
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of [Re(η6-arene)2]+ substituents in electro- and photocatalytic water reduction with [Co(terpy)2]3+ catalysts†
Joshua
Csucker
 *,
Nora Sophie
Grundmann
,
Olivier
Blacque
,
Bernhard
Spingler
and
Roger
Alberto
*,
Nora Sophie
Grundmann
,
Olivier
Blacque
,
Bernhard
Spingler
and
Roger
Alberto
Department of Chemistry, University of Zurich, Winterthurstrasse 190, CH-8057 Zurich, Switzerland. E-mail: joshua.csucker@chem.uzh.ch
First published on 21st January 2025
Abstract
In this report, we assess the electronic influence of [Re(η6-arene)2]+ sandwich complexes conjugated to the water reducing catalyst [Co(terpy)2]2+/3+. We compare catalytic performances and catalytic activities of conjugates in which the catalyst and the sandwich complex are coupled via carbon–carbon bonds or via secondary amine linkers. For this purpose, the [Re(η6-arene)2]+ sandwich was bound to the 4′-position of 2,2′:6′,2′′-terpyridine (terpy) and to the corresponding [CoIII(4′-R-terpy)2]n+ complexes. Electro- and photocatalytic water reduction activities of the model catalysts indicated that direct conjugation of the two functional subunits has adverse effects on the catalytic performances, presumably due to irreversible reduction of the rhenium complexes. Connecting the two functional units through a secondary amine linker had essentially no impact on cyclic voltammogram shapes and the influence on the electrocatalytic hydrogen evolution was minimal. Such conjugates increased catalyst stabilities during photocatalytic hydrogen production, which was reflected by higher turnover numbers and longer catalytically active phases than the catalysts alone. Accordingly, a general approach of attaching catalysts to the [Re(η6-arene)2]+ framework is demonstrated.
Introduction
[Re(η6-arene)2]+ sandwich complexes have emerged over the last ten years as novel building blocks in bioorganometallic chemistry1–6 and catalysis.7,8 Applications in the respective fields are inherently linked to the many properties of these [Re(η6-arene)2]+ sandwiches, such as compatibility with water and high thermodynamic and electrochemical stabilities. Accordingly, their fundamental chemistry is well developed.3,9–11 A key advantage [Re(η6-arene)2]+ holds over related sandwich complexes like ferrocene is its water solubility, enabled by the overall cationic charge.Two comprehensive studies have been reported with [Re(η6-arene)2]+ complexes bound to known catalysts. Attaching electronically decoupled [Re(η6-C6H6)2]+ to cobalt catalysts for the hydrogen evolving reaction (HER) showed a neglectable impact on turnover frequencies (TOFs) and numbers (TONs) compared to phenyl analogs.7 Conjugation of [Ru(bipy)(CO)2Cl2] (bipy = bipyridyl) units via methylene linkers to the rhenium sandwich structures retained the original CO2 to CO reducing performances of the ruthenium catalyst.8 Interestingly, through space cooperativity between two catalytic units was achieved by attaching two ruthenium catalysts to the same [Re(η6-C6H6)2]+ host. The cooperativity was reflected in higher TON and TOF per ruthenium of conjugates with two versus one catalyst(s).
These studies provide an insight on how the [Re(η6-arene)2]+ framework may be used as functional group(s) on known catalysts. However, both studies do not further elaborate on the electronic influence of the rhenium sandwiches on the catalytic units and their performances. Equally, little is known about the role of linker groups between the two functional subunits, the catalyst and the sandwich complex. To fill this gap, [Re(η6-arene)2]+ units are conjugated in this report to [Co(terpy)2]2+/3+ complexes since substituent effects on its catalytic performance, e.g. the electrochemistry, are well understood.12 Inductive effects by electron donating or - withdrawing substituents on the 4′-position of the terpy chelators directly influence the redox behavior of the cobalt center and correlate linearly with the substituent's Hammett parameters. Moreover, [Co(terpy)2]3+ is a well-known electro- and photocatalyst for water to H2 (ref. 13 and 14) and CO2 to CO (ref. 15 and 16) reductions. The 4′-substituent steers substrate selectivity between CO2 and water. Electron withdrawing groups (positive Hammett parameter) favor CO2 to CO whereas donating groups (negative Hammett parameter) favor water to H2 reduction in direct substrate selectivity studies.17
We report herein the impact of [Re(η6-arene)2]+ substituents in [Co(4′-R-terpy)2]n+ complexes on the electronic structures and electro- and photocatalytic H2O to H2 reduction performances.
Results and discussion
An initial assessment of the electronic environment of the [Re(η6-arene)2]+ framework was made based on the pKa values of η6-coordinated benzoic acid and the Hammett parameter resulting from the coordination. Native benzoic acid has a pKa of 4.20.18 The model complex Rc-COOH+ (Rc = [Re(η6-C6H6)(η6-C6H5R)]+,1Fig. 1, top) lowered the pKa of the carboxylic acid group to 2.63 corresponding to a “coordination” Hammett parameter of 1.57. As a reference, p-nitrobenzoic acid (pKa = 3.42 (ref. 18)) is roughly one order of magnitude less acidic with a σp value of 0.78,19 in contrast to ferrocenyl carboxylic acid (Fc-COOH, pKa = 7.79)20 or oxidized ferrocenium carboxylic acid ([Fc-COOH]+, pKa = 4.54 (ref. 20)), which are considerably less acidic given the electron rich ferrocene motif. The redox potentials of [Co(4′-R-terpy)2]n+ is known to correlate linearly with the substituent Hammett σp values (Fig. S1†).12 However, for a quantitative comparison of the Hammett parameters derived from the coordination of the Rc (or ferrocene) motif, attachment of Rc/Fc to the para position of benzoic acid would be required to obtain the true σp values.19,21,22 Preparing such compounds with Rc is synthetically complex and goes beyond the scope of this work. Yet, in the electrochemical section of this report, we correlate known substituent effects with the reduction potential of the CoIII/II couple of [Co(4′-R-terpy)2]3+ complexes (vide infra). | ||
| Fig. 1 Top: Chemical structures and corresponding pKa values of [Rc-COOH]+, Fc-COOH and [Fc-COOH]+. Bottom: Chemical structures of the four 4′-R-terpy chelators studied in this work with the respective literature sources. | ||
Synthesis
We chose to attach the Rc motif [Re(η6-arene)2]+ to the 4′-position of terpyridine to assess its electronic influence on [Co(4′-R-terpy)2]n+. This was either done directly ([1a]PF6, Fig. 1, bottom) through Kröhnke pyridine synthesis as previously reported by our group8 or via an –NH linker ([1b]PF6) by Buchwald–Hartwig coupling of [Re(η6-aniline)(η6-C6H6)]+ and 4′-chloroterpyridine.23 Analogous reference chelators 4′-phenyl-terpy (1c)24 and 4′-aniline-terpy (1d)25 without the Rc motif are known in the literature. Choosing the combination of chelators [1a]+ and 1c or [1b]+ and 1d ensures comparability between the electro- or physico-chemical properties of the different [Co(4′-R-terpy)2]n+ complexes. Varying the linkers between the Rc analog and terpy allow for the direct assessment of electronic and perhaps steric influences.Coordination of [1a]+, [1b]+ and their corresponding organic analogs 1c and 1d to cobalt proceeded most effectively with [Co(dppe)Cl2] in 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetonitrile/methanol mixtures. The respective [Co(4′-R-terpy)2]n+ derivatives were obtained in moderate to very good yields (20–82%, Scheme 1). As described by Sonia et al.,14 performing the reactions under ambient atmosphere oxidizes the CoII to CoIIIin situ. The only exception was [2]4+ which is persistent as CoII under these conditions. Oxidation of [4]4+ to [4]5+ occurs within a day in aerated solvents. Complexes [2]4+–[5]3+ presented in Scheme 1 are fully characterized by NMR, FT-IR, HR-ESI-MS. Single crystal XRD analyses of all compounds except [3](PF6)3 formed an amorphous solid, which further confirmed their chemical structures. 1H NMR signals of paramagnetic [Co(4′-R-terpy)2]2+ complexes [2]4+ and [4]4+ ranged from 6–100 ppm (ESI,† Fig. S8 and S11); consistent with analogous compounds described by Dickenson and colleagues.26
:1 acetonitrile/methanol mixtures. The respective [Co(4′-R-terpy)2]n+ derivatives were obtained in moderate to very good yields (20–82%, Scheme 1). As described by Sonia et al.,14 performing the reactions under ambient atmosphere oxidizes the CoII to CoIIIin situ. The only exception was [2]4+ which is persistent as CoII under these conditions. Oxidation of [4]4+ to [4]5+ occurs within a day in aerated solvents. Complexes [2]4+–[5]3+ presented in Scheme 1 are fully characterized by NMR, FT-IR, HR-ESI-MS. Single crystal XRD analyses of all compounds except [3](PF6)3 formed an amorphous solid, which further confirmed their chemical structures. 1H NMR signals of paramagnetic [Co(4′-R-terpy)2]2+ complexes [2]4+ and [4]4+ ranged from 6–100 ppm (ESI,† Fig. S8 and S11); consistent with analogous compounds described by Dickenson and colleagues.26
 | ||
| Scheme 1 All [Co(4′-R-terpy)2]n+ complexes [2]4+–[5]3+ are prepared by the reaction between the respective terpy chelators and [Co(dppe)Cl2]. | ||
Crystallography
Single crystals suitable for XRD analyses were collected by slow evaporation of Et2O into analyte solutions in CH3CN. Ellipsoid displacement plots27 of [2]4+, [4]4/5+ and [5]3+ are presented in Fig. 2. All crystallized [Co(4′-R-terpy)2]n+ complexes adopted distorted octahedral geometries. Co–N bonds were shorter in compound [2]4+ where the [Re(η6-arene)2]+ unit is directly attached to the [Co(4′-R-terpy)2]2+versus [4]4+ with a secondary amine linker connecting the two. Similarly, Co–N bonds are elongated in [5]3+versus [4]5+. A clear trend between electron withdrawing substituents and decreased Co–N bond length is obvious (ESI,† Table S10). Directly attached [Re(η6-arene)2]+ groups in [2]4+ act as an electron withdrawing group. Its effect is slightly moderated by the secondary amine linker in [4]5+. However, quantitative evaluation of the electronic effects of the [Re(η6-arene)2]+ framework cannot be based on single crystal analysis alone. Cyclic voltammetry experiments with compounds [2]4+–[5]3+ allowed for further assessments of the substituent effects on cobalt and ligand-based redox potentials. | ||
| Fig. 2 Ellipsoid displacement plots of a): [2]4+; b): [4]4+; c) [4]5+; d) [5]3+. Thermal ellipsoids represent 30% probability. Hydrogen atoms and counter-ions were removed for clarity. Detailed crystallographic data is available in the ESI† in Fig. S22–S25 and Tables S3–S9. | ||
Electrochemistry
Electron withdrawing groups in the 4′-position of [Co(4′-R-terpy)2]n+ complexes shift cobalt-based reductions anodically while the opposite is the case for the donating groups.12 Along this rationale, the electronic influences of the [Re(η6-arene)2]+ substituents on [Co(4′-R-terpy)2]n+ can be assessed by cyclic voltammetry. Complexes with 4′-phenyl ([3]3+) and 4′-aniline ([5]+) substituents serve as references. For consistency, all complexes were analyzed as CoIII compounds. As [2]4+ was isolated as a CoII species, electrochemical oxidation with a prepotential of +0.5 V was required to form the respective CoIII species in situ. The persistence of [2]4+ as a CoII species is already a first qualitative indications for the [Re(η6-arene)2]+ unit being strongly electron withdrawing.All complexes show fully reversible CoIII/II and CoII/I reductions (Fig. 3). Additionally, the ligand framework of compounds [3]3+and [5]3+, where the 4′-substituents are purely organic, undergo reversible reductions. Dickenson and coworkers report identical behaviour for analogous 4′-phenyl-terpy compounds.26,28 They report that while L0/−I reductions are terpyridine-based, the L−I/−II processes is associated with reduction of the distal phenyl group in [3]3+. Reversible ligand based reductions are absent in [2]3+ where the Rc motif is the substituent. Decreasing or increasing scan rates between 0.1–1 V s−1 revealed no reversible processes in this region. It is known that [Re(η6-arene)2]+ undergoes irreversible one electron reduction at and below −2 V versus Fc/Fc+.29 Hence, the lack of reversible reduction features in [2]5+ may be explained by irreversible reduction and the resulting reductive decomposition of the rhenium sandwich substituent. Similarly, when comparing [4]5+ with [5]3+, a reversible ligand-based reduction is observed at −2.18 V in [5]3+ but not in [4]5+. However, irreversible reduction features were observed. Again, alteration of scan rates did not further elucidate them. Evidently, direct conjugation of the rhenium sandwiches to [Co(4′-R-terpy)2]n+ disallows reversible ligand-based reductions but instead irreversible reduction of the rhenium sandwich occurs.
 | ||
| Fig. 3 Cyclic voltammograms of [2]4+–[5]3+. Spectra were recorded at 1 mM analyte concentration in 0.1 M TBA[PF6] electrolyte in DMF at 0.5 V s−1. | ||
Cobalt-based reductions of compounds bearing the Rc motif ([2]4+ and [4]5+) are all anodically shifted compared to the compounds with analogous organic substituents ([3]3+ and [5]3+ respectively). CoII/I reductions were shifted by roughly −0.1 V while CoIII/II reductions were less affected by the introduction of the Rc motif with an average shift of about −0.05 V. Thus, the anodic shifts induced by the rhenium bis-arenes corresponds roughly to that of a 4′-methylsulphone group (ΔEred (CoIII/II) = −0.13 V vs. Fc/Fc+ in CH3CN).12 Introduction of the amine linker between rhenium sandwich and terpy chelator promoted a global cathodic shift of cobalt and ligand based reduction potentials. For instance, reversible reduction of [4]4+/3+ (Ered = −1.07 V) is cathodically shifted by −0.26 V compared to [2]4+/3+ (Ered = −1.16 V).
As briefly mentioned above, the effect of electron donating and withdrawing substituents on the CoIII/II redox couple correlates linearly with the substituent Hammett parameter (σp). Complexes [2]5+–[5]3+ fit well with the Ered to σp correlations of existing [Co(4′-R-terpy)2]n+ complexes (ESI,† Fig. S1). Accordingly, we are able to postulate a σp value for the [Re(η6-C6H6)(η6-C6H5R)]+ motif of roughly 0.45, indicating a similarly strong electron withdrawing tendency as BF2 (0.48).19 Impressively, linking the two functional subunits via an amine group changed the electronics sufficiently to render it a slight electron donating group with a calculated σp value of roughly −0.08, comparable to SiMe3 groups (σp = −0.07).19 Accordingly, the electron withdrawing effect of the Rc motif can be mediated effectively by simply introducing a secondary amine between the two functional subunits. Given that direct conjugation between the two subunits in [2]5+ suppresses the ligand-based reductions entirely, the strategy of linking the [Re(η6-arene)2]+ and [Co(4′-R-terpy)2]n+ units via a secondary amine appears favorable over a direct conjugation approach. Anodic shifts of cobalt-based reductions are about the same between [2]5+ and [3]3+ or [4]5+ and [5]3+. The amine linker between the Rc motif and terpy moderates the electronics rather than electronically disconnecting the two subunits. These points suggest that amine linkers are well suited in this system, especially so given the ease of preparing them versus i.e. methylene or ether linkers.
Electrocatalysis
Stepwise addition of protons in the form of HNEt3[BF4] [2]4+–[4]3+ induced catalytic waves at L0/−I reductions of −2 to −2.5 V (Fig. 4). This potential corresponds to electrocatalytic water to hydrogen conversion. Even if such a reversible L0/−I reduction was not observed in [2]5+ and [4]5+ without substrate, both compounds showed an electrocatalytic wave for H2 production. Consequently, ligand-based reductions not observed in regular cyclic voltammetry measurements are made accessible upon addition of protons. The potentials of peak catalytic currents were anodically shifted by the [Re(η6-arene)2]+ substituents in [2]4+ and [4]5+ compared to analogous catalysts [3]3+ and [5]3+. In fact, [Re(η6-arene)2]+ substituents lower the electrocatalytic overpotential of [Co(4′-R-terpy)2]n+ WRCs and impact catalytic wave shapes. This is clearest when comparing electrocatalysis of [2]5+ with [3]3+. Addition of 50 eq. HNEt3[BF4] to [2]5+ splits the catalytic wave into two features whereas the same proton equivalents added to [3]3+ induced a single wave. Thus, there is reasonable evidence that the direct attachment of [Re(η6-arene)2]+ into the 4′-position of [Co(4′-R-terpy)2]n+ alters the mechanism of electrocatalytic hydrogen evolution. Comparing the catalytic waves of [4]5+ with [5]3+, the wave shape broadens and higher kobs of the catalysis with [4]5+ than that with [5]3+ can be proposed. The shape of the catalytic wave of [4]5+ indicates substrate consumption, whereas [5]3+ has a closer resemblance to cyclic voltammograms with pure kinetic conditions and without substrate depletion.30,31 Consequently, direct conjugation between [Re(η6-arene)2]+ and the catalyst is not a favorable strategy for catalysis. A secondary amine linker, however, amplifies the electronic impact by the [Re(η6-arene)2]+ framework onto the attached catalyst.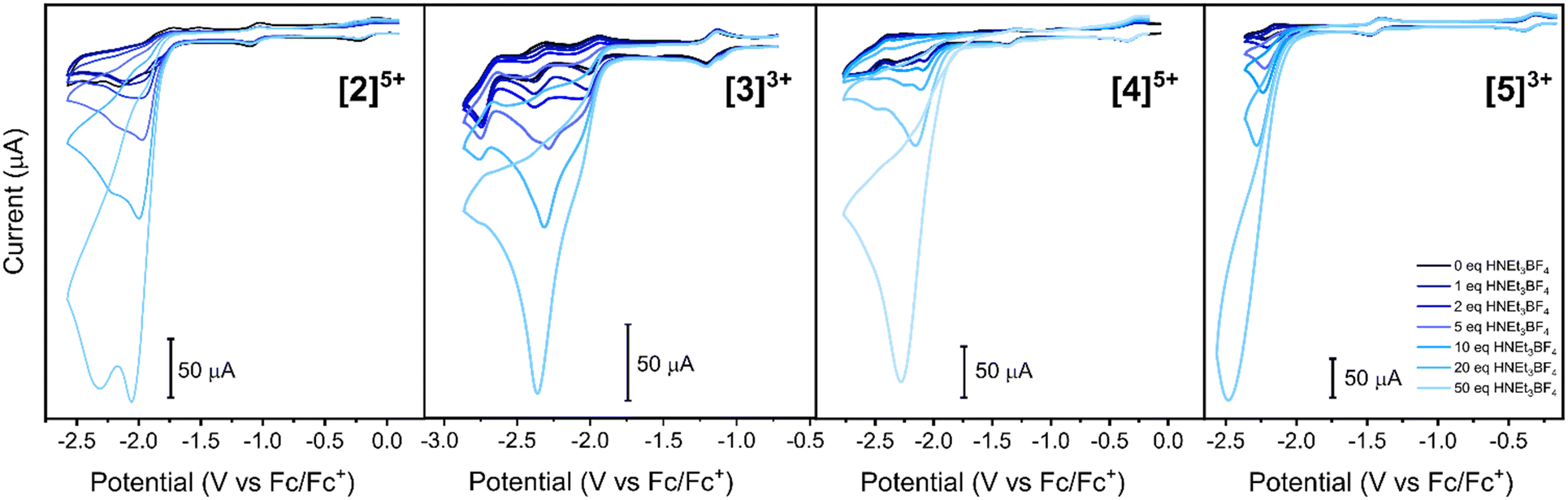 | ||
| Fig. 4 Electrocatalytic water to hydrogen reductions recorded by titration of compounds [2]4+–[5]3+ at a negative potential window by addition of increasing equivalents of HNEt3[BF4]. Voltammograms were recorded at 1 mM analyte concentrations in 0.1 M TBA[PF6] in DMF. The scan rate was 0.5 V s−1 at voltage steps of 0.005 V. | ||
Different mechanisms for the HER with cobalt polypyridyl WRCs are discussed in the literature. All consist of electronic (E) and chemical (C) elementary steps. The two pathways for [Co(terpy)2]n+ specifically discussed most often are alternating ECEC or sequential EECC type mechanisms.12,15,17,26,28,32,33 The CoIII/II reduction is considered recatalytic in ECEC type mechanisms.34,35 A model system chemically closest to compounds [2]5+–[5]3+ was reported by Panda and coworkers36 featuring ferrocene moieties in the 4′-position of the terpy ligands. They describe a CECE type mechanism with a dechelation and ligand protonation as the first step, presumably due to the high electron density delivered by the ferrocene substituents. Given the inverse electronic environment created by the rhenium sandwich in this study, we expect a ligand protonation to be unlikely. Mongal and colleagues reported mechanistic studies of the HER with electron rich thiophene substituents at the 4′-position, proposing a classical (E)ECEC type mechanism.13 Furthermore, Aroua et al. investigated mechanistic changes in the HER depending on the substituent pattern and derivatization of the terpy framework by electrocatalytic and computational methods.32 They propose two EECC pathways, depending on the aforementioned ligand alterations. The system presented herein differs from these literature reports as this system introduces electron withdrawing groups to slightly electron donating units. Thus, alterations in the reaction mechanism are expected. Plausible (E)ECEC and EECC mechanisms are depicted in Scheme S1.†
Given the different wave forms and wave currents observed with each catalyst, and the fact that [2]5+ is under an electron withdrawing regime while [4]5+ is under a slight electron donating regime, , it is therefore possible that each catalyst enables slightly different reaction pathways. To exclude one or the other mechanism beyond the fundamental molecular contribution is what we aimed at providing here. However, we find a cathodically shifted metal-based reductions favor an EECC type mechanism. In this regard, foot-of-the wave analysis as developed by Savéant and colleagues may give further insight into the kinetics of the electrocatalytic HER.37–39
Photocatalysis
Photocatalytic hydrogen productions with [4]Cl5, and [5]Cl3 as WRCs were recorded at varying catalyst concentrations. The oxidative half reaction of water splitting was replaced by a sacrificial electron donor (tris(2-carboxyethyl)phosphine (TCEP)) and an electron relay (sodium ascorbate). [Ru(bipy)3]Cl2 was chosen as photosensitizer (PS). A reaction scheme outlining the elementary steps of the photocatalytic setup is provided in the ESI† (Scheme S2). Hydrogen evolution was quantified by in line analyzing the headspace of capped reaction vessels by gas chromatography. A representative rate profile of H2 evolution is presented in Fig. 5a. Both WRCs achieved TONs above 2000 H2/Co at all concentrations (Fig. 5b). A spiking experiment with [4]5+ demonstrated that the WRC stability limits the performance of the photocatalytic system (Fig. 5c) at 5 μM WRC concentration. Hydrogen production resumed when new WRC was added whereas no recovery of H2 formation was observed upon re-addition of PS after catalytic breakdown. Thus, improving catalyst stability is crucial in the presented system. Remarkably, the [Re(η6-C6H6)2]+ substituted WRC [4]Cl5 is overall more stable than the 4′-aniline substituted analog [5]Cl3. This was reflected by the longer active phases of [4]Cl5versus [5]Cl3 and higher maximal turnover numbers (TONs) at 5, 10 and 20 μM WRC concentrations (Fig. 5b, Tables S1 and S2†). In fact, TONs of [4]Cl5 were consistently higher than those of [5]Cl3 by up to roughly one and a half fold at 10 μM. Only at low catalyst concentration (1 μM) did [5]Cl3 outperformed [4]Cl5. Our group has previously shown a linear relationship between maximum H2 evolution rate and photon flux.40 Accordingly, the higher TON observed for [4]5+ are the direct result of longer active catalysis given that e maximum evolution rates of [5]3+ tend to be marginally higher (Fig. S2 and S3,† Table S2). The photosystem becomes limiting at high WRC concentrations hence why the TON and TOFmax of [4]5+ and [5]3+ plateau at 50 μM and at around 2000 H2/Co.35,41,42 | ||
| Fig. 5 a) Rate profile of hydrogen evolution over time at 5 μM [4]Cl5 and [5]Cl3. b) TONs of [4]Cl5 and [5]Cl3versus cobalt concentration. c) Spike experiments with addition of fresh catalyst or photosensitizer after 8 h to photocatalytic H2 production with 5 μM [4]Cl5 as WRC. Conditions: 0.1 M TCEP, 0.1 M sodium ascorbate, 500 mM [Ru(bipy)3]Cl2, pH 5, 450 nm illumination. | ||
Evidently, linking the two subunits via the secondary amine masked the electron withdrawing effects of [Re(η6-C6H6)2]+ sufficiently and even delivered increased catalytic performance versus attaching aniline in the 4′-position.
Conclusions
In summary, a comprehensive study on the electronic role of [Re(η6-arene)2]+ sandwich complexes as a host for attached WRCs is presented. In general, rhenium bis-arenes act as strong electron withdrawing groups comparable to e.g. BF2. Direct connection of the two functional units through a C–C bond ([2]4+) greatly altered the electrochemical behavior of [Co(4′-R-terpy)2]3+, likely due to irreversible reductions of the rhenium sandwich. Introduction of a secondary amine linker ([4]5+) moderated the electronic effect to a degree, where a [Re(η6-C6H6)(η6-C6H5NH)]+ substituent becomes electron donating according to the correlation between the Hammett substituent parameter and its effect on the CoIII/II redox couple. Moreover, photocatalytic water to hydrogen reduction performances were enhanced over analogous catalysts with 4′-aniline substituents. Accordingly, the strategy of conjugating the [Re(η6-arene)2]+ motif via a secondary amine to [Co(4′-R-terpy)2]3+ complexes enhanced catalyst stability and lowered the overpotential required for H2 evolution. Given the large body of research on [M(terpy)2]n+, the presented work provides a simple and generalizable approach of combining them with the promising [Re(η6-arene)2]+ framework as hosts. No doubt, the strategy of linking the Rc motif via amine linkers may be expanded to other chelators and ligand classes in the future.Data availability
Experimental and spectroscopic data supporting this article have been included as part of the ESI.† Crystallographic data for [2](PF6)4 has been deposited at the name of repository, such as CCDC under 2391331. Crystallographic data for [4](PF6)4 has been deposited at the name of repository, such as CCDC under 2391334. Crystallographic data for [4](PF6)5 has been deposited at the name of repository, such as CCDC under 2391332. Crystallographic data for [5](PF6)3 has been deposited at the name of repository, such as CCDC under 2391333.Author contributions
JC synthesized and analysed all compounds and wrote the manuscript. NG performed all photocatalytic measurements. OB and BS performed the crystallographic work. RA supervised the project, secured funding and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the University of Zurich.Notes and references
- G. Meola, H. Braband, S. Jordi, T. Fox, O. Blacque, B. Spingler and R. Alberto, Dalton Trans., 2017, 46, 14631–14637 RSC.
- G. Meola, H. Braband, P. Schmutz, M. Benz, B. Spingler and R. Alberto, Inorg. Chem., 2016, 55, 11131–11139 Search PubMed.
- Q. Nadeem, G. Meola, H. Braband, R. Bolliger, O. Blacque, D. Hernández-Valdés and R. Alberto, Angew. Chem., Int. Ed., 2020, 59, 1197–1200 CrossRef CAS PubMed.
- N. F. Suremann, G. Meola, O. Blacque, H. Braband and R. Alberto, Organometallics, 2020, 39, 2713–2718 Search PubMed.
- C. Gotzmann, O. Blacque, T. Fox and R. Alberto, Eur. J. Inorg. Chem., 2021, 2021, 2493–2498 Search PubMed.
- F. Battistin, C. Fernandes, P. D. Raposinho, O. Blacque, A. Paulo and R. Alberto, Dalton Trans., 2023, 52, 15757–15766 Search PubMed.
- D. Hernández-Valdés, F. Avignon, P. Müller, G. Meola, B. Probst, T. Fox, B. Spingler and R. Alberto, Dalton Trans., 2020, 49, 5250–5256 RSC.
- D. Hernández-Valdés, R. Fernández-Terán, B. Probst, B. Spingler and R. Alberto, Helv. Chim. Acta, 2020, 103, e2000147 CrossRef.
- D. Hernández-Valdés, L. Wettstein, R. Fernández-Terán, B. Probst, T. Fox, B. Spingler, Q. Nadeem and R. Alberto, Chem. Commun., 2020, 56, 10658–10661 Search PubMed.
- D. Hernández-Valdés, G. Meola, H. Braband, B. Spingler and R. Alberto, Organometallics, 2018, 37, 2910–2916 CrossRef.
- L. Siebenmann, R. Bolliger, H. Braband, O. Blacque and R. Alberto, Inorg. Chem., 2022, 61, 3683–3689 CrossRef CAS PubMed.
- J. Chambers, B. Eaves, D. Parker, R. Claxton, P. S. Ray and S. J. Slattery, Inorg. Chim. Acta, 2006, 359, 2400–2406 CrossRef CAS.
- B. N. Mongal, S. Sk, A. Tiwari, S. Mehmood, Y. Soujanya, U. Pal and M. Chandrasekharam, Next Energy, 2024, 3, 100116 CrossRef.
- Sonia, A. K. Pandey, I. Verma and B. N. Mongal, Chem. Phys. Impact, 2024, 9, 100706 CrossRef.
- N. Elgrishi, M. B. Chambers, V. Artero and M. Fontecave, Phys. Chem. Chem. Phys., 2014, 16, 13635–13644 RSC.
- J. J. Leung, J. Warnan, K. H. Ly, N. Heidary, D. H. Nam, M. F. Kuehnel and E. Reisner, Nat. Catal., 2019, 2, 354–365 CrossRef CAS.
- N. Elgrishi, M. B. Chambers and M. Fontecave, Chem. Sci., 2015, 6, 2522–2531 RSC.
- Z. Rappoport, CRC Handbook of Tables for Organic Compound Identification, CRC Press, Inc., Boca Raton, Florid, 1984 Search PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- L. Fabbrizzi, ChemTexts, 2020, 6, 22 Search PubMed.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS.
- L. P. Hammett, Chem. Rev., 1935, 17, 125–136 CrossRef CAS.
- J. Csucker, M. Scarpi-Luttenauer, P. Mesdom, S. Hidalgo, O. Blacque, G. Gasser and R. Alberto, Inorg. Chem., 2024, 63, 18154–18161 Search PubMed.
- F. H. Case and W. A. Butte, J. Org. Chem., 1961, 26, 4415–4418 Search PubMed.
- E. C. Constable, C. E. Housecroft and Y. Tao, Synthesis, 2004, 2004, 869–874 CrossRef.
- J. C. Dickenson, M. E. Haley, J. T. Hyde, Z. M. Reid, T. J. Tarring, D. A. Iovan and D. P. Harrison, Inorg. Chem., 2021, 60, 9956–9969 Search PubMed.
- L. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- H. Ferreira, M. M. Conradie and J. Conradie, Inorg. Chim. Acta, 2019, 486, 26–35 CrossRef CAS.
- M. Benz, H. Braband, P. Schmutz, J. Halter and R. Alberto, Chem. Sci., 2015, 6, 165–169 RSC.
- K. J. Lee, N. Elgrishi, B. Kandemir and J. L. Dempsey, Nat. Rev. Chem., 2017, 1, 0039 CrossRef CAS.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 Search PubMed.
- S. Aroua, T. K. Todorova, P. Hommes, L.-M. Chamoreau, H.-U. Reissig, V. Mougel and M. Fontecave, Inorg. Chem., 2017, 56, 5930–5940 CrossRef CAS PubMed.
- S. Aroua, T. K. Todorova, V. Mougel, P. Hommes, H.-U. Reissig and M. Fontecave, ChemCatChem, 2017, 9, 2099–2105 CrossRef CAS.
- N. Queyriaux, R. T. Jane, J. Massin, V. Artero and M. Chavarot-Kerlidou, Coord. Chem. Rev., 2015, 304-305, 3–19 CrossRef CAS PubMed.
- A. Rodenberg, M. Orazietti, B. Probst, C. Bachmann, R. Alberto, K. K. Baldridge and P. Hamm, Inorg. Chem., 2015, 54, 646–657 CrossRef CAS PubMed.
- S. Kumar Padhi, E. Ahmad, S. Rai and B. Panda, Polyhedron, 2020, 187, 114677 CrossRef CAS.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS PubMed.
- C. Costentin and J.-M. Savéant, Nat. Rev. Chem., 2017, 1, 0087 CrossRef CAS.
- C. Costentin and J.-M. Savéant, ChemElectroChem, 2014, 1, 1226–1236 CrossRef CAS.
- E. Joliat-Wick, M. Mosberger, N. Weder, B. Spingler, B. Probst and R. Alberto, Chimia, 2021, 75, 180 CrossRef CAS PubMed.
- B. Probst, M. Guttentag, A. Rodenberg, P. Hamm and R. Alberto, Inorg. Chem., 2011, 50, 3404–3412 CrossRef CAS PubMed.
- B. Probst, A. Rodenberg, M. Guttentag, P. Hamm and R. Alberto, Inorg. Chem., 2010, 49, 6453–6460 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, spectroscopic (including SCXRD data), electro- and photocatalytic data. CCDC 2391331–2391334. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cy01282h |
| This journal is © The Royal Society of Chemistry 2025 |