
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and catalytic performance investigation of the Ni–N–C catalyst for CO2RR: a theoretical study†
Yiming
Sun
,
Xiaoyu
Wang
,
Zhuofan
Wu
,
Anmin
Liu
 and
Xuefeng
Ren
*
and
Xuefeng
Ren
*
School of Chemical Engineering, Ocean and Life Sciences, Dalian University of Technology, Panjin 124221, China. E-mail: renxuefeng@dlut.edu.cn
First published on 24th January 2025
Abstract
The combustion of fossil fuels is increasingly contributing to global warming. The recycling of CO2 plays a crucial role, and the creation of a highly efficient electrocatalyst is essential for enhancing the efficiency of the reaction. This work focused on the theoretical design of Ni–N–C catalysts with different coordination environments of Ni through quantum chemical calculations and analyzed the differences between the coordination environments of pyridine N and pyrrole N on the performance of catalytic CO2 reduction to CO in order to identify the most efficient catalyst configuration. The Ni–N bonding energy of the catalyst with a vacancy was greater than that of the catalyst without a vacancy, and the activation ability of Ni-pyridine N2C1–C was the best. Ultimately, examining various catalysts for converting CO2 into CO revealed that Ni-pyridine N2C1–C exhibited the most effective catalytic impact. In contrast to the energy barrier ΔG = 2.9903 eV in the absence of a catalyst, the energy barrier ΔG = −1.4029 eV during the CO2 to CO catalytic reaction decreased by 4.3932 eV. This decrease was the largest among all the catalysts mentioned above, and the reaction could be spontaneous from a thermodynamic perspective. The research results provide a theoretical reference for the experimental preparation of catalysts for CO2 to CO conversion and the resource utilization of CO2.
1 Introduction
Post industrial revolution, swift advancements in fossil fuel technology have emitted substantial quantities of carbon dioxide (CO2) for energy, intensifying the urgency of achieving carbon neutrality.1,2 Consequently, the primary emphasis is on carbon capture and utilization, making the advancement of CO2 reduction a widespread issue in both academic circles and society at large.In CO2RR, the electrocatalyst promotes the conversion of carbon dioxide into various “high-value” products with the help of electricity,3 involving the transfer of six or more electrons during the electrocatalytic reaction. In these CO2RR processes, C1 products (such as methane (CH4) and methanol (CH3OH)), C2 products (such as ethylene (C2H4), acetic acid (CH3COOH), and ethanol (C2H5OH)), and C3 products (such as n-propanol (C3H8O) and acetone (C3H5O)) are formed. The activity and selectivity of the electrocatalyst for reduction products are mainly oriented towards C1 products, mainly CO and HCOO−, rather than the highly reduced C1 and polycarbon (C2+) products. CO is the main component of syngas and gas, the main part of C1 compounds, which can also be used as a reducing agent in the metal smelting industry. Therefore, catalysts that catalyze CO2RR to produce CO are explored in this paper. Compared to photocatalysis,4 electrocatalytic CO2 reduction avoids the critical problem of catalyst deactivation due to electron–hole complexation. Currently, scientists have explored a variety of catalysts that perform well,5 such as pure metal catalysts,6–10 alloy catalysts,11 porous material catalysts,12–17 atomically dispersed metal-based catalysts,18–22 and metal oxide catalysts.23 However, monometallic catalysts have limited performance.24–26 The adsorption properties of alloy catalysts are influenced by interactions between neighboring metal atoms, and porous material catalysts can regulate electrolyte diffusion and generate a volume gradient, thus affecting local alkalinity and improving the selectivity of CO2RR. Still, they suffer from problems, such as low conductivity, poor stability, and lack of mesoporosity. The M–N–C catalysts have a lower cost than precious metal catalysts, and the metal sites generate a charge accumulation effect, which is favorable for CO2 chemisorption and provides good selectivity for CO generation. Different central metal ions have distinct electronic structures, geometrical configurations and d-orbital energy levels, so selecting suitable central metal elements is crucial for improving the electrocatalytic ability of M–N–C materials by optimizing surface adsorption. Among them, transition metal elements and main group elements exhibit good electrocatalytic ability when acting as center metal ions (Fe, Co, Ni, Cu, Zn, Mn, Sn and Sb). Integrating transition metal nickel into the N–C substance markedly enhances the catalytic efficiency of CO2RR. Computational models indicate that incorporating single-atom Ni lowers the energy barrier for reactions in CO2RR. Once the atomically scattered Ni was secured to the carbon substance, the single-atom Ni demonstrated peak atomic usage, augmented the concentration of active sites, and enhanced the catalytic efficiency of CO2RR. Alterations in the d-orbital energy state of the central metal ion occurred as it was encircled by distinct neighboring ligands, suggesting that modifying the coordination setting of the central metal ion might control the electronic configuration of the M–N–C substance. Changes in the coordination environment can alter the nitrogen atom species by changing the coordination number of the nitrogen atoms. Good catalytic activity for CO2RR compared to other M–Nx structures, such as M–N2, M–N3, and M–N5,27,28 most M–N4 structures, were more selective for CO in CO2RR.29–31
Nitrogen atoms can cause positive charge aggregation,32 making C adjacent to N the active site for electrocatalytic CO2RR. Guo33 conducted a methodical study on the activation capabilities and specificity of N-doped carbon catalysts in electrocatalytic CO2RR processes. Numerous M–N–C catalysts are known to be employed in the electrocatalytic process of CO2RR. Jia et al.34 synthesized a single-atom Ni catalyst SA-NiNG-NV with N vacancies possessing higher CO2RR selectivity. Cheng et al.35 synthesized a large number of Ni–N–C catalytic materials at the edges of the sites by applying microwave stripping, which exhibited extremely high activity in the electrocatalytic CO2RR process. Wang et al.36 systematically investigated single-atom catalysts with typical transition metals as active sites, and they found that the selectivity in CO2–CO conversion was ranked as Ni > Co > Fe, and the reactivity was ranked as Co > Ni > Fe. Ni-doped catalysts had high catalytic efficiency, good stability and low price, so this study was aimed at Ni–N–C catalysts. Altering the electron configuration of the coordination center M consequently impacts its location on the active site and the catalyst's specificity. Zhao's team37 discovered through DFT analysis that pyrrole N predominantly generated HCOOH during CO2RR catalysis, exhibiting the minimal overpotential of 0.24 V compared to other N-doped graphene types, while Wu38 ascertained that pyridinium N, as the primary catalyst, effectively converted CO2 into CO, demonstrating superior catalytic efficiency in theoretical terms.
Consequently, this study conducts theoretical analyses of the stability, CO2 adsorption stability, and catalytic efficiency of each catalyst using the DMol3 module in Materials Studio (MS) software. This was performed to evaluate the impact of various coordination settings, including the quantity and nature of nitrogen atom coordination and adsorption type, on the CO2 reduction efficiency of the Ni–N–C catalysts, and to identify catalysts with superior performance that could inform the use of Ni–N–C catalysts in CO2RR. DFT calculations can predict and guide experimental design, thereby conserving human and material resources while more effectively screening for catalysts with superior performance. Additionally, DFT enables a deeper understanding of reaction mechanisms.
2 Models and methods
Quantum chemical calculations can be used to design the required catalysts directly on MS software and calculate and predict their catalytic performance, which can reduce the time and workload required for experiments. Many repetitive tasks can be avoided, research efficiency can be improved, and the experimental cost can be reduced, which can provide references and guidance for experiments.The 5 × 4 graphene was intercepted for reconfiguration, which retained the structural characteristics and reduced the computation. The establishment of the Ni–N–C catalyst model was completed, and geometry optimization was performed by selecting calculation in the DMol3 module, selecting geometry optimization in the task module in the set up tab, and selecting customized in the quality module. Under the generalized gradient approximation (GGA), the functional chosen was selected as the Perdew–Burke–Ernzerhof (PBE) function, and the basis set with a double-valued atomic orbital plus polarization function (DNP) was chosen to describe the atomic orbitals. The energy convergence criteria, maximum force, and maximum displacement were set during geometry optimization at 2 × 10−5 Ha, 0.004 Ha Å−1, and 0.005 Å, respectively. To accelerate electron convergence, the smearing was set to 0.005 Ha. The maximum number of iterations was set to 1000 in more, with medium selected for overall accuracy. The max SCF cycle was set to 1000, and the COSMO solution environment was used with water selected as the solvent. The energy calculation was also done using the DMol3 module. The binding energy EB of the catalyst was calculated according to the following formula to select the catalyst configuration with the best stability.
| EB = Etotal − Ecarrier+Ni, | (1.1) |
| Eb = EB ÷ n, | (1.2) |
Next, the structure optimization of the Ni–N–C–CO2 structure was carried out. The structure-optimized CO2 was attached to the active center, and the parameters were set for structure optimization considering single-site adsorption, double-site adsorption and different atoms connected. The specific equations for calculating adsorption energy are as follows:
| Eads = Ea − ECO2 − Etotal. | (1.3) |
Ultimately, the Gibbs free energies for the CO2 reduction to CO reaction intermediates, both catalyzed and uncatalyzed, were determined.
3 Results and discussion
3.1 Ni–N–C catalyst design
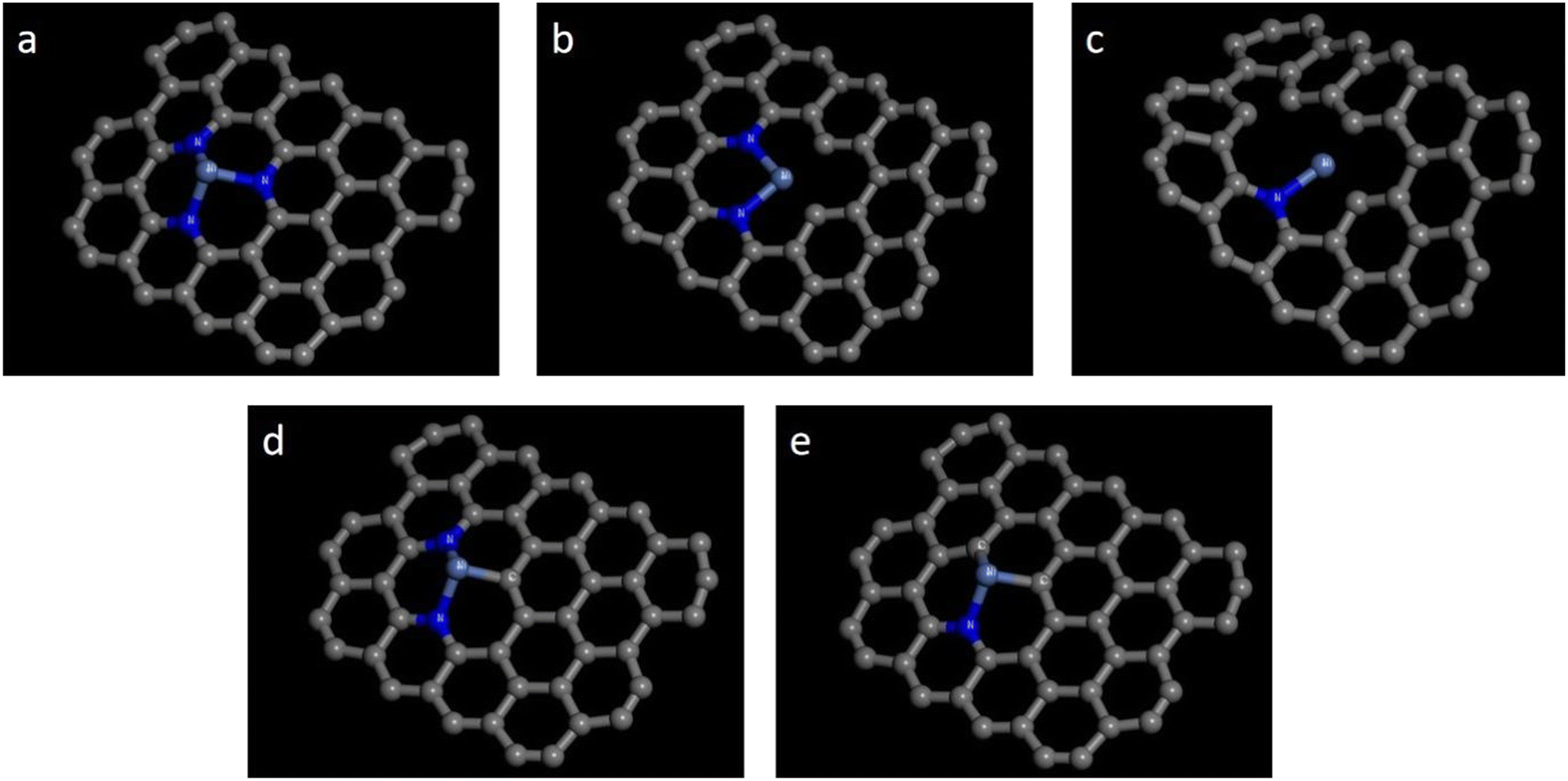 | ||
| Fig. 1 Five configurations of Ni-pyridine NnV3−n–C catalysts: (a) Ni-pyridine N3–C; (b) Ni-pyridine N2V1–C; (c) Ni-pyridine N1V2–C; (d) Ni-pyridine N2C1–C; and (e) Ni-pyridine N1C2–C. | ||
Ni-Pyridine NnV4−n–C catalysts were Ni–N–C catalysts consisting of four pyridine N atoms anchored to Ni atoms and containing vacancies, and each N atom formed a six-membered ring with graphene C in a pyridine structure. Fig. 2 illustrates that the Ni-pyridine NnV4−n–C catalysts can be categorized into seven configurations.
 | ||
| Fig. 2 Seven structures of Ni-pyridine NnV4−n–C catalysts: (a) Ni-pyridine N4–C; (b) Ni-pyridine N3V1–C; (c) Ni-pyridine N2V2–C; (d) Ni-pyridine N2V1–C; (e) Ni-pyridine N3C1–C; (f) Ni-pyridine N2C2–C; and (g) Ni-pyridine N1C3–C. | ||
 | ||
| Fig. 3 Seven structures of Ni-pyrrole NnV4−n–C catalysts: (a) Ni-pyrrole N4–C; (b) Ni-pyrrole N3V1–C; (c) Ni-pyrrole N2V2–C; (d) Ni-pyrrole N2V1–C; (e) Ni-pyrrole N3C1–C; (f) Ni-pyrrole N2C2–C; and (g) Ni-pyrrole N1C3–C. | ||
3.2 Stability studies of Ni–N–C catalysts
The Ni-pyridine NnV3−n–C catalyst was taken as an example, and the study calculated the average bond energies of the Ni–N and Ni–C bonds for each configuration. The results are presented in Table 1. Among the five structures, the one with the largest bond energy was the Ni-pyridine N2V1–C catalyst, which had an average bond energy of −0.0898 eV of the Ni–N bond, indicating that the catalyst had good stability. The largest total bond energy was the d structure, the Ni-pyridine N2C1–C catalyst, which was anchored by two N atoms and one C atom, with a total summed energy of EB = −0.1796 eV, showing good stability. In summary, the stable structures of the Ni-pyridine NnV3−n–C catalysts were Ni-pyridine N2V1–C and Ni-pyridine N2C1–C.| Isomers | E carrier+Ni/eV | E total/eV | E B/eV | E b/eV |
|---|---|---|---|---|
| a | −3536.73 | −3536.87 | −0.1481 | −0.0494 |
| b | −3481.98 | −3482.16 | −0.1796 | −0.0898 |
| c | −3427.27 | −3427.31 | −0.0500 | −0.0500 |
| d | −3520.02 | −3520.20 | −0.1798 | −0.0599 |
| e | −3503.39 | −3503.54 | −0.1511 | −0.0504 |
3.3 Effect of the number of vacancies on the performance of Ni–N–C catalysts
Subsequently, the catalysts underwent examination for the most occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). Both HOMO and LUMO symbolize the primary levels of molecular orbital energy. HOMO symbolizes the most filled molecular orbitals, while LUMO denotes the least occupied molecular orbital. For the energy band gap ΔE = ELUMO − EHOMO, the rate of electron movement within the catalyst can be ascertained using ΔE. A reduced ΔE indicates a quicker electron transfer rate in the front molecular orbitals and a more catalytic catalyst.The frontline molecular orbital energy levels and ΔE of the three previously selected Ni–N–C catalysts are illustrated in Fig. 4–6. Compared to the catalysts without vacancies, the catalysts containing vacancies were significantly larger for ΔE. The ΔE of the Ni-pyridine N3V1–C catalyst with vacancies was 0.872 eV, which was more than three times larger than that of the Ni-pyridine N4–C catalyst without vacancies (ΔE = 0.278 eV).
 | ||
| Fig. 4 Frontline molecular orbital energy levels and ΔE for Ni-pyridine NnV4−n–C catalysts: (a) Ni-pyridine N4–C; (b) Ni-pyridine N3V1–C; (c) Ni-pyridine N2V2–C; (d) Ni-pyridine N2V1–C; (e) Ni-pyridine N3C1–C; (f) Ni-pyridine N2C2–C; and (g) Ni-pyridine N1C3–C. | ||
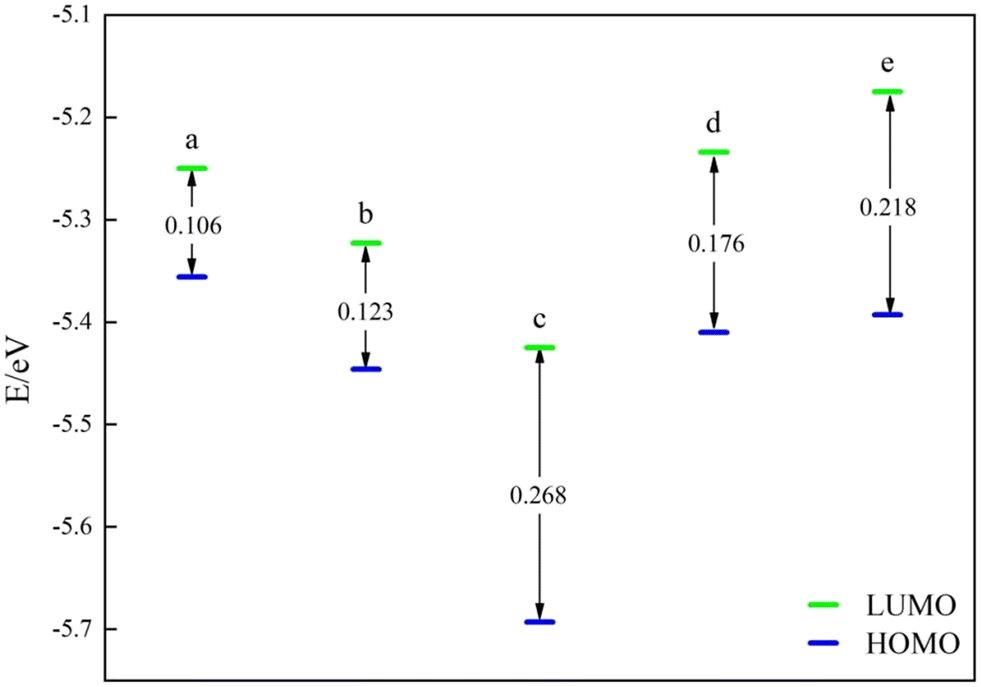 | ||
| Fig. 5 Frontline molecular orbital energy levels and ΔE for Ni-pyridine NnV3−n–C catalysts: (a) Ni-pyridine N3–C; (b) Ni-pyridine N2V1–C; (c) Ni-pyridine N1V2–C; (d) Ni-pyridine N2C1–C; and (e) Ni-pyridine N1C2–C. | ||
 | ||
| Fig. 6 Frontline molecular orbital energy levels and ΔE for Ni-pyrrole NnV4−n–C catalysts: (a) Ni-pyrrole N4–C; (b) Ni-pyrrole N3V1–C; (c) Ni-pyrrole N2V2–C; (d) Ni-pyrrole N2V1–C; (e) Ni-pyrrole N3C1–C; (f) Ni-pyrrole N2C2–C; and (g) Ni-pyrrole N1C3–C. | ||
3.4 Study of CO2 adsorption capacity of Ni–N–C catalysts
All atoms of the Ni-pyridine N2V1–C catalyst, Ni-pyrrole N2V2–C catalyst and Ni-pyridine N1V3–C catalyst were in the same plane, so there was no need to consider the direction of CO2 binding to the catalyst during structure optimization. Only single-site and double-site adsorption, as well as Ni–C bonding and Ni–O bonding, must be considered for a total of three cases.The Ni atoms of the Ni-pyridine N2C1–C catalyst and the Ni-pyridine N1V3–C catalyst were not in the same plane as the carrier graphene. Therefore, not only single-site and double-site adsorption but also the direction of CO2 molecule adsorption had to be considered, so there were 6 cases.
Table 2 demonstrates the Fukui functions for each Ni–N–C catalyst Ni atom as well as the N and C atoms attached to the Ni atom. From the table, it can be observed that the f(+) and f(−) of the Ni atoms of the six catalysts were the maximum values within the catalyst, indicating that this site could react with both Lewis acids and Lewis bases. Taking catalyst Ni-pyridine N2C1–C Catalyst Ni–C N–O as a case study, a periodic cell (Fig. S4†) was constructed, and the Fukui indices of all atoms were summarized in Table S1.† Therefore, it was the most suitable catalyst site.
| f(+) | f(−) | |||||
|---|---|---|---|---|---|---|
| Atom | Mulliken | Hirshfeld | Atom | Mulliken | Hirshfeld | |
| Ni-Pyridine N2V1–C | Ni(26) | 0.050 | 0.030 | Ni(26) | 0.055 | 0.030 |
| N(25) | 0.002 | 0.011 | N(25) | 0.002 | 0.010 | |
| N(31) | −0.000 | 0.008 | N(31) | 0.000 | 0.008 | |
| Ni-Pyridine N2C1–C | Ni(26) | 0.044 | 0.036 | Ni(26) | 0.055 | 0.030 |
| N(25) | 0.005 | 0.013 | N(25) | 0.005 | 0.013 | |
| N(31) | 0.000 | 0.007 | N(31) | 0.000 | 0.007 | |
| N(53) | 0.011 | 0.013 | N(53) | 0.011 | 0.013 | |
| Ni-Pyridine N1V3–C | Ni(39) | 0.053 | 0.030 | Ni(39) | 0.049 | 0.030 |
| N(19) | 0.002 | 0.012 | N(19) | 0.001 | 0.012 | |
| C(7) | 0.002 | 0.012 | C(7) | 0.017 | 0.012 | |
| C(20) | 0.015 | 0.015 | C(20) | 0.016 | 0.015 | |
| Ni-Pyrrole N2V2–C | Ni(40) | 0.076 | 0.039 | Ni(40) | 0.069 | 0.040 |
| N(19) | 0.001 | 0.013 | N(19) | 0.000 | 0.013 | |
| N(20) | 0.002 | 0.014 | N(20) | 0.002 | 0.014 | |
| Ni-Pyrrole N1V3–C | Ni(39) | 0.067 | 0.036 | Ni(39) | 0.065 | 0.037 |
| N(19) | 0.003 | 0.014 | N(19) | 0.003 | 0.014 | |
| C(7) | 0.0018 | 0.014 | C(7) | 0.018 | 0.014 | |
| C(20) | 0.015 | 0.014 | C(20) | 0.015 | 0.014 | |
Table 3 showed the Fukui index of CO2, from which it could be observed that the C atom of CO2 had the largest f(+) and a greater positron density, so in single-site adsorption, it could be assumed that the stability of the C atom connected to the Ni atom of the catalyst was greater than that of the O atom connected to the Ni atom. Therefore, the Ni–O connection was not considered for the time being.
| f(+) | |||
|---|---|---|---|
| Atom | Mulliken | Hirshfeld | |
| Ni-Pyrrole N1C3–C | C(1) | 0.462 | 0.384 |
| O(2) | 0.269 | 0.308 | |
| O(3) | 0.269 | 0.308 | |
(1) Ni-Pyridine N2V1–C catalysts. The structure of the Ni–N2V1–C–CO2 intermediate was designated (Fig. S1†). The bond lengths, bond angles and adsorption energies of the Ni–N2V1–C–CO2 structure were shown in Table 4.
| Isomers | E ads/eV | d C–O1/Å | d C–O2/Å |
|---|---|---|---|
| a | −0.0092 | 1.178 | 1.179 |
| b | −0.0087 | 1.178 | 1.178 |
| c | 0.0045 | 1.178 | 1.178 |
As shown in Table 4, the binding energies of (a) and (b) were negative and (c) was positive. Therefore, (c) CO2 could not be stabilized for adsorption. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond lengths of (a) were 1.178 Å and 1.179 Å, respectively, which were not much changed compared to the CO2 bond length of 1.178 Å. The CO bond lengths of (c) were both 1.178 Å, and there was no change in the bond lengths, so (a) and (b) had a low activation effect on CO2.
O bond lengths of (a) were 1.178 Å and 1.179 Å, respectively, which were not much changed compared to the CO2 bond length of 1.178 Å. The CO bond lengths of (c) were both 1.178 Å, and there was no change in the bond lengths, so (a) and (b) had a low activation effect on CO2.
(2) Ni-Pyridine N2C1–C catalysts. The bond lengths, bond angles and adsorption energies of the Ni-pyridine N2C1–C–CO2 structure (Fig. 7) are presented in Table 5.
 | ||
| Fig. 7 Isomers of the Ni-pyridine N2C1–C–CO2 intermediate: (a) Ni-pyridine N2C1–C–CO2 single-site adsorption A; (b) Ni-pyridine N2C1–C–CO2 single-site adsorption B; (c) Ni-pyridine N2C1–C–CO2 Ni–C N–O double-site adsorption A; (d) Ni-pyridine N2C1–C–CO2 Ni–C N–O double-site adsorption B; (e) Ni-pyridine N2C1–C–CO2 Ni–O N–C double-site adsorption A; and (f) Ni-pyridine N2C1–C–CO2 Ni–O N–C double-site adsorption B. | ||
| Isomers | E ads/eV | d C–O1/Å | d C–O2/Å |
|---|---|---|---|
| a | −0.0171 | 1.166 | 1.188 |
| b | −0.0088 | 1.178 | 1.178 |
| c | −0.0204 | 1.166 | 1.186 |
| d | −0.0089 | 1.177 | 1.178 |
| e | −0.0198 | 1.166 | 1.186 |
| f | −0.0085 | 1.177 | 1.178 |
As shown in Table 5, the adsorption energies of the six structures were negative, so all were stable. The adsorption energies of the three structures where CO2 was adsorbed from the side close to the Ni atom were significantly more stable. In terms of bond lengths, the structures in which CO2 was adsorbed from the side close to the Ni atom had a larger change in dC–O, which had a good activation effect on CO2 molecules.
(3) Ni-Pyridine N1V3–C catalysts. The structure of the Ni-pyridine N1V3–C–CO2 intermediate is shown in Fig. S2.† The bond lengths, bond angles and adsorption energies of the Ni-pyridine N1V3–C–CO2 intermediate are illustrated in Table 6.
| Isomers | E ads/eV | d C–O1/Å | d C–O2/Å |
|---|---|---|---|
| a | −0.0084 | 1.178 | 1.179 |
| b | −0.0008 | 1.178 | 1.178 |
| c | −0.0093 | 1.176 | 1.180 |
| d | 0.0039 | 1.178 | 1.178 |
| e | 0.0027 | 1.177 | 1.179 |
| f | 0.0018 | 1.177 | 1.178 |
In terms of binding energy, the binding energies Eads of (a), (b) and (c) were lower than the energy required for CO2 to be adsorbed stably, and the adsorption of CO2 was more stable from the side close to Ni atoms than that from the side far away from Ni atoms. In terms of bond lengths, the CO bond lengths of (a) and (b) were almost unchanged compared to CO2, so the catalytic activity of CO2RR might be very low. The CO bond lengths of (c) were 1.176 Å and 1.180 Å, which had a certain activation effect on CO2 molecules.
(4) Ni-Pyrrole N2V2–C catalysts. The structure of Ni-pyrrole N2V2–C–CO2 is shown in Fig. 8 as follows: (a) single-site adsorption, with Ni atoms attached to C atoms; (b) double-site adsorption, with Ni–C and N–O bonded; and (c) double-site adsorption, with N–O and N–C bonded. The binding energies and bond lengths of the Ni-pyrrole N2V2–C–CO2 intermediates are illustrated in Table 7.
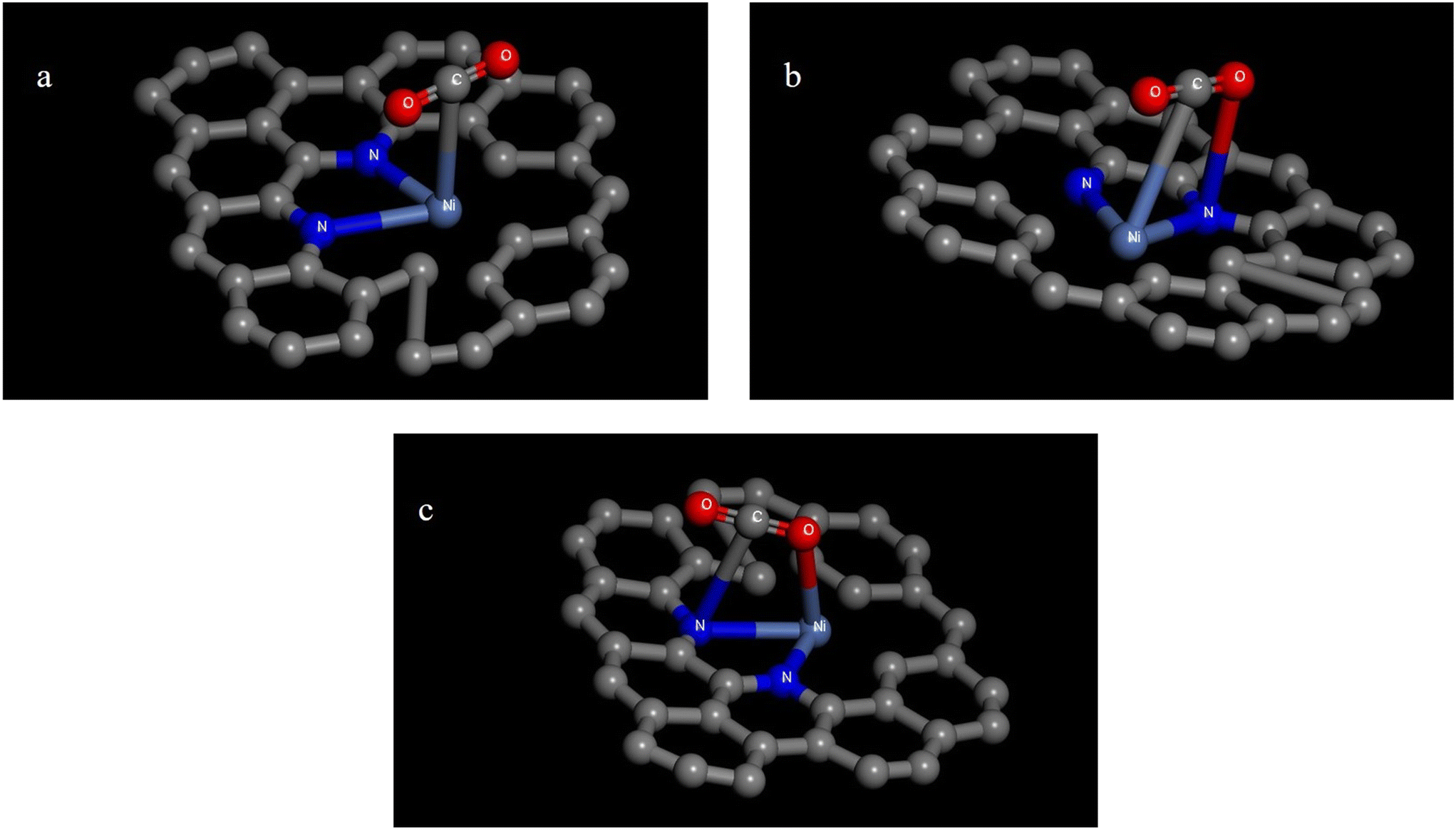 | ||
| Fig. 8 Isomers of the Ni-pyrrole N2V2–C–CO2 intermediate: (a) Ni-pyrrole N2V2–C–CO2 single-site adsorption; (b) Ni-pyrrole N2V2–C–CO2 Ni–C N–O double-site adsorption; and (c) Ni-pyrrole N2V2–C–CO2 Ni–O N–C double-site adsorption. | ||
| Isomers | E ads/eV | d C–O1/Å | d C–O2/Å |
|---|---|---|---|
| a | −0.0093 | 1.177 | 1.179 |
| b | −0.0096 | 1.177 | 1.178 |
| c | −0.0094 | 1.178 | 1.178 |
In terms of binding energy, the binding energies of all three intermediates were negative, so all CO2 could be stably adsorbed on the catalyst. According to the CO bond lengths, the three intermediates did not change much compared with CO2. Therefore, the activation of CO2 molecules of the Ni-pyrrole N2V2–C catalysts was low.
(5) Ni-Pyrrole N1V3–C catalysts. Fig. S3† depicts the isomers of the Ni-pyrrole N1V3–C–CO2 intermediate as follows: (a) single-site adsorption, with the Ni atom attached to the C atom; (b) double-site adsorption, with Ni–C and N–O bonded; and (c) double-site adsorption, with N–O–N–C bonded. The binding energies and bond lengths of the Ni-pyrrole N1V3–C–CO2 intermediate are shown in Table 8.
| Isomers | E ads/eV | d C–O1/Å | d C–O2/Å |
|---|---|---|---|
| a | −0.0090 | 1.178 | 1.178 |
| b | −0.0101 | 1.178 | 1.178 |
| c | −0.0099 | 1.178 | 1.178 |
As illustrated in Table 8, the binding energies of all three structures were negative, so CO2 could be stably adsorbed on the catalysts. However, the CO bond lengths of (a), (b) and (c) were all 1.178 Å, which was the same as that of CO2. Therefore, the activation of CO2 molecules by the three catalysts was low.
3.5 Study of the catalytic activity of catalysts for CO2RR catalyzation
 | ||
| Fig. 9 Reaction path diagram for catalysts in the case of single-site adsorption (where A represents adsorption from the proximal side and B represents adsorption from the distal side). | ||
Based on the magnitude of the energy barriers, it could be observed that the decisive step for the uncatalyzed and Ni-pyridine N1V3–C catalyst B was the formation of *CO, and the rest of the decisive step was the formation of *COOH in the first step.
The catalytic ability of the Ni-pyridine N2C1–C catalyst for two-site adsorption of CO2 from the proximal side with Ni–C, N–O bonding was first investigated. The adsorption energy data were summarized in Table S2.†
Five cases of catalyst two-site adsorption of CO2 were illustrated in Fig. 10. As observed from the figure, the Ni-pyridine N2C1–C catalyst Ni–C N–O A was the best catalyst with a reaction energy barrier of ΔG = −1.4029 eV, which was a decrease of 4.3932 eV compared to the energy barrier of ΔG = 2.9903 eV in the absence of the catalyst, and the reaction was exothermic and thermodynamically capable of proceeding spontaneously. The energy barrier for the reaction under Ni-pyridine N2C1–C catalyst Ni–O N–C B was 0.4084 eV, and the energy barrier of the reaction under Ni-pyridine N2C1–C catalyst Ni–C N–O B was 1.9764 eV. There was a substantial decrease in the energy barrier compared to the reaction without the catalyst, so both had good catalytic activity; the rest of the catalysts had limited catalytic performance.
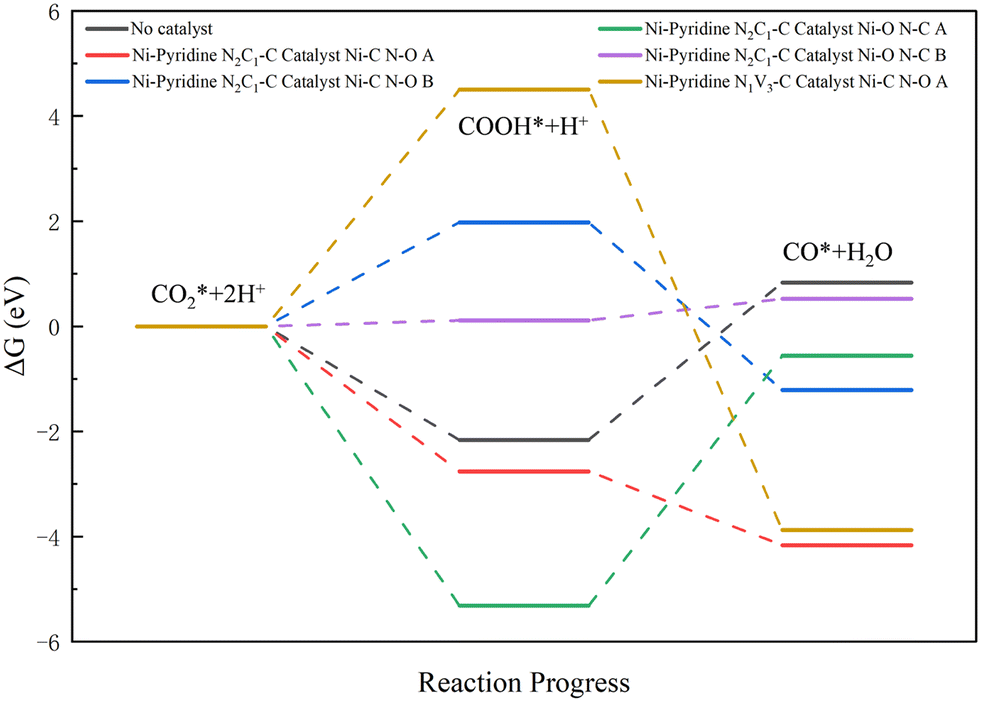 | ||
| Fig. 10 Reaction path diagram for catalysts in the case of two-site adsorption (where A represents adsorption from the near side and B represents adsorption from the far side). | ||
4 Conclusions
In this study, the stability, activation and catalytic performance of the catalysts were analyzed and screened for better catalysts by performing energy calculations and structure optimization of the constructed structures based on the theoretical basis of quantum chemistry. The following conclusions were obtained.(1) Vacancies could significantly affect the catalyst properties. The Ni–N–C catalyst with vacancies had a higher Ni–N bond energy and good catalyst stability. However, its ΔE was larger, and the electron conduction rate was slower than that of the catalyst without vacancies. The ΔE of the Ni-pyridine N3V1–C catalyst was 0.872 eV, which was more than three times that of the catalyst without the Ni-pyridine N4–C catalyst with ΔE = 0.278 eV.
(2) The catalysts with excellent activation performance for CO2 were screened as Ni-pyridine N2C1–C catalyst, Ni-pyridine N1V3–C catalyst and Ni-pyrrole N2V2–C catalyst. Among them, the best CO2 activation ability was the Ni-pyridine N2C1–C catalyst.
(3) In single-site adsorption, the best catalytic effect was achieved by Ni-pyridine N1V3–C catalyst A, with reaction ΔG = 0.2714 eV. In two-site adsorption, the best catalytic effect was achieved by Ni-pyridine N2C1–C Ni–C N–O A, with reaction ΔG = −1.4029 eV, and the reaction was able to proceed thermodynamically and spontaneously.
(4) Both single-site adsorption and dual-site adsorption reduced the reaction energy barrier compared to no catalyst. Overall, most of the catalytic activities of single-site adsorption were better than those of dual-site adsorption.
Data availability
The data used to support the findings of this paper are included within the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Supports of the Hefei Advanced Computing Center for this work are gratefully acknowledged.References
- L. Tang, R. Ii and K. Tokimatsu, et al., Development of human health damage factors related to CO2 emissions by considering future socioeconomic scenarios, Int. J. Life Cycle Assess., 2018, 23, 2288–2299 CrossRef CAS.
- J. Zhang, J. Shen and L. Xu, et al., The CO2 emission reduction path towards carbon neutrality in the Chinese steel industry: A review, Environ. Impact Assess. Rev., 2023, 99, 107017 CrossRef.
- Y. Zhang, X. Fan and X. He, et al., Ambient electrosynthesis of urea from carbon dioxide and nitrate over Mo2C nanosheet, Chin. Chem. Lett., 2024, 35(8), 109806 CrossRef CAS.
- X. Gao, H. He and W. Zhu, et al., Continuously Flow Photothermal Catalysis Efficiently CO2 Reduction Over S-Scheme 2D/0D Bi5O7I-OVs/Cd0.5Zn0.5S Heterojunction with Strong Interfacial Electric Field, Small, 2023, 19(12), 2206225 CrossRef CAS PubMed.
- S. Jin, Z. Hao and K. Zhang, et al., Advances and Challenges for the Electrochemical Reduction of CO2 to CO: From Fundamentals to Industrialization, Angew. Chem., Int. Ed., 2021, 60(38), 20627–20648 CrossRef CAS PubMed.
- B. Mohanty, S. Basu and B. K. Jena, Transition metal-based single-atom catalysts (TM-SACs); rising materials for electrochemical CO2 reduction, J. Energy Chem., 2022, 70, 444–471 CrossRef CAS.
- Z. Wang, K. Sun and C. Liang, et al., Synergistic Chemisorbing and Electronic Effects for Efficient CO2 Reduction Using Cysteamine-Functionalized Gold Nanoparticles, ACS Appl. Energy Mater., 2019, 2(1), 192–195 CrossRef CAS.
- E. L. Clark, S. Ringe and M. Tang, et al., Influence of Atomic Surface Structure on the Activity of Ag for the Electrochemical Reduction of CO2 to CO, ACS Catal., 2019, 9(5), 4006–4014 CrossRef CAS.
- J. Bonin, A. Maurin and M. Robert, Molecular catalysis of the electrochemical and photochemical reduction of CO2 with Fe and Co metal based complexes. Recent advances, Coord. Chem. Rev., 2017, 334, 184–198 CrossRef CAS.
- J. E. Pander, J. W. J. Lum and B. S. Yeo, The importance of morphology on the activity of lead cathodes for the reduction of carbon dioxide to formate, J. Mater. Chem. A, 2019, 7(8), 4093–4101 RSC.
- Z. B. Hoffman, T. S. Gray and K. B. Moraveck, et al., Electrochemical Reduction of Carbon Dioxide to Syngas and Formate at Dendritic Copper–Indium Electrocatalysts, ACS Catal., 2017, 7(8), 5381–5390 CrossRef CAS.
- J. Wu, M. Liu and P. P. Sharma, et al., Incorporation of Nitrogen Defects for Efficient Reduction of CO2 via Two-Electron Pathway on Three-Dimensional Graphene Foam, Nano Lett., 2016, 16(1), 466–470 CrossRef CAS PubMed.
- N. Yang, S. R. Waldvogel and X. Jiang, Electrochemistry of Carbon Dioxide on Carbon Electrodes, ACS Appl. Mater. Interfaces, 2016, 8(42), 28357–28371 CrossRef CAS PubMed.
- Q. Li, J. Fu and W. Zhu, et al., Tuning Sn-Catalysis for Electrochemical Reduction of CO2 to CO via the Core/Shell Cu/SnO2 Structure, J. Am. Chem. Soc., 2017, 139(12), 4290–4293 CrossRef CAS PubMed.
- Y. Lum, Y. Kwon and P. Lobaccaro, et al., Trace Levels of Copper in Carbon Materials Show Significant Electrochemical CO2 Reduction Activity, ACS Catal., 2016, 6(1), 202–209 CrossRef CAS.
- Q. Li, W. Zhu and J. Fu, et al., Controlled assembly of Cu nanoparticles on pyridinic-N rich graphene for electrochemical reduction of CO2 to ethylene, Nano Energy, 2016, 24, 1–9 CrossRef CAS.
- R. Daiyan, W. H. Saputera and Q. Zhang, et al., 3D Heterostructured Copper Electrode for Conversion of Carbon Dioxide to Alcohols at Low Overpotentials, Adv. Sustainable Syst., 2019, 3(1), 1800064 CrossRef.
- J. Kim, J. E. Dick and A. J. Bard, Advanced Electrochemistry of Individual Metal Clusters Electrodeposited Atom by Atom to Nanometer by Nanometer, Acc. Chem. Res., 2016, 49(11), 2587–2595 CrossRef CAS PubMed.
- T. Imaoka and K. Yamamoto, Wet-Chemical Strategy for Atom-Precise Metal Cluster Catalysts, Bull. Chem. Soc. Jpn., 2019, 92(5), 941–948 CrossRef CAS.
- Z. Luo, A. W. Castleman and S. N. Khanna Jr., Reactivity of Metal Clusters, Chem. Rev., 2016, 116(23), 14456–14492 CrossRef CAS PubMed.
- N. Thiyagarajan, D. Janmanchi and Y.-F. Tsai, et al., A Carbon Electrode Functionalized by a Tricopper Cluster Complex: Overcoming Overpotential and Production of Hydrogen Peroxide in the Oxygen Reduction Reaction, Angew. Chem., Int. Ed., 2018, 57(14), 3612–3616 CrossRef CAS PubMed.
- G. Li and R. Jin, Atomic level tuning of the catalytic properties: Doping effects of 25-atom bimetallic nanoclusters on styrene oxidation, Catal. Today, 2016, 278, 187–191 CrossRef CAS.
- S. Guo, S. Zhao and X. Wu, et al., A Co3O4-CDots-C3N4 three component electrocatalyst design concept for efficient and tunable CO2 reduction to syngas, Nat. Commun., 2017, 8(1), 1828 CrossRef PubMed.
- X. Wang, Z. Chen and X. Zhao, et al., Regulation of Coordination Number over Single Co Sites: Triggering the Efficient Electroreduction of CO2, Angew. Chem., Int. Ed., 2018, 57(7), 1944–1948 CrossRef CAS PubMed.
- S. Liang, L. Huang and Y. Gao, et al., Electrochemical Reduction of CO2 to CO over Transition Metal/N-Doped Carbon Catalysts: The Active Sites and Reaction Mechanism, Adv. Sci., 2021, 8(24), 2102886 Search PubMed.
- W. Ju, A. Bagger and G.-P. Hao, et al., Understanding activity and selectivity of metal-nitrogen-doped carbon catalysts for electrochemical reduction of CO2, Nat. Commun., 2017, 8(1), 944 Search PubMed.
- F. Pan, H. Zhang and K. Liu, et al., Unveiling Active Sites of CO2 Reduction on Nitrogen-Coordinated and Atomically Dispersed Iron and Cobalt Catalysts, ACS Catal., 2018, 8(4), 3116–3122 Search PubMed.
- J. Feng, H. Gao and L. Zheng, et al., A Mn-N3 single-atom catalyst embedded in graphitic carbon nitride for efficient CO2 electroreduction, Nat. Commun., 2020, 11(1), 4341 CrossRef PubMed.
- L. Bai, Z. Duan and X. Wen, et al., Atomically dispersed manganese-based catalysts for efficient catalysis of oxygen reduction reaction, Appl. Catal., B, 2019, 257, 117930 Search PubMed.
- X. Wen, H. Qi and Y. Cheng, et al., Cu Nanoparticles Embedded in N-Doped Carbon Materials for Oxygen Reduction Reaction, Chin. J. Chem., 2020, 38(9), 941–946 Search PubMed.
- L. Bai, Z. Duan and X. Wen, et al., Atomically dispersed manganese-based catalysts for efficient catalysis of oxygen reduction reaction, Appl. Catal., B, 2019, 257, 117930 Search PubMed.
- P. P. Sharma, J. Wu and R. M. Yadav, et al., Nitrogen-Doped Carbon Nanotube Arrays for High-Efficiency Electrochemical Reduction of CO2: On the Understanding of Defects, Defect Density, and Selectivity, Angew. Chem., Int. Ed., 2015, 54(46), 13701–13705 CrossRef CAS PubMed.
- G.-L. Chai and Z.-X. Guo, Highly effective sites and selectivity of nitrogen-doped graphene/CNT catalysts for CO2 electrochemical reduction, Chem. Sci., 2016, 7(2), 1268–1275 RSC.
- C. Jia, S. Li and Y. Zhao, et al., Nitrogen Vacancy Induced Coordinative Reconstruction of Single-Atom Ni Catalyst for Efficient Electrochemical CO2 Reduction, Adv. Funct. Mater., 2021, 31(51), 2107072 Search PubMed.
- Y. Cheng, S. Zhao and H. Li, et al., Unsaturated edge-anchored Ni single atoms on porous microwave exfoliated graphene oxide for electrochemical CO2, Appl. Catal., B, 2019, 243, 294–303 Search PubMed.
- C. Wang, X. Hu and X. Hu, et al., Typical transition metal single-atom catalysts with a metal-pyridine N structure for efficient CO2 electroreduction, Appl. Catal., B, 2021, 296, 120331 Search PubMed.
- Y. Liu, J. Zhao and Q. Cai, Pyrrolic-nitrogen doped graphene: a metal-free electrocatalyst with high efficiency and selectivity for the reduction of carbon dioxide to formic acid: a computational study, Phys. Chem. Chem. Phys., 2016, 18(7), 5491–5498 RSC.
- J. Wu, M. Liu and P. P. Sharma, et al., Incorporation of Nitrogen Defects for Efficient Reduction of CO2 via Two-Electron Pathway on Three-Dimensional Graphene Foam, Nano Lett., 2016, 16(1), 466–470 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy01394h |
| This journal is © The Royal Society of Chemistry 2025 |