
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of HCHO, CO and H2 by methane oxidation with O2 over Cu catalysts stabilized in silicoaluminophosphates†
Mana
Shimakawa
a,
Rieko
Nagase
a,
Ryoya
Kugo
a,
Junya
Ohyama
 b and
Sakae
Takenaka
*a
b and
Sakae
Takenaka
*a
aDepartment of Science and Engineering, Doshisha University, Tatara-miyakodani 1-3, Kyotanabe, Kyoto 610-0321, Japan. E-mail: stakenak@mail.doshisha.ac.jp
bFaculty of Advanced Science and Technology, Kumamoto University, 2-39-1 Kurokami, Chuo-ku, Kumamoto, 860-8555, Japan
First published on 13th February 2025
Abstract
The catalytic performance of Cu catalysts supported on SAPO34 (denoted as Cu/SAPO) for methane oxidation with O2 has been investigated in detail. Cu/SAPO catalysts formed H2 in addition to CO and HCHO during methane oxidation in the temperature range from 773 and 923 K, while the other 3d transition metal catalysts such as V, Cr, Mn, Fe, Co and Ni were less active for the formation of these products. The formation of HCHO, H2 and CO was also confirmed during the reaction over Cu catalysts supported on zeolites different from SAPO34, such as mordenite, faujasite, chabazite, and beta-zeolite, but the yields of these products, especially the H2 yield in the reaction over the Cu/SAPO catalyst, were significantly higher than those over other zeolite-supported Cu catalysts. In the oxidation over Cu/SAPO catalysts at 923 K, the yields of H2, CO, and HCHO reached 2.2, 5.4, and 0.7%, respectively, at a methane conversion of 8.2%. These Cu catalysts were characterized by XRD patterns and in situ UV-vis spectra. Highly dispersed Cu2+ species stabilized in SAPO34 were the active sites for methane oxidation, which were reduced to Cu+ species by methane and oxidized to Cu2+ by O2 to facilitate the reaction.
1. Introduction
Methane is distributed worldwide as a main component of natural gas and methane hydrate and it has been recognized as an abundant and clean energy resource.1,2 Thus, methane has been utilized as an energy source for the supply of heat and the generation of electricity. On the other hand, the utilization of methane as a feedstock in the chemical industry has been limited. Methane has been converted into a synthesis gas composed of a mixture of CO and H2 by reforming with H2O or CO2 (steam reforming or dry reforming) or partial oxidation with O2.3–5 Subsequently, synthesis gas can be converted to high value-added products such as methanol and hydrocarbons.6–8 While steam reforming can produce synthesis gas with a composition that is effective for producing methanol, dry reforming can contribute to reducing greenhouse gas emissions. Huge energy input is required for the production of H2 and CO through the reforming of methane with H2O and CO2 because their reactions are highly endothermic. Additionally, the reforming of methane has been operated at temperatures higher than 1073 K since methane is chemically inactive and has strong C–H bonds. The processes to produce methanol and hydrocarbons through the synthesis gas from methane thus need multiple steps inevitably. The direct conversion of methane by the oxidation with O2 into methanol, formaldehyde, CO and H2 is an ideal process from the viewpoint of green chemistry, because the reaction is exothermic and proceeds at low temperatures compared to steam reforming.9,10 Many research groups have developed catalysts for the conversion of methane into oxygenates such as methanol and formaldehyde.11–13 Methanol is formed by the oxidation of methane with O2 over Cu catalysts stabilized in various zeolites at low temperatures (at around 550 K).14–17 Atomically dispersed Cu oxides in zeolites such as mordenite and chabazite work as active sites for methanol formation. The reactions have been performed step by step, i.e., Cu catalysts supported on zeolites are brought into contact with O2 to form active oxygen species on the active sites, followed by being in contact with methane to form methoxy intermediates. Finally, the catalysts with intermediates are treated with water vapor to form methanol. Recently, it was demonstrated that methanol could be catalytically formed by contact of the Cu catalysts with mixed gases of methane, O2 and water vapor, but the yield of methanol was very low.18,19 Recently, it was reported that a Ni catalyst supported on silicoaluminophosphate zeolite (SAPO-5) exhibited excellent catalytic performance for syngas production, and the performance was improved by the addition of Sr oxide.20As for the partial oxidation of methane into formaldehyde, metal oxides such as V, Mo, Fe, and Cu supported on silica have been utilized as catalytically active components.21 Atomically dispersed metal oxide clusters supported on silica work as active sites for formaldehyde formation by methane oxidation with O2.22 For example, a formaldehyde selectivity of 16% was reported at a methane conversion of 10% in the oxidation of methane over V2O5/SiO2 at 903 K.23 Some metal phosphates also show excellent catalytic performance for the formation of formaldehyde by methane oxidation with O2.24 Crystallized FePO4 is the active catalyst for formaldehyde formation in methane oxidation at 673–823 K, showing a formaldehyde yield of around 0.3% at 823 K.25,26 We have also reported that copper phosphate Cu2P2O7 works as an active catalyst for the formation of formaldehyde through methane oxidation with O2.27 A formaldehyde yield of 1.0% could be obtained by methane oxidation over the Cu2P2O7 catalyst at 923 K. Additionally, the catalytic performance of Cu2P2O7 was improved by dilution with Al2O3 or deposition onto a SiO2 support, due to the formation of smaller Cu2P2O7 crystallites.28 The Cu2P2O7 catalysts modified with Al2O3 or SiO2 showed higher formaldehyde yields and excellent durability for methane oxidation at 923 K, compared to the pure Cu2P2O7 catalyst. We believe that highly dispersed Cu oxides surrounded with phosphate anions work as efficient active sites for methane oxidation with O2 to form formaldehyde. In the present study, Cu oxides are supported on silicoaluminophosphate zeolite (SAPO34) (denoted as Cu/SAPO, hereafter), in order to form atomically dispersed Cu oxides which interact with phosphates. Because SAPO34 is composed of tetrahedral SiO4, AlO4, and PO4 units, Cu cations should be surrounded with these units. We would report the formation of H2 in addition to formaldehyde and CO with high yields by methane oxidation with O2 over Cu/SAPO catalysts.
2. Experimental section
2.1 Preparation of catalysts
Supported Cu catalysts were prepared by a conventional impregnation method. Support materials were impregnated into an aqueous solution of Cu(CH3COO)2 at 363 K for 12 h and then dried up at 363 K. The samples were further dried at 363 K for 24 h in air. Finally, samples thus obtained were calcined at 973 K for 5 h in air. Mordenite (denoted as MOR, JRC-Z-HM20, SiO2/Al2O3 = 18), beta-zeolite (denoted as *BEA, JRC-Z-B25, SiO2/Al2O3 = 25), faujasite zeolite (FAU, JRC-Z-HY5.5, SiO2/Al2O3 = 5.6), silica (SiO2, JRC-SIO13), alumina (Al2O3, JRC-ALO8), chabazite (denoted as CHA, SiO2/Al2O3 = 26) and SAPO34 (denoted as SAPO, SiO2/Al2O3/P2O5 = 10/42/48) were used as supports. All the zeolite supports used in the present study were impregnated into an aqueous solution of NH4Cl, followed by calcination at 973 K, in order to obtain H+-exchanged zeolites. V, Cr, Mn, Fe, Co, Ni, Pd or Rh catalysts supported on SAPO were also prepared with a similar method described earlier by using (NH4)VO3, Cr(CH3COO)2, Mn(CH3COO)2·4H2O, Fe(CH3COO)2, Co(CH3COO)2·4H2O, Ni(NO3)2·6H2O, Pd(NO3)2 or RhCl3·3H2O, respectively. The loading of these metal cations in the supported catalysts was adjusted to 0.5 wt%, if otherwise noted.2.2 Methane oxidation with O2
Methane oxidation with O2 was carried out with a conventional gas flow system with a fixed catalyst bed at atmospheric pressure. Catalyst powder (0.050 g) diluted with quartz sand (1.0 g) was loaded on the catalyst bed in a quartz reactor (inner diameter = 6 mm). Temperatures of the catalyst bed in the reactor were monitored using the thermocouples installed in the reactor, which were in contact with the catalyst powder directly. The catalysts were treated with O2 (P(O2) = 20 kPa) diluted with Ar at 973 K for 1 h prior to methane oxidation. Mixed gases composed of CH4, O2 and Ar (CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2:Ar = 2:1:5 as a volume ratio, the total flow rate = 70 mL min−1) were introduced into the reactor and brought into contact with the catalysts for methane oxidation. Outlet gases from the reactor were passed through two traps cooled at ca. 273 and 200 K to condense a part of the products (HCHO and H2O). Gases through the traps (H2, CO and CO2) were analyzed by GC with Porapak Q and active carbon columns for the evaluation of their formation rates. The concentration of HCHO condensed in the two traps was evaluated using UV-vis spectroscopy (UV/vis: V-650 spectrometer, Jasco). The products in the cold traps were added to mixed aqueous solutions of ammonium acetate, acetic acid and acetylacetone at 353 K, to form 3,5-diacetyl-1,4-dihydrolutidine prior to the measurement of the UV-vis spectra.29 The concentration of HCHO was evaluated from the absorption at 413 nm due to 3,5-diacetyl-1,4-dihydrolutidine formed from HCHO. The liquid products were also analyzed by NMR, but the formation of methanol was not confirmed in the oxidation of methane with O2 over the catalysts tested in the present study.
:O2:Ar = 2:1:5 as a volume ratio, the total flow rate = 70 mL min−1) were introduced into the reactor and brought into contact with the catalysts for methane oxidation. Outlet gases from the reactor were passed through two traps cooled at ca. 273 and 200 K to condense a part of the products (HCHO and H2O). Gases through the traps (H2, CO and CO2) were analyzed by GC with Porapak Q and active carbon columns for the evaluation of their formation rates. The concentration of HCHO condensed in the two traps was evaluated using UV-vis spectroscopy (UV/vis: V-650 spectrometer, Jasco). The products in the cold traps were added to mixed aqueous solutions of ammonium acetate, acetic acid and acetylacetone at 353 K, to form 3,5-diacetyl-1,4-dihydrolutidine prior to the measurement of the UV-vis spectra.29 The concentration of HCHO was evaluated from the absorption at 413 nm due to 3,5-diacetyl-1,4-dihydrolutidine formed from HCHO. The liquid products were also analyzed by NMR, but the formation of methanol was not confirmed in the oxidation of methane with O2 over the catalysts tested in the present study.
Conversion of CH4(O2) was defined as follows,
| Conversion of CH4(or O2)/% = (reaction rate of CH4(or O2)) × 100/(flow rate of CH4(or O2) introduced into the reactor) |
| Selectivity to HCHO based on C/% = (formation rate of HCHO) × 100/(reaction rate of CH4) |
| Selectivity to HCHO based on H/% = (formation rate of HCHO × 2) × 100/(reaction rate of CH4 × 4) |
Selectivity to H2O based on H was evaluated stoichiometrically from the formation rates of CO, CO2, HCHO and H2 and the reaction rates of CH4 and O2, since it was difficult to accurately evaluate the amount of H2O using GC. The yield of each product can be calculated as the product of the conversion of CH4 and the selectivity to the corresponding product.
2.3 Characterization of catalysts
Powder X-ray diffraction (XRD) patterns of the catalysts were measured at room temperature (Rigaku MiniFlex, Cu Kα radiation, λ = 1.5418 Å). The crystal phases of the catalysts were identified using the powder diffraction file (PDF) database of the International Centre for Diffraction Data (ICDD). In situ UV-vis diffuse reflectance spectra were measured using a UV-vis spectrometer (V-750, Jasco) with an in situ flow cell (VHR-764B, Jasco) having a quartz cell window.30 Catalyst samples or BaSO4 powder for the measurement of the background spectrum were loaded on a stainless pan (inner diameter of 7.1 mm, height of 4.1 mm). Cu catalysts were treated under an O2 flow at 973 K for 30 min and then cooled to 773 K under N2 prior to the measurement of UV-vis spectra. Diffuse reflectance UV-vis spectra of the catalyst samples were obtained after exposing the samples into mixed gases composed of CH4, O2 and N2 at 773 K.3. Results and discussion
3.1 Methane oxidation over various catalysts
Table 1 shows the results of the oxidation of methane with O2 over different metal catalysts supported on SAPO. Aqueous solutions containing V, Cr, Mn, Fe, Co, Ni, Cu, Pd or Rh cations were impregnated into SAPO followed by calcination in air at 973 K. XRD patterns of these catalysts are shown in Fig. S1.† In the XRD patterns of all the catalysts, strong diffraction lines due to SAPO34 were observed at 2θ = 9, 13, 16, 21 and 31 degrees (PDF card No. 00-047-0630). Any diffraction lines assignable to compounds containing metals added into SAPO were not found in these XRD patterns. Metal species in these catalysts are thus suggested to be highly dispersed on SAPO.| Metal | Temp./K | Conv./% | Sel. C-base/% | Sel. H-base/% | Yield/% | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CH4 | O2 | HCHO | CO | CO2 | HCHO | H2 | H2O | HCHO | CO | H2 | ||
| Cu | 773 | 0.9 | 2.2 | 21 | 54 | 24 | 11 | 28 | 62 | 0.2 | 0.5 | 0.3 |
| 823 | 2.2 | 5.1 | 15 | 55 | 30 | 8 | 31 | 62 | 0.3 | 1.2 | 0.7 | |
| 873 | 4.9 | 10.2 | 11 | 60 | 29 | 6 | 29 | 66 | 0.6 | 2.9 | 1.4 | |
| 923 | 8.2 | 21.5 | 9 | 66 | 25 | 5 | 26 | 69 | 0.7 | 5.4 | 2.2 | |
| None (SAPO) | 773 | 0.1 | 0.2 | 0 | 100 | 0 | 0 | 6 | 94 | 0.0 | 0.1 | 0.0 |
| 823 | 0.2 | 0.4 | 0 | 67 | 33 | 0 | 10 | 90 | 0.0 | 0.1 | 0.0 | |
| 873 | 0.2 | 0.6 | 1 | 31 | 68 | 1 | 13 | 86 | 0.0 | 0.1 | 0.0 | |
| 923 | 0.5 | 1.9 | 1 | 11 | 88 | 1 | 6 | 93 | 0.0 | 0.1 | 0.2 | |
| V | 773 | 0.2 | 0.5 | 0 | 100 | 0 | 0 | 2 | 97 | 0.0 | 0.2 | 0.0 |
| 823 | 0.1 | 0.5 | 2 | 66 | 32 | 1 | 8 | 91 | 0.0 | 0.1 | 0.0 | |
| 873 | 0.2 | 0.7 | 5 | 29 | 66 | 2 | 11 | 87 | 0.0 | 0.1 | 0.0 | |
| 923 | 0.7 | 2.5 | 5 | 6 | 89 | 3 | 6 | 92 | 0.0 | 0.0 | 0.0 | |
| Cr | 773 | 0.9 | 3.2 | 1 | 67 | 32 | 0 | 3 | 96 | 0.0 | 0.6 | 0.0 |
| 823 | 2.1 | 7.5 | 1 | 57 | 42 | 0 | 3 | 96 | 0.0 | 1.2 | 0.1 | |
| 873 | 5.4 | 19.5 | 1 | 54 | 45 | 0 | 2 | 97 | 0.0 | 2.9 | 0.1 | |
| 923 | 11.6 | 44.4 | 0 | 56 | 43 | 0 | 2 | 98 | 0.0 | 6.5 | 0.2 | |
| Mn | 773 | 0.4 | 1.4 | 2 | 71 | 27 | 1 | 3 | 96 | 0.0 | 0.3 | 0.0 |
| 823 | 0.4 | 1.4 | 4 | 34 | 62 | 2 | 6 | 92 | 0.0 | 0.1 | 0.0 | |
| 873 | 0.6 | 2.4 | 4 | 17 | 80 | 2 | 9 | 90 | 0.0 | 0.1 | 0.1 | |
| 923 | 1.2 | 4.5 | 3 | 7 | 89 | 2 | 8 | 90 | 0.0 | 0.1 | 0.1 | |
| Fe | 773 | 0.1 | 0.3 | 3 | 56 | 41 | 2 | 19 | 79 | 0.0 | 0.1 | 0.0 |
| 823 | 0.2 | 0.5 | 4 | 29 | 67 | 2 | 27 | 70 | 0.0 | 0.0 | 0.0 | |
| 873 | 0.4 | 1.2 | 4 | 12 | 84 | 2 | 25 | 73 | 0.0 | 0.0 | 0.1 | |
| 923 | 0.9 | 3.1 | 4 | 5 | 91 | 2 | 20 | 78 | 0.0 | 0.0 | 0.2 | |
| Co | 773 | 0.4 | 0.2 | 1 | 57 | 42 | 0 | 23 | 76 | 0.0 | 0.2 | 0.1 |
| 823 | 0.5 | 1.8 | 1 | 11 | 88 | 0 | 15 | 85 | 0.0 | 0.1 | 0.1 | |
| 873 | 0.8 | 2.8 | 1 | 5 | 93 | 1 | 13 | 86 | 0.0 | 0.0 | 0.1 | |
| 923 | 1.2 | 4.5 | 2 | 6 | 92 | 1 | 15 | 84 | 0.0 | 0.1 | 0.2 | |
| Ni | 773 | 0.1 | 0.3 | 3 | 41 | 56 | 2 | 18 | 81 | 0.0 | 0.0 | 0.0 |
| 823 | 0.3 | 0.9 | 3 | 20 | 77 | 2 | 20 | 79 | 0.0 | 0.1 | 0.1 | |
| 873 | 0.7 | 2.5 | 3 | 7 | 90 | 1 | 15 | 83 | 0.0 | 0.1 | 0.1 | |
| 923 | 1.7 | 5.3 | 3 | 39 | 58 | 2 | 25 | 73 | 0.1 | 0.6 | 0.4 | |
| Pd | 623 | 0.2 | 0.9 | 0 | 6 | 94 | 0 | 0 | 100 | 0.0 | 0.0 | 0.0 |
| 673 | 1.1 | 4.4 | 0 | 2 | 98 | 0 | 0 | 100 | 0.0 | 0.0 | 0.0 | |
| 723 | 2.4 | 9.5 | 0 | 1 | 99 | 0 | 0 | 100 | 0.0 | 0.0 | 0.0 | |
| 773 | 23.5 | 99.0 | 0 | 17 | 83 | 0 | 12 | 88 | 0.0 | 4.0 | 2.7 | |
| Rh | 673 | 2.1 | 8.3 | 0 | 4 | 96 | 0 | 0 | 100 | 0.0 | 0.1 | 0.0 |
| 723 | 3.9 | 15.9 | 0 | 1 | 99 | 0 | 0 | 100 | 0.0 | 0.0 | 0.0 | |
| 773 | 21.1 | 91.9 | 0 | 7 | 93 | 0 | 3 | 97 | 0.0 | 1.4 | 0.7 | |
| 823 | 25.9 | 99.4 | 0 | 26 | 74 | 0 | 25 | 75 | 0.0 | 6.6 | 6.5 | |
The catalytic activity of SAPO for methane oxidation was significantly low as shown in Table 1. Metal cations added to SAPO should work as active sites for the reaction. Pd and Rh catalysts catalyzed methane oxidation to form CO2 and H2O in lower temperature ranges compared to the other catalysts shown in Table 1, indicating the higher activity of the former catalysts for the total oxidation of methane. CO and H2 were also formed in the reaction over Pd and Rh catalysts at high temperatures. As described below in detail, CO and H2 should be formed by reforming of methane with H2O and/or CO2 over these catalysts. In contrast, HCHO and CO in addition to CO2 were formed in the temperature range from 773 to 923 K over the 3d transition metal catalysts tested in the present study. Cr and Cu on SAPO showed higher catalytic activity for the oxidation of methane with O2, compared to the other 3d transition metal catalysts, as shown in Fig. 1(a). Although the catalytic activity of Cu/SAPO was similar to that of Cr/SAPO, the selectivity to HCHO over the former catalysts was significantly higher than that over the latter ones (Fig. S2†). As the reaction temperature increased in the reaction over the Cu/SAPO catalyst, the selectivity to HCHO decreased, and that to CO increased instead, suggesting that HCHO was converted into CO over the Cu/SAPO catalyst. It should be noted that the HCHO yield in the reaction over the Cu/SAPO catalyst was extremely higher than those over the other catalysts, as shown in Fig. 1(b). The HCHO yield for Cu/SAPO was as high as that in the reaction over crystallized Cu2P2O7 catalysts, which were one of the most active catalysts for HCHO formation by methane oxidation with O2.27 Cu cations supported on SAPO should interact with PO4 units in SAPO, similar to those in Cu2P2O7.27,28 Highly dispersed Cu cations chemically interacting with phosphates (PO4) would be effective for the formation of HCHO in methane oxidation with O2.
 | ||
| Fig. 1 Methane oxidation with O2 over different metal catalysts supported on SAPO catalysts. a) CH4 conversion, b) yield of HCHO, c) yield of CO and d) yield of H2. | ||
Surprisingly, the formation of H2 was confirmed during the reaction over the Cu/SAPO catalyst, although a substantial amount of gaseous O2 remained in the reactants due to low O2 conversion (Table 1). The selectivity to H2 was relatively high (28%) in the reaction over the Cu/SAPO catalyst at 773 K, and it did not decrease with the reaction temperature (Fig. S2d†). In contrast, in the case of the reaction over the Pd and Rh catalysts, H2 and CO were only observed once nearly all gaseous O2 was consumed (Table 1). The reaction behavior over the Pd and Rh catalysts aligns with that over previously reported conventional catalysts active for the production of synthesis gas such as supported Ni catalysts.31–35 In these catalysts, H2 and CO are produced via the indirect path: O2 is completely consumed at the upper parts of the catalyst bed in the flow reactors to form CO2 and H2O and then the remaining methane is converted into CO and H2 by reforming of methane with H2O and/or CO2 at the lower parts of the catalyst bed. Additionally, CO and H2O are formed from CO2 and H2 (reverse water gas shift reaction). The reaction behavior over the Cu/SAPO catalyst differs significantly from that over the conventional catalysts. The formation of H2 and CO at low O2 conversions over the Cu/SAPO catalyst indicates that these products are formed through a different mechanism than the reforming of methane.
The catalytic performance of Cu/SAPO for methane oxidation with O2 was compared with those of V, Cr, Mn, Fe, Co or Ni catalysts supported on SAPO in detail. The results are also shown in Table 1, Fig. 1 and S2.† The catalytic activity of Cr/SAPO was similar to that of Cu/SAPO, while the activity of the other catalysts was very low. All the 3d transition metal catalysts in Table 1 also formed HCHO, CO, CO2 and H2 during the reactions, but the selectivity to each product was strongly dependent on the type of metal which worked as the catalytically active site. Selectivities to HCHO, CO and H2 in the reaction over the Cu/SAPO catalyst were relatively higher in the whole temperature range (773–923 K) compared to those over the other catalysts. The selectivity to HCHO was also higher in the reaction over V/SAPO and the selectivities to H2 and CO were higher in the reactions over Co/SAPO and Ni/SAPO catalysts. However, these catalysts had much lower activity for methane oxidation, compared to the Cu/SAPO catalyst as shown in Fig. 1(a). When compared at the same methane conversion, the selectivities to HCHO and H2 over the Cu/SAPO catalyst were higher than those over the other metal catalysts (Fig. S3†). Thus, the yields of HCHO, CO and H2 were appreciably higher in the reaction over the Cu/SAPO catalyst than those over the other catalysts (Fig. 1). The yield of HCHO, CO and H2 was evaluated to be 0.7, 5.4 and 2.2%, respectively, at the methane conversion of 8.2% in the reaction over the Cu/SAPO catalyst at 923 K. It is meaningful that 74% of C atoms and 31% of H atoms in CH4 molecules consumed by the oxidation with O2 are utilized for the production of HCHO, CO and H2 in the reaction over the Cu/SAPO catalyst. We concluded that Cu cations stabilized in SAPO showed excellent catalytic performance for the partial oxidation of methane with O2 to form HCHO, CO and H2. Recently, Murata and Hosokawa et al. investigated the catalytic performance of various metal oxides for the partial oxidation of methane with O2.36 The ZrO2 catalyst showed the highest yield of synthesis gas in the reaction among 31 metal oxides tested in the study. ZrO2 catalyzed the partial oxidation of methane into synthesis gas through the direct path. Kobayashi also reported the partial oxidation of methane with O2 into H2 and CO over Rh–Re catalysts supported on Al2O3 through the direct path.37 They proposed that formates were formed on ZrO2 or low-valent ReOx species during methane oxidation with O2 and the intermediates were decomposed into CO, CO2 and H2. Additionally, Kobayashi et al. demonstrated that CO was formed by the partial oxidation of methane with O2 over the Re catalyst supported on mordenite to form CO through the direct path.38 In these previous studies, the formation of HCHO was not confirmed during methane oxidation with O2, while a high HCHO yield was obtained in the reaction over the Cu/SAPO catalyst. It is likely that different intermediates on the ZrO2 or Re catalyst are formed on Cu cations stabilized on SAPO during methane oxidation.
In order to clarify the origin of superior catalytic performance of Cu/SAPO for the formation of HCHO, H2 and CO in methane oxidation with O2, the catalytic performance of Cu catalysts supported on different zeolites such as mordenite (denoted as MOR, hereafter), chabazite (CHA), beta-zeolite (*BEA) and faujasite zeolite (FAU) was evaluated. The results for the reactions are listed in Table 2. The Cu loading was adjusted to be 0.5 wt% for all the Cu catalysts in Table 2. XRD patterns for these Cu catalysts are shown in Fig. S4.† Only diffraction lines due to the corresponding zeolites were observed in the XRD patterns for these catalysts, suggesting the presence of highly dispersed Cu species for all the Cu catalysts in Table 2. All the Cu catalysts supported on these zeolites catalyzed methane oxidation with O2 to form HCHO, CO, CO2, H2 and H2O, similar to the Cu/SAPO catalyst, but the conversion of methane and the selectivity to each product strongly depended on the type of zeolite. No CH3OH formation was observed at high temperatures such as 773 K during methane oxidation over any of the Cu catalysts in Table 2. The catalytic activity for the reaction was higher in the order of Cu/SAPO > Cu/MOR ≈ Cu/CHA ≫ Cu/*BEA > Cu/FAU as shown in Fig. 2. Cu/SAPO and Cu/MOR which had higher activity for methane oxidation tended to show lower selectivity to HCHO and higher selectivity to CO2 as shown in Fig. S5.† In contrast, the selectivity to H2 was the highest in the reaction over the Cu/SAPO catalyst although the catalyst had the highest activity among all the catalysts shown in Table 2. Thus, the yields of HCHO, CO and H2 were relatively higher in the reaction over the Cu/SAPO catalyst compared to their yields over the other Cu catalysts. In particular, the H2 yield for the Cu/SAPO catalyst was significantly higher than those for the other Cu catalysts. It is likely that interaction of atomically dispersed Cu cations with PO4 units in zeolites results in the formation of active sites for H2 production in the methane oxidation with O2.
| Support | Temp./K | Conv./% | Sel. C-base/% | Sel. H-base/% | Yield/% | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CH4 | O2 | CO | CO2 | HCHO | H2 | HCHO | H2O | HCHO | CO | H2 | ||
| SAPO | 773 | 0.9 | 2.2 | 54 | 24 | 21 | 28 | 11 | 62 | 0.2 | 0.5 | 0.3 |
| 823 | 2.2 | 5.1 | 55 | 30 | 15 | 31 | 8 | 62 | 0.3 | 1.2 | 0.7 | |
| 873 | 4.9 | 10.2 | 60 | 29 | 11 | 29 | 6 | 66 | 0.6 | 2.9 | 1.4 | |
| 923 | 8.2 | 21.5 | 66 | 25 | 9 | 26 | 5 | 69 | 0.7 | 5.4 | 2.2 | |
| *BEA | 773 | 0.2 | 0.5 | 57 | 9 | 35 | 17 | 17 | 66 | 0.1 | 0.1 | 0.0 |
| 823 | 0.3 | 0.8 | 38 | 27 | 35 | 0 | 17 | 83 | 0.1 | 0.1 | 0.0 | |
| 873 | 0.7 | 2.1 | 30 | 41 | 30 | 5 | 15 | 80 | 0.2 | 0.2 | 0.0 | |
| 923 | 1.7 | 4.9 | 46 | 32 | 22 | 6 | 11 | 83 | 0.8 | 0.4 | 0.1 | |
| MOR | 773 | 0.7 | 1.8 | 56 | 28 | 17 | 22 | 8 | 70 | 0.1 | 0.4 | 0.2 |
| 823 | 1.8 | 4.1 | 52 | 33 | 15 | 19 | 7 | 73 | 0.3 | 0.9 | 0.4 | |
| 873 | 3.6 | 8.4 | 49 | 40 | 11 | 17 | 6 | 78 | 0.4 | 1.8 | 0.6 | |
| 923 | 5.7 | 16.0 | 62 | 29 | 9 | 16 | 5 | 79 | 0.5 | 3.5 | 0.9 | |
| FAU | 773 | 0.1 | 0.3 | 70 | 0 | 30 | 0 | 15 | 85 | 0.0 | 0.0 | 0.0 |
| 823 | 0.1 | 0.5 | 54 | 0 | 35 | 0 | 17 | 83 | 0.0 | 0.1 | 0.0 | |
| 873 | 0.3 | 1.2 | 49 | 11 | 35 | 7 | 17 | 76 | 0.0 | 0.1 | 0.1 | |
| 923 | 0.7 | 2.9 | 57 | 16 | 24 | 7 | 12 | 81 | 0.0 | 0.4 | 0.2 | |
| CHA | 773 | 0.4 | 0.8 | 63 | 0 | 37 | 17 | 18 | 64 | 0.1 | 0.2 | 0.1 |
| 823 | 1.2 | 2.2 | 57 | 15 | 28 | 23 | 14 | 63 | 0.3 | 0.7 | 0.3 | |
| 873 | 2.9 | 6.1 | 67 | 14 | 19 | 24 | 10 | 67 | 0.6 | 2.0 | 0.7 | |
| 923 | 5.4 | 12.4 | 73 | 11 | 16 | 24 | 8 | 68 | 0.9 | 3.9 | 1.3 | |
| Al2O3 | 773 | 0.8 | 1.4 | 63 | 37 | 0 | 24 | 0 | 76 | 0.0 | 0.5 | 0.2 |
| 823 | 2.1 | 6.2 | 52 | 48 | 0 | 15 | 0 | 85 | 0.0 | 1.1 | 0.3 | |
| 873 | 5.2 | 15.6 | 46 | 54 | 0 | 10 | 0 | 90 | 0.0 | 2.4 | 0.5 | |
| 923 | 11.8 | 39.2 | 42 | 58 | 0 | 7 | 0 | 93 | 0.0 | 4.9 | 0.8 | |
| SiO2 | 773 | 0.4 | 2.1 | 27 | 66 | 6 | 0 | 3 | 97 | Tr | 0.1 | 0.0 |
| 823 | 0.7 | 3.2 | 17 | 48 | 36 | 2 | 18 | 80 | 0.3 | 0.1 | Tr | |
| 873 | 0.7 | 3.5 | 33 | 56 | 11 | 3 | 5 | 92 | 0.1 | 0.3 | Tr | |
| 923 | 1.2 | 5.2 | 38 | 41 | 21 | 3 | 11 | 86 | 0.3 | 0.5 | Tr | |
 | ||
| Fig. 2 Methane oxidation with O2 over Cu catalysts supported on different zeolites. a) CH4 conversion, b) yield of HCHO, c) yield of CO and d) yield of H2. | ||
Methane oxidation with O2 was also performed over Cu catalysts supported on SiO2 and Al2O3. The loading of Cu for both catalysts was adjusted to 0.5 wt% similar to that for the Cu/SAPO catalyst. The results for the reactions over these Cu catalysts are also shown in Table 2. In the XRD patterns for both catalysts, very weak peaks due to CuO were observed at 2θ = 35 and 39 degrees in addition to broad peaks due to the supports as shown in Fig. S6.† Cu species in these catalysts are present as small crystallites of CuO in addition to highly dispersed Cu oxides.39 The Cu/Al2O3 catalyst showed a similar activity for the reaction to the Cu/SAPO catalyst, whereas the catalytic activity of Cu/SiO2 was very low. The selectivity to HCHO was high in the reaction over the Cu/SiO2 catalyst, but that to H2 was extremely low. Wang et al. reported that highly dispersed Cu oxides on mesoporous silica catalyzed the partial oxidation of methane with O2 into HCHO.40 Highly dispersed Cu oxides on silica should form HCHO through methane oxidation, while CuO crystallites catalyzed the total oxidation of methane into CO2.27 In methane oxidation over the Cu/Al2O3 catalyst, the formation of HCHO was not observed but selectivities to CO and H2 were relatively high. From the results described earlier, we concluded that Cu cations stabilized on zeolites formed HCHO, CO and H2 with high yields by methane oxidation with O2.
Direct oxidation of methane with O2 into methanol over Cu catalysts supported on zeolites such as CHA, MOR and ZSM-5 has been investigated by many research groups. Monomers or dimers of Cu stabilized in zeolites are believed to be active sites for methanol formation in the reaction.30,41–43 Generally, highly dispersed Cu species stabilized in zeolites are activated with gaseous O2 to form oxygen species active for methanol formation before the catalysts come into contact with methane in the temperature range from 473 to 573 K.44 Formation of methanol is observed by the treatment of the catalysts with water vapor. High selectivity to methanol has been attained by the step-by-step reactions over these Cu catalysts, but the yield of methanol is extremely low due to low conversion of methane at low temperatures such as 473–573 K. In contrast, we performed methane oxidation with O2 without water cofed in a higher temperature range from 773 to 923 K, to form H2 in addition to HCHO, CO and CO2. Highly dispersed Cu species in zeolites should also work as active sites for methane oxidation with O2 to form HCHO, CO and H2 in the present study, because any diffraction lines due to crystallized Cu compounds were not observed in the XRD patterns for these Cu catalysts. Note that the crystal structure of SAPO (SAPO34) is the same as that of CHA, although the atomic component of SAPO (Si, Al, P and O) is different from that of CHA (Si, Al and O). As described hereafter, UV-vis spectra for Cu/SAPO and Cu/CHA showed the presence of atomically dispersed Cu2+. The Cu cations in Cu/SAPO should be surrounded with lattice oxygen atoms in SiO4, AlO4 and PO4 units, while Cu cations in the other zeolite-supported catalysts are in contact with SiO4 and AlO4 units. The interaction of Cu cations with PO4 units in SAPO should result in the excellent catalytic performance for the formation of HCHO, CO and H2 in methane oxidation with O2.
3.2 Catalytic performances of Cu/SAPO
The catalytic performance of Cu/SAPO for methane oxidation with O2 was examined in detail. Fig. 3 shows the results for methane oxidation with O2 over Cu/SAPO catalysts with different Cu loadings. Cu loadings in the catalysts were adjusted to be 0.3, 0.5, 1.0 or 1.5 wt%. The XRD patterns of these catalysts showed only diffraction lines due to SAPO34 as shown Fig. S7,† suggesting that Cu species were highly dispersed for all the catalysts irrespective of Cu loading. | ||
| Fig. 3 Methane oxidation with O2 over Cu/SAPO catalysts with different Cu loadings. a) CH4 conversion, b) yield of HCHO, c) yield of CO and d) yield of H2. | ||
The catalytic activity of Cu/SAPO for methane oxidation became higher with Cu loading. The amount of Cu cations in the catalyst bed was larger with the Cu loading in the catalysts since the same amount of the catalysts (0.050 g) was packed in the catalyst beds for all the reactions in Fig. 3. The selectivity to each product slightly depends on the Cu loading in the catalysts as shown in Fig. S8.† HCHO, CO and H2 in addition to CO2 and H2O were formed in the reaction over all the Cu/SAPO catalysts regardless of Cu loading. Selectivities to HCHO and H2 were decreased and instead those to CO2 and H2O were increased with Cu loading in the catalysts. On the other hand, the yields of HCHO, CO and H2 for the reactions over Cu/SAPO with Cu loadings of 0.5, 1.0 and 1.5 wt% were similar. It is likely that the number of active sites for the formation of these products was not significantly increased with Cu loading. It is easily expected that Cu dimers and Cu trimers are formed in the catalysts with Cu loading. Cu monomers stabilized in SAPO catalyze methane oxidation into HCHO, CO and H2, whereas Cu dimers and trimers preferentially form CO2 and H2O.
The stability of Cu/SAPO catalysts for methane oxidation at 923 K was evaluated. Fig. 4 shows the change of methane conversion and the yields of HCHO, CO and H2 as a function of time-on-stream in methane oxidation over Cu(0.5 wt%)/SAPO at 923 K. A slight increase in the selectivities to CO, HCHO and H2 was confirmed during the reaction over Cu/SAPO catalysts (Fig. S9†), while the methane conversion decreased slightly and gradually with time-on-stream. Thus, the yields of these products became slightly higher with time on stream of methane. These results indicate that Cu/SAPO catalysts are stable for the formation of H2, CO and HCHO in methane oxidation with O2 at 923 K in spite of high reaction temperatures.
 | ||
| Fig. 4 Kinetic curves for CH4 conversion (a) and yields of each product (b) in methane oxidation over the Cu(0.5 wt%)/SAPO catalyst at 923 K. | ||
The contact time of the reactants for methane oxidation over Cu/SAPO catalysts was changed in order to evaluate the contribution of successive oxidation of the primary products to product selectivity. The change of selectivity to each product was plotted as a function of W/F (W, weight of the catalyst (g); F, flow rate of the reactants (mL min−1)) in methane oxidation over the Cu(0.5 wt%)/SAPO catalyst at 923 K in Fig. 5. The selectivity based on C atoms to HCHO and CO decreased and that to CO2 instead increased with an increase in W/F values. Similar to the selectivity based on C atoms, the selectivity based on H atoms to HCHO and H2 also decreased and that to H2O increased as W/F values increased. These results strongly suggested that HCHO, CO and H2 are the primary products in methane oxidation with O2 over Cu/SAPO catalysts. Note that the selectivity to CO2 became low and it seemed to approach zero, as W/F values decreased. In contrast, the selectivity to H2O did not approach zero at low W/F values. CH4 would react with O2 to form H2O and some intermediates such as HCHO, which are adsorbed on active sites. The intermediates adsorbed on the active sites are desorbed and/or decomposed to form HCHO, H2 and CO.
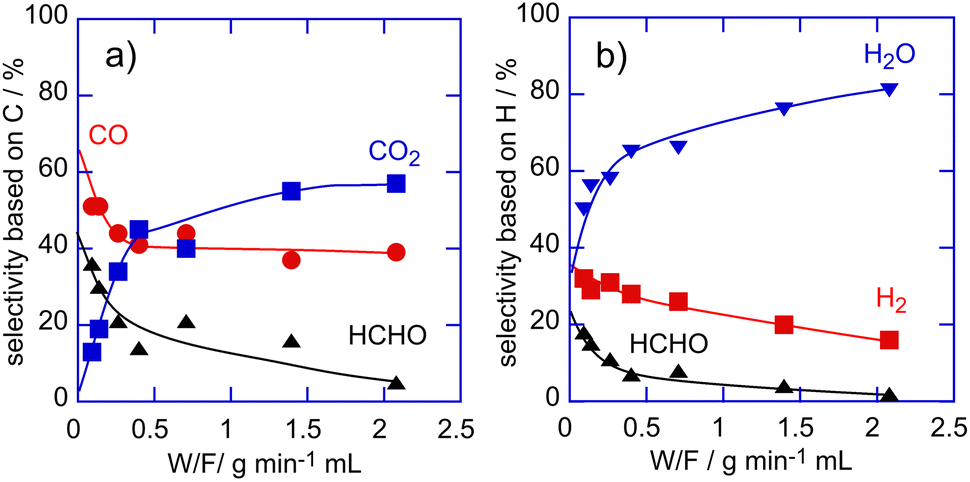 | ||
| Fig. 5 Change of selectivity to each product as a function of W/F in methane oxidation over the Cu/SAPO catalyst at 923 K. a) selectivity to each product based on C, b) selectivity to each product based on H. | ||
The dependence of the reaction rates of methane oxidation with O2 over the Cu(0.5 wt%)/SAPO catalyst on the partial pressures of CH4 or O2 was evaluated. Fig. 6 shows the reaction rates of CH4 against partial pressures of CH4 or O2. The reaction rate of CH4 increased with increasing partial pressures of CH4 and O2, but its increase appears to have saturated in the high partial pressure range, indicating that methane oxidation over Cu/SAPO proceeds through the Langmuir–Hinshelwood mechanism, i.e., both CH4 and O2 adsorb to form intermediates on the active sites on the Cu/SAPO catalyst. The reaction order of CH4 and O2 for the reaction rates of CH4 was evaluated to be 0.4 and 0.6, respectively. Thus, the activation of CH4 and O2 on the active sites would be related to the rate determining step for methane oxidation with O2 over the Cu/SAPO catalyst.
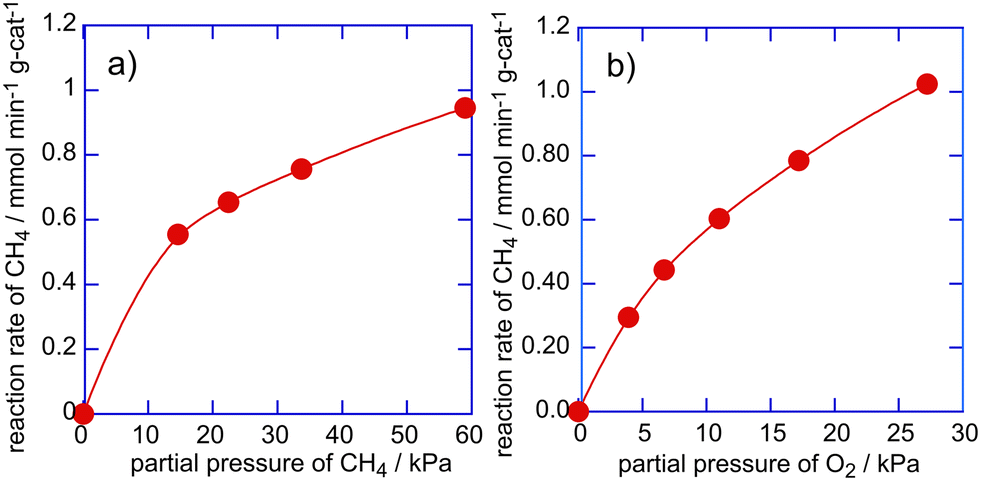 | ||
| Fig. 6 Dependence of the reaction rate of methane on the partial pressure of CH4 (a) and O2 (b) in methane oxidation over the Cu/SAPO catalyst at 823 K. | ||
Finally, the structures of Cu species stabilized in SAPO, *BEA and CHA were evaluated using in situ UV-vis spectra.45 The crystal structure of SAPO was the same as that of MOR. After these Cu catalysts were treated in O2 diluted with N2 (O2:N2 = 2:5 as the volume ratio, total flow rate = 70 mL min−1) at 973 K for 1 h, diffuse reflectance UV-vis spectra for these catalysts were measured at 773 K under diluted O2 stream. After the measurement of the spectra, mixed gases composed of CH4 and O2 diluted with N2 (CH4:O2:N2 = 2:1:4 as the volume ratio, total flow rate = 70 mL min−1) were brought into contact with these catalysts at 773 K and then the UV spectra for the catalysts were measured at 773 K under the mixed gases. Fig. 7 shows the UV-vis spectra for Cu(0.5 wt%)/SAPO, Cu(0.5 wt%)/CHA and Cu(0.5 wt%)/*BEA catalysts. A strong peak at around 300 nm and a broad absorption in the range of 600 to 850 nm were observed in the UV-vis spectra for all the fresh Cu catalysts. These peaks at around 300 nm are assigned to the charge transfer from ligands to metals (LMCT) for Cu2+, while the absorption in the range of 650 to 850 nm is due to the d–d transition of Cu2+.46,47 Thus, highly dispersed Cu2+ cations in these catalysts are stabilized in the framework of each zeolite. It should be noted that the position of the peak due to LMCT for Cu/SAPO is different from that for Cu/CHA and Cu/*BEA, indicating that the coordination of Cu2+ in the zeolite frameworks is different among these Cu catalysts. The peak position of LMCT for Cu2+ is reported to be sensitive to their coordination, i.e., the type and/or the number of anions (SiO4, AlO4 or PO4) around the cations.47 Cu2+ cations in Cu/SAPO are surrounded with phosphate PO4 units in addition to AlO4 and SiO4 units, whereas Cu2+ cations in Cu/CHA and Cu/*BEA interact with SiO4 and AlO4 units. The intensity for the absorption due to LMCT of Cu2+ and the d–d transition of Cu2+ slightly decreased by changing the gas from O2 to mixed gases of CH4 and O2. This spectral change is assignable to the reduction of a part of Cu2+ in the catalysts into Cu+ during methane oxidation at 773 K.30 The redox between Cu2+ and Cu+ is related to the oxidation of methane with O2. Cu2+ cations in the zeolites are reduced to Cu+ with CH4 molecules, and the Cu+ cations are oxidized with O2 to Cu2+ to form products such as CO and HCHO. Cu2+ cations dispersed in the zeolites should work as active sites for the formation of HCHO, H2 and CO in methane oxidation with O2. It is likely that the Cu catalysts supported on zeolites form HCHO intermediates adsorbed on the active sites. The behavior for the desorption and/or decomposition of the intermediates depends on the type of zeolite, due to differences in the number and/or strength of acid property.
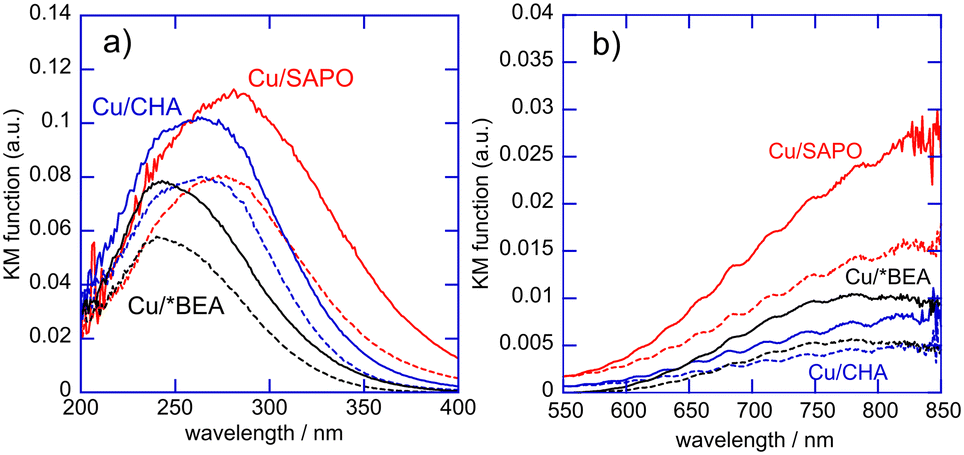 | ||
| Fig. 7 In situ diffuse reflectance UV-vis spectra for Cu/SAPO, Cu/CHA and Cu/*BEA catalysts (a) with wavelengths from 200 to 400 nm, and (b) from 550 to 850 nm. Solid lines for the catalysts before the reactions, and dotted lines for the catalysts during methane oxidation. | ||
Conclusions
The catalytic performance of Cu/SAPO for methane oxidation with O2 was evaluated in the temperature range of 773–923 K. Cu/SAPO formed H2 in addition to HCHO and CO during the reaction, and the yields of these products in the reaction over Cu/SAPO were significantly higher than those over other 3d transition metal catalysts (V, Cr, Mn, Fe, Co, Ni) supported on SAPO. In addition, Cu catalysts supported on zeolites different from SAPO (*BEA, MOR, FAU, CHA) also formed H2, CO and HCHO during methane oxidation and the H2 yield in the reaction over Cu/SAPO was the highest among the 3d metal/SAPO catalysts tested in this study. The interaction of Cu2+ and PO4 in SAPO provides excellent catalytic performance for the partial oxidation of methane with O2 to H2, CO and HCHO.Data availability
We confirm that the data supporting the findings of this study are available within the article and/or its ESI.†Author contributions
Mana Shimakawa: conceptualization, data curation and investigation. Reiko Nagase: investigation and data curation. Ryoya Kugo: investigation and data curation. Junya Ohyama: investigation and writing – review & editing. Sakae Takenaka: conceptualization, project administration, funding acquisition, formal analysis, investigation and writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 23K26459.References
- A. Demirbas, Energy Convers. Manage., 2010, 51, 1547–1561 CrossRef
.
- M. C. Alvarez-Galvan, N. Mota, M. Ojeda, S. Rojas, R. M. Navarro and J. L. G. Fierro, Catal. Today, 2011, 171, 15–23 CrossRef
- T. V. Choudhary and V. R. Choudhary, Angew. Chem., Int. Ed., 2008, 47, 1828–1847 CrossRef PubMed
- I. V. Zagaynov, Energy Fuels, 2021, 35, 9124–9136 CrossRef
- A. S. Ai-Fatesh, N. Patel, A. H. Fakeeha, M. F. Alotibi, S. B. Alreshaidan and R. Kumar, Catal. Rev.:Sci. Eng., 2024, 66, 2209–2307 CrossRef
- H. Schulz, Appl. Catal., A, 1999, 186, 3–12 CrossRef
- P. Tian, P. Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef
- J. Bao, G. Yang, Y. Yoneyama and N. Tsubaki, ACS Catal., 2019, 9(4), 3026–3053 CrossRef
- A. I. Olivos-Suarez, A. Szecsenyi, E. J. M. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, ACS Catal., 2016, 6, 2965–2981 CrossRef
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef PubMed
- P. Kumar, T. A. Al-Attas, J. Hu and M. G. Kibria, ACS Nano, 2022, 16, 8557–8618 CrossRef PubMed
- Y. Tang, Y. Li and F. Tao, Chem. Soc. Rev., 2022, 51, 376–423 RSC
- N. F. Dummer, D. J. Willock, Q. He, M. J. Howard, R. J. Lewis, G. Qi, S. H. Taylor, J. Xu, D. Bethell, C. J. Kiely and G. J. Hutchings, Chem. Rev., 2023, 123, 6359–6411 CrossRef PubMed
- M. N. Newton, A. J. Knorpp, V. L. Sushkevich, D. Palagin and J. A. van Bokhoven, Chem. Soc. Rev., 2020, 49, 1449–1486 RSC
- L. Tao, I. Lee and M. S. Sanchez, Catal. Sci. Technol., 2020, 10, 7124–7141 RSC
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef PubMed
- H. M. Rhoda, A. J. Heyer, B. E. R. Snyder, D. Plessers, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Chem. Rev., 2022, 122, 12207–12243 CrossRef PubMed
- J. Ohyama, A. Hirayama, Y. Tsuchimura, N. Kondou, H. Yoshida, M. Machida, S. Nishimura, K. Kato, I. Miyazato and K. Teranishi, Catal. Sci. Technol., 2021, 11, 3437–3446 RSC
- K. T. Dinh, M. M. Sullivan, K. Narsimhan, P. Serna, R. J. Meyer, M. Dincă and Y. Román-Leshkov, J. Am. Chem. Soc., 2019, 141, 11641–11650 CrossRef PubMed
- A. Al-Anazi, O. Bellahwel, K. C., S. Santhosh, A. E. Abasaeed, J. Abu-Dahrieh, A. H. Fakeeha, A. A. Ibrahim and A. S. Al-Fatesh, Catalysts, 2024, 14, 316 CrossRef
- M. J. G. Fait, A. Ricci, M. Holena, J. Rabeah, M.-M. Pohl, D. Linke and E. V. Kondratenko, Catal. Sci. Technol., 2019, 9, 5111–5121 RSC
- F. Arena, N. Giordano and A. Parmaliana, J. Catal., 1997, 167, 66–76 CrossRef
- R. G. Herman, Q. Sun, C. Shi, K. Klier, C. Wang, H. Hu, I. E. Wachs and M. M. Bhasin, Catal. Today, 1997, 37, 1–14 CrossRef
- A. Matsuda, T. Aihara, S. Kiyohara, Y. Kumagai, M. Hara and K. Kamata, ACS Appl. Nano Mater., 2024, 7, 10155–10167 CrossRef
- Y. Wang and K. Otsuka, J. Catal., 1995, 155, 256–267 CrossRef
- A. Matsuda, H. Tateno, K. Kamata and M. Hara, Catal. Sci. Technol., 2021, 11, 6987–6998 RSC
- T. Akiyama, M. Shimakawa and S. Takenaka, Chem. Lett., 2022, 51, 511–514 CrossRef
- M. Shimakawa and S. Takenaka, Catal. Sci. Technol., 2023, 13, 3859–3866 RSC
- T. Nash, Biochem. J., 1953, 55, 416–421 CrossRef PubMed
- J. Ohyama, Y. Tsuchimura, A. Hirayama, H. Iwai, H. Yoshida, M. Machida, S. Nishimura, K. Kato and K. Takahashi, ACS Catal., 2022, 12, 2454–2462 CrossRef
- G. Yang, X. Du, J. Ran, X. Wang, Y. Chen and L. Zhang, J. Phys. Chem. C, 2018, 122, 21468–21477 CrossRef
- E. L. Uzunova, F. Göltl, G. Kresse and J. Hafner, J. Phys. Chem. C, 2009, 113, 5274–5291 CrossRef
- L. Shi, J. Zhang, G. Shen, D. Fan, Y. Wen, Y. Zhao, R. Chen, M. Shen and B. Shan, Catal. Sci. Technol., 2019, 9, 1309–1316 RSC
- M. Prettre, C. Eichner and M. Perrin, Trans. Faraday Soc., 1946, 42, 335–339 RSC
- A. P. E. York, T. Xiao and M. L. H. Green, Top. Catal., 2003, 22, 345–358 CrossRef
- K. Murata, K. Arai, N. Kondo, R. Manabe, T. Yumura and S. Hosokawa, Catal. Sci. Technol., 2024, 14, 3253–3264 RSC
- L. Li, N. H. M. Dostagir, A. Shrotri, A. Fukuoka and H. Kobayashi, ACS Catal., 2021, 11, 3782–3789 CrossRef
- H. Kobayashi, A. Shrotri, K. Kato, A. Fukuoka and H. Kobayashi, Catal. Sci. Technol., 2023, 13, 5190–5196 RSC
- L. Fu, X. Li, Mi. Liu and H. Yang, J. Mater. Chem. A, 2013, 1, 14592–14605 RSC
- Y. Li, D. An, Q. Zhang and Y. Wang, J. Phys. Chem. C, 2008, 112, 13700–13708 CrossRef
- D. K. Pappas, M. Martini, M. Dyballa, K. Kvande, S. Teketel, K. A. Lomachenko, R. Baran, P. Clatzel, B. Arstad, G. Berlier, C. Labberti, S. Bordiga, U. Olsbye, S. Svelle, P. Beato and E. Borfecchia, J. Am. Chem. Soc., 2018, 140, 15270–15278 CrossRef PubMed
- H. M. Rhoda, D. P. Plessers, A. J. Heyer, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, J. Am. Chem. Soc., 2021, 143, 7531–7540 CrossRef PubMed
- A. Brenig, J. W. A. Fischer, D. Klose, G. Jeschke, J. A. van Bokhoven and V. L. Sushkevich, Angew. Chem., 2024, 63, e202411662 CrossRef PubMed
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, J. Am. Chem. Soc., 2005, 127, 1394–1395 CrossRef PubMed
- Y. Tsuchimura, H. Yoshida, M. Machida, S. Nishimura, K. Takahashi and J. Ohyama, Energy Fuels, 2023, 37, 9411–9418 CrossRef
- H. Li, C. Paolucci, I. Khurana, L. N. Wilcox, F. Göltl, J. D. Albarracin-Caballero, A. J. Shih, F. H. Ribeiro, R. Gounder and W. F. Schneider, Chem. Sci., 2019, 10, 2373–2384 RSC
- F. Göltl, S. Bhandari, E. A. Lebrón-Rodríguez, J. I. Gold, S. I. Zones, I. Hermans, J. A. Dumesic and M. Mavrikakis, Catal. Sci. Technol., 2022, 12, 2744–2748 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy01469c |
| This journal is © The Royal Society of Chemistry 2025 |