
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insights into the structure of CoCu bimetallic catalysts for CO2 hydrogenation into formate†
Ruijing
Dong
ab,
Chao
Wu
bc,
Truong-Giang
Vo
 b,
San Hua
Lim
b,
Xun
Cao
b,
Jia'E
Zheng
b,
Xin
Xiao
a,
Wei
Chu
*a and
Yan
Liu
*b
b,
San Hua
Lim
b,
Xun
Cao
b,
Jia'E
Zheng
b,
Xin
Xiao
a,
Wei
Chu
*a and
Yan
Liu
*b
aSchool of Chemical Engineering, Sichuan University, No. 24 South Section 1, Yihuan Road, Chengdu, 610065, China. E-mail: chuwei1965@scu.edu.cn
bInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology, and Research (A*STAR), 1 Pesek Road, Jurong Island, Singapore 627833, Republic of Singapore. E-mail: liu_yan@isce2.a-star.edu.sg
cCollege of Materials Science and Engineering, Sichuan University, No. 24 South Section 1, Yihuan Road, Chengdu, 610065, China
First published on 18th March 2025
Abstract
Direct hydrogenation of CO2 to valuable chemicals and fuels is a promising pathway for the valorization of detrimental CO2. This study delves into the mechanistic insights of CO2 hydrogenation into formate over bimetallic cobalt–copper on silica support catalysts (CoxCu/SiO2, x: 0–2). CoCu/SiO2 with a Co and Cu molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 showed the best activity for CO2 hydrogenation into formate with a maximum yield of 2.3 mmol g−1 h−1, and it exhibited 100% selectivity for formate and a turnover frequency (TOF) of 625.2 h−1 in NaOH media. The formate formation rate over CoCu/SiO2 was 2.5 times higher than the sum of those over monometallic Cu and Co catalysts. Combining X-ray absorption spectroscopy and transmission electron microscopy results provided insights into the active sites at the interface between cobalt and copper in CO2 hydrogenation into formate. Theoretical calculations clarified and highlighted that the rate determining step was the formation of a carbonate intermediate on the CoCu bimetallic composite during the CO2 conversion into formate. This work provides a theoretical reference for designing an efficient and cost-effective CoCu bimetallic catalyst for producing formate from CO2.
:1 showed the best activity for CO2 hydrogenation into formate with a maximum yield of 2.3 mmol g−1 h−1, and it exhibited 100% selectivity for formate and a turnover frequency (TOF) of 625.2 h−1 in NaOH media. The formate formation rate over CoCu/SiO2 was 2.5 times higher than the sum of those over monometallic Cu and Co catalysts. Combining X-ray absorption spectroscopy and transmission electron microscopy results provided insights into the active sites at the interface between cobalt and copper in CO2 hydrogenation into formate. Theoretical calculations clarified and highlighted that the rate determining step was the formation of a carbonate intermediate on the CoCu bimetallic composite during the CO2 conversion into formate. This work provides a theoretical reference for designing an efficient and cost-effective CoCu bimetallic catalyst for producing formate from CO2.
1. Introduction
In recent decades, the excessive use of fossil energy has led to an evident burgeoning in anthropogenic carbon dioxide (CO2) emissions into the atmosphere.1,2 Recognizing the detrimental impact of CO2 emission on global climate change, it is important to deal with CO2via carbon capture, utilization, and storage (CCUS) strategies.1,3–5 Among the strategies available, CO2 hydrogenation stands out owing to its dual benefits of mitigating anthropogenic CO2 emissions and generating valuable carbon compounds that serve as precursors for manufacturing chemicals and fuels.6–11 Formic acid is extensively used in various industrial applications like leather tanning, textile treatment, etc. More importantly, formic acid is an appealing liquid energy carrier that can complete a carbon-neutral cycle through reduction/reoxidation combustion without emitting hazardous byproducts.12,13 Therefore, the conversion of CO2 into formate/formic acid, which is an atom-economical process, has attracted significant attention over the past decades.14Historically, noble metal-based catalysts, such as PdCuH4 (ref. 15) or Ru/Rh,16,17 have often been used for CO2 hydrogenation into formic acid. Our group18 explored the reaction mechanism of CO2 conversion over noble metal PdCu catalysts through in situ DRIFTs, which revealed that bidentate carbonate and monodentate carbonate species are intermediates during CO2 hydrogenation into formate with a maximal formate formation rate of 12.8 mmol h−1 gmetal−1. Despite the good hydrogenation capabilities and selectivity toward formic acid of these precious metals,13 their prohibitive costs limit their large-scale applications. Therefore, developing and utilizing a non-noble metal catalyst for the thermal conversion of CO2 into formate is a promising solution to enhance its economic value and scalability.
Alternatively, non-precious transition metals, such as Ni, Fe, and Cu, are excellent CO2 hydrogenation alternatives owing to their abundance and affordability.13 Copper (Cu)-based catalysts have demonstrated high catalytic activity for CO2 reduction owing to their good hydrogen activation performance on metallic Cu and good selectivity for producing alcohols over Cu(I) species.15,19,20 Alloying Cu with other metals, such as Pd and Fe,15 is quite popular for enhancing its performance and achieving results similar to noble metals owing to the tunable surface structure and electronic features of multimetallic catalysts.21 Cobalt (Co)-based catalysts have been demonstrated to play a certain role in enhancing the reactivity and target product selectivity in CO2 conversion processes most likely due to their good CO2 adsorption and activation ability.22 Guo et al. reported that the incorporation of a small amount of cobalt into spinel ZnFe2O4 facilitated the high-yield production of liquid fuels.23 Dai et al. reported a CuCo bimetallic catalyst prepared via the electrodeposition of cobalt on the surface of copper to improve the selectivity of formic acid from CO2 electroreduction.24 Previous studies have shown that the selection of a cobalt oxidation state can tune the activity and selectivity of hydrocarbon production from carbon dioxide.25–27 In another study, it was reported that a well-balanced coexistence of Co and CoO on SiO2 support exhibited high methanol selectivity in the CO2 hydrogenation reaction.28
Despite its widespread use in CO2 hydrogenation applications, the understanding of the structural evolution of the catalytically active sites and the related pathways of the bimetallic CoCu-based catalyst during the CO2 hydrogenation process remain to be elucidated.21,25,26,29,30 Therefore, to fill in this gap, in this paper, bimetallic cobalt–copper on silica support catalysts (CoxCu/SiO2) was prepared and employed as a model catalyst to obtain deeper mechanistic insights into the active sites, the synergistic effect between each component, and the reaction pathway of CO2 to formate using a combination of in situ X-ray absorption spectroscopy and density functional theory calculations. It was found that carbonate intermediate formation was the reaction rate determining step, which provided a design principle for the industrial development of effective CoCu bimetallic catalysts for the conversion of CO2 to formate.
2. Experimental sections
2.1 Chemicals
Copper nitrate trihydrate (Cu(NO3)2·3H2O) was purchased from Strem Chemicals Inc., USA. Sodium hydroxide (NaOH), sodium bicarbonate (NaHCO3), sodium carbonate (Na2CO3), and cobalt nitrate hexahydrate (Co(NO3)2·6H2O) were purchased from Sigma Aldrich Inc., USA. Formic acid (HCOOH) was purchased from Alfa Aesar, Inc., USA. Deuterium oxide (D2O) was purchased from Cambridge Isotope Laboratories, Inc. (USA). Amorphous silicon dioxide (SiO2) was supplied by Aerosil Co., Ltd. Deionized water was produced by applying a Milli-Q lab water system.2.2 Preparation of CoxCu/SiO2
A simple co-impregnation method was utilized to introduce Co and Cu on the SiO2 substrate. SiO2 powder was dispersed in deionized water and transferred to a 250 ml one-necked flask. Subsequently, an aqueous solution of cobalt nitrate is added dropwise into a solution containing dissolved copper nitrate (total metal content 5 wt% based on SiO2), resulting in the formation of a pink suspension liquid. The mixture solution was stirred at room temperature for 1 hour before being subjected to rotary evaporation at 60 °C, 40 rpm, and 90 mbar to remove the solvent. The obtained pink solid was dried at 100 °C in an oven overnight. The samples were then calcined at 500 °C for 4 hours and ground into a black powdery product named CoxCuOy/SiO2 (where x is the molar ratio of Co relative to Cu and Oy denotes oxides). CoxCuOy/SiO2 was reduced by 30 mL min−1 of 5 vol% H2/Ar mixed gas for 2 h at 350 °C with a heating rate of 5 °C min−1 to obtain bimetallic CoxCu/SiO2 catalysts.2.3 Material characterization of CoxCu/SiO2
The element content of CoxCuOy/SiO2 was analyzed via X-ray fluorescence (XRF) spectroscopy using a loose powder method (Bruker, XRF S8 TIGER Series). The crystal structures of CoxCuOy/SiO2 and CoxCu/SiO2 were obtained using powder X-ray diffraction (XRD) with a Cu-Kα source (Kα = 0.154 nm) on a Bruker D8 Advance X-ray diffractometer. Element valence and existing species in monumental or bimetal were collected by X-ray photoelectron spectroscopy utilizing an XSAM800 XPS with an Mg-Kα source. The morphology and lattice information were examined using high resolution transmission electron microscopy (HRTEM) carried out on JEM-2100F with energy dispersive X-ray spectroscopy (EDX), which was utilized to describe elemental distribution on CoxCu/SiO2. The Co K-edge and Cu K-edge X-ray absorption near-edge structure (XANES), along with the extended X-ray absorption fine structure (EXAFS), of the CoxCuOy/SiO2 composite sample were measured at the XAFCA beamline of the Singapore Synchrotron Light Source (SSLS). The storage ring at the SSLS was operated with an energy level of E = 700 MeV, alongside a maximum electron beam current of Imax = 120 mA. Co foil and Cu foil were employed as standard reference materials to perform the energy calibration.The storage ring at the SSLS was operated with an energy level of E = 700 MeV, alongside a maximum electron beam current of Imax = 120 mA. Co foil and Cu foil were employed as standard reference materials to perform the energy calibration. In situ X-ray absorption fine structure (XAFS) spectra of the Co K-edge and Cu K-edge for CoCu/SiO2 and Cu/SiO2 samples were collected to study the local structure evolution during the catalytic reaction. A 60 mg catalyst sample was mixed with 40 mg of boron nitride for the testing. The in situ reduction was carried out using linear heating in a 10 v% H2/Ar atmosphere, with a heating rate of 8 °C min−1 to 350 °C. The XAFS data were collected every 5 minutes during the reaction.
The H2-temperature programmed reduction (TPR) of CoxCu/SiO2 catalysts was performed using a Thermo Scientific TPDRO 1100 analyzer equipped with a thermal conductivity detector. In a typical experimental protocol, 100 mg of CoxCuOy/SiO2 underwent pretreatment under a continuous flow of argon (30 mL min−1) at 400 °C for 1 hour. After the cooling phase reached 50 °C, a gas mixture of 5 vol% H2 in argon was introduced at a flow rate of 30 mL min−1. The thermal conditions were then systematically elevated from 50 °C to 800 °C at a controlled rate of 10 °C min−1.
2.4 CO2 hydrogenation reaction
In general, a 100 mL Parr reactor comprises 30 mL of 1.5 M NaOH and 500 mg of the pre-reduced catalyst at 350 °C for 2 h under 5% H2–Ar mixed gas, and it is subjected to three cycles of purging with CO2 to eliminate any residual air prior to being pressurized with 10 bar of CO2 and 30 bar of H2. The reaction temperature was maintained at 200 °C; agitation velocity was set at 300 rpm, and the duration of the reaction was extended to 20 hours. Upon completion of the reaction, it is imperative to separate the liquid phase of the reaction mixture utilizing a nylon filtration apparatus. The liquid product underwent analysis via high-performance liquid chromatography (HPLC) and liquid nuclear magnetic resonance (NMR). This product was diluted 10-fold with deionized water and subsequently injected into HPLC (Shimadzu 2013, Japan) utilizing a Bio-Rad Aminex HPX-87 ion exclusion column (300 mm × 7.8 mm). The mobile phase employed was 0.5 mM H2SO4 at a flow rate of 0.6 mL min−1. NMR spectra for 1H and 13C were acquired using a Bruker NMR 500 DRX spectrometer operating at 500 MHz, referenced against the proton resonance from partially deuterated D2O-d2 (δ = 4.79).31The turnover frequency (TOF) was obtained using the following equation (eqn (1)):
 | (1) |
2.5 Mechanism of CO2 hydrogenation on CoCu/SiO2
The reaction mechanism and pathway of CO2 hydrogenation on CoCu bimetals were studied via density functional theory (DFT) calculations. All DFT simulations were performed using the DMol3 program package from Materials Studio utilizing spin unrestricted DFT.32 The generalized gradient approximation, along with the Perdew–Burke–Ernzerhof equation, is utilized as the exchange–correlation functional. The employed basis set comprises a dual-valued polarization basis set that encompasses orbital polarization functions. The core processing is configured to utilize DFT semi-core pseudopotentials.33 The Monkhorst–Pack grid was 2 × 2 × 1. For geometry optimization and performance calculations, the convergence criteria were set to an energy tolerance of 1 × 10−5 Ha, a maximum force of 0.002 Ha Å−1, a maximum displacement of 0.005 Å, and a self-consistent field tolerance of 1.0 × 10−5 Ha. In this study, we select the face-centered cubic (FCC) Cu(100) as the catalyst model surface because the (100) facet is the most frequently exposed crystal face and is more active in various chemical reactions owing to its enriched unsaturated coordination numbers compared to Cu(111).34,35 The Co-promoted CoCu(100) model surface is based on the reported CoCu(100) model surface, as reported by Yu et al.36 The Cu(100) and CoCu(100) surface models used in our study are composed of four atomic layers in a 4 × 4 supercell with a 20 Å vacuum layer. The preferred adsorption configurations of all relevant species on the Cu(100) and CoCu(100) model surfaces were fully optimized during the target reaction process. The meticulously optimized and remarkably stable adsorption configurations pertaining to both the reactants and the products function effectively as the respective initial and final states within the intricate framework of the chemical reaction under investigation. Following this critical step, a comprehensive and methodologically rigorous approach known as the complete linear synchronous transition/quadratic synchronous transition (LST/QST) method is subsequently employed to systematically calculate and identify the potential transition states that may occur throughout each step of the reaction process. Additionally, in our current DFT simulations, we assumed that the adsorptive dissociation of H2 is not the rate-determining step because it can occur easily on metallic catalysts, leading to the formation of metal hydride species.37–393. Results and discussion
3.1 Preparation and characterization of CoxCu/SiO2
Fig. 1a depicts a schematic representation of the synthesis process, illustrating the formation of the cobalt–copper bimetallic catalyst following co-impregnation, involving calcination and reduction steps. The crystal structures of CoxCuOy/SiO2 and CoxCu/SiO2 were characterized via XRD, as presented in Fig. S1† and 1b. As shown in Fig. S1,† the diffraction peaks at 32.9, 37.4, 44.8, 59.0 and 65.2° can be attributed to Co3O4 (JCPDS no. 18-0408), while peaks at 2θ 35.48, 38.74, 46.28, 61.58 and 66.24° correspond to the (002), (111), (−112), (−113) and (−311) crystal planes of CuO (JCPDS 45-0937), respectively.18,40 As the Co ratio in the samples increased, the diffraction peaks corresponding to Co3O4 gradually increased. As shown in Fig. 1b, the main diffraction peak of all CoxCu/SiO2 samples is located between the Cu(111) and Co(111) facets, indicating the successful synthesis of CoCu bimetal. As shown in the magnified section of Fig. 1c, there is a notable shift in peak position with varying amounts of Co. The crystalline size of the bimetal nanoparticle is approximately 0.308 nm according to the Debye–Scherrer equation. Table S1† presents the X-ray fluorescence (XRF) analysis results, which confirm that the Co to Cu elemental ratios in CoxCu/SiO2 closely matched the intended proportions from the initial preparation. This indicates the successful synthesis of CoCu bimetallic catalysts through a straightforward co-impregnation method. Furthermore, adjusting the Co precursor ratios enables fine-tuning of the atomic ratio between Co and Cu in bimetallic catalysts.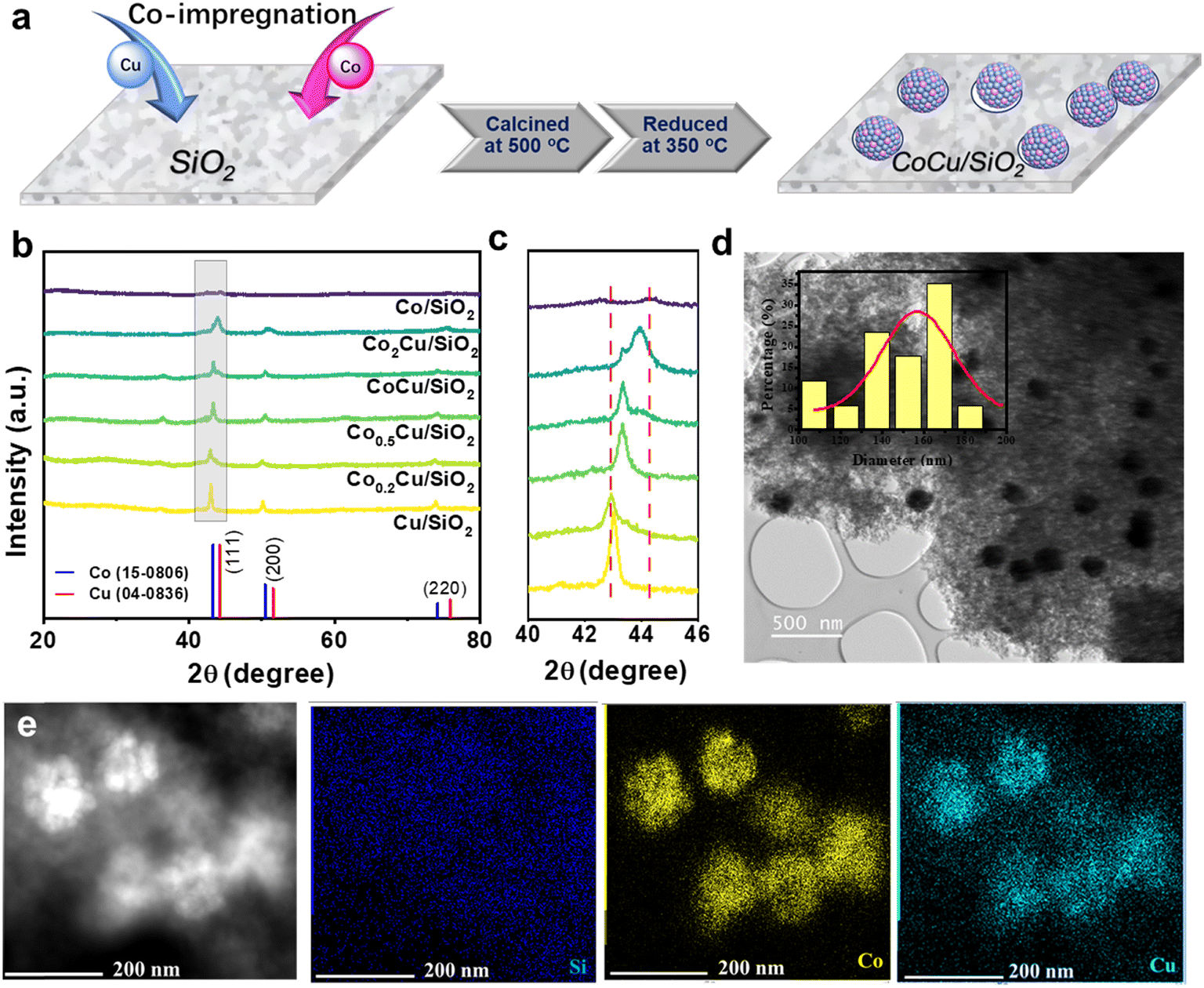 | ||
| Fig. 1 (a) Schematic of the synthesis of the CoCu/SiO2 catalyst, (b) XRD patterns of CoxCu/SiO2 catalysts, (c) enlarged XRD spectra in the selected grey region from (b), and (d) TEM image of CoCu/SiO2. The inset shows the particle size distribution of CoCu/SiO2. (e) High-angle annular dark-field scanning TEM image of CoCu/SiO2 and the corresponding element mapping images. | ||
The micromorphology of Cu/SiO2, Co/SiO2, and CoCu/SiO2 was analyzed by transmission electron microscopy (TEM), as depicted in Fig. S2† and 1d. The CoCu particles are intricately embedded within an amorphous SiO2 matrix, showing a well-defined nano-particle structure. The electron diffraction pattern of CoCu/SiO2, depicted in Fig. 1d, appears indistinct owing to the diminutive size of the crystal particles and the presence of an amorphous silica coating on the crystal surface. Additionally, TEM mapping images (Fig. 1e) show that both Co and Cu elements are dispersed evenly within the particles, demonstrating a uniform distribution of Co and Cu in bimetallic CoCu particles throughout a simple co-impregnation synthesis.
The HRTEM images of Cu/SiO2 and CoCu/SiO2 were acquired to study the lattice structure of the CoCu bimetallic catalyst. As shown in Fig. 2a, the HR-TEM of Cu/SiO2 showed a distinct lattice fringe with an interplanar distance of 0.205 nm, which corresponded to the (111) plane of pure metallic Cu. Meanwhile, an obvious moiré pattern was observed for CoCu/SiO2 samples, suggesting the high surface energy of CoCu/SiO2. The orange square marked in Fig. 2b is magnified, as displayed in Fig. 2c. A clear grain boundary can be observed, which demonstrates that there are different crystal zone axes. The dislocations and lattice strain resulted in a higher surface energy at the interface. The Fast Fourier Transform (FFT) patterns of the square regions in Fig. 2c are presented in respective colours, as shown in Fig. 2(d1–d4). The FFT diagram and IFFT taken revealed that the d-spacing of the adjacent lattice fringes were 0.21 nm (in the red region) and 0.22 nm (in the blue region), which matched that of the (111) plane of Cu and the (100) plane of Co, respectively.41
 | ||
| Fig. 2 (a) HR-TEM images of Cu/SiO2. The inset shows an inverse FFT profile. (b) HR-TEM images of CoCu/SiO2 and (c) FFT and (d) IFFT in the selected red and blue regions. | ||
H2 temperature-programmed reduction (H2-TPR), which can reveal the reducibility property, was conducted, and the corresponding profiles are presented in Fig. S3.† The numerous distinct peaks observed in the analysis correspond to various physicochemical properties inherent to the CuO particles. For instance, it can be noted that the peaks associated with lower temperatures indicate that the particles possess smaller dimensions, while the peaks observed at higher temperatures indicate either larger particles or those particles that are situated within the supporting structural matrix. The multiple peaks correspond to the different physicochemical properties of the CuO particles. For example, the lower temperature peak is associated with particles of smaller sizes, while the high-temperature peaks are related to larger particles or inside the support structure.42 Meanwhile, the reduction profile of the CoO/SiO2 catalyst shows a broad H2 consumption zone at 350–550 °C, which is attributed to the reduction of CoO to metallic Co.43 Interestingly, CoCu/SiO2 showed a single peak at the same temperature as Cu/SiO2 at 260 °C, suggesting the presence of a well-distributed bimetallic phase with high homogeneity in particle size.
In situ XANES and EXAFS of Cu and Co K-edges were employed to characterize the transformation of Cu and Co species during reduction. As shown in Fig. 3a, the Cu K edge XANES spectra indicate that the rising edges possess a red shift, while the white line gradually diminishes with an increase in temperature. The final state of Cu resembles that of Cu foil. A similar phenomenon is observed in the Co K-edge XANES, as shown in Fig. 3b, with the spectra pattern converging towards metallic Co as the temperature rises. This observation suggests that the active catalytic species of the reduced catalyst comprise a bimetal of Co and Cu. In Fig. 3c, the Cu K-edge EXAFS provides a more intuitive depiction, indicating that the primary peaks below 260 °C correspond to the Cu–O scattering path in the first shell and the Cu–Cu scattering path in the second shell. Beyond 260 °C, distinct Cu–Cu bonds of metallic Cu become prominent. This finding aligns with the conclusions drawn from the H2-TPR analysis, which suggests that the reduction in Cu occurs at around 260 °C.
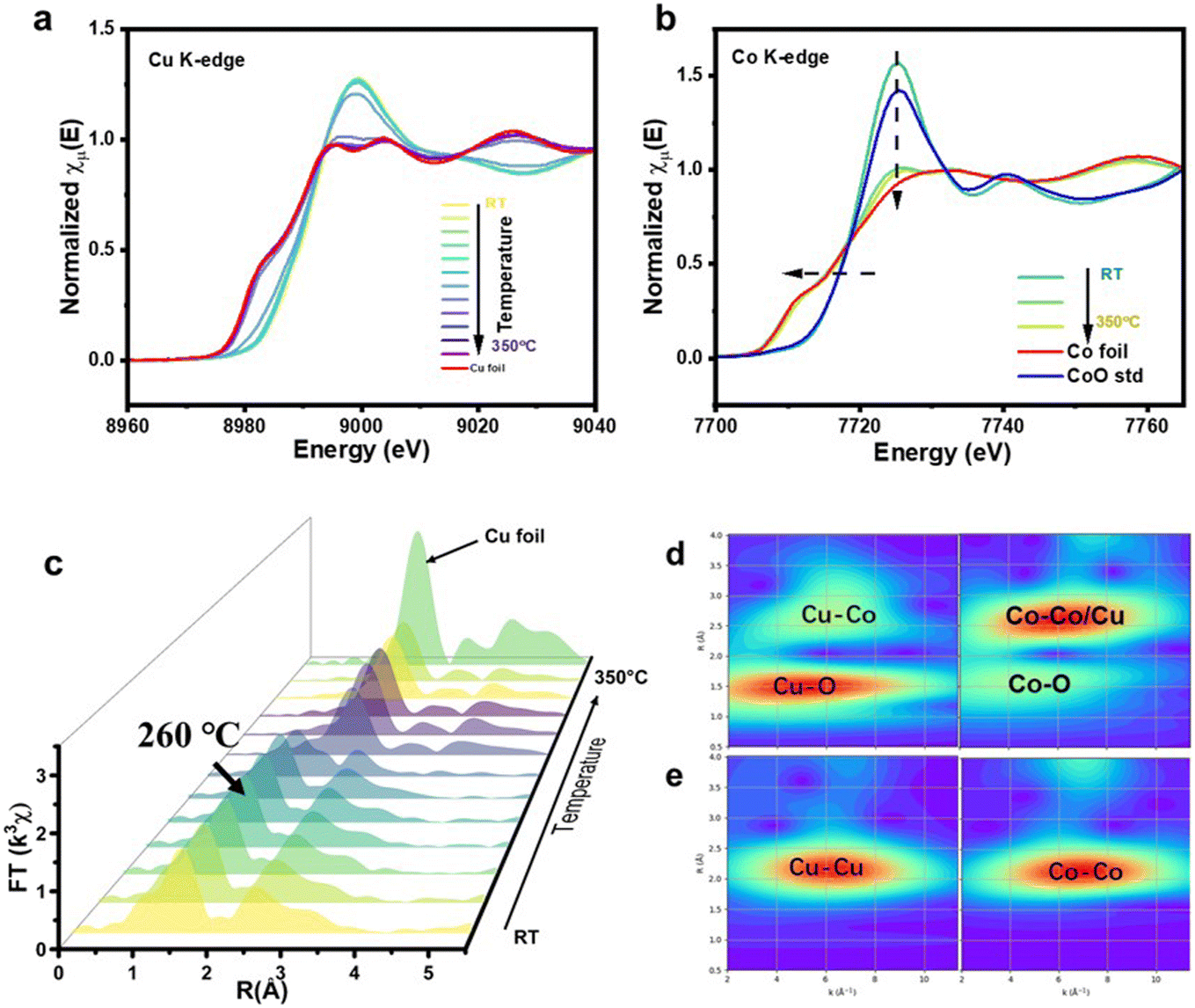 | ||
| Fig. 3 (a) Normalized K-edge XANES spectra of Cu at different temperatures ranging from 25 °C to 350 °C (in 40° increments). (b) Normalized K-edge XANES spectra of Co at 25 °C, 265 °C, and 350 °C. (c) Fourier transform of k2-weighed EXAFS spectra of Cu at different temperatures. (d and e) Wave transform of Cu (left) and Co (right) K-edge with k2 weight at (d) room temperature and (e) 350 °C. | ||
3.2 Catalytic CO2 hydrogenation performance of CoxCu/SiO2 catalyst
To determine the optimal reaction conditions for maximizing product yield, the difference, reaction pressure, and reaction temperature as well as Co and Cu molar ratios were first investigated. The resulting liquid products were qualitatively and quantitatively analyzed using HPLC. As shown in Fig. S5,† formate was found to be the only product under all the conditions investigated. Fig. S6† provides the 1H and 13C NMR spectra illustrating the chemical composition of the liquid product after the hydrogenation reaction. Using deuterium oxide (D2O) as the solvent, it was discerned that a solitary peak emerged, which was characterized by a chemical shift value of 171.08 in the 13C NMR spectrum. This specific peak can be confidently attributed to the presence of the carbon peak inherent to the formate. In the 1H NMR spectrum, a single peak at a chemical shift of 8.37 is evident, which can be unequivocally associated with the hydrogen atom within the carboxylate group present in formate. The results of the NMR spectra underscore the superior selectivity demonstrated by the CoCu/SiO2 catalyst in facilitating the conversion of CO2 into formate through the process of hydrogenation. Fig. S7† suggests that less than 0.1% of CH4 was generated.In Fig. 4a, when using CoCu/SiO2 as the catalyst, it was found that a H2:CO2 pressure ratio of 30:10 at a total pressure of 40 bar (30 bar of H2 and 10 bar of CO2) produced the highest formate yield. Meanwhile, as the reaction temperature increases, a higher yield is observed, as shown in Fig. 4b. This is attributed to thermodynamic constraints, where increasing the temperature is beneficial for the reaction. Based on this preliminary investigation, CoCu/SiO2 (Co:Cu ratio of 1:1), with reaction conditions of 30 bar of H2 and 10 bar of CO2 at 200 °C, gave the highest formate yield (2.3 mmol g−1 h−1) and the turn-over frequency (TOF) of 625.2 h−1. The impact of base additives, specifically NaOH, Na2CO3, and NaHCO3, was also studied using CoCu/SiO2 as the representative catalyst under two conditions: (1) with only pure H2 supplied, and (2) with a H2:CO2 ratio of 3:1. As shown in Fig. 4c, it is evident that no formate can be produced in the presence of NaOH when only pure H2 is supplied. However, with Na2CO3 and NaHCO3 under pure H2 conditions, a moderate amount of formate was obtained. This interesting finding suggests that carbonate or bicarbonate species could potentially act as carbon sources in the hydrogenation process. The relatively lower formate yield in NaHCO3 (0.39 mmol g−1 h−1) than that in Na2CO3 (1.23 mmol g−1 h−1) under pure hydrogen conditions is potentially due to the low saturated concentration of NaHCO3 at 1.1 M (at 20 °C), so a 1.5 M concentration could be applied. It is widely recognized that in heterogeneous catalytic CO2 hydrogenation systems, CO2 reacts with an aqueous dissolved base, such as KOH or NaOH, to form (bi)carbonate ions, which then act as the actual precursors for further hydrogenation into formate.44 When a mixture of H2 and CO2 was introduced, the formate production rate followed the order NaOH > Na2CO3 > NaHCO3 in this work. This stark contrast indicates that the hydrogenation of CO2 in the gas phase is more favored and feasible than hydrogenation processes involving carbonate or bicarbonate species under identical reaction conditions. This is likely due to the thermodynamic barrier encountered in hydrogenation through bicarbonate intermediates. The more stable nature of the carbonate/bicarbonate species compared to CO2 and the final formate makes it less favorable for further hydrogenation.44 These data provide valuable insights into the intricate nature of hydrogenation reactions and the role of different base additives in influencing reactivity.
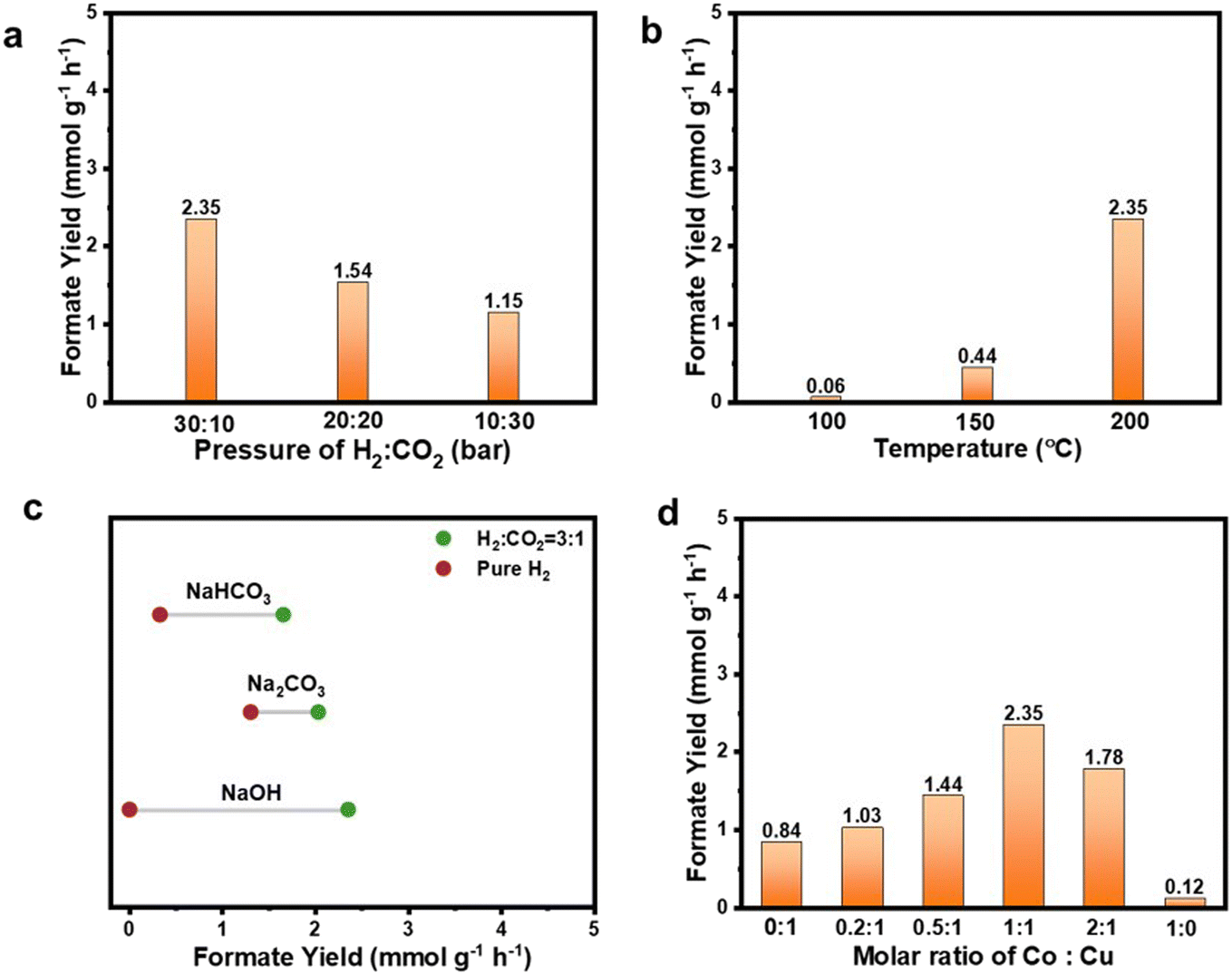 | ||
| Fig. 4 (a) Formate production rate on CoCu/SiO2 with (a) different pressure ratios of CO2/H2 at 200 °C in NaOH, (b) different reaction temperatures in NaOH with H2:CO2 of 3:1, and (c) different base additives at 200 °C with H2:CO2 of 3:1 or pure H2. (d) Formate production rate on CoxCu/SiO2 with different Co and Cu molar ratios at 200 °C in NaOH with H2:CO2 of 3:1. | ||
The effect of different Co:Cu molar ratios on productivity is also investigated. As shown in Fig. 4d, the CO2 hydrogenation products of Cu/SiO2 and Co/SiO2 were primarily trace amounts of formate with rates of 0.12 and 0.8 mmol g−1 h−1, respectively. Compared with pure Cu/SiO2 and Co/SiO2, all CoxCu/SiO2 improved hydrogenation activity. Formate yield increased with higher concentrations of Co but started to decline once the Co to Cu ratio surpassed 1:1. The highest formate yield of 2.3 mmol g−1 h−1 achieved with the Co to Cu molar ratio was 1:1 (CoCu/SiO2).
To understand the underlying different formate yields when the Co:Cu ratio varies, the surface chemical properties of the mono- and bi-metallic CoxCu/SiO2 catalysts were analyzed by XPS. Fig. 5a shows the Cu 2p spectra for Cu/SiO2 and CoxCu/SiO2. The Cu 2p3/2 peak at 934.5 eV can be assigned to Cuδ+ species. With the increase in Co content, the Cu peak shifted to a lower binding energy, suggesting that the Cu species were in an electron-rich environment induced by the interfacial electron transfer from Co to Cu.45 However, this effect became less pronounced for CoCu/SiO2 and Co2Cu/SiO2 likely owing to weaker Co–Cu interactions as Co became the predominant component.43 Meanwhile, the Co 2p in CoxCu/SiO2 was not detected by XPS until the Co content was increased high enough in CoCu/SiO2. The peak appearing at 781 eV can be attributed to Coδ+ (Fig. S4†), further confirming its electron-deficient environment.
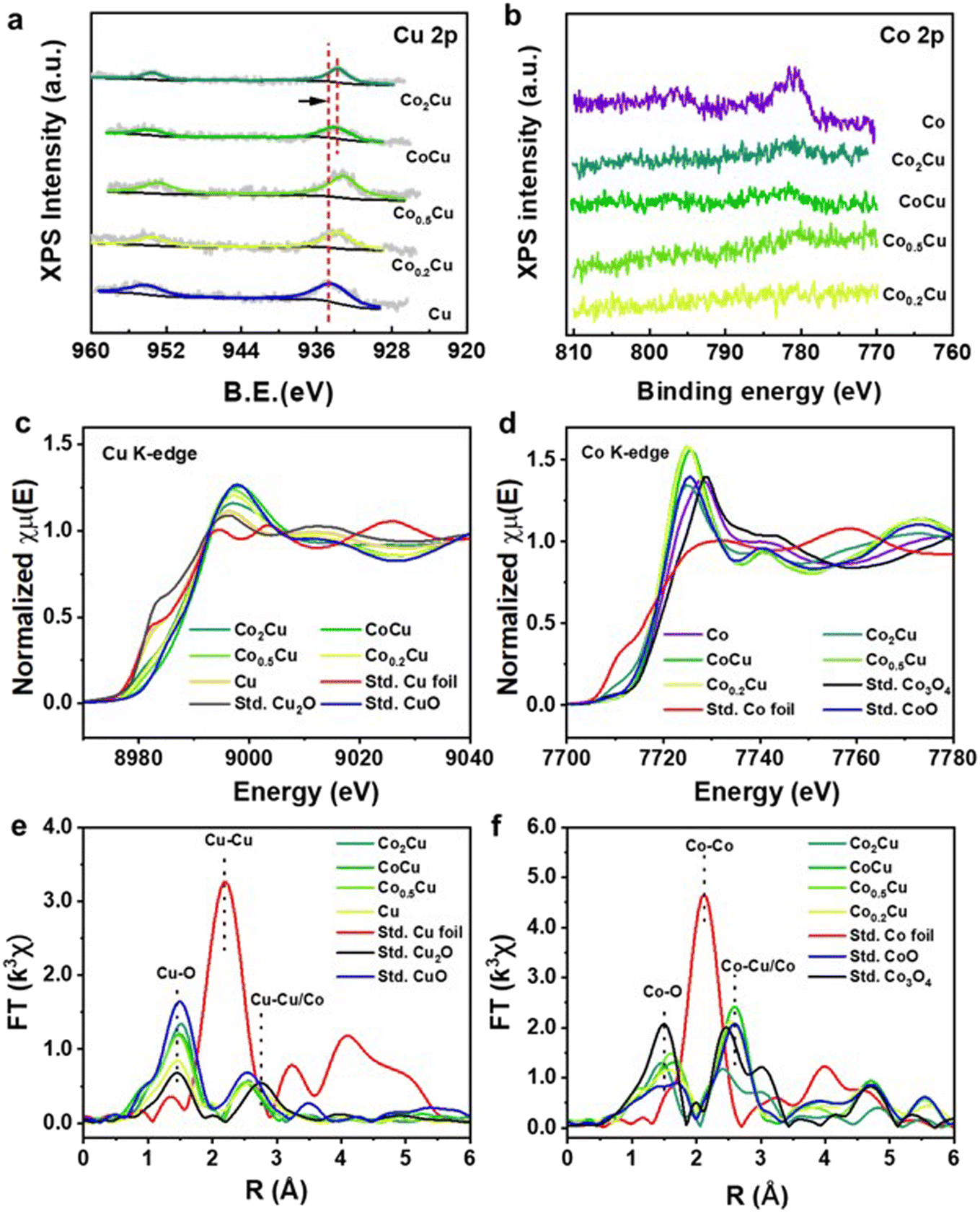 | ||
| Fig. 5 XPS spectra for (a) Cu 2p and (b) Co 2p of CoxCu/SiO2 with different Co contents. Normalized K-edge XANES spectra of (c) Cu and (d) Co. Fourier transform of k3-weighed EXAFS spectra of (e) Cu and (f) Co for CoxCu/SiO2 with different Co:Cu ratios. | ||
Ex situ XANES and EXAFS of Cu and Co K-edge were used to explore further the coordination environment of the Co and Cu species in CoxCu/SiO2 catalysts. Fig. 5c compares CoxCu/SiO2, CuxO and Cu foil. The K edge XANES of Cu in CoCu/SiO2 possesses a higher energy rising edge and is close to the CuO reference, suggesting that the valence of Cu in CoCu/SiO2 is close to +2. The white line (8998 eV) intensity of Cu K-edge XANES in CoCu/SiO2 was the highest among all the CoxCu/SiO2, suggesting that the Cu species in this sample is relatively electron-rich and has a higher valence state compared to the others.46 As shown in Fig. 5d, the rising edge of the Co K-edge XANES for Co in CoCu/SiO2 is similar to that of CoO and lies between those of Co3O4 and Co foil, suggesting that the average valence of Co in CoCu/SiO2 is below +2.7 and close to +2. This conclusion aligns with the results obtained from XPS analysis. As shown in Fig. 5e, the Fourier transform (FT) of the Cu K-edge EXAFS of the CuO reference shows a dominant peak of the Cu–Cu scattering path at 2.75 Å. With the introduction of Co, the peak of Cu–Cu/Co shifted to a lower R, implying that Co–Cu bimetallic oxide was formed. Furthermore, combining the Cu K-edge and Co K-edge XAFS spectra, it becomes evident that there is a transfer of electrons occurring between the copper and cobalt species, highlighting the intricate interplay between different metal species and their respective valence states. We speculate that the high electron density at Cu centers aids in the improvement of CO2 hydrogenation into formic acid. Such Cu sites enhance the electronegativity of dissociated hydride species. This, in turn, enhances the reactivity of nucleophilic attack on the carbon center of CO2.44,47,48 A similar observation has also been reported by Mori et al., who demonstrated that neighbouring Ag atoms in a PdAg catalyst help create an electron-rich Pd center, leading to an increase in the electronegativity of dissociated hydride species, thereby enhancing the reaction performance.48 Meanwhile, Xu et al. explicitly pointed out that owing to the electron-rich surface of MoC, the activation energy was reduced considerably, facilitating CO2 hydrogenation into formic acid.49
Next, the DFT methodology was systematically utilized to investigate the intricate reaction energetics associated with the process of carbon dioxide hydrogenation, specifically aimed at the synthesis of formate occurring over the catalytic surfaces of both Cu/SiO2 and the optimized CuCo/SiO2 configurations. Furthermore, the resultant potential energy diagram, along with the schematic representations of the transition states relevant to this reaction, is meticulously illustrated in Fig. 6. Typically, under alkaline conditions, the reduction of HCO3-bicarbonate species to formate was reported as the rate-determining step that determines the overall rate of CO2 hydrogenation reactions.39 In this process, the active H atom on the metal catalysts attacks the C atom of the HCO3-species rather than the O atom, resulting in the generation of chemisorbed formate. As shown in Fig. 6, cobalt promoters lowered the barrier energy of Cu(100) by ∼58%, from 279.1 to 162.3 kJ mol−1 for the hydrogenation of adsorbed HCO3-bicarbonates. Additionally, the cobalt promoters of the CoCu(100) model resulted in an exothermic reaction energy of −9.6 kJ mol−1 in contrast to the endothermic reaction energy of pristine Cu(100) at 44.9 kJ mol−1. Thus, the CO2 hydrogenation under alkaline conditions was thermodynamically more favorable at the CoCu(100) than at Cu(100) catalyst surfaces. Hence, our DFT calculations showed that Co-promoted Cu catalysts can enhance the catalytic hydrogenation of CO2 by reducing the barrier energy of the hydrogenation of bicarbonate species.
 | ||
| Fig. 6 Reaction pathway for the hydrogenation of bicarbonate into formate species: (a) Cu(100) model catalyst surface and (b) CoCu(100) model catalyst surface, as determined using the LST/QST method. “Reactant” comprised the model catalyst surface with adsorbed bicarbonate and 2 dissociated H-atoms. “TS” refers to the transition state. “Product” comprised the model catalyst surface with adsorbed formate and 1 H-atom. White ball = hydrogen, red ball = oxygen, purple ball = cobalt and brown ball = copper. The figures are not drawn to scale. | ||
In addition to reducing the barrier energy of hydrogenation of bicarbonate intermediates, a previous work reported that a Co-promoted Cu catalyst is beneficial for stabilizing CO2 and dissociating H2 on CoCu(100) surfaces. For example, Qiu et al.50 reported that the adsorption energies of CO2 on the Cu(100) surface increased from −49.4 kJ mol−1 to 37.7 kJ mol−1 when it was doped with Co atoms. In other words, CO2 can be stabilized much better on the Co-doped Cu(100) than on the pure Cu(100) surfaces and can improve the stability of the chemisorption of CO2. When H2 adsorbs on Cu(100), it typically dissociates into individual hydrogen atoms.51 Furthermore, the addition of Co-dopant onto the Cu(100) surfaces reduced the hydrogen dissociation energy barrier by 0.15 eV (from 0.54 to 0.39 eV),52 and making H2 molecules easier to dissociate as atomic H on bimetallic CoCu(100) surfaces.
Conclusion
In this work, bimetallic CoCu on a silica support was prepared and employed as a catalyst for CO2 hydrogenation. The effects of metal ratio and reaction parameters, including temperature, pressure, and base additives, were also thoroughly investigated. Formate, an important energy carrier, was found to be the sole product under all the conditions. It is found that the CoCu/SiO2 with a molar ratio of Co to Cu at 1:1 exhibited the highest formate production rate of 2.3 mmol g−1 h−1 at 200 °C, 40 bar (H2:CO2 of 3:1) and NaOH as the base additive. Investigation into electronic properties reveals that cobalt acts as a promoter to enhance electron density around Cu sites, which in turn facilitates the overall hydrogenation performance. Furthermore, the bimetallic catalyst possesses a lower adsorption energy barrier than the monometallic Cu, which provides a reliable catalytic mechanism for the conversion of CO2 to formate on the CoCu bimetallic catalyst. This study elucidates the catalytic mechanism, clarifies the reaction pathway of CO2 hydrogenation, and provides advice for the development of non-precious metal catalysts for the conversion of CO2 into formate.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors thank the China Scholarship Council (CSC) for providing the PhD scholarship and ISCE2 (A-STAR, Singapore) for providing financial support.References
- T. M. Gür, Carbon Dioxide Emissions, Capture, Storage and Utilization: Review of Materials, Processes and Technologies, Prog. Energy Combust. Sci., 2022, 89, 100965 CrossRef
.
- C. Song, Global challenges and strategies for control, conversion and utilization of CO2 for sustainable development involving energy, catalysis, adsorption and chemical processing, Catal. Today, 2006, 115, 232 CrossRef
- S. Ahn, K. Park, K. R. Lee, A. Haider, C. V. Nguyen, H. Jin, S. J. Yoo, S. Yoon and K.-D. Jung, Atomically dispersed Ru(III) on N-doped mesoporous carbon hollow spheres as catalysts for CO2 hydrogenation to formate, Chem. Eng. J., 2022, 442, 136185 CrossRef CAS
- M. Ding, R. W. Flaig, H. L. Jiang and O. M. Yaghi, Carbon capture and conversion using metal-organic frameworks and MOF-based materials, Chem. Soc. Rev., 2019, 48(10), 2783–2828 RSC
- A. Banerjee, G. R. Dick, T. Yoshino and M. W. Kanan, Carbon dioxide utilization via carbonate-promoted C-H carboxylation, Nature, 2016, 531(7593), 215–219 CrossRef CAS PubMed
- R. Li, J. Hu, M. Deng, H. Wang, X. Wang, Y. Hu, H. L. Jiang, J. Jiang, Q. Zhang, Y. Xie and Y. Xiong, Integration of an inorganic semiconductor with a metal-organic framework: a platform for enhanced gaseous photocatalytic reactions, Adv. Mater., 2014, 26(28), 4783–4788 CrossRef CAS
- D. Wang, R. Huang, W. Liu, D. Sun and Z. Li, Fe-Based MOFs for Photocatalytic CO2 Reduction: Role of Coordination Unsaturated Sites and Dual Excitation Pathways, ACS Catal., 2014, 4(12), 4254–4260 CrossRef CAS
- R. Senthil Kumar, S. Senthil Kumar and M. Anbu Kulandainathan, Highly selective electrochemical reduction of carbon dioxide using Cu based metal organic framework as an electrocatalyst, Electrochem. Commun., 2012, 25, 70–73 CrossRef CAS
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst, Nat. Chem., 2017, 9(10), 1019–1024 CrossRef CAS
- A. Y. Khodakov, W. Chu and P. Fongarland, Advances in the development of novel cobalt Fischer-Tropsch catalysts for synthesis of long-chain hydrocarbons and clean fuels, Chem. Rev., 2007, 107(5), 1692–1744 CrossRef CAS PubMed
- X. Xi, F. Zeng, H. Zhang, X. Wu, J. Ren, T. Bisswanger, C. Stampfer, J. P. Hofmann, R. Palkovits and H. J. Heeres, CO2 Hydrogenation to Higher Alcohols over K-Promoted Bimetallic Fe–In Catalysts on a Ce–ZrO2 Support, ACS Sustainable Chem. Eng., 2021, 9(18), 6235–6249 CrossRef CAS
- K. Lee, H. Yan, Q. Sun, Z. Zhang and N. Yan, Mechanism-Guided Catalyst Design for CO2 Hydrogenation to Formate and Methanol, Acc. Mater. Res., 2023, 4(9), 746–757 CrossRef CAS
- J. Zhao, J. Wang, Y. Bai, H. Du, J. Yang, B. Yin, B. Jiang and H. Li, Mesoporous Ru(Co, Ni)B bimetallic amorphous alloy for CO2 hydrogenation to formic acid, J. CO2 Util., 2023, 74, 102528 CrossRef CAS
- K. Schuchmann and V. Muller, Direct and Reversible Hydrogenationof CO2 to Formate by a BacterialCarbon Dioxide Reductase, Science, 2013, 342(6164), 1382–1385 CAS
- G. Liu, P. Poths, X. Zhang, Z. Zhu, M. Marshall, M. Blankenhorn, A. N. Alexandrova and K. H. Bowen, CO2 Hydrogenation to Formate and Formic Acid by Bimetallic Palladium-Copper Hydride Clusters, J. Am. Chem. Soc., 2020, 142(17), 7930–7936 CrossRef CAS PubMed
- A. Jaleel, A. Haider, C. V. Nguyen, K. R. Lee, S. Choung, J. W. Han, S.-H. Baek, C.-H. Shin and K.-D. Jung, Structural effect of Nitrogen/Carbon on the stability of anchored Ru catalysts for CO2 hydrogenation to formate, Chem. Eng. J., 2022, 433, 133571 CAS
- S. Ghoshal, P. Roy, A. Pramanik and P. Sarkar, Ru/Rh catalyzed selective hydrogenation of CO2 to formic acid: a first principles microkinetics analysis, Catal. Sci. Technol., 2022, 12(23), 7219–7232 CAS
- X. Xiao, J. Gao, S. Xi, S. H. Lim, A. K. W. Png, A. Borgna, W. Chu and Y. Liu, Experimental and in situ DRIFTs studies on confined metallic copper stabilized Pd species for enhanced CO2 reduction to formate, Appl. Catal., B, 2022, 309, 121239 CAS
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. Fernandez Sanz and J. A. Rodriguez, Catalysis. Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2, Science, 2014, 345(6196), 546–550 CAS
- Y. Liu, Y. Yang, Q. Sun, Z. Wang, B. Huang, Y. Dai, X. Qin and X. Zhang, Chemical adsorption enhanced CO2 capture and photoreduction over a copper porphyrin based metal organic framework, ACS Appl. Mater. Interfaces, 2013, 5(15), 7654–7658 CAS
- Z. Sun, X. Nie and Z. Zhao, Computational Design of a CoCu-Modified Indium Oxide Catalyst Promoting CO2 Activation and Hydrogenation through Electronic Regulation, Energy Fuels, 2022, 36(14), 7915–7920 CAS
- T. Fan, H. Liu, S. Shao, Y. Gong, G. Li and Z. Tang, Cobalt Catalysts Enable Selective Hydrogenation of CO2 toward Diverse Products: Recent Progress and Perspective, J. Phys. Chem. Lett., 2021, 12(43), 10486–10496 CrossRef CAS PubMed
- L. Guo, X. Gao, W. Gao, H. Wu, X. Wang, S. Sun, Y. Wei, Y. Kugue, X. Guo, J. Sun and N. Tsubaki, High-yield production of liquid fuels in CO2 hydrogenation on a zeolite-free Fe-based catalyst, Chem. Sci., 2022, 14(1), 171–178 RSC
- C. Dai, L. Sun, J. Song, H. Liao, A. C. Fisher and Z. J. Xu, Selective Electroreduction of Carbon Dioxide to Formic Acid on Cobalt-Decorated Copper Thin Films, Small Methods, 2019, 3(11), 1900362 CrossRef CAS
- I. C. T. Have, J. J. G. Kromwijk, M. Monai, D. Ferri, E. B. Sterk, F. Meirer and B. M. Weckhuysen, Uncovering the reaction mechanism behind CoO as active phase for CO2 hydrogenation, Nat. Commun., 2022, 13(1), 324 CrossRef CAS PubMed
- M. Wang, G. Zhang, J. Zhu, W. Li, J. Wang, K. Bian, Y. Liu, F. Ding, C. Song and X. Guo, Unraveling the tunable selectivity on cobalt oxide and metallic cobalt sites for CO2 hydrogenation, Chem. Eng. J., 2022, 446, 137217 CrossRef CAS
- S. Liu, Y. He, W. Fu, J. Ren, J. Chen, H. Chen, R. Sun, Z. Tang, C. Mebrahtu and F. Zeng, Synergy of Co0-Co2+ in cobalt-based catalysts for CO2 hydrogenation: Quantifying via reduced and exposed atoms fraction, Appl. Catal., A, 2024, 670, 119549 CrossRef CAS
- L. Wang, E. Guan, Y. Wang, L. Wang, Z. Gong, Y. Cui, X. Meng, B. C. Gates and F. S. Xiao, Silica accelerates the selective hydrogenation of CO2 to methanol on cobalt catalysts, Nat. Commun., 2020, 11(1), 1033 CrossRef CAS PubMed
- Z. Wang, C. Yang, X. Li, X. Song, C. Pei, Z.-J. Zhao and J. Gong, The role of CO2 dissociation in CO2 hydrogenation to ethanol on CoCu/silica catalysts, Nano Res., 2022, 16(5), 6128–6133 Search PubMed
- S. Liu, C. Yang, S. Zha, D. Sharapa, F. Studt, Z. J. Zhao and J. Gong, Moderate Surface Segregation Promotes Selective Ethanol Production in CO2 Hydrogenation Reaction over CoCu Catalysts, Angew. Chem., Int. Ed., 2022, 61(2), e202109027 CrossRef CAS PubMed
- B. An, Z. Li, Y. Song, J. Zhang, L. Zeng, C. Wang and W. Lin, Cooperative copper centres in a metal–organic framework for selective conversion of CO2 to ethanol, Nat. Catal., 2019, 2(8), 709–717 CrossRef CAS
- B. Delley, An all-electron numerical method for solving the local density functional for polyatomic molecules, J. Chem. Phys., 1990, 92(1), 508–517 CAS
- B. Delley, Hardness conserving semilocal pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155125 CrossRef
- L. Vitos, A. V. Ruban, H. L. Skriver and J. Kollár, The surface energy of metals, Surf. Sci., 1998, 411(1–2), 186–202 CrossRef CAS
- G.-C. Wang and J. Nakamura, Structure Sensitivity for Forward and Reverse Water-Gas Shift Reactions on Copper Surfaces: A DFT Study, J. Phys. Chem. Lett., 2010, 1(20), 3053–3057 CrossRef CAS
- Y. Yu, J. Zhang, X. Sun and M. Zhang, Carbon chain growth mechanism of higher alcohols synthesis from syngas on CoCu(100): A combined DFT and kMC study, Surf. Sci., 2020, 691, 121513 CrossRef CAS
- G. A. Filonenko, W. L. Vrijburg, E. J. M. Hensen and E. A. Pidko, On the activity of supported Au catalysts in the liquid phase hydrogenation of CO2 to formates, J. Catal., 2016, 343, 97–105 CrossRef CAS
- S. Masuda, K. Mori, Y. Futamura and H. Yamashita, PdAg Nanoparticles Supported on Functionalized Mesoporous Carbon: Promotional Effect of Surface Amine Groups in Reversible Hydrogen Delivery/Storage Mediated by Formic Acid/CO2, ACS Catal., 2018, 8(3), 2277–2285 CrossRef CAS
- K. Mori, S. Masuda, H. Tanaka, K. Yoshizawa, M. Che and H. Yamashita, Phenylamine-functionalized mesoporous silica supported PdAg nanoparticles: a dual heterogeneous catalyst for formic acid/CO2-mediated chemical hydrogen delivery/storage, Chem. Commun., 2017, 53(34), 4677–4680 RSC
- R. Nazir, A. Khalfani, O. Abdelfattah, A. Kumar, M. A. Saleh Saad and S. Ali, Nanosheet Synthesis of Mixed Co3O4/CuO via Combustion Method for Methanol Oxidation and Carbon Dioxide Reduction, Langmuir, 2020, 36(42), 12760–12771 CrossRef CAS PubMed
- J. R. Deka, D. Saikia, N.-F. Lu, K.-T. Chen, H.-M. Kao and Y.-C. Yang, Space confined synthesis of highly dispersed bimetallic CoCu nanoparticles as effective catalysts for ammonia borane dehydrogenation and 4-nitrophenol reduction, Appl. Surf. Sci., 2021, 538, 148091 CAS
- L. H. Vieira, M. A. Rossi, L. F. Rasteiro, J. M. Assaf and E. M. Assaf, CO2 Hydrogenation to Methanol over Mesoporous SiO2-Coated Cu-Based Catalysts, ACS Nanosci. Au, 2024, 4(4), 235–242 CAS
- Y. Wang, J. Yang, Y. Sun, D. Ye, B. Shan, S. C. E. Tsang and X. Tu, Engineering Ni-Co bimetallic interfaces for ambient plasma-catalytic CO2 hydrogenation to methanol, Chem, 2024, 10(8), 2590–2606 CAS
- Q. Liu, X. Yang, L. Li, S. Miao, Y. Li, Y. Li, X. Wang, Y. Huang and T. Zhang, Direct catalytic hydrogenation of CO2 to formate over a Schiff-base-mediated gold nanocatalyst, Nat. Commun., 2017, 8(1), 1407 Search PubMed
- Y. Sheng, X. Lin, S. Yue, Y. Liu, X. Zou, X. Wang and X. Lu, Highly efficient non-noble metallic NiCu nanoalloy catalysts for hydrogenation of nitroarenes, Mater. Adv., 2021, 2(20), 6722–6730 CAS
- P. Verma, S. Zhang, S. Song, K. Mori, Y. Kuwahara, M. Wen, H. Yamashita and T. An, Recent strategies for enhancing the catalytic activity of CO2 hydrogenation to formate/formic acid over Pd-based catalyst, J. CO2 Util., 2021, 54, 101765 CAS
- B. Jin, X. Ye, H. Zhong, F. Jin and Y. H. Hu, Enhanced photocatalytic CO2 hydrogenation with wide-spectrum utilization over black TiO2 supported catalyst, Chin. Chem. Lett., 2022, 33(2), 812–816 CAS
- K. Mori, T. Sano, H. Kobayashi and H. Yamashita, Surface Engineering of a Supported PdAg Catalyst for Hydrogenation of CO2 to Formic Acid: Elucidating the Active Pd Atoms in Alloy Nanoparticles, J. Am. Chem. Soc., 2018, 140(28), 8902–8909 CAS
- D. Xu, S.-Y. Xia, Q.-Y. Li, J.-S. Chen and X.-H. Li, Hydrogen-rich surface of MoC catalysts for efficient CO2 hydrogenation induced by a coupled hydrogen donator, Nano Res., 2024, 17(8), 7762–7767 CAS
- M. Qiu, Z. Fang, Y. Li, J. Zhu, X. Huang, K. Ding, W. Chen and Y. Zhang, First-principles investigation of the activation of CO2 molecule on TM/Cu (TM=Fe, Co and Ni) surface alloys, Appl. Surf. Sci., 2015, 353, 902–912 CAS
- P. B. Rasmussen, P. M. Holmblad, H. Christoffersen, P. A. Taylor and I. Chorkendorff, Dissociative adsorption of hydrogen on Cu(100) at low-temperatures, Surf. Sci., 1993, 287, 78–93 Search PubMed
- Y. J. Li, X. J. Liu, L. Li, Z. J. Zhang and X. J. Shen, Theoretical new insights into hydrogen interaction with single-atom Zn- and Co-doped copper metal catalysts, Appl. Surf. Sci., 2021, 551, 149365 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00017c |
| This journal is © The Royal Society of Chemistry 2025 |