Maximizing methanol selectivity over the microporous FeS-1 catalyst via aqueous-phase partial oxidation of methane with H2O2†
Received
17th September 2024
, Accepted 9th January 2025
First published on 18th February 2025
Abstract
The conversion of methane to methanol faces challenges in liquid-phase systems due to lower methanol selectivity, often resulting in higher formic acid production. Previous studies have shown that iron-exchanged MFI zeolites, i.e., Fe–ZSM-5, tend to favor formic acid over methanol due to the indiscriminate decomposition of the oxidant. To address this, our study aims to identify the active sites responsible for such over-oxidation and develop methods to suppress these sites, thereby enhancing methanol selectivity. Hence, we have utilized hydrothermally synthesized microporous iron silicalite-1 (FeS-1) with an MFI structure and conducted a systematic comparison of its catalytic performance with FeZSM-5 and Fe–ZSM-5, which contain framework and extra-framework iron sites, respectively. This comparison highlights the relationship between active site distribution and methanol selectivity. Additionally, the analysis using DRUV-VIS and EPR spectroscopic techniques suggests that the yield of methanol and formic acid is found to vary monotonically with the amount of iron in framework and extra-framework sites, respectively, in the zeolitic matrix of fresh FeS-1. Therefore, selectively removing extra-framework iron and/or partially dissolving framework iron in MFI-based catalysts results in a significant reduction in formic acid yield, with only a small effect on methanol yield. Interestingly, in contrast to Fe–ZSM-5, both FeZSM-5 and FeS-1 maintain a significant amount of framework iron in the framework sites which results in a prominent enhancement in methanol selectivity (65%). Further investigation into FeS-1, FeZSM-5, and Fe–ZSM-5 underscored the importance of framework Si–O–Fe linkages in enhancing methanol selectivity.
1. Introduction
The utilization of methane, a key constituent in natural gas, for methanol production is a significant area of interest due to its underexploited potential. Research on the partial oxidation of methane has predominantly focused on two approaches, i.e., either in the gas phase at elevated temperatures or in the liquid phase at lower temperatures.1–43 Conventional methods involving high-temperature conversion to syn-gas followed by methanol synthesis are energy-intensive and inefficient. An alternative approach, direct catalytic oxidation to methanol, presents itself but faces challenges such as low methanol selectivity. In the gas-phase partial oxidation of methane over various iron-containing zeolites, such as Fe–MFI, Fe–MOR, Fe–CHA, and other similar iron–zeolites systems and metal–organic frameworks (MOFs), using different oxidants like N2O, O2, etc., studies are still being pursued for improving methanol protection strategies leading to both methanol selectivity and yield.6,7,13–23 Similarly, copper based zeolites were also explored in the gas-phase system for understanding the copper speciation, nuclearity of the active sites, and different methanol protection strategies for improving the selectivity.24–37 However, in this gas phase system, methanol productivity is lower compared to liquid phase methane oxidation.38–40 There have been investigations utilizing iron-containing zeolites, with notable studies of aqueous phase oxidation of methane with hydrogen peroxide as an oxidant conducted by Hutchings and coworkers,2,9–12 which has emerged as a promising solution. These authors have exclusively developed a method for converting methane into oxygenates using iron-modified MFI-based zeolite catalysts like FeS-1, FeZSM-5, and Fe–ZSM-5, among others. In the meantime, several other research groups3–5,42,43 have also conducted experiments using similar types of iron-containing zeolite catalysts for the oxidation of methane.
At this juncture, it is noteworthy that the hydrothermally synthesized iron silicalite-1 (FeS-1) catalyst results in a considerable enhancement in activity, achieving approximately 96% selectivity towards the partially oxygenated products, viz., methanol and formic acid.12 However, an important challenge in selectively hydroxylating methane is the prevention of over-oxidation of methanol by controlling the concentration of active oxidizer which possibly eliminates the potential impact of excessive peroxide concentration on methanol production.2,9–12 Nevertheless, based on the various in-depth characterization data, Hutchings and coworkers12 have proposed that the active site involves a μ-oxo-bridged di-iron complex at the extra-framework position (Fe–ZSM-5). In other words, the augmented presence of octahedral (extra-framework) iron favours an enhancement of catalytic efficacy. Additionally, the presence of non-framework Lewis acid sites such as FexOy is also known for decomposing the oxidant homolytically which results in selective formation of formic acid or complete oxidation to CO2.2,44–47
Indeed, selectively eliminating weakly bound trivalent iron such as extra-framework and/or non-framework species by partial dissolution or leaching or deferration will reduce over-oxidation thereby enhancing methanol selectivity. To target specific species within the silicate and/or aluminosilicate matrix, we utilized a modified method involving ammonium nitrate solution to selectively remove extra-framework and/or non-framework iron from the MFI matrix. This approach is analogous to the use of ammonium acetate for eliminating secondary phases in transition element-incorporating zeolitic matrices.47 Thus, deferration will be performed for all the catalysts under investigation, and the resulting catalysts will be subjected to in-depth characterization to assess the local environment of trivalent iron in the silicalite framework structure. Thus, in this investigation, we present a systematic study on the utilization of hydrothermally synthesized iron silicalite (FeS-1), as well as hydrothermally synthesized FeZSM-5 and ion-exchanged Fe–ZSM-5 catalysts for the liquid-phase oxidation of methane to oxygenates. A preliminary account of this work was presented elsewhere.48
2. Experimental
2.1. Starting materials
Various chemicals, viz., tetraethyl orthosilicate (TEOS ≥99.0%), fumed silica (BET surface area: 370–420 m2 g−1), tetra n-propyl ammonium hydroxide (TPAOH, 40% solution in water), iron nitrate nonahydrate (99.95%), iron(III) chloride (97%), oxalic acid dihydrate (>99%), aluminum nitrate nonahydrate (99.997% trace metals basis), ammonium nitrate and D2O (99.95%) were all purchased from Sigma Aldrich and used without further treatment. Commercial H–ZSM-5 (SiO2/Al2O3 = 30) and H–ZSM-5 (SiO2/Al2O3 = 86) were procured from Sud Chemie India. H2O2 (35% w/w in water) and NaOH (97%) were procured from Merck Chemicals and methane gas (99.99% purity) was purchased from Indo Gas Agencies, Chennai.
2.2. Fe-modified MFI-based catalysts
The isomorphous substitution of tetravalent silicon with trivalent iron in the silicalite-1 framework results in the formation of an iron silicalite-1 (FeS-1) zeolitic structure.49–52 The incorporation of Fe3+ species in such a framework generates a negative charge, which is balanced by protons (Brønsted) that exhibit an acidic strength comparable to that of H–ZSM-5. Although the Brønsted acid sites may exhibit similar strength as in H–FeS-1 and H–ZSM-5, it has been documented that the structural stability of the two frameworks differs significantly.49–54 Calcination of H-FeS-1 causes the migration of framework iron into extra-framework and/or non-framework positions, consequently, a decrease in framework iron is noticed. To validate these, we have used samples prepared by the aqueous-phase ion exchange method to get iron-exchanged ZSM-5, i.e., Fe–ZSM-5, catalysts with various iron loadings. Further, a systematic investigation was performed to understand the interplay of these different sites upon various treatments.
Synthesis of FeS-1 (FC).
The high-silica iron MFI catalysts with varying SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe2O3 ratios were synthesized by the conventional hydrothermal route as per the reported procedure with a modified iron nitrate to oxalic acid ratio.12 Accordingly, 50 mmol of TEOS was stirred vigorously at RT for one hour, followed by dropwise addition of TPAOH (16 mmol) in 12 ml water with vigorous stirring at RT. The colorless gel was subsequently stirred at 60 °C for 2 h. Thereafter, 10 ml aqueous solution of iron nitrate nonahydrate (0.15 g) and oxalic acid dihydrate (0.15 g) was added dropwise to obtain a clear gel with the molar composition: SiO2:Fe2O3:H2C2O4:TPAOH = 1:0.00362:0.013:0.312. The resulting clear gel was stirred at 60 °C for 5 h, transferred to a Teflon-lined stainless-steel autoclave and crystallized at 175 °C for 120 h. The as-synthesized material was recovered by filtration, washed with deionized water, and dried (110 °C, 16 h). The dried sample was calcined (for 8 h at 550 °C with a heating rate of 1 °C min−1) in flowing nitrogen (20 ml min−1, 5 h) and air (20 ml min−1, 3 h). The calcined H-FeS-1 or simply FeS-1 (276) sample is referred to as the fresh catalyst (FC), whereas FeS-1 samples with different molar ratios, such as 200 and 134, were prepared by adjusting the quantities of Fe(NO3)3·9H2O and oxalic acid while maintaining the remaining steps of the procedure constant.
:Fe2O3 ratios were synthesized by the conventional hydrothermal route as per the reported procedure with a modified iron nitrate to oxalic acid ratio.12 Accordingly, 50 mmol of TEOS was stirred vigorously at RT for one hour, followed by dropwise addition of TPAOH (16 mmol) in 12 ml water with vigorous stirring at RT. The colorless gel was subsequently stirred at 60 °C for 2 h. Thereafter, 10 ml aqueous solution of iron nitrate nonahydrate (0.15 g) and oxalic acid dihydrate (0.15 g) was added dropwise to obtain a clear gel with the molar composition: SiO2:Fe2O3:H2C2O4:TPAOH = 1:0.00362:0.013:0.312. The resulting clear gel was stirred at 60 °C for 5 h, transferred to a Teflon-lined stainless-steel autoclave and crystallized at 175 °C for 120 h. The as-synthesized material was recovered by filtration, washed with deionized water, and dried (110 °C, 16 h). The dried sample was calcined (for 8 h at 550 °C with a heating rate of 1 °C min−1) in flowing nitrogen (20 ml min−1, 5 h) and air (20 ml min−1, 3 h). The calcined H-FeS-1 or simply FeS-1 (276) sample is referred to as the fresh catalyst (FC), whereas FeS-1 samples with different molar ratios, such as 200 and 134, were prepared by adjusting the quantities of Fe(NO3)3·9H2O and oxalic acid while maintaining the remaining steps of the procedure constant.
Synthesis of FeS-1 (100).
We have also employed yet another procedure for the synthesis of Na–FeS-1 (100) using fumed silica as the silica source as per the procedure reported earlier.55,56 Accordingly, 1.2 g of NaOH was added to a mixture of 8 g of TPABr in 50 ml water taken in a Teflon vessel. After 0.5 h of stirring, 1.8 g of fumed silica was added slowly for complete dissolution. After 4 h, FeCl3 solution containing 0.1 g FeCl3 in 4 ml of water was added to the above mixture and pH around 11–12 was adjusted by using a few drops of conc. HCl. This mixture was further stirred for 12 h and then transferred to an autoclave for aging. The aging process was carried out at 175 °C for 8 days in a muffle furnace. The resulting sample was then washed with distilled water to remove impurities and then calcined at 400 °C for 5 h in an inert atmosphere. As before, the calcined Na–FeS-1 or simply FeS-1 (100) is referred to as the fresh catalyst (FC).
Synthesis of FeZSM-5 (FC).
The method outlined in the preceding section for preparing H-FeS-1 was appropriately adjusted to produce FeZSM-5, facilitating the simultaneous isomorphous substitution of both iron and aluminum within the MFI framework structure.12 Accordingly, a mixture of TEOS (50 mmol) and TPAOH (16 mmol) was stirred to obtain a clear gel. Thereafter, 0.40 g of aluminum nitrate nonahydrate was added to the gel, before adding the mixture of 0.15 g of iron nitrate and 0.15 g of oxalic acid at 60 °C. The resulting clear gel was stirred at 60 °C for 5 h, transferred to a Teflon-lined stainless-steel autoclave and crystallized at 175 °C for 120 h. The as-synthesized material was recovered by filtration, washed with deionized water, and dried (110 °C, 16 h). The dried sample was calcined (for 8 h at 550 °C with a heating rate of 1 °C min−1) in flowing nitrogen (20 ml min−1, 5 h) and air (20 ml min−1, 3 h). The calcined H-FeZSM-5 or simply FeZSM-5 (138) is designated as the fresh catalyst (FC), i.e., FeZSM-5 (FC).
Preparation of Fe–ZSM-5 (FC).
For comparison, we have prepared Fe–ZSM-5 using commercial H–ZSM-5 (30) with a molar ratio of SiO2:Al2O3 = 30 via an aqueous-phase ion exchange method.11,12,57 A solution of Fe(NO3)3 was prepared in 20 ml of water. Subsequently, 0.1 g of H–ZSM-5 was added to this solution, and the mixture was vigorously stirred at 85 °C for 8 h. The catalyst was then dried at 80 °C for 6 h. The dried sample was calcined at 550 °C for 3 h with a ramp rate of 5 °C min−1 in flowing nitrogen, resulting in the formation of the Fe–ZSM-5 catalyst.
2.3. Deferration of Fe-modified MFI catalysts
Synthesis of FeS-1 (DC).
To eliminate the excess trivalent iron outside the framework, aqueous phase ion exchange using an ammonium nitrate solution was employed. Approximately 0.1 g of the FeS-1 sample underwent vigorous stirring in a 50 ml aqueous solution containing 0.1 M ammonium nitrate at room temperature overnight. Following this, the solution was filtered, washed with water, and subjected to drying at 80 °C for 6 hours. The resulting material is labeled as deferrated catalysts, specifically FeS-1 (DC).
Synthesis of FeZSM-5 (DC) and Fe–ZSM-5 (DC).
The deferration process for the specified samples was conducted similarly to that outlined above, utilizing a 0.2 M and 0.05 M ammonium nitrate solution, and the resultant samples are denoted as FeZSM-5 (DC) and Fe–ZSM-5 (DC), respectively.
2.4. Catalyst characterization
All the catalysts were systematically characterized by various analytical, spectroscopic, and imaging techniques. The powder X-ray diffraction (XRD) patterns were recorded using a Bruker D8 focus advance diffractometer with Cu Kα radiation (λ = 1.5418 Å) operating at 40 kV and recorded between 5 and 70° with a 2θ step size of 0.02. The refinements of the diffraction patterns are performed using Xpert high-score software for structural data. During the refinements, the unit cell parameters are adjusted, and the background and peak shape are modeled by a 3-term polynomial and pseudo-Voigt functions, respectively. The morphology of the materials was analyzed using a Hitachi S-4800 scanning electron microscope (SEM). The textural properties of the catalysts were obtained using nitrogen adsorption–desorption isotherms measured at liquid nitrogen temperature (77 K) using a Quantachrome ASIQwin gas sorption analyzer. The samples were initially degassed at 250 °C for 8 h before the measurements. The specific surface area was calculated using the BET (Brunauer–Emmett–Teller) method, and the Dubinin–Astakhov model and t-plot model were used to calculate the micropore size distribution and external surface area of the samples.
Diffuse reflectance ultra-violet visible spectroscopy (DRUV-VIS) measurements were performed in a UV-2600 Shimadzu spectrophotometer equipped with a diffuse reflectance accessory. The spectra were deconvoluted using Origin Pro software 2023 using the Gaussian function having the bands centered at 220 nm for the symmetrical framework (FeFW-S), 246 nm for the distorted tetrahedral framework (FeFW-D), and 280 nm for the extra-framework (FeEFW). Since the absorption band beyond 400 nm is negligible, the sub-bands are obtained within the 200–400 nm region for the FeS-1 catalysts. Electron Paramagnetic Resonance (EPR) spectra were recorded using a JEOL JES FA200 (X-band) spectrometer both at room temperature (298 K) and 77 K.
The Fe content of the synthesized zeolites was estimated using ICP-MS (Perkin-Elmer NexION 300X spectrometer). Approximately 0.1 g of the Fe–MFI samples were weighed accurately and digested with 0.5 mL of concentrated hydrofluoric acid (48%) by heating at 100 °C for 30 min. After evaporating the acid, 0.25 mL each of concentrated nitric acid (68%) and conc. hydrochloric acid (37%) were added. It is then digested for another 30 min. and diluted to 5 mL with deionized water. 2 mL of boric acid (2%) was further added to neutralize the unreacted HF. It is then filtered, and the known amount is added into a 2 M nitric acid solution before analysis. Temperature-programmed desorption of ammonia (NH3-TPD) was performed in a Micrometrics Autochem II, with 150 mg samples placed in the reaction tube. After pre-treating at 300 °C for 2 h under a helium flow of 50 mL min−1, the sample was cooled to 170 °C and dosed for 30 min with 15% ammonia in helium (balance), at 50 mL min−1 to ensure complete adsorption of ammonia onto all available acid sites. TPD samples were recorded in the range of 50 to 600 °C at a ramp rate of 5.0 °C min−1, under a helium flow of 50 mL min−1. The TPD traces were initially adjusted for baseline by subtracting the TPD of the blank-from the NH3-TPD.
2.5. Catalyst testing
The catalyst activity testing for aqueous-phase oxidation of methane with H2O2 as an oxidant was analyzed under conditions used by other authors.5,12,57,58 A stainless-steel autoclave (Parr Instrument Company) containing a 50 mL Teflon liner vessel was used as a stirred batch reactor. The reactor was charged with a 10 mL solution of 0.5 M H2O2 and 25 mg of catalyst. After sealing, the reactor was charged with CH4 to a fixed pressure of 30 bar after a series of purges (5 times with CH4 at 30 bar) to remove the contaminant gases. The reactor was then heated to the reaction temperature (typically 50 °C) and stirred at 570 rpm once the desired temperature was reached. The reactions were carried out for the desired time (typically 30 minutes), after which the vessel was cooled in ice to quench the reaction and minimize the loss of the product. The resultant solution was filtered using a micro filter and analyzed by 1H NMR in a 500 MHz Bruker NMR using D2O (Sigma Aldrich, 99.95% purity) as a solvent, and 1% sodium salt of DSS as a reference. The gas-phase products were analyzed using an Agilent gas chromatograph (GC-7890A) equipped with a PORAPAK Q column, a methanizer, and a flame ionization detector (FID).
3. Results and discussion
3.1. FeS-1 (FC) catalysts
The fresh catalysts (FeS-1 (FC)) include no sodium or alkali since they were synthesized without alkali in the starting gel. Fig. 1 depicts the powder XRD patterns of FeS-1 (FC) samples with varying SiO2/Fe2O3 ratios. The samples have shown characteristic reflections typical of MFI topology with good crystallinity53,54 and the Rietveld refined parameters for the orthorhombic unit cell parameters are listed in Table 1. It can be seen from this table that, as expected, the unit cell parameters increase linearly with iron content (Fig. S1†) in the silicalite matrix.49,50 The observed increase in the unit cell dimension could be attributed to the isomorphous substitution of larger cations such as trivalent iron (rFe3+ = 0.63 Å) in the tetravalent silicon (rSi4+ = 0.40 Å) framework structure.59 Likewise, a similar trend was also observed for FeZSM-5, and the same is reflected in the lattice constants due to the simultaneous presence of both trivalent iron and trivalent aluminum (rAl3+ = 0.53 Å) in the S-1 framework (see also Fig. S2† and Table 1). Indeed, a similar unit cell expansion was also observed for the heteroatom-incorporating microporous MFI-based60 as well as mesoporous MCM-41-based56 structures leading to a noticeable change in the unit cell volume owing to the distinctive differences in crystal radii of the constituent ions59 and/or in the bond lengths of Al–O (1.74 Å), Si–O (1.63 Å), and Fe–O (1.85 Å).58 Therefore, the increase in the dimensions of the unit cell unequivocally suggests the isomorphous substitution of trivalent iron into the silicalite matrix, and the complementary data are presented in Fig. S1.† Further, as shown in Table 1, the ICP-OES analyses reveal that the experimentally achieved iron content in Sil–Fe synthesized zeolites closely matches the theoretically expected values and spectroscopically deduced values.
 |
| | Fig. 1 Powder XRD patterns of FeS-1 (FC): (a) S-1 (∞), (b) FeS-1 (276), (c) FeS-1 (200) and (d) FeS-1 (134). | |
Table 1 Structural, textural, and compositional properties of various MFI-type materials
| Catalysta |
Iron content (wt%) |
Unit cell parameters |
S
BET (m2 g−1) |
V
P (cm3 g−1) |
| Nominal composition |
ICP-MS |
DRUV-VIS |
| FeFWb |
FeEFW |
FeNFW |
a (Å) |
b (Å) |
c (Å) |
V (Å3) |
| The following information is important to note: Number in parentheses indicates the nominal SiO2/Fe2O3 ratio, [SiO2 + Al2O3]/Fe2O3 or SiO2/Al2O3 ratio. The trivalent iron can either be ion-exchanged or isomorphously substituted. The framework trivalent iron content can be calculated as: (FeICP-MS − FeEFW). Ion-exchanged trivalent iron. Isomorphously substituted trivalent iron. Prepared under different synthesis conditions – for details see the experimental section; additionally, the sample contains non-framework Fe2O3. |
| S-1 (∞) |
— |
— |
— |
— |
— |
20.1031 |
19.8955 |
13.3815 |
5352.10 |
273 |
0.078 |
| H–ZSM-5 (30) |
— |
— |
— |
— |
— |
20.0986 |
19.9121 |
13.3952 |
5360.82 |
285 |
0.085 |
| Fe–ZSM-5 (30)c,e |
1.50 |
— |
0.16 |
0.30 |
|
20.0554 |
19.9732 |
13.4141 |
5373.29 |
240 |
0.075 |
| FeZSM-5 (138)d,e |
0.75 |
0.69 |
0.34 |
0.21 |
0.14 |
20.1246 |
19.9324 |
13.4182 |
5382.37 |
395 |
0.139 |
| FeS-1 (276)d |
0.50 |
0.47 |
0.28 |
0.12 |
0.07 |
20.1101 |
19.9179 |
13.3999 |
5367.34 |
435 |
0.183 |
| FeS-1 (200)d |
0.75 |
0.62 |
0.41 |
0.15 |
0.06 |
20.1065 |
19.9581 |
13.4044 |
5379.04 |
439 |
0.184 |
| FeS-1 (134)d |
1.00 |
0.75 |
0.53 |
0.17 |
0.05 |
20.1051 |
19.9795 |
13.4485 |
5402.12 |
406 |
0.170 |
| FeS-1 (100)d,e |
1.75 |
1.60 |
0.51 |
0.22 |
0.87 |
20.1179 |
19.9592 |
13.4174 |
5387.58 |
377 |
0.154 |
The morphology and the crystal size of the various samples were elucidated by SEM and the images are displayed in Fig. 2. It can be from this figure that the SEM images of FeS-1 (FC) catalysts with different SiO2/Fe2O3 ratios exhibit a characteristic coffin-shaped morphology with a crystal size of about 50 × 20 μm (Fig. 2a–c) which is a typical characteristic of MFI framework structures. Furthermore, the morphology and crystal size of the zeolites remain consistent across various SiO2/Fe2O3 ratios, suggesting that these characteristics are unaffected by changes in the zeolite framework. On the other hand, the catalyst FeS-1 (100) synthesized using different starting materials also exhibited the coffin-type ellipsoid/spherical morphology having serrated outgrowths with smaller dimensions of about 2 μm (Fig. 2d). However, owing to the Na-form of FeS-1 and the presence of iron oxide clusters/oligomers, a detailed investigation of this material is not considered here, however, the key results are given in the ESI† for completeness. Fig. S3† depicts the N2 absorption–desorption isotherms of the various samples, which show representative type-I isotherms with a sharp rise at a relative pressure (p/p0) < 1, indicating the characteristic zeolitic microporosity. The typical BET surface area of the materials is around 400 m2 g−1 with a micropore volume of about 0.020 cm3 g−1. The textural properties of all the various materials are presented in Table 1.
 |
| | Fig. 2 SEM images of FeS-1 (FC) with varied SiO2/Fe2O3 ratios: (a) 276; (b) 200; (c) 134; (d) 100. | |
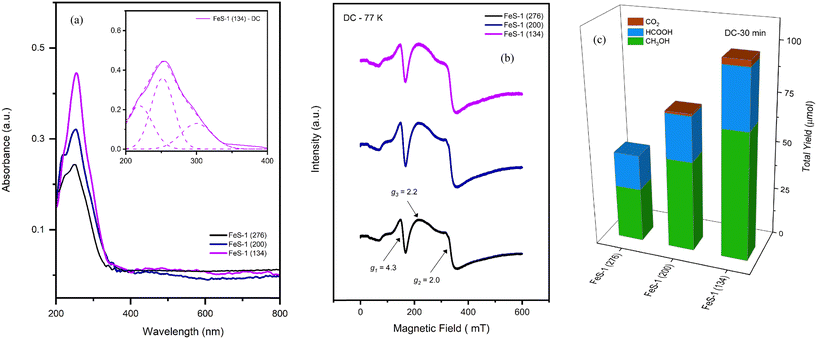
The DRUV-VIS method is valuable for examining the electronic configuration of isolated transition metal ions in the silicate matrix. Fig. 3(a) displays the DRUV-VIS spectra of FeS-1 (FC) catalysts. It can be seen from this figure that the intense band featured at 220 nm and 250 nm could be attributed to the presence of two types of framework sites, viz., symmetrical and distorted framework Fe3+ in the tetrahedral environment in a silicate matrix.58,61,62 The distinct bands observed in the UV region, particularly the strong band around 250 nm accompanied by a shoulder at 220 nm, are attributed to Laporte-allowed ligand-to-metal charge-transfer transitions involving trivalent iron in tetrahedral [FeO4]− geometry. It can also be noticed from Fig. 3(a)-inset and S4† that the deconvoluted spectral bands corresponding to various species at different sites within the silicate matrix are centered at 220 nm for the symmetrical framework (FeFW-S), 246 nm for the distorted tetrahedral framework (FeFW-D), and 280 nm for the extra-framework (FeEFW).61,62 Since the absorption band beyond 400 nm is negligible, the sub-bands are obtained within the 200–400 nm region for the FeS-1 catalysts. At this juncture, it is interesting to note that, unlike many aluminosilicate matrices, the incorporation of trivalent iron in silicate framework structures does not display any significant absorption beyond 350 nm, indicating the absence of isolated nanoclusters and/or non-framework octahedral Fe3+ nanoclusters such as iron hydroxide, iron oxy-hydroxide, or iron oxide. However, the weak shoulder containing a charge transfer band that appears at around 300 nm (see Fig. 3(a) inset) could be attributed to the formation of extra-framework Fe3+ ([Fe(OH)2]+)56 and/or distorted framework Fe3+ ([FeO4]−)55,56 in the silicate matrix. On the other hand, the variation in the band intensities for the different samples is ascribed to an increase in iron content indicating the increased formation of framework Fe sites in the silicate matrix. In contrast, it can be seen from Fig. S5† that the DRUV-VIS spectrum of FeS-1 (100) exhibits a broad transition in the range of 350–700 nm. This broadening of spectra with increasing iron concentration implies the possible presence of non-framework octahedral iron species, consistent with the existing literature.56,58,61–65
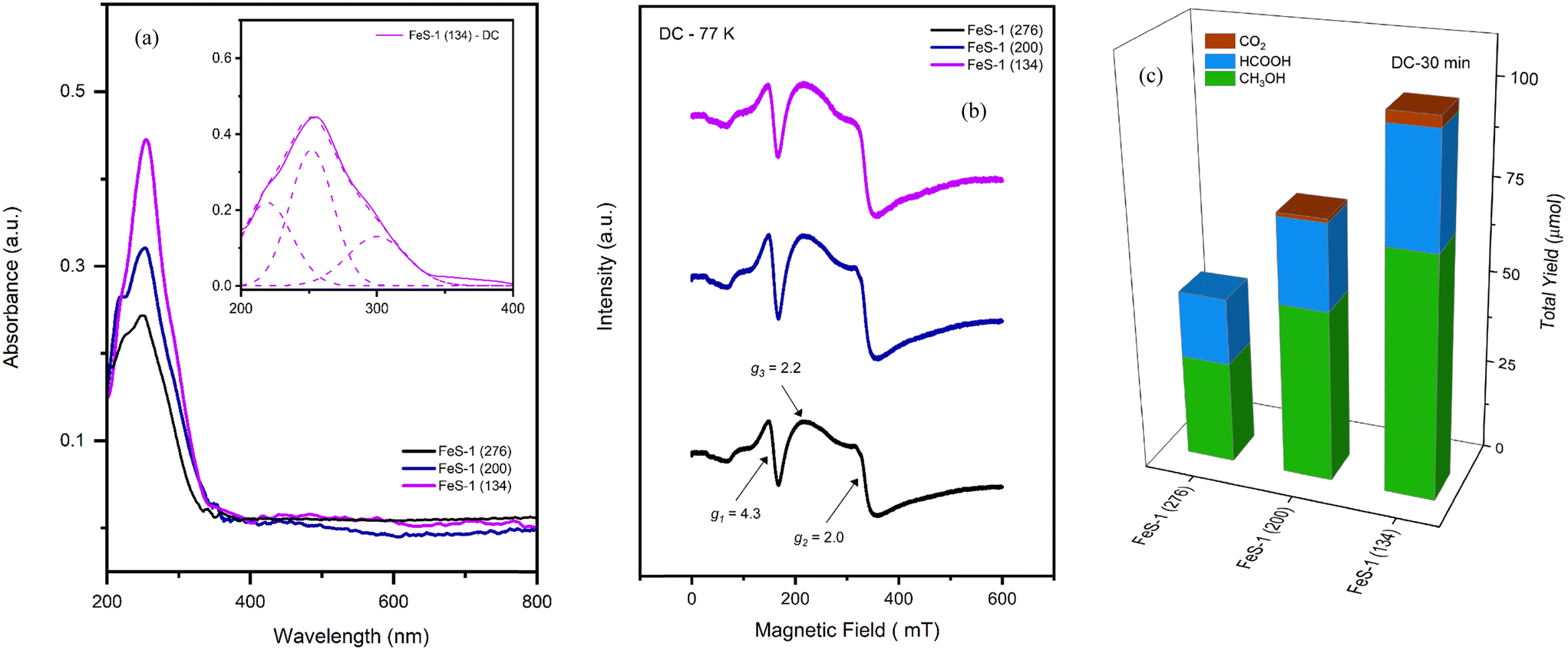
 |
| | Fig. 3 (a) DRUV-vis spectra of FeS-1 (FC). (b) EPR spectra of fresh FeS-1 (FC). (c) Catalytic activity of FeS-1 (FC). Reaction conditions: CH4 pressure 30 bar, 10 ml of 0.5 M H2O2 (aq), 25 mg catalyst, 570 rpm, and 50 °C. The inset in Fig. 3(a) is the deconvoluted DRUV-VIS spectrum which indicate the presence of various trivalent iron species in the microporous iron silicalite-1 structure. | |
Fig. 3(b) depicts the X-band EPR spectra of various FeS-1 (FS) catalysts recorded at 77 K which portray an isotropic narrow signal at g1 = 4.31 and a broad signal at g2 = 1.99–2.01. The former (g1) corresponds to the isolated tetrahedral (high-spin) trivalent iron strongly rhombic distorted in the framework position of the zeolitic matrix (D ≫ hν, E/D = 1/3) and the latter (g2) indicates high symmetry sites that can arise from both an isolated trivalent iron present in the symmetrical framework and/or the extra-framework sites (D ≫ hν, E/D = 0).51,64 Further, it is interesting to note that the g1-signal intensity sharply increased at liquid nitrogen temperature (Curie–Weiss behavior) compared to the corresponding room temperature (RT) spectra (Fig. S6†) suggesting that the isormorphously substituted ions are in the distorted tetrahedral environment,55 and that it is possible only for isolated paramagnetic species.64 On the other hand, the weak signal around g3 = 2.3 suggests the formation of iron oxide nanoclusters upon heat treatment. Similar dislodgement of framework iron species was also observed for mesoporous FeHMA and FeMCM-41 samples upon calcination.55,56 Thus, the examination of the EPR signal revealed the presence of trivalent iron occupying two distinct tetrahedral sites in addition to octahedral coordination within the structure. When the template is removed by calcination (cf. Fig. S5†), some of the tetrahedral Fe3+ is displaced from the framework, particularly for the higher SiO2/Fe2O3 ratio sample, viz., FeS-1 (100), and relocates to octahedral sites both within (extra-framework) and outside (non-framework) the matrix.55,56 In contrast, the spectra of FeS-1 with higher SiO2/Fe2O3 ratios (134–276) are intact and change insignificantly. This suggests that the Fe3+ ions in these samples did not dislodge from the matrix even after calcination or indicates the absence of non-framework iron oxide clusters/oligomers. These findings are well aligned with the results of DRUV-VIS studies.
Fig. 3(c) highlights the catalytic performance of various FeS-1 (FC) catalysts as a function of the SiO2/Fe2O3 ratio. As can be seen from this figure, the catalyst FeS-1 (276) shows the lowest activity due to low acidity, while catalysts FeS-1 (200) and FeS-1 (134) show higher activity. The total yield, which includes the oxygenates as well as CO2, increased from 86 to 294 μmol with an increase in iron content in the silicate matrix. Although the amount of methanol increased, its selectivity dropped from 30 to 20% as the formic acid selectivity increased owing to an increase in the extra-framework sites resulting in over-oxidation.4,66 However, the anticipated undesired product CO2 could be averted by the absence of non-framework iron oxide in these catalysts which are known to over-oxidize the oxygenates.67 Control experiments, including those conducted without a catalyst (blank experiments) and reactions carried out with silicalite-1, showed no catalytic activity. This suggests that the presence of hetero-ions such as trivalent iron is responsible for the observed activity. Owing to the observed higher intrinsic activity in FeS-1 samples with varying SiO2/Fe2O3 ratios (cf.Fig. 3(c)), we chose these samples to investigate their acid strength through NH3-TPD analysis as well as to determine the acid density. However, a significant limitation of this method is that the adsorption of ammonia is not exclusive to Brønsted acid sites. The acidity level played a crucial role in activating hydrogen peroxide during the partial oxidation of methane under mild conditions. The rate of formic acid production and the overall formation of oxygenated compounds were directly linked to the total acidity. Conversely, samples with lower acidity demonstrated significantly reduced production of oxygenates and higher methanol selectivity which is in line with the literature.5
Considering this, the NH3 desorption profiles depicted in Fig. 4 cannot be straightforwardly used to conclusively attribute the desorbed ammonia solely to Brønsted or Lewis acid sites. Nonetheless, they also offer crucial insights into the general characteristics and differences of acid sites such as weak or strong in zeolite structures, along with quantifying the total acid site density. As depicted in Fig. 4, all the profiles exhibit two peaks in agreement with the literature, the one centered within 180–230 °C and the other between 290 and 320 °C, attributed to weak and strong acid sites, respectively.5,54 Table S1† shows that the quantity of acidic sites is determined by the ammonia desorption area. With an increase in the SiO2/Fe2O3 ratio, there is a decrease in the total acid quantity, as well as in the amounts of strong and weak acids. This decline in acidity stems from the decrease in the iron content within the framework as the SiO2/Fe2O3 ratio rises, leading to a decrease in Si–O(H)–Fe bonds. Furthermore, as the SiO2/Fe2O3 ratio increases, both the low-temperature (LT) and high-temperature (HT) peaks tend to shift towards lower temperatures. This shift may be attributed to the decrease in iron content in the sample, thereby weakening its acidity. The FeS-1 (276) sample demonstrates significantly lower peak intensities, indicating fewer and weaker acidic sites (85 μmol g−1), whereas FeS-1 (200) and FeS-1 (134) display higher acidity levels, namely 111 and 181 μmol g−1, respectively. Studies have indicated that the acidity of FeS-1 closely resembles that of ZSM-5 and that the identification of the low-temperature (LT) peak is still a topic of discussion, with some attributing it to weak Lewis acid sites primarily originating from extra-framework metal species, while others associate it with weak Brønsted acid sites formed by bridged hydroxyl groups, Si–O(H)–Fe, i.e., the distorted octahedral sites.54,56 However, it is interesting to note that such sites are directly responsible for the exclusive formation of formic acid and it correlates well with the data presented in Table 2.
 |
| | Fig. 4 NH3-TPD profiles of FeS-1 (FS) catalysts with different iron contents. | |
Table 2 Methane to methanol conversion data for various MFI-type materials
| Catalyst |
Total Fe contenta (wt%) |
Oxygenate yieldb (μmol) |
CH3OH selectivityd (%) |
H2O2 conversion (%) |
Ref. |
| ICP-MS |
CH3OH |
HCOOH |
CO2 |
Total |
| Reaction conditions: catalyst amount = 25 mg; [H2O2] = 0.5 M; T = 50 °C; t = 30 min; 570 rpm. ICP-MS data. Oxygenate yield is quantified using 1H-NMR spectroscopy with D2O as a reference and CO2 using a GC-FID coupled with a methaniser. Fresh catalyst (FC). SCH3OH = YCH3OH/(YCH3OH + YHCOOH + YCO2). Deferrated catalyst (DC). Recycled catalyst (RC). Trivalent iron (Fe3+)-exchanged samples. Non-templated ZSM-5. The following code, viz., FeZSM-5 or (Fe,Al)S-1, is interchangeably used in the literature. |
| FeS-1 (134)c |
0.75 |
61 |
204 |
29 |
294 |
21 |
17 |
This work |
| FeS-1 (134)e |
0.64 |
66 |
30 |
5 |
101 |
65 |
13 |
This work |
| FeS-1 (134)f |
0.62 |
60 |
28 |
6 |
94 |
64 |
11 |
This work |
| FeS-1 (200)c |
0.62 |
40 |
114 |
17 |
171 |
23 |
12 |
This work |
| FeS-1 (200)e |
0.53 |
43 |
23 |
2 |
68 |
63 |
8 |
This work |
| FeS-1 (276)c |
0.47 |
25 |
51 |
10 |
86 |
29 |
8 |
This work |
| FeS-1 (276)e |
0.37 |
28 |
18 |
0 |
46 |
61 |
5 |
This work |
| FeZSM-5 (80)c |
0.69 |
97 |
208 |
17 |
322 |
30 |
24 |
This work |
| FeZSM-5 (80)e |
0.47 |
80 |
92 |
15 |
187 |
43 |
11 |
This work |
| Fe–ZSM-5 (30)c |
0.86 |
39 |
101 |
23 |
163 |
24 |
58 |
This work |
| Fe–ZSM-5 (30)e,g |
0.65 |
41 |
80 |
27 |
148 |
28 |
46 |
This work |
| FeS-1 (276) |
0.50 |
15 |
61 |
6 |
82 |
18 |
— |
5
|
| FeS-1 (254) |
0.52 |
18 |
56 |
11 |
85 |
19 |
— |
6
|
| FeZSM-5 (84) |
0.49 |
21 |
158 |
16 |
197 |
11 |
— |
6
|
| Fe–ZSM-5 (70)g |
0.40 |
17 |
182 |
16 |
215 |
8 |
— |
20
|
| Fe–ZSM-5 (35)g,h |
0.40 |
34 |
595 |
10 |
640 |
5 |
— |
20
|
| (Fe,Al)MFI (100)i |
1.10 |
43 |
530 |
40 |
613 |
7 |
78 |
4
|
| (Fe,Ga)MFI (50)i |
1.63 |
36 |
648 |
25 |
709 |
5 |
80 |
4
|
Several reports in the literature emphasized the role of FeEFW in the ion-exchanged Fe–ZSM-5,2,4,5,57,58 though the importance of FeFW sites for enhancing the methanol selectivity is yet to be explored. Following the work of Nikolopoulos et al.,62 we have calculated the amount of FeFW and FeEFW by deconvolution of the DRUV-VIS spectra (Fig. S4†), and the results are listed in Table 1. Fig. 5 illustrates a linear correlation between the yields of methanol and formic acid with different Fe species in the framework (FeFW) and extra-framework (FeEFW) positions, respectively. It can be seen from this figure that the amount of FeEFW correlates linearly with the formic acid yield. Further, such a correlation between FeEFW and formic acid yield and the proportionality between FeFW and methanol yield indicates the need to further investigate the importance of manipulating the amount of FeFW and FeEFW to vary methanol selectivity. Thus, the trade-off between the total oxygenate yield and methanol selectivity can be potentially decided by a balance between these sites.4,5,10,66 Likewise, the area of the EPR signal intensity (Fig. 3(b)) using the peak-to-peak method was also calculated using the approximate relative intensities obtained from the expression, Iret = A × ΔB2 where A is the peak-to-peak amplitude and ΔB is the peak-to-peak width, and the corresponding values are given in Table S2.† These values are used to correlate the amount of methanol and formic acid with these two types of iron sites, and it is shown in Fig. 6. The correlation between framework iron and methanol motivates us to selectively remove extra-framework iron sites by deferration which will be discussed in the following section.
 |
| | Fig. 5 A correlation between oxygenate yield and Fe content deduced from DRUV-VIS spectral data (see Table 1). | |
 |
| | Fig. 6 A correlation between oxygenate yield and EPR data (see Table S1†). | |
3.2. FeS-1 (DC) catalysts
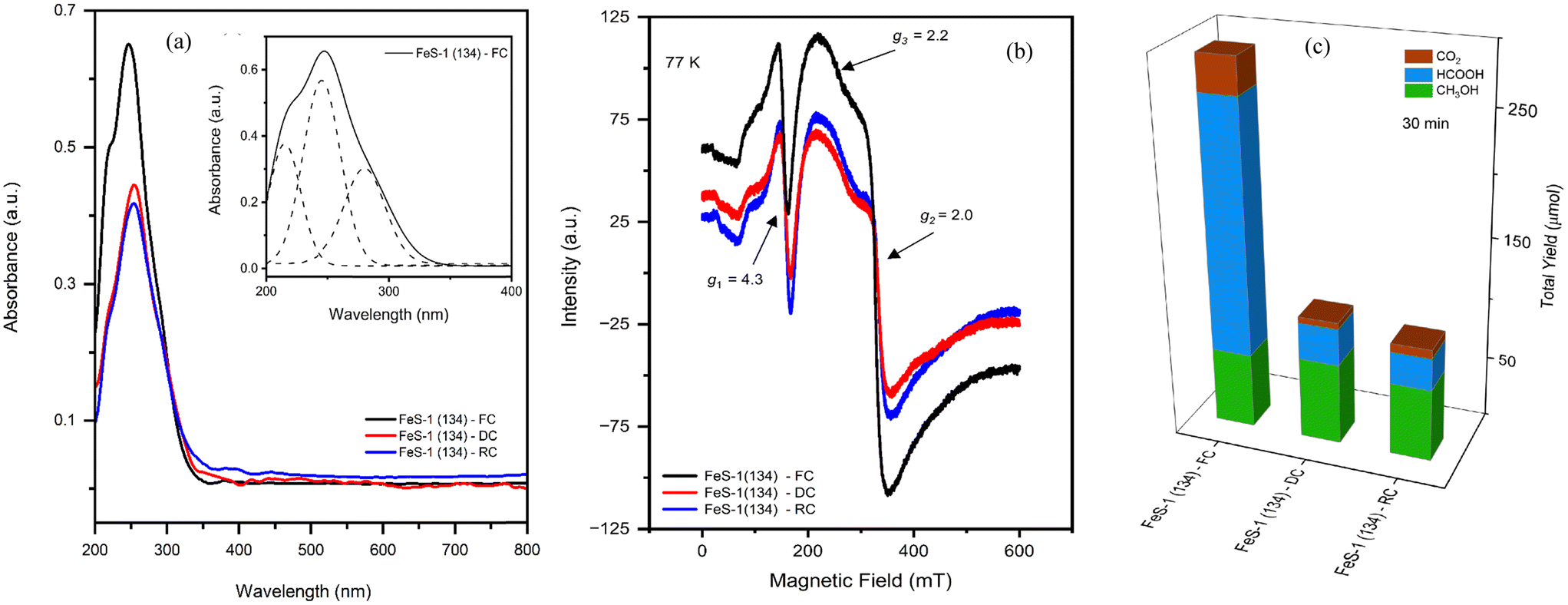
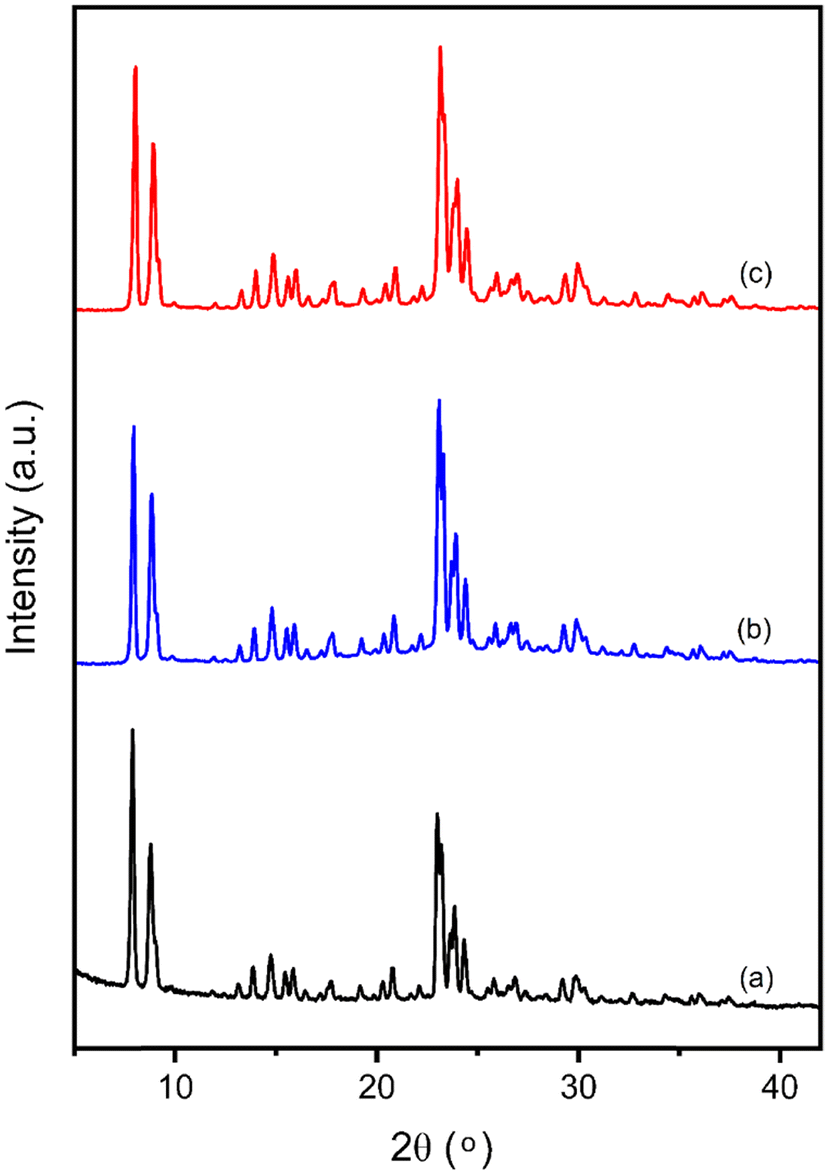
Fig. 7 presents the powder XRD patterns of FeS-1 (DC) samples across different SiO2/Fe2O3 ratios. These samples exhibit characteristic reflections indicative of MFI topology, displaying good crystallinity. It is worth noting here that the diffraction patterns remain nearly the same even after the deferration experiment or washing treatment indicating the intactness of the structure (see Fig. 1) and this is further confirmed by the HR-SEM images (Fig. 8) which show no visible change in the morphology compared to the FeS-1 (FC) samples (cf.Fig. 2). Fig. 9(a) and 10 depict the DRUV-VIS spectra of the deferrated FeS-1 catalysts. It can be seen from this figure that the absorption band that corresponds to FeEFW species decreases significantly, whereas a relatively minor decrease is observed for the corresponding FeFW. A similar observation was also made from EPR, see Fig. 9(b) and 10 where, as expected, a noticeable decrease in signal intensity for g′ = 1.99 could be attributed to the partial removal of extra-framework Fe species after deferration.51 The loss of signal intensity of the extra-framework Fe species aligns well with the observation of reduced intensity in the absorption band of the extra-framework Fe region in the DRUV-VIS spectra. Fig. 9(c) displays the catalytic performance of the deferrated FeS-1 catalysts. This histogram reveals a notable decrease in formic acid yield, while the methanol yield remains relatively stable or shows a slight increase. Thus, deferrated FeS-1 catalysts exhibit a significant increase in methanol selectivity by approximately 60% without a significant decline in methanol yield (Table 2), with a marked decrease in CO2 yield.
 |
| | Fig. 7 XRD patterns of various FeS-1 (DC) catalysts: (a) 276; (b) 200; (c) 134. | |
 |
| | Fig. 8 SEM images of various FeS-1 (DC) catalysts: (a) 276; (b) 200; (c) 134. | |
 |
| | Fig. 9 (a) DRUV-vis spectra of FeS-1 (DC). (b) EPR spectra of FeS-1 (DC). (c) Catalytic activity of FeS-1 (DC). Reaction conditions: CH4 pressure 30 bar, 10 ml of 0.5 M H2O2 (aq), 25 mg catalyst, 570 rpm, and 50 °C. The inset in Fig. 9(a) is the deconvoluted DRUV-VIS spectrum which indicate the presence of various trivalent iron species in the microporous iron silicalite-1 structure. | |
 |
| | Fig. 10 EPR spectra of fresh FeS-1 (FC) and deferrated FeS-1 (DC) at 77 K, with (a) FeS-1 (276), (b) FeS-1 (200), (c) FeS-1 (134) and (d) FeS-1 (100). The fresh catalysts were treated with 0.1 M NH4NO3 solution to obtain the corresponding deferrated catalysts. | |
Fig. 11 illustrate the correlation between the formation of oxygenates and the role of framework and extra-framework iron in the zeolitic matrix, obtained through the deconvoluted DRUV-VIS (Fig. 11(a)) and EPR (Fig. 11(b)) spectral data (Tables 1 and S3†). Further, unlike the fresh catalysts, it can be seen from these figures that the deferrated catalysts show a decrease in the slope of FeEFW indicating the removal of extra-framework iron and/or the symmetrical framework iron from the FeS-1 matrix, which therefore results in the selective formation of methanol. In this context, it is interesting to note that the loss of formic acid yield is proportional to the loss of extra-framework iron species, as implied by the relationship between the loss of formic acid yield (Fig. 11(c)) on deferration versus the difference in extra-framework iron species using both DRUV-VIS and EPR spectral data (Tables S3 and S4†). For all three catalysts, only a minor loss in methanol yield is observed which correlates with both DRUV-VIS and EPR spectral data for framework Fe, i.e., deferration results in the removal of extra-framework Fe without a loss of framework Fe sites. This is well supported by both XRD (see Fig. 7) and SEM (see Fig. 8) images of the deferrated FeS-1 catalysts. In other words, the catalysts that have undergone deferration maintain their original structure closely, albeit with some leached iron present outside the framework. This leads to a notable reduction in formic acid production, while the methanol output remains relatively unaffected.
 |
| | Fig. 11 (a) Correlation between oxygenate yield and Fe content was deduced from DRUV-VIS spectral data. (b) Correlation between oxygenate yield and EPR data. (c) Correlation between formic acid loss and extra-framework iron content deduced from DRUV-VIS and EPR spectral data. | |
3.3. FeZSM-5 and Fe–ZSM-5 catalysts
Fresh and deferrated catalysts.
Earlier, it was demonstrated that the deferrated FeS-1 catalyst exhibited a decreased amount of extra-framework Fe sites that led to the selective formation of methanol as a major product. Since FeS-1 catalysts contain ion-exchange sites only through framework Fe, the deferration was also tested on the FeZSM-5 matrix that has two different types of ion-exchanging sites (framework Fe and Al). The correlation established for FeS-1 (Fig. 11) is also observed in Fe–ZSM-5 (Fig. S10b†). Further, the commercial ion-exchanged catalyst, Fe–ZSM-5, was kept for comparison as it is known to also contain non-framework Fe-oxides sites. Fig. 12 shows the DRUV-VIS spectra of the fresh and deferrated FeZSM-5 and Fe–ZSM-5 catalysts. The spectra exhibit characteristic absorption bands corresponding to the FeFW sites with an additional broadening extending to higher wavelengths in comparison to the FeS-1 catalysts. The individual trivalent iron entities found within the FeZSM-5 matrix, arranged in tetrahedral coordination, may transform either additional active FeEFW species located within the pores and/or form FeNFW FexOy clusters. Consistent with the existing literature,68 the trivalent iron present in FeZSM-5 is incorporated into the ZSM-5 framework via isomorphic substitution, thereby enhancing Brønsted acidity. These iron ions are also attached to cationic exchange sites located within the pores of the zeolite. On the other hand, the non-framework iron sites are predominantly prevalent in Fe–ZSM-5 catalysts, and it consists of a wider range of Fe-species including extra-framework Fe at exchange sites, oligomeric FexOy clusters, and Fe2O3 nanoparticles at the non-framework sites. This is well supported by both XRD (Fig. S1 and S2†), SEM (Fig. 13), and EPR data (Fig. 14(a)).
 |
| | Fig. 12 DRUV-VIS spectra of: (a) fresh and (b) deferrated Fe/MFI catalysts. The insets are the deconvoluted DRUV-VIS spectra which indicate the presence of various trivalent iron species in the ZSM-5 and S-1 matrices. | |
 |
| | Fig. 13 SEM images of: (a) FeZSM-5 (FC); (b) FeZSM-5 (DC); (c) Fe–ZSM-5 (FC); (d) Fe–ZSM-5 (FC). | |
 |
| | Fig. 14 (a) EPR spectra of fresh and deferrated FeZSM-5 (isomorphous Fe) and Fe–ZSM-5 (ion-exchanged Fe) at 77 K. (b) Catalytic activity of Fe–ZSM-5 and FeZSM-5 (138). Reaction conditions: CH4 pressure 30 bar, 10 ml of 0.5 M H2O2 (aq), 25 mg catalyst, 570 rpm, and 50 °C. | |
The catalytic activity of the fresh and deferrated FeZSM-5 (138) catalysts for the same reaction conditions is shown in Fig. 14(b). The fresh FeZSM-5 (138) exhibited a higher total oxygenate yield than the corresponding FeS-1 (134) catalyst due to the introduction of Al3+ in the gel. However, after the deferration, both methanol and formic acid yields dropped, although the drop in formic acid yield was very significant. As in FeS-1 catalysts, the methanol selectivity is higher in deferrated FeZSM-5, though it was accompanied by a loss in methanol yield. Since the extra-framework Fe sites are usually higher in FeZSM-5 (138) – (FC) compared to FeS-1 (134) – FC under the identical iron content, deferration with higher concentration around 0.2 M ammonium nitrate solution retained a similar methanol selectivity of 65%. The catalytic performance of the various iron loadings of Fe–ZSM-5 is tabulated in Table S4† for comparison. This reflects the different amounts of extra-framework Fe sites and also potentially the removal of additional extra-framework Lewis acid sites such as Fe–O–Al sites that are known for the unselective decomposition of peroxide.44–47 The presence of both Al3+ and Fe3+ in the framework therefore brings out the further complexities in the catalyst design due to the formation of extra-framework Lewis acid sites that are known for the homolytic decomposition of peroxide. Consistent with the literature, we also report that the iron-exchanged samples (Fe–ZSM-5) with different loading were also prepared using iron chloride as the iron source. The preliminary reaction results of these various catalysts are given in Fig. S8† where the oxygenate yield goes through a maximum with iron content for Fe–ZSM-5 (30) and Fe–ZSM-5 (86). This observation is in line with the DRUV-VIS data wherein the spectra broaden with an increase in iron content in the samples (Fig. S9†).
To the best of our knowledge, the methanol selectivity of 68% obtained with deferrated FeS-1 (134) is the only instance of an Fe-based catalyst that yields more than twice as much methanol as formic acid, without the usage of secondary metal as a promoter which is also depicted in Table 2.9,12,67,69 Hence, we further investigated the activity of deferrated FeS-1 by doubling the reaction time to 60 min in Fig. 15. It was interesting to observe that a negligible amount of CO2 was detected, the yield of both methanol and formic acid increased, and methanol selectivity remained the same as that with 30 min reaction time for the deferrated catalysts. The methanol yield for deferrated FeS-1 (134), for example, was around 140 μmol with a selectivity of 65% and no CO2 formation. This also suggests the importance of the deferration process on removing the unselective sites which could be potentially the extra-framework Lewis acid sites, for improving the methanol selectivity. This forms a crucial part of catalyst design for effective H2O2 utilization for the formation of methanol from methane.
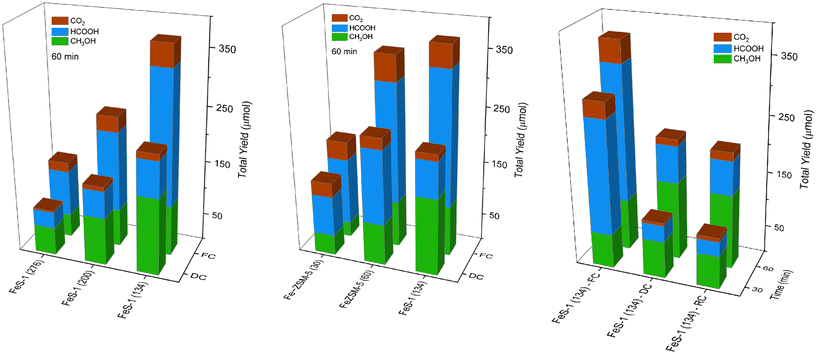 |
| | Fig. 15 The catalytic activity of FeZSM-5 and FeS-1 samples in a batch reactor at 60 min. Reaction conditions: CH4 pressure 30 bar, 10 ml of 0.5 M H2O2 (aq), 25 mg catalyst, 570 rpm, and 50 °C. | |
Previous literature indicates that, under similar conditions, the reaction proceeds in the intrinsic kinetic regime without notable diffusion limitations. As shown in Fig. S11a,† the yield of oxygenates is directly proportional to catalyst mass, consistent with previous studies.11 Additionally, under our specific reaction conditions, methane—due to its limited solubility in water—serves as the limiting reagent, with an approximate [CH4]:[H2O2] ratio of 1:30, estimated using Henry's law constant and assuming equilibrium between methane in the liquid and gas phases. Our reaction conditions match these established parameters.5,11,57,67 The reaction is considered to be in a differential regime for methane, as its low conversion and limited solubility ensure negligible changes in its concentration during the reaction. At these methane pressures and excess oxidant concentration, the system operates under the kinetically limited regime for methane, and we consider it reasonable to assume first-order kinetics for H2O2. Moreover, as illustrated in Fig. S11(b),† there is minimal variation in the product distribution pattern within the range of stirring speeds used in this study. Similar behaviour has also been reported in the literature, even at significantly higher stirring speeds (approximately 1500 rpm), suggesting that the process is independent of stirring speed.9,10
3.4. Recycling studies
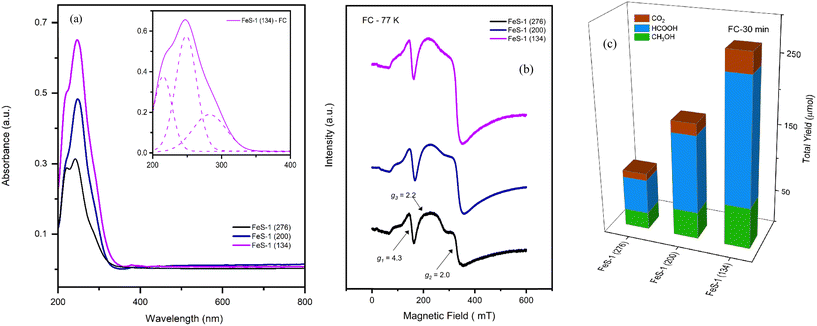
Finally, Fig. 16(c) compares the activity of the fresh/deferrated/recycled FeS-1 (134) catalysts. Specifically, the deferrated FeS-1 (134) catalyst was filtered after the reaction and reused once again under the same reaction conditions. It can be seen from this figure that for the 30 min and 60 min experiments the activity of the catalyst is retained. This observation is well supported by the DRUV-VIS (Fig. 16(a)), EPR (Fig. 16(b)), XRD (Fig. 17), and SEM (Fig. 18) investigation which confirms that the crystallinity and morphology of the original batch, FeS-1 (DC), were retained. Further, the ICP analysis indicates the amount of iron-ions in the reaction filtrate being less than 10 ppm. This confirms that the active species, iron, remains intact in the solid catalyst.
 |
| | Fig. 16 (a) EPR spectra of fresh and deferrated and recycled deferrated FeS-1 at 77 K. (b) DR UV-vis spectra of fresh and deferrated and recycled deferrated FeS-1. (c) The catalytic activity of fresh and deferrated and recycled deferrated FeS-1 at 77 K, in a batch reactor at 30 min. Reaction conditions: CH4 pressure 30 bar, 10 ml of 0.5 M H2O2 (aq), 25 mg catalyst, 570 rpm, and 50 °C. The inset in Fig. 16(a) is the deconvoluted DRUV-VIS spectrum which indicate the presence of various trivalent iron species in the microporous iron silicalite-1 structure. | |
 |
| | Fig. 17 Powder XRD patterns of FeS-1 (134): (a) FC; (b) DC; (c) RC. | |
 |
| | Fig. 18 HR-SEM images of various FeS-1 (134) catalysts. (a) FC; (b) DC; (c) RC. | |
4. Conclusion
We explored the application of FeS-1, FeZSM-5, and Fe–ZSM-5 catalyst systems systematically for the aqueous phase oxidation of methane under mild conditions employing hydrogen peroxide as the oxidant. Our investigation clearly elucidated the role of framework, extra-framework, and non-framework iron sites in catalytic activity efficiency. Remarkably, FeS-1 emerged as exceptionally active, achieving methane oxidation with a methanol selectivity of up to 65%, marking a record high in the reported literature. Our findings, therefore, reveal that specific active sites, in particular, the framework iron species, are crucial for achieving higher methanol selectivity. The individual oxygenates such as methanol and formic acid are found to increase monotonically with increasing framework iron and extra-framework iron, respectively. This finding is well supported by DRUV-VIS and EPR spectral data. On the other hand, interestingly, the deferration of FeS-1, under relatively milder conditions to remove the extra-framework iron sites, resulted in a significant drop in formic acid yield while retaining the methanol yield. Indeed, the formic acid loss was proportional to the disappearance of both the DRUV-VIS absorption band and the EPR signal corresponding to the extra-framework iron species of the FeS-1 matrix. This could potentially be due to the absence of both extra-framework iron and non-framework iron or iron-oxide Lewis acid sites in FeS-1. Furthermore, the high methanol selectivity of 65% and decreased oxidant consumption were retained even on doubling the batch reaction time. In contrast, FeZSM-5 (having both framework Fe and Al) showed higher total oxygenates than the corresponding FeS-1. Although, as observed with FeS-1, the oxygenate yield dropped for deferrated FeZSM-5, at the cost of methanol yield. The results thus indicate that the beneficial role of framework iron towards the methanol selectivity cannot be ignored. In the ion-exchanged Fe–ZSM-5 catalysts, formic acid yield decreases monotonically after deferration which brings out the importance of hydrothermal synthesis towards fine iron dispersion for the methanol selectivity.
Analysis of various measurements revealed that paramagnetic trivalent iron is isomorphously incorporated into the silicate framework of the MFI structure. These ions display both distorted and symmetrical tetrahedral geometries. However, the substitution of the larger trivalent iron ions for the smaller tetravalent silicon atoms results in a degree of thermal instability at the tetrahedral trivalent iron sites. During calcination, this instability leads to partial displacement, causing some trivalent iron to form finely dispersed superparamagnetic non-framework iron oxide species. The increased activity was thus attributed to the presence of trivalent iron in both framework and extra-framework sites. Conversely, non-framework iron species, such as iron oxide clusters and particles, contribute to the complete oxidation of CO2. Our findings demonstrate that iron silicalite-1 (FeS-1) serves as an outstanding platform for developing model catalysts capable of simulating a complete enzymatic cycle, thereby opening new opportunities for fundamental research into the direct activation of methane. Additionally, acid sites play a pivotal role in influencing catalytic activity. Samples with high iron loading, up to a SiO2/Fe2O3 ratio of 134, displayed exceptional catalytic performance and the highest intrinsic activity among all tested materials. Future studies integrating advanced operando spectroscopic techniques for site quantification, along with theoretical modeling, could offer a more comprehensive understanding of the roles of various iron sites and other parameters in enhancing methanol yield during aqueous phase oxidation. Additionally, investigating the nature of extra-framework iron sites and studying their local geometry and coordination through methods such as EXAFS/XANES, chemisorption, and titration techniques, combined with mechanistic studies, would provide valuable insights.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
PS and NSK conceived and supervised the project. PS and BV designed and developed the catalysts. BV synthesized and characterized the materials and performed catalytic experiments. PS, NSK and BV played a key role in analyzing the data, engaging in discussions, and contributing to manuscript writing. The final manuscript was reviewed and edited by PS and NSK.
Conflicts of interest
“There are no conflicts to declare”.
Acknowledgements
The authors thank the Science and Engineering Research Board (SERB), Department of Science and Technology (DST), New Delhi for funding NCCR, IIT-Madras under grant number – IR/S1/CU-01/2002. The authors are also grateful to Professor B. Viswanathan and Professor Shinya Hayami for their kind support, constant encouragement, and fruitful discussion.
References
- Y. Tang, Y. Li and F. Tao, Chem. Soc. Rev., 2022, 51, 376–423 RSC.
- S. Sun, A. J. Barnes, X. Gong, R. J. Lewis, N. F. Dummer, T. Bere, G. Shaw, N. Richards, D. J. Morgan and G. J. Hutchings, Catal. Sci. Technol., 2021, 11, 8052–8064 RSC.
- J. Kang and E. D. Park, Catalysts, 2020, 10, 299 CrossRef CAS.
- H. Zuo and E. Klemm, Appl. Catal., A, 2019, 583, 117121 CrossRef CAS.
- M. Shahami and D. F. Shantz, Catal. Sci. Technol., 2019, 9, 2945–2951 RSC.
- Y. K. Chow, N. F. Dummer, J. H. Carter, C. Williams, G. Shaw, D. J. Willock, S. H. Taylor, S. Yacob, R. J. Meyer, M. M. Bhasin and G. J. Hutchings, Catal. Sci. Technol., 2018, 8, 154–163 RSC.
- M. V. Parfenov, E. V. Starokon, L. V. Pirutko and G. I. Panov, J. Catal., 2014, 318, 14–21 CrossRef CAS.
- N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521–8573 CrossRef CAS PubMed.
- M. M. Forde, M. Armstrong, R. McVicker, P. P. Wells, N. Dimitratos, Q. He, L. Lu, R. L. Jenkins, C. Hammond, J. A. Lopez-Sanchez, C. J. Kiely and G. J. Hutching, Chem. Sci., 2014, 5, 3603–3616 RSC.
- C. Hammond, N. Dimitratos, J. A. Lopez-Sanchez, R. L. Jenkins, G. Whiting, S. A. Kondrat, M. H. Ab Rahim, M. M. Forde, A. Thetford, H. Hagen, E. E. Stangland, J. M. Moulijn, S. H. Taylor, D. J. Willock and G. J. Hutchings, ACS Catal., 2013, 3, 1835–1844 CrossRef CAS.
- C. Hammond, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, M. H. Ab Rahim, M. M. Forde, A. Thetford, D. M. Murphy, H. Hagen, E. E. Stangland, J. M. Moulijn, S. H. Taylor, D. J. Willock and G. J. Hutchings, Chem. – Eur. J., 2012, 18, 15735–15745 CrossRef CAS PubMed.
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS PubMed.
- G. I. Panov, G. A. Sheveleva, A. S. Kharitonov, V. N. Romannikov and L. A. Vostrikova, Appl. Catal., A, 1992, 82, 31–36 CrossRef CAS.
- M. V. Parfenov, E. V. Starokon, S. V. Semikolenov and G. I. Panov, J. Catal., 2009, 263, 173–180 CrossRef CAS.
- E. V. Starokon, M. V. Parfenov, S. S. Arzumanov, L. V. Pirutko, A. G. Stepanov and G. I. Panov, J. Catal., 2013, 300, 47–54 CrossRef CAS.
- E. V. Starokon, M. V. Parfenov, L. V. Pirutko, S. I. Abornev and G. I. Panov, J. Phys. Chem. C, 2011, 115, 2155–2161 CrossRef CAS.
- G. Zhao, E. Benhelal, A. Adesina, E. Kennedy and M. Stockenhuber, J. Phys. Chem. C, 2019, 123, 27436–27447 CrossRef CAS.
- G. Zhao, K. Chodyko, E. Benhelal, A. Adesina, E. Kennedy and M. Stockenhuber, J. Catal., 2021, 400, 10–19 CrossRef CAS.
- B. R. Wood, J. A. Reimer, A. T. Bell, M. T. Janicke and K. C. Ott, J. Catal., 2004, 225, 300–306 CrossRef CAS.
- I. Gokce, M. O. Ozbek and B. Ipek, J. Catal., 2023, 427, 115113 CrossRef CAS.
- D. Y. Osadchii, A. I. Olivos-Suarez, Á. Szécsényi, G. Li, M. A. Nasalevich, I. A. Dugulan, P. S. Crespo, E. J. M. Hensen, S. L. Veber, M. V. Fedin, G. Sankar, E. A. Pidko and J. Gascon, ACS Catal., 2018, 8, 5542–5548 CrossRef CAS.
- P. Xiao, K. Nakamura, Y. Lu, J. Huang, L. Wang, R. Osuga, M. Nishibori, Y. Wang, H. Gies and T. Yokoi, ACS Catal., 2023, 13, 16168–16178 CrossRef CAS.
- G. Zhao, P. Yan, K. Procter, A. Adesina, Y. Jin, E. Kennedy and M. Stockenhuber, J. Catal., 2023, 417, 140–152 CrossRef CAS.
- J. Zheng, J. Ye, M. A. Ortuño, J. L. Fulton, O. Y. Gutiérrez, D. M. Camaioni, R. K. Motkuri, Z. Li, T. E. Webber, B. L. Mehdi, N. D. Browning, R. L. Penn, O. K. Farha, J. T. Hupp, D. G. Truhlar, C. J. Cramer and J. A. Lercher, J. Am. Chem. Soc., 2019, 141, 9292–9304 CrossRef CAS PubMed.
- N. V. Beznis, B. M. Weckhuysen and J. H. Bitter, Catal. Lett., 2010, 138, 14–22 CrossRef CAS.
- S. E. Bozbag, P. Sot, M. Nachtegaal, M. Ranocchiari, J. A. Van Bokhoven and C. Mesters, ACS Catal., 2018, 8, 5721–5731 CrossRef CAS.
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, J. Am. Chem. Soc., 2005, 127, 1394–1395 CrossRef CAS PubMed.
- P. Xiao, Y. Wang, K. Nakamura, Y. Lu, J. N. Kondo, H. Gies and T. Yokoi, Catal. Sci. Technol., 2023, 13, 5831–5841 RSC.
- P. J. Smeets, M. H. Groothaert and R. A. Schoonheydt, Catal. Today, 2005, 110, 303–309 CrossRef CAS.
- M. Dyballa, D. K. Pappas, K. Kvande, E. Borfecchia, B. Arstad, P. Beato, U. Olsbye and S. Svelle, ACS Catal., 2019, 9, 365–375 CrossRef CAS.
- B. Ipek, M. J. Wulfers, H. Kim, F. Göltl, I. Hermans, J. P. Smith, K. S. Booksh, C. M. Brown and R. F. Lobo, ACS Catal., 2017, 7, 4291–4303 CrossRef CAS.
- A. J. Knorpp, M. A. Newton, V. L. Sushkevich, P. P. Zimmermann, A. B. Pinar and J. A. van Bokhoven, Catal. Sci. Technol., 2019, 9, 2806–2811 RSC.
- H. V. Le, S. Parishan, A. Sagaltchik, C. Göbel, C. Schlesiger, W. Malzer, A. Trunschke, R. Schomäcker and A. Thomas, ACS Catal., 2017, 7, 1403–1412 CrossRef CAS.
- M. A. Newton, A. J. Knorpp, V. L. Sushkevich, D. Palagin and J. A. Van Bokhoven, Chem. Soc. Rev., 2020, 49, 1449–1486 RSC.
- V. L. Sushkevich and J. A. van Bokhoven, ACS Catal., 2019, 9, 6293–6304 CrossRef CAS.
- H. Zhang, J. Guo and Y. Cao, Chem. Commun., 2023, 60, 228–231 RSC.
- K. Nakamura, P. Xiao, R. Osuga, Y. Wang, S. Yasuda, T. Matsumoto, J. N. Kondo, M. Yabushita, A. Muramatsu, H. Gies and T. Yokoi, Catal. Sci. Technol., 2023, 13, 2648–2651 RSC.
- M. Ravi, V. L. Sushkevich, A. J. Knorpp, M. A. Newton, D. Palagin, A. B. Pinar, M. Ranocchiari and J. A. van Bokhoven, Nat. Catal., 2019, 2, 485–494 CrossRef CAS.
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef CAS PubMed.
- K. T. Dinh, M. M. Sullivan, P. Serna, R. J. Meyer, M. Dincă and Y. Román-Leshkov, ACS Catal., 2018, 8, 8306–8313 CrossRef CAS.
- P. Xiao, Y. Wang, K. Nakamura, Y. Lu, J. N. Kondo, H. Gies and T. Yokoi, Catal. Sci. Technol., 2023, 13, 5831–5841 RSC.
- Z. Fang, H. Murayama, Q. Zhao, B. Liu, F. Jiang, Y. Xu, M. Tokunaga and X. Liu, Catal. Sci. Technol., 2019, 9, 6946–6956 RSC.
- T. Yu, Z. Li, W. Jones, Y. Liu, Q. He, W. Song, P. Du, B. Yang, H. An, D. M. Farmer, C. Qiu, A. Wang, B. M. Weckhuysen, A. M. Beale and W. Luo, Chem. Sci., 2021, 12, 3152–3160 RSC.
- K. Sun, H. Xia, Z. Feng, R. van Santen, E. Hensen and C. Li, J. Catal., 2008, 254, 383–396 CrossRef CAS.
- M. Ravi, V. L. Sushkevich and J. A. van Bokhoven, Nat. Mater., 2020, 19, 1047–1056 CrossRef CAS PubMed.
- Z. Zhu, H. Ma, W. Liao, P. Tang, K. Yang, T. Su, W. Ren and H. Lü, Appl. Catal., B, 2021, 288, 120022 CrossRef CAS.
- A. Sakthivl and P. Selvam, J. Catal., 2002, 211, 134–143 CrossRef CAS; P. Selvam and S. E. Dapurkar, J. Catal., 2005, 229, 64–71 CrossRef; P. Selvam and S. K. Mohapatra, J. Catal., 2005, 233, 276–287 CrossRef.
-
B. Venugopal, N. S. Kaisare and P. Selvam, 9th World Congress on Oxidation Catalysis: Oxidation for a Sustainable Future and Clean Environment, Cardiff, September 4-8, 2022 Search PubMed.
- R. Szostak, V. Nair and T. L. Thomas, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 487 RSC.
- A. Maegher, V. Nair and R. Szostak, Zeolites, 1988, 8, 3 CrossRef.
- A. Brückner, R. Lück, W. Wieker, B. Fahlke and H. Mehner, Zeolites, 1992, 12, 380–385 CrossRef.
- S. Bordiga, R. Buzzoni, F. Geobaldo, C. Lamberti, E. Giamello, A. Zecchina, G. Leofanti, G. Petrini, G. Tozzola and G. Vlaic, J. Catal., 1996, 158, 486 CrossRef CAS.
- D. S. Bhange and V. Ramaswamy, Mater. Res. Bull., 2007, 42, 851–860 CrossRef CAS.
- A. S. Al-dughaither and H. De Lasa, Ind. Eng. Chem. Res., 2014, 53, 15303–15316 CrossRef CAS.
- P. Selvam and S. K. Mohapatra, J. Catal., 2006, 238, 88–99 CrossRef CAS.
-
(a) S. K. Badamali, A. Sakthivel and P. Selvam, Catal. Today, 2000, 63, 291–295 CrossRef CAS;
(b) S. K. Badamali, A. Sakthivel and P. Selvam, Catal. Lett., 2000, 65, 153–157 CrossRef CAS.
- A. Oda, K. Aono, N. Murata, K. Murata, M. Yasumoto, N. Tsunoji, K. Sawabe and A. Satsuma, Catal. Sci. Technol., 2022, 12, 542–550 RSC.
- Q. Cheng, G. Li, X. Yao, L. Zheng, J. Wang, A. H. Emwas, P. Castaño, J. Ruiz-Martínez and Y. Han, J. Am. Chem. Soc., 2023, 145, 5888–5898 CrossRef CAS PubMed.
- R. D. Shannon and C. T. Prewitt, Acta Crystallogr., Sect. B, 1969, 25, 925 CrossRef CAS.
- R. Khairova, S. Komaty, A. Dikhtiarenko, J. L. Cerrillo, S. K. Veeranmaril, S. Telalović, A. A. Tapia, J.-L. Hazemann, J. Ruiz-Martinez and J. Gascon, Angew. Chem., Int. Ed., 2023, 62, e202311048 CrossRef CAS PubMed.
- G. Centi, S. Perathoner, F. Pino, R. Arrigo, G. Giordano, A. Katovic and V. Pedulà, Catal. Today, 2005, 110, 211–220 CrossRef CAS.
- N. Nikolopoulos, R. G. Geitenbeek, G. T. Whiting and B. M. Weckhuysen, J. Catal., 2021, 396, 136–147 CrossRef CAS.
- J. W. Park and H. Chon, J. Catal., 1992, 133, 159 CrossRef CAS.
- D. Goldfab, M. Bernardo, K. G. Strohmaier, D. E. W. Vaughan and H. Thomann, J. Am. Chem. Soc., 1994, 116, 6344 CrossRef.
- I. P. Pozdnyakov, E. M. Glebov, V. F. Plyusnin, V. P. Grivin, Y. V. Ivanov, D. Y. Vorobyev and N. M. Bazhin, Pure Appl. Chem., 2000, 72, 2187–2197 CrossRef CAS.
- M. S. Kim, K. H. Park, S. J. Cho and E. D. Park, Catal. Today, 2021, 376, 113–118 CrossRef CAS.
- C. Hammond, N. Dimitratos, R. L. Jenkins, J. A. Lopez-Sanchez, S. A. Kondrat, M. Hasbi Ab Rahim, M. M. Forde, A. Thetford, S. H. Taylor, H. Hagen, E. E. Stangland, J. H. Kang, J. M. Moulijn, D. J. Willock and G. J. Hutchings, ACS Catal., 2013, 3, 689–699 CrossRef CAS.
- Y. Liu, A. Bolshakov, M. Ćoza, V. Drozhzhin, E. J. M. Hensen and N. Kosinov, ACS Catal., 2023, 13, 8128–8138 CrossRef CAS.
- T. Yu, Z. Li, L. Lin, S. Chu, Y. Su, W. Song, A. Wang, B. M. Weckhuysen and W. Luo, ACS Catal., 2021, 11, 6684–6691 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a and
Parasuraman
Selvam
a and
Parasuraman
Selvam