
Fluorinated covalent organic frameworks for visible-light driven CO2 reduction†
Wei-Jia
Wang
*a,
Bin
Li
b,
Jing
Gao
a and
Kaihong
Chen
 *c
*c
aCollege of Biological and Chemical Engineering, Zhejiang University of Science and Technology, Hangzhou 310023, China. E-mail: wjwang@zust.edu.cn
bHangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, No.1 Xiangshanzhi Lane, Hangzhou, 310024, China
cThe Institute of Green Chemistry and Engineering, School of Chemistry and Chemical Engineering, Nanjing University, Suzhou, Jiangsu 215163, China. E-mail: kchen@nju.edu.cn
First published on 2nd December 2024
Abstract
The metal-free visible-light-driven CO2 reduction reaction was achieved by using a fluorine-atom-modified COF, i.e. N3F4-COF, which exhibited over a 5-fold enhancement compared to the pristine COF for syngas production. The activity can be further improved by incorporating a Ru-based photosensitizer, and the syngas ratio could be regulated from 2.57 to 0.14 (CO/H2) by altering the hydrophilicity of the photosensitizers.
Introduction
Carbon dioxide (CO2) is a prominent greenhouse gas with significant implications for the climate and atmospheric temperatures.1–3 Currently, the selective conversion of CO2 into solar fuels and chemicals via photocatalysis is a crucial transformation for both the chemical industry and environmental sustainability.4–9 The photocatalytic CO2 reduction reaction (CO2RR) to produce syngas, an essential industrial feedstock for the Fischer–Tropsch process and methanol synthesis, represents a promising yet challenging approach. This challenge arises from difficulties in controlling the selectivity of CO and H2 and in enhancing the overall catalytic activity.10–14 In general, the CO2RR requires light absorption by a semiconductor to generate photoexcited electron–hole pairs, followed by the separation of these pairs and their subsequent migration to the semiconductor surface to catalyze redox reactions.15–23 Therefore, developing a strategy to engineer photocatalysts with strong light absorption capabilities, efficient photogenerated charge separation and transport, and high CO2 adsorption ability is highly desirable.24–26Covalent organic frameworks (COFs) are a class of crystalline materials with well-defined and periodic structures that allow for precise chemical design and versatile applications.27–30 This design flexibility has been exploited in various architectures to enhance photocatalytic activity. For example, anchoring functional moieties to the backbones of the COF can regulate the electron push–pull effect to narrow the bandgap.31–33 Moreover, designing a COF with a donor–acceptor (D–A) structure is a prevalent strategy to facilitate the separation and migration of photoexcited pairs.34–37 However, only a few of the COFs can perform the photocatalytic CO2RR under real metal-free conditions, considering the potential for trace metal residues from the synthesis process.38–41
Herein, we demonstrate that fluorination is a versatile strategy for regulating COF morphology, narrowing the bandgap, facilitating the separation of photoexcited pairs, and improving CO2 adsorption ability. Specifically, the fluorine atom-modified COF, i.e. N3F4-COF, demonstrates over a 5-fold enhancement in the metal-free photocatalytic CO2RR compared to the pristine COF, i.e. N3-COF. Furthermore, activity enhancement and tunable CO/H2 selectivity can be achieved by adding different Ru-based photosensitizers.
Results and discussion
The 2D COFs, N3-COF and N3F4-COF, were synthesized via metal-free Schiff base condensation, as illustrated in Scheme 1. N3F4-COF was prepared by reacting 2,3,5,6-tetrafluoroterephthalaldehyde (PTA-F4) with 2,4,6-tris(4-aminophenyl)-triazine (TAT) (see the ESI† for further details). The structure and synthesis of N3-COF, which serves as a comparative structure, have been reported in a previous work.42 CP/MAS 13C NMR of N3F4-COF (Fig. 1a) shows six peaks in the aromatic benzene ring region (δ = 117 to 138 ppm) and signals corresponding to imine functionalities at ∼154 ppm. Similar chemical shifts are observed for N3-COF (Fig. S1†). Fourier-transform infrared (FTIR) spectra of the materials reveal characteristic stretching bands (Fig. 1b and S2†), with bands at 1579 cm−1 and 1503 cm−1 corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N Schiff base bonds and aromatic triazine CN bonds, respectively. The band at 1020 cm−1 corresponds to C–F bonds, providing evidence for the incorporation of fluorine atoms into the N3F4-COF structure. The aldehyde band at 1708 cm−1, associated with unreacted terminal groups, is significantly reduced or absent in N3F4-COF, indicating a higher degree of polymerization. Powder X-ray diffraction (PXRD) analysis shows good crystallinity for both COFs (Fig. 1c and S3†). Compared to that in N3-COF, the X-ray photoelectron spectroscopy (XPS) spectrum of C 1s in N3F4-COF displays additional C–F peaks, further indicating the presence of the F atom (Fig. S4b and S5d†). Contact-angle measurement (Fig. 1d and S7†) reveals that both COFs have a contact angle greater than 120°, highlighting their hydrophobic nature.
N Schiff base bonds and aromatic triazine CN bonds, respectively. The band at 1020 cm−1 corresponds to C–F bonds, providing evidence for the incorporation of fluorine atoms into the N3F4-COF structure. The aldehyde band at 1708 cm−1, associated with unreacted terminal groups, is significantly reduced or absent in N3F4-COF, indicating a higher degree of polymerization. Powder X-ray diffraction (PXRD) analysis shows good crystallinity for both COFs (Fig. 1c and S3†). Compared to that in N3-COF, the X-ray photoelectron spectroscopy (XPS) spectrum of C 1s in N3F4-COF displays additional C–F peaks, further indicating the presence of the F atom (Fig. S4b and S5d†). Contact-angle measurement (Fig. 1d and S7†) reveals that both COFs have a contact angle greater than 120°, highlighting their hydrophobic nature.
 | ||
| Scheme 1 Design and synthesis of modified covalent organic frameworks through Schiff base condensation. | ||
 | ||
| Fig. 1 Structural and hydrophobicity characterization. (a) CP/MAS 13C NMR spectrum of N3F4-COF. (b) FT-IR spectra of monomers and N3F4-COF. (c) PXRD pattern of N3F4-COF. (d) Contact angle measurement of N3F4-COF. | ||
Scanning electron microscopy (SEM) images (Fig. 2a and b) show that N3F4-COF has a uniform morphology resembling sea anemones, with “tentacles” measuring 100–200 nm in length. In contrast, N3-COF has a smaller particle size (∼1 μm) and an agglomerate structure (Fig. S5†). The morphology change in N3F4-COF is attributed to the introduction of fluorine atoms. Energy dispersive spectroscopy (EDS) mapping images in Fig. 2c show a uniform distribution of C, N, and F, indicating the high elemental uniformity on N3F4-COF. Transmission electron microscopy (TEM) further confirms a uniform surface and good crystallinity (Fig. S9†), attributable to successful condensation.
 | ||
| Fig. 2 Morphological characterization of N3F4-COF. (a) and (b) SEM images, and (c) elemental mapping images. | ||
Surface area analysis using the Brunauer–Emmett–Teller (BET) method (Table 1 and Fig. 3) reveals a significantly larger specific surface area for N3F4-COF (4801.8 m2 g−1) compared to N3-COF (253.8 m2 g−1). The isotherms (Fig. 3a and b) for both COFs exhibit type I characteristics, indicative of microporous structures with saturation adsorption corresponding to micropore filling. CO2 uptake capacities (Table 1 and Fig. S10†) show that N3F4-COF has higher CO2 adsorption abilities than N3-COF at both 273 K and 298 K. Then, using the Clausius–Clapeyron relationship, the isosteric heat of adsorption (Qst) was calculated from the 273 and 298 K isotherms, with Qst values of 8.9 kJ mol−1 for N3-COF and 16.2 kJ mol−1 for N3F4-COF. The higher CO2 affinity of N3F4-COF is attributed to the interaction between fluorine atoms and CO2, indicating that N3F4-COF, with its optimal electron-withdrawing group content, maximizes the reactivity.43
 | ||
| Fig. 3 N2 adsorption–desorption isotherms collected at 77 K for (a) N3F4-COF and (b) N3-COF, respectively; pore size distribution for (c) N3F4-COF and (d) N3-COF, respectively. | ||
UV-vis diffuse reflectance spectra (Fig. 4a) demonstrate that N3F4-COF, enriched with fluorine atoms, exhibits enhanced visible-light absorption ability compared to N3-COF. The bandgaps (Eg) calculated from the Tauc plots are 2.3 eV and 2.6 eV for N3F4-COF and N3-COF, respectively. The conduction band minimum (CBM) potentials derived from the Mott–Schottky plots are −1.06 V and −0.91 V (vs. NHE) for N3F4-COF and N3-COF, respectively (Fig. 4c). The energy band structure diagram (Fig. 4d) indicates that both COFs are suitable for visible-light absorption in the photocatalytic reduction of CO2 and proton to CO and H2, respectively. Clearly, the substitution of H atoms with fluorine atoms enhances visible absorbance, resulting in changes to the composition and band edge.44
 | ||
| Fig. 4 (a) UV/vis DR spectra and (b) schematic band structure diagram of N3-COF and N3F4-COF; Mott–Schottky plots of (c) N3F4-COF and (d) N3-COF. | ||
Photogenerated charge separation was further investigated through transient photocurrent examined in the presence and absence of light. As illustrated in Fig. 5, the photocurrent density of N3F4-COF is higher than that of N3-COF, suggesting more efficient separation of photogenerated carriers in N3F4-COF. This observation aligns with electrochemical impedance spectroscopy (EIS) measurements, which show that N3F4-COF displays a smaller semicircle in the Nyquist plots, indicating low charge transfer resistance. This enhanced mobility is attributed to the higher fluorine content in N3F4-COF, which form a D (PTA-F4 linker)-A (TAT unit) structure that facilitate the separation and migration of photoexcited carriers.
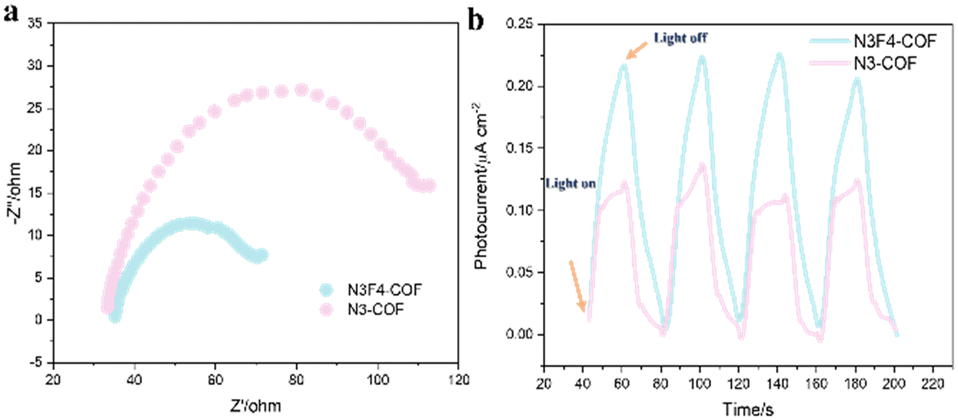 | ||
| Fig. 5 (a) Nyquist plots and (b) transient photocurrent response of N3-COF and N3F4-COF. | ||
Collectively, fluorination significantly impacts the morphologies of COFs and improves CO2 adsorption ability. Additionally, the narrowed bandgap and enhanced separation and migration of photoexcited pairs are achieved through anchoring fluorine atoms into the COF structure.
Photocatalytic experiments were conducted using N3-COF and N3F4-COF as catalysts in 3 mL acetonitrile solution containing 0.5 mL triethanolamine (TEOA), 1 mL water as a proton source, and 0.05 mM BIH as the electron donor, under a CO2 atmosphere (1.0 atm, 298 K). It should be mentioned that only CO and H2 were detected as products, and no HCOOH or CH3OH was observed in the liquid phase by 1H NMR. N3F4-COF demonstrated a maximum activity of 440 μmolCO g−1 and 115 μmolH2 g−1 after 4 h of irradiation with a xenon lamp (300 W, λ ≥ 420 nm) (Fig. 6a). In contrast, N3-COF showed poor activity for producing either CO or H2. This high activity under metal-free conditions for N3F4-COF should be ascribed to the introduction of fluorine atoms.
 | ||
| Fig. 6 (a) Catalytic performances over N3F4-COF and N3-COF; (b) control experiments by using N3F4-COF in 2 h CO2 photoreduction; (c) time curve of CO/H2 formation; (d) durability measurements of N3F4-COF; and [Ru(phen)3](PF6)2 was added after 4th cycle. | ||
As shown in Fig. 6b and c, the addition of [Ru(phen)3](PF6)2 as a photosensitizer enhanced the CO2RR activity for N3F4-COF, achieving 9400 μmol g−1 CO and 3650 μmol g−1 H2. This performance is comparable to other catalyst systems used for the photocatalytic CO2RR to produce syngas (Tables S1 and S2†). In contrast, [Ru(phen)3](PF6)2 alone exhibited a much lower catalytic activity. Control experiments, depicted in Fig. 6b, confirmed that no CO was formed in a N2 atmosphere, proving that CO originates from CO2. Furthermore, control exclusion experiments (excluding the light source, water, or electron donors) or replacing the photosensitizer with dyes45 (e.g., eosin Y or methylene blue) showed no CO2RR activity.
Interestingly, when a hydrophobic photosensitizer (Ru(bpy)3Cl2·6H2O) was introduced, N3F4-COF exhibited 88% selectivity towards H2, while the hydrophilic photosensitizer ([Ru(phen)3](PF6)2) resulted in 72% selectivity towards CO. This unexpected result can be attributed to two factors: (1) the hydrophobic nature of N3F4-COF critically enhances CO2RR performance by increasing the interfacial CO2 concentration. Molecular dynamics simulations revealed that the hydrophobic microenvironment prevents hydrogen bond formation on the carbonaceous co-catalyst surface.46 (2) A hydrophilic photosensitizer, which dissolves easily in the aqueous phase, may contribute to the formation of “oil-photocatalyst-water” layers,47,48 potentially alleviating CO2 solubility limitations and sluggish diffusion in water, thereby accelerating CO2-to-CO conversion while reducing HER rates.
Then, the long-term stability and reusability of N3F4-COF were also evaluated (Fig. 6d). After 4 h, the photocatalytic activity began to decline, and syngas production became nearly negligible after 8 h. However, the CO/H2 selectivity remained stable for at least 20 h, indicating that the decomposition process does not significantly affect the CO and H2 production ratio. To confirm the reason for the decreased activity, the system was evacuated and CO2 gas was refilled every 2 h. The photocatalytic activity decreased after the 4th cycle but could be restored by adding [Ru(phen)3](PF6)2, suggesting that the decreased activity originated from the decomposition of the photosensitizer. In addition, no significant changes were observed in the XPS spectra of the reused N3F4-COF (Fig. S6†).
Based on the above results, a proposed mechanism for N3F4-COF is shown in Fig. 7a. Upon visible-light excitation, photogenerated electrons transfer from the valence band maximum (VBM) (PTA-F4 linker) to the CBM (triazine unit), facilitating surface reduction reactions. Moreover, [Ru(phen)3]2+* is generated under visible light irradiation, then the photogenerated electron is transferred to the triazine unit on the N3F4-COF catalyst. The holes in [Ru(phen)3]2+ and N3F4-COF are consumed by the electron donor, i.e. BIH. Furthermore, in the three-phase interface photocatalysis shown in Fig. 7b, the hydrophilic [Ru(phen)3](PF6)2 dissolves in the aqueous layer, while electronic sacrificial agents (i.e. BIH and TEOA) dissolve in the organic layer, with the hydrophobic N3F4-COF dispersed between these layers. This setup demonstrated that the hydrophobic catalyst plays a critical role in regulating CO2RR performance by increasing the interfacial CO2 concentration and alleviating CO2 mass transfer limitations in the aqueous layer. In short, efficient electron transfer and CO2 concentration in the aqueous phase are key factors controlling the CO2RR and HER activity. Higher electron transfer efficiency promotes CO formation, while a hydrophobic microenvironment prevents hydrogen formation.
 | ||
| Fig. 7 (a) Schematic illustration of the charge-transfer process under light irradiation. (b) Possible mechanism of the three-phase photocatalysis for CO2 reduction over N3F4-COF and inside proposed photoactive electron transfer and reaction pathway in the photoreduction of CO2. | ||
Conclusions
In summary, fluorine-atom-modified COFs, i.e. N3F4-COF, have been successfully designed and developed as efficient and durable photocatalysts for the CO2RR to yield syngas. The introduction of fluorine atoms enables regulation of morphology and improves surface area, CO2 adsorption capacity, visible light absorption, and photoinduced electron–hole separation efficiency. Consequently, N3F4-COF exhibits significantly higher activity in the metal-free photocatalytic CO2RR compared to N3-COF. Additionally, the activity can be further enhanced by incorporating a Ru-based photosensitizer, and the selectivity between CO and H2 can be easily tuned by altering the hydrophilicity of the photosensitizers. These findings provide a foundation for designing novel catalysts that could advance CO2 reduction technologies.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the General Research Projects of the Zhejiang Provincial Education Department (Y202454316), the National Natural Science Foundation of China (No. 22005154), and the AI & AI for Science Project of Nanjing University (Fundamental Research Funds for the Central Universities, No. 14380006) for the financial support.References
- K. Kleijne, M. A. J. Huijbregts, F. Knobloch, R. Zelm, J. P. Hilbers, H. Coninck and S. V. Hanssen, Nat. Energy, 2024, 9, 1139–1152 CrossRef.
- M. Meinshausen, N. Meinshausen, W. Hare, S. C. B. Raper, K. Frieler, R. Knutti, D. J. Frame and M. R. Allen, Nature, 2009, 458, 1158–1162 CrossRef CAS.
- T. R. Karl and K. E. Trenberth, Science, 2003, 302, 1719–1723 CrossRef CAS PubMed.
- X. Ding, W. Liu, J. Zhao, L. Wang and Z. Zou, Adv. Mater., 2024, 2312093 CrossRef.
- J. Yu, A. Muhetaer, Q. Li and D. Xu, Small, 2024, 20, 2402952 CrossRef CAS PubMed.
- M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- F.-Y. Ren, K. Chen, L.-Q. Qiu, J.-M. Chen, D. J. Darensbourg and L.-N. He, Angew. Chem., Int. Ed., 2022, 61, e202200751 CrossRef CAS PubMed.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- S. Qi, K. Zhu, T. Xu, H. Zhang, X. Guo, J. Wang, F. Zhang and X. Zong, Adv. Mater., 2024, 36, 2403328 CrossRef CAS PubMed.
- Y.-N. Gong, S. Wang, H.-J. Dong, J.-H. Mei, D.-C. Zhong and T.-B. Lu, Appl. Catal., B, 2024, 357, 124310 CrossRef CAS.
- Z. B. Fang, T. T. Liu, J. X. Liu, S. Y. Jin, X. P. Wu, X. Q. Gong, K. C. Wang, Q. Yin, T. F. Liu, R. Cao and H. C. Zhou, J. Am. Chem. Soc., 2020, 142, 12515–12523 CrossRef CAS PubMed.
- X. B. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- J. X. Fan, X. X. Yue, Y. N. Liu, D. Q. Li and J. T. Feng, Chem Catal., 2022, 2, 531–549 CrossRef CAS.
- W.-J. Wang, K.-H. Chen, Z.-W. Yang, B.-W. Peng and L.-N. He, J. Mater. Chem. A, 2021, 9, 16699–16705 RSC.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr., Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- Y.-N. Gong, S.-Q. Zhao, H.-J. Wang, Z.-M. Ge, C. Liao, K.-Y. Tao, D.-C. Zhong, S. Ken and T.-B. Lu, Angew. Chem., Int. Ed., 2024, e202411639 CAS.
- W. Lin, J. Lin, X. Zhang, L. Zhang, R. A. Borse and Y. Wang, J. Am. Chem. Soc., 2023, 145, 18141–18147 CrossRef CAS.
- S. Yoshino, T. Takayama, Y. Yamaguchi, A. Iwase and A. Kudo, Acc. Chem. Res., 2022, 55, 966–977 CrossRef CAS PubMed.
- H. Tada, M. Fujishima and H. Kobayashi, Chem. Soc. Rev., 2011, 40, 4232–4243 RSC.
- Y.-N. Gong, X. Guan and H.-L. Jiang, Coord. Chem. Rev., 2023, 475, 214889 CrossRef CAS.
- Y. Yang, H.-Y. Zhang, Y. Wang, L.-H. Shao, L. Fang, H. Dong, M. Lu, L.-Z. Dong, Y.-Q. Lan and F.-M. Zhang, Adv. Mater., 2023, 35, 2304170 CrossRef CAS PubMed.
- H. Li, R. Li, G. Liu, M. Zhai and J. Yu, Adv. Mater., 2023, 35, 2301307 Search PubMed.
- Q. Zhang, S. Gao, Y. Guo, J. Huiyong Wang, X. Wei, H. Su, Z. Zhang and J. Wang Liu, Nat. Commun., 2023, 14, 1147 CrossRef CAS.
- J. Li, G. Chen, Y. Zhu, Z. Liang, A. Pei, C.-L. Wu, H. Wang, H. R. Lee, K. Liu, S. Chu and Y. Cui, Nat. Catal., 2018, 1, 592–600 CrossRef CAS.
- J. Li, J. Zhou, X.-H. Wang, C. Guo, R.-H. Li, H. Zhuang, W. Feng, Y. Hua and Y.-Q. Lan, Angew. Chem., Int. Ed., 2024, e202411721 CAS.
- T. H. M. Pham, J. Zhang, M. Li, T. H. Shen, Y. Ko, V. Tileli and W. Luo, Adv. Energy Mater., 2022, 12, 2103663 CrossRef CAS.
- T. He, Z. Zhao, R. Liu, X. Liu, B. Ni, Y. Wei, Y. Wu, W. Yuan, H. Peng, Z. Jiang and Y. Zhao, J. Am. Chem. Soc., 2023, 145, 6057–6066 CrossRef CAS.
- W. Zhang, L. Chen, S. Dai, C. Zhao, C. Ma, L. Wei, M. Zhu, S. Y. Chong, H. Yang, L. Liu, Y. Bai, M. Yu, Y. Xu, X.-W. Zhu, Q. Zhu, S. An, R. S. Sprick, M. A. Little, X. Wu, S. Jiang, Y. Wu, Y.-B. Zhang, H. Tian, W.-H. Zhu and A. I. Cooper, Nature, 2022, 604, 72–79 CrossRef CAS.
- C. Gropp, T. Ma, N. Hanikel and O. M. Yaghi, Science, 2020, 370, eabd6406 CrossRef CAS.
- K. Sun, O. J. Silveira, Y. Ma, Y. Hasegawa, M. Matsumoto, S. Kera, O. Krejčí, A. S. Foster and S. Kawai, Nat. Chem., 2023, 15, 136–142 CrossRef CAS PubMed.
- C. Li, J. Liu, H. Li, K. Wu, J. Wang and Q. Yang, Nat. Commun., 2022, 13, 2357 CrossRef CAS PubMed.
- K. Wang, Z. Jia, X. Bai, S. E. Hodgkiss, L. Chen, S. Y. Chong, X. Wang, H. Yang, Y. Xu, F. Feng, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2020, 142, 11131–11138 CrossRef CAS PubMed.
- L. Hao, R. Shen, C. Huang, Z. Liang, N. Li, P. Zhang, X. Li, C. Qin and X. Li, Appl. Catal., B, 2023, 330, 122581 CrossRef CAS.
- J. Yang, S. Ghosh, J. Roeser, A. Acharjya, C. Penschke, Y. Tsutsui, J. Rabeah, T. Wang, S. Y. D. Tameu, M.-Y. Ye, J. Grüneberg, S. Li, C. Li, R. Schomäcker, R. V. D. Krol, S. Seki, P. Saalfrank and A. Thomas, Nat. Commun., 2022, 13, 6317 CrossRef CAS PubMed.
- S. Barman, A. Singh, F. A. Rahimi and T. K. Maji, J. Am. Chem. Soc., 2021, 143, 16284–16292 CrossRef CAS PubMed.
- M. Zhang, P. Huang, J.-P. Liao, M.-Y. Yang, S.-B. Zhang, Y.-F. Liu, M. Lu, S.-L. Li, Y.-P. Cai and Y.-Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202311999 CrossRef CAS.
- Y. Wang, W. Hao, H. Liu, R. Chen, Q. Pan, Z. Li and Y. Zhao, Nat. Commun., 2022, 13, 100 CrossRef CAS PubMed.
- J. Yang, Z. Chen, L. Zhang and Q. Zhang, ACS Nano, 2024, 18, 21804–21835 CrossRef CAS.
- L. Qin, C. Ma, J. Zhang and T. Zhou, Adv. Funct. Mater., 2024, 34, 2401562 CrossRef CAS.
- S. Biswas, F. A. Rahimi, R. K. Saravanan, A. Dey, J. Chauhan, D. Surendran, S. Nath and T. K. Maji, Chem. Sci., 2024, 15, 16259–16270 RSC.
- F. Meng, J. Wang, M. Chen, Z. Wang, G. Bai and X. Lan, ACS Catal., 2023, 13, 12142–12152 CrossRef CAS.
- D. Mullangi, S. Shalini, S. Nandi and B. Choksi, J. Mater. Chem. A, 2017, 5, 8376–8384 RSC.
- C. S. Diercks, S. Lin, N. Kornienko, E. A. Kapustin, E. M. Nichols, C. Zhu, Y. Zhao, C. J. Chang and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 1116–1122 CrossRef CAS.
- T. Li, N. Tsubaki and Z. Jin, J. Mater. Sci. Technol., 2024, 169, 82–104 CrossRef CAS.
- Z. Zhou, H. Yao, Y. Wu, T. Li, N. Tsubaki and Z. Jin, Acta Phys.-Chim. Sin., 2024, 40, 2312010 Search PubMed.
- X. B. Chen, S. H. Shen, L. J. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS.
- Q.-S. Huang, C. Chu, Q. Li, Q. Liu, X. Liu, J. Sun, B.-J. Ni and S. Mao, ACS Catal., 2023, 13, 11232–11243 CrossRef CAS.
- Y. Li, Z. Pei, D. Luan and X. W. D. Lou, J. Am. Chem. Soc., 2024, 146, 3343–3351 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy01276c |
| This journal is © The Royal Society of Chemistry 2025 |