
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing activity and selectivity of palladium catalysts in ketone α-arylation by tailoring the imine chelate of pyridinium amidate (PYA) ligands†
Esaïe
Reusser
 ,
Michael
Aeschlimann
and
Martin
Albrecht
*
,
Michael
Aeschlimann
and
Martin
Albrecht
*
Department of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: martin.albrecht@unibe.ch
First published on 18th December 2024
Abstract
Even though α-arylation of ketones is attractive for direct C–H functionalization of organic substrates, the method largely relies on phosphine-ligated palladium complexes. Only recently, efforts have focused on developing nitrogen-based ligands as a more sustainable alternative to phosphines, with pyridine-functionalized pyridinium amidate (pyr-PYA) N,N′-bidentate ligands displaying good selectivity and activity. Here, we report on a second generation set of catalyst precursors that feature a 5-membered N-heterocycle instead of a pyridine as chelating unit of the PYA ligand to provide less steric congestion for the rate-limiting transmetalation of the enolate. To this end, new heterocycle-functionalized PYA palladium(II) complexes containing an oxazole (5b), N-phenyl-triazole (5c), N-methyl pyrazole (5d), N-phenyl-pyrazole, (5e), N-xylyl-pyrazole (5f), and N-isopropyl-pyrazole (5g) were synthesized compared to the parent pyr-PYA complex 5a. Less packing of the palladium coordination sphere was evidenced from solid state X-ray diffraction analysis. While the catalytic activity of the oxazole system was lower, all other complexes showed higher activity. In particular, complex 5g comprised of an electron-donating and sterically demanding iPr-pyrazole chelating PYA ligand is remarkably stable towards air and moisture and shows outstanding catalytic activity with complete selectivity (>99% yield) and turnover frequencies up to 1200 h−1, surpassing that of parent 5a by one order of magnitude and rivalling the most active phosphine-based palladium systems. Kinetic studies demonstrate a first order rate-dependence on palladium and the substrate. Some deviation of linearity together with poisoning experiments suggest a mixed homogeneous/heterogeneous pathway, though the reproducible kinetics of in situ catalyst recycling experiments strongly point to a molecularly defined active species, demonstrating the high potential of PYA-based ligands.
Introduction
Transition-metal-catalyzed cross-coupling methodologies have become a fundamental strategy in the synthesis of pharmaceuticals, natural products, and fine chemicals.1–3 In the late 1990s, Buchwald and Hartwig broadened the scope of cross-coupling reactions to include the direct α-arylation of enolizable ketones (Fig. 1a).4–7 This expansion offers access to versatile chemical intermediates and to a structural motif that is abundant in many active pharmaceutical ingredients.8–11 The previously used stoichiometric methods often suffered from narrow scope and selectivity, whereas transition metal catalysts overcome these problems and additionally make the process considerably more sustainable by reducing the amount of (hazardous) waste.12,13 Early work demonstrated the relevance of palladium for the catalytic α-arylation of ketones, especially when employed in conjunction with sterically hindered, strongly electron-donating phosphine ligands.14–16 Specifically, wide bite angle diphosphine ligands such as I or II (Fig. 1b) impart high steric hindrance and therefore disfavor the β-H elimination side reaction.4,5,17 However, phosphine-based catalysts often suffer from low thermal stability and high sensitivity towards oxidation. Furthermore, due to their highly apolar nature, these ligands are often challenging to separate from the reaction mixture. As alternative ligands to phosphines, N-heterocyclic carbenes (NHCs) have emerged as valuable ligands to palladium for α-arylation catalysis and have shown remarkable potency in activating aryl chloride (complex III).18–21 They also offer access to molecularly well-defined pre-catalysts, in contrast to first-generation catalytic systems that were generated in situ.22–26 Notably, though, catalysts based on NHCs generally exhibit moderate turnover frequencies (TOFs) compared to chelating phosphine systems.21 Furthermore, earlier investigations on carbene-based ligands in alkene oligomerization showed that those ligands tend to reductively eliminate from the hydride intermediate via β-hydrogen elimination, yielding the corresponding imidazolium salt and ensuing deactivation, although such processes might be prevented under basic conditions.27–29 | ||
| Fig. 1 a) General reaction scheme of ketone α-arylation; b) previously reported catalytic systems for the ketone α-arylation reaction and their optimized performance; c) limiting resonance structures of ortho-pyridinium amidate (PYA) ligand; d) structural modification of the precatalyst targeted in this work and depiction of the α-angle. | ||
Similar to NHCs, pyridinium amidates (PYAs) are characterized by strong σ-donation that is beneficial in stabilizing metal complexes under harsh conditions. Indeed, initial investigations suggested that the donor strength of these ligands is comparable to that of traditional NHC ligands.30–32 Furthermore, a typical feature of PYA ligands is their ability to adopt different resonance structures that exhibit either π-basic or π-acidic properties, implying electronic donor flexibility that responds to the electronic environment of the coordinated metal center (Fig. 1c).33 This adaptability may be particularly beneficial in palladium-catalyzed cross-coupling processes involving both oxidative addition and reductive elimination steps. Ligands capable of flexibly adjusting their donor properties are expected to enhance the accessibility of both zero-valent and +II oxidation states, thus contributing to the efficiency of the catalytic system. The beneficial role of PYA ligands has been demonstrated in previous studies on palladium-catalyzed cross-coupling reactions, such as Suzuki–Miyaura reactions.34 These PYA ligands are readily synthesized from cheap aminopyridine, and offer vast opportunities for introducing potentially chelating donor sites.33,35 We recently exploited the facile accessibility by introducing a set of air- and moisture-stable palladium(II) complexes featuring a N,N′-bidentate chelating ligand based on electronically flexible PYA ligands linked to pyridine (IV, Fig. 1b)36 and showed their activity in catalytic α-arylation of ketones.37 Their performance is remarkably high for nitrogen-based ligands, with turnover numbers reaching up to 7300 and turnover frequencies approaching 9000 h−1, placing these catalysts on a competitive level with the best NHC and phosphine-based systems. Mechanistic investigations indicated that both oxidative addition and reductive elimination proceed swiftly in PYA-based catalysts, likely facilitated by the donor flexibility of the PYA ligand, while enolate coordination was identified as the turnover-limiting step. The previously observed ligand effects strongly support a catalytically active species that maintains coordination with the pyridyl–PYA ligand.37 These observations suggest that further customization of the imine donor, such as replacing the pyridyl unit by other donor units holds promise for enhancing catalytic performance. Specifically, the introduction of 5-membered heterocyclic ligand scaffolds affects the space available for coordination at the cis-position by alleviating the steric impact of substituents, therefore strongly modulating activity and selectivity of the catalyst. The more open α angle (Fig. 1d) might suppress the deleterious effect of bulky ligand substituents on the catalyst selectivity while retaining the high activity.
Here we demonstrate that palladium(II) complexes with a chelating PYA ligand comprised of a pyrazole or triazole donor constitute precursors to highly active and selective ketone α-arylation catalysts, enabling full conversions in less than 10 min. This work illustrates the usefulness of ligand design to tailor selectivity and activity of existing catalysts.
Results and discussion
Synthesis and characterization of PYA palladium complexes
A series of novel PYA palladium complexes were prepared starting from commercially available 2-amino-3-methylpyridine in four straightforward synthetic steps (Fig. 2a). In the presence of benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) as coupling agent and NEt3, the aminopyridine reacted with the appropriate heterocyclic carboxylic acid 1a–1d to afford the corresponding amides 2a–2d in good yields (62–79%). The reaction was followed by 1H NMR spectroscopy, since the appearance of the NH resonance at δH ∼10 ppm together with the disappearance of the broad singlet at δH = 5.8 ppm due to the aniline NH2 group were diagnostic for the formation of the corresponding amides 2. Subsequent methylation of the pyridine was selective even in the presence of an excess of methyl iodide, indicated by the appearance of a new N-methyl resonance around 4.2 ppm in the 1H NMR spectrum together with a marked downfield shift of the aromatic pyridine signals. Upon precipitation with Et2O, iodide salts 3a–3d were isolated in 60–78% yield. Deprotonation of these pyridinium salts proceeded smoothly in the presence of an excess aqueous KOH and yielded the neutral PYA ligands 4a–4d in excellent yields (79–94%), indicated by the diagnostic upfield shift of the pyridyl N-methyl resonance from 4.3 to 3.8 ppm together with the disappearance of the NH signal. Coordination of palladium using [PdCl2(cod)] was essentially quantitative and afforded complexes 5a–5d as yellow solids (Fig. 2b). Upon palladium coordination, the PYA N–CH3 signal shifted back to ∼4.3 ppm suggesting a similar electronic influence of a proton on the PYA unit as the palladium(II) fragment. All the palladium complexes were stable towards air, light and moisture for weeks. Crystals suitable for X-ray diffraction were grown for complexes 5b and 5d by slow diffusion of Et2O into a complex solution in CH2Cl2 and MeOH, respectively (Fig. 2c). | ||
| Fig. 2 (a) General synthetic scheme for the preparation of the Pd-PYA complexes 5a to 5d; (b) schematic drawings of complexes 5a–d; (c) molecular structures of complexes 5b and 5d determined by X-ray diffraction analysis (30% probability ellipsoids, hydrogen atoms and co-crystallized solvent molecules omitted for clarity). | ||
Both complexes displayed a palladium center in a slightly distorted square planar geometry (τ4 = 0.094 and 0.034 for 5b and 5d, respectively)38 similar to the configuration of 5a (Table 1). For all complexes Pd–Cltrans was slightly longer compared to Pd–Clcis, indicating a stronger trans influence of the PYA donor site compared with the heterocyclic imine donor. Ligand bite angles, which are known to be highly relevant in cross-coupling processes,39 are similar for 5a, 5b and 5d (Nhet–Pd–NPYA 79.8 ± 0.6°), despite the transition from a 6- to a 5-membered heterocyclic scaffold. Analysis of the buried volume (%Vbur)40 induced by the different ligands revealed that switching from 6- to 5-membered heterocyclic coordination sites effectively reduces the %Vbur (38.6% for 5bvs. 40.2% for 5a). The steric hindrance of the pyridine ligand was restored upon adding a methyl substituent at the pseudo-ortho position of the heterocycle, despite its distal orientation from the Pd center (%Vbur = 40.6 for 5d). The wider α angle in 5b and 5d (128.8(13)° and 123.3(15)° vs. 119.1(1)° in 5a) illustrates the larger space created by the 5- vs. 6-membered heterocycle. Consequently, the distance between the (pseudo-)ortho proton of the heterocycle and the adjacent chloride Cltrans is some 2.69 Å for 5a, yet markedly longer at 3.08 Å in 5b.
| 5a (Het = py) | 5b (Het = ox) | 5d (Het = prz) | |
|---|---|---|---|
| a Data from ref. 37. b Percentage buried volume %Vbur determined according to ref. 40, see ESI† for details c The α angle was determined as Npy–Cpy–H for 5a, Nox–Cox–H for 5b, and Nprz–Nprz2–CMe for 5d (see also Fig. 1d). d Tetrahedral distortion parameter τ4 determined according to ref. 38. | |||
| Pd–NHet/Å | 2.013(3) | 1.9979(15) | 2.0369(14) |
| Pd–NPYA/Å | 2.006(3) | 2.0479(15) | 2.0253(15) |
| Pd–Cltrans/Å | 2.3030(9) | 2.3086(5) | 2.2990(5) |
| Pd–Clcis/Å | 2.2935(11) | 2.2977(5) | 2.2861(5) |
| NPYA–Cα/Å | 1.395(4) | 1.392(2) | 1.392(2) |
| NPYA–CCO/Å | 1.340(4) | 1.356(2) | 1.349(2) |
| NPYA–Pd–NHet /° | 80.35(10) | 80.33(6) | 79.20(6) |
| %Vburb | 40.2 | 38.6 | 40.6 |
| α/°c | 119.1(3) | 128.8(13) | 123.3(15) |
| τ 4 | 0.084 | 0.094 | 0.034 |
Evaluation in the catalytic α-arylation of propiophenone
The catalytic activity of complexes 5b–5d was probed in the α-arylation of ketones using propiophenone and bromobenzene as model substrates (Table 2).37 Comparison to complex 5a as benchmark catalyst precursor revealed a substantial influence of the heterocyclic ligand on the catalytic activity. Substitution of pyridine with the oxazole moiety (5b) had a negative effect on the reaction rate, reducing the TOFmax from 130 to 85 h−1 as well as a reduced yield at full conversion of ArBr from 87% to 70% (entries 1,2). In contrast, insertion of either a triazole or a pyrazole unit (5c and 5d) improved the activity, affording TOFmax = 270 and 830 h−1, respectively (entries 3, 4). However, triazole complex 5c suffered from a diminished selectivity as it only reached a moderate yield of 77% at full conversion, while the pyrazole-substituted analogue 5d gave 91% yield after 1 h. The higher activity of N-methyl pyrazolyl-functionalized complex 5d might arise from the stronger donor properties of pyrazole compared to the oxazole and triazole analogues. Indeed, the lower pKa of the parent N-methyl pyrazole heterocycle (pKa 2.06) suggests a higher Lewis basicity than that of oxazole (pKa 0.8) or triazole (pKa 1.25).41–44 Steric arguments may also play a role, especially due to the methyl substituent on the pyrazole unit, though the %Vbur of 5d is similar to 5a (see above).|
|
|||||
|---|---|---|---|---|---|
| Entry | [Pd] | Optimized yieldb (time) | Yield | TOFmax/h−1 | k ini/h−1 |
| a Reaction conditions: PhBr (1.0 mmol), propiophenone (1.2 mmol), [Pd] (0.01 mmol), NaOtBu (1.32 mmol), 1,4-dioxane (2.0 mL), 105 °C under N2, yield determined by GC analysis using hexamethylbenzene as internal standard; for TOFmax evaluations see Fig. S63;† for calculation of initial rate constants kini see Fig. S64;† b Optimized yields determined without sampling, using identical conditions except propiophenone (1.0 mmol) NaOtBu (1.1 mmol), dioxane (1.0 mL). | |||||
| 1 | 5a | 87% (2 h) | 82% | 130 | 1.7 |
| 2 | 5b | 70% (2 h) | 67% | 85 | 1.0 |
| 3 | 5c | 77% (1 h) | 77% | 270 | 3.3 |
| 4 | 5d | 91% (1 h) | 84% | 830 | 8.3 |

Kinetic and mechanistic investigations
Due to its enhanced activity, complex 5d containing a pyrazole donor was investigated in more detail as a catalyst. Running the ketone arylation at different reaction temperatures allowed for an Eyring analysis of the initial reaction rates (Fig. S79†) and furnished activation parameters ΔH‡ = 109 ± 3 kJ mol−1 and ΔS‡ = −34 ± 8 J K−1 mol−1. These values are very similar to those established for 5a (ΔH‡ = 105 ± 10 kJ mol−1 and ΔS‡ = −89 ± 26 J K−1 mol−1),37 suggesting a similar catalytic cycle and corroborating an associative turnover-limiting step.45 Variation of the catalyst concentration indicated a first-order rate-dependence in catalyst, both for 5a and 5d (Fig. 3, S67–S69†).46 These observations demonstrate the involvement of the catalyst in the turnover-limiting step and strongly point towards a molecularly defined catalytically active species. Notably, the linear fit for 5d deviates significantly from the expected zero y-intercept, pointing to some curvature and a partial order in catalyst at higher concentrations. Such a behavior may be caused by an off-cycle monomer-dimer equilibrium, or by the involvement of nanoparticles, with ligand dissociation as a fast pre-equilibrium. In agreement with a more intricate role of the catalyst, log–log plots revealed a broken order in [5d], yet clear first order in [5a] (Fig. S74†). | ||
| Fig. 3 Rate-dependence with respect to catalyst concentration a) for complex 5a, and b) for complex 5d, indicating a first-order dependence for both catalytic systems. | ||
Modulating the concentration of bromobenzene suggests a pseudo zero order dependence of the rate (Fig. 4a, S76†), corroborating previous conclusions that oxidative addition is not rate-limiting with these PYA palladium catalysts.37,47 The slightly negative slope indicates an inhibitory effect, which may be linked to the formation of radical species or the involvement of nanoparticles.48–50
 | ||
| Fig. 4 Rate-dependence of the 5d-catalyzed α-arylation on a) bromobenzene, and b) propiophenone, revealing essentially zero-order dependence on the aryl halide, and different regimes of first-order dependence on the ketone. | ||
In contrast, variation of the propiophenone concentration gave a less clear picture. At low concentration (<0.65 M), a linear correlation and first-order reaction kinetics were established (Fig. 4, S75†), corroborating the involvement of the ketone in the rate-limiting step as previously deduced from Hammett studies.37,51 However, at higher propiophenone concentrations (>0.65 M), the correlation is less obvious and the slope different. This deviation might be attributed to the changed conditions, as higher ketone concentrations require larger amounts of NaOtBu. Alternatively, the data may be correlated to a sigmoidal curve, which would point to a departure from a homogeneous model with a molecular catalyst towards the formation of heterogeneous aggregates.
In an attempt to distinguish a homogeneous from a heterogeneous mode of action, poisoning experiments were performed. To this end, catalytic runs were carried out under standard conditions,52 yet at approximately 30% conversion, either PPh3 (10 eq. relative to Pd) or Hg (>70 eq. relative to Pd) was added to the reaction mixture.53 The pertinent conversion profiles indicate that either of the two additives effectively inhibit the catalytic activity of both 5a and 5d (Fig. S77 and S78†). This outcome seems controversial at first sight as an excess of phosphine is supposed to competitively bind to the metal center and thus inhibit substrate coordination in a homogeneous system, whereas mercury is assumed to act on a heterogeneous catalyst through the formation of amalgams and ensuing passivation of reactive surfaces.45 Both poisoning tests have their limitations,54,55 and furthermore, we noted that the activity was not immediately suppressed, but only some 30 min after addition of the additive for both PPh3 and Hg, suggesting some residual activity even after poisoning. These observations therefore suggest mixed homo- and heterogeneous mechanisms, in which colloidal—possibly ligand-stabilized56—palladium(0) constitutes a resting state, from which PYA–palladium species can dissociate and operate as homogeneous catalysts, a mode of action that has sometimes been referred to as pseudo-homogeneous mechanism,57–60 It also hints to higher complexity than the simplistic oxidative addition–transmetalation–reductive elimination cycle generally portrayed in textbooks.61
The robustness of the catalytically active species derived from 5d was further probed by a set of in situ catalyst recycling experiments. When adding a new batch of substrates to the reaction mixture every 30 min, no significant loss of activity nor any modification of the conversion profile was noted for up to five additional cycles (Fig. 5). These runs indicate a persistently active species. This conclusion was further reinforced by a filtration experiment, in which the reaction mixture was filtered through a 200 nm membrane after 30 min and charged with a new batch of substrates. Conversion of this new batch was essentially identical to the conversion before filtration (Fig. S80†), suggesting either small colloids or molecular palladium species as catalyst resting state. Notably, when the filtration was performed under air rather than a N2 atmosphere, the catalytic activity markedly decreased (Fig. S80†), which points to a palladium(0) resting state and thus corroborates the catalyst poisoning data.
 | ||
| Fig. 5 Catalyst recycling experiment for the arylation of propiophenone with complex 5d. Left: turnover increase upon addition of new batches of substrates every 30 min (each addition point marked with an arrow); right: superimposed time-yield profiles of the different catalyst recycling runs indicating no substantial change in reaction kinetics. Initial reaction conditions: PhBr (1.0 mmol), propiophenone (1.0 mmol), 5d (0.01 mmol), NaOtBu (1.1 mmol), 1,4-dioxane (2.0 mL), 105 °C under N2. At each addition point, i.e. every 30 min, a pre-heated solution of PhBr (1.0 mmol) and NaOtBu (1.1 mmol) in 1,4-dioxane (1.0 mL) followed by propiophenone (1.0 mmol) was added. Yields determined by GC analysis using hexamethylbenzene as internal standard. | ||
Catalyst activation pathways
While the reduction of PdII catalyst precursors in the presence of phosphine ligands has been extensively studied,62–66 much less is known about phosphine-free reduction pathways.67–70 We therefore investigated two specific scenarios for palladium(0) generation from complex 5d, namely i) enolate coordination to palladium(II) via a ligand substitution process followed by reductive C–Cl bond elimination, and ii) β hydrogen elimination from the coordinated enolate with formation of a meta-stable palladium hydride intermediate (Fig. S81†). In a series of stoichiometric experiments, addition of either propiophenone or NaOtBu immediately afforded a new species according to the downfield shift of the PYA resonances in the 1H NMR spectrum, which was attributed to coordination of the additive (Fig. S82†).71 When using 4-fluoropropiophenone, the new species was very similar to the one formed upon propiophenone addition, with slight 1H NMR shift differences attributed to the electronic effect of the fluorine substituent. For instance, the PYA N-methyl signal moved from 3.48 ppm in 5d to 3.66 and 3.64 upon addition of propiophenone and 4-fluoropropiophenone, respectively.72 These solutions were stable also when heated to 80 °C. However, when mixing 5d with stoichiometric amounts of the ketone and NaOtBu, an immediate reaction occurred, resulting in the formation of a species characterized by broad signals in the 1H NMR spectrum, along with the appearance of a precipitate. MS analysis did not show any chlorinated propiophenone, thus discarding a C–Cl reductive elimination process as the predominant catalyst activation step. Remarkably, when using 4-fluoropropriophenone as the ketone together with the base and complex 5d, all ketone was consumed within 10 min according to 1H NMR spectroscopy, and the 19F NMR spectrum did not reveal any signal, suggesting either precipitation of the product or massive broadening of the signal (Fig. S83 and S84†). This loss of 19F NMR signal and the broadened 1H NMR resonances might concur with β-hydrogen elimination as catalyst activation pathway, if the formed unsubstituted enone is unstable.Pyrazole functionalization for catalyst optimization
The mechanistic experiments strongly suggest a molecularly defined active species, also supported by conversion rates that are strongly influenced by the ligand design. For this reason, the best-performing ligand 4d was further modified to improve the catalytic activity. Variation of the pyrazole wingtip substituent from Me in 5d to phenyl (5e), 3,5-dimethylphenyl (5f), and iso-propyl (5g) aimed at probing both the electronic and steric implications of this substituent on the catalytic performance. The corresponding ligand precursors were prepared starting from commercially available ethyl 1H-pyrazole-3-carboxylate by nucleophilic substitution (R = alkyl) or copper-catalyzed Ullmann coupling (R = aryl),73 and subsequent ester hydrolysis to yield the carboxylic acids 1e–1g (Fig. 6a). Subsequent coupling with aminopyridine followed by pyridine methylation and palladation was performed according to methodologies used for 5d and afforded complexes 5e–5g (Fig. 6b).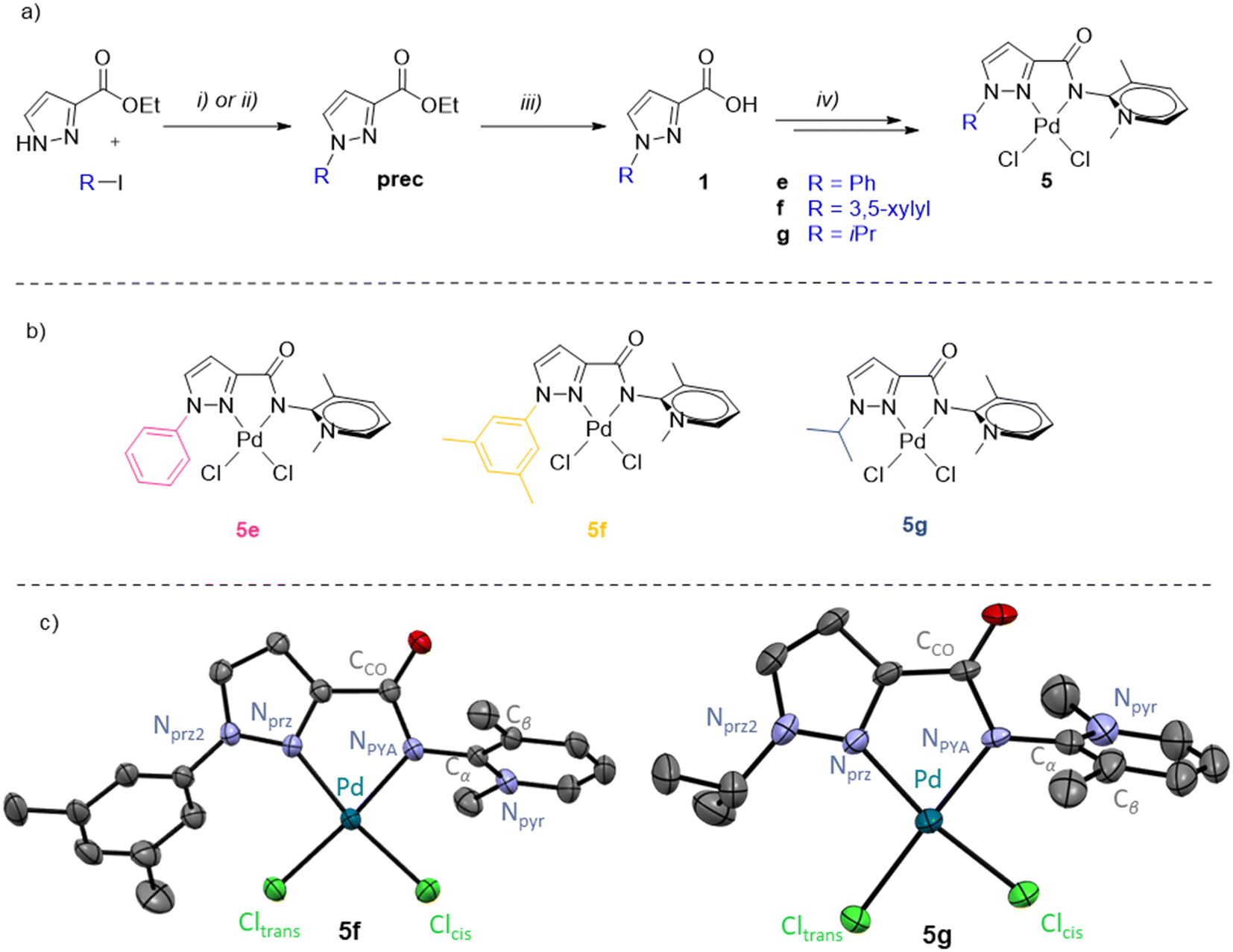 | ||
| Fig. 6 a) Synthetic scheme for the preparation of the carboxylic acids 1e–1g and complexes 5e–5g. Reactions and conditions: i) DMEDA (0.2 eq.), CuI 0.1 eq., K2CO3 1.2 eq., 1,4-dioxane; ii) Cs2CO3 2 eq., MeCN; iii) first LiOH 2.0 eq. MeOH/H2O, then HClaq. 2.2 eq.; iv) according to Fig. 2a. Prec-g and 1g were not isolated and directly used for the next step; b) schematic drawing of pyrazole-functionalized PYA palladium complexes 5e–5g; c) molecular structures of complexes 5f and 5g determined by X-ray diffraction analysis (30% probability ellipsoids, hydrogen atoms and co-crystallized solvent molecules omitted for clarity). | ||
While the NMR spectra of 5e and 5f are unremarkable, 1H NMR spectroscopy of 5g showed a marked downfield shift of the isopropyl CH proton upon complexation from δH = 4.53 in ligand 4g to 5.96 ppm in the complex. Such strong deshielding is indicative of an electrostatic interaction with the chloride ligand.33,74 Crystals suitable for X-ray diffraction of complexes 5f and 5g were obtained by slow diffusion of Et2O into a CH2Cl2 solution of the complexes (Fig. 6c). Similarly to complexes 5a and 5d, the molecular structures of 5f and 5g feature a square planar palladium center with the Pd–Cltrans bond consistently longer than Pd–Clcis due to the higher trans influence of the PYA donor (Table 3). The steric impact of the bidentate PYA ligand in complexes 5f and 5g is moderately higher than in the pyridyl–PYA system of 5a (%Vbur = 42.3% vs. 40.2%). For comparison, the introduction of an aryl-substituent at the pyridine 6-position increases the %Vbur by >8%.37 The structure of 5g features a close contact between the iPr CH hydrogen and Cltrans (H⋯Cl 2.525(3) Å), in agreement with the marked downfield shift of this resonance in solution NMR spectroscopy.
| 5f | 5g | |
|---|---|---|
| a Percentage buried volume %Vbur determined according to ref. 40. b The α angle was determined as angle Nprz–Nprz2–CR (see also Fig. 1d). c Tetrahedral distortion parameter τ4 determined according to ref. 38. | ||
| Pd–Nprz/Å | 2.0375(17) | 2.033(11) |
| Pd–NPYA/Å | 2.044(2) | 2.006(10) |
| Pd–Cltrans/Å | 2.2942(7) | 2.299(3) |
| Pd–Clcis/Å | 2.2812(6) | 2.2794(9) |
| NPYA–Cα/Å | 1.385(3) | 1.377(10) |
| NPYA–CCO/Å | 1.362(3) | 1.383(11) |
| %Vbura | 42.3 | 42.3 |
| α/°b | 123.44(18) | 121.0(8) |
| τ 4 | 0.086 | 0.074 |
The functionalized pyrazolyl complexes 5e–5g were evaluated in catalytic propiophenone arylation applying the same conditions that were used for complex 5d (Table 4, Fig. S65†). All pyrazole-substituted complexes 5d–f exhibited higher catalytic activity compared to the parent pyridyl–PYA precatalyst 5a, with TOFmax values ranging from 420 to 1180 h−1 as opposed to 130 h−1 for 5a. However, this increased activity did not always result in improved selectivity. Only the alkyl-substituted pyrazolyl complexes 5d and 5g demonstrated superior selectivity, achieving 91% and >99% yield, respectively, compared to 87% for 5a (Table 4vs.Table 2, entry 1). While there is no steric correlation between the pyrazolyl substituent and catalytic activity nor yield, there is an obvious correlation with the electronic properties of the R group with TOFmax increasing along the series 5e < 5f < 5d < 5g. Thus, electron-donating alkyl groups induce higher activity than aryl substituents, and within both the aryl and the alkyl substituents, the more donating xylyl and iPr groups impart better performance than Ph and Me, respectively. The most active catalyst derived from 5g reached full conversion in just 6 min at 105 °C and afforded quantitative yields, which is rather rare in ketone α-arylation catalysis75–77 and indicates a very effective suppression of side reactions by the iPr substituted pyrazolyl–PYA ligand.14,15,78–80 The beneficial role of strong donor ligands corroborates the conclusions from studies focusing on modulation of the PYA unit of 5a.30 Mechanistically, this behavior may be rationalized with the higher trans effect of stronger donor ligands, which labilizes the bromide ligand after ArBr oxidative addition81 and hence facilitates enolate binding.
| Entry | Catalyst | R | Optimized yieldb | Yield | TOFmax/h−1 | k ini/h−1 |
|---|---|---|---|---|---|---|
| a Reaction conditions: PhBr (1.0 mmol), propiophenone (1.2 mmol), [Pd] (0.01 mmol), NaOtBu (1.32 mmol), 1,4-dioxane (2.0 mL), 105 °C under N2, 60 min; yield determined by GC analysis using hexamethylbenzene as internal standard; for TOFmax evaluations see Fig. S65;† for calculation of initial rate constant kini see Fig. S66.† b Optimized yields determined without sampling, using identical conditions except propiophenone (1.0 mmol) NaOtBu (1.1 mmol), dioxane (1.0 mL). | ||||||
| 1 | 5d | Me | 91% | 84% | 830 | 8.3 |
| 2 | 5e | Ph | 79% | 72% | 420 | 4.2 |
| 3 | 5f | Xyl | 83% | 74% | 730 | 8.1 |
| 4 | 5g | iPr | >99% | 91% | 1200 | 15 |
Interestingly, the improved selectivity derived from steric shielding of the imine side contrasts with the reactivity trend observed when substituents were introduced to the pyridine in 5a.37 Introduction of a bulky xylyl group at the pyridyl 6-position resulted in a more active yet less selective catalyst. A reduced selectivity may be rationalized by less efficient enolate transmetalation due to the more prominent orientations of the pyridyl substituents into the catalytically relevant space, as juxtaposed by the pertinent α-angles.
To assess the potential of the most active catalyst 5g, the model reaction was performed under various conditions that are typically posing challenges for palladium-catalyzed α-arylation (Table 5). For example, catalysis under an atmosphere of air instead of N2 gave still decent activity with complex 5g (82% conversion vs. 99% under N2, entries 1, 2).82 In contrast, complex 5a is only poorly active under these conditions (27%, entry 3).37 Running the reaction in bench-grade dioxane that was stored in air and contained 0.3% H2O according to a Karl-Fischer titration gave 80% product (entry 4), similar to reactions performed in air and indicating a high robustness of the catalytic system towards air and moisture. This robustness is particularly relevant when considering that dry solvents and anaerobic conditions contribute significantly to the overall costs of a synthetic process. Reducing the reaction temperature from 105 to 60 °C afforded almost quantitative yields, albeit with a considerable extension of the reaction time to 72 h (entry 5). While complex 5a reached low TONs with (activated) aryl chlorides, complex 5g was inactive towards these substrates, even upon increasing the reaction temperature to 125 °C (entries 6, 7). A competition experiment using equimolar quantities of bromo- and chlorobenzene revealed almost full conversion of the aryl bromide exclusively (entry 8), indicating that aryl chlorides are catalytic bystanders without any significant inhibition effect. Such behavior is further confirmed by the chemoselective transformation of 1-bromo-4-chlorobenzene with full retention of the chloride (entry 9, Fig. S85†), which might be a desirable feature for late-stage functionalization and synthetic diversification.83–86 Notably, phosphine- and NHC-based palladium catalysts lack such selectivity between aryl bromides and chlorides.21,87 Reducing the catalyst loading to 100 ppm resulted in a lowered 50% yield (entry 10). While this outcome corresponds to a respectable 5000 TON with a TOFmax = 2300 h−1, the incomplete 69% conversion of PhBr suggests gradual deactivation of the catalyst.
|
|
|||
|---|---|---|---|
| Entry | Deviation from above | Yield | Reaction time/h |
| a General reaction conditions: aryl halide (1.0 mmol), propiophenone (1.0 mmol), 5g (0.01 mmol), NaOtBu (1.1 mmol), dry 1,4-dioxane (2.0 mL) in a 10 mL vial, 105 °C under N2, yield determined by GC analysis using hexamethylbenzene as internal standard. b Determined by Karl-Fischer titration. c ArCl (1.2 mmol), no conversion also at 125 °C. | |||
| 1 | None | >99% | 1.0 |
| 2 | Air instead of N2 atmosphere | 82% | 1.5 |
| 3 | Air instead of N2, 5a instead of 5g | 27% | 1.5 |
| 4 | Moist dioxane (0.3% H2O)b | 80% | 1.5 |
| 5 | 60 °C instead of 105 °C | 92% | 72 |
| 6 | PhCl instead of PhBrc | <1% | 1.5 |
| 7 | 4-CF3–C6H4Cl instead of PhBrc | <1% | 1.5 |
| 8 | PhCl (100 mol%) as additive | 80% | 1.5 |
| 9 | 4-Cl–C6H4Br instead of PhBr | 88% | 1.5 |
| 10 | 100 ppm 5g, 125 °C | 50% | 24 |

Moreover, the enhanced activity of 5g compared to 5a was confirmed by comparing the catalytic performance with challenging substrates (Fig. 7). Under standard conditions and using PhBr as the arylating agent, 5g gave consistently higher product yields than 5a. For example, phenyl acetophenone was arylated by 5a in a modest 24% yield, while 5g induced 88% of α-arylation within 2 h. This enhanced activity also reinforces enolate transmetalation as critical step on the catalytic cycle.
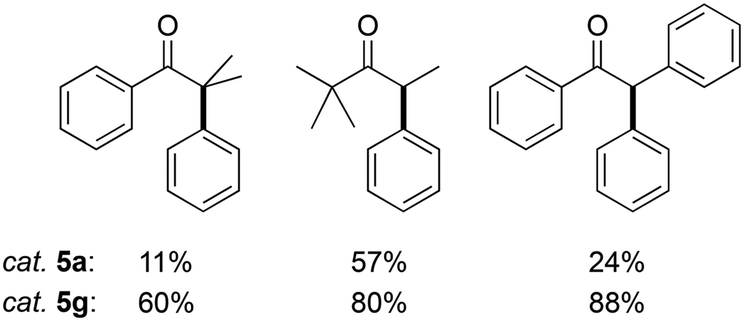 | ||
| Fig. 7 Products from α-arylation of challenging ketones under standard reaction conditions, i.e., phenyl bromide (1.0 mmol), ketone (1.0 mmol), 5g (0.01 mmol), NaOtBu (1.1 mmol), dry 1,4-dioxane (2.0 mL), 105 °C under N2, 2 h. | ||
Conclusions
By using the synthetic versatility of PYA functionalization, a set of new palladium(II) complexes were prepared containing a N,N′-bidentate chelating PYA ligand substituted with a N-heterocyclic imine donor. While oxazole functionalization of the PYA ligand results in lower catalytic activity in ketone α-arylation, triazole and especially pyrazole donors increase the activity. Tailoring of the N-substituent of the pyrazole provided complex 5g featuring catalytic activity that is about 10 times higher than that of the parent complex with a pyridine–PYA ligand. Moreover, the selectivity of the arylation approaches an ideal pathway with quantitative yields, and therefore provides a catalyst precursor that is competitive to some of the best systems based on phosphine or NHC ligands. Mechanistic work including kinetic experiments, catalyst poisoning studies, and catalyst recycling supports a homogeneous reaction trajectory with a molecularly defined species as active catalyst and an oxygen-sensitive catalyst resting state. These conclusions underpin the relevance of ligand design and advocate for further optimization of the chelating PYA ligand scaffold in order to address limitations of 5g such as the inactivity towards aryl chlorides and the increase of turnover numbers. Notably, these principles also warrant the development of new N-based ligand systems for improving other challenging C–C bond formation catalysis.Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data for compounds 5b, 5d, 5f, and 5g has been deposited at the CCDC under No. 2394912–2394915 and can be obtained from https://www.ccdc.cam.ac.uk free of charge.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank Crystallography Service of the DCBP for all X-ray analyses, and the Swiss National Science Foundation (grant no. 200020_212863) for generous financial support of this work.References
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef PubMed.
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef PubMed.
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef PubMed.
- J. M. Fox, X. Huang, A. Chieffi and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 1360–1370 CrossRef.
- M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 1473–1478 CrossRef.
- B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1997, 119, 12382–12383 CrossRef.
- M. Palucki and S. L. Buchwald, J. Am. Chem. Soc., 1997, 119, 11108–11109 CrossRef.
- P. Novak and R. Martin, Curr. Org. Chem., 2011, 15, 3233–3262 CrossRef.
- F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 1082–1146 CrossRef PubMed.
- S. T. Sivanandan, A. Shaji, I. Ibnusaud, C. C. C. J. Seechurn and T. J. Colacot, Eur. J. Org. Chem., 2015, 38–49 CrossRef.
- K. J. Swathy, P. V. Saranya and G. Anilkumar, Appl. Organomet. Chem., 2024, 38, e7508 CrossRef CAS.
- T. Mino, T. Matsuda, K. Maruhashi and M. Yamashita, Organometallics, 1997, 16, 3241–3242 CrossRef CAS.
- J. H. Ryan and P. J. Stang, Tetrahedron Lett., 1997, 38, 5061–5064 CrossRef CAS.
- B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1997, 119, 12382–12383 CrossRef CAS.
- M. Palucki and S. L. Buchwald, J. Am. Chem. Soc., 1997, 119, 11108–11109 CrossRef CAS.
- C. C. C. Johansson and T. J. Colacot, Angew. Chem., Int. Ed., 2010, 49, 676–707 CrossRef CAS PubMed.
- D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234–245 CrossRef CAS PubMed.
- M. S. Viciu, R. F. Germaneau and S. P. Nolan, Org. Lett., 2002, 4, 4053–4056 CrossRef PubMed.
- V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef PubMed.
- E. Marelli, M. Corpet, S. R. Davies and S. P. Nolan, Chem. – Eur. J., 2014, 20, 17272–17276 CrossRef PubMed.
- S. Ostrowska, T. Scattolin and S. P. Nolan, Chem. Commun., 2021, 57, 4354–4375 RSC.
- O. Navarro, N. Marion, Y. Oonishi, R. A. Kelly and S. P. Nolan, J. Org. Chem., 2006, 71, 685–692 CrossRef PubMed.
- S. Wolf and H. Plenio, J. Organomet. Chem., 2009, 694, 1487–1492 CrossRef.
- G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC.
- O. Kühl, Coord. Chem. Rev., 2009, 253, 2481–2492 CrossRef.
- S. Díez-González and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874–883 CrossRef.
- D. McGuinness, Dalton Trans., 2009, 6915–6923 RSC.
- V. Khlebnikov, A. Meduri, H. Mueller-Bunz, T. Montini, P. Fornasiero, E. Zangrando, B. Milani and M. Albrecht, Organometallics, 2012, 31, 976–986 CrossRef.
- M. Heckenroth, V. Khlebnikov, A. Neels, P. Schurtenberger and M. Albrecht, ChemCatChem, 2011, 3, 167–173 Search PubMed.
- M. E. Doster and S. A. Johnson, Angew. Chem., Int. Ed., 2009, 48, 2185–2187 CrossRef CAS PubMed.
- R. J. Thatcher, D. G. Johnson, J. M. Slattery and R. E. Douthwaite, Chem. – Eur. J., 2012, 18, 4329–4336 CrossRef CAS PubMed.
- H. V. Huynh and J. T. Vossen, Inorg. Chem., 2020, 59, 12486–12493 Search PubMed.
- J. J. Race and M. Albrecht, ACS Catal., 2023, 13, 9891–9904 CrossRef CAS.
- P. D. W. Boyd, L. J. Wright and M. N. Zafar, Inorg. Chem., 2011, 50, 10522–10524 Search PubMed.
- M. Navarro, C. Segarra, T. Pfister and M. Albrecht, Organometallics, 2020, 39, 2383–2391 Search PubMed.
- G. M. O'Maille, A. Dall'Anese, P. Grossenbacher, T. Montini, B. Milani and M. Albrecht, Dalton Trans., 2021, 50, 6133–6145 Search PubMed.
- E. Reusser and M. Albrecht, Dalton Trans., 2023, 52, 16688–16697 RSC.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- M.-N. Birkholz, Z. Freixa and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099–1118 RSC.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- A. Santoro, L. J. Kershaw Cook, R. Kulmaczewski, S. A. Barrett, O. Cespedes and M. A. Halcrow, Inorg. Chem., 2015, 54, 682–693 CrossRef PubMed.
- P. Comba, A. Eisenschmidt, L. R. Gahan, G. R. Hanson, N. Mehrkens and M. Westphal, Dalton Trans., 2016, 45, 18931–18945 RSC.
- V. Cinà, M. Russo, G. Lazzara, D. Chillura Martino and P. Lo Meo, Carbohydr. Polym., 2017, 157, 1393–1403 CrossRef PubMed.
- The pKa value 1.25 was measured for the methyl-triazole, and therefore the pKa of phenyl-triazole should be even lower. Calculations for the phenyl-triazole by ACD labs resulted in pKa = 0.2 ± 0.7.
- H. Eyring, J. Chem. Phys., 2004, 3, 107–115 CrossRef.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 2028–2031 CrossRef PubMed.
- P. Sambasiva Rao, C. Kurumurthy, B. Veeraswamy, G. Santhosh Kumar, Y. Poornachandra, C. Ganesh Kumar, S. B. Vasamsetti, S. Kotamraju and B. Narsaiah, Eur. J. Med. Chem., 2014, 80, 184–191 CrossRef PubMed.
- The slightly negative slope may originate from catalyst inhibition through radical species generated from the excess ArBr at elevated temperatures. Such radical formation may lead to catalyst deactivation and side reactions, such as homocoupling, see ref. 49 and 50.
- H. Lakmini, I. Ciofini, A. Jutand, C. Amatore and C. Adamo, J. Phys. Chem. A, 2008, 112, 12896–12903 CrossRef PubMed.
- S. N. S. Vasconcelos, J. S. Reis, I. M. de Oliveira, M. N. Balfour and H. A. Stefani, Tetrahedron, 2019, 75, 1865–1959 CrossRef.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef.
- For complex 5d, the reaction was performed at a lower temperature of 90 °C to facilitate sampling in the linear regime.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A:Chem., 2003, 198, 317–341 CrossRef.
- V. M. Chernyshev, A. V. Astakhov, I. E. Chikunov, R. V. Tyurin, D. B. Eremin, G. S. Ranny, V. N. Khrustalev and V. P. Ananikov, ACS Catal., 2019, 9, 2984–2995 CrossRef.
- I. C. Chagunda, T. Fisher, M. Schierling and J. S. McIndoe, Organometallics, 2023, 42, 2938–2945 CrossRef.
- A. Y. Kostyukovich, A. M. Tsedilin, E. D. Sushchenko, D. B. Eremin, A. S. Kashin, M. A. Topchiy, A. F. Asachenko, M. S. Nechaev and V. P. Ananikov, Inorg. Chem. Front., 2019, 6, 482–492 RSC.
- N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef.
- D. B. Eremin and V. P. Ananikov, Coord. Chem. Rev., 2017, 346, 2–19 CrossRef.
- (a) D. Canseco-Gonzalez, A. Gniewek, M. Szulmanowicz, H. Müller-Bunz, A. M. Trzeciak and M. Albrecht, Chem. – Eur. J., 2012, 18, 6055–6062 CrossRef PubMed; (b) E. Bulatov, E. Lahtinen, L. Kivijärvi, E. Hey-Hawkins and M. Haukka, ChemCatChem, 2020, 12, 4831–4838 CrossRef.
- B. Sun, L. Ning and H. C. Zeng, J. Am. Chem. Soc., 2020, 142, 13823–13832 CrossRef PubMed.
- G. E. Clarke, J. D. Firth, L. A. Ledingham, C. S. Horbaczewskyj, R. A. Bourne, J. T. W. Bray, P. L. Martin, J. B. Eastwood, R. Campbell, A. Pagett, D. J. MacQuarrie, J. M. Slattery, J. M. Lynam, A. C. Whitwood, J. Milani, S. Hart, J. Wilson and I. J. S. Fairlamb, Nat. Commun., 2024, 15, 3968 CrossRef PubMed.
- Z. Csákai, R. Skoda-Földes and L. Kollár, Inorg. Chim. Acta, 1999, 286, 93–97 CrossRef.
- G. A. Grasa and T. J. Colacot, Org. Lett., 2007, 9, 5489–5492 CrossRef PubMed.
- C. S. Wei, G. H. M. Davies, O. Soltani, J. Albrecht, Q. Gao, C. Pathirana, Y. Hsiao, S. Tummala and M. D. Eastgate, Angew. Chem., Int. Ed., 2013, 52, 5822–5826 CrossRef PubMed.
- H. Li, G. A. Grasa and T. J. Colacot, Org. Lett., 2010, 12, 3332–3335 CrossRef PubMed.
- C. Palladino, T. Fantoni, L. Ferrazzano, B. Muzzi, A. Ricci, A. Tolomelli and W. Cabri, ACS Catal., 2023, 13, 12048–12061 CrossRef.
- P. R. Melvin, D. Balcells, N. Hazari and A. Nova, ACS Catal., 2015, 5, 5596–5606 CrossRef.
- J. A. Molina de la Torre, P. Espinet and A. C. Albéniz, Organometallics, 2013, 32, 5428–5434 CrossRef.
- M. C. D'Alterio, È. Casals-Cruañas, N. V. Tzouras, G. Talarico, S. P. Nolan and A. Poater, Chem. – Eur. J., 2021, 27, 13481–13493 CrossRef PubMed.
- V. Semeniuchenko, S. Sharif, N. Rana, N. Chandrasoma, W. M. Braje, R. T. Baker, J. M. Manthorpe, W. J. Pietro and M. G. Organ, J. Am. Chem. Soc., 2024, 146, 29224–29236 CrossRef PubMed.
- N. F. Both, J. Thaens, A. Spannenberg, H. Jiao, K. Junge and M. Beller, ACS Catal., 2024, 14, 4082–4092 CrossRef.
- In line with only a weak interaction of the ketone, the 19F NMR resonance of the substrate shifts only marginally from δF = −106.68 to −106.71 upon exposure to 5d.
- While arylations proceeded in good yields (79% and 90%), nucleophilic substitution with iPrI was compromised by poor selectivity between pyrazole N1 and N2 alkylation and therefore resulted in only modest 47% yield.
- G. Canil, V. Rosar, S. Dalla Marta, S. Bronco, F. Fini, C. Carfagna, J. Durand and B. Milani, ChemCatChem, 2015, 7, 2255–2264 CrossRef.
- M. S. Viciu, R. A. Kelly, E. D. Stevens, F. Naud, M. Studer and S. P. Nolan, Org. Lett., 2003, 5, 1479–1482 CrossRef PubMed.
- G. A. Grasa and T. J. Colacot, Org. Process Res. Dev., 2008, 12, 522–529 CrossRef.
- A. Bugarin and B. T. Connell, Chem. Commun., 2011, 47, 7218–7220 RSC.
- J. M. Fox, X. Huang, A. Chieffi and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 1360–1370 CrossRef.
- S. Doherty, J. G. Knight, C. H. Smyth, R. W. Harrington and W. Clegg, Organometallics, 2008, 27, 1679–1682 CrossRef.
- G. Adjabeng, T. Brenstrum, C. S. Frampton, A. J. Robertson, J. Hillhouse, J. McNulty and A. Capretta, J. Org. Chem., 2004, 69, 5082–5086 CrossRef PubMed.
- B. Pinter, V. V. Speybroeck, M. Waroquier, P. Geerlings and F. D. Proft, Phys. Chem. Chem. Phys., 2013, 15, 17354–17365 RSC.
- In agreement with its enhanced air-stability, complex 5g also performed well after filtration in air and reached 65% conversion after 30 min in the second cycle, while 5a stalled around 10% conversion in the same period (Fig. S80b†).
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- B. Liégault, I. Petrov, S. I. Gorelsky and K. Fagnou, J. Org. Chem., 2010, 75, 1047–1060 CrossRef PubMed.
- I. Kalvet, T. Sperger, T. Scattolin, G. Magnin and F. Schoenebeck, Angew. Chem., Int. Ed., 2017, 56, 7078–7082 CrossRef PubMed.
- K.-H. Liu, G.-Q. Hu, C.-X. Wang, F.-F. Sheng, J.-W. Bai, J.-G. Gu and H.-H. Zhang, Org. Lett., 2021, 23, 5626–5630 CrossRef PubMed.
- A. Ehrentraut, A. Zapf and M. Beller, Adv. Synth. Catal., 2002, 344, 209–217 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures for all new compounds including NMR spectra, crystallographic details, buried volume calculations, and catalytic procedures (pdf). CCDC 2394912–2394915. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cy01337a |
| This journal is © The Royal Society of Chemistry 2025 |