivative. Kinetic modeling and parameter fitting were successfully performed for the hydrogenation of CBD, which incorporated catalyst deactivation. Catalytic static mixer (CSM) technology is therefore demonstrated to be an industrially viable solution for the hydrogenation of cannabinoid derivatives.
Introduction
Phytocannabinoids are a large class of terpenoid compounds naturally derived from Cannabis sativa, an indigenous plant to Central Asia.1 These compounds can bind to specific cannabinoid receptors in the body, which are responsible for psychotropic and physiological effects.2 Among the many natural cannabinoids, cannabidiol (CBD, 1) and tetrahydrocannabinol (THC), with its two isomers Δ8-THC (6) and the psychoactive regioisomer Δ9-THC (5), are the best known.3,4 In recent years CBD, along with its synthetic hydrogenated derivatives, 8,9-dihydrocannabidiol (H2CBD, 2) and tetrahydrocannabidiol (H4CBD, 3 and 4), and THC, with its hydrogenated derivative, hexahydrocannabinol (HHC, 7 and 8), have received considerable attention due to their interesting biological activity.5–7 Trace amounts of HHC have been rarely identified as a component in Cannabis sativa. These compounds have shown promise in the treatment of inflammation, neurodegenerative diseases and cancer.8,9 H2CBD and H4CBD (cis and trans) compounds can be prepared by the acid-catalyzed coupling of inexpensive α-phellandrene with bulk chemicals olivetol or citronellal in reasonable yield.10–12
Catalytic hydrogenation is a cost-effective and viable synthetic strategy to access H2CBD, H4CBD and HHC from their corresponding naturally derived cannabinoids (Scheme 1).12 Typically, the preparative methods reported in the available literature are performed on only milligram scales under batch conditions. One of the main challenges associated with the catalytic hydrogenation of cannabinoid derivatives is the formation of mixtures, due to their inherent complex chemical structures.
 |
| | Scheme 1 a) Hydrogenation of CBD (1) into H2CBD (2), cis-H4CBD (3) and trans-H4CBD (4). b) Hydrogenation of Δ9-THC (5) and Δ8-THC (6) into (9R)HHC (7) and (9S)HHC (8). R and S stereochemistry in HHC refers to the carbon atom in position 9. | |
Ben-Shabat and co-workers synthesized H2CBD from CBD in ethyl acetate (EtOAc) using Adam's catalyst (PtO2, 10%) at ambient temperature, 4 bar pressure and 4 h reaction time, providing full conversion and H2CBD as the main product component (86%, by GC-MS analysis), with the remaining mass balance attributed to impurities.13 Zi and co-workers obtained H2CBD in 64% yield (20 mg) when using Pd/C (5%) in CH2Cl2 at room temperature and 2 h reaction time.14 Merli and co-workers synthesized H2CBD from CBD (on a 50 mg scale) in a very successful manner using Lindlar's catalyst (Pd–CaCO3, 15% w/w, poisoned) in a solvent mixture of hexane and EtOAc (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to afford the product in 97% yield.15 The reaction was performed at room temperature, under atmospheric pressure and with a reaction time of 23 h. The same group achieved the preparation of H4CBD on a similar scale using Pd/C (20% w/w) as catalyst, giving the product in 98% yield with a 9:1 cis–trans mixture. Very recently, Singh and co-workers reported the synthesis of H4CBD using 3 equiv. of Pd/C (THF:MeOH 1:9, rt, 1 atm, 5–6 h) to afford the product in 73% yield (99% purity).16
:1) to afford the product in 97% yield.15 The reaction was performed at room temperature, under atmospheric pressure and with a reaction time of 23 h. The same group achieved the preparation of H4CBD on a similar scale using Pd/C (20% w/w) as catalyst, giving the product in 98% yield with a 9:1 cis–trans mixture. Very recently, Singh and co-workers reported the synthesis of H4CBD using 3 equiv. of Pd/C (THF:MeOH 1:9, rt, 1 atm, 5–6 h) to afford the product in 73% yield (99% purity).16
Cruces and colleagues reported a gram-scale procedure for the preparation of H4CBD and HHC from CBD and THC, respectively. The conditions used were Pd/C (0.1 mol%) in ethanol (EtOH) at 25 °C and 1 bar. The diastereomers were successfully separated by supercritical fluid chromatography (SFC).9 Razdan and co-workers observed that the hydrogenation of Δ9-THC over PtO2 afforded a 94:6 S:R mixture, while Pd/C reversed the ratio to 35:65.17 Gaoni and Mechoulam found that over Pt catalysts, the hydrogenation of Δ9-THC produced mainly the R form (2:1), whereas Δ8-THC favored the formation of the S isomer (3:1).18 These results show that the distribution of isomers is very sensitive to the catalysts and conditions used in the synthesis.
The batch conditions highlighted above typically used mild reaction conditions (i.e., room temperature, atmospheric pressure and the reaction times were on the timescale of hours). Sustainable chemical processes rely not only on effective chemistry but also on the implementation of reactor technologies that enhance reaction performance, process intensify and improve overall safety.19 Thus, we were interested in the fact that there are scarce results that pertain to the use of continuous processing for the synthesis of these hydrogenated compounds. Continuous flow catalytic hydrogenation processes are usually performed using trickle-bed or packed-bed reactors.20 Cannazza, Citti and co-workers presented a continuous flow preparation of HHC.21 The team performed cyclization of CBD under batch conditions by using two different acids, each selected to give preferential selectivity toward either the Δ8-THC or Δ9-THC isomer.22 The crude reaction mixtures were then neutralized and then subjected to flow hydrogenation. The crude mixtures were hydrogenated in a ThalesNano H-Cube flow reactor with the following conditions: 3 mm 10% Pd/C cartridge, 30 °C, 20 bar and 1 mL min−1. The crude mixture containing Δ9-THC as the main isomer produced mainly S-HHC (57:43), whereas Δ8-THC produced mainly R-HHC (61:39). An alternative continuous hydrogenation method is to utilize a membrane flow microreactor. In this approach, the catalyst is supported or coated to the surface of a gas-permeable membrane.23,24 This approach gives very precise control over the gas permeation during the reaction. However, these reactors can be difficult to scale, as highlighted by Jensen and co-worker.25
Another recently developed alternative to packed-bed reactors are catalytic static mixers (CSMs), where the catalyst is coated on 3D-printed metal inserts, that can be housed within a shell-and-tube reactor.26–32 CSMs have been shown to provide high surface areas, high heat transfer, low metal leaching and a low pressure drop when compared to packed bed solutions.30 Since their development, CSMs have been applied to a variety of hydrogenation processes.33–35 Perhaps most relevant to our study is the work by Hornung, Xie and co-workers.31 The team used Pt, Pd, Ni and Ru on alumina (Al2O3) CSMs to hydrogenate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C unsaturated compounds of relevance for the fragrance industry.
C unsaturated compounds of relevance for the fragrance industry.
Despite showing great promise for organic synthesis, the use of CSMs in processes for the preparation of active pharmaceutical ingredients and key intermediates is not without its challenges. For instance, Hornung and co-workers observed that selectivity has a strong dependence on the history of the catalyst.32 Kappe, Williams and co-workers observed deactivation in the hydrogenation of nitro compounds.36 To our knowledge there are no previous reports using CSMs for the hydrogenation of cannabinoid derivatives. We were interested in testing the applicability of the CSMs to the hydrogenation of cannabinoid derivatives. In this work we focus on the development of the continuous hydrogenation of CBD and THC, showcasing the potential preparative-scale application of continuous processing to this class of important compounds.
Experimental
Material and synthetic procedures
Chemicals.
CBD (CBD Brothers Germany, >99%, with minor impurity cannabidivarine <1%), Δ9-THC (Gatt-Koller Dronabinol, >99%), ethanol (VWR, >95%), isopropanol (VWR, >95%), ethyl acetate (VWR, >99.5%), petroleum ether (VWR, >99.5%), acetonitrile (VWR, HPLC grade), water (VWR, HPLC grade), ethanol (VWR, >99.7%), acetonitrile-d3 (Eurisotop D021EAS, 99.8%, water <0.05%) and hexadecane (Alfa Aesar, 99%) were used without further purification. Δ8-THC was prepared from CBD, see ESI,† section 1.8. CSMs (Pd-electroplated, Pd/alumina, Pt/alumina, Ru/alumina) were purchased from Precision Catalysts. Prior to their insertion into the reactor, the CSMs were washed with acetone, dried and weighed. Upon removal, the same procedure was performed. The feed solutions were freshly prepared on the day of testing.
Testing methodologies
Experimental flow setup.
A schematic of the flow system used in this study is shown in Scheme 2. The liquid reservoir feed either comprised of a carrier solvent for the screening experiments, or a solution of the substrate in the case of the long runs and kinetic experiments. The liquid stream was introduced by using a HPLC pump (Knauer Azura). A six-port valve was fitted with a sample loop (4 mL or 10 mL volume) for the introduction of a small volume of the cannabinoid derivative solution for the screening experiments. The liquid stream was mixed with the H2 stream in a Y-mixer. H2 gas was produced using a commercial H2 generator (ThalesNano, H-Genie), equipped with an integrated mass flow controller (MFC) for the controlled introduction of the gas into the flow system. After mixing, the reaction stream entered a shell-in-tube reactor (Ehrfeld Miprowa) equipped with catalytic static mixer (CSM) inserts (11.8 mm × 150 mm). Either 2 or 4 CSMs were used in the experiments. The temperature of the reactor was controlled by a thermostat (Huber CC-304). A detailed description of the Miprowa system and configuration is provided in the ESI.† A similar setup was also described previously.29,36 The outlet stream was cooled by passing it through a coil submerged in a water bath. The system was pressurized with a backpressure regulator (Equilibar HC 276). The gas and liquid stream were then separated within a gas liquid separator and the liquid phase was collected in a flask for analysis.
 |
| | Scheme 2 Schematic representation of the continuous flow hydrogenation CSM configuration. Note: MFC = mass flow controller, p = pressure gauge, T = temperature sensor, and BPR = back pressure regulator. | |
Start-up, collection and switch-off procedure for the screening tests.
The reactor was set to the desired temperature and pressure, whilst isopropanol (iPrOH) and H2 were fed into the reactor, and the system was then left to reach the desired conditions for at least 30 min. No other pre-activation of the catalyst was performed. After reaching stable conditions, iPrOH was switched to the reaction solvent and left for at least 10 min. For the screening experiments, the feed solution, containing the cannabinoid derivative in the reaction solvent, was introduced via a sample loop by using a lure-lock syringe. The six-port valve was operated in manual fashion to introduce the cannabinoid derivative feed solution into the reactor and the time was set to 0 min. The outlet stream from the reactor was collected in a fractionated manner. For the long runs, the feed solution was introduced through the pump and no sample loop was used. At the end of the experiment run, the system was flushed with the reaction solvent, and then with isopropanol whilst the system was allowed to cool down.
Testing procedure for the kinetic investigation.
Fitting of kinetic models and rate parameters were performed for the hydrogenation of CBD over Pt/alumina CSMs. Two different sets of CSMs (labeled A and B), each comprising of two CSMs were used for the reaction profiling. These CSMs were inserted into the top channel of the reactor, to minimize the volume of feed solution required to equilibrate the system. The residence time was varied at fixed points (8 per temperature) from 34 to 408 s by changing the liquid flow rate in the range from 6 to 0.5 mL min−1, respectively (based on a channel volume of 3.4 mL). Three times the reactor volume was processed to reach steady state prior to sampling each experimental point. After the collection of a series of points at 50 °C, the temperature was then increased to 80 °C. The reactor was then cooled down, the CSMs were washed with acetone and a new test at two sequential temperatures (60 and 70 °C) was performed.
Analysis and spectroscopic methods
GC-FID, GC-MS, 1H-NMR and 13C-NMR.
We used a GC-FID (Shimadzu GC FID 230), with an RTX-5MS column (30 m × 0.25 mm ID × 0.25 μm) and helium as carrier gas (40 cm s−1). The samples were diluted in acetonitrile (MeCN) prior to the measurement. We assumed that the cannabinoid derivatives possessed the same response factor due to their similar structures. This assumption was successfully validated by 1H-NMR (300 MHz, Bruker Advanced III): for a mixture of CBD, H2CBD and H4CBD the differences between the two analysis approaches was <3.2%. For R-HHC, S-HHC and Δ8-THC, the difference was larger, but we only had a limited number of samples for comparison (ESI,† section 1.3). For the kinetic testing, the GC-FID was calibrated for CBD using a 0.1 M solution of hexadecane as internal standard. The concentrations of the other species used the same calibration, assuming equality of the response factors. NMR samples were analyzed on a Bruker Advanced III (300 MHz for 1H-NMR, 75 MHz for 13C-NMR) using MeCN-d3 as deuterated solvent. Identification of unknown peaks was performed using a GC-MS (Shimadzu GC-MSQP2010 SE) with a similar column and injection profile to that of GC-FID. The detector was a quadrupole with pre-rods and electron impact ionization.
Scanning electron microscopy (SEM) and energy dispersive X-ray (EDS) spectroscopy imaging.
SEM and EDS analysis were performed using a Zeiss Gemini DSM 982 field emission SEM. The SEM and EDS imaging of different CSMs showed a homogeneous distribution of the coating and the active metal on the surface. Care must be taken when installing the CSMs into the reactor, since removal of the coating does occur. The average weight-based content of the noble metal for the respective CSMs were measured to be 5.2% for Pt, 9.9% for Pd and 5.5% for Ru. No carbon was observed on the used catalysts which showed that there was no coke formation. The images are provided in the ESI† (section 1.6).
Results and discussion
Hydrogenation of CBD
Catalyst screening.
We commenced our investigation with the examination of the hydrogenation of CBD to H2CBD and H4CBD. We tested the reaction over four different CSM types: Pd-electroplated (Pd/EP), Pd/alumina, Pt/alumina and Ru/alumina at different temperatures (Table 1). Low conversion was observed for Pd/EP, probably due to the comparatively low catalyst surface area. In addition, many low-level impurities were observed (entries 1–3). Pd/alumina (entries 4–6) was more active and relatively selective to H4CBD at low temperatures than its electroplated counterpart, but it formed a similar level of impurities. Trost and co-workers reported that the hydrogenation of double bonds over Pd occurs with a π-allyl absorption mechanism that could promote isomerization, and thus the formation of impurities.37 This behavior has been observed in the hydrogenation of limonene, which contains the same structural motif as CBD.38,39 Pd CSMs were therefore not further considered.
Table 1 Results from the catalyst screening for the hydrogenation of CBD (1). Conditions: C1,0 = 0.1 M (in EtOAc), p = 20 bar, tres = 102 s, H/S = 11 and 2 CSMs
| N |
CSM |
T
|
H/S |
Conv. |
Selectivity |
cis:trans |
|
1
|
2
|
3 + 4 |
Oth. |
| (°C) |
(−) |
(%) |
(−) |
| 1 |
Pd/EP |
60 |
11.4 |
18.2 |
81.2 |
8.2 |
10.6 |
2.0 |
| 2 |
Pd/EP |
100 |
11.4 |
51.6 |
70.6 |
16.9 |
12.4 |
2.5 |
| 3 |
Pd/EP |
140 |
12.0 |
89.7 |
36.1 |
51.2 |
12.7 |
2.3 |
| 4 |
Pd/Al2O3 |
60 |
11.3 |
64.5 |
44.3 |
31.6 |
15.5 |
2.2 |
| 5 |
Pd/Al2O3 |
100 |
11.3 |
95.0 |
14.9 |
68.5 |
14.9 |
2.2 |
| 6 |
Pd/Al2O3 |
140 |
11.3 |
99.2 |
4.1 |
85.1 |
9.9 |
2.2 |
| 7 |
Pt/Al2O3 |
60 |
11.4 |
94.7 |
57.1 |
42.2 |
0.7 |
7.8 |
| 8 |
Pt/Al2O3 |
140 |
11.4 |
100 |
13.4 |
86.0 |
0.6 |
3.7 |
| 9 |
Ru/Al2O3 |
60 |
11.2 |
82.7 |
89.8 |
9.8 |
0.4 |
Only cis |
| 10 |
Ru/Al2O3 |
100 |
11.2 |
95.7 |
68.6 |
30.7 |
0.8 |
10.4 |
| 11 |
Ru/Al2O3 |
140 |
11.2 |
98.7 |
39.0 |
58.6 |
2.5 |
5.3 |
Pt/alumina (entries 7–8) displayed higher activity than Pd/alumina for the conversion of CBD, but less activity in the subsequent overreaction of H2CBD to H4CBD. In addition, the number of impurities formed was low. This observation was in-line with the observation by Hornung, Xie and co-workers regarding the preference of Pt for terminal CC bonds over internal CC bonds.31 Most importantly Pt/alumina formed significantly fewer impurities than Pd/alumina. The reaction displayed a preference for the cis isomer (3.7:1), cf. Pd/alumina (2.2:1). These results indicated that Pt/alumina could be a suitable catalyst for the synthesis of H2CBD at low temperatures and potentially H4CBD at high temperatures. Ru/alumina (entries 9–11) displayed moderate activity, performing better than Pd/alumina, but lower than Pt/alumina. In terms of selectivity, Ru/alumina was the most selective catalyst to H2CBD under milder conditions (entry 9). At higher temperatures, the conversion of CBD was similar to Pd/alumina and Pt/alumina, but Ru/alumina produced mixtures of H2CBD and H4CBD. Interestingly, Ru/alumina displayed almost selective preference for cis-H4CBD, which suggests a stronger interaction of the molecule with the surface, or a different orientation of the molecule compared to the other metals. Unfortunately, Ru/alumina displayed poor reproducibility and strong catalyst deactivation during stability testing. Thus, we decided not to investigate it further and focused our optimization and robustness testing with the Pt/alumina CSMs.
Continuous parameters
Previous batch hydrogenation studies showed that EtOH and EtOAc were commonly used for this chemistry. We found that EtOAc performed the best out of the two solvents, whereas EtOH resulted in the formation of more impurities (Table S6†). The influence of H2 to substrate ratio (H/S) was also assessed (Table 2). The testing regime was checked by changing the hydrogen to substrate molar ratio (H/S) at fixed liquid flow rates (range 0.75 to 2 mL min−1) and at different gas flows (range 50 to 200 mL min−1). The results (entry 1–5) showed that at 2 mL min−1 of liquid flow no significant differences could be observed for H/S ratios to as low as 5.7 (H2 gas = 25 mLn min−1). For a lower liquid flow rate (0.75 mL min−1), the threshold for a kinetically limited regime was 119 (200 mLn min−1), as a higher gas velocity appeared to be necessary to achieve good mixing. Thus, most of the runs in this study were performed at 200 mLn min−1. The impact of pressure resulted in only a small change in conversion (entries 6–8). We performed a two-factor, two-level design of experiments (DoE) to assess the influence of pressure and temperature on the formation of H4CBD over Pt/alumina (ESI,† section 2.4.4). Pressure was found to be non-significant, therefore we fixed its value to 20 barg throughout the remaining study. In contrast, temperature had a significant influence over the conversion and the product selectivity. The absence of catalyst surface saturation was checked by investigating the conversion at different substrate concentrations (entries 8–9). Both 0.1 M and 0.3 M provided similar conversion values.
Table 2 Results from the catalyst screening for the hydrogenation of CBD (1) over Pt/alumina (2 CSMs) in EtOAc
| N |
C
0
|
T
|
p
|
Liquid |
H/S |
Conv. |
Selectivity |
|
1
|
2
|
3 + 4 |
Oth. |
| (M) |
(°C) |
(barg) |
(mL min−1) |
(−) |
(%) |
| 1 |
0.1 |
80 |
20 |
2 |
5.7 |
98.1 |
7.6 |
73.9 |
18.1 |
| 2 |
0.1 |
80 |
20 |
2 |
17.2 |
98 |
7.3 |
74.8 |
17.4 |
| 3 |
0.1 |
80 |
20 |
0.75 |
59.0 |
98 |
53.8 |
40.2 |
6.50 |
| 4 |
0.1 |
80 |
20 |
0.75 |
119.0 |
98.7 |
48.9 |
44.9 |
7.4 |
| 5 |
0.1 |
80 |
20 |
0.75 |
178.0 |
98.7 |
48.8 |
46.3 |
7.7 |
| 6 |
0.1 |
60 |
5.5 |
1.9 |
11.7 |
94.5 |
94.6 |
5.1 |
0.2 |
| 7 |
0.1 |
60 |
15.8 |
1.9 |
11.7 |
92.8 |
94.6 |
5.1 |
0.2 |
| 8 |
0.1 |
60 |
11 |
1.9 |
23.4 |
98.7 |
69.9 |
29.3 |
0.8 |
| 9 |
0.3 |
60 |
11 |
1.9 |
7.8 |
97.8 |
71.8 |
27.4 |
0.8 |
An experiment using CBD over Pt/alumina was run at 80 °C in the absence of H2. The experiment showed neither conversion of CBD nor side product formation. This result indicated that there was unlikely to be any background reaction occurring either in solution or with the catalyst/catalyst support (e.g., cyclization of CBD).
Catalyst operating range and stability.
Previously, we mentioned that the deactivation of CSMs is a key challenge, therefore we were interested in studying long-term stability under different reaction conditions. We performed long runs with CBD over Pt/alumina at 60 °C, 80 °C, 100 °C and 140 °C, to assess the stability of the catalyst (Fig. 1). Above 80 °C the catalyst showed a strong and linear deactivation trend for the formation of H4CBD. The formation of cis-H4CBD was more influenced by deactivation than trans-H4CBD. The formation of H2CBD was less affected, as seen by its increase over operation time. To counteract the catalyst deactivation observed in the hydrogenation of nitrobenzoic acid, Hornung and co-workers suggested that increasing temperature, reaction time, hydrogen content or pressure could temporarily increase the conversion.40 However, this provided no guarantee of improving the long-term catalyst durability at high temperatures, which usually decreases with increasing temperature.
 |
| | Fig. 1 a) Catalyst performance for CBD over Pt/alumina (4 CSMs) at 140 °C. b) Slope of deactivation for H2CBD (k1), cis-H4CBD (k2) and trans-H4CBD (k3) at different temperatures. | |
Previously, we proposed that water could be used as cosolvent to minimize catalyst deactivation, as this could increase the wettability of the catalyst, thus helping to remove any compound deposited on the surface.41 Water might help to solubilize the species responsible for causing catalyst deactivation. Moreover, the wettability of the catalyst surface can influence the diffusion rate of reactants or products, including the ability of H2 to reach the surface. However, this approach was impractical in our system due to the low miscibility of H2O and EtOAc, and the insolubility of CBD and its derivatives in H2O. Thus, we decided to limit the temperature to a maximum of 80 °C. A proposed mechanism for deactivation will be described later in the manuscript.
CSM catalyst deactivation and the synthesis of H2CBD.
The deactivation of CSMs due to exposure to high temperature could be turned in our favor. In the investigation we observed a strong difference between the selectivity of fresh Pt/alumina CSMs tested at 60 °C (Table 3, entry 1), with that being exposed to high temperatures (140 °C, entry 3) and tested in a long run at 60 °C (entry 5). The catalyst, initially producing about 42% of H4CBD, became highly selective towards H2CBD. This deactivation could be exploited for the synthesis of H2CBD. H2CBD is typically synthesized in batch with low activity catalysts (e.g. Adams' or Lindlar's catalyst). We assessed the long-term stability of the poisoned Pt/alumina CSMs in the synthesis of H2CBD in a 55 min long run at 60 °C, 215 s, 11 barg. A stable conversion of 97% was achieved, with a selectivity of 93% for H2CBD (Fig. 2). After purification, we obtained H2CBD in a yield of 90% yield, corresponding to a throughput of 3.3 g h−1.
Table 3 Conversion and selectivity in the hydrogenation of CBD over Pt/alumina (4CSMs), showing deactivation due to high temperature cycling
| N |
T
|
Conv. |
Selectivity |
cis:trans |
|
1
|
2
|
3 + 4 |
| (°C) |
(%) |
(−) |
| 1 |
60 |
94.7 |
57.1 |
42.2 |
7.8 |
| 2 |
90 |
98.8 |
61.7 |
36.5 |
6.0 |
| 3 |
140 |
100 |
13.4 |
86.0 |
3.7 |
| 4 |
100 |
96.4 |
73.7 |
25.2 |
5.0 |
| 5 |
60 |
74.2 |
98.1 |
1.2 |
5.8 |
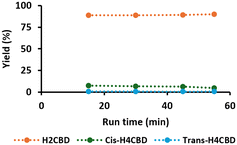 |
| | Fig. 2 Long run performance for the hydrogenation of 0.1 M CBD to H2CBD over deactivated Pt/alumina (4 CSMs). Conditions: T = 60 °C, tres = 215 s, p = 11 bar, H/S = 11.7 and 55 min run time. | |
Hydrogenation of CBD to H4CBD.
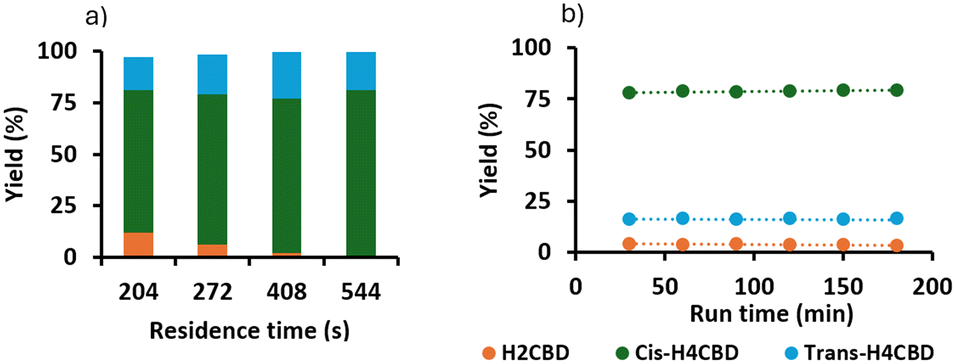
In the initial screening experiments, we showed that Pt/alumina converted CBD into a mixture of H2CBD and H4CBD at low temperatures. To achieve full conversion, we performed the reaction at our limit temperature of 80 °C, as at lower temperatures long residence times would be required, which would reduce productivity. At 80 °C and a residence time of 272 s, we could obtain good conversion of CBD to H4CBD, with less than 6% of H2CBD (Fig. 3a). We performed a 90 min long run at 20 barg, 272 s, H/S 59 and we observed stable performance and full conversion of CBD, but with 12% H2CBD remaining. Thus, we performed another run and extended the residence time to 544 s (Fig. 3b). This run gave >99% conversion, 95% selectivity for H4CBD with a cis:trans ratio of 4.8 (by GC), or 5.5 (by NMR). There was only 4% of H2CBD remaining. This corresponded to a throughput of 1.23 g h−1. We speculated that the low volumes used in the screening tests could be responsible for the higher formation of H4CBD observed at shorter residence times, which resulted in higher back mixing.
 |
| | Fig. 3 a) Influence of the tres on the hydrogenation of a 0.1 M solution of CBD over Pt/alumina (4 CSMs) at 80 °C and 20 bar. b) Long run for the hydrogenation of 0.1 M CBD to H4CBD over Pt/alumina CSMs. Conditions: T = 80 °C, tres = 544 s, p = 20 bar, H/S = 119 and 180 min run time. | |
Hydrogenation of H2CBD to cis- and trans-H4CBD.
To gain further insight into the conversion of H2CBD to H4CBD, we hydrogenated a 0.1 M solution of H2CBD over Pt/alumina at three temperatures (60 °C, 80 °C and 100 °C), see Fig. 4b. As a comparison, we also processed a 0.1 M solution of CBD under the same conditions (Fig. 4a). CBD converted quickly into both H2CBD and H4CBD, whereas the conversion of H2CBD to H4CBD was not enhanced by the absence of the external double bond, yielding a similar amount of H4CBD to that obtained for CBD, with a similar cis to trans ratio (7.6). This result agreed with a σ-coordination between the metal and the double bonds, as proposed by Augustine and co-workers.42 The results show a facile hydrogenation of the external double bond, with a more challenging hydrogenation of the internal double bond. The predominance of cis-H4CBD despite the coordination mode suggested, indicated that other parameters could control the stereochemistry, such as the presence of the bulky aromatic fragment. Interestingly, fewer impurities were formed when H2CBD was used as starting material, suggesting that CBD was the source of most of the impurities.
 |
| | Fig. 4 Yields for the hydrogenation processes over Pt/alumina (4 CSMs) at three different temperatures: (a) CBD; and (b) H2CBD. Conditions: 0.1 M substrate, p = 20 bar, tres = 102 s and H/S = 11.2. | |
Hydrogenation of Δ8-THC and Δ9-THC to R-HHC and S-HHC
Hydrogenation of Δ9-THC.
We screened the reaction of Δ9-THC over Pt/alumina at three different temperatures using a fixed residence time (408 s) and then subsequently at three different residence times at a constant temperature of 80 °C (Fig. 5). At 80 °C and 408 s, a conversion of 98% was obtained with a R to S ratio of 2.2. These conditions (80 °C, 20 bar and 408 s) were tested in a 25 min long run and we achieved stable and full conversion of Δ9-THC to HHC, >99% conversion and 98% product selectivity (R:S ratio of 2.4), which corresponded to a productivity of 1.6 g h−1. Interestingly, a significant amount (9%) of Δ8-THC was formed at 60 °C (9%). There was only a residual amount at 90 °C, probably as it was rapidly hydrogenated to HHC. Previously, we assessed that no isomerization of Δ9-THC to Δ8-THC occurs on the support without hydrogen, so we can assume that the isomerization to Δ8-THC is either a side reaction or a possible intermediate step in the conversion of Δ9-THC to HHC.
 |
| | Fig. 5 Yields for the hydrogenation of a 0.1 M solution of Δ9-THC over Pt/alumina (4 CSMs) at: (a) different temperatures at a constant tres of 408 s; and (b) at different tres at 80 °C. | |
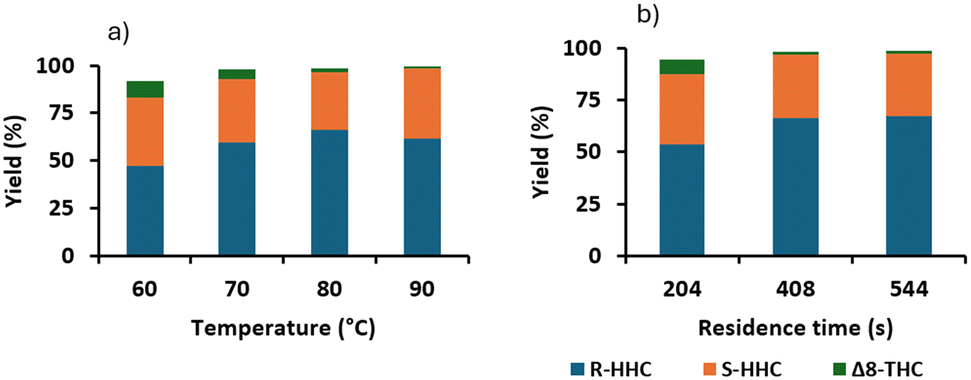
Hydrogenation of Δ8-THC.
The reaction of Δ8-THC was tested over Pt/alumina at three different temperatures (60 °C, 70 °C and 80 °C) and at a constant residence time of 408 s. We observed >95% conversion at 70 °C (Fig. 6a), reaching >98% conversion at 80 °C, the same conditions that had been used for the hydrogenation of Δ9-THC. The robustness of these conditions was assessed in a 55 min long run experiment (Fig. 6b). The process was stable and 98% conversion and >99% selectivity to HHC were achieved, corresponding to a productivity of 1.7 g h−1. The R:S ratio was 1.7, which was lower for the hydrogenation of Δ9-THC (2.4).
 |
| | Fig. 6 Hydrogenation of a 0.1 M solution of Δ8-THC over Pt/alumina (4 CSMs) a) screening of different temperatures for the hydrogenation. Conditions: p = 20 bar and tres = 408 s. b) Long run at 80 °C, 408 s, 20 bar and 55 min run time. | |
Kinetics of hydrogenation of CBD over Pt/alumina
General methodology.
The presence of internal mass transfer diffusional limitation was assessed by calculating the diffusional Damköhler number (DaII) (see ESI† section 5). Under the conditions explored, a DaII ≪ 1 was obtained for all scenarios, which indicated that internal mass transfer limitations were minimal. The lumped kinetics for the hydrogenation of CBD, over two different sets of Pt/alumina CSMs, were defined using reactions (1)–(3):| |  | (1) |
| |  | (2) |
| |  | (3) |
Earlier we showed that CSMs underwent linear deactivation over time. We accounted for deactivation using an activity factor (1 − kd·t), where kd is the rate constant of deactivation and t is the residence time.43,44 Different scenarios were assessed, including no deactivation, deactivation influencing only step(s) (1), (2), (2) and (3), and (1), (2) and (3). The lowest value for the residual sum of squares was achieved when deactivation was assumed to influence all the reactions, leading to the set of eqn (4)–(7) (see ESI,† section 3.2). We found that the hydrogenation of H2CBD to cis-H4CBD was more affected than the other reactions. We selected to opt for one deactivation constant for all the reactions, as we knew that deactivation was occurring in all the steps.| |  | (4) |
| |  | (5) |
| |  | (6) |
| |  | (7) |
In the model we assumed that the amount of hydrogen remained constant during the investigation, due to the large excess used. The kinetic constants were fitted in Matlab R2022b using the unconstrained minimization suite, fminsearch, applied on the norm of the normalized concentrations, obtained by solving the system of ODEs with the ode15s suite. The errors were calculated using the Jacobian and the covariance matrix, computed with the nlinfit and the nlparci suites. The values of the kinetic constants were then used for the calculation of the Arrhenius parameters.
We also fitted the kinetics with Dynochem. This software fits the Arrhenius parameters using the whole dataset at a reference temperature (Tref = 50.5 °C). Dynochem enabled the implementation of a more sophisticated model that included the gas to liquid mass transfer coefficient (kLa), which was fixed to 0.5 1 s−1, based on values reported for similar static mixer systems.45 Henry's constant was also included to account for the solubility of hydrogen at different temperatures. Limited variations were observed when these two parameters were varied, an indication that our assumption of the absence of external mass transfer limitations was correct. To account for deactivation in the model structure fitted in Dynochem, we added reaction (8) to the set (1)–(3). The input concentration of Pt was computed by multiplying the mass of supported material per CSM stated by the manufacturer (350 mg) to the platinum weight percent (5.2%) measured by SEM.
| |  | (8) |
Kinetic results.
The Matlab fitting of the kinetic data for the set (B) of platinum CSMs, with and without deactivation (only at 50 °C) are reported in Fig. 7. The activation energies (Ea) computed in Matlab and Dynochem are reported in Table 4. The values display good agreement. The full set of parameters is reported in the ESI† (section 3.2). With respect to kd, it should be emphasized that the value of the obtained deactivation constant could be potentially misleading. During the kinetic studies each point was collected after a steady state was achieved. This implied that the exact time elapsed for each point increased along the kinetic profile, equaling the sum of times elapsed during the collection of the previous samples. This means that the deactivation constants found were approximately one order of magnitude higher than those computed considering the real time elapsed, as shown in the ESI (section 3.2.3). Detailed modeling of deactivation is a challenging problem and requires more in-depth studies. The obtained values should be considered more as fitting parameters, rather than true measured reaction rate parameters.
 |
| | Fig. 7 Kinetic profiles for the hydrogenation of a 0.1 M solution of CBD over Pt/alumina 2 CSMs (set B), at different temperatures. Lines = fitted model; points = measured data. | |
Table 4 Values for the activation energies (Ea) for the two sets of Pt/alumina CSMs (A and B) fitted with Matlab and Dynochem
| CSM |
Step |
E
a (kJ mol−1) |
| Matlab |
Dynochem |
| Pt/alumina (A) |
1 |
17 ± 13 |
18 ± 4 |
| 2 |
38 ± 11 |
40 ± 5 |
| 3 |
52 ± 11 |
54 ± 5 |
| d |
14 ± 9 |
16 ± 8 |
| Pt/alumina (B) |
1 |
21 ± 5 |
21 ± 4 |
| 2 |
33 ± 10 |
38 ± 6 |
| 3 |
43 ± 15 |
52 ± 6 |
| d |
22 ± 7 |
23 ± 12 |
Validation of the kinetics results.
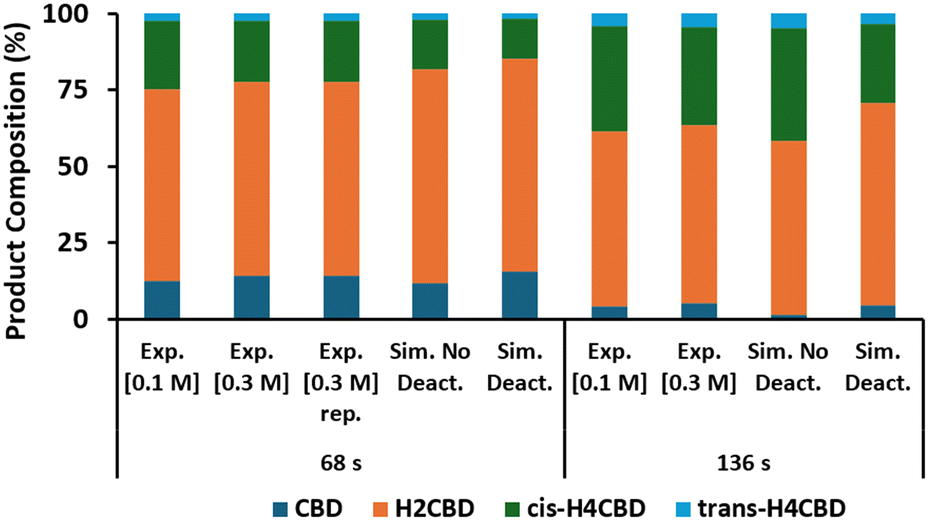
The kinetic model was validated using CSMs set (B), at 65 °C, two residence times (68 s and 136 s) and two concentrations of CBD (0.1 M and 0.3 M). The results are displayed in Fig. 8, where the model with and without deactivation was considered. The experimental data at similar residence times display good agreement. There was relatively little deactivation occurring. We were pleased with this finding, as this meant the conditions were appropriate for our long-run preparative scale synthesis. Overall, there is good agreement between the experimental and the simulated data. This agrees with the original finding that deactivation is slower at lower temperatures.
 |
| | Fig. 8 Comparison between the experimental (exp.) results in validation runs and the simulated results obtained by using the fitted kinetic constants (sim.), and with (deact.) or without deactivation (no deact.). | |
Investigation of CSM deactivation
Deactivation in the hydrogenation of CBD.
Throughout this project we observed a linear deactivation trend at elevated temperatures for the CSMs. Linear deactivation can be linked to metal leaching, coke formation, or irreversible adsorption of organic species. Initially, we assessed metal leaching as a possible cause of catalyst deactivation, by inductively coupled plasma mass spectrometry (ICP-MS) for several of the long runs. All the samples from the collected solutions showed negligible amounts of metal (<1 ppb). At these conditions (80 °C and 20 bar H2), we speculated that coke was unlikely to be formed. This assumption was confirmed by SEM of the used catalyst, with no coke formation observed. Moreover, the activity of the catalyst could be recovered by external washing, which would be unlikely to work if carbon was formed on the catalyst. Lastly, we investigated the loss of active sites due to the adsorption of organic molecules on the surface of the catalyst.46 Interestingly, we found that an ‘inline’ cleaning of the catalyst by using EtOAc or iPrOH did not help recover the activity. Removal of the CSMs and external washing with acetone provided close to full recovery, thus suggesting that surface covering was indeed occurring on the CSMs. A similar finding was reported by Zhang and Zhou.47 They observed the deactivation of the catalyst used in the hydrogenation of phenol to cyclohexanone. The deactivation could be linked to pore blocking, which could be partially solved by using a series of sequential washing steps with H2O and EtOH.47 A possible identification of which species could be responsible for the deactivation was provided by the work of Jentoft and co-workers, who investigated the hydrogenation of phenol to cyclohexanol, observing a strong deactivation in the presence of alcohol.48 They claimed that phenols displayed a preference for planar adsorption onto the surface of Pt, thus blocking active sites for other processes. The high adsorption energy of phenols made this process more likely to occur at higher temperatures. Similarly, Sajiki and co-workers observed deactivation in the processing of phenolic compounds over Ru/C, due to a coordination of either the lone pairs of the OH groups, or the π-electron systems of the aromatic ring to the metal.49 To better understand the role of the phenol and the terpenoid ring on the deactivation, we studied a simpler system comprising limonene and resorcinol.
Deactivation in the hydrogenation of limonene and resorcinol as surrogates of CBD.
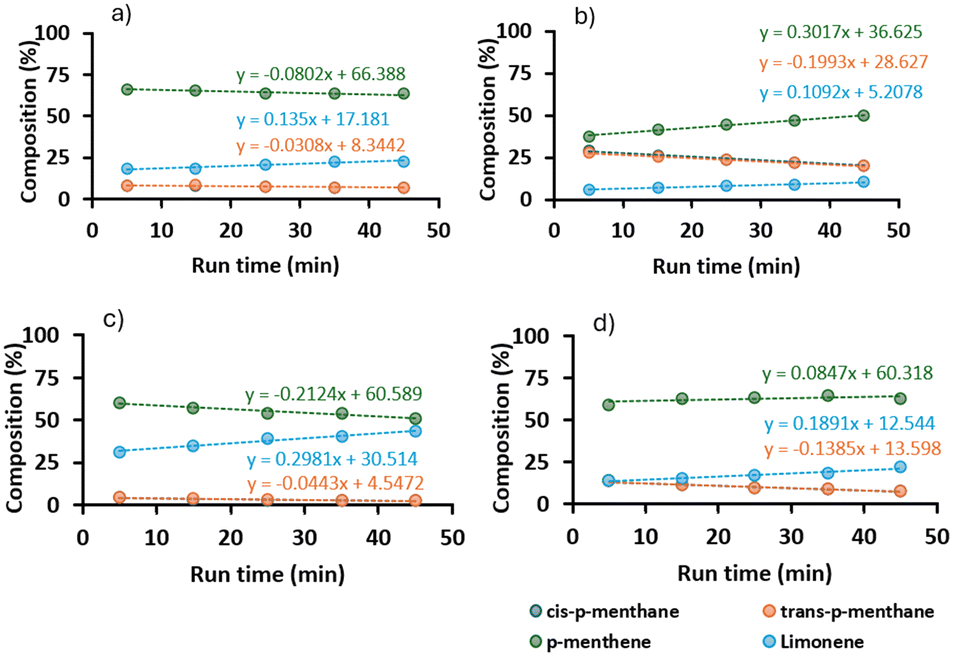
To assess the possible influence on deactivation of the two constituents of CBD, i.e. the aromatic and the terpenoid fragments, we utilized the additive approach pioneered by Collins and Glorius, where the constituents of a complex molecule can be studied using simpler surrogate compounds, so-called additives, to decouple their impact.50,51 We selected limonene and resorcinol as our two additives (Scheme 3).52 A solution of limonene and one containing both limonene 10 and resorcinol 14 were hydrogenated over a Pt/alumina CSM at 60 °C and 100 °C, 20 barg and within a short residence time (51 s), thereby avoiding full conversion (Fig. 9). Resorcinol remained unaffected during the process. Limonene instead converted preferentially into mixtures of p-menthene and p-menthane, with the two isomers of p-menthane forming in equal amount. These results were in good agreement with those obtained by others.53,54 A cis to trans ratio of 1, when compared to the larger preference for cis-H4CBD in the hydrogenation of CBD, suggested that the presence of the aromatic ring either sterically hindered the interaction of CBD with the catalyst surface, or promoted a phenol-mediated adsorption of CBD onto the surface that favored the cis isomer. This result would be compatible with the faster decrease in the concentration of cis-H4CBD compared to that of trans-H4CBD, which was earlier described.
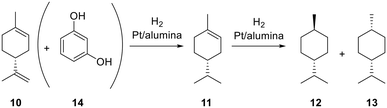 |
| | Scheme 3 Hydrogenation of limonene (10) to p-menthene (11), trans-p-menthane (12) and cis-p-menthane (13), with or without resorcinol (14). | |
 |
| | Fig. 9 Composition profiles for the hydrogenation of limonene over Pt/alumina (1 CSM). (a) No resorcinol, 60 °C. b) No resorcinol, 100 °C. c) With resorcinol, 60 °C. d) With resorcinol, 100 °C. | |
Interestingly, for the hydrogenation of a solution of limonene alone, linear deactivation was observed at both low and high temperatures. This finding suggested that the aromatic ring was not solely responsible for deactivation and that most likely internal mass transfer limitations influenced the catalyst at high temperatures. When the temperature rose, the hydrogenation of limonene occurred mostly at the outlet of the pores, thus involving a smaller surface of the catalyst than that available at lower temperatures. A stronger coordination of the molecule with the surface increased the covering and therefore slowed down the conversion, thus deactivating the catalyst. This deactivation therefore affected the slower conversion of p-menthene to p-menthane more than that of limonene to p-menthene as can be seen from Fig. 9. This resembled the fast deactivation of H2CBD to H4CBD we observed for CBD. When resorcinol was added to the solution of limonene, we observed that the conversion of limonene to p-menthene was more affected than that of p-menthene to p-menthane. This observation was compatible with a competition between limonene and resorcinol for the catalytic sites, as suggested by Sajiki and co-workers.49 In the presence of resorcinol, the amount of p-menthane formed dropped on the fresh catalyst at 100 °C more than it occurred when resorcinol was not inserted (from 29% to 14%). This suggested that the coordination of resorcinol with the catalytic sites increases at higher temperatures and therefore impacts on the conversion of p-menthene to p-menthane. At lower temperature this effect was less pronounced enabling a larger availability of the catalyst surface. We can conclude that, for CBD, deactivation was caused by a lower usage of the catalyst surface at higher temperatures, by a concomitant decrease in the active surface due to the accumulation of starting material and product through the phenol ring onto the surface over time and by a coordination of CBD with the surface through the aromatic ring, which disfavors the conversion of H2CBD to H4CBD.
Conclusions
We investigated the hydrogenation of CBD, Δ8-THC and Δ9-THC over different metal-coated CSMs, namely Pd-electroplated, Pd/alumina, Pt/alumina and Ru/alumina. We observed that Pd-electroplated and Pd/alumina, despite their high activity for the conversion of CBD into H4CBD, resulted in the formation of impurities. Ru/alumina provided moderate activity, but was prone to deactivation and the reproducibility was poor. Pt/alumina displayed the highest activity and selectivity for both CBD and THC hydrogenation, providing that the temperature did not exceed 80 °C. The optimized conditions and performance metrics for the hydrogenation of CBD to H2CBD and H4CBD, and for Δ8-THC and Δ9-THC to (9R)-HHC and (9S)-HHC are shown in Scheme 4. We observed that a linear deactivation rate for the catalyst was observed at temperatures above 80 °C for CBD hydrogenation. The deactivation was linked to a lower use of the catalyst surface at higher temperatures due to internal mass transfer limitation. In addition, a decrease in the available catalyst surface was observed due to the accumulation of starting material and product through the phenol ring onto the surface. However, we identified that the removal of the CSM and washing of the catalyst surface with acetone provided close to full recovery in the catalytic activity. We showed that it was possible to use partially deactivated Pt/alumina CSMs to selectively and consistently produce H2CBD. We also fitted the multistep reaction kinetics for the hydrogenation of CBD in flow, including consideration of the catalyst deactivation. Overall, the results showed that Pt/alumina CSMs are a viable technology for the continuous-flow selective preparative-scale synthesis of hydrogenated cannabinoids. We believe these results will increase interest in utilizing such CSM technology in active pharmaceutical ingredient synthesis and manufacture. Even though the conditions must be finely tuned to avoid either the formation of side products or deactivation of the catalysts.
 |
| | Scheme 4 Summary of the optimized conditions for the hydrogenation of the cannabinoid derivatives: a) CBD to H2CBD and (cis and trans) H4CBD; and b) Δ8-THC and Δ9-THC to (9R and 9S) HHC. | |
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The Research Center Pharmaceutical Engineering (RCPE) is funded within the framework of COMET – Competence Centers for Excellent Technologies by BMK, BMAW, Land Steiermark, and SFG. The COMET program is managed by the FFG. This work was partly funded through the Austrian Research Promotion Agency (FFG) as part of the “Twin4Pharma” project within the COMET Module program. Gerald Auer (Department of Earth Science at the University of Graz, NAWI Graz Geocenter) is acknowledged for performing the SEM and EDS spectroscopy imaging. Walter Gössler (Institute for Analytical Chemistry, University of Graz) is acknowledged for the ICPMS analysis.
Notes and references
- L. O. Hanuš, S. M. Meyer, E. Muñoz, O. Taglialatela-Scafati and G. Appendino, Nat. Prod. Rep., 2016, 33, 1357 RSC.
- R. Mechoulam, Science, 1970, 168, 1159 CrossRef CAS PubMed.
- M. Tagen and L. E. Klumpers, Br. J. Pharmacol., 2022, 179, 3915 CrossRef CAS PubMed.
- S. D. Banister, J. C. Arnold, M. Connor, M. Glass and I. S. McGregor, ACS Chem. Neurosci., 2019, 10, 2160 CrossRef CAS PubMed.
- E. A. Aguillón, R. A. C. Leão, L. S. M. Miranda and R. O. M. A. de Souza, Chem. – Eur. J., 2021, 27, 5577 CrossRef PubMed.
- X. Wang, H. Zhang, Y. Liu, Y. Xu, B. Yang, H. Li and L. Chen, Bioorg. Chem., 2023, 140, 106810 CrossRef CAS PubMed.
- P. Morales, P. H. Reggio and N. Jagerovic, Front. Pharmacol., 2017, 8 DOI:10.3389/fphar.2017.00422.
- S. Burstein, Bioorg. Med. Chem., 2015, 23, 1377–1385 CrossRef CAS PubMed.
- A. Collins, G. Ramirez, T. Tesfatsion, K. P. Ray, S. Caudill and W. Cruces, Nat. Prod. Commun., 2023, 18 DOI:10.1177/1934578X231158910.
- Q. Wu, M. Guo, L. Zou, Q. Wang and Y. Xia, Molecules, 2023, 28, 445 CrossRef CAS PubMed.
- M. Mascal, N. Hafezi and D. Wang, Sci. Rep., 2019, 9, 7778 CrossRef PubMed.
- M. L. Docampo-Palacios, G. A. Ramirez, T. T. Tesfatsion, A. Okhova, M. Pittiglio, K. P. Ray and W. Cruces, Molecules, 2023, 28, 6434 CrossRef CAS PubMed.
- S. Ben-Shabat, L. O. Hanuš, G. Katzavian and R. Gallily, J. Med. Chem., 2006, 49(3), 1113 CrossRef CAS PubMed.
- C. T. Zi, Y. R. Xie, Y. Niu, Z. H. Liu, L. Yang, Y. K. Xi, Z. J. Li, F. M. Zhang, Z. M. Xiang and J. Sheng, Phytochem. Lett., 2022, 51, 97 CrossRef CAS.
- P. Seccamani, C. Franco, S. Protti, A. Porta, A. Profumo, D. Caprioglio, S. Salamone, B. Mannucci and D. Merli, J. Nat. Prod., 2021, 84, 2858 CrossRef CAS PubMed.
- P. S. Cham, Deepika, R. Bhat, D. Raina, D. Manhas, P. Kotwal, D. P. Mindala, N. Pandey, A. Ghosh, S. Saran, U. Nandi, I. A. Khan and P. P. Singh, ACS Infect. Dis., 2024, 10, 64 Search PubMed.
- D. B. Uliss, G. R. Handrick, H. C. Dalzell and R. K. Razdan, Tetrahedron, 1978, 34, 1885 CrossRef CAS.
- Y. Gaoni and R. Mechoulam, Tetrahedron, 1966, 22, 1481 CrossRef CAS.
- R. P. Utikar and V. V. Ranade, ACS Sustainable Chem. Eng., 2017, 5, 3607 Search PubMed.
- E. Masson, E. M. Maciejewski, K. M. P. Wheelhouse and L. J. Edwards, Org. Process Res. Dev., 2022, 26, 2190 CrossRef CAS.
- F. Russo, M. A. Vandelli, G. Biagini, M. Schmid, L. Luongo, M. Perrone, F. Ricciardi, S. Maione, A. Laganà, A.
L. Capriotti, A. Gallo, L. Carbone, E. Perrone, G. Gigli, G. Cannazza and C. Citti, Sci. Rep., 2023, 13, 11061 CrossRef CAS PubMed.
- Rather than prepare these compounds batchwise, our group recently described a continuous flow synthesis for the reaction of CBD to THC, see: B. Bassetti, C. A. Hone and C. O. Kappe, J. Org. Chem., 2023, 88, 6227–6231 CrossRef CAS PubMed.
- M. Liu, X. Zhu, R. Chen, Q. Liao, H. Feng and L. Li, Chem. Eng. J., 2016, 301, 35 CrossRef CAS.
- B. Venezia, L. Panariello, D. Biri, J. Shin, S. Damilos, A. N. P. Radhakrishnan, C. Blackman and A. Gavriilidis, Catal. Today, 2021, 362, 104 CrossRef CAS.
- L. Yang and K. F. Jensen, Org. Process Res. Dev., 2013, 17, 927 CrossRef CAS.
- A. Avril, C. H. Hornung, A. Urban, D. Fraser, M. Horne, J.-P. Veder, J. Tsanaktsidis, T. Rodopoulos, C. Henry and D. R. Gunasegaram, React. Chem. Eng., 2017, 2, 180 RSC.
- J. Gardiner, X. Nguyen, C. Genet, M. D. Horne, C. H. Hornung and J. Tsanaktsidis, Org. Process Res. Dev., 2018, 22, 1448 CrossRef CAS.
- M. Kundra, Y. Zhu, X. Nguyen, D. Fraser, C. H. Hornung and J. Tsanaktsidis, React. Chem. Eng., 2022, 7, 284 RSC.
- R. Lebl, Y. Zhu, D. Ng, C. H. Hornung, D. Cantillo and C. O. Kappe, Catal. Today, 2022, 338, 55 CrossRef.
- Y. Zhu, B. B. M. Sultan, X. Nguyen and C. Hornung, J. Flow Chem., 2021, 11, 515 CrossRef CAS.
- M. Kundra, T. Grall, D. Ng, Z. Xie and C. H. Hornung, Ind. Eng. Chem. Res., 2021, 60, 1989 CrossRef CAS.
- C. H. Hornung, X. Nguyen, A. Carafa, J. Gardiner, A. Urban, D. Fraser, M. D. Horne, D. R. Gunasegaram and J. Tsanaktsidis, Org. Process Res. Dev., 2017, 21, 1311 CrossRef CAS.
- X. Fan, A. A. Lapkin and P. K. Plucinski, Catal. Today, 2009, 147, S313 CrossRef CAS.
- L. Vanoye, B. Guicheret, C. Rivera-Cárcamo, R. Castro Contreras, C. de Bellefon, V. Meille, P. Serp, R. Philippe and A. Favre-Réguillon, Chem. Eng. J., 2022, 441, 135951 CrossRef CAS.
- G. Vilé, D. Ng, Z. Xie, I. Martinez-Botella, J. Tsanaktsidis and C. H. Hornung, ChemCatChem, 2022, 14, e202101941 CrossRef.
- R. Lebl, S. Bachmann, P. Tosatti, J. Sedelmeier, K. Püntener, J. D. Williams and C. O. Kappe, Org. Process Res. Dev., 2021, 25, 1988 CrossRef CAS.
- B. M. Trost, P. E. Strege, L. Weber, T. J. Fullerton and T. J. Dietsehe, J. Am. Chem. Soc., 1978, 100, 3407 CrossRef CAS.
- R. J. Grau, P. D. Zgolicz, C. Gutierrez and H. A. Taher, J. Mol. Catal. A: Chem., 1999, 148, 203 CrossRef CAS.
- E. Bogel-Łukasik, R. Bogel-Łukasik and M. N. da Ponte, Monatsh. Chem., 2009, 140, 1361 CrossRef.
- R. Legg, C. Zhang, M. Bourchier, S. Cole, I. Martinez-Botella, X. Nguyen, Y. Zhu, W. Liew, S. Saubern, J. Tsanaktsidis and C. H. Hornung, Chem. Ing. Tech., 2022, 94, 1017 CrossRef CAS.
- S. Martinuzzi, M. Mex, J. Milic, C. A. Hone and C. O. Kappe, Org. Process Res. Dev., 2025, 29, 363 CrossRef CAS.
- R. L. Augustine, F. Yaghmaie and J. F. Van Peppen, J. Org. Chem., 1984, 49, 1865 CrossRef CAS.
- For an overview of mathematical deactivation models, see: Z. M. Shakor and E. N. Al-Shafei, RSC Adv., 2023, 13, 22579 RSC.
- M. Usman, D. Cresswell and A. Garforth, Ind. Eng. Chem. Res., 2012, 51, 158 CrossRef CAS.
- A. M. Al Taweel, J. Yan, F. Azizi, D. Odedra and H. G. Homaa, Chem. Eng. Sci., 2005, 60, 6378 CrossRef CAS.
- P. Forzatti and L. Lietti, Catal. Today, 1999, 52, 165 CrossRef CAS.
- Y. Zhang and J. Zhou, ChemistrySelect, 2022, 7, e202104214 CrossRef CAS.
- Z. Liu, I. A. Hamad, Y. Li, Y. Chen, S. Wang, R. E. Jentoft and F. C. Jentoft, Appl. Catal., A, 2019, 585, 117199 CrossRef.
- T. Maegawa, A. Akashi, K. Yaguchi, Y. Iwasaki, M. Shigetsura, Y. Monguchi and H. Sajiki, Chem. – Eur. J., 2009, 15, 6953 CrossRef CAS PubMed.
- K. Collins and F. Glorius, Nat. Chem., 2013, 5, 597 CrossRef CAS PubMed.
- K. D. Collins and F. Glorius, Acc. Chem. Res., 2015, 48, 619 CrossRef CAS PubMed.
- For our flow development of the additive approach, see: K. Simon, P. Sagmeister, R. Munday, K. Leslie, C. A. Hone and C. O. Kappe, Catal. Sci. Technol., 2022, 12, 1799 RSC.
- G. Rubulotta, K. L. Luska, C. A. Urbina-Blanco, T. Eifert, R. Palkovits, E. A. Quadrelli, C. Thieuleux and W. Leitner, ACS Sustainable Chem. Eng., 2017, 5, 3762 CrossRef CAS.
- F. J. N. Moutombi, A. S. Fabiano-Tixier, O. Clarisse, F. Chemat and M. Touaibia, J. Chem., 2020, 5946345 CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Christopher A.
Hone
c,
Christopher A.
Hone