
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient green synthesis of ammonia: from mechanistic understanding to reactor design for potential production
Zhenhui
Kou
a,
Dong
Shi
a,
Bin
Yang
a,
Zhongjian
Li
 a,
Qinghua
Zhang
a,
Jianguo
Lu
e,
Tao
Zhang
c,
Lecheng
Lei
a,
Yuanyuan
Li
f,
Liming
Dai
*d and
Yang
Hou
*ab
a,
Qinghua
Zhang
a,
Jianguo
Lu
e,
Tao
Zhang
c,
Lecheng
Lei
a,
Yuanyuan
Li
f,
Liming
Dai
*d and
Yang
Hou
*ab
aKey Laboratory of Biomass Chemical Engineering of Ministry of Education, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: yhou@zju.edu.cn
bZhejiang University Hydrogen Energy Institute, Hangzhou 310027, China
cKey Laboratory of Marine Materials and Related Technologies, Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo 315201, China
dARC Centre of Excellence for Carbon Science and Innovation, Australian Carbon Materials Centre, School of Chemical Engineering, The University of New South Wales Sydney, Sydney, NSW 2052, Australia. E-mail: l.dai@unsw.edu.au
eState Key Laboratory of Silicon and Advanced Semiconductor Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, China
fWallenberg Wood Science Center, Department of Fiber and Polymer Technology, KTH Royal Institute of Technology, Stockholm, 10044, Sweden
First published on 8th October 2025
Abstract
Ammonia (NH3), one of the world's most vital chemicals and energy carriers, has attracted wide attention. Currently, NH3 is mainly produced using the traditional, energy-intensive Haber–Bosch (H–B) technology, which has a large impact on the environment. Therefore, developing a low-cost, high-efficiency, and eco-friendly way to produce NH3 is highly desirable. Photo-, electro-, photoelectro-, and alkali–metal-mediated catalytic reactions powered by renewable and clean energy under ambient conditions offer alternatives to the H–B process and have recently gained significant interest. However, efficient nitrogen reduction is a key requirement, limiting the selectivity and activity for the green synthesis of NH3 because the N2 activation process in a green catalytic system is difficult to complete due to its thermodynamic instability and chemical inertness. Compared to the reduction of N2, the catalytic reduction of some soluble and harmful high-valent sources (e.g., NO, NO2−, and NO3−) is considered an effective method for increasing NH3 synthesis efficiency. This review article focuses on the important features of the green catalytic conversion of multiple nitrogen resources into NH3 by summarizing the fundamental mechanistic understanding, catalytic descriptors, and current advances, along with the various catalysts used for these conversion strategies and their structure–activity relationships. Meanwhile, opportunities and prospects for reactor design and construction for potential NH3 production at high current densities are also discussed, focusing on achieving a high yield rate, Faraday efficiency, and energy efficiency. This will provide valuable guidance for constructing catalysts and optimizing reaction systems that can meet the needs of practical applications.
 Zhenhui Kou | Zhenhui Kou is completing his PhD degree under Prof. Yang Hou at the College of Chemical and Biological Engineering at Zhejiang University. His current interests focus on electrocatalytic reduction reaction technology. |
 Liming Dai | Prof. Liming Dai is a Professor at the School of Chemical Engineering at the University of New South Wales. His current research mainly focuses on the synthesis, functionalization, and device fabrication of conjugated polymers and carbon nanomaterials for energy-related and biomedical applications. |
 Yang Hou | Prof. Yang Hou is a Professor at the College of Chemical and Biological Engineering at Zhejiang University. His current research mainly focuses on the design and synthesis of carbon-based materials for energy and environmental applications. |
1. Introduction
Ammonia (NH3) is an essential raw material for fertilizers, chemicals, and medical products, playing a important role in the development and progress of society.1–3 Specifically, a sufficient amount of NH3 is required to produce nitrogen-based fertilizers like NH4NO3 and urea, as well as other nitrogenous compounds, leading to a seven- to eight-fold increase in food production.4–6 Beyond its primary uses, NH3 serves as a critical precursor in diverse sectors, including national defense (explosives), plastics, dyes, synthetic fibers, and resins.7–10 Furthermore, NH3 is also an ideal eco-friendly energy storage species and a carbon-free energy carrier11–13 because of its large hydrogen content (17.6 wt%) and high energy density (4.32 kWh L−1 or 6.25 kWh kg−1)14 and the ease with which it can be liquefied for storage and transportation.10,15 Therefore, research into NH3 preparation pathways is attractive.Currently, the Haber–Bosch (H–B) process is the primary method for large-scale NH3 production, converting N2 and H2 into NH3.16–18 However, the H–B process still has numerous problems. First, the sluggish exothermic reaction (N2 + 3H2 → 2NH3, ΔH298K = −45.9 kJ mol−1, ΔG298K = −16.4 kJ mol−1) between inert N2 and H2 requires harsh conditions.19–23 Second, the H–B technique consumes about 1% of the world's energy supply and leaves a major carbon footprint.24,25 Third, the hydrogen produced through steam methane reforming or coal gasification has led to more than 420 million tons of carbon dioxide emissions.26,27 These negative factors make the H–B method an energy-intensive industrial technique. Additionally, the efficiency of the H–B technology is low, with an NH3 yield of about 15–20% in a single pass, although a 97% yield can be achieved by recycling the gas reactants multiple times.28 Therefore, it is very important to overcome these limitations. Considering the increasing global energy demand and environmental concerns, the scientific community should focus on how sustainable energy can support reactions at mild temperatures and pressures and how to synthesize NH3 on a large scale using this green conversion pathway. Therefore, developing green synthesis technologies is important.
Inspired by biological N2 fixation,29 where many plants use nitrogenase to convert N2 from the air into NH3 fertilizer under sunlight for self-growth,30 recent studies show that nitrogenase displays photocatalytic conversion activity and plays a significant role in NH3 synthesis.31 However, due to the unsatisfactory conversion rate of photo-assisted N2 fixation, recent work has mainly focused on developing more efficient strategies. Using renewable electricity instead of fossil fuels has been widely accepted.16 An efficient technology for obtaining green NH3 is to use the electrocatalytic water splitting process, which produces the solvent and proton source.32 Its raw materials are H2O and N2, and the overall reaction is 3H2O + N2 → 2NH3 + 3/2O2, driven only by renewable electricity.31 The efficient transformation of renewable energy and the conversion of clean electricity into chemical energy are critical for energy and environmental protection.33–35 Therefore, this has aroused widespread interest among scientists worldwide. More importantly, photoelectrochemical, alkali metal-mediated, and other conversion technologies have also been proposed to further improve or optimize the Faraday efficiency (FE), energy efficiency (EE), current density, reaction speed, and yield rate of NH3.36–42
Furthermore, to facilitate the practical applications of green catalytic conversion methods, the United States Department of Energy has set key parameters as benchmarks. The current FE remains at 60%, a value below the target of over 90%. Meanwhile, the current activity indexes for electrocatalytic NH3 synthesis are two to three orders of magnitude below the target standards.20,43,44
Therefore, despite ongoing research, green NH3 synthesis is far from large-scale applications for the following reasons: (a) the thermodynamic energy barrier of the competing hydrogen evolution reaction (HER) is lower than that of the NH3 synthesis reaction.45 (b) The chemically inert N2 molecule has a strong N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond energy (941 kJ mol−1), requiring a massive energy input to cleave the first bond. (c) N2 molecules are electronically stable due to a high ionization energy of 1525 kJ mol−1 and a large energy gap of 1044 kJ mol−1, which makes transferring an electron to N2 molecules kinetically difficult. (d) N2 protonation is the rate-determining step (RDS) because N2 has a lower proton affinity compared to reaction intermediates like diazene (N2H2) and hydrazine (N2H4).6,20,46 (e) The limited solubility of N2 molecules in aqueous solutions generates a low current density and a low production rate.47,48 Thus, building suitable catalysts to activate N2 molecules, through methods such as doping engineering, defect engineering, crystal facet engineering, and dimensional engineering,49–51 is the first step for high-efficiency NH3 production.
N bond energy (941 kJ mol−1), requiring a massive energy input to cleave the first bond. (c) N2 molecules are electronically stable due to a high ionization energy of 1525 kJ mol−1 and a large energy gap of 1044 kJ mol−1, which makes transferring an electron to N2 molecules kinetically difficult. (d) N2 protonation is the rate-determining step (RDS) because N2 has a lower proton affinity compared to reaction intermediates like diazene (N2H2) and hydrazine (N2H4).6,20,46 (e) The limited solubility of N2 molecules in aqueous solutions generates a low current density and a low production rate.47,48 Thus, building suitable catalysts to activate N2 molecules, through methods such as doping engineering, defect engineering, crystal facet engineering, and dimensional engineering,49–51 is the first step for high-efficiency NH3 production.
In addition to constructing efficient catalysts, using alternative nitrogen sources, such as nitric oxide or nitrogen dioxide (NO or NO2) with low dissociation energy and nitrate or nitrite (NO3−, NO2−) with high solubility, is considered an effective way to decrease energy consumption and increase the production rate and selectivity of NH3.7,21,52,53 Among these, NO is one of the major pollutants produced by the burning of fossil fuels.54,55 As its concentration increases, it causes serious ecological problems such as acid rain, ozone depletion, and haze,56 with potential risks about 300 times greater than those associated with CO2 emissions. Meanwhile, the high solubility of NOx− makes it difficult to remove from water, resulting in a series of physiological diseases.58,59 The contaminant NO2− can also cause cancer,60 while NO3− can disrupt the human endocrine system. The World Health Organization has recommended a maximum contaminant level of 50 mg L−1 in drinking water to minimize the adverse health effects of nitrogen oxides.57 Thus, in addition to alleviating energy and resource shortages, reducing NO, NO2−, and NO3− to NH3 is very beneficial for controlling wastewater pollution, as these processes involve the low dissociation energy (204 kJ mol−1) of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds.61 Moreover, NO3− and NO2− usually tend to be adsorbed and activated by catalytic materials, which effectively settle the insoluble difficulty of N2, setting the fundamental for large-scale NH3 synthesis.21 However, there are also certain limitations. Take the NO3− reduction reaction (NO3RR) (NO3− + 9H+ + 8e− → NH3 + 3H2O) as an example. Undesired by-products like NO2−, N2 and N2H4 are inevitably produced during the process.62,63 The selectivity of the NO3RR depends on the overpotential and current density.9,64 Therefore, a timely summary of various catalysts in NH3 synthesis will not only improve the understanding of current progress but also inspire the construction of advanced catalysts, achieving the effect of ‘killing two birds with one stone’. Meanwhile, the innovation of catalytic materials and reactors also plays an important role in reaction system engineering, and further optimization of synthetic NH3 systems requires innovation in reactor engineering. Thus, an advanced synthetic NH3 system can only be achieved through joint optimization and innovation. This article will focus on developing high-performance reaction units to minimize the mass transfer limit of the reactants, thereby significantly improving key parameters such as system current density, FE, EE, and running time. Finally, the transformation of green synthetic NH3 technology from the laboratory to an industrial scale is expected to be realized.
O bonds.61 Moreover, NO3− and NO2− usually tend to be adsorbed and activated by catalytic materials, which effectively settle the insoluble difficulty of N2, setting the fundamental for large-scale NH3 synthesis.21 However, there are also certain limitations. Take the NO3− reduction reaction (NO3RR) (NO3− + 9H+ + 8e− → NH3 + 3H2O) as an example. Undesired by-products like NO2−, N2 and N2H4 are inevitably produced during the process.62,63 The selectivity of the NO3RR depends on the overpotential and current density.9,64 Therefore, a timely summary of various catalysts in NH3 synthesis will not only improve the understanding of current progress but also inspire the construction of advanced catalysts, achieving the effect of ‘killing two birds with one stone’. Meanwhile, the innovation of catalytic materials and reactors also plays an important role in reaction system engineering, and further optimization of synthetic NH3 systems requires innovation in reactor engineering. Thus, an advanced synthetic NH3 system can only be achieved through joint optimization and innovation. This article will focus on developing high-performance reaction units to minimize the mass transfer limit of the reactants, thereby significantly improving key parameters such as system current density, FE, EE, and running time. Finally, the transformation of green synthetic NH3 technology from the laboratory to an industrial scale is expected to be realized.
To our knowledge, many review papers have been published on this popular topic, covering NH3 synthesis methods, types of advanced catalysts, structure–activity relationships, reaction pathways, and mechanisms.20,28,44,65–70 However, most of these reviews focus on a specific topic, limited to a single catalytic conversion strategy (e.g., photocatalytic, electrocatalytic, or thermocatalytic), a specific electrocatalyst (e.g., two-dimensional monoelemental materials, transition metal dichalcogenides, MXenes, or molecular catalysts), or one type of nitrogen source (e.g., nitrogen, nitric oxide, nitrite, or nitrate). In recent decades, much research has been done to develop promising green NH3 synthesis by designing high-performance catalysts using theoretical calculations and advanced synthesis technologies, which requires a timely, comprehensive review of this rapidly developing field. Therefore, instead of focusing on a single topic as in previous reviews, this article presents a comprehensive review covering recent research progress in value-added NH3 production through photo-, electro-, photo/electrochemical, lithium-mediated, calcium-mediated and other advanced green pathways. We will provide an overview of the history and fundamental science behind traditional NH3 synthesis technology and its existing problems, along with the outlook on green NH3, given the high global demand for ammonia. Special emphasis will be placed on the state-of-the-art catalytic technology, material design and preparation from different nitrogen sources (e.g., nitrogen, nitric oxide, nitrite, or nitrate), structural characterization, and performance evaluation, as well as the main factors influencing selectivity and activity, the development of reactors for green NH3 synthesis, and the associated theoretical calculations on reaction mechanisms for different catalysts. Finally, the outlook on NH3 synthesis using multiple catalysts, including challenges and opportunities for developing new catalysts, novel catalytic methods, and reactor devices, provided in recently reported operando fundamental studies, and potential industrial applications will be summarized and discussed (Fig. 1).
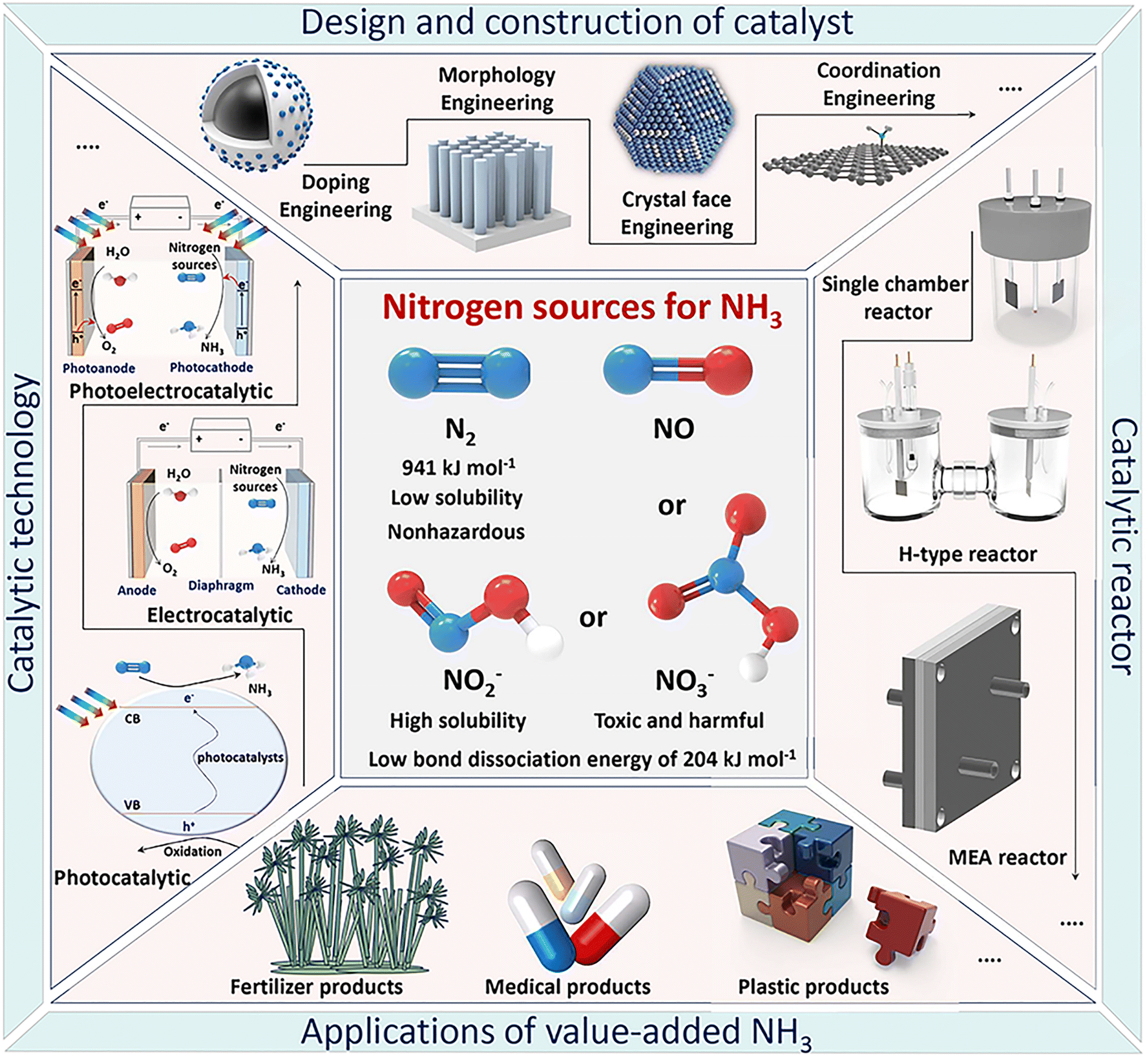 | ||
| Fig. 1 Schematic illustration of efficient value-added NH3 product synthesis by various green catalytic conversion technologies from different nitrogen sources. | ||
2. Fundamentals of ammonia synthesis
To provide comprehensive and systematic information about green NH3 synthesis, a thorough and fundamental summary of the topic is necessary. In this section, we briefly summarize the reaction mechanisms and specific pathways for different nitrogen resources (N2, NO, NO2−, and NO3−), including the multiple intermediates, conversion steps, and reaction pathways. After explaining the basic mechanism, we will comprehensively summarize various methods for NH3 synthesis, including the design and construction of reactors, testing methods for NH3 products, error removal strategies, and performance evaluation criteria. Finally, we will describe in detail the various catalytic activity descriptors, such as FE, NH3 yield, EE, and stability, used to evaluate catalytic performance in real time to further optimize the experiments.2.1 Mechanism of ammonia synthesis
N bonds break at high temperature and pressure to produce NH3. In contrast, in the associative mechanism, the NN bonds are not completely broken until the first NH3 molecule is released. Specifically, the associative pathway includes distal, alternating, and enzymatic hydrogenation. In the distal and alternating pathways, N2 molecules bind end-on to the catalyst's surface. In the distal mechanism, the distant N atom first combines with H to form *NH3, while the other N atom does not combine with H until the first NH3 molecule is released. In contrast, in the alternating pathway, the two N atoms are hydrogenated in turn before both NH3 molecules are released at the same time. In the enzymatic pathway, the linear N2 molecules are parallel to the electrode surface, where they react with H to produce the target products alternately or concurrently. Recently, the MvK pathway has also been proposed. In general, the production of NH3via transition metal nitrides (TMNs) usually follows the MvK pathway. In the MvK mechanism, N atoms from the TMNs are reduced to NH3 and external N2 fills the vacancy to complete the recovery of the TMNs and begin a new NH3 synthesis process.73 Compared to other pathways, the MvK pathway greatly improves mass transfer kinetics and overcomes the poor solubility of N2 molecules because the ‘gas–liquid–solid’ three-phase reaction is divided into ‘liquid–solid’ and ‘gas–solid’ reactions.10 Currently, work investigating the catalytic mechanism usually depends on theoretical calculations, with a few actual experiments. Therefore, more in situ electrochemical characterization techniques can be used to determine the key conversion steps from N2 to NH3, such as in situ electrochemical scanning tunneling microscopy, in situ electrochemical atomic force microscopy, in situ Fourier transform infrared spectroscopy, in situ Raman spectroscopy, and in situ mass spectrometry, to track real-time changes in the surface structure of electrocatalysts and surface electrocatalytic processes. Furthermore, combining experiments with theoretical calculations can clearly explain the mechanism for a specific catalytic reaction process, which will have a major impact on the development of NH3 synthesis.
| NO + 5H+ + 5e− → NH3 + H2O | (1) |
| 2NO + 4H+ + 4e− → N2 + 2H2O | (2) |
| 2NO + 2H+ + 2e− → N2O + H2O | (3) |
Similar to the nitrogen reduction reaction (NRR), the NORR mechanism also includes dissociative and associative pathways.76 In the dissociative mechanism, the NO bond is first broken to produce *N and *O, and then the hydrogenation process begins. In contrast, during the associative pathway, NO is first hydrogenated to HxNOHy intermediates, which are then further protonated to NH3. In these reaction paths, hydrogenation occurs through either the Tafel process, which involves the initial formation of adsorbed *H followed by surface hydrogenation, or the Heyrovsky process, where NO molecules and intermediates are directly protonated. Hence, there are four types of NORR mechanisms: Tafel combined with dissociative, Tafel combined with associative, Heyrovsky combined with dissociative, and Heyrovsky combined with associative. Among these, the hydrogenation steps in the associative Tafel and associative Heyrovsky mechanisms are further classified into distal or alternating pathways. In the distal pathway, O or N atoms are completely hydrogenated to H2O or NH3. In the alternating pathway, O or N atoms are alternately hydrogenated. The specific reaction processes are as follows in eqn (4)–(9):78
| NO → *N + *O | (4) |
| Tafel-type: H+ + e− → *H | (5) |
| *N + *H → *NH | (6) |
| Heyrovsky-type: *N + H+ + e− → *NH | (7) |
| *NH + *H → *NH2 | (8) |
| *NH2 + *H → NH3 | (9) |
| NO3− + 9H+ + 8e− → NH3 + 3H2O | (10) |
Specifically, taking the NO3RR as an example, there are two different pathways: indirect autocatalytic reduction and direct external-field-assisted catalytic reduction. A peracidic environment (pH < 0) and a high concentration (>1.0 mol L−1) of NO3− favor the indirect autocatalytic pathway, where NO3− does not participate in the electron transfer process.67,81–84 In contrast, direct external-field-assisted catalytic reduction, which usually operates at low concentrations (<1.0 mol L−1),76 involves an electron transfer process and includes two pathways: the first involves adsorbed *H and the second involves electrons from the catalysts. The first pathway involves reducing adsorbed H2O molecules to produce *H, followed by the direct reduction of NO3− to NH3 through a series of intermediate changes, such as *NO2−, *NO, *N, *NH, and *NH2.85 Notably, forming N–N bonds is kinetically less favorable than forming N–H bonds.86 Therefore, the favorable adsorption of *H can boost NH3 production. In the second pathway, the first step is the reduction of the intermediate *NO3− to *NO2−.87–89 Next, the *NO2− can be hydrogenated to produce *NO, which is then converted into NH3 through a series of protonation processes. In this process, *HNO is formed first, which then converts90 to *H2NO and *NH2OH and finally, the adsorbed *NH2OH is reduced to NH3.67,84
2.2 Research methods for ammonia synthesis
First, it is essential to thoroughly examine the reported overall NH3 production, especially in aqueous systems, to confirm that it originates from N2. Contamination from sources like catalysts, electrolytes, and reactants may result in false positives for NH3 synthesis performance. At the same time, quantification techniques may not provide sufficient detection limits or consistency, since the NH3 synthesis performance can easily be biased when NH3 concentrations are not significantly higher than impurities. The high variance is likely a result of this.91–93 Since NH3 synthesis experiments can be ambiguous in identifying nitrogen sources, rigorous testing procedures have been proposed,94–96 including performing NH3 synthesis with non-catalyst electrodes, switching reactants from N2 to Ar, and using quantitative isotope-labeled tests.46 Second, other aspects of the reaction system may also compromise NH3 production. A further study of catalytic reactor design and operation is especially vital, as this is essential for achieving a rational reaction system design and avoiding false positives. Under ideal operating conditions, modified reactor components have been shown to increase activity.97,98 To minimize false positives, contamination-prone reactor components are removed, and rigorous reactor operation protocols are implemented.91,92,99 Consequently, developing a reliable reactor along with strict and correct testing methods will improve the development of NH3 synthesis.To avoid errors from nitrogen impurities, necessary purification measures must be taken, such as using sulfuric acid as an adsorbent to remove impurities from the raw reactants. Soluble nitrogen-containing impurities and poorly soluble nitrogen oxides also need to be removed via washing and selective reduction methods, respectively, followed by using H2SO4 to remove excess NH3 species.100 Poorly soluble impurities can also be oxidized to soluble impurities like NO using a H2O2/S2O82− solution and then removed. If the reaction solvents or catalysts contain nitrogen impurities, they can be removed via heat treatment or alkali treatment. For ion exchange membranes used in a reactor, in addition to standard pretreatment, the treated membranes can be further sonicated in deionized water and dilute H2SO4.101
Besides, to precisely determine the catalytic activity and avoid false positive test results, the NH3 produced must also be detected again using a reliable method. The most common detection methods include chemical detection using Nessler's reagent, indophenol blue, and salicylate, fluorometric detection, conductivity measurements, titrimetric methods, isotope labeling, 1H NMR, electrode-based detection, ultraviolet and visible spectrophotometry, ion chromatography, and gas chromatography.67,68 Notably, the detection results can be influenced by many kinds of metal ions, hydrazine, and carbonyl compounds. To minimize interference and prevent the formation of turbid samples when using the Nessler's reagent method,102,103 Rochelle salt solution is usually added.68 For the indophenol blue method, a blue product, indophenol, is produced through the reaction between NH3, phenol, and hypochlorite in an alkaline solvent.104 This method is less efficient when there is an excess of NH3. In the salicylate method, sodium salicylate replaces phenol, which avoids producing toxic and volatile reaction products. Due to the low reactivity of salicylate, a higher concentration is needed to achieve sensitivity like the other methods.105,106
The fluorometric method mainly involves the interaction between NH3, o-phthalaldehyde, and sulfite to produce a strongly fluorescent compound.107 When the NH3 concentration exceeds 1.0 μmol L−1, the measured concentration and fluorescence intensity are not linearly related. Also, amines and amino acids can affect the results in aqueous solutions.108,109
Additionally, the conductivity method uses the reaction of NH3 with acid, where the resulting salts reduce the solution's conductivity. A standard curve created with known NH3 concentrations can be used to determine the exact NH3 concentration in the solution based on the conductivity decrease.110 However, the solution's conductivity can affect the detection.
For detecting NH3 concentrations higher than 5.0 mg L−1, the titrimetric method is often used. The method involves distilling the reaction solution and then titrating the NH3 in the distilled products with an H2SO4 solution until the indicator changes color.111 To determine if the prepared NH3 comes from the N2 reactants, especially for catalysts containing nitrogen, 15N2 isotopic experiments are necessary. Additionally, 1H NMR methods are required to detect and calibrate 15NH4+.68,112
Using a specific electrode to measure the NH3 concentration is quick and simple,113,114 including NH3 sensing electrodes and NH4+ selective electrodes. The NH3 sensing electrode consists of a membrane, a pH electrode, and a reference electrode, with an NH4Cl solution used as the electrode solution. The pH of the solution being tested is increased by adding a base to convert NH4+ into NH3 and the NH3 then diffuses through the membrane until both sides have the same partial pressure. A standard curve generated with known NH3 concentrations can be used to determine the NH3 concentration in the internal solution, as the NH3 concentration in the tested solution is proportional to the pH change in the electrode solution. The NH4+ selective electrode has a polyvinyl chloride film containing an NH4+ carrier and the pH of the solution being tested is decreased to convert NH3 into NH4+. When a solution containing NH4+ meets the NH4+ selective electrode, a potential is generated on the membrane. Following this mechanism, a standard curve produced with known concentrations of NH4+ can be used to determine the concentration in the unknown solution.6,109
In addition to the description of the electrochemical system above, specific performance measurements for the electrosynthesis of NH3 usually involve linear sweep voltammetry (LSV), chronoamperometry (CA), cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS), multi-potential steps, and multi-current steps. LSV curves display the change in current density across a potential range for a given electrocatalytic process. By comparing the LSV curves measured in an Ar-saturated versus a N2 saturated electrolyte, one can determine whether nitrogen reduction occurs at a certain potential range based on the difference in current density, in which this difference can be regarded as a contribution from the target reduction process. Regarding the CA technique, the current is monitored as a function of reaction time at a controlled potential. The obtained CA data can be used to calculate the FE of the electroreduction process and to evaluate the stability of catalysts under long-term electrolysis. The stability of the electrochemical reaction system can also be evaluated by comparing the LSV curves before and after repeated CV measurements. Besides this, a nearly constant conversion, selectivity, and FE for NH3 can also illustrate that the electrocatalyst has good stability.84 In addition, the CV technique can also be used to determine the double-layer capacitance for estimating the electrochemically active surface area of catalysts, which can be used to normalize the surface area of the electrode, thereby excluding the influence of surface roughness. EIS measurements can be used to evaluate the electrochemical reaction rate by analyzing the electrolyte resistance and charge-transfer resistance.68
2.3 Catalytic activity descriptors of ammonia synthesis
The conversion rate of the nitrogen source is calculated using the below formulas (11)–(13):84
Conversion = Δcnitrogen![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) source/c0 × 100% source/c0 × 100% | (11) |
The selectivity for the NH3 product is calculated using the formula below:
| Selectivity = c/Δcnitrogensource × 100% | (12) |
The FE for NH3 is calculated using the formula below:
| FE = [(n × 96485 × c × V)/(M × Q)] × 100% | (13) |
source is the change in the concentration of the nitrogen source before and after electrolysis; c is the concentration of the produced NH3; V is the volume of the electrolyte in the cathode compartment; n is the number of electrons transferred in the cathode reduction reaction; M is the molar mass of NH3; and Q is the total amount of charge passed through the cathode during the reaction.
| Yield = (c × V)/(t × S) | (14) |
| Yield = (c × V)/(t × m) | (15) |
Based on the analysis above, the NH3 yield rate is the amount of NH3 produced per unit of time and per unit of catalyst mass (or electrode surface area), which reflects the reaction rate of NH3 synthesis. It is also important that when using mg−1 as the standardized unit, some studies use the catalyst's total mass as the baseline, while others use the actual active sites for standardization. This can lead to unreliable comparisons of the NH3 yield rate. Therefore, comparing performance using unified units is very important for progress in NH3 synthesis.10
| EE = Energy required/Energy input = ΔG0n/EcellIt | (16) |
| ΔG0n = zE0Fn = E0Q = E0ItFE (E0 = Eanode − Ecathode) | (17) |
| Ecell = Eanode − Ecathode + ηcathode + ηanode + Eloss + ηmasstransfer | (18) |
transfer represents the external overpotentials from mass transfer limitations; and Eloss is the ohmic dissipation, equal to the current multiplied by the resistance of the entire reaction system.
As seen in the formulas above, to achieve a high EE, the Ecell should be reduced, and the FE should be improved as much as possible. On the other hand, it is recognized that the oxygen evolution reaction has thermodynamic and kinetic difficulties, which consume a significant portion of the supplied electrical energy.118,119 According to systematic analysis, the anode oxidation reaction, which produces low-value products, consumes more than 90% of the electrical energy, compared to less than 10% for the production of the target products at the cathode.120 Therefore, optimizing the anode reaction in the NH3 synthesis system can further reduce energy consumption and improve the system's economic efficiency.
3. Photochemical synthesis of ammonia
Inspired by how plant nitrogenases convert N2 into NH3 using solar energy, efficiently using renewable energy and converting it into chemical energy is crucial for protecting the environment and conserving energy. In this section, we first analyze the mechanism of photocatalytic NH3 synthesis. Specifically, the photoreduction process includes two steps: (a) photoexcited electrons transfer from the valence band to the conduction band, leaving photogenerated holes in their original positions; (b) free photogenerated electrons and holes migrate to the photocatalyst surface to participate in redox reactions, while some electrons and holes recombine. Photocatalytic NH3 synthesis is a six-electron, six-proton process that includes N2 adsorption, activation, and cleavage of the NN bond, combined with a hydrogenation process, and the production and desorption of NH3.31 However, most reported photocatalysts face key problems, such as severe photogenerated charge recombination, weak N2 adsorption, difficult NN bond dissociation, the poor proton affinity of the N2 molecule preventing electron transport and Lewis acid–base reactions, and the difficult N2 reaction caused by its large 10.82 eV energy gap,121 all of which result in low photocatalytic NH3 synthesis efficiency. Therefore, the rational design and preparation of efficient photocatalysts for N2 reduction still present great challenges. We will first describe the photo-response of photocatalysts and the evaluation criteria for photocatalytic performance and then summarize engineering strategies for improving photocatalytic performance. Then, specific examples for each type of photocatalyst will be discussed in terms of catalyst design strategies, performance, and structure–activity relationship (Table 1).
| Catalysts | Nitrogen source | Experimental conditions | Yield rate | Reactor | Ref. |
|---|---|---|---|---|---|
| Al-PMOF(Fe) | N2 | Visible light, λ > 420 nm, 100 mW cm−2 | 127 μg h−1 g−1 cat. | A custom-designed quartz reactor with magnetic stirring | 122 |
| FeS2@VN-CN | N2 | A doped Hg immersion lamp, 350 W | 800 μg h−1 | A water-cooled quartz-glass inlay | 123 |
| MIL-100(Fe)-200W | N2 | 300 W xenon lamp with a 420 nm cutoff filter, simulate visible light, 420–800 nm, 130 mW cm−2 | 115.1 μmol g−1 h−1 | A reactor equipped with a circulating water outer jacket to maintain a temperature of 25 °C | 124 |
| ZnCr-LDH nanosheets | N2 | Simulated solar light irradiation, 300 W xenon lamp as the excitation source, 200–800 nm, 5.0 W cm−2 | 33.19 μmol g−1 h−1 | A quartz reactor equipped with a circulating water outer jacket to maintain a constant temperature of 25 °C | 125 |
| P–Fe/W18O49 | N2 | A xenon lamp used as a simulated solar light source, 100 mW cm−2 | 187.6 μmol g−1 h−1 | A self-made mobile phase reaction cell with a circulating thermostatic water bath to maintain a constant temperature of 25 °C | 126 |
| Ru/RuO2/C-TiOx | N2 | 300 W xenon lamp equipped with a 395 nm or 420 nm cut-off filter | 109.3 μmol g−1 h−1 | A quartz reactor maintained below 25 °C | 127 |
| Ru-K2Ta2O6−x | N2 | UV irradiation or visible light irradiation (>400 nm) from a 300 W xenon lamp equipped with cut-off filters | 335 μg g−1 h−1 | An airtight quartz reactor with a vacuum environment | 128 |
| Au@UiO-66 | N2 | Visible light (>400 nm, 100 mW cm−2) | 18.9 mmol gAu−1 h−1 | A homemade outer irradiation-type gas-flow cell | 129 |
| GDY@Fe–B | N2 | Visible light irradiation (λ ![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) 400 nm), using a 300 W xenon lamp 400 nm), using a 300 W xenon lamp |
1762.35 ± 153.71 μmol h−1 gcat.−1 | A gas recycling set-up | 130 |
| CuCr-LDH | N2 | Visible light irradiation (λ 400 nm), using a 300 W xenon lamp |
184.8 μmol L−1 | A quartz reactor maintained below 25 °C | 131 |
| FePc-POF | N2 | A 300 W xenon lamp equipped with a 400 nm cutoff filter, 200 mW cm−2 | 1820.7 μmol g−1 h−1 | A solvent-in-gas reactor | 132 |
| TiO2-B-2 | N2 | A 300 W xenon lamp, λ > 300 nm | 3.35 mg h−1 g−1 | A self-made quartz reactor | 139 |
| WO3/CdS | N2 | A 300 W xenon lamp | 35.8 μmol h−1 g−1 | A gas–solid reaction system | 140 |
| BCN | N2 | Light irradiation (with a cut-off filter, λ > 400 nm, a light intensity of 0.5 W cm−2, 250 W Xe lamp) | 313.9 μmol g−1 h−1 | A CEL-HPR100S + photocatalytic reactor | 141 |
| LiH | N2 | A xenon lamp with the UV cut-off filter (300–420nm), 136.5 mW cm−2 |
518 μmol g−1 h−1 | A quartz tube as a fixed-bed reactor | 142 |
| BaONCs-TNS | NO3− | 300 W xenon lamp | 89.79 mmol gcat.−1 h−1 | An MC-GF250 photocatalytic reactor | 143 |
The evaluation criteria for photocatalytic ammonia synthesis catalysts include two main aspects: (1) the evaluation of photo-response includes (a) light absorption ability, determined by the absorption edge position, absorption intensity, and bandgap width to assess the visible-light response; (b) charge carrier separation efficiency, measured through photocurrent density and photoluminescence spectroscopy to determine charge separation effectiveness, with transient fluorescence lifetime tests further revealing carrier lifetime and migration ability; and (c) band structure alignment, where the valence and conduction band positions of the photocatalyst must match the required redox potentials to ensure the reaction proceeds efficiently. (2) The evaluation of photocatalytic activity involves key parameters including ammonia production rate, stability, turnover frequency, and quantum efficiency. The ammonia production rate, measured as the product yield per unit of time per unit of catalyst mass or area, is a metric used to compare different photocatalysts, with better performers showing higher yields. Stability is demonstrated by comparing ammonia production rates after multiple cycling tests or prolonged illumination. Faradaic efficiency, calculated as the ratio of electrons used for ammonia synthesis to the total electrons consumed, reflects the catalyst's selectivity for the target product, where high values indicate effective suppression of the competing hydrogen evolution reaction and preferential use of electrons for ammonia production. Turnover frequency measures the number of ammonia molecules produced per active site per unit of time and serves as an indicator of intrinsic catalytic activity. Quantum efficiency directly measures the proportion of incident photons converted into electrons that participate in ammonia synthesis, effectively representing the catalyst's ability to harvest light and separate charges. When combined with turnover frequency, quantum efficiency helps evaluate catalytic performance issues. For instance, high light absorption and charge separation efficiencies coupled with a low quantum efficiency may suggest slow surface N2 activation or proton transfer, requiring an evaluation of the intrinsic activity through turnover frequency analysis. Furthermore, the correlation between quantum efficiency and faradaic efficiency reveals how electrons are used. A high quantum efficiency with a low faradaic efficiency indicates that the hydrogen evolution side reaction is dominant, while high values for both parameters show the preferential use of electrons for ammonia synthesis.
To create efficient photocatalytic materials, inspired by the biological molecule chlorophyll, which has a favorable binding site for the N2 atom, a porphyrin-based metal–organic framework (PMOF) was developed that contains atomically dispersed Al and Fe metal centers to promote the adsorption and activation of N2 (Fig. 2(a) and (b)) (Al-PMOF(Fe)).122 Al-PMOF(Fe) showed a remarkable production rate of 127 μg h−1 gcat.−1 (Fig. 2(c)), outperforming the Al-PMOF, which had a rate of 84.5 μg h−1 gcat.−1. The porphyrin ligand efficiently harvested light, and the active Fe–N sites lowered the difficulty of the rate-determining step, N2 activation, thus improving the photocatalytic activity. Furthermore, inspired by the high optical absorption and high charge carrier mobility of biological enzymes, carbon nitride (CN) heterogeneous materials containing N defects and loaded with FeS2 were created by high-temperature calcination and alkali treatment (Fig. 2(d)). Using the favorable light absorption and N2 activation properties of the Fe–S system, as well as the high activity of the prepared CN (Fig. 2(e)), an NH3 yield of up to 800 μg L−1 was obtained, which was about 400% higher than that of pristine CN.123 FeS2 acted like a cocatalyst, increasing the NH3 yield through π-back-donation from Fe centers to the imine nitrogen of the defect-rich CN, thereby activating the structure and increasing NH3 generation from cyano groups (Fig. 2(f)).
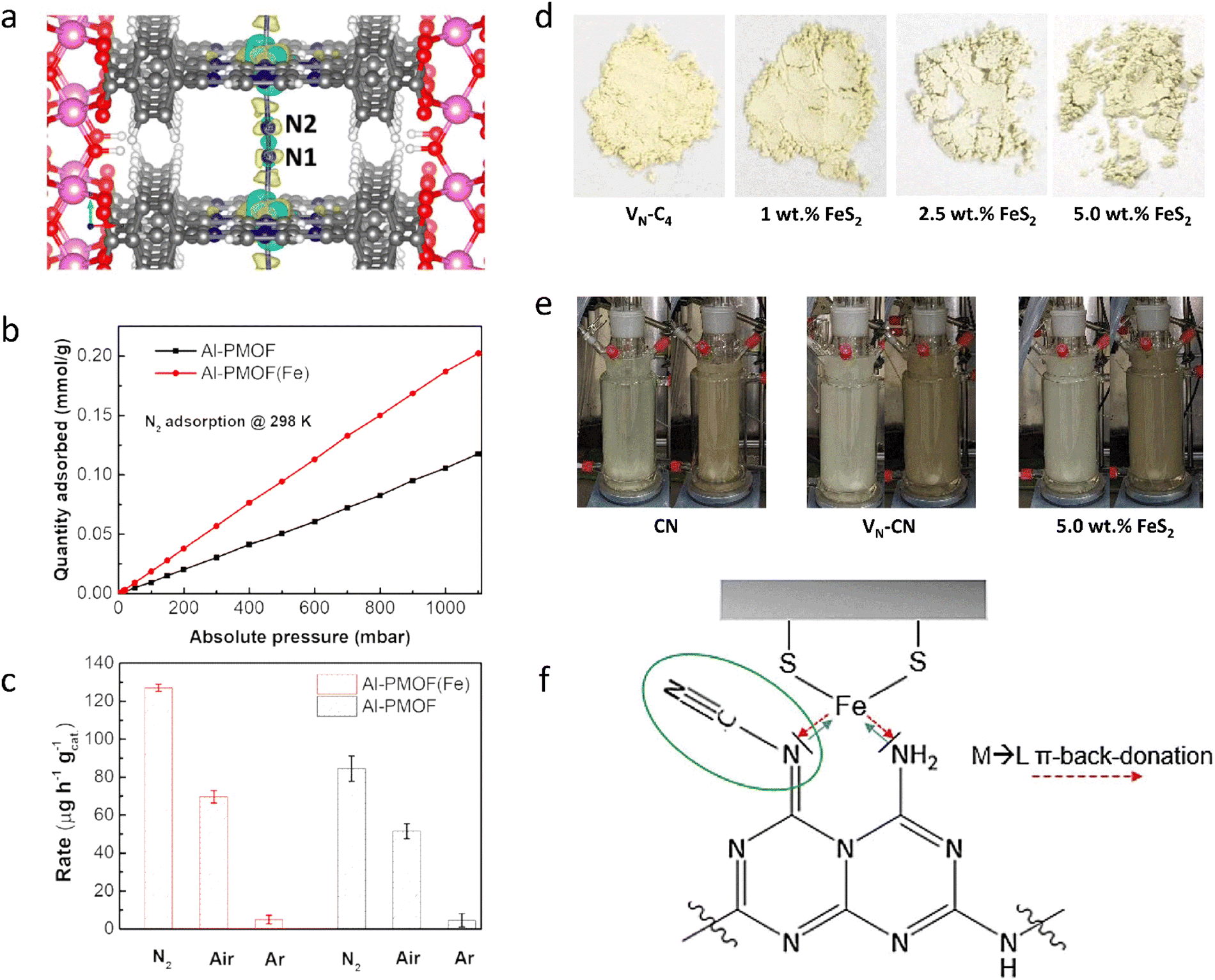 | ||
| Fig. 2 (a) Adsorption model of the N2 molecule for Al-PMOF(Fe). (b) N2 gas adsorption isotherms for Al-PMOF and Al-PMOF(Fe) at 298 K. (c) NH3 yield rates for Al-PMOF and Al-PMOF(Fe). (d) Photographs of VN–C4 and VN–C4 containing different amounts of FeS2. (e) Photocatalyst process over different catalysts. (f) Activity enhancement mechanism diagram of the Fe–S system. Reproduced with permission from ref. 122 Copyright 2021, American Chemical Society. Ref. 123 Copyright 2022, Wiley-VCH. | ||
To address the key problems of adsorbing and activating inert N2 molecules in photocatalytic NH3 synthesis, a trinuclear Fe–O cluster-based MOF (MIL-100(Fe)) catalyst with dual defects was synthesized using a non-thermal plasma-assisted strategy (Fig. 3(a)).124 By regulating the Fe–O coordination number with plasma power, the terminal ligand defects in the MIL-100(Fe)-200W catalyst were 7.7 times higher than that in the pristine catalyst under visible light irradiation (Fig. 3(b)). By accurately controlling the types and amounts of defects in MIL-100(Fe), the NH3 yield rate was remarkably increased. This was due to the formation of abundant exposed, coordinatively unsaturated Fe sites, which injected more d-electrons into the N2 molecules to produce the key intermediate *NNH, thereby decreasing the energy barrier of the RDS and efficiently activating N2 (Fig. 3(c)). These methods could also be applied to common inorganic semiconductor photoelectrodes, improving their NH3 synthesis performance by an order of magnitude or more through the rational manipulation of defect concentrations. Along these lines, ZnCr-Layered double hydroxide (LDH) nanosheets were produced by a simple coprecipitation method combined with an alkali etching strategy (Fig. 3(d)).125 The etched nanosheets displayed a higher performance for the photo-fixation of N2 to NH3 compared to the pristine nanosheets. The etched ZnCr-LDH nanosheets achieved an NH3 yield rate of 33.19 μmol g−1 h−1, which was 10 times higher than that of the pristine ZnCr-LDH nanosheets. The alkali method created a high concentration of oxygen vacancies and low-coordination metal centers, in which the unsaturated active sites promoted the adsorption and activation of N2, thus lowering the reaction energy barrier to NH3 and significantly increasing photocatalytic activity. In addition to introducing defect sites, heteroatom doping has similar effects. For example, Fe-doped W18O49 nanowires (Fe/W18O49) were obtained by an agent-modified solvothermal method and then mixed with black phosphorus quantum dots in an aqueous solution under vigorous stirring to form the final product, P–Fe/W18O49. The as-prepared P–Fe/W18O49 significantly promoted photocatalytic activity for fixing N2 into NH3. Specifically, an NH3 yield rate of up to 187.6 μmol g−1 h−1 was achieved, nearly an order of magnitude higher than that of the pristine W18O49 (18.9 μmol g−1 h−1).126 The increased photocatalytic N2 fixation activities were mainly attributed to the bulk Fe-doping, which elevated the conduction band position and the d-band center, thereby increasing the N2 reduction ability and suppressing charge recombination (Fig. 3(e) and (f)). Moreover, the anchored black phosphorus quantum dots could remarkably increase surface N2 adsorption and the cleavage of the NN bond through the W–P dimer adsorption mode.
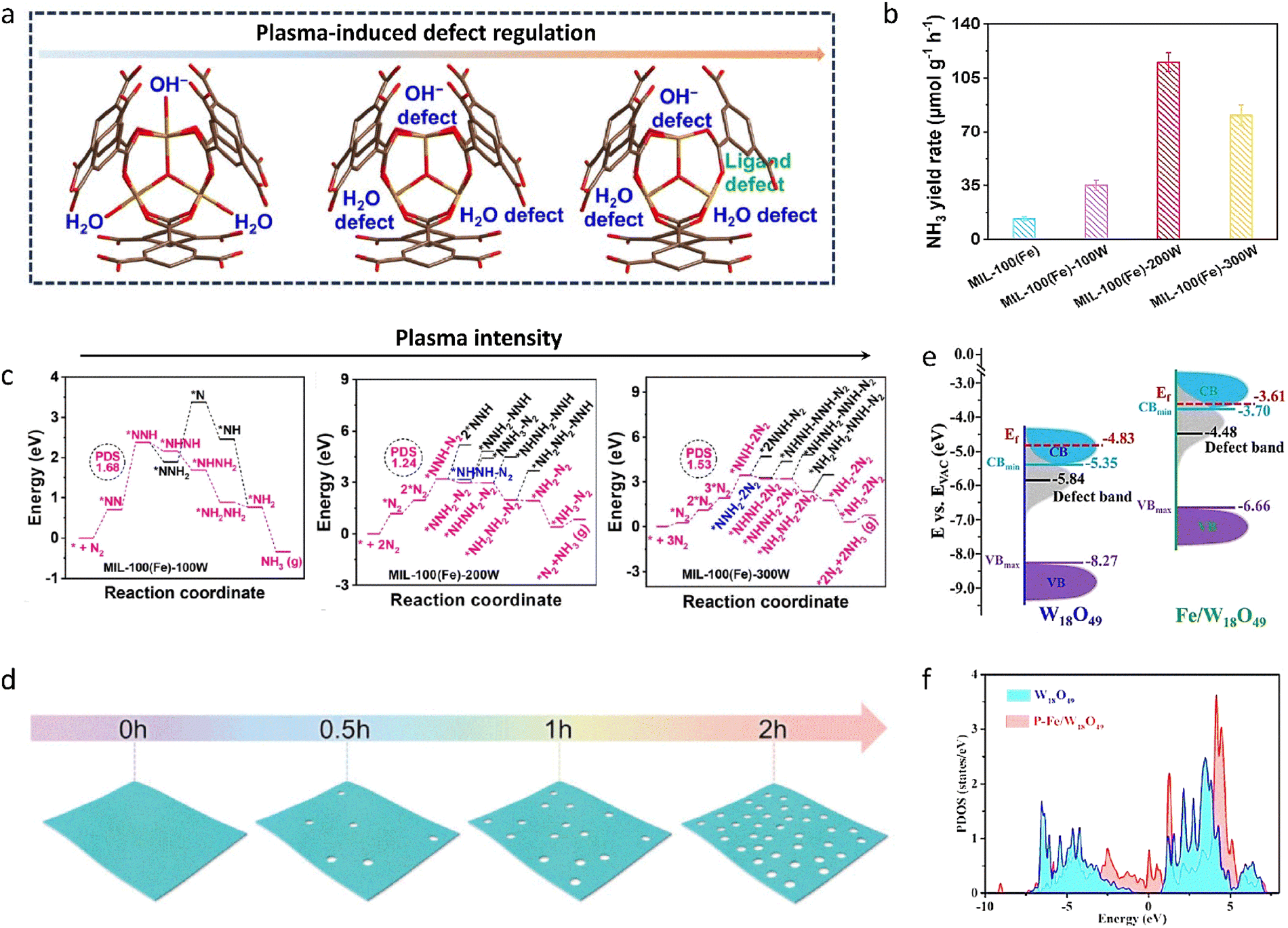 | ||
| Fig. 3 (a) Scheme of the plasma-assisted synthesis of MIL-100(Fe) with defects. (b) Photocatalytic NH3 yield rate for different catalysts. (c) Free energy diagrams of MIL-100(Fe) synthesized at different plasma intensities. (d) Scheme of the etching synthesis of ZnCr-LDH nanosheets at different alkali-treatment times. (e) The band structures of W18O49 and Fe/W18O49 samples. (f) The projected density of state diagram of W18O49 and P–Fe/W18O49 samples. Reproduced with permission from ref. 124 Copyright 2024, Wiley-VCH. Ref. 125 Copyright 2020, Wiley-VCH. Ref. 126 Copyright 2022, Wiley-VCH. | ||
Following the innovative ideas above, it was found that immobilizing or doping with precious metals can also address the high activation energy barrier and difficult adsorption of the N2 reactant. C-doped anatase titanium oxide (C-TiOx) nanosheets with a high concentration of Ti3+ species were fabricated using an H2O2-assisted thermal-oxidation etching (H2O2-TOE) strategy. The prepared C-TiOx nanosheets were further combined with Ru/RuO2 (Ru/RuO2/C-TiOx) and subsequently used as a photocatalyst for NH3 synthesis. A remarkable NH3 yield rate of 109.3 μmol g−1 h−1 was achieved.127 Benefiting from the high concentration of Ti3+ and the large surface area of the synthesized C-TiOx, the material could significantly chemisorb and activate N2 molecules and provide plenty of multi-electron transfer channels (Fig. 4(a)). Moreover, coupling this with a strong synergistic effect that simultaneously enhanced charge separation and transfer after introducing Ru species further boosted the photocatalytic performance of NH3 synthesis. Following this line of research, a variety of perovskite- and pyrochlore-structured tantalates with immobilized Ru species were prepared (Ru–K2Ta2O6−x) (Fig. 4(b)), followed by processing with a high-temperature solid-state reduction to introduce abundant oxygen vacancies and low-valent Ta. The resulting pyrochlore tantalates exhibited a higher conduction band energy level with a stronger reducing ability of excited electrons (Fig. 4(c)).128 The constructed catalyst, with its interface Schottky barrier, enhanced the adsorption of N2 molecules and the efficiency of photo-induced electron transfer from the photocatalyst to the N2 molecules (Fig. 4(d)). Compared with perovskite-type KTaO3−x, the NH3 yield rate of Ru–K2Ta2O6−x showed a 3.7-fold increase (335 μg g−1h−1). The Schottky barrier and spontaneous electron transfer between K2Ta2O6−x and Ru species improved photoexcited charge separation and accumulated energetic electrons to facilitate N2 activation (Fig. 4(e)), ultimately boosting the photocatalytic activity of NH3 synthesis. From the study above, the interaction between the active sites and the carrier materials is crucial for enhancing the activity of NH3 synthesis. Therefore, the development of novel carrier materials is expected to further modify the behavior of reactants. MOFs can serve as multifunctional substrates, not only to anchor active species with high dispersion but also to provide interconnected nanoreactors with ultra-high surface areas for the diffusion and collision of N2 molecules towards the active sites. In this context, Au nanoparticles (AuNPs) were immobilized within UiO-66 particles by impregnation and reduction to create a gas-membrane-solution reaction interface (Fig. 4(f) and (g)). Gaseous N2 molecules could directly enter the porous Au@UiO-66 membrane, effectively increasing the availability of N2 near the AuNPs, resulting in an NH3 yield rate of 18.9 mmol gAu−1 h−1 under visible light.129 The multifunctional AuNPs functioned as a photosensitizer, cocatalyst, and plasma accelerator to generate hot electrons, accelerating the conversion process from N2 to NH3 and reducing the activation energy barrier, ultimately enhancing the photocatalytic activity.
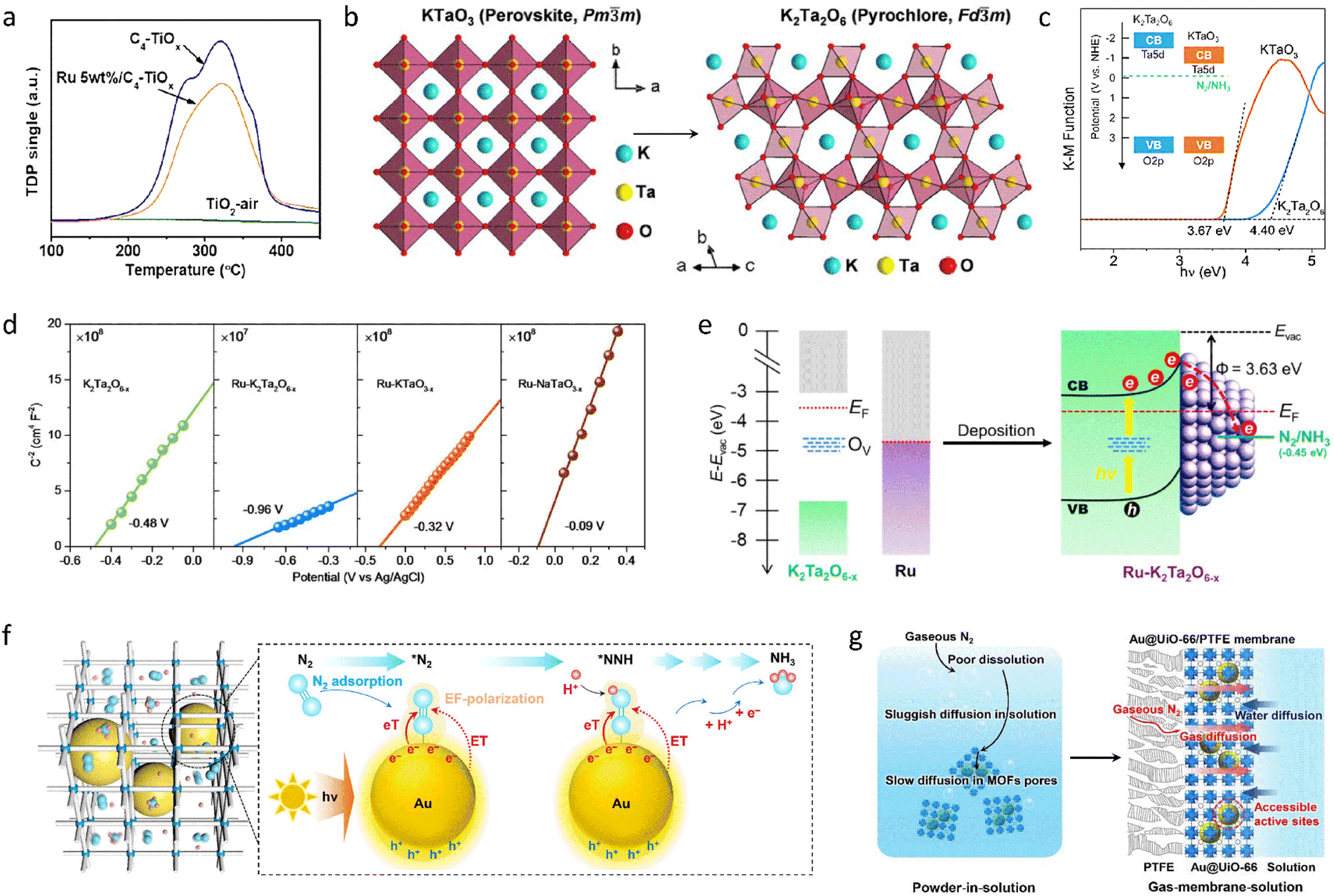 | ||
| Fig. 4 (a) Temperature-programmed desorption of N2 for different catalysts. (b) Crystal structure images of perovskite KTaO3 and pyrochlore K2Ta2O6. (c) Band gap of KTaO3 and K2Ta2O6 calculated from the Kubelka–Munk function versus photon energy. (d) Mott–Schottky plots of different catalysts. (e) The mechanism diagram of photocatalytic performance improvement for Ru–K2Ta2O6−x. (f) The nanostructure and photocatalytic N2 reduction step diagram of Au@UiO-66. (g) The diagram of a traditional homogeneous powder-in-solution system and a gas–membrane–solution reaction system. Reproduced with permission from ref. 127 Copyright 2021, Wiley-VCH. Ref. 128 Copyright 2023, Wiley-VCH. Ref. 129 Copyright 2021, American Chemical Society. | ||
Inspired by the uneven surface charge distribution, large network plane, and large cavity structure of graphdiyne (GDY), a highly active GDY heterojunction photocatalyst (GDY@Fe–B) was produced through a modified Glaser–Hay coupling reaction combined with co-precipitation.130 The prepared GDY@Fe–B catalyst showed remarkable performance for photocatalytic NH3 synthesis, with a very high NH3 yield of 1762.35 ± 153.71 μmol h−1 gcat.−1 and 100% selectivity. Adding GDY effectively changed the coordination environment and valence state of the Fe atoms. This atomic-level change in the metal atoms’ immediate environment adjusted the reaction mode and improved the photocatalytic activity. This strategy took full advantage of the structure and properties of GDY and showed that the new aggregate structure could control the form, coordination environment, and valence state of the Fe oxide, opening a new direction for the atomic-scale modification of photocatalysts. To further promote the chemisorption and activation of N2 at normal temperature and pressure, an MIIMIII-LDH (MII = Mg, Zn, Ni, Cu; MIII = Al, Cr) nanosheet photocatalyst was produced by a simple coprecipitation method (Fig. 5(a)).131 Among these, the CuCr-LDH photocatalyst had a significant NH3 yield rate of 184.8 μmol L−1 under UV-vis illumination. The introduction of oxygen-rich defects and Cu(II) ions resulted in a clear structural distortion, which increased the adsorption and activation of N2 molecules and accelerated the transport of electrons generated by visible light from the photoelectrode to N2 (Fig. 5(b) and (c)), thus promoting the NH3 synthesis.
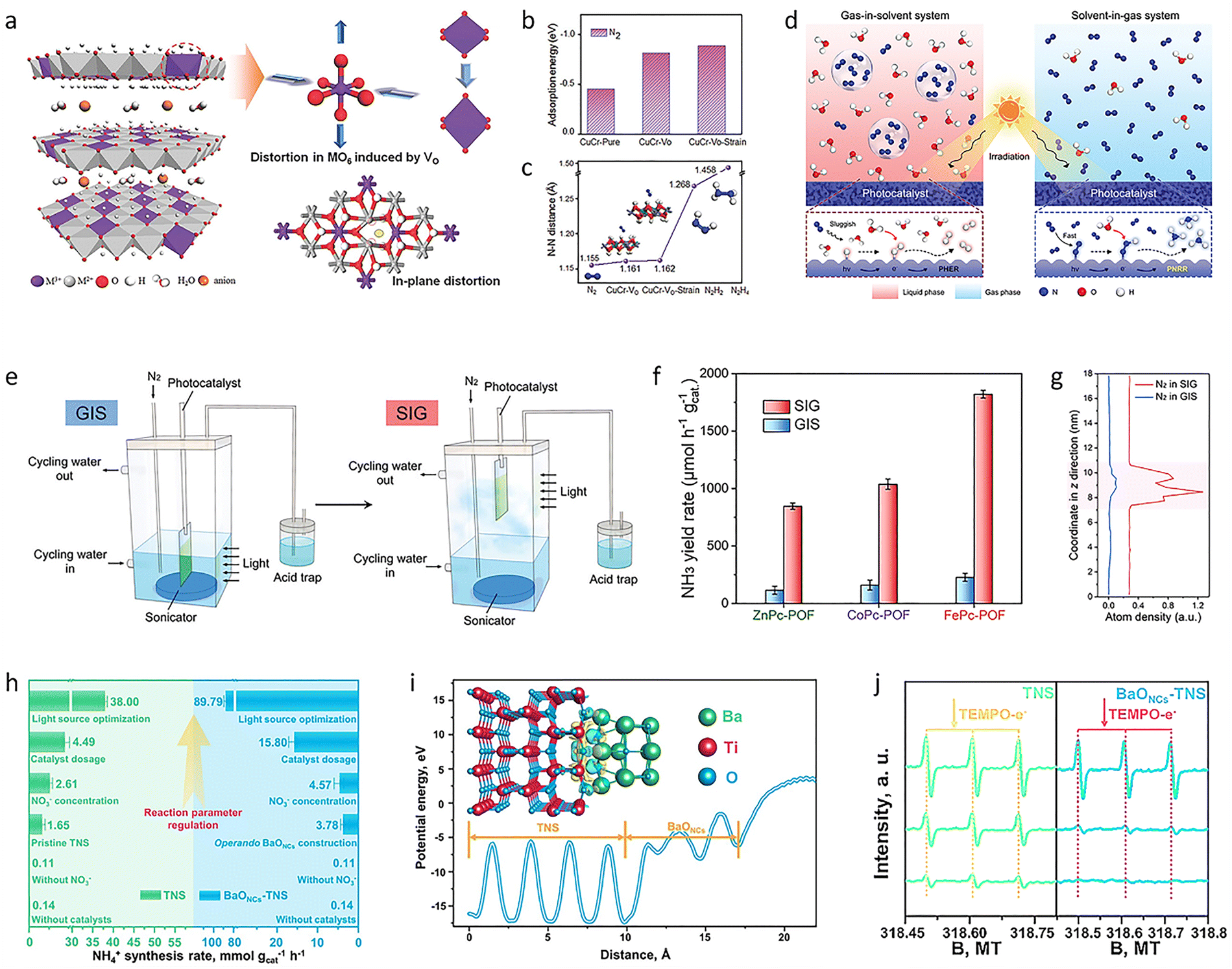 | ||
| Fig. 5 (a) Structure scheme of MIIMIII-LDH nanosheets. (b) Adsorption energies of N2 on different samples. (c) N–N distance of different samples and N2, N2H2, and N2H4 molecules. (d) Schematic illustration of a gas-in-solvent system and a solvent-in-gas system mechanism. (e) Reaction devices of a gas-in-solvent system and a solvent-in-gas system. (f) NH3 yield rate of different catalysts in a gas-in-solvent system and a solvent-in-gas system. (g) Distribution of N2 in a gas-in-solvent system and a solvent-in-gas system along the change of the z direction. (h) Photosynthesis NH4+ performance of TNS and BaONCs–TNS. (i) Charge difference distribution at the BaONCs–TNS interface for illustrating the charge transfer. (j) Electron paramagnetic resonance spectra of TNS and BaONCs–TNS. Reproduced with permission from ref. 131 Copyright 2017, Wiley-VCH. Ref. 132 Copyright 2023, Wiley-VCH. Ref. 143 Copyright 2022, Springer Nature. | ||
Additionally, it is well known that the low thermodynamic energy barrier of the competing HER and the very low solubility of N2 gas also led to inefficient NH3 synthesis in a solvent-heavy environment. Therefore, optimizing the conventional gas-in-solvent system is also expected to solve this fundamental reaction problem. For this reason, constructing a solvent-in-gas system, combined with porous organic framework materials, offers a great opportunity for NH3 production (Fig. 5(d) and (e)). Based on this, MPc-POF (M = Fe, Co, Zn) photocatalysts were prepared by the Schiff base reaction of tetraamino-metal-phthalocyanine with terephthalaldehyde under solvothermal conditions.132 The prepared catalyst, combined with a solvent-in-gas system, showed a continuous, ultra-high NH3 generation rate of 1820.7 μmol g−1 h−1, which is more than eight times higher than the conventional gas-in-solvent system (226.2 μmol g−1 h−1) (Fig. 5(f)). In the solvent-in-gas system, the MPc-POF operated directly in an N2 environment. This not only accumulated a high concentration of N2 in the framework (Fig. 5(g)) but also limited the contact between the solvent and the photocatalyst, efficiently inhibiting the competitive HER process and increasing the efficiency of photocatalytic NH3 synthesis.
In addition to optimizing the behavior of N2 molecules, the problem of photogenerated charge recombination, which leads to low NH3 synthesis efficiency, has also recently been addressed. TiO2 has been widely used in the photocatalytic synthesis of NH3.133–135 Until now, TiO2-based photocatalysts have been limited by their wide bandgap (3.2 eV for anatase and 3.0 eV for rutile) and high electron–hole recombination rate.136 Therefore, defects and nonmetal doping have been widely used to narrow the bandgap and shift its optical response to the visible light region.137,138 Following this idea, an anatase TiO2 (101) catalyst modified by boron doping was synthesized through a calcination method.139 The prepared B-doped TiO2 nanosheet achieved an NH3 yield rate of 3.35 mg h−1 g−1 under simulated sunlight irradiation, which was 1.74 times higher than that of pure TiO2. B-doping remarkably improved the photocurrent response, generating more electrons and accelerating the separation and transfer of photogenerated electron–hole pairs, which increased the photocatalytic NH3 synthesis performance. In addition, creating photocatalytic materials with heterogeneous interfaces is also expected to optimize the behavior of photogenerated charges and carriers. For instance, reduced CdS nanoparticles were grown in place on an oxidized WO3 surface through a hydrothermal method to produce a redox heterojunction catalyst. In this heterojunction, interfacial charge redistribution can effectively promote the selective accumulation of photogenerated electrons and holes in the CdS and WO3 components.140 Thus, N2 molecules could be activated and reduced to NH3 through a multi-electron process with a yield of 35.8 μmol h−1 g−1. Due to the tight interface and the built-in electric field created between the heterojunction components, the resulting internal electric field and Coulomb force could provide the driving force to induce the separation and migration of photogenerated carriers. Therefore, strongly reducing photoelectrons and strongly oxidizing photogenerated holes accumulated in the CdS and WO3 components, respectively, which overcame the thermodynamic barrier for converting N2 into NH3. To minimize the recombination of photogenerated carriers, a B-atom-doped g-C3N4 nanosheet (BCN) was prepared by a simple heat treatment method.141 The NH3 yield rate of the prepared BCN reached 313.9 μmol g−1 h−1, which was about 10 times that of pure g-C3N4. The created B–N–C bonds could not only stably anchor the exposed active N atoms on the surface of g-C3N4 but also effectively inhibit photogenerated carrier recombination by forming p–n junctions at the nanoscale. Meanwhile, the B atom also served as an active site for the adsorption and activation of N2 molecules. Photon-driven chemical processes are usually mediated by metal oxides, nitrides, and sulfides, but their light conversion efficiency is limited by carrier recombination. Therefore, early work has mainly focused on optimizing photogenerated carrier recombination.
Recently, a lithium hydride (LiH) catalyst has been developed for the photochemical synthesis of NH3. The photolysis of LiH is fundamentally different from the behavior of common oxide and nitride semiconductors. The photolysis of LiH under ultraviolet irradiation can produce long-lived, photon-produced electrons that reside in hydrogen vacancies, called F-centers. The generation of light-driven F-centers can provide an alternative method for carrier separation to support chemical transformations that are kinetically or thermodynamically challenging.142 Specifically, the LiH catalyst is easily photolyzed into H2 under ultraviolet irradiation, and the photolysis of LiH can produce localized ‘active’ electrons, forming an electron-rich state on the surface, which provides a favorable environment for the reduction of N2. Photoactivated LiH could split NN bonds to form N–H bonds under mild conditions. The yield rate was 518 μmol g−1 h−1. This work shows the development potential of hydride semiconductors in the photo-fixation N2 reaction, expanding the understanding of ‘hydride N2 fixation chemistry’ and providing ideas for developing a new generation of light-driven NH3 synthesis technology.
As an alternative feedstock, NO3−, with its high solubility and proton affinity, can be easily dissociated for sustainable NH3 production. Therefore, a general strategy was developed to construct subnanometric alkaline-earth oxide clusters (MONCs, where M = Mg, Ca, Sr, or Ba) anchored on TiO2 nanosheets (TNS) during the reaction. After the construction of BaONCs-TNS, a high NH4+ photosynthesis rate of 89.79 mmol gcat.−1 h−1 was reached with nearly 100% selectivity (Fig. 5(h)). A total NH4+ yield of 0.78 mmol was achieved within 72 hours.143 The local interfacial structure was precisely tailored to strengthen charge transfer at the BaONCs/TNS interface (Fig. 5(i)). It was then revealed that the eight-electron transfer reaction for NO3− reduction was notably accelerated, achieving a high rate for sustainable NH4+ photosynthesis (Fig. 5(j)). To further achieve high-selectivity NO3− reduction for NH3 synthesis, it is necessary to improve the overall photocatalytic efficiency, because both oxidation and reduction reactions together determine the redox efficiency. Under light irradiation, the catalyst produces an equal number of electrons (e−) and holes (h+) at the same time and carries out a reduction half-reaction and an oxidation half-reaction, respectively. However, most studies only focus on one half of the reactions. The precise regulation of the oxidation reaction can be used as an effective strategy to directly adjust the efficiency and selectivity of the reduction reaction for NH3 synthesis. In this regard, a redox system was formed by combining NO3− reduction with ethylene glycol oxidation. Specifically, Cu–O–Ti clusters anchored on TiO2 nanosheets (CuOx@TNS) were designed to regulate the selective reduction of NO3−. The NH3 photosynthesis rate of CuOx@TNS was 16.04 ± 0.40 mmol gcat.−1 h−1, and the selectivity for NH3 was close to 100%.144 The molecular mechanism of the synergistic reaction between NO3− reduction and ethylene glycol oxidation showed that introducing ethylene glycol oxidation could effectively promote the consumption of h+ and generate alkoxy radicals (˙R). At the active site of the Cu–O–Ti clusters, photogenerated h+ preferentially oxidized ethylene glycol to produce ˙R, which efficiently inhibited the formation of strongly oxidizing hydroxyl radicals. Meanwhile, the construction of Cu–O–Ti clusters increased the production of e−, thus promoting NH3 synthesis efficiency. The overall photocatalytic redox efficiency was improved by matching the oxidation half-reactions and constructing high-performance catalytic sites, and the directed regulation of the reduction half-reactions at the active sites was realized, leading to efficient and highly selective photocatalytic NO3− reduction.
4. Electrochemical synthesis of ammonia
4.1 Nitrogen source with a zero oxidation state, N2
The traditional H–B approach for NH3 synthesis involves reacting high-purity N2 and H2 under harsh, high-temperature and high-pressure conditions,145 resulting in low efficiency, significant energy consumption, and large greenhouse gas emissions. The chemical inertness of N2 makes the reduction of N2 at room temperature and atmospheric pressure a great challenge.28 Fortunately, the electrochemical reduction of N2 offers a sustainable method to overcome this bottleneck.146–148 In this section, the types of catalytic materials and the performance of electrocatalytic NH3 synthesis are summarized in Table 2. Specifically, a single-atom dispersed FeSA–N–C catalyst was prepared by carbonizing a polypyrrole–Fe coordination compound at 600 °C. The single-atom Fe catalyst enabled the electroreduction of N2 to NH3 with a superior FE of 56.55% and a large yield rate of 7.48 μgh−1mg−1 at 0 V vs. RHE, far surpassing the metal-free N–C catalyst (a FE of 9.34% and a yield rate of 4.75 μgh−1mg−1). The Fe active sites could lower the free energy barrier for *N2 adsorption and suppress H2 production. The central Fe sites also decreased the energy barrier of the rate-determining step, *H2O–N2 + H+ + e− → *H2O–NNH, in which protons and electrons tend to attack the *N2 to facilitate hydrogenation, thus boosting the NRR performance.149 Besides metal single-atom catalysts that can efficiently separate the NRR from the HER, modulating the electron density near the catalyst's active sites is also an effective strategy. For instance, a catalyst of Cu NPs anchored on a polyimide (PI) surface with different conjugating degrees was prepared by a wet impregnation method. A standard NRR test over the Cu/PI-300 electrode with a Cu content of 5% resulted in the best NH3 FE of 6.56% at −0.3V vs. RHE, with an NH3 yield rate of 12.4 μg h−1 cm−2, outperforming the catalyst of Cu NPs fixed on a carbon substrate, which had an FE of only 0.17% and a very low yield rate of 0.7 μg h−1 cm−2. The electron redistribution at the interface of PI and Cu NPs efficiently modulated the electron density of the Cu NPs, increasing the adsorption capacity for N2 and activating the N2 molecules. At the same time, the dissociation step from *NNH4 to *NH2 was also tailored by the electron-deficient Cu surface, thus boosting the NH3 generation yield and selectivity.150 The present result provides guidance in the search for catalysts to optimize this difficult reaction, from impossible to high yield. In addition to optimizing the electronic structure of catalysts by using the interaction between the supports and the active metals, changing the structural configurations of the catalyst itself also has a similar effect on improving NRR activity. For example, MoS2 catalysts with different structural configurations, including 2H, 1T′, and 1T′′ crystal structures, were synthesized by the deintercalation of potassium from a KMoS2 crystal.151 Among them, the 1T′′ MoS2 catalyst exhibited the best NRR performance, with the highest NH3 yield rate of 9.09 μg h−1 mg−1 at −0.3 V vs. RHE, which was roughly 2 times that of 1T′ MoS2 and 9 times that of 2H MoS2. The NH3 FE of 13.6% for 1T′′ MoS2 also surpassed that of the 2H and 1T′ MoS2 counterparts. The formation of zigzag and diamond-like Mo–Mo chains in the 1T′′ phases can efficiently change the local electron density and enhance N2 adsorption, thereby improving the NRR kinetics. Compared with only boosting catalytic performance, metal-free materials offer the clear advantage of avoiding metal ion release, thereby reducing the environmental impact. Recently, a type of B4C nanosheet was synthesized by the liquid-phase exfoliation method, which showed competitive NRR activity while being environmentally friendly.152 This metal-free B4C was capable of achieving an NH3 yield rate and an FE as high as 26.57 μg h−1 mgcat.−1 and 15.95% at −0.75 V vs. RHE, respectively, outperforming most reported aqueous-based NRR catalysts, such as Au nanorods (an NH3 yield rate of 1.65 μg h−1 cm−2 and a FE of 3.88%)15 and Pd/C (an NH3 yield rate of 4.5 μg h−1 mg−1 and a FE of 8.2%).153 This study opens an exciting new avenue for the rational design of metal-free electrocatalysts for NH3 synthesis with robust performance.
| Catalysts | Nitrogen sources | Faraday efficiency (%) | Yield rate | Ref. |
|---|---|---|---|---|
| FeSA–N–C | N2 | 56.55 | 7.48 μgh−1 mg−1 |
149 |
| Cu/PI-300 | N2 | 6.56 | 3.44 μg h−1 mg−1 | 150 |
| 1T′′ MoS2 | N2 | 13.6 | 9.09 μgh−1 mg−1 |
151 |
| B4C | N2 | 15.95 | 26.57 μg h−1 mg−1 | 152 |
| Ru-LCN | NO | 65.96 | 765.34 μg h−1 mg−1 | 172 |
| RuGa | NO | 72.3 | 5450.2 μg h−1 mg−1 | 178 |
| *Cu foam | NO | 93.5 | 8790.7 μg h−1 cm−2 | 78 |
| *Cu electrode | NO | 90 | 30702 μg h−1 cm−2 |
167 |
| CoS1−x | NO | 53.62 | 1492.41 μg h−1 mg−1 |
180 |
| MoS2/GF | NO | 76.6 | 411.8 μg h−1 mg−1 |
55 |
| Co1/MoS2 | NO | 87.7 | 7398.4 μg h−1 mg−1 | 168 |
| NiFe LDH | NO | 29.6 | 634.67 μg h−1 mg−1 | 184 |
| Bi NDs | NO | 89.2 | 1194 μg h−1 mg−1 | 52 |
| *a-B2.6C@TiO2/Ti | NO | 87.6 | 3678.6 μg h−1 cm−2 | 187 |
| *Ru–Cu NW/CF | NO2− | 94.1 | 211730 μg h−1 cm−2 |
189 |
| Pd/CuO | NO2− | 91.8 | 906.4 μg h−1 mg−1 | 192 |
| *Cu2O/CF | NO2− | 94.21 | 7510.73 μg h−1 cm−2 | 207 |
| Ni3B@NiB2.74 | NO2− | 59.0 | 8427.75 μg h−1 mg−1 | 210 |
| Ni@JBC-800 | NO2− | 83.4 | 4117.3 μg h−1 mg−1 | 211 |
| C/Co3O4 | NO2− | 100 | 8198 μg h−1 mg−1 | 196 |
| Ru-ST-12 | NO3− | — | 94520 μg h−1 mg−1 |
64 |
| Ru-CuNW | NO3− | 96 | 191250 μg h−1 mg−1 |
228 |
| Rh@Cu-0.6% | NO3− | 93 | 43220 μg h−1 mg−1 |
229 |
| Pd (111) | NO3− | 35 | 306.8 μg h−1 mg−1 | 199 |
| *a-RuO2/CF | NO3− | 97.46 | 1968.6 μg h−1 cm−2 | 231 |
| *CoP NAs/CF | NO3− | ∼100 | 16252 μg h−1 cm−2 |
214 |
| Fe/Ni2P | NO3− | 94.3 | 2085 μg h−1 mg−1 | 239 |
| *Cu0/GDYNA | NO3− | 81.25 | 262650 μg h−1 cm−2 |
243 |
| Cu MNC-x | NO3− | 94.8 | 92922 μg h−1 mg−1 |
244 |
| Fe SAC | NO3− | 75 | 20000 μgh−1mg−1 |
242 |
| FeMo–N–C SAC | NO3− | 94 | 612 μg h−1 mg−1 | 225 |
4.2 Nitrogen source with a plus two oxidation state, NO
Despite its natural abundance, N2 is kinetically inert,19,154 and at the same time, the competitive HER has faster kinetics. Therefore, scaling up this transformation technology to an industrial level is challenging. Currently, NO is a major environmentally harmful compound produced by the burning of fossil fuels in industrial processes.55 Its increased concentrations can cause serious environmental problems, such as ozone depletion, smog, acid rain, and eutrophication, and eventually contribute to global warming,52,56,155–157 which poses a potential risk to human health.75,158 Currently, the selective catalytic reduction of NO is used to convert it into harmless N2.159 However, this method usually suffers from secondary pollution and high costs,55,160 and many selective catalysts only show good activity at high temperatures.159 Through this process, it can be seen that NO, with its low bond dissociation energy, is more reactive than N2 molecules.7,161 Meanwhile, the end products of NO reduction are often high-value products, such as NH3, hydroxylamine (NH2OH), and nitrous oxide (N2O), which can be used in the production of caprolactam for the nylon industry.162,163 A mild reaction environment and simplified reaction facilities contribute to the practical appeal of electrochemical techniques driven by renewable electricity. Taken together, the electrochemical NORR is an encouraging concept that shows great potential for both high-value NH3 synthesis and NO elimination.164 However, the reduction potential of the NORR process, which involves a five-electron transfer, is approximately 0.77 V vs. RHE,165 and it also exhibits slow reaction kinetics and multiple intermediates, leading to an unsatisfactory NH3 yield rate and FE.78,166–168 Therefore, an in-depth investigation into the NORR mechanism and effective design strategies for highly active electrocatalysts will be discussed in this section.A large number of previous studies have shown that two-dimensional materials have an ultra-high specific surface area with abundant defect sites and a controllable electronic structure, which can promote the adsorption of reactive small molecules and provide a large number of active sites, while also offering an atomic-layer-thick platform that makes it easy to study the structure–activity relationship.169 Therefore, they are considered to be powerful catalysts for the electrosynthesis of NH3.31 Meanwhile, according to relevant reports, a series of precious metals have been shown to have excellent NORR activity.170,171 Therefore, the excellent properties of dimensional engineering coupled with precious metals are expected to promote the research and development of the NORR. For example, low-coordination-number Ru nanosheets synthesized by a hydrothermal method were treated with Ar/H2 plasma.172 The prepared electrocatalyst exhibited excellent NORR performance; at a low NO concentration of 1%, it showed a high FE of 65.96% and a high production rate of 45.02 μmol h−1 mg−1, clearly outperforming its counterpart of high-coordination-number Ru nanosheets, which had a low FE of 37.25% and a yield rate of 25.57 μmol h−1 mg−1. The excellent NORR activity came from the decrease in the coordination number of Ru, which led to an upward shift of the d-band center and a decrease in the potential-determining step from *HNO to *HNOH; the adsorption of NO and the activation of the NO bond were also promoted. Unlike dimensional engineering, an electron-rich environment also helps to improve the intrinsic NORR activity of Ru-based catalysts. Examples include anchoring Ru atoms on an electron-rich substrate or metallizing Ru species using metal substrates with lower electronegativity.173–177 Specifically, the body-centered cubic RuGa intermetallic compound, in which Ga atoms occupy the vertices of the unit cell with Ru atoms fixed at the body center, was synthesized by a substrate-anchored thermal annealing method.178 The RuGa intermetallic compound exhibited an excellent NH3 FE of 72.3% and production rate of 320.6 μmol h−1 mg−1 for NH3 synthesis at −0.2 V vs. RHE, which was higher than that of hexagonal close-packed Ru NPs, with a lower FE of 59.8% and a yield rate of 235.4 μmol h−1 mg−1. After the introduction of Ga species, the RuGa intermetallic compound showed metallic characteristics with a band gap of 0. At the same time, the bimetallic compound displayed a lower energy barrier for the RDS step (adsorbing and activating the *HNO intermediate), which decreased from the original 0.88 eV to 0.49 eV. These factors together accelerated the transformation of NO by speeding up the activation of N–O bonds via electron supply from the Ru atoms.
Considering that the low abundance and high price of noble metals and their alloys hinder their industrial applications,52 the electrocatalysts made from earth-abundant elements are inevitable. Recent DFT calculations show that Cu exhibits excellent activity for the NORR as well as superior selectivity for NH3 over H2. Therefore, a commercial Cu foam was directly used as an independent, self-supporting catalyst for NORR reactions, which can even reach the level of the H–B process, displaying a record-high yield rate of 517.1 μmol h−1 cm−2 and an NH3 FE of 93.5% in 0.25 M Li2SO4 at −0.9 V vs. RHE.78 Thus, numerous Cu-based catalysts have been explored for facilitating the NORR process. For example, the working electrode of an NORR cell was produced by depositing a commercial Cu ink (Cu particle size <100 nm) on a gas diffusion layer with a loading of 0.25 mg cm−2. This prepared Cu electrode exhibited a high NH3 FE of roughly 90% and a yield rate of 1806 μmol cm−2 h−1, which was superior to electrodes formed by deposition of other metals, including Fe, Co, Ni, Pd, Ag, and Pt. Furthermore, the relationship between NO concentration and N–N coupling was discovered through the NORR process using this Cu electrode at different NO concentrations. N–N coupling and single N products were preferred at high and low NO coverage rates, respectively. In addition, by exploring the local pH of the reaction environment, it was found that the Cu-based catalyst could reach a nearly 100% NH3 FE of 100% under acidic conditions, while the FE of NH3 dropped to about 80% under alkaline conditions, which provided guidance for optimizing the selectivity of NORR products.167 Aiming to reduce NO in gaseous pollutants at a low cost, transition-metal dichalcogenide materials also exhibit admirable electrocatalytic ability for facilitating the NORR process.55,179,180 For example, CoS containing S defects (CoS1−x) was prepared via plasma treatment for 2 minutes at 300 W in an Ar atmosphere. The as-synthesized CoS1−x showed excellent NORR activity in 0.2 M Na2SO4, with a high NH3 FE of 53.62% and a large NH3 yield rate of 44.67 μmol cm−2 h−1, significantly outperforming natural CoS, which had a lower FE of 36.68% and a small NH3 yield rate of 27.02 μmol cm−2 h−1. Due to the localization of electrons near the S defects, the activation process of NO was promoted. Meanwhile, the *NO + H → *NOH step was also accelerated, thus showing remarkable NORR performance.180
Compared with defect engineering to accelerate the NORR process, doping engineering for transition-metal dichalcogenide materials also shows excellent activity. Recently, MoS2 has been reported as an effective NORR catalyst. Benefiting from a large number of positively charged Mo sites, which could facilitate NO adsorption and activation via an ‘acceptance–donation’ mechanism and disfavor the binding of protons as well as the coupling of N–N bonds, the MoS2 electrocatalyst attained a high FE of 76.6% and an NH3 yield rate of 99.6 μmol cm−2 h−1 in an acidic electrolyte.55 The results above indicate that the single MoS2 catalyst is expected to improve its catalytic activity through further modification. For instance, a Co1/MoS2 catalyst with Co1–S3 active sites was successfully constructed by an impregnation strategy in a Co(Ac)2·4H2O solution, in which a Co atom was introduced onto the MoS2 surface supported on carbon cloth. The as-synthesized Co1/MoS2 catalyst exhibited an extremely high FE of 87.7% and an outstanding NH3 yield of 217.60 μmol cm−2 h−1, substantially outperforming pristine MoS2. In the Co1–S3 active sites formed by the doping of foreign metal atoms, Co and S species jointly activated NO and improved its coverage, while the adsorption of *H was thermodynamically prohibited, clearly indicating the advantage of doping engineering.168
In addition, it should also be clear that the formation of N2 and N2O by-products in the NORR process is very thermodynamically favorable.181,182 Therefore, to further achieve high selectivity of the NORR to NH3, it is necessary to construct a suitable catalyst that can effectively prevent both N–N bond coupling and *N diffusion.164 At the same time, based on the discussion above, we believe that the results of combining dimensional engineering and doping engineering to regulate the catalyst will be very interesting and fascinating. Along these lines, based on high-throughput first-principles calculations, by confining 27 transition metal atoms (including all 3d, 4d, and 5d transition metal elements, except Zn, Cd, and Hg) on an MoS2 platform with an S defect, it was found that Ti, Fe, Co, Ni, Cu, and La@MoS2−x showed excellent NORR performance in theory.183 By anchoring these transition metal atoms through the S defects, the confined space of the S defect sites effectively prevents the approach of two NO molecules by strongly binding the N atoms, thus hindering N–N bond coupling to form N2O2. The under-coordinated Mo edges as active sites would thus guarantee the high selectivity of NO-to-NH3.183
In addition to optimizing the NORR by regulating the adsorption and activation of NO and reducing the occurrence of by-products, decreasing the hydrogen evolution kinetics of the catalysts is also a powerful means. For instance, NiFe LDH was synthesized on Ni foam by a one-step hydrothermal strategy at 120 °C with the assistance of ethylene glycol. The produced NiFe LDH displayed an excellent FE of 82% and a large NH3 yield rate of 112 μmol cm−2 h−1, beyond the reported Ru-based catalysts with an NH3 FE of 29.6%. The Ni foam with its porous structure could prevent the accumulation of catalysts, exposing numerous active sites to adsorb or transport reactants, and the NiFe-LDH nanosheets effectively inhibited the HER, thus showing excellent NORR activity.184 Furthermore, due to the potential environmental contamination caused by the extensive use of metal-based catalysts, a non-toxic and environmentally friendly metal, Bi, has been reported as a promising alternative for high-efficiency electrochemical applications.185,186 Therefore, highly active Bi nanodendrites were prepared by electrochemical deposition on the surface of a Ti film for 1200 s in a 1.0 mM BiCl3 solution at −0.5 to −0.8 V vs. Ag/AgCl. They attained a high FE of 89.2% at −0.5 V vs. RHE and a remarkable NH3 yield rate of 1194 μg h−1 mgcat.−1, which clearly outperformed Bi powder. The excellent catalytic activity of the NORR came from the spiky structure of the Bi nanodendrites, which could amplify the local electric field near the tip and promote the adsorption and activation of NO molecules through an effective charge redistribution mechanism.52
In the past, various precious metal- and non-precious metal-based catalysts have been used to accelerate the NORR process. The development of non-metallic materials with high electrical conductivity, excellent electrochemical stability, and good corrosion resistance is inevitable for environmental sustainability and large-scale industrial applications.
Recently, for instance, a kind of amorphous boron carbide (B2.6C), as a free-standing electrode, was successfully synthesized through the magnetron sputtering technique onto a TiO2/Ti substrate.187 It showed sustained and superb NORR activity for synthesizing NH3, with a high FE of 87.6% and an amazing yield rate of 3678.6 μg cm−2 h−1, superior to the pristine TiO2/Ti plate, which had an FE of 42.6% and an NH3 yield rate of 563.5 μg cm−2 h−1. Benefiting from the enlarged electrochemical active area and the B–C bonds over the B2.6C layer, external electrons were effectively injected into the NO molecules, and the bond length of N–O was adjusted from 1.15 Å to 1.41 Å, jointly activating the NO molecules and thus exhibiting excellent NORR performance.
4.3 Nitrogen source with a plus three oxidation state, NO2−
Currently, the high dissociation energy of the NN triple bond (941 kJ mol−1), the extremely low solubility of N2 in aqueous media, and the overwhelming competition from the HER are the main reasons preventing the widespread application of the NRR.21,188,189 Therefore, it is urgent to find a suitable N source for efficient electrosynthesis. Currently, large quantities of nitrite (NO2−) flow into surface water and seep into groundwater as a result of agricultural production and industrial discharges, which not only damages ecosystems but also poses a serious threat to human health.92,190–192 Specifically, the ingestion of NO2− can lead to the production of N-nitrosoamine byproducts, which have been shown to be carcinogens. It also damages hemoglobin, leading to cell degradation and even brain damage in infants.193 Therefore, the recovery and transformation of NO2− are of great significance to human health and the nitrogen cycle.
The conversion of environmentally harmful NO2− to high-value NH3 through electrochemical reduction is a promising green and sustainable solution.194,195 In fact, NO2− is an attractive source of nitrogen because of its lower dissociation energy and better solubility compared to N2.192 Meanwhile, compared with the nitrogen resource NO, NO2− exhibits a higher reduction potential of 0.897 V vs. RHE. However, the electrocatalytic conversion process from NO2− to NH3 (NO2RR) usually faces slow kinetics and a complex distribution of products, which greatly hinders the progress of NH3 synthesis.197 Therefore, this section mainly summarizes the research on high-efficiency NO2RR electrocatalysts and provides guidelines for future research and design methods in this field.
At present, among various transition metal-based catalysts, Cu-based materials have become the most attractive catalysts for the electrochemical conversion of NOx to value-added NH3.189,198 For example, a Cu nanowire anchoring Ru atom (Ru–Cu NW/CF) electrocatalyst was synthesized by a cation exchange strategy, in which Cu foam covered with Cu(OH)2 nanowires was immersed in a 5.0 mmol L−1 RuCl3 solution for 2 hours, followed by annealing at 180 °C to complete the Ru loading. The synthesized Ru–Cu NW/CF displayed an outstanding FE of 94.1% and a large NH3 yield rate of up to 211.73 mg h−1 cm−2 at −0.6 V vs. RHE, which was approximately 5.0 times higher than that of the Cu NW/CF. By introducing single-atomic Ru into the Cu lattice, the adsorption capacity of the catalyst for NO2− was improved. Meanwhile, the Ru–Cu NW/CF had a faster RDS from *NO to *NOH compared with the Cu NW/CF, thus displaying excellent NO2RR performance.189 Besides, the modulation of morphology and structure and the introduction of 4d-block atoms like Pd are attractive strategies to improve the activity of Cu-based catalysts.192,199,200 For example, CuCl2 was reacted with a Na2PdCl4 solution in a water bath at 60 °C to prepare two-dimensional Pd/CuO catalysts with heterogeneous interfaces and porosity. The obtained Pd/CuO catalyst exhibited an excellent NH3 yield rate of 906.4 μg mg−1 h−1 and a high FE of 91.8%, which was significantly better than Pd NPs (an FE of 29% and an NH3 yield rate of 298.4 μg mg−1 h−1) and CuO (an FE of 86% and an NH3 yield rate of 920.4 μg mg−1 h−1). A built-in electric field was spontaneously constructed at the Pd/CuO heterointerface, which optimized the electronic structure of Pd and Cu, thereby enhancing the electrical conductivity. Meanwhile, Pd NPs could also increase the selectivity for NH3 by forming adsorbed H, which jointly boosted the NO2RR performance.192
In addition to transition metals in the 4d region, 5d region transition metals have the potential to further improve the selectivity of the NO2RR. For instance, based on DFT calculations, it was found that compared to precious metals in the 4d region (e.g., Ru, Pd), precious metals in the 5d region could have higher theoretical H surface coverages.193,201 Considering the economic problems in the catalyst preparation process and that alloying engineering could optimize NO2RR activity,202–204 a 5d region noble metal alloy is expected to further improve the selectivity and formation rate of NH3. Recently, a CuxIr1−x (x = 10, 25, and 100) nano-alloy was successfully prepared in a microwave reactor at 125 °C. The developed CuxIr1−x exhibited 100% selectivity for NH3 in a near-neutral environment, with performance similar to that of commercial Pd NPs for the NO2RR in industry.193 The H-coverage of the Ir atom was higher than that of the Pd atom, indicating that Ir could thermodynamically accommodate more adsorbed H on its surface, thus showing higher NH3 selectivity. Although noble metal-based catalysts with excellent activity have been constructed using single-atom and alloying engineering, non-noble catalysts are still the most promising candidates for the industrialized production of NH3.205,206 For example, a simple electrode of Cu2O particles supported by Cu foam (CF@Cu2O) was synthesized by an anodic oxidation process at 5.0 V for 5.0 minutes, combined with an annealing treatment at 550 °C. The CF@Cu2O catalyst displayed a remarkable FE of 94.21% combined with a high production rate of 7510.73 μg h−1 cm−2, superior to the Cu foam electrode (an FE of 55.1% and an NH3 yield rate of 1450.12 μg h−1 cm−2). Compared with Cu (111), Cu2O (111) had a lower energy barrier for the RDS from *NO to *NOH, and it could inhibit the HER process, thereby facilitating the NO2RR.207 Besides, considering that an acceptable FE and NH3 yield rate were usually obtained at high overpotentials, another 3d metal, Ni, with a strong adsorption capacity for NO2−, is expected to solve these difficulties.208 However, it should be noted that Ni atoms have very high activity for the HER.209 Recently, a type of Ni-based catalyst of surface B-rich core–shell nickel boride NPs (Ni3B@NiB2.74) was prepared by a wet chemistry method, in which Ni(CH3CO2)2·4H2O was stirred with NaBH4 and annealed at 300 °C (Fig. 6(a) and (b)). The prepared Ni3B@NiB2.74 NPs exhibited about 100% conversion efficiency of NO2− to NH3, with a significant NH3 yield rate of 198.3 μmol cm−2 h−1 at −0.3 V vs. RHE. The NH3 FE of the control Ni NPs, 59.0%, was about 1.6 times lower than that of Ni3B@NiB2.74 (Fig. 6(c)). The surface-enriched, electron-deficient B sites promoted the adsorption of liquid NO2−, which also transferred electrons to the Ni sites to prevent their oxidation and diluted the Ni sites available to participate in the HER (Fig. 6(d) and (e)), thus improving the catalytic activity of the NO2RR (Fig. 6(f) and (g)).210 In addition to modification methods involving non-metallic element doping, the selection of a suitable catalyst support is also an effective strategy. For example, juncus soaked in a Ni salt solution was annealed at a high temperature for about 2.0 hours to successfully produce a kind of Ni NP-based catalyst on juncus-derived biomass carbon (Ni@JBC-800).211 When tested for the NO2RR, the Ni@JBC-800 catalyst was capable of exhibiting a remarkable NH3 yield rate of 4117.3 μg h−1 mg−1 and a high FE of 83.4% at −0.5 V vs. RHE, surpassing most reported Ni-based catalysts, such as Ni2P nanosheets (an FE of 90.2% and an NH3 yield rate of 2692.2 μg h−1 cm−2)212 and Ni–NSA–VNi (an FE of 88.9% and an NH3 yield rate of 4011.6 μg h−1 cm−2).213 Benefiting from biomass-derived carbon as a carrier, it could not only prevent the agglomeration of catalysts but also improve conductivity. Meanwhile, the empty tubular structure of juncus in the 3D interconnect also facilitated gas and electrolyte diffusion, increasing NO2RR activity. In addition, promoting the transport of ions or electrons by inducing the redistribution of charges near the doping sites is also a powerful means to improve catalytic activity.214–216 Recently, carbon-doped hollow nanotubes were prepared via the in situ topotactic conversion of C2O42−-doped CoC2O4 to C4+-doped Co3O4 at 400 °C (C/Co3O4). Such C/Co3O4 exhibited an ultrahigh FE of about 100% from −0.1 V to −0.6 V vs. RHE and a large NH3 yield rate of 4.10 mg h−1 cm−2 at −0.6 V vs. RHE, significantly outperforming the Co3O4 catalyst, which had a poor NH3 yield rate of 1.78 mg h−1 cm−2. C-doping enhanced the adsorption and activation ability for NO2− ions and also led to a fast charge transfer process. At the same time, this, coupled with the lower energy barrier required for the potential-determining step (*N + e− + H2O → *NH + OH−) jointly promoted the NO2RR process.196 In addition to optimizing the morphology and electronic structure of catalysts, optimizing the reaction conditions can also be used as a strategy to enhance the activity of the NO2RR. For example, a continuous and scalable plasma–electrochemical hybrid technology was developed for NH3 production (Fig. 6(h)), which coupled a plasma reactor with an electrochemical H-cell to activate air and improve the reaction rate and energy efficiency (Fig. 6(i)–(m)). The working electrode was Cu foam covered with Cu nanowires, which exhibited an excellent FE of 100% and a yield rate of 45 nmol s−1 cm−2.217 Compared with the traditional NH3 electrosynthesis process, it was worth noting that the plasma-assisted conversion system was characterized by lower energy consumption (253 kW h kg−1 NH3) (Fig. 6(n)). Therefore, this hybrid technology facilitates on-site, decentralized production of NH3 that can be powered by cheap, surplus renewable resources.
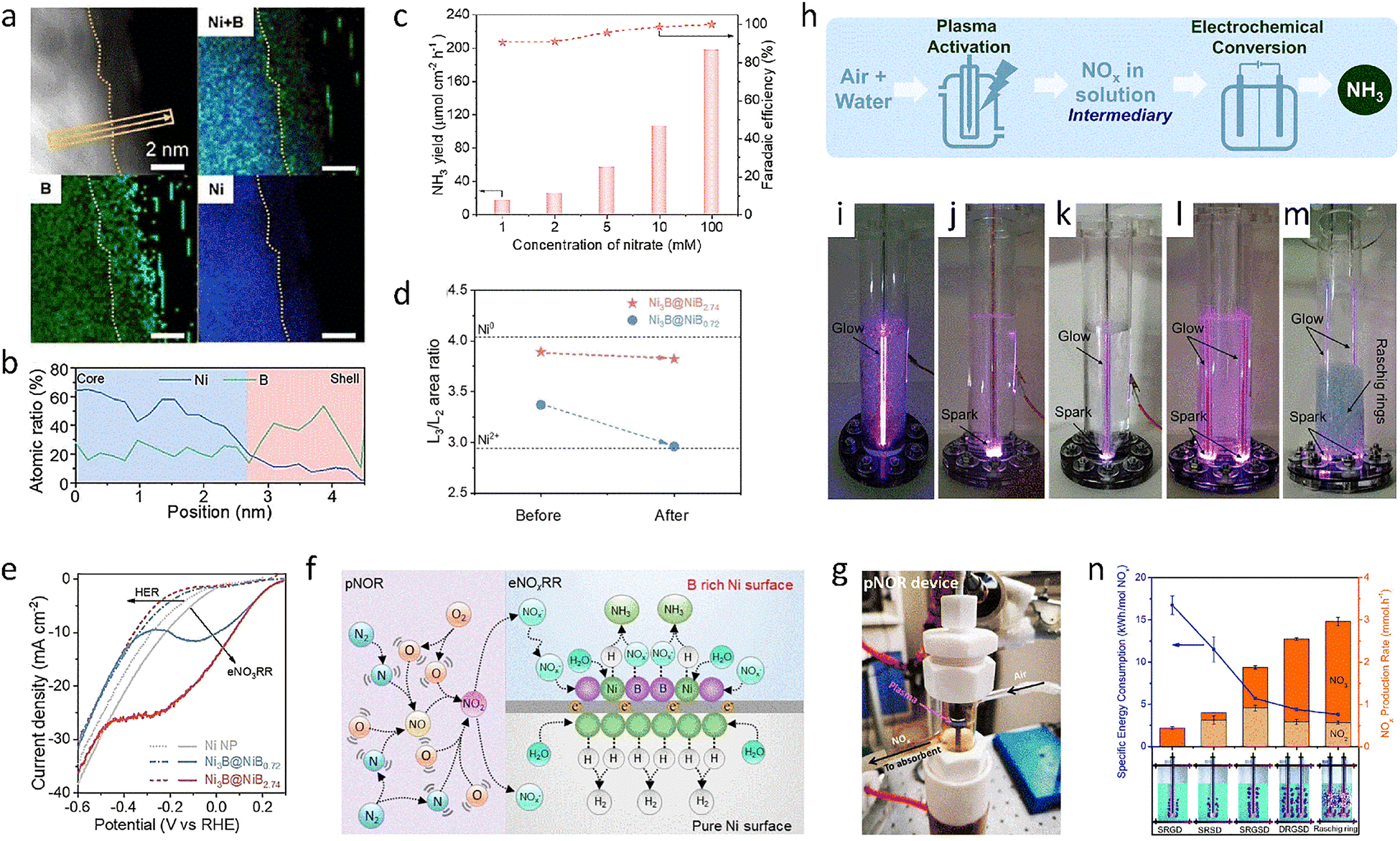 | ||
| Fig. 6 (a) EDX elemental mapping of Ni3B@NiB2.74. (b) Linear scan of Ni3B@NiB2.74. (c) NH3 yield rate and FE of Ni3B@NiB2.74. (d) L3/L2 area ratio in the EELS spectrum of different samples for illustrating the Ni species effectively protected, thus preventing Ni sites from participating in the HER process. (e) LSV curves of different samples with and without NO3− for illustrating dominance of the HER and the NO3RR. (f)–(g) Diagram of the mechanism about boosted NH3 synthesis performance and related device. (h) Schematic illustration of plasma combined with electrocatalytic technology for NH3 synthesis. (i)–(m) Digital photos of different plasma reactors. (n) Energy consumption and NOx yield rate of different plasma reactors. Reproduced with permission from ref. 210 Copyright 2021, Wiley-VCH. Ref. 217 Copyright 2021, The Royal Society of Chemistry. | ||
4.4 Nitrogen source with a plus five oxidation state, NO3−
To further overcome the challenges of slow NRR dynamics, discovering and using new nitrogen sources is key. NO3− is the second most abundant form of nitrogen and is present at high concentrations as an environmental pollutant in industrial waste streams and water runoff.218,219 The electrochemical NO3RR is attractive due to the large solubility of NO3− in aqueous electrolytes and its relatively low bond dissociation energy (204 kJ mol−1). Meanwhile, the NO3RR avoids the activation of the NN bond and the complex gas–liquid–solid interfacial reactions, providing a more practical route.67,220,221 In previous studies, the NO3RR has been extensively studied for denitrification and water purification.222,223
Recently, significant attention has shifted to the selective reduction of NO3− to NH3via the eight electron transfer pathway (E0 = 0.82 V vs. RHE).221 The eight-electron transfer pathway is very complex, involving many potential reaction intermediates (NO2, NO2−, NO, N2O, N2, NH3, NH2OH, and N2H4), in which the initial reduction from NO3− to NO2− is considered the RDS.224 Further, depending on the electrolyte conditions and the adsorption strength of the intermediates on the catalyst surface, the reduced *NO is selectively shunted to either NH3 or N2.225 It is worth noting that, according to thermodynamic theory, the most stable product of the NO3RR is N2, which has the highest equilibrium potential of all possible products in the NO3RR process (E0 = 1.246 V vs. SHE).67 Therefore, challenges still exist in the NO3RR, such as slow reaction kinetics and low product selectivity, which greatly restrict its further application.
Specifically, it is well known that the NO3RR process involves the transfer of eight electrons coupled with nine protons, during which nitrogen oxyanions and dinitrogen are unavoidably generated as undesired byproducts.64 For traditional electrocatalysts, high selectivity for NH3 could be obtained only under low overpotential conditions, as the HER dominated under high current conditions.62,226,227 Therefore, catalysts that inhibit the HER process are expected to achieve the efficient synthesis of NH3. For instance, an Ru/O-doped-Ru core/shell nanocluster with tensile strain was successfully constructed by a modified sol–gel route (Fig. 7(a)–(c)), in which the strained Ru-ST-12 nanocluster was synthesized by an electrochemical redox process of amorphous ruthenium oxychloride nanoclusters. The synthesized electrocatalyst displayed an excellent NH3 yield rate of 5.56 mol gcat.−1 h−1 (Fig. 7(d)), in contrast to the NH3 yield rate of Ru nanoclusters with a close-to-zero strain (0.10 mol gcat.−1 h−1), which was much lower than that of Ru-ST-12. The main contributor to this excellent NO3RR performance came from the stretched lattice strain inhibiting the HER, while the lattice strain favored the formation of *H for promoting the hydrogenation of key intermediates to form NH3.64 To inhibit the competitive HER, constructing the catalyst with a suitable substrate is also a favorable strategy. Recently, a Cu nanowire catalyst with highly dispersed Ru atoms (Ru–CuNW) was obtained by a cation exchange process for Cu foam containing Cu(OH)2 nanowires in a RuCl3 solution for 12 hours.228 The Ru–CuNW reached a maximum NH3 FE of 96% at 0.04 V vs. RHE and showed a large NH3 generation rate of 76500 μg h−1 cm−2 (Fig. 7(e)), which significantly outperformed the CuNW (31% FE) and Ru NPs (52% FE). The introduction of highly dispersed Ru atoms provided additional adsorption sites for NO3− and inhibited the desorption of the *NO2− intermediate (Fig. 7(f) and (g)). The N–N coupling process was concurrently prevented by limiting the movement of N* species on the catalyst surface (Fig. 7(h)). Meanwhile, the surrounding Cu sites inhibited the progression of the HER (Fig. 7(i)), thus increasing the activity of the NO3RR. Recently, it has been reported that improving the surface hydrogenation capabilities of the catalyst also contributes to the generation of NH3. For example, Rh single atoms and clusters were loaded on Cu substrates by using the difference in reduction potentials of the Cu and Rh elements (Rh@Cu-0.6%), in which a 1.0 cm−2 Cu foil supporting Cu nanowires was soaked in RhCl3.229 This Rh@Cu-0.6% catalyst showed an NH3 FE of 93% at −0.2 V vs. RHE and reached a record yield rate of 21.61 mg h−1 cm−2 at −0.4 V vs. RHE. The performance of Rh@Cu-0.6% was obviously improved compared with Cu NWs (an FE of 72% at −0.2 V vs. RHE) and Rh NPs (an FE of 12% at −0.2 V vs. RHE). The synergistic effect between the Cu and Rh species enhanced the activity of the NO3RR, in which the adsorbed hydrogen on the Rh sites transferred to the *NO intermediate, which facilitated hydrogenation and the formation of NH3. In addition to inhibiting the competitive HER during the NO3RR, finding suitable strategies to preferentially form *NO and *N2O intermediates is also critical to the synthesis of NH3.199,230 Recently, nanocubes, cuboctahedra, octahedra, and concave nanocubes of Pd catalysts with controlled lattice planes were synthesized to explore the relationship between shape-controlled catalysts and NO3RR activity. According to the electrochemical NO3RR test, the order of NO3RR activity was Pd (111) > Pd (100). The cuboctahedron sample with both Pd (111) and Pd (100) planes demonstrated the highest NH3 yield rate of 306.8 μg h−1 mgPd−1 with an FE of 35%. Benefiting from the eight (111) facets and six (100) facets constructing the cuboctahedron Pd, the (111) facets had fast dynamics for activating and reducing NO3− to NO2− and the (100) facets were good at reducing NO2− to NH3. Thus, controlling the nanostructures and facets of Pd could boost the NO3RR performance.199 In addition to doping engineering and the regulation of structures with specific lattice planes, the degree of structural disorder of catalysts is also one of the strategies to improve the activity of the NO3RR. For example, amorphous (a-RuO2), low crystallinity (lc-RuO2), and high crystallinity RuO2 nanosheets (hc-RuO2) covered on carbon paper were synthesized by a molten salt method by tailoring the calcination temperature and time. Among them, compared with lc/hc-RuO2, a-RuO2 exhibited the highest NH3 FE of 97.46%, far outperforming the counterparts of lc/hc-samples, which had FEs of 55.27% and 7.03%, respectively. The results above revealed that amorphous structures with numerous vacancies could regulate the d-band center of Ru and suppress the HER process, thus resulting in a low energy barrier for the RDS (*NH2 → *NH3) and promoting catalytic performance.231 Considering the rarity of precious metals and the economics of the catalyst preparation process, the use of non-precious-metal-based electrocatalysts is inevitable. Inspired by a related report that showed that Co-based materials can selectively produce NH3 through the NO3RR like Rh-based materials,232 a CoP nanosheet array fixed on carbon fiber (CoP NAs/CF) was produced by an annealing treatment of Co(OH)2 NAs/CF in NaH2PO2 at 300 °C, in which an electrodeposition method was used to synthesize Co(OH)2 NAs/CF. The NO3RR performance was recorded at −0.3 V vs. RHE, and the CoP NAs/CF exhibited an NH3 generation rate of 9.56 mol h−1 m−2 with a high FE of ∼100%, which significantly outperformed the H–B method. The introduction of P species accelerated the RDS from *HNO3 to *NO2, and the surface self-reconstruction of CoP to form Co(OH)2 could promote water splitting to release active hydrogen to hydrogenate the adsorbed NO3−, improving the production efficiency of NH3.214 Apart from this, other first-row transition metal phosphides (e.g., Mn, Fe, Ni, and Cu) have also been proven not only to be excellent alternatives for the HER,233,234 but also to exhibit some hydrogenation-treating abilities, such as hydrodesulfurization, hydrodeoxygenation, and hydrodenitrogenation.235–238 For example, an Fe-doped Ni2P (Fe/Ni2P) catalyst was constructed by an annealing treatment of the compounds of NiFe LDH and NaH2PO2 at 300 °C. The as-prepared electrocatalyst exhibited prominent NO3RR performance, with an FE of 94.3% and an excellent NH3 yield rate of 4.17 mg h−1 cm−2 at −0.4 V vs. RHE. Owing to the Fe doping, the d-band center and electronic structure of the Ni species were altered, and the RDS was also changed from *NO + 2H+ + 2e− → *N + H2O to *NO2 + H2O + 2e− → *NO + 2OH−, indicating more favorable kinetics for the NO3RR on Fe/Ni2P. This work highlighted the prospect of bimetallic phosphides for the NO3RR.239 As the NO3RR involves many possible intermediates and products,221,240–242 the rational design and construction of catalysts at the atomic level could further optimize this complex reduction process. For instance, a zero-valent atomic Cu catalyst immobilized on graphdiyne (Cu0/GDY) was produced by electrochemical deposition at 10 mA cm−2 for 200 s.243 The highly monodispersed Cu0 catalyst achieved a high FE of 81.25% and a large NH3 yield rate of 15.45 mmol h−1 cm−2, which was better than the pristine GDY, with a negligible NH3 yield rate and FE. Due to the incomplete charge transfer between GDY and the zero-valent Cu atoms, the Cu0/GDY catalyst was endowed with extremely high activity, which showed that zero-valent atomic-based electrocatalysts could be used as a goodplatform
to convert NO3− to NH3. In addition, adjusting the coordination environment of a single metal site can also result in a favorable thermodynamic process for the electrosynthesis of NH3. For instance, an atomic-Cu anchored N-doped carbon (NC) catalyst with Cu(I)–N3C1 sites was prepared by the pyrolysis of a Cu ZIF-8 precursor at 950 °C. NO3RR experiments demonstrated that this catalyst displayed a large NH3 yield rate of 5466 mmol gCu−1 h−1 and a conversion efficiency of 94.8% for NO3− at −0.64 V vs. RHE, which was obviously superior to the traditional Cu NC, with a 72.2% conversion efficiency. The coordination environment of Cu(I)–N3C1 localized the charge around the Cu sites and adsorbed the intermediates *NO3 and *H onto neighboring Cu/C sites, stabilizing the adsorbed *H for hydrogenation and suppressing the HER. It could also boost the RDS from *HNO3 to *NO2, thus leading to a thermodynamically favorable process for the NO3RR.244 In addition to the development of single-atomic Cu-based materials, Fe-based compounds have been used in the Haber–Bosch process for highly efficient NH3 synthesis at an industrial grade.28,49,92 Therefore, the reasonable design of single-atomic Fe-based catalysts is expected to achieve a breakthrough in the performance of the NO3RR. Recently, an Fe single-atom catalyst (Fe SAC) was synthesized by a transition-metal-assisted carbonization strategy using silicon dioxide as a template; specifically, pyrolysis of a mixture including a metal salt, o-phenylenediamine, and silicon dioxide. The catalyst delivered a high FE of 75% at −0.66 V vs. RHE and an impressive NH3 production rate of about 20000 μg h−1 mgcat.−1, whereas the NH3 yield rate of Fe NPs was only half that of the Fe SAC, at about 11500 μg h−1 mgcat.−1. More importantly, the lack of neighboring metal sites in the Fe SAC could effectively prevent the N–N coupling step, combined with a favorable RDS including *NO to *HNO and *HNO to *N, thereby boosting the selectivity and yield rate of NH3. In addition, based on this high performance, to further optimize the NO3RR process for obtaining a controllable reaction path and high selectivity for the ideal target products, bi-single-atomic electrocatalysts with multiple active sites can be used as a general design strategy for heterogeneous catalysis.242 For example, an FeMo–N–C SAC catalyst was synthesized via the sacrificial support method. The activity of the bimetallic catalyst FeMo–N–C SAC for the NO3RR was obviously improved compared with its monometallic counterparts of Fe or Mo SAC. Specifically, the FeMo–N–C SAC showed an increased FE of up to 94% at −0.45 V vs. RHE, with the NH3 yield rate improved to 18.0 μmol cm−2 h−1, which was 1.8 times greater than Fe–N–C and 3.5 times greater than Mo–N–C. By integrating individual Fe and Mo sites on a single catalyst, unique reaction pathways were synergized, in which Mo sites could promote the dissociative–adsorption process of *NO3−, and then the Fe sites exhibited fast kinetics for reducing *NO2− to NH3, thus achieving high performance for the NO3RR. This cascade design provided guidelines for future sustainable NH3 synthesis.225
 | ||
| Fig. 7 (a)–(c) Diagram of an Ru/oxygen-doped-Ru core/shell nanocluster with different degrees of tensile strains. (d) NH3 yield rate of an Ru/oxygen-doped-Ru core/shell nanocluster with different degrees of tensile strains. (e) NH3 yield rate of the Ru–CuNW sample. (f) Electron paramagnetic resonance spectra of different catalysts. (g) Free energy diagram for different catalysts during the NO3RR process. (h) Free energy diagram for NO3− adsorption and the HER process of different catalysts. (i) Free energy diagram for adsorbed N behaviour. Reproduced with permission from ref. 64 Copyright 2020, American Chemical Society. Ref. 228 Copyright 2022, Springer Nature. | ||
5. Photoelectrochemical synthesis of ammonia
Various strategies have been explored to promote NH3 synthesis, including biochemical, photocatalytic, electrochemical, and photoelectrochemical (PEC) methods.245–247 The PEC approach is a promising technology for NH3 production because it combines the advantages of both photocatalysis and electrocatalysis to facilitate N2 conversion.248,249 This approach not only allows for the possibility of NH3 production under mild conditions, such as ambient temperature and atmospheric pressure, but can also be powered by sunlight.250 The PEC process includes the following specific steps: (i) the absorption of light and the excitation of charge carriers; (ii) the separation and transport of photoexcited carriers to the surface of the photoelectrode; and (iii) reactions involving oxidation or reduction on the catalyst's surface.251 Hence, designing and constructing photoelectrode materials with broad solar spectral absorption, fast charge carrier generation and separation, and a large and effective catalytically active surface, while also adsorbing enough N2 molecules, is crucial for achieving a high-efficiency PEC process, especially at low overpotentials.252,253 The relevant prepared photoelectrode materials and their PEC activities are summarized in Table 3.| Catalysts | Nitrogen source | Experimental conditions | Faraday efficiency (%) | Yield rate | Reactor | Ref. |
|---|---|---|---|---|---|---|
| Cu2O | N2 | 100 mW cm−2, 0.1 M KOH | 20 | 7.2 μg cm−2 h−1 | An undivided three-electrode system equipped with a 300 W Xe lamp, an IR (water) filter | 254 |
| CoCu/TiO2/Sb2Se3 | NO3− | 100 mW cm−2, 0.1 M KNO3 and 10 mM H2SO4 | 88.01 | 15.91 μmol h−1 cm−2 | A standard three-electrode configuration in an air-tight H-cell | 37 |
| TiOx-250/CdS/CZTS | NO3− | 100 mW cm−2, 0.1 M KNO3 and 10 mM H2SO4 | 89.1 | 8.21 μmol h−1 cm−2 | A standard three-electrode configuration in an air-tight H-cell. | 255 |
| O_SiNW/Au | NO3− | 100 mW cm−2, 0.5 M K2SO4 with 10 mM KNO3 | 95.6 | 7.8 μg cm−2 h−1 | A potentiostat in a gas-sealed, undivided, three-electrode cell | 256 |
| BP | N2 | 100 mW cm−2, 0.1 M HCl which was purged with high purity N2 (99.999%) | 23.3 | 102.4 μg h−1 mgcat.−1 | A sealed H-type two-compartment silica glass cell separated by a 25.4 μm thick Nafion 211 membrane | 257 |
| Au-PTFE/TS | N2 | 100 mW cm−2, 0.05 M aqueous H2SO4 with 0.05 M Na2SO3 | 37.8 | 18.9 μg cm−2 h−1 | A three-electrode sealed cell | 250 |
| Si photocathode with Li ions | N2 | 400 mW cm−2, 0.2 M LiBF4 in 99 vol% THF and 1 vol% EtOH | 95 | 52.4 μg cm−2 h−1 | A customized 3-electrode single compartment polyether ether ketone cell | 258 |
| PdCu/TiO2/Si with Li ions | N2 | 100 mW·cm−2, 1 M LiClO4 of propylene carbonate mixed with proton sources | 46.15 | 43.09 μg cm−2 h−1 | A conventional three-electrode configuration | 36 |
| Cs0.05(FA0.83MA0.17)0.95Pb(Br0.17I0.83)3 coupled with glycerol oxidation | NO3− | 100 mW cm−2, 1.0M KOH with various KNO3 concentrations |
99.5 ± 0.8 |
1744.9 ± 20.6 μg cm−2 h−1 | A half-cell reactor with a conventional three-electrode system | 259 |
| n+np+-Si photocathode with Au/PCN | N2 | 100 mW cm−2, N2-saturated 0.05 M H2SO4 | 61.8 | 13.8 μg h−1 cm−2 | A standard air-tight H-shaped quartz reactor | 253 |
A few studies have recently been reported on the PEC conversion of N2 to NH3. For example, a Cu2O photocathode was constructed by an electrochemical deposition strategy to study PEC N2 reduction. Using isotopically labeled 15N2 in a 0.1 M KOH solution under simulated solar illumination, the FE of the Cu2O photocathode to produce 15NH3 was 20% at 0.4 V vs. RHE. This potential was significantly more positive than the thermodynamic reduction potential of N2, suggesting that using photoexcited electrons in the Cu2O photocathode could reduce the energy required for NH3 production (Fig. 8(a)).254 This work strongly showed the effectiveness of PEC technology for NH3 synthesis. However, it is worth noting that the photoelectrodes used for PEC NH3 production usually face problems such as low light absorption ability, limited carrier transport, low selectivity, and a low yield rate. As a potential material, antimony selenide (Sb2Se3) has the advantages of a narrow band gap of ∼1.2 eV, strong light absorption, and fast charge transfer capacity. Recently, a photocathode was synthesized by a spin-coating method, followed by annealing at 350 °C. Furthermore, a TiO2 layer was deposited on the as-prepared Sb2Se3 photocathode and then coated with a CoCu co-catalyst by electrodeposition (Fig. 8(b)), resulting in a final photocathode named CoCu/TiO2/Sb2Se3.37 The CoCu/TiO2/Sb2Se3 photocathode exhibited a high FE of 88.01% and a yield rate of 15.91 μmol h−1 cm−2 (Fig. 8(c)), which was 20 times higher than that of TiO2/Sb2Se3. Due to its excellent light absorption capacity, carrier transfer capability, and charge separation efficiency (Fig. 8(d)), the CoCu/TiO2/Sb2Se3 photocathode effectively adsorbed the NO3− reactant and intermediates, while the CoCu co-catalyst optimized the thermodynamic energy barrier of the NO3RR and HER processes, thus jointly resulting in a high PEC activity. In addition to the design of heterostructures, the modification of the photoelectrode's own nanostructure is also expected to solve the problems of low PEC performance. A kesterite (Cu2ZnSnS4 (CZTS)) photocathode was synthesized by loading a defect-engineered TiOx cocatalyst on a CdS/CZTS photocathode via spray-coating. The TiOx-250/CdS/CZTS achieved an 89.1% NH3 FE at 0.1 V vs. RHE and the highest yield rate of 8.21 μmol h−1 cm−2 at −0.2 V vs. RHE.255 This work implemented a defect engineering strategy on a kesterite-based photocathode that effectively enhanced the adsorption of the reactant NO3− and *NO2 intermediate, while a full surface coverage of TiOx strongly suppressed the generation of the by-product NO2−, thus boosting PEC NH3 production efficiency (Fig. 8(e)). In addition, photoelectrodes with efficient mass transfer are also expected to break the performance bottleneck. For instance, an ordered silicon nanowire array photocathode (O_SiNW) modified with Au NPs (O_SiNW/Au) was fabricated by a modified metal-assisted chemical etching method (Fig. 8(f)). The O_SiNW/Au array photocathode demonstrated a remarkable FE of 95.6% and a high production rate of 7.8 μg cm−2 h−1.256 Among them, the optical and electrochemical properties of the Si electrode were modified by constructing the O_SiNW array. The O_SiNW allowed for smoother mass transport and reduced charge recombination compared to planar or randomly grown Si electrodes, which exhibited an improved onset potential for the NO3RR. Meanwhile, enabling the uniform distribution of Au NPs and facilitating mass transport during the reaction greatly improved the PEC performance for the NO3RR. It is worth noting that compared with metal-based catalysts for NH3 production, metal-free catalysts have great advantages in terms of environment and construction cost. For example, a black phosphorus (BP) photoelectrode was fabricated by the layer-by-layer assembly of BP nanosheets on an indium tin oxide (ITO) substrate, in which the BP nanosheets were synthesized by electrochemical exfoliation (Fig. 9(a)).257 Without a sacrificial agent added, the PEC NRR activity in the N2 saturated aqueous electrolyte was outstanding, with a high yield rate of 102.4 μg h−1 mgcat.−1, which displayed the highest performance among non-metal catalysts applied in photo- or electrocatalysis. Benefiting from the hydrogen-desorption-favorable surface of the BP photoelectrode as well as the synergistic effect from the coupling of a light-excitation-enhanced electrocatalytic process and an external-voltage-promoted photocatalytic process, the system above showed excellent PEC NH3 activity (Fig. 9(b)).
 | ||
| Fig. 8 (a) Schematic illustration of electrochemical and PEC NRR. (b) SEM image of CoCu/TiO2/Sb2Se3. (c) PEC NH3 performance of different catalysts. (d) Time-resolved photoluminescence decay spectroscopy of different samples. (e) Schematic illustration of TiOx with different vacancies. (f) SEM images of O_SiNW and O_SiNW/Au. Reproduced with permission from ref. 254 Copyright 2020, American Chemical Society. Ref. 37 Copyright 2024, Wiley-VCH. Ref. 255 Copyright 2022, Wiley-VCH. Ref. 256 Copyright 2022, Wiley-VCH. | ||
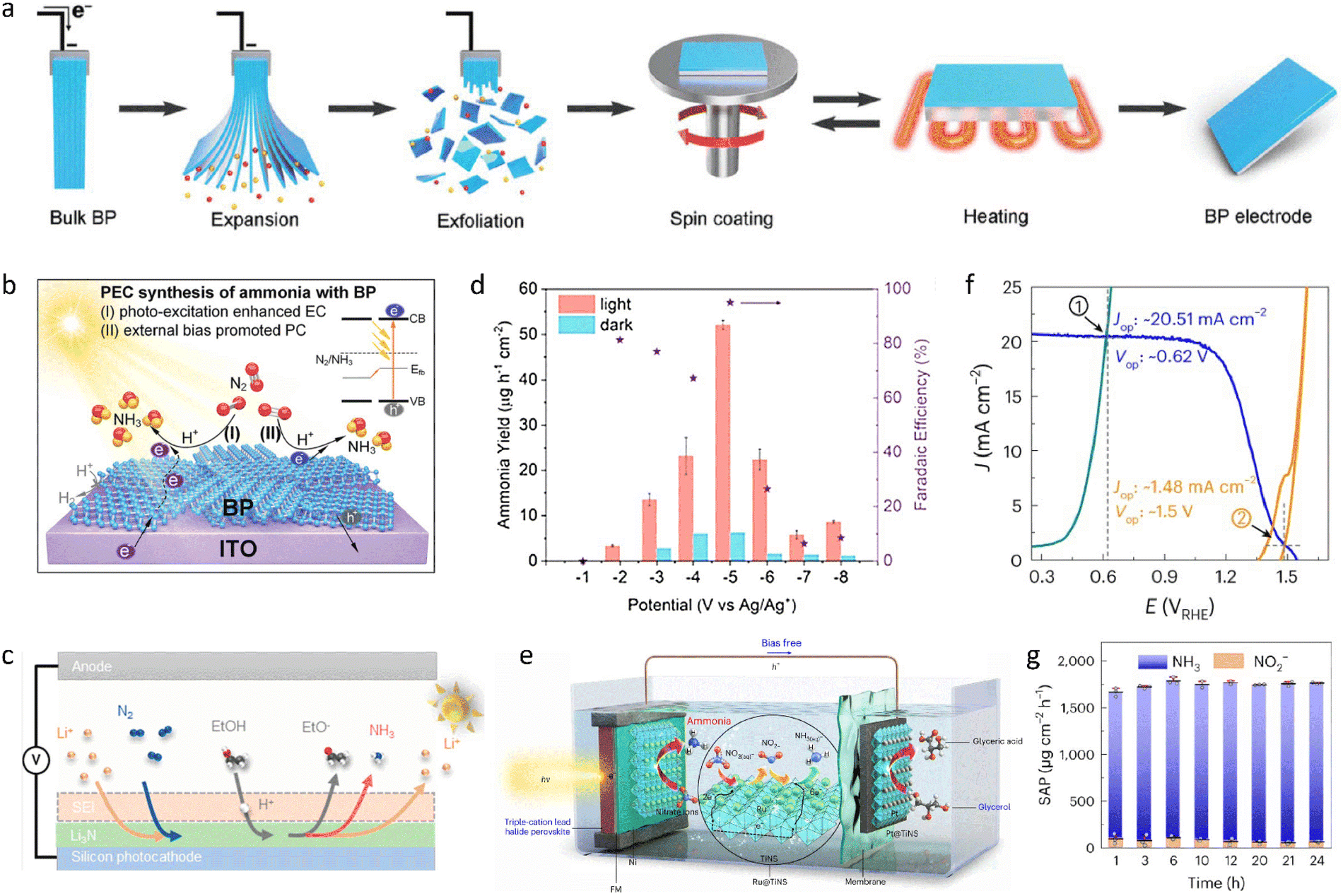 | ||
| Fig. 9 (a) Schematic illustration of the exfoliation process of BP nanosheets and production of electrodes. (b) PEC performance enhancement mechanisms of the BP electrode. (c) Schematic illustration of an Li-mediated PEC NH3 synthesis process. (d) PEC NH3 yield rate under light and dark conditions. (e) Schematic illustration of cathodic NH3 production combined an anodic glycerol oxidation system. (f) Polarization curves of cathodic NH3 production combined an anodic glycerol oxidation system or a water oxidation system. (g) NH3 yield rate along with the change of time. Reproduced with permission from ref. 257 Copyright 2020, Wiley-VCH. Ref. 258 Copyright 2023, American Chemical Society. Ref. 259 Copyright 2024, Springer Nature. | ||
The NH3 synthesis processes mentioned above all occurred in aqueous solvents. As we all know, an aqueous reaction system is unfavorable under mild conditions, with a conversion efficiency of ∼10% or lower, mainly due to the poor selectivity for NH3 and the strong competitive HER. Therefore, it is urgent to simultaneously satisfy the stringent requirements for gas N2 diffusion, electron transport, and proton production. In this context, a unique aerophilic–hydrophilic Si-based heterostructure photocathode was designed for the conversion of N2 to NH3 in an acid electrolyte under mild conditions. The system containing a photo-absorber of Si, a gas-diffusion layer of poly(tetrafluoroethylene) (PTFE), and active sites of Au NPs displayed an excellent yield rate of ∼18.9 μg cm−2 h−1 and an FE of 37.8.250 A functional aerophilic-hydrophilic heterostructure was constructed via the porous PTFE frameworks with highly dispersed Au NPs, which created a layer with enriched N2 and controlled proton activity in an aqueous solvent, thus boosting NH3 synthesis activity. Apart from aqueous electrolytes, lithium (Li)-containing non-aqueous electrolytes have also been used for enhancing the NH3 yield. For example, a PEC Li-mediated NRR (Li-NRR) system composed of a p-type Si photocathode as the photoabsorber and Li ions as the reduction mediator reduced N2 into NH3 in non-aqueous tetrahydrofuran (THF) solvents with ethanol as the proton donor (Fig. 9(c)).258 Thus, the as-synthesized photocathode resulted in a high NH3 yield rate of 52.4 μg cm−2 h−1 and an excellent FE of 95% (Fig. 9(d)). The light illumination on the Si photocathodes improved the NH3 yield dramatically because more photogenerated electrons from the light-excited Si photocathode induced the reduction of Li+ ions into metallic Li. Based on this research idea, a Si-based photocathode with a hierarchical structure (PdCu/TiO2/Si) was produced, achieving a record yield of 43.09 μg cm−2 h−1 and a remarkable FE of 46.15% under mixed gas conditions (0.12 MPa O2 and 3.88 MPa N2).36 The PdCu/TiO2/Si photocathode facilitated the conversion process from the formation of Li3N and hydrogenation to produce NH3, to the release of Li+ to restart a new PEC Li-NRR. At the same time, the PEC Li-NRR process was further enhanced by introducing a small amount of O2 or CO2 under pressure, which accelerated the decomposition of Li3N. In addition to the development of non-aqueous reaction systems, the current NH3 production process also faces a large additional bias due to the high thermodynamic potential for water oxidation (1.23 V). Moreover, inorganic-based photoelectrodes also have difficulties such as poor utilization of solar flux (wide bandgaps of 2.1–3.0 eV) and slow charge transport. Therefore, developing a new redox system for PEC NH3 synthesis is particularly important. For example, a high-performance PEC system included NH3 production and a glycerol oxidation reaction (Fig. 9(e) and (f)).259 Among them, the perovskite-based material Cs0.05(FA0.83MA0.17)0.95Pb(Br0.17I0.83)3 was used as the photocathode, and a Pt-loaded TiNS electrocatalyst (Pt@TiNS) was used for the anode reaction. The perovskite-based photocathode combined with the glycerol oxidation anode showed a photocurrent density of 21.2 ± 0.7 mA cm−2 with an FE of 99.5 ± 0.8% for NH3 production. Moreover, bias-free solar NH3 production with a solar-to-NH3 productivity of 1744.9 ± 20.6 μg cm−2 h−1 was achieved (Fig. 9(g)). Benefiting from a lower voltage for glycerol oxidation compared with water oxidation (Fig. 9(f)), while the as-prepared catalysts had a relatively narrow bandgap and good charge transport properties, a high system efficiency and productivity were obtained.
After optimizing the catalysts, reaction environment, and system, the next step in developing green transformation technology is to focus on the reactor. Currently, PEC devices face challenges in synergistically managing optical, electrical, and catalytic properties. By using a cascade n+np+-Si photocathode that decouples light-harvesting and electrocatalysis, it was possible to improve light-harvesting, carrier separation and transport, and catalytic reactions. Impressively, at −0.10 V vs. RHE, an n+np+-Si photocathode integrated with Au/porous carbon nitride (Au/PCN) displayed remarkable PEC performance for NH3 synthesis, with a high FE of 61.8% and a yield rate of 13.8 μg h−1 cm−2. This was the best performance ever reported for the PEC NRR at such a low applied potential.253 Specifically, the low-coupling photoelectronic system based on the Au/PCN with its porous structure improved light absorption, carrier transport, charge transfer, and catalytic activity. Meanwhile, the structural design of the integrated catalyst was favorable for adsorbing a high concentration of N2, significantly promoting NH3 synthesis.
6. Alkali metal mediated synthesis of ammonia
In the past few years, various catalysts and catalytic conversion technologies have been used to study green NH3 synthesis. However, due to the competitive HER process in aqueous systems, only 1% of electrons are used to produce NH3, leading to low catalytic performance.188,260 Besides, even with benchmarking protocols to eliminate interference from contamination and reliably quantify the NH3 produced, NH3 synthesis in aqueous solutions faces various problems, such as low current density for synthesizing NH3, FE, yield rate, and EE, and environmental pollution.14,91,93,261–263 Based on these concerns, a series of research articles on aqueous electrocatalytic NH3 synthesis found that NH3 production was too low to determine if it came from N2 reduction. Therefore, the feasibility of the electroreduction path for NH3 synthesis in aqueous systems remains uncertain.14 Furthermore, quantitative isotope and rigorous gas purification experiments have proven that the Li-NRR method is a reliable way to synthesize NH3 at room temperature.91The history of the Li-NRR began in the 1930s with the electrochemical deposition of lithium, followed by nitridation and protolysis in alcohol-based electrolytes.264 Within an autoclave-type electrolysis cell, an initial current of 0.5 A was applied to deposit lithium metal for 5.0 h. The Li was electrodeposited in an electrolyte of lithium chloride in ethanol. Although negligible NH3 was measured at atmospheric pressure, the FE increased by up to 10% at a high-pressure of 1000 bar. The low NH3 selectivity was thought to be due to the formation of a surface coating on the electrode.265 The Li-NRR was then revisited in the 1990s. Researchers first reported using THF as the electrolyte solvent in the Li-NRR.266,267 The experiments took place in a single-compartment autoclave cell where lithium metal was plated onto a copper electrode in a 0.2 M lithium perchlorate, 1% ethanol in THF electrolyte. The process used 50 bar of N2 gas to produce NH3 with an FE of 48.7%. Further screening of working-electrode metals, lithium salts, and proton-donor additives led to an NH3 FE of 59.8% under optimized experimental conditions. To further optimize the Li-NRR technology, researchers developed a three-step strategy of separate electrolysis, nitridation, and protonation, which achieved an NH3 FE of 88.5%.268 In this process, LiOH was first electrolyzed at 450 °C, and then the molten salt was exposed to N2 gas at temperatures up to 100 °C. The lithium nitride then reacted exothermically with water to produce NH3. Unlike a continuous process, batch processes require extra separation and purification steps and a large Li source, which can increase system cost and complexity. Therefore, compared to NH3 synthesis in aqueous systems and under traditional reaction conditions, the single-process electrochemical Li-NRR under ambient conditions has been shown to be commercially promising.269 Specifically, Li metal can dissociate N2 under ambient conditions, and its high enthalpy of hydration and small nucleus make it the strongest reducing agent of all metals (−3.04 V vs. SHE).270 Electrodeposition of Li metal occurs in non-aqueous and aprotic electrolytes, which are thermodynamically unstable with Li metal but kinetically stable because of a surface passivation layer.271 This surface film called the solid–electrolyte interphase (SEI) reduces further reactions between the metallic Li and the electrolyte. The Li-NRR is a unique technique because N2 reduction and protonation depend on and occur within the SEI layer, which is essential for mediating the catalysis process. In this system, the electrochemical deposition of metallic Li with N2 gas forms lithium nitride, which then reacts with available protons to produce NH3.269 Thus, Li is capable of producing NH3 at ambient pressure because it has a low work function and high reactivity.
Therefore, Li can break the strong NN bonds and fix N2 as Li3N, which is then converted into NH3. Furthermore, the disruption of water molecules by Li+ ions slows water splitting.272 The Li-NRR reaction process involves the reduction of Li+ ions to metallic Li at the working electrode,273 where the Li metal reacts with N2 to produce Li3N. This Li3N is then protonated with a source like alcohol to form NH3.263 In this process, freshly plated Li, produced by electroreducing Li+ ions in the electrolyte, is believed to dissociate N2; the surface N is then reduced through a series of electron and proton transfers to form NH3. In the Li-NRR system, the decomposition of organic electrolytes during Li deposition on the cathode forms a significant part of the SEI layer, which is an ionically conductive, porous passivation layer on the electrode surface. However, its exact composition and mechanistic role in the Li-NRR remain unclear, since it is sensitive to air. Even so, this technology typically achieves an FE between 5% and 20%,91,274 with a recent breakthrough reaching 69% (20 bar N2).275 To optimize the three-phase interface reaction rate, the Li-NRR process recently used a gas diffusion electrode (GDE) configuration, which showed an excellent FE of 35%. However, the updated system only operated stably for a few minutes and had a high potential.98 A potential cycling method recently reduced this instability.276 As a result, the FE increased from 20% to 37% with cycling at an EE of 7% over several days. Nevertheless, the Renewable Energy to Fuels Through Utilization of Energy-Dense Liquids (REFUEL) program of the US Department of Energy set the following performance targets for the commercialization of NH3 synthesis: a NH3 yield rate of 10−6 mol s−1 cm−2, an FE of 90% at a current density of 300 mA cm−2, and durable catalyst stability, with catalytic efficiency decreasing by no more than 0.3% after 1000 hours of electrolysis.277 The Li-NRR is far from this goal. A major problem in Li-NRR technology is currently the EE, since Li plating generally requires a negative potential (−3.04 V vs. SHE). Assuming an FE of 80% and minimizing all overpotentials at the anode by using another anode reaction like the hydrogen oxidation reaction (HOR), the EE would increase to 26%. To address this, small amounts of O2 were added to the feed gas to improve the FE and stability of the Li-NRR process (Fig. 10(a)).274 A high FE of 78.0 ± 1.3% can be achieved by mixing in a certain amount of O2, which results in an EE of 11.7 ± 0.5%. The addition of O2 can modify Li+ diffusion through the SEI layer while maintaining the optimal concentration of protons and N2 at the surface, thereby influencing the deposition behavior and the stability of the deposited layers. More importantly, this research offers substantial advantages for the scale-up of synthetic NH3 processes, as the Li-NRR does not require ultra-pure N2.
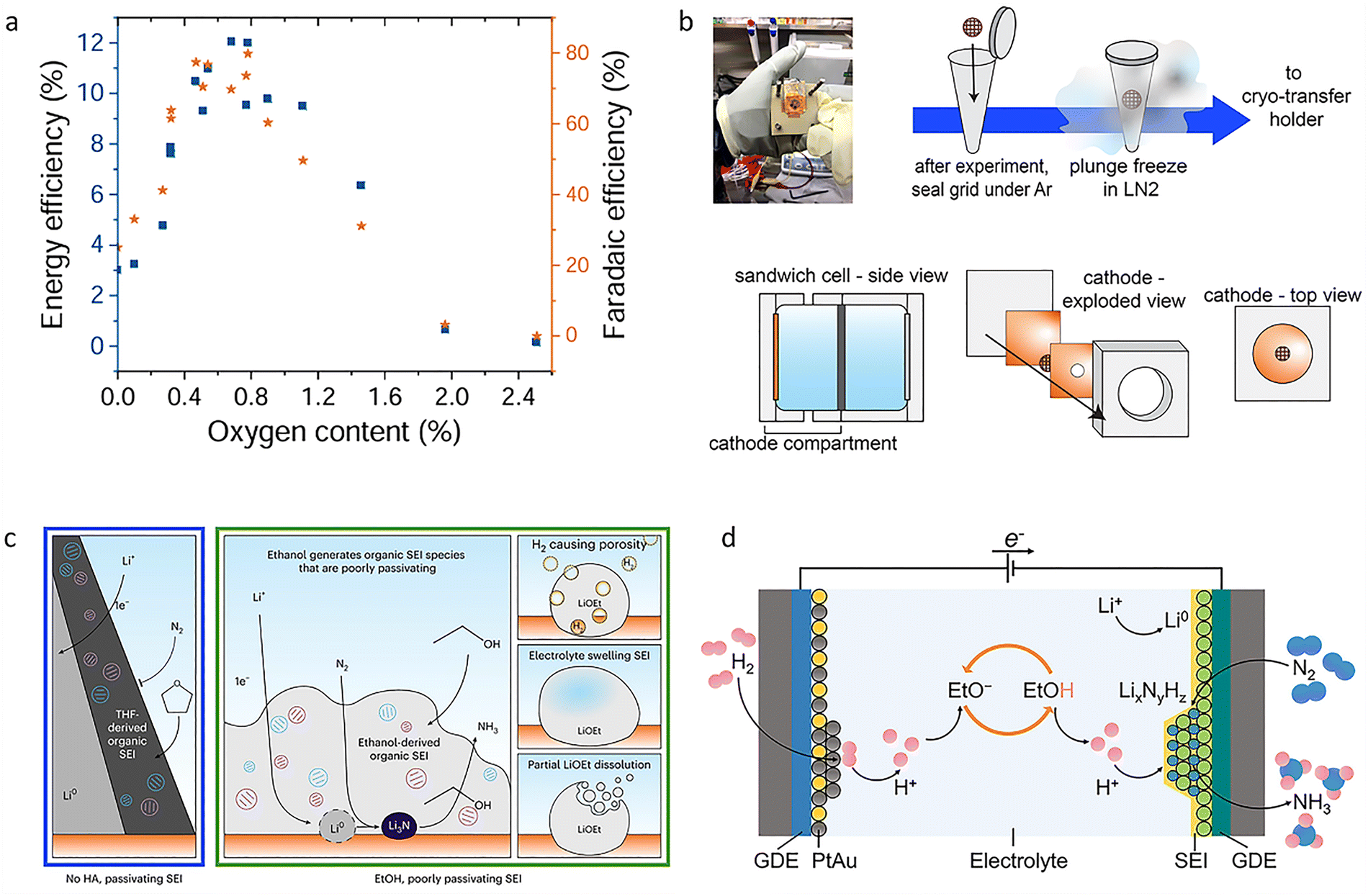 | ||
| Fig. 10 (a) Diagram of the change of FE and EE with oxygen content. (b) The picture and diagram of cryo-electron microscopy. (c) Schematic illustration of the Li-NRR mechanism. (d) Schematic process of the Li-NRR combined with the HOR process. Reproduced with permission from ref. 274 Copyright 2021, American Association for the Advancement of Science. Ref. 278 Copyright 2023, Springer Nature. Ref. 280 Copyright 2023, American Association for the Advancement of Science. | ||
To further optimize the activity of the Li-NRR, exploring its fundamental mechanism has become key. Recently, relevant studies were performed for the first time to analyze the entire process of the Li-NRR reaction using cryo-electron microscopy (Fig. 10(b)). The proton donor, ethanol, is the main driving force of the Li-NRR. In the absence of a proton donor, the decomposition products of the fluoroborate ion and tetrahydrofuran form a passivated SEI. N2 and the electrolyte cannot react with Li because they cannot penetrate and pass through the SEI. At the same time, Li ions can diffuse into the SEI, resulting in the continuous formation of metallic Li dendrites, which is not conducive to the subsequent reaction. With the addition of ethanol, the amorphous SEI becomes dominated by ethanol decomposition products, and N2 and the electrolyte can penetrate the interface, thereby continuously consuming the deposited Li metal to form NH3 gas and driving the reaction forward. Therefore, for the Li-NRR, an SEI with poor passivation effectively enhances NH3 synthesis, so it is necessary to introduce proton donors to promote the reaction (Fig. 10(c)). However, if there is an excessive reaction between the Li metal and the proton donor, a large amount of hydrogen gas will be generated, resulting in an irreversible loss of Li metal. Therefore, in the subsequent design and improvement of the reaction, it is necessary not only to select a suitable combination of lithium salt and solvent but also a highly stable proton donor to achieve SEI activation while avoiding the loss of Li metal and improving reaction safety and production efficiency.278
Further understanding of the Li-NRR mechanism is also expected through innovations in the components of the reaction system. In this regard, a reference electrode (LiFePO4) was prepared for measuring potentials and estimating sources of EE loss. Specifically, it contained LiFePO4 discs and a negative electrode of Li metal separated by 1.0 M lithium bis(trifluoromethylsulfonyl)imide wetted glass fiber. After an eight-millimeter hole was punched in the center of the LiFePO4 disc, a sandwich cell was assembled. A phase of fast (de)intercalation of Li ions characterizes LiFePO4 after partial lithiation. It also possessed low polarizability, and its redox equilibrium potential tended to remain stable relative to Li metal for a long time, which could reveal the relationship between partial current density and potential. The LiFePO4/Li+ equilibrium was also used as a tool to detect changes in Li ion activity in situ. This study is expected to move the field toward a more definitive system to fully understand the Li-NRR process.279
Typically, most Li-NRR reports have used sacrificial solvents as proton sources. However, to be feasible in real production, protons must come from hydrogen (or water). Recently, the hydrogen oxidation reaction (HOR) was proposed as an anode reaction for Li-NRR systems to provide a sustainable hydrogen source (Fig. 10(d)).280 The PtAu alloy catalyst greatly reduced the anode potential and avoided the oxidation of the solvent. Under normal temperature and pressure conditions, an FE of 61% and an EE of 13% were achieved for NH3 production, and an NH3 FE of nearly 100% was achieved in intermittent reactors with 15 bar or 20 bar pressures, at a high current density of 1.0 A cm−2. Specifically, Li+ ions diffused from the bulk electrolyte through the SEI to the cathode electrode surface for electroreduction to Li metal, which then reacted with N2 to form Li3N species. The Li3N species were further protonated by a proton shuttle agent to continuously produce NH3. The Li-NRR combined with anode hydrogen oxidation could effectively avoid using a sacrificial organic solvent as a proton source via the recyclability of the ethanol proton shuttle, while further decreasing the cell voltage.
The proton shuttle has a decisive effect on the performance of Li-NRR systems because it changes the properties of the SEI. Therefore, based on the studies above, screening proton shuttles in the Li-NRR coupled with the HOR system is expected to further improve the performance of the Li-NRR (Fig. 11(a)). To address this challenge, proton shuttle screening experiments were performed in a continuous-flow reactor in the presence of the HOR at the anode. After passing a charge of 700 C, phenol (PhOH) achieved the highest NH3 FE of 72 ± 3% and an EE of 15 ± 1% at ambient pressure and temperature, which exceeds the state-of-the-art ethanol (Fig. 11(b)).281 Specifically, the deprotonated form (PhO−) of the PhOH was proven to be the primary species responsible for transferring protons during the Li-NRR process (Fig. 11(c)). Moreover, theoretical mass transport modeling clarified the dependence of the Li-NRR performance on the acid dissociation constant and diffusion coefficient of the proton shuttles.281 This discovery provides a comprehensive framework for rational design principles for efficient proton shuttles in the Li-mediated electrosynthesis of NH3. Following this pioneering work, the performance of different Li salts was compared in a specific reactor (Fig. 11(d)–(g)). LiBF4 achieved the highest FE of 95% and an NH3 yield rate of 2.5 μmol s−1 cm−2 (Fig. 11(h)–(i)). The excellent performance can be ascribed to the formation of a uniform SEI on the electrode.282 At the same time, related work reported a Li-NRR system with almost 100% current-to-NH3 efficiency, enabled by a compact ionic layering generated by LiNTf2 at the interface, although at an elevated pressure of 15 bar.283 Besides the proton donor, Li salts have also received massive attention from the community due to their great impact on the performance of the Li-NRR.284
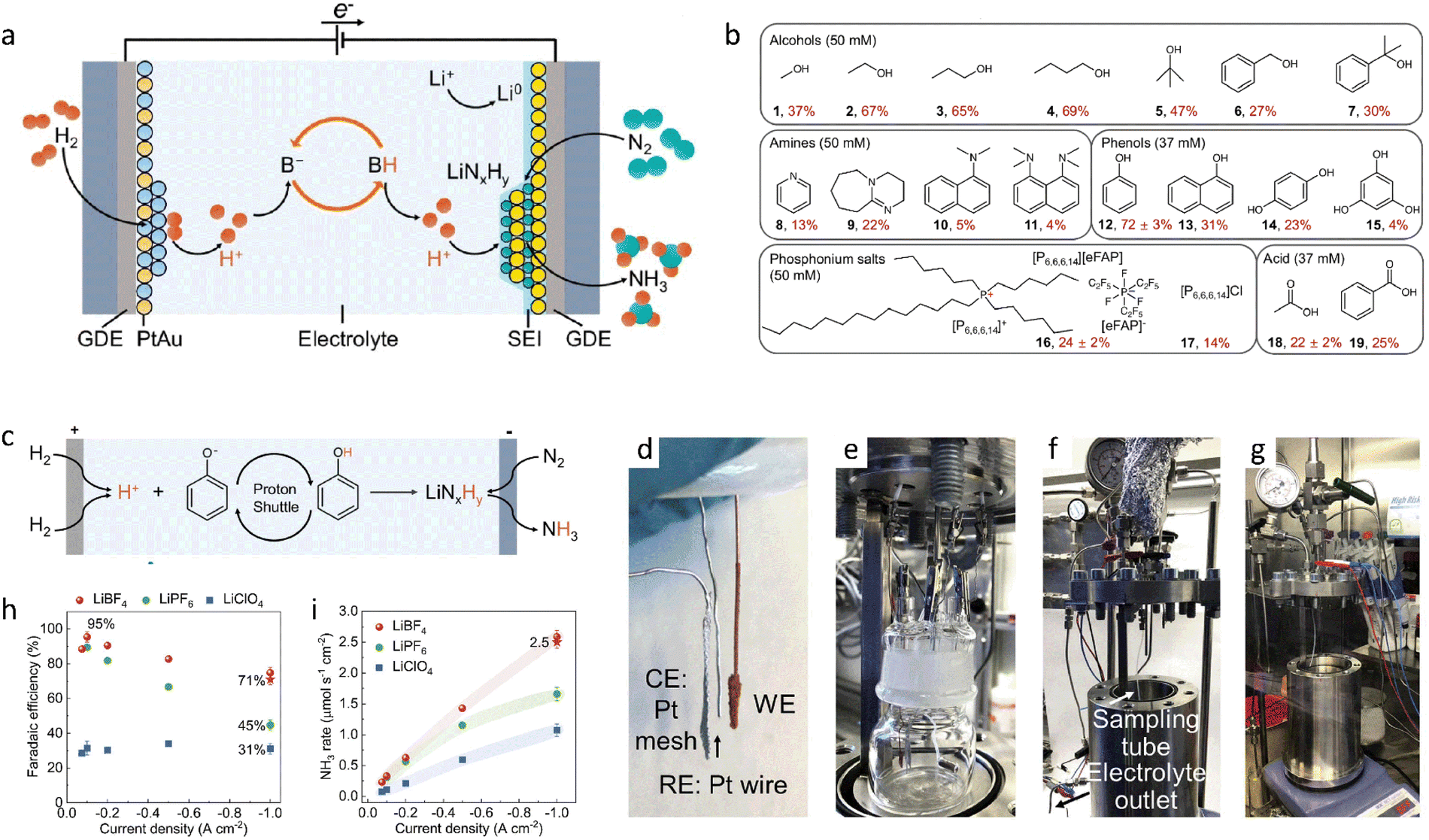 | ||
| Fig. 11 (a) Schematic illustration of the proton-shuttling process for the Li-NRR. (b) NH3 FE for different proton shuttles. (c) Schematic illustration of the phenol shuttling process for the Li-NRR. (d)–(g) Digital photos of the Li-NRR pressure reactor. (h) NH3 FE of different Li salts. (i) NH3 yield rate of different Li salts. Reproduced with permission from ref. 281 Copyright 2024, Springer Nature. Ref. 282 Copyright 2022, Cell Press. | ||
As we all know, the electrolyte solution for NH3 synthesis via the Li-NRR mainly includes the main electrolyte, such as THF, the proton shuttle, and a Li salt. Numerous experimental results show that all these factors will affect the performance of the reaction system.284 Therefore, in addition to the development of proton shuttles and Li salts, improving the other components of the electrolyte is also important. Recently, a series of solvents, including 2-methyltetrahydrofuran (2-MeTHF), tetrahydropyran (THP), dimethoxyethane (DME), diglyme (DG), and tetraethylene glycol dimethyl ether (TEGDME), were studied systematically in the Li-NRR process (Fig. 12(a)). The assessments of solvent candidates are based on their conductivity, parasitic reactions, product distribution, and FE. Meanwhile, promising candidates should have the following characteristics such as good solubility for lithium salts, the ability to provide sufficient conductivity, reliable stability to endure reactive metallic Li at the cathode and the oxidation potential at the anode, and the capability to induce a favorable SEI layer for N2 reduction. These characteristics directly dictate the performance of the Li-NRR, such as the NH3 yield rate, FE, EE, and stability. Specifically, compared with cyclic ether-based electrolytes, the SEI in linear ether-based electrolytes has a higher proportion of inorganics and heavier organics, giving it a better ability to resist ethanol corrosion and thus trapping more NH3 in deposits. Notably, an impressive NH3 FE of 58.5 ± 6.1% was obtained in the THF-based electrolyte (0.11 M ethanol), while a decent FE of 30.0 ± 4.2% was achieved in the DME-based electrolyte (0.11 M ethanol), but at a significantly lower overpotential compared with THF, verifying its potential in the Li-NRR.284
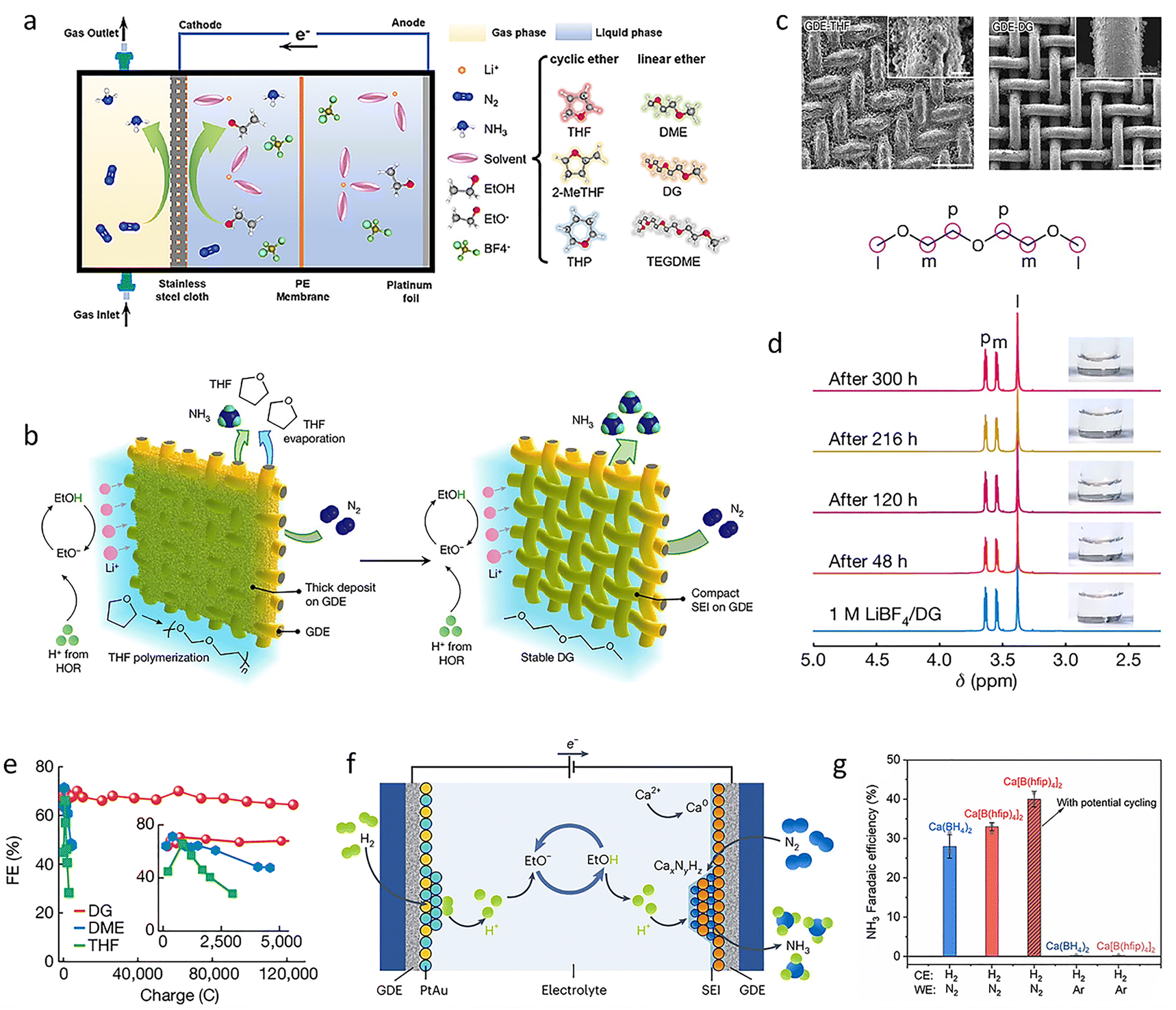 | ||
| Fig. 12 (a) Schematic illustration of the gas diffusion cell with different solvents. (b) NH3 synthesis system with ring-ether-based and chain-ether-based solvents. (c) SEM images of the GDE after long-time NH3 production using THF and DG solvents. (d) NMR spectra of the DG-based Li-NRR system. (e) NH3 FE of different solvent-based Li-NRR systems. (f) Schematic illustration of the Ca-mediated NH3 synthesis process. (g) NH3 FE of the Ca-mediated NH3 synthesis system with different Ca salts. Reproduced with permission from ref. 284 Copyright 2023, American Chemical Society. Ref. 285 Copyright 2024, Springer Nature. Ref. 286 Copyright 2024, Springer Nature. | ||
Based on the explorations above, including the introduction of the HOR on the anode, screening proton shuttles and suitable solvents can make the Li-NRR more sustainable. However, it is worth noting that the problems of solvent polymerization and the volatility of THF in the Li-NRR process make it difficult to achieve long-term operation and mass production. At the same time, to ensure excellent ionic conductivity and smooth plating of Li, the solvent must allow for noticeable solubility of the Li salts. In addition to being compatible with metallic Li to produce an SEI layer, it also needs to be matched with proton shuttles to facilitate the delivery of protons. Therefore, based on the research above on chain-ether-based solvents, recently, the ring-ether-based solvent THF was also replaced by the chain-ether-based solvent diethylene glycol dimethyl ether (DG) to achieve long-term continuous electrosynthesis of NH3 (Fig. 12(b)).285 The DG solvent did not polymerize easily under reaction conditions, has a high boiling point (162 °C vs. 66 °C for THF), and formed a dense and uniform SEI layer (Fig. 12(c) and (d)), thus boosting the release of NH3 and ensuring electrolyte stability. Specifically, the Li-NRR system with the DG solvent achieved 300 hours of continuous operation in a flow electrolyzer with a 25 cm2 electrode at 1.0 bar pressure and mild conditions, displaying an NH3 FE of 64 ± 1% (Fig. 12(e)). These results strongly indicate that long-term, stable, continuous NH3 electrosynthesis via the Li-NRR can be achieved by using more stable chain-ether-based solvents. To further solve the problems in the industrial application of the Li-NRR, for example, developing high-current-density processes requires the study of more suitable GDE configurations. Therefore, future research on the Li-NRR should focus on achieving remarkable FE and EE at industrial current densities while maintaining long-term stability.
As of now, Li-NRR systems face numerous challenges, such as high input voltage, the volatilization of toxic organic solvents, gas–liquid mass transfer limitations, and ambiguity in the fundamental mechanism.263 The following perspectives are proposed based on these challenges: (1) metals with a lower deposition potential should be used to replace Li metal or be alloyed with Li metal to reduce the power consumption of the metal-mediated NH3 synthesis system, and the ability of the deposited metal to activate N2 should also be tested and verified. (2) Many liquid electrolytes from the field of Li-ion batteries are expected to be alternative solvents for Li-NRR systems, such as 1,2-dimethoxypropane, dimethoxymethane, propylene carbonate, ethylene carbonate, dimethyl carbonate, diethyl carbonate, and ethyl methyl carbonate. (3) To eliminate mass transfer limitations and the high internal resistance of the reactor, the Li-NRR system can be run in a flow cell, a slim flow cell, or a membrane electrode assembly (MEA) reactor. The GDE configuration will be able to directly transport the gaseous reactants to the catalytic interface, and the zero-gap characteristics can greatly eliminate the high internal resistance caused by the distance between the traditional working electrode and the reference electrode, finally helping the Li-NRR technology move toward practical applications. (4) The development and use of new in situ/operando technologies, such as electrochemical quartz crystal microbalance with dissipation monitoring, X-ray diffraction, X-ray absorption fine structure, Raman spectroscopy, and transmission electron microscopy, coupled with artificial intelligence and advanced ab initio molecular dynamics for real-time detection and simulation of the entire reaction process, will help determine the emergence and transformation of key reaction intermediates and monitor key components such as the SEI layer, ultimately leading to an understanding of the internal mechanism.
Based on the outlook above for the future of the Li-NRR, metals other than Li remain to be explored for potential advantages in terms of EE, reaction speed, stability, reactor design, and cost. Recently, a calcium salt, Ca[B(hfip)4]2, was synthesized by the reaction of Ca(BH4)2 with hexafluoroisopropanol. Ca[B(hfip)4]2 dissolved in THF functioned as an electrolyte, and ethanol was used as a proton shuttle agent (Fig. 12(f)). The Ca-NRR process was successfully performed in a continuous-flow electrolytic cell. At normal temperature and pressure, Ca[B(hfip)4]2 achieved an NH3 FE of 40 ± 2% (Fig. 12(g)). The dissociation energy barrier of N2 on the surface of metallic Ca was relatively low, which indicated that the dissociation rate of N2 on the surface of Ca was very fast at normal temperature and pressure. Compared to Li metal, Ca is a promising alternative. This result opened the door to using metals with high abundance for the mediated synthesis of NH3, such as Mg, Ba, Sr, and Na, which could be used, thus allowing for the selection of efficient, highly selective, stable, cost-effective, and scalable NH3 synthesis technologies.286
In addition to optimizing various parameters and the mediating metals in Li-NRR technology, a significant increase in yield rates per geometric surface area is required to make the metal-mediated NH3 production strategy competitive for large-scale applications. For example, a high-surface-area Cu electrode was synthesized using a hydrogen bubble templating method on a Ni foam substrate (HBTCu). At a high current density (100 mA cmgeo−2) and 20 bar N2, the developed HBTCu catalyst achieved an NH3 yield rate of 46.0 ± 6.8 nmol s−1 cmgeo−2, which was 50 times higher than that of a flat copper foil. The high current density was attributed to the use of a high-specific-surface-area substrate.287
7. Design criteria for green ammonia synthesis catalysts
7.1 Photocatalytic ammonia synthesis
The low efficiency of photocatalytic ammonia synthesis is caused by several factors: (1) it is difficult to balance light absorption and charge separation. For instance, narrow-bandgap semiconductors like CdS (2.4 eV) can absorb visible light, but their charge recombination rate is high (over 80%). Wide-bandgap materials such as TiO2 (3.2 eV) have good charge separation but only respond to ultraviolet light. These problems lead to the photogenerated electron–hole pair lifetime of the photocatalyst being much shorter than the time required for N2 reduction. (2) N2 activation and proton transfer kinetics are limited. High-energy electrons are needed to break the NN bonds, while the reduction potential of photogenerated electrons is limited. The low solubility of N2 in aqueous solutions leads to a low N2 coverage rate on the catalyst surface. At the same time, the high proton concentration in acidic media easily triggers the hydrogen evolution reaction, which involves a 2e− transfer with relatively fast reaction kinetics, while the proton transfer rate is low in neutral or alkaline media. (3) Photocatalysts can suffer from photocorrosion and chemical dissolution. The strong oxidizing property of photogenerated holes leads to the self-decomposition of the catalysts. For instance, metal sulfides like CdS and MoS2 are oxidized to SO42− by holes, and the active metal centers of single-atom catalysts can dissolve in acidic environments. Therefore, to achieve highly active photocatalytic ammonia synthesis, the design of photocatalysts should consider the following aspects: (i) select narrow-bandgap semiconductors to ensure a response to visible light (400–700 nm) and even near-infrared light (over 700 nm), such as metal oxides like BiVO4 and nitrates like g-C3N4. On this basis, nanoparticles of precious metals like Au and Ag can be introduced to enhance light capture. Constructing heterostructured photocatalysts, such as Z-type and S-type, promotes efficient charge separation and transport while retaining strong reducing electrons and strong oxidizing holes. In addition, a co-catalyst can be introduced as an electron trap to accelerate the transfer of electrons to the N2 molecules. (ii) Design catalysts with single atoms of transition metals to promote the breaking of NN bonds. N2 adsorption can also be enhanced by introducing oxygen or sulfur vacancies and nitrophilic groups. Introducing hydrophobic functional groups at the catalyst interface can inhibit the competitive hydrogen evolution reaction. (iii) Extending the stability of photocatalysts enhances their photocatalytic efficiency. Strategic stabilization approaches focus on the following modifications: depositing an epitaxial protective layer to physically isolate the active sites and precisely engineering the coordination environment of the metal centers to thermodynamically disfavor dissolution. The protective layers typically exhibit critical characteristics: chemical inertness against hole oxidation, high transparency to maintain light penetration, and controlled porosity to permit reactant diffusion while blocking oxidative species. Concurrently, modifying the primary coordination sphere of the active metal sites through nitrogen or phosphorus coordination or strain engineering alters the electronic structure.
7.2 Electrocatalytic ammonia synthesis
The design of high-performance electrocatalysts for ammonia synthesis requires a clear understanding of the key challenges in this process. First, N2 has a low HOMO energy level and a high LUMO energy level, making electron injection difficult and resulting in the high stability of the NN bond. Second, N2 and protons (H+) compete for adsorption sites on the catalyst surface; the overlapping potential windows required for electrocatalytic ammonia synthesis and the hydrogen evolution reaction (HER) result in the dominance of the kinetically easier HER. Taking N2 as an example, its relatively low solubility in the aqueous phase, combined with mass transfer resistance at the gas–liquid–solid three-phase interface, results in an insufficient local reactant concentration. In summary, the design of electrocatalysts for ammonia synthesis should consider the following aspects: (1) nanostructure engineering: strategies such as constructing two-dimensional nanosheets, interface defect engineering, and designing porous structures can significantly increase the number of active sites in the catalyst while accelerating reactant mass transfer through three-dimensional porous active interfaces. Furthermore, introducing a second metal or a non-metal (e.g., B, P, and S) can alter the charge distribution of the catalyst. By regulating the electronic structure of the catalyst or using synergistic effects between different sites, rapid adsorption and activation of reactants at active sites can be achieved, providing the necessary protons and electrons for the reaction and promoting ammonia synthesis. (2) Support engineering: introducing carbon-based supports, metal oxide/chalcogenide supports, and organic framework supports can enhance the catalyst's conductivity. Meanwhile, the d-band center of the metal sites can be modulated by regulating metal-support interactions to optimize N2 adsorption strength. Besides, N, O, or S sites or vacancies on the support surface can anchor highly dispersed active metal atoms (e.g., Fe, Mo, and Ru), improving active site utilization while preventing agglomeration and enabling rapid reactant conversion. (3) Interface engineering: hydrophobic modification of the catalyst surface can regulate proton adsorption strength and suppress the competing HER. Additionally, modifying the electrode surface with nitrophilic groups can increase the concentration of reactants for ammonia synthesis.
7.3 Photoelectrocatalytic ammonia synthesis
Photoelectrocatalytic ammonia synthesis includes the following specific steps: (i) the absorption of light and the excitation of charge carriers; (ii) the separation and transport of photoexcited carriers to the surface of the photoelectrode; and (iii) reduction reactions on the catalyst's surface. Hence, designing and constructing photoelectrode materials with broad solar spectral absorption, fast charge carrier generation and separation, and a large and effective catalytically active surface, while also adsorbing enough N2 molecules, is crucial for achieving a high-efficiency photoelectrocatalytic ammonia synthesis process, especially at low overpotentials. Based on the discussion above, the design of catalysts typically proceeds from two key perspectives: (1) from a photochemical standpoint, catalysts must exhibit high efficiency in photogenerated charge carrier excitation and rapid electron–hole separation. To address the challenges of carrier generation and electron–hole recombination, band structure and electronic structure modulation have proven to be effective approaches. Specifically, band structure engineering involves combining semiconductor materials with different band structures (such as TiO2/CdS and BiVO4/Cu2O systems) to form type-II heterojunctions, Schottky junctions, and other configurations. These heterostructures effectively promote the separation of photogenerated electrons and holes, thereby enhancing charge carrier utilization and photocatalytic efficiency. Among them, type-II heterojunctions drive the spatial separation of electrons and holes through built-in electric fields, while Schottky junctions optimize carrier migration and separation processes through band alignment between two semiconductor materials. Besides, electronic structure modulation can be divided into non-metal doping and metal doping. Non-metal doping (e.g., N or S doping in TiO2) enables directional charge transfer and adjusts the catalyst's Fermi level, while metal doping (e.g., Bi doping in Cu2O) introduces intermediate energy levels; both approaches enhance the visible light response. (2) From an electrochemical perspective, catalysts must possess appropriate adsorption and desorption kinetics for reactants and intermediates to improve electrocatalytic activity and selectivity. Therefore, active site design and surface structure engineering represent two effective strategies. Specifically, active site design generally includes defect engineering and atomic site optimization. Defect engineering involves creating oxygen vacancies and metal vacancies, where oxygen vacancies provide electron-rich regions to promote N2 adsorption and activation, and exposed unsaturated metal sites enhance N2 binding energy. Atomic site optimization strategies include both single-atom and dual-atom site designs. Well-defined single-atom coordination environments increase active site utilization and ammonia selectivity, while dual-atom sites can cooperatively adsorb and activate N2, reducing the energy barrier of the rate-determining step. Furthermore, surface structure design includes morphology control and crystal lattice engineering. Morphology control exposes more active sites and increases the electrochemical active surface area to enhance the mass transport of reactants and intermediates, accelerating their conversion. Crystal lattice engineering effectively improves electron transport, and well-defined lattice structures can enhance selectivity for the target product.7.4 Alkali metal mediated ammonia synthesis
In non-aqueous lithium-mediated ammonia synthesis systems, the reaction proceeds through three key steps: (1) the electrochemical reduction of Li+ to metallic Li (Li+ + e− → Li0) at the working electrode, (2) the chemical reaction between Li0 and N2 to form lithium nitride (6Li + N2 → 2Li3N), and (3) the protonation of Li3N to generate ammonia (Li3N + 3H+ → 3Li+ + NH3). A critical component of this process is the solid–electrolyte interphase (SEI) layer formed through electrolyte decomposition during Li deposition. This ionically conductive yet electronically insulating porous layer has selective permeability; excessive passivation can hinder N2 and electrolyte penetration, preventing essential reactions with metallic Li. Based on this reaction mechanism, current research mainly focuses on key components in non-aqueous systems, such as organic electrolyte formulation, lithium salt selection, proton source design, proton shuttle agents, and metals with lower deposition potentials. Therefore, while most reported systems use cost-effective stainless-steel cloth electrodes, advanced electrode designs should include 3D nanostructured architectures to enhance the specific surface area, optimize Li+ diffusion pathways, and provide confined spaces for homogeneous Li deposition. These designs should also incorporate nucleation modifiers, such as Ag nanoparticles, to reduce the Li nucleation overpotential, guide uniform Li plating and stripping, and maintain SEI layer stability during cycling.These design strategies aim to simultaneously address three critical challenges: (i) improving N2 accessibility to active Li sites, (ii) preventing dendrite formation, and (iii) maintaining stable SEI properties during prolonged operation, which are key requirements for scaling green ammonia synthesis technology.
7.5 N2 for ammonia synthesis
The activation and conversion of N2 molecules is the central challenge in this reaction. Catalyst design must focus on the efficient dissociation or protonation of the NN triple bonds. Specifically, the catalyst should have: (i) high-efficiency N2 activation by providing electrons to populate N2's antibonding pi orbitals, typically achieved using transition metals like Fe, Ru, and Mo; (ii) three-dimensional porous nanostructures to increase N2 adsorption sites and reactant concentration at the catalytic interface; (iii) the ability to suppress the competing hydrogen evolution reaction through the selection of catalysts with weak H* adsorption. Furthermore, metal-nonmetal or bimetallic site catalysts can be designed to modify the atomic coordination environment of the metal centers and increase electron localization. Introducing electron-deficient nonmetal sites can polarize N2 molecules, which synergistically promotes N2 activation and the following hydrogenation steps.
7.6 NO for ammonia synthesis
This reaction faces several challenges: (a) NO may be over-reduced to N2O or N2; (b) NO tends to strongly bind with metal centers, blocking active sites; (c) hydrogenation steps require precise control to avoid byproducts like NH2OH. Therefore, catalyst design should focus on: (i) selecting appropriate metal sites and modulating supports to prevent excessive NO adsorption while weakening the NO bonds; (ii) designing dual metal sites (e.g., metal–acid or metal–sulfur) to cooperatively promote reactant adsorption and hydrogenation and facilitate oxygen removal; (iii) using steric confinement effects (e.g., metal organic framework micropores) to suppress N2O formation; (iv) adding electronegative elements to regulate the d-orbital electrons of metal atoms, preventing excessive NO hydrogenation.
7.7 NO2− and NO3− for ammonia synthesis
The reduction of NO2−/NO3− to ammonia faces several challenges: (a) complex proton-coupled electron transfer processes with various intermediates and products (e.g., NO2, NO2−, NO, N2O, N2, NH3, NH2OH, and N2H4) can lead to side reactions through kinetic competition, reducing ammonia selectivity; (b) high overpotentials are needed for NO2−/NO3− reduction, particularly at proton-coupled electron transfer steps, which increases energy consumption; (c) the hydrogenation pathways for NO2− strongly depend on pH, where NO is favored under acidic conditions and NH3 under alkaline conditions. Thus, developing catalysts with both high activity and selectivity is crucial. Design strategies include: (i) creating efficient nitrogen source adsorption and activation sites; (ii) adding secondary metal components to form heterointerfaces with built-in electric fields that improve the metal's electronic structure and promote proton adsorption for selective ammonia production; (iii) creating multicomponent catalysts (e.g., 5d noble metals) to increase surface H* coverage and improve ammonia selectivity and formation rates; (iv) developing non-precious metal catalysts (e.g., Cu and Ni) with electron-deficient nonmetal sites (e.g., B) to suppress hydrogen evolution. Notably, engineering the interfacial microenvironment, for example, through oxide coatings, –OH group functionalization, or restructuring the interfacial water network, can also increase ammonia production efficiency.8. Reactors for synthesis of ammonia
8.1 Electrocatalytic ammonia synthesis reactors
In previous studies, mainstream research on NH3 synthesis has mainly focused on designing catalysts and catalytic technology routes, rather than on innovating the engineering of the catalytic conversion device. Therefore, the development of electrocatalytic reactors has been slow. It is well known that reactors are critical to catalytic performance. Their design and development involve integrated knowledge from other disciplines, such as material forming and processing, the fluid mechanics of gas–liquid transfer, chemical processes, and analog simulation. Although great strides have been made in the field, major breakthroughs in basic research and practical applications are still needed to bridge the gap between experimental platforms and industrial applications. In addition to the fundamental drivers that promote catalytic performance, engineering aspects such as device architecture and process coupling deserve to be studied and highlighted, as they could indeed facilitate the deployment of this fundamental research on an industrial scale. In this regard, more effort should be devoted to developing high-performance electrolytic devices to minimize the mass transfer limits of reactants, which can significantly improve energy efficiency. Among these, the rapid development of reactors can accelerate the transformation of green ammonia synthesis technology from a laboratory platform to industrial scale production.Ideal electrolytic reactors should have a good function of promoting electron transfer, ion transfer, and controlled gas diffusion, while preventing the mixing of the electrolyte and products. The rational design of the reactor is very vital for achieving a high EE for NH3 synthesis. In this section, the setup of each reactor and the associated experimental procedures will be briefly described. The advantages and disadvantages of each reactor will also be discussed.
It is well known that a reactor can use input energy to drive non-spontaneous reactions. At present, various reactors have been designed for the synthesis of NH3, such as single-chamber cells, H-type cells, flow cells, and MEA reactors, which consist of an electrode, an electrolyte, a GDE, and a diaphragm. Taking the three-electrode electrolyzer of a single-chamber cell as an example, it is composed of a measuring circuit and a polarization circuit, forming a closed-loop circuit between the CE and the WE, with the potential of the WE determined by the RE. In an actual electrocatalytic reaction process, when an external voltage is applied to the reaction system, the ions in the electrolyte will be driven by an electrode with opposite charges, and a reduction or oxidation reaction will occur at the cathode or anode to produce the target products.
In addition, the H-type cell, consisting of two chambers (anode and cathode), is separated by ion exchange membranes. A reduction reaction occurs in the cathode chamber and an oxidation reaction happens on the other side. The ion exchange membranes can effectively block the crossing of different ions and avoid the loss of products. The flow cell device consists of a gaseous cathode chamber, a liquid cathode chamber, and an anode chamber (Fig. 13(a)). A hydrophobic GDE is used to separate the gas–liquid cathode chamber, and the target reaction occurs within the three-phase boundary of the GDE. The GDE as the cathode is completely immersed in the electrolyte, while the configuration exposes one side to the feed gas, allowing the liquid and gas phases to coexist within the catalyst layer. Recently, emerging GDEs combined with flow cell devices have efficiently boosted NH3 synthesis performance and pushed the current density to practical levels by reducing the mass transport limitation of the gas phase. After iterative updating of the reactor, the latest high-performance MEA devices consist of catalysts pressed directly onto both sides of the ion exchange membrane. The MEA configuration takes advantage of the GDE configuration in the flow cell while minimizing the use of electrolyte through direct contact between the catalyst layer and the ion exchange membrane (Fig. 13(b)). This configuration can significantly reduce the full-cell resistance, greatly increasing the current density. Despite the suitability of H-cells and GDE configurations for basic research, such as catalyst development, MEA configurations are highly attractive for industrial applications, while further optimization of components is a promising direction. However, the development of MEAs and catalysts has been carried out separately so far. Therefore, new opportunities exist to translate the catalytic strategies developed in the H-type/flow cell regime into MEA devices to improve the energy efficiency of the whole cell. At the same time, when a catalyst is considered for commercial applications, its important performance indicators at high current densities should be carefully reported. It is also worth noting that catalytic stability tests are typically conducted over a few hours, rather than the hundreds to thousands of hours required for industrial applications. Moreover, many catalyst deactivation mechanisms, such as surface reconstruction, contamination, and mechanical failure, are likely to occur during long-term electrolysis; these phenomena must be carefully examined. In addition, the accurate quantification of reactor energy efficiency and related conversion products is particularly important. These parameters will help to evaluate and standardize the industrialization process of electrochemical NH3 synthesis more strictly.288
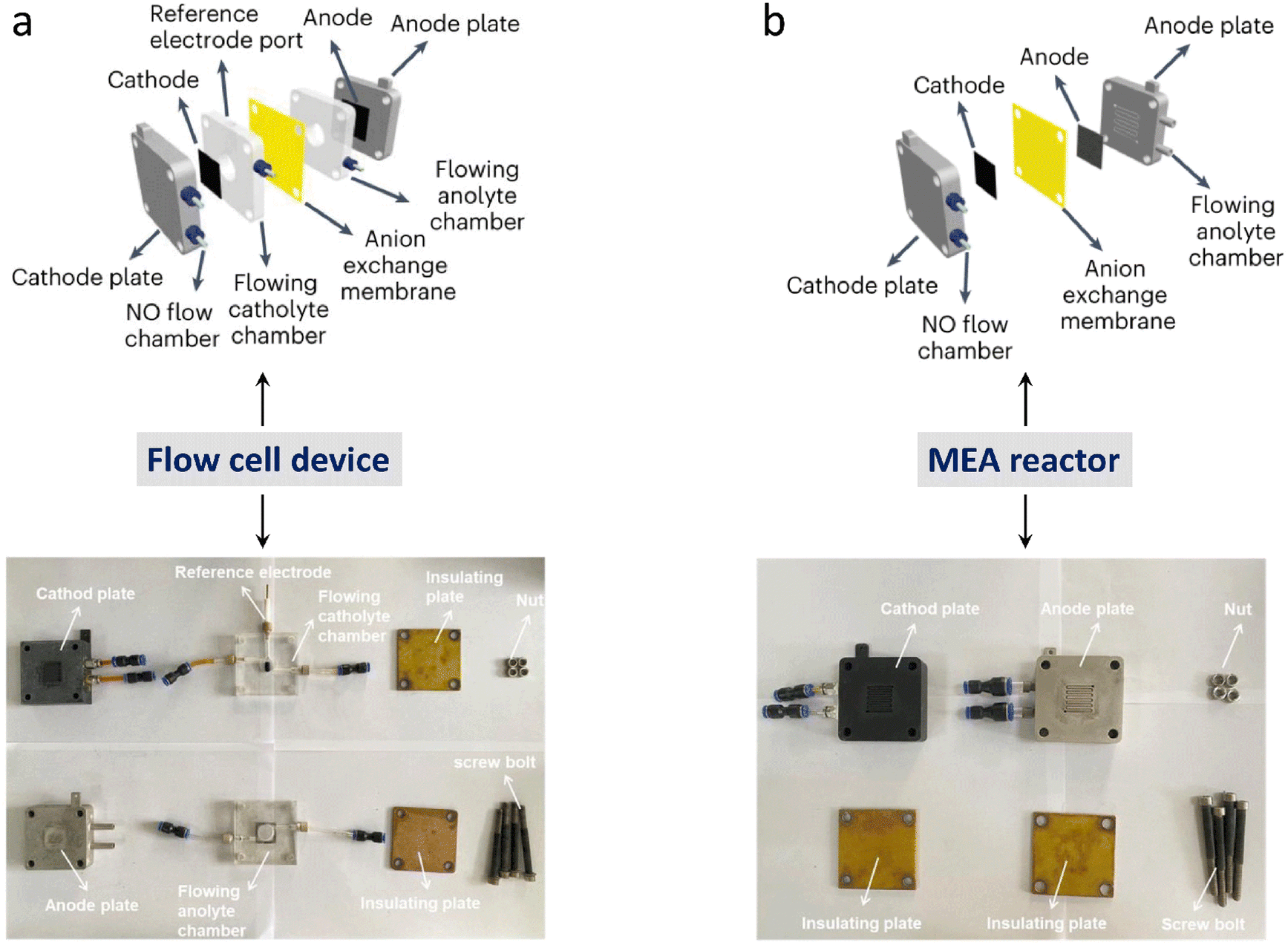 | ||
| Fig. 13 Schematic illustration and digital photos of a (a) flow cell device and (b) MEA reactor. Reproduced with permission from ref. 292 Copyright 2023, Springer Nature. | ||
Based on the various types of reaction devices, a corresponding summary is made. Specifically, the structure of the traditional three-electrode device is a single-chamber reactor, where reduction and oxidation reactions are carried out in the same electrolyte environment. For example, NH3 products were produced using a three-electrode single cell, with a Pt mesh and an Ag/AgCl electrode as the counter and reference electrodes, respectively. 5.0 mg of the catalyst was dispersed into 1000 μL of a solvent consisting of 400 μL of distilled water, 500 μL of ethanol, and 100 μL of Nafion to achieve a homogeneous catalyst ink by ultrasonication. Then, 50 μL of an ink of sulfur-and-nitrogen-codoped carbon matrix loading single Fe atoms (FeSA-NSC) was loaded on carbon paper (1 × 1 cm2) to prepare the working electrode. Before NRR tests, the electrolyte was purged with N2 gas for 30 minutes. The three-electrode reaction system based on FeSA-NSC exhibited excellent performance, with a yield rate of 30.4 μg h−1 mgcat.−1 and an NH3 FE of 21.9% at −0.4 V, surpassing those of most reported Fe-based electrocatalysts developed to date.289 Besides, a single-reaction cell with a metal–nitrogen–carbon catalyst was also used to promote the electrosynthesis of NH3 under mild conditions, illustrating the reliability of the single-chamber reactor, with a remarkable yield rate of 31.9 μg h−1 mgcat.−1 and NH3 FE of 11.8% at −0.4 V.5
The H-type cell is currently widely used in reactions involving product upgrading, such as the carbon dioxide reduction reaction, the oxygen reduction reaction, and the NH3 synthesis reaction. In the H-type cell, the redox reaction chambers are usually separated by ion exchange membranes of different types and models, making the reaction environment of the anode and cathode relatively independent so that the two chambers can be controlled more flexibly. Recently, a typical two-compartment H-type cell was adopted, separated by a Nafion 211 membrane and containing Ag/AgCl as the reference electrode and a Pt wire as the counter electrode, to conduct NH3 electrosynthesis, achieving a high yield rate of 16.1 μg h−1 mgcat.−1 and an FE of 11.8% in alkaline media.12 Currently, single-chamber and H-type cells are the two most widely used reactors in electrocatalysis technology. However, it is worth noting that although the single-chamber reactor has a simple structure and is convenient to operate, the NH3 generated on the cathode can easily diffuse to the anode in a narrow and connected environment and be further oxidized, resulting in a low yield rate and wasted electrical energy. Therefore, to solve this problem, without increasing the complexity of the reactor structure, the H-type cell uses a membrane to separate the oxidation and reduction reaction chambers, which are relatively independent, making it difficult for the prepared products to cross over. As a result, the H-type device has a more obvious advantage in improving the production yield. This effectively shows that the electrolyzer configuration indirectly determines the performance advantage of the catalytic system to a certain extent. Therefore, to promote the activity of the NH3 synthesis system to the industrial standard, it is necessary to conduct an in-depth analysis and rationally design and optimize the electrolyzer structure, according to the key scientific issues causing the low activity index. For example, the distance between the WE and the RE leads to a large internal resistance in the traditional three-electrode single-chamber and H-type cells, resulting in a big gap between the electrochemical test performance and the commercial performance requirements. Furthermore, due to the slow dissociation step of the highly stable NN bond, the NH3 formation rate or NH3 partial current density of the N2 reduction reaction is extremely low (∼0.1 mA cm−2). The low productivity is not only far from industrial requirements, but even the influence of trace impurities makes the experimental results unreliable.
At the same time, due to the limited catalyst activity and the fierce competition of the cathode HER, the NH3 production rate and FE achieved are still very limited. According to the performance targets required for the commercialization of the electrosynthesis of NH3 published by the REFUEL program, most currently reported performance is far from satisfactory, and the road to industrialization and commercialization is still long. Therefore, it is necessary to further optimize the reactor, decrease its internal resistance, and construct a high-current-density and high-selectivity NH3 electrosynthesis device.
To build efficient catalytic reactors, we should start from the essence of the synthesis reaction. The extremely low solubility of N2 gas in aqueous solutions has two effects on electrochemical NH3 production in aqueous solution: (a) the lower N2 concentration in the electrolyte is not conducive to increasing the N2 reduction to NH3, compared to proton donors like water molecules and protons; (b) the NH3 production rate can be limited by the mass transfer of N2 molecules. To solve these problems, a reactor with a fast and efficient contact interface of gas–liquid–solid may be a promising approach. The three-phase boundary is a particularly significant concept in the field of fuel cells; it, which is the complex contact region that includes the reactant gas, the solvent, and the catalyst. In this case, the gas–liquid ratio in a conventional reactor can be effectively modified from gas-in-solvent to solvent-in-gas, so that mass transfer will not be a vital factor. In addition, to solve the surge of internal resistance in the reactor caused by excessive electrode spacing, polymer-based ion exchange membranes were introduced to construct MEA devices. The membrane can be a proton exchange membrane (PEM) or an anion exchange membrane (AEM). By constructing a near-zero-gap MEA reactor,20 the internal resistance of the reactor can be reduced by orders of magnitude.
Based on the reactor optimization strategies understood above, the N2 reduction activity was evaluated using a continuous flow cell with a 25 cm2 GDE that has recently been produced. A three-chamber continuous flow cell was first constructed, where the GDE (stainless-steel cloth (SSC)) was placed between the reactant gas flow plate and the electrolyte flow plate (Fig. 14(a)). The N2 and the electrolyte could be supplied to different sides of the GDE, in which the injected N2 can directly participate in the reaction, breaking the mass transfer limitation (Fig. 14(b) and (c)). The design of the continuous flow reactor effectively improved NH3 synthesis, with an FE of 61 ± 1% and an EE of 13 ± 1% at a current density of 6 mA cm−2 (Fig. 14(d)), under 1.0 bar and an effective area of 25 cm2. Meanwhile, benefiting from more than half of the gas phase product being NH3, the separation of the gas and liquid phases would cause low reactor resistance, further improving performance (Fig. 14(e)). Although the three-compartment, continuous flow reactor achieved high FE and EE for NH3 electrosynthesis, it does not solve all the problems at the industrial application level.280 Future studies should focus on boosting the current density, facilitating the mass transfer process, and effectively regulating the pressure gradient between the gas and the electrolyte. The main goal is to achieve high FE and EE under industrial conditions. To solve the technical bottlenecks mentioned above, such as limited gas mass transfer and significant voltage loss, an MEA configuration based on a polymer electrolyte film was recently proposed. The cathode of Li-deposited stainless steel, the polymer electrolyte of a Li salt-doped polyethylene oxide film, and the anode of carbon paper containing Pt/C particles were pressed to form a ‘three-in-one’ MEA structure (Fig. 15(a)–(c)), which can be used in combination with a conventional gas diffusion electrolytic cell to significantly improve the efficiency of N2 mass transfer. Due to the ultrathin membrane electrode, the ohmic impedance of the reactor was significantly reduced. Specifically, a yield rate of 2.41 ± 0.14 μmol h−1 cmgeo−2 and an FE of 8.9 ± 1.7% were obtained with a large catalytic area of 5.0 cmgeo−2. With its advantages of efficient gas transfer, reduced solvent consumption, and compact design, the proposed MEA configuration will enable the most promising Li-NRR process to be applied in a practical manner (Fig. 15(d)). The advantages of an MEA device with a GDE have been clearly shown. In the following, the two types of reactors were also compared to inspire the optimal reaction configuration. An electrode with catalysts directly in contact with an ion exchange membrane was the first type of reactor, while in the second reactor, an additional GDE was inserted between the electrocatalyst layer and the Nafion membrane (Fig. 15(e) and (f)). The additional GDE serves two purposes: (i) it significantly reduces the amount of NH3 that crosses the Nafion membrane; using an additional layer of GDE will eliminate the penetration of NH3. (ii) The additional GDE hinders the proton transfer rate, thereby inhibiting the HER and ultimately improving the FE by reducing proton availability. Compared with the single GDE configuration based on an Fe2O3/CNTs catalyst, when the additional GDE configurations were used for NH3 synthesis, the related NH3 yield rate and FE increased by 158% and 571%, respectively, confirming the effectiveness of the strategy above.291
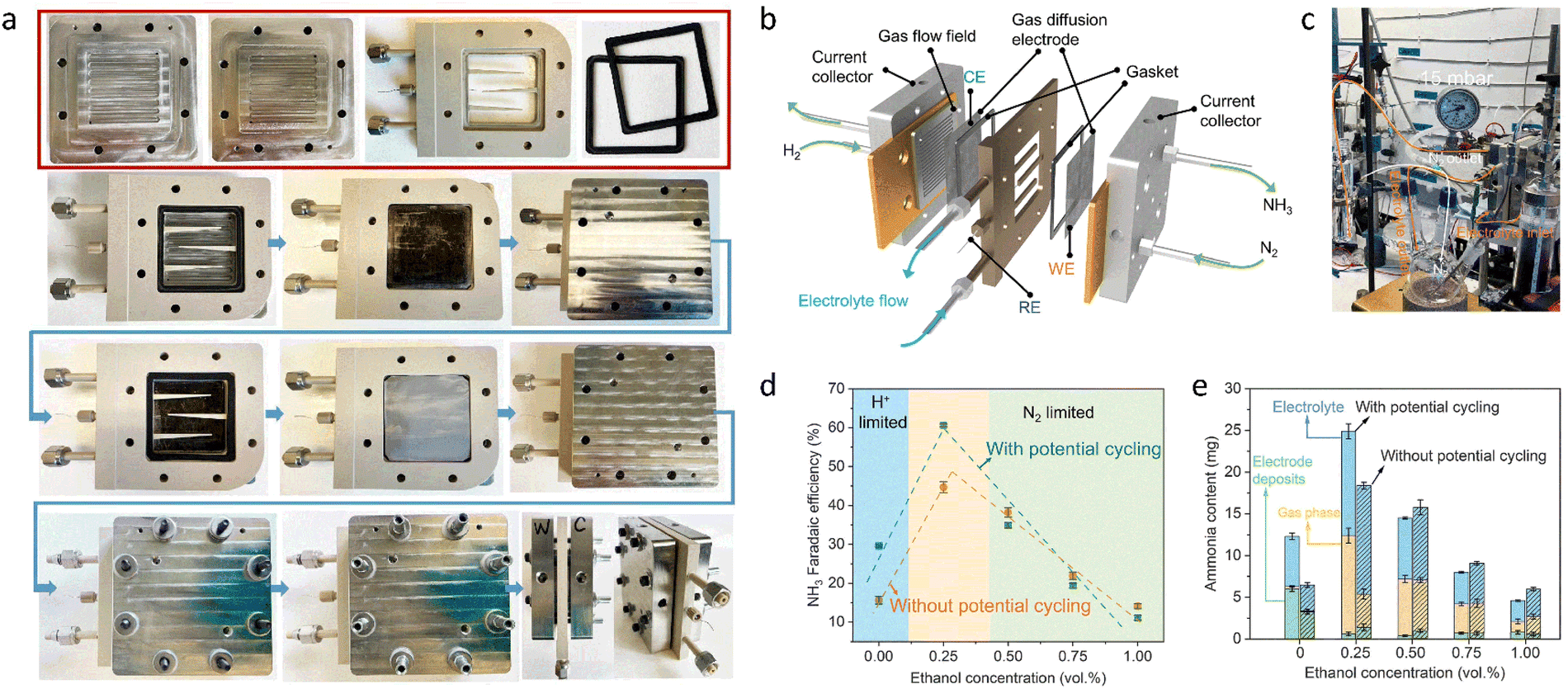 | ||
| Fig. 14 (a) Digital photos of assembling a three-compartment continuous-flow reactor. (b) Schematic illustration of continuous-flow electrolyzer configurations. (c) Digital photo of the continuous-flow electrolyzer setup. (d) FE of NH3 for a continuous-flow electrolyzer. (e) Produced NH3 amounts for a continuous-flow electrolyzer. Reproduced with permission from ref. 280 Copyright 2023, American Association for the Advancement of Science. | ||
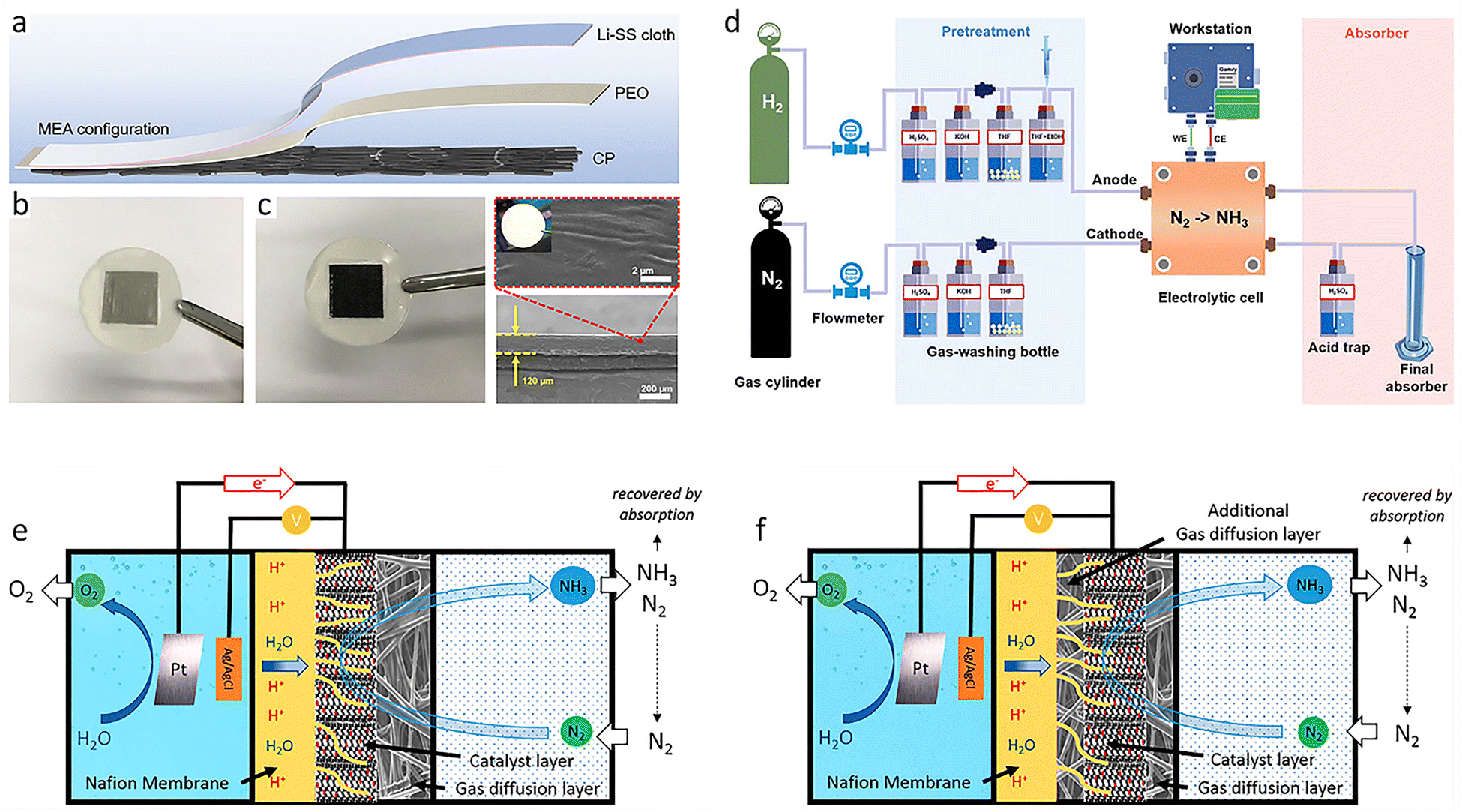 | ||
| Fig. 15 (a) Schematic illustration of “three-in-one” MEA. Digital photos (b) and SEM images (c) of a “three-in-one” MEA loading catalyst. (d) Complete flow chart of a synthetic NH3 device. Schematic illustration of (e) the three-phase reactor without additional GDE, and (f) the upgraded three-phase reactor with additional GDE. Reproduced with permission from ref. 290 Copyright 2023, The Royal Society of Chemistry. Ref. 291 Copyright 2017, American Chemical Society. | ||
Based on previous technology development, optimizing the GDE and upgrading and scaling up the MEA reactor are expected to further advance NH3 electrosynthesis technology to an industrial platform. Recently, a new type of GDE was developed that is different from commercial GDEs. In this new GDE, the microporous layer is first prepared by brushing a suspension of carbon black and polytetrafluoroethylene onto carbon paper. The mass ratio of PTFE in the microporous layer was 40 wt% and the carbon black loading was 1.0 mg cm−2. Furthermore, the scaled-up MEA electrolyzer contained the prepared GDE loaded with a Cu6Sn5 catalyst with an active area of 100 cm2 as the cathode and an iridium black-coated foam as the anode (Fig. 16(a)). A piece of quaternary ammonium poly(N-methylpiperidine-co-p-terphenyl) membrane served as the anion exchange membrane. The flow rate of the 0.5 M KOH electrolyte solution was maintained at 125 mL min−1, and the flow rate of NO was controlled at 1.875 L min−1. Benefiting from the upgrade of the MEA device, the NH3 yield rate in a scaled-up electrolyzer based on the Cu6Sn5 material achieved ∼2.5 mol h−1 with an NH3 FE of about 80% at an ultra-high current of 400 A (Fig. 16(b) and (c)). The results presented here indicate that the sustainable production of NH3 is feasible in a reverse nitrogen cycle driven by renewable electricity.292
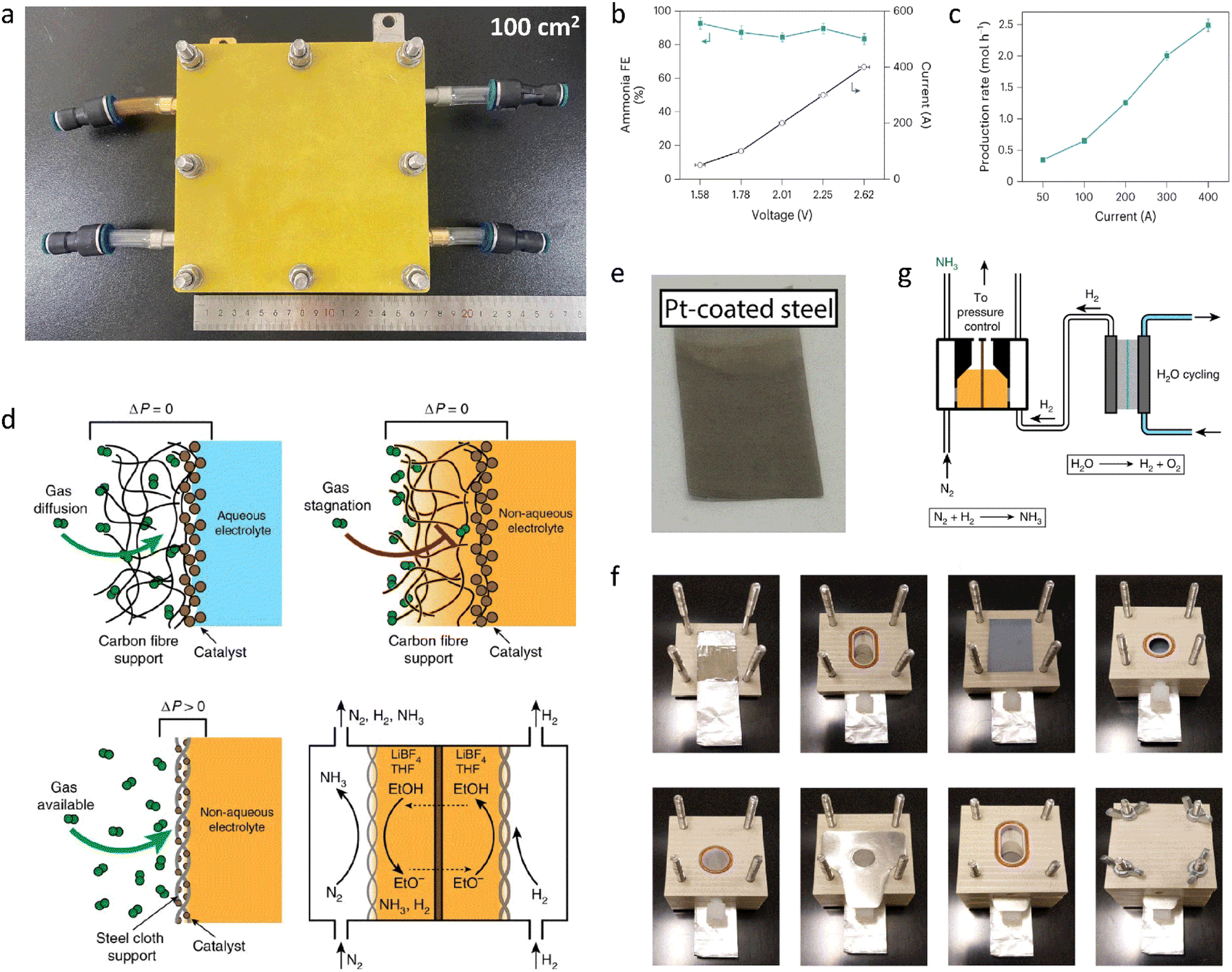 | ||
| Fig. 16 (a) Digital photo of the large area MEA electrolyzer. (b) NH3 FE, and (c) NH3 yield rate at high current. (d) Carbon fiber supported GDE in aqueous and non-aqueous environments and upgraded steel cloth supported GDE applied in non-aqueous environments. (e) Photo of Pt-coated steel cloth. (f) Digital photos of assembling a three-compartment cell with a steel cloth supported GDE. (g) Pressure controlled NH3 electrosynthesis reactor. Reproduced with permission from ref. 292 Copyright 2023, Springer Nature. Ref. 98 Copyright 2020, Springer Nature. | ||
In addition, it is well known that the most important function of the GDE is to bring the reaction gas, electrolyte, and catalyst into close contact, minimizing the diffusion distance of the reaction gas and thus greatly increasing the diffusion rate. Therefore, the GDE is usually subjected to a hydrophobic treatment to prevent the electrolyte from penetrating and wetting its fiber structure. Unfortunately, if the main component of the electrolyte is a non-aqueous solvent, the hydrophobic interaction between the GDE carrier and the electrolyte will be lost. In this case, the electrolyte will enter the fiber structure of the GDE and flood the catalyst, leading to a greatly increased diffusion distance and a reduced current density for the reaction. Therefore, for a non-aqueous NH3 synthesis system, optimizing the internal components of the reactor is also important. Following this idea, GDE-like functionality is achieved by controlling material wetting and electrolyte penetration into the support in a non-aqueous system (Fig. 16(d)).98 Specifically, a stainless steel cloth (SSC) was used as a substrate for catalyst deposition, and electrolyte penetration was controlled by maintaining a pressure gradient (Fig. 16(e)–(g)). This method was used for an efficient HOR on Pt-coated SSC at a current density of 25 mA cm−2 in THF and propylene carbonate-based electrolytes. In addition, Li metal loaded on an SSC substrate was able to produce NH3 using the Li-NRR method. The partial current density of NH3 was 8.8 ± 1.4 mA cm−2, and the FE was 47.5 ± 4.0%. The two electrodes were coupled together to build an NH3 synthesis reactor, which had a high FE of 30 ± 2%.98
Although these electrocatalysts have made progress in highly selective NH3 production, their current density and NH3 yield rate are far below practical application requirements. A large part of the problem stems from the fact that current reactors lack short-term ion balance and stability. For high-efficiency NH3 synthesis, a suitable reactor remains to be developed. Based on this challenge, a reactor equipped with a bipolar membrane (BPM) and a Cu-activated Co electrode was used to synthesize NH3.293 Specifically, the catalyst in the BPM intermediate layer effectively dissociates H2O molecules, and the resulting H+/OH− moves from the intermediate layer to the poles, ensuring ionic equilibrium.208,294 The target optimized reactor based on the BPM membrane delivered an excellent FE of 96.45% and a high NH3 yield rate of 279.44 mg h−1 cm−2, as well as a high current density of 2.64 A cm−2. To facilitate the practical application of scalable NH3 synthesis, a BPM reactor with a large-area electrode (10 × 10 cm2) was constructed for the gram-scale synthesis of NH3. Under a constant current condition of ∼40 A, the yield rate and FE can reach 4.11 g·h−1 and 97.19%, respectively, thus showing great potential for industrial NH3 production.293
Based on the important breakthrough in the field of BPM reactors for NH3 electrosynthesis, optimizing the BPM interface is expected to further promote the electrosynthesis of NH3 toward large-scale applications. The ionic selectivity of bipolar membranes is formed by the principle of electrostatic repulsion. Although H+/OH− ions produced in situ can adjust the acid–base microenvironment, the structure or performance of the ionic membranes will be degraded. Therefore, to solve this problem, physically interlocked bipolar membranes were prepared according to the traditional ‘mortise-tenon joint’ structure. The membranes with a 3D physically interlocked interface can efficiently dissociate water, and the imbalance of ion migration can be eliminated in the in situ acid–base environment, thus realizing the continuous, stable operation of the NH3 electrosynthesis process (Fig. 17(a) and (b)). Among them, the innovative design of the bipolar membrane with an interlocking structure not only increases the sites for water splitting but also cleverly uses the ‘self-swelling’' effect of the polymer in water to achieve a ‘self-locking’ effect, improving the stability of the bipolar membrane (Fig. 17(c)). The electrosynthesis of NH3 was carried out using a reactor based on a BPM with a ‘mortise-tenon joint’ structure, which achieved an FE of 86.2% and an NH3 production rate of 68.4 mg h−1 cm−2 using merely 2000 ppm NO3− in alkaline electrolytes at a high current density of 1000 mA cm−2. Moreover, a >100-hour operation at 1000 mA cm−2 also endorsed the confidence of using the as-prepared BPM reactor at a high FE and yield rate (Fig. 17(d)).295
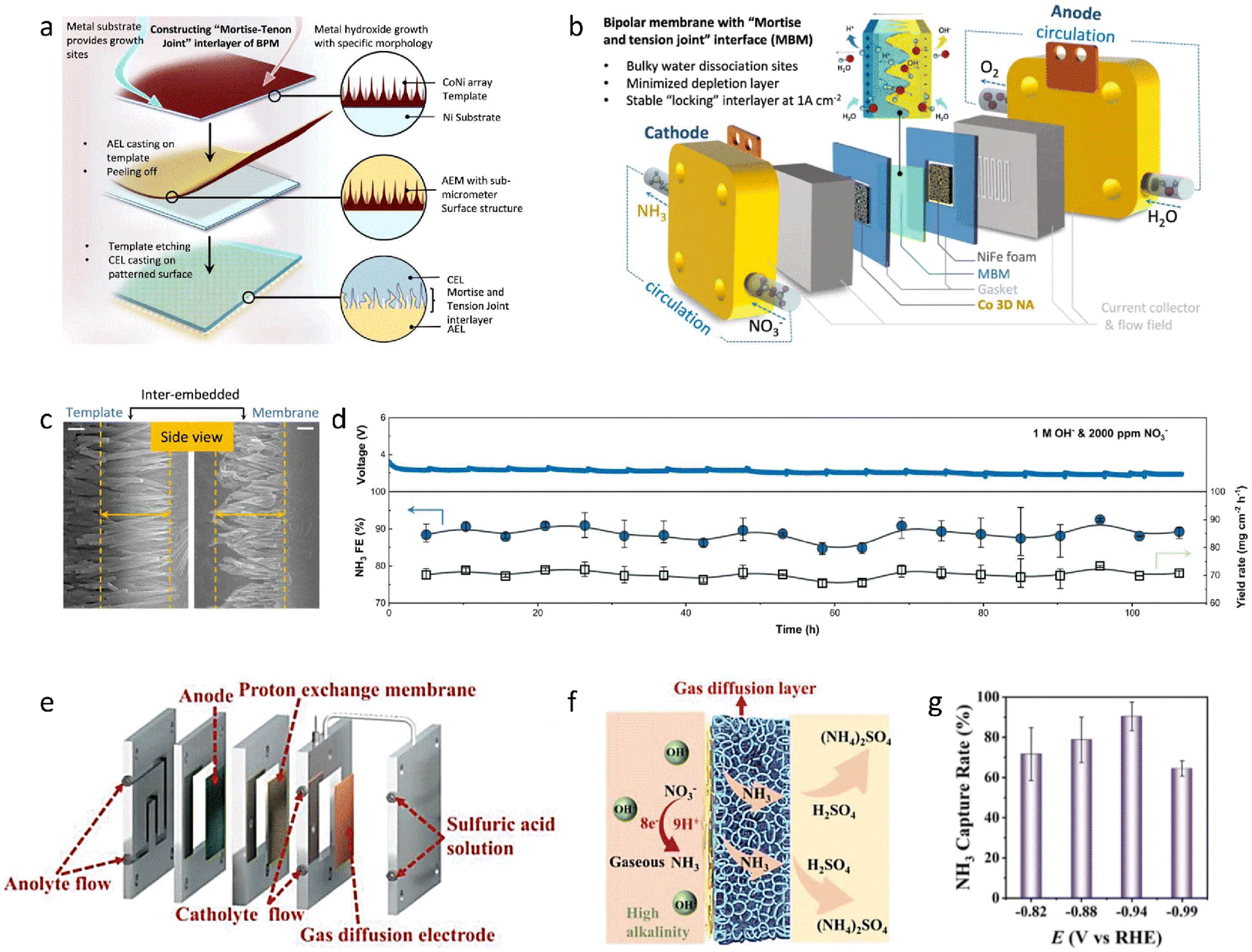 | ||
| Fig. 17 (a) Schematic illustration of the “Mortise–Tenon Joint” structure of BPM. (b) Schematic illustration of the BPM reactor for NH3 synthesis. (c) SEM images of the “Mortise–Tenon Joint” structure of BPM. (d) FE and yield rate of NH3 for a long-term BPM synthesis system at a high current density of 1000 mA cm−2. (e) Schematic illustration of a “two-in-one” reactor integrating the electrolytic and NH3 capture chamber. (f) Schematic illustration of capturing NH3 using a gas diffusion layer. (g) Diagram of the NH3 capture rate. Reproduced with permission from ref. 295 Copyright 2023, Springer Nature. Ref. 296 Copyright 2022, Wiley-VCH. | ||
Through the development of high efficiency conversion technology, high activity catalysts, and high-performance reactors, NH3 synthesis technology has made great progress. However, in the advance of synthetic NH3 technology, although NH3 production and capture are equally critical for practical applications, not enough attention has been paid to capturing it from the electrolyte. Based on the fact that almost all NH3 generated in the low-current NH3 synthesis process is in a gaseous state on the electrode surface, a ‘two-in-one’ electrolytic cell was recently designed to integrate the electrolytic chamber and the NH3 capture chamber through a commercial GDE, aiming to achieve simultaneous NH3 production and capture (Fig. 17(e)).296 The prepared electrolyzer promoted NH3 synthesis by rapidly moving products away from the three-phase interface. Specifically, by using a sulfuric acid solution to capture the synthesized NH3, the two liquid flow plates were separated by a commercial GDE (Fig. 17(f)). The gaseous NH3 generated on the other side of the GDE loaded with catalyst spontaneously passed through the GDE and directly came into contact with the plate of flowing H2SO4 solution to achieve real-time preparation and capture of NH3 products. Consequently, through a carefully designed synthesize-and-capture monolithic reactor, a yield rate of 2.1 mmol h−1 cm−2 was obtained at a high current density of 528.0 mA cm−2, along with a high capture efficiency of 90.4% (Fig. 17(g)).296
8.2 Photocatalytic ammonia synthesis reactors
In photocatalytic systems, quartz glass reactors are commonly used (Fig. 18(a)),297 equipped with magnetic stirring devices, a Xe lamp (with cut-off filters for spectral selection), a solar simulator, or actual sunlight illumination, along with gas bubbling apparatus. The photocatalyst powder is uniformly dispersed in the reactor containing the reactants and electrolyte solution. The reaction temperature is maintained at 25 °C (unless otherwise specified) through circulating cooling water. While such slurry-phase photocatalytic reactors offer operational simplicity and a large catalyst-reactant contact area, they present challenges including difficult catalyst recovery and low light utilization efficiency.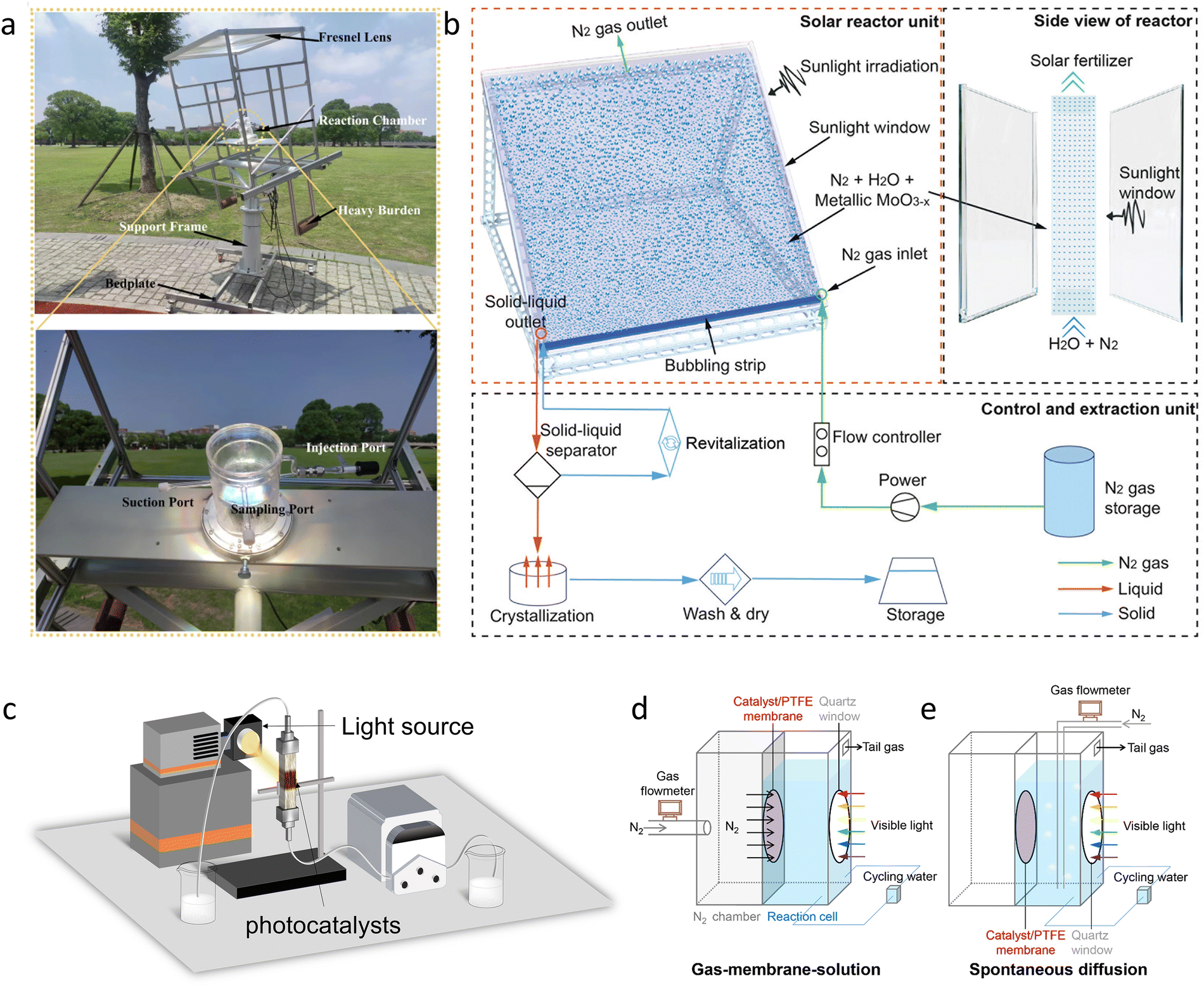 | ||
| Fig. 18 (a) Digital photos of a quartz glass photocatalytic reactor. (b) Flat plate photocatalytic ammonia synthesis reactor. (c) Fixed-bed photocatalytic ammonia synthesis reactor. (d) Gas-membrane-solution photocatalytic ammonia synthesis reactor. (e) Spontaneous diffusion photocatalytic ammonia synthesis reactor. Reproduced with permission from ref. 297 Copyright 2024, Springer Nature. Ref. 298 Copyright 2025, Wiley-VCH. Ref. 129 Copyright 2021, American Chemical Society. | ||
Recent advancements have led to the development of flat-plate and fixed-bed photocatalytic reactors (Fig. 18(b) and (c)).298 In these systems, catalysts are immobilized on supports (e.g., glass fibers, porous ceramics, and conductive substrates) as thin films or coatings. Illumination occurs either from one side or through the center of the catalyst-loaded support to drive photocatalytic ammonia synthesis. These reactors facilitate catalyst recovery and enable continuous-flow operation, although light penetration depth remains a limitation. Future designs could use three-dimensional porous supports to enhance both gas mass transfer and light harvesting. Additionally, a gas-membrane-solution reactor configuration has been developed (Fig. 18(d)).129 In this design, fabricated catalyst/polytetrafluoroethylene (PTFE) membranes are sandwiched and sealed between gas chambers and reaction cells, with the catalyst side immersed in the reaction solution. Light irradiation through the reactor window illuminates the catalytic electrode while N2 gas continuously diffuses through the PTFE side into the reaction cell for photocatalytic ammonia production. In another configuration, the spontaneous diffusion reactor immerses catalyst/PTFE membranes directly in the reaction solution (Fig. 18(e)).129 During operation, high-purity N2 gas is continuously bubbled through the solution while light irradiates the catalyst/PTFE membranes to drive the photocatalytic reaction.
The fundamental objective of photocatalytic ammonia synthesis reactors is to achieve efficient, stable, and scalable ammonia production. Based on the analysis above, future reactor optimizations should focus on: (1) enhancing light utilization efficiency through reflective internal surfaces to minimize light loss; (2) integrating low-temperature condensation and other separation technologies for timely product extraction; (3) improving reactor designs to maximize both photon capture and mass transport while maintaining practical scalability.
8.3 Photoelectrocatalytic ammonia synthesis reactors
Currently, photoelectrocatalytic ammonia synthesis systems primarily use three typical reactor configurations including the single-chamber cell, the H-type cell, and the flow cell, each with distinct structural characteristics and applications. Specifically, (1) the single-chamber cell reactor (Fig. 19(a))299 consists of five key components: photoelectrocatalysts as the working electrode, a counter electrode, a reference electrode, an electrolyte, and a light source. The photoelectrode serves as the core functional unit, simultaneously performing light harvesting, charge separation, and catalytic reduction. The counter electrode maintains charge balance by completing the electrical circuit, while the reference electrode (e.g., Ag/AgCl or Hg/HgO) enables precise control of the working electrode potential. The electrolyte system not only provides ionic conduction but also participates in proton-coupled electron transfer processes. In typical operation, light penetrates through a quartz window to illuminate the photoelectrode, driving ammonia synthesis under an applied bias. However, this simple design has significant drawbacks: ammonia products generated at the cathode readily diffuse to the anode region, leading to product oxidation and consequently severe reduction in reaction selectivity and faradaic efficiency. (2) The H-type cell reactor (Fig. 19(b)):300 to address the limitations above, the H-type cell uses a dual-chamber structure physically separated by an ion-exchange membrane, isolating the cathode chamber (containing the photoelectrode) from the anode chamber (containing the counter and reference electrodes). This separation effectively prevents ammonia from diffusing to the anode side, significantly increasing the ammonia production rate. Nevertheless, the H-type cell introduces new technical challenges: (i) the large electrode-membrane spacing substantially increases internal resistance; (ii) the static reactor design severely limits the mass transport of nitrogen sources; (iii) the batch operation mode cannot meet industrial-scale continuous production requirements. These inherent limitations ultimately constrain the maximum achievable current density and practical applicability. Therefore, the flow cell reactor successfully overcomes these constraints through a revolutionary structural design. (3) The flow-cell reactor (Fig. 19(c) and (d)):301,302 distinct from conventional configurations, flow cells use highly integrated modular designs with core components including bipolar plates, photoelectrode pairs (photocathode/anode), peristaltic pump systems, gas diffusion layers (GDL), and ion-exchange membranes. The bipolar plates provide mechanical support and sealing while serving as efficient current collectors to construct compact electron conduction networks. Notably, the precisely engineered microfluidic channels (e.g., serpentine or parallel patterns) on the bipolar plate surfaces enable the directional transport of reactants and products, effectively mitigating concentration polarization. Coupled with pump-driven forced convection and GDL-optimized gas transport, this design dramatically enhances reaction efficiency at triple-phase boundaries. Light irradiation through specialized transparent conductive windows (e.g., FTO glass) activates the photocathode surface, enabling efficient ammonia synthesis under optimized potential control. Furthermore, integrating an acid solution absorption device enables real-time product collection, significantly improving overall production efficiency (Fig. 19(e)).302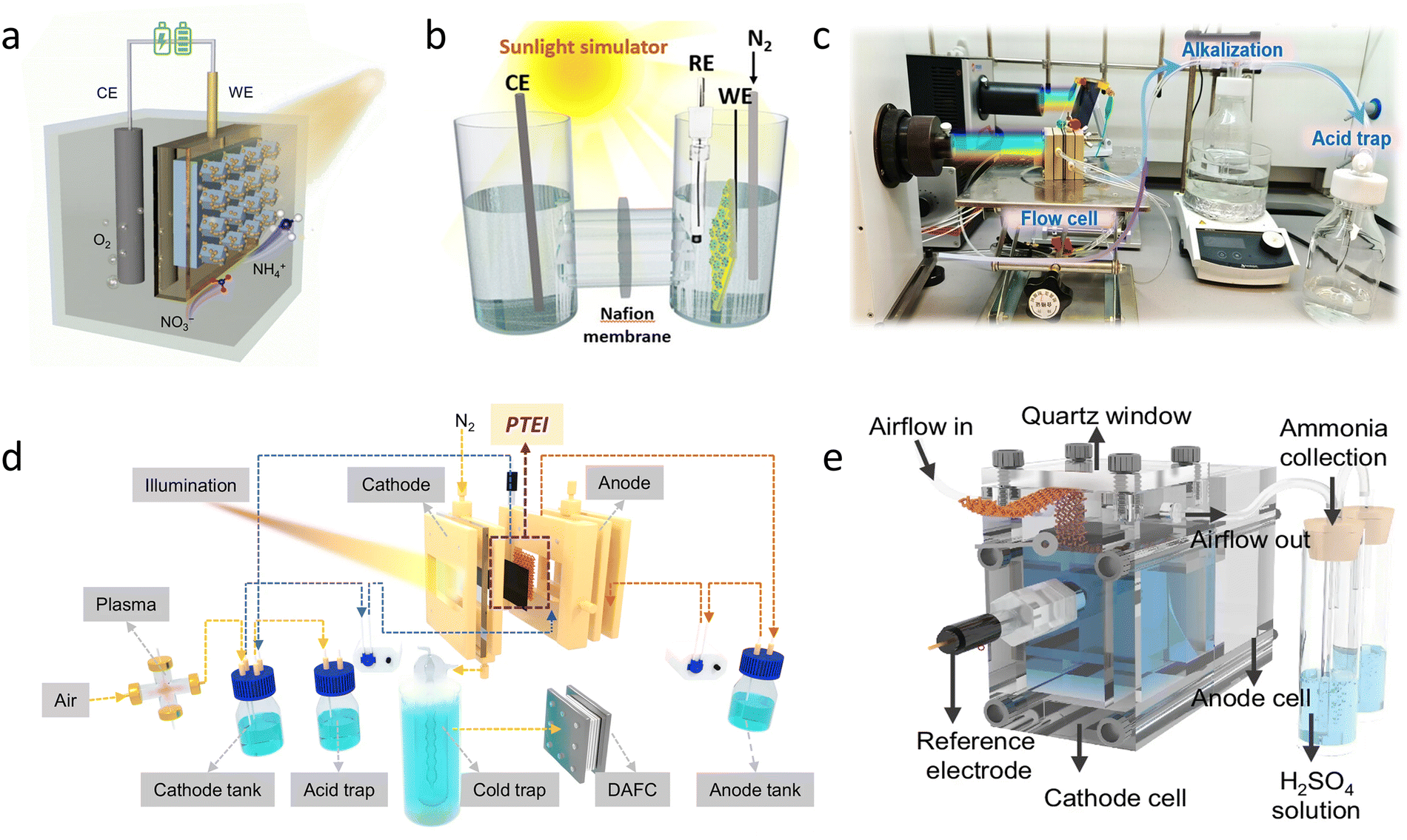 | ||
| Fig. 19 (a) Schematic illustration of a single chamber photoelectrocatalytic ammonia synthesis reactor. (b) H-type photoelectrocatalytic ammonia synthesis reactor. (c) Digital photo of a flow-cell photoelectrocatalytic ammonia synthesis reactor. (d) Schematic illustration of a flow-cell photoelectrocatalytic ammonia synthesis reactor. (e) Schematic of harvesting ammonia products from the reactor. Spontaneous diffusion photocatalytic ammonia synthesis reactor. Reproduced with permission from ref. 299 Copyright 2024, The Royal Society of Chemistry. Ref. 300 Copyright 2023, American Chemical Society. Ref. 301 Copyright 2024, Wiley-VCH. Ref. 302 Copyright 2025, Springer Nature. | ||
Based on this analysis, the future development of photoelectrochemical ammonia synthesis reactors should focus on the advanced optimization of flow-cell systems, including: (1) multi-scale channel designs for enhanced mass transport; (2) the multifunctional integration of bipolar plates for electrical conduction, thermal management, and mechanical stability; and (3) synergistic regulation strategies for photo-electro-thermal field coupling. These innovations will accelerate the transition of photoelectrochemical ammonia synthesis toward industrial-scale applications.
8.4 Alkali metal mediated ammonia synthesis reactors
The reactor serves as the pivotal platform in lithium-mediated ammonia synthesis, enabling both nitrogen activation and lithium cycling. Its structural design and operational parameters fundamentally govern the reaction kinetics and ammonia selectivity. The technology has evolved through three progressive reactor configurations: primitive single-chamber reactors addressing the direct interaction of lithium and N2, optimized continuous-flow reactors for precise control of the gas–liquid–solid interface, and advanced solid-state electrolyte reactors overcoming challenges in proton transport and lithium cycling stability. Specifically, (1) the single-chamber reactor is composed of an autoclave vessel, a single-chamber glass cell, a three-electrode system (a Pt wire as the reference electrode, a Pt net as the counter electrode, and stainless-steel cloth as the working electrode), a lithium salt, and an organic electrolyte. This reactor, with its simple structure, can effectively maintain direct contact between the lithium metal deposited on the working electrode and the nitrogen reactants under pressurized conditions and achieve the activation and reduction of the nitrogen reactants. However, this system is confronted with the challenges of lithium dendrite growth and the insufficient proton supply capacity of organic electrolytes, especially the severe electrolyte depletion that limits long-term stability and large-scale continuous ammonia synthesis. (2) Continuous-flow reactors effectively overcome the limitations of the single-chamber reactor. They adopt a three-chamber structure design, including a central flowing electrolyte chamber (promoting lithium-ion transport), a cathodic N2 reduction chamber (achieving lithium-mediated nitrogen activation), and an anodic H2 oxidation chamber (providing protons). This innovative configuration uses the physical isolation of reaction zones to achieve critical improvements: substantial suppression of lithium dendrite formation and parasitic side reactions, precise control over the gas–liquid–solid triple-phase boundary, and optimized synchronization between proton delivery and nitrogen reduction kinetics. The architectural design consequently enhances both system stability and faradaic efficiency, establishing a novel paradigm for selective ammonia synthesis. However, three persistent challenges remain: (i) limited proton mobility in organic media, (ii) the progressive decomposition of organic electrolytes under reductive potentials, and (iii) spatially heterogeneous lithium deposition and dissolution behavior leading to current density variations across the electrodes. (3) Solid-state electrolyte reactors mainly use solid-state lithium-ion conductors to replace liquid organic electrolytes. These solid electrolytes offer fundamental advantages: they integrate proton and lithium-ion dual conduction characteristics, construct directional transport channels, and significantly reduce mass transfer resistance. In addition, they eliminate the problem of liquid organic electrolyte loss, as they can withstand high voltage and strong reducing environments, thereby extending reactor stability. The rigid solid-state electrolytes can mechanically inhibit the penetration of lithium dendrites, and surface modifications (such as artificial SEI layers) can induce the uniform deposition and dissolution of lithium. This system further decouples and optimizes the three functional zones of nitrogen reduction, proton supply, and lithium cycling through a modular stacking design, providing a possible path for achieving the efficient and stable industrialization of ammonia synthesis.8.5 Optimization strategies and directions for ammonia synthesis reactors
Recently, reactors for ammonia synthesis have been updated from single-chamber cells, H-type cells, and flow cells to MEA electrolyzers. Among these, the mass transfer limit of the reactants and the internal resistance of the reactors have been significantly reduced, achieving efficient ammonia synthesis. However, further optimization and improvement of the reactors will contribute to the industrialization of green ammonia synthesis technology. Therefore, considering the composition of advanced MEA electrolyzers (including cathode/anode electrodes, cathode/anode plates, GDEs, ion-exchange membranes, and flow channels) as well as the subsequent product separation and purification steps, we think that the following aspects should be carried out in the design of future reactors: (1) ion-exchange membranes are some of the core components, and their properties directly affect proton conduction efficiency, ammonia selectivity, system stability, and energy consumption. Therefore, new short-side-chain perfluorosulfonic acid membranes or non-fluoropolymers should be developed and designed to reduce membrane resistance. Alternatively, proton jumping sites should be introduced by doping heteropoly acids or ionic liquids to reduce the internal resistance and accelerate proton mobility, thereby improving reactor performance. In addition, by introducing basic groups and other means to enhance the corrosion resistance of the proton exchange membrane, coupled with strengthening its mechanical strength, the stability of the proton exchange membrane is improved, realizing the industrial application potential of green ammonia synthesis. The gradient structure of the ion-exchange membrane is equally important. It can use the asymmetric pore distribution and hydrophilic–hydrophobic gradient of the membrane itself to regulate gas permeation and drying phenomena, while also promoting the supply of protons to the cathode and preventing water flooding. Besides, we should integrate bipolar membranes into reactors, where water molecules dissociate within the bipolar membrane structure, rapidly providing protons for the cathode chamber. Meanwhile, asymmetric acid/base reactions are achieved at the anode and cathode, optimizing the thermodynamics of the system and ultimately enhancing the performance of the ammonia synthesis reactors. (2) The preparation methods for electrodes in MEA electrolyzers mainly include catalyst-coated membrane (CCM) and catalyst-coated substrate (CCS). Among them, the CCS strategy directly coats the catalyst ink on the gas diffusion layer (GDL) to form a GDE. Then, it is combined with the ion-exchange membrane through hot pressing and assembled into MEA electrolyzers. The CCS technology will lead to a relatively high interface resistance. The CCM technology directly coats the catalyst ink on both sides of the ion-exchange membrane to form anode and cathode catalyst layers, which are then combined with the GDL through hot pressing. This technology can achieve zero-gap MEA electrolyzers, significantly reducing the internal interface resistance of the reactor and realizing high-performance ammonia synthesis at the device level. It is also worth noting that renewable energy has intermittent characteristics. During the start-up and shutdown of MEA electrolyzers, the electrode potential can change, reducing the yield of the target products. Therefore, developing a cathode electrode that can operate stably at a wide current density is of great significance for enhancing the product yield. (3) Various flow channel configurations are constructed by multi-scale simulation technology, including serpentine, leaf-vein, interdigitated, and composite flow channels, to optimize the flow channels of MEA electrolyzers, reducing local concentration polarization and enhancing mass transfer within the reactors.In addition, to promote the industrialization of green ammonia synthesis technology, megawatt-level MEA electrolyzers are achieved by connecting standardized reactor units in parallel or series, while avoiding uneven performance caused by overly large single tank sizes within MEA electrolyzers. It is also necessary to pay attention to the overheating problem of the reactor under high current density. Built-in cooling channels can extend the operating stability of the reactors and electrodes. At the same time, the hydrophobic/hydrophilic regulation of the GDE can control the mass transfer of the reaction gas and the water immersion of the electrodes. To enhance the economic efficiency of the overall system, ammonia separation, capture, and collection devices can be coupled with the reaction system. In conclusion, through multi-disciplinary collaborative innovation, such as in computational science, materials science, and artificial intelligence science, the development of reactors with high current density, high FE, and durable stability is expected to become the core technology for green ammonia production.
9. Summary and outlook
Recent advances in green ammonia synthesis have been systematically reviewed. First, we outline the reaction mechanisms and fundamental research aspects of using multiple nitrogen sources (N2, NO, NO2−, and NO3−) for ammonia synthesis, covering detection methods, reactor design, measurement protocols, and catalytic activity descriptors. Next, we comprehensively analyze the critical factors governing different green synthesis approaches—including photocatalytic, electrocatalytic, photoelectrochemical, lithium-mediated, and calcium-mediated methods—with an emphasis on activity metrics such as faradaic efficiency and NH3 yield rate, reaction mechanisms, and optimization strategies. Additionally, we explain the structure–activity relationships of various catalysts to establish design principles for enhanced ammonia performance. Finally, we discuss future research directions, such as the development of advanced catalysts and reactor systems, operando characterization techniques for mechanistic insights, and scalability challenges for industrial adoption.Specifically, we first explain the reaction pathways and reduction mechanisms of various nitrogen sources, including dissociative, associative, Mars–van Krevelen, Tafel-dissociative, Tafel-associative, Heyrovsky-dissociative, Heyrovsky-associative, indirect autocatalytic, and direct catalytic reduction pathways. We then detail ammonia detection methods, such as chemical detection using Nessler's reagent, indophenol blue, and salicylate, fluorometric detection, and titrimetric assays, as well as advanced techniques like isotope labeling, 1H NMR, electrode-based sensing, UV-vis spectrophotometry, ion chromatography, and gas chromatography. Additionally, we outline key electrochemical experimental protocols, covering electrode preparation and functionalization, standard potential calibration, linear sweep voltammetry, chronoamperometry, cyclic voltammetry, electrochemical impedance spectroscopy, and multi-potential step measurements. Critical activity metrics, including faradaic efficiency, NH3 yield rate, energy efficiency, and stability, are also systematically evaluated to provide readers with a foundational understanding of green catalytic ammonia synthesis. Subsequently, the discussion is organized into five sections, each focusing on advanced catalyst synthesis and performance optimization based on distinct catalytic technologies.
Among these, we provide an in-depth analysis of high-profile green catalytic ammonia synthesis technologies, including photocatalytic, electrocatalytic, photoelectrocatalytic, and alkali metal-mediated approaches. By addressing critical scientific challenges—such as photogenerated charge recombination, poor reactant solubility, weak N2 adsorption, inefficient NN bond dissociation, limited proton affinity, the large energy gap of N2 molecules, dominant competitive reactions, and unfavorable rate-determining steps—we systematically evaluate catalyst modification strategies and reaction system optimizations to enhance performance and explain structure–activity relationships. Furthermore, we identify key approaches for improving faradaic efficiency, energy efficiency, and NH3 yield rates, offering a foundational framework and future directions for advancing green ammonia synthesis research.
However, we note that despite significant advancements in innovative electrocatalysts driving progress in green ammonia synthesis, this emerging field remains hindered by critical challenges—including low current density, insufficient catalytic activity, and suboptimal faradaic efficiency and energy efficiency, which collectively impede commercialization. A key yet often overlooked factor is the electrolyzer, an essential component of the reaction system, whose synergistic role with the catalyst is pivotal for efficient ammonia production. In the final section, we comprehensively review the evolution of reactor designs over the past year, beginning with the basic configuration and components of single-chamber cells, progressing to H-type cells with diaphragms to prevent reactant–product crossover and further to the gas diffusion electrode, which optimizes gas–liquid–solid triple-phase contact. We culminate with advanced membrane electrode assembly devices, which minimize internal resistance and enable zero-gap operation to enhance current density. Additionally, we analyze critical reactor components, including gas diffusion layers, diaphragm materials and architectures, and integrated gas capture-conversion systems. We firmly believe these insights will accelerate the transition of green ammonia synthesis from laboratory-scale research to industrial-scale applications.
Beyond providing newcomers with a foundational understanding of this research field and its core scientific challenges, this comprehensive review offers experienced researchers critical insights into accelerating large-scale practical applications. Despite significant interdisciplinary advancements, significant breakthroughs in both fundamental research and practical applications are still essential to achieve industrial-scale economic viability. Key considerations must include:
(1) From the catalyst's point of view, universal and robust synthesis strategies are essential for translating lab-scale technologies to industrial application. (2) From the reactor design point of view, reaction systems require careful optimization of electrolytes, membranes, and reactor configurations to maximize catalyst performance by mitigating nitrogen diffusion limitations, thereby meeting the stringent requirements of high NH3 yield rates and faradaic efficiency for future ammonia synthesis systems. (3) From the theoretical simulation point of view, cutting-edge computational techniques—including density functional theory, artificial intelligence, and ab initio molecular dynamics—should be employed to predict catalytic enhancement mechanisms and guide the rational design of catalysts and reaction systems. (4) Some advanced operando characterization methods, such as in situ XRD, FTIR, XPS, Raman, and XAS, should also be used to further explore the actual active sites, reaction intermediates, and basic mechanism. (5) Experimental details should also be paid attention to to ensure reliable, reproducible results and eliminate false positives. (6) As research scales from nanomaterials to industrial reactors, biosafe and physically safe systems must be developed. Additionally, the design of cost-effective, high-performance components like catalysts and membranes will be one of the keys to the technology.
From the discussion above, although the research into the extensive construction of catalysts and the efficient production of large-scale reactors is challenging, it is also encouraging to explore in-depth the fundamental reaction mechanism, theoretical investigations, and robust characterization techniques coupled with trustworthy detection strategies, and to create an illuminating pathway for the future development and deployment of a value-added, clean-energy economy. In summary, there are both challenges and opportunities for the synthesis of ammonia through green catalytic pathways. Therefore, with further research and development and the continued efforts around the world, this very important field will make great contributions to the progress of science, technology, and society.
Author contributions
Z. K. completed writing – original draft, review, and editing. Z. K. and Y. H. carried out investigation and summarized the methodology. Z. K., D. S., T. Z., Y. L., L. D., and Y. H. contributed to helpful discussion during the writing of the manuscript. L. D. and Y. H. performed supervision. Y. H., B. Y., Z. L., J. L., Q. Z., and L. L. provided resources. Y. H. performed funding acquisition and reviewing and editing of the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included, and no new data were generated or analyzed as part of this review.Acknowledgements
We acknowledge the financial support from the National Key Research and Development Program of China (2022YFB4002100), the National Natural Science Foundation of China (22425805, 22278364, U22A20432, 22211530045, 22178308, 22238008, and 22578393), the development project of Zhejiang Province's “Jianbing” and “Lingyan” (2023C01226), the Key Technology Breakthrough Program of Ningbo “Science and Innovation Yongjiang 2035” (2024H024), the Fundamental Research Funds for the Central Universities (226-2024-00060), and Zhejiang Provincial Natural Science Foundation of China under grant no. LD25B060001, the Fundamental Research Funds for the Zhejiang Provincial Universities (226-2025-00224). L. D. acknowledges partial financial support from the Australian Research Council (ARC, FL190100126 and CE230100032).References
- L. Hollevoet, M. D. Ras, M. Roeffaers, J. Hofkens and J. A. Martens, ACS Energy Lett., 2020, 5, 1124–1127 CrossRef CAS.
- S. Han, H. Li, T. Li, F. Chen, R. Yang, Y. Yu and B. Zhang, Nat. Catal., 2023, 6, 402–414 CrossRef CAS.
- P. H. V. Langevelde, I. Katsounaros and M. T. M. Koper, Joule, 2021, 5, 290–294 CrossRef.
- X. Yang, S. Mukherjee, T. O'Carroll, Y. Hou, M. R. Singh, J. A. Gauthier and G. Wu, Angew. Chem., Int. Ed., 2023, 62, e202215938 CrossRef CAS PubMed.
- Y. Li, J. Li, Dr. J. Huang, J. Chen, Y. Kong, B. Yang, Z. Li, L. Lei, G. Chai, Z. Wen, L. Dai and Y. Hou, Angew. Chem., Int. Ed., 2021, 60, 9078–9085 CrossRef CAS PubMed.
- X. Cui, C. Tang and Q. Zhang, Adv. Energy Mater., 2018, 8, 1800369 CrossRef.
- V. Rosca, M. Duca, M. T. Groot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- R. Schlögl, Angew. Chem., Int. Ed., 2003, 42, 2004–2008 CrossRef PubMed.
- X. Liang, H. Zhu, X. Yang, S. Xue, Z. Liang, X. Ren, A. Liu and G. Wu, Small Struct., 2023, 4, 2200202 CrossRef CAS.
- X. Zhao, G. Hu, G. Chen, H. Zhang, S. Zhang and H. Wang, Adv. Mater., 2021, 33, 2007650 CrossRef CAS PubMed.
- S. Wang, F. Ichihara, H. Pang, H. Chen and J. Ye, Adv. Funct. Mater., 2018, 28, 1803309 CrossRef.
- Y. Kong, Y. Li, X. Sang, B. Yang, Z. Li, S. Zheng, Q. Zhang, S. Yao, X. Yang, L. Lei, S. Zhou, G. Wu and Y. Hou, Adv. Mater., 2021, 34, 2103548 CrossRef PubMed.
- X. Peng, L. Zeng, D. Wang, Z. Liu, Y. Li, Z. Li, B. Yang, L. Lei, L. Dai and Y. Hou, Chem. Soc. Rev., 2023, 52, 2193–2237 RSC.
- J. Choi, B. H. R. Suryanto, D. Wang, H. Du, R. Y. Hodgetts, F. M. F. Vallana, D. R. MacFarlane and A. N. Simonov, Nat. Commun., 2020, 11, 5546 CrossRef CAS PubMed.
- D. Bao, Q. Zhang, F. Meng, H. Zhong, M. Shi, Y. Zhang, J. Yan, Q. Jiang and X. Zhang, Adv. Mater., 2017, 29, 1604799 CrossRef PubMed.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed.
- J. N. Galloway, A. R. Townsend, J. W. Erisman, M. Bekunda, Z. Cai, J. R. Freney, L. A. Martinelli, S. P. Seitzinger and M. A. Sutton, Science, 2008, 320, 889–892 CrossRef CAS PubMed.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- H. Jia and E. A. Quadrelli, Chem. Soc. Rev., 2014, 43, 547–564 RSC.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C. Chen, M. R. Smith III and T. W. Hamann, Chem. Rev., 2020, 120, 5437–5516 CrossRef CAS PubMed.
- J. Wang, T. Feng, J. Chen, V. Ramalingam, Z. Li, D. M. Kabtamu, J. He and X. Fang, Nano Energy, 2021, 86, 106088 CrossRef CAS.
- K. Wang, D. Smith and Y. Zheng, Carbon Resour. Convers., 2018, 1, 2–31 Search PubMed.
- K. Zhang, A. Cao, L. H. Wandall, J. Vernieres, J. Kibsgaard, J. K. Nørskov and I. Chorkendorff, Science, 2024, 383, 1357–1363 CrossRef CAS PubMed.
- G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef CAS PubMed.
- T. A. Bazhenova and A. E. Shilov, Coord. Chem. Rev., 1995, 144, 69–145 CrossRef CAS.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 CrossRef CAS.
- C. J. M. Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- B. H. R. Suryanto, H. Du, D. Wang, J. Chen, A. N. Simonov and D. R. Macfarlane, Nat. Catal., 2019, 2, 290–296 CrossRef CAS.
- S. J. Leigh, Science, 1998, 279, 506–507 CrossRef.
- G. Zhang, Y. Li, C. He, X. Ren, P. Zhang and H. Mi, Adv. Energy Mater., 2021, 11, 2003294 CrossRef CAS.
- L. Wang, M. Xia, H. Wang, K. Huang, C. Qian, C. T. Maravelias and G. A. Ozin, Joule, 2018, 2, 1055–1074 CrossRef CAS.
- F. Chen, A. Elgazzar, S. Pecaut, C. Qiu, Y. Feng, S. Ashokkumar, Z. Yu, C. Sellers, S. Hao, P. Zhu and H. Wang, Nat. Catal., 2024, 7, 1032–1043 CrossRef CAS.
- X. Chen, Y. Cheng, B. Zhang, J. Zhou and S. He, Nat. Commun., 2024, 15, 6278 CrossRef CAS PubMed.
- Y. Wang, F. Hao, M. Sun, M. Liu, J. Zhou, Y. Xiong, C. Ye, X. Wang, F. Liu, J. Wang, P. Lu, Y. Ma, J. Yin, H. Chen, Q. Zhang, L. Gu, H. Chen, B. Huang and Z. Fan, Adv. Mater., 2024, 36, 2313548 CrossRef CAS PubMed.
- X. Zhang, Y. Lyu, H. Zhou, J. Zheng, A. Huang, J. Ding, C. Xie, R. D. Marco, N. Tsud, V. Kalinovych, S. Jiang, L. Dai and S. Wang, Adv. Mater., 2023, 35, 2211894 CrossRef CAS PubMed.
- S. Ren, R. Gao, N. T. Nguyen and L. Wang, Angew. Chem., Int. Ed., 2024, 63, e202317414 CrossRef CAS PubMed.
- H. Jiang, G. Chen, O. Savateev and H. Wang, Joule, 2023, 7, 253–256 CrossRef.
- S. Feng, W. Gao, J. Guo, H. Cao and P. Chen, ACS Energy Lett., 2023, 8, 1567–1574 CrossRef CAS.
- L. Gao, Y. Cao, C. Wang, X. Yu, W. Li, Y. Zhou, B. Wang, Y. Yao, C. Wu, W. Luo and Z. Zou, Angew. Chem., Int. Ed., 2021, 60, 5257 CrossRef CAS PubMed.
- N. H. Deissler, J. B. V. Mygind, K. Li, V. A. Niemann, P. Benedek, V. Vinci, S. Li, X. Fu, P. C. K. Vesborg, T. F. Jaramillo, J. Kibsgaard, J. Drnecc and I. Chorkendorff, Energy Environ. Sci., 2024, 17, 3482–3492 RSC.
- X. Fu, S. Li, N. H. Deissler, J. B. V. Mygind, J. Kibsgaard and I. Chorkendorff, ACS Energy Lett., 2024, 8, 3790–3795 CrossRef.
- Y. Ren, C. Yu, X. Tan, Q. Wei, Z. Wang, L. Ni, L. Wang and J. Qiu, Energy Environ. Sci., 2022, 15, 2776–2805 RSC.
- A. J. Martín, T. Shinagawa and J. P. Ramírez, Chem, 2019, 5, 263–283 Search PubMed.
- Y. Ren, C. Yu, X. Tan, H. Huang, Q. Wei and J. Qiu, Energy Environ. Sci., 2021, 14, 1176–1193 RSC.
- W. Bi, N. Shaigan, A. Malek, K. Fatih, E. Gyenge and D. P. Wilkinson, Energy Environ. Sci., 2022, 15, 2259–2287 RSC.
- A. R. Singh, B. A. Rohr, J. A. Schwalbe, M. Cargnello, K. Chan, T. F. Jaramillo, I. Chorkendorff and J. K. Nørskov, ACS Catal., 2016, 7, 706–709 CrossRef.
- E. Skúlason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. A. Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- Y. Wan, J. Xu and R. Lv, Mater. Today, 2019, 27, 69–90 CrossRef CAS.
- Y. Zhai, Z. Zhu, C. Zhu, K. Chen, X. Zhang, J. Tang and J. Chen, Mater. Today, 2020, 38, 99–113 CrossRef CAS.
- X. Chen, Y. Guo, X. Du, Y. Zeng, J. Chu, C. Gong, J. Huang, C. Fan, X. Wang and J. Xiong, Adv. Energy Mater., 2019, 10, 1903172 CrossRef.
- Y. Lin, J. Liang, H. Li, L. Zhang, T. Mou, T. Li, L. Yue, Y. Ji, Q. Liu, Y. Luo, N. Li, B. Tang, Q. Wu, M. S. Hamdy, D. Ma and X. Sun, Mater. Today Phys., 2022, 22, 100611 CrossRef CAS.
- Z. Zhang, X. Feng, Z. Zhang, L. Chen, W. Liu, L. Tong, X. Gao and J. Zhang, J. Am. Chem. Soc., 2024, 146, 14898–14904 CrossRef CAS PubMed.
- H. Huo, Q. Zhang, F. Liu and K. He, Environ. Sci. Technol., 2013, 47, 1711–1718 CrossRef CAS PubMed.
- L. Zhang, J. Liang, Y. Wang, T. Mou, Y. Lin, L. Yue, T. Li, Q. Liu, Y. Luo, N. Li, B. Tang, Y. Liu, S. Gao, A. A. Alshehri, X. Guo, D. Ma and X. Sun, Angew. Chem., Int. Ed., 2021, 60, 25263–25268 CrossRef CAS PubMed.
- N. Gruber and J. N. Galloway, Nature, 2008, 451, 293–296 CrossRef CAS PubMed.
- Y. Zeng, C. Priest, G. Wang and G. Wu, Small Methods, 2020, 4, 2000672 CrossRef CAS.
- G. F. Mcisaac, M. B. David, G. Z. Gertner and D. A. Goolsby, Nature, 2001, 414, 166–167 CrossRef CAS PubMed.
- D. E. Canfield, A. N. Glazer and P. G. Falkowski, Science, 2010, 330, 192–196 CrossRef CAS PubMed.
- J. O. Lundberg, E. Weitzberg, J. A. Cole and N. Benjamin, Nat. Rev. Microbiol., 2004, 2, 593–602 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- J. Liu, D. Richards, N. Singh and B. R. Goldsmith, ACS Catal., 2019, 9, 7052–7064 CrossRef CAS.
- D. P. Butcher Jr and A. A. Gewirth, Nano Energy, 2016, 29, 457–465 CrossRef.
- J. Li, G. Zhan, J. Yang, F. Quan, C. Mao, Y. Liu, B. Wang, F. Lei, L. Li, A. W. M. Chan, L. Xu, Y. Shi, Y. Du, W. Hao, P. K. Wong, J. Wang, S. Dou, L. Zhang and J. C. Yu, J. Am. Chem. Soc., 2020, 142, 7036–7046 CrossRef CAS PubMed.
- Y. Xiong, Y. Wang, J. Zhou, F. Liu, F. Hao and Z. Fan, Adv. Mater., 2024, 36, 2304021 CrossRef CAS PubMed.
- Y. Shi, Z. Zhao, D. Yang, J. Tan, X. Xin, Y. Liu and Z. Jiang, Chem. Soc. Rev., 2023, 52, 6938–6956 RSC.
- H. Xu, Y. Ma, J. Chen, W. Zhang and J. Yang, Chem. Soc. Rev., 2022, 51, 2710–2758 RSC.
- Y. Pang, C. Su, G. Jia, L. Xu and Z. Shao, Chem. Soc. Rev., 2021, 50, 12744–12787 RSC.
- C. Guo, J. Ran, A. Vasileff and S. Qiao, Energy Environ. Sci., 2018, 11, 45–56 RSC.
- X. Zhang, C. Pei, Z. Zhao and J. Gong, Energy Environ. Sci., 2024, 17, 2381–2405 RSC.
- S. Wang, X. Hai, X. Ding, K. Chang, Y. Xiang, X. Meng, Z. Yang, H. Chen and J. Ye, Adv. Mater., 2017, 29, 1701774 CrossRef PubMed.
- J. G. Howalt, T. Bligaard, J. Rossmeislb and T. Vegge, Phys. Chem. Chem. Phys., 2013, 15, 7785–7795 RSC.
- J. Rittle and J. C. Peters, J. Am. Chem. Soc., 2016, 138, 4243–4248 CrossRef CAS PubMed.
- J. Lim, C. A. Fernández, S. W. Lee and M. C. Hatzell, ACS Energy Lett., 2021, 6, 3676–3685 CrossRef CAS.
- S. P. Eckel, Z. Zhang, R. Habre, E. B. Rappaport, W. S. Linn, K. Berhane, Y. Zhang, T. M. Bastain and F. D. Gilliland, Eur. Respir. J., 2016, 47, 1348–1356 CrossRef PubMed.
- L. Ooyang, J. Liang, Y. Luo, D. Zheng, S. Sun, Q. Liu, Md. S. Hamdy, X. Sun and B. Ying, Chin. J. Catal., 2023, 50, 6–44 CrossRef.
- X. Peng, Y. Mi, H. Bao, Y. Liu, D. Qi, Y. Qiu, L. Zhuo, S. Zhao, J. Sun, X. Tang, J. Luo and X. Liu, Nano Energy, 2020, 78, 105321 CrossRef CAS.
- J. Long, S. Chen, Y. Zhang, C. Guo, X. Fu, D. Deng and J. Xiao, Angew. Chem., Int. Ed., 2020, 59, 9711–9718 CrossRef CAS PubMed.
- P. Liu, J. Liang, J. Wang, L. Zhang, J. Li, L. Yue, Y. Ren, T. Li, Y. Luo, N. Li, B. Tang, Q. Liu, A. M. Asiri, Q. Kong and X. Sun, Chem. Commun., 2021, 57, 13562–13565 RSC.
- R. Miao, D. Chen, Z. Guo, Y. Zhou, C. Chen and S. Wang, Appl. Catal., B, 2024, 344, 123662 CrossRef CAS.
- M. T. Groot and M. T. M. Koper, J. Electroanal. Chem., 2004, 562, 81–94 CrossRef.
- R. Lange, E. Maisonhaute, R. Robin and V. Vivier, Electrochem. Commun., 2013, 29, 25–28 CrossRef CAS.
- D. Sicsic, F. B. Célérier and B. Tribollet, Eur. J. Inorg. Chem., 2014, 6174–6184, DOI:10.1002/ejic.201402708.
- Y. Wang, C. Wang, M. Li, Y. Yu and B. Zhang, Chem. Soc. Rev., 2021, 50, 6720–6733 RSC.
- D. Xu, Y. Li, L. Yin, Y. Ji, J. Niu and Y. Yu, Front. Environ. Sci. Eng., 2018, 12, 9, DOI:10.1007/s11783-018-1033-z.
- J. Gao, B. Jiang, C. Ni, Y. Qi and X. Bi, Chem. Eng. J., 2020, 382, 123034 CrossRef CAS.
- A. R. Cook, N. Dimitrijevic, B. W. Dreyfus, D. Meisel, L. A. Curtiss and D. M. Camaioni, J. Phys. Chem. A, 2001, 105, 3658–3666 CrossRef CAS.
- Y. Li, Y. K. Go, H. Ooka, D. He, F. Jin, S. H. Kim and R. Nakamura, Angew. Chem., Int. Ed., 2020, 59, 9744–9750 CrossRef CAS PubMed.
- R. Jia, Y. Wang, C. Wang, Y. Ling, Y. Yu and B. Zhang, ACS Catal., 2020, 10, 3533–3540 CrossRef CAS.
- M. D. Bartberger, W. Liu, E. Ford, K. M. Miranda, C. Switzer, J. M. Fukuto, P. J. Farmer, D. A. Wink and K. N. Houk, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10958–10963 CrossRef CAS PubMed.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. Mcenaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- C. Tang and S. Qiao, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- L. Li, C. Tang, D. Yao, Y. Zheng and S. Qiao, ACS Energy Lett., 2019, 4, 2111–2116 CrossRef CAS.
- H. Iriawan, S. Z. Andersen, X. Zhang, B. M. Comer, J. Barrio, P. Chen, A. J. Medford, I. E. L. Stephens, I. Chorkendorff and Y. Shao-Horn, Nat. Rev. Methods Primers, 2021, 1, 56 CrossRef CAS.
- H. Liu, N. Guijarro and J. Luo, J. Energy Chem., 2021, 61, 149–154 CrossRef CAS.
- L. Shi, Y. Yin, S. Wang, X. Xu, H. Wu, J. Zhang, S. Wang and H. Sun, Appl. Catal., B, 2020, 278, 119325 CrossRef CAS.
- N. C. Kani, A. Prajapati, B. A. Collins, J. D. Goodpaster and M. R. Singh, ACS Catal., 2020, 10, 14592–14603 CrossRef CAS.
- N. Lazouski, M. Chung, K. Williams, M. L. Gala and K. Manthiram, Nat. Catal., 2020, 3, 463–469 CrossRef CAS.
- F. Hanifpour, A. Sveinbjörnsson, C. P. Canales, E. Skúlason and H. D. Flosadóttir, Angew. Chem., Int. Ed., 2020, 132, 23138–23142 CrossRef.
- W. Shan, F. Liu, H. He, X. Shi and C. Zhang, ChemCatChem, 2011, 3, 1286–1289 CrossRef CAS.
- H. Shen, C. Choi, J. Masa, X. Li, J. Qiu, Y. Jung and Z. Sun, Chem, 2021, 7, 1708–1754 CAS.
- S. H. Yuen and A. G. Pollard, J. Sci. Food Agric., 1952, 3, 441–447 CrossRef CAS.
- ASTM D1426-15, Standard Test Methods for Ammonia Nitrogen in Water, 2021, ASTM International, 100 Barr Harbor Drive, PO Box C700, West Conshohocken, PA 19428–2959, United States.
- W. T. Bolleter, C. J. Bushman and P. W. Tidwell, Anal. Chem., 1961, 33, 592–594 CrossRef CAS.
- C. E. Bower and T. H. Hansen, Can. J. Fish. Aquat. Sci., 1980, 37, 794–798 CrossRef CAS.
- H. Verdouw, C. J. A. V. Echteld and E. M. J. Dekkers, Water Res., 1978, 12, 399–402 CrossRef CAS.
- N. Amornthammarong and J. Zhang, Anal. Chem., 2008, 80, 1019–1026 CrossRef CAS PubMed.
- X. Gao, Y. Wen, D. Qu, L. An, S. Luan, W. Jiang, X. Zong, X. Liu and Z. Sun, ACS Sustainable Chem. Eng., 2018, 6, 5342–5348 CrossRef CAS.
- L. Zhou and C. E. Boyd, Aquaculture, 2016, 450, 187–193 CrossRef CAS.
- P. Wang, F. Cchang, W. Gao, J. Guo, G. Wu, T. He and P. Chen, Nat. Chem., 2017, 9, 64–70 CrossRef CAS PubMed.
- E. W. Rice, B. Baird, A. D. Eaton and L. S. Clesceri,Standard Methods for the Examination of Water and Wastewater, American Water Works Association, 2012 Search PubMed.
- F. Zhou, L. M. Azofra, M. Ali, M. Kar, A. N. Simonov, C. M. Worth, C. Sun, X. Zhang and D. R. MacFarlane, Energy Environ. Sci., 2017, 10, 2516–2520 RSC.
- D. Borden and R. F. Giese, Clays Clay Miner., 2001, 49, 444–445 CrossRef CAS.
- A. Leduy and R. Samson, Biotechnol. Lett., 1982, 4, 303–306 CrossRef CAS.
- R. Peters, M. Baltruweit, T. Grube, R. C. Samsun and D. Stolten, J. CO2 Util., 2019, 34, 616–634 CrossRef CAS.
- X. Li, P. Anderson, H. R. M. Jhong, M. Paster, J. F. Stubbins and P. J. A. Kenis, Energy Fuels, 2016, 30, 5980–5989 CrossRef CAS.
- W. Lai, Y. Qiao, J. Zhang, Z. Lin and H. Huang, Energy Environ. Sci., 2022, 15, 3603–3629 RSC.
- J. Na, B. Seo, J. Kim, C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H. S. Oh and U. Lee, Nat. Commun., 2019, 10, 5193 CrossRef PubMed.
- L. Gao, X. Cui, C. D. Sewell, J. Li and Z. Lin, Chem. Soc. Rev., 2021, 50, 8428–8469 RSC.
- S. Verma, S. Lu and P. J. A. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- X. Chen, N. Li, Z. Kong, W. Ong and X. Zhao, Mater. Horiz., 2018, 5, 9–27 RSC.
- S. Shang, W. Xiong, C. Yang, B. Johannessen, R. Liu, H. Y. Hsu, Q. Gu, M. K. H. Leung and J. Shang, ACS Nano, 2021, 15, 9670–9678 CrossRef CAS PubMed.
- J. Zander, J. Timm, M. Weiss and R. Marschall, Adv. Energy Mater., 2022, 12, 2202403 CrossRef CAS.
- X. Wang, G. Fan, S. Guo, R. Gao, Y. Guo, C. Han, Y. Gao, J. Zhang, X. Gu and L. Wu, Angew. Chem., Int. Ed., 2024, 63, e202404258 CrossRef CAS PubMed.
- Y. Zhao, L. Zheng, R. Shi, S. Zhang, X. Bian, F. Wu, X. Cao, G. I. N. Waterhouse and T. Zhang, Adv. Energy Mater., 2020, 10, 2002199 CrossRef CAS.
- G. Dong, X. Huang and Y. Bi, Angew. Chem., Int. Ed., 2022, 61, e202204271 CrossRef CAS PubMed.
- Q. Han, C. Wu, H. Jiao, R. Xu, Y. Wang, J. Xie, Q. Guo and J. Tang, Adv. Mater., 2021, 33, 2008180 CrossRef CAS PubMed.
- Z. Zhao, R. Tan, Y. Kong, Z. Zhang, S. Qiu, X. Mu and L. Li, Angew. Chem., Int. Ed., 2023, 62, e202303629 CrossRef CAS PubMed.
- L. Chen, Y. Hao, Y. Guo, Q. Zhang, J. Li, W. Gao, L. Ren, X. Su, L. Hu, N. Zhang, S. Li, X. Feng, L. Gu, Y. Zhang, A. Yin and B. Wang, J. Am. Chem. Soc., 2021, 143, 5727–5736 Search PubMed.
- Y. Fang, Y. Xue, L. Hui, H. Yu and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 3170–3174 CrossRef CAS PubMed.
- Y. Zhao, Y. Zhao, G. I. N. Waterhouse, L. Zheng, X. Cao, F. Teng, L. Wu, C. H. Tung, D. O'Hare and T. Zhang, Adv. Mater., 2017, 29, 1703828 CrossRef PubMed.
- S. Liu, M. Wang, H. Ji, L. Zhang, J. Ni, N. Li, T. Qian, C. Yan and J. Lu, Adv. Mater., 2023, 35, 2211730 CrossRef CAS PubMed.
- G. N. Schrauzer and T. D. Guth, J. Am. Chem. Soc., 1977, 99, 7189–7193 CrossRef CAS.
- Z. Zhao, D. Wang, R. Gao, G. Wen, M. Feng, G. Song, J. Zhu, D. Luo, H. Tan, X. Ge, W. Zhang, Y. Zhang, L. Zheng, H. Li and Z. Chen, Angew. Chem., Int. Ed., 2021, 60, 11910–11918 CrossRef CAS PubMed.
- Y. Zhao, Y. Zhao, R. Shi, B. Wang, G. I. N. Waterhouse, L. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2019, 31, 1806482 CrossRef PubMed.
- S. Tan, H. Feng, Q. Zheng, X. Cui, J. Zhao, Y. Luo, J. Yang, B. Wang and J. G. Hou, J. Am. Chem. Soc., 2020, 142, 826–834 CrossRef CAS PubMed.
- C. Xu, Y. Zhang, J. Chen, S. Li, Y. Zhang and G. Qin, Sci. China Mater., 2021, 64, 128–136 CrossRef CAS.
- W. Zhang, H. He, H. Li, L. Duan, L. Zu, Y. Zhai, W. Li, L. Wang, H. Fu and D. Zhao, Adv. Energy Mater., 2021, 11, 2003303 CrossRef CAS.
- C. Liu, D. Hao, J. Ye, S. Ye, F. Zhou, H. Xie, G. Qin, J. Xu, J. Liu, S. Li and C. Sun, Adv. Energy Mater., 2023, 13, 2204126 CrossRef CAS.
- P. Xia, X. Pan, S. Jiang, J. Yu, B. He, P. M. Ismail, W. Bai, J. Yang, L. Yang, H. Zhang, M. Cheng, H. Li, Q. Zhang, C. Xiao and Y. Xie, Adv. Mater., 2022, 34, 2200563 CrossRef CAS PubMed.
- W. Wang, H. Zhou, Y. Liu, S. Zhang, Y. Zhang, G. Wang, H. Zhang and H. Zhao, Small, 2020, 16, 1906880 CrossRef CAS.
- Y. Guan, H. Wen, K. Cui, Q. Wang, W. Gao, Y. Cai, Z. Cheng, Q. Pei, Z. Li, H. Cao, T. He, J. Guo and P. Chen, Nat. Chem., 2024, 16, 373–379 CrossRef CAS PubMed.
- J. Li, R. Chen, J. Wang, Y. Zhou, G. Yang and F. Dong, Nat. Commun., 2022, 13, 1098 CrossRef CAS PubMed.
- R. Chen, S. Shen, K. Wang and F. Dong, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2312550120 CrossRef CAS PubMed.
- Y. Wang and T. J. Meyer, Chem, 2019, 5, 496–497 CAS.
- Y. Yang, S. Wang, H. Wen, T. Ye, J. Chen, C. Li and M. Du, Angew. Chem., Int. Ed., 2019, 58, 15362–15366 CrossRef CAS PubMed.
- B. Cui, J. Zhang, S. Liu, X. Liu, W. Xiang, L. Liu, H. Xin, M. J. Leflerc and S. Licht, Green Chem., 2017, 19, 298–304 RSC.
- N. Cao and G. Zheng, Nano Res., 2018, 11, 2992–3008 CrossRef CAS.
- M. Wang, S. Liu, T. Qian, J. Liu, J. Zhou, H. Ji, J. Xiong, J. Zhong and C. Yan, Nat. Commun., 2019, 10, 341 CrossRef CAS PubMed.
- Y. Lin, S. Zhang, Z. Xue, J. Zhang, H. Su, T. Zhao, G. Zhai, X. Li, M. Antonietti and J. Chen, Nat. Commun., 2019, 10, 4380 CrossRef PubMed.
- G. Lin, Q. Ju, X. Guo, W. Zhao, S. Adimi, J. Ye, Q. Bi, J. Wang, M. Yang and F. Huang, Adv. Mater., 2021, 33, 2007509 CrossRef CAS PubMed.
- W. Qiu, X. Xie, J. Qiu, W. Fang, R. Liang, X. Ren, X. Ji, G. Cui, A. M. Asiri, G. Cui, B. Tang and X. Sun, Nat. Commun., 2018, 9, 3485 CrossRef PubMed.
- J. Wang, L. Yu, L. Hu, G. Chen, H. Xin and X. Feng, Nat. Commun., 2018, 9, 1795 CrossRef PubMed.
- B. A. MacKay and M. D. Fryzuk, Chem. Rev., 2004, 104, 385–402 CrossRef CAS PubMed.
- H. Qian, S. Xu, J. Cao, F. Ren, W. Wei, J. Meng and L. Wu, Nat. Sustain., 2021, 4, 417–425 CrossRef.
- K. Li, D. J. Jacob, H. Liao, J. Zhu, V. Shah, L. Shen, K. H. Bates, Q. Zhang and S. Zhai, Nat. Geosci., 2019, 12, 906–910 CrossRef CAS.
- S. Ozgen, S. Cernuschi and S. Caserini, Renewable Sustainable Energy Rev., 2021, 135, 110113 CrossRef CAS.
- L. Pagalan, C. Bickford, W. Weikum, B. Lanphear, M. Brauer, N. Lanphear, G. E. Hanley, T. F. Oberlander and M. Winters, JAMA Pediatr., 2019, 173, 86–92 CrossRef PubMed.
- L. Han, S. Cai, M. Gao, J. Y. Hasegawa, P. Wang, J. Zhang, L. Shi and D. Zhang, Chem. Rev., 2019, 119, 10916–10976 CrossRef CAS PubMed.
- A. M. Beale, F. Gao, I. L. Gonzalez, C. H. F. Peden and J. Szanyi, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC.
- C. Liu, Y. Wang and B. Zhang, Sci. China: Chem., 2020, 63, 1173–1174 CrossRef CAS.
- K. Otsuka, H. Sawada and I. Yamanaka, J. Electrochem. Soc., 1996, 143, 3491–3497 CrossRef CAS.
- P. D. Schepper, V. A. Danilov and J. F. M. Denayer, Int. J. Energy Res., 2016, 40, 1355–1366 CrossRef.
- M. Tursun and C. Wu, Inorg. Chem., 2022, 61, 17448–17458 CrossRef CAS PubMed.
- J. Choi, B. H. R. Suryanto, D. Wang, H. Du, R. Y. Hodgetts, F. M. F. Vallana, D. R. Macfarlane and A. N. Simonov, Nat. Commun., 2020, 11, 5546 CrossRef CAS PubMed.
- S. S. Markandaraj, T. Markandaraj and S. Shanmugam, Adv. Sci., 2022, 9, 2201410 CrossRef PubMed.
- B. H. Ko, B. Hasa, H. Shin, Y. Zhao and F. Jiao, J. Am. Chem. Soc., 2022, 144, 1258–1266 CrossRef CAS PubMed.
- X. Li, K. Chen, X. Lu, D. Ma and K. Chu, Chem. Eng. J., 2023, 454, 140333 CrossRef CAS.
- Z. Kou, K. Wang, Z. Liu, L. Zeng, Z. Li, B. Yang, L. Lei, C. Yuan and Y. Hou, Small Struct., 2022, 3, 2100153 CrossRef CAS.
- J. Liang, Q. Zhou, T. Mou, H. Chen, L. Yue, Y. Luo, Q. Liu, M. S. Hamdy, A. A. Alshehri, F. Gong and X. Sun, Nano Res., 2022, 15, 4008–4013 CrossRef CAS.
- H. J. Chun, V. Apaja, A. Clayborne, K. Honkala and J. Greeley, ACS Catal., 2017, 7, 3869–3882 CrossRef CAS.
- Y. Li, C. Cheng, S. Han, Y. Huang, X. Du, B. Zhang and Y. Yu, ACS Energy Lett., 2022, 7, 1187–1194 CrossRef CAS.
- Y. Li, S. Zhang, W. Cheng, Y. Chen, D. Luan, S. Gao and X. (David) Lou, Adv. Mater., 2022, 34, 2105204 CrossRef CAS PubMed.
- Y. Yan, B. Huang, J. Wang, H. Wang and W. Cai, J. Catal., 2007, 249, 311–317 CrossRef CAS.
- P. Zhou, N. Li, Y. Chao, W. Zhang, F. Lv, K. Wang, W. Yang, P. Gao and S. Guo, Angew. Chem., Int. Ed., 2019, 58, 14184–14188 CrossRef CAS PubMed.
- J. Mao, C. He, J. Pei, W. Chen, D. He, Y. He, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 4958 CrossRef PubMed.
- H. Zhang, P. An, W. Zhou, B. Guan, P. Zhang, J. Dong and X. (David) Lou, Sci. Adv., 2018, 4, eaao6657 CrossRef PubMed.
- H. Zhang, Y. Li, C. Cheng, J. Zhou, P. Yin, H. Wu, Z. Liang, J. Zhang, Q. Yun, A. Wang, L. Zhu, B. Zhang, W. Cao, X. Meng, J. Xia, Y. Yu and Q. Lu, Angew. Chem., Int. Ed., 2023, 62, e202213351 CrossRef CAS PubMed.
- H. Liu, K. Xiang, B. Yang, X. Xie, D. Wang, C. Zhang, Z. Liu, S. Yang, C. Liu, J. Zou and L. Chai, Environ. Sci. Pollut. Res., 2017, 24, 14249–14258 CrossRef CAS PubMed.
- L. Zhang, Q. Zhou, J. Liang, L. Yue, T. Li, Y. Luo, Q. Liu, N. Li, B. Tang, F. Gong, X. Guo and X. Sun, Inorg. Chem., 2022, 61, 8096–8102 CrossRef CAS PubMed.
- A. Clayborne, H. J. Chun, R. B. Rankin and J. Greeley, Angew. Chem., Int. Ed., 2015, 127, 8373–8376 CrossRef.
- S. Deshpande and J. Greeley, ACS Catal., 2020, 10, 9320–9327 CrossRef CAS.
- B. He, P. Lv, D. Wu, X. Li, R. Zhu, K. Chu, D. Ma and Y. Jia, J. Mater. Chem. A, 2022, 10, 18690–18700 RSC.
- G. Meng, T. Wei, W. Liu, W. Li, S. Zhang, W. Liu, Q. Liu, H. Bao, J. Luog and X. Liu, Chem. Commun., 2022, 58, 8097–8100 RSC.
- Y. Feng, Q. Shao, F. Lv, L. Bu, J. Guo, S. Guo and X. Huang, Adv. Sci., 2020, 7, 1800178 CrossRef CAS.
- L. Li, C. Tang, B. Xia, H. Jin, Y. Zheng and S. Qiao, ACS Catal., 2019, 9, 2902–2908 CrossRef CAS.
- J. Liang, P. Liu, Q. Li, T. Li, L. Yue, Y. Luo, Q. Liu, N. Li, B. Tang, A. A. Alshehri, I. Shakir, P. O. Agboola, C. Sun and X. Sun, Angew. Chem., Int. Ed., 2022, 61, e202202087 CrossRef CAS PubMed.
- G. Chen, X. Cao, S. Wu, X. Zeng, L. Ding, M. Zhu and H. Wang, J. Am. Chem. Soc., 2017, 139, 9771–9774 CrossRef CAS PubMed.
- N. Q. Tran, L. T. Duy, D. C. Truong, B. T. N. Le, B. T. Phan, Y. Cho, X. Liu and H. Lee, Chem. Commun., 2022, 58, 5257–5260 RSC.
- E. J. Zakem, A. A. Haj, M. J. Church, G. L. Dijken, S. Dutkiewicz, S. Q. Foster, R. W. Fulweiler, M. M. Mills and M. J. Follows, Nat. Commun., 2018, 9, 1206 CrossRef PubMed.
- D. He, Y. Li, H. Ooka, Y. K. Go, F. Jin, S. H. Kim and R. Nakamura, J. Am. Chem. Soc., 2018, 140, 2012–2015 CrossRef CAS PubMed.
- S. Liu, L. Cui, S. Yin, H. Ren, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2022, 319, 121876 CrossRef CAS.
- H. Li, C. Yan, H. Guo, K. Shin, S. M. Humphrey, C. J. Werth and G. Henkelman, ACS Catal., 2020, 10, 7915–7921 CrossRef CAS.
- Y. Arikawa, Y. Otsubo, H. Fujino, S. Horiuchi, E. Sakuda and K. Umakoshi, J. Am. Chem. Soc., 2018, 140, 842–847 CrossRef CAS PubMed.
- Y. Guo, J. R. Stroka, B. Kandemir, C. E. Dickerson and K. L. Bren, J. Am. Chem. Soc., 2018, 140, 16888–16892 CrossRef CAS PubMed.
- R. Zhang, S. Zhang, Y. Guo, C. Li, J. Liu, Z. Huang, Y. Zhao, Y. Li and C. Zhi, Energy Environ. Sci., 2022, 15, 3024–3032 RSC.
- W. Qiu, X. Chen, Y. Liu, D. Xiao, P. Wang, R. Li, K. Liu, Z. Jin and P. Li, Appl. Catal., B, 2022, 315, 121548 CrossRef CAS.
- Y. Wang, W. Zhou, R. Jia, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5350–5354 CrossRef CAS PubMed.
- J. Lim, C. Liu, J. Park, Y. Liu, T. P. Senftle, S. W. Lee and M. C. Hatzell, ACS Catal., 2021, 11, 7568–7577 CrossRef CAS.
- H. Shin, S. Jung, S. Bae, W. Lee and H. Kim, Environ. Sci. Technol., 2014, 48, 12768–12774 CrossRef CAS PubMed.
- C. Fierro, J. Phys. Chem., 1988, 92, 4401–4405 CrossRef CAS.
- S. Seraj, P. Kunal, H. Li, G. Henkelman, S. M. Humphrey and C. J. Werth, ACS Catal., 2017, 7, 3268–3276 CrossRef CAS.
- H. Li, S. Guo, K. Shin, M. S. Wong and G. Henkelman, ACS Catal., 2019, 9, 7957–7966 CrossRef CAS.
- S. García, L. Zhang, G. W. Piburn, G. Henkelman and S. M. Humphrey, ACS Nano, 2014, 8, 11512–11521 CrossRef PubMed.
- S. E. Braley, J. Xie, Y. Losovyj and J. M. Smith, J. Am. Chem. Soc., 2021, 143, 7203–7208 CrossRef CAS PubMed.
- J. R. Stroka, B. Kandemir, E. M. Matson and K. L. Bren, ACS Catal., 2020, 10, 13968–13972 CrossRef CAS.
- Q. Chen, X. An, Q. Liu, X. Wu, L. Xie, J. Zhang, W. Yao, M. S. Hamdy, Q. Kong and X. Sun, Chem. Commun., 2022, 58, 517–520 RSC.
- Y. Wang, A. Xu, Z. Wang, L. Huang, J. Li, F. Li, J. Wicks, M. Luo, D. H. Nam, C. S. Tan, Y. Ding, J. Wu, Y. Lum, C. T. Dinh, D. Sinton, G. Zheng and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 5702–5708 CrossRef CAS PubMed.
- L. Mattarozzi, S. Cattarin, N. Comisso, P. Guerriero, M. Musiani, L. V. Gómez and E. Verlato, Electrochim. Acta, 2013, 89, 488–496 CrossRef CAS.
- L. Li, C. Tang, X. Cui, Y. Zheng, X. Wang, H. Xu, S. Zhang, T. Shao, K. Davey and S. Qiao, Angew. Chem., Int. Ed., 2021, 60, 14131–14137 CrossRef CAS PubMed.
- X. Li, Z. Li, L. Zhang, D. Zhao, J. Li, S. Sun, L. Xie, Q. Liu, A. A. Alshehri, Y. Luo, Y. Liao, Q. Kong and X. Sun, Nanoscale, 2022, 14, 13073–13077 RSC.
- G. Wen, J. Liang, L. Zhang, T. Li, Q. Liu, X. An, X. Shi, Y. Liu, S. Gao, A. M. Asiri, Y. Luo, Q. Kong and X. Sun, J. Colloid Interface Sci., 2022, 606, 1055–1063 CrossRef CAS PubMed.
- C. Wang, W. Zhou, Z. Sun, Y. Wang, B. Zhang and Y. Yu, J. Mater. Chem. A, 2021, 9, 239–243 RSC.
- S. Ye, Z. Chen, G. Zhang, W. Chen, C. Peng, X. Yang, L. Zheng, Y. Li, X. Ren, H. Cao, D. Xue, J. Qiu, Q. Zhang and J. Liu, Energy Environ. Sci., 2022, 15, 760–770 RSC.
- T. Ling, D. Yan, H. Wang, Y. Jiao, Z. Hu, Y. Zheng, L. Zheng, J. Mao, H. Liu, X. Du, M. Jaroniec and S. Qiao, Nat. Commun., 2017, 8, 1509 CrossRef PubMed.
- F. Lei, L. Zhang, Y. Sun, L. Liang, K. Liu, J. Xu, Q. Zhang, B. Pan, Y. Luo and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 9266–9270 CrossRef CAS PubMed.
- J. Sun, D. Alam, R. Daiyan, H. Masood, T. Zhang, R. Zhou, P. J. Cullen, E. C. Lovell, A. Jalili and R. Amal, Energy Environ. Sci., 2021, 14, 865–872 RSC.
- I. Katsounaros, M. Dortsiou and G. Kyriacou, J. Hazard. Mater., 2009, 171, 323–327 CrossRef CAS PubMed.
- R. Chauhan and V. C. Srivastava, Chem. Eng. J., 2020, 386, 122065 CrossRef CAS.
- M. Teng, J. Ye, C. Wan, G. He and H. Chen, Ind. Eng. Chem. Res., 2022, 61, 14731–14746 CrossRef CAS.
- G. Chen, Y. Yuan, H. Jiang, S. Ren, L. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 5, 605–613 CrossRef CAS.
- D. Xu, Y. Li, L. Yin, Y. Ji, J. Niu and Y. Yu, Front. Environ. Sci. Eng., 2018, 12, 9, DOI:10.1007/s11783-018-1033-z.
- H. Liu, J. Park, Y. Chen, Y. Qiu, Y. Cheng, K. Srivastava, S. Gu, B. H. Shanks, L. T. Roling and W. Li, ACS Catal., 2021, 11, 8431–8442 CrossRef CAS.
- M. Duca and M. T. M. Koper, Energy Environ. Sci., 2012, 5, 9726–9742 RSC.
- E. Murphy, Y. Liu, I. Matanovic, S. Guo, P. Tieu, Y. Huang, A. Ly, S. Das, I. Zenyuk, X. Pan, E. Spoerke and P. Atanassov, ACS Catal., 2022, 12, 6651–6662 CrossRef CAS.
- S. Guo, K. Heck, S. Kasiraju, H. Qian, Z. Zhao, L. C. Grabow, J. T. Miller and M. S. Wong, ACS Catal., 2018, 8, 503–515 CrossRef CAS.
- S. E. Bae, K. L. Stewart and A. A. Gewirth, J. Am. Chem. Soc., 2007, 129, 10171–10180 CrossRef CAS PubMed.
- F. Chen, Z. Wu, S. Gupta, D. J. Rivera, S. V. Lambeets, S. Pecaut, J. Y. T. Kim, P. Zhu, Y. Z. Finfrock, D. M. Meira, G. King, G. Gao, W. Xu, D. A. Cullen, H. Zhou, Y. Han, D. E. Perea, C. L. Muhich and H. Wang, Nat. Nanotechnol., 2022, 17, 759–767 CrossRef CAS PubMed.
- H. Liu, X. Lang, C. Zhu, J. Timoshenko, M. Rüscher, L. Bai, N. Guijarro, H. Yin, Y. Peng, J. Li, Z. Liu, W. Wang, B. R. Cuenya and J. Luo, Angew. Chem., Int. Ed., 2022, 61, e202202556 CrossRef CAS PubMed.
- R. Zhang, D. Shuai, K. A. Guy, J. R. Shapley, T. J. Strathmann and C. J. Werth, ChemCatChem, 2013, 5, 313–321 CrossRef CAS.
- Y. Wang, H. Li, W. Zhou, X. Zhang, B. Zhang and Y. Yu, Angew. Chem., Int. Ed., 2022, 61, e202202604 CrossRef CAS PubMed.
- X. Deng, Y. Yang, L. Wang, X. Fu and J. Luo, Adv. Sci., 2021, 8, 2004523 CrossRef CAS PubMed.
- R. Amaral and N. Y. Dzade, Mater. Today Commun., 2024, 38, 107824 CrossRef CAS.
- J. F. Callejas, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Chem. Mater., 2016, 28, 6017–6044 CrossRef CAS.
- R. Prins and M. E. Bussell, Catal. Lett., 2012, 142, 1413–1436 CrossRef CAS.
- Y. Yang, C. O. Hernández, V. A. P. O’Shea, J. M. Coronado and D. P. Serrano, ACS Catal., 2012, 2, 592–598 CrossRef CAS.
- W. R. A. M. Robinson, J. N. M. Gestel, T. I. Korányi, S. Eijsbouts, A. M. van der Kraan, J. A. R. van Veen and V. H. J. Beer, J. Catal., 1996, 161, 539–550 CrossRef CAS.
- L. Wei, D. Liu, B. A. Rosales, J. W. Evans and J. Vela, ACS Catal., 2020, 10, 3618–3628 CrossRef CAS.
- R. Zhang, Y. Guo, S. Zhang, D. Chen, Y. Zhao, Z. Huang, L. Ma, P. Li, Q. Yang, G. Liang and C. Zhi, Adv. Energy Mater., 2022, 12, 2103872 CrossRef CAS.
- I. Katsounaros, D. Ipsakis, C. Polatides and G. Kyriacou, Electrochim. Acta, 2006, 52, 1329–1338 CrossRef CAS.
- M. Dortsiou and G. Kyriacou, J. Electroanal. Chem., 2009, 630, 69–74 CrossRef CAS.
- Z. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F. Chen, J. Y. Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 12, 2870 CrossRef CAS PubMed.
- Z. Zheng, L. Qi, Y. Xue and Y. Li, Nano Today, 2022, 43, 101431 CrossRef CAS.
- Y. Xue, Q. Yu, Q. Ma, Y. Chen, C. Zhang, W. Teng, J. Fan and W. Zhang, Environ. Sci. Technol., 2022, 56, 14797–14807 CrossRef CAS PubMed.
- J. Liu, M. S. Kelley, W. Wu, A. Banerjee, A. P. Douvalis, J. Wu, Y. Zhang, G. C. Schatz and M. G. Kanatzidis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5530–5535 CrossRef CAS PubMed.
- Y. Song, D. Johnson, R. Peng, D. K. Hensley, P. V. Bonnesen, L. Liang, J. Huang, F. Yang, F. Zhang, R. Qiao, A. P. Baddorf, T. J. Tschaplinski, N. L. Engle, M. C. Hatzell, Z. Wu, D. A. Cullen, H. M. Meyer III, B. G. Sumpter and A. J. Rondinonoe, Sci. Adv., 2018, 4, e1700336 CrossRef PubMed.
- C. Li, T. Wang, Z. Zhao, W. Yang, J. Li, A. Li, Z. Yang, G. A. Ozin and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 5278–5282 CrossRef CAS PubMed.
- B. Zhou, P. Ou, N. Pant, S. Cheng, S. Vanka, S. Chu, R. T. Rashid, G. Botton, J. Song and Z. Mi, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1330–1338 CrossRef CAS PubMed.
- H. Fu, P. Varadhan, M. Tsai, W. Li, Q. Ding, C. Lin, M. Bonifazi, A. Fratalocchi, S. Jin and J. He, Nano Energy, 2020, 70, 104478 CrossRef CAS.
- J. Zheng, Y. Lyu, M. Qiao, R. Wang, Y. Zhou, H. Li, C. Chen, Y. Li, H. Zhou, S. Jiang and S. Wang, Chem, 2019, 5, 617–633 CAS.
- M. G. Walter, E. L. Warren, J. R. Mckone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- J. Hou, M. Yang and J. Zhang, Nanoscale, 2020, 12, 6900–6920 RSC.
- K. Peramaiah, V. Ramalingam, H. Fu, M. M. Alsabban, R. Ahmad, L. Cavallo, V. Tung, K. Huang and J. He, Adv. Mater., 2021, 33, 2100812 CrossRef CAS PubMed.
- Y. J. Jang, A. E. Lindberg, M. A. Lumley and K. S. Choi, ACS Energy Lett., 2020, 5, 1834–1839 CrossRef CAS.
- S. Zhou, K. Sun, C. Y. Toe, J. Yin, J. Huang, Y. Zeng, D. Zhang, W. Chen, O. F. Mohammed, X. Hao and R. Amal, Adv. Mater., 2022, 34, 2201670 CrossRef CAS PubMed.
- H. E. Kim, J. Kim, E. C. Ra, H. Zhang, Y. J. Jang and J. S. Lee, Angew. Chem., Int. Ed., 2022, 61, e202204117 CrossRef CAS PubMed.
- D. Liu, J. Wang, S. Bian, Q. Liu, Y. Gao, X. Wang, P. K. Chu and X. Yu, Adv. Funct. Mater., 2020, 30, 2002731 CrossRef CAS.
- H. Huang, W. Tu, L. Fang, Y. Xiao, F. Niu, H. Zhu, X. Zhu, L. Wang, Y. Xiong, J. Feng, H. Li, M. Zhang, Y. Zhang, Y. Yao, Y. Zhou, H. Li and Z. Zou, ACS Energy Lett., 2023, 8, 4235–4241 CrossRef CAS.
- A. Tayyebi, R. Mehrotra, M. A. Mubarok, J. Kim, M. Zafari, M. Tayebi, D. Oh, S. H. Lee, J. E. Matthews, S. W. Lee, T. J. Shin, G. Lee, T. F. Jaramillo, S. Y. Jang and J. W. Jang, Nat. Catal., 2024, 7, 510–521 CrossRef CAS.
- M. S. Iqbal, Y. Ruan, R. Iftikhar, F. Z. Khan, W. Li, L. Hao, A. W. Robertson, G. Percoco and Z. Sun, Ind. Chem. Mater., 2023, 1, 563–581 RSC.
- J. Kibsgaard, J. K. Nørskov and I. Chorkendorff, ACS Energy Lett., 2019, 4, 2986–2988 CrossRef CAS.
- L. F. Greenlee, J. N. Renner and S. L. Foster, ACS Catal., 2018, 8, 7820–7827 CrossRef CAS.
- X. Fu, J. Zhang and Y. Kang, Chem. Catal., 2022, 2, 2590–2613 CAS.
- F. Fichter, P. Girard and H. Erlenmeyer, Helv. Chim. Acta, 1930, 13, 1228–1236 CrossRef CAS.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047 CrossRef CAS.
- A. Tsuneto, A. Kudo and T. Sakata, Chem. Lett., 1993, 851–854 CrossRef CAS.
- A. Tsuneto, A. Kudo and T. Sakata, J. Electroanal. Chem., 1994, 367, 183–188 CrossRef CAS.
- J. M. McEnaney, A. R. Singh, J. A. Schwalbe, J. Kibsgaard, J. C. Lin, M. Cargnello, T. F. Jaramillo and J. K. Nørskov, Energy Environ. Sci., 2017, 10, 1621–1630 RSC.
- W. Chang, A. Jain, F. Rezaie and K. Manthiram, Nat. Catal., 2024, 7, 231–241 CrossRef CAS.
- E. F. Mcfarlane and F. C. Tompkins, Trans. Faraday Soc., 1962, 58, 997–1007 RSC.
- A. N. Dey, Thin Solid Films, 1977, 43, 131–171 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K. C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- N. Lazouski, Z. J. Schiffer, K. Williams and K. Manthiram, Joule, 2019, 3, 1127–1139 CrossRef CAS.
- K. Li, S. Z. Andersen, M. J. Statt, M. Saccoccio, V. J. Bukas, K. Krempl, R. Sažinas, J. B. Pedersen, V. Shadravan, Y. Zhou, D. Chakraborty, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Science, 2021, 374, 1593–1597 CrossRef CAS PubMed.
- B. H. R. Suryanto, K. Matuszek, J. Choi, R. Y. Hodgetts, H. Du, J. M. Bakker, C. S. M. Kang, P. V. Cherepanov, A. N. Simonov and D. R. Macfarlane, Science, 2021, 372, 1187–1191 CrossRef CAS PubMed.
- S. Z. Andersen, M. J. Statt, V. J. Bukas, S. G. Shapel, J. B. Pedersen, K. Krempl, M. Saccoccio, D. Chakraborty, J. Kibsgaard, P. C. K. Vesborg, J. Nørskov and I. Chorkendorff, Energy Environ. Sci., 2020, 13, 4291–4300 RSC.
- G. Soloveichik, in 2019 AIChE Annual Meeting (AIChE, 2019). https://arpa-e.energy.gov/technologies/programs/refuel.
- K. Steinberg, X. Yuan, C. K. Klein, N. Lazouski, M. Mecklenburg, K. Manthiram and Y. Li, Nat. Energy, 2023, 8, 138–148 CrossRef CAS.
- R. Tort, O. Westhead, M. Spry, B. J. V. Davies, M. P. Ryan, M. M. Titirici and I. E. L. Stephens, ACS Energy Lett., 2023, 8, 1003–1009 CrossRef CAS PubMed.
- X. Fu, J. B. Pedersen, Y. Zhou, M. Saccoccio, S. Li, R. Sažinas, K. Li, S. Z. Andersen, A. Xu, N. H. Deissler, J. B. V. Mygind, C. Wei, J. Kibsgarrd, P. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Science, 2023, 379, 707–712 CrossRef CAS PubMed.
- X. Fu, A. Xu, J. B. Pedersen, S. Li, R. Sažinas, Y. Zhou, S. Z. Andersen, M. Saccoccio, N. H. Deissler, J. B. V. Mygind, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Nat. Commun., 2024, 15, 2417 CrossRef CAS PubMed.
- S. Li, Y. Zhou, K. Li, M. Saccoccio, R. Sažinas, S. Z. Andersen, J. B. Pedersen, X. Fu, V. Shadravan, D. Chakraborty, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Joule, 2022, 6, 2083–2101 CrossRef CAS PubMed.
- H. Du, M. Chatti, R. Y. Hodgetts, P. V. Cherepanov, C. K. Nguyen, K. Matuszek, D. R. MacFarlane and A. N. Simonov, Nature, 2022, 609, 722–727 CrossRef CAS PubMed.
- X. Cai, X. Li, J. You, F. Yang, Z. Shadike, S. Qin, L. Luo, Y. Guo, X. Yan, S. Shen, G. Wei, Z. J. Xu and J. Zhang, J. Am. Chem. Soc., 2023, 145, 25716–25725 CrossRef CAS PubMed.
- S. Li, Y. Zhou, X. Fu, J. B. Pedersen, M. Saccoccio, S. Z. Andersen, K. E. Rasmussen, P. J. Kempen, C. D. Damsgaard, A. Xu, R. Sažinas, J. B. V. Mygind, N. H. Deissler, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Nature, 2024, 629, 92–97 CrossRef CAS PubMed.
- X. Fu, V. A. Niemann, Y. Zhou, S. Li, K. Zhang, J. B. Pedersen, M. Saccoccio, S. Z. Andersen, K. E. Rasmussen, P. Benedek, A. Xu, N. H. Deissler, J. B. V. Mygind, A. C. Nielander, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Nat. Mater., 2024, 23, 101–107 CrossRef CAS PubMed.
- K. Li, S. G. Shapel, D. Hochfilzer, J. B. Pedersen, K. Krempl, S. Z. Andersen, R. Sažinas, M. Saccoccio, S. Li, D. Chakraborty, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, ACS Energy Lett., 2022, 7, 36–41 CrossRef CAS.
- C. Chen, Y. Li and P. Yang, Joule, 2021, 5, 737–742 CrossRef.
- Y. Li, Y. Ji, Y. Zhao, J. Chen, S. Zheng, X. Sang, B. Yang, Z. Li, L. Lei, Z. Wen, X. Feng and Y. Hou, Adv. Mater., 2022, 34, 2202240 CrossRef CAS PubMed.
- X. Cai, Z. Shadike, X. Cai, X. Li, L. Luo, L. An, J. Yin, G. Wei, F. Yang, S. Shen and J. Zhang, Energy Environ. Sci., 2023, 16, 3063–3073 RSC.
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, ACS Sustainable Chem. Eng., 2017, 5, 7393–7400 CrossRef CAS.
- J. Shao, H. Jing, P. Wei, X. Fu, L. Pang, Y. Song, K. Ye, M. Li, L. Jiang, J. Ma, R. Li, R. Si, Z. Peng, G. Wang and J. Xiao, Nat. Energy, 2023, 8, 1273–1283 CrossRef CAS.
- D. Liu, Z. Meng, Y. Zhu, X. Sun, X. Deng, M. Shi, Q. Hao, X. Kang, T. Dai, H. Zhong, J. Yan and Q. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202315238 CrossRef CAS PubMed.
- J. C. Bui, E. W. Lees, D. H. Marin, T. N. Stovall, L. Chen, A. Kusoglu, A. C. Nielander, T. F. Jaramillo, S. W. Boettcher, A. T. Bell and A. Z. Weber, Nat. Chem Eng., 2024, 1, 45–60 CrossRef.
- Z. Xu, L. Wan, Y. Liao, M. Pang, Q. Xu, P. Wang and B. Wang, Nat. Commun., 2023, 14, 1619 CrossRef CAS PubMed.
- L. Mi, Q. Huo, J. Cao, X. Chen, H. Yang, Q. Hu and C. He, Adv. Energy Mater., 2022, 12, 2202247 CrossRef CAS.
- J. Li, B. Sheng, Y. Chen, J. Yang, P. Wang, Y. Li, T. Yu, H. Pan, L. Qiu, Y. Li, J. Song, L. Zhu, X. Wang, Z. Huang and B. Zhou, Nat. Commun., 2024, 15, 7393 CrossRef CAS PubMed.
- L. Mi, Q. Huo, J. Cao, X. Chen, H. Yang, Q. Hu, C. He, J. Wang, G. Ran, J. Gao, D. Li, G. I. N. Waterhouse, R. Shi, W. Zhang, J. Tang, L. Wu, Y. Zhao and T. Zhang, Adv. Mater., 2025, 37, 2420199 CrossRef PubMed.
- Y. Li, Q. Zhang, H. Dai, D. He, Z. Ke and X. Xiao, Energy Environ. Sci., 2024, 17, 9233–9243 RSC.
- A. Zabelina, E. Miliutina, J. Dedek, A. Trelin, D. Zabelin, R. Valiev, R. Ramazanov, V. Burtsev, D. Popelkova, M. Stastny, V. Svorcik and O. Lyutakov, ACS Catal., 2023, 13, 10916–10926 CrossRef CAS PubMed.
- Y. Fang, M. Li, Y. Gao, Y. Wen and B. Shan, Angew. Chem., Int. Ed., 2025, 64, e202415729 CrossRef CAS PubMed.
- P. Wang, P. Li, Z. Pan, K. Liu, M. Xie, L. Zhou, M. Zhou, G. Yu and Z. Jin, Nat. Commun., 2025, 16, 5581 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |