Tailoring the glucose oxidation activity of anodized copper thin films†
Received
18th October 2024
, Accepted 25th March 2025
First published on 1st April 2025
Abstract
Glucose oxidation is a fundamental reaction in biosensing and energy conversion, and anodized copper electrodes have emerged as promising catalysts for its enhancement. This paper systematically investigates the link between anodization parameters and glucose oxidation on thin copper films, unraveling crucial insights into their optimization. By careful control of the anodization parameters, distinct species can be favoured, such as Cu2O, Cu(OH)2, and CuO, with varying catalytic activities. Applying a polarization in 1 M KOH at 0 V (vs. Ag|AgCl) results in the formation of a highly active surface CuO layer, which delivers significant performance improvements compared to the bare copper electrode. Namely, a 55% sensitivity gain in the 0.1–0.5 mM range, and a remarkable 73% gain in the 0.75–2 mM range. Furthermore, the manufactured electrodes display an extremely low limit of detection – only 0.004 mM. Such an exceptional result can be ascribed to the scalable and reproducible manufacturing done directly on cleanroom-compatible platforms. These insights not only clarify the effect of thin copper film anodization on glucose oxidation, but also chart a practical path towards improving the real-world efficiency of copper-based integrated systems for biosensing and energy conversion.
Introduction
Glucose oxidation is crucial in various domains such as biosensing, biofuel cells, and energy storage. Among these, diabetes monitoring currently has the greatest economic and societal relevance, with a market value of $11.71 billion in 2021 (ref. 1) and more than 537m people affected by the disease.2 Enzymes, such as glucose oxidase, are commonly used in glucose sensing technologies, exhibiting good sensitivity and selectivity.3 However, their storage and usage conditions require careful control due to their sensitivity to pH and temperature.4 The limited stability of these sensors, typically lasting a few weeks, is considered their primary weakness.5,6 Compared to their enzymatic counterparts, non enzymatic sensors not only are more durable, but they also exhibit greater sensitivities and can be more easily miniaturized, as they are not limited by macro-sized molecules.7 Over the last decades, a plethora of non enzymatic glucose electrocatalysts has been developed and investigated. Early works highlighted how noble metals (e.g., Pt,8,9 Pd,10 and Au (ref. 11 and 12)) can directly oxidize glucose in neutral solutions. Nevertheless, the high cost and low resistance towards chloride poisoning is still limiting their application for glucose sensing.13,14 As a result, transition metal oxides (e.g., Ni(OH)2,15,16 CuxO,17 CO3O4 (ref. 18) and mixed oxides19) have also been explored, sometimes with carbon-based materials as supports (e.g., carbon nanotubes20 and boron-doped diamond21). Among these, copper oxides are particularly appealing due to their exceptional catalytic activity and resistance to poisoning.22–24 Substantial research effort has been devoted to synthesizing CuxO-based electrodes in diverse forms, such as nanoparticles,25 nanocubes,26 nanoneedles,27 nanowires,28 and nanoflowers.29 A common fabrication approach involves directly immobilizing copper oxide catalysts onto a supporting material such as glassy carbon. However, the weak adhesion of CuxO structures to the surface often leads to unreliable results. To address this issue, the use of a polymer binder, such as Nafion®, has been proposed.30,31 Alternatively, copper oxides can be synthesized on the surface of Cu substrates through wet-chemical methods28 or thermal oxidation.32 Although these techniques offer improved adhesion of the oxide to the substrate surface, achieving large-scale and reproducible synthesis of uniform materials remains challenging. In contrast, electrochemical oxidation methods, such as potentiostatic, potentiodynamic or galvanostatic anodization, have been employed as direct fabrication methods for highly sensitive and stable CuxO-based glucose sensors.33–37 Compared to traditional synthesis methods, anodization offers several advantages. It is a relatively simple and low-cost technique that can be conducted under ambient conditions without the need for high temperatures or toxic chemicals. Moreover, it allows for precise control over the thickness and morphology of the surface oxide/hydroxide layer, enabling the reproducible design of remarkably active surfaces. Depending on the chosen electrolyte and applied potential/current density, different morphologies can be produced. Notable mentions include the galvanostatic production of nanowires by Wu et al.38 in KOH solution, the controlled nanowire diameter by potentiostatic anodization (10–50 V) in carbonate solution by Stępniowski,39 nanoporous structures in aqueous/nonaqueous electrolytes at high voltage (≈10 V) by Allam et al.40 Although promising, the research has solely investigated the properties of thick copper foils which don't necessarily translate to microfabricated sensors. This discrepancy arises from the generally higher resistivity of thin films compared to bulk electrodes;41,42 a challenge further exacerbated by the complexity of removing O2 impurities in widespread PVD systems such as sputtering.43–45 Moreover, the lack of consensus on the mechanism glucose oxidation on copper combined with the mixed phase composition of most anodized surfaces, requires the validation of each technique for glucose oxidation. To address this issue, researchers have directly evaluated the ability of differently anodized copper foils to oxidize glucose. For a comprehensive summary of the challenges, approaches, and advantages associated with this method, we refer to the excellent review by Giziński et al.37 Nonetheless, most of these studies33,36,46,47 have focused on specific sets of anodization parameters without providing a justification for their selection. Recently, the group of Noda48 investigated the effect of the hydroxide ion strength in the anodization solution for methanol oxidation. However intriguing, their work studied the effect of the OH− concentration at a single potential, thus leaving many questions still unanswered. This lack of exhaustive understanding and rationalization hinders the progress of CuxO-based catalysts. Therefore, the objective of this paper is to provide a comprehensive outline of the potentiostatic anodization process applied to thin film copper electrodes, specifically for glucose oxidation applications. By increasing in small steps the anodization potential together with the KOH concentration, we aim to evaluate the glucose sensitivity for each individual condition. In this way, we can establish a distinct correlation encompassing the cyclic voltammetric curve, overall sensitivity, and the surface physico-chemical properties generated through each set of anodization parameters. The results of this work will help to fill the knowledge gaps and facilitate the development of highly efficient and integrated CuxO-based glucose sensors and electrocatalysts more broadly.
Experimental section
Electrode preparation
In this work, 7 × 7 mm Cu/Pt/glass electrodes were fabricated starting from 4′′ borosilicate glass wafers. Through RF magnetron sputtering, a 300 nm thick layer of Pt was deposited, acting as a current collector. Pt was chosen over Au due to its greater compatibility with the fabrication of integrated electrochemical sensors, where the counter electrode is commonly Pt. Afterwards, 300 nm layer of Cu was deposited on top of the Pt and through careful control of base pressure (2.5 × 10−5 mBar) and working pressure (1 × 10−2 mBar) a well adherent and non-oxidized layer was obtained, as confirmed by GI-XRD (Fig. 4D). The deposition parameters are reported in Table S1.† The thickness of the sputtered Cu thin films was assessed using a Dektak Profilometer (Veeco Instruments INC.). Three repeats were collected along different regions of each film. The average measured thickness was 290 nm, close to the 300 nm expected from sputtering rate calculations. The electrodes after dicing were 7 × 14 mm. Through the use of PVC tape a 7 × 7 mm geometric area was defined. Lastly, the copper deposition on the electrical contact was removed by the use of 67% HNO3, thus exposing the Pt underneath. This allowed to avoid undesired fluctuations in potential due to copper oxide/hydroxide formation by accidental contact with the highly alkaline electrolytic solution. Before each anodization the electrode was polarized at −1 V for 50 s, to reduce the natural passivation layer on top of the exposed copper.
Electrochemical characterization
All electrochemical measurements were made with a multichannel potentiostat (PalmSens MultiPalmSens4) in a 3-electrode configuration. The reference electrode was a triple junction Ag|AgCl|KCl electrode (Sigma Aldrich) with a bridge filled with 3.8 M KCl solution. The bridge was emptied and refilled between each measurement. The counter electrode was a Pt coil (Sigma Aldrich). The working electrode was 7 mm × 7 mm Cu/Pt/glass electrode and the electrical connection to the plate was achieved with a tantalum clip (Redoxme) on the exposed Pt. The experiments were conducted in air at ambient conditions under a fume hood. The cyclic voltammetry and chronoamperometry experiments were performed in 50 mL solutions in borosilicate glass beakers. For all of the cyclic voltammetry experiments, the third cycle was taken, and unless otherwise specified, the scan rate used was 100 mV s−1. The potential window was selected from 0 V to 0.8 V to ensure the stability of the electrodes. Chronoamperometric calibrations were conducted at 0.5 V with constant stirring at 350 rpm. All electrochemical calibrations were done in triplicate. To evaluate the reproducibility of our process, we replicated the anodizations at −0.2 V, −0.1 V, and 0 V in 1 M KOH three times. The chronoamperometric curves were very similar, with only slight differences in current densities (S1†). This difference may be attributed to the non-uniform thickness of the RF sputtering system49 and the minimal potential instabilities of the reference electrode.
Morphological characterization
The morphological characterization of the samples was done with a Helios Nanolab G3 CX DualBeam FIB-SEM. Copper tape was used to ground the conductive topside of the electrode with the SEM sample holder, thus avoiding unwanted sample charging.
X-ray photoelectron spectroscopy
XPS was employed to investigate the surface chemical composition of the as-grown and the polarized copper films. XPS was performed in a FlexMod SPECS system equipped with a monochromatic 100 W Al source (E = 1486.7 eV). The base pressure of the SPECS system is in the 10−10 mbar range. XPS spectra were recorded with a step of 0.05 eV and a pass energy of 20 eV. The XPS data were analyzed with the CasaXPS software, and a Shirley function was used to subtract the background radiation. Spectra are energy-calibrated relative to the C 1s peak of adventitious carbon (284.8 eV).
X-ray diffraction
The grazing incidence X-ray diffraction (GIXRD) and X-ray reflectometry (XRR) analysis was performed with PANalytical X'Pert Pro diffractometer with a Cu Kα source. All GIXRD patterns are acquired without a monochromator and with an incident angle of 0.5°.
Chemicals
All chemicals were purchased from Sigma Aldrich. Glucose had a purity of >99.5% and KOH pellets (reagent grade) were used to make all the alkaline solutions. L-Ascorbic acid and acetaminophen had a purity of 99%, and the latter was kept at −8 °C. All chemicals were used as received, without additional purification steps. Stock solutions were prepared fresh daily in 0.1 M KOH.
Statistical analysis
In the course of cyclic voltammetry and chronoamperometry experiments, the recording of current, potential, and time was undertaken. Subsequently, the current values were normalized by the geometric area of the electrode (0.49 cm2) to determine the current density. No additional normalization procedures were applied. Each calibration experiment, whether via chronoamperometry or cyclic voltammetry, had a sample size of 3. When a linear interpolation was performed, the corresponding coefficient of determination R2 is provided. The data is presented as mean ± standard deviation.
Results and discussion
Electrochemical reactions for thin Cu electrode anodization
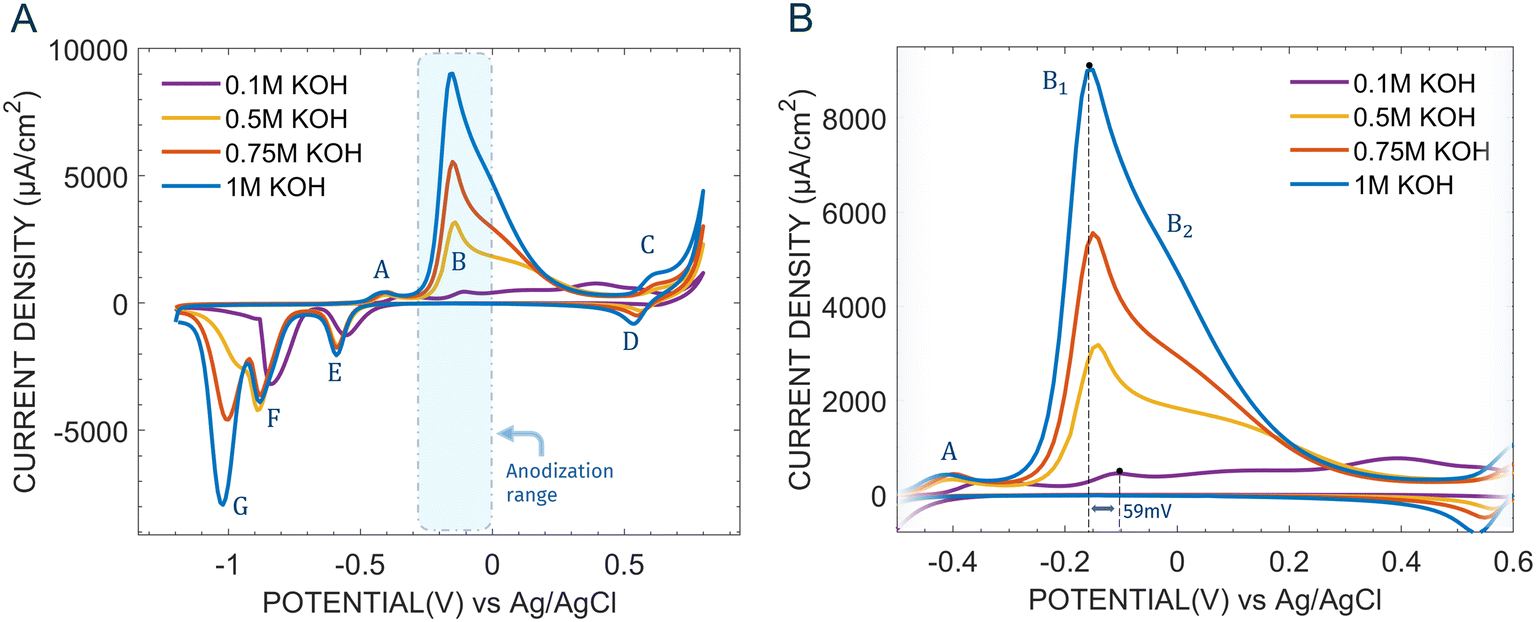
Prior to the selection of the anodization potential, we recorded the cyclic voltammograms of microfabricated Cu electrodes between −1.2 V and 0.8 V at different KOH concentrations Fig. 1A. The main features of the voltammograms match well with those reported by several researchers for polished Cu electrodes.50 Three anodic peaks (A, B, C) and four cathodic peaks (D, E, F, G) can be discerned, associated with redox transitions of Cu(0), Cu(I), Cu(II) and – allegedly51 – Cu(III) for peaks C, D. A small shoulder before A is present, although a consensus on the exact nature of this reaction is not well established.52 On this, Ambrose et al.53 proposed that the species Cu(OH)2− is formed by the direct electrooxidation of Cu. Based on the revised Pourbaix diagram of Cu by Beverskog et al.,54 Giri et al.52 later proposed that the formation of soluble Cu(OH)2− may be preceded by an intermediate step of Cu to Cu2O oxidation (reactions (1) and (2)).| | | 2Cu + 2OH− → Cu2O + H2O + 2e− | (1) |
| | | Cu2O + 2OH− + H2O ⇔ 2Cu(OH)2− | (2) |
As the potential is increased the rate of formation of Cu2O dominates over that of dissolution, giving rise to a peak (peak A). As the potential is further increased the current increases sharply, leading to peak B (Fig. 1A). This peak can be further subdivided in two, B1 and B2 (Fig. 1B), the former occurring at more cathodic potentials. Mainly two mechanistic descriptions of the reactions underlying peak B have been advanced. One, by Haleem et al.55 involves the formation of soluble or insoluble hydroxides such as Cu(OH)42− and Cu(OH)2 respectively by the oxidation of Cu or Cu2O. However, this model supposes the formation of the thermodynamically unfavoured Cu(OH) and permits only one peak instead of two. Another reaction scheme was proposed by Ambrose et al.,53 where the reactions underlying B1 and B2 differ depending on pH of the solution. Accordingly, peak B1 could be attributed to the oxidation of Cu and Cu2O to soluble Cu(OH)42− or insoluble Cu(OH)2 through reactions (3)–(5). Clearly, the greater magnitude of peak B1 compared to peak A suggests that reaction (4) will dominate over 3, since the latter requires Cu2O formed by reaction (1).
 |
| | Fig. 1 (A) Cyclic voltammograms for Cu electrodes at different KOH concentration (0.1 M, 0.5 M, 0.75 M, 1 M), taken between −1.2 V and 0.8 V at a scan of 100 mV s−1. (B) Zoomed-in cyclic voltammograms, highlighting the position of A, B1, B2 and the 59 mV shift going from 0.1 M KOH to 1 M KOH (here shown for B1). | |
• At 1 M KOH:
| | | Cu2O + 6OH− + H2O → 2Cu(OH)42− + 2e− | (3) |
| | | Cu + 4OH− → Cu(OH)42− + 2e− | (4) |
• At 0.1 M KOH
| | | Cu2O + 2OH− + H2O → 2Cu(OH)2 + 2e− | (5) |
Given enough time to reach supersaturation, the build up of Cu(OH)
42− near the surface produced by
reactions (3) and
(4) can lead to the homogeneous/heterogeneous nucleation of Cu(OH)
2 microneedles (
reaction (6)). However, as the potential is increased close to peak B
2 passivation reactions start to compete with the dissolution process (
reactions (3) and
(4)) that feeds
reaction (6). Specifically, the oxidation of metallic copper by OH
− ions, giving Cu(OH)
2 and CuO, as per
reactions (7) and
(8) respectively.
| | | Cu(OH)42− ⇔ Cu(OH)2 + 2OH− | (6) |
| | | Cu + 2OH− → Cu(OH)2 + 2e− | (7) |
| | | Cu + 2OH− → CuO + H2O + 2e− | (8) |
As a result, the chemical species on the electrode's surface in proximity to the B peaks are likely to be Cu(OH)
2 closer to B
1 and CuO closer to B
2.
52 Therefore, by applying a potential between the end of peak A and peak B it is possible to study the gradual change from Cu
2O, to Cu(OH)
2 and CuO. The presence of a potential shift of 59 mV between 1 M KOH and 0.1 M KOH for B
1 (
Fig. 1B) is particularly relevant for copper anodization research, given that significantly different species are formed in a range of approximately 300 mV. Furthermore, as previously mentioned, the possible oxidative pathways are also affected by the solution's pH, thus requiring careful consideration.
Fig. 2 shows the chronoamperometric curves, each lasting 150 s, for microfabricated Cu electrodes obtained by applying 0 V, −0.1 V and −0.2 V at different KOH concentrations. As illustrated, a change of 100 mV in anodization potential can lead to significantly different chronoamperometric curves. For an applied potential of −0.2 V and KOH concentration lower than 1 M, the current density drops rapidly due to the formation of Cu2O, usually of no more than 6 nm.56,57 This process of Cu(I) oxide layer formation can be described by reaction (1). Instead, at −0.2 V for 1 M KOH concentration a completely different current time transient is obtained. The curve is characterized by an initial current drop until an inflection point is reached, then the current rises again and after reaching a maximum it slowly decays close to zero. This particular current–time response for copper has been traditionally attributed to a process of nucleation and growth of Cu(OH)2/CuO nanowires,58–60 in analogy with aluminum anodization.61 Based on the general mechanism proposed by Shoesmith and Ambrose,53,59,62 we can explain the chronoamperometric curve at −0.2 V in 1 M KOH (Fig. 2) as follows: (i) current density decay to a local minimum at around 4 s due to Cu2O formation,62 as per reaction (1). The insulating layer decreases the charge transport between the electrode and the electrolyte. (ii) Dissolution of the insulating film by conversion into Cu(OH)42− (reaction (3)) with the formation of channels in the oxide that expose fresh Cu; (iii) dissolution of Cu by reaction (4) with the formation of Cu(OH)42−. (iv) Formation high Cu(OH)42− concentration regions, that convert into Cu(OH)2 (reaction (6)), growing into nanowires. The nucleation of hydrous Cu(OH)2 requires supersaturated Cu(OH)42− near the electrode's surface, as shown by previous work where upon electrode rotation the reaction is inhibited.59 (v) The current increases until a maximum is reached at around 20 s, corresponding to the point where the Cu(OH)2 crystals start to impinge upon one another, thereby progressively impeding reactions (3) and (4). As a result, the current gradually falls close to zero, with nanowire growth halting at t = 120 s. A simplified illustration of the mechanism of nanowire formation is shown in Fig. 3. The change in nanowire morphology at different points in the current–time curve for an anodization at −0.2 V (vs. Ag|AgClsat) and 1 M KOH has been studied by Stepniowski et al.58 The authors noted a progressive increase in nanowire thickness and length for longer anodization times. On the other hand, the anodizations at 0 V (Fig. 2) show dramatically lower current densities while still maintaining the same nucleation and growth profile. This can be explained by noting that 0 V in 0.5 M, 0.75 M and 1 M KOH is closer to B2 than B1. Hence, at polarization the Cu surface transforms into Cu2O (reaction (1)) but also into CuO (reaction (8)). This limits the formation of Cu(OH)42−, as reactions (3) and (4) can happen only on the available Cu or Cu2O sites. As a result, under these conditions the predominant surface species will be CuO, instead of Cu(OH)2 or Cu2O. In general, all anodizations done in 0.1 M KOH show a sharp decline current densities independently of the applied potential. This is because, as reported by Ambrose et al.,53reaction (4) is replaced by reactions (1) and (5). Therefore, the process of dissolution is severely limited, likely resulting in the formation of a Cu2O layer. Lastly, at an applied potential of −0.1 V we can observe (Fig. 2) that the current–time transients are also typical of a nucleation and growth process (except in 0.1 M KOH). Here, an intermediate behavior can be noted between the anodizations at 0 V and −0.2 V, where at −0.1 V in 0.5 M KOH the curve resembles that at −0.2 V and 1 M KOH, and that at −0.1 V in 1 M KOH resembles that at 0 V in 0.5 M KOH. This change the shape of the curves at −0.1 V, associated with a reduction of current density for increasing KOH concentration, can be attributed to the progressively more favourable formation of CuO which impedes further etching and thus competes with the process of nanowire formation.60
 |
| | Fig. 2 Chronoamperometric curves at 0 V, −0.1 V, −0.2 V (from left to right) at different concentrations of KOH, each performed for 150 s. | |
 |
| | Fig. 3 Simplified mechanism of nanowire formation in 1 M KOH and at −0.2 V. (1) Formation of insulating Cu2O layer, by reaction of Cu with OH−. (2) Dissolution of Cu2O with formation of channels which expose fresh Cu. (3) Dissolution of both Cu2O and Cu. (4) The formation of Cu(OH)2 nanowires from supersaturated regions of Cu(OH)42−. | |
Electrodes' morphological and compositional characterization
After anodizing via chronoamperometry the copper electrodes under different potentials (−0.2 V, −0.1 V, 0 V) and KOH molarities (0.1 M, 0.5 M, 0.75 M, 1 M), we selected 4 configurations whose chemical and morphological state was expected to differ significantly. Specifically: −0.1 V in 0.5 M, −0.2 V in 0.75 M, 0 V in 1 M and a bare Cu electrode. To simplify the nomenclature we will henceforth refer to them by (S + applied potential), with the bare referred to as SBare. All the morphological, chemical and crystallographic characterizations were performed after 3 cycles in 0.1 M KOH ranging from 0 V to 0.8 V. This approach allowed us to consider the stable state conditions of use.
Scanning electron microscopy (SEM) was conducted to examine the surface morphology, as illustrated in (Fig. 4A). SBare exhibits a flat surface, devoid of significant features, suggesting that cycling between 0 V and 0.8 V promotes the formation of a passivating film, which effectively shields the surface from corrosion. Similarly, S0V maintains the predominantly flat morphology observed in SBare, albeit with the presence of short, sparse nanowires. In stark contrast, S-0.1V displays a dense network of nanowires, each with a diameter of approximately 10 nm, and these nanowires have formed in clusters with an average cluster diameter of 122 ± 21 nm. A detailed comparison between S-0.1V and SBare at higher magnification (50k×) is provided in S4.† Finally, S-0.2V is distinguished by a porous structure, likely resulting from a competitive interplay between reactions (1) and (2). The samples were analyzed using X-ray photoelectron spectroscopy (XPS) to evaluate their surface chemical states. A survey spectrum of SBare (S2†) reveals prominent core-level lines associated with copper, including a Cu 2p doublet (Cu 2p1/2 and 2p3/2) at 930–955 eV, Cu 3s at 123 eV, Cu 3p at 76 eV, and Cu 3d at 3 eV. Peaks corresponding to oxygen (O 1s) at 530 eV and adventitious carbon (C 1s) at 285 eV are also present. Fig. 4B shows the Cu 2p3/2 signals for electrodes biased at various potentials, with the binding energies of relevant copper species superimposed. By comparing the main emission band's peak positions with literature values for Cu(0), Cu2O, CuO, and Cu(OH)2, qualitative insights can be drawn. All samples display a well-defined shakeup satellite, indicating the presence of Cu(II) species.63–66 The main peak of SBare is at a binding energy of 933.3 eV, close to that of pure CuO at 933.8 ± 0.2 eV.67 Additionally, a small shoulder at lower binding energies corresponds to either metallic Cu or Cu2O, found at 932.6 ± 0.2 eV and 932.4 ± 0.2 eV, respectively.67 This shoulder disappears in S0V, with the peak maximum shifting to 933.7 eV, suggesting an increase in the CuO component at the expense of Cu(0) and Cu2O. The peak maximum in the L3M4,5M4,5 Auger spectrum for S0V is at a kinetic energy of 917.5 eV, close to the reference value of 917.6 eV for CuO,68 and slightly lower than SBare at 917.9 eV. S-0.1V shows a peak maximum at 934.6 eV, closely matching Cu(OH)2 at 934.5 ± 0.2 eV.67 A similar shift is seen in the L3M4,5M4,5 Auger spectrum, with its maximum at a kinetic energy of 916.4 eV, near 916.25 eV for pure Cu(OH)2. Moreover, the shakeup feature in S-0.1V differs from all other samples, exhibiting the typical shape of Cu(OH)2 reported in the literature.68 As expected, the O 1s spectrum of S-0.1V shows a dominant Cu–OH component at a binding energy of 531.1 eV (S3†). In contrast, the shoulder at lower binding energy observed in SBare is more pronounced in S-0.2V, shifting the peak maximum to 932.6 eV. This suggests that the polarization at −0.2 V increases the Cu(0) or Cu2O fraction at the expense of CuO, consistent with thermodynamic predictions.69 Despite the increased Cu(0) or Cu2O fraction, the presence of a dominant CuO component means that the L3M4,5M4,5 Auger spectrum peak position does not significantly shift from SBare to S-0.2V, with only a slight shoulder emerging at higher kinetic energies. Quantifying the relative contributions of different species inmixed-oxidation-state copper electrodes is challenging due to significant peak overlap. To address this, the well-established fitting procedure developed by Biesinger et al.68 was employed. This method fits the entire Cu 2p3/2 peak shape using a set of peaks for each species, constrained in binding energy, full width at half maximum (FWHM), and relative areas. Peak positions and relative areas were referenced to the first peak of each species, with variations of ±0.1 eV allowed to account for the imprecision due to the C 1s calibration. Similarly, the FWHM was allowed to vary by ±0.1 eV. This approach, while similar to using actual line shapes from pure compounds, offers greater flexibility in its application. Fig. 4C shows the fitted Cu 2p3/2 spectra of SBare, S0V, S-0.1V, and S-0.2V. The fitting parameters, along with the fractional areas of each component, are detailed in the ESI† (Tables S2–S5). Consistent with the previous qualitative analysis, SBare is primarily composed of CuO (89%), followed by Cu(0) + Cu2O (11%). In S0V, the CuO fraction increases to 93%, with some Cu(OH)2 (5%) and only trace amounts of Cu(0) + Cu2O (1%). S-0.1V is dominated by Cu(OH)2 (55%), followed by CuO (45%). S-0.2V, compared to SBare, has a lower CuO fraction (84%) but an increased Cu(0) + Cu2O component (15%).
 |
| | Fig. 4 (A) SEM images of SBare, S0V, S-0.1V and S-0.2V. (B) Cu 2p3/2 XPS spectra combined, overlayed with reference binding energies (C) Cu 2p3/2 XPS spectra of SBare, S0V, S-0.1V, S-0.2V samples with corresponding fit. (D) XRD diffraction patterns for SBare (D.1), S-0.1V (D.2), S-0.2V (D.3) and S0V (D.4). | |
X-ray diffraction (XRD) analysis was also conducted to gain additional insights, with the corresponding diffractograms shown in Fig. 4D. Regardless of polarization potential, the anodization does not appear to affect the entire volume of the thin copper film, as evidenced by the presence of diffraction peaks for Cu and Pt in all samples (SBare, S0V, S-0.2V, S-0.1V). For SBare, S0V, and S-0.2V, the diffraction pattern is dominated by the Pt/Cu bilayer signal, suggesting that the thickness of the produced anodic oxides is too small to be reliably detected by XRD. Copper diffraction peaks are observed at 2θ values of 43.3, 50.4, and 74.1 degrees, with corresponding Miller indices (hkl) of (111), (200), and (220). Platinum diffraction peaks are noted at 2θ values of 39.9, 46.4, and 67.4 degrees, with corresponding Miller indices of (111), (200), and (220). These planes indicate that a cubic phase structure can be assigned to both the copper (ICSD01-089-2838) and platinum (ICSD01-087-0640) layers. In contrast, the XRD pattern of S-0.1V indicates that the produced nanowires are crystalline and primarily composed of Cu(OH)2. Diffraction peaks of Cu(OH)2 are observed at 2θ values of 23.8, 34.1, 35.9, 38.2, 39.8, 53.2, 53.4 and 64.6 degrees, with corresponding Miller indices of (021), (002), (111), (041), (022), (130), (150), (132), and (152). These planes suggest an orthorhombic crystal structure for Cu(OH)2 (ICSD01-072-0140). It is possible that some peaks associated with CuO may also be present, but they might be too weak to be distinguished from adjacent Cu(OH)2 peaks. The average crystallite size was estimated using the Scherrer formula.70
| |  | (9) |
where ‘
λ’ is wave length of X-ray (0.1541 nm), ‘
β’ is FWHM (full width at half maximum), ‘
θ’ is the diffraction angle and ‘
d’ is the mean crystallite diameter. To estimate the crystallite size for the Cu(OH)
2 nanowires of S-0.1V we considered the three most intense peaks (2
θ = 23.8°, 34.1°, 53.4°) and obtained a value of 12 ± 2 nm. For the Cu and Pt layers on SBare, the three most intense peaks were considered for each. For Pt (2
θ = 39.9°, 46.4°, 67.4°) the mean crystallite size was 14 ± 2 nm; for Cu (2
θ = 43.3°, 50.4°, 74.1°) it was 11 ± 4 nm.
Effect of process parameters on glucose oxidation at anodized Cu electrodes
Once the microfabricated electrodes were anodized under different potentials (−0.2 V, −0.1 V, 0 V) and KOH concentrations (0.1 M, 0.5 M, 0.75 M, 1 M), we proceeded to evaluate their electrocatalytic performance towards glucose electrooxidation. The selection of a potentiodynamic method, such as cyclic voltammetry, over a potentiostatic one aimed at providing additional metrics to assess the oxidation process. Of these, the shape of the CVs and the current density at different potentials can point to different mechanisms.71,72 Thus, we acquired the CVs of the bare and the anodized electrodes in a solution of 0.1 M KOH with 8 mM of glucose.
Fig. 5A and B compare respectively the obtained CVs at the third cycle as a function of the KOH molarity of the anodization solution and of the applied anodization potential. Here we show that anodizations done on a reduced copper surface introduce stable chemical and morphological changes that can either favour and disfavour glucose oxidation. The CV of the bare Cu in a glucose solution (red curve in Fig. 5C) presents the typical plateau profile which has been traditionally ascribed to a Cu(II) to Cu(III) transition.7 Additionally, the symmetric shape of the CV,73 combined with the decreasing sensitivity from cycle to cycle (S6†) would suggest the presence of an adsorption step of oxidation products. Xie et al.24 advanced the existence of two limiting mechanisms for glucose oxidation on copper electrodes: on the left side of the peak the hydroxide adsorption is limiting, whereas on the right side the adsorption of glucose is limiting. Inspired by the study of Fleischmann et al.74 on nickel oxide, the authors proposed the adsorbed hydroxyl radical to be the main oxidizer, with the rate determining step being the H abstraction from the Cα and the formation of a bridged cyclic intermediate. In their model, as the electrode is polarized anodically, Cu2O would transform to CuO according to the following reaction:
| | | Cu2O + 2OH− → 2CuO + H2O + 2e− | (10) |
Subsequently, Cu(
III) species would form and mediate the oxidation of glucose. However, this is still a contested issue, given that thermodynamic predictions indicate its formation potential to be more anodic than OER. Lately, Barragan
et al.51 further put into question the formation of Cu(
III) species, proposing instead that a partial charge transfer occurs between the vacancies in the CuO and the adsorbed hydroxide ions.
Reaction (11) represents the coupling of the vacancies h
+ and the adsorbed OH, and
reaction (12) shows the formation of OH radical through effective charge transfer. The latter is expected to happen to a lesser extent, according to the authors.
51| |  | (11) |
| |  | (12) |
| | | (OH−ads)(h+) + RCH3OH → H2Oads + RCH2OH | (13) |
The energy accumulated in the pairing between the vacancies h
+ and OH
− catalyses the oxidation of glucose through
reaction (13). Through a two electron transfer, gluconolactone and two water molecules are expected to form, succeeded by additional oxidation reactions. Within this framework, the initial step of glucose oxidation doesn't require the formation of a Cu(
III)–glucose complex but leverages the semiconductive properties of CuO. This would imply that CuO formation is a necessary step to produce the active (OH
−ads)(h
+) sites. Our results support this model: for 1 M KOH we observe a progressive increase in the glucose oxidation current for increasing anodization potential (
Fig. 5A). At the same time, the peak current (excluded the component from oxygen evolution) shifts cathodically, indicative of a decreasing energetic barrier to glucose electrooxidation. This is due to a greater fraction of CuO to Cu(OH)
2, as the anodization is performed at potentials gradually closer to B
2 and further to B
1 (
Fig. 1). Similarly, for a 0 V polarization the KOH molarity has a slight positive effect on the glucose oxidation current density – albeit not as dramatic as the applied potential. This may be due to a 59 mV cathodic shift going from 0.1 M to 1 M, as previously mentioned. Consequently, the most performing electrode is obtained by polarizing at 0 V in 1 M KOH (S0V), as it maximizes the fraction of the catalytically active cupric oxide.
 |
| | Fig. 5 Cyclic voltammograms in 0.1 M KOH and 8 mM glucose at 100 mV s−1 scan rate. (A) Effect of the anodization potential for a fixed concentration of KOH. (B) Effect of the KOH concentration for a fixed anodization potential. (C) Cyclic voltammograms of the samples selected for morphological, chemical and crystallographic analysis. | |
The CVs at −0.1 V for varied KOH concentrations (Fig. 5B) show very distinct profiles. As this potential is at the cross point of A, B1, B2, the description of all the represented CVs allows to cover all the observed variability within the [KOH molarity-potential] space. At −0.1 V for 0.1 M of KOH, peak A is favoured and the current densities are similar to that of the bare Cu (SBare) until they drop at 0.6 V. Its shape strongly resembles that of sample S-0.2V (Fig. 5C), which as expected presented a greater Cu2O fraction (Table S5†). The reduced currents at >0.6 V may be due to a lower affinity for oxygen evolution or for successive glucose oxidation steps. Although Cu2O is expected to convert to CuO through reaction (10), the quick scanning of potentials in a CV is likely to affect only a superficial layer. Hence, the amount of (OH−ads)(h+) is limited by the extent of Cu2O being transformed into CuO. Conversely, at M KOH the voltammogram rises more slowly than the bare Cu, until reaching the same current densities at 0.8 V. Under these conditions B1 is favoured and Cu(OH)2/CuO nanowires form, as highlighted by our characterization of sample S-0.1V (Fig. 4). The presence of Cu(OH)2 nanowires appears to introduce an overpotential, slowing down the electrooxidation process. Barragan et al.,51 reported for Cu(OH)2 an intermediate activity between Cu and CuO. However, our findings indicate the copper hydroxide sample (S-0.1V) to be the least performing. This could be attributed to a morphological difference, as our samples had a dense layer of Cu(OH)2 nanowires, instead of a film obtained through chemical means. Nonetheless, improved catalytic activity was reported going from Cu(OH)2 to CuO. S. Zhou et al.75 in their Cu anodization work found Cu(OH)2 to be more active for glucose sensing than CuO. Nevertheless, the study lacked a comparison with a Cu electrode, and their CuO electrode may have under performed due to a significantly longer anodization process, resulting in a thicker oxide layer, as compared to their Cu(OH)2 electrode.
The effect of the anodization parameters (KOH concentration and anodization potential) on the glucose oxidation current in CV is summarized by the a contour plot in Fig. 6. The graph was produced by recording the peak current density of each CV at 8 mM glucose in 0.1 M KOH for a total of 15 unique combinations of anodization parameters (Table S6†). Afterwards, a cubic 2D interpolation was performed in order to transform a discrete set of datapoints into a surface. Accordingly, applying a potential of 0 V leads to the greatest performance improvements, with 1 M KOH being the best electrolyte concentration. Moreover, at potentials between −0.1 V and −0.2 V and KOH concentrations between 0.4 M and 0.8 M a region of impaired catalytic activity is observed, where the response is worse than for a bare Cu electrode. Within this range Cu(OH)2 nanowires are formed, as confirmed by our characterization of S-0.1V (Fig. 4), the chronoamperometry curves (Fig. 2) and the clear resemblance in shape of the CVs in 8 mM glucose (Fig. 5).
 |
| | Fig. 6 Contour plot of the effect of process parameters on the glucose oxidation current. | |
Sensing performance towards glucose via cyclic voltammetry and chronoamperometry
Our systematic study on the effect of the anodization parameters on the electrocatalytic activity towards glucose clearly highlighted that an anodization step at 0 V in 1 M KOH led to the greatest current density for 8 mM glucose in 0.1 M KOH (Fig. 6, Table S6†). Although a single concentration CV can be used as a useful screening method, the different response towards oxygen evolution superimposing to the glucose oxidation could inflate the actual sensitivity increase. Consequently, we performed a full calibration with cyclic voltammetry and chronoamperometry of sample S0V and the bare copper sample SBare.
Fig. 7 shows the CVs at the third cycle for S0V and SBare, with the corresponding calibration plot. Both samples show very good linearity in the 1–10 mM range, with S0V having a sensitivity of 916 ± 17 μA cm−2 mM−1, against a sensitivity of 723 ± 9 μA cm−2 mM−1 for SBare – equivalent to a 26% increase. Although it is not always specified, the cycle number has a significant effect on the sensor's response. For S0V, a loss of ≈25% of sensitivity is observed from the first to the third cycle, after which it stabilizes (S6†). Whereas for SBare, this sensitivity drop is only ≈17%. As a result, the sensitivity gain between S0V and SBare reduces from 40% to 26% between the first and third cycle. Given that CuO and Cu electrodes are generally considered to be stable within this potential range,7 the observed current reduction is likely associated with the adsorption of oxidation products,73 of which S0V appears to be more susceptible. S0V might favour additional electrooxidation steps whose products tend to temporarily block the active sites. The limit of detection (LOD) was calculated using the following formula:76
| |  | (14) |
where
σblank is the standard deviation of the blank and
S is the sensitivity. Here, we obtained
σblank by evaluating the current densities of three blanks at 0.46 V, corresponding to the potential of the peak at the lowest concentration (1 mM). For S0V and SBare respectively, the LOD in the 1–10 mM range is 1.35 mM and 0.81 mM. We further complemented the potentiodynamic study by recording the CVs for S0V and SBare with a glucose concentration of 2 mM in 0.1 M KOH at different scan rate (from 25 mV s
−1 until 200 mV s
−1 in steps of 25 mV s
−1). The peak glucose current was observed to be linearly dependent on the square root of the scan rate for both samples (S7
†), indicative of diffusion limited process.
72 Here we considered the current density at 0.6 V to minimize the interference of oxygen evolution.
 |
| | Fig. 7 (A) Cyclic voltammograms in 0.1 M KOH for the S0V sample with a glucose concentration between 0–10 mM. (B) Cyclic voltammograms in 0.1 M KOH for the SBare sample with a glucose concentration between 0–10 mM. (C) Calibration plot of the glucose oxidation peak for S0V and SBare, third cycle considered. All data is expressed as a mean ± SD (n = 3). | |
Fig. 8A presents the chronoamperometric curves for S0V and SBare with the addition of 0.1 mM of glucose until 0.5 mM and then in steps of 0.25 mM until 2 mM. Fig. 8B includes the calibration curves obtained from three independent chronoamperometry experiments each, with the calculated sensitivities in the bar plot in Fig. 8C. To obtain a calibration curve we averaged the value of the current density in the middle 10 s for each 30 s step, in order to minimize fluctuations. The sensitivity in the 0.1 mM to 0.5 mM range for sample S0V and SBare respectively is 955 ± 11 μA cm−2 mM−1 and 616 ± 9 μA cm−2 mM−1, equivalent to a 55% gain over SBare. Whereas, in the 0.75 mM to 2 mM S0V had a sensitivity of 767 ± 18 μA cm−2 mM−1 and SBare of 443 ± 10 μA cm−2 mM−1, with a corresponding gain of 73% over SBare. The LOD was calculated by using eqn (14), where σblank corresponds to the standard deviation for three samples of the mean current density from 40 s to 45 s after polarization, prior to the first glucose addition. For S0V and SBare respectively, the LOD in the 0.1–0.5 mM range is 0.0041 mM and 0.0597 mM, while in the 0.75–2 mM range is 0.0051 mM and 0.08 mM. Consequently, cyclic voltammetry may not be the most appropriate electroanalytical technique for glucose sensing on microfabricated copper electrodes due to the deactivation process observed from the first to the second cycle, combined with its relatively high LOD. In contrast, chronoamperometry allows to obtain an order of magnitude lower LOD and greater gains in sensitivity (73% and 55% in the two linear ranges compared to 26% at the third cycle in CV). Anodized bulk copper electrodes in the literature (refer to Table 3 in Giziński et al.37) perform similarly with respect to glucose sensing. However, such a comparison would not be fair for several reasons. First, the resistivity of thin Cu films surpasses that of bulk Cu.41 Secondly, although highly scalable, the RF magnetron sputtering in the absence of ultra-high vacuum can lead to oxygen impurities in the film that increase its overall resistivity.77 Lastly, a Pt sub-layer was introduced to ensure a dependable electrical connection. Thus, the electron transfer is further hampered by the inherently worse conduction of Pt (compared to Cu) and the existence of a the Pt–Cu interface. Nonetheless, moving from bulk to film, together with some associated drawbacks, is a necessary step to achieve fully integrated and compact devices.
 |
| | Fig. 8 (A) Chronoamperometric curves of S0V (dark blue) and SBare (light blue) in 0.1 M KOH at 0.5 V showing additions of 0.1 mM of glucose until 0.5 mM and then of 0.25 mM until 2 mM. (B) Calibration plot for S0V (dark blue) and SBare (light blue) for an applied potential of 0.5 V. (C) Corresponding sensitivities in the 0.1–0.5 mM and 0.75–2 mM ranges for S0V (dark blue) and SBare (light blue). All data is expressed as a mean ± SD (n = 3). | |
Interference resistance
The presence of interferents in the blood serum, such as the easily oxidizable endogenous compounds like ascorbic acid (AA) and exogenous ones like paracetamol (PCM), may complicate the accurate estimation of glucose concentration.17 Thus, we evaluated the selectivity of our thin copper film electrodes to glucose by chronoamperometry at an applied potential of 0.5 V. We added 2 mM of glucose to a 0.1 M KOH solution and then observed the change in current density at the addition of 0.1 mM of ascorbic acid first and of 0.1 mM acetaminophen thereafter (Fig. 9). The relative concentrations were chosen to mimick blood serum values.5 The addition of 0.1 mM of PCM caused a current density increase of 3% and 4% for S0V and SBare, respectively. Whereas, the addition of 0.1 mM of AA led to a 5% and a 6% increase for S0V and SBare, respectively. In conclusion, both electrodes showed satisfactory selectivity towards AA and PCM, with the anodized electrode having between 30% to 40% lower relative current change compared to the bare.
 |
| | Fig. 9 Interference study at a potential of 0.5 V, with additions of 2 mM glucose, 0.1 mM ascorbic acid, 0.1 mM paracetamol. SBare is compared with S0V. | |
Conclusions
This study systematically delved into the relationship between anodization parameters of thin copper film electrodes and their catalytic performance towards glucose oxidation. By precisely controlling the anodization potential and solution molarity, we selectively favor specific copper oxide species—Cu(OH)2, Cu2O and CuO—each exhibiting distinct catalytic activities. Notably, the formation of copper hydroxide nanowires at −0.1 V in 0.5 M KOH does not enhance the catalytic activity toward glucose, despite a substantial increase in surface area. Similarly, anodizing at −0.2 V in less than 0.75 M KOH will mostly result in a Cu2O layer, which we observed to underperform. In contrast, applying a polarization at 0 V in 1 M KOH produces a highly active CuO surface layer, leading to a remarkable sensitivity enhancement compared to bare copper electrodes. Chronoamperometry measurements show a 55% increase in the 0.1–0.5 mM range, yielding a sensitivity of 955 ± 11 μA cm−2 mM−1, and a 73% increase in the 0.75–2 mM range, with a sensitivity of 767 ± 18 μA cm−2 mM−1. Our findings provide compelling evidence supporting Barragan's model, where the semiconductive properties of CuO, rather than the Cu(II)/Cu(III) couple, drive glucose oxidation activity. These insights pave the way for more efficient (bio)sensors and energy conversion devices by integrating a simple anodization step in the fabrication process. High surface area copper-based electrodes, such as thin films on nanostructured supports, will particularly benefit from these findings. Future work will focus on validating the electrodes in real sample solutions and exploring a broader range of anodization potentials (>0 V) and KOH molarities (>1 M) to further enhance glucose oxidation efficiency.
Data availability
Electrochemical data, XPS and XRD available at the Open Science Framework database: https://osf.io/y9tb7/?view_only=9b007c883b694cada807dd1bd79b8a7e.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge funding from Internal Funds of KU Leuven (IDN/22/005). F. F. and C. F. acknowledge the support of the Research Foundation Flanders (FWO) for the PhD fellowships with number 1S61723N and 1S58823N, respectively.
References
- S. K. Das, K. K. Nayak, P. R. Krishnaswamy, V. Kumar and N. Bhat, Review—Electrochemistry and Other Emerging Technologies for Continuous Glucose Monitoring Devices, ECS Sens. Plus, 2022, 1, 031601 CrossRef CAS.
-
https://www.who.int/health-topics/diabetes
.
- H. Teymourian, A. Barfidokht and J. Wang, Electrochemical glucose sensors in diabetes management: an updated review (2010–2020), Chem. Soc. Rev., 2020, 49, 7671–7709 RSC.
- G. Wang, X. He, L. Wang, A. Gu, Y. Huang, B. Fang, B. Geng and X. Zhang, Non-enzymatic electrochemical sensing of glucose, Microchim. Acta, 2013, 180, 161–186 CrossRef CAS.
- M. Wei, Y. Qiao, H. Zhao, J. Liang, T. Li, Y. Luo, S. Lu, X. Shi, W. Lu and X. Sun, Electrochemical non-enzymatic glucose sensors: recent progress and perspectives, Chem. Commun., 2020, 56, 14553–14569 RSC.
- Y. Jing, S. J. Chang, C.-J. Chen and J.-T. Liu, Review—Glucose Monitoring Sensors: History, Principle, and Challenges, J. Electrochem. Soc., 2022, 169, 057514 CrossRef CAS.
- T. T. Aun, N. M. Salleh, U. F. M. Ali and N. S. A. Manan, Non-Enzymatic Glucose Sensors Involving Copper: An Electrochemical Perspective, Crit. Rev. Anal. Chem., 2021, 1–57 Search PubMed.
- I. Taurino, G. Sanzò, R. Antiochia, C. Tortolini, F. Mazzei, G. Favero, G. D. Micheli and S. Carrara, Recent advances in Third Generation Biosensors based on Au and Pt Nanostructured Electrodes, TrAC, Trends Anal. Chem., 2016, 79, 151–159 CrossRef CAS.
- Y. Vassilyev, O. Khazova and N. Nikolaeva, Kinetics and mechanism of glucose electrooxidation on different electrode-catalysts Part I. Adsorption and oxidation on platinum, J. Electroanal. Chem. Interfacial Electrochem., 1985, 196, 105–125 CrossRef.
- C.-L. Yang, X.-H. Zhang, G. Lan, L.-Y. Chen, M.-W. Chen, Y.-Q. Zeng and J.-Q. Jiang, Pd-based nanoporous metals for enzyme-free electrochemical glucose sensors, Chin. Chem. Lett., 2014, 25, 496–500 CrossRef CAS.
- M. Pasta, R. Ruffo, E. Falletta, C. M. Mari and C. D. Pina, Alkaline glucose oxidation on nanostructured gold electrodes, Gold Bull., 2010, 43, 57–64 CrossRef CAS.
- M. W. Hsiao, R. R. Adžić and E. B. Yeager, Electrochemical Oxidation of Glucose on Single Crystal and Polycrystalline Gold Surfaces in Phosphate Buffer, J. Electrochem. Soc., 1996, 143, 759–767 CrossRef CAS.
- V. Bagotzky, Y. Vassilyev, J. Weber and J. Pirtskhalava, Adsorption of anions on smooth platinum electrodes, J. Electroanal. Chem. Interfacial Electrochem., 1970, 27, 31–46 CrossRef.
- M. Pasta, F. L. Mantia and Y. Cui, Mechanism of glucose electrochemical oxidation on gold surface, Electrochim. Acta, 2010, 55, 5561–5568 CrossRef CAS.
- F. Franceschini and I. Taurino, Nickel-based catalysts for non-enzymatic electrochemical sensing of glucose: A review, Phys. Med., 2022, 14, 100054 CrossRef.
- H. Jia, N. Shang, Y. Feng, H. Ye, J. Zhao, H. Wang, C. Wang and Y. Zhang, Facile preparation of Ni nanoparticle embedded on mesoporous carbon nanorods for non-enzymatic glucose detection, J. Colloid Interface Sci., 2021, 583, 310–320 CrossRef CAS PubMed.
- G. A. Naikoo,
et al., Fourth-generation glucose sensors composed of copper nanostructures for diabetes management: A critical review, Bioeng. Transl. Med., 2022, 7, e10248 CrossRef CAS PubMed.
- Y. Ding, Y. Wang, L. Su, M. Bellagamba, H. Zhang and Y. Lei, Electrospun Co3O4 nanofibers for sensitive and
selective glucose detection, Biosens. Bioelectron., 2010, 26, 542–548 CrossRef CAS PubMed.
- X. Wang, L. Hao, R. Du, H. Wang, J. Dong and Y. Zhang, Synthesis of unique three-dimensional CoMn2O4@Ni(OH)2 nanocages via Kirkendall effect for non-enzymatic glucose sensing, J. Colloid Interface Sci., 2024, 653, 730–740 CrossRef CAS PubMed.
- P. Gupta, V. K. Gupta, A. Huseinov, C. E. Rahm, K. Gazica and N. T. Alvarez, Highly sensitive non-enzymatic glucose sensor based on carbon nanotube microelectrode set, Sens. Actuators, B, 2021, 348, 130688 CrossRef CAS.
- Q. Gao, W. Zhang, C. Zhao, W. Yan, S. Han, Y. Li, Q. Liu, X. Li and D. Liu, Laser-Engraved Boron-Doped Diamond for Copper Catalyzed Non-Enzymatic Glucose Sensing, Adv. Mater. Interfaces, 2022, 9, 2200034 CrossRef CAS.
- C. Li, Y. Su, S. Zhang, X. Lv, H. Xia and Y. Wang, An improved sensitivity nonenzymatic glucose biosensor based on a Cu x O modified electrode, Biosens. Bioelectron., 2010, 26, 903–907 CrossRef CAS PubMed.
- L. A. Colon, R. Dadoo and R. N. Zare, Determination of carbohydrates by capillary zone electrophoresis with amperometric detection at a copper microelectrode, Anal. Chem., 1993, 65, 476–481 CrossRef CAS.
- Y. Xie and C. O. Huber, Electrocatalysis and amperometric detection using an electrode made of copper oxide and carbon paste, Anal. Chem., 1991, 63, 1714–1719 CrossRef CAS.
- A. Singh, A. Hazarika, L. Dutta, A. Bhuyan and M. Bhuyan, A fully handwritten-on-paper copper nanoparticle ink-based electroanalytical sweat glucose biosensor fabricated using dual-step pencil and pen approach, Anal. Chim. Acta, 2022, 1227, 340257 CrossRef CAS PubMed.
- A. Jiménez-Rodríguez, E. Sotelo, L. Martínez, Y. Huttel, M. U. González, A. Mayoral, J. M. García-Martín, M. Videa and J. L. Cholula-Díaz, Green synthesis of starch-capped Cu 2 O nanocubes and their application in the direct electrochemical detection of glucose, RSC Adv., 2021, 11, 13711–13721 RSC.
- S. Yang, G. Li, D. Wang, Z. Qiao and L. Qu, Synthesis of nanoneedle-like copper oxide on N-doped reduced graphene oxide: A three-dimensional hybrid for nonenzymatic glucose sensor, Sens. Actuators, B, 2017, 238, 588–595 CrossRef CAS.
- Y. Zhang, L. Su, D. Manuzzi, H. V. E. de lo Monteros, W. Jia, D. Huo, C. Hou and Y. Lei, Ultrasensitive and selective non-enzymatic glucose detection using copper nanowires, Biosens. Bioelectron., 2012, 31, 426–432 CrossRef CAS PubMed.
- K. Yuan, Y. Zhang, S. Huang, S. Yang, S. Zhao, F. Liu, Q. Peng, Y. Zhao, G. Zhang, J. Fan and G. Zang, Copper Nanoflowers on Carbon Cloth as a Flexible Electrode Toward Both Enzymeless Electrocatalytic Glucose and H2O2, Electroanalysis, 2021, 33, 1800–1809 CrossRef CAS.
- S. K. Meher and G. R. Rao, Archetypal sandwich-structured CuO for high performance non-enzymatic sensing of glucose, Nanoscale, 2013, 5, 2089–2099 RSC.
- A. Ashok, A. Kumar and F. Tarlochan, Highly efficient nonenzymatic glucose sensors based on CuO nanoparticles, Appl. Surf. Sci., 2019, 481, 712–722 CrossRef CAS.
- X. Cao, CuO Nanowires Fabricated by Thermal Oxidation of Cu Foils towards Electrochemical Detection of Glucose, Micromachines, 2022, 13, 2010 CrossRef PubMed.
- T. S. Babu and T. Ramachandran, Development of highly sensitive non-enzymatic sensor for the selective determination of glucose and fabrication of a working model, Electrochim. Acta, 2010, 55, 1612–1618 CrossRef CAS.
- K. Dhara, J. Stanley, T. Ramachandran, B. G. Nair and T. G. S. Babu, Cupric Oxide Modified Screen Printed Electrode for the Nonenzymatic Glucose Sensing, J. Nanosci. Nanotechnol., 2016, 16, 8772–8778 CrossRef CAS.
- L. Wang, J. Fu, H. Hou and Y. Song, A facile strategy to prepare Cu2O/Cu electrode as a sensitive enzyme-free glucose sensor, Int. J. Electrochem. Sci., 2012, 7, 12587–12600 CrossRef CAS.
- H.-H. Fan, W.-L. Weng, C.-Y. Lee and C.-N. Liao, Electrochemical Cycling-Induced Spiky Cu x O/Cu Nanowire Array for Glucose Sensing, ACS Omega, 2019, 4, 12222–12229 CrossRef CAS PubMed.
- D. Giziński, A. Brudzisz, J. S. Santos, F. Trivinho-Strixino, W. J. Stępniowski and T. Czujko, Nanostructured Anodic Copper Oxides as Catalysts in Electrochemical and Photoelectrochemical Reactions, Catalysts, 2020, 10, 1338 CrossRef.
- X. Wu, H. Bai, J. Zhang, F. Chen and G. Shi, Copper Hydroxide Nanoneedle and Nanotube Arrays Fabricated by Anodization of Copper, J. Phys. Chem. B, 2005, 109, 22836–22842 CrossRef CAS PubMed.
- W. J. Stępniowski, D. Paliwoda, Z. Chen, K. Landskron and W. Z. Misiołek, Hard anodization of copper in potassium carbonate aqueous solution, Mater. Lett., 2019, 252, 182–185 CrossRef.
- N. K. Allam and C. A. Grimes, Electrochemical fabrication of complex copper oxide nanoarchitectures via copper anodization in aqueous and non-aqueous electrolytes, Mater. Lett., 2011, 65, 1949–1955 CrossRef CAS.
- X. Cui, D. A. Hutt and P. P. Conway, Evolution of microstructure and electrical conductivity of electroless copper deposits on a glass substrate, Thin Solid Films, 2012, 520, 6095–6099 CrossRef CAS.
- W. Zhang, S. Brongersma, O. Richard, B. Brijs, R. Palmans, L. Froyen and K. Maex, Influence of the electron mean free path on the resistivity of thin metal films, Microelectron. Eng., 2004, 76, 146–152 CrossRef CAS.
- E. Schmiedl, P. Wissmann and H.-U. Finzel, The Electrical Resistivity of Ultra-Thin Copper Films, Z. Naturforsch., A: Phys. Sci., 2008, 63, 739–744 CAS.
- J. Su, J. Zhang, Y. Liu, M. Jiang and L. Zhou, Parameter-dependent oxidation of physically sputtered Cu and the related fabrication of Cu-based semiconductor films with metallic resistivity, Sci. China Mater., 2016, 59, 144–150 CAS.
- R. Chandra, A. K. Chawla and P. Ayyub, Optical and Structural Properties of Sputter-Deposited Nanocrystalline Cu2O Films: Effect of Sputtering Gas, J. Nanosci. Nanotechnol., 2006, 6, 1119–1123 CrossRef CAS PubMed.
- X. Li, C. Wei, J. Fu, L. Wang, S. Chen, P. Li, H. Li and Y. Song, Electrolyte-controllable synthesis of Cu x O with novel morphology and their application in glucose sensors, RSC Adv., 2014, 4, 52067–52073 CAS.
- L. Xu, Q. Yang, X. Liu, J. Liu and X. Sun, One-dimensional copper oxide nanotube arrays: biosensors for glucose detection, RSC Adv., 2013, 4, 1449–1455 RSC.
- S. Anantharaj, H. Sugime, S. Yamaoka and S. Noda, Pushing the Limits of Rapid Anodic Growth of CuO/Cu(OH)2 Nanoneedles on Cu for the Methanol Oxidation Reaction: Anodization pH Is the Game Changer, ACS Appl. Energy Mater., 2021, 4, 899–912 CrossRef CAS.
- S. Swann, Film thickness distribution in magnetron sputtering, Vacuum, 1988, 38, 791–794 CrossRef CAS.
- H.-H. Strehblow, V. Maurice and P. Marcus, Initial and later stages of anodic oxide formation on Cu, chemical aspects, structure and electronic properties, Electrochim. Acta, 2001, 46, 3755–3766 CrossRef CAS.
- J. T. C. Barragan, S. Kogikoski, E. T. S. G. da Silva and L. T. Kubota, Insight into the Electro-Oxidation Mechanism of Glucose and Other Carbohydrates by CuO-Based Electrodes, Anal. Chem., 2018, 90, 3357–3365 CrossRef CAS PubMed.
- S. D. Giri and A. Sarkar, Electrochemical Study of Bulk and Monolayer Copper in Alkaline Solution, J. Electrochem. Soc., 2016, 163, H252–H259 CrossRef CAS.
- J. Ambrose, R. Barradas and D. Shoesmith, Investigations of copper in aqueous alkaline solutions by cyclic voltammetry, J. Electroanal. Chem. Interfacial Electrochem., 1973, 47, 47–64 CAS.
- B. Beverskog and I. Puigdomenech, Revised Pourbaix Diagrams for Copper at 25 to 300°C, J. Electrochem. Soc., 1997, 144, 3476–3483 CAS.
- S. A. e. Haleem and B. G. Ateya, Cyclic voltammetry of copper in sodium hydroxide solutions, J. Electroanal. Chem. Interfacial Electrochem., 1981, 117, 309–319 Search PubMed.
- J. Kunze, V. Maurice, L. H. Klein, H.-H. Strehblow and P. Marcus, In situ STM study of the duplex passive films formed on Cu(111) and Cu(001) in 0.1 M NaOH, Corros. Sci., 2004, 46, 245–264 CAS.
- B. Miller, Split-Ring Disk Study of the Anodic Processes at a Copper Electrode in Alkaline Solution, J. Electrochem. Soc., 1969, 116, 1675–1680 CAS.
- W. J. Stepniowski, H. Yoo, J. Choi, P. Chilimoniuk, K. Karczewski and T. Czujko, Investigation of oxide nanowires growth on copper via passivation in NaOH aqueous solution, Surf. Interfaces, 2019, 14, 15–18 CAS.
- D. W. Shoesmith, T. E. Rummery, D. Owen and W. Lee, Anodic Oxidation of Copper in Alkaline Solutions: I. Nucleation and Growth of Cupric Hydroxide Films, J. Electrochem. Soc., 1976, 123, 790–799 CAS.
- J. G. Becerra, R. Salvarezza and A. Arvia, The influence of slow Cu(OH)2 phase formation on the electrochemical behaviour of copper in alkaline solutions, Electrochim. Acta, 1988, 33, 613–621 CrossRef CAS.
- V. P. Parkhutik and V. I. Shershulsky, Theoretical modelling of porous oxide growth on aluminium, J. Phys. D: Appl. Phys., 1992, 25, 1258 CrossRef CAS.
- D. Shoesmith, S. Sunder, M. Bailey, G. Wallace and F. Stanchell, Anodic oxidation of copper in alkaline solutions Part IV. Nature of the passivating film, J. Electroanal. Chem. Interfacial Electrochem., 1983, 143, 153–165 CrossRef CAS.
- J. Otamiri, S. Andersson and A. Andersson, Ammoxidation of toluene by YBa2Cu3O6+x and copper oxides Activity and XPS studies, Appl. Catal., 1990, 65, 159–174 CrossRef CAS.
- S. Poulston, P. M. Parlett, P. Stone and M. Bowker, Surface Oxidation and Reduction of CuO and Cu2O Studied Using XPS and XAES, Surf. Interface Anal., 1996, 24, 811–820 CrossRef CAS.
- P. Salvador, J. Fierro, J. Amador, C. Cascales and I. Rasines, XPS study of the dependence on stoichiometry and interaction with water of copper and oxygen valence states in the YBa2Cu3O7-x compound, J. Solid State Chem., 1989, 81, 240–249 CrossRef CAS.
- D. Wang, A. C. Miller and M. R. Notis, XPS Study of the Oxidation Behavior of the Cu3Sn Intermetallic Compound at Low Temperatures, Surf. Interface Anal., 1996, 24, 127–132 CrossRef CAS.
- W. Kautek and J. G. Gordon, XPS Studies of Anodic Surface Films on Copper Electrodes, J. Electrochem. Soc., 1990, 137, 2672–2677 CrossRef CAS.
- M. C. Biesinger, Advanced analysis of copper X-ray photoelectron spectra, Surf. Interface Anal., 2017, 49, 1325–1334 CrossRef CAS.
-
M. Pourbaix, Atlas d’équilibres électrochimiques, Gauthier-Villars, 1963 Search PubMed.
- P. Scherrer, Nachr Ges wiss goettingen, Math. Phys., 1918, 2, 98–100 Search PubMed.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, A Practical Beginner's Guide to Cyclic Voltammetry, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
-
A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, 1980 Search PubMed.
- L. Ostervold, S. I. P. Bakovic, J. Hestekin and L. F. Greenlee, Electrochemical biomass upgrading: degradation of glucose to lactic acid on a copper(ii) electrode, RSC Adv., 2021, 11, 31208–31218 RSC.
- M. L. Fleischmann, K. A. Korinek and D. Pletcher, The kinetics and mechanism of the oxidation of amines and alcohols at oxide-covered nickel, silver, copper, and cobalt electrodes, J. Chem. Soc., Perkin Trans. 1, 1972, 1396–1403 CAS.
- S. Zhou, X. Feng, H. Shi, J. Chen, F. Zhang and W. Song, Direct growth of vertically aligned arrays of Cu(OH)2 nanotubes for the electrochemical sensing of glucose, Sens. Actuators, B, 2013, 177, 445–452 CAS.
- A Statistical Overview of Standard (IUPAC and ACS) and New Procedures for Determining the Limits of Detection and Quantification, De Gruyter, 1997.
- S. Lee, J. Y. Kim, T.-W. Lee, W.-K. Kim, B.-S. Kim, J. H. Park, J.-S. Bae, Y. C. Cho, J. Kim, M.-W. Oh, C. S. Hwang and S.-Y. Jeong, Fabrication of high-quality single-crystal Cu thin films using radio-frequency sputtering, Sci. Rep., 2014, 4, 6230 CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Catarina
Fernandes
b,
Koen
Schouteden
a,
Jon
Ustarroz
*a,
Catarina
Fernandes
b,
Koen
Schouteden
a,
Jon
Ustarroz