
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical power sources enabled by multi-ion carriers
Yu
Zhang
ab,
Pingping
Wu
b,
Chunxiao
Chen
b,
YangJie
Liu
b,
Xiaoqi
Cai
b,
Wenli
Liang
b,
Minghao
Li
bc,
Xinyu
Zhuang
b,
Yujie
Li
b,
Xipeng
Chen
b,
Mengyuan
Sun
bc,
Lan
Wei
a,
Xiang
Hu
 *bc and
Zhenhai
Wen
*bc
*bc and
Zhenhai
Wen
*bc
aCollege of Chemistry and Chemical Engineering, Xinyang Normal University, Xinyang 464000, China
bState Key Laboratory of Structural Chemistry, and Fujian Provincial Key Laboratory of Materials and Techniques Toward Hydrogen Energy, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China. E-mail: huxiang@fjirsm.ac.cn; wen@fjirsm.ac.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 18th September 2025
Abstract
The pursuit of high-performance, sustainable, and adaptable energy storage systems stands at the forefront of addressing the ever-growing demands of our modern world. Among the most compelling frontiers in this endeavour are electrochemical technologies empowered by multi-ion carriers, which transcend the intrinsic limitations of conventional single-ion systems. By harmonizing the transport and redox behaviour of diverse cations and anions, these systems give rise to novel mechanisms of charge balance, extended electrochemical stability windows, and cooperative redox pathways. This review offers a panoramic exploration of recent advances in multi-ion carrier-enabled electrochemical energy technologies, with a particular focus on hybrid batteries, capacitors, fuel cells, and redox flow batteries. Through these case studies, we elucidate how the interplay of multiple ions governs structure–function relationships and enhances overall electrochemical performance. Central to this discussion are the underlying working principles, representative device architectures, and the latest innovations in electrode and electrolyte materials. Special attention is devoted to the way multi-ion transport phenomena unlock new electrochemical landscapes, accelerating ion kinetics, stabilizing interphases, and enabling emergent pathways unavailable to single-ion systems. We further highlight forward-looking trends in hybrid ionic configurations, such as the integration of cations, co-transport of cation–anion pairs, and the engineering of aqueous–nonaqueous hybrid systems. In closing, we provide a critical assessment of the electrochemical advantages, scalability prospects, and practical challenges that lie ahead, ranging from kinetic harmonization across multiple ions to scalable device fabrication and the mitigation of complexity-driven safety concerns. By weaving together insights from materials science, electrochemistry, and systems engineering, this review lays a foundation for the rational design of next-generation multi-ion electrochemical energy devices that promise to redefine the limits of performance and versatility.
 Yu Zhang | Yu Zhang is an Associate Professor at Xinyang Normal University. She received her PhD degree from Northwest University in 2016. After graduation, she joined Xinyang Normal University as a faculty member. Her primary research focuses on the design and synthesis of functional nanomaterials for energy storage and conversion, with particular emphasis on their applications in electrochemical energy conversion, storage systems and electrocatalysis. |
 Xiang Hu | Xiang Hu is an Associate Professor at the Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences (FJIRSM, CAS). He received his PhD degree in materials science and engineering from Fuzhou University in 2021. He was a postdoctoral fellow in the laboratory of Prof. Zhenhai Wen at the FJIRSM from 2021 to 2023. His research interests focus on the design of functional nanomaterials, interfacial engineering, and the elucidation of reaction mechanisms for electrochemical energy storage systems, such as metal-ion batteries and metal-ion hybrid capacitors. |
 Zhenhai Wen | Zhenhai Wen is a Professor at the Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences. He currently directs the Center for Applied Chemistry and the Fujian Key Laboratory of Hydrogen Energy Materials. He received his PhD in 2008 and conducted postdoctoral research at the Max Planck Institute and the University of Wisconsin-Milwaukee. He is a recipient of the National Science Fund for Distinguished Young Scholars. His research focuses on advanced electrode materials and hybrid electrochemical energy devices. He has published over 300 papers (h-index: 104, 35 |
1. Introduction
Electrochemical energy systems are devices that enable the reversible conversion between electrical and chemical energies through redox reactions at the interface of electrodes and electrolytes.1–3 Electrolyzers and batteries represent two fundamental forms of electrochemical systems, operating in opposite directions of energy conversion. While electrolyzers use electrical energy to drive non-spontaneous reactions for the production of target chemical products, batteries harness energy from spontaneous electrochemical reactions to deliver usable electrical power.4–6 Therefore, electrochemical energy technologies play a pivotal role in powering portable electronics, electric vehicles, and stationary energy storage systems. More importantly, in the context of global decarbonization and energy transition, these devices serve as foundational technologies for the efficient utilization, storage, and high-quality integration of intermittent renewable energy sources such as solar, wind, and hydropower. These technologies encompass batteries, fuel cells, supercapacitors, and electrolyzers, all of which rely on electrochemical reactions to store, convert, or utilize energy in a controllable and sustainable manner.A defining feature of electrochemical energy devices is their reliance on the coordinated transport of two distinct types of charge carriers (i.e. electrons and ions) to complete a closed electrical circuit. During operation, electrons are generated at the anode through oxidation reactions, travel through the external circuit to the cathode, and are consumed via reduction reactions. Simultaneously, ions migrate through the internal electrolyte media, such as liquid, polymeric, or solid-state electrolytes, to maintain charge neutrality and sustain the electrochemical reactions at the electrode interfaces. The interplay between electronic and ionic charge carriers is mediated by electrochemical reactions at the electrode/electrolyte interfaces. In this context, the electrochemical reaction can be regarded as a medium that conducts both electrons and ions, effectively bridging the external and internal segments of the circuit, thus closing the loop and enabling continuous operation of the electrochemical energy devices.
What makes these systems particularly complex and tunable is the fundamentally different nature of the two types of charge carriers. Electrons are uniform in nature, possessing identical charge, mass, and behaviour across all systems, and are transported primarily through electronic conductors or first-class conductors in external circuits. By contrast, ions exhibit a high degree of chemical and physical diversity. This diversity includes variations in charge polarity and magnitude, ionic radius, hydration energy, mobility, solvation behaviour, and electrochemical reactivity. Such diversity renders ionic charge carriers inherently more complex and dynamic than electrons, and their behaviour is highly sensitive to the surrounding electrolyte composition, temperature, and the structure and chemistry of the electrode/electrolyte interface. Ionic charge carriers play a fundamental role in governing the efficiency, energy and power density, reversibility, safety, and durability of electrochemical power devices. In fact, the selection and precise control of ionic species can profoundly influence both device design and overall performance.
In electrochemical devices, ionic charge carriers encompass both the ions involved in redox reactions at the anode and cathode, as well as those responsible for charge transport within the electrolyte. Although ions exhibit remarkable diversity in terms of charge, size, mobility, and solvation behaviour, some conventional electrochemical systems have predominantly relied on a single ionic species to fulfill all these roles. That is, the same ion is responsible for both participating in electrode reactions and migrating through the electrolyte to maintain charge neutrality and complete the internal circuit. For example, Li+ and Na+ function as the dominant charge carriers in lithium-ion and sodium-ion batteries (LIBs and SIBs), respectively.7–10 Likewise, protons (H+) are the sole ion current carriers in proton exchange membrane fuel cells (PEMFCs), while hydroxide ions (OH−) act as the primary carriers in alkaline fuel cells and batteries.11–13
Although this single-ion carrier system offers advantages in terms of simplified design, streamlined material selection, and ease of theoretical modelling, it also imposes fundamental limitations, including material compatibility, limited ion transport efficiency, sluggish reaction kinetics, reduced system tunability, and ultimately, compromised overall performance. Relying on a sole ionic carrier narrows the range of viable electrode and electrolyte chemistries, which can lead to suboptimal charge-transfer processes, a constrained electrochemical stability window, and diminished adaptability to varying operational or environmental conditions.
In an electrochemical system, any oxidizable reaction can theoretically serve as the anodic process, while any reducible reaction can be used at the cathode. This inherent flexibility offers a broad reaction space for the design of electrochemical systems. On this basis, employing multiple ionic species as charge carriers for both anodic and cathodic reactions not only breaks the conventional reliance on single-ion systems, but also opens up diverse opportunities for developing novel and multifunctional electrochemical devices. For instance, the cooperative migration of different ions can enable the coupling of energy storage with value-added chemical synthesis, thereby advancing the design of electrochemical systems featuring multi-energy integration and multi-scale regulation. Hence, electrochemical systems enabled by multi-ion carriers represent a paradigm shift from single-ion designs by decoupling or diversifying the functional roles of ionic species within the device. Rather than relying on single-ion carriers to mediate both electrode reactions and ionic conduction, these systems harness the complementary properties of multiple ionic species, which may vary in valence, size, mobility, or redox activity, to fulfill distinct functional roles. This emerging strategy enables one ion species to primarily engage in redox reactions, while another is optimized for rapid ionic transport or enhanced interfacial stability within the electrolyte, or vice versa, depending on the system design.
There is growing interest in such multi-ion electrochemical architectures, where distinct ions either activate separate electrochemical pathways or synergistically cooperate to enhance charge transport across electrodes and electrolytes. By decoupling these roles, multi-ion systems offer significantly greater design flexibility, enabling the use of previously incompatible materials, tailoring of interfacial chemistries, and fine-tuning of reaction environments.14,15 Moreover, the incorporation of diverse ion species expands the range of accessible redox potentials, enhances the electrochemical stability window, and opens avenues for new charge compensation mechanisms, ultimately leading to improved reaction kinetics, higher energy density, and better operational stability under diverse conditions. This shift toward cooperative ionic functionality holds great promise for the next generation of energy storage and conversion devices, including hybrid batteries, capacitors, and advanced fuel cells.
Despite the growing momentum in this field, a comprehensive understanding of multi-ion electrochemical systems remains fragmented, with relevant studies often isolated across distinct device types, chemistries, or ionic configurations. While traditional multi-ion systems, such as supercapacitors with electrolyte ion pairing or inorganic redox flow batteries, have been extensively reviewed in earlier works, these discussions are often constrained to specific device classes and well-established mechanisms. However, recent breakthroughs in electrochemical technologies have given rise to a new generation of multi-ion energy devices, wherein multi-ion carriers are intentionally integrated to perform distinct and cooperative functions, ranging from charge compensation and redox activity to transport enhancement and interface stabilization. These include, for example, hybrid-ion batteries utilizing both monovalent and multivalent cations, dual-anion systems, pH-gradient modulators, and emerging devices that combine aqueous and non-aqueous electrolyte environments to enable broader electrochemical tunability.
Despite the growing interest in multi-ion carrier-enabled electrochemical systems, the current body of literature remains highly fragmented, with existing discussions often limited to specific device types, such as redox flow batteries or hybrid supercapacitors, or narrowly focused on particular ionic species.16–19 To date, no systematic review has holistically captured the design principles, ion-specific functionalities, and broad material strategies that underpin this emerging class of electrochemical architecture. As a result, there remains a lack of integrated understanding regarding how multiple ionic species can be synergistically utilized to optimize electrochemical performance across diverse platforms.
This review aims to address that critical knowledge gap by providing a comprehensive and forward-looking analysis of multi-ionic carrier-enabled electrochemical energy devices, with a particular emphasis on newly developed, reimagined, or underexplored systems that go beyond traditional implementations. These include devices that exploit decoupled redox-transport ion roles, hybrid ion electrolytes, asymmetric ionic configurations, and interface-modulating ions, all of which represent a deviation from conventional single-ion paradigms. To this end, we adopt a cross-disciplinary perspective, bridging advances in electrochemistry, materials science, and systems engineering, to deliver a unifying framework for understanding and advancing multi-ion strategies. Our goal is not only to highlight current progress but also to provide design guidelines, mechanistic insights, and emerging challenges that will inform the rational development of next-generation energy storage and conversion technologies.
This review provides a comprehensive and logically structured overview of electrochemical power sources (EPSs) enabled by multi-ion carriers. We begin by establishing a fundamental framework that classifies multi-ion EPSs based on ionic functionality, device architecture, and synergistic ion interactions, laying the groundwork for understanding how multi-ion strategies enhance energy storage and conversion. Building on this foundation, we systematically examine various device types, including hybrid batteries (cationic, anionic, mixed-ion, and multi-cathode systems), hybrid battery-capacitors employing mono- and multi-valent ions, and a diverse range of fuel cells, such as hybrid acid–alkali systems, solid oxide fuel cells with mixed conduction, and metal–air or H2O2-based configurations. Flow battery systems are also discussed, with emphasis on redox-active species design, membrane engineering, and system-level ion transport optimization. Finally, we explore key challenges and opportunities in multi-ion EPSs, covering material design, interfacial chemistry, performance metrics, and emerging applications from grid-scale storage to advanced fuel cells and integrated electrosynthesis.
By synthesizing insights across these sections, this review offers a multidimensional perspective on the design, mechanisms, and future directions of multi-ion electrochemical systems. Our analysis not only consolidates foundational understanding but also highlights emerging trends, critical challenges, and promising opportunities that are reshaping the field. Designed to serve as both a comprehensive reference and a strategic guide, this article aims to bridge the gap between fundamental science and practical engineering, empowering the advancement of multi-ion carrier-enabled energy technologies. Ultimately, we aspire to inspire interdisciplinary collaboration among researchers, engineers, and industry stakeholders by fostering a deeper understanding of ion transport mechanisms, interfacial dynamics, and sustainable material strategies. Through this integrated approach, we aim to accelerate the development of efficient, adaptable, and environmentally responsible electrochemical energy devices that meet the complex demands of the future energy landscape.
2. Fundamentals of multi-ion EPSs
2.1. Concept
The operation of EPSs fundamentally depends on the coupled transport of electrons and ions, forming a closed-loop circuit essential for charge conservation and sustained energy conversion. During charge and discharge processes, electrons flow through the external circuit, while ions migrate through the electrolyte to maintain electroneutrality within the cell. To accommodate this dual transport mechanism, electrochemical reactions at the interface between electrodes and electrolytes serve as the critical junction where electronic and ionic charge carriers converge, acting as a dual-conduction gateway that enables electron flow through the external circuit and ion transport within the cell. Ion carriers in electrochemical systems may serve as transport-mediating ions within the electrolyte, redox-active ions and product ions involved in electrochemical redox reactions, or interfacial modulators.While traditional systems typically employ a single ionic species to perform all these roles, multi-ion EPSs are energy storage or conversion systems that intentionally employ two or more distinct ionic species to fulfill a specific electrochemical function. For instance, one ion may engage in redox reactions or be produced as a reaction product, while others ensure efficient ionic transport, buffer charge imbalances, or stabilize electrode/electrolyte interfaces. By decoupling these functions across multiple ions, multi-ion EPSs allow independent tuning of redox chemistry, transport dynamics, and interfacial behaviour, leading to enhanced system performance.
The identity, mobility, and redox activity of each ionic species significantly influence key performance metrics such as reaction kinetics, operating voltage, energy density, and long-term stability. To support these differentiated roles, multi-ion systems often incorporate hybrid electrolytes or multi-channel ionic conductors, paired with electrodes engineered for selective responsiveness to specific ions. During operation, this configuration enables cooperative behaviour. For instance, one ion may undergo intercalation or conversion-type redox reactions, while another concurrently regulates interfacial properties or maintains charge neutrality, cooperatively improving system efficiency and stability.
This collaborative approach to ion transport yields several compelling advantages over conventional single-ion systems. First, enriched redox mechanisms allow for tandem or simultaneous redox processes, potentially enhancing energy efficiency and capacity. Second, broader material compatibility enables the use of a wider range of electrode and electrolyte materials, accelerating innovation and optimization. Third, improved charge/discharge kinetics result from synergistic ion transport, reduced interfacial impedance and mitigated ion concentration gradients. Fourth, tailored interfacial chemistry can be achieved by leveraging the interplay of multiple ions, promoting the controlled formation of stable and functional solid–electrolyte interphases (SEI) or cathode electrolyte interphases (CEI), thereby enhancing cycling life and safety. Altogether, the structural and mechanistic flexibility of multi-ion EPSs enables precise control over ion transport, redox processes, and interfacial dynamics, making them highly adaptable for a wide range of applications, including grid-scale energy storage, high-power batteries, and flexible electronics.
2.2. Historical perspective
In the dynamic landscape of electrochemical energy storage and conversion, multi-ion carrier power supply devices stand as a cornerstone of innovation, with their development intricately tied to the manipulation of diverse ionic species. The timeline of these devices, categorized into hybrid batteries, hybrid battery capacitors, fuel cells, and redox flow batteries (Fig. 1), vividly illustrates how multi-ion carriers underpin advancements in energy technology, enabling breakthroughs in performance, efficiency, and application versatility. | ||
| Fig. 1 A brief development history of representative multi-ion EPSs. | ||
Hybrid batteries mark an early and pivotal chapter in the exploration of multi-ion carriers. The journey began in 1800 when the Italian scientist Alessandro Volta invented the Voltaic Pile. In this pioneering battery, zinc ions (Zn2+) migrated toward the Cu cathode, while protons (H+) moved toward the Zn anode, maintaining charge balance in the solution. Then exploration advanced in 1966 with the emergence of Li–Cl2 batteries, where the interplay of Zn2+ and Cl− ions drives energy storage and release. A major milestone followed in 1996 with the advent of Li-based dual-ion batteries (Li-DIBs), which employed both Li+ and PF6− ions, marking a critical advancement in understanding dual-ion electrochemistry. As research progressed, the ionic landscape expanded with the emergence of Li–Na hybrid ion batteries (Li–Na HIBs) in 2006 and Zn–Mn hybrid ion batteries (Zn2+/Mn2+) in 2011, broadening the spectrum of multi-ion configurations. These hybrid batteries utilize multi-ion carriers, including different metal cations and, in some cases, accompanying anions, to orchestrate electrochemical reactions. By combining ions with distinct properties, hybrid batteries push beyond the limitations of single-ion systems, aiming for higher energy densities, wider voltage windows, and improved cycle stability. A representative example is the Zn–Al HIBs developed in 2015, where the synergistic behavior of Zn2+ and Al3+ facilitates unique redox pathways and interfacial dynamics, enabling performance metrics that are difficult to attain in traditional single-ion counterparts.
Building on the foundations laid by hybrid batteries, hybrid battery capacitors further exemplify the power of multi-ion carriers. The emergence of sodium-ion hybrid capacitors (SIHCs) in 2012 marked a significant step forward, where the combination of Na+ and ClO4− ions enabled the integration of battery-like and capacitor-like behaviours within a single device. As the field progressed, the development of aluminum-ion hybrid capacitors (AIHCs) based on Al3+ and NO3− ions in 2014, followed by zinc-ion hybrid capacitors (Zn2+/SO42−) in 2016, highlighted the importance of controlled ion transport and synergistic ion interactions. These systems exploit the complementary electrochemical properties of multiple ionic species to simultaneously deliver high-energy and high-power outputs. The continued evolution of ion chemistries, exemplified by the emergence of ammonium-ion hybrid capacitors (AmIHCs) based on NH4+ and SO42− in 2022, highlights how multi-ion strategies drive functional diversification and performance enhancement across a broad spectrum of electrochemical energy storage applications.
Fuel cells represent a critical frontier in multi-ion carrier-driven energy conversion, where the efficient transfer of ions dictates power generation. The Zn–air batteries (ZABs) reported in 1878 pioneered this path, with hydroxide ions (OH−) and Zn related species (e.g., Zn(OH)42−) facilitating the electrochemical oxidation of Zn and O2 reduction. As technology advances, systems like Al–air batteries (Al(OH)4−/OH−) reported in 1981 and dual-ion Li–air batteries (Li+/OH−) reported in 2011 expand the ion-mediated reactions, each leveraging specific cations and anions to drive fuel oxidation and oxygen reduction reactions (ORRs). Modern developments, such as the anticipated hybrid acid–alkali formaldehyde fuel cell (HCOO−, H+, and OH−) reported in 2025, highlight the growing complexity of multi-ion carriers. In fuel cells, protons (H+), OH−, or other ionic species must be precisely shuttled to enable the electrochemical conversion of fuels like hydrogen (H2) and formaldehyde into electricity. The multi-ion dynamics here are not just about energy storage but about sustained energy conversion, where the movement and reaction of ions at electrodes and through electrolytes determine the efficiency, durability, and scalability of fuel cell systems. Thus, fuel cells demonstrate how multi-ion carriers are indispensable for unlocking the potential of electrochemical energy conversion, bridging the gap between chemical fuels and electrical power.
Redox flow batteries (RFBs) close this evolutionary loop, emphasizing the sustained role of multi-ion carriers in large-scale energy storage. The iron–chromium flow batteries (Fe–Cr FBs) reported in 1974 initiated this journey, with Fe2+/Fe3+ and Cr2+/Cr3+ ions shuttling between tanks to store and release energy. Subsequent RFBs, like the zinc bromine flow batteries (Zn–Br2 FBs) based on Zn2+/Br− reported in 1977 and zinc iron flow batteries (Zn–Fe FBs) based on Zn2+/Fe2+/Fe3+ reported in 1979, continue to rely on ion shuttling, each introducing unique cation–anion combinations to tailor energy density, solubility, and reaction kinetics. The aqueous organic redox flow batteries (AORFBs) further illustrated the adaptability of multi-ion carriers in 2014, using H+ and Br− to enable organic-based, potentially more sustainable energy storage. In RFBs, the separation of redox-active electrolytes and the continuous flow of multi-ion-laden solutions allow for scalable energy storage, making them ideal for grid-level applications. The multi-ion carriers here dictate the battery's capacity (through ion concentration), voltage (via redox potential differences), and cycle life (influenced by ion stability). As such, redox flow batteries exemplify how mastering multi-ion carrier dynamics is key to developing reliable, large-scale energy storage solutions that can support the transition to renewable energy grids.
Across hybrid batteries, hybrid battery capacitors, fuel cells, and redox flow batteries, the common thread is the centrality of multi-ion carriers. These ionic species are not just passive components but active enablers of performance optimization, functional diversification, and technological scalability. From tailoring electrochemical reactions in hybrid batteries to facilitating energy conversion in fuel cells and enabling large-scale storage in redox flow batteries, multi-ion carriers underpin the past, present, and future of multi-ion carrier power supply devices. As research continues to push boundaries, the deliberate design and manipulation of multi-ion systems will remain pivotal in unlocking next-generation energy technologies, meeting global demands for efficient, sustainable, and versatile energy storage and conversion.
2.3. Classification and categories
Multi-ion EPSs encompass a broad class of energy storage and conversion systems that strategically incorporate two or more distinct ionic species to perform complementary roles. These systems can be classified from various perspectives, including ion types or function, configuration, origin, and dynamic behavior, each reflecting a different aspect of their operational complexity and design philosophy.From a function-based perspective, multi-ion EPSs can be categorized according to the roles fulfilled by each ion type. In complementary-ion systems, the ionic species are assigned distinct functions: ions participate directly in or may be produced during energy conversion via electron transfer; transport-mediating ions support ionic conductivity and maintain charge balance; and interface-modulating ions enhance interfacial stability or regulate reaction kinetics. Alternatively, some systems utilize multi-redox-ion configurations, where two or more ionic species are electrochemically active. These systems often exhibit broad electrochemical windows or novel redox mechanisms, as exemplified by hybrid acid/alkali batteries or fuel cells involving both OH− and H+ in aqueous environments.20–23
A configuration-based classification focuses on how ionic species are spatially arranged and interact within the system. In spatially separated ion systems, different ions operate in distinct compartments, such as the anode and cathode regions, typically separated by ion-selective membranes. This configuration is common in dual-ion batteries and some flow batteries. Conversely, in mixed-ion systems, all ionic species coexist in a single electrolyte and operate simultaneously, often resulting in complex but synergistic ion–ion and ion-electrode interactions that enhance overall performance.
Multi-ion EPSs can also be differentiated based on the origin of their ionic diversity. Intrinsically multi-ion systems arise from materials or electrolytes that naturally host multiple mobile ions, such as mixed-cation solid-state electrolytes or multi-component ionic liquids.24–27 In contrast, extrinsically engineered systems involve the deliberate introduction of secondary or tertiary ionic species, either through additives, co-electrolytes, or electrode functionalization, to achieve targeted performance enhancements such as improved cycling stability or interfacial protection.
Finally, a dynamics-based classification considers the temporal role of each ion during device operation. In sequential-ion systems, different ions dominate at different stages of charge/discharge or under specific operating conditions. For example, a primary redox ion may be active during normal operation, while a secondary ion stabilizes the interface during rest or overpotential events. In simultaneous-ion systems, multiple ions participate concurrently throughout the electrochemical cycle, often requiring finely tuned ion transport and reaction coordination to avoid interference and maximize synergy.
This multi-dimensional classification not only reflects the growing complexity of multi-ion EPS architectures but also provides a useful framework for guiding material design, mechanistic understanding, and system-level optimization in next-generation electrochemical technologies.
3. Hybrid batteries
Hybrid batteries have recently emerged as a distinct family of electrochemical energy-storage devices that transcend the intrinsic limitations of single-ion “rocking-chair” chemistries by orchestrating the concerted transport of multiple ionic species. By mobilizing multiple types of charge carriers either simultaneously or sequentially, these systems leverage complementary redox potentials, diffusion kinetics, and solvation behaviors, thereby opening new avenues toward enhanced energy density, improved rate performance, and extended cycle life. Architecturally, multi-ion systems encompass a diverse array of configurations, ranging from purely cationic (e.g., Li+, Na+, K+, Ca2+, Mg2+, Zn2+, Al3+, NH4+, and H+) designs that shuttle two or more metal ions to mixed cation–anion (e.g., ClO4−, PF6−, TFSI−, Cl−, Br−, and I−) systems, wherein oppositely charged species operate cooperatively, and further to more complex triple or quadruple ion frameworks that integrate multiple cations and anions within a unified electrochemical circuit.28 Each configuration enables a dynamic interplay of intercalation, alloying, conversion, and capacitive processes, thereby offering unprecedented flexibility in both material selection and cell architecture design.A practical framework for understanding these architectures is to classify them based on the charge type (cationic or anionic) and the number of principal mobile species, as illustrated schematically in Fig. 2. In dual-cation systems, two distinct metal ions, referred to as “M1” and “M2”, may originate from either the electrodes or the electrolyte. The most straightforward configuration operates analogously to a two-lane shuttle (Fig. 2a). During the charging process, M1 is de-intercalated from the cathode, whereas M2 undergoes plating, alloying, or intercalation at the anode. These processes are reversed during discharge. Since many multivalent ions (typical M2 species such as Mg2+, Zn2+, or Al3+) exhibit sluggish diffusion kinetics or unfavourable intercalation thermodynamics in conventional cathode materials, the simultaneous participation of a fast-diffusing monovalent ion (e.g., Li+ or Na+) can help mitigate kinetic limitations and stabilize the cathode host structure.29,30 A more synergistic design involves the co-insertion of both cations into a single electrode, while the counter electrode accommodates only M2 (Fig. 2b). This co-intercalation strategy has been demonstrated to alleviate volume strain, extend the accessible redox potential window, and markedly enhance rate capability compared to single-cation systems.
 | ||
| Fig. 2 Schematic illustration of representative hybrid battery configurations based on different charge carrier types: (a) and (b) dual-cation systems; (c) cation–anion dual-ion system; (d) triple-ion system with two cations and one anion; (e) triple-ion system with one cation and two anions; and (f) quadruple-ion system comprising two cations and two anions. | ||
When one mobile species is a cation (M1) and the other an anion (N1), the system constitutes a cation–anion dual-ion battery (Fig. 2c).31 During charging, M1 migrates toward the anode while N1 moves toward the cathode, which is typically composed of graphite. This configuration allows for the replacement of costly transition-metal oxides with low-cost carbonaceous hosts, while also enabling a broader electrochemical window determined by the high insertion potential of the anion. Since both ionic species vacate their respective host structures during discharge, the overall electrode frameworks remain largely intact, leading to excellent cycling stability and enhanced material sustainability.
Advancing beyond the dual-ion concept, triple-ion strategies introduce a second cation (M2) alongside the cation–anion pair (Fig. 2d). Co-alloying of M1 and M2 at a Sn or Al anode can alleviate the acute volume expansion that plagues single-metal alloying, while the simultaneous intercalation of N1 into graphite upshifts the average discharge voltage. Analogously, one can envisage a configuration that couples a single cation (M1) to two distinct anions (N1 and N2) (Fig. 2e), or the fully fledged quadruple-ion architecture in which two cations and two anions cycle concertedly (Fig. 2f). In theory, each additional mobile species adds a new degree of freedom for tailoring reaction thermodynamics, transport pathways, and mechanical buffering, paving the way for devices that simultaneously maximize power, energy, and durability.
By integrating disparate charge carriers, hybrid batteries can effectively compensate for the limitations associated with any single ion, such as electrode degradation, narrow electrochemical windows, or sluggish kinetics, while simultaneously harnessing their collective strengths.32 This versatility positions multi-ion systems as a powerful platform for balancing performance, cost, and resource sustainability across applications ranging from wearable electronics to grid-scale storage. As research pushes towards increasingly complex but high-performance chemistries, hybrid batteries lie at the forefront of multi-ion energy-storage innovation. The sections that follow will survey recent advances in cationic hybrids and then extend to mixed cation–anion devices, highlighting how the deliberate orchestration of multiple ionic species defines the cutting edge of next-generation electrochemical technology.
3.1. Dual-cation hybrid batteries
Dual-cation hybrid batteries (DCHBs) represent a prominent subclass of hybrid energy storage systems, in which multiple positively charged ions are co-employed as active charge carriers. These include both monovalent ions such as Li+, Na+, and K+, and multivalent ions such as Mg2+, Zn2+, Ca2+, and Al3+. Based on the physical and chemical properties of these elements themselves (Fig. 3a and b), Li+ possesses the smallest ionic radius and lowest atomic mass among the monovalent cations, facilitating rapid and deep intercalation, and enabling high gravimetric and volumetric energy densities. In contrast, the larger radii of Na+ and especially K+ result in greater steric hindrance within host structures, which hinders solid-state diffusion and reduces specific capacities. Multivalent cations such as Mg2+, Zn2+, Ca2+, and Al3+ offer the potential for multi-electron transfer per ion and, in some cases, possess ionic radii comparable to or even smaller than Na+.33,34 However, their higher charge density intensifies electrostatic interactions with host lattices, which significantly impedes ion mobility and often leads to interfacial polarization or structural passivation. | ||
| Fig. 3 (a) and (b) Comparison of ionic radii, atomic weights, gravimetric and volumetric specific capacities, and standard electrode potentials vs. SHE for various elements commonly used in hybrid battery systems. (c) Schematic illustration of the working mechanisms of conventional single-ion batteries compared with dual-cation hybrid battery systems. | ||
The dual-cation strategy is intended to combine the complementary strengths of each ion type, namely the high intrinsic energy density of lithium, the abundance and low cost of Na, the rapid transport kinetics of K, the dendrite-free deposition behavior of Mg, the low standard electrode potential versus standard hydrogen electrode (SHE) and aqueous compatibility of Zn, and the high volumetric capacity of Al. By strategically integrating the complementary properties of different metal ions, DCHBs facilitate cooperative redox reactions and customized ion transport pathways. This approach improves energy density, accelerates charge transfer, enhances structural stability, and prolongs cycle life, all of which are performance metrics typically constrained in conventional single-ion systems (Fig. 3c and d). The performance metrics of various dual-cation HIBs are compiled and presented in Table S1. In addition, the inclusion of secondary cations mitigates supply-chain risks by partially substituting critical elements with earth-abundant alternatives.
Continued progress in electrode architecture, electrolyte formulation, and interfacial engineering is rapidly elevating DCHBs' performance, underscoring their promise as high-efficiency, resource-conscious, and durable energy-storage platforms. The subsections that follow systematically review the principal cationic hybrid chemistries, focusing on their reaction mechanisms, material innovations, and practical advantages.
Recent advancements in this field have focused on engineering electrode architectures and electrolyte formulations that optimize concurrent Li+/Na+ intercalation dynamics. A representative study performed by Wei et al.36 demonstrated this principle through Li-rich layered oxide cathodes (Li1.18Ni0.15Co0.15Mn0.52O2), where systematic electrochemical activation induced a dual-cation migration mechanism. Post-activation characterization revealed Na+ participation in charge compensation processes alongside Li+, achieving remarkable rate performance (90 mAh g−1 at 10C) and cycling stability (105 mAh g−1 retention over 200 cycles at 5C). This work highlights the potential of multi-ion systems to overcome the classic dilemma between energy density and cost, while also revealing challenges related to the long-term structural evolution caused by mixed-ion transport. Interfacial interactions between hetero-ionic species and host lattices, such as intercalation-induced stress and phase transitions, demand deeper mechanistic insights to realize the full benefits of multi-ion designs. Based on the concept of integrating sodium metal anodes with lithium-based cathodes to optimize energy density and cycling stability, Zhang et al.37 developed a LSHIB configuration comprising a sodium metal anode and a lithium iron phosphate (LFP) cathode, achieving an impressive coulombic efficiency of 99.2% over 100 cycles. This dual-cation design harnesses the simultaneous involvement of both Li+ and Na+ ions in electrochemical reactions (Fig. 4a). The hybrid system exhibits significantly lower voltage hysteresis, with a reduction of 247 mV, and displays sharper redox peaks in cyclic voltammetry, indicating enhanced electrochemical reversibility (Fig. 4b), reflecting improved charge transfer kinetics and more efficient Na+ de-solvation. Notably, while lithium–metal counterparts experience rapid capacity degradation, the LSHIB retains both high capacity and coulombic efficiency over prolonged cycling (Fig. 4c), underscoring how multi-ion architectures synergistically stabilize electrochemical interfaces and optimize energy efficiency through complementary ion transport dynamics.
 | ||
| Fig. 4 (a) Schematic illustration of the Li–Na HIBs. (b) Free energy profiles and optimized geometries calculated for the solvation of Li+ and Na+. (c) CVs of the Li metal batteries and the Li–Na hybrid batteries. Reproduced with permission from ref. 37. Copyright 2018, Wiley. (d) A strategy of locally targeting anchor (LTA) with self-healing properties for all-solid-state sodium metal batteries. (e) Evolution process of interface during cycling process without LTA treatment. (f) Evolution process of interface during cycling steps via LTA treatment. Reproduced with permission from ref. 46. Copyright 2023, Wiley. (g) Electro-active species involved during charging a hybrid battery made of a Na3V2(PO4)3 cathode, a Mg anode, and an electrolyte of 0.2 M [Mg2Cl2][AlCl4]2 and 0.4 M NaAlCl4 in DME. (h) Illustration of the operating principle of a Mg–Na HIBs. (i) Cyclic voltammograms of a pure Mg electrolyte and a hybrid Mg–Na electrolyte in a three-electrode cell. Inset shows the accumulated charge during Mg deposition–dissolution cycle. Reproduced with permission from ref. 53. Copyright 2017, Elsevier. | ||
Structural engineering breakthroughs further optimized ion-host compatibility. Song et al.38 engineered sodium manganese oxide (NMO) cathodes via reduction-driven ion exchange, achieving 195 mAh g−1 over 50 cycles. Na+ acted as both a charge carrier and a structural stabilizer, yet precise control of ion-exchange kinetics remained challenging. Expanding this approach, Ma et al.39 developed high-entropy doped Na3V2(PO4)3@C (HENVP@C) by incorporating five cations (Ti4+, Mn2+, Fe2+, Zr4+, and Mo6+). This induced lattice distortion, expanding Na+ diffusion channels and activating V4+/V5+ redox (3.8/3.9 V). The material delivered 76.8 mAh g−1 at 50C in SIBs, showcasing enhanced Li+/Na+ co-storage kinetics through entropy-driven stabilization. Composite cathode designs have emerged as an effective strategy to balance energy density and cost in hybrid battery systems. Xie et al.40 developed a practical Li–Na hybrid battery featuring a blended cathode composed of 80 wt% Na3V2(PO4)2F3 (NVPF) and 20 wt% LiNi0.8Co0.1Mn0.1O2 paired with a hard carbon anode. This composite cathode improved the discharge voltage by approximately 0.1 V through modulation of phase transitions, resulting in a high capacity of 147 mAh g−1 and an energy density of 543 Wh kg−1 at a 0.1C rate. Electrochemical impedance spectroscopy revealed a substantial reduction in charge transfer resistance to 12.2 Ω, which is only 1/15th of that observed for pure NVPF cathodes. In addition, the diffusion coefficients for Na+ and Li+ increased markedly to 4.7 × 10−13 cm2 s−1 and 9.5 × 10−12 cm2 s−1, respectively. These enhancements collectively contribute to improved kinetics and overall battery performance.
Electrolyte and interfacial engineering proved pivotal for kinetic enhancement. Wang et al.41 employed sodium difluoro(oxalato)borate (NaODFB) as a novel salt, enabling Li1.2Ni0.13Co0.13Mn0.63O2 cathodes to retain 151 mAh g−1 after 200 cycles. This tailored formulation modulated solvation structures to accommodate mixed-ion transport. Xia et al.42 advanced sodium anode stability by constructing a lithium-containing hybrid SEI (LSEI) through electrochemical modification. This LSEI incorporated Li3N, LiF, and Li2CO3, which collectively reduced the Na+ diffusion barrier; for example, LiF exhibited a barrier of 0.28 eV compared to 0.48 eV for NaF. Additionally, it significantly increased the Young's modulus from 2.38 to 6.88 GPa. These improvements enabled stable cycling for 500 hours at a current density of 30 mA cm−2 in symmetric cells, as well as 97.9% capacity retention under a high cathode loading of 39.3 mg cm−2.
Taken together, these innovations firmly establish LSHIBs as a paradigm-shifting platform that combines lithium's high energy density with sodium's cost-effectiveness and sustainability. However, despite these promising advances, critical challenges persist - chief among them are the need to optimize the coordination of Li+ and Na+ transport, to mitigate cross-ion interference at electrode–electrolyte interfaces, and to address capacity fading associated with prolonged mixed-ion cycling.
3.1.2.1. Na–K HIBs. In Na–K hybrid systems, Na+ offers superior thermodynamic stability, attributed to its relatively lower redox potential and stronger coulombic interaction with host materials. In contrast, K+ exhibits faster diffusion kinetics, facilitated by its larger ionic radius, lower charge density, and weaker solvation effect in electrolytes. The synergistic participation of both ions in SPHIBs not only enables more efficient ion transport but also broadens the electrochemical window and enhances overall electrochemical reversibility.
To fully harness the benefits of this multi-ion carrier mechanism, rational material design is crucial, with a particular emphasis on cathode architecture. Among various candidates, potassium Prussian blue analogues (K-PBAs) have garnered significant attention due to their open-framework structures, large ionic channels, and chemically tunable lattice. These features make them ideal hosts for accommodating both Na+ and K+.44 Recent studies have highlighted the critical role of defect engineering in optimizing Na+/K+ co-intercalation dynamics. Specifically, the controlled introduction of anionic (vacancy-type) defects can modulate the local electrostatic environment and ion diffusion pathways. For instance, Wei et al.45 introduced ∼20% controlled vacancies into K-PBAs, which expanded K+ diffusion channels while suppressing excessive Na+ insertion. This not only optimized ion transport kinetics and mitigated lattice strain, but also enabled a high initial discharge capacity of 128 mAh g−1 with 81% capacity retention over 300 cycles, demonstrating a level of performance that remains unattainable in conventional single-ion systems. Importantly, the advantages of multi-ion carriers extend into solid-state battery systems. Incorporating trace K+ from cathodes such as K2MnFe(CN)6 promotes the in situ formation of a Na–K alloy interphase at the anode when used with natrium super ionic conductor (NASICON)-type solid electrolytes. This semi-liquid interphase not only enhances interfacial wettability and mechanical compliance but also serves as a self-healing layer to mitigate dendrite growth. Despite the larger ionic radius of K+, Ni et al.46 proposed a local targeting anchor strategy that effectively utilizes the dendrite-free nature of the Na–K alloy and the electrostatic shielding effect provided by a K+-rich layer originating from the cathode. This dual-function approach significantly enhances the interfacial stability of solid-state Na metal batteries. Within solid electrolytes such as Na3Zr2Si2PO12 (NZSP), the presence and migration of K+ subtly modulates Na+ transport behavior by reshaping ionic conduction pathways, lowering energy barriers for migration, and mitigating localized field intensities at deposition sites, thereby enabling more uniform Na metal plating (Fig. 4d–f).
Beyond cathodes, extending the multi-ion carrier concept to anodes has also proven highly effective. Semi-solid Na–K alloy anodes demonstrate a harmonious balance between Na+/K+ co-transport kinetics and interfacial stability.47 This architecture leverages the strong affinity between solid Na and the liquid Na–K phase to confine the liquid within a stable interface, thereby overcoming issues of structural instability and parasitic reactions observed in purely liquid systems. When paired with NaClO4-based electrolytes, the system spontaneously forms a high-modulus SEI (10.3 GPa) enriched in KClO4 crystals, which effectively suppress dendrite growth - validated through both computational simulations and in situ microscopy. As a result, the symmetric cells demonstrate stable cycling for over 1000 hours at 0.5 mA cm−2, while full cells with Na3V2(PO4)3 cathodes exhibit excellent rate capability (93 mAh g−2 at 20C) and long-term stability (89.18% capacity retention over 1000 cycles). Notably, they maintain robust performance even in solid-state configurations, highlighting the critical role of multi-ion carriers in balancing interfacial rigidity with efficient ion diffusion. Parallel innovations in electrolyte design further amplify the benefits of multi-ion carrier systems. A notable example is the use of 4 Å molecular sieves (Na2O·Al2O3·2SiO2) in a novel ion-exchange strategy to construct Na+/K+ hybrid electrolytes.48 These sieves simultaneously introduce Na+ ions and dehydrate the system, resulting in significantly reduced charge-transfer resistance (5.9 Ω vs. 35 Ω in KIBs without Na+). At a 25 wt% sieve loading (corresponding to 9 wt% Na+), the hybrid electrolyte enables rapid co-alloying on Sn foil anodes. Ex situ XRD and density functional theory (DFT) calculations reveal the preferential formation of NaSn, which facilitates subsequent KSn formation. This pre-sodiated phase enlarges ion transport pathways and lowers K+ migration barriers from 0.88 eV to 0.35 eV, ultimately yielding outstanding rate performance (74% capacity retention at 30C) and stable cycling (98% over 500 cycles at 5C), across a wide temperature range (−20 to 55 °C).
Collectively, these advances in cathodes, anodes, electrolytes, and solid-state interfaces highlight how SPHIBs address the inherent trade-offs between energy density and reaction kinetics. By strategically combining the structural-stabilizing role of Na+ with the fast transport properties of K+, multi-ion carrier systems offer a unified design strategy for realizing durable, high-performance, and thermally robust energy storage solutions.
3.1.2.2. Na–Al HIBs. With a theoretical specific capacity of 2980 mAh g−1 and a volumetric capacity of 8040 mAh cm−3, Al3+ offers a significantly higher energy density than monovalent ions such as Li+ and Na+, making it a highly attractive component for high-energy hybrid systems.49 In addition, the natural abundance and low cost of aluminum further enhance the feasibility of SAHIBs for large-scale energy storage applications. Despite these advantages, the practical implementation of SAHIBs faces major obstacles due to low reduction potential (−1.68 V vs. SHE) of Al3+, which leads to severe hydrogen evolution in aqueous electrolytes. This issue necessitates the use of stable non-aqueous electrolytes. However, the design of such electrolytes presents a complex challenge, as it requires an optimal balance between ionic conductivity, electrochemical stability, and interfacial compatibility with the anode further complicated by the metal's high reactivity.
To address these barriers, Sun et al.50 developed a pioneering SAHIB system composed of an aluminum anode, a sodium vanadium phosphate (Na3V2(PO4)3, NVP) cathode, and a hybrid electrolyte of NaAlCl4 and 1-ethyl-3-methylimidazolium chloride (EMImCl) with aluminum chloride. This system leverages the complementary electrochemical roles of Al3+ and Na+ ions. At the anode, Al3+ undergoes reversible deposition and stripping, while Na+ ions are simultaneously extracted and inserted at the cathode. The specific electrochemical reactions are as follows.
Cathode:
| 3Na3V2(PO4)3–6e− ↔ 3NaV2(PO4)3 + 6Na+ | (1) |
Anode:
| 8NaAl2Cl7 + 6Na+ + 6e− ↔ 2Al + 14NaAlCl4 | (2) |
The coordinated operation of these two processes enables a discharge voltage of 1.25 V, a cathode capacity of 99 mAh g−1, and a coulombic efficiency of 98% over extended cycling. By integrating high-capacity multi-electron redox chemistry of Al3+ with stable intercalation behavior of Na+ ions, this SAHIB design effectively balances high energy density with electrochemical stability, achieving a performance synergy that is difficult to realize in conventional single-ion systems. Moreover, the customized hybrid electrolyte plays a crucial role in maintaining interfacial stability and suppressing degradation, demonstrating the importance of electrolyte formulation in facilitating efficient and compatible multi-ion transport.
3.1.2.3. Na–Mg HIBs. Magnesium is an appealing anode material for hybrid-ion systems due to its high volumetric capacity (3833 mAh cm−3) and low reduction potential (−2.37 V vs. SHE). In SMHIBs, the dual-cation strategy leverages the high theoretical capacity and dendrite-free deposition behavior of Mg2+ alongside the fast intercalation kinetics and redox stability of Na+, yielding a balanced and efficient multi-ion storage system.51 Despite these synergistic advantages, the development of SMHIBs faces critical challenges. The inherently sluggish solid-state diffusion of Mg2+ in most cathode materials and the limited anodic stability of existing electrolytes result in rapid capacity fading and poor rate capability, ultimately restricting the practical viability of these systems.
In early efforts to address these issues, Dong et al.52 reported a hybrid system consisting of a Mg metal anode, a Berlin Green (FeFe(CN)6) cathode, and a dual-salt electrolyte containing both Mg and Na salts. The cell design exploited the 0.36 V thermodynamic redox potential difference between the Mg2+/Mg and Na+/Na couples, which allowed preferential Mg deposition at the anode while enabling reversible Na deintercalation at the cathode. The specific reaction of this study is as follows:
Cathode:
| FeFe(CN)6 + 2Na+ + 2e− ↔ Na2FeFe(CN)6 | (3) |
Anode:
| Mg + AlCl4− ↔ ½[Mg2Cl2]2+ + AlCl3 + 2e− | (4) |
The resultant SMHIBs delivered an average discharge voltage of 2.2 V, a reversible capacity of 143 mAh g−1, and an energy density of 135 Wh kg−1, demonstrating the feasibility of dual-cation transport. Building on this foundation, Li et al.53 advanced SMHIB design with a Na-intercalated NaV2(PO4)3 cathode and a hybrid Mg–Na electrolyte (Fig. 4g). During discharge, Na+ intercalation into NaV2(PO4)3 synergized with magnesium dissolution (Fig. 4h), achieving a maximum operating voltage of 2.6 V vs. Mg and 86% capacity retention at 10C. The tailored electrolyte formulation, free of excess Cl− ions, effectively prevented NaCl formation and improved Mg plating/stripping reversibility, as confirmed by cyclic voltammetry (Fig. 4i). These advances highlight the pivotal role of electrolyte design in enhancing multi-ion transport efficiency and interfacial stability.
Building upon previous work, Wang et al.54 developed a Mg2+/Na+ hybrid electrolyte system designed to enable synergistic co-intercalation in a Mn-NVO//PTCDI full cell. In this design, the fast diffusion kinetics of Na+ effectively compensate for the sluggish solid-state transport of Mg2+, thereby enhancing both structural stability and rate performance. As a result, the battery delivers a high specific capacity of 249.9 mAh g−1, operates stably at 1.75 V, and achieves an impressive cycle life exceeding 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, effectively overcoming key challenges that have long impeded the development of Mg-based batteries. Moving beyond conventional polyanionic frameworks, researchers have increasingly focused on transition metal chalcogenides, such as sulfides (e.g., FeS2) and selenides (e.g., FeSe2), due to their inherently large interlayer spacing and robust structural resilience. For instance, Zhou et al.55 demonstrated a ternary CoSe/NiSe2/CuSe2 cathode paired with a sodium bis(trifluoromethanesulfonyl)imide (NaTFSI)-based electrolyte in triglyme, achieving a reversible capacity of 115.5 mAh g−1 after 2000 cycles. In a related approach, a layered FeSe2 cathode operated with a bi-ionic [Mg2Cl2][AlCl4]2/NaTFSI electrolyte in 1,2-dimethoxyethane (DME) delivered a specific capacity of 154 mAh g−1, corresponding to an energy density of 260.9 Wh kg−1 after 800 cycles.56 This system also exhibited outstanding Mg|Mg symmetric cell performance, maintaining an overvoltage below 50 mV for more than 500 hours, and demonstrated practical applicability in pouch cells, delivering 347.7 mAh at 0.2 A g−1 over 160 cycles.
000 cycles, effectively overcoming key challenges that have long impeded the development of Mg-based batteries. Moving beyond conventional polyanionic frameworks, researchers have increasingly focused on transition metal chalcogenides, such as sulfides (e.g., FeS2) and selenides (e.g., FeSe2), due to their inherently large interlayer spacing and robust structural resilience. For instance, Zhou et al.55 demonstrated a ternary CoSe/NiSe2/CuSe2 cathode paired with a sodium bis(trifluoromethanesulfonyl)imide (NaTFSI)-based electrolyte in triglyme, achieving a reversible capacity of 115.5 mAh g−1 after 2000 cycles. In a related approach, a layered FeSe2 cathode operated with a bi-ionic [Mg2Cl2][AlCl4]2/NaTFSI electrolyte in 1,2-dimethoxyethane (DME) delivered a specific capacity of 154 mAh g−1, corresponding to an energy density of 260.9 Wh kg−1 after 800 cycles.56 This system also exhibited outstanding Mg|Mg symmetric cell performance, maintaining an overvoltage below 50 mV for more than 500 hours, and demonstrated practical applicability in pouch cells, delivering 347.7 mAh at 0.2 A g−1 over 160 cycles.
These advancements demonstrate that multi-ion carrier strategies - through the integration of tailored cathode architectures, well-designed hybrid electrolytes, and coordinated Mg2+/Na+ transport mechanisms - can effectively balance energy density, rate capability, and cycling stability, thereby overcoming the inherent limitations of single-ion systems.
To overcome these limitations, potassium-based hybrid-ion batteries (PHIBs) have emerged as a promising alternative, offering a transformative approach that integrates multiple types of charge carriers. By harnessing the synergistic interactions among different ions, PHIBs can accelerate charge transfer kinetics, broaden the operational voltage window, and improve overall ion transport efficiency.58 This multi-ion carrier strategy not only alleviates the diffusion-related constraints of K+ ions but also unlocks new opportunities for engineering advanced energy storage systems. A landmark advancement demonstrating the potential of multi-ion carrier systems is the development of dual-metal hybrid-ion architectures. For example, Wang et al.59 reported a potassium–zinc hybrid-ion battery (PZHIB) employing a rhombohedral Fe0.35Mn0.65[Fe(CN)6] (FeMnHCF) cathode paired with a zinc metal anode. This innovative configuration achieved a low overpotential of just 0.2 V and a high charge voltage exceeding 1.9 V, resulting in an energy efficiency greater than 89%, which was further improved to 93% through K+-mediated voltage plateau stabilization. The FeMnHCF cathode exhibited rapid Zn2+ storage kinetics and excellent charge retention, enabling stable cycling over 1000 cycles. This work exemplifies how the multi-ion approach can synergistically combine the fast kinetics of divalent ions with the interfacial benefits of K+, effectively reconciling high energy density with long-term durability. Beyond dual-metal systems, multi-ion carrier strategies are also reshaping the design landscape of KIBs in aqueous environments. Hua et al.60 developed an aqueous K+/H+ hybrid-ion alkaline battery (PHHIB) using a 30% KOH electrolyte, where K+ and H+ function as co-charge carriers. The battery employed a potassium nickel hexacyanoferrate (KNi[Fe(CN)6]) cathode and nitrogen-doped mesoporous carbon sheet anode, achieving a high cathode capacity of 232.7 mAh g−1 at 100 mA g−1, minimal self-discharge, and superior rate capability. The concentrated alkaline environment effectively mitigated ion competition, allowing the full cell to deliver 93.6 mAh g−1 at 100 mA g−1 while retaining 82.4% of its capacity after 1000 cycles at 2000 mA g−1. These results further underscore the versatility and promise of multi-ion strategies in enhancing reaction kinetics, cycling performance, and scalability of potassium-based energy storage systems.
These representative studies underscore the transformative potential of PHIBs, in which the strategic integration of multiple ionic species effectively overcomes the intrinsic diffusion and interfacial limitations of conventional monovalent-ion batteries. By strategically harmonizing diverse ionic species, PHIBs exemplify how multi-ion carrier systems can redefine electrochemical mechanisms, optimize electrode–electrolyte interfaces, and pave the way for sustainable, high-efficiency alternatives to conventional battery chemistries.
3.1.4.1. Mg–Li HIBs. Magnesium lithium hybrid ion batteries (MLHIBs) represent a promising class of multi-ion energy storage systems that synergistically integrate the high energy density and fast ion transport kinetics of LIBs with the intrinsic safety, high volumetric capacity, and dendrite-free cycling of MIBs. Although LIBs currently dominate the energy storage market, their reliance on limited lithium resources and growing safety concerns underscore the urgency of developing alternative technologies. In contrast, MIBs offer clear advantages in terms of cost, abundance, and safety. However, their application is limited by the sluggish diffusion and poor reversibility of divalent Mg2+ ions, which result in low power density and restricted material compatibility.
As early as 2006, Gofer et al. provided key experimental evidence that adding lithium chloride (LiCl) to magnesium electrolyte solutions can significantly enhance their ionic conductivity.63 This finding was pivotal in addressing a fundamental limitation of magnesium-based systems, namely the low ionic mobility of bulky and strongly coordinated Mg2+ species in conventional non-aqueous solvents. It also laid an important foundation for the development of MLHIBs, which aim to combine the fast redox kinetics of Li+ with the high capacity and stable storage behavior of Mg2+ within a single electrochemical system. Further advancing multi-ion carrier systems, Yagi et al. proposed a polyvalent-metal storage battery system based on a dual-salt electrolyte containing both a lithium salt and a Mg, Ca, or Al salt (Fig. 5a).64 In this system, positive electrodes commonly used in lithium-ion batteries, such as LiFePO4 or LiCoO2, are combined with polyvalent metal anodes, enabling the design of high-voltage cells operating in the range of 3–4 V. In recent years, advances in MLHB technology have been driven by three critical research fronts: (i) the design of cathode materials that can accommodate both Li+ and Mg2+ while maintaining structural stability and rapid ion diffusion; (ii) the development of compatible anode materials capable of enabling reversible Mg2+/Li+ storage and mitigating interfacial passivation; and (iii) the engineering of hybrid electrolyte systems that ensure efficient dual-cation transport, suppress side reactions, and stabilize electrode–electrolyte interfaces. The following sections will provide an in-depth overview of recent progress in these three key areas.
 | ||
| Fig. 5 (a) Schematic diagram of a Mg–Li HIB based on a dual-salt electrolyte system. Reproduced with permission from ref. 64. Copyright 2014, Royal Society of Chemistry. (b) Energetic stabilities of the occupations of available interstitial sites in Mo6S8 by Mg2+ and Li+ along various Mg2+/Li+ (mixed)-insertion paths calculated by DFT. Reproduced with permission from ref. 67. Copyright 2014, American Chemical Society. (c) Schematic diagram of Mg–K HIBs structure. (d) Typical charge–discharge profiles of KMHCF using different electrolytes. (e) Cycling stability of KMHCF-Mg HIBs using different electrolytes at the current density of 20 mA g−1. Reproduced with permission from ref. 80. Copyright 2024, Springer Nature. (f) Schematic diagram of Mg–Al HIBs structure. Reproduced with permission from ref. 82. Copyright 2019, American Chemical Society. (g). Scheme depicting the speciation in the as-prepared and conditioned magnesium aluminum chloride complex electrolyte solution. Reproduced with permission from ref. 416. Copyright 2015, American Chemical Society. | ||
3.1.4.1.1. Advances in cathode materials. Cathode development in MLHIBs has strategically aimed to overcome the intrinsic kinetic limitations of Mg2+ ions - particularly their sluggish solid-state diffusion and strong electrostatic interactions with host lattices while harnessing the fast intercalation kinetics of Li+ to simultaneously achieve high power and energy densities. Early efforts to directly adopt conventional LIB cathodes, such as layered transition metal oxides, proved largely incompatible with Mg2+ due to poor ion mobility and limited reversibility. This challenge prompted a pivotal shift toward the exploration of nanostructured architectures and hybrid materials designed to facilitate multi-ion transport.
A key advancement was demonstrated by Cen et al.65 who employed ultrathin VO2(B) nanosheets as cathodes within an MLHIB configuration consisting of a Mg anode, a Mo current collector, and an all-phenyl complex/LiCl electrolyte. The VO2(B) cathode delivered a high reversible capacity of 275 mAh g−1 and excellent cycling stability. This study not only improved Li+ insertion kinetics but also addressed Mg2+-related kinetic bottlenecks through intelligent current collector selection, demonstrating the potential of VO2(B) as a viable hybrid cathode material for high-performance MLHIBs. Simultaneously, Chen et al.66 investigated NH4V4O10 microflower-like nanostructures as cathode materials. Benefiting from high ionic conductivity and favorable Li+ mobility, this material delivered 228 mAh g−1 capacity and retained 72.3% after 100 cycles at 100 mA g−1. Its nanostructured design effectively mitigated the common issues of poor cycling stability and slow ion diffusion, reinforcing the critical role of nanoscale engineering in enhancing MLHIB performance. To directly tackle the sluggish diffusion of Mg2+, Si et al. introduced a dual-cation co-insertion strategy using Cu3VS4, allowing simultaneous incorporation of both Li+ and Mg2+ into the host lattice. This approach significantly suppressed structural degradation typically associated with Mg2+ intercalation and improved rate capability, highlighting the effectiveness of deliberate multi-ion accommodation within a robust host framework.
Beyond vanadium oxides, transition metal sulfides and selenides have emerged as promising cathode candidates. For example, Cho et al.67 systematically explored the electrochemical behavior of hybrid Mg2+/Li+ rechargeable batteries through a combination of theoretical modeling and experimental validation across both atomistic and macroscopic scales. Using DFT calculations, they elucidated the thermodynamic landscape of Mg2+ and Li+ co-insertion into Mo6S8 cathode materials, identifying a critical Li+ activity threshold at which lithiation becomes more favorable than magnesiation in pristine Mo6S8 (Fig. 5b). This fundamental insight into ion insertion preference provides a theoretical foundation for tuning the electrochemical response of Chevrel phase hosts. Moreover, by tailoring the insertion chemistry via a dual-salt electrolyte formulation, the study achieved remarkable rate performance, with a capacity retention of 93.6% at 20C and 87.5% at 30C under ambient conditions. These results highlight the potential of electrolyte engineering and ion-chemistry control in enabling high-power MLHIBs. Diem et al.68 developed a self-supporting, binder-free V2O5 nanofiber cathode that simplified electrode fabrication while simultaneously reducing internal resistance, addressing longstanding challenges commonly encountered in hybrid systems. While the material's electronic conductivity remained suboptimal, proposed enhancements such as graphene/rGO integration or doping strategies offered a clear path for further optimization. Pushing further into interfacial engineering, Shu et al.69 constructed VS4/MoS2 heterojunctions as cathodes. This design significantly enhanced both structural robustness and electron/ion conductivity, leading to improved cycling stability and rate performance. Most recently, Zhang et al.70 achieved notable progress with the development of a VMS@CC-2 cathode, which delivered exceptional energy density, rapid charge–discharge capability, and long-term cycling stability. This work exemplifies the integration of rational design principles, including defect modulation and interface optimization, toward realizing high-voltage, high-energy MLHIB systems.
Collectively, these studies underscore the critical importance of hybrid material architectures, nanoscale engineering, multi-ion diffusion strategies, and interfacial tuning in overcoming the intertwined challenges of sluggish Mg2+ kinetics, structural degradation, and achieving high-voltage stability in multi-ion carrier systems.
3.1.4.1.2. Advances in anode materials. Achieving high-performance anodes in MLHIBs requires innovative strategies to bridge the kinetic mismatch between fast Li+ intercalation and sluggish Mg2+ diffusion, while simultaneously preventing structural degradation during cycling. Conventional graphite anodes are fundamentally unsuitable because Mg2+ has a high de-solvation energy exceeding 2 eV and experiences strong electrostatic repulsion within the graphite's narrow interlayer spacing of 3.35 Å, which necessitates the development of alternative materials with engineered nanoarchitectures that facilitate ion transport.
MXenes have emerged as versatile ion-hosting matrices through their tunable surface chemistry and gallery spacing. Byeon et al.71 were among the first to explore Ti3C2Tx MXenes (Tx = –O, –OH, –F) as dual-cation anodes. Their metallic conductivity (∼6000 S cm−1) and hydrophilic surfaces enabled synchronous Li+/Mg2+ co-intercalation with minimal lattice distortion, as confirmed by in situ XRD. The system delivered 90% capacity retention over 500 cycles at 0.5C in a MgCl2/AlCl3–LiCl hybrid electrolyte. This work revealed the dynamic interlayer expansion of MXenes during multi-ion storage and suggested that selective surface functionalization (e.g., O-rich terminals) could enhance Mg2+ affinity and guide ion transport hierarchies. Building on this foundation, Li et al.72 introduced a composite strategy by incorporating cetyltrimethylammonium bromide (CTAB) into Ti3C2 MXenes via electrostatic self-assembly. The introduction of CTAB expanded the interlayer spacing from 12.8 Å to 22.3 Å, while concurrent nitrogen doping (5.2 at%) enhanced the electronic conductivity by approximately 300%. The resulting dual-modified anode delivered a specific capacity of 115.9 mAh g−1 at 0.1 A g−1 and exhibited exceptional cycling stability, with a minimal capacity decay rate of only 0.02% per cycle over 1000 cycles. DFT calculations further revealed a substantial reduction in the Mg2+ diffusion barrier, from 1.2 eV in graphite to 0.38 eV in the modified MXene structure. This approach underscores the importance of synergistic spatial and electronic modulation in enabling efficient multivalent ion storage and suggests that similar design principles could be extended to emerging MBenes, such as Mo2B, which are theoretically projected to achieve capacities exceeding 200 mAh g−1 due to the high multivalent ion affinity of boron-based frameworks.
Heterointerface engineering has also proven effective in overcoming Mg2+ transport limitations. Gao et al.73 constructed epitaxial Cu2Se/CoSe heterojunctions via hydrothermal selenization, where a 0.5 nm lattice mismatch between CoSe nanosheets and Cu2Se nanoparticles created an internal electric field (0.8 eV offset). Operando X-ray absorption near edge structure (XANES) revealed charge redistribution that accelerated Mg2+ de-solvation kinetics, enabling a high capacity of 320 mAh g−1 at 0.1 A g−1, with 91% of the charge storage attributed to pseudocapacitive behavior. This architecture enabled full-cell MLHIBs to achieve 143 Wh kg−1 energy density. The strategy opens avenues for gradient heterostructures to homogenize ion flux and suppress interfacial strain. In parallel, electrolyte–anode interactions have been increasingly recognized as a decisive factor in enabling multi-ion reversibility. Tsai et al.74 designed a deep eutectic solvent (DES) composed of urea–MgCl2–LiClO4 (3:1:1 molar ratio), with 0.5% H2O as an additive. Raman spectroscopy revealed strong Mg2+/urea (160 cm−1 shift) and Li+/ClO4− coordination, leading to high ionic conductivity (8.7 mS cm−1). When coupled with a perylene diimide/reduced graphene oxide anode, the system achieved a capacity of 139.8 mAh g−1 at 0.03 A g−1 with nearly 100% coulombic efficiency, outperforming conventional electrolyte systems. Their findings suggest that ligand-exchange mechanisms and redox-active anions (e.g., closo-B12Cl122−) can simultaneously enhance ion conductivity and contribute to anion-based storage. System-level integration has further validated the promise of multi-ion designs. Pei et al.75 constructed full MLHIBs using a Mg foil anode and a VO2 cathode in an all-phenyl complex (APC)/LiCl electrolyte. In situ NMR confirmed Li+-facilitated Mg2+ plating/stripping, while the VO2 cathode maintained a stable 1.75 V discharge plateau through V4+/V3+ redox reactions. This configuration achieved a record energy density of 427 Wh kg−1 at 55 °C. However, Mg anode passivation remains a key challenge. The development of artificial hybrid SEI incorporating LiF–MgF2 nanocomposites may provide a viable route to synchronize interfacial multi-ion kinetics.
Collectively, these advances converge on three key pillars of innovation that include atomic-scale ion-channel engineering at interlayer and interface levels to address kinetic asymmetry, electrolyte solvation environment design to regulate ion transfer behavior, and integrated optimization of electrodes, electrolytes, and device architecture, which together pave the way toward achieving multi-ion batteries with energy densities approaching 500 Wh kg−1.
3.1.4.1.3. Advances in electrolyte materials. Electrolyte innovation is a cornerstone in advancing MLHIBs, where the primary challenge is to simultaneously achieve rapid Mg2+ and Li+ transport, maintain interfacial compatibility with Mg anodes, and ensure stable operation at high voltages. Cheng et al.76 pioneered a dual-salt electrolyte strategy tailored for Mg/LiFePO4 and Mg/LiMn2O4 systems. Their electrolyte formulation leveraged the high redox potential of Mg2+ (2.4 V versus Mg/Mg2+) alongside the fast kinetics of Li+, resulting in cell voltages exceeding 2.5 V, which represents an increase of more than 30% compared to conventional Mg-based electrolytes. Raman spectroscopy analysis confirmed the suppression of magnesium anode passivation through the formation of a [Mg2(μ-Cl)3·6THF]+ complex, which contributed to achieving a specific energy density of 246 Wh kg−1. This study revealed the delicate balance between high-voltage stability and interfacial kinetics, suggesting that optimized salt ratios could further reduce concentration polarization and improve reversibility.
To further address kinetic limitations, Pei et al.75 developed a MLHIB system that employed VO2 as the cathode, magnesium metal as the anode, and an all-phenyl complex (APC) combined with LiCl as the electrolyte. Operando electrochemical impedance spectroscopy (EIS) measurements revealed a 60% reduction in charge-transfer resistance at VO2 cathodes compared to APC-only systems. This enhancement, attributed to Li+ acting as a “kinetic mediator”, led to a record energy density of 427 Wh kg−1 with over 95% capacity retention after 100 cycles at 55 °C. The results highlight the underexplored potential of leveraging Li+ transport to compensate for sluggish Mg2+ kinetics and suggest future exploration of anion-functionalized Li salts (e.g., BF4−) to simultaneously improve conductivity and suppress Cl−-induced corrosion. Expanding on interfacial engineering, Ding et al.77 introduced a corrosion-resistant APC–LiTFSI hybrid electrolyte, where substituting Cl− with TFSI− reduced Mg anode overpotential by 220 mV and increased ionic conductivity to 8.2 mS cm−1 – 40% higher than APC–LiCl. Synchrotron XANES analysis at NiCo2S4 cathodes revealed that TFSI− promoted the formation of a stable Li+-conductive SEI, which facilitated cooperative Mg2+/Li+ insertion. The result was a highly stable system: symmetric Mg|Mg cells showed <50 mV polarization over 500 hours, and full cells delivered 204.7 mAh g−1 for 2600 cycles with only 0.003% decay per cycle. This study underscores the importance of anion electronegativity in tuning multi-ion interfacial behavior and opens the door to exploring fluorinated anions (e.g., FTFSI−) for further improving oxidative stability above 3.5 V.
Pushing electrolyte concentration boundaries, Yang et al.78 developed an asymmetric water-in-salt electrolyte (WiSE) featuring a high Mg2+ to Li+ molar ratio imbalance of 1 to 10 at 20 M total salt concentration. This formulation expanded the electrochemical stability window to 3.2 V, exceeding the typical limit of conventional aqueous batteries. Operando XRD and DFT simulations revealed a coupled ion-exchange mechanism, where Mg2+ displaced Li+ from the cathode (ΔG = −1.8 eV), while Li+ rapidly diffused through a perylene-based anode. This “Li+-for-Mg2+ relay” enabled a high reversible capacity of 144.1 mAh g−1 at 1 A g−1 and 92% capacity retention after 500 cycles. Such asymmetric designs present exciting opportunities for integrating ion-selective membranes in quasi-solid-state systems to decouple Mg2+/Li+ transport pathways.
Collectively, these advances underscore electrolyte design in MLHIBs as a multidimensional optimization challenge. Dual-salt chemistries offer tunable voltage profiles, anion selection critically influences interfacial charge-transfer kinetics, and asymmetric ion concentrations enable the creation of ion-specific transport and reaction pathways. Looking ahead, achieving further breakthroughs will require atomic-level control of solvation environments, potentially by integrating fluorinated co-solvents or metal–organic frameworks. This approach aims to harmonize the transference numbers of Li+ and Mg2+ while simultaneously stabilizing hybrid solid–electrolyte interphase layers under dynamic multi-ion flux conditions.
3.1.4.2. Mg–K HIBs. Magnesium potassium hybrid ion batteries (MPHIBs) represent a significant advancement in multi-ion carrier battery technology, as they effectively overcome the key limitations of conventional magnesium-based systems. Traditional magnesium ion electrolytes, such as Mg[B(HFIP)4]2, offer advantages like cost efficiency, high energy density, and safety. However, their narrow electrochemical windows (less than 3.2 V) and inherent corrosivity limit the operating voltages of hybrid systems. This limitation is reflected in the relatively low output voltages of Mg–Li and Mg–Na HIBs, which achieve only about 2.5 V and 1.4 V, respectively. In contrast, MPHIBs overcome these challenges by leveraging the synergistic interaction between Mg2+ and K+ ions. Specifically, K+ ions provide rapid diffusion kinetics and enable high-voltage stability, significantly enhancing the overall performance of the hybrid system.
The development of Mg–K systems was initially explored in 2020; Wang et al.79 reported a pioneering aqueous MPHIB, utilizing a mixed electrolyte containing both Mg and K ions alongside a novel porous cathode structure. This study introduced potassium ions into the Mg-based battery system, aiming to address the inherent sluggish kinetics of multivalent Mg2+ transport. By leveraging the synergistic effect between Mg2+ and K+ carriers, the battery demonstrated significantly enhanced rate capability and cycling stability, highlighting the improvements in electrolyte stability and cathode materials. The use of potassium manganese hexacyanoferrate (KMHCF) cathodes marked a notable step forward, as they enabled the co-migration of Mg2+ and K+ ions in ether-based electrolytes, contributing to improved voltage output and cycling stability in MPHIBs. Building on this foundation, Fu et al.80 recently achieved a significant leap forward by pairing KMHCF cathodes with tantalum (Ta) current collectors (Fig. 5c). This strategic design yielded a high discharge plateau of 3.0 V and an impressive energy density of 360 Wh kg−1, surpassing the performance of comparable Mg–Li and Mg–Na systems. A key factor in this performance improvement was the use of KTFSI-enhanced electrolytes, which optimized both voltage output and reversibility, as demonstrated by comparative voltage profiles (Fig. 5d and e). These results suggest that multi-ion carrier strategies, by combining the high theoretical capacity of Mg2+ with the favorable ionic mobility of K+, offer an effective approach to addressing the voltage limitations and cycling instability commonly observed in conventional single-ion systems.
3.1.4.3. Mg–Al HIBs. Magnesium aluminum hybrid ion batteries (MAHIBs) represent a promising class of multivalent ion hybrid energy storage systems that harness the synergistic transport and storage behavior of both Mg2+ and Al3+ ions. These two multivalent metals are highly attractive candidates for next-generation rechargeable batteries because of their high theoretical capacities of 3579 mAh g−1 for Al3+/Al and 2205 mAh g−1 for Mg2+/Mg, along with low standard reduction potentials of −1.66 volts for Al3+/Al and −2.37 V for Mg2+/Mg versus the SHE.81 However, the practical deployment of Mg- and Al-based batteries has been significantly hindered by the lack of compatible electrolytes that can simultaneously enable efficient and reversible metal plating/stripping while suppressing side reactions such as passivation, dendrite formation, and electrolyte decomposition.
A growing body of research on MAHIBs has underscored the importance of electrolyte speciation in governing electrochemical performance. In particular, adjusting the stoichiometric ratio of AlCl3 to MgCl2 to 1:1 in ether-based solvents such as tetraethylene glycol dimethyl ether (TEGDME) effectively suppresses the formation of electrochemically unstable AlCl2+ cations. This adjustment prevents parasitic cementation reactions, in which aluminum is spontaneously deposited at the expense of magnesium metal, thereby mitigating magnesium corrosion, enhancing coulombic efficiency, and eliminating capacity fading without the need for conditioning cycles (Fig. 5f).82 These findings reveal that Al3+ plays a dual role in multi-ion systems: beyond acting as an active charge carrier, it modulates interfacial chemistry and improves Mg2+ transport by altering local solvation and coordination environments.
Building on this understanding, earlier studies conducted using MgCl2/AlCl3 complexes in ether solvents like tetrahydrofuran (THF) and TEGDME further revealed that electrochemical reversibility is highly sensitive to electrolyte composition. For instance, systems with a Mg:Al molar ratio of 2:1 required conditioning cycles to remove inactive [AlCl4]− and [Al2Cl7]− species, which otherwise led to parasitic Al deposition. Raman spectroscopy and pair distribution function analysis confirmed the in situ formation of [Mg2(μ-Cl)3·6THF]+ clusters during conditioning (Fig. 5g), which not only restored active Al3+ availability but also promoted uniform Mg2+ plating by enhancing Cl− surface coverage and modifying the electrode–electrolyte interface.83 These complementary findings demonstrate how precise electrolyte engineering in MAHIBs enables the transformation of interionic complexity into functional synergy, reinforcing the critical role of controlled speciation in optimizing multi-ion carrier battery systems.
Multi-ion carrier systems address these limitations by exploiting the synergistic interplay between Mg2+ and Al3+ ions, as demonstrated by Pagot et al.84 through their innovative ionic liquid-based Al/Mg hybrid electrolyte [Pyr14Cl/(AlCl3)1.5]/(δ-MgCl2)x. The incorporation of Mg2+ into the system shifted the Al3+ reduction potential from −1.66 V to −2.37 V, representing a negative shift of 0.71 V. This shift facilitated alloy-type Al plating and stripping at substantially lower overpotentials. This dual-cation effect not only enhances energy density but also reduces kinetic and thermodynamic barriers traditionally limiting the individual use of Mg2+ or Al3+. Electrochemical testing in symmetric Mg|electrolyte|Mg cells showed remarkable cycling stability with a coulombic efficiency of 99.66% and an overvoltage of <50 mV over 700 cycles. Stripping-deposition profiles on Pt electrodes further confirmed the anodic stability of the electrolyte up to +2.35 V vs. Mg/Mg2+. Structural characterization revealed a complex ionic network comprising Pyr14+, Cl− AlCl4−, Al2Cl7−, and solvated Mg2+ species, which collectively enable efficient, stable transport of both multivalent ions.
By integrating the complementary physicochemical characteristics of Mg2+ and Al3+, such as their distinct ionic radii, charge densities, and solvation behaviours, MAHIBs establish a synergistic ion transport mechanism. This multi-ion strategy not only enhances energy density through high-valence charge storage but also improves ionic conductivity and structural stability, thereby achieving a well-balanced performance in terms of energy output, rate capability, and long-term cycling stability.
At the core of these strategies lies the deliberate introduction of auxiliary charge carriers, both cationic (such as Li+, Na+, and K+) and anionic (such as PF6−, BF4−, BH4−), which dynamically modulate the local electrochemical environment to enhance the overall behavior of Ca2+. These secondary ions do not serve merely as supporting species; rather, they play active roles in tailoring electrolyte solvation structures, stabilizing interfacial phases, and improving redox reaction kinetics. For instance, small and fast-diffusing cations like Li+ can restructure the solvation sheath around Ca2+, reducing its effective de-solvation energy and thereby improving ionic mobility. Concurrently, carefully selected anions contribute to the formation of a stable, ionically conductive SEI, mitigating calcium surface passivation. More importantly, these auxiliary ions can also facilitate redox-active processes at the cathode, especially in complex conversion-type systems, by participating in co-intercalation or catalytic pathways.
Several representative systems have demonstrated the effectiveness of multi-ion synergy in overcoming the intrinsic limitations of Ca-based batteries. A particularly notable example is the development of a carbon-confined covalent sulfur cathode, synthesized via plasma-enhanced C/S co-deposition.86 This nanostructured cathode architecture not only immobilized soluble polysulfide intermediates but also provided a conductive matrix to facilitate redox reactions. It was coupled with a dual-salt electrolyte system composed of LiPF6 and Ca(BF4)2 dissolved in carbonate solvents. The resulting Ca–S battery exhibited a high reversible capacity of 824.6 mAh g−1 along with excellent cycling stability over 145 cycles. Mechanistically, this performance was enabled by the cooperative roles of the two cationic species. Li+ ions actively participated in the sulfur redox process, improving reaction kinetics and promoting more complete and reversible sulfur conversion. In parallel, Ca2+ served as the primary charge carrier, supporting overall ionic conductivity and contributing to the system's energy density.
In a related study, Meng et al.87 reported a Ca–FeS2 battery utilizing a hybrid electrolyte composed of Ca(BH4)2 and LiBH4 dissolved in tetrahydrofuran (THF). In this system, Li+ ions acted as the catalyst for the FeS2 conversion reaction and reduced the de-solvation barriers of Ca2+, while BH4− anions effectively suppressed interfacial resistance, thereby facilitating improved Ca ion transport. This combination enabled a reversible capacity of 303 mAh g−1 with a coulombic efficiency of 96.7% over 200 cycles. The results underscored the cooperative functionality of both cationic and anionic species within the electrolyte in enhancing redox kinetics and interface compatibility. The impact of multi-ion strategies extends beyond the bulk electrolyte to interfacial engineering. Song et al.88 developed an artificial hybrid SEI composed of nanocrystalline Na+/Ca2+ carbonates and CaH2/Ca3N2 species. This engineered interlayer acted as an ion-conductive yet electron-insulating interface, effectively stabilizing the calcium metal surface. When combined with an electrolyte enriched in PF6− and K+ ions, the system achieved ultra-stable Ca plating/stripping behaviour over 1400 hours with minimal polarization, demonstrating the power of multi-ion coordination in constructing robust interfacial chemistries. Further demonstrating the power of reciprocal ion synergy, Zhou et al.89 introduced a dual-cation Ca/Na–S battery system based on a Ca(PF6)2/NaPF6 electrolyte. In this configuration, Na+ ions enhanced the conversion kinetics of calcium polysulfides at the cathode, while Ca2+ played a complementary role in buffering Na+ flux at the anode, thus mitigating dendrite formation. This cooperative interaction resulted in a high reversible capacity of 947 mAh g−1 and excellent cycling stability at ambient temperature.
Collectively, these pioneering advancements compellingly illustrate the central and indispensable role of multi-ion carrier strategies in unlocking the full electrochemical potential of Ca-ion hybrid batteries. By meticulously tailoring electrolyte compositions to incorporate synergistic ion pairs (e.g., Ca2+, Li+, Na+, K+, PF6−, and BH4−), researchers have systematically addressed core limitations pertaining to ionic mobility, redox kinetics, and interface stability. Looking ahead, the continued exploration of cooperative ion interactions, potentially guided by advanced computational modelling and in situ characterization techniques, combined with the development of ion-selective membranes and chemically stable hybrid electrolytes, is expected to accelerate progress toward high-voltage, high-capacity, and long-life multivalent energy storage systems.
In this context, researchers have strategically introduced auxiliary cations such as Mn2+, Li+, Na+, K+, Ca2+, Mg2+, and Al3+, along with functional anions like I−, SO42−, and [Zn(OH)4]2−, to construct advanced ZIHB systems. These additional ionic species do not merely act as inert co-solvates but instead engage actively in regulating redox dynamics, restructuring electrode–electrolyte interfaces, and modulating solvation structures. Their cooperative interactions facilitate synergistic ion transport, promote interfacial stability, and accelerate charge-transfer kinetics. By enabling multi-dimensional control over electrochemical processes, such multi-ion configurations significantly surpass the capabilities of conventional single-ion systems. They enhance ionic conductivity, stabilize cycling behaviour, and mitigate degradation pathways, thereby improving the overall performance of ZIHBs. Within this framework, Zn serves not only as the primary charge carrier but also as a central element orchestrating complex ionic synergies. The following subsections will explore representative ZIHB chemistries where these multi-ion strategies have delivered transformative improvements.
3.1.6.1. Zn–Mn HIBs. Zn–Mn hybrid ion batteries (ZMHIBs) represent one of the most historically entrenched and technically versatile battery chemistries, with roots extending back to the seminal Leclanché cell (Zn–MnO2–NH4Cl) introduced in 1868 and subsequently evolved into dry cell formats by Hellesen in 1886.92 Originally developed as primary (non-rechargeable) systems, ZMHIBs underwent a significant transformation with the advent of alkaline secondary battery technologies, enabling their deployment in rechargeable applications. In conventional aqueous Zn–Mn configurations, metallic Zn functions as the anode, oxidizing to Zn2+ during discharge, while MnO2 at the cathode undergoes reduction accompanied by Zn2+ intercalation.93 This electrochemical interplay is reversed during charging, where Zn2+ deintercalated from the cathode and is electrodeposited back onto the anode. While these systems have demonstrated admirable safety and cost-effectiveness, challenges such as sluggish Zn2+ kinetics, Mn dissolution, and interfacial incompatibility continue to restrict their energy density and long-term cycling stability.
To address these intrinsic limitations, researchers have explored multi-ion carrier strategies by incorporating additional active ionic species such as H+, Mn2+, I−, and SO42−, which work in concert with the native Zn2+ transport mechanism.94–97 This multidimensional ionic orchestration serves not merely to support ion conduction but actively modifies redox mechanisms, stabilizes electrode interfaces, and decouples energy storage from rate performance. The integration of auxiliary ions within the Zn–Mn framework exemplifies the broader paradigm shift toward hybridized ion chemistries, wherein multiple carriers operate synergistically to resolve longstanding bottlenecks in electrochemical reversibility and performance degradation.
A compelling demonstration of this principle is embodied in the design of a polyvinylpyrrolidone (PVP)-intercalated MnO2 hybrid superlattice featuring expanded (001) planes, which selectively accommodate H+via the Grotthuss mechanism.98 This structural innovation not only facilitates fast H+ transport via a hydrogen-bonded network, but also leverages the hybrid superlattice configuration to enhance charge delocalization and electronic entropy. Within this framework, the simultaneous intercalation of Zn2+ and H+ plays distinct yet complementary roles: H+ contributes to ultrafast charge transfer kinetics, while Zn2+ ions serve as the principal charge carriers, enabling high energy storage capacity. This decoupling of electrochemical functions, in which H+ enhances power density while Zn2+ contributes to energy density, effectively overcomes the conventional trade-off between high-rate capability and large capacity. Building on the notion that redox activity itself can be mediated through auxiliary species, a hybrid cathode combining Mn2+ and I− ions has been developed, leveraging the redox couple I3−/I− to facilitate the Mn2+/MnO2 conversion process in a buffered acetate electrolyte.99 The redox sequence proceeds as follows:
Cathode:
| I2 + 2e− → 2I− | (5) |
Anode:
| MnO2 + 3I− + 4H+ → Mn2+ + 2H2O + I3− | (6) |
| I3 + 3e− → 3I− | (7) |
| Zn–2e− → Zn2+ | (8) |
The concept of ionic asymmetry is further explored in a hybrid Zn–Mn redox flow battery, which integrates an acidic Mn2+/MnO2 redox couple with an alkaline Zn/[Zn(OH)4]2− pair.100 In this architecture, the anolyte and catholyte are physically separated but electrochemically coupled, allowing for independent optimization of redox kinetics and ionic stability. The introduction of [Zn(OH)4]2− effectively suppresses Zn dendrite formation, while the use of Ni–Mg catalysts and Bi-embedded carbon felt electrodes enhances the reversibility of the Mn redox reaction. This configuration delivers an impressive operating voltage of 2.75 V and achieves an energy efficiency of 89.8% over 150 cycles, establishing a strong case for redox-asymmetric, multi-ion electrolyte systems in flow battery platforms. Complementary advances in quasi-solid-state electrolytes further underscore the role of solvation engineering in multi-ion systems. Incorporating kaolinite, a layered clay mineral with strong water-binding capacity, into the hybrid electrolyte matrix not only confines free H2O but also facilitates the enrichment of SO42− anions within the solvation sheath, thereby enhancing the ionic environment for multi-ion transport.101 This tailored solvation structure mitigates side reactions and dendrite growth on the Zn anode, enhances Zn2+ flux uniformity, and improves interfacial compatibility. Paired with an α-MnO2 cathode, the resulting battery delivers a high specific capacity of 348 mAh g−1 at 0.2 A g−1 and retains 87% of its capacity after 500 cycles at 0.5 A g−1, with minimal degradation over 100 cycles, demonstrating the benefits of controlled ionic microenvironments in hybrid systems.
A major obstacle for aqueous Zn systems is the stark mismatch between the pH environments favored by their two electrodes. MnO2 delivers its highest reversible capacity through proton-coupled conversion reactions that proceed most efficiently in mildly acidic electrolytes. In contrast, metallic Zn corrodes rapidly in acid, evolving hydrogen and forming soluble Zn salts; it deposits smoothly and with a low overpotential only in alkaline media. To resolve this conflict, researchers have devised pH-decoupled architectures that allow each electrode to reside in its preferred medium while still sharing a common external circuit.102 A representative design employs three liquid compartments: an acidic catholyte in contact with the MnO2 cathode, an alkaline anolyte bathing the Zn metal, and a neutral potassium-sulfate reservoir sandwiched between them (Fig. 6a).103 Cation-and anion-selective membranes flank the central reservoir, permitting only targeted ion migration. During discharge, K+ ions drift toward the anolyte to balance the electrons consumed at the Zn electrode, whereas SO42− ions move toward the catholyte to neutralize the protons consumed by MnO2. This bidirectional ion shuttle maintains electroneutrality without allowing corrosive species to cross over. By keeping each electrode in its ideal pH window, the three-compartment cell achieves a thermodynamic voltage of 2.83 V and an energy density of about 1622 Wh kg−1 based on the mass of MnO2, far surpassing conventional single-electrolyte Zn batteries. Performance is further enhanced by integrating a recirculation loop that continuously refreshes each electrolyte stream, thereby suppressing concentration gradients at the electrode surfaces and enabling fast charge–discharge operation with minimal polarization.104
 | ||
| Fig. 6 (a) Working mechanisms of a three-chamber decoupled Zn–MnO2 battery system. Reproduced with permission from ref. 103. Copyright 2020, Springer Nature. (b) Comparison of operating voltage and specific capacity for various cathode materials employed in ZIHBs. Reproduced with permission from ref. 105. Copyright 2020, Wiley. (c) Schematic illustration of the redox process for the NiHCF//Zn battery during the charge–discharge processes. Reproduced with permission from ref. 119. Copyright 2016, Elsevier (d) schematic illustrations of the energy storage mechanism of Na(VO)2(PO4)1.5F0.5. Reproduced with permission from ref. 117. Copyright 2022, Royal Society of Chemistry. | ||
In summary, the strategic integration of multiple charge carriers in ZMHIBs has emerged as a transformative approach for overcoming the limitations traditionally associated with Zn–MnO2 chemistries. The cooperative roles of H+, Zn2+, Mn2+, I−, [Zn(OH)4]2−, and SO42− in these systems enhance both redox kinetics and energy storage capacity, while simultaneously enabling exceptional architectural versatility across a wide range of configurations, including solid-state, flow, and hybrid electrolyte formats. Through these synergistic interactions, multi-ion systems unlock performance metrics that are unachievable through single-ion pathways alone.
To address the challenge of achieving reversible Zn2+ intercalation and extraction in cathode materials, which is primarily hindered by strong electrostatic interactions between Zn2+ ions and the host crystal lattice in conventional ZIBs, novel rechargeable Zn-based hybrid batteries have been developed. These systems typically pair a Zn metal anode with traditional cathode materials used in other metal-ion batteries. In such configurations, Zn2+ ions undergo reversible plating and stripping at the anode, while alternative metal ions are reversibly intercalated and deintercalated at the cathode. Based on the nature of the participating metal ions, reported ZIHBs can be broadly classified into six categories: Zn–Li hybrid batteries, Zn–Na hybrid batteries, Zn–K hybrid batteries, Zn–Mg hybrid batteries, Zn–Ca hybrid batteries, and Zn–Al hybrid batteries (Fig. 6b).105 In the following sections, we systematically introduce and discuss each of these dual-cation Zn-based hybrid battery systems.
3.1.6.2. Zn–Li HIBs. Despite significant progress in ZMHIBs, the pursuit of higher energy density, broader operational stability, and improved electrochemical reversibility has spurred interest in alternative hybrid configurations. Among them, Zn–Li hybrid ion batteries (ZLHIBs) have emerged as a promising platform that leverages the complementary strengths of Zn and Li-based chemistries. Zn provides high theoretical capacity, low cost, and natural abundance, while Li+ enables access to high-voltage cathodes with well-established intercalation mechanisms.106 The coexistence of Zn2+ and Li+ as a dual charge carrier not only expands the electrochemical window but also mitigates key challenges associated with aqueous Zn-based systems, such as hydrogen evolution at the Zn anode, limited ionic mobility in aqueous media, and poor low-temperature performance. Importantly, the incorporation of Li+ introduces access to higher-voltage cathodes and more diverse redox chemistries, while Zn2+ maintains system safety and cost-effectiveness.
Recent advances in ZLHIBs have centered around three representative lithium-based cathode materials: LiFePO4 (LFP), LiMn2O4 (LMO), and Li3V2(PO4)3 (LVP). Across these systems, electrolyte engineering has proven critical to unlocking the full potential of the Zn2+/Li+ dual-cation framework. Strategies such as modulating water activity, tailoring solvation structures, extending electrochemical stability windows, and facilitating selective ion transport have been key to enabling stable and efficient operation. This section highlights how the integration of multi-ion coordination with precisely engineered electrolyte environments can effectively guide the development of next-generation, high-performance ZLHBs.
A particularly illustrative case of such hybridization is found in ZLHIBs employing LFP as the cathode. Here, the challenge lies in harmonizing the dual-cation transport mechanisms while maintaining the stability of both electrodes in an aqueous environment. Electrolyte engineering has proven central to this endeavour, particularly in controlling water activity and solvation behaviour. For instance, Su et al.107 introduced a polyacrylamide (PAM)-modified aqueous electrolyte, which reduces the activity of free water molecules, thereby suppressing parasitic hydrogen evolution at the Zn anode and stabilizing the interfacial environment. In this system, Li+ serves as the primary intercalating ion at the LFP cathode, while Zn2+ cycles at the anode. The tailored electrolyte facilitated 82% capacity retention after 350 cycles at 0.5 A g−1, demonstrating the efficacy of water regulation in sustaining dual-cation transport. Building on this approach, Ciurduc et al.108 employed molecular crowding electrolytes (MCEs) using polyethylene glycol (PEG400) to restructure the solvation environments of both Zn2+ and Li+, leading to substantial reduction in self-discharge rates and improved cycling stability. Recognizing the vulnerability of aqueous systems at low temperatures, Yan et al. designed a hygroscopic bilayer PEG/bacterial cellulose (BC) electrolyte.109 This innovative electrolyte architecture serves to reduce water molecule activity and simultaneously improve ionic conduction for both Li+ and Zn2+ at sub-zero temperatures. Their Zn–LFP hybrid cells employing this electrolyte retained 85.14% of their initial capacity after 300 cycles at −20 °C, while achieving near 100% coulombic efficiency, underscoring the benefits of dual-cation configurations in extreme environments.
The implementation of LMO cathodes in ZLHIBs presents additional challenges due to intrinsic instability of LMO under aqueous or alkaline conditions, often resulting in cathode dissolution and Zn anode passivation. To address these issues, a phase-separated electrolyte (PSE) system composed of immiscible aqueous and organic phases was developed, incorporating ionic liquids such as Py14TFSI in fluoroether (HFE) solvents.110 This biphasic system significantly extended the electrochemical stability window (ESW) to 4.6 V by physically segregating water from the LMO cathode. As a result, Zn–LMO batteries achieved high-voltage platforms up to 3.12 V and discharge voltages of 2.95 V and 2.75 V, ultimately delivering energy densities exceeding 360 Wh kg−1. Moreover, the strategy proved adaptable to other lithium-based cathodes, including LiNi0.5Mn1.5O4 and LiCoMnO4, where average working voltages reached 3.41 V and 3.37 V, respectively. These results underscore how selective phase partitioning can be employed to spatially manage redox species and stabilize dual-cation transport in high-voltage aqueous systems.
Complementary innovations in solvation structure engineering have further enhanced Zn–LMO hybrid systems. Jaumaux et al.111 utilized a solvent system based on N-methylformamide (NMF), co-dissolving lithium triflate and zinc triflate to create a uniform solvation environment conducive to both Li2+ and Zn2+ transport. This hybrid electrolyte expanded the electrochemical stability window to 3.25 V and enabled highly stable cycling with 99.7% coulombic efficiency over 400 cycles. In parallel, efforts to address anode-side passivation led Tao et al.112 to develop carbon-doped porous Zn anodes that enhance ion diffusion and reduce interfacial resistance, improving initial discharge capacities to 150 mAh g−1 and retaining 60 mAh g−1 over 210 cycles.
Further extending the multi-ion carrier concept, recent attention has turned to LVP-based cathodes, where polyanionic frameworks traditionally suffer from poor kinetics. To mitigate this, Jiang et al.113 employed Mn2+ doping to enhance both conductivity and structural stability within the LVP host, reporting a Zn//Li3V1.94Mn0.06(PO4)3 hybrid system with an initial capacity of 106.5 mAh g−1 and excellent rate and cycling performance. In a more architecturally advanced approach, Liu et al.114 developed a hierarchical “scarf-blanket” structure in LiVPO4F (LVPF) cathodes, combining carbon nanotube networks with polypyrrole coatings to improve electron transport and structural cohesion. The specific reaction of this study is as follows:
Cathode:
| 2LiVPO4F–2e−–2Li+ → 2VPO4F | (9) |
Anode:
| Zn–2e− → Zn2+ | (10) |
Collectively, these advances underscore the growing potential of Zn–Li hybrid systems as a fertile ground for exploiting multi-ion carrier mechanisms. By partitioning ionic functionalities across coordinated electrolyte–electrode architectures and fine-tuning solvation dynamics for both Zn2+ and Li+, ZLHBs have begun to transcend the conventional limitations of aqueous and non-aqueous battery systems alike. Through innovations in electrolyte formulation, cathode stabilization, anode surface engineering, and interfacial control, these systems achieve a sophisticated orchestration of multiple charge carriers, enabling high-voltage operation, broad temperature adaptability, and long-term cycling robustness.
3.1.6.3. Zn–Na HIBs. Zn Na hybrid ion batteries (ZNHIBs) have emerged as a promising strategy to overcome the inherent limitations of conventional aqueous ZIBs by harnessing the complementary electrochemical characteristics of Zn2+ and Na+ ions. In these systems, a Zn metal anode is paired with a sodium-based cathode, enabling a dual-cation mechanism in which each ion plays a distinct functional role. Zn2+ serves as the primary charge carrier at the anode, offering high theoretical capacity and low redox potential, while Na+ participates in the cathodic redox process, contributing to improved structural reversibility and reaction kinetics (Fig. 6c). Specifically, the Na+ shuttle enhances cathode stability and supports faster ion transport, while the presence of Zn2+ ensures high energy density and compatibility with aqueous systems. Altogether, the integration of these two ion species not only expands the electrochemical stability window but also enhances overall battery efficiency.
A defining feature of ZNHIBs is the use of Na-hosting cathodes, which critically determine the electrochemical behaviour of the full cell. PBAs, NASICON-type compounds, and other Na+-intercalation hosts have been widely explored for this role. PBAs, such as FeFe(CN)6, NiHCF (nickel hexacyanoferrate), and ZnHCF (zinc hexacyanoferrate), have garnered particular interest due to their robust open-framework structures that accommodate reversible Na+ intercalation. However, their performance is often constrained by structural defects, such as lattice vacancies and interstitial water, as well as low specific surface areas. Moreover, the strong coulombic interaction between divalent Zn2+ and water molecules can interfere with Na+ diffusion or trigger undesirable side reactions, undermining long-term cycling stability.
Cathode:
| C–FeFe(CN)6 + 1.65Na+ + 1.65e− → C–Na1.65FeFe(CN)6 | (11) |
Anode:
| 0.82Zn → 0.82Zn2+ + 1.65e− | (12) |
Overall:
| C–FeFe(CN)6 + 1.65Na+ + 0.82Zn → C–Na1.65FeFe(CN)6 + 0.82Zn2+ | (13) |
To mitigate these limitations, considerable efforts have been directed toward structural refinement and defect suppression in PBAs cathodes. A notable example involves the synthesis of C–FeFe(CN)6via a citric acid chelation approach, which significantly reduced defect density and improved surface area, leading to enhanced ion transport and electrochemical stability.115 This system achieved a high-capacity retention of 88% after 500 cycles, with the redox reaction governed by a dual-cation mechanism in which Na+ intercalates into the cathode while Zn undergoes reversible stripping and plating at the anode. The specific reaction of this study is as follows:
Similarly, monoclinic NiHCF has demonstrated a near-theoretical capacity along with enhanced long-term reversibility, further validating the feasibility of its application in hybrid Zn–Na systems.116
Beyond PBAs, NASICON-type materials offer an attractive platform for Zn–Na hybridization, owing to their intrinsically high Na+ mobility, robust three-dimensional frameworks, and structural resilience during repeated intercalation/deintercalation cycles. Despite these advantages, typical NASICON materials often operate at relatively low voltages (below 0.8 V vs. Zn/Zn2+), and aqueous instability becomes a concern, particularly for high-voltage vanadium-based NASICON cathodes. Strategies to overcome these limitations have included introducing high-voltage inductive centers, such as PO43−, F−, and PO4F4−, and adopting alternative compositions like monoclinic Li3V2(PO4)3 or rhombohedral Na3V2(PO4)3. These approaches have successfully raised redox potentials while maintaining structural integrity. A recent study on sodium fluorophosphate oxovanadium materials (Nax(VO)2(PO4)yFz), synthesized via solvothermal routes, exemplified the benefits of this strategy (Fig. 6d).117 These cathodes exhibited a specific capacity of 87.2 mAh g−1 and exceptional rate performance. XRD analyses during charge–discharge cycles confirmed the reversible intercalation of both Na+ and Zn2+, validating the co-storage mechanism fundamental to ZNHIBs. Complementing these efforts, NASICON-type M3V2(PO4)3 (where M = Li, Na) cathodes have also demonstrated dual-cation conduction behaviour when paired with Zn anodes, achieving capacity retention above 84% after 200 cycles.118 The coexistence of Na+ and Zn2+ within the electrolyte enables a dual-cation conduction mechanism, further reinforcing the multi-ion carrier concept as a transformative strategy for achieving enhanced electrochemical stability and performance.
In addition to cathode optimization, electrolyte engineering plays an equally critical role in advancing ZNHIB performance. Lu et al.119 demonstrated that modifying the electrolyte composition by introducing ZnSO4 markedly improved Na+ intercalation kinetics and significantly stabilized the electrochemical interface, especially when paired with a graded porous Zn anode. Cyclic voltammetry results revealed pronounced cathodic responses when Na+ and Zn2+ coexist, highlighting their cooperative redox activity in the hybrid system. A novel solvation structure engineered within concentrated aqueous electrolytes was shown to drastically suppress hydrogen evolution and inhibit dendrite formation on the Zn surface.120 This electrolyte optimization extended the Zn plating/stripping lifespan to 1600 hours with an impressive coulombic efficiency of 99.96% sustained over 700 cycles. Such advancements underscore the crucial role of electrolyte design in multi-ion carrier systems, as effective control over solvation structures directly impacts the stability and reversibility of both Na+ and Zn2+ redox processes.
By capitalizing on the distinct electrochemical properties of alkali metal ions like Na+ in conjunction with Zn2+, the multi-ion carrier strategy offers a viable pathway for addressing Zn anode challenges while simultaneously enhancing energy density and cycling stability in ZNHIBs. The incorporation of PBA and NASICON-type materials as cathodes has yielded significant improvements in energy density, rate performance, and long-term durability.
3.1.6.4. Zn–K HIBs. Zinc potassium hybrid batteries (ZPHIBs) have attracted growing interest for large-scale energy storage due to their dual-cation mechanism. By coordinating the transport of Zn2+ and K+ ions, ZPHIBs overcome limitations of conventional systems, offering improved rate capability, greater voltage stability, and longer cycle life. These advantages help mitigate challenges such as slow ion diffusion, voltage fluctuations during operation, and long-term material degradation in traditional battery technologies.
Despite these intrinsic benefits, challenges persist in fully realizing the potential of ZPHIBs, including the need to enhance ionic conductivity within electrode materials for both Zn2+ and K+, suppress parasitic reactions such as hydrogen evolution at the Zn anode, and minimize cathode dissolution or structural degradation that can cause capacity fade. The design and optimization of multi-ion carrier cathodes, particularly those based on PBAs, have emerged as pivotal to advancing ZPHIB performance. PBAs such as copper hexacyanoferrate (CuHCFe), potassium manganese ferrocyanide (KCM), Fe0.35Mn0.65[Fe(CN)6] (FeMnHCF), and Ni–Zn hexacyanoferrate (Ni3Zn1HCF) offer open-framework crystal structures ideally suited for reversible K+ intercalation, thereby facilitating efficient charge storage and ion diffusion. Nonetheless, intrinsic challenges such as capacity limitations due to Mn3+ dissolution in KCM and structural defects in Ni3Zn1HCF continue to constrain their electrochemical performance.
Research has therefore focused on strategic modifications, including cation substitution within the PBA lattice to improve stability, electrolyte formulation tuning to optimize ion transport and suppress side reactions, and synthesis control aimed at enhancing crystallinity and reducing defects. A representative advancement was demonstrated by Liu et al.121 who developed a thermally assisted ZPHIB utilizing a CuHCFe cathode and Zn metal anode in a hybrid K/Zn-ion aqueous electrolyte. This system achieved a notable discharge voltage of 1.75 V and exhibited a temperature coefficient of −0.97 mV K−1, indicative of stable and efficient cycling even under thermal fluctuations. The enhanced ion mobility of K+ within the electrolyte was key to maintaining electrochemical performance at low temperatures, though elevated temperatures still posed stability challenges. In a parallel effort, Yu et al.122 engineered a partially copper-substituted KCM cathode, effectively mitigating Mn3+ dissolution and elevating the cathode's working potential. Their ZPHIB displayed an operating voltage of 1.75 V and superior rate performance, alongside a marked suppression of Zn dendrite formation. This dendrite inhibition was attributed to an electrostatic shielding effect imparted by K+ ions, which likely adsorb onto or accumulate near the Zn anode surface, modulating local electric fields to homogenize Zn2+ deposition and prevent localized current density spikes responsible for dendrite nucleation. This illustrates an important role for auxiliary ions in multi-ion carrier systems beyond charge storage; K+ here exerts a non-redox, physical stabilization effect critical for anode longevity.
Further illuminating the multi-ion interaction mechanism, Wang et al.59 explored the rhombohedral FeMnHCF cathode, which exhibited excellent Zn2+ and K+ storage capabilities. The FeMnHCF framework facilitates the intercalation of both guest ions, enabling hybrid charge storage behavior. Their investigations, including cyclic voltammetry in both pure Zn2+ and mixed Zn2+/K+ ions electrolytes, revealed that K+ acted as the primary charge carrier within the FeMnHCF framework. This was confirmed by the superior rate performance and decreased ion diffusion barriers observed when K+ was present, resulting in an impressive energy efficiency of 93%. The reduced overpotential and enhanced reversibility associated with K+ intercalation were central to this outcome, demonstrating how judicious multi-ion carrier selection can substantially elevate battery efficiency.
These studies collectively underscore the transformative potential of ZPHIB systems, where the synergistic interaction between Zn2+ and K+ ions leads to markedly enhanced battery performance. Zn2+ contributes high capacity and benefits from reduced dendrite formation due to the presence of K+, while K+ offers fast diffusion kinetics and low intercalation overpotential, resulting in improved overall electrochemical behavior. The strategic use of multi-ion carriers, exemplified by the Zn2+/K+ combination, provides substantial improvements in energy efficiency, voltage stability, and cycling durability.
3.1.6.5. Zn–Mg HIBs. Zinc magnesium hybrid batteries (ZMgHIBs) combine the complementary electrochemical properties of Zn2+ and Mg2+. In this system, Zn2+ offers fast redox kinetics and high reversibility in aqueous electrolytes, while Mg2+ contributes a high volumetric capacity, abundant natural reserves, and low cost. However, the ZMgHIBs face notable challenges, including the poor reversibility of Mg2+ intercalation, the sluggish diffusion kinetics due to the divalent nature and high charge density of Mg2+, and the dissolution of active materials that can degrade long-term performance. Addressing these limitations requires coordinated advances in both electrolyte formulation and cathode material design. In particular, mixed ZnSO4–MgSO4 electrolytes have garnered attention as a practical and scalable solution for enabling dual-cation transport while stabilizing electrode interfaces, thereby facilitating co-intercalation of Zn2+ and Mg2+ into cathode frameworks.
A representative demonstration of this concept was presented by Zhang et al., who synthesized MgxV2O5·nH2O (MgVO) nanobelts and systematically investigated their electrochemical behavior across electrolytes with varying Zn2+ to Mg2+ molar ratios.123 Among the tested formulations, the 1.0 M ZnSO4–1.0 M MgSO4 electrolyte yielded the best performance, achieving a specific capacity of 374 mAh g−1 at 100 mA g−1 and retaining 90.3% of its initial capacity after 200 cycles at 1 A g−1. Structural analysis revealed that both Zn2+ and Mg2+ were co-intercalated into the MgVO cathode. Notably, Mg2+ ions acted as structural “pillars” within the layered V2O5 host, enhancing the mechanical integrity of the lattice during repeated cycling. This finding illustrates a quintessential advantage of multi-ion carrier systems, where one ion (Mg2+) primarily serves a stabilizing, structural role, while the other (Zn2+) contributes more actively to charge storage and fast transport.
Cathode:
| Mg(1−x)Mn2O4 + yMg2+ + (x − y)Zn2+ → Mg[(1−x)+y]Zn(x−y)Mn2O4 + xe− | (14) |
Anode:
| Zn → Zn2+ + 2e− | (15) |
Building upon this multi-ion paradigm, further progress has been made using spinel-type cathodes such as MgMn2O4 (MMO), a tetragonal derivative of the widely studied ZnMn2O4. While MMO offers the potential for high Mg2+ storage, its practical performance has been constrained by two critical factors: the slow lattice diffusion of Mg2+ and the structural instability associated with Jahn–Teller distortions of Mn3+. To circumvent these issues, Soundharrajan et al.124 introduced Mn2+ directly into the Zn2+/Mg2+ dual-salt electrolyte. This modification not only promoted reversible co-insertion of Mg2+ and Zn2+ into the MMO cathode but also contributed to structural stabilization through in situ Mn2+ participation. The overall reaction mechanism of the ZMgHIBs involves Zn oxidation at the anode and coupled dual-cation insertion into the MMO lattice:
This ZMgHIB system delivered a remarkable energy density of 370 Wh kg−1, representing the highest value reported at the time for any spinel cathode across both aqueous and non-aqueous ZIB systems.
These examples from ZMgHIB research underscore the multidimensional benefits of employing a multi-ion carrier strategy in aqueous battery systems. The simultaneous presence of Zn2+ and Mg2+, along with stabilizing ions such as Mn2+ in the electrolyte, leads to synergistic improvements in cathode structural stability, ion transport efficiency, and interfacial integrity. Mg2+ intercalation helps maintain the integrity of the host lattice and suppresses side reactions, while Zn2+ facilitates rapid charge transport and delivers high cycling reversibility, resulting in enhanced overall battery performance.
3.1.6.6. Zn–Ca HIBs. Zinc calcium hybrid batteries (ZCHIBs) have recently gained attention as a promising aqueous energy storage system, primarily by aiming to leverage the complementary electrochemical characteristics of Zn2+ and Ca2+ ions. Although Ca2+ is a divalent ion, it exhibits certain favorable transport properties in aqueous media, such as possessing the smallest Stokes radius and the lowest polarization strength among common multivalent cations. These intrinsic traits can afford high ionic mobility and rapid diffusion kinetics, making Ca2+ potentially well-suited for fast charge transport processes.
However, its practical application as a primary charge carrier in batteries is significantly hindered by its relatively large crystallographic ionic radius. This large size can induce substantial volume changes and consequent structural degradation in traditional host materials, such as layered oxides and PBAs, during repeated intercalation/deintercalation cycles. Additionally, calcium metal itself exhibits poor electrochemical reversibility when used as an anode in aqueous electrolytes, further limiting the development of stable Ca-based anodes for aqueous systems.
To address these inherent challenges, Zhao et al.125 developed a ZCHIB system using a NASICON-type Na3V2(PO4)3 (NVP) cathode. The NVP particles were surface-engineered with amorphous carbon and reduced graphene oxide (denoted as NVP@AC@rGO) to improve electrical conductivity and structural stability. This tailored cathode was paired with a Zn metal anode and operated in an aqueous electrolyte containing both Zn2+ and Ca2+, typically introduced as Zn(OTF)2 and Ca(OTF)2 salts. The electrochemical mechanism of the ZCHB involves the coordinated transport of Zn2+, Ca2+, and triflate (OTF−) anions. During charging, Zn is oxidized at the anode and plates back during discharge. Meanwhile, Ca2+ ions reversibly intercalate into and deintercalate from the NVP framework, as described by the redox reaction. The reaction can be described as:
Cathode:
| NaV2(PO4)3 + xCa2+ + 2xe− → CaxNaV2(PO4)3 | (16) |
Anode:
| xZn → xZn2+ + 2xe− | (17) |
Overall:
| NaV2(PO4)3 + xCa2+ + xZn → CaxNaV2(PO4)3 + xZn2+ | (18) |
The robust three-dimensional NASICON framework of NVP offers spacious diffusion channels alongside strong structural rigidity, which together enable effective accommodation of divalent Ca2+ ions while minimizing volumetric strain. The system demonstrated excellent cycling performance exceeding 1000 cycles, with fast kinetics arising from the dual-cation transport mechanism. In addition to its role in the cathode redox process, Ca2+ also plays a critical interfacial function at the Zn anode. Electrochemical characterization methods, such as cyclic voltammetry and rate capability tests, demonstrated that the inclusion of Ca2+ in the electrolyte substantially prolongs the lifespan of the Zn anode, increasing it by more than 20 times compared to systems without Ca2+. This enhancement is not due to faradaic involvement at the anode but rather to the formation of a Ca2+-enriched interfacial layer. Owing to its larger ionic radius and lower charge density relative to Zn2+, Ca2+ preferentially adsorbs onto the Zn surface, creating an electrostatic shielding layer. This layer effectively suppresses dendritic Zn growth and promotes uniform Zn deposition during repeated cycling, thereby enhancing both the safety and longevity of the battery. The dual role of Ca2+, acting both as a redox-active species at the cathode and as a non-redox stabilizing agent at the anode, exemplifies the versatility of auxiliary ions within multi-ion systems. Importantly, Zn2+ remains the primary charge carrier, ensuring high energy output, while Ca2+ enhances interfacial stability and structural reversibility.
Despite these advances, further optimization of ZCHIBs faces several challenges. Although NASICON-type cathodes provide a relatively stable structural framework, the repeated insertion and extraction of Ca2+ can still generate structural stress during extended cycling, particularly under high mass loading. In addition, competitive intercalation between Zn2+ and Ca2+ at the cathode introduces complexities in ion selectivity, diffusion dynamics, and long-term capacity retention.
3.1.6.7. Zn–Al HIBs. Aqueous Al ion batteries (AIBs) have garnered growing interest due to aluminum's earth abundance, low cost, and exceptional theoretical capacities, both gravimetric and volumetric. Al3+ possesses a highly negative standard potential (−1.67 V) and an extremely large hydration free energy (−4525 kJ mol−1), underscoring its high thermodynamic stability in aqueous solutions. The trivalent nature and strong hydration of Al3+ cause inherently sluggish electrochemical kinetics and pronounced parasitic hydrogen evolution reactions, especially at the anode. These issues result in rapid capacity fading and unstable electrode–electrolyte interfaces, limiting the practical application of AIBs as standalone systems.
To circumvent these intrinsic bottlenecks, a multi-ion carrier strategy has emerged as a powerful approach most notably realized in the form of zinc aluminum hybrid batteries (ZAHIBs). In these systems, the complementary roles of Zn2+ and Al3+ ions are deliberately harnessed to achieve a synergistic interplay of fast kinetics, interfacial stabilization, and enhanced cycling reversibility. Zn2+ ions, owing to their fast transport and weak solvation, serve as the main charge carriers, whereas Al3+ ions fine-tune interfacial chemistry, mitigate side reactions, and influence ion transport behaviour. This dual-cation approach illustrates the core principle of multi-ion carrier batteries, which is to overcome the limitations of single-ion chemistries by harnessing cooperative interactions between different ionic species.
One of the most illustrative implementations of this strategy is the use of an aluminum–urea hydrated eutectic electrolyte (AUHEE), prepared by mixing Al2(SO4)3·18H2O and urea in specific stoichiometric ratios.126 In this formulation, urea functions as a high-dielectric Lewis base that forms coordination complexes with Al3+, yielding solvated species of the form [Al(Urea)x(H2O)y]3+. This coordination environment weakens the strong Al3+–H2O interactions that typically drive HER and passivation layer (Al(OH)3) formation. Consequently, AUHEE not only mitigates gas evolution but also promotes electrochemical reversibility by altering the hydration structure of Al3+. When employed in a full-cell configuration comprising a V2O5 cathode and a Zn anode, the AUHEE electrolyte facilitates the in situ formation of a dynamic Al–Zn alloy layer at the anode–electrolyte interface during the charging process. In this dual-cation system, both Zn2+ and Al3+ ions actively participate in the redox reactions. At the cathode, Zn2+ and Al3+ are sequentially intercalated into the V2O5 lattice, contributing to multivalent charge storage. Meanwhile, the anodic reaction involves the reversible dissolution and reformation of the Al–Zn alloy. The overall electrochemical process can be described as follows:
Cathode:
| V2O5 + 0.32Al3+ + 0.96e− → Al0.32V2O5 | (19) |
| V2O5 + 0.29Zn2+ + 0.58e− → Zn0.29V2O5 | (20) |
Anode:
| Al–Zn alloy→ xAl3+ + yZn2+ + (3x + 2y)e− | (21) |
This dynamically evolving alloy interface plays a crucial role in reducing interfacial polarization, increasing ionic conductivity, and facilitating charge transfer across the electrolyte–anode boundary. The resulting ZAHIB achieves a specific capacity of 250 mAh g−1 with excellent stability over 1500 cycles. Importantly, this system exemplifies a key feature of multi-ion operation in which Zn2+ serves as the main charge carrier while Al3+ modulates solvation chemistry and interfacial behaviour, effectively decoupling the electrochemical functions of the coexisting ions. This represents a sophisticated form of in situ interfacial engineering driven by a multi-ion environment, wherein the system self-optimizes under electrochemical conditions to favor performance-enhancing interfacial transformations.
Despite these advances, Al3+ hydrolysis remains a concern. The formation of Al(OH)3 and H+ not only consumes active species but also leads to increased resistance and reduced coulombic efficiency. To address these interfacial instabilities, especially those occurring at the Zn anode, advanced electrode design strategies have been actively developed. One notable example is the development of a dual-layer anode comprising ZnCr2O4 coated with polyvinylidene fluoride (PVDF), denoted as PVDF@ZnCr2O4@Zn (PV@ZC).127 This bilayer architecture is engineered to achieve a balanced interfacial environment by combining the hydrophilic, nucleation-guiding properties of ZnCr2O4 with the hydrophobic, corrosion-resistant nature of PVDF. Compared to unmodified Zn or ZnCr2O4 (ZC) electrodes, the PV@ZC configuration exhibited enhanced thermodynamic stability, as indicated by a higher corrosion potential, and demonstrated an extended cycling lifespan of over 6100 cycles.
These findings reinforce the potential of ZAHIBs as a versatile platform for high-performance aqueous batteries. By combining the high-capacity and fast-kinetic contributions of Zn2+ with the interfacial modulation and solvation control enabled by Al3+, ZAHIBs offer a finely tuned electrochemical environment that addresses the core challenges of aqueous battery systems.
 | ||
| Fig. 7 (a) Schematic illustration and (b) current–voltage curves of the cathode and anode in an ammonium-ion hybrid battery. (c) Comparison of hydrated ionic radii for various ions. (d) Molecular model of the of NH4+. (e) Galvanostatic charge/discharge curves of the NH4+-ion hybrid battery and the assembled Zn2+-ion battery. Reproduced with permission from ref. 138. Copyright 2024, Royal Society of Chemistry. Panels illustrate representative configurations of proton hybrid batteries, in which H+ is co-stored or co-inserted with (f) Mn2+, reproduced with permission from ref. 142. Copyright 2022, Wiley. (g) Zn2+, reproduced with permission from ref. 146. Copyright 2024, Elsevier and (h) K+. Reproduced with permission from ref. 149. Copyright 2024, Royal Society of Chemistry. | ||
Unlike conventional metal-ion batteries, the electrochemical behavior of AmIBs is governed by the unique physicochemical properties of NH4+, which include a relatively small hydrated radius (Fig. 7c), fast diffusion kinetics in aqueous media, and the unprecedented ability to form directional hydrogen bonds (Fig. 7d). These hydrogen-bonding interactions occur both with surrounding water molecules and with oxygen- or nitrogen-containing groups within host electrode frameworks, enabling alternative modes of ion-host binding that go beyond simple coulombic attraction.130 This behavior introduces novel redox chemistry, facilitates tailored solvation structures, and unlocks specialized ion transport pathways, potentially including Grotthuss-like mechanisms, unavailable to traditional metal ions. The integration of NH4+ into multi-ion systems introduces a new dimension of ionic versatility, enabling the modulation of interfacial charge transfer dynamics, redox equilibria, and output voltage in ways that synergize with coexisting cationic or anionic species (Fig. 7b). Given this reversible ion-electron transfer mechanism, the electrochemical performance of AmIBs is primarily governed by the structural and chemical properties of the electrode materials.
Among cathode materials, polymorphs of MnO2 have been extensively investigated due to their structural versatility and favorable NH4+ intercalation behavior. Notably, α-MnO2 has demonstrated superior performance relative to its β- and γ-counterparts. Liu et al.131 systematically compared these polymorphs in both standard ammonium-ion batteries (AmIBs) and ammonium–sodium hybrid ion batteries (ASHIBs), confirming that α-MnO2 enables more efficient NH4+ intercalation and exceptional cycling stability. The system delivered a high specific capacity of 161.8 mAh g−1 across a 1.8 V voltage window and retained 99.7% capacity after 10000 cycles. Crucially, this performance arose from the cooperative redox involvement of NH4+ and Na+, while SO42− served as an electrostatic stabilizer within the electrolyte. The interfacial coordination and multi-ion charge transport processes demonstrated here highlight the potential of synergistic ion mechanisms to enhance electrochemical reversibility and energy output. Building on this multi-ion design principle, Gong et al.132 fabricated a free-standing VO2/rGO aerogel that exploits the interfacial electric field between VO2 and graphene to accelerate both charge transfer and ionic diffusion. The 3D porous structure supplies abundant active sites, and importantly, the field effect facilitates the reversible co-storage of NH4+ and H3O+, enabling an ultrahigh mass energy density. Similarly, Zhang et al.133 synthesized a layered Ni-BTA/VOH composite in which one-dimensional conductive MOFs (Ni-BTA) intercalate between VOH layers. This structural integration increases the interlayer spacing (from 12.9 Å to 13.8 Å), boosts conductivity, and stabilizes the layered framework. VOH contributes via reversible valence transitions of vanadium (V5+ ↔ V4+) and dynamic hydrogen bonding with NH4+, while Ni-BTA actively participates through reversible C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N to C–N bond conversion that electrostatically attracts NH4+. These cooperative multi-ion interactions effectively combine electronic and ionic processes for improved capacity and structural integrity.
N to C–N bond conversion that electrostatically attracts NH4+. These cooperative multi-ion interactions effectively combine electronic and ionic processes for improved capacity and structural integrity.
On the anode side, Bao et al.134 identified a V2CTx MXene as a high-performance material whose capacity is unlocked through a multi-ion-driven mechanism within an NH4Ac electrolyte. Here, NH4+ acts as the primary charge carrier while Ac− ions regulate the storage process. In situ electrochemical quartz crystal microbalance measurements reveal a two-step mechanism involving initial electrostatic adsorption of NH4+ followed by redox reactions between d-V2CTx and coordinated [NH4+(HAc)3] species. The Ac− anions mediate this process by forming transient [NH4+(HAc)3]⋯O coordination bonds, which enable reversible valence transitions and sustained pseudocapacitive behavior. This nuanced interplay between multiple ionic species underpins the enhanced energy storage capabilities of the system. In another compelling example of phase-engineered multi-ion optimization, Qi et al.135 developed 1T/2H-MoS2 nanoflowers as an anode material, demonstrating superior NH4+ storage performance. The metallic 1T phase improves electronic conductivity, while the stable 2H phase maintains structural robustness. DFT calculations confirm that the hybrid phase widens interlayer spacing (0.68 nm vs. 0.615 nm in 2H-MoS2) and lowers the diffusion energy barrier (0.84 eV vs. 0.96 eV), resulting in enhanced ionic mobility and stable cycling (76.22% retention over 5000 cycles at 1 A g−1). This study highlights how structural duality and multi-ion dynamics collectively contribute to advanced charge storage behavior.
To further boost energy output, Pan et al.136 reported the design of an amorphous nickel–cobalt double hydroxide (A-NiCo-DH) cathode prepared via electrochemical activation, which created abundant hydrogen vacancies and removed inactive interlayer anions. These structural modifications significantly improved NH4+ and H+ co-insertion, thereby enhancing both ionic conductivity and electronic transport. When paired with a Zn anode in a dual-cation configuration, the system delivered a high specific capacity of 280.6 mAh g−1 and achieved an energy density of 306 Wh kg−1 at a power density of 745.8 W kg−1. These performance metrics underscore the critical role of optimized multi-ion interactions in pushing the boundaries of aqueous battery technologies. Liu et al.137 introduced a covalent organic framework exhibiting 141 mAh g−1 specific capacity and 90% capacity retention over 8000 cycles. The charge storage mechanism is governed by reversible hydrogen bonding between carbonyl groups and NH4+, coupled with dynamic electron transfer within the framework. The well-defined porous channels facilitate rapid NH4+ transport, with capacitive surface-controlled processes dominating the charge–discharge behavior. This exemplifies the synergy of structural design and ion-specific interactions enabled by multi-ion participation.
Parallel to electrode material development, electrolyte engineering has played a critical role in amplifying the performance advantages of NH4+-based systems. In particular, concentrated ammonium salt electrolytes have been leveraged to fine-tune solvation structures and minimize interfacial energy barriers. Meng et al.138 demonstrated that increasing the molarity of ammonium acetate (CH3COONH4) in solution directly altered the solvation shell of NH4+, likely by decreasing its hydration number or modifying ion–solvent interactions. This change in the solvation environment reduced de-solvation energy barriers at the electrode interface and promoted faster NH4+ intercalation kinetics. Based on this principle, a hybrid NH4+–Zn2+ battery was constructed using a high-concentration electrolyte composed of 15 M CH3COONH4 + 2 M Zn(CH3COO)2. The charge/discharge profiles and corresponding differential capacity curves of the hybrid device reveal a prominent discharge plateau around 1.76 V, attributed to the high insertion potential of NH4+ relative to the Zn2+/Zn couple (Fig. 7e). This discharge voltage is significantly higher than that observed in conventional Zn-ion batteries, highlighting the electrochemical advantage conferred by NH4+ participation. In this configuration, NH4+ serves as the primary cathodic ion, while Zn2+ acts as the anodic charge carrier, and acetate anions provide ionic balance, ensuring stable electrochemical operation. Expanding on electrolyte innovation, Zou et al.139 developed high-entropy electrolytes by adding 1 mM each of Cu2+, Co2+, Ni2+, Zn2+, and Mn2+ to a 1 M (NH4)2SO4 solution, used in combination with a high-entropy Prussian blue analogue (N-HEPBA) cathode. These multivalent cations were shown to reversibly insert into N-HEPBA, selectively activating Fe, Cu, Co, and Mn redox sites to augment NH4+ storage. The system exhibited remarkable cycling stability over 1700 cycles at 0.2 A g−1 and 3500 cycles at 1 A g−1, validating the robustness of multi-ion-assisted energy storage pathways.
Collectively, these advancements in AmIBs and their hybrid variants affirm the immense potential of non-metallic multi-ion carrier systems in transforming aqueous energy storage. The unique hydrogen-bonding capabilities and rapid diffusion kinetics of NH4+, when synergistically paired with metallic ions such as Na+, Zn2+, and H+, enable the modulation of redox processes, solvation dynamics, and interfacial stability in ways not accessible to single ion chemistries.
In Mn2+–proton hybrid batteries, the use of H+ addresses the fundamental kinetic challenges posed by Mn2+, whose large ionic radius and high charge density impede rapid diffusion and intercalation dynamics. To exploit the fast proton transport pathway, Yang et al.142 systematically designed a series of quinone-based anode materials and evaluated their electrochemical behavior in acidic electrolytes, with a particular focus on proton-coupled charge storage mechanisms (Fig. 7f). Among these, tetramethylbenzoquinone (TMBQ) demonstrated a high proton diffusion coefficient (∼10−6 cm2 s−1) and a low migration energy barrier of 0.26 eV, indicating highly efficient proton transport within the TMBQ framework. To validate its practical applicability, a full cell was assembled using a TMBQ/reduced graphene oxide (rGO) composite anode and a MnO2@graphite felt (GF) cathode, paired with a mixed electrolyte of 0.5 M H2SO4 and 1 M MnSO4. The resulting TMBQ//MnO2 device achieved an impressive power density exceeding 20000 W kg−1 and demonstrated remarkable cycling stability, retaining 77% of its capacity after 4000 cycles at a current rate of 5C. These results emphasize the promise of molecularly engineered quinone materials for high-performance proton-based hybrid energy storage systems. Complementing this work, Li et al.143 developed a monoclinic VO2 nanorod-based system featuring a tunnel structure that permits reversible co-insertion of Mn2+ and H+ ions. They identified a major degradation mechanism stemming from high H+ activity, which accelerated vanadium dissolution. To mitigate this, a hydrogen-bonding reconstruction strategy was employed, wherein the electric double-layer structure at the VO2 interface was modulated using ethylene glycol (EG) as an additive. EG not only altered the solvation environment but also suppressed the surface accumulation of H+ and water molecules, thereby stabilizing the vanadium framework. The electrochemical reactions involved were:
Cathode:
| VO2 + xH+ + yMn2+ + (2y + x)e− → HxMnyVO2 | (22) |
Anode:
| HxMnyVO2 → VO2 + xH+ + yMn2+ + (2y + x)e− | (23) |
A similar co-storage concept has been demonstrated in Zn2+–proton systems, wherein the sluggish kinetics of hydrated Zn2+ ions (e.g., Zn(H2O)62+) are effectively offset by the presence of mobile H+. Ghosh et al.144 introduced 1,4,5,8-naphthalenetetracarboxylic dianhydride (NTCDA) as a redox-active anode host capable of reversibly accommodating both Zn2+ and H+ ions. When paired with an MnO2 cathode, the resulting hybrid batteries achieved a specific capacity of 41 mAh and an average output voltage of 0.80 V. Notably, the presence of H+ was critical in maintaining redox stability within the organic framework and enabling enhanced ion transport across the electrolyte–electrode interface.
The cooperative function of H+ also extends to inorganic electrode systems, particularly those involving engineered hydrogen-bond networks. Li et al.145 employed NH4+, which possesses structural similarities to H+ and the ability to form extensive hydrogen bonds, to pre-establish hydrogen-bonding networks within cathode materials such as α-V2O5 and MnO2. By modulating the NH4+ content, the resulting materials exhibited enhanced capacity and maintained favorable electrochemical performance even under high-rate conditions, highlighting the critical role of hydrogen-bonding networks in promoting both capacity and rate capability. Ex situ XRD analysis further confirmed a reversible structural evolution during cycling, as reflected by a shift of the (001) peak to a lower angle during discharge. This shift indicates interlayer expansion caused by the co-intercalation of H+ and Zn2+, which gradually returned to its original position upon charging. The resulting electrodes showed significant improvements in capacity retention and power density, reinforcing the advantages of engineering ion interactions at the molecular level.
Beyond tunnel-structured oxides, H+ co-storage has also been applied to heteroatom-doped bismuth chalcogenides. Xie et al.146 synthesized Cl-doped Bi2Te3 (CBT) via hydrothermal treatment and showed that chlorine incorporation improved electronic conductivity and enhanced Zn2+/H+ adsorption (Fig. 7g). DFT calculations supported the experimental results, indicating enhanced ion binding and electronic transport, which collectively yielded a high capacity of 305 mAh g−1 and excellent rate performance. Similarly, Chen et al.147 introduced sulfur into Bi2Te3 to produce S-doped Bi2Te3−x (SBT), which benefited from enhanced Zn2+ adsorption, improved conductivity, and a synergistic Zn2+/H+ co-storage mechanism. Layered MnO2 systems with large interlayer spacings (∼7 Å) further exemplify the utility of co-insertion architectures. Ding et al.148 designed a pre-intercalated phase, K0.36H0.26MnO2·0.28H2O (K36), that facilitated Zn2+ and H+ co-insertion, enabling a structural phase transition into mixed-layered KxHyZnzMnO2c2O and spinel ZnMn2O4 nanodomains during electrochemical cycling. These transformations revealed the capacity of multi-ion systems to dynamically reconfigure electrode structures for enhanced performance.
Extending the H+ co-insertion concept into alkaline media, recent efforts have explored K+–H+ hybrid systems (Fig. 7h), particularly through the integration of Ni-based Prussian blue analogues. Hua et al.149 synthesized Zn–Ni hexacyanoferrates (NixZnyHCF) with varied Ni:Zn ratios via a simple co-precipitation method. Unlike neutral electrolytes, in which the voltage output typically scales with the Zn content, only those materials featuring an optimal nickel-to-zinc ratio of three to one demonstrated significantly enhanced redox potentials when tested in 30% KOH. When coupled with an activated carbon anode, the K+/H+ hybrid batteries maintained excellent capacity over extended cycling and exhibited robust performance at elevated current densities, reflecting the complementary transport roles of K+, H+, and OH− ions in enhancing electrochemical dynamics.
Altogether, H+ hybrid batteries underscore the pivotal role of H+ as agile co-transporters in multi-ion battery systems. Whether functioning as kinetic enhancers, structural stabilizers, or facilitators of complex redox equilibria, H+ ions offer an unparalleled degree of functional tunability when synergistically coupled with metal cations across various electrode chemistries and electrolyte environments. Proton hybrid batteries, in particular, stand as a compelling embodiment of the multi-ion carrier strategy, bridging the high energy densities typically associated with fuel cells and the operational simplicity of rechargeable batteries.
3.2. Cation–anion hybrid batteries
Cation–anion hybrid batteries represent an advanced embodiment of the multi-ion carrier strategy, wherein both cations and anions as active charge carriers actively participate in charge storage processes. In these systems, cations typically undergo intercalation or redox reactions at the anode, while anions are reversibly accommodated at the cathode. This multi-ion mechanism not only expands the number of available ion transport pathways but also broadens the electrochemical voltage window. Representative platforms include configurations in which anions such as PF6−, TFSI−, ClO4−, Cl−, Br−, and I− operate in tandem with cations such as Li+, Na+, K+, NH4+, Ca2+, Mg2+, Zn2+, or Al3+. The cooperative migration of these ionic species accelerates redox kinetics, enables wider voltage ranges, and opens up new opportunities for designing tailored electrodes and electrolytes. Such coordination helps mitigate side reactions and improves overall electrochemical reversibility. As a result, cation–anion hybrid batteries have garnered significant interest for applications requiring high voltage, long cycle life, and enhanced safety, including grid storage and electric transportation, marking them as a cornerstone of the evolving multi-ion energy storage landscape. | ||
| Fig. 8 (a) Schematic comparison of charge transfer energy barriers at the cathode between conventional “rocking-chair” batteries and dual-ion batteries. (b) Comparative evaluation of the practical performance of representative energy storage systems (supercapacitors, lead–acid batteries, sodium-ion batteries, lithium-ion batteries, lithium metal batteries, and dual-ion batteries) under both room temperature and low-temperature conditions. Reproduced with permission from ref. 150. Copyright 2024, Wiley. (c) Overview of various DIB configurations, highlighting the diversity of employed cationic and anionic charge carriers. (d) Theoretical specific capacities and corresponding operating voltages of different anode materials. Reproduced with permission from ref. 153. Copyright 2023, American Chemical Society. (e) Summary of potential strategies for electrolyte modification to enhance electrochemical performance. | ||
Recent research has therefore focused on the systematic optimization of the three core components, namely the cathode, the anode and the electrolyte, in order to fully realize the potential of the DIB concept (Fig. 8c). At the cathode side, carbonaceous materials, such as expanded graphite (EG), have been widely employed due to their high chemical stability, large interlayer spacing, and favorable redox potentials for reversible anion intercalation.151,152 Anode materials in DIBs span a broad range of chemistries, including intercalation-type (e.g., graphite and Ti-based oxides), conversion-type (e.g., metal sulfides and oxides), and alloy-type (e.g., Si, Sn) systems.153 Each class offers unique advantages in terms of capacity, redox potential, and reaction reversibility, enabling tailored optimization for specific ionic carriers (Fig. 8d). Electrolyte design is equally critical because it determines ionic conductivity, voltage window, interfacial compatibility, and overall safety.154–156 Formulations with high oxidative stability, including highly concentrated “water in salt” systems, quasi-solid-state water-in-gel electrolytes, and additive-enabled organic solvents, have been shown to broaden the electrochemical window beyond 3 V while effectively suppressing parasitic side reactions (Fig. 8e). Among these strategies, electrolyte additives play a particularly important role by tailoring the solvation structure of ions and facilitating the formation of robust interphases. These interphases are essential for ensuring long-term cycling stability and maintaining compatibility with both anion- and cation-storing electrodes.
DIB chemistries can be broadly categorised by the charge carrier combination employed. Monovalent systems (Li+, Na+, K+, and NH4+) generally provide higher rate capability, whereas multivalent configurations (Mg2+, Ca2+, Zn2+, and Al3+) promise greater volumetric capacity but require tailored host structures to overcome sluggish diffusion kinetics. The following sections provide a comprehensive overview of each type of DIB, highlighting its underlying reaction mechanisms, recent progress in electrode and electrolyte materials, corresponding electrochemical performances (as summarized in Table S2), and the key challenges that need to be overcome for practical implementation.
3.2.1.1. Monovalent metal DIBs (Li/Na/K). With the increasing demand for sustainable and high-performance energy storage, monovalent metal-based DIBs have garnered significant research interest. These batteries operate through a dual-ion redox mechanism, where monovalent metal cations (Li+, Na+, and K+) are inserted into the anode while anions (e.g., PF6−) intercalate into the cathode during charging and discharging. The overall electrochemical performance depends on the synergy between the cathode, anode, and electrolyte, which collectively influence key metrics such as capacity, efficiency, and cycling stability. Take the dual-graphite DIBs as a representative example, their fundamental charge–discharge reactions involve the simultaneous intercalation of cations and anions into graphite electrodes. The electrochemical processes can be described as follows:
Cathode:
| C + xA− → AxC + xe− | (24) |
Anode:
| xLi+ + xe− + C → LixC | (25) |
Despite these advantages, several technical challenges persist. Anion intercalation is generally hindered by sluggish diffusion kinetics and the limited availability of active intercalation sites on the cathode. Furthermore, the stability of the electrolyte under high-voltage operation and its compatibility with both electrodes are critical yet unresolved issues that can compromise the long-term cycling performance and safety of the battery. Addressing these challenges requires the rational design of electrode architectures that offer improved ion accessibility, expanded interlayer spacing, and tailored surface chemistry to promote both cation and anion transport. In parallel, electrolyte formulations must be optimized to ensure high oxidative stability, reduced viscosity, and adequate solvation dynamics. Therefore, the following section will systematically review recent optimization strategies designed to enhance the performance of DIBs.
3.2.1.1.1. Intercalation-type cathodes. Among the cathode materials explored for dual-ion batteries, EG has attracted considerable interest because it can reversibly host large anions such as PF6− and TFSI−. Nevertheless, its practical use is constrained by the intrinsically sluggish diffusion of these bulky anions, whose large ionic radii and low diffusion coefficients result in poor rate capability, limited cycling stability, and overall sluggish kinetics. Additional limitations arise from solvent co-intercalation and the finite number of available galleries within the graphite lattice, both of which restrict the attainable capacity.
Intercalation in graphite proceeds through a well-defined “staging” sequence in which anions occupy every nth graphene layer rather than inserting uniformly. As illustrated in Fig. 9a, the stage number corresponds to the count of graphene sheets separating adjacent intercalated ions. While staging helps accommodate large anions, it amplifies diffusion constraints because each stage transition requires significant lattice expansion and contraction. The anode presents a complementary challenge. Graphite and related carbon materials suffer from slow de-solvation of incoming cations, which in turn limits cation reaction kinetics. Consequently, the overall cell performance is often governed by the mismatch between rapid anion intercalation at the cathode and sluggish cation kinetics at the anode.
 | ||
| Fig. 9 (a) Schematic illustration of the various intercalation stages in a graphite cathode. (b) Schematic representation of possible charge storage mechanisms at the anode side in DIBs, including ion adsorption/insertion, alloying, and metal plating processes. | ||
To overcome these challenges, researchers have explored structural modifications to enhance the intercalation dynamics. For instance, Zhang et al.157 introduced carboxylic anhydride groups between graphite layers to regulate interlayer spacing and particle size. This approach preserved intercalation sites, maintained voltage stability, and significantly improved cycling performance. The resulting graphite cathode delivered a reversible capacity of 91.2 mAh g−1 at 2C and retained 80% capacity after 1000 cycles, offering a promising route to high-performance anion-intercalating cathodes. Beyond chemical functionalization, graphite morphology and electrode formulation have also proven critical to performance enhancement. Chan et al.158 systematically compared different graphite types and binders in LiPF6-based electrolytes. Among these, KS6-type graphite, characterized by its high specific surface area of 20.0 m2 g−1, demonstrated superior electrochemical performance. It delivered a capacity of 92 mAh g−1 at a low current density of 10 mA g−1 and maintained 90% of this capacity at a higher rate of 500 mA g−1, while also retaining 94% of its initial capacity after 200 cycles. Notably, the use of a fluorine-free carboxymethyl cellulose (CMC) binder reduced electrostatic repulsion with PF6− anions, thereby enhancing ion migration and improving rate performance. In addition, graphite's capacity can be further modulated by adjusting surface area, oxidation level, and composite composition. Ghosh et al.159 demonstrated that a composite of graphite and high-surface-area carbon (HSC) at a 75:25 ratio nearly doubled the discharge capacity compared to pure graphite, achieving 54 mAh g−1 at 100 mA g−1. This hybrid structure also showed enhanced rate performance and cycling stability, retaining 70.2% of its capacity after 625 cycles, whereas pure graphite retained only about 50%. These findings underscore the importance of rational design in composite cathodes to overcome graphite's intrinsic limitations.
While graphite remains a foundational material in DIB research, alternative anion-hosting cathodes have expanded the design landscape and addressed limitations in capacity and tunability. Fan et al.160 developed a potassium-based DIB employing a polytriphenylamine (PTPAn) cathode and graphite anode, where PF6− anions reversibly interacted with nitrogen-rich redox-active sites in the polymer matrix. The cell delivered a discharge capacity of 60 mAh g−1 at 3.23 V and maintained 75.5% capacity retention after 500 cycles, highlighting the potential of organic cathodes for achieving sustainable and modular DIB designs. Further expanding the cathode design space, Yu et al.161 reported the first application of a transition-metal cathode in a non-aqueous DIB, using copper powder as the active material. The Cu cathode delivered a remarkable reversible capacity of 762 mAh g−1 at 3.2 V (vs. Li+/Li), attributed to a distinct multi-step redox mechanism rather than conventional intercalation. To suppress Cu2+ crossover and enhance reversibility, an anion-exchange membrane and asymmetrical electrolyte configuration were employed, effectively mitigating polarization and improving cell performance. This work opens new avenues for transition-metal-based cathodes and multi-ion systems beyond traditional intercalation chemistries.
In parallel with cathode innovations, the selection and design of anode materials are equally critical for optimizing overall DIB performance. Anode materials typically fall into three categories which include intercalation-type, conversion-type, and alloy-type (Fig. 9b). Each category is characterized by unique structural features, reaction mechanisms, and electrochemical properties, which collectively influence the charge storage dynamics and cycling stability of the battery. A thorough understanding of these anode types, including their advantages and limitations, is essential not only for balancing the kinetics of dual-ion systems but also for guiding the development of next-generation multi-ion storage technologies.
3.2.1.1.2. Intercalation-type anodes. Intercalation-type anodes act as reversible hosts for metal ions during charge–discharge cycles, playing a fundamental role in monovalent metal DIBs. Carbon-based materials, especially graphite, have been extensively studied due to their layered structure that facilitates smooth metal ion insertion at low potentials. However, their limited theoretical capacity and relatively slow ion diffusion have motivated the exploration of alternative carbon materials with enhanced electrochemical performance. For instance, Fan et al.162 introduced soft carbon (SC) as an anode for sodium-based DIBs, achieving higher discharge capacity and superior cycling stability compared to graphite. The SC anode maintained 81.8% capacity retention after 800 cycles at 1000 mA g−1, indicating improved sodium-ion storage kinetics and durability. Further improvements in energy density and rate capability have been pursued through structural modifications such as nitrogen doping and graphite expansion, both aimed at accelerating ion diffusion and enhancing charge storage efficiency.
To mitigate volume expansion and mechanical degradation during cycling, “zero-strain” materials like lithium titanate (Li4Ti5O12, LTO) and sodium titanate (NTO) have attracted attention. These materials exhibit a negligible volume change during ion intercalation, resulting in excellent cycling stability and safety. Yan et al.163 reported perovskite-structured NaTaO3 nanocrystals as lithium-based DIB anodes with high-capacity retention via a hybrid pseudocapacitive conversion/intercalation mechanism. Similarly, Qiu et al.164 developed a composite of lamellar sodium titanium silicate (NTSO) on nitrogen-rich 3D carbon (NTSO/3DC-N), which enhanced sodium-ion storage by promoting electron transfer and increasing defect density.
Recent advances in two-dimensional materials, especially MXenes, underscore their potential as intercalation hosts owing to their unique layered architectures, tunable surface chemistries, and superior electrical conductivity.165–167 Unlike the conventional “rocking-chair” mechanism, which solely relies on cation migration, anion-based storage strategies enable the intercalation of anions into the cathode. This approach effectively circumvents the high energy cost associated with cation de-solvation during charge transfer. Compared to traditional energy storage systems, DIBs exhibit significant promise for operation in low-temperature environments. In this context, Xia et al.150 explored the use of the Ti3C2 MXene as an anode material and discovered unusually fast K+ diffusion even at sub-zero temperatures. This behavior was attributed to the preferential solvation of K+ by solvent molecules rather than anions, which suppressed the formation of a resistive SEI. As a result, the MXene-based system achieved rapid charging and excellent cycling stability at temperatures as low as −60 °C, underscoring the potential of MXenes for K-based DIB applications under extreme environmental conditions.
Heterostructured materials further enhance ion diffusion, conductivity, and mechanical robustness. For example, Zhang Tao's team168 developed lithium-enriched graphite (LEG) combined with nano-silicon powders (NSPs) to form a lithium-enriched silicon/graphite (LESG) anode. The LESG anode showed an exceptional initial coulombic efficiency (ICE) of 116%, stability in air and moisture without gas evolution, and durable cycling over 400 cycles. This underscores the promise of pre-lithiated silicon-based anodes for high-energy-density lithium-ion batteries.
In summary, intercalation-type anodes, which include modified carbon structures, titanate-based materials, two-dimensional MXenes, and heterostructured composites, continue to play a pivotal role in advancing monovalent metal dual-ion batteries by facilitating higher capacities, faster ion transport, and improved stability.
3.2.1.1.3. Conversion-type anodes. Conversion-type anodes store ions and facilitate charge transfer through redox reactions that involve changes in the valence states of metal ions. During charging, metal ions in the anode material are reduced from higher to lower valence states or to their metallic form, creating vacancies that accommodate inserted ions. During discharging, this process reverses: the reduced metals are oxidized back to higher valence states, releasing ions that migrate back to the cathode, completing the charge–discharge cycle.
A representative example of a conversion-type anode is molybdenum disulfide (MoS2), which is capable of accommodating both intercalation and conversion reactions for sodium and potassium ion storage. Despite its high theoretical capacity, MoS2 suffers from inherent drawbacks such as substantial volume expansion and poor cycling stability during the conversion process. These limitations necessitate further optimization through structural engineering and compositional modification, particularly via nano-structuring and carbon integration strategies aimed at improving mechanical integrity and electrochemical performance. For instance, Cui et al.169 developed a DIB employing a hierarchical (MoS2/CF)@MoS2@C composite as the anode and graphite as the cathode. The 3D carbon framework not only improved electrical conductivity and mechanical robustness but also facilitated high mass loading and uniform dispersion of MoS2. This composite architecture delivered high initial capacity and excellent cycling stability in SIBs, and when incorporated into a Na-based DIB configuration, maintained a high reversible capacity even after 500 cycles under high-voltage operation. Building upon this foundational work, Liu et al.170 adopted a source-template approach to construct MoS1.5Te0.5@C nanocables with a core–shell nanowire–nanotube morphology. further advancing the design of high-performance Na-based DIB anodes. The incorporation of tellurium (Te) into the MoS2 matrix not only enhanced the electronic conductivity but also synergistically accelerated the kinetics of the conversion reaction. The unique tubular morphology alleviated mechanical strain associated with volume expansion, while simultaneously exposing abundant active sites and enabling efficient ion diffusion pathways. This dual benefit resulted in an average working voltage of approximately 3.1 V, facilitated by the characteristic dual-ion storage mechanism of Na+ intercalation at the anode and PF6− intercalation at the graphite cathode. The Te doping synergistically accelerated conversion reaction kinetics, while the unique morphology mitigated volume expansion and provided abundant active sites and rapid ion transport pathways. Ex situ XRD and Raman spectroscopy further confirmed the reversible expansion and contraction of the graphite interlayer spacing during PF6− intercalation and deintercalation, validating the stability of the cathodic reaction. Additionally, the conformal carbon coating on the MoS1.5Te0.5 surface effectively suppressed active material dissolution and mitigated parasitic side reactions, leading to further improvement in cycling stability.
In a different approach, the Hao research team171 developed ultrasmall manganese silicate nanosheets anchored on reduced graphene oxide (RGO) as conversion-type anodes for Li-based DIBs. This design forms an in situ Li4SiO4 ion-conducting coating, shortens ion diffusion paths, and utilizes pseudocapacitive effects to boost ion diffusion kinetics, effectively addressing the sluggish kinetics typical of conversion-type anodes. The resulting battery delivers a discharge capacity of 191 mAh g−1 at 0.5C and retains 128 mAh g−1 after 150 cycles at 2C, outperforming many existing Li-based DIBs. The hydrothermal synthesis method prevents nanosheet aggregation, offering a novel route to high-performance conversion-type anodes. For Na+ ion storage, Wu's team172 encapsulated SnP2O7 nanoparticles within N/P-doped graphene to form a conversion-type anode material for both SIBs and Na-based DIBs. This composite exhibits high reversible capacity, excellent cycling stability, and outstanding rate performance. In sodium-ion half-cells, it achieves 412.9 mAh g−1 at 0.05 A g−1, maintains 86.2 mAh g−1 after 1000 cycles at 2 A g−1, and delivers 182.3 mAh g−1 at 2 A g−1. Moreover, this is the first demonstration of SnP2O7/NPG anodes paired with graphite cathodes to assemble high-performance Na-based DIBs, which operate at a median discharge voltage of 2.5 V and retain 63.2% capacity after 100 cycles. The study also clarifies the Na-ion intercalation/deintercalation mechanism of SnP2O7 through experimental and theoretical analyses, providing insight into its superior electrochemical behavior.
Despite the diversity in material systems and structural designs, these studies converge on improving battery performance by enhancing ion diffusion rates and cycling stability of conversion-type anode materials. MoS2, as a prototypical conversion-type anode, shows excellent capacity but is limited by volume expansion and instability. Structural designs such as 3D carbon frameworks and nanowire/nanotube morphologies have effectively mitigated these issues by improving conductivity, ion transport, and mechanical stability. Similarly, composite strategies like anchoring active materials on RGO or N/P-doped graphene have further boosted electrochemical kinetics and cycling performance in Li- and Na-based dual-ion batteries.
These advancements illustrate that although Li-, Na-, and K-ion batteries differ in materials and mechanisms, high-performance conversion-type anodes can be achieved through thoughtful material design and composite engineering. Future research can continue to explore diverse composite strategies to optimize electrode materials, aiming for enhanced energy density, rate capability, and long-term cycling stability.
3.2.1.1.4. Alloy-type anodes. In monovalent metal DIBs, alloy-type anodes undergo reversible alloying with alkali metal ions (e.g., Li+, Na+ or K+), enabling concurrent ion storage and charge transfer. During charging, alkali metal ions from the electrolyte diffuse into the anode and react with the host metal to form intermetallic alloy phases; upon discharge, the alloy de-alloys and releases the metal ions back into the electrolyte for subsequent intercalation into the cathode. A classic example is the use of silicon-based anodes in LIBs, where lithium ions alloy with silicon to form Li15Si4 during charging and revert to elemental Si upon discharge.173,174 While offering exceptionally high theoretical capacities, such alloy-type materials are plagued by severe volume expansion, leading to mechanical degradation and unstable cycling performance.
To mitigate these issues and adapt alloying-type anodes for DIBs, Jiang et al.175 designed a flexible silicon-based anode supported on a nylon fabric substrate, incorporating a conductive Cu–Ni transition layer between the silicon active material and the underlying fabric. This hierarchical “nylon-Cu–Ni–Si” architecture imparts high elasticity, robust mechanical resilience, and enhanced electronic conductivity. Critically, the Cu–Ni interlayer serves as a mechanical buffer that accommodates silicon's substantial volume fluctuations during lithiation and de-lithiation, thus preserving structural integrity and ensuring continuous electronic pathways under high-rate operation. On the cathode side, PF6− anions undergo intercalation and deintercalation within an EG framework, as evidenced by distinct stage transitions and characteristic high-voltage plateaus. The compatibility between the PF6− intercalation kinetics and the flexible silicon anode was further confirmed through operando XRD analysis, which revealed reversible evolution of diffraction peaks associated with Li12Si7 and other alloy phases. The resulting silicon-graphite DIBs (SGDIBs) demonstrated remarkable rate capability, maintaining approximately 70% of its capacity even at ultrahigh charge/discharge rates of up to 150C. The high working voltage (∼4.0 V at 2C, with an average of ∼3.4 V) arises from the high intercalation potential of PF6− anions into graphite, enabling not only high energy density but also improved power output.
Expanding on alloy anode concepts, Ji et al.176 proposed the use of metal foils including Sn, Pb, K, and Na that serve dual roles as both anodes and current collectors. This approach simplifies battery architecture, reduces inactive material weight, and significantly improves energy density. In K-based DIBs, a Sn foil anode undergoes reversible alloying/dealloying with K+ ions. Experiments showed that the Sn foil retained 93% capacity after 300 cycles at 50 mA g−1, demonstrating excellent long-term stability. Additional advantages include environmental friendliness, low cost, and high safety. Due to efficient reaction mechanisms and a high operating voltage window (3.0–5.0 V), K-based DIBs with Sn foil anodes achieve an energy density of 155 Wh kg−1, comparable to commercial LIBs. Importantly, Sn foil anodes maintain structural integrity after extensive cycling with no significant dendrite formation, ensuring battery safety. Building on this, Wei's team177 developed a self-supported alloy anode for K-based DIBs using Sn-based materials. They synthesized binder-, conductive agent-, and current collector-free SnSb@PCNWs (porous carbon nanowires) via electrospinning. This design simplifies battery structure and increases active material content. The porous carbon nanowires facilitate rapid electron/ion transport and buffer volume expansion of SnSb nanoparticles during alloying/dealloying, greatly enhancing cycling stability. The dual-metal composition (Sn and Sb) reacts sequentially with K+ ions, reducing stress concentration and volume changes. The SnSb@PCNW anode delivers a high reversible capacity of 255.3 mAh g−1 at 2C, significantly outperforming traditional carbon-based anodes (<100 mAh g−1). This innovative design, leveraging the porous carbon matrix, substantially improves capacity, rate capability, and cycle life, offering promising avenues for next-generation energy storage devices.
The development of intercalation-, conversion-, and alloy-type anodes in monovalent DIBs highlights the critical role of multi-ion carrier mechanisms in next-generation energy storage. Synergistic interactions among various ion storage pathways enhance charge transport, structural stability, and electrochemical performance, enabling high energy density and long cycle life. Through deliberate material design strategies such as multi-element doping, nanoscale structural optimization, and interfacial modification, multi-ion carrier power supply devices can be engineered to deliver enhanced efficiency and long-term durability.
3.2.1.2. Monovalent nonmetal DIBs (NH4+). As the demand for sustainable energy storage solutions intensifies, traditional metal-ion batteries face growing challenges related to resource scarcity, environmental concerns, and high costs. This has driven extensive exploration of alternative energy storage systems that reduce or eliminate reliance on critical metals. Among these, monovalent nonmetal dual-ion batteries have emerged as a promising class, employing both cations and anions as charge carriers to enable efficient multi-ion EPSs. The incorporation of multiple ionic species not only broadens the electrochemical design space but also allows tunable electrochemical properties through careful selection of ion pairs and electrolyte environments.
A representative example is the ammonium-ion dual-ion batteries (AmDIBs), which utilizes NH4+ as the primary cationic carrier. In these systems, NH4+ reversibly intercalates and de-intercalates at the anode, while anions undergo charge compensation at the cathode.178 This dual-ion transport mechanism distinguishes AmDIBs from conventional single-ion batteries and enhances ionic conductivity, especially in aqueous electrolytes. Compared to lithium-ion batteries, AmDIBs offer several advantages, including the abundance and low cost of ammonium-based materials, high ionic mobility, and improved safety of NH4+ in aqueous environments, positioning them as attractive candidates for metal-free energy storage.
3.2.1.2.1. Electrolytes in AmDIBs. Aqueous electrolytes are predominantly used in AmDIBs due to their inherent safety and cost-effectiveness; however, their narrow electrochemical stability window limits battery voltage and long-term cycling performance. To overcome this challenge, electrolyte modifications such as the addition of ionic liquids have been investigated to regulate interfacial properties and enhance stability. For example, Li et al.179 developed two distinct electrolyte systems: one based on a 7 mol per kilogram NH4Cl aqueous solution, and another composed of a mixed electrolyte containing 3.5 mol kg−1 EmimCl and NH4Cl. These were evaluated in full-cell configurations featuring I4 as the cathode and 3,4,9,10-perylenetetracarboxylic dianhydride (PTCDA) as the anode. Their combined molecular dynamics simulations and experiments showed that imidazole-based ionic liquids significantly suppress water decomposition, improving battery stability. This effect arises from steric hindrance and π–π interactions of ionic liquid cations, which modulate interfacial water content, prevent excessive NH4+ hydration, and mitigate parasitic side reactions. Consequently, the ionic liquid-containing battery demonstrated markedly enhanced cycling stability, retaining significantly higher capacity after 150 cycles compared to the ionic liquid-free system.
Despite these advantages, NH4+ faces limitations due to its susceptibility to reduction at around −1 V versus the SHE, restricting its electrochemical window. To address this, Zhao et al.180 developed an AmDIB system using NH4+ and PF6− as dual charge carriers, with NH4+ intercalating into perylenetetracarboxylic diimide (PTCDI) at the anode and PF6− into graphite at the cathode. A non-aqueous electrolyte composed of 1 M NH4PF6 in a 1:1 mass ratio mixture of adiponitrile (ADN) and ethyl methyl carbonate (EMC) was designed to suppress NH4+ reduction. Spectroscopic analyses using NMR, Raman, and IR techniques revealed that ADN exhibits a preferential interaction with PF6− anions, whereas EMC predominantly coordinates with NH4+ cations, collectively forming a stable and well-defined solvation structure. This tailored solvation environment extends the electrochemical stability window beyond 4.5 V, significantly enhancing both electrolyte stability and overall battery performance.
Many studies have explored the electrochemical differences between concentrated and dilute electrolytes. Han et al.181 compared highly concentrated electrolytes (HCEs) and dilute electrolytes (LCEs) in AmDIBs, revealing notable benefits of HCEs. In HCEs, strengthened interactions between NH4+ and water molecules, as well as between SO42− and NH4+, reduce free water content and promote stronger ion pairing. Small- and wide-angle X-ray scattering (SWAXS) experiments confirmed new scattering peaks in HCEs, attributed to charge–charge correlations and ion channel formation. Although electrolyte conductivity decreases, concentrated (NH4)2SO4 solutions exhibit a wider electrochemical window than dilute counterparts. Using these HCEs, the PTCDI//N-CuHCF full cell demonstrated outstanding electrochemical performance, maintaining stable cycling with strong capacity retention over prolonged operation, high coulombic efficiency, and a consistent average voltage output of approximately 1.0 V. This configuration also achieved a favorable balance between energy and power output, delivering high specific energy and power density, thereby highlighting the efficacy of HCEs in enabling durable and efficient dual-ion systems. Similarly, Yan et al.182 developed an aqueous AmDIB operating between −40 °C and 80 °C using a “water-in-salt” electrolyte. This concentrated electrolyte broadens the electrochemical stability window by suppressing water electrolysis and side reactions, while also preventing H+ co-intercalation, ensuring NH4+ as the dominant charge carrier and improving cyclability. Additionally, the electrolyte lowers the freezing point and raises the boiling point, enhancing battery performance across a wide temperature range. By disrupting the water's hydrogen-bond network, the electrolyte reduces water decomposition and improves safety, while simultaneously boosting electrochemical performance through enhanced ionic mobility and optimized electrode/electrolyte interfaces.
Electrolyte optimization is key to improving AmDIBs. While aqueous electrolytes offer safety and low cost, their narrow stability limits performance. Incorporating ionic liquids and highly concentrated electrolytes expands the voltage window, suppresses water decomposition, and enhances interfacial stability. Tailored solvation structures prevent side reactions and extend cycle life, enabling wide-temperature operation. These advances address fundamental electrolyte challenges and promote the practical use of AmDIBs.
3.2.1.2.2. Electrode materials in AmDIBs. The choice of electrode materials is crucial for determining the electrochemical performance of AmDIBs. Transition metal oxides are widely studied due to their structural tunability and abundant redox-active sites, which facilitate reversible NH4+ intercalation and deintercalation. Among these, Mn3O4 exhibits high specific capacity and excellent cycling stability, largely attributed to its amorphous nanostructure that enhances ion diffusion kinetics.
However, structural degradation under high voltage conditions remains a challenge, limiting long-term stability. To better understand the NH4+ storage mechanism, Yu Song et al.128 investigated NH4+ interactions in manganese oxides using electrodeposited δ-MnO2. They found that NH4+ forms dynamic hydrogen bonds (1.69–1.96 Å) with lattice oxygen and water molecules, resulting in a lower insertion energy (−4.12 eV) compared to K+ (−3.22 eV). These interactions improve NH4+ reversibility and contribute to a high capacity of 176 mAh g−1 in 0.5 M NH4Ac electrolyte, with 94.7% capacity retention after 10000 cycles. Based on this, Wu et al.183 assembled an aqueous ammonium-ion full cell with an α-MnO2 cathode and a PTCDA anode, introducing a novel electrode combination that advances AmDIB development. The α-MnO2 cathode delivered about 190 mAh g−1 at 0.1 A g−1 and maintained excellent performance over 50000 cycles in 1 M (NH4)2SO4, outperforming most reported ammonium-ion hosts in cycling stability. The full cell also showed strong rate performance, retaining 83.2 mAh g−1 at a high current density of 10 A g−1, indicating superior rate performance and the material's ability to function well during high current charging/discharging. Wu et al.184 also pioneered the use of oxygen-deficient vanadium dioxide as an anode material. Oxygen vacancies expanded the lattice, creating wider channels for NH4+ migration, lowering diffusion barriers, and enhancing electrochemical performance. The d-VO electrode exhibited a reversible capacity of about 200 mAh g−1, good cycling stability (72.9% capacity retention after 1000 cycles), and excellent rate capability. This work highlights how defect engineering and structural control can unlock unique NH4+ storage mechanisms and improve non-metal ion battery performance.
PBAs are among the most extensively developed electrode materials for AmDIBs, thanks to their robust and interconnected three-dimensional open framework. This unique structure provides abundant active sites and significantly shortens the diffusion paths of ammonium ions, thereby enhancing the battery's rate performance. The first report on PBAs in AmDIBs was by Cui and colleagues,185 who synthesized copper hexacyanoferrate (CuHCF) and nickel hexacyanoferrate (NiHCF) nano-powders via a simple co-precipitation method using copper or nickel nitrate and potassium ferricyanide. Both CuHCF and NiHCF showed electrochemical activity in aqueous NH4+ electrolytes, achieving a specific capacity of about 60 mAh g−1 at low rates. Impressively, they retained 75% of their discharge capacity even at a high rate of 41.7C, demonstrating excellent fast-charging and discharging capability. CuHCF maintained 77% of its initial capacity after 500 cycles in ammonium-ion electrolyte, outperforming its lithium and sodium ion counterparts. NiHCF showed even better cycling stability, retaining 88% capacity after 500 cycles. Li and colleagues186 further advanced PBAs by synthesizing NaFeHCF nanoparticles with a unique truncated cube morphology (∼500 nm) controlled by stirring speed. As a cathode, NaFeHCF delivered a high discharge capacity of 62 mAh g−1 and retained 77.4% capacity at 2 A g−1, attributed to fast charge transfer and ion diffusion. The material exhibited exceptional cycling stability, with no capacity decay after 50000 cycles, mainly due to the stable redox behavior of high-spin nitrogen-coordinated Fe2+/Fe3+ pairs. This work optimized synthesis and provided theoretical insights for performance improvement, broadening the material's application prospects. More recently, Du and his team187 introduced Fe substitution into manganese-based PBAs, creating FeMnHCF with dual Mn and Fe metal active sites. This design enabled a high-potential Mn3+/Mn2+–N redox reaction, increasing both voltage and capacity, thus overcoming the limited storage capacity seen in traditional PBAs such as NiHCF and CuHCF. To complement the high-potential FeMnHCF cathode, a high-concentration NH4CF3SO3 electrolyte was developed, featuring a low-solvation NH4+ ion structure and restricted water molecules. This electrolyte weakened water interactions and widened the voltage window. When paired with 24 M NH4CF3SO3 electrolyte, FeMnHCF demonstrated high average discharge voltage, capacity, energy density, and excellent cycling performance, outperforming many aqueous AmDIBs. The dual-metal active sites provide significant advantages for NH4+ storage compared to traditional PBAs. These studies continuously optimize the synthesis processes and performance of PBAs, paving the way for their widespread application in AmDIBs and making them a promising key cathode material for such batteries.
Altogether, AmDIBs are multi-ion carrier batteries fabricated by using both NH4+ cations and anions, enhancing ionic conductivity and electrochemical performance beyond traditional single-ion systems. Innovations in electrolyte design, including the development of ionic liquids, concentrated solutions, and non-aqueous solvents, have significantly contributed to the expansion of electrochemical stability windows and the enhancement of cycling stability. Meanwhile, transition metal oxides and PBAs function as effective cathode materials, offering high capacity and stable cycling through their robust structures and optimized NH4+ storage. Collectively, the coordinated optimization of multi-ion transport electrolytes and electrode materials drives significant progress in AmDIBs, highlighting their potential as safe, sustainable, and economically viable multi-ion carrier energy storage systems.
3.2.1.3. Multivalent metal DIBs (Mg/Ca/Zn/Al). Multivalent metal dual-ion batteries have emerged as a compelling alternative to traditional monovalent systems such as Li+, Na+, and K+ because of their distinctive charge storage mechanisms, higher theoretical energy densities, and potential cost advantages. Unlike conventional dual-ion batteries that rely on monovalent cations for charge compensation, multivalent ion dual-ion batteries utilize divalent or trivalent cations such as Zn2+, Mg2+, and Al3+, which can transfer two or more electrons per ion during redox reactions.188,189 This multi-electron transfer capability significantly boosts charge storage efficiency. In addition, the natural abundance and low cost of multivalent metals, particularly Mg and Al, make these systems attractive for large-scale and sustainable energy storage applications.
However, despite these potential benefits, the practical implementation of multivalent ion DIBs remains limited by several intrinsic challenges. The high charge density of multivalent ions leads to strong electrostatic interactions with both host materials and surrounding solvent molecules, which impedes ion transport, increases interfacial resistance, and negatively impacts the overall reaction kinetics. Moreover, the sluggish diffusion behavior and poor mobility of multivalent ions in conventional electrolytes further constrain power performance and cycle life.
Recent research has primarily focused on two critical aspects to address these limitations, namely electrode architecture and electrolyte design. On the electrode side, materials featuring open-framework structures, including layered or tunnel-structured metal oxides and sulfides, have been investigated to promote ion intercalation and accommodate volume changes during cycling.154,190,191 On the electrolyte side, efforts have centered on developing highly conductive, low-viscosity electrolytes with strong chemical compatibility, aiming to enhance ion transport and mitigate interfacial degradation.
Given both the advantages and challenges of multivalent DIBs, significant research efforts have been devoted to unlocking their full potential through the design of advanced materials and deeper understanding of underlying electrochemical processes. In the following sections, we present a comprehensive overview of recent advances in multicarrier transport mechanisms within various multivalent dual-ion battery systems, focusing on Mg-, Ca-, Zn-, and Al-based configurations. Special emphasis is placed on the cooperative behavior of multiple ionic species, interfacial engineering strategies, and electrochemical performance optimization, with the goal of offering guidance for the rational design of next-generation multivalent energy storage devices.
3.2.1.4. Mg DIBs. The absence of dendritic growth in Mg metal anodes reduces safety concerns common in LIBs, such as thermal runaway. Additionally, the divalent nature of Mg2+ allows for greater charge transfer per ion, offering the potential for higher energy storage capacity. Despite these advantages, practical implementation of MIBs faces significant challenges. Slow solid-state diffusion of Mg2+ ions, strong Lewis's acidity, and poor reversibility at metal interfaces collectively limit both rate capability and long-term cycling stability.
To overcome these intrinsic limitations, recent efforts have increasingly turned toward multi-ion carrier strategies, especially in the design of Mg-based dual-ion batteries (MDIBs). By decoupling cationic and anionic transport pathways, MDIBs enable more flexible and dynamic redox architectures, improving both ion accessibility and reaction reversibility. One illustrative example was reported by Zhu et al.,192 who developed a water-based MDIBs featuring a 4.5 M Mg(NO3)2 aqueous electrolyte paired with polymer-based redox-active electrodes. This system utilized a liquid-phase charge storage mechanism to bypass the sluggish solid-state diffusion of Mg2+, achieving enhanced rate performance and improved electrochemical stability. The formation of water-shared ion pairs between Mg2+ and NO3− simultaneously reduced water activity and expanded the electrolyte's stability window, thereby extending the cycle life and enhancing energy efficiency. Beyond polymer cathodes, the use of multielectron redox centers further enriches the electrochemical landscape of MDIBs. Morag et al.189 reported a MDIB based on a four-electron Te0/Te4+ conversion mechanism, enabled by blending Mg(TFSI)2 into a classical magnesium–aluminum chloride complex (MACC) electrolyte. This carefully engineered dual-salt electrolyte not only facilitated the dissociation of Cl− from the MACC complex, alleviating ion aggregation, but also served as a buffer that mitigated Mg passivation and solvent consumption during cycling. The resulting system achieved a high reversible capacity of 543 mAh g−1 and an energy density of 850 Wh kg−1, marking a significant advancement for multivalent chemistries. The multifunctional role of Mg(TFSI)2 in simultaneously modulating solvation dynamics and enhancing redox stability in MDIBs exemplifies how multi-ion coordination can open electrochemical pathways that remain inaccessible in traditional single-ion systems.
In parallel, Cheng et al.193 introduced an unconventional strategy to tackle electrolyte degradation and anode instability, long-standing issues in MDIBs. Rather than suppressing anodic side reactions, they proposed using an active metal as both the anode and current collector to replenish M+ ions in the electrolyte, thereby compensating for the anions deintercalated from the graphite cathode. In the specific case of Mg//graphite configurations, the low redox potential of Mg (−2.35 V vs. SHE) offers higher cell voltage output when paired with graphite, compared to transition metals such as Fe, Co, Cu, Zn, etc. Moreover, the high de-solvation energy of Mg2+ (9.06 eV vs. 3.08 eV for Li+) makes it less prone to reduction, facilitating stable cation replenishment during extended cycling. This strategy enables the gradual transformation of Mg into a Li3Mg7 alloy, which suppresses dendritic lithium growth and promotes uniform deposition. The Mg//graphite cell exhibited remarkable cycling stability, maintaining 94.6% of its capacity after 1700 cycles at 2C, significantly surpassing the performance of both Cu//graphite and graphite//graphite cells. This approach introduces a broadly applicable framework for multi-ion regulation at the anode, combining high energy density (196.8 Wh kg−1) with practical scalability, as demonstrated by pouch cell operation for small electronic devices.
Electrolyte design remains central to the continued advancement of MDIBs. Radi et al.194 developed a novel electrolyte system by integrating commercial Mg(TFSI)2 with dibutyl magnesium (Mg(butyl)2), enhancing both the deposition–stripping reversibility and interfacial compatibility with magnesium metal. By using a titanium current collector, selected for its compatible lattice structure with magnesium, the authors were able to lower nucleation overpotentials and promote uniform magnesium plating. Importantly, this study demonstrated that the structure of the solvation shell, the Mg2+ transference number, and the interfacial resistance are key determinants of redox kinetics in multivalent systems. Given that anion mobility often surpasses that of Mg2+ in such systems, effective multi-ion transport design requires a nuanced balance between ionic species distribution and interfacial dynamics in MDIBs, rather than reliance solely on redox potential tuning.
3.2.1.5. Ca DIBs. Ca dual ion batteries (CDIBs) have recently transcended their early reputation as “difficult-to-activate” calcium-ion systems and are now recognized as a model platform for illustrating how a multi-ion-carrier strategy can re-engineer every facet of multi-ion EPSs. In conventional calcium ion batteries (CIBs), the divalent Ca2+ ion offers theoretical advantages due to its near-Li+ reduction potential and dual-charge transfer capability. However, practical devices have long been limited by operating voltages below 2 V, sluggish diffusion kinetics, and severe interfacial instabilities that typically lead to rapid capacity fading within just a few dozen cycles.195 By sharing the task of charge compensation between fast, highly polarisable anions (PF6−, FSI−, and TFSI−) and the cation (Ca2+), the CDIBs simultaneously raise their electrochemical window beyond 4 V and mitigate the kinetic bottleneck that once defined Ca chemistry.196
The foundational configuration of CDIBs was reported by Wu et al.,197 who designed a system featuring mesocarbon microbeads (MCMB) as the anode and EG as the cathode, immersed in a 0.7 M Ca(PF6)2 carbonate electrolyte. Operando studies demonstrated reversible Ca2+ intercalation into MCMB and PF6− anion intercalation into EG, yielding an average working voltage of 4.6 V and 94% capacity retention over 300 cycles, a level of performance previously deemed unachievable for calcium-based systems. The system's strength lies in its symmetric architecture, wherein both electrodes utilize carbon frameworks with complementary ionic intercalants, forming a self-regulating platform that mitigates structural degradation. Subsequent work has shown that electrolyte design is equally critical to sustaining multi-ion choreography. Prabakar et al.198 replaced volatile carbonates with a pyrrolidinium-based ionic liquid doped with a single-molecule Ca-tetraglyme complex, thereby eliminating parasitic cation intercalation and stabilising both [Ca·G4]2+ and TFSI− migration through graphite hosts for hundreds of cycles. Li et al.195 pushed the concept further with a 3.5 M Ca(FSI)2 “super-concentrated” electrolyte that locks Ca2+ inside a tight solvation sheath, suppresses solvent breakdown and enables concerted Ca2+–FSI− shuttling between an organic PTCDA anode and graphite cathode with 85% capacity retention over 350 cycles. These studies collectively highlight how manipulating the solvation micro-environment can fine-tune the relative mobilities of cations and anions, an insight that is uniquely accessible in a multi-ion framework.
Beyond carbon hosts, alloying and polyanionic insertion chemistries are now being co-opted to accommodate divalent charge without sacrificing kinetics. Wang et al.199 demonstrated that a Sn foil anode forms a ductile Ca7Sn6 alloy under an operating voltage of 4.45 V in CDIBs, while maintaining 95% capacity retention after 350 cycles. In situ stress analysis attributed this excellent cycling stability to the generation of compressive stress during alloy formation, which effectively inhibits crack propagation and contributes to the formation of a stable CaF2-rich SEI. Xu et al.196 introduced Na0.5VPO4.8F0.7, a material with a rigid polyanionic framework that accommodates Ca2+ insertion with minimal structural distortion, exhibiting only a 1.4% volume change and low diffusion barriers below 0.6 eV. This CDIB design enabled 90% capacity retention over 500 cycles at an average voltage of approximately 3.2 V. These findings, together with advances in alloy-type anodes, highlight the potential of multi-component ion storage systems to achieve high capacity while maintaining mechanical robustness, provided that the host lattice is precisely engineered to support reversible multivalent ion insertion.
By distributing the electrochemical workload between Ca2+ and a co-operative anion, CDIBs have overcome the longstanding challenges of low voltage, sluggish kinetics, and rapid degradation. The emerging consensus is that future progress in calcium systems, and more broadly in other multivalent chemistries, will depend on our ability to orchestrate the transport and interaction of multiple charge carriers within well-coordinated electrode–electrolyte architectures.
3.2.1.6. Zn DIBs. Zinc dual-ion batteries (ZDIBs) operate based on the reversible shuttling of Zn2+ ions between the cathode and anode, enabling bidirectional ion transport that underpins highly reversible electrochemical cycling. The inherent properties of Zn, including its low cost, natural abundance, and compatibility with aqueous electrolytes, support the potential applicability of ZDIBs in large-scale energy storage systems. However, the practical deployment of these systems is still hindered by inherent challenges, including electrolyte instability, interfacial degradation, and the uncontrolled growth of zinc dendrites. These issues adversely affect the reversibility and long-term reliability of Zn2+ transport within multi-ion environments.
Recent advances in cathode material development, electrolyte formulation, and electrode architecture have significantly improved the performance and durability of ZDIBs. Particular focus has been placed on optimizing the interaction and synergy between multiple ionic species. For instance, Kushwaha et al.200 introduced a redox-active covalent organic framework (COF) as a cathode in conjunction with a ZnSO4 aqueous electrolyte, leveraging nitrogen- and oxygen-rich functional groups capable of Zn2+ chelation. This strategic design resulted in a significant increase in specific capacity, rising from 208 to 690 mAh g−1, marking the highest capacity reported for COF-based Zn-ion systems to date. This result underscores the importance of tuning ion-host interactions to facilitate efficient Zn2+ storage within a multi-ion framework. In parallel, to overcome the limitations of conventional polymer cathodes such as limited accessibility of redox-active sites and hindered ion diffusion, innovative material processing strategies have been developed. Zhang et al.201 employed KBr as a porogen to engineer the morphology of poly(triphenylamine) (PTPA), resulting in a microporous, nitrogen-rich polymer (m-PTPA) with a three-dimensional conjugated structure, pore sizes of 6 Å and 12 Å, and a high surface area of 655 m2 g−1. This unique architecture enabled highly reversible Cl− storage through redox-active amine/imine sites and delivered a pseudocapacitive contribution exceeding 99%, with exceptional conductivity and insolubility that circumvented the common pitfalls of small-molecule organic electrodes. The interplay of Zn2+ and Cl− ions within such porous polymer frameworks exemplifies how coordinated multi-ion transport can be optimized through molecular and morphological design. Architectural innovations in electrode design have further propelled the development of high-capacity and long-life ZDIBs. Guo and co-workers202 devised a core–shell Ag@C cathode by pyrolyzing glucose in the presence of silver particles, producing a robust composite for pairing with a metallic Zn anode and ZnCl2 electrolyte. Within this system, the redox transition between Ag and AgCl was efficiently stabilized by the carbon shell, which buffered volume changes and preserved structural integrity during cycling. Simultaneously, the use of a neutral ZnCl2 electrolyte played a critical role in suppressing Zn dendrite formation, effectively mitigating one of the most persistent issues in aqueous Zn-based batteries. As a result, the system delivered a volumetric capacity of 1301.5 mAh cm−3 and retained 96.1% of its capacity after 1700 cycles at a 20C rate, highlighting the synergistic benefits of combining ion-specific electrolyte formulations with dual-ion electrode architectures. Efforts to balance cost, performance, and scalability have also inspired the development of novel carbon-based cathodes. Guo et al.202 introduced an aqueous ZDIB employing nitrogen-doped few-layer graphene-like carbon (NFG) as the cathode material. In a ZnCl2-based water-in-salt electrolyte, the NFG electrode enabled reversible intercalation of complex [ZnClx]2−x species, achieving a specific capacity of 134 mAh g−1 with a voltage plateau of 1.85 V and long-term cycling stability. Notably, despite the use of concentrated salt, the intrinsic affordability of ZnCl2 preserved the cost-effectiveness of the system, presenting a viable path toward practical, high-performance multi-ion configurations.
In addition to conventional DIBs configurations, the emerging concept of reverse dual-ion batteries (RDIBs) presents a compelling alternative by inverting the traditional ion storage paradigm, wherein anions are stored at the negative electrode and cations at the positive. A representative example is the pioneering work by Wu et al.,203 who implemented a rotaxane-functionalized carbon (Fc/C) anode coupled with a Prussian blue analogue (Zn3[Fe(CN)6]2) cathode in a highly concentrated 30 M ZnCl2 “water-in-salt” electrolyte. This reversed configuration achieved a reversible capacity of 30 mAh g−1 with an average voltage of 0.9 V and demonstrated 58% capacity retention after 1000 cycles. The performance was largely attributed to the electrolyte's high ionic strength, which stabilized the electrochemical interface and facilitated multi-ion shuttling under the reversed polarity conditions. Expanding on this concept, Placke and co-workers204 introduced a ZDIB utilizing a water-in-bisalt electrolyte composed of NaFSI and Zn(TFSI)2. This system allowed for high-voltage anion intercalation into the natural graphite cathode, achieving a discharge voltage of ∼2.25 V and a specific energy approaching 200 Wh kg−1. These results not only demonstrate the compatibility of abundant, low-cost materials with advanced electrolyte formulations, but also exemplify the potential of carefully tuned multi-ion chemistries in high-energy aqueous Zn2+ systems. Sustainability considerations have further propelled research in this domain, particularly around electrolyte safety and environmental impact. Sun et al.205 proposed a hydrogel-based water-in-salt electrolyte composed of polyvinyl alcohol (PVA) and gelatin. This electrolyte formulation offered broad electrochemical stability, effective Zn dendrite suppression, and rapid biodegradability, reflecting how multi-functional electrolyte design can simultaneously meet electrochemical and ecological requirements in multi-ion systems. Further advancing the concept of electrolyte-driven optimization, Sethi et al.206 compared two Prussian blue analogues as cation hosts, with their larger framework and lower hydration level, facilitated higher deintercalation potentials (1.14 V vs. Ag/AgCl). Their use of a 10 M ZnCl2 electrolyte not only improved the redox potential alignment via the Nernst effect but also enhanced the cycling stability of the Fc/C anode. The full cell delivered 23 Wh kg−1 with 80% capacity retention after 50 cycles at 5C, highlighting how electrolyte concentration and ion activity critically modulate multi-ion transport and energy output.
To overcome the inherent energy density limitations of aqueous DIBs, recent efforts have turned toward exploring multivalent anion intercalation mechanisms. Zheng et al.207 conducted a systematic investigation of ClO4−, SO42−, and PO43− intercalation into graphite using DFT simulations and spectroscopic techniques. Their findings revealed that ClO4− and SO42− achieved deeper and more reversible intercalation due to favorable charge-to-size ratios and diffusion dynamics, while PO43− suffered from strong electrostatic interactions that impeded ion mobility. The Zn-graphite DIBs constructed using ClO4− and SO42− exhibited energy densities of 106 and 112 Wh kg−1, respectively, at ∼1.85 V, underscoring the significance of anion valency and structural compatibility in tuning voltage and capacity through multi-ion design. Complementary to these advances, Wang et al.208 developed an aqueous ZDIB system using a (CH3)4NCl + Na2CO3 electrolyte in combination with graphite and Zn electrodes. By carefully tuning the pH and electrolyte composition, particularly using a 5 M (CH3)4NCl solution with supersaturated Na2CO3, they achieved a high discharge voltage of 2.6 V and a specific capacity of 226.2 mAh g−1, maintaining stable performance over 3200 cycles. Cyclic voltammetry and in situ XRD analysis provided mechanistic insight into the reversible anion intercalation processes, while the use of industrially accessible materials demonstrated the system's commercial viability.
To circumvent the reliance on pre-deposited Zn metal anodes and mitigate dendrite-induced failures that commonly plague aqueous Zn-based systems, Wang et al.209 proposed an anode-free ZDIB architecture, employing a silver-coated copper substrate to direct uniform Zn plating and stripping. This design enabled controlled Zn nucleation and substantially prolonged the cycling life, achieving over 80% capacity retention after 1000 cycles. During the charge process, the graphite cathode exhibited a distinct phase evolution from Stage-III to Stage-I’, accompanied by a contraction in interlayer spacing. Zhou et al.210 developed a fluorinated electrolyte system for high-performance anode-free ZDIBs by integrating fluorinated carbonate solvents with an ethoxy(pentafluoro)cyclotriphosphazene (PFPN) additive. This electrolyte exhibits a high oxidative stability with a decomposition voltage of 2.75 V, significantly expanding the electrochemical window compared to conventional aqueous or organic systems. The fluorinated solvents not only confer non-flammability and enhanced safety but also exhibit reduced Zn2+ solvation ability, which promotes the incorporation of anions into the Zn2+ solvation shell. This shift in solvation structure facilitates the formation of an anion-derived SEI on the Zn anode surface, which in turn guides the uniform, planar deposition of Zn and suppresses dendritic growth. As a result, the Zn//Zn symmetric cell achieves a remarkable Zn utilization of 91% over 140 hours of cycling, a performance rarely reported in the literature. Furthermore, the PFPN additive contributes to the formation of a stable CEI film that enhances the oxidative stability of the electrolyte, enabling prolonged cycling of Zn//graphite dual-ion batteries up to 1000 cycles. Notably, the system also supports an anode-free configuration, which demonstrates stable cycling over 100 cycles and delivers a high average discharge midpoint voltage of 1.84 V. Therefore, this anode-free configuration more than doubled the energy density relative to conventional ZDIBs, underscoring its potential for next-generation aqueous systems where eliminating excess Zn can significantly improve energy efficiency and volumetric compactness (Fig. 10). Such strategies also reflect the broader utility of multi-ion transport, as Zn2+ shuttling remains central to system function while interface optimization governs reversibility and cycling performance. Beyond bulk electrode engineering, interfacial modulation has emerged as a crucial dimension for advancing multi-ion carrier systems. Yang et al.211 developed cationic two-dimensional polymer films synthesized through Knoevenagel condensation, yielding well-ordered 1D channels with intrinsic anion selectivity. These 2D polymers exhibited strong affinity toward bis(trifluoromethanesulfonyl)imide (TFSI−) anions, enabling selective ion transport and enhanced interfacial compatibility. When integrated into high-voltage ZDIBs, the polymer films not only stabilized ion transport pathways but also significantly prolonged cycle life by suppressing parasitic reactions and enhancing the robustness of the electrode–electrolyte interface.
 | ||
| Fig. 10 (a) Schematic illustration of the fundamental working principles of traditional and anode-free DIBs. (b) Representative configurations of various battery architectures, with emphasis on the energy and design advantages of anode-free DIBs. | ||
In summary, ZDIBs leverage the coordinated migration of multiple ionic species, including Zn2+ along with Cl−, TFSI−, or complex anions such as [ZnClx]2−x. This multi-ion transport mechanism helps overcome the inherent limitations of conventional aqueous batteries with respect to energy density, voltage output, and cycling stability. In addition to enhancing interfacial kinetics and expanding the electrochemical stability window, it also enables new strategies for optimizing electrolyte composition and tuning electrode–electrolyte interactions.
3.2.1.7. Al DIBs. Al dual ion batteries (ADIBs) operate based on a dual-ion mechanism that involves both multivalent aluminum cations (Al3+) and mobile anions (e.g., PF6−, ClO4−, or AlCl4−). During the charging process, Al3+ ions are electrochemically deposited onto the aluminum metal anode, while the corresponding anions are intercalated into the cathode structure. Upon discharging, the Al metal is oxidized to release Al3+ ions, and the intercalated anions are simultaneously deintercalated from the cathode. The spatial separation of charge storage between cation deposition at the anode and anion intercalation at the cathode enhances charge-transfer kinetics and helps maintain stable voltage profiles, which are essential for high-power energy storage systems.
The foundational concept of ADIBs was first introduced by Tang et al.,212 who reported an Al–graphite dual-ion configuration using Al foil as the anode and graphite as the cathode, with PF6− anions serving as the primary charge carrier at the cathode. During the charge process, the graphite cathode undergoes a sequential staging mechanism of PF6− intercalation, while the Al anode forms a Li–Al alloy protected by a SEI. These dynamics established the core functionality of ADIBs without relying on costly transition metal cathodes, thereby promoting both material sustainability and economic viability. The system operated at an average voltage of 4.2 V and demonstrated high energy density with excellent cycling performance, providing a benchmark that has catalyzed ongoing advances in the field. Building upon this work, Zhang et al.213 developed a novel ADIB by incorporating a three-dimensional graphene foam cathode with an aluminum metal anode and an Al(ClO4)3-based electrolyte in a mixed solvent of propylene carbonate and fluoroethylene carbonate. The battery achieved high reversibility through ClO4− intercalation/deintercalation at the cathode and Al plating/stripping at the anode. Notably, it exhibited a discharge voltage of approximately 1 V, could be fully charged in just 13 minutes, and sustained discharge durations exceeding 73 minutes. Furthermore, the cell maintained a specific capacity of 101 mAh g−1 at a high-rate of 2000 mA g−1 over 400 cycles, underscoring its potential for high-rate applications. Nevertheless, despite these encouraging results, long-term stability under practical operating conditions remains a challenge that warrants further investigation. Progress in cathode materials has played a critical role in improving ADIB performance. For instance, M. Latha et al.214 introduced a WS2/graphene composite cathode synthesized via an ultrasonic-assisted method. The layered WS2 structure offered abundant intercalation sites for anions, while the conductive graphene network facilitated efficient electron transport. This synergistic configuration delivered a high specific capacity of 152 mAh g−1 and stable cycling over 150 cycles, outperforming pure WS2 electrodes. These results highlight the importance of nanocomposite engineering in promoting dual-ion transport dynamics and improving electrochemical efficiency in multi-ion systems.
Beyond inorganic and carbonaceous frameworks, polymer-based cathodes have demonstrated notable potential in fine-tuning ionic and electronic transport in ADIBs. Ma et al.215 developed redox-active dihydrophenazine (Pz)-based materials by coupling Pz with either pyrene (Py) or biphenyl (Ph), resulting in two conjugated microporous polymers, PyPz and PhPz. These polymers were designed to serve as AlCl4−-hosting cathodes for ADIBs, enabling efficient ion storage through their extended conjugation and porous frameworks. PyPz, in particular, exhibited excellent redox activity due to its extended π-conjugated structure and high surface area, which enabled efficient AlCl4− intercalation into its porous network. The redox-active dihydrophenazine units contributed to high areal capacities (2.53 mAh cm−2), ultra-long cycling life (over 100000 cycles), and robust low-temperature performance (−30 °C). Importantly, the insoluble and structurally stable polymer framework effectively suppressed dissolution of active materials, a common degradation pathway in organic cathodes, thereby ensuring long-term durability under practical conditions. Expanding on polymer design strategies, Luo et al.216 introduced a donor–acceptor (D–A) conjugated polymer, PPTZ-AQ, composed of benzothiazine (PTZ) donor and anthraquinone (AQ) acceptor units. This molecular architecture enabled overlapping redox potentials and promoted charge delocalization along the polymer backbone, significantly enhancing electrochemical kinetics. Comparative studies with homopolymers PAQ and PPTZ confirmed that PPTZ-AQ delivered superior specific capacity, rate capability, and cycling durability. These findings illustrate the potential of D–A type bipolar conjugated polymers as next-generation cathode materials for multi-ion systems, offering a high degree of tunability and structural flexibility that surpass conventional inorganic or carbon-based alternatives. These findings underscore the potential of D–A conjugated polymers as structurally versatile and redox-tunable cathode materials tailored for multi-ion storage in DIBs.
Overall, the development of ADIBs demonstrates that the coordinated transport of both cations and anions can facilitate decoupled charge storage, accelerate reaction kinetics, and allow for more versatile material design. Advances in inorganic, carbon-based, and polymeric cathode architectures have enabled ADIBs to address the inherent limitations of conventional single-ion systems. Moreover, the broad selection of electrode materials, including graphite, layered dichalcogenides, and conjugated polymers, provides abundant opportunities for precise structural and chemical customization.
The physicochemical diversity of chlorine, bromine and iodine dictates markedly different cell chemistries. Chlorine offers the highest redox potential but suffers from extreme volatility; bromine provides a balance between voltage and chemical stability yet endures serious shuttle loss via soluble polybromides; iodine is a solid under ambient conditions, giving superior safety and very high specific capacity, but its polyiodide intermediates still dissolve and migrate. Understanding these contrasts has steered research toward tailored confinement matrices, redox-mediating additives, and hybrid-ion electrolytes that simultaneously suppress halogen escape and accelerate charge transfer.
In the era beyond lithium-based technologies, aqueous rechargeable ZIBs have attracted widespread interest owing to their inherent safety, cost-effectiveness, and environmental friendliness. Among these systems, aqueous Zn–halogen batteries have emerged as a promising subclass, leveraging the high redox activity of halogen species and the abundant availability of Zn metal. Taking metallic Zn as a representative example, its electrochemical behavior in aqueous media is notably influenced by the pH of the electrolyte (Fig. 11a).220 As an amphoteric metal, Zn exhibits distinct deposition and dissolution characteristics depending on the acidity or alkalinity of the electrolyte. In particular, because halogen species undergo spontaneous disproportionation reactions in strongly alkaline environments, most Zn–halogen batteries are designed to operate under neutral or mildly acidic conditions. This electrolyte strategy differs from that of traditional alkaline Zn-based batteries and serves to suppress Zn passivation, thereby facilitating more reversible Zn stripping and plating processes. During battery discharge, Zn at the anode undergoes oxidation, releasing electrons and forming Zn2+ ions, which migrate through the electrolyte toward the cathode. Upon charging, the process is reversed as Zn2+ ions are reduced and re-deposited on the anode surface under the influence of an external power source. At the cathode, halogen-based redox reactions take place, which can generally be categorized into two types: X−/X0 redox and X−/X0/X+ multivalent redox.
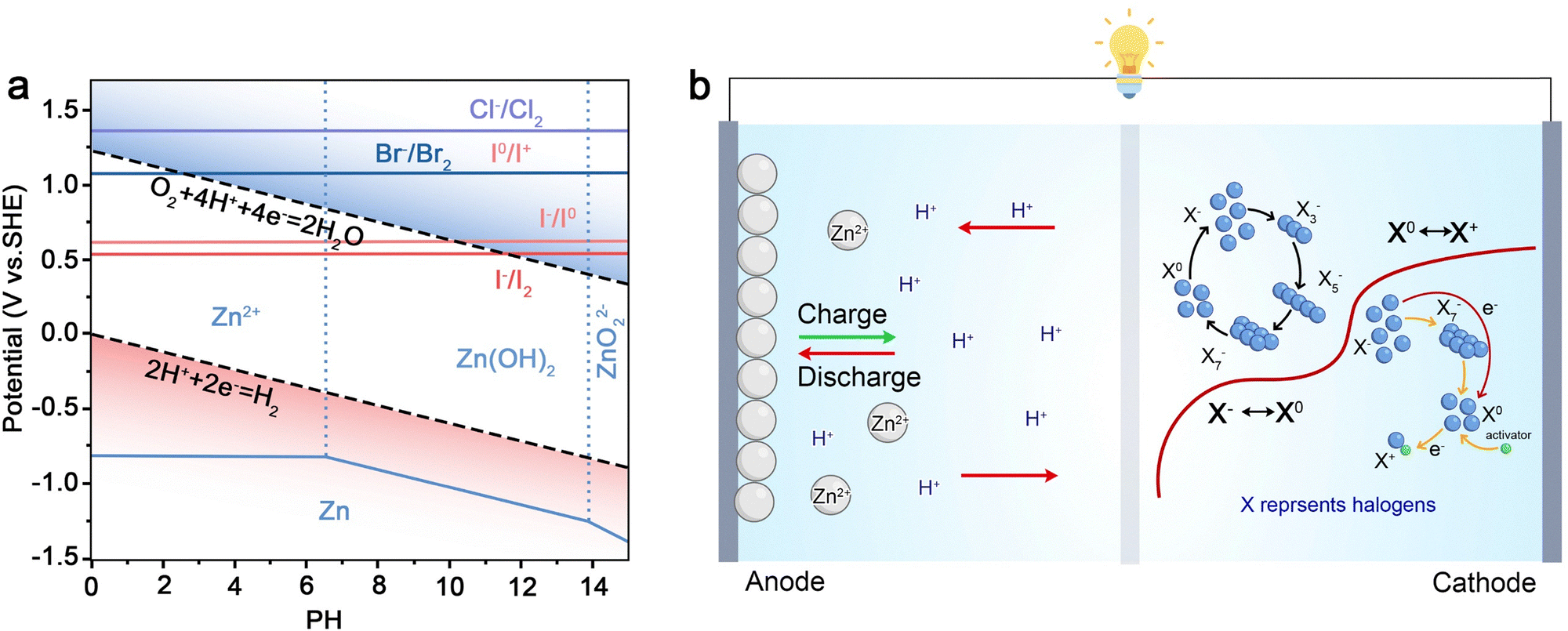 | ||
| Fig. 11 (a) Pourbaix diagram of water overlaid with the standard electrode potentials of Mz+/M redox couples, where M represents Cl, Br, I, and Zn. (b) Schematic illustration of representative reaction mechanisms occurring at metal Zn anodes and halogen cathodes. Reproduced with permission from ref. 221. Copyright 2024, Royal Society of Chemistry. | ||
In the X−/X0 redox pathway, halogen molecules (X2) are reduced to halide ions (X−) during discharge, while the reverse oxidation occurs upon charging (Fig. 11b). However, this process often leads to the formation of polyhalide intermediates (such as X3−, X5−, and X7−, except in the case of Cl), resulting from the complexation of molecular halogens with halide ions. These intermediates tend to trigger parasitic reactions, contributing to self-discharge and compromising long-term battery stability. In more advanced systems, the X−/X0/X+ redox process has been explored, particularly in the case of iodine. Here, iodine molecules (I2) can be further oxidized to I+, forming interhalogen compounds in the presence of highly electronegative halide ions such as F−, Cl−, and Br−. This expanded redox window (I− → I0 → I+) introduces new opportunities for the design of high-voltage Zn–I2 batteries by leveraging tailored electrolyte compositions containing such halide additives.
The MHBs enables multi-electron transfer processes that contribute additional capacity. By decoupling charge transport between cations and anions and promoting multi-electron redox activity, MHBs offer a promising pathway toward high-performance, cost-effective, and scalable battery technologies. In the following sections, we systematically discuss the redox mechanisms, fundamental challenges, electrochemical performances (Table S3), and recent advancements in representative MHBs, with a focus on illustrative examples and the strategies devised to overcome their specific limitations.
3.2.2.1. Li halogen batteries. Lithium halogen batteries (LHBs) are an emerging battery technology that combines the high theoretical energy density of lithium-based chemistry with the rich redox activity of halogen species. In these systems, lithium ions undergo reversible plating and stripping at the anode, while halogen molecules participate in multi-electron redox reactions at the cathode (Fig. 12a). This dual redox mechanism enables high output voltages and energy densities, making LHBs attractive for grid-level storage, electric vehicles, and aerospace applications. Practical realisation, however, is hampered by the volatility of halogen species, parasitic shuttle effects, and sluggish ion transport, calling for precisely engineered electrodes and electrolytes that can confine halogens and promote fast, stable multi-ion transport.
 | ||
| Fig. 12 (a) Schematic diagram of metal halogen batteries. (b) Energy density, discharge platform, maximum cut-off specific capacity, energy efficiency, operation temperature and (c) reversible mechanisms among Na–Br(+) batteries, Na–Br2 batteries, and Na–Cl2 batteries. Reproduced with permission from ref. 232. Copyright 2023, Wiley. | ||
3.2.2.1.1. Li–Cl2 batteries. Within the LHB family, lithium chlorine (Li–Cl2) batteries exploit the reversible Cl−/Cl2 couple and therefore offer one of the highest theoretical energy densities among halogen chemistries. During charging, LiCl oxidises to Cl2, which is physically confined in a porous cathode host; during discharge, Cl2 is reduced back to Cl−, regenerating LiCl. Prototype devices, however, have long been limited by Cl2 leakage, severe capacity fade and safety concerns. Recent research tackles these shortcomings through the coordinated use of multi-ion carriers, catalytic interfaces and tailored electrolytes, thereby uniting high energy density with practical cyclability.
The multi-ion carrier mechanism, which leverages the synergistic transport of Li+, Cl−, and Al3+/F− ions, is demonstrated in a Li–Cl2 battery employing CO2-activated graphite as the cathode.221 Coin cells with a Li metal anode and a CO2-activated graphite cathode employ a neutral electrolyte comprising 1.8 M LiCl and 1.8 M AlCl3 in SOCl2 with a 2 wt% LiFSI additive (≈0.047 M in solution). During the first discharge to 2 V, Li+ migration from the anode combines with Cl− (from SOCl2 reduction) to deposit LiCl on CO2-activated graphite (DGr-ac), delivering ∼3.37 V and ∼1911 mAh g−1 due to enhanced pore sites/defects from CO2 activation, while unmodified DGr-ac yields only ∼3.18 V and ∼1,91 mAh g−1. Auger electron spectroscopy confirms uniform LiCl coating post-discharge. Al3+ (from AlCl3) optimizes ionic conduction, while F− (from LiFSI-derived SEI) stabilizes cycling. Charging releases trapped Cl2 (via Cl− oxidation, mass spectrometry-verified), enabling reversible redox via pore-hosted LiCl storage exceeding intercalation limits. To further address the kinetic limitations inherent in Cl− de-solvation and LiCl decomposition, Li et al.222 designed atomically dispersed Co–N4 catalytic sites through controlled pyrolysis. DFT calculations revealed that these Co–N4 centers significantly lower the energy barrier for LiCl decomposition by 1.2 eV relative to unmodified carbon, catalytically accelerating both Cl− oxidation and Li+ migration. The resulting electrochemical system achieved a high discharge voltage of 3.4 V and a specific capacity of 2500 mAh g−1, maintaining 92% capacity retention after 200 cycles even at −20 °C. These results highlight the dual catalytic function of Co–N4 centers in facilitating concerted multi-ion transfer, enabling fast and stable operation across challenging thermal conditions.
Complementary to these electrode-level advancements, the design of novel electrolytes capable of supporting dual-ion conduction has proven equally critical. Sun et al.223 developed a methyl chlorodifluoroacetate (MCDFA)-based electrolyte as a safer, non-corrosive alternative to SOCl2. This system not only ensures a broad operational temperature range (−40 to 120 °C), but also enables simultaneous conduction of Li+ and Al3+ ions. Raman and NMR spectroscopy confirmed weak solvent–cation interactions and suppressed side reactions, resulting in a high ionic conductivity (6.57 mS cm−1 at 25 °C; 2.38 mS cm−1 at −40 °C). More importantly, the inclusion of AlCl3 additives facilitated the reversible reduction of MCDFA and enhanced the stability of Cl−/Cl2 redox cycling. In practical pouch cell configurations, the platform delivered an energy density of 800 mAh g−1, representing a 40% enhancement over conventional SOCl2-based systems.
These advances illustrate how multi-ion carrier strategies can systematically overcome the longstanding trade-offs between energy density, cyclability, and rate capability in halogen-based batteries. In particular, Li–Cl2 systems demonstrate how the coordinated design of functional components can collectively overcome the intrinsic limitations of conventional single-ion architectures. This includes the use of porous graphite to efficiently confine Cl2 gas, the incorporation of Co–N4 catalytic centers to accelerate Li+/Cl− interfacial kinetics, and the implementation of MCDFA-based electrolytes that enable stable multi-ion conduction across a wide temperature range.
3.2.2.1.2. Li–Br2 batteries. Lithium bromine (Li–Br2) batteries epitomize the paradigm-shifting capabilities of multi-ion carrier systems, where the synergistic interplay of Li+, Br−, Cl−, and Al3+ ions unlocks exceptional theoretical energy densities (∼1273 Wh kg−1) and operating voltages (∼4.12 V), far surpassing conventional single-ion designs. However, the practical deployment of Li–Br2 batteries has long been hindered by intrinsic challenges such as the bromine shuttle effect and the dissolution of polybromide intermediates, which compromise coulombic efficiency, capacity retention, and long-term stability. These limitations have catalyzed the development of advanced strategies that harness the principles of multi-ion carrier chemistry to stabilize redox kinetics and facilitate efficient interfacial charge transfer.
Recent breakthroughs in Li–Br2 batteries highlight the critical role of multi-ion dynamics in achieving high reversibility and energy density. The carbonized MOF cathode material (ZIF-8/MIL-53) developed by Benjamin B. Peterson's team224 achieved a highly reversible Li–Br2 battery through multi-ion synergy. The nitrogen-doped porous carbon matrix has the following characteristics: the microporous structure (<2 nm) chemically anchors Br−/Br3− species through C–Br/N–Br bonds, effectively inhibiting the bromine shuttle effect; the mesoporous network promotes rapid Li+ transport and ensures efficient redox reaction (∼3.5 V). Among them, the ZIF-8 cathode exhibits better performance (273 mAh g−1, 88% capacity after 100 cycles), thanks to its higher nitrogen doping (enhanced bromine adsorption), more uniform 3nm pore size distribution (compared to 2–30 nm of MIL-53) and higher LiBr loading (22.7 wt%). This study provides new ideas for the development of practical high energy density (>900 Wh kg−1) halogen battery systems. The evolution of multi-electron redox chemistry was further exemplified in the development of a static Li–Br battery platform, which employed a TBABr3 catholyte and a Cl−-enriched electrolyte to facilitate a novel Br−/Br+ redox couple.225 This configuration produced a stable discharge plateau at 3.8 V and achieved a high capacity of 653 mAh g−1 with 98% coulombic efficiency over 100 cycles. Critically, the system exhibited low charge-transfer resistance (149.6 Ω) and minimal polarization (28 mV), which reflected the enhanced interfacial dynamics enabled by synergistic Li+/Br−/Cl− transport. Parallel progress in aqueous Li–Br systems has reinforced the centrality of multi-ion transport in unlocking high-performance metrics. For instance, the development of a highly concentrated 26 m Li–B5–C15–O6 electrolyte comprising LiBr, LiCl, and LiOAc facilitated a wide electrochemical stability window of 2.5 V. When paired with nitrogen-doped activated carbon (NAc) cathodes, the system achieved enhanced Br redox kinetics with a capacity of 376 mAh g−1 and 97% retention over 7800 cycles.226 The incorporation of LiOAc additives served to reduce water activity, effectively suppressing parasitic side reactions. When paired with an LTP@C/CNT anode, this approach enabled long-term full-cell operation, achieving a capacity of 158.2 mAh g−1 after 10000 cycles. These results collectively highlight the effectiveness of multi-ion modulation in maintaining electrode stability and prolonging cycle life under aqueous conditions.
These advances underscore the critical importance of multi-ion carrier architectures in reconciling energy density, voltage stability, and cycling durability. By enabling coordinated transport of Li+, Br−, Cl−, and Al3+ ions, Li–Br2 batteries surpass the limitations of single-ion systems. This multi-ion synergy enhances redox kinetics and ionic conductivity, while strategies such as physical confinement, chemical anchoring, redox mediation, and ion–solvent interaction optimization effectively suppress bromine crossover and stabilize interfaces.
3.2.2.1.3. Li–I2 batteries. Lithium iodine (Li–I2) batteries address the fundamental limitations inherent to halogen-based energy storage systems, particularly the challenge of balancing high energy density with electrochemical stability. Despite their impressive theoretical capacity approaching 2118 mAh g−1 and intrinsically fast redox kinetics, these systems have historically been hindered by persistent issues such as polyhalide dissolution and irreversible active material loss, which have restricted their practical application.
Recent advances have highlighted the essential role of coordinated multi-ion transport and stabilization mechanisms in overcoming these obstacles. Ding et al.227 introduced a pioneering triple-confinement strategy that synergistically combines mesoporous carbon matrices, nitrogen-doped chemical anchoring sites, and iodine–bromine complexes (I2Br3−). This comprehensive approach simultaneously addresses physical confinement, chemical binding, and solvation energy modulation, resulting in a CMK-3/LiI/LiBr cathode architecture that notably triples the cycling lifespan compared to conventional designs, while maintaining a high capacity of 463.4 mAh g−1. Building upon this foundation, Li et al.228 further advanced the field by developing a Cl−-mediated three-electron transfer system involving both I−/I+ and Cl−/Cl0 redox couples. This multi-ion redox configuration achieved outstanding performance metrics, including a capacity of 631 mAh g−1, an energy density of 2013 Wh kg−1, and a stable discharge plateau at 3.85 V, with less than 10% capacity degradation over 600 cycles. In situ Raman and XPS spectroscopies elucidated the complex conversion pathway from I− to I3− to oxidized I+Clx species, alongside the activation of Cl−/Cl0 redox, providing fundamental insight into how multi-ion coordination mitigates the typical trade-off between stability and reactivity in high-energy halogen systems.
Importantly, the multi-ion carrier concept extends beyond cathode material design to encompass electrolyte engineering. Gu et al.229 developed a quasi-solid-state electrolyte featuring amorphous metal–organic framework (MOF)/aramid interfacial networks that simultaneously enhance Li+ conductivity (1.1 × 10−3 S cm−1) and immobilize polyiodides through electron-mismatch-induced adsorption. This dual-function electrolyte effectively suppresses shuttle effects, enabling ultra-long cycle stability with a capacity retention of 94.1% after 3000 cycles at 20C, while maintaining compatibility with diverse cathode materials.
By integrating multi-level confinement architectures, orchestrated multi-electron and multi-ion redox pathways, and quasi-solid-state electrolytes with optimized ion transport and polyhalide stabilization, Li–I2 batteries have achieved significant advancements in capacity retention, voltage stability, and long-term cycling performance.
3.2.2.2. Na halogen batteries. Sodium halogen batteries constitute a promising category of secondary batteries that utilize metallic sodium as the anode and halogens or their derivatives as the cathode. The electrochemical operation of these systems is governed by reversible redox reactions involving sodium ions (Na+) and halogen species across multiple oxidation states (X−/X0/X+), enabling efficient charge storage and release during cycling. In reverse, halide ions are oxidized back into halogen molecules, and sodium ions reintegrate into the anode under an applied electric field. However, their practical deployment faces critical challenges, such as polyhalide dissolution (e.g., Br− shuttle effects causing capacity fade), dendritic sodium growth that can lead to short circuits, and relatively low operating voltages (e.g., ∼2.5 V for Na–I2 systems). As such, current research efforts are focused on extending cycle life (>300 cycles), suppressing parasitic side reactions, and developing electrolytes that offer both halogen compatibility and stable sodium plating/stripping behavior.
3.2.2.2.1. Na–Cl2 batteries. Sodium chlorine (Na–Cl2) batteries epitomize a promising class of multi-ion carrier electrochemical systems, where the coupled transport and redox activity of sodium cations (Na+) and chlorine-based anions (Cl−/Cl2) enables access to exceptional theoretical energy densities approaching 2500 Wh kg−1. In these systems, energy storage and release occur through reversible electrochemical reactions where sodium metal is oxidized at the anode and chlorine molecules are reduced at the cathode, with the opposite reactions taking place during charging. This dual-ion transport mechanism not only broadens the electrochemical landscape compared to traditional single-ion systems but also provides a unique platform for exploiting abundant and low-cost elements such as sodium and chlorine.
Despite their conceptual advantages, early Na–Cl2 batteries, which commonly utilized molten NaAlCl4-based electrolytes and operated at temperatures above 150 °C, faced several critical challenges.
These included the volatilization of chlorine gas, corrosive degradation of internal components, and limited reversibility of the Cl−/Cl2 redox couple. Altogether, these issues severely constrained their cycling stability, often limiting performance to fewer than 100 cycles. In addition, the passivation of cathode surfaces by electronically insulating NaCl and the propensity for sodium dendrite formation posed formidable barriers to practical applications. These issues underscored the urgent need for new strategies to regulate the behavior of both cationic and anionic species across electrodes and interfaces.
Recent breakthroughs have decisively demonstrated that the rational design of multi-ion carrier pathways can overcome these limitations by enhancing ionic conductivity, redox kinetics, and interfacial stability in tandem. Xiang et al.219 introduced a class of nitrogen-doped carbon cubosomes featuring a 3D bicontinuous mesoporous architecture with ∼39 nm pores and ∼34 nm walls. This framework not only facilitates efficient Na+ transport and electron conduction but also enhances Cl2 chemisorption and storage. As a result, the system achieved remarkable cycle stability of 250 cycles, providing compelling evidence that a carefully tuned electrode microstructure can simultaneously accommodate multiple ion fluxes. In parallel, Feng et al.230 developed an iodide-mediated redox shuttle (I−/I3−) that actively participates in the cathode reaction environment by dynamically dissolving the passivating NaCl layer and promoting Cl− oxidation. This redox mediator not only minimizes polarization but also improves the reversibility of Cl2 electrochemistry over extended cycling, highlighting the potential of redox intermediates as dynamic regulators of multi-ion transport. Expanding further on ion accessibility, Ma et al.231 designed a freestanding vertical-channel graphene cathode, which reduces tortuosity and enables fast SOCl2 vapor diffusion, thereby preventing pore clogging and ensuring homogeneous NaCl deposition. This architecture exemplifies how structural alignment at the nanoscale can accelerate anion transport and maintain electrochemical uniformity, a principle that may be generalized to other alkali metal–halogen systems.
To overcome the limitations associated with traditional Na–Cl2 systems, particularly the safety concerns related to toxic and volatile chlorine gas, recent efforts have explored the use of liquid Br2 as a cathode material in Na–Br2 batteries. While this substitution mitigates gas-related risks, it introduces new challenges, including sluggish redox kinetics and volatility of bromine at elevated temperatures. In response to these issues, Feng et al.232 introduced interhalogen polyanions (BrCl2−) into the electrolyte to regulate bromine redox behavior, enabling more efficient reaction kinetics at low temperatures. This led to the development of a novel Na–Br+ battery architecture, where Cl− anions play a crucial role in stabilizing and activating Br+ species through a reversible Br−/BrCl2− redox mechanism. The presence of BrCl2− polyanions facilitates additional electron transfer pathways and significantly improves the reversibility of bromine redox reactions. As a result, the Na–Br+ battery exhibits low polarization (0.18 V), a high energy density of 872 Wh kg−1, and remarkable temperature resilience across a broad range from −60 °C to 60 °C (Fig. 12b and c). Furthermore, the system demonstrates stable cycling over 400 cycles, highlighting the potential of interhalogen chemistry and halide-regulated redox strategies for developing high-performance, wide-temperature sodium–halogen batteries.
These synergistic advancements highlight how the deliberate orchestration of multi-ion carrier pathways, involving Na+ conduction, Cl−/Cl2 redox mediation, and interfacial ion regulation, can effectively overcome the inherent kinetic limitations and stability challenges that have historically hindered the development of Na–Cl2 batteries.
3.2.2.2.2. Na–Br2 batteries. Sodium–bromine (Na–Br2) batteries represent an emerging and versatile platform within the family of multi-ion carrier electrochemical systems, distinguished by their reliance on the coupled transport and redox behavior of sodium (Na+) and bromine-based species (Br−/Br3−/Br2). Operating through the reversible migration of Na+ ions between a sodium-based anode and a bromine-based cathode, Na–Br2 batteries undergo unique redox transformations involving Br2 reduction and the formation of soluble tribromide intermediates, which enable rapid kinetics and facilitate energy storage through both faradaic and surface charge processes.
Recent innovations in device architecture and interfacial regulation have underscored the central role of multi-ion coordination in advancing Na–Br2 battery performance. Wang et al.233 designed a dual-stimuli-responsive Na//Br2 system that exemplifies the multifunctionality achievable through strategic ion management. Their design integrates a sodium@carbon-fiber (Na@CF) composite anode with an AC-based bromine cathode, leveraging the high surface area of the AC (∼1800 m2 g−1) to promote double-layer capacitance in parallel with Br2 redox activity. This synergy yields a hybrid faradaic-capacitive behavior, enabling a power density of 1200 W kg−1 and a reversible capacity of 231 mAh g−1. Impressively, the system delivers a full-cell energy density of 103 Wh kg−1, maintains 98% capacity retention over 50 cycles, and exhibits thermally modulated, voltage-dependent electrochromism, with switching speeds accelerating from 1 hour to just 10 minutes as current density increases. In situ Raman spectroscopy further elucidates the Br−/Br3− redox mechanism responsible for both energy storage and optical modulation, affirming the system's electrochemical and multifunctional coherence. Complementing this work, Wang et al.234 advanced Na–Br2 chemistry in the aqueous regime through the development of a hybrid-ion battery coupling an aqueous Br2 redox couple with a sodium ion embedding anode. This design capitalizes on the high activity and ionic mobility of aqueous-phase redox species while mitigating the instability and low capacity that typically afflict aqueous batteries.
These advances highlight that precise regulation of Na+ and Br−/Br3−/Br2 transport plays a pivotal role in enabling Na–Br2 batteries to combine high energy density with rapid redox kinetics and diverse functionalities. Such functionalities include electrochromic behavior and the seamless integration of faradaic and capacitive charge storage, highlighting the transformative potential of multi-ion coordination in advancing scalable and intelligent energy storage technologies.
3.2.2.2.3. Na–I2 batteries. The operation of sodium–iodine (Na–I2) batteries relies on the coupled redox behavior of Na+ and iodine species (I2, I−, and I3−), enabling dual-ion electrochemical processes that offer high theoretical capacities.235,236 The practical utilization of iodine-based systems has been constrained by the concurrent dissolution of intermediate species and sluggish I2/2I− redox kinetics, both of which contribute to rapid capacity decay and compromised long-term stability. Recent developments have demonstrated that treating this system through the lens of multi-ion carrier strategies, particularly by coordinating Na+ ion transport with controlled iodine speciation dynamics, can lead to substantial performance improvement. This strategy involves the integration of structural confinement, catalytic enhancement of redox reactions, and precise interfacial control, collectively mitigating iodine dissolution and accelerating redox kinetics to achieve more stable and efficient Na–I2 energy storage.
The electrochemical performance of Na–I2 batteries is fundamentally governed by the coordinated regulation of multiple ionic species, where Na+ cations and iodine anions (I−/I3−) engage in intricate synergistic interactions. At the cathode interface, dual-ion co-regulation is critical, as demonstrated by Gong's237 quantum-confined iodine system, where Na+ intercalation kinetics and I3− redox activity are simultaneously optimized through graphene-mediated ion channels, achieving a remarkable 141 mAh g−1 capacity. The ion-selective interface concept represents a breakthrough in multi-ion management. Tian's238 NaI-rich SEI layer creates a kinetic ion filter, offering a low 0.02 eV barrier for Na+ diffusion while effectively blocking I3− crossover through strong chemical interactions. This selective ion transport enables an unprecedented cycle stability of 2200 h by precisely controlling the movement of different ionic species. Molecular-scale ion anchoring strategies have emerged as powerful tools for polyiodide control. Wang's239 MOF design employs Fe–O4 sites as multi-ion hubs, simultaneously coordinating I3− anions while maintaining pathways for Na+ transport. This delicate balance between ion confinement and mobility results in both high capacity (208 mAh g−1) and exceptional cycling stability. The ion transport architecture in 2D materials reveals fascinating size-charge selectivity. Qian's240 iodinene nanosheets create vertically aligned ion highways, where the 0.07 eV Na+ migration barrier and I− adsorption sites work in concert to enable ultrafast (10 A g−1) yet stable operation. By facilitating a direct I2 to NaI redox pathway, Zhang's241 catalytic strategy simplifies ionic processes within the system. This not only suppresses the generation of unstable polyiodide intermediates but also exemplifies how targeted catalyst design can enhance multi-ion reaction selectivity and suppress competing side reactions. Advanced ion-sieving matrices like Zhang's242 γ-Mo2N/N-doped carbon demonstrate spatially resolved ion management, with mesopores optimized for Na+ flux and chemical traps for I3− retention. Feng's230 hydrogen-bonding strategy introduces a novel ion stabilization mechanism, where I+–Cl− intermediates are stabilized through multiple non-covalent interactions. The ultimate frontier lies in system-level ion orchestration, as showcased by Xu's243 separator engineering, which establishes gradient ion regulation across the entire cell. This approach creates distinct transport environments for different ionic species, from electrostatic barriers for polyiodides to low-resistance paths for Na+.
In summary, by simultaneously regulating Na+ ion transport and iodine speciation, recent strategies involving nanoconfinement, catalytic interfaces, two-dimensional materials, and multifunctional membranes have markedly improved energy density, rate capability, and cycling stability. Continued progress will require precise control of host–guest interactions, a deeper understanding of multi-ion migration mechanisms, and the scalable construction of hierarchical architectures, all of which are essential for realizing the full potential of Na–I2 systems in practical energy storage applications.
3.2.2.3. K halogen batteries. Potassium halogen batteries represent a promising frontier in multi-ion carrier systems, capitalizing on the abundant availability of potassium and the high electronegativity of halogen cathodes such as iodine and bromine to offer a cost-effective alternative to lithium-based energy storage. These systems harness the synergistic electrochemical interplay between K+ and halogen redox couples (X2/X−), facilitating reversible energy storage through coupled multi-ion migration and redox reactions. The fundamental electrochemical process involves the oxidation of potassium metal at the anode, releasing K+ ions and electrons, while the halogen molecules at the cathode undergo reduction to form halide ions, as described by the overall reaction. Despite their theoretical advantages, practical applications are hindered by the large K+ ionic radius, which slows ion diffusion, and by cathode instability caused by halogen dissolution and shuttle effects. As a result, research has focused on material and electrolyte innovations to improve ion transport, reduce active material loss, and enhance cycling stability.
3.2.2.3.1. K–Br2 batteries. Within this family, potassium bromide (K–Br) batteries have recently gained considerable attention as an alternative energy storage technology poised to alleviate lithium resource constraints. This system operates on a redox reaction between liquid bromine (Br2) and K+ ions, utilizing a hollow carbon sphere cathode paired with a K metal anode. A critical advancement is the formation of a crystalline KBr film during discharge, which serves as a reversible phase crucial to achieving a high specific capacity of approximately 400 mAh g−1 at a stable voltage of 3.72 V.244 This nanostructured KBr layer not only stabilizes the electrode interface but also facilitates efficient multi-ion coordination, thereby promoting enhanced electrochemical reversibility and cycle life. Overall, the K–Br batteries demonstrate the feasibility of potassium-based systems, with the potential to contribute significantly to the future of large-scale energy storage technologies.
3.2.2.3.2. K–I2 batteries. Similarly, potassium–iodine (K–I2) batteries have emerged as compelling candidates for sustainable energy storage, combining high power density and environmental compatibility with the potential for rapid charge/discharge capability. Yet, these batteries encounter intrinsic limitations including the dissolution of polyiodide intermediates and unstable redox chemistry, which undermine cycling stability and capacity retention. Addressing these issues, Zhao et al.217 developed a scalable “solution-adsorption” approach to fabricate flexible, free-standing electrodes by wrapping carbon nanotube paper (CNP) with the Ti3C2(OH)x MXene. This hybrid architecture exploits the strong affinity of CNP for the I3−/I− redox couple alongside MXene's surface Ti–OH groups, which form robust halogen bonds (I⋯H–O) to effectively suppress polyiodide dissolution. The resulting electrode exhibits highly reversible redox behavior, delivering a specific capacity of 226 mAh g−1 at 0.2C, excellent cycling stability with 83% capacity retention over 1000 cycles at 10C, and ultrafast charge/discharge within 10 minutes. Notably, a 100 mAh pouch cell prototype achieves an energy density of 130 Wh kg−1, surpassing conventional lithium-ion cathodes and underscoring the commercial viability of K–I2 systems for grid-scale energy storage.
3.2.2.4. Ca halogen batteries. The Ca halogen batteries integrate the high volumetric capacity of Ca metal anodes (2075 mAh cm−3) with the high redox potential of halogen cathodes (typically exceeding 2.5 V), offering a promising route toward high-energy-density storage. A key strategy involves suppressing the shuttle effect of halogen species through the porous confinement of bromine or iodine, which effectively limits active material loss and enhances cycling stability. Complementing this, the use of a borohydride–bromide mixed electrolyte enables reversible Ca2+ deposition and dissolution, addressing a longstanding challenge in calcium-based systems. The synergy between physical confinement and electrolyte design stabilizes redox reactions and facilitates the dual-ion electrochemical mechanism, in which Ca2+ cations and halide (X−) anions undergo coupled migration and redox processes that are essential for energy storage.
3.2.2.4.1. Ca–Cl2 batteries. Among these systems, calcium–chlorine (Ca–Cl2) batteries have recently drawn attention for addressing fundamental issues limiting conventional calcium metal batteries, such as insufficient energy density and poor performance at low temperatures. A recent breakthrough employed a specially engineered electrolyte based on lithium difluoro(oxalate)borate (LiDFOB), which facilitates the reversible CaCl2/Cl2 redox couple.245 This electrolyte not only promotes high specific capacity (up to 1000 mAh g−1) but also enables excellent rate capability (500 mA g−1), while maintaining a remarkable capacity retention of 96.5% over 30 days. Critically, the LiDFOB-mediated electrolyte enhances the dissociation and transport of Ca2+ ions and Cl-based species, thereby improving multi-ion carrier dynamics and electrochemical kinetics. This strategic electrolyte design mitigates the sluggish ion diffusion typically associated with divalent carriers and complex halide species, illustrating how deliberate multi-ion carrier engineering can overcome intrinsic electrochemical challenges.
By harnessing the synergistic transport and redox behaviors of Ca2+ and halogen anions through carefully engineered electrode architectures and tailored electrolytes, calcium–halogen batteries present a promising pathway toward high-capacity, long-life, and structurally robust energy storage systems. However, the realization of their full potential in large-scale, high-energy applications remains hindered by critical challenges, particularly the sluggish diffusion of multiple halide species and the inherently limited kinetics of solid-state Ca2+ conduction.
3.2.2.5. Mg halogen batteries. Mg–halide batteries function through the coupled processes of Mg2+ ion transport and halogen-based redox reactions, capitalizing on magnesium's high volumetric capacity (∼3833 mAh cm−3) and intrinsic safety due to its dendrite-free deposition behavior. While the electrochemical framework of Mg–halogen systems is conceptually simple, their practical realization is hindered by several intrinsic challenges. These include sluggish Mg2+ diffusion kinetics due to its strong solvation and high charge density, instability of conventional electrolytes in the presence of reactive halogens, and the shuttle effect of dissolved halogen species, all of which impair coulombic efficiency and cycling life.
To overcome these obstacles, recent studies have centered on the development of corrosion-resistant electrolytes with enhanced chemical compatibility, carbon–halogen composite cathodes that improve redox reversibility, and solid-state or quasi-solid-state architectures that provide better interfacial contact and controlled ion transport. These innovations not only alleviate Mg2+ transport limitations but also enhance the reversibility of halogen redox reactions, both of which are essential for optimizing the performance of multi-ion systems in magnesium–halide batteries.
3.2.2.5.1. Mg–Br2 batteries. An exemplary strategy was reported by Guo et al.,246 who proposed a bond-weakening approach in a Mg–Br2 battery by incorporating succinimide (SN−) ions to disrupt the strong Mg–Br bonding interactions. The introduction of these soft anionic species led to the formation of a flexible, weakly coordinated environment around magnesium, facilitating both efficient Mg2+ de-solvation and accelerated Br2 conversion kinetics. This tailored coordination chemistry enabled rapid multi-ion transport and ultrafast redox kinetics, delivering a high discharge voltage of 2.7 V, a reversible specific capacity of 326 mAh g−1, and remarkable cycling stability even under extreme conditions (10C rates and −55 °C operation). Crucially, this system highlights the transformative impact of deliberately engineered multi-ion transport environments, which simultaneously enhance ion mobility, stabilize interfacial processes, and facilitate efficient halogen redox reactions.
3.2.2.6. Zn halogen batteries. Zn halogen batteries constitute a pivotal subclass within multi-ion carrier power supply systems, employing Zn metal as a high-capacity anode and halogen-based cathodes to facilitate reversible redox reactions. This dual-ion process, involving both Zn2+ cations and halide anions (X−), defines the multi-ion carrier characteristic of Zn–halogen batteries. Despite these benefits, Zn–halogen batteries face several challenges that limit practical applications. These include halogen sublimation and volatility, slow reaction kinetics at the halogen cathode, shuttle effects caused by soluble halogen species, and dendritic growth of Zn that threatens battery safety and cycle life. To overcome these issues, research has focused on electrolyte optimization, physical and chemical confinement of halogens, and interface engineering to improve redox reversibility, suppress shuttle effects, and control Zn deposition.
3.2.2.6.1. Zn–Cl2 batteries. Within this family, Zn–Cl2 batteries typically employ aqueous electrolytes, where Zn oxidation occurs at the anode and chlorine reduction at the cathode. However, the high redox potential of chlorine often exceeds the electrochemical stability window of water, leading to parasitic reactions such as chlorine gas evolution and electrolyte decomposition, which pose significant challenges for long-term operation. Instead of relying on a single ionic species, multi-ion batteries utilize the synergistic coordination between multiple charge carriers to improve redox reversibility, ionic transport, and electrode stability. For example, Liang et al.247 developed a high-energy aqueous Zn–Cl2 battery that incorporates chlorine-based redox activity within a concentrated ZnCl2 electrolyte, enabling a reversible three-electron transfer process and dramatically enhancing energy density. This design capitalizes on the co-existence and interplay of Zn2+, Cl−, and I− ions, exemplifying the functional advantages of multi-species participation in aqueous systems. Further advancing this concept, Wang and co-workers208 introduced a Zn–Cl2 battery comprising a graphite paper cathode, a Zn anode, and an aqueous electrolyte containing tetramethylammonium chloride and Na2CO3. The optimized electrolyte pH enabled stable chlorine redox reactions while maintaining high voltage and long cycle life. This system not only demonstrates the practicality of halide coordination with organic and inorganic ions but also reveals the potential for cost-effective, scalable, and environmentally friendly manufacturing via multi-ion chemistry.
Mechanistic insights into halogen storage behavior were offered by Xu et al.,248 who investigated ZnCl2/ZnBr2 hydrate-based aqueous dual-ion batteries using graphite electrodes. The study revealed a dynamic competition between intercalation and conversion processes at the cathode, driven by solvation states and interfacial ion interactions. These processes are fundamentally determined by the multi-ion composition of the electrolyte, underscoring its critical role in shaping electrochemical behavior. These findings underscore the importance of understanding not only the redox chemistry but also the structural and solvation environment surrounding active ions. To further overcome the limitations of sluggish Cl2 cathodic kinetics and limited coulombic efficiency, Chen's group249 designed a Zn–Cl2 battery in which MnO2 serves as a redox adsorbent, regulating chlorine storage and release (Fig. 13a–c). This architecture enabled a discharge voltage of 2.0 V at 2.5 mA cm−2 and demonstrated outstanding cycling stability over 1000 cycles with an average coulombic efficiency of 91.6%, far surpassing conventional Zn–Cl2 systems.
 | ||
| Fig. 13 (a) Schematic diagram of Zn–Cl2@MnO2 battery with the addition of Mn2+ in the electrolyte. (b) CV curves of Zn–Cl2@MnO2 battery and conventional Zn–Cl2 battery. (c) Discharge curves of conventional Zn–Cl2 batteries and Zn–Cl2@MnO2 batteries. Inset: Time–current curves in the corresponding case. The batteries were charged to 0.4 mAh cm−2 at 2.3 V and discharged to 1.4 V at 2.5 mA cm−2. Reproduced with permission from ref. 249. Copyright 2023, Wiley. (d) Reaction mechanism, challenges, and optimization strategies of zinc-bromine batteries. (e) Cyclic voltammetry (CV) curves of the Zn//I2, Zn//IBr, and Zn//Br2 cells. Reproduced with permission from ref. 250. Copyright 2023, Wiley. (f) Illustration of the reactions of iodine batteries. Reproduced with permission from ref. 254. Copyright 2021, Royal Society of Chemistry. (g) and (h) Schematic comparison of charge–discharge processes in conventional and high-energy iodine electrodes. Reproduced with permission from ref. 263. Copyright 2023, Wiley. | ||
The integration of multi-ion carrier mechanisms in Zn–Cl2 batteries offers a rational strategy to address persistent challenges such as halogen shuttle effects, Zn dendrite formation, and electrolyte decomposition. By harnessing the synergistic interplay between multiple ionic species, these systems promote fast ion transport and enhance interfacial stability, thereby improving both electrochemical performance and long-term durability.
3.2.2.6.2. Zn–Br2 batteries. Zn–Br2 batteries function through a redox mechanism between Zn and bromine in an aqueous ZnBr2 electrolyte, where Zn undergoes reversible plating and stripping at the anode, while bromine species are stabilized at the cathode through complexation reactions (Fig. 13d). However, their broader adoption has been limited by persistent issues such as self-discharge, bromine volatility, and Zn anode degradation, which stem largely from unregulated ion transport and interfacial instability.
Recent breakthroughs have underscored the pivotal role of multi-ion carrier strategies in addressing these challenges and unlocking the full potential of Zn–Br2 batteries. Notably, Chen et al.250 have recently advanced Zn–halogen chemistry by introducing an all-active solid inter-halogen compound, IBr, as the cathode material. In this architecture, the neutral bromine species (Br0) generated during oxidation are coordinatively immobilised by iodine, thereby suppressing the cross-diffusion of Br2/Br3− that typically plagues bromine-based cells. This mitigation of active species crossover significantly reduces self-discharge and enhances coulombic efficiency. As a result, the Zn–IBr battery achieved a high energy density of 385.8 Wh kg−1. Notably, the solid IBr participates in redox processes as an integrated interhalogen compound, rather than undergoing separate redox reactions of I− and Br− (Fig. 13e). This unique behavior gives rise to two distinct pairs of redox peaks, indicating complex but well-coordinated redox activity. Yu et al.251 developed a mixed alkali–acid electrolyte system that achieved a record-high average operating voltage of 2.1 V, which stands as the highest reported for aqueous Zn-based batteries to date. This advancement also led to significant improvements in specific capacity and cycling durability. This work exemplifies how rational electrolyte design can manipulate ionic environments to broaden the operational window of multi-ion systems. Complementing this, Hu et al.252 tackled the long-standing issue of polybromide shuttling and Zn dendrite formation by incorporating methylpyridinium bromide (MPIBr) as an electrolyte additive. This molecular-level engineering effectively suppressed polybromide crossover and promoted uniform, dendrite-free Zn deposition, thereby highlighting the importance of interfacial ion regulation in stabilizing complex multi-ion dynamics. In addition to electrolyte innovations, catalytic strategies have been employed to address the inherent sluggishness and solubility of bromine redox intermediates. For example, Chen et al.253 developed a FeSAC-CMK catalyst featuring atomically dispersed Fe–N5 sites, which concurrently immobilized bromine species, inhibited the formation of Br3− intermediates, and accelerated the Br−/Br0 redox conversion. This multifunctional electrocatalyst not only improved redox kinetics but also enhanced overall energy density and cycling stability in Zn–Br2 batteries by facilitating efficient multi-ion interactions.
These synergistic developments in electrolyte engineering, interfacial modification, and catalytic mediation collectively illustrate the transformative potential of multi-ion carrier strategies in advancing Zn–Br2 batteries technology, offering new pathways to overcome the intrinsic limitations of single-ion systems while achieving superior energy density, voltage output, and long-term cycling performance.
3.2.2.6.3. Zn–I2 batteries. The operation of Zn–I2 batteries relies on the cooperative interaction between the highly reversible Zn plating and stripping processes at the anode and the complex redox behavior of iodine species at the cathode, which involves multiple electron transfer steps and phase transitions among I2, I5−, I3−, and I−. This combined mechanism underpins their notably high theoretical energy density. However, the electrochemical performance of Zn–I2 batteries hinges on the precise coordination of multiple ionic species. Although the shuttling of soluble polyiodides enables high capacities, it simultaneously promotes parasitic reactions and active-material loss; likewise, uneven Zn2+ flux can trigger dendrite formation and side reactions. Consequently, progress in this chemistry hinges on a fundamental understanding of multi-ion transport, coupled with judicious electrolyte formulation and interfacial engineering.
Recent studies underscore how electrolyte regulation can unlock otherwise inaccessible high-valent iodine redox processes. Zhi et al. showed that introducing fluoride or chloride ions activates and stabilises the I0 → I+ transition in addition to the conventional I−/I+ couple at ∼0.54 V, yielding two stable discharge plateaus at 1.70 V and 1.45 V and markedly raising the cell's voltage output. Complementary advances in host architecture and electrolyte design further demonstrate the power of coordinated multi-ion control (Fig. 13f).254 Bai et al.'s255 work on nanoporous carbon cloth demonstrated that rational host structure design can confine iodine species while maintaining redox accessibility, boosting both energy density and cycling stability. Building on this, Shang et al.256 developed an alginate-based hydrogel electrolyte with ion-selective properties that effectively suppressed both triiodide shuttle and Zn side reactions. A major leap forward was made by Zou et al.,257 who pioneered a four-electron redox process involving I2/I+ coupling. Enabled by a finely tuned electrolyte composition, this approach significantly boosted energy density through the precise modulation of multi-electron transfer pathways. In a complementary approach, Li et al.258 developed a ternary eutectic electrolyte system incorporating nicotinamide ligands. The resulting [I(NA)2]+ complex, combined with stabilized Zn deposition, improved reversibility at both electrodes by orchestrating multi-ion interactions at the molecular level.
Beyond bulk-phase regulation, recent studies have expanded the frontier of multi-ion management to interfacial engineering. Chai et al.259 introduced halogen bonding strategies that significantly strengthen iodine–host interactions at the atomic level, establishing a versatile platform applicable to various metal–halogen battery systems. Extending this concept into solid-state domains, Liu et al.260 developed a dual-network hydrogel electrolyte that simultaneously regulated Zn2+ and I− transport, thereby improving both mechanical integrity and electrode/electrolyte interfacial stability in Zn–I2 batteries. Further innovations have explored new avenues of material functionality. Wu et al.261 polyaniline binder technology provided chemical anchoring for polyiodides while preserving ionic conductivity, offering a robust strategy for electrode architecture optimization. Meanwhile, Xu et al.262 addressed the challenge of low-temperature performance by employing a hydrated eutectic electrolyte. This electrolyte, with its precisely engineered solvation structures, effectively suppressed polyiodide disproportionation, thereby enabling stable multi-ion transport and reliable Zn–I2 batteries operation even in harsh, low-temperature environments.
Recent breakthroughs in multi-ion carrier systems demonstrate that the strategic regulation of multiple ionic species can significantly enhance battery performance. In addition to their role as charge carriers, halide ions such as chloride (Cl) and fluoride (F−) have been shown to play a pivotal role in stabilizing reactive iodine species through interhalogen interactions, thereby enhancing the electrochemical reversibility of iodine-based redox processes. Electrolyte engineering strategies incorporating Cl− and F− salts have proven particularly effective in stabilizing high-valence I+ species, which are typically unstable in conventional aqueous environments (Fig. 13g and h). For instance, in a representative Zn–I2 battery system employing a Cl−-containing electrolyte, two distinct discharge voltage plateaus at 1.65 V and 1.30 V vs. Zn/Zn2+ were observed, which can be attributed to the activation of the I0/I+ redox couple.263 This activation not only extended the electrochemical window but also resulted in a remarkable 108% increase in specific capacity and a 231% enhancement in energy density. These improvements were further supported by the use of MXene-based host materials, which contributed to improved reaction kinetics and effective confinement of iodine species. Spectroscopic analyses confirmed the essential role of Cl− and F− in stabilizing the I0/I+ redox transition, underscoring the importance of multi-ion coordination in expanding the accessible redox chemistry of iodine. Specifically, in bismuth-iodide perovskite cathodes, the synergistic interplay among the redox states I−, I0, and I+, combined with Cl− mediators and BAD+ stabilizers, is stabilized through Bi–I–I halogen bonds and CN–H–I hydrogen bonds. This well-orchestrated ionic network greatly improves cycling stability, enabling the cathode to retain 92% of its capacity even after 30000 cycles.264 For aqueous Zn–I2 systems, a gelatinized starch binder selectively confines I−, I3−, and I2 through its helical structure, suppressing shuttling while accommodating volume changes via hydrogen-bonded networks, enabling 89.6% capacity retention over 48000 cycles.265 Further advancing this concept, a ternary eutectic electrolyte leverages the Br−-mediated solvation structure [Zn(H2O)3(Ace)2Br]+ to stabilize iodine redox chemistry while directing uniform Zn2+ deposition, achieving near-100% iodine reutilization and dendrite suppression.266 These Zn–I2 batteries highlight the transformative potential of multi-ion coordination, where targeted interactions between redox-active species, mediators, and stabilizers unlock enhanced stability, capacity, and kinetics in next-generation energy storage.
Collectively, these advancements highlight the potential of Zn–I2 batteries as a model platform for multi-ion carrier energy storage, wherein the coordinated regulation of Zn2+ cations, I− anions, and polyiodide intermediates plays a critical role in achieving superior electrochemical performance. Through the integration of host–guest chemistry, coordination interactions, and interfacial engineering, researchers have progressively elevated Zn–I2 batteries from conceptual frameworks to practically viable energy storage systems.
3.3. Multi-cation and multi-anion batteries
Multi-ion carrier batteries are transforming EPSs by establishing a new paradigm based on synergistic cation and anion co-redox (CCAR), in which both cations such as Li+, Na+, and Mg2+ and anions actively engage in redox reactions. Unlike conventional single-ion systems that are often constrained by charge compensation bottlenecks, unidirectional ion transport, or polarization effects, CCAR systems enable the concurrent intercalation of cations at the anode and incorporation or coordination of anions at the cathode during charging, with the process fully reversing upon discharge. This synchronized ionic movement not only enhances total charge storage capacity but also facilitates more efficient ion transport and interfacial stability, as anions often contribute to stabilizing solvation structures or electrode–electrolyte interfaces. Crucially, this multi-ion mechanism decouples redox potentials, enabling flexible electrode selection and mitigating voltage hysteresis or capacity fading commonly seen in single-ion systems. The CCAR model therefore illustrates how multi-ion cooperation can fundamentally transcend the classical electrochemical limitations of battery chemistry, opening avenues toward higher capacity, longer life, and more adaptable storage architectures.The conceptual roots of multi-ion redox can be traced to the seminal work of Rouxel267 in 1996, who first proposed the possibility of coupled cationic and anionic redox reactions within lithium–sulfur compounds in lithium-ion systems. This early recognition of anion involvement in reversible redox behavior laid the theoretical foundation for the modern CCAR framework, demonstrating that the inclusion of multiple charge carriers could unlock additional redox-active sites and boost theoretical capacities beyond what is achievable through cation storage alone. Building on this foundation, recent research has brought multi-ion carrier chemistry from theoretical postulation to the practical design strategy. Notable progress in this area includes the work by Lv et al.,268 who pioneered a quasi-solid-state Zn-dual halogen battery system. By enabling the concurrent activation of I−/I0/I+ and Br−/Br0 redox couples, this configuration achieves a triple continuous electron transfer pathway. This multi-ion activation dramatically enhances specific capacity while preserving the safety profile and environmental benignity of the aqueous system. Extending this concept, Xie269 developed an isohalogen electrolyte strategy wherein bromine mediates iodine redox activity. The formation of IBr/Br2 intermediates facilitates multi-electron transfer with reduced voltage polarization, yielding a substantial energy density improvement and redefining the thermodynamic landscape of halogen-based systems (Fig. 14a and b). These innovations clearly highlight how multi-ion carrier systems can bridge the gap between the favorable redox potentials of halogen chemistry and the kinetic constraints of aqueous Zn-based devices.
 | ||
| Fig. 14 (a) Schematic of an assembled battery with a multielectron transfer cathode and cadmium metal as the anode. (b) The charge–discharge curves of the IBA and IA electrolytes at 20 mA cm−2 (IBA electrolyte: 1 M HI + 1 M HBr + 1 M H2SO4 + 1 M CdSO4; IA electrolyte: 1 M HI + 1 M H2SO4 + 1 M CdSO4). Reproduced with permission from ref. 269. Copyright 2024, Springer Nature. (c) CV curve of dual-host shows individual redox process of viologen and triazine block. (d) Comparison of the amplified GITT curves to show voltage relaxation process during discharge. Reproduced with permission from ref. 270. Copyright 2023, Wiley. (e) Schematic representation illustrating the transformation of the cathode during the charging and discharging processes for a Zn8CaNaVO-44@CS cell employing 3 M Zn(OTF)2 and 0.1 M Na2SO4 as the electrolyte. Reproduced with permission from ref. 271. Copyright 2024, Royal Society of Chemistry. | ||
A paradigm shift is also emerging through multi-ion co-storage in porous organic frameworks, which effectively overcomes the intrinsic limitations of single-ion storage mechanisms. For example, Sun et al.270 designed a dual-host system featuring sequential storage stages: initial viologen reduction involving anion extraction, followed by simultaneous triazine/viologen reduction via cation–anion co-insertion, and finally, triazine-based cation storage. This A−/M+ (PF6/Li+, OTF/Mg2+, and OTF/Zn2+) co-storage mechanism significantly enhances reaction kinetics, as evidenced by galvanostatic intermittent titration technique (GITT) analysis showing a reduced voltage relaxation of 342 mV compared to single-ion hosts. Moreover, the advanced triple-mode operation involving A+, A−, and M+ achieved outstanding performance, delivering a capacity of 348 mAh g−1 across a wide voltage range from 1.5 to 4.5 V, with energy and power densities reaching 878 Wh kg−1 and 28 kW kg−1, respectively (Fig. 14c and d). These breakthroughs stem from finely tuned coulombic interactions that lower the Li+ diffusion barrier by 73%, while maintaining robust cycling stability over 20000 cycles. This establishes a new benchmark for hybrid systems that transcend the traditional battery-supercapacitor performance tradeoff. Equally compelling are recent advances in aqueous systems, where multi-ion regulation plays a pivotal role in achieving high performance under challenging conditions. Zhang et al.271 demonstrated that dual-cation doping with Na+ and Ca2+, combined with the in situ formation of a stable cathode–electrolyte interphase, enabled adaptive Zn2+/H+ intercalation in aqueous ZIBs. This design achieved a capacity retention of 99.4% after 120 cycles, even in harsh operational environments (Fig. 14e). Their findings highlight the broad applicability of multi-ion strategies not only in non-aqueous systems but also in aqueous batteries, where the presence of competing ionic species often complicates both interfacial dynamics and bulk transport processes.
These studies collectively illustrate that multi-ion carrier engineering represents more than just an incremental improvement over single-ion strategies; it reflects a fundamental shift in the conceptual framework of multi-ion EPSs. By enabling multiple ionic species to actively and cooperatively participate in redox reactions, reinforce structural integrity, and regulate interfacial dynamics, this approach provides a comprehensive solution to the limitations inherent in conventional battery systems.
4. Hybrid battery-capacitors
The evolution from conventional capacitors to advanced supercapacitors marks a pivotal shift in the landscape of multi-ion EPSs, driven by the transformation from purely electrostatic mechanisms to interfacial and bulk ion-involved charge storage. At the heart of this transition lies the increasingly central role of ion carriers, not merely as charge balancers but as active participants and reaction substrates in energy storage processes. Early dielectric capacitors, dating back to the mid-18th century, operated purely through electron-based electrostatic storage across vacuum or dielectric materials, with no ionic involvement.272 These systems offered ultrahigh power densities (in the MW kg−1 range) due to instantaneous charge-displacement but suffered from extremely low energy densities (≪0.1 Wh kg−1) and were limited by their lack of tunable chemistry. The advent of electrolytic capacitors in the 1930s introduced thin metal oxide dielectrics and conductive electrolytes, where ions began to play a passive electrostatic role in forming interfacial charge balancing layers.273 Although energy density improved slightly (∼0.01–0.1 Wh kg−1), the fundamental storage mechanism remained predominantly electrostatic, with the primary carriers still being electrons and bound charges within the dielectric layer.A transformative leap occurred with the introduction of electric double-layer capacitors (EDLCs), grounded in Helmholtz's interfacial capacitance model,274 and exemplified by Becker's early porous carbon systems.272 These first-generation supercapacitors leveraged high-surface-area carbon materials to physically adsorb electrolyte cations/anions (e.g., K+, SO42−) at the electrode/electrolyte interface, forming an electric double layer (Fig. 15a).275 This breakthrough redefined ion carriers as the primary energy storage medium, as reversible ion migration and surface adsorption replaced static electrostatics. The resulting devices delivered moderate energy densities (5–10 Wh kg−1), exceptionally high power densities (1–10 kW kg−1), and cycle life exceeding 500000 cycles.276 However, the performance remained constrained by the surface-confined nature of the charge storage, pore-ion size mismatch, and limited voltage windows.
 | ||
| Fig. 15 (a)–(c) Representative electrochemical charge storage mechanisms, including electric double-layer capacitance, pseudo-capacitance, and battery-type storage. (d) Summary of characteristic features associated with each mechanism, including cyclic voltammetry profiles, galvanostatic charge–discharge behavior. | ||
The discovery of pseudo-capacitance in 1962 by Conway marked a second milestone, enabling faradaic charge transfer processes at or near electrode surfaces.277 Here, a highly reversible Faraday charge transfer reaction occurs at the electrode/electrolyte interface, accompanied simultaneously by the insertion/extraction or adsorption/desorption of electrolyte ions to maintain charge balance. Multi-ion participation was no longer auxiliary but essential to the Faraday reaction, involving mechanisms such as surface redox reactions in conductive polymers and metal oxides (e.g., RuO2 and MnO2),278 and ion intercalation into layered hosts like MXenes and δ-MnO2 (Fig. 15b).279 These systems exploited the synergistic interplay between electrons and ions, especially H+ and small cations, which facilitated fast redox transitions and significantly boosted energy density (up to 20–50 Wh kg−1) compared to EDLCs.280 While power density was generally lower than EDLCs due to ion diffusion limitations, pseudo-capacitors bridged the gap between capacitive and battery-like behaviors, underscoring the versatility and importance of tailoring ionic species and transport pathways.
Further innovations led to hybrid battery-capacitors, which combine a battery-type electrode (capable of bulk ion intercalation or deep redox reactions) with a capacitive electrode (typically carbon-based) (Fig. 15c). In battery systems, the appearance of distinct redox peaks in cyclic voltammetry (CV) profiles and well-defined voltage plateaus in galvanostatic charge–discharge (GCD) curves reflects the reversible insertion and extraction of ions (Fig. 15d).281,282 By contrast, supercapacitors exhibit nearly rectangular CV traces and linear GCD curves with constant slopes, signatures of surface-controlled charge storage. Hybrid battery-capacitor devices combine these two electrochemical paradigms, seeking to deliver both the high energy density of batteries and the rapid charge–discharge capability of supercapacitors. Consequently, their CV curves often retain a quasi-rectangular shape, while their GCD profiles lack pronounced voltage plateaus. These features confirm that hybrid systems can sustain swift ion transport for high power output without sacrificing the energy storage advantages intrinsic to battery-type reactions.
Hybrid battery-capacitors are generally classified into three mechanistic categories based on the distinct ion migration pathways involved during electrochemical cycling: the “accordion” mechanism, the “rocking-chair” mechanism, and a mixed mechanism. In the accordion-type configuration, battery-type materials typically serve as the anode, while capacitor-type materials function as the cathode (Fig. 16a).283,284 During charging, metal cations are inserted into the anode, whereas anions are electrostatically adsorbed onto the cathode surface. Upon discharge, the reverse process occurs, with metal ions extracted from the anode and anions desorbed from the cathode. However, due to irreversible adsorption or trapping of ions at the electrode interfaces, progressive changes in electrolyte concentration may occur over long-term cycling, potentially affecting device stability. The rocking-chair mechanism features a reversed configuration in which the cathode comprises battery-type materials, and the anode adopts capacitor-like characteristics (Fig. 16b). During charging, metal cations are fully extracted from the cathode and migrate through the electrolyte to adsorb onto the anode surface. Discharge prompts their desorption from the anode and subsequent reinsertion into the cathode, which is analogous to LIBs. Unlike the accordion-type, the rocking-chair architecture maintains a relatively constant electrolyte composition throughout the cycle, offering better chemical balance and improved long-term electrolyte stability. In contrast, the mixed mechanism integrates battery-type and capacitor-type characteristics within at least one electrode (Fig. 16c). For instance, a hybrid cathode may simultaneously undergo metal-ion extraction and anion adsorption during charging, while a capacitor-type anode adsorbs the corresponding metal ions. Upon discharge, metal ions are desorbed from the anode and reinserted into the cathode, accompanied by the desorption of anions. This configuration leverages both faradaic and non-faradaic processes within a single electrode, enabling synergistic energy and power performance. The coexistence of redox reactions and physical charge adsorption/desorption provides additional flexibility for device optimization.
 | ||
| Fig. 16 (a)–(c) Different work mechanisms include the “accordion” mechanism (electrochemical intercalation (−)//physical adsorption (+)), “rocking chair” mechanism (physical adsorption (−)//electrochemical intercalation (+)), and hybrid mechanism (electrochemical intercalation (−)//physical adsorption + electrochemical intercalation (+)), respectively. (d) Ragone plots of different energy storage devices. (e) Performance comparison among a commercial SC, a Li4Ti5O12‖AC LIHC, and an 18650 rechargeable LiCoO2‖graphite LIBs. Reproduced with permission from ref. 285. Copyright 2024, Elsevier. (f) The number of publications on various hybrid capacitor systems according to the Web of Science database (June 2025). | ||
Significant advances have been achieved in this field, beginning with the work of G. G. Amatucci in 2001, who developed Li4Ti5O12//AC systems. This was followed by progress from Fuji Heavy Industries, whose “Li+ capacitors” reached energy densities of up to 13 Wh kg−1 and operated at voltages around 3.7 V by pre-lithiating the anode.285 These hybrid systems exhibit dual-ion dynamics, whereby cations such as Li+, Na+, or Zn2+ intercalate into the battery-type electrode, while anions undergo surface adsorption within the capacitive electrode. The delicate balance between the storage mechanisms demands kinetic matching, structural optimization, and careful electrolyte formulation. By combining the high energy density characteristic of battery with the superior power density and long cycle life typical of supercapacitors, hybrid devices have emerged as promising platforms for high-efficiency energy storage (Fig. 16d).285 Lithium-ion hybrid capacitor (LIHC) technology, in particular, has reached a relatively advanced stage of development and is now approaching the “double-high” performance target of approximately 100 Wh kg−1 in energy density and 100 kW kg−1 in power density, creating favourable conditions for near-term commercialization (Fig. 16e).285
Multi-ion strategies have also expanded to include multivalent carriers (e.g., Mg2+, Ca2+, Zn2+, and Al3+), which offer higher theoretical capacities due to their greater charge per ion, yet face challenges related to size, solvation, diffusion kinetics, and host material stability. To overcome the limitations of conventional electrolytes, advanced systems now employ ionic liquids, water-in-salt electrolytes, and solid-state conductors to widen voltage windows, enhance safety, and tune ion mobility. Simultaneously, the precision design of electrode materials, through pore size tuning, surface functionalization, gradient pore architectures, and interlayer spacing modulation, has enabled tailored ion-material interactions. In situ techniques such as operando XRD and Raman spectroscopy, combined with molecular dynamics simulations, are playing an increasingly vital role in elucidating the complex transport and solvation behaviors of ion carriers under realistic working conditions.
This evolution outlines a clear trajectory in supercapacitor development, from systems with no ion-carrier involvement to those where ion-carriers play a direct, multifaceted role in charge storage. The progression moves from negligible energy density to moderate (ion-assisted) to higher values enabled by physical adsorption, redox reactions, and finally deep intercalation processes. As ion-carriers from passive spectators to active charge storage media and chemical participants, their characteristics, including size, valence, solvation structure, and transport kinetics, become critical design parameters. Meanwhile, the trade-off between energy and power becomes increasingly dictated by the speed of ion transport across interfaces and within solids.
Against this backdrop, the multi-ion carrier systems, hybrid battery-capacitors, emerge as a transformative strategy, offering synergistic pathways for high-capacity, fast-response, and long-life energy storage. Their potential is already evident in commercial Li+ capacitors for automotive start-stop systems and grid frequency regulation, as well as in emerging Zn-based and ammonium-ion systems for flexible electronics.275 As global demand surges, hybrid capacitors featuring multi-ion mechanisms, ranging from monovalent (e.g., Li+, Na+, and K+) to multivalent (e.g., Mg2+, Ca2+, Zn2+, and Al3+), and even non-metallic cations (e.g., NH4+), are attracting intense research attention. Over the last decade, research activity in the field of hybrid battery-capacitors has grown substantially. Bibliometric analyses of publications from 2012 to 2024 reveal a particularly rapid rise in interest, reflecting the accelerating momentum of this technology (Fig. 16f). This sustained growth underscores both the broad scientific engagement and the significant potential of hybrid capacitor systems as promising candidates for next-generation energy storage applications (Tables S4 and S5). This chapter focuses on these rapidly evolving hybrid systems, dissecting the transport, storage, and reaction behaviors of diverse ion carriers and their precise regulation. By highlighting these advances, we aim to provide guidance for the next generation of high-performance, multi-ion-driven supercapacitor technologies.
In this context, these characteristics highlight the transformative potential of HBCs in redefining multi-ion EPSs. By leveraging the cooperative dynamics of diverse ion species and coupling them with a wide range of material chemistries, HBCs provide a compelling blueprint for achieving the elusive combination of high energy and power densities, long cycle life, and operational versatility. The sections that follow classify representative HBC configurations according to their ionic species, highlighting the distinct roles of monovalent and multivalent ions in shaping the performance landscape of this rapidly evolving technology.
4.1. Monovalent ion hybrid capacitors
000 cycles. Moreover, their adaptability to a wide range of environmental conditions further underscores their technological potential.
Kinetic mismatches between anode and cathode processes, disparities in specific capacity, and incompatibilities between electrode materials and electrolyte formulations can lead to suboptimal performance and long-term degradation. These limitations are exacerbated under extreme temperature or cycling conditions, where ionic transport and electrode stability become increasingly critical. Addressing these issues necessitates a holistic design strategy, wherein the chemical, structural, and electrochemical properties of all components are co-optimized.
To this end, recent research has focused on the rational design of electrode materials that align with the dynamic requirements of multi-ion systems. Tailoring materials with high specific capacity, stable cycling behavior, and suitable electrochemical windows has proven essential to unlocking the full potential of AMIHCs. The intrinsic properties of alkali metal ions, especially variations in their ionic radii, charge densities, and solvation behaviors, present a dual effect in the optimization process. On one hand, these differences offer valuable opportunities for tuning transport kinetics and interfacial reactions; on the other hand, they introduce additional complexities in balancing ionic mobility, electrode compatibility, and electrolyte stability within multi-ion carrier systems. Therefore, advances in atomic-level engineering, nanostructured morphologies, and interfacial modulation are pivotal in enabling efficient ion transport and reversible charge storage.
The following sections will delve deeper into the material design strategies employed for AMIHCs, focusing on how cathode and anode optimization contributes to performance enhancement. Through this exploration, the transformative role of multi-ion carrier engineering will be further elucidated, underscoring its central importance in the development of hybrid energy storage systems with superior electrochemical characteristics.
4.1.1.1. Cathode materials. In AMIHCs, cathode material design is critical to achieving a balance between high energy and power densities. Commonly employed materials such as activated carbon (AC), carbon nanotubes (CNTs), and graphene possess high surface areas, which facilitate rapid physical adsorption and desorption of alkali metal ions including Li+, Na+, and K+.290–292 This characteristic promotes efficient charge transport while simultaneously reducing internal resistance, contributing to improved electrochemical performance. These characteristics are particularly advantageous for high-power output. However, conventional EDLCs, which rely solely on non-faradaic charge storage, are inherently limited in terms of specific capacitance and energy density.
To address these limitations, recent efforts have focused on engineering carbon-based architectures with optimized porosity, surface chemistry, and redox activity to enhance ion accessibility and exploit multi-ion interactions. AC, despite its widespread use due to low cost and a high surface area (>1000 m2 g−1), remains constrained by its EDLC-dominated storage mechanism. In response, Han et al.293 incorporated 3D graphene into AC, forming a composite with a larger surface area (1402 m2 g−1), which improved volumetric capacitance by 137% (to 45 mAh cm−3). This enhancement enabled LIHCs to achieve energy and power densities of 45 Wh L−1 and 19 kW L−1, respectively. Expanding on this strategy, Yi et al.294 developed a nitrogen-doped hierarchical activated carbon (ANC) cathode featuring an exceptionally high surface area (2651 m2 g−1) and well-defined mesoporosity (2.4–2.8 nm). The incorporation of pyrrolic-N functionalities transformed physical adsorption into quasi-chemisorption, significantly accelerating K+ transport. When paired with a homologous anode and a concentrated ether-based electrolyte (4 M KFSI/DME), the ANC cathode acted as an “ion-buffering reservoir”, harmonizing ion flux across the full cell and delivering a specific energy of 49 Wh kg−1 at 5 A g−1 while maintaining 80% capacity after 3000 cycles.
Renowned for their exceptional electrical conductivity (>104 S cm−1) and mechanical stability, CNTs present significant potential. However, challenges like high cost and a tendency towards agglomeration must be addressed. In SIHCs, Cui et al.295 demonstrated that multi-ion storage, involving Na+ at the Sn anode and PF6− at a N/S co-doped hierarchical carbon nanosheet (HCN) cathode, can dramatically boost electrochemical performance. This dual-ion mechanism yielded an impressive energy density of 250 Wh kg−1 and a cycle life exceeding 10000 cycles, illustrating the vital role of cathode-specific ion storage in unlocking multi-ion synergies. Complementing this, the integration of CNTs into electrode architectures offers further opportunities for optimization. For instance, Li et al.296 demonstrated the efficacy of nitrogen-doped CNTs embedded in mesoporous carbon (N-doped CNTs@MC). The resulting material exhibited a high specific surface area (1806.3 m2 g−1) and robust pseudocapacitive properties, enabling impressive performance in PIHCs with 90% capacity retention after 10000 cycles at a high current density (10 A g−1). This demonstrates the feasibility of employing CNTs in multi-ion storage devices.
Graphene, with its theoretical surface area of 2630 m2 g−1 and high electronic conductivity (∼104 S cm−1), also holds great promise as a cathode material. However, challenges such as restacking and chemical inertness hinder its practical implementation. To counter these issues, oxygen-containing surface functional groups (e.g., carboxyls) have been introduced to improve hydrophilicity and facilitate ion transport. Jin et al.297 developed a hierarchical porous graphene (MP-G) cathode capable of retaining over 80% of its capacity at a high current density of 30 A g−1 without degradation over 5000 cycles. When paired with an edge-carboxylated graphene nanosheet (G-COOH) anode in an LIHC system, the full device achieved an energy density of 120.8 Wh kg−1 and a remarkable power density of 53550 W kg−1, with a 98.9% retention rate after 50000 cycles. Further advancing multi-ion carrier optimization, Qin et al.298 designed a potential-regulated nitrogen-doped carbon nanosheet cathode that achieved a specific capacity of 150 mAh g−1 at 0.2 A g−1, which is nearly four times higher than that of conventional AC. This performance arose from a synergistic mechanism that combined EDLC behavior with Li+-targeted pseudo-capacitance, resulting in a 17-fold enhancement in energy density and negligible decay after 10000 cycles.
Extending beyond traditional carbon-based materials, 2D transition metal carbides and nitrides (MXenes), particularly Ti3C2Tx, have emerged as promising cathode candidates due to their metallic conductivity (>10000 S cm−1) and tunable surface chemistries (e.g., –O, –F, and –OH), which can host various alkali ions (Li+, Na+, and K+). However, their tendency to restack due to van der Waals interactions limits ion accessibility and active site exposure. To enhance multi-ion (Na+/K+) transport in MXenes, Li et al.299 constructed a 3D MXene/acid-activated carbon (AAC) framework, where the AAC acted as a structural spacer that expanded interlayer distances and established a hierarchical porous network. This configuration enhanced the kinetics of Na+/K+ transport and promoted synergistic interactions between ions and oxygen-functionalized MXene surfaces. The resulting composite achieved a high capacitance of 378 F g−1 at 0.5 A g−1 and 97.4% retention after 10000 cycles, affirming its capacity to support stable, high-rate multi-ion storage.
Collectively, these advancements illustrate the central role of surface chemistry engineering, hierarchical porosity, and multi-ion-specific interaction mechanisms in the rational design of AMIHC cathodes. Through the strategic incorporation of functional groups, optimized pore architectures, and synergistic ion-hosting features, cathode materials can be tailored to simultaneously accommodate multiple alkali ions.
4.1.1.2. Anode materials. In tandem with cathodic design, the anode serves as a crucial component in AMIHCs, where it governs charge balance, structural integrity, and the efficient accommodation of diverse alkali ions. For such systems, ideal anode materials must combine high specific capacity and low redox potential with the chemical and structural adaptability required to host multiple ion species.300,301 This adaptability is particularly essential in AMIHCs, where the coordination of cations such as Li+, Na+, and K+ necessitate host frameworks capable of withstanding varied ionic radii, coordination environments, and transport dynamics. As a result, substantial research efforts have been devoted to engineering intercalation-, conversion-, and alloy-type anodes that can support stable and reversible multi-ion storage. The overarching goal in these designs is to achieve synergistic electron-ion transport and mitigate mechanical or structural degradation under multivalent electrochemical cycling.
Intercalation-type materials have garnered significant attention due to their inherent structural reversibility and favorable ion diffusion kinetics. Traditional candidates such as graphite, hard carbon, and titanium oxides often display well-defined voltage profiles indicative of stable redox mechanisms.302–305 Among them, carbon-based materials are particularly promising owing to their good electrical conductivity, high surface area, tunable porosity, and low cost. However, while graphite offers excellent Li+ intercalation performance, its narrow interlayer spacing (∼0.335 nm) renders it suboptimal for the larger Na+ and K+ ions. To address this constraint, structural engineering strategies have been deployed to expand interlayer spacing and enhance pore connectivity. For example, Yang et al.306 synthesized a 3D porous carbon derived from sodium citrate featuring hierarchical porosity and expanded interlayer gaps, which enabled improved Na+ accessibility and exceptional cycling stability, retaining 75.6% capacity after 15000 cycles. Similarly, Shi et al.307 employed nitrogen-doped graphene fibers with an enlarged interlayer spacing of 0.378 nm in a dual-carbon, achieving an energy density of 142 Wh kg−1 and ultrafast charging capability (full charge within 8.5 seconds), demonstrating excellent rate performance. Furthermore, this device significantly improved structural stability and cycling reversibility through the synergistic effect of K+ adsorption/intercalation dual mechanisms in high and low voltage regions, further validating the great potential of potassium storage structural engineering. Further tailoring of carbonaceous structures through doping and defect engineering has also proven essential in optimizing ion-host interactions across various carrier types. For instance, Li et al.308 fabricated highly graphitized and heteroatom-doped carbon nanosheets for Li+ storage, enabling superior conductivity and ion transport. Their LIHCs delivered 112 Wh kg−1 at a power density of 19600 W kg−1 with long-term cycling stability. Complementary to interlayer modulation, 3D architectures further aid in hosting multiple ions by shortening diffusion pathways and buffering structural changes. Notably, porous aromatic framework-templated carbon with interlayer distances reaching 0.428 nm supported high-performance Li-ion micro-capacitors with excellent volumetric energy density and robust cycling.309
The implementation of heteroatom doping (e.g., N, B, P, S, and O) introduces electronic structure modulation, defects, and additional redox-active sites, further tuning the anode's electrochemical response in multi-ion systems.310 For example, Shen et al.311 developed LiV3O4 nanoparticles encapsulated within N-doped carbon nanowires, achieving a blend of intercalation and pseudocapacitive behavior with a high energy density of 136.4 Wh kg−1 in LIHCs (Fig. 17a). However, excessive doping may lead to structural disorder and reduced conductivity, underscoring the need for controlled dopant distribution in multi-ion environments. This challenge is adeptly addressed by precision doping architectures: Hu et al.312 demonstrated that phosphorus/nitrogen co-doped hierarchical porous carbon nanofibers (PN-HPCNFs) reduce K+ diffusion barriers to 0.12 eV and enhance K+ adsorption energy (−2.30 eV) via DFT calculations, enabling a potassium ion hybrid capacitor with 191 Wh kg−1 at 7560 W kg−1 and 82.3% capacity retention over 8000 cycles. Extending this paradigm to diverse ion carriers, the same group implanted Mn single atoms in N,F-codoped carbon nanosheets (MnSAs/NF-CNs), where Mn–N4 sites simultaneously minimize Na+ diffusion barriers (0.11 eV) and maximize ClO4− adsorption energy (−0.98 eV). This dual-ion optimization yielded SHICs with 197 Wh kg−1 at 9350 W kg−1 and 85.2% capacity retention after 10000 cycles (Fig. 17b). Collectively, these advances underscore how atomic-scale doping and hierarchical design concertedly regulate divergent ion carriers (cations/anions), unlocking high-energy, high-power, and durable hybrid storage.
 | ||
| Fig. 17 (a) Voltage profiles and electrochemical reaction mechanism of novel LICs. Reproduced with permission from ref. 311. Copyright 2017, Wiley. (b) Long-term cycle performance of the SIHCs at 1 A g−1. Inset: Photograph of the miniature twin-engined aircraft and LED arrays powered by our SIHC device. Reproduced with permission from ref. 312. Copyright 2021, Royal Society of Chemistry. (c) STEM mapping images of 3S-Nb2O5-HoMSs. (d) CV curves at different scan rates of the 3S-Nb2O5-HoMSs//AC LIC device. (e) Galvanostatic charge–discharge curves at different current densities. Reproduced with permission from ref. 319. Copyright 2020, Wiley. (f) CV curves at 2 mV s−1 of the VOx@N-MXene HSs//AC device. Reproduced with permission from ref. 320. Copyright 2024, Wiley. (g) Schematic illustration of an SIHCs. (h) In situ XRD patterns during charge and discharge, and the corresponding charge/discharge profile of the SIHCs at 40 mA g−1. (i) Energy and power densities of the SIHCs in comparison with other energy storage systems based on total mass of active materials. Reproduced with permission from ref. 328. Copyright 2018, American Chemical Society. | ||
Transitioning beyond carbon systems, titanium-based materials such as TiO2 and Li4Ti5O12 (LTO) offer robust frameworks and pseudocapacitive-intercalation hybrid behavior across multiple alkali ions.313 Despite their intrinsic low conductivity, innovative composite architectures have overcome this limitation. Li et al.314 utilized a MOF-templated approach to synthesize TiO2/C heterostructures with 3D conductive networks, elevating Na+ pseudocapacitive contributions to 82% and delivering 142.7 Wh kg−1 at 250 W kg−1 with over 90% retention across 10000 cycles by optimizing Na+ and electron co-transport. Similarly, Zhao et al.315 regulated K+ carrier kinetics in carbon-coated K2Ti2O5 microspheres (S-KTO@C), where a 10-nm carbon layer expedited electron transfer while porous microstructures shortened solid-state K+ diffusion paths, transforming storage to surface-dominated pseudo-capacitance (48.6% at 0.1 mV s−1). This balanced dual-carrier flow yielded 57.4 Wh kg−1 at 1.74 kW kg−1 and maintained 80.3% capacity after 3000 cycles in PIHCs. Additional carrier-specific optimization was demonstrated by Li's group,308 who embedded nanoscale LTO in mesoporous carbon frameworks (n-LTO@MC), facilitating a 100-fold increase in Li+ diffusion through hierarchical pores spanning 2–100 nm. Meanwhile, Xiao et al.316 designed FeS2/TiO2 heterostructures derived from ilmenite, which significantly reduced the Na+ diffusion barrier from 0.78 eV to 0.42 eV through interfacial carrier modulation. This strategy not only improved ion transport kinetics but also achieved a remarkable cost reduction of five orders of magnitude, underscoring the economic viability of engineered multi-ion carrier architectures for large-scale energy storage applications.
Beyond conventional anodes, emerging 2D transition metal carbides and nitrides (MXenes), particularly Ti3C2Tx, have demonstrated exceptional promise as multi-ion carrier hosts due to their metallic conductivity (>6000 S cm−1), surface-rich chemistry, and tunable interlayer spacing. However, restacking tendencies hinder their ion-accessible surface area, necessitating deliberate morphological control. Fan et al.317 addressed this by combining nitrogen doping with 3D-printed porous frameworks (5–50 μm), where the N dopants optimized surface charge states for ion adsorption and the macro-porous architecture facilitated rapid transport. This design tripled areal energy density to 1.18 mWh cm−2 while maintaining 75% capacity after 3500 cycles through balanced multi-ion flux. Furthermore, Fang's group318 fabricated MXene-based three-dimensional sphere-tube superstructures using a spray-freeze-drying technique. In this architecture, curvature-induced strain with radii below 10 nm effectively suppressed layer restacking and significantly enhanced K+ transport by shortening ion diffusion pathways by approximately 60%. As a result, the device exhibited outstanding cycling stability over 10000 cycles with negligible capacity degradation. This work illustrates the critical role of geometric confinement and interface engineering in enabling long-term, high-rate performance in multi-ion energy storage systems.
In the context of multi-ion carrier power supply devices, conversion-type anode materials such as metal oxides, sulfides, and selenides have gained significant attention for their ability to store charge through multi-electron transfer mechanisms. These processes typically involve the breaking and reformation of chemical bonds, enabling higher specific capacities compared to intercalation-type materials. This mechanism affords them exceptionally high theoretical capacities, a key advantage in devices where energy density is a crucial parameter. However, their practical application remains significantly hindered by intrinsic limitations, including drastic volume expansion (>200%), low electronic conductivity, and sluggish ion diffusion kinetics, all of which are exacerbated in multi-ion systems where simultaneous cation and anion transport must be finely balanced.
To tackle these challenges, structural engineering strategies have been adopted to facilitate efficient multi-ion transport while preserving mechanical and electrochemical integrity. A representative example is the triple-shelled Nb2O5 hollow microspheres (Fig. 17c) developed by Bi and co-workers,319 featuring 20–50 nm inter-shell spacing that enhances Li+ diffusivity by fivefold. The LIHCs achieved capacitive-pseudocapacitive hybrid storage through a dual-carrier synergy mechanism (Fig. 17d and e), where rapid PF6− adsorption at the cathode synergized with slow Li+ intercalation at the anode, maintaining 89% capacity retention over 10000 cycles at 1 A g−1. However, the system still exhibited a relatively high charge transfer resistance (∼148 Ω), highlighting the need for further optimization of interfacial ion transport pathways within multi-ion architectures. In response to this limitation, subsequent research has focused on reducing interfacial resistance and improving ionic/electronic coupling. For instance, Yuan et al.320 developed amorphous vanadium oxide hollow spheres encapsulated in an N-doped MXene (VOx@N-MXene HSs). Guided by machine learning-assisted molecular simulations, the study revealed that amorphization induces abundant structural defects and short-range Na+ diffusion pathways, significantly enhancing Na+ mobility (Fig. 17f). The resulting SIHCs delivered a high energy density of 198.3 Wh kg−1 along with outstanding long-term cycling stability, retaining 81.8% of its capacity after 8000 cycles. Liu et al.321 addressed this by coupling MnO with reduced graphene oxide (rGO) and introducing oxygen vacancies, effectively lowering interfacial resistance by 83% and accelerating Li+ adsorption kinetics. Nevertheless, the system still experienced substantial volumetric strain (∼20%), which compromised initial coulombic efficiency. To mitigate such mechanical instability, Cheng et al.322 introduced a zero-strain CdNb2O6 anode, leveraging a robust Nb–O framework to confine structural expansion to <1%, achieving a high coulombic efficiency of 97.4%. This development marks a critical benchmark in the realization of high-rate, durable anodes for alkali-ion hybrid capacitors, where strain minimization is essential to accommodate repeated multi-ion fluxes.
Beyond oxides, transition metal sulfides and selenides present another class of conversion-type materials with favorable conductivity and cost-effectiveness.323,324 Yet, their structural degradation upon cycling presents a persistent barrier, especially under the demanding conditions of dual-ion transport. To address this, advanced architecture and heterointerface designs have been adopted. Zhao et al.325 engineered a two-dimensional MoSe2/graphene heterostructure with spatially decoupled electron and ion channels, where the enlarged interlayer spacing enhanced Na+ diffusion and the conductive graphene backbone (>103 S cm−1) supported rapid charge transport. This hybrid architecture delivered stable operation with an energy density of 43 Wh kg−1 at an ultra-high-power density of 10752 W kg−1 over 5000 cycles, demonstrating how spatial ion-electron separation can optimize multi-ion dynamics. Further progress has been achieved in potassium-based systems, where the large ionic radius of K+ poses significant diffusion and strain challenges. Ge et al.326 addressed this by constructing a three-dimensional FeSe2/N-doped carbon composite, where quantum tunneling pathways facilitated rapid electron transport and defect engineering lowered the K+ adsorption barrier, sustaining a high capacity of 158 mAh g−1 at 2 A g−1 despite enduring ∼200% volume expansion. Chen et al.327 advanced the design by creating one-dimensional aligned NbSe2/NSe carbon nanofibers (NSeCNFs), where 2.1 nm carbon nanochannels and delocalized charge distributions enabled directional K+ diffusion. Meanwhile, strong Se–C bonds buffered lattice strain, ensuring an impressive 83% capacity retention after 10000 cycles in PIHCs.
Alloy-type anodes have garnered significant attention in the development of multi-ion carrier power supply devices due to their capability to undergo multi-electron alloying reactions, thereby delivering exceptionally high theoretical capacities. In the context of multi-ion hybrid capacitors, these materials hold promise for bridging the energy-power gap by facilitating simultaneous accommodation of multiple charge carriers. However, their practical deployment is impeded by inherent mechanical and interfacial instabilities. The substantial volume fluctuations associated with repeated alloying/dealloying reactions disrupt both electronic percolation networks and ion transport pathways, leading to loss of electrical contact, hindered ion diffusion, and eventual device failure. To address these limitations, Yuan et al.328 introduced a presodiated porous NaBi anode paired with an AC cathode in a diglyme-based electrolyte (Fig. 17g). In situ XRD and SEM characterization methods revealed that the dynamically evolving 3D porous network effectively accommodated the substantial volume expansion, reaching up to 256% during the Bi to Na3Bi phase transition, while simultaneously facilitating rapid Na+ diffusion through continuous ion pathways and maintaining structural integrity during cycling (Fig. 17h). As a result, the resulting SIHCs achieved a high specific capacity of 298 mAh g−1 at 2 A g−1, a power density of 11.1 kW kg−1, an energy density of 106.5 Wh kg−1 (Fig. 17i), and outstanding cycling stability with 98.6% capacity retention over 1000 cycles. These findings underscore the effectiveness of porous alloy architectures in synchronizing Na+ transport with capacitive PF6− adsorption processes. Despite these advances, mechanical instability during long-term cycling remains a key concern. To further improve cycling durability, Chojnacka et al.329 developed a holistic optimization strategy based on Sn4P3 alloy anodes. Their approach combined three key design elements into a unified framework. First, a continuous three-dimensional conductive network was constructed using glucose-derived hydrothermal carbon, which improved electronic conductivity and structural integrity. Second, the anode-to-cathode capacity ratio (Q−/Q+) was precisely regulated to ensure balanced charge distribution and mitigate localized overpotentials. Third, controlled pre-sodiation of the Sn4P3 anode was employed to tailor the initial sodiation kinetics and promote stable interfacial formation. This integrated strategy successfully limited volume expansion to 67%, suppressed the formation of metallic sodium, and facilitated the development of a robust SEI during extended cycling. As a result, the optimized SIHCs achieved a high-capacity retention rate of 80% after 10000 cycles, underscoring the crucial role of simultaneously regulating electronic and ionic transport channels in alloy-based AMIHCs to enable stable long-term energy storage.
Building on this foundation, Lin et al.330 proposed a dual-carrier synergy strategy using a bismuth-based anode to further unlock the potential of alloy-type materials in multi-ion configurations. By encapsulating Bi nanoparticles within a lignin-derived, three-dimensional nitrogen-doped porous carbon matrix (Bi@LNPC), they achieved a highly interconnected electronic framework and ample ion-accessible surfaces. The hierarchical porosity reduced Na+ diffusion distances, while nitrogen doping introduced abundant defect sites that enhanced interfacial ion adsorption and charge transfer. This rational design enabled Bi@LNPC//AC SIHCs to operate efficiently over a wide voltage range (1.0–3.8 V), delivering an energy density of 63 Wh kg−1 at a power density of 680 W kg−1 and maintaining 87% capacity retention over 3000 cycles.
Altogether, the development of anode materials for AMIHCs highlights the critical need to regulate multi-ion transport within structurally and chemically adaptable host frameworks. As systems increasingly exploit multiple alkali ions to optimize the energy-power trade-off, the anode must function not merely as a storage site, but as an active interface where electron-ion coupling, mechanical robustness, and interfacial kinetics are synergistically engineered. Achieving high-performance AMIHCs thus relies not only on the intrinsic properties of anode materials, but also on their ability to dynamically accommodate diverse ionic species under complex electrochemical conditions. These insights redefine the anode as a tunable, ion-responsive domain essential to unlocking the full potential of multi-ion carrier power supply devices.
The charge-storage processes in AmIHCs can involve four primary mechanisms that have been identified to date: (i) NH4+ intercalation–deintercalation, (ii) cooperative NH4+/H+ co-insertion–extraction, (iii) surface adsorption–desorption, and (iv) conversion reactions. A representative intercalation-type cell is illustrated in Fig. 18a, in which layered δ-MnO2 serves as the cathode, activated carbon cloth (ACC) as the anode, and (NH4)2SO4 as the electrolyte.331 During discharge, NH4+ ions insert into the interlayer galleries of δ-MnO2, while SO42− anions are electrostatically adsorbed on the ACC surface. The charge process proceeds in the reverse direction, with NH4+ de-intercalation from δ-MnO2 and desorption of SO42− from the ACC anode. Throughout cycling, NH4+ shuttles reversibly between the electrolyte and the δ-MnO2 host; its insertion is accompanied by reversible formation and rupture of hydrogen bonds within the MnO2 layers. Fig. 18b summarizes the proposed NH4+-storage mechanism. In the initial discharge, pristine δ-MnO2 accommodates NH4+ ions and likely co-intercalated water molecules, within its interlayer space. During subsequent charging, most of these NH4+ ions are extracted, whereas a small fraction, together with interlayer water, remains hydrogen-bonded to adjacent MnO sheets. These residual species stabilize the metastable δ phase and facilitate the highly reversible NH4+ intercalation–deintercalation observed in later cycles, thereby underpinning the excellent cyclability of the AmIHCs.
 | ||
| Fig. 18 (a) Schematic illustration of the designed aqueous δ-MnO2//ACC AmIHCs; (b) schematic illustration of the NH4+ ion storage mechanism in the layered δ-MnO2. Reproduced with permission from ref. 331. Copyright 2022, Wiley. (c) GCD profiles at different current densities; (d) cycling stability at current density of 20 A g−1 demonstrate bending destructive tests of PTCDI//MnOx/MnS2-400 pouch-type HSC by driving electric fans at different bending angles. Reproduced with permission from ref. 332. Copyright 2025, Elsevier. (e) CV profiles of MC700 with various electrolytes; (f) CV curves at various potential windows; (g) GCD curves at various potential windows. Reproduced with permission from ref. 336. Copyright 2022, Wiley. | ||
The practical realization of high-performance AmIHCs hinges on the development of electrode materials capable of reversibly hosting NH4+ ions while maintaining structural integrity and facilitating fast electron/ion transport. Intercalation-type materials, particularly those with layered structures, have attracted significant attention owing to their tunable interlayer spacing, pseudocapacitive charge storage capability, and the potential for structural modulation to enhance electrochemical performance. Nonetheless, many of these materials suffer from low intrinsic conductivity and limited rate capability, prompting efforts toward compositional engineering and interface optimization.
Among transition metal oxides, manganese dioxide (MnO2) is a widely studied positive electrode material due to its low cost, high theoretical capacity, and favorable 2D ion diffusion channels. The dynamic formation and disruption of hydrogen bonds within its interlayers facilitate NH4+ intercalation while buffering volume changes and maintaining ion transport pathways. For instance, Chen et al.331 addressed these challenges by constructing a δ-MnO2/activated carbon cloth composite, which achieved an areal energy density of 861.2 μWh cm−2 and retained 72.2% capacity after 5000 cycles, demonstrating the effectiveness of combining pseudocapacitive oxides with conductive frameworks. Complementary strategies have also emerged. Liu et al.332 constructed a MnOx/MnS2 p–n junction cathode, where the built-in electric field (BIEF) at the junction interface accelerated NH4+ migration and enabled in situ stabilization. This design achieved both high capacities, with values of 838.56 F g−1 or 186.35 mAh g−1 at 1 A g−1, and exceptional long-term stability, retaining 96.42% of its capacity over 40000 cycles (Fig. 18c and d). In addition, the system delivered an energy density of 79.57 Wh kg−1, demonstrating the effectiveness of interfacial field modulation in multi-ion storage systems. Vanadium oxides offer a different advantage by supporting multiple charge storage modes, including intercalation and pseudo-capacitance. Nevertheless, their limited electronic conductivity and phase instability remain major obstacles. Researchers have tackled these issues through strategies such as Co2+ pre-intercalation in VO2 nanorods to enhance conductivity and inhibit vanadium dissolution, achieving improved areal capacitance and prolonged cycling.333 Another effective approach involved synthesizing R-V10O24 nanowires, which utilize hydrogen bonding to stabilize NH4+ intercalation, enabling high capacity (203.1 mAh g−1 at 0.3 A g−1) and 97.6% retention after 20000 cycles.334
Tungsten trioxide (WO3), known for its robust NH4+ intercalation and high theoretical capacity, is similarly constrained by sluggish ion transport and low electronic conductivity. These issues were addressed by Chen et al.335 through the pre-intercalation of NH4+ into hexagonal WO3 (h-WO3). First-principles calculations indicated that the presence of hydrogen bonding within the structure significantly reduces the diffusion barrier, thereby facilitating more efficient ion transport. This resulted in enhanced areal capacitance and superior durability over 13600 cycles, further confirming the feasibility of structural pre-intercalation strategies in optimizing NH4+ kinetics. Similarly, molybdenum-based materials such as MoO3 and MoS2 offer high capacities and flexible interlayer tunability. MoS2's intrinsic conductivity supports rapid charge transport, yet its accessibility to NH4+ ions is limited by interlayer constraints. MoO3, although chemically compatible, suffers from solubility issues and structural instability. Dai et al.336 mitigated these drawbacks by fabricating a thermally treated MoO3@C composite enriched with oxygen vacancies, which improved ion accessibility and promoted fast NH4+ diffusion, achieving stable cycling and excellent rate performance (Fig. 18e–g). The strategy was further extended to a MoS2@PANI composite featuring a core–shell structure, whose high conductivity and porous morphology enabled exceptional capacitance and long-term durability over 10000 cycles.337 These advances illustrate the pivotal role of interface design and heterostructure engineering in tailoring AmIHC electrode behavior.
Transition metal sulfides (TMSs) have garnered significant interest as electrode materials for AISCs due to their high electrical conductivity, diverse redox states, and structural tunability. However, their tendency toward structural degradation impairs cycling performance. Wu et al.338 overcame these limitations using pseudocapacitive CuCo2S4 nanowires grown on carbon paper. NH4+ storage in this system is facilitated by strong hydrogen-bonding interactions (N–H⋯S) and directional charge transfer to Co sites. DFT simulations confirmed a high NH4+ adsorption energy (−3.48 eV), leading to exceptional electrode capacity (1512 C g−1 at 1 A g−1), an energy density of 74.17 Wh kg−1, and 83.28% capacitance retention after 10000 cycles. In addition to metal oxides and sulfides, layered double hydroxides (LDHs) are recognized for their multi-electron redox behavior and anion exchange capacity. Nonetheless, low conductivity and structural instability limit their use. Wang et al.339 addressed this by electrochemically reconstructing Co–Fe LDHs, introducing oxygen vacancies and hierarchical porosity. The resulting material delivered a threefold increase in reversible NH4+ storage, with high specific capacitance, energy density, and cycling durability in AmIHC devices.
Beyond conventional intercalation hosts, emerging materials such as polyoxometalates (POMs) are being explored for their surface-mediated pseudocapacitive behavior. While bulk intercalation is limited by poor structural stability and small diffusion channels, surface-functionalization strategies offer a viable alternative. To address these challenges, Chen et al.337 introduced phosphate-functionalized α-MoO3 anodes, which promote NH4+ storage through surface phosphate–oxygen coordination. This design bypasses the limitations of bulk intercalation by enabling interfacial pseudo-capacitance, thereby enhancing both ion accessibility and electrochemical performance. The resulting AmIHCs achieved an exceptional areal capacitance of 15.3 F cm−3 at a current density of 2 mA cm−2, representing a threefold improvement over pristine α-MoO3. In addition, the system demonstrated an energy density of 2.48 mWh cm−2 and retained 82% of its initial capacity after 3000 charge–discharge cycles, indicating strong cycling stability. While this strategy significantly improved capacity, the moderate cycle stability indicates that further refinement is needed for long-term operation. Liang et al.340 advanced POM-based electrode design by employing Keggin-type polyoxometalates with exposed Mo–Ox surface sites capable of forming hydrogen bonds with NH4+. This strong interfacial interaction minimizes structural distortion during cycling and enables dominant pseudocapacitive behavior. As a result, the AmIHC achieved an ultrahigh specific capacity of 619.4 mAh g−1 at 1 A g−1, exceptional cycling stability with 100% capacity retention over 20000 cycles at 10 A g−1, and a high energy density of 125.3 Wh kg−1 in asymmetric configurations.
Metal–organic frameworks (MOFs), often explored for EDLC behavior, can also be tailored for pseudocapacitive NH4+ storage through structural and electronic modifications. However, their practical application is often constrained by suboptimal NH4+ storage capacity and sluggish ion diffusion kinetics. Gao et al.341 developed a two-dimensional conjugated MOF cathode, namely336 iodine-embedded Cu-HHB (Cu-HHB/I2). DFT calculations indicated a strong affinity between polyiodide species and the Cu–O4 coordination environment, with an adsorption energy of 1.68 eV, underscoring the material's capacity to stably accommodate iodine species and facilitate efficient redox activity. This design facilitated reversible polyiodide redox reactions and promoted efficient NH4+ storage, leading to a significant enhancement in cathodic capacity. However, optimizing a single electrode, especially the cathode, proved inadequate for achieving well-balanced and high-performance energy storage, highlighting the need for coordinated electrode and electrolyte engineering. The device's limited voltage window and overall energy density remained bottlenecks. Recognizing this, Huang et al.342 advanced the anode design by constructing hierarchical porous carbon nanofibers embedded with one-dimensional conjugated Ni-BTA MOFs (HPCNFs@Ni-BTA). Their DFT results showed that the symmetric NiN4 coordination units exhibit balanced electron distribution, enabling enhanced NH4+ adsorption and fast kinetics through reversible CN/C–N redox reactions at the anode. By pairing a high-capacity cathode with a redox-active anode, this dual-electrode optimization approach effectively overcomes the performance asymmetry seen in earlier MOF-based AmIHCs. This work exemplifies a rational design strategy that goes beyond traditional EDLC mechanisms, leveraging pseudocapacitive behavior in MOFs to maximize overall device efficiency within multi-ion carrier frameworks.
Collectively, these studies highlight the central importance of multi-ion carrier design in AmIHCs, where NH4+ serves as both a performance-enhancing and structure-defining component. The broad range of host materials, encompassing oxides, sulfides, MOFs, and POMs, highlights the intricate interdependence among ion transport pathways, redox processes, and structural evolution. Crucially, optimizing AmIHC performance requires not only high-capacity electrode materials but also a deep understanding of how NH4+ interacts at the molecular and interfacial levels. As such, AmIHCs offer a rich platform for exploring multi-ion transport mechanisms and represent a vital branch of multi-ion carrier power supply devices poised for sustainable and high-power energy applications.
4.2. Multivalent metal ion hybrid capacitors
Multivalent metal ion hybrid capacitors (MMIHCs) have emerged as a pivotal subclass within the broader family of hybrid capacitive energy storage devices. By incorporating multivalent cations such as Zn2+, Mg2+, Al3+, and Ca2+ as charge carriers, these systems capitalize on the multi-electron redox capabilities intrinsic to higher-valency ions.79,343–345 Each of these multivalent ions can transfer more than one electron per redox event, significantly increasing the theoretical capacity and volumetric energy density compared to their monovalent counterparts. When integrated into asymmetric architectures that combine battery-type and capacitor-type components, MMIHCs can harness both faradaic and non-faradaic processes to yield electrochemical systems with hybridized kinetics and enhanced energy-power synergies.However, the integration of multivalent ions introduces complex challenges that underscore the need for refined material and interface engineering. These ions often exhibit sluggish diffusion kinetics due to their high charge density, leading to elevated de-solvation energy barriers and increased interfacial charge-transfer resistance. Furthermore, mismatched kinetics between the slow faradaic reactions at battery-type electrodes and the rapid non-faradaic responses at capacitive electrodes complicate system-level optimization. To overcome these intrinsic limitations, the development of high-performance electrode materials with engineered ion transport pathways, as well as electrolytes capable of stabilizing and shuttling multivalent carriers efficiently, is imperative. This section provides a comprehensive examination of the design principles and electrochemical characteristics of representative MMIHC systems based on Zn2+, Mg2+, Ca2+, and Al3+, with a focus on how multi-ion carrier strategies have been leveraged to mitigate these issues and enhance overall device performance.
 | ||
| Fig. 19 Schematic illustrations of energy storage mechanisms in ZIHCs: (a) a configuration comprising a capacitive cathode (based on electric double-layer capacitance or pseudocapacitance) coupled with a zinc deposition/stripping anode. (b) An alternative design employing a battery-type cathode (involving Zn2+ intercalation/deintercalation) paired with a capacitive anode (EDLC or pseudocapacitive behavior). (c) Schematic of AC‖ZnMg-0.1‖Zn energy storage devices. Reproduced with permission from ref. 346. Copyright 2021, Wiley. | ||
Despite these advantages, ZIHCs face persistent challenges arising from the complex behavior of multivalent Zn2+ ions in concert with co-transported species. One of the foremost issues is the formation of Zn dendrites during repeated plating/stripping cycles, which can lead to internal short circuits and severely limit cycle life. Moreover, cathode degradation, electrolyte depletion, and parasitic side reactions (such as hydrogen evolution and Zn corrosion) further compromise the long-term stability and efficiency of the system. These degradation pathways are intimately connected to the complexities of multi-ion transport and interfacial reactivity, especially under conditions of high current density or extended cycling. This underscores the critical importance of precise regulation of ion flux and the interactions between electrodes and electrolytes.
Recent advancements in multi-ion carrier engineering have unveiled sophisticated mitigation strategies, including the design of three-dimensional zincophilic matrices with tailored nucleation sites, the development of dual-ion selective membranes to regulate carrier flux, and the formulation of eutectic electrolytes with optimized solvation structures to enhance Zn2+ transference numbers while suppressing parasitic side reactions. Particularly noteworthy are emerging cathode architectures employing heteroatom-doped carbon scaffolds and MXene-based composites, which not only provide expanded ion transport channels for multi-carrier systems but also introduce redox-active sites for synergistic Zn2+/H+ co-storage, thereby amplifying both capacity and rate performance. These material innovations, coupled with advanced operando characterization techniques, are progressively unraveling the complex interphasial phenomena governing multi-ion carrier behavior, charting a course toward next-generation energy storage devices that fully exploit the thermodynamic and kinetic advantages of coordinated multi-ion transport mechanisms.
4.2.1.1. Capacitor-type cathode materials. Capacitive electrode materials serve as a foundational component in the development of ZIHCs, where their ability to regulate multi-ion carrier dynamics plays a pivotal role in balancing high-power output with sustained energy storage. In these hybrid systems, the electrochemical performance of the cathode fundamentally depends on its ability to synchronize rapid interfacial ion processes, including adsorption and desorption, with reversible redox reactions occurring within the bulk material. This coupling of surface-driven capacitive behavior and faradaic charge storage is essential for fully exploiting the benefits of multi-ion participation.
Typically, ZIHC cathode materials are categorized into carbon-based frameworks and non-carbon pseudocapacitive compounds. Regardless of the classification, their effectiveness depends on how efficiently they can accommodate multiple ionic species (e.g., Zn2+, H+, or anions) through a combination of rapid ion-accessible surface reactions and deeper structural redox activity. Such synergistic mechanisms not only enhance charge storage kinetics but also promote the comprehensive utilization of all available charge carriers, thereby optimizing the performance metrics of ZIHCs in both energy and power dimensions.
Among various capacitive architectures, carbon-based materials have emerged as the most extensively explored class due to their inherent conductivity, large specific surface area, and structural tunability. Traditional AC, while effective in facilitating EDLC, is constrained by single-ion mechanisms, thereby limiting its energy density. To overcome this bottleneck, recent research has focused on pseudocapacitive carbon frameworks engineered with heteroatom doping (N, O, P, and B) and hierarchical porosity. These modifications not only preserve high-rate performance but also introduce redox-active sites that accommodate multi-ion interactions. For example, Dong et al.347 reported AC cathodes delivering 84 Wh kg−1 at 14.9 kW kg−1, though ion diffusion limitations emerged at high rates due to insufficient multi-ion engagement. To address such kinetic barriers, Sun et al.348 developed a multifunctional electrolyte additive based on erythritol (Eryt), which effectively reconstructs the solvation structure of Zn2+, lowers the de-solvation energy barrier, suppresses Zn dendrite growth and side reactions, and disrupts the hydrogen-bond network of water molecules, thereby reducing the electrolyte's freezing point to below −50 °C and enabling excellent low-temperature performance. As a result, a ZIHC utilizing AC in ZnSO4 + 0.8 M Eryt retained 98.2% of its capacity after 30000 cycles at room temperature and maintained 94.8% even at −20 °C. Further refinement through multi-heteroatom doping has proven especially effective. Lee et al.349 developed activated carbon cathodes co-doped with phosphorus and boron, where the synergistic redistribution of charge notably improved both electrical conductivity and affinity for the electrolyte. This resulted in excellent capacity retention (>30000 cycles) and a high-rate performance of 84.0 mAh g−1 at 10 A g−1, clearly illustrating the potential of atomic-level tailoring to optimize multi-ion adsorption energetics. These breakthroughs underscore a paradigm shift toward multi-ion responsive electrode engineering, in which deliberate atomic-level modifications transform otherwise inert carbon matrices into dynamic platforms capable of bidirectional ion regulation.
Porous carbons (PCs), featuring finely engineered micro-, meso-, and macroporous structures, have emerged as highly effective platforms for accommodating multiple charge carriers in ZIHCs. Their hierarchical porosity enables both rapid ion diffusion and abundant active sites, making them well-suited for facilitating multi-ion storage mechanisms involving Zn2+, H+, or anions. Among various strategies, oxygen-rich PCs have demonstrated particular promise. For example, Yin et al.350 showed that introducing oxygen-containing functional groups, combined with a carbon coating to suppress Zn dendrite formation, enabled stable H+/O-based redox reactions and delivered superior energy densities. To overcome the inherent drawbacks of some biomass precursors, including low specific surface area and insufficient pore formation, Lu et al.351 designed a biomass-derived porous carbon through a combined approach involving hydrothermal modification, carbonization, and activation. The resulting material exhibited enhanced porosity and achieved a high specific capacitance of 339.8 F g−1 at 0.5 A g−1 in 6 M KOH. In parallel, Li et al.352 successfully developed a 3D tremella-like PC-825 with a hollow structure via a NaCl/K2C2O4-assisted one-step oxidative activation strategy. The key innovation of this material lies in its hierarchical porosity (coexistence of micro-, meso-, and macropores) and an expanded interlayer spacing of 0.384 nm, enabling a high specific capacity of 193.1 mAh g−1 when applied as a cathode in ZIHCs. Sustainable synthesis routes have further diversified the PC design space. For instance, lignosulfonate-derived nitrogen/oxygen co-doped porous carbons (LNPCs), prepared via supramolecular self-assembly, achieved 266 F g−1.353 Similarly, ZIF-8-derived hierarchical carbon microspheres developed by Liu et al.354 combined heteroatom-rich surfaces with efficient ion transport pathways, delivering 171.9 mAh g−1. Wang et al.355 leveraged rice husks to fabricate a nitrogen/phosphorus/sulfur tri-doped hierarchical porous carbon (NPS-HPC) through potassium bicarbonate activation and in situ doping. The resulting ZIHCs exhibited a high specific capacity of 198.56 mAh g−1 at 0.1 A g−1, alongside an energy density of 167.69 Wh kg−1 and a power density of 19.64 kW kg−1. DFT calculations confirmed that the N/P/S co-doping significantly improved Zn2+ adsorption energy (0.987 eV), contributing to an outstanding capacity retention of 99.98% after 23000 cycles at 5 A g−1. Moreover, hollow carbon architectures have shown additional benefits in ion accessibility and structural robustness. Fei et al.'s356 bowl-shaped carbon structures and Du et al.'s344 double-shell carbon spheres exemplify this approach, offering improved ion diffusion and mechanical stability under long-term cycling.
Beyond carbon systems, non-carbonaceous pseudocapacitive cathodes have broadened the capabilities of ZIHCs by exploiting robust faradaic interactions. Silicon, although conventionally hampered by sluggish ion mobility and limited electronic conductivity, has recently undergone a resurgence due to advances in defect engineering and electronic structure modulation. Guo et al.357 employed silicene cathodes in conjunction with water-in-salt electrolyte, achieving 10000-cycle stability and even 112% capacity retention. These performance gains were attributed to surface-dominated ion interactions, although intrinsic transport limitations persisted. To address these barriers, Deng et al.358 introduced Mg-doped porous silicon, which enhanced electrical conductivity and ion transport by altering the electronic structure. This design yielded an energy density of 135.8 mJ cm−2 and 92% retention over 20000 cycles, underscoring the effectiveness of defect engineering in expanding the ZIHC cathode landscape.
MXenes have emerged as a cornerstone material for advanced ZIHCs, offering a unique combination of high electrical conductivity, redox-active surface chemistry, and tunable interlayer spacing, which are all essential attributes for promoting efficient multi-ion transport. Li et al.359 developed 3D H+-intercalated Ti3C2Tx frameworks that facilitated efficient Zn2+ intercalation while significantly minimizing self-discharge, a critical requirement for ensuring stable operation in wearable and flexible electronic devices. Expanding MXene's utility, Jalal et al.360 designed a photo-rechargeable ZIHC using Te-modified Ti3C2Tx photocathodes. Here, the MXene served dual roles: as a charge storage medium and a light-absorbing material. The hybrid structure achieved a 50.86% capacitance increase under illumination due to improved interlayer spacing and enhanced photoresponsivity. Further leveraging MXene's multifunctionality, Fang et al.361 constructed a composite cathode by integrating NiMn-hexacyanoferrate with Ti3C2Tx, enabling a dual-ion storage mechanism involving Zn2+ and H+. The electrolyte-driven transformation between Zn4SO4(OH)6·xH2O (ZSH) and ZnxMnO(OH)2 (ZMO) significantly boosted energy density and charge/discharge kinetics. In an alternative application, Zhang et al. employed an MXene as a catalytic initiator in fast free radical polymerization (FRP) to prepare quasi-solid-state gel polymer electrolytes (GPEs) in situ. This design regulated Zn2+ flux, suppressed dendrite formation, and facilitated high-rate performance. The resulting ZIHC delivered 105.5 Wh kg−1 energy density, 9231 W kg−1 power density, and 86% capacity retention after 50000 cycles.362
Transition metal oxides, owing to their multivalent redox chemistry, have emerged as promising candidates for enhancing multi-ion transport and storage in ZIHCs. Manganese and vanadium oxides, in particular, exhibit rich oxidation states conducive to fast and reversible faradaic reactions. For instance, Wang et al.363 reported a MnO2–CNT cathode paired with a MXene anode, delivering an impressive energy density of 98.6 Wh kg−1 and excellent rate capability. To enhance Zn2+ transport, Zhang et al.364 introduced Fe3+ into MnO2, effectively expanding interlayer spacing. The Fe–MnO2 cathode exhibited a high capacitance of 1190 mF cm−2 at 2 mA cm−2 and demonstrated excellent durability by retaining 97.33% of its capacity after 10000 cycles. Impressively, the flexible ZIHCs incorporating a PVA/CMC dual-network gel electrolyte maintained 93.75% capacity retention over 15000 cycles even under low-temperature conditions (−20 °C) and mechanical bending. Vanadium oxides have likewise enabled novel functionalities: Boruah et al.365 integrated a V2O5 photoanode with an AC cathode to fabricate a photo-rechargeable ZIHC, achieving a 63% increase in capacity under illumination and enabling solar-driven self-charging. Xu et al.366 further expanded the versatility of vanadium oxides by co-intercalating Co2+ and polyaniline (PANI) into V2O5 to form CoVO–PANI, where Co2+ pillars expanded the interlayer spacing to 1.41 nm, and PANI enhanced electron transport. This synergy reduced the Zn2+ diffusion barrier and charge transfer resistance, resulting in a high areal capacitance of 847.3 mF cm−2 and 81.37% retention after 20000 cycles, with excellent flexibility under mechanical stress.
Electrolyte engineering plays a foundational role in advancing ZIHCs by enabling effective regulation of multiple ionic species, particularly Zn2+, H+, and SO42−. This regulation is essential for optimizing ion transport, improving interfacial compatibility, and enhancing electrochemical performance. By tailoring the solvation environment, ionic conductivity, and electrode–electrolyte interactions, multi-ion systems can be fine-tuned to overcome traditional trade-offs between energy density, cycling stability, and operational flexibility. A representative breakthrough comes from Wang et al.,367 who developed a ZnCl2-based water-in-salt hydrogel electrolyte embedded within a polyacrylamide (PAM) matrix. The PAM network features abundant layered and interconnected pores, which facilitate electrolyte infiltration and ion diffusion. The incorporation of Cl− anions was particularly effective in inducing Zn2+ de-solvation, forming compact [ZnCl(H2O)5]+ complexes that promoted uniform Zn plating/stripping behavior. This facilitated stable interface formation and improved ion accessibility to microporous carbon electrodes. As a result, the assembled ZIHCs delivered a high energy density of 217 Wh kg−1 and an exceptional cycling life of 100000 cycles. Remarkably, the device also demonstrated excellent low-temperature performance, retaining 92.9% of its capacitance after over 40000 cycles at −20 °C, with a high ionic conductivity of 17.9 mS cm−1. Similarly, Huang et al. employed a phosphorene-assisted, high-concentration water-in-salt electrolyte to address the challenge of narrow electrochemical windows in aqueous systems.368 The electrolyte's unique coordination environment suppressed water decomposition, extended the voltage window beyond the theoretical 1.23 V limit, and mitigated self-discharge through ion–solvent structure regulation. These studies collectively highlight the importance of solvation control and coordinated ion structuring in enhancing both thermodynamic stability and kinetic performance. Beyond solvation engineering, fine-tuning the anionic composition and electrode–electrolyte affinity offers another route to regulate multi-ion transport. In a Zn–TiN system, TiN's strong anion-binding capability enabled the formation of a dual-locking mechanism that stabilized SO42− distribution within ZnSO4 electrolytes. This synergy between Zn anodes and the conductive TiN framework resulted in a capacitance of 489.8 F g−1 with 83.92% retention after 500 hours. Notably, TiN's high conductivity and fibrous morphology also allowed for scalable cathode loading (0.6–5.0 mg cm−2), yielding a linear increase in discharge capacitance and a volumetric energy density of 47.86 Wh L−1. When paired with a PAM-based gel electrolyte, the flexible device maintained 354.3 F g−1 over 3500 cycles under mechanical deformation, demonstrating excellent adaptability for wearable electronics.
Altogether, these advances underscore the transformative potential of electrolyte–electrode co-design in enabling high-performance ZIHCs. By harmonizing multi-ion flux regulation with conductive scaffold architectures, these systems not only suppress interfacial degradation and dendrite formation but also achieve unprecedented integration of energy density, rate capability, and mechanical resilience.
4.2.1.2. Battery-type anode materials. In ZIHCs, battery-type anodes offer the advantage of high capacity through Zn2+ plating and stripping, yet they are inherently constrained by kinetic bottlenecks associated with competing ionic transport and unstable electrode–electrolyte interfaces. Unlike their capacitive cathode counterparts, which leverage rapid surface redox or ion adsorption processes, battery-type anodes engage in bulk electrochemical transformations. These reactions are inherently prone to challenges such as slow Zn2+ diffusion, interfacial degradation, and competition among coexisting ionic species, including H+, electrolyte anions (e.g., SO42−, Cl−, and Ac−), and other cations present in dual-ion systems.369–371 This intricate interplay within the anode microenvironment not only challenges charge transport and deposition uniformity but also amplifies the risk of dendritic growth and parasitic reactions, highlighting the critical need for finely tuned multi-ion regulation.
A foundational demonstration of these challenges and opportunities was provided by Wang et al.,372 who employed a Zn anode paired with oxidized carbon nanotube cathodes in a ZnSO4 electrolyte. This system achieved stable cycling over 5000 cycles at an operating voltage of 1.8 V. However, despite its promising durability, the study also revealed persistent issues such as inefficient Zn2+ redistribution and uncontrolled dendrite formation, highlighting the need for further interfacial and morphological optimization. These issues exemplify the broader difficulties in managing multiple ionic fluxes within battery-type electrodes. Recognizing the limitations imposed by mechanical and electrochemical instability, researchers have since advanced toward structurally adaptive anode architectures. For instance, Wang's group373 recently developed a stretchable Zn anode via a swelling-induced wrinkling approach. By electroplating Zn onto a self-healing polyurethane substrate, they created a mechanically robust and deformable electrode, capable of maintaining 95.1% capacitance over 12000 cycles under extreme strain conditions (300% tensile strain, 180° torsion), while delivering a high volumetric energy density of 39.86 mWh cm−3. This system exhibited electrochemical stability even under multi-axis bending, underscoring how mechanical compliance and ion transport must be co-optimized in multi-ion carrier devices to ensure performance continuity in flexible or wearable formats. These advancements underscore the critical importance of co-optimizing electrode morphology, ion transport pathways, and interfacial chemistry to ensure stable multi-ion dynamics under diverse operating conditions. By concurrently tackling critical challenges such as ionic competition, structural degradation, and phase boundary instability, these integrated approaches demonstrate the transformative potential of multi-ion carrier engineering to surpass traditional limitations in energy density, power delivery, and cycle longevity within EPS systems.
Beyond physical design, electrolyte engineering has emerged as a pivotal strategy in resolving the multidimensional challenges associated with battery-type anodes in ZIHCs. Multi-ion carrier electrolytes, when properly modulated, can dramatically reshape ion transport behavior, deposition thermodynamics, and interface chemistry. A seminal example reported by Wang et al.374 utilized a chitosan-based quasi-solid electrolyte with mesoporous channels, which enabled planar Zn plating through coordinated stress redistribution and electrostatic regulation, yielding 95% capacity retention over 20000 cycles. Building further on this paradigm, Shin et al.375 introduced a Zn–Cu dual-ion electrolyte system, wherein competitive cation interactions effectively inhibited dendritic Zn growth while enhancing anode utilization. This approach achieved an energy density of 41 Wh kg−1 along with an ultralong cycle life exceeding 22000 cycles, delivering performance that rivals and, in some instances, exceeds that of conventional lithium– and sodium-ion systems. Similarly, Chen et al.376 employed a carboxyl-functionalized cellulose hydrogel electrolyte, which imposed molecular-level regulation on Zn2+ migration and deposition. This tailored electrolyte achieved an unprecedented 70000-cycle lifespan at 5 A g−1, reaffirming the critical role of ion-specific interactions and interface stabilization in maximizing battery-type anode longevity. Innovations have also progressed through the use of chemical additives. Shi et al.377 introduced diphenylsulfone imide into the electrolyte, which effectively stabilized the electrode–electrolyte interface and enabled cycling beyond 300000 cycles. This work demonstrates how molecular-level modifications can significantly reduce multi-ion cross-talk and cumulative degradation. Even more compellingly, Wang et al.346 reported a Zn2+/Mg2+ dual-cation electrolyte system that unveiled a novel electrochemical mechanism (Fig. 19c). In this system, Mg2+ ions act through electrostatic shielding to suppress Zn dendrite formation, while simultaneously facilitating the reversible co-adsorption of Zn2+, Mg2+, and SO42− ions at the activated carbon cathode. Raman spectroscopy confirmed weakened M–OSO32− (M = Zn2+, Mg2+) bonds, which enabled dynamic phase transitions involving Zn4SO4(OH)6·xH2O, culminating in 98.7% capacity retention after 10000 cycles. This exemplifies how the cohabitation of diverse charge carriers can catalyze self-regulating interfacial behavior, balancing structural adaptability and long-term reversibility. To further enhance ionic coordination and transport, Lu et al.378 pioneered the use of deep eutectic electrolytes comprising heavy water/dimethyl sulfoxide and Zn salts, forming low-viscosity media conducive to fast Zn2+ mobility and controlled de-solvation. These systems delivered nearly 100% capacitance retention over 30000 cycles. Li et al.370 refined this approach by developing a “self-decoupling” quasi-eutectic electrolyte, wherein vehicle-type and hopping-type ion transport were synergistically combined to lower kinetic barriers and regulate multi-ion fluxes. The result was a durable and efficient energy storage system that captures the essence of multi-ion convergence for superior electrochemical performance.
Collectively, these advances underscore a fundamental principle that the intentional integration of multiple ion carriers, achieved through tailored electrolyte design, strategic electrode architecture, and precise additive engineering, is crucial for fully realizing the capabilities of hybrid energy storage systems. As ZIHCs continue to evolve, both capacitor-type cathodes and battery-type anodes emerge as pivotal platforms for harnessing the synergistic effects of diverse ionic species. By enabling complementary charge storage mechanisms and enhancing interfacial kinetics, these dual-electrode systems exemplify how multi-ion carrier strategies can transcend traditional limitations, paving the way for high-performance hybrid capacitors capable of operating reliably under dynamic and high-demand conditions.
Early contributions, such as the work conducted by Asmara et al.,379 laid the groundwork by developing magnesium-ion gel polymer electrolytes capable of mitigating ionic mobility limitations in electrochemical double-layer capacitors. Since then, research has increasingly focused on cathode engineering to enhance MIHC performance, particularly by integrating structural confinement with optimized multi-ion transport dynamics. For example, Mg-OMS-2/graphene composites took advantage of the open 2 × 2 tunnel structures in manganese oxide molecular sieves to enable reversible Mg2+ intercalation.380 This design achieved a stable discharge capacity of 44.1 mAh g−1 and maintained 95.8% capacity retention over 500 cycles, demonstrating the effectiveness of integrating structural accessibility with optimized ion transport kinetics. Concurrently, progress in anode design has also contributed to performance improvements. Zhang et al.381 demonstrated that carbon molecular sieves with preserved mesoporosity ensured efficient Mg2+ accessibility and delivered over 800 stable cycles. On the cathode side, low-cost Mn3O4 materials underwent in situ transformation into Birnessite-type nanosheets during cycling, which, despite inherent rate limitations, provided enhanced charge storage via dynamic structural reconfiguration.382
The versatility of multi-ion strategies in MIHCs is exemplified by diverse material innovations spanning morphology design, defect chemistry, phase engineering, and external stimulus coupling. For instance, Li et al.383 reported β-MnO2 nanoflowers with optimized Mn3+ content, which unlocked capacitive-dominated charge storage behavior in β-MnO2//AC asymmetric configurations. This design achieved an energy density of 60.8 Wh kg−1 and 74.1% capacity retention over 3500 cycles, highlighting the benefits of tailoring electronic structure and morphology to enhance ion accessibility. Beyond monolithic morphologies, complex heterostructured cathodes such as Mn2O3@TiO2@MXene further demonstrate how internal electric field engineering and increased surface area can accelerate Mg2+ diffusion. This configuration delivered a specific capacitance of 292.4 F g−1 and exhibited excellent cycling stability over 2400 cycles.384 Complementing these physical design strategies, defect chemistry has emerged as a powerful approach to modulate multi-ion transport dynamics. For example, Liu et al.353 engineered dual-ion defects, including sulfur vacancies and manganese deficiencies, within MnS/MnO heterostructures. This synergistic defect strategy significantly improved Mg2+ transport, resulting in a high capacity of 237.9 mAh g−1 and excellent cycling stability with 94.3% retention after 10000 cycles.
The material landscape of MIHCs has also been expanded by advanced coordination compounds. Lanthanide-based MOFs, such as Park et al.'s385 Ho-HHTP nanorods, offer both high electronic conductivity and strong chelation capabilities. Mg2+ ions are reversibly stored via coordination with catechol ligands and insertion into Ho vacancies, yielding superior electrochemical stability and robust cycling performance. Simultaneously, phase engineering has emerged as a key strategy for improving Mg2+ accommodation. A Cs-induced phase transformation of Mn3O4 produced Mn-oxide phases with enhanced capacitive characteristics, achieving an energy density of 124.91 Wh kg−1. Ethylenediamine-functionalized Mn2O3 cathodes further demonstrated dynamic Mg2+ chelation behavior, delivering a reversible capacity of 188.97 mAh g−1 and 98.83% retention over 2000 cycles (Fig. 20a–c).386,387 Moving beyond passive material modifications, light-assisted MIHCs have introduced photonic activation as an innovative enhancement mechanism. Park et al.388 demonstrated a VO2-based photoresponsive cathode that significantly boosted capacitance (by 56%) and energy density (by 21%) under solar illumination. This work illustrates the potential of integrating photonic and ionic pathways within a single device architecture to surpass conventional electrochemical limitations.
 | ||
| Fig. 20 (a) Magnesium ion capacitor assembly diagram. (b) GCD curves of EDA-Mn2O3//AC. (c) Comparison of the cycling performances of Mn2O3//AC and EDA-Mn2O3//AC; the inset shows an alarm clock image. Reproduced with permission from ref. 386. Copyright 2024, Wiley. (d) Cycling performances of the assembled CaxNHVO-H@GO@CNT//AC CIHCs full cells at 50 mA g−1. (e) In situ ATR-FTIR spectra of NHVO-H@GO@CNT and the corresponding GCD profiles. (f) Schematic illustration depicting the dynamic behavior of the electrolyte at the electrode interface during cycling. Reproduced with permission from ref. 390. Copyright 2024, Wiley. (g) Schematic of the AIHCs (Al//AC) cell during discharge and (h) working mechanism of the cell in thevoltage range of 0–2.0 V. (i) Gravimetric energy and power densities of the SHC900 AIHCs full-cell. Reproduced with permission from ref. 393. Copyright 2022, the Electrochemical Society. | ||
Collectively, the integration of solvation dynamics, phase engineering, interfacial defect modulation, and light-responsive strategies demonstrates the unique potential of multi-ion coordination in MIHCs. By utilizing the synergistic interactions among multiple charge carriers, these systems demonstrate performance improvements that remain challenging for conventional single-ion frameworks.
To overcome these limitations, recent efforts have shifted toward amorphous and heterostructured materials, where disordered atomic arrangements and interfacial tailoring offer new degrees of freedom for tuning ion dynamics. For instance, Wang et al.389 demonstrated that amorphous MnOx (A-MnOx) nanospheres, synthesized via a simple solution-phase method, can significantly enhance Ca2+ storage performance in calcium-ion asymmetric supercapacitors (CASC). The defect-rich and isotropic structure of A-MnOx facilitates efficient ion diffusion and accommodates volume expansion during cycling, yielding a specific capacitance of 175 F g−1 across a wide voltage window (0–2 V) and an energy density of 96.8 Wh kg−1 at a power density of 500 W kg−1. These findings highlight the importance of structural disorder in amplifying ionic conductivity and promoting robust charge transport pathways in multi-ion systems. In parallel, architectural innovations have focused on engineering ion transport channels and stabilizing interfacial processes through pre-intercalation and composite design strategies. A notable example is the (NH4)2V6O16·1.35H2O@graphene oxide@carbon nanotube (NHVO-H@GO@CNT) composite cathode developed by Wang et al.,390 which employs a hierarchically integrated heterointerface to facilitate the synchronized co-migration of Ca2+ and TFSI− anions. This structural configuration not only enhances ionic conductivity but also buffers the structural framework during repeated cycling. The full cell constructed using CaxNHVO-H@GO@CNT as the cathode and activated carbon as the anode delivered a discharge capacity of 62.8 mAh g−1 over 190 cycles (Fig. 20d). Operando ATR-FTIR spectroscopy provided critical insights into the interfacial processes involved (Fig. 20e and f), revealing that Ca2+ de-solvation at the electrode surface is closely coupled with the redistribution of TFSI− in the electrolyte. This dynamic interaction plays a pivotal role in maintaining ionic flux balance and ensuring interfacial stability throughout the charge and discharge cycle.
These advances highlight the critical role of multi-ion coordination in governing the electrochemical performance and long-term stability of CIHCs. The interplay between Ca2+ de-solvation kinetics, anion mobility, and defect-engineered electrodes is essential for achieving high-capacity and durable operation. By precisely tuning ionic speciation, interfacial charge distribution, and bulk transport dynamics, CIHCs are evolving from early conceptual designs into a transformative platform for next-generation multi-ion energy storage, combining high capacity, fast kinetics, and structural adaptability to meet the growing demands of sustainable, high-power applications.
Initial breakthroughs in AIHCs demonstrated the feasibility of Al3+ intercalation in host frameworks. Li et al.391 investigated Al3+ storage using a Prussian blue analog (Al0.2CuFe-PBA), achieving a reversible capacity of 55 mAh g−1 in aqueous electrolytes. While this work laid a conceptual foundation for Al3+-based energy storage, it also revealed intrinsic kinetic limitations associated with the strong hydration shell of multivalent ions. These challenges highlighted the necessity for advanced multi-ion carrier strategies that could facilitate efficient de-solvation, ion transport, and interfacial charge transfer.
Subsequent studies have significantly expanded the operational scope of AIHCs through innovations in electrode architecture, electrolyte formulation, and cell configuration. A landmark contribution by Ma et al.392 introduced an asymmetric aqueous rocking-chair AIHC featuring highly adaptive, pore-structured electrodes engineered for dynamic compatibility with hydrated Al3+ ions. This design enabled a broadened voltage window of up to 2.0 V, resulting in a volumetric energy density of 112 Wh L−1 and a remarkable power density of 30000 W L−1, with excellent cycling stability exceeding 10000 cycles. These results underscore the critical role of synchronized multi-ion transport and stable interfacial double-layer formation in sustaining high-performance operation. Complementary advances in non-aqueous systems were reported by Kim et al.,393 who developed an ionic liquid-based AIHC using AlCl3 paired with pore-engineered activated carbon electrodes. As illustrated in Fig. 20g and h, the Al/AC device operates via dual faradaic and capacitive processes, with the cathode functioning as an EDLC and the anode as an Al-ion battery-type electrode across a 0–2.0 V range. This hybrid configuration achieved an energy density of 51 Wh kg−1 while maintaining 97.9% of its capacitance after 10000 cycles, thereby outperforming conventional EDLC and nearing the performance standards of lithium ion technologies. A comparative Ragone plot (Fig. 20i) further highlights the superior energy-power profile of the SHC900-based AIHCs, positioning it favorably against EDLCs, lithium-ion batteries, and lithium-ion capacitors.
The evolution of AIHCs has also been propelled by environmentally conscious materials design that aligns with the principles of green chemistry. Feng et al.394 developed a solvent-free synthesis strategy to produce porous carbon spheres (PCSs), which not only minimized the ecological footprint of electrode fabrication but also delivered a record-high double-layer capacitance of 200 mAh g−1. This work demonstrated that sustainable material synthesis can be effectively integrated with Al3+ storage chemistry to improve overall device performance. Building on this, He et al. developed a composite electrode consisting of polypyrrole-coated molybdenum disulfide (PPy@MoS2/CC). The MoS2 component, with its naturally layered architecture, facilitated efficient Al3+ intercalation when coupled with an optimized electrochemical deposition method. The resulting electrode achieved a high capacitance of 815 F g−1 at 2 A g−1, with 71.3% of the total charge storage arising from diffusion-controlled processes. This performance notably surpassed that of similar systems based on monovalent (K+, Na+) or divalent (Mg2+) ions, highlighting the unique advantages of trivalent ion carriers in AIHCs.395 A particularly compelling demonstration of multi-ion synergy was reported by Tang et al.,396 who constructed a symmetric AIHC using benzoquinone-preintercalated layered vanadium oxide (BQ-VO). The expanded interlayer spacing (13.3 Å) not only facilitated Al3+ intercalation but also enabled a reversible redox process involving ClO4−/Cl− anions in an Al(ClO4)3/acetonitrile electrolyte. This dual-ion storage mechanism delivered a high specific capacity of 152 mAh g−1 and exemplified how the cooperative interaction between multivalent cations and complementary anions can significantly enhance both energy storage capacity and cycling reversibility.
Collectively, these advances demonstrate that the electrochemical performance of AIHCs is determined not only by the properties of Al3+, but by the coordinated interaction of multiple charge carriers, interfacial dynamics, and structural design. Key performance limits are defined by the interplay among hydration shell regulation, charge redistribution at electrode interfaces, and mechanical strain accommodation within host materials. While challenges such as Al3+-induced polarization and interfacial degradation persist, ongoing improvements in pore architecture, electrolyte formulation, and green synthesis are steadily addressing these issues. Looking ahead, the integration of operando spectroscopy, multiscale modeling, and machine learning-driven material discovery is expected to deepen understanding of ion transport mechanisms and enhance the synergistic behavior between Al3+ and Co-ions.
Although HBCs have garnered increasing attention due to their ability to bridge the gap between high-energy batteries and high-power capacitors, several intrinsic challenges continue to hinder their practical competitiveness relative to DIBs. A primary limitation lies in the kinetic mismatch between the battery-type and capacitor-type electrodes, which often leads to asymmetric charge–discharge behavior and underutilization of electrode capacity. Additionally, the poor interfacial compatibility between electrodes and electrolytes contribute to issues such as rapid capacity fading and limited cycling stability. The necessity of managing ion transport across dissimilar electrode materials further complicates cell architecture and impairs power density. In contrast, DIBs often operate at higher voltages due to the intercalation-based reaction mechanism at the cathode. When paired with appropriately matched ion-host materials, this feature enables higher energy density, broader electrochemical tunability, and increased design flexibility.
To bridge the performance gap between HBCs and DIBs, future research should focus on developing electrode materials with harmonized charge storage kinetics and compatible ion diffusion dynamics. The design of multifunctional electrolytes capable of stabilizing both faradaic and non-faradaic processes is also essential. In addition, interfacial engineering strategies that improve electrolyte wettability, suppress parasitic reactions, and enhance structural integrity will be key to improving HBC longevity. Coupling these advances with in situ/operando diagnostic techniques and data-driven material discovery platforms could accelerate the rational optimization of HBC systems. Collectively, these efforts are expected to push HBC technologies toward more competitive performance and broader application potential.
5. Fuel cells
Fuel cells have long been regarded as a cornerstone of clean energy technologies due to their high energy conversion efficiency and zero carbon emissions and are emerging as critical platforms in the transition to a low-carbon energy future. From Schoenbein's seminal work in 1838 to Grove's gas battery prototype in 1839,397 the evolution of fuel cells has been underpinned by their ability to directly convert chemical energy into electricity through electrochemical redox reactions, circumventing the thermodynamic losses inherent in combustion-based power generation. A typical fuel cell consists of an anode, a cathode, a membrane, and an electrolyte. During operation, fuels such as H2, carbon monoxide, or small organic molecules are oxidized at the anode, while oxidants like O2 or hydrogen peroxide (H2O2) are reduced at the cathode (Fig. 21a). Although H2 remains the most commonly used fuel, hydrocarbon-derived fuels including hydrazine (N2H4), sodium borohydride (NaBH4), alcohols, and aldehydes have gained increasing attention. Unlike primary batteries, fuel cells are not limited by internal reactant storage and can provide continuous and stable electricity generation as long as fuel and oxidants are supplied. By bypassing combustion, fuel cells achieve higher energy efficiency, enhanced environmental compatibility, structural flexibility, and fuel adaptability. | ||
| Fig. 21 (a) A comparison diagram of various fuel cells and their corresponding ionic carriers. (b) Scheme of hybrid acid–alkali fuel cells (taking the bipolar membrane as an example). | ||
Fuel cells are traditionally categorized based on their electrolyte composition and ion transport characteristics in alkaline fuel cells (AFCs), proton exchange membrane fuel cells (PEMFCs), phosphoric acid fuel cells (PAFCs), molten carbonate fuel cells (MCFCs), and solid oxide fuel cells (SOFCs).397 These conventional systems rely predominantly on monovalent ionic conduction, either H+ in acidic or polymer-based membranes, or oxygen ions (O2−) in ceramic electrolytes, to mediate charge transfer between electrodes (Fig. 21a).22,398–400 However, challenges such as catalyst poisoning, insufficient electrolyte stability, limited humidity tolerance, and high temperature requirements have spurred a paradigm shift to incorporating multiple ionic species fuel cells for enhanced transport dynamics and reactivity.
Among these, PEMFCs and SOFCs currently represent the most widely implemented technologies. PEMFCs operate below 100–300 °C and typically rely on platinum (Pt) based electrocatalysts, leading to high material costs and sensitivity to CO poisoning.398 Furthermore, their proton conductivity degrades in low-humidity environments, and non-uniform fuel distribution can corrode catalyst layers. In contrast, SOFCs utilize high-temperature (typically >700 °C) O2− conduction, offering broader fuel flexibility but suffering from long startup times, thermal mismatch-induced degradation, and elevated system costs.401
To overcome these limitations, the development of multi-ion carrier systems has emerged as a promising strategy. Hybrid acid–alkali fuel cells, for instance, incorporate spatially separated acidic and basic electrolytes, enabling concurrent H+ and OH− conduction. Ionic equilibrium is maintained through selective migration of counter-ions (e.g., K+, Cl−), facilitating efficient charge transport while expanding the range of viable fuels, including N2H4, borohydride, alcohols, formic acid, and formaldehyde (Fig. 21b). In parallel, the evolution of SOFCs has been accelerated through the design of mixed ionic-electronic conductor (MIEC) electrodes capable of simultaneously conducting H+, O2−, and e−, thus enlarging reaction zones and suppressing interfacial polarization.402 Strategies such as A-site deficiency and B-site doping have been shown to induce synergistic oxygen vacancy and proton defect formation, enabling co-migration of O2−/H+/e− and improving power density and fuel adaptability at intermediate temperatures.
And by replacing the H2-rich fuels with metals (e.g., Li, Na, K, Zn, Mg, Fe, Al, etc.) as the anode, and using open air (e.g., O2, N2, CO2, etc.) as the cathode oxidant, metal–air battery batteries (MABs) systems are constructed. Once constrained by single-ion chemistry (e.g., Li+ or Na+), these systems now exploit dynamic coordination between metal cations and oxygen-derived species (e.g., H2O, OH−), yielding superior discharge kinetics and tailored product evolution.403,404 This has led to the rational design of hybrid electrolytes such as Li+/OH−, which decouple traditional ionic transport limitations, enhancing both energy density and reversibility. MABs can theoretically achieve energy densities exceeding 1000 Wh kg−1, making them attractive for lightweight and portable energy systems.
As for the cathode, H2O2 is a liquid oxidant that can replace O2 for the cathode reaction. Metal–H2O2 fuel cells have been revitalized by integrating multi-ion transport schemes. Initially developed by the U.S. Naval Research Laboratory (NRL) in the 1960s,405 these systems pair metals such as Zn, Mg, or Al as anodes with H2O2 as the liquid oxidant at the cathode. The energy conversion process involves not only the migration of metal cations (Mn+) and reactive oxygen intermediates (e.g., HO2− and O2), but also pH-coupled carriers (H+/OH−), allowing tunable redox kinetics, high output power, and corrosion control. These cells are particularly advantageous for fast-start applications and low-weight systems due to the liquid-phase catholyte and modular design.
A novel direction in aqueous fuel cells involves metal–H2O systems, where H2O, rather than atmospheric O2, serves as the cathodic reactant. The spontaneous oxidation of active metals (e.g., Al, Mg, Zn) triggers hydrogen evolution from water, which is subsequently utilized electrochemically with air or other oxidants. This water-centric design bypasses the need for gaseous O2, enabling sealed operation in marine or subterranean environments.13,406
The historical trajectory of fuel cell development, from single-ion (H+ or O2−) conduction to strategically engineered multi-ion systems, demonstrates a clear evolution in charge transport complexity. Modern advances in multi-ion design not only extend the operational temperature and fuel flexibility of fuel cells but also enhance redox efficiency, durability, and energy density across a wide range of application scenarios. This section will explore recent breakthroughs in hybrid acid–alkali fuel cells, hybrid H+/O2−/e− conducting SOFCs, MABs, and metal–H2O2 and metal–H2O fuel cells, highlighting how the synergistic interplay between diverse ionic carriers fundamentally redefines interface dynamics and electrochemical performance.
5.1. Hybrid acid–alkali fuel cells
Traditional PEMFCs operate on a principle that can be likened to the “reverse” process of water electrolysis, comprising an anode, a cathode, and a proton exchange membrane.12,407,408 The anode serves as the site for the hydrogen oxidation reaction (HOR), while the cathode facilitates the oxygen reduction reaction (ORR).11,409 Both electrodes in PEMFCs are equipped with catalysts to facilitate electrochemical reactions, while the proton exchange membrane (PEM) serves as the electrolyte.456,457 PEMFCs typically operate using H2 fuel under either acidic or alkaline conditions, referred to as acidic or alkaline fuel cells, respectively. In these systems, H+ and OH− function as the primary ionic carriers. It is well known that acid–base neutralization (H+ + OH− → H2O) is a spontaneous and exothermic process with a standard molar Gibbs free energy change (ΔGθ) of 79.9 kJ mol−1. When this neutralization energy is converted into electricity, it is referred to as electrochemical neutralization energy (ENE).458 In a conventional alkaline fuel cell (pH = 14), H2 is oxidized to H2O at the anode (Eθ = 0.828 V), while O2 is reduced to OH− ions at the cathode (Eθ = 0.401 V), yielding a theoretical cell voltage of 1.229 V (Fig. 22a).458In contrast, an acidic fuel cell (pH = 0) operates with hydrogen oxidation at the anode to generate protons (Eθ = 0 V), and oxygen reduction at the cathode to form water (Eθ = 1.229 V), also resulting in a theoretical voltage of 1.229 V (Fig. 22b). However, since both the HOR and ORR are pH-dependent, decoupling the acid and alkali environments, where the ORR occurs in acidic media (Eθ = 1.229 V) and HOR in alkaline media (Eθ = 0.828 V), enables a significantly enhanced theoretical voltage of 2.057 V due to the additional contribution from ENE (Fig. 22c).458 The corresponding electrochemical reactions and electrode potentials vary with electrolyte pH, as outlined below:
HOR at the anode:
| H2 → 2H+ + 2e− (Eθ = 0 V, pH = 0) | (26) |
| H2 + 2OH− → 2H2O + 2e− (Eθ = −0.828 V, pH = 14) | (27) |
ORR at the cathode:
| 4H+ + O2 + 4e− → 2H2O (Eθ = 1.229 V, pH = 0) | (28) |
| 2H2O + O2 + 4e− → 4OH− (Eθ = 0.401 V, pH = 14) | (29) |
Overall:
Acidic HOR and alkaline ORR:
| 2H2 + O2 + 2H2O → 4H+ + 4OH− (Eθ = 0.401 V) | (30) |
Alkaline HOR and acidic ORR (Fig. 22c):
| 2H2 + O2 + 4H+ + 4OH− → 6H2O (Eθ = 1.229 V + 0.828 V = 2.057 V) | (31) |
 | ||
| Fig. 22 (a)–(c) Comparison of polarization curves for ORR and HOR, along with theoretical cell voltages, in alkaline, acidic, and hybrid acid–alkali H2/O2 fuel cell systems, respectively. Reproduced with permission from ref. 458. Copyright 2024, Wiley. (d) Side view of a planar microfluidic membraneless fuel cell. Reproduced with permission from ref. 410. Copyright 2005, American Chemical Society. (e) Schematic of the operation mechanism for hybrid fuel cells integrating alkaline HOR at the anode and acidic ORR at the cathode. Reproduced with permission from ref. 411. Copyright 2009, American Chemical Society. (f) Configuration of a CF-FC comprising an alkaline HOR chamber (NaOH), a neutral ion transport chamber (Na2SO4), and an acidic ORR chamber (H2SO4), separated by AEM and CEM. Reproduced with permission from ref. 21. Copyright 2024, Wiley. (g) Reaction scheme based on an acid–alkaline patterned cathode catalyst layer. Reproduced with permission from ref. 412. Copyright 2023 Elsevier. (h) Schematic of a membraneless continuous-flow acid–alkali bipolar electrolyte DHzFC using graphite plates sealed with polymeric materials. Reproduced with permission from ref. 416. Copyright 2014, the Korean Electrochemical Society. (i) Schematic of a hybrid acid–alkali DHzFC replacing the sluggish ORR with a more facile HER. Reproduced with permission from ref. 417. Copyright 2023, Elsevier. | ||
This principle has inspired the use of asymmetric acid–alkali electrolytes to extend the potential window in aqueous electrochemical devices, improve energy and power efficiency, and eliminate the reliance on noble metal catalysts, thereby reducing costs. Recent studies have demonstrated the applicability of hybrid acid–alkali fuel cells to a wide range of fuels, including H2, N2H4, sodium borohydride, formaldehyde, alcohols, and ascorbic acid. This section provides a comprehensive overview of these systems, with a particular focus on hybrid acid–alkali H2 fuel cells, direct hydrazine fuel cells (DHzFCs), direct borohydride fuel cells (DBFCs), formaldehyde hydrogen fuel cells (FHFCs), direct alcohol fuel cells (DAFCs), and direct ascorbic acid fuel cells (DAAFCs).
Building on this concept, bipolar membrane (BPM) fuel cells emerged as a structurally robust alternative, integrating cation-exchange membrane (CEM, e.g., Nafion) and anion-exchange membrane (AEM) into a single unit to orchestrate directional H+ and OH− transport (Fig. 22e).411 Interestingly, the balance between electroosmotic drag of water and diffusion of H+/OH− could address the chronic water-management challenges of traditional PEMFCs. Nonetheless, challenges remain with physically bonded CEM–AEM assemblies. Potential safety risks such as membrane delamination and electrochemical instability at the hot-pressed interface may result in water accumulation, phase separation, and structural degradation over time.412
To circumvent these limitations, Dai et al.21 proposed a coupled-flow fuel cell (CF-FC) with spatially segregated acidic (ORR), neutral (ion-exchange), and alkaline (HOR) chambers, separated by an AEM and a CEM (Fig. 22f). This architecture leveraged the electrochemical neutralization energy of H+ and OH− (theoretical OCV = 2.057 V) while enabling Na+ and SO42− to serve as auxiliary charge carriers, achieving an experimental OCV of 1.81 V and stable discharge at 1.16 V (50 mA cm−2 for 110 h). Further innovation came from Qiao et al.,412 who hybridized the catalyst layer itself by embedding alkaline ionic conductors (e.g., quaternary ammonium hydroxides) into a traditional Nafion-based acidic catalyst layer (CL). This “mixed-matrix” approach localized H+/OH− recombination at the nanoscale, creating self-humidified microenvironments that maintained membrane hydration without flooding (Fig. 22g). The resulting interfacial charge equilibration not only improved proton conductivity under low humidity but also demonstrated how multi-ion carrier engineering could be scaled down to the molecular level.
To address this challenge, Durga et al.416 devised a membraneless continuous-flow acid–alkali bipolar electrolyte fuel cell using graphite plates molded with poly(dimethylsiloxane) (PDMS) and sealed with poly(methylmethacrylate) (PMMA) (Fig. 22h). In this configuration, N2H4 as the fuel undergoes oxidation in the alkaline compartment, while H2O2 is reduced at the acidic cathode, the specific reaction equation is as follows:416
Anode:
| N2H4 + 4OH− → N2 + 4H2O + 4e− (Eθ = −1.17 V) | (32) |
| Cathode: |
| 3H2O2 + 6H+ + 6e− → 6H2O (Eθ = 1.78 V) | (33) |
The spatial separation of the two reactions allows for the direct utilization of the pH gradient and enables the simultaneous conduction of H+ and OH− across the liquid–liquid interface. This multi-ion carrier system yields an OCV of 1.72 V and a peak power density of 27.2 mW cm−2, underscoring the benefits of coupling hydrazine oxidation with a highly reactive cathodic process under asymmetric pH conditions.
Recognizing that the ORR remains a bottleneck in such configurations, researchers have turned to alternative cathodic reactions with superior kinetics. In particular, Wang's team417 proposed a hybrid acid–alkali DHzFC that replaces the sluggish ORR with the more facile HER, thereby enabling concurrent power generation and H2 production (Fig. 22i). Utilizing a 3D nanoporous Ni–Co selenide (NixCo1−xSe2) catalyst, the system facilitates hydrazine oxidation (HzOR) at the alkaline anode and the HER at the acidic cathode, denoted as:
Anodic reaction:
| N2H4 + 4OH− → N2 + 4H2O + 4e− | (34) |
Cathodic reaction:
| 2H+ + 2e− → H2 | (35) |
This reaction pairing capitalizes on the fast 2e− HER pathway compared to the 4e− ORR and significantly lowers the voltage barrier for hydrogen production. The bidirectional migration of OH− and H+ across the BPM leverages the substantial pH gradient (ΔpH ≈ 14.6), converting ENE into enhanced OCV (theoretical 1.16 V). The integrated system achieves a peak power density of 13.3 mW cm−2, near-100% faradaic efficiency (FE) for hydrogen generation (>180 μmol h−1), and stable cycling over 105 cycles. This design demonstrates how multi-ion conduction through BPMs not only facilitates charge balance but also boosts energy efficiency, providing a dual-functional energy solution that merges hydrogen production with sustainable power output. Expanding upon this dual-ion strategy, Liu et al.413 developed a similarly configured hybrid acid–alkali system using Pd-based bifunctional electrocatalysts to further optimize HER and HzOR performance, which pushed performance boundaries to 18.2 mW cm−2 with 120 h stability. The atomic-level design of Pd clusters optimized the Gibbs free energy landscape for both N2H4 oxidation and hydrogen evolution, enabling tunable H2 production rates (0.184–0.932 mmol h−1) while maintaining nearly-100% FE.
Conventional DBFCs typically operate in alkaline environments due to the instability of BH4− in neutral or acidic media (Fig. 23a).399 In this context, the electrochemical oxidation of BH4− generates 8e− at the anode, while the ORR at the cathode, as described by the follow reaction:399
 | ||
| Fig. 23 Schematic representations of various hybrid or multi-ion direct liquid fuel cells: (a) a typical DBFC utilizing O2, air, or H2O2 as oxidants. Reproduced with permission from ref. 399. Copyright 2010, Elsevier. (b) Configuration and operating principles of a DBFC. Reproduced with permission from ref. 420. Copyright 2010 Royal Society of Chemistry. (c) Schematic of a bifunctional BOR-HER fuel cell. Reproduced with permission from ref. 401. Copyright 2025, Elsevier. (d) An h-AAFHFC for simultaneous electricity and H2 production. Reproduced with permission from ref. 422. Copyright 2025, Wiley. (e) Theoretical voltage profiles of DEFCs. Reproduced with permission from ref. 426. Copyright 2011, Elsevier. (f) A hybrid acid–alkali DEFC employing an alkaline anode and acidic cathode configuration. Reproduced with permission from ref. 426. Copyright 2011, Elsevier. (g) A hybrid acid–alkali DGFC integrated with the GOR-ORR and the GOR-HER. Reproduced with permission from ref. 429. Copyright 2024, Wiley. (h) Operating principle of a DAAFC containing VO2+/VO2+ in an acidic catholyte. Reproduced with permission from ref. 432. Copyright 2022, Elsevier. | ||
Anodic reaction:
| BH4− + 8OH− → BO2− + 6H2O + 8e− (Eθ = −1.24 V vs. SHE) | (36) |
Cathodic reaction:
For O2 as the oxidant:
| O2 + 2H2O + 4e− → 4OH− (Eθ = 0.40 V vs. SHE) | (37) |
For H2O2 as the oxidant:
| H2O2 + 2e− → 2OH− (Eθ = 0.87 V vs. SHE) for alkaline media | (38) |
| H2O2 + 2H+ + 2e− → 2H2O (Eθ = 1.77 V vs. SHE) for acidic media | (39) |
In all these cases, the ionic conduction involves the co-migration of BH4−, BO2−, H+, or OH−, indicating the presence of a multi-ion carrier environment essential for charge transport and electrochemical conversion. However, several fundamental issues plague traditional DBFCs. The low redox potential of the cathodic ORR (0.4 V vs. SHE) in alkaline media restricts the achievable cell voltage (theoretical 1.64 V; practical OCV ∼1 V), while the poor selectivity of the borohydride oxidation reaction (BOR) often leads to hydrolysis rather than electron transfer, thus reducing the actual electron yield below the theoretical 8e− and lowering energy conversion efficiency, whereas combining anode and cathodic reactions leads to the net cell reaction of a typical acid–alkali DBFC, with a theoretical OCV of 1.64–3.01 V.399
A transformative approach to these limitations emerged through the integration of hybrid acid–alkali electrolytes, effectively introducing additional ionic species and enhancing reaction kinetics via pH gradient-driven energetics.419 For example, Wang et al.420 pioneered an acid–alkali DBFC utilizing a ceramic lithium super-ionic conductor (LISICON) as the separator (Fig. 23b). In this design, BH4− oxidation occurs in the alkaline compartment, while H2O2 reduction takes place in the acidic compartment, mediated by Li+ migration across the LISICON membrane, written as:
At the alkaline anode:
| Zn + 4OH− → ZnO22− + 2H2O + 2e− (Eθ = −1.3 V) | (40) |
| BH4− + 2OH− → BH3OH− + H2O + 2e− (Eθ = 1.24 V) | (41) |
At the acid cathode:
| 4H2O2 + 8H+ + 8e− → 8H2O (Eθ = 1.77 V) | (42) |
This multi-ion conduction framework, where Li+ migrates from the alkaline anode to the acidic cathode while maintaining a suppressed H+ backflow, sustains a stable cell voltage of 2.1 V, significantly higher than traditional DBFCs. The dual contribution from BH4− and Zn oxidation at the anode and H2O2 reduction at the cathode theoretically enables voltages up to 3 V. Nonetheless, practical performance was constrained by slow H2O2 reduction kinetics and modest Li+ conductivity (10−4 S cm−1), highlighting the critical role of ion transport optimization in hybrid systems.
Further advancements in multi-ion carrier management were demonstrated by Liu et al.22 who replaced the traditional ORR with HER at the acidic cathode, coupled with an 8e− BOR at the alkaline anode, represented as (Fig. 23c):
At the alkaline anode:
| BH4− + 8OH− → BO2− + 6H2O + 8e− | (43) |
At the acidic cathode:
| 2H+ + 2e− → H2 | (44) |
Their bifunctional catalyst created an OH−-enriched microenvironment on Pd active sites, effectively suppressing BH4− hydrolysis while promoting electrochemical oxidation. The system harnessed the pH gradient-driven ENE through directional OH−/H+ migration, generating a 0.86 V built-in potential that enabled self-powered operation. This design achieved a peak power density of 19.9 mW cm−2 and near-quantitative FE for H2 production (1.86 mmol h−1 at 100 mA cm−2), showcasing how intelligent ion flux control could simultaneously address energy conversion and storage challenges.
Expanding on this concept, Zhu et al.401 developed a Cu-based catalyst (Cu–CoP/CF) for a bifunctional BOR-HER system. The in situ reduction of Cu+ to Cu0 induces a localized OH−-rich microenvironment on the catalyst surface, inhibiting H+ adsorption and thereby suppressing BH4− hydrolysis (Fig. 23c). Concurrently, the thermodynamic barrier for the BOR is lowered, boosting the oxidation selectivity to 85%. The resulting device achieves a peak power density of 114 mW cm−2, over twice that of conventional DBFCs, and a H2 production rate of 40 mol h−1 m−2.
These developments exemplify how integrating acid–alkali fuel cells with carefully engineered catalysts and exploiting multi-ion carrier transport, comprising BH4−, OH−, H+, BO2−, and even Li+, can dramatically enhance the efficiency and controllability of H2 conversion processes. These strategies represent a paradigm shift in DBFC design, enabling new avenues for practical and sustainable hydrogen energy systems.
At the heart of this innovation lies a sophisticated multi-ion carrier mechanism operating across pH gradients. The alkaline anode compartment facilitates formaldehyde oxidation to formate, while the acidic cathode promotes H+ reduction, with OH− and H+ migration through ion-selective membranes creating an additional ENE contribution. This carefully orchestrated ion transport h-AAFHFC system achieves remarkable performance metrics, an OCV of 1.11 V, a peak power density of 94 mW cm−2, and an extraordinary ≈200% FE for simultaneous hydrogen production at both electrodes. Such an extraordinary FE reflects not only the utilization of formaldehyde as a dual-purpose fuel and hydrogen source but also the efficient coupling of redox processes facilitated by the multi-ion flux. In addition, the co-generation of electricity and acetic acid as a value-added product further enhances the system's sustainability and economic viability. Moreover, the catalyst design and system integration underscore the importance of precise electrochemical interface engineering, where the orchestrated transport of multiple ion species plays a central role in regulating reaction environments, charge balance, and overall energy efficiency.
To address these inherent trade-offs, recent strategies have focused on leveraging the advantages of both acidic and alkaline systems through spatial pH separation and the introduction of multi-ion carrier mechanisms. As shown in Fig. 23e, when comparing conventional PEMs and AEMs, DEFCs using O2 as the oxidant show an equivalent theoretical voltage of 1.14 V. However, the lower anodic and cathodic overpotentials in AEM-DEFCs suggest more favorable kinetics in alkaline media. Upon switching the oxidant to H2O2, both systems benefit from an elevated cathodic potential, yielding higher theoretical voltages of 1.69 V for PEM-DEFCs and 1.61 V for AEM-DEFCs (Fig. 23f). To further enhance cell output, a hybrid system comprising an alkaline anode and acidic cathode with H2O2 as the oxidant was introduced, integrating multi-ion transport pathways and establishing a strong pH gradient. NaOH enables the ethanol oxidation reaction (EOR) at the anode (pH = 14), while H2SO4 supports H+-mediated H2O2 reduction at the cathode (pH = 0), written as:
Anode:
| CH3CH2OH + 12OH− → 2CO2 + 9H2O + 12e− (Eθ = −0.74 V) | (45) |
Cathode:
| 6H2O2 + 12H+ + 12e− → 12H2O (Eθ = +1.78 V) | (46) |
An et al. first realized this configuration,426 achieving an OCV of 1.60 V and a peak power density of 240 mW cm−2, significantly outperforming conventional DEFCs by 2–4 fold. These improvements derive from higher thermodynamic potential (2.52 V), reduced cathodic overpotential, and enhanced ethanol oxidation kinetics. The system's operation hinges on the coordinated migration of OH− from the alkaline anode and H+ at the acidic cathode, while Na+/SO42− serves as charge-balancing species.
Despite these advances, challenges remain. H2O2 decomposition on Pt cathodes and ethanol crossover generate mixed potentials that reduce effective cathode voltage. Nevertheless, this hybrid strategy has been extended to other alcohols. Pan et al.424 developed a split-pH DEGFC achieving a power density of 115 mW cm−2 and an OCV of 1.42 V, comparable to O2-fed alkaline DEGFCs. By pairing alkaline anode chambers (with 1-propanol, glycerol, and 2-propanol in NaOH) with acidic peroxide cathodes (in H2SO4), these systems maintained >100 mW cm−2 power densities while utilizing cost-effective carbon black cathodes.427 This design flexibility stems from the decoupled optimization of ion-specific environments: OH−-rich anodes facilitate alcohol dehydrogenation through nucleophilic attack, while H+-dominant cathodes accelerate peroxide reduction kinetics, with the interfacial pH gradient contributing an additional 0.059 V/ΔpH unit through ENE. Notably, this design eliminates the need for Pt, validating the feasibility of cost-effective cathode materials while supporting a wide spectrum of liquid fuels.
Most recently, our group428,429 has elevated this concept to new heights through innovative catalyst and system designs. The high-entropy sulfide anode (FeCoNiCrMnS2/CC) in hybrid alkali–acid DGFCs demonstrated how multi-element synergy could overcome precious metal limitations while maintaining 50.1 mW cm−2 power output and 120 h stability. The configuration exploits the synergistic migration of OH− and H+ to respectively drive the glycerol oxidation reaction (GOR), ORR, and HER, expressed as follows (Fig. 23g):
Anode:
| C3H8O3 + 11OH− → 3HCOO− + 8H2O + 8e− | (47) |
Cathode:
ORR:
| O2 + 4H+ + 4e− → 2H2O | (48) |
HER:
| 2H+ + 2e− → H2 | (49) |
This architecture not only suppresses precious metal dependence via multi-element synergy but also achieves chemical production, formate generation at 120 μmol h−1 cm−2, while maintaining excellent stability (<0.1% metal dissolution). This represents a significant step toward a dual-function “energy-chemical coproduction” platform and exemplifies the potential of high-entropy materials in electrochemical systems. Notably, the integrated GOR-ORR and GOR-HER system established a closed-loop, self-powered biorefinery paradigm mediated by precisely controlled multi-ion carriers dynamics.429 The breakthrough intermetallic PtZn@N–C catalyst system further pushed boundaries, achieving a peak power density of 286.8 mW cm−2 at an industrial-relevant current density (100 mA cm−2), through optimized OH−/H+ flux balance.318
In summary, hybrid acid–alkali fuel cells provide an innovative path forward for high-efficiency, low-cost energy systems. By leveraging the spatial separation of redox environments and enabling synergistic multi-ion transport (e.g., reactive ions OH−/H+, and charge-balancing species Na+/SO42−), these systems overcome the kinetic and thermodynamic constraints of conventional DAFCs. The development of multi-ion carrier mechanisms not only enhances voltage output and fuel utilization but also enables integrated chemical production, offering a transformative framework for sustainable electrochemical energy conversion.
Anode:
| C6H8O6 → C6H6O6 + 2H+ + 2e− | (50) |
Cathode:
| O2 + 4H+ + 4e− → 2H2O | (51) |
Overall:
| 2C6H8O6 + O2 → 2C6H6O6 + 2H2O | (52) |
These reactions, particularly favored in acidic environments, are notable for their environmental compatibility, safety, and cost-effectiveness, positioning DAAFCs as attractive candidates for portable electronics, medical applications, and biosensors. Nonetheless, conventional DAAFCs have been constrained by limitations including low energy density, sluggish reaction kinetics, high catalyst cost, and limited durability, which collectively impede their broader implementation.
A split-pH configuration has been proposed as a strategy to enhance electrochemical performance through dual-ion coordination and improved reaction kinetics. In a pioneering study, Muneeb et al.431 designed an DAAFC employing an alkaline fuel stream composed of ascorbate and KOH at the anode, and an acidic oxidant stream of hydrogen peroxide in H2SO4 at the cathode. This pH-gradient-enabled device achieved an OCV of 1.31 V and a peak power density of 158 mW cm−2 at 60 °C using 30% H2O2. Notably, this performance was attained using cost-effective carbon black electrodes, demonstrating how intelligent ion management could circumvent precious metal dependencies.
To efficiently convert ascorbic acid (H2A)/ascorbate (A2−) into electricity, Wang et al.432 developed an innovative DAAFC system through their innovative vanadium-mediated system. In the alkaline anode, A2− oxidizes to dehydroascorbic acid (DHA) at a much lower potential, approximately 0.6 V lower than that under acidic conditions, thereby reducing the activation energy for e− release. Simultaneously, the acidic catholyte containing VO2+/VO2+ enables efficient ORR, facilitated by both direct VO2+ oxidation and indirect catalysis via HNO3. This multi-ion synergy (OH−/VO2+/VO2+/H+) yielded exceptional performance metrics, a maximum power density of 449.4 mW cm−2 and a short-circuit current density of 987 mA cm−2 at 90 °C, while maintaining full compatibility with non-precious graphite electrodes.
These collective developments highlight how hybrid acid–alkali architectures transcend the limitations of conventional fuel cells by: (1) exploiting pH gradients to enhance thermodynamic potentials; (2) enabling the use of alternative fuels through kinetic pathway optimization; and (3) integrating energy conversion with valuable chemical production. Such strategies not only elevate the energy conversion efficiency but also introduce multifunctionality, enabling simultaneous power generation and H2 production within a unified, scalable platform.
5.2. Hybrid H+/O2−/e− conducting SOFCs
SOFCs have long been recognized for their superior energy conversion efficiency (60–85%) and broad fuel flexibility, enabling the direct utilization of H2, methane, and methanol in both centralized and distributed energy systems.433 Their compatibility with renewable and fossil energy carriers, coupled with low emissions and noise-free operation, underlines their promise as sustainable power solutions. However, traditional SOFCs predominantly rely on O2− as charge carriers and require high operating temperatures (600–1000 °C), which not only induce material degradation (e.g., thermal expansion mismatch and electrode sintering) and slow system start-up but also elevate costs and limit widespread applicability (Fig. 24a and d). These issues are largely attributed to the high activation energy of O2− migration, which drastically suppresses ionic conductivity at intermediate temperatures (<600 °C).397 | ||
| Fig. 24 Schematic illustration of ion/e− transport mechanisms in (a) conventional SOFCs Reproduced with permission from ref. 397. Copyright 2024, Springer Nature, (b) H-SOFCs Reproduced with permission from ref. 434. Copyright 2023, Wiley and (c) hybrid H+/O2−/e− conducting SOFCs Reproduced with permission from ref. 402. Copyright 2021, Springer Nature. Depiction of bulk and surface transport pathways and electrode reaction sites within a single particle for (d) H+/e− conductors. Reproduced with permission from ref. 397. Copyright 2024, Springer Nature, (e) O2−/e− conductors, reproduced with permission from ref. 434. Copyright 2023, Wiley and (f) H+/O2−/e− triple-conducting materials. Reproduced with permission from ref. 402. Copyright 2021, Springer Nature. | ||
To overcome these limitations, H+-conducting solid oxide cells (H-SOFCs) have emerged as an alternative architecture, leveraging electrolytes such as BaZr0.1Ce0.7Y0.2O3 (BZCY) that facilitate efficient H+ conduction (Fig. 24b and e).434 Compared to O2−, protons offer significantly lower activation energy, enabling effective operation at intermediate temperatures (400–700 °C) and thereby improving system durability and stability. Moreover, the cathodic formation of H2O from H+ ions mitigates fuel dilution effects, enhancing fuel utilization efficiency. Recent developments in H+-conducting materials and electrode interfaces have driven H-SOFC operation toward even lower temperatures (<500 °C), marking a pivotal shift in fuel cell technology.
A critical factor that governs cathode performance in these systems is the nature of charge transport pathways. In pure electronic conductors, the ORR is constrained to the narrow triple-phase boundary (TPB), where gas, electrode, and electrolyte converge.397 This severely limits the density of active sites. Single ion-electron conductors, such as H+/e− or O2−/e− systems, can modestly expand the reaction zone to the electrode surface or bulk, yet their catalytic efficacy remains bounded by the unidirectionality of ion conduction (Fig. 24c). In contrast, mixed ionic–electronic conductors that support concurrent transport of H+/O2−/e− markedly broaden the electrochemically active zone, enabling both surface and bulk ORR activity (Fig. 24f).402 These materials serve as ideal cathode candidates for H-SOFCs, effectively reducing polarization losses and enhancing power output.
Pioneering work conducted by Zhu et al.435 introduced ceria-based dual-phase composites that integrate oxygen ion conductors (e.g., Gd/Sm-doped ceria) with H+-conducting salts (e.g., Li/NaCO3), achieving hybrid O2−/H+/e− co-conduction between 400 and 650 °C. This synergistic conduction supports simultaneous ion migration through the O2− and H+, enabling electrochemical reactions at both electrodes. The resulting increase in ion flux not only enhances current output but also improves electrode reaction kinetics, offering a fundamentally distinct charge transport paradigm from conventional single-ion systems. Power densities ranging from 200 to 1000 mW cm−2 under H2 and 200–700 mW cm−2 with methane or methanol fuels highlight the superior temperature tolerance and fuel versatility of this multi-ion approach. Kim et al.411 developed NdBa0.5Sr0.5Co0.5O5+δ (NBSCF), a layered perovskite cathode with triple conductivity (H+/O2−/e−), in conjunction with a BaZr0.1Ce0.7Y0.1−xYbxO3−δ (BZCYYb) electrolyte. The alternately stacked [LnO] and [CoO] layers form rapid O2− channels, while H+ conduction and electronic transport are simultaneously enabled.436 The synergy among all three carriers significantly improves electrochemical kinetics at the electrode–electrolyte interface, yielding a power density of 1.61 W cm−2 with stable performance over 500 h at 1023 K. This material overcomes the limitations of traditional unifunctional cathodes and mitigates interfacial polarization by enabling concurrent multi-ion transport.
Building on this, Duan et al.437 engineered BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY), a Co/Fe-doped derivative of yttrium-doped barium zirconate (BZY), which introduced electronic conductivity and established a triple conduction pathway. Their innovative solid-state reactive sintering (SSRS) process enabled one-step co-sintering of dense electrolytes and porous electrodes at 1400 °C. This design reduced polarization resistance to 0.1 Ω cm2 at 500 °C and demonstrated excellent durability (>1000 h) and low-temperature performance (e.g., 290 mW cm−2 at 600 °C using methane), underscoring the practical viability of multi-ion strategies in commercial low-temperature fuel cells. To further enhance ionic transport, Ren et al.438 introduced A-site Ba deficiencies into BCFZY, creating additional oxygen vacancies and achieving simultaneous optimization of H+/O2−/e− conductivity. The Ba0.9CFZY cathode achieved 668.64 mW cm−2 at 600 °C, 38% higher than the stoichiometric counterpart, offering a novel route for defect engineering in H-SOFCs. Liang et al.439 advanced this concept by B-site doping 5% Mg into BCFZY, yielding Ba(Co0.4Fe0.4Zr0.1Y0.1)0.95Mg0.05O3−δ (BCFZYM). The Mg2+ dopant created additional oxygen vacancies, enhanced surface exchange, and stabilized the oxidation states of Co/Fe. A peak power density of 850 mW cm−2 at 600 °C was maintained without degradation for 300 h, providing both high catalytic activity and operational robustness.
Recognizing the limitations of traditional perovskites, which suffer from poor hydration and high thermal expansion, researchers have turned to Ruddlesden–Popper (R–P) type structures. Wang et al.440 developed PrBaNiMnO7−δ, a bilayer R–P perovskite with A/B site doping that synergistically enhances both oxygen vacancy concentration and proton transport. The resulting cells demonstrated outstanding low-temperature performance (259 mW cm−2 at 500 °C, 135 mW cm−2 at 400 °C), low polarization resistance (0.084 Ω cm2), and stable operation over 100 h, setting new benchmarks for H-SOFC cathodes. Further advances by Huan et al.441 yielded a triple-layer R–P cathode, Sr3EuFe2.5Co0.5O10−δ, with co-doped Eu and Co to improve structural and catalytic properties. The material reached 900 mW cm−2 at 700 °C with a polarization resistance of 0.030 Ω cm2, establishing new records for R–P cathode performance and confirming the role of oxygen vacancies in facilitating the ORR. Complementarily, Song et al.442 developed a nanocomposite bifunctional air electrode incorporating perovskite, R–P phase, CeO2, and NiO nanoparticles. This design offered both excellent PCFC and electrolysis functionality, achieving 531 mW cm−2 in fuel cell mode and −364 mA cm−2 at 1.3 V in electrolysis mode, demonstrating the practical utility of multi-ion cooperative designs in reversible energy systems.
Beyond electrodes, electrolyte innovation is central to enabling multi-ion conduction. Zhu's team443 engineered the Gd0.1Ce0.9O2−δ (GDC) electrolyte via surface/interface engineering, where oxygen vacancies and amorphous layer distribution were tuned to enhance the cooperative transport of H+ and O2−. A H+-conductivity of 0.17 S cm−1 at 500 °C and a peak power density nearing 1000 mW cm−2, demonstrate the potential of interface-engineered hybrid electrolytes.
Altogether, these innovations highlight a critical evolution in fuel cell design, from single-ion systems toward sophisticated multi-ion conductors capable of synergistic H+/O2−/e− transport. By orchestrating diverse ionic carriers in both electrodes and electrolytes, hybrid H+/O2−/e− conducting SOFCs can operate efficiently across a broad temperature range, with enhanced kinetics, durability, and fuel flexibility.
5.3. Metal–air batteries
MABs, distinguished by their open-structured cathodes that eliminate the need for preloaded active materials, have emerged as a cornerstone of next-generation energy storage technologies due to their exceptional theoretical energy densities (e.g., 5928 Wh kg−1 for Li–O2 systems), environmental compatibility, and cost-effectiveness compared to conventional batteries (Fig. 25a).403 Tracing their origins to Maiché's pioneering zinc-based system in 1878, featuring porous air cathodes and Pt-coated carbon electrodes, early MABs were hindered by low output voltages (<1 V), corrosive acidic electrolytes, and leakage issues.403 A transformative breakthrough arrived in 1932 with Heise and Schumacher's alkaline ZABs, which elevated operational voltages above 1.4 V and established the foundation for modern electrolyte architectures.444 The 21st century has witnessed a renaissance in MAB research, driven by advances in materials science and the urgent demand for sustainable energy solutions (Fig. 25b).404 Crucially, this evolution has shifted from single-ion-dominated systems (e.g., Zn2+ and Li+) to multi-ion carrier mechanisms, where synergistic interactions between metal cations (e.g., Zn2+ and Li+) and oxygen-derived species (HO2−, O2−, and OH−) govern interfacial charge transfer. | ||
| Fig. 25 (a) Schematic of aqueous/non-aqueous MABs mechanisms. (b) Key performance metrics of representative MABs (Date from). | ||
Fundamentally, MABs store and convert energy through redox processes involving a metallic anode (Mg, Al, Zn, Fe, etc.) and an air cathode that facilitates gas-phase reactions involving O2, N2, CO2, or H2.403,445 Energy storage is achieved via metal oxidation/deposition at the anode and the ORR/OER at the cathode, with representative pathways for O2-based systems detailed in Table 1. Although this electrochemical architecture is universal across MABs, the ion transport mechanisms vary significantly between aqueous and non-aqueous systems, revealing the critical role of multi-ion carrier design in determining performance outcomes.
| Metal anode | Electrolyte | Anode reaction | Cathode reaction | Overall reaction | Ref. |
|---|---|---|---|---|---|
| Li/Na/K | Nonaqueous | M ⇌ M+ + e− | 2M+ + O2 + 2e− ⇌ M2O2 | O2 + M ⇄ M2O2 | 448 |
| 4M+ + O2 + 4e− ⇌ 2M2O | 4M + O2 ⇌ 2M2O | ||||
| Aqueous | O2 + 2H2O + 4e− ⇄ 4OH− | 4M + O2 + 2H2O ⇄ 4MOH | 404 | ||
| Zn | Aqueous | Zn + 4OH− ⇄ Zn(OH)42− + 2e− | O2 + 2H2O + 4e− ⇄ 4OH− | O2 + 2Zn ⇄ 2ZnO | 449 |
| Zn(OH)42− ⇄ ZnO + 2OH− + H2O | |||||
| Mg | Aqueous | Mg ⇄ Mg2+ + 2e− | O2 + 2H2O + 4e− ⇄ 4OH− | 2Mg + O2 + 2H2O ⇄ 2Mg(OH)2 | 450 |
| Nonaqueous | Mg2+ + O2 + 2e− ⇄ MgO2 | Mg + O2 ⇄ MgO2 | 451 | ||
| 2Mg2+ + O2 + 4e− ⇄ 2MgO | 2Mg + O2 ⇄ 2MgO | ||||
| Sn | Aqueous | Sn + 4OH− ⇄ SnO22− + 2H2O + 2e− | O2 + 2H2O + 4e− ⇄ 4OH− | 2Sn + O2 ⇄ 2SnO2 | 452 |
| Si | Aqueous | Si + 4OH− ⇄ Si(OH)4 + 4e− | O2 + 2H2O + 4e− ⇄ 4OH− | Si + O2 + 2H2O ⇄ Si(OH)4 | 453 |
| Al | Aqueous | Al + 4OH− ⇄ Al(OH)4− + 3e− | O2 + 2H2O + 4e− ⇄ 4OH− | 3O2 + 4Al ⇄ 2Al2O3 | 454 |
| Fe | Aqueous | Fe + 2OH− ⇄ Fe(OH)2 + 2e− | O2 + 2H2O + 4e− ⇄ 4OH− | 2O2 + 3Fe ⇄ Fe3O4 | 455 |
| 3Fe(OH)2 + 2OH− ⇄ Fe3O4 + 4H2O + 2e− |
In non-aqueous MABs, particularly those utilizing reactive metals like Li or Na, the use of organic carbonates or ionic liquids as electrolytes restricts ionic conduction primarily to metal cations (Li+ and Na+).445,446 This singular ion dependence, however, introduces limitations in interfacial stability and kinetic efficiency, due to the absence of complementary ionic species that can assist in modulating surface reactions and stabilizing intermediate products. Then, hybrid electrolyte systems have been developed where the air cathode operates under alkaline conditions, enabling a cooperative Li+/OH− or Na+/OH− transport mechanism that enhances ionic conductivity, reduces polarization, and broadens electrochemical compatibility.447
Conversely, in aqueous systems, particularly in ZABs and Al–air configurations, the use of alkaline electrolytes naturally promotes multi-ion participation, where hydroxide ions (OH−) and metal–hydroxide complexes (e.g., Zn(OH)42− and Al(OH)4−) participate actively in interfacial redox processes. OH− not only accelerates ORR/OER kinetics but also stabilizes soluble metal intermediates, mitigating issues like anode passivation and polarization losses. These dual roles exemplify the intrinsic advantages of multi-ion coordination in achieving high reversibility and long-term operational stability. More complex electrolyte schemes, such as hybrid acid–alkali systems, further demonstrate how coordinated transport involving Mn+/OH−/H+ species can decouple competing interfacial processes and offer finely tunable control over nucleation, product morphology, and redox potential distributions.
Altogether, the diverse ionic environments across different MABs chemistries, whether Mn+/OH− coupling in alkaline media or Mn+/H+/OH− interplay in hybrid systems, dictate nucleation kinetics, discharge product morphology, and long-term stability. This section dissects these mechanisms across monovalent (Li+, Na+, and K+), divalent (Zn2+, Mg2+, Sn2+, etc.), and polyvalent (Al3+ and Fe3+) systems, proposing advanced strategies to actively engineer ion interactions. By framing carrier multiplicity as a deliberate design axis rather than a passive consequence, we chart a path toward next-generation MABs where tailored ion coordination redefines the limits of energy density and durability.
 | ||
| Fig. 26 Schematic representation of monovalent MABs using (a) a single organic electrolyte and (b) a hybrid organic/aqueous electrolyte. (c) Galvanostatic discharge profiles of Na–O2 batteries operated in pure and humidified air with hybrid electrolytes. (d) First discharge and charge curves at 0.5 mA cm−2. Reproduced with permission from ref. 462. Copyright 2015, Elsevier. (e) Discharge–charge curves of a non-aqueous Mg–O2 battery with NaI additive. Reproduced with permission from ref. 463. Copyright 2015, Royal Society of Chemistry. (f) Initial discharge–charge profiles of a Na–air battery using liquid electrolytes with and without Fe(C5H5)2 at 500 mA g−1. Reproduced with permission from ref. 464. Copyright 2015, Royal Society of Chemistry. | ||
To address these issues, a solid–liquid hybrid electrolyte architecture that integrates multiple ion carriers has emerged, enabling novel ionic synergies. Zhou et al.461 introduced a paradigm wherein a Li metal anode in organic 1 M LiClO4 in ethylene carbonate/dimethyl carbonate electrolyte is separated from an aqueous LiOH-based air cathode by a LISICON solid-state electrolyte (Fig. 26b). In this design, Li+ generated during anodic oxidation migrates across LISICON and reacts with OH− in the aqueous phase to form soluble LiOH, while O2 is reduced to OH− at the cathode (O2 + 2H2O + 4e− → 4OH−). This architecture prevents solid Li2O2 precipitation and associated parasitic reactions by leveraging OH− solubility, thus enhancing reaction reversibility and elevating theoretical energy density to 5698 Wh kg−1. Inspired by this approach, similar multi-ion strategies have been adapted for Na–O2 batteries. Hashimoto and Hayashi462 constructed a Na–O2 battery incorporating hybrid organic/aqueous electrolytes and NASICON separators (Fig. 26b). The galvanostatic discharge profiles measured in pure and humidified air demonstrated a theoretical discharge capacity of 1200 mAh g−1 for the hybrid setup (Fig. 26c). The resulting Na+/OH− dual-ion transport mitigated interfacial bottlenecks, reducing overpotential to 0.6 V, increasing discharge capacity 57-fold (503 mAh g−1vs. 8.8 mAh g−1 in aqueous electrolytes), and enhancing power density 10-fold (12.4 vs. 1.4 mW cm−2 in aqueous electrolytes) (Fig. 26d).
The aqueous phase also offers enhanced humidity tolerance and suppresses side reactions, extending battery lifespan. Senthilkumar et al.447 further refined this concept with a NASICON-NaOH configuration, enabling low polarization (0.59 V), high round-trip efficiency (∼83%), and capacity (500 mAh g−1) surpassing even nonaqueous Li–O2 analogs. These studies underline the transformative role of multi-ion carrier design, where phase-segregated pathways for Na+ and OH− substantially enhance kinetics, interfacial stability, and energy output in Na–O2 batteries.
Beyond solid–liquid phase hybridization, the inclusion of soluble redox mediators introduces another dimension of multi-ion coordination. These redox mediators accelerate sluggish O2 redox reactions while enabling cooperative ion-e− transfer pathways. Glusac and co-workers459 reported that triarylmethyl cations (R+) in Li–air batteries are electrochemically reduced to form RO-OR peroxides via outer-sphere mechanisms, synergizing with Li+/O2 inner-sphere reactions. This hybrid mechanism redirects Li2O2 formation into the solution phase, mitigating electrode passivation and boosting discharge capacity by up to 35-fold (2.3 mAh cm−2). Similarly, in SABs, the introduction of NaI promotes a redox-active environment wherein Na+, I−, and I3− interact dynamically.463 During discharge, Na+ combines with O2 to form Na2O2, while charging oxidizes I− to I3−, which decomposes Na2O2 catalytically to regenerate NaI and O2. This ionically rich environment supports a reversible capacity of ∼1000 mAh g−1(Fig. 26e). However, the volatility of iodine species prompted the exploration of alternative mediators such as ferrocene (Fe(C5H5)2+), which facilitates stepwise catalytic decomposition of Na2O2 while maintaining low charge voltages (<3.0 V) (Fig. 26f).464 These redox mediators not only introduce new charge transfer channels but also act as auxiliary ion carriers, enhancing conductivity and selectivity in both discharge and recharge processes.
To further suppress side reactions and improve both the ORR and the OER, multifunctional soluble mediators like Ir(acac)3 have been developed. In Li–O2 systems, iridium(III) acetylacetonate (Ir(acac)3) forms reversible Ir(acac)3-O2− complexes during the ORR, stabilizing reactive intermediates in solution and directing Li2O2 precipitation away from the cathode. During the OER, Ir(acac)3 promotes Li2O2 decomposition via an electrochemical-chemical sequence, reducing overpotentials and suppressing nucleophilic attack by O2− radicals.465 These multifunctional complexes exemplify how orchestrating multi-ion interactions (e.g., Li+, O2−, and redox mediator complexes) can resolve trade-offs between reactivity, stability, and energy density.
Multi-ion dynamics also govern product selectivity, as demonstrated by studies on environmental factors and gas-phase composition in Na–O2 batteries.466,467 Under dry air, Na2O2 predominates,468–470 while humidity facilitates OH−-driven NaOH formation.471 CO2 co-reduction generates carbonates (Na2CO3, Na2C2O4) via Na+/C2O62−/CO42− co-migration. Das et al.470,472 showed that in a Na–O2/CO2 hybrid cell, the presence of 40–63% CO2 leads to a 2.6-fold capacity enhancement. Two proposed mechanisms elucidate these complex transformations: one featuring stepwise O2− and CO2 coupling to form oxalate, and another based on direct O2−–CO2 reduction in organic media, which can be described as follows:
Mechanism 1:
| 4O2 + 4e− → 4O2˙− | (53) |
| 2O2˙− + CO2 → 2CO4˙− | (54) |
| 2CO4˙− + 2CO2 → 2C2O6˙− | (55) |
| C2O6˙− + O2˙− → C2O62− + O2 | (56) |
| C2O62− + 2O2˙− + 4Na+ → 2Na2CO3 + 2O2 | (57) |
| Mechanism 2 (the reduction path of O2 in organic solvent): |
| O2 + e− → O2− or O2 + 2e− → O22− | (58) |
| CO2 + O22− → CO42− | (59) |
| CO42− + CO2 + 2Na+ → Na2C2O4 + O2 | (60) |
These ion-specific interactions are further exemplified in electrolyte-dependent product selectivity: ionic liquids favor Na2C2O4 formation, while tetraethylene glycol promotes Na2CO3/Na2C2O4 co-existence, underscoring how multi-ion carrier environments tailor reaction products.
In contrast, K–O2 batteries uniquely benefit from the formation of KO2, which offers high conductivity, thermodynamic stability, and round-trip efficiencies exceeding 90% under anhydrous conditions.473 These advantages arise from the hard–soft acid–base (HSAB) compatibility between soft acid K+ and soft base O2−, enabling selective 1e− reduction and stable KO2 cycling.473,474 However, moisture triggers parasitic side reactions yielding KOH and K2CO3, while the formation of K2O2 compromises efficiency. These effects necessitate precise K+/O2− flux control and advanced moisture-barrier or solid-state electrolyte designs to maintain single-phase KO2 pathways.474–476
Ultimately, advances in Li+/OH− and Na+/OH− dual-ion transport frameworks have revealed new opportunities to overcome kinetic limitations and enhance cycling stability in MABs.466,467,477 Architectures that spatially decouple ionic functions (e.g., using Na+ to optimize anodic transport and I−/I3− or OH− to promote cathode ORR/OER activity and product solubility) represent a critical design frontier. Yet, analogous advancements in K–O2 systems remain scarce, demanding systematic exploration of ion-coupled interfaces (e.g., K+/O2−/OH− coordination) and stable electrolyte architectures to unlock their full potential.
The energy storage mechanisms in ZABs, whether utilizing O2, CO2, or N2 as cathodic reactants, are universally mediated by synergistic ion transport phenomena. In conventional aqueous alkaline ZABs, the discharge process involves the oxidation of Zn at the anode coupled with the ORR at the cathode, creating a closed loop of Zn(OH)42−/OH− dual-carrier exchange. This interplay is not merely incidental but engineered: the tetrahedral Zn(OH)42− complex stabilizes dissolved Zn2+ intermediates, while OH− ions simultaneously mediate ORR kinetics and prevent anode passivation. The multi-ion coordination is further exemplified in hybrid acid–alkali ZABs configurations, where selective ionomer membranes or solid-state electrolytes spatially segregate Zn2+ and OH− transport to optimize interfacial charge transfer while suppressing dendrite formation.480
Expanding beyond O2-based systems, emerging CO2 or N2-fed ZABs demonstrate even more complex multi-ion carrier synergies. In Zn–CO2 batteries, the cathodic reduction of CO2 generates carbon-containing species (e.g., CO and HCOOH) that couple with Zn2+ to form soluble intermediates (e.g., Zn(OH)42−), leveraging Zn(OH)42−/H+/OH− co-migration to enhance reaction homogeneity.481 Similarly, Zn–N2 batteries exploit Zn2+/NH4+ interactions during the cathodic N2 reduction reaction (NRR) to NH3 or NH4+.482 Thus, ZABs serve not only as an archetype of MAB systems but also as a compelling demonstration of how multi-ion transport underpins advanced electrochemical functionality.
5.3.2.1. Zn–O2 batteries. In Zn–O2 batteries, the incorporation of zinc acetate (Zn(Ac)2) into alkali electrolytes has emerged as a crucial strategy to mitigate dendrite formation and surface passivation, which simultaneously modulates Zn electrode position morphology through preferential crystal facet adsorption and maintains electrolyte stability by sequestering CO2-derived carbonate species before they can precipitate and block air cathode pores. This dual-action mechanism exemplifies how deliberate multi-ion carrier engineering (Zn2+/CH3COO−/OH− coordination) can overcome intrinsic limitations of conventional ZAB systems.
Aqueous alkaline electrolytes (KOH, KOH + Zn(Ac)2, 7 ≤ pH < 14) remain the cornerstone of high-performance Zn–O2 batteries due to their exceptional ORR kinetics (200–300 mV overpotential reduction) and superior ionic conductivity (>100 mS cm−1).483 The discharge process involves complex multi-ion interactions: Zn oxidation generates soluble Zn(OH)42− at the anode, while the cathode facilitates the ORR through competing pathways. The ideal 4e− transfer route (O2 + 2H2O + 4e− → 4OH−) is often compromised by the parasitic 2e− pathway (O2 + H2O + 2e− → HO2− + OH−, HO2− + H2O + 2e− → 3OH−, 2HO2− → 2OH− + O2), which not only reduces energy efficiency but also generates oxidative species that degrade cell components. Crucially, both pathways are governed by multi-ion carrier dynamics, Zn(OH)42− migration from the anode intersects with OH− and oxygen-derived intermediates (O2−, HO2−) at the cathode, demonstrating how ion transport networks dictate reaction selectivity and system stability. Further, to overcome the sluggish kinetics of the multi-e− ORR and to enhance overall efficiency, researchers have developed advanced cathode materials, ranging from heteroatom-doped carbon nanostructures and transition metal oxides to atomically dispersed catalysts, to modulate intermediate adsorption/desorption energies and facilitate multi-ion transport. Comparative performance metrics for these catalysts are detailed in Table S6, highlighting their contributions to enhancing Zn–O2 battery performance.
Pushing beyond conventional energy and power limitations, our group pioneered an asymmetry-electrolyte Zn–air battery (AEZAB) in which a BPM separates an alkaline NaOH anolyte from an acidic H2SO4 catholyte.484 The AEZAB elevates the theoretical cell voltage to 2.55 V by coupling Zn oxidation in alkaline media with the acidic ORR (Fig. 27a) and delivers a high maximum power density of 270 mW cm−2, 1.88-fold higher than that of conventional ZABs (143 mW cm−2) (Fig. 27b). The mechanism is proposed as follows:
 | ||
| Fig. 27 (a) Schematic diagram of the as-assembled AEZAB. (b) Discharge polarization and corresponding power density curves of AEZAB and ZAB. Reproduced with permission from ref. 484. Copyright 2019, Springer Nature. (c) Schematic illustration of hybrid acid–alkaline ZABs employing AEM and CEM. Reproduced with permission from ref. 480. Copyright 2024, Wiley. (d) Schematic of hybrid acid–alkali ZABs based on a ZnSTM. Reproduced with permission from ref. 485. Copyright 2021, Elsevier. (e) Voltage profile of an AQDS-mediated ZAB, where the solid line denotes the first cycle and the dashed line indicates constant–current cycling following full anode replacement after Zn depletion. Reproduced with permission from ref. 487. Copyright 2022, Wiley. (f) Schematic of the cell configuration and working mechanism of a rechargeable seawater-based ZAB. Reproduced with permission from ref. 488. Copyright 2020, Elsevier. | ||
Alkaline anode:
| Zn + 2OH− → H2O + ZnO + 2e− | (61) |
Acidic cathode:
| 4H+ + O2 + 4e− → 2H2O | (62) |
Extending this concept, they further developed a hybrid acid–alkali ZAB,480 wherein a desalination chamber bridges the acidic and alkaline compartments with the AEM and the CEM, enabling concurrent Na+/Cl− migration and electricity generation (375 mW cm−2), while removing salts from seawater (Fig. 27c). This multifunctional configuration leverages a rich palette of coexisting ions, H+, OH−, Na+, Cl−, and Zn(OH)42−, demonstrating the potential of hybrid electrolyte systems to integrate energy conversion with water treatment.
Building upon this electrolyte decoupling strategy, asymmetric MABs incorporating zinc-based solid transmembrane membranes (ZnSTM) have been introduced to achieve selective Zn2+conduction while co-mediating H+, OH−, and Zn(OH)42− transport (Fig. 27d).485 By physically isolating KOH and H2SO4 electrolytes yet enabling selective ion exchange, ZnSTM suppresses neutralization reactions and interfacial instability, common challenges in pH-asymmetric systems. This configuration achieves an OCV of 1.96 V, an energy density of 1354 Wh kg−1 (based on Zn), and a round-trip efficiency of 76.6% over 100 h, further highlighting the role of tailored multi-ion pathways in stabilizing high-performance Zn–O2 architectures.
Recent paradigm-shifting insights into ORR/OER mechanisms have challenged the long-standing assumption that zinc peroxide (ZnO2) formation is irreversible.486 It was shown that ZnO2 can reversibly form and decompose by constructing a hydrophobic, anion-rich inner Helmholtz layer at the air cathode. This design creates a water-deficient interface, which effectively stabilizes the 2e− ORR pathway. Using hydrophobic anions such as CF3SO3− to exclude H2O from the reaction site, the team enabled Zn+-mediated ZnO2 cycling with minimal parasitic hydrolysis, achieving a remarkable 1600 h cycle life at 0.1 mA cm−2. In parallel, Wang et al.487 advanced the 2e− ORR by introducing the anthraquinone-2,7-disulfonic acid disodium salt (AQDS) as a soluble redox mediator in alkaline media. Through electrochemical-chemical coupling, AQDS mediates the reduction of O2 to HO2−, bypassing heterogeneous catalysis and enabling high-efficiency ORR (Fig. 27e). This AQDS2−/HO2−/OH−/H+ co-transport mechanism lowers activation barriers and reduces overpotentials, yielding 85% energy efficiency at 10 mA cm−2, and reinforcing the advantage of redox-mediated multi-ion synergy in boosting Zn–O2 battery performance.
These advances have extended Zn–O2 battery capabilities into extreme operational domains. Zhang et al.488 developed seawater-derived electrolytes (6 M KOH + 0.2 M Zn(Ac)2 dissolved in seawater) that exploit OH−/Zn(OH)42− dual-ion transport to buffer Cl− interference (Fig. 27f). Further innovation yielded a CsOH-based electrolyte that sustained 500 cycles with a narrow 0.8 V voltage gap at 5.0 mA cm−2, even at −10 °C, showcasing exceptional low-temperature adaptability.489 Modern Zn–O2 systems are increasingly multifunctional, extending beyond energy storage into applications such as H2O purification, hydrogen evolution, and H2O2 synthesis via a 2e− ORR, all of which are orchestrated by carefully tuned fluxes of H+, OH−, and Zn(OH)42−.486 By engineering ionic gradients, hybrid electrolytes, and redox-active interfaces, multi-ion carrier strategies have transformed Zn–O2 batteries into integrated energy-environmental platforms. These systems transcend historical trade-offs between energy density, reversibility, and operational versatility, firmly positioning ZABs as a critical node in the future of sustainable energy technologies.
5.3.2.2. Zn–CO2 batteries. Zn–CO2 batteries, an emerging subclass of MABs, uniquely integrate electrochemical energy storage with carbon capture and utilization, offering a dual-function platform that capitalizes on the dynamic interplay of multiple ionic species (Table S6). Structurally analogous to Zn–O2 systems yet distinct in their cathodic chemistry, these batteries employ BPMs to maintain critical pH gradients between alkaline anolyte (KOH/Zn(Ac)2) and near-neutral catholyte (KHCO3 ± HCOO−), enabling selective ion transport while preventing electrolyte crossover.490 This architectural innovation creates a dynamic multi-ion environment where zinc speciation varies dramatically with local pH: neutral conditions favor Zn2+ as the primary charge carrier (Zn → Zn2+ + 2e−), while alkaline media promote zincate formation (Zn + 4OH− → Zn(OH)42− + 2e−) followed by dehydration to ZnO (Zn(OH)42− → ZnO + 2OH− + H2O). This pH-dependent Zn2+/OH− interplay directly influences anode morphology and cycling stability, demonstrating how electrolyte engineering can steer phase evolution through controlled ion coordination.
Concurrently, the cathode mediates CO2 reduction (CO2RR) through multiple competing pathways, which are similarly governed by electrolyte composition and ion flux. In neutral media, CO2 is electrochemically reduced to formic acid (CO2 + 2H+ + 2e− → HCOOH), whereas in alkaline environments, the pathway shifts toward formate production (CO2 + 2e− + H2O → HCOO− + OH−).491 These divergent pathways illustrate the modulatory roles of H+, OH−, and HCOO− ions in tuning product selectivity and stabilizing reactive intermediates. Collectively, this complex ionic choreography, spanning Zn2+/H+ in neutral media and Zn(OH)42−/HCOO−/OH− in alkali media, addresses intrinsic limitations of nonaqueous CO2 batteries, such as low CO2 solubility and parasitic reactions, by leveraging the high ionic conductivity and buffering capabilities of aqueous electrolytes (>100 mS cm−1).
To systematically address CO2RR selectivity challenges and round-trip efficiency, Wang et al. developed a breakthrough reversible system employing a Pd-based bifunctional cathode within a BPM architecture (Fig. 28a).492 By isolating alkaline anolyte (1 M KOH + 0.02 M Zn(Ac)2) from neutral catholyte (1 M NaCl + 0.1 M HCOONa), this design harnesses Zn(OH)42−/OH−/H+ multi-ion carrier dynamics to achieve exceptional performance: 89% FE for HCOOH production, and 81.2% energy efficiency with 788 Wh kg−1 theoretical density. The system maintains 73.5% efficiency over 100 cycles, demonstrating how precise ion flux control can stabilize both energy storage and CO2 conversion processes. Expanding this paradigm, the same group later introduced an Ir@Au bimetallic catalyst system that mimics photosynthetic CO2 fixation within a pH-decoupled system (anolyte: KOH + Zn(Ac)2; catholyte: CO2-saturated KHCO3).493 Here, optimized Zn2+ migration drives CO2-to-CO conversion with 90% FE at 1.14 V, while Zn2+/OH−/H+ co-transport enables efficient charging through the ORR and Zn deposition (Fig. 28b).
 | ||
| Fig. 28 (a) Schematic illustration of a reversible Zn–CO2 battery operating in a hybrid acid-salt electrolyte. Reproduced with permission from ref. 492. Copyright 2018, Wiley. (b) Schematic of an aqueous rechargeable Zn–CO2 electrochemical cell employing a hybrid alkali-salt electrolyte. Reproduced with permission from ref. 493. Copyright 2019, Wiley. (c) Schematic representation of a hybrid acid–alkali Zn–CO2 battery. Reproduced with permission from ref. 495. Copyright 2022, the American Chemical Society. (d) Schematic of a Zn–N2 battery featuring a FeSA-N-C-900 cathode. (e) Proposed reaction mechanism for N2-to-NH3 conversion (blue and white spheres representing N and H atoms, respectively). Reproduced with permission from ref. 489. Copyright 2021, Wiley. (f) Schematic diagram of the flow-cell device for NRR. (g) Illustration of the NRR reaction pathway. (h) In situ ATR-FTIR spectra of FeSA-N-C-900 at −0.2, −0.4, and −0.6 V. Reproduced with permission from ref. 497. Copyright 2022, Wiley. | ||
Building on these advances, Bi-based electrocatalysts have also shown promise. Bi cluster-anchored hollow carbon spheres (BiC/HCS) used as cathodes in a BPM configuration (acidic catholyte: 0.8 M KHCO3; alkaline anolyte: 6 M KOH + 0.2 M Zn(Ac)2) achieved 92% FE for HCOOH production, an OCV of 0.94 V, and a peak power density of 7.2 mW cm−2, with stable operation over 200 cycles.494 Complementarily, Liu et al.495 introduced a BiPdC alloy catalyst in a similar bipolar system (anolyte: 1 M KOH + 0.02 M Zn(Ac)2; catholyte: 0.1 M KHCO3 + 0.1 M HCOONa), which maintained 1.1 V over 45 h at 0.5 mA cm−2 and achieved 52.6% FE for HCOOH (Fig. 28c). In these systems, multi-ion transport is again pivotal: Zn(OH)42−, OH−, and H+ collectively support the redox interplay between Zn stripping/plating and CO2 conversion. The following reactions illustrate this synergy:
Anode:
| Zn–2e− ⇌ Zn2+ | (63) |
| Zn2+ + 4OH− ⇌ Zn(OH)42− | (64) |
| Zn(OH)42− ⇌ ZnO + 2OH− + H2O | (65) |
Cathode:
| CO2 + 2H+ + 2e− ⇌ HCOOH | (66) |
Overall:
| Zn + CO2 + 2H+ + 2OH− ⇌ HCOOH + ZnO + H2O | (67) |
The success of these architectures underscores the critical role of these multi-ion carriers (e.g., Zn2+, OH−, H+, and HCOO−) in synchronizing the CO2RR with Zn plating/stripping. By decoupling pH environments and tailoring ion flux through bipolar membranes, such systems mitigate crossover-induced neutralization while enhancing interfacial kinetics. Despite progress, challenges persist in achieving high-rate performance and controlling reactive intermediates (e.g., *COOH and *CO). Nevertheless, the integration of bifunctional catalysts, pH-decoupled electrolytes, and multi-ion carrier engineering positions Zn–CO2 batteries as transformative platforms for sustainable energy storage and carbon neutrality, bridging renewable energy conversion with CO2 valorization through precisely regulated ion dynamics.
5.3.2.3. Zn–N2 batteries. The electrochemical nitrogen reduction reaction (NRR) has emerged as a transformative alternative to the century-old Haber–Bosch process, offering a sustainable pathway for ammonia (NH3) synthesis under ambient conditions powered by renewable energy. Despite this promise, the practical deployment of electrochemical NRR remains impeded by intrinsically low NH3-yield and FE, primarily attributed to sluggish reaction kinetics, poor selectivity, and the competing HER. These limitations are being overcome through innovative Zn–N2 battery architectures that leverage sophisticated multi-ion carrier dynamics to simultaneously achieve energy storage and N2 fixation.496
A notable example is the pH-decoupled Zn–N2 battery developed by Hou et al.,496 which employs metallic NbS2 nanosheets as a cathode catalyst. This system utilizes a BPM to spatially segregate an alkaline Zn anolyte from an acidic N2-saturated catholyte, thereby leveraging differential ion flux to enhance performance. During operation, Zn at the anode undergoes oxidation, with the released Zn2+ contributing to charge transport. Simultaneously, at the cathode, N2 is reduced through an 8H+/6e− pathway to form NH4+, while OH− generated at the anode migrates across the BPM to maintain electroneutrality. The mechanism is proposed as follows:
Anode:
| Zn–2e− → Zn2+ | (68) |
Cathode:
| N2 + 8H+ + 6e− → 2NH4+ | (69) |
This orchestrated Zn2+/H+/OH−/NH4+ multi-ion carrier mechanism achieves a discharge power density of 0.31 mW cm−2 and an energy density of 714 mAh g−1 at 0.05 mA cm−2, sustaining stable operation over 10 h. The strategic decoupling of pH environments not only inhibits proton-hydroxide neutralization but also facilitates directional ion flow, improving both selectivity and efficiency of nitrogen fixation.
Building upon this framework, a Zn–N2 hybrid system incorporating Fe-based single-atom catalysts further demonstrates the utility of multi-ion carrier dynamics. In this architecture, a BPM separates an acidic 0.1 M HCl catholyte from an alkaline 4.0 M NaOH anolyte (Fig. 28d).489 During discharge, the NRR proceeds on the Fe single-atom active sites in the acid environment, while metallic Zn undergoes anodic oxidation in the basic phase; the reactions are shown as follows (Fig. 28e):
Anode:
| 3Zn + 6OH−–6e− → 3ZnO + 3H2O | (70) |
Cathode:
| N2 + 8H+ + 6e− → 2NH4+ | (71) |
Importantly, the BPM masterfully coordinates the transport of Na+, Cl−, H+, OH−, and NH4+ ions, maintaining crucial pH gradients while enabling efficient N2-to-NH3 conversion (31.9 μg h−1 mg−1, 11.8% FE at −0.4 V) and Zn stripping/plating. The system achieves a peak power density of 4.5 mW cm−2 and energy density of 815 Wh kg−1, significantly outperforming conventional single-electrolyte designs.
Further enhancements in catalytic performance were realized by tailoring the local electronic environment of the active site. Sulfur-coordinated Fe single-atom catalysts (FeSA-NSC-900) enabled improved NRR activity with a FE of 21.9% at a high current density of 10 mA cm−2 (Fig. 28f).497 This research underscores the critical importance of local electronic structure tuning in optimizing NRR kinetics within multi-ion carrier environments.
These advancements highlight how multi-ion carrier engineering, spanning Zn2+, H+, OH−, and NH4+, enables precise control over reaction microenvironments, mitigates parasitic HER, and stabilizes NRR intermediates. However, challenges persist in enhancing catalytic selectivity (e.g., suppressing competing HER), stabilizing reactive species (e.g., *N2Hx), and elucidating ion-coupled reaction mechanisms (Fig. 28g). While, the enlarged ATR-FTIR spectra clearly reveal that FeSA-NSC-900 effectively facilitates the kinetic transformation of *N2 → *NNH in the NRR (Fig. 28h).
Future efforts must integrate operando characterization and computational modeling to decode carrier-specific interactions at electrified interfaces, ultimately bridging the gap between lab-scale innovation and industrial deployment.
The breakthrough realization that multi-ion carrier engineering could circumvent these thermodynamic constraints has revolutionized MgAB development. Leung et al.450 introduced a dual-electrolyte architecture consisting of 3 M H2SO4 catholyte and 5 M NaNO3 anolyte, separated by a Nafion N211 membrane (Fig. 29a). This pH-decoupled design enables co-migration of Mg2+ and H+: Mg is oxidized at the anode, while the cathodic ORR proceeds via a 4e− H+-driven pathway. The corresponding reactions are shown as follows:
 | ||
| Fig. 29 (a) Schematic illustration of a dual-electrolyte MgAB employing an H2SO4 catholyte and NaNO3 anolyte. (b) Comparison of polarization curves between dual- and single-electrolyte MgABs. Reproduced with permission from ref. 450. Copyright 2021, Elsevier. (c) Working principle of an “all-in-one” MgAB integrating a unified Mg anode system. Reproduced with permission from ref. 501. Copyright 2025, Elsevier. (d) Proposed catalytic mechanism during the charging process using an I2-DMSO electrolyte at the air cathode. (e) Discharge–charge profiles of a non-aqueous Mg–O2 battery with iodine additives at 60 °C (black, pink, green, and red curves represent the first to fourth cycles; the blue curve represents performance without iodine). Reproduced with permission from ref. 469. Copyright 2013, Royal Society of Chemistry. (f) Schematic of the discharge mechanism of the ML11 anode in MgABs. Reproduced with permission from ref. 504. Copyright 2025, Royal Society of Chemistry. | ||
Anode:
| Mg → Mg2+ + 2e− | (72) |
Cathode:
| O2 + 4H+ + 4e− → 2H2O | (73) |
The acidic environment not only elevates the cathode potential, increasing the cell voltage (OCV = 2.23 V), but also suppresses OH−-induced Mg(OH)2 formation, leading to an energy density of 2043 mWh g−1 at 20 mA cm−2, and a discharge capacity of 1851 mAh g−1 with 84% coulombic efficiency at 50 mA cm−2 (Fig. 29b).
To simultaneously address cathodic sluggishness and anode passivation, Wang et al.501 engineered an “all-in-one” Mg anode system incorporating a Na+-selective ion-exchange membrane and a Mg–alginate/MgCl2/NaCl-based solid-state electrolyte (Fig. 29c). This platform supports Mg2+/Na+/Cl−/OH− multi-ion transport: Mg2+ migrates through the solid electrolyte, Na+ facilitates charge compensation at the cathode and effectively blocks Mg2+/OH− crossover, mitigating passivation, while Cl− disrupts interfacial oxide layers and reduces polarization resistance. The system achieves a remarkable 506 h cycle life at 1 mA cm−2 with 69.7% energy efficiency.
In non-aqueous systems, multi-ion carrier transport mechanisms can also be designed. Shiga et al.469,471 pioneered a catalytic cycling architecture employing I2-dimethyl sulfoxide (DMSO) and TEMPO-anion complexes at the air cathode (Fig. 29d). This system introduces Mg2+/I− dual-carrier dynamics, wherein discharge yields MgO via the ORR (Mg2+ + 2O2− + 2e− → MgO), while charging leverages oxidative decomposition of MgO by I2 (MgO + I2 → Mg2+ + 2I− + 1/2O2 + 2e−), regenerating MgI2 and reactivating the redox mediator. I− thus acts as a secondary ion carrier, synergizing with Mg2+ to facilitate reversible oxygen redox chemistry, reducing kinetic overpotentials and overcoming MgO's thermodynamic barrier. As a result, the discharge capacity of a non-aqueous Mg–O2 battery is significantly improved upon the introduction of iodine additives, compared to iodine-free systems (Fig. 29e). Additionally, the use of hydrophobic gas diffusion layers facilitates O2 transport and alleviates mass transfer limitations.502
Alloying strategies further enhance multi-ion synergies. Mg–xIn alloys (e.g., Mg–1In)503 leverage In3+/Mg2+ interactions to suppress self-corrosion (i0, HER ≈ 10−10 A cm−2) while stabilizing discharge pH, delivering 2100.2 mWh g−1 at 10 mA cm−2 and 780 h durability at 2.5 mA cm−2. Similarly, Mg–Li–Zn–Y alloys (e.g., ML11)504 exploit Li+-dominated reactions to form LiOH preferentially, reducing Mg corrosion and achieving 1727 mWh g−1 at 20 mA cm−2 through β-Li phase uniformity (Fig. 29f).
The most transformative applications of multi-ion carrier principles emerge in hybrid Mg–O2/CO2 systems. These architectures harness Mg2+/C2O42− dual-ion interactions to simultaneously achieve energy storage and carbon capture. The stepwise carboxylation pathway (O2 + 2e− → O22−, CO2 + O22− → CO42−, CO42− + CO2 → C2O42− + O2) demonstrates how carefully engineered ion cascades can convert thermodynamic challenges into opportunities, delivering 1441.38 Wh kg−1 (7.77-fold higher than single alkaline batteries) while producing recyclable MgC2O4.
Taken together, these developments underscore the transformative potential of multi-ion carrier engineering in MgABs. By integrating species such as Mg2+, H+, Na+, Cl−, OH−, and C2O42−, these strategies address long-standing barriers including anode passivation, poor reversibility, and low cathodic kinetics. Ion-specific control over transport, redox mediation, and interfacial environments across dual-electrolyte systems, solid-state conductors, and hybrid alloys allows MgABs to transcend the limitations of conventional single-ion systems. Looking forward, the exploration of hybrid electrolytes (e.g., Mg2+/TFSI−), dual-ion electrocatalysts, and dynamic interface tuning holds promise for further enhancing MgABs' robustness.
To address these limitations, Chao et al.507 introduced a reconfigured reaction pathway based on stannite ions (SnO22−) as the primary charge carriers (Fig. 30a). In this system, Sn undergoes oxidation at the alkaline anode to form SnO22−, while OH− ions generated at the cathode via the ORR sustain ionic conductivity and electrochemical continuity (Fig. 30b). The reduction energy for the SnO22− → Sn transformation is as low as 181 kJ mol−1, nearly half that required for SnO32− → Sn (342.96 kJ mol−1), indicating a more favorable reduction pathway (Fig. 30c). Substituting SnO32− with SnO22− not only lowers the activation energy significantly (by 171.48 kJ mol−1), but also optimizes the solvation structure (coordination number = 4.37), and enhanced diffusion kinetics (1.72 × 10−5 cm2 s−1), thus enabling efficient SnO22−/OH− multi-ion transport. The result is a highly efficient discharge–charge process with 99.5% Sn utilization, 420 Wh kg−1 energy density, and over 1900 h of dendrite-free cycling stability, underscoring the critical role of rational ion-pathway design in driving electrochemical performance.
 | ||
| Fig. 30 (a) Schematic illustration of Sn electrodeposition from a KOH electrolyte containing SnO22− species. (b) Schematic of the mechanism of a Snair battery utilizing SnO22− as the active species. (c) Calculated Gibbs free energy and potentials for the reduction of SnO32− and SnO22− to metallic Sn. Reproduced with permission from ref. 507. Copyright 2023, American Chemical Society. (d) Schematic illustration of the fabrication process of the Cu-PVDF@Sn electrode. (e) Polarization and power density curves of the Sn-air battery based on the Cu-PVDF@Sn electrode at various operating temperatures. Reproduced with permission from ref. 508. Copyright 2025, Elsevier. (f) Schematic of a SiAB employing an asymmetric alkaline anolyte (3 M KOH) and acidic catholyte (1 M H2SO4). (g) Polarization curves of SiABs operated in asymmetric (KOH-H2SO4) vs. symmetric (single KOH) aqueous electrolytes. Reproduced with permission from ref. 512. Copyright 2025, Wiley. | ||
To further enhance interfacial uniformity and long-term stability, Hu et al.508 developed a flexible, corrosion-resistant hierarchical anode composed of Cu-PVDF@Sn microchannels (Fig. 30d). This architecture effectively homogenizes the SnO22−/Sn redox interface, achieving 92.61% coulombic efficiency over 1200 cycles, 800 h operation at 5 mA cm−2, and boosting energy density to 474 Wh kg−1. Complementary to this, the integration of polymer-based gel electrolytes has enabled flexible Sn–air batteries to operate across a broad temperature window (−15 to 60 °C), achieving an energy density of 504 Wh kg−1 and a peak power density 80 mW cm−2 at 5 mA cm−2, thus meeting the practical demands of flexible electronics (Fig. 30e).
Notably, the fusion of photocatalysis with Sn–air chemistry has introduced an additional dimension to ion-electron synergy. For example, the incorporation of Fe2O3@TiO2/Ti heterojunctions as bifunctional photoelectrodes facilitates solar-assisted charging, with light-induced electron–hole separation significantly reducing the overpotential to just 40 mV while simultaneously accelerating ORR/OER kinetics.509 This solar-coupled strategy expands the functional landscape of Sn–air batteries beyond traditional electrochemical boundaries, highlighting a compelling energy-chemical hybridization approach.
All these advances, ranging from reengineering charge carriers (SnO22−vs. SnO32−), optimizing interface stability via microstructured anodes and gel electrolytes, to integrating photocatalytic components, demonstrate how multi-ion carrier orchestration lies at the core of Sn–air battery innovation. By coordinating SnO22−, OH−, and photogenerated species within controlled electrochemical microenvironments, these systems achieve enhanced energy density, cycling durability, and environmental adaptability.
Subsequent designs employed alkaline aqueous systems in which Si anode oxidation and cathodic ORRs in alkaline media are as follows:511
Anode:
| Si + 4OH− ⇄ Si(OH)4 + 4e− | (74) |
Cathode:
| O2 + 2H2O + 4e− ⇄ 4OH− | (75) |
Overall:
| Si + O2 + 2H2O ⇄ Si(OH)4 | (76) |
However, the exclusive use of OH− as the sole charge carrier introduced critical drawbacks, most notably, the formation of passivating SiO2(OH)22− layers via side reactions (Si + 2OH− + 2H2O → SiO2(OH)22− + 2H2), which severely impede charge transport and reduce system efficiency. The inability to decouple oxidation and reduction environments hindered the long-term stability and performance scalability of conventional alkaline SiABs.
A significant breakthrough was realized with the development of an asymmetric quasi-solid-state SiAB architecture, which introduced a BPM to separate an alkaline anolyte (3 M KOH) from an acidic catholyte (1 M H2SO4), thereby enabling H+/OH− dual-ion carrier dynamics (Fig. 30f).512 In this design, Si oxidation at the anode is mediated by OH−, while H+ at the cathode facilitates the ORR, resulting in a spatially resolved ion flux that effectively suppresses SiO2 passivation and promotes higher voltage operation. The reaction mechanism can be described as:
Anode:
| Si + 12(HF)2F− → SiF4 + 8(HF)3F−+ 4e− | (77) |
Cathode:
| O2 + 4H+ + 4e− → 2H2O | (78) |
This asymmetric configuration achieves an OCV of 1.87 V, substantially exceeding that of single-alkaline systems (1.48 V), and delivers a power density of 621 μW cm−2, nearly 2-fold higher than the reported for conventional alkaline SiABs (205 μW cm−2) (Fig. 30g). Importantly, this H+/OH− co-regulation not only enhances interfacial kinetics but also ensures environmentally friendly operation by producing Si(OH)4 as a benign byproduct. The system maintains stable operation for 188 h with a practical energy density of 77.3 Wh kg−1, outperforming Pt/C-based benchmarks and confirming its suitability for long-term, low-toxicity applications in portable energy systems.
SiABs have undergone the development process from OH−-dominated alkaline systems to H+/OH− co-regulated asymmetric architectures, by tailoring ionic environments through bipolar membranes and quasi-solid-state electrolytes, parasitic reactions such as Si corrosion and H2 evolution are minimized, while ORR/OER kinetics are simultaneously enhanced. Future innovations in hybrid electrolytes (e.g., gel polymers for H+/OH− stabilization) and dual-ion catalysts promise to further bridge the gap between theoretical promise and real-world applications, solidifying SiABs as cornerstone technologies in sustainable energy storage.
 | ||
| Fig. 31 Schematic representations of reaction pathways and ion transport mechanisms in (a) single-electrolyte and (b) dual-electrolyte Al–air batteries, and (c) hybrid acid–alkali Al–air battery. Reproduced with permission from ref. 518. Copyright 2024, Wiley. (d) Polarization and power density curves comparing single- and dual-electrolyte Al–air cells at 1.5 mL min−1 flow rate. (e) Corresponding electrode-specific polarization profiles. Reproduced with permission from ref. 515. Copyright 2021, Elsevier. (f) Schematic of Fe–air battery employing a Li-SSE. Reproduced with permission from ref. 527. Copyright 2017, American Chemical Society. | ||
Anode:
| Al + 4OH− ⇄ Al(OH)4− + 3e− | (79) |
Cathode:
| O2 + 2H2O + 4e− ⇄ 4OH− | (80) |
Overall:
| 3O2 + 4Al ⇄ 2Al2O3 | (81) |
Despite the high ionic conductivity (>1 S cm−1) and rapid kinetics of aqueous systems, the electrochemical window is fundamentally constrained by parasitic hydrogen evolution (Al + OH− + 3H2O → Al(OH)4− + 3/2H2) and anode passivation via Al2O3 film formation, significantly undermining both energy efficiency and coulombic reversibility.514 In recent years, researchers have conducted in-depth studies on the development of cathode catalysts to enhance the performance of Al–O2 batteries (Table S6).
To circumvent these limitations, dual-electrolyte strategies have been employed to spatially decouple the anodic and cathodic microenvironments, enabling tailored ion flux and pH-gradient-driven voltage amplification. For instance, microfluidic fuel cells (MFCs) that partition alkaline anolytes (1 M KOH) and acidic catholytes (1 M H2SO4) via porous carbon membranes introduce a complex but beneficial interplay of H+/OH−/Al(OH)4− ions transport (Fig. 31b). This design delivers a peak power density of 690 mW cm−2 by optimizing pH gradient (ΔpH ≈ 13) and ionic coordination, representing a 142% improvement over the single-electrolyte counterpart.515 Polarization curves further reveal that O2 reduction is more favorable in acidic media (Fig. 31d and e).
Building upon this, our group516 designed a hybrid acid–alkali cell with a BPM separating 4 M NaOH and 2 M H2SO4 electrolytes, where Al undergoes oxidative dissolution (Al + 4OH− → Al(OH)4− + 3e−) at the anode while H+ at the cathode enable H2 evolution (2H+ + 2e− → 2H2), yielding a 99% FE at 300 mA cm−2 and producing 448.9 L m−2 h−1 of H2. Further enhancement was achieved through the integration of Pt@CoN4-G bifunctional catalysts that facilitate the ORR/OER by supporting Na+/SO42−/OH−/H+/Al(OH)4− co-migration, delivering 222 mW cm−2 power output at 1.95 V.517
Most notably, our group's518 latest architecture leveraged ENE (ΔpH ≈ 14) to couple anodic Al(OH)4− formation (Al + 4OH− → Al(OH)4− + 3e−) and cathodic acidic ORR (O2 + 4H+ + 4e− → 2H2O) with CEM, achieving an unprecedented OCV of 2.72 V and a peak power density of 827 mW cm−2, further boosted to 1029 mW cm−2 under pure O2 conditions, demonstrating how multi-ion orchestration (Al(OH)4−/Na+/OH−/H+) amplifies voltage through pH-driven potential superposition (Fig. 31c).
Beyond aqueous systems, ionic liquid-based Al–air batteries have introduced new paradigms for corrosion inhibition and reversibility. Electrolytes such as EMIMCl/AlCl3 support reversible Al plating/stripping via complex equilibria (Al + 7AlCl4− → 4Al2Cl7− + 3e−), while also preventing the HER.519,520 Fluorinated ionic liquids like [EMIM(HF)2.3F] generate Al–O–F protective films that suppress passivation and stabilize interfacial redox reactions.521,522 Solid-state innovations have further expanded the design space, such as agarose-based electrolytes,523 further highlighting the advantages of controlling OH− and Al(OH)4− mobility at solid–liquid boundaries, achieving energy densities of 2148.5 mAh g−1 and 2766.9 Wh kg−1, respectively, while enhancing mechanical integrity and safety.
Collectively, these innovations consolidate the foundational importance of multi-ion carrier engineering in Al–air battery development. From manipulating pH-dependent ion gradients in dual-electrolyte systems to optimizing Al3+ coordination chemistry in ionic liquids and solid gels, the fine-tuning of carrier species such as Al(OH)4−, OH−, H+, Na+, SO42−, and AlCl4− is vital for simultaneously mitigating corrosion, enhancing charge transport, and maximizing electrochemical reversibility.
The field experienced a transformative breakthrough with the introduction of multi-ion carrier engineering. Deyab and Mohsen's526 innovative use of 1-ethyl-3-methylimidazolium lactate (EML) ionic liquid as an electrolyte additive established a paradigm-shifting dual-ion carrier mechanism (C3H5O3−/EML+). The lactate anions (C3H5O3−) coordinate with Fe2+ to form soluble Fe(C3H5O3)2 complexes (solubility ∼0.1 M), effectively preventing oxide precipitation and enabling reversible iron dissolution/deposition. Simultaneously, EML cations adsorb onto electrode surfaces, providing 97% HER suppression through electrostatic shielding. This synergistic ion interaction enhances discharge capacity to 312 mAh g−1 and extends cycle life to 1000 cycles with 94% capacity retention, exemplifying how tailored ion interactions resolve interfacial instability.
Expanding on this multi-ion paradigm, Yu and Manthiram527 designed a hybrid Fe–air battery employing solid-state alkali-ion conducting membranes to spatially separate the alkaline anolyte (LiOH/NaOH) from the acidic catholyte (H3PO4 or LiH2PO4/NaH2PO4) (Fig. 31f). This architecture enables synergistic OH−/Li+/Na+/H+ transport, in which Fe undergoes oxidation in the alkaline chamber (Fe + 2OH− → Fe(OH)2 + 2e−), while H+ drives the ORR in the acidic cathode (O2 + 4H+ + 4e− → 2H2O). The membrane-regulated migration of Li+/Na+ ions preserves electrochemical neutrality and stabilizes the pH gradient (ΔpH ≈ 7), thereby minimizing the HER and preventing Fe3+ passivation. This pH-decoupled strategy yields a high theoretical cell voltage of 2.11 V, with 92% coulombic efficiency and zero capacity fade across 50 cycles at ±1.0 mA cm−2, illustrating the efficacy of ion-selective transport in bridging kinetic and thermodynamic disparities between redox processes.
Briefly, Fe–air systems by orchestrating ion-specific interactions, whether through lactate coordination chemistry, electrostatic modulation by ionic liquids, or pH-gradient-driven cation shuttling, harness the nuanced roles of C3H5O3−, EML+, OH−, Li+, Na+, and H+ in optimizing charge transport and interfacial stability. By engineering multi-ion carrier networks, future MAB systems could transcend current energy density ceilings while achieving unprecedented operational robustness under realistic environmental conditions.
5.4. Metal–H2O2 fuel cells
The development of metal–H2O2 fuel cell systems traces its origins to pioneering work in the 1960s by the U.S. NRL and other institutions, where early prototypes utilizing Al, Mg, and Zn anodes paired with H2O2 oxidants were explored as high-energy power sources (Table S7).528 While these systems were initially classified as “metal-oxidant” fuel cells due to their consumable anodes and redox-based cathodes, contemporary understanding recognizes them as sophisticated multi-ion carrier platforms where the interplay of metal cations (Zn2+, Mg2+, Al3+), peroxide derivatives (HO2−, O2), and pH-dependent H+/OH− transport governs their electrochemical performance (Fig. 32a). | ||
| Fig. 32 Schematic diagram of multi-ion carrier in (a) metal–H2O2 fuel cell architectures employing acidic, alkaline, and neutral electrolytes; (b) metal–H2O systems including Zn–H2O, Mg–H2O, and Al–H2O batteries. | ||
The unique advantage of metal–H2O2 systems lies in their cathode chemistry, where concentrated H2O2 decomposes into reactive oxygen species (OH−, H2O, or O2) through three primary pathways: spontaneous decomposition, alkaline electrochemical reduction, or acidic reduction-process that enable exceptional power densities unattainable in conventional MABs configurations. Advanced dual-electrolyte architectures have been developed for Al and Mg systems to mitigate anode corrosion, physically separating the alkaline anolyte (where metal oxidation occurs) from catholyte compartments containing pH-optimized H2O2 reduction environments. In contrast, Zn–H2O2 systems leverage established ORR/OER bifunctional catalysts from ZAB technology, demonstrating the versatility of multi-ion carrier design principles across different metal platforms.
H2O2 decomposition:
| H2O2 → H2O + ½O2 | (82) |
In alkaline medium:
| H2O2 + 2e− → 2OH− | (83) |
In acidic media:
| H2O2 + 2H+ + 2e− → 2H2O | (84) |
Beyond their electrochemical novelty, metal–H2O2 fuel cells offer a compelling combination of attributes, substantially higher theoretical energy densities than LIBs, material abundance, environmental compatibility, and system flexibility, positioning them as strong candidates for next-generation energy solutions. These advantages are further amplified when viewed through the lens of multi-ion carrier dynamics, which play a central role in optimizing charge transport, regulating interfacial reactions, and stabilizing redox kinetics across varied electrolytic environments. This section provides a comprehensive overview of the energy storage mechanisms and recent progress in Al-, Mg-, and Zn-based H2O2 fuel cell systems, with particular emphasis on how the intelligent coordination of multiple ionic species enables the advancement of these promising technologies.
The growing interest in Al–H2O2 fuel cells stems from the synergistic integration of Al's high electrochemical reactivity and H2O2's strong oxidizing ability. H2O2 not only serves as a dense oxygen carrier but also enables flexible cathodic reaction pathways. Optimization of electrocatalysts, electrolyte formulations, and anode architecture has progressively enhanced the power density, energy efficiency, and cycling durability of Al–H2O2 systems. Mechanistically, the cell relies on the redox coupling between aluminum oxidation and H2O2 reduction, where OH− and AlO2− serve as the principal charge carriers, which is as follows:
Anode:
| Al + 4OH− → AlO2− + 2H2O + 3e− | (85) |
Cathode in alkaline:
| H2O2 + 2e− → 2OH− | (86) |
Cathode in acidic media:
| H2O2 + 2H+ + 2e− → 2H2O | (87) |
However, in alkaline Al–H2O2 systems, Al anodes suffer from parasitic side reactions and significant overpotentials that limit the achievable energy output. To address these challenges, researchers have developed strategies including cathode catalyst enhancement,529 anode surface modification,530 electrolyte composition tuning,531 and overpotential reduction. Anode engineering suppresses unwanted corrosion, optimized electrolytes mitigate side reactions, and advanced cathode materials substantially improve catalytic performance, all contributing to higher system efficiency.
Recently, dual-electrolyte designs strategically segregate concentrated alkaline anolyte (3–4 M NaOH/KOH) from acid/neutral catholyte using ion-selective membranes (e.g., Nafion), thereby mitigating AlO2−/H+ crossover while optimizing the respective electrode microenvironments.530,531 This compartmentalization not only suppresses parasitic corrosion but also enhances the mobility of critical charge carriers, addressing the historical limitation of energy output falling significantly below theoretical predictions due to overpotential effects and side reactions.
Subsequently, the breakthroughs in cathode engineering have propelled the system's performance to new heights through nanoarchitectured catalysts that redefine multi-ion interfacial dynamics. Chang et al.532 demonstrated the efficacy of silver nanowire-decorated carbon cloth (Ag NW//CC) cathodes in microfluidic cells, achieving 43 W m−2 power density at 0.1 M H2O2 through optimized OH−/AlO2− transport, while subsequent work on 3D Ag nanowire aerogels532 delivered a staggering 271 W m−2 (28.1 mW cm−2), a 8-fold enhancement over conventional silver rod cathodes, by maximizing active site accessibility and reactant flux. Parallel advancements by Li et al.533 with Pd- and CoOx-loaded graphene oxide/nickel foam (rGO/NF) cathodes (267–240 mW cm−2) and Sun et al.534 with hierarchical Pd/SnO2/NF structures (168 mW cm−2) further underscore how tailored catalyst supports can amplify the synergy between electron transfer and ion migration. These systems universally exploit the dual roles of OH− (charge carrier and reaction participant) and AlO2− (anodic product and ionic conductor), with Na+ acting as a supplementary charge-balancing species in alkaline environments.
Beyond energy storage, the multi-ion functionality of Al–H2O2 cells has been leveraged for simultaneous power generation and water purification. Cervantes et al.535 constructed a galvanic cell using pure Al anodes and graphite cathodes immersed in an electrolyte containing KCl and kaolin-simulated suspended solids, with H2O2 added as the cathodic oxidant. During operation, the anodic oxidation of Al generated Al3+ and Al(OH)3/AlO2− species, which acted as coagulants to remove 96.6% of water turbidity, while cathodic H2O2 reduction (to HO2− and OH−) sustained the alkaline environment:
Anodic:
| 2Al + 6H2O → 2Al3+ + 6OH− + 6H+ + 6e− | (88) |
| 2Al3+ + 6OH− → 2Al(OH)3 | (89) |
| 2Al3+ + 8OH− → 2AlO2− + 4H2O | (90) |
Cathodic:
| 6H+ + 6e− → 3H2 | (91) |
| H2O2 + OH− → HO2− + H2O | (92) |
| 3HO2− + 3H2O + 6e− → 9OH− | (93) |
Overall:
| 2Al + 6H2O → 2Al(OH)3 + 3H2 | (94) |
| 2Al + 3HO2− → 2AlO2− + OH− + H2O | (95) |
This integrated system demonstrated an average cell voltage of 0.613 V and a current output of 157 μA, while reducing turbidity by 96.6% without any external energy input. The electrochemical network established here is sustained not only by e− and OH− transport but also by the dynamic migration and conversion of Al3+, AlO2−, and H+, forming a complex multi-ion pathway. This configuration exemplifies how a multi-ion conduction mechanism enables the dual functions of energy delivery and environmental remediation within a single device platform.
In summary, Al–H2O2 fuel cells present a compelling case for future energy technologies, especially in scenarios demanding lightweight, high-energy-density solutions such as underwater propulsion or portable power systems. Their architecture and function embody the core principles of multi-ion carrier devices, where ionic diversity is not a by-product but a designed advantage, enabling synchronized e−-ion processes, enhancing electrochemical kinetics, and broadening system functionalities. As such, Al–H2O2 fuel cells offer a vivid illustration of how multi-ion frameworks can bridge energy conversion with value-added environmental applications, further solidifying their relevance in power supply technologies.
Anode:
| Mg → Mg2+ + 2e− | (96) |
Cathode:
| H2O2 + 2H+ + 2e− → 2H2O | (97) |
Overall:
| Mg + H2O2 + 2H+ → Mg2+ + 2H2O | (98) |
Despite these advantages, Mg–H2O2 fuel cells face challenges at high current densities due to cathodic polarization and mass transport limitations, particularly in conventional planar cathodes where concentration gradients and reactant depletion zones accelerate voltage decay. A transformative breakthrough came with the development of three-dimensional cathodes, beginning with Patrissi et al.'s537 carbon microfibrous arrays (CMA) fabricated via directed electrospinning. This architecture utilizing Mg foils as the anode and a Nafion 115 membrane for ion separation, the catholyte comprised 0.06 M H2O2, 0.2 M H2SO4, and 40 g L−1 NaCl in a flow-type, when loaded with Pd:Ir catalysts, reduced concentration polarization by enhancing turbulent mixing and thinning diffusion layers, achieving an 84 mW cm−2 peak power density (58% higher than planar cathodes) while maintaining 88% H2O2 utilization. The CMA's success underscored how 3D designs could optimize the interplay between electron transfer (via Pd:Ir sites), proton migration (through Nafion), and Mg2+/H+ transport in the anolyte.
Further innovations addressed material stability in acidic environments. Sun et al.538 implemented a dual-chamber Mg–H2O2 fuel cell integrating corrosion-resistant material design with optimized architecture. This system used a titanium foam-supported palladium nanoparticle cathode (Pd/Ti), effectively replacing corrosion-prone Ni/C substrates, which withstood 50 h operation at 1.5–2.0 V in 1.0 M H2SO4/0.5 M H2O2, delivering 168 mW cm−2 power density. This work revealed the delicate balance between H+ concentration (>1.0 M for kinetics) and H2O2 levels (>0.5 M to suppress parasitic decomposition), highlighting how multi-ion competition (H+/Mg2+/Na+/Cl−) governs efficiency. The latest advancement by Sun et al.539 features a cathode, termed Pd/rGO/CP, in which Pd nanoparticles dispersed on crumpled graphene sheets synergize with carbon paper, achieving a power density of 215 mW cm−2 at 60 °C. The rGO's 3D structure not only enhanced H+/e− coupling but also mitigated intermediate adsorption, enabling stable 50 h discharge with ultralow Pd loading (0.025 mg cm−2), underscoring its high energy efficiency and potential for extended underwater missions.
These advancements underscore the pivotal role of multi-ion interactions in shaping the electrochemical behavior of Mg–H2O2 fuel cells. Beyond electron flow and H+ conduction through the PEM, these systems harness the coordinated migration and transformation of Mg2+, Na+, and Cl− ions, forming a complex ionic network that fundamentally dictates device performance. Such multi-ion synergy enhances energy efficiency, operational stability, and application versatility, particularly in decentralized and marine energy scenarios. Through the integration of rational material selection, structural optimization, and electrolyte engineering, Mg–H2O2 cells have emerged as promising candidates for robust, high-power-density, and corrosion-resistant energy systems under harsh environmental conditions. Future research should aim to scale these principles toward industrial deployment while further elucidating the kinetic interplay among charge carriers under extreme operating conditions.
Anode:
| Zn + 4OH− → Zn(OH)42− + 2e− | (99) |
Cathode:
| 2H2O2 → 2H2O + O2 | (100) |
| ½O2 + H2O + 2e− → 2OH− | (101) |
Overall:
| Zn + H2O2 + 2OH− → Zn(OH)42− | (102) |
Further advancements in cathode engineering have dramatically enhanced power density and stability. Liu's team541 designed a bifunctional catalyst, ZnCo–NC-DMEA-900, achieving 442 mW cm−2 at 150 mA cm−2 and 966 Wh kg−1 energy density in 6 M KOH/0.2 M Zn(OAc)2 electrolyte. The catalyst's hierarchical porosity optimized the interplay between OH− transport and O2 diffusion at the TPB, while Zn2+/K+ migration ensured charge balance. This work was extended by a biomass-derived ZIF-67/nori-800 catalyst,542 which reached 476 mW cm−2 and 964 Wh kg−1 by mitigating H2O2-induced degradation through a novel decomposition chamber design. Moreover, it featured an impressive OCV of 1.44 V and a discharge voltage of 1.29 V at 20 mA cm−2. These systems underscore how tailored ion-transport pathways, governed by OH−, Zn(OH)42−, and K+, can overcome the <100 mW cm−2 ceiling of conventional ZABs.543
The pinnacle of this evolution is the edge-enriched catalyst (ZnCo–NC-S-900),544 which delivered 510 mW cm−2 and 953 Wh kg−1 by synchronizing ORR/OER kinetics with Zn(OH)42− ↔ ZnO precipitation dynamics. Here, the H2O2-derived O2 undergoes reduction at the cathode (generating OH−), while Zn2+/OH− coordination at the anode forms soluble Zn(OH)42−via the reactions:
Anode:
| Zn + 4OH− → Zn(OH)42− + 2e− | (103) |
| Zn(OH)42− → ZnO + H2O + 2OH− | (104) |
Cathode:
| 2H2O2 → O2 + 2H2O | (105) |
| O2 + 4e− + 2H2O → 4OH− | (106) |
Overall:
| Zn + H2O2 → ZnO + H2O | (107) |
Pushing the paradigm further, Nagaiah et al.545 recently proposed an innovative “two birds-one stone”. In this strategy, the cathode performs a 2e− ORR to generate H2O2 during discharge in an acidic environment, serving as both an energy carrier and a chemical product. The system employed MnWO4 as a bifunctional electrocatalyst, offering both high selectivity for H2O2 production and excellent OER activity, while mitigating overpotentials and enhancing reversibility. During discharge, O2 is reduced to H2O2 at the acidic cathode while Zn is oxidized at the alkaline anode. Upon charging, H2O undergoes the OER at the cathode, and Zn2+ is reduced and redeposited at the anode, enabling a closed-loop oxygen cycle, described by the reactions:
Anode:
| Zn ⇄ Zn2+ + 2e− | (108) |
| Zn2+ + 4OH− ⇄ Zn(OH)42− | (109) |
Cathode:
Discharge process:
| O2 + 2H+ + 2e− ⇄ H2O2 | (110) |
Charge process:
| 2H2O ⇄ O2 + 4H+ + 4e− | (111) |
In addition to electrocatalytic advancements, the integration of photoelectrocatalytic materials into Zn–H2O2 battery systems has opened new dimensions in the dual conversion of solar energy and chemical fuels. Zhang et al.'s pioneering work546 demonstrated this paradigm by developing a bias-free photocathode system (PDS-Fe2O3@Ov-NiFe2O4-HR-Ti), where oxygen vacancies and heterojunction coupling synergistically enhanced e− transfer kinetics while stabilizing Fe3+/Fe2+ and Ni3+/Ni2+ redox cycles for H2O2 generation. During discharge, e− released from the Zn anode reduce cathodically generated H2O2 through ORR pathways, aided by the redox shuttles, while Zn2+ and OH− concurrently form intermediate species such as Zn(OH)42−. The system achieved a high OCV of 1.3 V and a peak power density of 8.5 mW cm−2. In neutral PBS electrolyte, the overall cell reaction proceeds with the generation of value-added byproducts like NaZnPO4, as represented by:
Photocathode reaction:
| O2 + 2H+ + 2e− → H2O2 | (112) |
Anode reaction:
| Zn + Na+ + PO43− → NaZnPO4 + 2e− | (113) |
Overall reaction:
| Zn + Na+ + PO43− + O2 + 2H+ → NaZnPO4 + H2O2 | (114) |
Simultaneously, the development of functional molecular catalysts, particularly metalloporphyrins, metallophthalocyanines, and their analogues, has shown great promise in modulating ORR pathways under both dark and illuminated conditions.405 These MOFs can selectively catalyze the 2e− ORR to generate H2O2 or the 4e− pathway to produce H2O, depending on the central metal ion and ligand field environment. When integrated into photoelectrochemical systems, their unique electronic structures facilitate efficient e−–H+ transfer during the ORR while resisting degradation in acidic media, a critical advantage for integrating photogenerated H2O2 directly into fuel cells. When deployed in Zn–H2O2 fuel cells, these molecular catalysts enhance both power density (via improved ORR kinetics) and cycling stability (through mitigated radical-induced decomposition), with their π-conjugated systems serving as electron highways that synergize with OH−/Zn(OH)42− transport networks.
In conclusion, metal–H2O2 fuel cells have evolved into multifunctional energy systems, with performance fundamentally driven by the coordinated transport of multiple ions such as Zn2+, Mg2+, AlO2−, HO2−, OH−, and H+. By engineering catalysts, electrodes, and electrolytes to regulate multi-ion fluxes and redox selectivity, these systems have evolved from simple chemical cells to multifunctional devices capable of performing in O2-free environments and producing valuable chemical coproducts. Future efforts should focus on scaling up ion-selective interfaces and developing modular platforms where electrochemical, photochemical, and catalytic domains synergistically operate within a multi-ion framework to further push the boundaries of energy storage technologies.
5.5. Metal–H2O fuel cells
Unlike traditional MABs, metal–H2O fuel cells generate energy through the oxidation of metal anodes and the catalytic reduction of H2O, typically via the HER, eliminating the need for external O2 or air cathodes.445 This intrinsic design simplification, coupled with the use of abundant and low-cost metal anodes and environmentally benign reaction products, renders metal–H2O systems highly suitable for specialized scenarios such as underwater operations, remote-area power supplies, and portable or unmanned devices. By bypassing the reliance on atmospheric O2 and reducing system complexity, these cells offer high energy density, excellent environmental adaptability, and enhanced practicality for decentralized applications.Recent advancements in metal–H2O fuel cells, including Zn–H2O, Mg–H2O, and Al–H2O systems, have demonstrated notable progress (Table S8). Among these, Zn–H2O fuel cells have achieved remarkable performance improvements through optimized catalyst design and structural engineering, enabling high energy and power outputs without requiring O2 feed.451 This makes them especially promising for submerged and O2-limited environments. However, broader commercialization remains hindered by the high cost and scarcity of efficient cathode catalysts, particularly noble metals like Pt/C. Thus, developing low-cost, high-performance alternatives is a critical research focus.
Multi-ion carrier engineering further enhances the performance of metal–H2O fuel cells by regulating ion transport dynamics (Fig. 32b). During operation, ion carriers facilitate not only e− and ion transfer but also optimize electrolyte ion flux, significantly improving energy conversion efficiency. For example, in Zn–H2O systems, strategically managing the migration of OH−, H+, and Zn(OH)42− ions can stabilize electrode reactions across varying pH gradients, leading to higher power density and extended cycle life.450 This multi-ion synergistic transport mechanism mitigates energy losses common in conventional battery systems, offering a promising pathway for commercializing metal–H2O fuel cells.
Anode:
| Zn + 2OH−–2e− → ZnO + H2O | (115) |
Cathode:
| 2H2O + 2e− → 2OH− + H2 | (116) |
During charging, ZnO undergoes reduction at the cathode while oxygen evolves at the anode, as represented by:
Anode:
| ZnO + H2O + 2e− → Zn + 2OH− | (117) |
Cathode:
| 2OH−–2e− → 1/2O2 + H2O | (118) |
Notably, in such alkaline systems, OH− serves as the sole ionic carriers, highlighting a limitation in ion diversity compared to other multi-ion configurations. Instead, it has driven focused material and interface engineering efforts to optimize ionic conductivity, reaction kinetics, and system reversibility.547–549
The true potential of multi-ion carrier engineering became apparent in hybrid electrolyte systems, where our group550 demonstrated that spatial segregation of NaOH anolyte and H2SO4 catholyte could harness the synergistic OH−/H+ coupling, achieving an OCV of 1.25 V and a power density of 80 mW cm−2 alongside continuous H2 production (0.166 mL s−1). The specific energy storage mechanism of this system is as follows:
Zn oxidation in alkaline anolyte:
| Zn + 2OH− → ZnO + H2O + 2e− | (119) |
H2 evolution in acidic catholyte:
| 2H+ + 2e− → H2 | (120) |
Collectively, the multi-ion carrier engineering, spanning OH−, H+, Na+, SO42−, and Zn(OH)42−, resolves the trade-offs between energy density, efficiency, and cost in Zn–H2O systems. By tailoring ion flux across pH gradients and optimizing catalyst–electrolyte interfaces, such designs transcend traditional electrolysis limitations, positioning Zn–H2O fuel cells as versatile platforms for renewable energy storage and green hydrogen economies.
Anode:
| Mg → Mg2+ + 2e− | (121) |
Cathode:
| 2H2O + 2e− → 2OH− + H2 | (122) |
Overall:
| Mg + 2H2O → Mg(OH)2 + H2 | (123) |
The strategic utilization of seawater as a natural electrolyte underscores the sustainability and cost-efficiency of Mg–seawater batteries, offering a compelling alternative to traditional LIBs in marine environments. The continuous optimization of anode alloys, catalytic cathodes, and electrolyte formulations has significantly enhanced power density and cycling stability, expanding their applications from deep-sea exploration to portable power and emergency energy systems (Table S8).
Modern advancements have focused on overcoming three critical challenges: (1) anode passivation, (2) sluggish hydrogen evolution kinetics, and (3) electrolyte optimization. Hu et al.559 developed MOF-derived gradient carbon–cobalt monolith cathodes achieving 45 mW cm−2 power density with 168 h stability in natural seawater, while Fasmin's558 2D modeling provided critical insights into ion transport mechanisms.
The most transformative development came through photoelectrochemical integration. Behm et al.560 introduced a photo-assisted Mg–H2O battery (PAMHB) with a CuSCN/Cu2O heterojunction photocathode. This system harnessed photoelectrochemical synergy, where Mg anodic oxidation and corrosion released e−, while the heterojunction's type-II band alignment facilitated charge separation, photogenerated electrons drove the HER, and holes were efficiently extracted via CuSCN. The built-in electric field (0.35 eV) at the heterointerface reduced the HER energy barrier, achieving a photocurrent density of 4.32 mA cm−2, an OCV of 1.48 V, and a peak power density of 1.18 mW cm−2 with an H2 production rate of 0.56 mL cm−2 min−1. This innovative integration of photoelectrochemistry and multi-ion carrier dynamics (Mg2+/Na+/OH−) exemplifies the potential of hybrid energy conversion systems for sustainable power generation. All in all, these advancements highlight how Mg–H2O fuel cells, through sophisticated multi-ion carrier engineering, are transitioning from conceptual prototypes to practical energy solutions for marine and portable applications.
Anode:
| Al + 3OH− → Al(OH)3 + 3e− | (124) |
Cathode:
| 2H2O + 2e− → H2 + 2OH− | (125) |
Overall:
| 2Al + 6H2O → 3H2 + 2Al(OH)3 | (126) |
Despite its thermodynamic promise, practical implementation also faces passivating oxide/hydroxide layers on metal Al anodes, parasitic hydrogen evolution reducing coulombic efficiency, and kinetically sluggish cathodic HER.561 Recent advances in electrolyte engineering, such as polyacrylic acid (PAA)-modified alkaline seawater (pH = 12), demonstrate how macromolecular additives can suppress anode corrosion while maintaining ionic conductivity.562
The carrier dynamics become more complex in acidic systems, where Al3+, H+, and OH− collectively participate in charge transport. Fontaine et al.557 revealed that an Al–H2SO4 (0.5 M) cell with Pt/C cathode achieves an OCV of 1.45 V and a peak power of 75 mW cm−2 at 365 mA cm−2 through the coupled reactions:
Anode:
| 2Al + 6H2O ↔ 2Al3+ + 6OH− + 6H+ + 6e− | (127) |
Cathode:
| 6H+ + 6e− ↔ 3H2 | (128) |
Overall:
| 2Al + 6H2O ↔ 2Al(OH)3 + 3H2 | (129) |
Substituting Pt/C with MoS2/RGO heterostructures maintained 25 mW cm−2 power output at 115 mA cm−2 with 57 mL h−1 H2 evolution, albeit with reduced HER turnover frequency.557 This trade-off between cost and performance highlights the need for advanced catalysts that balance economic viability with multi-ion interfacial kinetics. The system's ability to produce fuel-cell-grade H2 without purification underscores its potential as a decentralized energy-hydrogen cogeneration platform. Future breakthroughs will require holistic optimization of carrier transport across the solid–liquid interface while mitigating anode passivation and cathode overpotentials.
The evolution of metal–H2O fuel cells represents a transformative step in aqueous energy conversion, redefining traditional corrosion-prone chemistries into tunable, high-performance systems. Central to this progress is multi-ion carrier engineering, where coordinated transport of OH−, H+, Na+, Cl−, Zn(OH)42−, Mg2+, and Al3+ ensures charge balance, pH regulation, and redox stability. These ions are not passive participants but active enablers of synchronized hydrogen production and electricity generation. Innovations in hybrid electrolytes, bifunctional catalysts, and photo-assisted designs have further amplified system efficiency and environmental adaptability, especially in O2-free or marine settings. By integrating energy storage with clean H2 output, metal–H2O fuel cells showcase the strategic value of multi-ion pathways in developing decentralized and sustainable power solutions. Future research should prioritize the design of ion-selective membranes, pH-coupled catalytic interfaces, and interfacial transport control to unlock the full potential of these energy platforms.
6. Redox flow batteries
RFBs exemplify multi-ion carrier EPSs, utilizing reversible redox reactions of active ionic species dissolved in liquid electrolytes at both electrodes. A defining characteristic of these systems is the spatial separation between the energy storage medium (i.e. the flowing electrolyte) and the power-generating unit (i.e. the electrochemical cell stack), enabling independent scaling of energy capacity and power output. The integration of multiple ionic species within these electrolytes allows for enhanced tunability of redox potentials, improved ionic conductivity, and the potential for multi-electron transfer pathways, further elevating their versatility and performance. As illustrated in Fig. 33a.563 a typical flow battery consists of two external electrolyte tanks connected to a central cell stack and operates based on the dynamic transport of ionic carriers between compartments. In this context, the presence and interplay of multiple ionic species are fundamental to ensuring efficient charge transfer and overall system performance. Central to this architecture is the ion-selective membrane, which ensures continuous ionic conductivity while minimizing cross-contamination between the anolyte and catholyte. The aqueous electrolyte formulations widely used in these systems further enhance safety by eliminating flammable solvents, reducing thermal runaway risk and broadening operating conditions.564 These benefits, combined with demonstrated lifespans exceeding 20000 cycles and the feasibility of electrolyte regeneration, render flow batteries a compelling solution for long-duration, stationary energy storage.565
 | ||
| Fig. 33 (a) Schematic diagram of the typical AORFBs device. Organic molecular of AORFBs in three types: (b) H+-carrier systems: HydBPyMeCl, reproduced with permission from ref. 596. Copyright 2025, American Chemical Society. CSFA-Cl, reproduced with permission from ref. 597. Copyright 2024, Wiley. DMAMPO, reproduced with permission from ref. 598. Copyright 2024, Wiley. PTO-PTS, reproduced with permission from ref. 599. Copyright 2024, American Chemical Society. HATNTA; reproduced with permission from ref. 600. Copyright 2023, Elsevier. (c) OH-carrier systems: 4C7SFL, reproduced with permission from ref. 578. Copyright 2021, the American Association for the Advancement of Science. Cys-DHAQ, reproduced with permission from ref. 602. Copyright 2025, Springer Nature. DBEP; reproduced with permission from ref. 603. Copyright 2024, American Chemical Society. (d) Organic-ion-based systems: TMAP-bisV, reproduced with permission from ref. 592. Copyright 2025, Wiley. N + TEMPOD, reproduced with permission from ref. 573. Copyright 2025, Springer Nature. NDI, reproduced with permission from ref. 605. Copyright 2023, the Chinese Chemical Society. dex-NDI, reproduced with permission from ref. 606. Copyright 2024, Wiley. HSPC. Reproduced with permission from ref. 607. Copyright 2025, Wiley. | ||
Historically, the evolution of flow battery technology can be traced back to the 19th century, with early concepts of electrochemical energy storage emerging alongside foundational work in redox chemistry. However, practical development began in the 1970s when NASA and other research institutions explored RFBs as scalable solutions for long-duration energy storage. The first modern flow battery, i.e. the Fe–Cr system, was demonstrated during this period. Despite technical limitations, such as crossover and poor efficiency, it established the core principles of flow-based energy storage. The subsequent decades witnessed significant advancements, especially with the introduction of the all-vanadium redox flow batteries (VRFBs) in the 1980s, in which the redox pairs of VO2+/VO2+ and V3+/V2+ serve as the respective redox couples in the positive and negative electrolytes.566–570 Since then, flow batteries have diversified into various chemistries, including Zn–Br2 FBs, Zn–Fe FBs, polysulfide-bromine (PSB), and all-iron flow batteries (AIFBs), driven by demands for grid-scale energy storage, renewable energy integration, and improved cost-performance ratios.571–575
While VRFBs have dominated the landscape of commercial flow battery technologies due to well-established chemistry and scalability, the limited availability and fluctuating cost of vanadium have significantly constrained their widespread deployment. These challenges have spurred the development of AORFBs as a promising alternative. AORFBs utilize structurally tunable organic molecules as redox-active species, dissolved in separate aqueous anolyte and catholyte reservoirs.576,577 Crucially, these systems rely on the migration of multiple ionic carriers, such as H+, alkali metal cations (e.g., K+ and Na+), anions (e.g., Cl− and OH−), and organic ions (e.g., quaternary ammonium ion, imidazolium ion), through ion-exchange membranes (IEMs), which plays a vital role in maintaining charge neutrality, optimizing ionic conductivity, and ensuring long-term cycling stability. The inherent flexibility of AORFBs also enables asymmetric configurations, where organic redox species are paired with inorganic redox couples (e.g., Fe(CN)64−/Fe(CN)63− or V2+/V3+), further highlighting the strategic utility of multi-ion carriers in tuning redox potential windows, minimizing crossover, and enhancing overall device performance (Fig. 33a).578
Because this review focuses on recent advances in emerging RFBs enabled by multiple ionic carriers, we do not cover conventional inorganic redox flow batteries (e.g., VRFB or Fe–Cr systems), which have been comprehensively reviewed elsewhere. Instead, our discussion highlights innovative multi-ion flow architectures that utilize cooperative or sequential ionic transport, including organic-ion redox chemistries, asymmetric ion-exchange systems, and hybrid H+-organic ion-coupled platforms. Readers interested in the fundamentals and technological evolution of traditional inorganic flow batteries are referred to several excellent review articles on the subject.563,579,580
To meet the demands of high-performance devices, current organic molecular design strategies prioritize three key features: π-conjugated structures for redox-state stabilization, high aqueous solubility (typically >1.5 M), and strong electrochemical reversibility across a wide pH range.581 Guided by these principles, significant progress has been made in developing organic redox-active molecules, such as anthraquinone, fluorene, pyridine, pyrene, naphthalene, and phenazine derivatives. A critical determinant of AORFB performance is the role of multiple ionic carriers, including organic ions, H+ or OH−, which collectively influence ion transport, pH stability, and redox kinetics across ion-selective membranes. Based on their ion transport characteristics, AORFBs can be broadly classified into three types: (i) H+-carrier systems, where H+ serves as the primary charge carrier; (ii) OH−-carrier systems, using OH− under alkaline conditions; and (iii) organic-ion-based systems, in which functionalized redox molecules bearing sulfonate, ammonium, or carboxylate groups function simultaneously as redox centers and mobile ionic carriers.
Representative advances include anthraquinone-based systems enabling reversible 2e−/2H+ reactions in acidic media,582,583 hydroxylated anthraquinones paired with ferricyanide in alkaline media,584 and recent innovations in redox regeneration mechanisms (e.g., DHAQ/DHA/DHAL cycles).585 Beyond anthraquinones, a range of redox cores, such as bipyridiniums,586–589 phenazines,590,591 fluorenone,578 nitroxide radicals (e.g., TEMPO),592–594 and ferrocene derivatives,588,595 have been modified using electron-withdrawing or -donating groups to tune redox potential, solubility, and stability. While earlier reviews have covered the broad evolution of AORFB technology,563,579,580 this analysis focuses specifically on cutting-edge developments in the past two years, with particular emphasis on: (1) advances in multi-ion transport mechanisms, (2) breakthroughs in membrane design, and (3) system-level optimization.
6.1. Aqueous organic redox flow batteries
Recent developments have focused on increasing e−-transfer capacity while improving aqueous compatibility through molecular design (Fig. 33b). A representative example is the azo-functionalized bipyridinium, 4,4′-hydrazobis(1-methylpyridinium) dichloride (HydBPyMeCl).596 This 2e−/2H+ catholyte operates reversibly at +0.64 V vs. Ag/AgCl in 2 M HCl and, when paired with a V2+/V3+ anolyte, delivers exceptional stability (99.997% capacity retention per cycle over 70 days), high utilization (87.2%), and a volumetric capacity of 47.1 Ah L−1 at 80 mA cm−2.
Wang et al.597 introduced a more advanced 4e− system based on chlorinated spirobifluorenyl ammonium (CSFA) salts, leveraging an in situ chlorination mechanism with Cl− to stabilize oxidized states. The twisted spiro-structure improves solubility and membrane selectivity by suppressing π-conjugation. In pairing with silicotungstic acid (SWO), the 1.4 M CSFA-Cl catholyte maintained >99.4% coulombic efficiency and >95% utilization over 250 cycles. Remarkably, the first charge involved a 12e− process coupled with the release of 12H+, showcasing its role as a potent H+-generating catholyte. Li's group598 pioneered an in situ electrochemical synthesis approach using pyrene-based molecules, such as 2-((dimethylamino)methyl)-1-hydroxypyrene (DMAMPO), which undergoes reversible 4e−/4H+ cycling in H2SO4. The resulting cell achieved >50 Ah L−1 capacity with 96% utilization and stable cycling over 7500 cycles (∼70 days). To further enhance redox density, they developed pyrene-4,5,9,10-tetraone-1-sulfonate (PTO-PTS), a sulfonated, thermally stable 4e−/4H+ redox molecule, achieving 59.6 Wh L−1 energy density and outstanding stability over 5200 cycles.599 The extended π-conjugation and optimized electrostatic potential were critical to its performance.
Pushing e−-transfer boundaries further, Song et al.600 reported a 6e−/6H+ catholyte, hexaazatrinaphthalene triacid (HATNTA). Although solubility is currently limited by π–π stacking (1.4 M), the system delivered a specific capacity of 37.2 Ah L−1 and peak power density of 238 mW cm−2, with minimal capacity fade (0.021% per cycle), highlighting the promise of π-expanded structures for high H+ throughput. Efforts to improve air stability without compromising redox activity have also been successful. Li's team601 developed 2,4,6,8-tetramethylamine methylene-1,5-naphthalene diol (TAND), a water-soluble, air-tolerant naphthalene derivative. Synthesized via a scalable, low-toxicity route, 1.5 M TAND demonstrated strong ambient durability and delivered up to 50 Ah L−1 with negligible decay over 40 days in full cells paired with the SWO anolyte.
These advancements demonstrate the growing complexity and importance of proton management in AORFBs. From 2e− systems like HydBPyMeCl to 6e− candidates like HATNTA, progress in proton-coupled redox chemistries is increasingly dependent on integrating multi-H+/e− transfer mechanisms, solubility tuning, and ionic coordination. The strategic design of H+-generating redox centers, synergized with multi-ion transport pathways, forms a foundation for scalable, high-energy-density AORFBs suitable for grid-scale energy storage.
Breakthroughs in this field were achieved with 2e− transfer systems, where Wang and Zhang578 established fluorenone (FL) derivatives as robust, catalyst-free redox mediators. Their work demonstrated that strategic incorporation of e−-withdrawing sulfonate and carboxylate groups could stabilize critical FL− and FL2− intermediates, enabling reversible ketone-alcohol interconversion on carbon electrodes. The optimized 4-carboxylic-7-sulfonate fluorenol (4C7SFL) exhibited remarkable solubility exceeding 1.5 M in NaOH, facilitating a high-capacity flow system when paired with the [Fe(CN)6]3−/[Fe(CN)6]4− catholyte. During charging, FL molecules undergo electrochemical reduction to radical anions, which then disproportionate to form FL and FL-OH, showcasing a well-controlled 2e− transfer process with coordinated hydroxide ion management.
Based on these foundational 2e− systems, Jin et al.602 introduced a bioinspired approach by grafting 1,5-dihydroxyanthraquinone (1,5-DHAQ) with cysteine, yielding Cys-DHAQ with significantly enhanced solubility (0.63 M) and electrochemical reversibility. At pH = 14, this system operated via a 2e−, 0H+ reduction pathway, accompanied by the generation of 2OH−. The hydrated, highly reduced form (Cys-DHAQ6−) exhibited remarkable stability, enabling ultralong cycling with a capacity decay rate as low as 0.00038% per cycle over 1100 cycles, a 100-fold improvement over unmodified 1,5-DHAQ.
Beyond 2e− systems, efforts have focused on increasing redox complexity without compromising aqueous solubility or chemical robustness. Jin's group603 addressed the solubility-stability dilemma in phenazine-based AORFBs by incorporating flexible, hydrophilic O-alkyl carboxylic acid side chains into the redox core. The resulting molecule, phenazine-(2,3-diyl)bis(oxy)diacetic acid (DBEP), supported a stable multi-electron process in 1.0 M KOH with high reversibility. When paired with K4Fe(CN)6, the DBEP-based flow battery delivered an open circuit voltage of 1.17 V and demonstrated minimal capacity fade (0.003% per cycle) over 1000 cycles at 50 mA cm−2. These results exemplify how molecular functionalization, tuning both π-electronic resonance and ion-dipole interactions, can coordinate hydroxide ion release with reversible redox processes, thereby achieving efficient charge transport and superior alkaline compatibility.
Altogether, these advances delineate a clear trend: increasing e− transfer capacity necessitates more sophisticated ion coordination. The coupling between redox activity and OH− generation sets the performance limits of alkaline AORFBs. Future progress will hinge on developing multi-e− redox molecules, beyond 2e−, that retain precise hydroxide control, high solubility, and long-term stability. Hybrid structures integrating features from quinones, phenazines, and other cores may offer a pathway to simultaneously enhance energy density, efficiency, and OH− transport fidelity.
A paradigm shift came with work by Scherman and Grey,604 who demonstrated that π-dimer formation, rather than promoting degradation, can suppress ROS-induced decay when optimized via singlet–triplet energy gap tuning. This insight laid the foundation for air-tolerant AORFBs by thermodynamically trapping radical intermediates. Building on this, Xu et al.592 designed bis(bipyridinium) molecules (M-bisV, TMAP-bisV) with propylene linkers that enable intramolecular π–π stacking, forming stabilized π-dimers upon reduction. DFT calculations revealed a drastic reduction in dimerization energy barriers (from −14.4 to −103.9 kJ mol−1), leading to radical confinement and enhanced stability. When paired with a trimethylolpropane triacrylate (TEMPTMA) catholyte, TMAP-bisV achieved 66.5 Ah L−1 volumetric capacity and an ultra-low capacity fade of just 0.014% per day.
While progress in anolyte design is notable, catholyte development remains challenging, particularly in balancing high redox potential with membrane selectivity. TEMPO-based catholytes, though highly reversible at >0.8 V vs. SHE, suffer from molecular crossover due to their small size. To address this, Feng and Li573 introduced ionic liquid-mimicking dimers (i-TEMPODs) such as N + TEMPOD, designed for bulkier molecular size and stable operation under WIS conditions. In pairing with a highly soluble viologen (Dex-EtOH-Vi), the system achieved 4 M e− concentration, 101 Ah L−1 capacity, and 47.3 Wh L−1 energy density over 96 days, demonstrating the efficacy of steric hindrance and internal charge balance. Further expanding molecular diversity, naphthalene diimide (NDI) derivatives have shown promise with a “one-step, two-electron” redox cascade (NDI2+ ↔ NDI+ ↔ NDI0). When paired with the FcNCl catholyte, NDI-based systems retained 98.44% of capacity over 350 cycles, the most stable 2e− AORFBs reported to date, although concentration limits and synthesis costs remain hurdles.605
To address scalability, He et al.606 synthesized highly conjugated, functionalized NDI derivatives (dex-NDI) via a low-cost hydrothermal route (∼$ 0.16 g−1). Featuring hydrophilic groups for self-assembly via π–π stacking and hydrogen bonding, dex-NDI demonstrated enhanced solubility (1.85 M), stabilized 2e− redox transitions, and enabled 2.4 M e− concentration. In full cells with the MiAcNH-TEMPO catholyte, the system reached 54.4 Ah L−1 capacity and 318 mW cm−2 power density, establishing a new benchmark for cost-effective, high-performance AORFBs.
Despite these advances, redox instability under multi-e− conditions remains a critical bottleneck. Even rigid π-conjugated frameworks such as anthraquinones or phenazines may suffer from irreversible degradation due to localized electron density. To mitigate this, Wang's team607 proposed a resonance-stabilized phenazine dication (HSPC) with 2,3-dihydroxy substitution. The resulting 2e− process (HSPC ↔ re-HSPC) benefits from extensive delocalization across phenazine and quinoid moieties, enabling a highly reversible redox platform with minimal fade (0.0009% per cycle) at 1.4 M e− concentration and a peak power density of 0.11 W cm−2, coupled with K4Fe(CN)6.
Taken together, these developments highlight the central role of organic radical stabilization in advancing organic ion-conducting AORFBs. Through strategies such as controlled π-dimerization, intramolecular association, extended conjugation, and resonance delocalization, researchers have enhanced the electrochemical reversibility, air stability, and scalability of these systems. Continued molecular engineering of functional organic ions capable of multi-e− transfer and intrinsic charge balancing offers a promising pathway toward next-generation flow batteries with high energy density and cost-effective scalability.
6.2. Ion-exchange membranes
The critical role of IEMs in AORFBs has driven significant innovations in membrane design to address the fundamental challenge of simultaneously achieving high ion selectivity and conductivity. While conventional perfluorosulfonic acid membranes like Nafion have been widely adopted for their excellent proton conductivity and chemical stability. Yet, in multi-ion AORFB environments, their inherent trade-off between ion transport and selectivity leads to substantial crossover of redox-active species, resulting in capacity fade and reduced energy efficiency. This limitation stems from the nanoscale phase-separated morphology of traditional IEMs, where ill-defined hydrophilic/hydrophobic domains create a persistent conductivity-selectivity dilemma.A conceptual breakthrough was realized by Schubert et al.,586 who demonstrated that inexpensive dialysis membranes could serve as functional ion-selective barriers in NaCl-based AORFBs, relying on size exclusion and charge repulsion mechanisms rather than traditional ion-exchange functionalities. This seminal work underscored the feasibility of managing multiple ionic species using non-ionic membranes and prompted a wave of innovation focused on engineering membranes with well-defined, nano-confined transport pathways. Among the most promising of these approaches is the development of polymers of intrinsic microporosity (PIMs). Unlike conventional IEMs that rely on hydrated channels, PIMs use micropores created by inefficient packing of rigid and contorted polymer backbones, allowing the combination of size-sieving selectivity with enhanced ionic permeability. Song's team436,608 pioneered this direction through strategic functionalization of PIMs with Tröger's base and amidoxime groups, demonstrating how tailored side chains could optimize both pore dimensions and local chemical environments to enhance specific ion transport while minimizing organic species crossover. These innovations were complemented by sulfonic,609 and carboxylic acid-functionalized variants610 that achieved unprecedented combinations of ion selectivity and conductivity through precisely engineered polymer-ion interactions.
Recently, a landmark advancement by Xu's team611 introduced a self-supporting covalently bonded polymer framework membrane based on rigid triazine structures (Fig. 34a and b), sub-nanometer channels that enable near-frictionless ion transport (Na+ diffusion coefficient: 1.18 × 10−9 m2 s−1) with ultralow resistance (0.17 Ω cm2). These structural innovations allowed AORFBs to achieve unprecedented current densities (500 mA cm−2) while maintaining exceptional cycling stability, overcoming the longstanding conductivity-selectivity trade-off through precisely engineered pore confinement effects. Then, Xu's group611 further developed branched microporous AEM incorporating twisted spirobifluorene monomers and all-carbon poly(aryl piperidine) backbones that form interconnected 3D networks (Fig. 34c), achieving remarkable Cl− conductivity (120 mS cm−1 at 80 °C) and stable operation at 400 mA cm−2. Subsequent refinements through covalent triazine framework membranes demonstrated selective anion transport tuning via methylation, enhancing Cl− conductivity while maintaining F− permeability, a critical advancement for managing charge asymmetry in multi-ion systems (Fig. 34d).612
 | ||
| Fig. 34 Schematic illustrations of advanced ion-selective polymer membranes with tunable ion transport pathways: (a) pore architecture and chemical environment tailored to enable fast and selective ion conduction. (b) Ion-conductive moieties embedded within membrane pores and a covalently bonded network constructed from rigid triazine units. (c) Semiflexible 3D network architecture derived from spiral-branched polymer design strategy, facilitating interconnected ion pathways. Reproduced with permission from ref. 611. Copyright 2023, Springer Nature. (d) Left: Schematic of a 3D interconnected microporous network enabling efficient anion transport. Right: Molecular structure and synthetic route of a covalent triazine framework-based membrane. Reproduced with permission from ref. 612. Copyright 2025, Springer Nature. | ||
Despite the promise of such high-performance microporous membranes, their synthesis often involves complex procedures and expensive monomers, posing challenges for large-scale applications. Addressing this limitation, Xu's group613 integrated the advantages of conventional microphase-separated ion-exchange membranes (IEMs) and polymers of intrinsic microporosity (PIMs) by converting dense IEMs into microporous structures (Fig. 35a and b). This was achieved through an in situ hypercrosslinking strategy applied to commercially available quaternized poly(2,6-dimethyl-1,4-phenylene oxide) (QPPO), yielding hypercrosslinked QPPO (HC-QPPO) membranes. These membranes demonstrated a twofold increase in ionic conductivity and a tenfold improvement in ion selectivity relative to their pristine counterparts. When employed in neutral AORFBs with BTMAP-Vi/TEMPTMA electrolytes, the HC-QPPO membranes dramatically suppressed capacity decay, offering a cost-effective pathway to integrate high selectivity, chemical robustness, and low resistance within a scalable platform, an essential criterion for real-world deployment of multi-ion carriers systems.
 | ||
| Fig. 35 (a) Schematic of charged microporous membranes derived from the hypercrosslinking of conventional IEMs (green spheres denote fixed charged groups). (b) Illustration of the facile hypercrosslinking process and the resulting microporous polymer network structure. Reproduced with permission from ref. 613. Copyright 2024, Wiley. (c) Chemical structure and chain segment of conventional sPEEK. (d) Schematic depiction of ion channel morphology in sPEEK membranes. (e) Chemical structure of sPEEK-Trip incorporating triptycene units that introduce internal free volume. (f) Schematic illustration of hourglass-shaped interconnected ion channels in sPEEK-Trip membranes, facilitating rapid and selective ion transport, particularly enabling the conduction of both cations (black arrows) and hydroxide ions (red arrows) in alkaline media. Reproduced with permission from ref. 614. Copyright 2025, Elsevier. | ||
Further emphasizing the potential of dual-ion conductive membranes, Song et al.614 re-engineered sulfonated poly(ether ether ketone) (sPEEK) and sPEEK-Trip membranes by incorporating triptycene units into the polymer backbone to induce intrinsic microporosity (Fig. 35c–f). Unlike conventional linear sPEEK, this twisted, rigid architecture formed well-connected water channels and supported efficient transport of both K+ and OH− ions. Sulfonation via silicon-protected reagents further refined the channel morphology, enabling electrostatic-driven cation conduction alongside Grotthuss-type OH− hopping mechanisms. This dual-ion conduction strategy delivered superior performance in alkaline AORFBs systems based on quinone, achieving high energy efficiency and remarkable current densities up to 700 mA cm−2. The structural design not only ensured stability under aggressive pH conditions but also maximized ion mobility, setting new benchmarks in membrane-enabled flow battery performance.
Altogether, through rational control over polymer topology, pore architecture, and functional group distribution, these membranes not only resolve the long-standing conductivity-selectivity dilemma but also enable synchronized cation–anion transport, critical for achieving high coulombic efficiency, stable cycling, and scalable power output. As flow battery systems evolve toward higher energy densities and faster kinetics, multi-ion carrier membranes will remain indispensable to bridging the gap between laboratory advances and grid-scale energy storage applications.
6.3. System-level optimization
Additionally, system-level optimization, such as the transition toward pH-neutral electrolytes, has significantly mitigated corrosion and side reactions linked to extreme pH environments, enabling more stable and sustainable operation under practical conditions.577 In recent years, the development of pH-decoupled AORFBs represents a significant advancement in multi-ion carrier systems, where strategic management of proton and hydroxide ion transport enables cell voltages exceeding the thermodynamic water-splitting limit of 1.23 V.615 While early pH-decoupled systems suffered from severe proton/hydroxide crossover and associated efficiency losses, the innovative work by Aziz et al.616 demonstrated how controlled pH differentials coupled with regeneration systems could mitigate these challenges. Their mild pH-decoupling approach, utilizing AEM and CEM to separate the acidic and alkaline sides, carefully matched redox pairs (benzo[a]hydroxyphenazine-7/8-carboxylic acid (BHPC) in mild alkaline and tris(4,4′-bis(hydroxymethyl)-2,2′-bipyridine)iron(III) dichloride (Fe(Bhmbby)3) in mild acid) (Fig. 36b), achieved breakthrough performance including an OCV > 1.7 V, a peak power density of 140 mW cm−2, coulombic efficiency of 99%, and daily capacity decay <0.07%, while maintaining 85% round-trip efficiency, a testament to the power of Na+/Cl−/OH−/H+ multi-ion carrier optimization in balancing electrochemical potential and ionic crossover (Fig. 36a). | ||
| Fig. 36 (a) Schematic illustration of ion concentration gradients and transport directions in a three-chamber flow cell. (b) Comparison of LSV curves between a BPM sub-cell and the integrated three-chamber system, highlighting enhanced voltage response via spatial ion management. Reproduced with permission from ref. 616. Copyright 2024, Springer Nature. (c) Construction of an aqueous membrane-free flow battery based on (SPr2)V0/− couple with [Fe(CN)6]3−/4− redox species. (d) Cyclic voltammograms of the anolyte and catholyte at pH = 7, measured at 0.1 M active species and a scan rate of 10 mV s−1. Reproduced with permission from ref. 20. Copyright 2025, Wiley. | ||
Parallel to these advancements, the emergence of membraneless flow batteries presents a distinct yet complementary trajectory for cost-effective energy storage. By utilizing immiscible biphasic electrolytes to achieve spontaneous phase separation of redox-active species, these systems eliminate the need for ion exchange membranes altogether, potentially reducing system complexity and cost. As demonstrated by Marcilla's team,20 by combining LiTFSI and (NH4)2SO4, they engineered a low-viscosity (<3.2 mPa s), high-conductivity (158 mS cm−1) immiscible electrolyte pair, where [Fe(CN)6]3−/4− localizes in the sulfate-rich top phase and a sulfonated viologen derivative ((SPr2)V0/−) partitions into the TFSI-rich bottom phase (Fig. 36c). Operating under near-neutral conditions, this configuration achieves a distribution coefficient (K) exceeding 10, promoting sharp concentration gradients and efficient phase-specific ion transport. The resulting multi-ion environment extends the OCV to 1.09 V and increases power density by 3.5-fold relative to PEG-based biphasic systems (Fig. 36d). Impressively, the battery retained 98.5% of its capacity over 400 cycles (33 days) under flow conditions, establishing a foundation for durable, membrane-free, multi-ion carrier systems.
Despite significant advancements in redox-active molecule design, electrolyte optimization, and ion transport regulation, the energy density and long-term stability of AORFBs still lag behind commercial VRFBs. To overcome these limitations, future efforts must focus on the development of novel organic redox species capable of stable multi-e− transfer (>2e−), alongside electrolyte systems that balance high ionic conductivity, low viscosity, and broad electrochemical windows. Hybrid electrolytes and multi-ion formulations may offer new synergies, enabling improved compatibility and enhanced redox reversibility. Simultaneously, membrane materials must be tailored to support selective transport of multivalent ions (e.g., Zn2+ and Al3+) through precise molecular sieving and high chemical stability.
Beyond the molecular and materials scale, system-level innovations are equally vital. Adaptive flow control, real-time sensing, and AI-assisted battery management strategies will be critical for maintaining ionic balance and suppressing degradation. The integration of bioinspired membrane structures or stimuli-responsive transport mechanisms may further improve selectivity and operational flexibility. Addressing these challenges will require cross-disciplinary collaboration and a paradigm shift that views complexity not as a limitation, but as a functional advantage. Such a holistic strategy is essential to position multi-ion AORFBs as viable, next-generation technologies for scalable, long-duration and sustainable energy storage.
7. Concluding remarks and perspectives
7.1. Challenges
The promise of multi-ion EPSs lies in the orchestration of diverse ionic species, each performing a complementary role in redox reactions, charge transport, and interfacial stabilization. Yet, the very richness that makes these systems powerful also renders them exquisitely complex. The deliberate inclusion of multiple ionic species introduces a multidimensional design landscape, one that touches every layer of the device, from materials and interfaces to system integration and scale-up, which introduces a host of scientific and practical challenges that must be addressed with equal sophistication (Fig. 37). | ||
| Fig. 37 Challenges, future directions, and opportunities of multi-ion EPs. | ||
One of the most fundamental scientific hurdles is the precise discrimination and coordination of multiple ions within shared electrolytic and interfacial environments. Ions of differing valence states, sizes, charge densities, solvation structures, and chemical reactivities do not always coexist peacefully. Their interactions can lead to antagonistic phenomena such as cation–anion pairing, ion-solvent competition, and active-site rivalry, disrupting redox equilibria, degrading electrochemical stability, and triggering side reactions. Designing a compatible electrolyte in this context becomes an act of chemical diplomacy, requiring a finely tuned balance of ionic preferences and the mitigation of cross-species interference to preserve both performance and longevity.
Multi-ion systems require the precise co-optimization of ionic flux, electric field distribution, and redox synchronization. In such environments, ion transport resembles a congested highway, where multiple species compete for mobility and selective access through membranes or interphases. Interionic crosstalk can impair ionic conductivity, disrupt charge balance, and compromise transport selectivity. Overcoming these challenges calls for the development of ion-selective membranes and adaptive separators capable of regulating multi-species ion flux with molecular-level precision, demanding advances at the intersection of electrostatics, nanostructured materials, and interfacial chemistry.
In systems where multiple ion species coexist, the formation of stable and functional interphases, such as SEI or CEI, becomes increasingly challenging. Conventional membranes like Nafion often lack the selectivity needed to distinguish between monovalent and multivalent ions, leading to ionic crossover, contamination, and capacity degradation. While solid-state and gel electrolytes may provide improved compartmentalization, they frequently suffer from low ionic conductivity and poor interfacial contact in mixed-ion environments. There is an urgent need for advanced electrolyte platforms capable of molecular-level ion recognition, tailored solvation environments, and high ionic mobility. Promising candidates include hybrid ionic liquids, zwitterionic polymers, and bio-inspired chelating systems designed to reconcile selectivity with transport efficiency.
From a manufacturing and scalability standpoint, multi-ion EPSs pose challenges that extend far beyond the laboratory. High-purity precursors, functional separators, and custom battery management systems inflate the levelized cost of storage, often exceeding that of mature single-ion technologies. Materials that perform well in coin-cell prototypes may falter in large-format devices, where ionic cross-talk and degradation effects become magnified over time. The need to reproducibly synthesize complex electrolytes, fabricate ion-tolerant electrodes, and ensure long-term system integrity under dynamic operating conditions highlights the importance of manufacturability and durability as co-equal goals with performance.
Engineering these systems for real-world use further increases the demand for precision control and system-level coordination. Flow rates, ionic gradients, membrane selectivity, and electrode kinetics must be continuously monitored and dynamically tuned to avoid performance drift or failure. Yet this sophistication must be achieved without prohibitive cost, energy overhead, or fragility. Expanding the electrochemical operating window while maintaining safety and reliability will require creative combinations of tailored solvents, redox-buffering strategies, and hybrid electrolyte systems that balance both thermodynamic stability and kinetic control.
Finally, safety considerations become increasingly urgent as complexity grows. The inclusion of multiple reactive species broadens the landscape of potential failure modes, from thermal instability and gas evolution to dendrite formation and parasitic reactions. As multi-ion EPSs are adapted for critical applications such as grid storage, electric vehicles, and wearable electronics, ensuring their resilience under stress, abuse, and long-term cycling conditions becomes essential.
These challenges do not diminish the potential of multi-ion EPSs; they on the contrary define its frontier. Each obstacle marks a call for innovation: membranes engineered to distinguish ions by geometry or charge, solvent systems that reconcile competing redox chemistries, and interfaces that self-organize in response to local ionic environments. In this landscape, complexity is not a barrier but a new scientific language. As we look ahead, the path forward lies not in simplifying these systems, but in mastering their intricacies. What follows is a roadmap for future research, outlining how the mastery of complexity can reimagine electrochemical power sources as systems that are not merely more powerful, but profoundly more intelligent, adaptable, and resilient.
7.2. Roadmap for future research
The transition from single-ion to multi-ion EPSs represents a paradigm shift that transcends materials innovation, fundamentally redefining the principles of electrochemical design. To fully harness this potential, a targeted research roadmap must integrate advances across four interdependent domains: ion selection, transport regulation, interface engineering, and system architecture, each meticulously tailored to the operational requirements of specific energy storage technologies.Hybrid batteries exemplify this approach, where the strategic integration of multivalent (e.g., Mg2+ and Zn2+) and monovalent ions (e.g., Li+ and Na+) enables synergistic performance: the former provides high-capacity storage while the latter ensures rapid transport and interfacial stabilization. However, this advantage hinges on overcoming critical challenges, including the synchronization of disparate redox kinetics and the prevention of ionic cross-talk through novel electrolyte formulations. Complementary innovations in ion-selective transport channels, spatially graded electrodes, and dynamic interphases will be essential to maintain electrochemical coherence within these hybrid architectures.
This principle of functional ion partitioning extends to HBCs, where multi-ion strategies must reconcile contradictory demands, fast ionic mobility for capacitor-like power delivery versus controlled redox activity for battery-like energy storage. Achieving this balance requires dual-ion pathways: one optimized for rapid electric double-layer formation and another dedicated to faradaic processes. Success in this domain will depend on breakthroughs in bifunctional electrolytes with segregated ionic domains, hierarchically porous electrodes capable of selective ion sieving, and adaptive interfaces that dynamically reconfigure under varying operational loads. Notably, nanoscale control over solvation structures and interfacial field distributions emerges as a universal requirement for sustaining performance across extended cycles.
The versatility of multi-ion systems is particularly evident in fuel cells, where decoupled protonic/anionic transport (e.g., coexisting H+/OH−) can simultaneously broaden operational windows and mitigate catalyst degradation. Yet this advantage introduces new complexities, including inter-ion competition and interfacial instability under pH gradients. Addressing these challenges demands co-conductive membranes with angstrom-level ion selectivity, mixed-ion-tolerant catalytic surfaces, and self-regulating interfaces capable of real-time ionic flux modulation. Similarly, RFBs, arguably the most natural platform for multi-ion implementations, leverage modular architectures to enable voltage-staggered redox couples and even co-mediated chemical synthesis (e.g., H2 evolution coupled with energy storage). However, these systems require meticulous design of stable multivalent redox pairs, advanced separators with molecular-level crossover suppression, and flow fields engineered for kinetic synchronization of multiple ionic species.
Across all technologies, interface engineering emerges as a critical theme. Multi-ion systems often produce chemically diverse and dynamically evolving interfaces that govern device stability and performance. Future directions include the creation of ion-specific interphases, self-restructuring solid–liquid boundaries, and functional coatings that modulate ion flux at the molecular level. These features must accommodate asymmetric ion reactivity, variable hydration shells, and fluctuating electric fields without compromising long-term cyclability.
Understanding such complex phenomena requires high-resolution analytical tools and multi-scale modeling frameworks. In situ and operando techniques, such as cryo-electron microscopy, isotope tracing, and synchrotron spectroscopy, should be coupled with machine learning-assisted data analytics and multi-physics simulations. These will enable the identification of ion-specific transport barriers, emergent interfacial behaviors, and cooperative transport phenomena in real-time operating environments.
From lab-scale development to real-world deployment, key engineering considerations include manufacturability, safety, and integration with renewable sources. Strategies such as 3D-printed flow architectures, modular stacked cell configurations, and ion-gradient-driven compartmentalization will support flexible scale-up. Sustainability must be embedded throughout, favoring non-toxic, earth-abundant elements, closed-loop recycling, and lifecycle-informed material selection.
Economically, the levelized cost of storage (LCOS) remains a critical benchmark for evaluating the feasibility of emerging energy storage systems. The deliberate inclusion of multivalent and organic ionic species, hybrid electrolytes, and ion-selective membranes in multi-ion EPSs introduces significant cost variability. These systems often require high-purity salts, advanced polymers, and engineered interfaces, which elevate capital expenditures relative to conventional single-ion technologies. Additionally, the synthesis of such tailored components frequently involves low-throughput, non-scalable processes. To address these challenges, future efforts should prioritize the use of earth-abundant, non-toxic, and recyclable materials and develop modular synthesis strategies compatible with roll-to-roll or 3D printing techniques, enabling cost-effective scale-up.
From a systems perspective, the ability of multi-ion EPSs to partition electrochemical functions among distinct ionic species offers unprecedented opportunities for design flexibility and performance optimization. However, this complexity also necessitates advanced battery management systems, real-time monitoring, and adaptive control algorithms to maintain ionic balance and operational stability. This calls for the development of low-cost, high-resolution sensing technologies and AI-assisted control strategies to ensure reliable, scalable operation. While current multi-ion systems may not yet match the LCOS of mature lithium-ion technologies due to material and integration costs, they show long-term potential in scenarios where traditional systems fall short, such as modular energy storage, long-duration operation, and harsh environmental conditions. In grid-scale flow batteries or hybrid systems coupling energy storage with CO2 reduction or hydrogen production, the multifunctionality of multi-ion EPSs can deliver additional value streams, enhancing energy return on investment. Ultimately, realizing their promise requires a shift toward application-driven design rooted in fundamental science, treating ionic complexity as a strategic asset for next-generation energy systems.
7.3. Opportunities
While the roadmap outlines the challenges that must be met, the opportunities offered by multi-ion EPSs are no less compelling. These systems are not merely enhancements of existing technologies; they represent a fundamental reimagining of what electrochemical platforms can achieve across functionality, architecture, and application. At their core, multi-ion EPSs allow for tandem, sequential, or co-mediated redox processes, enabling electrochemical reactions that are inaccessible in conventional single-ion systems. This unlocks the potential for voltage-staggered energy storage, coordinated energy, chemical conversion, and redox-coupled electrosynthesis, vastly expanding the operating landscape. By delegating distinct electrochemical roles of different ions for charge storage, transport, or interfacial stabilization, multi-ion systems alleviate traditional bottlenecks including ion concentration gradients, interfacial instability, and sluggish kinetics, resulting in faster charge–discharge rates, higher power density, and greater design flexibility.Thanks to their tunable composition and reaction dynamics response, multi-ion EPSs are well suited to pair with variable renewable inputs like wind or solar. Their modular and dynamic response capabilities make them excellent candidates for grid balancing, off-grid storage, and hybrid energy-conversion systems. Beyond energy storage, multi-ion EPSs offer avenues for integrated energy-chemical systems. These may include simultaneous hydrogen production and waste valorization, CO2 capture and conversion, or electrosynthetic routes to value-added chemicals. Such coupling transforms EPSs from static energy devices into multifunctional electrochemical factories.
From wearable electronics to implantable medical devices, the versatility of multi-ion systems offers energy solutions that are flexible, biocompatible, and even self-healing. Their tailored voltage profiles and chemical resilience make them ideal for powering electronics under diverse environmental conditions, meeting the demands of wearable technologies, implantable devices, and smart sensing platforms.
The transportation sector, particularly electric vehicles, presents another high-impact frontier. the integration of systems like Li–Na hybrid batteries provides a dual advantage: higher energy density and improved low-temperature performance. For instance, a recent study590 reported Na-based dual-ion batteries achieving 380 Wh kg−1, significantly reducing Li demand CATL's 2023 commercialization of a Li–Na all-battery (AB) pack represents a milestone in hybrid energy storage. By spatially arranging sodium-ion cells in low-temperature zones of the battery pack, this system overcomes cold-weather limitations, achieving a 5% range improvement at sub-zero temperatures while leveraging sodium's thermal resilience. This strategic integration illustrates how multi-ion design can yield a synergistic performance greater than the sum of its parts, a true realization of “1 + 1 > 2” in practical electrochemistry.
Multi-ion redox flow batteries offer yet another path forward, uniquely suited for large-scale grid integration. Their scalable, long-duration, and environmentally friendly characteristics, paired with the ability to incorporate multiple redox couples, provide a highly customizable platform for stationary storage. Their modularity allows system designers to balance power and capacity independently, tailoring performance for the fluctuating demands of renewable-powered grids.
Finally, the rise of multi-ion EPSs represents more than an evolution in materials, signifying a convergence of disciplines. At the crossroads of electrochemistry, materials science, nanotechnology, and data-driven design, these systems offer an opportunity to rethink energy devices as intelligent, responsive, and multifunctional platforms. Their development encourages a holistic approach to system design, from the atomic scale to the grid, and from laboratory discovery to real-world deployment.
7.4. Conclusion
The emergence of multi-ion carrier systems marks a paradigm shift in electrochemical energy technologies, offering new strategies for charge storage, ion transport, and interfacial modulation. In contrast to conventional single-ion systems, multi-ion EPSs exploit the complementary properties of a diverse range of ionic species, including monovalent, multivalent, organic, and anionic carriers, to enable a finely tuned yet robust coordination of redox activity, ionic conductivity, and structural adaptability. This review highlights recent advances across a range of technological platforms, including hybrid batteries, HBCs, fuel cells, and RFBs, illustrating how the deliberate orchestration of multiple ionic species can overcome the intrinsic limitations of single-ion transport.However, the intricate advantages of multi-ion EPSs also bring forth significant scientific and engineering challenges. Ion-specific discrimination, redox synchronization, and selective transport must all occur within shared electrolytic and interfacial environments, a demanding task due to competitive solvation, ion-pairing, and cross-reactivity. The formation of robust SEI/CEI, suppression of ionic crossover, and mitigation of parasitic reactions are further complicated by the coexistence of chemically and physically distinct ions. Moreover, most conventional membranes, electrolytes, and electrode materials lack the selectivity and structural compatibility needed to support multi-species transport without compromising electrochemical performance.
To navigate these challenges, future research must adopt a holistic and application-driven design philosophy. This includes the development of ion-selective membranes, adaptive separators, and solvation-engineered electrolytes that regulate ion flux with molecular precision. Advanced interfacial architectures capable of dynamic self-restructuring will be key to maintaining long-term electrochemical stability in complex ionic environments. Integrating these materials innovations with real-time sensing, AI-assisted control algorithms, and modular system architectures will also be essential for scalable and intelligent energy storage solutions.
Opportunities for multi-ion EPSs extend well beyond conventional battery applications. Their tunable redox dynamics and ionic modularity make them uniquely suited for grid-scale storage, coupling with renewable energy sources, and hybrid energy-chemical systems that enable CO2 conversion, H2 production, or electrosynthesis of valuable chemicals. In wearable electronics and electric vehicles, multi-ion configurations offer tailored voltage profiles, enhanced thermal stability, and resilience under diverse operating conditions.
In sum, multi-ion EPSs are not simply an evolutionary step; they represent a conceptual reimagining of electrochemical systems as adaptive, multifunctional, and highly engineered platforms. Unlocking their full potential will require breakthroughs in electrolyte chemistry, membrane science, interfacial engineering, and computational design, supported by operando diagnostics and machine-learning-guided discovery. As we move toward a decarbonized and electrified energy future, mastering ionic complexity is not a hindrance but a key enabler, transforming electrochemical power systems into intelligent, responsive, and resilient energy technologies for the decades ahead.
Author contributions
Y. Zhang, X. Hu and Z. Wen conceived the project and coordinated the overall structure and review logic; P. Wu and C. Chen conducted the literature survey and contributed to the drafting of the manuscript; Y. Liu was responsible for figure preparation; X. Cai, W. Liang, and M. Li assisted in mechanism analysis and the comparative discussion of electrochemical performance; X. Zhuang, Y. Li, and X. Chen contributed to the design strategies of materials and electrolyte systems; M. Sun and L. Wei managed the references and assisted with formatting. All authors discussed the results and approved the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Data availability
The data supporting this article have been included as part of the SI. See DOI: https://doi.org/10.1039/d5cs00785bAcknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 22225902, U22A20436, 22209185, and 22409171), the National Key Research & Development Program of China (2022YFE0115900, 2023YFA1507101, and 2021YFA1501500), the Self-deployment Project Research Program of Haixi Institutes, Chinese Academy of Sciences (no. CXZX-2022-GH04, CXZX-2023-JQ08), and Science and Technology Program of Fuzhou (2023-P-009), the China National Postdoctoral Program for Innovative Talents (BX20230365), and the China Postdoctoral Science Foundation (2023M743494).Notes and references
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed
.
- J. Y. Hwang, S. T. Myung and Y. K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed
- B. Li, J. Zheng, H. Zhang, L. Jin, D. Yang, H. Lv, C. Shen, A. Shellikeri, Y. Zheng, R. Gong, J. P. Zheng and C. Zhang, Adv. Mater., 2018, 30, e1705670 CrossRef PubMed
- Y. Wang, X. Yang, Z. Zhang, X. Hu, Y. Meng, X. Wang, D. Zhou, H. Liu, B. Li and G. Wang, eScience, 2022, 2, 573–590 CrossRef
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, e1800561 CrossRef PubMed
- N. Zheng, H. Ji, J. Wang, M. Zhang, L. Wei, R. Shi, K. Jia, X. Wu, X. Xiao, Z. Zhuang, B. Li, H. M. Cheng and G. Zhou, J. Am. Chem. Soc., 2024, 146, 27819–27829 CrossRef CAS PubMed
- H. Li, H. Liu, S. Luo, J. Arbiol, E. Suard, T. Bergfeldt, A. Missyul, V. Baran, S. Mangold, Y. Zhang, W. Hua, M. Knapp, H. Ehrenberg, F. Pan and S. Indris, Nat. Commun., 2025, 16, 2203 CrossRef CAS PubMed
- Y. Tang, Q. Zhang, W. Zuo, S. Zhou, G. Zeng, B. Zhang, H. Zhang, Z. Huang, L. Zheng, J. Xu, W. Yin, Y. Qiu, Y. Xiao, Q. Zhang, T. Zhao, H.-G. Liao, I. Hwang, C.-J. Sun, K. Amine, Q. Wang, Y. Sun, G.-L. Xu, L. Gu, Y. Qiao and S.-G. Sun, Nat. Sustainable, 2024, 7, 348–359 CrossRef
- F. Wu, J. Maier and Y. Yu, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC
- S. Liu, S. Liu, J. Bao, Z. Huang, L. Wei, N. Chen, Z. Hu, W. H. Huang, C. W. Pao, Q. Kong, J. Han, L. Li and X. Huang, Angew. Chem., Int. Ed., 2025, 64, e202421013 CrossRef CAS PubMed
- R. Haider, Y. Wen, Z. F. Ma, D. P. Wilkinson, L. Zhang, X. Yuan, S. Song and J. Zhang, Chem. Soc. Rev., 2021, 50, 1138–1187 Search PubMed
- K. Jiao, J. Xuan, Q. Du, Z. Bao, B. Xie, B. Wang, Y. Zhao, L. Fan, H. Wang, Z. Hou, S. Huo, N. P. Brandon, Y. Yin and M. D. Guiver, Nature, 2021, 595, 361–369 CrossRef CAS PubMed
- L. She, H. Cheng, Z. Yuan, Z. Shen, Q. Wu, W. Zhong, S. Zhang, B. Zhang, C. Liu, M. Zhang, H. Pan and Y. Lu, Adv. Sci., 2024, 11, e2305061 CrossRef PubMed
- Z. Zhao and H. N. Alshareef, Adv. Mater., 2024, 36, e2309223 CrossRef PubMed
- D. Sun, D. Guo, Y. Lu, J. Chen, Y. Lu, X. Han, X. Feng, L. Lu, H. Wang and M. Ouyang, Energy Environ. Sci., 2024, 17, 7512–7542 RSC
- S. Ma, W. Yan, Y. Dong, Y. Su, L. Ma, Y. Li, Y. Fang, B. Wang, S. Wu, C. Liu, S. Chen, L. Chen, Q. Huang, J. Wang, N. Li and F. Wu, Mater. Today, 2024, 75, 334–358 CrossRef CAS
- T. Hu, J. Li, Y. Wang, S. Chen, T. Yu, H.-M. Cheng, Z. Sun, Q. Xu and F. Li, Joule, 2023, 7, 1176–1205 CrossRef CAS
- W. Zhang, Y. Liu and Z. Guo, Sci. Adv., 2019, 5, eaav7412 CrossRef CAS PubMed
- P. Navalpotro, C. S. Santos, M. L. Alcantara, V. Munoz-Perales, S. E. Ibanez, A. Martinez-Bejarano, N. Jiyane, C. Neves, R. Rubio-Presa, T. Quast, W. Schuhmann, J. A. P. Coutinho and R. Marcilla, Angew. Chem., Int. Ed., 2025, 64, e202424650 CrossRef CAS PubMed
- L. Shi, D. Liu, X. Lin, R. Cheng, F. Liu, C. Kim, C. Hu, J. Qiu, R. Amal and L. Dai, Adv. Mater., 2024, 36, e2314077 CrossRef PubMed
- X. Liu, W. Sun, J. Chen and Z. Wen, Angew. Chem., Int. Ed., 2024, 63, e202317313 CrossRef CAS PubMed
- J. Yang, H. Xu, J. Li, K. Gong, F. Yue, X. Han, K. Wu, P. Shao, Q. Fu, Y. Zhu, W. Xu, X. Huang, J. Xie, F. Wang, W. Yang, T. Zhang, Z. Xu, X. Feng and B. Wang, Science, 2024, 385, 1115–1120 CrossRef CAS PubMed
- Z. Cheng, H. Liu, M. Zhang, H. Pan, C. Sheng, W. Li, M. Wagemaker, P. He and H. Zhou, Nat. Commun., 2025, 16, 1723 CrossRef CAS PubMed
- O. Alshangiti, G. Galatolo, C. Di Mino, T. F. Headen, J. Christianson, S. Merotto, G. J. Rees, Y. Delavoux, M. Swadźba-Kwaśny and M. Pasta, ACS Energy Lett., 2024, 9, 6104–6108 CrossRef CAS PubMed
- K. Shimoda, Y. Morita, K. Noi, T. Fukunaga, Z. Ogumi and T. Abe, ACS Energy Lett., 2023, 8, 2570–2575 CrossRef CAS
- X. Zhao, Z. Zhao-Karger, M. Fichtner and X. Shen, Angew. Chem., Int. Ed., 2020, 59, 5902–5949 CrossRef CAS PubMed
- Z. Yang, X. H. Liu, X. X. He, W. H. Lai, L. Li, Y. Qiao, S. L. Chou and M. Wu, Adv. Funct. Mater., 2020, 31, 2006457 CrossRef
- L. Mu, D. Hou, E. E. Foley, M. Dai, J. Zhang, Z. Jiang, M. M. Rahman, Y. Fu, L. Ma, E. Hu, S. Sainio, D. Nordlund, J. Liu, J. M. Hu, Y. Liu, R. J. Clement and F. Lin, J. Am. Chem. Soc., 2024, 146, 26916–26925 CrossRef CAS PubMed
- Y. Sun, J. Weng, P. Zhou, W. Yuan, Y. Pan, X. Wu, J. Zhou and F. Cheng, Adv. Mater., 2024, 36, e2410575 CrossRef PubMed
- W. Xu, L. Li, Y. Zhao, S. Li, H. Yang, H. Tong and Z. Wang, Energy Environ. Sci., 2025, 18, 2686–2719 RSC
- S. Wang, S. Jiao, D. Tian, H. S. Chen, H. Jiao, J. Tu, Y. Liu and D. N. Fang, Adv. Mater., 2017, 29, 1606349 CrossRef PubMed
- X. Geng, X. Hou, X. He and H. J. Fan, Adv. Energy Mater., 2024, 14, 2304094 CrossRef CAS
- X. Gao, H. Wu, C. Su, C. Lu, Y. Dai, S. Zhao, X. Hu, F. Zhao, W. Zhang, I. P. Parkin, C. J. Carmalt and G. He, Energy Environ. Sci., 2023, 16, 1364–1383 RSC
- S. Jin, X. Gao, S. Hong, Y. Deng, P. Chen, R. Yang, Y. L. Joo and L. A. Archer, Joule, 2024, 8, 746–763 CrossRef CAS
- Z. Wei, Y. Gao, L. Wang, C. Zhang, X. Bian, Q. Fu, C. Wang, Y. Wei, F. Du and G. Chen, Chemistry, 2016, 22, 11610–11616 CrossRef CAS PubMed
- Q. Zhang, Y. Lu, L. Miao, Q. Zhao, K. Xia, J. Liang, S. L. Chou and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 14796–14800 CrossRef CAS PubMed
- J. Song, J. Gim, S. Park and J. Kim, Electrochim. Acta, 2018, 261, 42–48 CrossRef CAS
- B. Ma, Y. Liang, X. Ma, W. Wang, C. Miao, G. Yu, Q. Wang, C. Xu and X. Cui, J. Colloid Interface Sci., 2025, 696, 137824 CrossRef CAS PubMed
- Y. Xie, Y. Yang, S. Chen, Y. Huang, Z. Chen, X. Wu, L. Zhu, R. Zeng and Z. Yuan, J. Power Sources, 2025, 639, 236659 CrossRef CAS
- S. Q. Wang, J. Zhang, Z. X. Xu and J. L. Wang, ChemistrySelect, 2021, 6, 12288–12294 CrossRef CAS
- L. Xia, L. Lin, J. Li, Y. Zhang, H. Zheng, X. Chang, J. Liang, B. Sa, L. Wang, J. Lin, D. L. Peng, K. Amine and Q. Xie, Angew. Chem., Int. Ed., 2025, 64, e202423090 CrossRef CAS PubMed
- C. Zhao, Q. Wang, Z. Yao, J. Wang, B. Sanchez-Lengeling, F. Ding, X. Qi, Y. Lu, X. Bai, B. Li, H. Li, A. Aspuru-Guzik, X. Huang, C. Delmas, M. Wagemaker, L. Chen and Y. S. Hu, Science, 2020, 370, 708–711 CrossRef CAS PubMed
- H. Zhang, J. Li, J. Liu, Y. Gao, Y. Fan, X. Liu, C. Guo, H. Liu, X. Chen, X. Wu, Y. Liu, Q. Gu, L. Li, J. Wang and S. L. Chou, Nat. Commun., 2025, 16, 2520 CrossRef CAS PubMed
- R. Z. Wei, X. W. Zhai, H. R. Tinker, P. He, C. A. F. Nason, Y. P. Han, V. Celorrio, G. Sankar, M. Zhou and Y. Xu, Adv. Funct. Mater., 2023, 33, 2308227 CrossRef CAS
- Q. Ni, Y. Xiong, Z. Sun, C. Sun, Y. Li, X. Yuan, H. Jin and Y. Zhao, Adv. Energy Mater., 2023, 13, 2300271 CrossRef CAS
- Z. Chen, L. Wang, Z. Chen, Y. Zhong, X. Wang and J. Tu, Adv. Funct. Mater., 2025, 2502682 CrossRef
- K. Shin, Y. Zheng, F. Zhang, S. Wu and Y. Tang, ACS Appl. Energy Mater., 2020, 3, 7030–7038 CrossRef CAS
- S. Wang, Y. Guo, X. Du, L. Xiong, Z. Liang, M. Ma, Y. Xie, W. You, Y. Meng, Y. Liu and M. Liu, Nat. Commun., 2024, 15, 6476 CrossRef CAS PubMed
- X.-G. Sun, Z. Zhang, H. Y. Guan, C. A. Bridges, Y. Fang, Y.-S. Hu, G. M. Veith and S. Dai, J. Mater. Chem. A, 2017, 5, 6589–6596 RSC
- M. Mao, T. Gao, S. Hou and C. Wang, Chem. Soc. Rev., 2018, 47, 8804–8841 RSC
- H. Dong, Y. Li, Y. Liang, G. Li, C.-J. Sun, Y. Ren, Y. Lu and Y. Yao, Chem. Commun., 2016, 52, 8263–8266 RSC
- Y. Li, Q. An, Y. Cheng, Y. Liang, Y. Ren, C.-J. Sun, H. Dong, Z. Tang, G. Li and Y. Yao, Nano Energy, 2017, 34, 188–194 CrossRef CAS
- X. Wang, T. Li, X. Zhang, Y. Wang, H. Li, H.-F. Li, G. Zhao and C. Han, J. Energy Chem., 2024, 96, 79–88 CrossRef CAS
- T. Zhou, Y. Zhu, Y. Shen, H. Qiu, T. Han, J. Li and J. Liu, Chem. Commun., 2024, 60, 5338–5341 RSC
- T. Zhou, T. Han, X. Lin, J. Liu, X. Zeng, P. Zhan, J. Liu and J. Niu, Nano Lett., 2024, 24, 4400–4407 CrossRef CAS PubMed
- S. Dhir, S. Wheeler, I. Capone and M. Pasta, Chem, 2020, 6, 2442–2460 CAS
- Q. Zhou, H. K. Liu, S. X. Dou and S. Chong, ACS Nano, 2024, 18, 7287–7297 CrossRef CAS PubMed
- D. Wang, C. Li, Q. Li, H. Li, J. Rehman, C. Zhi and L. Zhu, Nano Energy, 2022, 104, 107990 CrossRef CAS
- R. Hua, C. Xu, H. Yang, D. Qu, R. Zhang, D. Liu, H. Tang, J. Li and D. Qu, ACS Appl. Mater. Interfaces, 2024, 16, 20520–20532 CAS
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed
- J. Zhang, J. Liu, M. Wang, Z. Zhang, Z. Zhou, X. Chen, A. Du, S. Dong, Z. Li, G. Li and G. Cui, Energy Environ. Sci., 2023, 16, 1111–1124 RSC
- Y. Gofer, O. Chusid, H. Gizbar, Y. Viestfrid, H. E. Gottlieb, V. Marks and D. Aurbach, Electrochem. Solid-State Lett., 2006, 9, A257 CrossRef CAS
- S. Yagi, T. Ichitsubo, Y. Shirai, S. Yanai, T. Doi, K. Murase and E. Matsubara, J. Mater. Chem. A, 2014, 2, 1144–1149 RSC
- Y. Cen, S. Li, Y. Zhou, X. Cai, X. Wang, Q. Xiang, B. B. Hu, D. M. Yu, Y. P. Liu and C. G. Chen, J. Electrochem. Soc., 2019, 166, A1660–A1667 CrossRef CAS
- Q. Q. Chen, Q. Xia, Y. X. Xu, P. F. Wang and Q. Q. Tan, Mater. Lett., 2019, 247, 178–181 CrossRef CAS
- J. H. Cho, M. Aykol, S. Kim, J. H. Ha, C. Wolverton, K. Y. Chung, K. B. Kim and B. W. Cho, J. Am. Chem. Soc., 2014, 136, 16116–16119 CrossRef CAS PubMed
- A. M. Diem, K. Hildenbrand, L. Raafat, J. Bill and Z. Burghard, RSC Adv., 2021, 11, 1354–1359 RSC
- Q. Shu, X. J. Hou, K. M. Hou, X. H. Ye, Q. H. Cao, D. T. Li and G. Q. Suo, J. Alloys Compd., 2023, 966, 171584 CrossRef CAS
- Y. Zhang, W. Xiao, Y. Zhao, J. Li, D. Yang, C. Zhu and Y. Chen, Small, 2024, 20, e2406683 CrossRef PubMed
- A. Byeon, M. Q. Zhao, C. E. Ren, J. Halim, S. Kota, P. Urbankowski, B. Anasori, M. W. Barsoum and Y. Gogotsi, ACS Appl. Mater. Interfaces, 2017, 9, 4296–4300 CrossRef CAS PubMed
- X. Li, Y. Tang, L. Liu, Y. Zhang, R. Sheng and Y. NuLi, J. Colloid Interface Sci., 2022, 608, 2455–2462 CrossRef CAS PubMed
- Z. H. Gao, R. Shi, Y. P. Zhao, J. G. Zhang, J. C. Liu, Y. F. Zhu, Y. N. Liu, J. Wang and L. Q. Li, Electrochim. Acta, 2023, 469, 13079–13082 CrossRef
- H. Y. Tsai, M. S. Kumar, B. Vedhanarayanan, H. H. Shen and T. W. Lin, Batteries, 2023, 9, 6245–6269 CrossRef
- C. Pei, F. Xiong, J. Sheng, Y. Yin, S. Tan, D. Wang, C. Han, Q. An and L. Mai, ACS Appl. Mater. Interfaces, 2017, 9, 17060–17066 CrossRef CAS PubMed
- Y. Cheng, Y. Shao, J. G. Zhang, V. L. Sprenkle, J. Liu and G. Li, Chem. Commun., 2014, 50, 9644–9646 RSC
- Y. Ding, T. Han, Z. Wu, Y. Guan, J. Hu, C. Hu, Y. Tian and J. Liu, ACS Nano, 2022, 16, 15369–15381 CrossRef CAS PubMed
- T. Yang, F. M. Ma, X. Q. Zhang, Y. A. Yang, J. S. Lv, Z. F. Jin, L. R. Wang, H. M. Gao, X. L. Wang and N. Chen, Appl. Surf. Sci., 2024, 660, 159995 CrossRef CAS
- Y. Tang, X. Li, H. Lv, W. Wang, Q. Yang, C. Zhi and H. Li, Angew. Chem., Int. Ed., 2021, 60, 5443–5452 CrossRef CAS PubMed
- Z. Fu, J. S. Tan, C. F. Sun and W. Z. Deng, Ionics, 2024, 30, 4055–4062 CrossRef CAS
- S. Guan, Q. Peng, X. Guo, Y. Zheng, E. Liao, S. Sun, K. Shin, B. Liu, X. Zhou, C. Zou and Y. Tang, Chem. Eng. J., 2024, 493, 152864 CrossRef CAS
- G. Bieker, M. Salama, M. Kolek, Y. Gofer, P. Bieker, D. Aurbach and M. Winter, ACS Appl. Mater. Interfaces, 2019, 11, 24057–24066 CrossRef CAS PubMed
- K. A. See, K. W. Chapman, L. Zhu, K. M. Wiaderek, O. J. Borkiewicz, C. J. Barile, P. J. Chupas and A. A. Gewirth, J. Am. Chem. Soc., 2016, 138, 328–337 CrossRef CAS PubMed
- G. Pagot, K. Vezzù, S. G. Greenbaum and V. Di Noto, J. Power Sources, 2021, 493, 229681 CrossRef CAS
- L. Ye, M. Liao, K. Zhang, M. Zheng, Y. Jiang, X. Cheng, C. Wang, Q. Xu, C. Tang, P. Li, Y. Wen, Y. Xu, X. Sun, P. Chen, H. Sun, Y. Gao, Y. Zhang, B. Wang, J. Lu, H. Zhou, Y. Wang, Y. Xia, X. Xu and H. Peng, Nature, 2024, 626, 313–318 CrossRef CAS PubMed
- S. Wu, Z. Jiang, C. Wu, H. Li, J. Huang, N. Li and S. Huang, Adv. Funct. Mater., 2024, 34, 2313441 CrossRef CAS
- Z. Meng, A. Reupert, Y. Tang, Z. Li, G. Karkera, L. Wang, A. Roy, T. Diemant, M. Fichtner and Z. Zhao-Karger, ACS Appl. Mater. Interfaces, 2022, 14, 54616–54622 CrossRef CAS
- H. Song and C. Wang, Energy Environ. Mater., 2022, 6, e12325 CrossRef
- D. Zhou, X. Tang, X. Zhang, F. Zhang, J. Wu, F. Kang, B. Li and G. Wang, Nano Lett., 2021, 21, 3548–3556 CrossRef CAS PubMed
- X. Guo, J. Lu, M. Wang, A. Chen, H. Hong, Q. Li, J. Zhu, Y. Wang, S. Yang, Z. Huang, Y. Wang, Z. Pei and C. Zhi, Chem, 2024, 10, 3607–3621 CAS
- L. Cao, D. Li, E. Hu, J. Xu, T. Deng, L. Ma, Y. Wang, X. Q. Yang and C. Wang, J. Am. Chem. Soc., 2020, 142, 21404–21409 CrossRef CAS PubMed
- D. D. Sarma and A. K. Shukla, ACS Energy Lett., 2018, 3, 2841–2845 CrossRef CAS
- F. Zhao, J. Li, A. Chutia, L. Liu, L. Kang, F. Lai, H. Dong, X. Gao, Y. Tan, T. Liu, I. P. Parkin and G. He, Energy Environ. Sci., 2024, 17, 1497–1508 RSC
- C. Li, H. Yuan, T. Liu, R. Zhang, J. Zhu, H. Cui, Y. Wang, D. Cao, D. Wang and C. Zhi, Angew. Chem., Int. Ed., 2024, 63, e202403504 CrossRef CAS PubMed
- X. Gao, C. Shen, H. Dong, Y. Dai, P. Jiang, I. P. Parkin, H. Zhang, C. J. Carmalt and G. He, Energy Environ. Sci., 2024, 17, 2287–2297 RSC
- J. L. Yang, T. Xiao, T. Xiao, J. Li, Z. Yu, K. Liu, P. Yang and H. J. Fan, Adv. Mater., 2024, 36, e2313610 CrossRef PubMed
- P. Hei, Y. Sai, C. Liu, W. Li, J. Wang, X. Sun, Y. Song and X. X. Liu, Angew. Chem., Int. Ed., 2024, 63, e202316082 CrossRef PubMed
- A. Zhang, R. Zhao, Y. Wang, J. Yue, J. Yang, X. Wang, C. Wu and Y. Bai, Angew. Chem., Int. Ed., 2023, 62, e202313163 CrossRef CAS PubMed
- J. Wu, J. Huang, X. Chi, J. Yang and Y. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 53627–53635 CrossRef CAS PubMed
- M. Kim, S. Lee, J. Choi, J. Park, J.-W. Park and M. Park, Energy Storage Mater., 2023, 55, 698–707 CrossRef
- Y. Pan, Z. Liu, S. Liu, L. Qin, Y. Yang, M. Zhou, Y. Sun, X. Cao, S. Liang and G. Fang, Adv. Energy Mater., 2023, 13, 2203766 CrossRef CAS
- C. Liu, X. Chi, Q. Han and Y. Liu, Adv. Energy Mater., 2020, 10, 1903589 CrossRef CAS
- C. Zhong, B. Liu, J. Ding, X. Liu, Y. Zhong, Y. Li, C. Sun, X. Han, Y. Deng, N. Zhao and W. Hu, Nat. Energy, 2020, 5, 440–449 CrossRef CAS
- J. Lei, Y. Yao, Z. Wang and Y.-C. Lu, Energy Environ. Sci., 2021, 14, 4418–4426 RSC
- Y. Shi, Y. Chen, L. Shi, K. Wang, B. Wang, L. Li, Y. Ma, Y. Li, Z. Sun, W. Ali and S. Ding, Small, 2020, 16, e2000730 CrossRef PubMed
- J. Hao, S. Zhang, H. Wu, L. Yuan, K. Davey and S. Z. Qiao, Chem. Soc. Rev., 2024, 53, 4312–4332 RSC
- Q. Su, Y. Song, Y. Qin, L. Liu, W. Chen, M. Mo, S. Guo, S. Liang and G. Fang, ACS Appl. Energy Mater., 2023, 6, 11299–11307 CrossRef CAS
- D. E. Ciurduc, C. D. L. Cruz, N. Patil, A. Mavrandonakis and R. Marcilla, Energy Storage Mater., 2022, 53, 532–543 CrossRef
- C. Yan, Y. Wang, Z. Chen and X. Deng, Batteries Supercaps, 2021, 4, 1627–1635 CrossRef CAS
- A. Chen, Y. Zhang, Q. Li, G. Liang, S. Yang, Z. Huang, Q. Yang, H. Hu, X. Li, Z. Chen, J. Fan and C. Zhi, Energy Environ. Sci., 2023, 16, 4054–4064 RSC
- P. Jaumaux, S. J. Wang, S. Q. Zhao, B. Sun and G. X. Wang, Energy Environ. Mater., 2023, 6, e12578 CrossRef CAS
- H. Tao, X. Tong, L. Gan, S. Zhang, X. Zhang and X. Liu, J. Alloys Compd., 2016, 658, 119–124 CrossRef CAS
- Y. Jiang, Y. Xiang, Q. Zou, B. Liu, S. Liu, H. Zeng, L. Chen, J. Li, X. Wu and L. Xiong, Ionics, 2022, 28, 3855–3864 CrossRef CAS
- Z. Liu, Q. Yang, D. Wang, G. Liang, Y. Zhu, F. Mo, Z. Huang, X. Li, L. Ma, T. Tang, Z. Lu and C. Zhi, Adv. Energy Mater., 2019, 9, 1902473 CrossRef CAS
- C. Yang, Y. Zhao, J. Fan, L. Li, J. Zhou, K. Wang, F. Lu and H. Sun, Mater. Adv., 2024, 5, 5885–5895 RSC
- F. Ma, X. Yuan, T. Xu, S. Zhou, X. Xiong, Q. Zhou, N. Yu, J. Ye, Y. Wu and T. van Ree, Energy Fuels, 2020, 34, 13104–13110 CrossRef CAS
- X. Zhao, L. Mao, Q. Cheng, F. Liao, G. Yang and L. Chen, Chem. Commun., 2022, 58, 7522–7525 RSC
- H. B. Zhao, C. J. Hu, H. W. Cheng, J. H. Fang, Y. P. Xie, W. Y. Fang, T. N. Doan, T. K. Hoang, J. Q. Xu and P. Chen, Sci. Rep., 2016, 6, 25809 CrossRef CAS PubMed
- K. Lu, B. Song, J. Zhang and H. Ma, J. Power Sources, 2016, 321, 257–263 CrossRef CAS
- H. Ao, W. Zhu, M. Liu, W. Zhang, Z. Hou, X. Wu, Y. Zhu and Y. Qian, Small Methods, 2021, 5, e2100418 CrossRef PubMed
- Y. Liu, C. Gao, J. Yun, Y. Kim, M. Kim, J. Li, X. Li and S. W. Lee, ACS Appl. Energy Mater., 2022, 5, 2780–2785 CrossRef CAS
- W. Yu, J. Ge, Y. Hu, D. Shen, W. Luo, S. Chen, L. Wu, Z. Liu, J. Zhou, H. Yang and B. Lu, Sci. China Mater., 2022, 66, 923–931 CrossRef
- Y. Zhang, H. Li, S. Huang, S. Fan, L. Sun, B. Tian, F. Chen, Y. Wang, Y. Shi and H. Y. Yang, Nano-Micro Lett., 2020, 12, 60 CrossRef CAS PubMed
- V. Soundharrajan, B. Sambandam, S. Kim, V. Mathew, J. Jo, S. Kim, J. Lee, S. Islam, K. Kim, Y.-K. Sun and J. Kim, ACS Energy Lett., 2018, 3, 1998–2004 CrossRef CAS
- S. Zhao, C. Li, X. Zhang, N. Li, T. Wang, X. Li, C. Wang, G. Qu and X. Xu, Sci. Bull., 2023, 68, 56–64 CrossRef CAS PubMed
- C. Lu, Z. Wang, Y. Zhang, G. Tang, Y. Wang, X. Guo, J. Li and L. Wei, Nano Energy, 2024, 120, 109158 CrossRef CAS
- C. Lu, Z. Wang, J. Gao, J. Li and L. Wei, Adv. Energy Mater., 2024, 14, 2304016 CrossRef CAS
- Y. Song, Q. Pan, H. Lv, D. Yang, Z. Qin, M. Y. Zhang, X. Sun and X. X. Liu, Angew. Chem., Int. Ed., 2021, 60, 5718–5722 CrossRef CAS PubMed
- K. Niu, J. Shi, L. Zhang, Y. Yue, S. Mo, S. Li, W. Li, L. Wen, Y. Hou, L. Sun, S. Yan, F. Long and Y. Gao, Adv. Sci., 2024, 11, e2305524 CrossRef PubMed
- J. Shi, K. Niu, L. Zhang, Q. Chen, M. Deng, L. Sun, S. Cheng and Y. Gao, ACS Energy Lett., 2024, 9, 2000–2006 CrossRef CAS
- Y. Liu, K. Xiang, W. Zhou, W. Deng, H. Zhu and H. Chen, Small, 2024, 20, e2308741 CrossRef PubMed
- J. n Gong, P. Bai, Y. Zhang, Z. Zhou, Q. Wang, H. Lv, T. Hu, X. Wu and C. Meng, Small, 2024, 21, 2408467 CrossRef PubMed
- Z. Zhang, Y. Zhang, Z. Gao, T. Lv, Y. Liu, T. Hu and C. Meng, J. Power Sources, 2024, 102, 114246 Search PubMed
- Z. Bao, C. Lu, Q. Liu, F. Ye, W. Li, Y. Zhou, L. Pan, L. Duan, H. Tang, Y. Wu, L. Hu and Z. Sun, Nat. Commun., 2024, 15, 1934 CrossRef CAS PubMed
- X. Qi, Y. Zhu, Y. Xu, W. Chen, Z. Hu, L. Xi, Y. Xie, H. Hou, G. Zou and X. Ji, Energy Storage Mater., 2025, 75, 104063 CrossRef
- Q. Pan, P. Hei, Y. Song, J. Meng, C. Liu and X.-X. Liu, Nano Res., 2022, 16, 2495–2501 CrossRef
- J. Liu, K. Guo, W. Guo, J. Chang, Y. Li and F. Bao, Angew. Chem., Int. Ed., 2025, 64, e202421266 CrossRef PubMed
- J. Meng, Y. Song, J. Wang, P. Hei, C. Liu, M. Li, Y. Lin and X. X. Liu, Chem. Sci., 2023, 15, 220–229 RSC
- J. Zou, C. Wang, B. Xu, Y. Shen, Z. Zeng, L. Fu and M. Zeng, Small, 2025, 21, 2501370 CrossRef CAS PubMed
- Z. Li, Z. Zhang, H. Liao, Y. Zheng and Y. Gao, ACS Nano, 2025, 19, 12680–12709 CrossRef CAS PubMed
- Z. Hu, L. Lin, Y. Jiang, L. Sun, W. Liu, Q. Wang and F. Wang, Energy Storage Mater., 2024, 73, 103820 CrossRef
- X. Yang, Y. Ni, Y. Lu, Q. Zhang, J. Hou, G. Yang, X. Liu, W. Xie, Z. Yan, Q. Zhao and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202209642 CrossRef CAS PubMed
- M. Li, C. Li, C. Zuo, J. Hu, C. Li, W. Luo, S. Luo, A. Duan, J. Wang, X. Wang, W. Sun and L. Mai, Adv. Mater., 2024, 36, e2407233 CrossRef PubMed
- M. Ghosh, V. Vijayakumar, M. Kurian, S. Dilwale and S. Kurungot, Dalton Trans., 2021, 50, 4237–4243 RSC
- M. Li, Y. X. Zhang, J. S. Hu, X. P. Wang, J. X. Zhu, C. J. Niu, C. H. Han and L. Q. Mai, Nano Energy, 2022, 100, 107539 CrossRef CAS
- X. Xie, X. Wang, Q. Cai, H. Xie and L. Chen, J. Energy Storage, 2024, 76, 109814 CrossRef
- L. Chen, X. Liang, X. Wang, G. Peng and H. Xie, Small, 2024, 20, e2306697 CrossRef PubMed
- S. Ding, L. Liu, R. Qin, X. Chen, A. Song, J. Li, S. Li, Q. Zhao and F. Pan, ACS Appl. Mater. Interfaces, 2021, 13, 22466–22474 CrossRef CAS PubMed
- R. Hua, C. Xu, D. Lin, D. Qu, X. Li, R. Zhang, Z. Xie, H. Tang, J. Li and D. Liu, New J. Chem., 2024, 48, 1108–1120 RSC
- Y. Xia, F. Yu, D. Nie, Y. Jiang, M. Sun, L. Que, L. Deng, L. Zhao, Q. Zhang and Z. Wang, Angew. Chem., Int. Ed., 2024, 63, e202406765 CrossRef CAS PubMed
- K. Yang, Q. Liu, Y. Zheng, H. Yin, S. Zhang and Y. Tang, Angew. Chem., Int. Ed., 2021, 60, 6326–6332 CrossRef CAS PubMed
- M. Zhang, M. Shoaib, H. Fei, T. Wang, J. Zhong, L. Fan, L. Wang, H. Luo, S. Tan, Y. Wang, J. Zhu, J. Hu and B. Lu, Adv. Energy Mater., 2019, 9, 1901663 CrossRef CAS
- S. Qiao, Q. Zhou, M. Ma, H. K. Liu, S. X. Dou and S. Chong, ACS Nano, 2023, 17, 11220–11252 CrossRef CAS PubMed
- H. Jiang, X. Han, X. Du, Z. Chen, C. Lu, X. Li, H. Zhang, J. Zhao, P. Han and G. Cui, Adv. Mater., 2022, 34, e2108665 CrossRef PubMed
- X. Tong, X. Ou, N. Wu, H. Wang, J. Li and Y. Tang, Adv. Energy Mater., 2021, 11, 2100151 CrossRef CAS
- Z. Yang, X. Z. Zhou, Z. Q. Hao, J. Chen, L. Li, Q. Zhao, W. H. Lai and S. L. Chou, Angew. Chem., Int. Ed., 2024, 63, e202313142 CrossRef CAS PubMed
- M. Zhang, Y. Pei, W. Liu, R. Liang, Y.-P. Deng, Z. Chen and A. Yu, Nano Energy, 2021, 81, 105643 CrossRef CAS
- C. Y. Chan, P.-K. Lee, Z. Xu and D. Y. W. Yu, Electrochim. Acta, 2018, 263, 34–39 CrossRef CAS
- S. Ghosh, N. Mp, S. Muduli, S. Bhowmik and S. K. Martha, Electrochim. Acta, 2023, 441, 141754 CrossRef CAS
- L. Fan, Q. Liu, Z. Xu and B. Lu, ACS Energy Lett., 2017, 2, 1614–1620 CrossRef CAS
- M. Yu, Y. Sui, S. K. Sandstrom, C. Y. Wu, H. Yang, W. Stickle, W. Luo and X. Ji, Angew. Chem., Int. Ed., 2022, 61, e202212191 CrossRef CAS PubMed
- L. Fan, Q. Liu, S. Chen, Z. Xu and B. Lu, Adv. Energy Mater., 2017, 7, 1602778 CrossRef
- T. Yan, R. Ding, D. Ying, Y. Huang, Y. Huang, C. Tan, X. Sun, P. Gao and E. Liu, J. Mater. Chem. A, 2020, 8, 4747–4752 RSC
- X. Qiu, Y. Duan, L.-Z. Fan and X. Wang, Chem. Eng. J., 2023, 477, 146737 CrossRef CAS
- M. Ghidiu, M. R. Lukatskaya, M. Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CrossRef CAS PubMed
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed
- P. Simon and Y. Gogotsi, Nat. Mater., 2020, 19, 1151–1163 CrossRef CAS PubMed
- Y. J. Gao, C. H. Cui, Z. K. Huang, G. Y. Pan, Y. F. Gu, Y. N. Yang, F. Bai, Z. Sun and T. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202404637 CrossRef CAS PubMed
- C. Cui, Z. Wei, J. Xu, Y. Zhang, S. Liu, H. Liu, M. Mao, S. Wang, J. Ma and S. Dou, Energy Storage Mater., 2018, 15, 22–30 CrossRef
- Y. Liu, X. Hu, J. Li, G. Zhong, J. Yuan, H. Zhan, Y. Tang and Z. Wen, Nat. Commun., 2022, 13, 663 CrossRef CAS PubMed
- S. M. Hao, J. Qu, W. Chang, Y. J. Zhang, Y. Tang and Z. Z. Yu, ChemElectroChem, 2019, 6, 1040–1046 CrossRef CAS
- Q. Wu, J. Wang, H.-G. Wang, Z. Si, C. Li and J. Bai, Electrochim. Acta, 2021, 369, 137657 CrossRef CAS
- H. Pan, L. Wang, Y. Shi, C. Sheng, S. Yang, P. He and H. Zhou, Nat. Commun., 2024, 15, 2263 CrossRef CAS PubMed
- H. Wang, J. Fu, C. Wang, J. Wang, A. Yang, C. Li, Q. Sun, Y. Cui and H. Li, Energy Environ. Sci., 2020, 13, 848–858 RSC
- C. Jiang, L. Xiang, S. Miao, L. Shi, D. Xie, J. Yan, Z. Zheng, X. Zhang and Y. Tang, Adv. Mater., 2020, 32, e1908470 CrossRef PubMed
- B. Ji, F. Zhang, X. Song and Y. Tang, Adv. Mater., 2017, 29, 1700519 CrossRef PubMed
- C. Wei, D. Gong, D. Xie and Y. Tang, ACS Energy Lett., 2021, 6, 4336–4344 CrossRef CAS
- C. Chen, C. S. Lee and Y. Tang, Nanomicro Lett., 2023, 15, 121 Search PubMed
- H. Li, Y. Zhang, S. Ying, G. Zhou, X. Tang and X. Liu, J. Mater. Chem. A, 2025, 704, 135496 CAS
- Z. Zhao, Y. Lei, L. Shi, Z. Tian, M. N. Hedhili, Y. Khan and H. N. Alshareef, Angew. Chem., Int. Ed., 2022, 61, e202212941 CrossRef CAS PubMed
- J. Han, M. Zarrabeitia, A. Mariani, M. Kuenzel, A. Mullaliu, A. Varzi and S. Passerini, Adv. Mater., 2022, 34, e2201877 CrossRef PubMed
- L. Yan, Y.-e Qi, X. Dong, Y. Wang and Y. Xia, eScience, 2021, 1, 212–218 CrossRef
- Y. Wu, Z. Xu, R. Ren, N. Lv, J. Yang, J. Zhang, H. Ren, S. Dong and X. Dong, ACS Appl. Mater. Interfaces, 2023, 15, 12434–12442 CrossRef CAS PubMed
- Y. Wu, S. Dong, N. Lv, Z. Xu, R. Ren, G. Zhu, B. Huang, Y. Zhang and X. Dong, Small, 2022, 18, 2204888 CrossRef CAS PubMed
- C. D. Wessells, S. V. Peddada, M. T. McDowell, R. A. Huggins and Y. Cui, J. Electrochem. Soc., 2011, 159, A98 CrossRef
- C. Li, W. Yan, S. Liang, P. Wang, J. Wang, L. Fu, Y. Zhu, Y. Chen, Y. Wu and W. Huang, Nanoscale Horiz., 2019, 4, 991–998 RSC
- L. Du, S. Bi, M. Yang, Z. Tie, M. Zhang and Z. Niu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2214545119 CrossRef CAS PubMed
- Q. Guo, K. i Kim, H. Jiang, L. Zhang, C. Zhang, D. Yu, Q. Ni, X. Chang, T. Chen, H. Xia and X. Ji, Adv. Funct. Mater., 2020, 30, 2002825 CrossRef CAS
- A. Morag, X. Chu, M. Marczewski, J. Kunigkeit, C. Neumann, D. Sabaghi, G. Z. Zukowska, J. Du, X. Li, A. Turchanin, E. Brunner, X. Feng and M. Yu, Angew. Chem., Int. Ed., 2024, 63, e202401818 CrossRef CAS PubMed
- Y. Ge, B. Liu, D. Wu, Y. Zhang, S. Tang, H. Jiang, J. Li, H. Zhang, X. Tian and J. Yang, ACS Energy Lett., 2025, 10, 1615–1622 CrossRef CAS
- D. Yu, L. Cheng, M. Chen, J. Wang, W. Zhou, W. Wei and H. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 45755–45762 CrossRef CAS PubMed
- Y. Zhu, J. Yin, A. H. Emwas, O. F. Mohammed and H. N. Alshareef, Adv. Funct. Mater., 2021, 31, 2107523 CrossRef CAS
- Z. Cheng, Q. Yao, C. Zheng, F. Zhang, K. Song, Q. Dong, J. Pan and J. Yang, Adv. Funct. Mater., 2023, 34, 2310449 CrossRef
- M. Radi, T. Purkait, D. S. Tchitchekova, A. R. Goñi, R. Markowski, C. Bodin, C. Courrèges, R. Dedryvère and A. Ponrouch, Adv. Energy Mater., 2024, 14, 2401587 CrossRef CAS
- J. Li, C. Han, X. Ou and Y. Tang, Angew. Chem., Int. Ed., 2022, 61, e202116668 CrossRef CAS PubMed
- Z. L. Xu, J. Park, J. Wang, H. Moon, G. Yoon, J. Lim, Y. J. Ko, S. P. Cho, S. Y. Lee and K. Kang, Nat. Commun., 2021, 12, 3369 CrossRef CAS PubMed
- S. Wu, F. Zhang and Y. Tang, Adv. Sci., 2018, 5, 1701082 CrossRef PubMed
- S. J. R. Prabakar, K. S. Sohn and M. Pyo, ACS Appl. Mater. Interfaces, 2020, 12, 16481–16489 CrossRef CAS PubMed
- M. Wang, C. Jiang, S. Zhang, X. Song, Y. Tang and H. M. Cheng, Nat. Chem., 2018, 10, 667–672 CrossRef CAS PubMed
- R. Kushwaha, C. Jain, P. Shekhar, D. Rase, R. Illathvalappil, D. Mekan, A. Camellus, C. P. Vinod and R. Vaidhyanathan, Adv. Energy Mater., 2023, 13, 2301049 CrossRef CAS
- H. Zhang, L. Zhong, J. Xie, F. Yang, X. Liu and X. Lu, Adv. Mater., 2021, 33, e2101857 CrossRef PubMed
- Q. Guo, K. i Kim, H. Jiang, L. Zhang, C. Zhang, D. Yu, Q. Ni, X. Chang, T. Chen, H. Xia and X. Ji, Adv. Funct. Mater., 2020, 30, 2002825 CrossRef CAS
- X. Wu, Y. Xu, C. Zhang, D. P. Leonard, A. Markir, J. Lu and X. Ji, J. Am. Chem. Soc., 2019, 141, 6338–6344 CrossRef CAS PubMed
- I. A. Rodríguez-Pérez, L. Zhang, J. M. Wrogemann, D. M. Driscoll, M. L. Sushko, K. S. Han, J. L. Fulton, M. H. Engelhard, M. Balasubramanian, V. V. Viswanathan, V. Murugesan, X. Li, D. Reed, V. Sprenkle, M. Winter and T. Placke, Adv. Energy Mater., 2020, 10, 2001256 CrossRef
- L. Sun, Y. Yao, L. Dai, M. Jiao, B. Ding, Q. Yu, J. Tang and B. Liu, Energy Storage Mater., 2022, 47, 187–194 CrossRef
- A. Sethi, U. A. Kumar and V. M. Dhavale, ChemPhysChem, 2023, 24, e202300098 CrossRef CAS PubMed
- Y. Zheng, X. Xie, H. Ueno, T. Deng and W. Zheng, Adv. Energy Mater., 2024, 14, 2401914 CrossRef CAS
- L. Wang, Q. Tian, M. Li, X. Duan, J. Li, H. Weng and S. Xu, Chem. Phys. Lett., 2023, 833, 140887 CrossRef CAS
- G. Wang, M. Zhu, G. Chen, Z. Qu, B. Kohn, U. Scheler, X. Chu, Y. Fu, O. G. Schmidt and X. Feng, Adv. Mater., 2022, 34, e2201957 CrossRef PubMed
- K. Zhou, G. Liu, X. Zhu, G. Liu, X. Yu, Z. Guo and Y. Wang, Angew. Chem., Int. Ed., 2025, 64, e202413959 CrossRef CAS PubMed
- Y. Yang, D. Sabaghi, C. Liu, A. Dianat, D. Mucke, H. Qi, Y. Liu, M. Hambsch, Z. K. Xu, M. Yu, G. Cuniberti, S. C. B. Mannsfeld, U. Kaiser, R. Dong, Z. Wang and X. Feng, Angew. Chem., Int. Ed., 2024, 63, e202316299 CrossRef CAS PubMed
- X. Zhang, Y. Tang, F. Zhang and C. S. Lee, Adv. Energy Mater., 2016, 6, 1502588 CrossRef
- E. Zhang, W. Cao, B. Wang, X. Yu, L. Wang, Z. Xu and B. Lu, Energy Storage Mater., 2018, 11, 91–99 CrossRef
- M. Latha and J. Vatsala Rani, J. Electrochem. Soc., 2019, 167, 070501 CrossRef
- W. Ma, L. W. Luo, X. Huang, P. Dong, Y. Chen, C. Zhang, F. Huang, J. X. Jiang and Y. Cao, Adv. Energy Mater., 2022, 13, 2205879 Search PubMed
- L. W. Luo, C. Zhang, W. Ma, C. Han, X. Ai, Y. Chen, Y. Xu, X. Ji and J. X. Jiang, Adv.
Mater., 2024, 36, e2406106 CrossRef PubMed
- S. Zhao, B. Zhang, L. Li, P. Zhang, G. Li, Z. Zhu, Y. Choi, L. Dong, M. Luo and S. Guo, J. Am. Chem. Soc., 2025, 147, 669–677 CrossRef PubMed
- M. Chen, G. Chen, C. Sun, X. Li, M. Zhang, H. Hua, J. Zhao and Y. Yang, Angew. Chem., Int. Ed., 2025, 64, e202502005 CrossRef CAS PubMed
- L. Xiang, Q. Xu, H. Zhang, S. Geng, R. Cui, T. Xiao, P. Chen, L. Wu, W. Yu, H. Peng, Y. Mai and H. Sun, Angew. Chem., Int. Ed., 2023, 62, e202312001 CrossRef CAS PubMed
- Z. Yan, Q.-H. Yang and C. Yang, J. Mater. Chem. A, 2024, 12, 24746–24760 RSC
- G. Zhu, P. Liang, C. L. Huang, C. C. Huang, Y. Y. Li, S. C. Wu, J. Li, F. Wang, X. Tian, W. H. Huang, S. K. Jiang, W. H. Hung, H. Chen, M. C. Lin, B. J. Hwang and H. Dai, J. Am. Chem. Soc., 2022, 144, 22505–22513 CrossRef CAS PubMed
- P. Li, C. Ma, Y. Wang, S. Zhai, G. Ma, D. Kong and Z. Li, Adv. Mater., 2025, 37, e2418990 CrossRef PubMed
- B. Yuan, Q. Xu, S. Tang, S. Geng, Q. Chen, Y. Wang, X. Zhao, C. Zhang, S. Wang, Z. Ouyang and H. Sun, Sci. China: Chem., 2025, 1–7 CAS
- B. B. Peterson, E. M. Andrews, F. Hung and J. C. Flake, J. Power Sources, 2021, 492, 229658 CrossRef CAS
- X. Li, Y. Wang, J. Lu, P. Li, Z. Huang, G. Liang, H. He and C. Zhi, Sci. Adv., 2024, 10, eadl0587 CrossRef CAS PubMed
- C. Peng, C. Zheng, J. Jia, Z. Huang, Y. Cao, Q. Wang, B. Shi, S. Hu, X. Liang, T. Yi, G. Li and W. Tang, ACS Appl. Mater. Interfaces, 2025, 17, 7773–7783 CrossRef CAS PubMed
- M. Ding, R. Shi, J. Qu and M. Tong, Chin. Chem. Lett., 2023, 34, 108248 CrossRef CAS
- X. Li, Y. Wang, J. Lu, S. Li, P. Li, Z. Huang, G. Liang, H. He and C. Zhi, Angew. Chem., Int. Ed., 2023, 62, e202310168 CrossRef CAS PubMed
- J. Gu, C. Dong, Y. Zhu, H. Liu, J. Ji, Y. Yu, C. Ma, C. Zhou, L. Mai and X. Xu, Angew. Chem., Int. Ed., 2025, e202507184 CAS
- L. Feng, Y. Gong and J. Lin, Chem. Eng. J., 2024, 493, 152612 CrossRef CAS
- C. Ma, W. Feng, D. Kong, X. Wei, X. Gong, J. Yang, J. Han and L. Zhi, Small, 2024, 20, e2310978 CrossRef PubMed
- W. Feng, J. Yang, X. Wei, C. Ma, J. Han, G. Ma, H. Wang, D. Kong and L. Zhi, Angew. Chem., Int. Ed., 2025, e202503752 CAS
- F. Wang, H. Yang, J. Zhang, P. Zhang, G. Wang, X. Zhuang, G. Cuniberti and X. Feng, Adv. Mater., 2018, 30, e1800028 CrossRef PubMed
- H. Wang, R. Wang, Z. Song, H. Zhang, H. Zhang, Y. Wang and X. Li, J. Mater. Chem. A, 2019, 7, 13050–13059 RSC
- P. Liang, G. Zhu, W. Wang, C. L. Huang, S. C. Wu, J. Zhou, X. Zhou, Y. Wu, S. Wang, M. Wang, L. Zhang, C. C. Ming, J. Li, F. Wang, M. Sun, Y. Y. Li, B. J. Hwang and H. Dai, J. Am. Chem. Soc., 2025, 147, 18541–18549 CrossRef CAS PubMed
- X. Dong, L. Chen, J. Liu, S. Haller, Y. Wang and Y. Xia, Sci. Adv., 2016, 2, e1501038 CrossRef PubMed
- D. Gong, B. Wang, J. Zhu, R. Podila, A. M. Rao, X. Yu, Z. Xu and B. Lu, Adv. Energy Mater., 2017, 7, 1601885 CrossRef
- H. Tian, H. Shao, Y. Chen, X. Fang, P. Xiong, B. Sun, P. H. L. Notten and G. Wang, Nano Energy, 2019, 57, 692–702 CrossRef CAS
- F. Wang, Z. Liu, C. Yang, H. Zhong, G. Nam, P. Zhang, R. Dong, Y. Wu, J. Cho, J. Zhang and X. Feng, Adv. Mater., 2020, 32, e1905361 CrossRef PubMed
- M. Qian, Z. Xu, Z. Wang, B. Wei, H. Wang, S. Hu, L. M. Liu and L. Guo, Adv. Mater., 2020, 32, e2004835 CrossRef PubMed
- H. Zhang, Z. Shang, S. Gao, B. Song, W. Zhang, R. Cao, S. Jiao, Y. Cheng, Q. Chen and K. Lu, J. Mater. Chem. A, 2022, 10, 11325–11331 RSC
- T. Zhang, F. Wei, Y. Wu, W. Li, L. Huang, J. Fu, C. Jing, J. Cheng and S. Liu, Adv. Sci., 2023, 10, e2301918 CrossRef PubMed
- J. Xu, Y. Qiu, J. Yang, H. Li, P. Han, Y. Jin, H. Liu, B. Sun and G. Wang, Adv. Funct. Mater., 2023, 34, 2306206 CrossRef
- M. Xia, Y. Feng, J. Wei, A. M. Rao, J. Zhou and B. Lu, Adv. Funct. Mater., 2022, 32, 2205879 CrossRef CAS
- S. Geng, X. Zhao, Q. Xu, B. Yuan, Y. Wang, M. Liao, L. Ye, S. Wang, Z. Ouyang, L. Wu, Y. Wang, C. Ma, X. Zhao and H. Sun, Nat. Commun., 2024, 15, 944 CrossRef CAS PubMed
- L. Guo, A. Chen, A. Wang, Z. Hu, H. Zhang and J. Luo, J. Am. Chem. Soc., 2024, 146, 26855–26862 CrossRef CAS PubMed
- G. Liang, B. Liang, A. Chen, J. Zhu, Q. Li, Z. Huang, X. Li, Y. Wang, X. Wang, B. Xiong, X. Jin, S. Bai, J. Fan and C. Zhi, Nat. Commun., 2023, 14, 1856 CrossRef CAS PubMed
- S. Xu, L. Shen, X. Wang, S. Gu, W. Sun and Y. Huang, Chem. Commun., 2024, 60, 1027–1030 RSC
- N. Chen, W. Wang, Y. Ma, M. Chuai, X. Zheng, M. Wang, Y. Xu, Y. Yuan, J. Sun, K. Li, Y. Meng, C. Shen and W. Chen, Small Methods, 2024, 8, e2201553 CrossRef PubMed
- S. Chen, Y. Ying, S. Wang, L. Ma, H. Huang, X. Wang, X. Jin, S. Bai and C. Zhi, Angew. Chem., Int. Ed., 2023, 62, e202301467 CrossRef CAS PubMed
- F. Yu, L. Pang, X. Wang, E. R. Waclawik, F. Wang, K. Ostrikov and H. Wang, Energy Storage Mater., 2019, 19, 56–61 CrossRef
- L. Hu, C. Dai, Y. Zhu, X. Hou, Z. Liu, X. Geng, H. Wang, J. Chen, N. Sun, Q. Rong, Y. Zhu, X. He and Y. Lin, Energy Environ. Sci., 2024, 17, 5552–5562 RSC
- S. Chen, C. Peng, D. Zhu and C. Zhi, Adv. Mater., 2024, 36, e2409810 CrossRef PubMed
- X. Li, M. Li, Z. Huang, G. Liang, Z. Chen, Q. Yang, Q. Huang and C. Zhi, Energy Environ. Sci., 2021, 14, 407–413 RSC
- C. Bai, F. Cai, L. Wang, S. Guo, X. Liu and Z. Yuan, Nano Res., 2018, 11, 3548–3554 CrossRef CAS
- W. Shang, J. Zhu, Y. Liu, L. Kang, S. Liu, B. Huang, J. Song, X. Li, F. Jiang, W. Du, Y. Gao and H. Luo, ACS Appl. Mater. Interfaces, 2021, 13, 24756–24764 CrossRef CAS PubMed
- Y. Zou, T. Liu, Q. Du, Y. Li, H. Yi, X. Zhou, Z. Li, L. Gao, L. Zhang and X. Liang, Nat. Commun., 2021, 12, 170 CrossRef CAS PubMed
- W. Li, H. Xu, H. Zhang, F. Wei, T. Zhang, Y. Wu, L. Huang, J. Fu, C. Jing, J. Cheng and S. Liu, Energy Environ. Sci., 2023, 16, 4502–4510 RSC
- S. Chai, J. Yao, Y. Wang, J. Zhu and J. Jiang, Chem. Eng. J., 2022, 439, 135676 CrossRef CAS
- Q. Liu, C. Xia, C. He, W. Guo, Z. P. Wu, Z. Li, Q. Zhao and B. Y. Xia, Angew. Chem., Int. Ed., 2022, 61, e202210567 CrossRef CAS PubMed
- W. Wu, C. Li, Z. Wang, H.-Y. Shi, Y. Song, X.-X. Liu and X. Sun, Chem. Eng. J., 2022, 428, 131283 CrossRef CAS
- H. Xu, R. Zhang, D. Luo, J. Wang, K. Huang, J. Chi, H. Dou, X. Zhang and G. Sun, Energy Storage Mater., 2023, 63, 103019 CrossRef
- S. Bi, H. Wang, Y. Zhang, M. Yang, Q. Li, J. Tian and Z. Niu, Angew. Chem., Int. Ed., 2023, 62, e202409071 Search PubMed
- J. Gong, H. Zhang, X. Liang, P. Li, Y. Liu, X. Li, C. Zhi, Z. Zhu, X. C. Zeng, N. Li and J. Xu, Adv. Funct. Mater., 2024, 34, 2411137 CrossRef CAS
- Z.-T. Yu, Z.-S. Gong, R.-H. Wen, Y.-J. Hou, Z.-Q. Luo, Z.-H. Yuan and N. Zhang, Rare Met., 2024, 43, 6351–6361 CrossRef CAS
- R. Zhao, K. Lu, M. Pasha, R. Kuang, H. Zhang and S. Lu, J. Mater. Chem. A, 2024, 12, 24468–24476 RSC
- J. Rouxel, Chem. – Eur. J., 2006, 2, 1053–1059 CrossRef
- S. Lv, T. Fang, Z. Ding, Y. Wang, H. Jiang, C. Wei, D. Zhou, X. Tang and X. Liu, ACS Nano, 2022, 16, 20389–20399 CrossRef CAS PubMed
- C. Xie, C. Wang, Y. Xu, T. Li, Q. Fu and X. Li, Nat. Energy, 2024, 9, 714–724 CrossRef CAS
- W. Sun, C. Zhou, Y. Fan, Y. He, H. Zhang, Z. Quan, H. Kong, F. Fu, J. Qin, Y. Shen and H. Chen, Angew. Chem., Int. Ed., 2023, 62, e202300158 CrossRef CAS PubMed
- C. Zhang, Y. Huang, X. Xu, Z. Chen, G. Xiao, Y. Zhong, X. Wang, C. Gu and J. Tu, Energy Environ. Sci., 2024, 17, 4090–4103 RSC
- J. Sun, B. Luo and H. Li, Adv. Energy Sustainable Res., 2022, 3, 2100191 CrossRef
- Q. Zhao, G. Zhang, S. Yang and Y. Li, J. Energy Storage, 2025, 108, 236168 Search PubMed
- Y. Shao, M. F. El-Kady, J. Sun, Y. Li, Q. Zhang, M. Zhu, H. Wang, B. Dunn and R. B. Kaner, Chem. Rev., 2018, 118, 9233–9280 CrossRef CAS PubMed
- D. Li, J. Qiu, Y.-J. Zhu, H. Zhang, M.-G. Ma, X. Liu and H. Li, Nano Energy, 2024, 132, 110345 CrossRef
- X. Chen, R. Paul and L. Dai, Natl. Sci. Rev., 2017, 4, 453–489 CrossRef CAS
- C. Xiao, H. Wang, R. Usiskin, P. A. van Aken and J. Maier, Science, 2024, 386, 407–413 CrossRef CAS PubMed
- Y. Wang, Y. Song and Y. Xia, Chem. Soc. Rev., 2016, 45, 5925–5950 RSC
- S. Fleischmann, Y. Zhang, X. Wang, P. T. Cummings, J. Wu, P. Simon, Y. Gogotsi, V. Presser and V. Augustyn, Nat. Energy, 2022, 7, 222–228 CrossRef CAS
- X. Wang, T. S. Mathis, K. Li, Z. Lin, L. Vlcek, T. Torita, N. C. Osti, C. Hatter, P. Urbankowski, A. Sarycheva, M. Tyagi, E. Mamontov, P. Simon and Y. Gogotsi, Nat. Energy, 2019, 4, 241–248 CrossRef CAS
- S. Fleischmann, J. B. Mitchell, R. Wang, C. Zhan, D. E. Jiang, V. Presser and V. Augustyn, Chem. Rev., 2020, 120, 6738–6782 CrossRef CAS PubMed
- J. Li, C. Liu, R. Momen, J. Cai, X. Hu, F. Zhu, H. Liu, L. Xu, W. Deng, H. Hou, G. Zou and X. Ji, Coord. Chem. Rev., 2024, 517, 216018 CrossRef CAS
- H. Wang, C. Zhu, D. Chao, Q. Yan and H. J. Fan, Adv. Mater., 2017, 29, 1702093 CrossRef PubMed
- J. Kim, M. S. Choi, K. H. Shin, M. Kota, Y. Kang, S. Lee, J. Y. Lee and H. S. Park, Adv. Mater., 2019, 31, e1803444 CrossRef PubMed
- S. Zhao, G. Li, B. Zhang, T. Li, M. Luo, B. Sun, G. Wang and S. Guo, Joule, 2024, 8, 922–943 CrossRef CAS
- S. Liu, L. Kang, J. Zhang, S. C. Jun and Y. Yamauchi, ACS Energy Lett., 2021, 6, 4127–4154 CrossRef CAS
- S. Wang, L. Li, W. He, Y. Shao, Y. Li, Y. Wu and X. Hao, Adv. Funct. Mater., 2020, 30, 2000350 CrossRef CAS
- X. Wang, Q. Li, L. Zhang, Z. Hu, L. Yu, T. Jiang, C. Lu, C. Yan, J. Sun and Z. Liu, Adv. Mater., 2018, 30, e1800963 CrossRef PubMed
- Y. Li, Y. Yang, P. Zhou, T. Gao, Z. Xu, S. Lin, H. Chen, J. Zhou and S. Guo, Matter, 2019, 1, 893–910 CrossRef
- X. Liu, G. A. Elia, B. S. Qin, H. Zhang, P. Ruschhaupt, S. Fang, A. Varzi and S. Passerini, ACS Energy Lett., 2019, 4, 2675–2682 CrossRef CAS
- Y.-Y. Wang, Z.-Y. Wang, Y.-J. Xu, W.-H. Chen, G.-S. Shao and B.-H. Hou, Energy Environ. Sci., 2024, 17, 6811–6820 RSC
- X. Hu, G. Zhong, J. Li, Y. Liu, J. Yuan, J. Chen, H. Zhan and Z. Wen, Energy Environ. Sci., 2020, 13, 2431–2440 RSC
- D. Han, Z. Weng, P. Li, Y. Tao, C. Cui, L. Zhang, W. Lin, Y. Gao, D. Kong and Q.-H. Yang, Energy Storage Mater., 2019, 18, 133–138 CrossRef
- Y. Yi, Z. Zeng, X. Lian, S. Dou and J. Sun, Small, 2022, 18, e2107139 CrossRef PubMed
- C. Cui, H. Wang, M. Wang, X. Ou, Z. Wei, J. Ma and Y. Tang, Small, 2019, 15, e1902659 CrossRef PubMed
- H. Li, Y. Gong, H. Zhou, J. Li, K. Yang, B. Mao, J. Zhang, Y. Shi, J. Deng, M. Mao, Z. Huang, S. Jiao, Y. Kuang, Y. Zhao and S. Luo, Nat. Commun., 2023, 14, 6407 CrossRef CAS PubMed
- L. Jin, X. Guo, R. Gong, J. Zheng, Z. Xiang, C. Zhang and J. P. Zheng, Energy Storage Mater., 2019, 23, 409–417 CrossRef
- H. Qin, H. Chao, M. Zhang, Y. Huang, H. Liu, J. Cheng, L. Cao, Q. Xu, L. Guan, X. Teng, Y. Li, K. Wang, H. Guo, H. Hu and M. Wu, Carbon, 2021, 180, 110–117 CrossRef CAS
- Y. Li, C. Pan, P. Kamdem and X.-J. Jin, Energy Fuels, 2020, 34, 10120–10130 CrossRef CAS
- X. Hu, Y. Liu, J. Chen, L. Yi, H. Zhan and Z. Wen, Adv. Energy Mater., 2019, 9, 1901533 CrossRef CAS
- T. Zhang, Z. Mao, X. Shi, J. Jin, B. He, R. Wang, Y. Gong and H. Wang, Energy Environ. Sci., 2022, 15, 158–168 RSC
- Y. Z. Li, H. W. Wang, L. B. Wang, R. Wang, B. B. He, Y. S. Gong and X. L. Hu, Energy Storage Mater., 2019, 23, 95–104 CrossRef
- C. Wang, W. Wu, C. Zhao, T. Liu, L. Wang and J. Zhu, Electrochim. Acta, 2022, 421, 140438 CrossRef CAS
- P. Wang, B. Yang, G. Zhang, L. Zhang, H. Jiao, J. Chen and X. Yan, Chem. Eng. J., 2018, 353, 453–459 CrossRef CAS
- Y. Song, Y. Peng, H. Li, X. Sun, L. Li, C. Zhang and F. Yin, Chem. Eng. J., 2022, 447, 137450 CrossRef CAS
- B. Yang, J. Chen, S. Lei, R. Guo, H. Li, S. Shi and X. Yan, Adv. Energy Mater., 2018, 8, 1702409 CrossRef
- X. Shi, H. Wang, Z. Xie, Z. Mao, T. Zhang, J. Jin, B. He, R. Wang, Y. Gong and H. J. Fan, Adv. Mater., 2024, 36, e2406794 CrossRef PubMed
- G. Li, Y. Huang, Z. Yin, H. Guo, Y. Liu, H. Cheng, Y. Wu, X. Ji and J. Wang, Energy Storage Mater., 2020, 24, 304–311 CrossRef
- X. Li, M. Sun, C. Xu, X. Zhang, G. Wang, Y. Tian and G. Zhu, Adv. Funct. Mater., 2023, 33, 2300460 CrossRef CAS
- Y. Yuan, Z. Chen, H. Yu, X. Zhang, T. Liu, M. Xia, R. Zheng, M. Shui and J. Shu, Energy Storage Mater., 2020, 32, 65–90 CrossRef
- S. Chen, L. Shen, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1605650 CrossRef PubMed
- X. Hu, G. Wang, J. Li, J. Huang, Y. Liu, G. Zhong, J. Yuan, H. Zhan and Z. Wen, Energy Environ. Sci., 2021, 14, 4564–4573 RSC
- S. Luo, T. Yuan, L. Soule, J. Ruan, Y. Zhao, D. Sun, J. Yang, M. Liu and S. Zheng, Adv. Funct. Mater., 2019, 30, 1908309 CrossRef
- H. Li, J. Lang, S. Lei, J. Chen, K. Wang, L. Liu, T. Zhang, W. Liu and X. Yan, Adv. Funct. Mater., 2018, 28, 1800757 CrossRef
- S. Zhao, L. Dong, B. Sun, K. Yan, J. Zhang, S. Wan, F. He, P. Munroe, P. H. L. Notten and G. Wang, Small, 2020, 16, e1906131 CrossRef PubMed
- X. Xiao, X. Duan, Z. Song, X. Deng, W. Deng, H. Hou, R. Zheng, G. Zou and X. Ji, Adv. Funct. Mater., 2022, 32, 2110476 CrossRef CAS
- Z. Fan, C. Wei, L. Yu, Z. Xia, J. Cai, Z. Tian, G. Zou, S. X. Dou and J. Sun, ACS Nano, 2020, 14, 867–876 CrossRef PubMed
- Y. Z. Fang, R. Hu, K. Zhu, K. Ye, J. Yan, G. Wang and D. Cao, Adv. Funct. Mater., 2020, 30, 2005663 CrossRef CAS
- R. Bi, N. Xu, H. Ren, N. Yang, Y. Sun, A. Cao, R. Yu and D. Wang, Angew. Chem., Int. Ed., 2020, 59, 4865–4868 CrossRef CAS PubMed
- J. Yuan, D. Pan, J. Chen, Y. Liu, J. Yu, X. Hu, H. Zhan and Z. Wen, Adv. Mater., 2024, 36, e2408923 CrossRef PubMed
- W. Liu, X. Zhang, Y. Xu, L. Wang, Z. Li, C. Li, K. Wang, X. Sun, Y. An, Z. S. Wu and Y. Ma, Adv. Funct. Mater., 2022, 32, 2202342 Search PubMed
- C. Cheng, D. Wu, T. Gong, Y. Yan, Y. Liu, W. Ji, L. Hou and C. Yuan, Adv. Energy Mater., 2023, 13, 2302107 CrossRef CAS
- J. Cui, S. Yao, Z. Lu, J.-Q. Huang, W. G. Chong, F. Ciucci and J.-K. Kim, Adv.
Energy Mater., 2018, 8, 1702488 CrossRef
- Y. Su, Y.-F. Li, S.-G. Gong, Y.-H. Song, B. Li, X.-L. Wu, J.-P. Zhang, D.-T. Liu, C.-L. Shao and H.-Z. Sun, Appl. Surf. Sci., 2023, 610, 155494 CrossRef CAS
- X. Zhao, W. Cai, Y. Yang, X. Song, Z. Neale, H.-E. Wang, J. Sui and G. Cao, Nano Energy, 2018, 47, 224–234 CrossRef CAS
- J. Ge, B. Wang, J. Wang, Q. Zhang and B. Lu, Adv. Energy Mater., 2019, 10, 1903277 CrossRef
- M. Chen, L. Wang, X. Sheng, T. Wang, J. Zhou, S. Li, X. Shen, M. Zhang, Q. Zhang, X. Yu, J. Zhu and B. Lu, Adv. Funct. Mater., 2020, 30, 2004247 CrossRef CAS
- Y. Yuan, C. Wang, K. Lei, H. Li, F. Li and J. Chen, ACS Cent. Sci., 2018, 4, 1261–1265 CrossRef CAS PubMed
- A. Chojnacka, X. Pan, C. Bachetzky, E. Brunner and F. Béguin, Energy Storage Mater., 2022, 51, 719–732 CrossRef
- Z.-H. Lin, X.-Q. Qiu, X.-H. Zu, X.-S. Zhang, L. Zhong, S.-R. Sun, S.-H. Hao, Y.-J. Sun and W.-L. Zhang, Rare Met., 2023, 43, 1037–1047 Search PubMed
- Q. Chen, J. Jin, M. Song, X. Zhang, H. Li, J. Zhang, G. Hou, Y. Tang, L. Mai and L. Zhou, Adv. Mater., 2022, 34, e2107992 CrossRef PubMed
- J.-C. Liu, K. Wang, Y. Sun, H. Li, X. Han, X. Duan, Z.-H. Huang and T. Ma, Nano Energy, 2025, 136, 110764 CrossRef CAS
- Q. Chen, Z. Tang, H. Li, W. Liang, Y. Zeng, J. Zhang, G. Hou and Y. Tang, ACS Appl. Mater. Interfaces, 2024, 16, 18824–18832 CrossRef CAS PubMed
- L. Chen, X. Zhang, Z. Wang, X. Kong, J. Zhang, J. Zeng and D. Wang, Chem. Eng. J., 2024, 500, 157472 CrossRef CAS
- Q. Chen, M. Song, X. Zhang, J. Zhang, G. Hou and Y. Tang, J. Mater. Chem. A, 2022, 10, 15614–15622 RSC
- J. Dai, X. Qi, L. Xia, Q. Xue, L. Luo, X. Wang, C. Yang, D. Li, H. Xie, A. Cabot, L. Dai and Y. Xu, Adv. Funct. Mater., 2022, 33, 2212440 CrossRef
- Q. Chen, H. Li, X. Lou, J. Zhang, G. Hou, J. Lu and Y. Tang, Adv. Funct. Mater., 2023, 33, 2214920 CrossRef CAS
- Q. Wu, Y. Zhang, G. Liu, X. Cui, S. Tao, H. Jiang, Y. Lin, R. Peng, X. Zhang, Z. Huang, Y. Song, Y. Ding, S. M. Akhlaq, Y. Wu, K. Tao, E. Xie, Z. Zhang and Z.-S. Wu, Energy Storage Mater., 2024, 70, 103474 CrossRef
- D. Wang, J. Sun and L. Chen, ChemSusChem, 2023, 16, e202300207 CrossRef CAS PubMed
- Y. Liang, H. Zhang, M. Huo, X. Zhang, K. Qin, H. Wang, Q. Li, X. Zhao, Z. Xing, J. Chang and G. Zhu, Adv. Mater., 2025, 37, e2415545 CrossRef PubMed
- M. Gao, Z. Wang, Z. Liu, Y. Huang, F. Wang, M. Wang, S. Yang, J. Li, J. Liu, H. Qi, P. Zhang, X. Lu and X. Feng, Adv. Mater., 2023, 35, e2305575 CrossRef PubMed
- Y. Huang, M. Gao, Y. Fu, J. Li, F. Wang, S. Yang, M. Wang, Z. Qian, X. Lu, P. Zhang and R. Wang, Energy Storage Mater., 2024, 70, 103522 CrossRef
- Q. Yang, F. Mo, Z. Liu, L. Ma, X. Li, D. Fang, S. Chen, S. Zhang and C. Zhi, Adv. Mater., 2019, 31, e1901521 Search PubMed
- J. Du, Q. Han, Y. Chen, M. Peng, L. Xie and A. Chen, Angew. Chem., Int. Ed., 2024, 63, e202411066 CrossRef CAS PubMed
- R. Kang, D. Zhang, H. Wang, B. Zhang, X. Zhang, G. Chen, Y. Du and J. Zhang, Energy Environ. Sci., 2024, 17, 7135–7146 RSC
- P. Wang, X. Xie, Z. Xing, X. Chen, G. Fang, B. Lu, J. Zhou, S. Liang and H. J. Fan, Adv. Energy Mater., 2021, 11, 2101158 CrossRef CAS
- L. Dong, X. Ma, Y. Li, L. Zhao, W. Liu, J. Cheng, C. Xu, B. Li, Q.-H. Yang and F. Kang, Energy Storage Mater., 2018, 13, 96–102 CrossRef
- M. Sun, Z. Zhang, S. Fu, Y. Zhang, R. Wang, H. Mu, C. Lian, W. Wang and G. Wang, J. Energy Chem., 2024, 94, 477–485 CrossRef CAS
- Y. G. Lee and G. H. An, ACS Appl. Mater. Interfaces, 2020, 12, 41342–41349 CrossRef CAS PubMed
- J. Yin, W. Zhang, W. Wang, N. A. Alhebshi, N. Salah and H. N. Alshareef, Adv. Energy Mater., 2020, 10, 2001705 CrossRef CAS
- Y. Lu, G. Zhou, Z. Zhang, C. Li, Q. Dong, Y. Su and W. Chen, Chem. Eng. J., 2025, 508, 161085 CrossRef CAS
- C. Li, L. Yan, X. Li, M. Wang, J. Kong, W. Bao and L. Chang, J. Energy Storage, 2024, 86, 111220 CrossRef
- W. Zhang, J. Yin, W. Jian, Y. Wu, L. Chen, M. Sun, U. Schwingenschlögl, X. Qiu and H. N. Alshareef, Nano
Energy, 2022, 103, 107827 CrossRef CAS
- H. Liu, D. Tong, L. Peng, L. Huang, Z. Gu, Z. Sheng, F. Zhang, Y. Zhang, Z. Hu, T. Qian and H. Zhu, ACS Appl. Energy Mater., 2023, 6, 6752–6759 CrossRef CAS
- S. Wang, F. Wei, W. Geng, Z. Ren and Y. Lv, Chem. Eng. J., 2025, 515, 163462 CrossRef CAS
- R. X. Fei, H. W. Wang, Q. Wang, R. Y. Qiu, S. S. Tang, R. Wang, B. B. He, Y. S. Gong and H. J. Fan, Adv. Energy Mater., 2020, 10, 2002741 CrossRef CAS
- Q. Guo, J. Liu, C. Bai, N. Chen and L. Qu, ACS Nano, 2021, 15, 16533–16541 CrossRef CAS PubMed
- S. Deng, J. Li, G. M. Zewdie, X. Jiang, M. Ji, J. Shen, G. Gao, G. Wu, Z. Bao and H. S. Kang, Adv. Funct. Mater., 2023, 34, 2311259 CrossRef
- F. Li, Y.-l Liu, G.-G. Wang, S.-Y. Zhang, D.-Q. Zhao, K. Fang, H.-Y. Zhang and H. Y. Yang, Chem. Eng. J., 2022, 435, 135052 CrossRef CAS
- J. Azadmanjiri, J. Regner, L. Dekanovsky, B. Wu, J. Luxa and Z. Sofer, Small, 2024, 20, e2305972 CrossRef PubMed
- K. Fang, J. Zhang, J. Yu, Y. Hu, J. Wang, Z. Wang and B. Zhao, Chem. Eng. J., 2025, 513, 162824 CrossRef CAS
- W. Zhang, C. Zeng, M. Zhang, C. Zhao, D. Chao, G. Zhou and C. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202413728 CrossRef CAS PubMed
- S. Wang, Q. Wang, W. Zeng, M. Wang, L. Ruan and Y. Ma, Nanomicro Lett., 2019, 11, 70 Search PubMed
- K. Zhang, Y. Bai, L. Wang, Y. Gao, X. Li, X. Yang and W. Lu, Langmuir, 2024, 40, 26561–26569 CrossRef CAS PubMed
- B. D. Boruah, B. Wen, S. Nagane, X. Zhang, S. D. Stranks, A. Boies and M. De Volder, ACS Energy Lett., 2020, 5, 3132–3139 CrossRef CAS
- Y. Xu, X. Yang, X. Li, Y. Gao, L. Wang and W. Lü, J. Power Sources, 2024, 623, 235399 CrossRef CAS
- C. Wang, Z. Pei, Q. Meng, C. Zhang, X. Sui, Z. Yuan, S. Wang and Y. Chen, Angew. Chem., Int. Ed., 2021, 60, 990–997 CrossRef CAS PubMed
- Z. Huang, T. Wang, H. Song, X. Li, G. Liang, D. Wang, Q. Yang, Z. Chen, L. Ma, Z. Liu, B. Gao, J. Fan and C. Zhi, Angew. Chem., Int. Ed., 2021, 60, 1011–1021 CrossRef CAS PubMed
- C. C. Hou, Y. Wang, L. Zou, M. Wang, H. Liu, Z. Liu, H. F. Wang, C. Li and Q. Xu, Adv. Mater., 2021, 33, e2101698 CrossRef PubMed
- J. Li, Y. Lou, S. Zhou, Y. Chen, X. Zhao, A. Azizi, S. Lin, L. Fu, C. Han, Z. Su and A. Pan, Angew. Chem., Int. Ed., 2024, 63, e202406906 CrossRef CAS PubMed
- W. Pan, Y. Wang, X. Zhao, Y. Zhao, X. Liu, J. Xuan, H. Wang and D. Y. C. Leung, Adv. Funct. Mater., 2021, 31, 2008783 CrossRef CAS
- Y. Tian, R. Amal and D.-W. Wang, Front. Energy Res., 2016, 4, 34 Search PubMed
- W. Wang, L. Gao, Z. Kong, B. Ma, M. Han, G. Wang and C. Li, Adv. Mater., 2023, 35, e2303353 CrossRef PubMed
- R. Wang, W. Wang, M. Sun, Y. Hu and G. Wang, Angew. Chem., Int. Ed., 2024, 63, e202317154 CrossRef CAS PubMed
- C. Shin, L. Yao, S.-Y. Jeong and T. N. Ng, Sci. Adv., 2024, 10, eadf9951 CrossRef CAS PubMed
- K. Chen, J. Huang, J. Yuan, S. Qin, P. Huang, C. Wan, Y. You, Y. Guo, Q. Xu and H. Xie, Energy Storage Mater., 2023, 63, 102963 CrossRef
- W. Shi, Z. Song, W. Sun, Y. Liu, Y. Jiang, Q. Li and Q. An, Small, 2024, 20, e2308282 CrossRef PubMed
- X. Lu, L. Tao, K. Qu, A. Amardeep and J. Liu, Adv. Funct. Mater., 2023, 33, 2211736 CrossRef CAS
- S. N. Asmara, M. Z. Kufian, S. R. Majid and A. K. Arof, Electrochim. Acta, 2011, 57, 91–97 CrossRef CAS
- H. Zhang, K. Ye, K. Zhu, R. Cang, X. Wang, G. Wang and D. Cao, ACS Sustainable Chem. Eng., 2017, 5, 6727–6735 CrossRef CAS
- H. Zhang, D. Cao, X. Bai, H. Xie, X. Liu, X. Jiang, H. Lin and H. He, ACS Sustainable Chem. Eng., 2019, 7, 6113–6121 CrossRef CAS
- X. Cao, L. Wang, J. Chen and J. Zheng, ChemElectroChem, 2018, 5, 2789–2794 CrossRef CAS
- S. Li, J.-G. Zhang, Y.-Y. Yan, L.-L. Yu and J.-T. Zhao, J. Energy Storage, 2023, 59, 106456 CrossRef
- M. Li, Y. Ding, S. Zhang, Y. Sun, M. Liu, J. Zhao, B. Yin and T. Ma, Small Struct., 2023, 5, 2300371 CrossRef
- H. R. Park, G. Jang, J. S. Byun, Y. Park, W. I. Kim, S. J. Lee, Y. G. Chung, Y. M. Jung and H. S. Park, Adv. Funct. Mater., 2024, 35, 2417288 CrossRef
- M. Li, Y. Ding, S. Zhang, M. Liu, J. Li, Y. Sun, L. Zhu, H. Li, Z. G. Yu, Y. W. Zhang, H. Pan, B. Yin and T. Ma, Angew. Chem., Int. Ed., 2024, 63, e202412735 CrossRef CAS PubMed
- M. Li, Y. Ding, S. Zhang, M. Liu, Y. Sun, Y. Zhang, B. Yin and T. Ma, J. Mater. Chem. A, 2024, 12, 10269–10278 RSC
- S. K. Park, B. D. Boruah, A. Pujari, B. M. Kim and M. De Volder, Small, 2022, 18, e2202785 CrossRef PubMed
- D. Wang, X. Han and X. Zhang, J. Power Sources, 2024, 599, 234215 CrossRef CAS
- J. Wang, Y. Zhang, F. Qiao, Y. Jiang, R. Yu, J. Li, S. Lee, Y. Dai, F. Guo, P. Jiang, L. Zhang, Q. An, G. He and L. Mai, Adv. Mater., 2024, 36, e2403371 CrossRef PubMed
- Z. Li, K. Xiang, W. Xing, W. C. Carter and Y. M. Chiang, Adv. Energy Mater., 2014, 5, 1401410 CrossRef
- H. Ma, H. Chen, Y. Hu, B. Yang, J. Feng, Y. Xu, Y. Sun, H. Cheng, C. Li, X. Yan and L. Qu, Energy Environ. Sci., 2022, 15, 1131–1143 RSC
- Y. I. Kim, B. Kim, J. Baek, J.-H. Kim and J. Yoo, J. Electrochem. Soc., 2022, 169, 120521 CrossRef CAS
- S. Feng, L. Xing, K. Li, H. Wang, Q. An, L. Zhou and L. Mai, Small Methods, 2023, 7, e2300150 CrossRef PubMed
- Y. He, X. Pan, Z. Zhang, Q. Long, Q. Liu, S. Azat, Q. Abbas, G. Qiu, Z. Supiyeva and X. Chen, J. Power Sources, 2025, 114, 115902 Search PubMed
- R. Tang, Y. Guo, Y. Li, K. Li and Y. Gong, J. Energy Storage, 2024, 90, 111731 CrossRef
- W. Zhang, X. Zhang, Y. Song and G. Wang, Next Sustainable, 2024, 3, 100028 CrossRef
- X. Liu, W. Sun, X. Hu, J. Chen and Z. Wen, Chem. Eng. J., 2023, 474, 145355 CrossRef CAS
- J. Ma, N. A. Choudhury and Y. Sahai, Renewable Sustainable Energy Rev., 2010, 14, 183–199 CrossRef CAS
- I. Belhaj, M. Faria, B. Sljukic, V. Geraldes and D. M. F. Santos, Membranes, 2023, 13, 730 CrossRef CAS PubMed
- L. Zhu, C. Chen, T. Wu, X. Yu, H. Tian, F. Kong, Z. Chang, W. Luo, X. Cui and J. Shi, Chem, 2025, 11, 102331 CAS
- M. Papac, V. Stevanovic, A. Zakutayev and R. O'Hayre, Nat. Mater., 2021, 20, 301–313 CrossRef CAS PubMed
- H.-F. Wang and Q. Xu, Matter, 2019, 1, 565–595 CrossRef CAS
- C. Wang, Y. Yu, J. Niu, Y. Liu, D. Bridges, X. Liu, J. Pooran, Y. Zhang and A. Hu, Appl. Sci., 2019, 9, 2787 CrossRef CAS
- J. Liu, M. Zhou, K. Jin, J. Li, F. Meng and X. Wei, Battery Energy, 2023, 3, 20230049 CrossRef
- S. Liu, C. Li, M. J. Zachman, Y. Zeng, H. Yu, B. Li, M. Wang, J. Braaten, J. Liu, H. M. Meyer, M. Lucero, A. J. Kropf, E. E. Alp, Q. Gong, Q. Shi, Z. Feng, H. Xu, G. Wang, D. J. Myers, J. Xie, D. A. Cullen, S. Litster and G. Wu, Nat. Energy, 2022, 7, 652–663 CrossRef CAS
- F. Zhang, B. Zu, B. Wang, Z. Qin, J. Yao, Z. Wang, L. Fan and K. Jiao, Joule, 2025, 9, 101853 CrossRef CAS
- J. Yang, H. Xu, J. Li, K. Gong, F. Yue, X. Han, K. Wu, P. Shao, Q. Fu and Y. Zhu, Science, 2024, 385, 1115–1120 CrossRef CAS PubMed
- B. Peng, Z. Liu, L. Sementa, Q. Jia, Q. Sun, C. U. Segre, E. Liu, M. Xu, Y.-H. Tsai, X. Yan, Z. Zhao, J. Huang, X. Pan, X. Duan, A. Fortunelli and Y. Huang, Nat. Catal., 2024, 7, 818–828 CrossRef CAS
- J. L. Cohen, D. J. Volpe, D. A. Westly, A. Pechenik and H. D. Abruña, Langmuir, 2005, 21, 3544–3550 CrossRef CAS PubMed
- J. Kim, S. Sengodan, G. Kwon, D. Ding, J. Shin, M. Liu and G. Kim, ChemSusChem, 2014, 7, 2811–2815 CrossRef CAS PubMed
- K. Qiao, H. Liu, S. Huang, X. Zeng and D. Cao, Int. J. Hydrogen Energy, 2024, 50, 209–220 CrossRef CAS
- X. Liu, W. Sun, X. Hu, J. Chen and Z. Wen, Chem. Eng. J., 2023, 474, 145355 CrossRef CAS
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC
- K. Tamura and T. Kahara, J. Electrochem. Soc., 1976, 123, 776 CrossRef CAS
- S. Durga, K. Ponmani, S. Kiruthika and B. Muthukumaran, J. Electrochem. Sci. Technol., 2014, 5, 73–81 CrossRef CAS
- G. Wang, J. Chen, P. Cai, J. Jia and Z. Wen, J. Mater. Chem. A, 2018, 6, 17763–17770 RSC
- S. C. Amendola, P. Onnerud, M. T. Kelly, P. J. Petillo, S. L. Sharp-Goldman and M. Binder, J. Power Sources, 1999, 84, 130–133 CrossRef CAS
- U. B. Demirci and P. Miele, Energy Environ. Sci., 2009, 2, 627–637 RSC
- Y. Wang, P. He and H. Zhou, Energy Environ. Sci., 2010, 3, 1437–1446 RSC
- Y. Yang, X. Wu, M. Ahmad, F. Si, S. Chen, C. Liu, Y. Zhang, L. Wang, J. Zhang, J. L. Luo and X. Z. Fu, Angew. Chem., Int. Ed., 2023, 62, e202302950 CrossRef CAS PubMed
- F. Hu, K. Chen, Z. Lu, J. Gao, S. Lan, J. Chen, S. Ci and Z. Wen, Angew. Chem., Int. Ed., 2025, 64, e202504894 CrossRef CAS PubMed
- C. Rice, S. Ha, R. Masel, P. Waszczuk, A. Wieckowski and T. Barnard, J. Power Sources, 2002, 111, 83–89 CrossRef CAS
- Z. F. Pan, L. An, T. S. Zhao and Z. K. Tang, Progr. Energy Combust. Sci., 2018, 66, 141–175 CrossRef
- B. C. Ong, S. K. Kamarudin and S. Basri, Int. J. Hydrogen Energy, 2017, 42, 10142–10157 CrossRef CAS
- L. An, T. S. Zhao, R. Chen and Q. X. Wu, J. Power Sources, 2011, 196, 6219–6222 CrossRef CAS
- I. Chino, K. Hendrix, A. Keramati, O. Muneeb and J. L. Haan, Appl. Energy, 2019, 251, 113323 CrossRef CAS
- P. Wang, G. Wang, K. Chen, W. Pan, L. Yi, J. Wang, Q. Chen, J. Chen and Z. Wen, Nano Energy, 2023, 118, 108992 CrossRef CAS
- P. Wang, K. Chen, J. Chen, G. Wang, W. Pan and Z. Wen, Adv. Funct. Mater., 2024, 34, 2309509 CrossRef
- R. Tan, Y. Qin, M. Liu, H. Wang, R. Su, R. Xiao, J. Li, L. Hu, W. Gu and C. Zhu, Adv. Funct. Mater., 2023, 33, 2305673 CrossRef CAS
- O. Muneeb, I. Chino, A. Saenz and J. L. Haan, J. Power Sources, 2019, 413, 216–221 CrossRef CAS
- F. Wang, D. Ouyang and X. Zhao, Energy Convers. Manage., 2022, 255, 115343 CrossRef CAS
- Z. Wu, P. Zhu, Y. Huang, J. Yao, F. Yang, Z. Zhang and M. Ni, Chem. Rev., 2025, 125, 2184–2268 CrossRef CAS PubMed
- F. He, Y. Zhou, T. Hu, Y. Xu, M. Hou, F. Zhu, D. Liu, H. Zhang, K. Xu, M. Liu and Y. Chen, Adv. Mater., 2023, 35, e2209469 CrossRef PubMed
- B. Zhu, Key Eng. Mater., 2004, 280, 413–418 Search PubMed
- R. Tan, A. Wang, R. Malpass-Evans, R. Williams, E. W. Zhao, T. Liu, C. Ye, X. Zhou, B. P. Darwich, Z. Fan, L. Turcani, E. Jackson, L. Chen, S. Y. Chong, T. Li, K. E. Jelfs, A. I. Cooper, N. P. Brandon, C. P. Grey, N. B. McKeown and Q. Song, Nat. Mater., 2020, 19, 195–202 CrossRef CAS PubMed
- C. Duan, J. Tong, M. Shang, S. Nikodemski, M. Sanders, S. Ricote, A. Almansoori and R. O’Hayre, Science, 2015, 349, 1321–1326 CrossRef CAS PubMed
- R. Ren, Z. Wang, C. Xu, W. Sun, J. Qiao, D. W. Rooney and K. Sun, J. Mater. Chem. A, 2019, 7, 18365–18372 RSC
- M. Liang, Y. Song, D. Liu, L. Xu, M. Xu, G. Yang, W. Wang, W. Zhou, R. Ran and Z. Shao, Appl. Catal., B, 2022, 318, 121868 CrossRef CAS
- Q. Wang, J. Hou, Y. Fan, X.-A. Xi, J. Li, Y. Lu, G. Huo, L. Shao, X.-Z. Fu and J.-L. Luo, J. Mater. Chem. A, 2020, 8, 7704–7712 RSC
- D. Huan, L. Zhang, X. Li, Y. Xie, N. Shi, S. Xue, C. Xia, R. Peng and Y. Lu, ChemSusChem, 2020, 13, 4994–5003 CrossRef CAS PubMed
- Y. Song, J. Liu, Y. Wang, D. Guan, A. Seong, M. Liang, M. J. Robson, X. Xiong, Z. Zhang, G. Kim, Z. Shao and F. Ciucci, Adv. Energy Mater., 2021, 11, 2101899 CrossRef CAS
- Y. Yu, M. A. K. Y. Shah, H. Wang, X. Cheng, L. Guo, J. Huang, P. Lund and B. Zhu, Energy Mater. Adv., 2024, 5, 0081 CrossRef CAS
- G. W. Heise and E. A. Schumacher, Trans. Electrochem. Soc., 1932, 62, 383 CrossRef
- Q. Wang, S. Kaushik, X. Xiao and Q. Xu, Chem. Soc. Rev., 2023, 52, 6139–6190 RSC
- Z. Zheng, C. Wu, Q. Gu, K. Konstantinov and J. Wang, Energy Environ. Mater., 2021, 4, 158–177 CrossRef CAS
- B. Senthilkumar, Z. Khan, S. Park, I. Seo, H. Ko and Y. Kim, J. Power Sources, 2016, 311, 29–34 CrossRef CAS
- T. M. Gür, Energy Environ. Sci., 2018, 11, 2696–2767 RSC
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC
- K. W. Leong, Y. Wang, W. Pan, S. Luo, X. Zhao and D. Y. C. Leung, J. Power Sources, 2021, 506, 230144 CrossRef CAS
- Y. Wang, Y. Sun, W. Ren, D. Zhang, Y. Yang, J. Yang, J. Wang, X. Zeng and Y. NuLi, Energy Mater., 2022, 2, 200024 CrossRef CAS
- H. Zhang, D. J. Liu, K. Xu and Y. S. Meng, Adv. Mater., 2025, e2417757 CrossRef PubMed
- S. Hu, Z. Wang, J. Wang, S. Pang, B. Wang and M. Zhu, Coord. Chem. Rev., 2024, 517, 216045 CrossRef CAS
- X. Bi, R. Wang, Y. Yuan, D. Zhang, T. Zhang, L. Ma, T. Wu, R. Shahbazian-Yassar, K. Amine and J. Lu, Nano Lett., 2020, 20, 4681–4686 CrossRef CAS PubMed
- R. D. McKerracher, C. Ponce de Leon, R. G. A. Wills, A. A. Shah and F. C. Walsh, ChemPlusChem, 2014, 80, 323–335 CrossRef
- X. X. Wang, D. H. Guan, C. L. Miao, D. C. Kong, L. J. Zheng and J. J. Xu, J. Am. Chem. Soc., 2023, 145, 5718–5729 CrossRef CAS PubMed
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed
- K. Abraham and Z. Jiang, J. Electrochem. Soc., 1996, 143, 1 CrossRef CAS
- E. J. Askins, M. R. Zoric, M. Li, R. Amine, K. Amine, L. A. Curtiss and K. D. Glusac, Nat. Chem., 2023, 15, 1247–1254 CrossRef CAS PubMed
- L. Qin, N. Xiao, S. Zhang, X. Chen and Y. Wu, Angew. Chem., Int. Ed., 2020, 59, 10498–10501 CrossRef CAS PubMed
- P. He, Y. Wang and H. Zhou, J. Power Sources, 2011, 196, 5611–5616 CrossRef CAS
- T. Hashimoto and K. Hayashi, Electrochim. Acta, 2015, 182, 809–814 CrossRef CAS
- W. W. Yin, Z. Shadike, Y. Yang, F. Ding, L. Sang, H. Li and Z. W. Fu, Chem. Commun., 2015, 51, 2324–2327 RSC
- S. Xu, Y. Lu, H. Wang, H. D. Abruña and L. A. Archer, J. Mater. Chem. A, 2014, 2, 17723–17729 RSC
- S. Xing, Z. Zhang, Y. Dou, M. Li, J. Wu, Z. Zhang and Z. Zhou, CCS Chem., 2024, 6, 1810–1820 CrossRef CAS
- C. L. Bender, P. Hartmann, M. Vračar, P. Adelhelm and J. Janek, Adv. Energy Mater., 2014, 4, 1301863 CrossRef
- P. Hartmann, C. L. Bender, J. Sann, A. K. Durr, M. Jansen, J. Janek and P. Adelhelm, Phys. Chem. Chem. Phys., 2013, 15, 11661–11672 RSC
- Z. Jian, Y. Chen, F. Li, T. Zhang, C. Liu and H. Zhou, J. Power Sources, 2014, 251, 466–469 CrossRef CAS
- W. Liu, Q. Sun, Y. Yang, J. Y. Xie and Z. W. Fu, Chem. Commun. (Camb.), 2013, 49, 1951–1953 RSC
- Q. Sun, Y. Yang and Z.-W. Fu, Electrochem. Commun., 2012, 16, 22–25 CrossRef CAS
- T. Shiga, Y. Hase, Y. Yagi, N. Takahashi and K. Takechi, J. Phys. Chem. Lett., 2014, 5, 1648–1652 CrossRef CAS PubMed
- S. K. Das, S. Xu and L. A. Archer, Electrochem. Commun., 2013, 27, 59–62 CrossRef CAS
- X. Ren and Y. Wu, J. Am. Chem. Soc., 2013, 135, 2923–2926 CrossRef CAS PubMed
- C. O. Laoire, S. Mukerjee, K. Abraham, E. J. Plichta and M. A. Hendrickson, J. Phys. Chem. C, 2010, 114, 9178–9186 CrossRef CAS
- C. O. Laoire, S. Mukerjee, K. Abraham, E. J. Plichta and M. A. Hendrickson, J. Phys. Chem. C, 2009, 113, 20127–20134 CrossRef CAS
- Z. Cui, G. Fu, Y. Li and J. B. Goodenough, Angew. Chem., Int. Ed., 2017, 56, 9901–9905 CrossRef CAS PubMed
- P. Hartmann, M. Heinemann, C. L. Bender, K. Graf, R.-P. Baumann, P. Adelhelm, C. Heiliger and J. Janek, J. Phys. Chem. C, 2015, 119, 22778–22786 CrossRef CAS
- S. Yang and H. Knickle, J. Power Sources, 2002, 112, 162–173 CrossRef CAS
- J. S. Lee, S. Tai Kim, R. Cao, N. S. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2010, 1, 34–50 CrossRef
- J. Gao, D. Pan, K. Chen, Y. Liu, J. Chen and Z. Wen, Adv. Energy Mater., 2024, 14, 2302862 CrossRef
- Y. Guo, R. Zhang, S. Zhang and C. Zhi, Nano Mater. Sci., 2022, 9, 004 Search PubMed
- F. Meng, X. Xiong, S. He, Y. Liu and R. Hu, Adv. Energy Mater., 2023, 13, 2103622 Search PubMed
- Y. Wang, L. Zhang and T. Hu, Acta Chim. Sin., 2015, 73, 2010022 Search PubMed
- P. Cai, X. Peng, J. Huang, J. Jia, X. Hu and Z. Wen, Sci. China: Chem., 2019, 62, 385–392 CrossRef CAS
- C. Lin, S.-H. Kim, Q. Xu, D.-H. Kim, G. Ali, S. S. Shinde, S. Yang, Y. Yang, X. Li, Z. Jiang and J.-H. Lee, Matter, 2021, 4, 1287–1304 CrossRef CAS
- J.-N. Liu, C.-X. Zhao, J. Wang, D. Ren, B.-Q. Li and Q. Zhang, Energy Environ. Sci., 2022, 15, 4542–4553 RSC
- S. Huang, H. Zhang, J. Zhuang, M. Zhou, M. Gao, F. Zhang and Q. Wang, Adv. Energy Mater., 2022, 12, 2103622 CrossRef CAS
- J. Yu, C.-X. Zhao, J.-N. Liu, B.-Q. Li, C. Tang and Q. Zhang, Green Chem. Eng., 2020, 1, 117–123 CrossRef
- C. X. Zhao, J. N. Liu, N. Yao, J. Wang, D. Ren, X. Chen, B. Q. Li and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 15281–15285 CrossRef CAS PubMed
- W. Guo, Y. Wang, Q. Yi, E. Devid, X. Li, P. Lei, W. Shan, K. Qi, L. Shi and L. Gao, Front. Energy Res., 2023, 11, 1194674 CrossRef
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu, Y. Li and Y. Li, Nat. Commun., 2018, 9, 1320 CrossRef PubMed
- J. Xie, X. Wang, J. Lv, Y. Huang, M. Wu, Y. Wang and J. Yao, Angew. Chem., Int. Ed., 2018, 57, 16996–17001 CrossRef CAS PubMed
- X. Wang, J. Xie, M. A. Ghausi, J. Lv, Y. Huang, M. Wu, Y. Wang and J. Yao, Adv. Mater., 2019, 31, e1807807 CrossRef PubMed
- M. Yang, S. Liu, J. Sun, M. Jin, R. Fu, S. Zhang, H. Li, Z. Sun, J. Luo and X. Liu, Appl. Catal., B, 2022, 307, 121145 CrossRef CAS
- S. Gao, S. Chen, Q. Liu, S. Zhang, G. Qi, J. Luo and X. Liu, ACS Appl. Nano Mater., 2022, 5, 12387–12394 CrossRef CAS
- H. Wang, J. Si, T. Zhang, Y. Li, B. Yang, Z. Li, J. Chen, Z. Wen, C. Yuan, L. Lei and Y. Hou, Appl. Catal., B, 2020, 270, 118892 CrossRef CAS
- Y. Li, Y. Ji, Y. Zhao, J. Chen, S. Zheng, X. Sang, B. Yang, Z. Li, L. Lei, Z. Wen, X. Feng and Y. Hou, Adv. Mater., 2022, 34, e2202240 CrossRef PubMed
- C. S. Li, Y. Sun, F. Gebert and S. L. Chou, Adv. Energy Mater., 2017, 7, 1700869 CrossRef
- J. G. Smith, J. Naruse, H. Hiramatsu and D. J. Siegel, Chem. Mater., 2016, 28, 1390–1401 CrossRef CAS
- G. Vardar, E. G. Nelson, J. G. Smith, J. Naruse, H. Hiramatsu, B. M. Bartlett, A. E. S. Sleightholme, D. J. Siegel and C. W. Monroe, Chem. Mater., 2015, 27, 7564–7568 CrossRef CAS
- M. Liu, Q. Zhang, X. Zhang, H. Fan, J. Gao, Z. Jing, M. Wang, Z. Wang and E. Wang, Chem. Eng. J., 2023, 472, 145154 CrossRef CAS
- W.-W. Yin, J.-L. Yue, M.-H. Cao, W. Liu, J.-J. Ding, F. Ding, L. Sang and Z.-W. Fu, J. Mater. Chem. A, 2015, 3, 19027–19032 RSC
- D. Li, L. Hou, H. Du, H. Wei, X. Liu, Q. Wang, C. Yang and Y. Wei, Nanoscale, 2025, 17, 4649–4658 RSC
- X. Cheng, B. Sun, T. Wei, J. Dong, X. Cao, J. Zhang, Y. Zhang, T. Wang, Y. Liu, F. Zhong, M. Liang and J. Li, J. Mater. Chem. A, 2025, 13, 8642–8653 RSC
- Y. Zhao, H. Hong, L. Zhong, J. Zhu, Y. Hou, S. Wang, H. Lv, P. liang, Y. Guo, D. Wang, P. Li, Y. Wang, Q. Li, S. C. Cao, H. Li and C. Zhi, Adv. Energy Mater., 2023, 13, 2300627 CrossRef CAS
- S. Hu and M. Zhu, Energy Storage Mater., 2023, 61, 102866 CrossRef
- W. Zhou, M. Song, P. Liang, X. Li, X. Liu, H. Li, T. Zhang, B. Wang, R. Zhao, Z. Zhao, W. Li, D. Zhao and D. Chao, J. Am. Chem. Soc., 2023, 145, 10880–10889 CrossRef CAS PubMed
- J. Wang, Z. Wang, Y. Chen, S. Gao and S. Hu, J. Energy Chem., 2025, 106, 823–831 CrossRef CAS
- M. Gao, R. Wang, X. Lu, Y. Fan, Z. Guo and Y. Wang, Angew. Chem., Int. Ed., 2024, 63, e202407856 CrossRef CAS PubMed
- G. Cohn and Y. Ein-Eli, J. Power Sources, 2010, 195, 4963–4970 CrossRef CAS
- Y. E. Durmus, Ö. Aslanbas, S. Kayser, H. Tempel, F. Hausen, L. G. J. de Haart, J. Granwehr, Y. Ein-Eli, R.-A. Eichel and H. Kungl, Electrochim. Acta, 2017, 225, 215–224 CrossRef CAS
- S. Pang, J. Wang, B. Wang, M. Zhu, G. Hu, H. Xie and S. Hu, Carbon Energy, 2025, 7, e661 CrossRef CAS
- S. Zaromb, J. Electrochem. Soc., 1962, 109, 1125 CrossRef CAS
- Y. Wang, W. Pan, H. Y. H. Kwok, H. Zhang, X. Lu and D. Y. C. Leung, J. Power Sources, 2019, 437, 226896 CrossRef CAS
- L. Wang, R. Cheng, W. Wang, G. Yang, M. K. H. Leung, F. Liu and S.-P. Feng, Electrochim. Acta, 2021, 388, 138584 CrossRef CAS
- M. Zhang, H. Li, P. Cai, K. Chen and Z. Wen, Adv. Funct. Mater., 2021, 31, 2103248 CrossRef CAS
- M. Zhang, H. Li, J. Chen, F. X. Ma, L. Zhen, Z. Wen and C. Y. Xu, Adv. Funct. Mater., 2023, 33, 2303189 CrossRef CAS
- K. Chen, Y. Liang, D. Pan, J. Huang, J. Gao, Z. Lu, X. Liu, J. Chen, H. Zhang, X. Hu and Z. Wen, Adv. Energy Mater., 2024, 15, 2303661 Search PubMed
- N. Bogolowski and J.-F. Drillet, Electrochim. Acta, 2018, 274, 353–358 CrossRef CAS
- R. Mori, RSC Adv., 2017, 7, 6389–6395 RSC
- D. Gelman, B. Shvartsev, I. Wallwater, S. Kozokaro, V. Fidelsky, A. Sagy, A. Oz, S. Baltianski, Y. Tsur and Y. Ein-Eli, J. Power Sources, 2017, 364, 110–120 CrossRef CAS
- B. Shvartsev, D. Gelman, D. Amram and Y. Ein-Eli, Langmuir, 2015, 31, 13860–13866 CrossRef CAS PubMed
- P. Sun, J. Chen, Y. Huang, J.-H. Tian, S. Li, G. Wang, Q. Zhang, Z. Tian and L. Zhang, Energy Storage Mater., 2021, 34, 427–435 CrossRef
- R. Qiu, J. Y. Zheng, H. G. Cha, M. H. Jung, K. J. Lee and Y. S. Kang, Nanoscale, 2012, 4, 1565–1567 RSC
-
W. K. Tan, G. Kawamura, H. Muto and A. Matsuda, Sustainable Materials for Next Generation Energy Devices, 2021, pp. 59–83 Search PubMed
- M. A. Deyab and Q. Mohsen, Renewable Sustainable Energy Rev., 2021, 139, 16832 CrossRef
- X. Yu and A. Manthiram, ACS Energy Lett., 2017, 2, 1050–1055 CrossRef CAS
- J. Liu, M. Zhou, K. Jin, J. Li, F. Meng and X. Wei, Battery Energy, 2024, 3, 20230049 CrossRef CAS
- Ø. Hasvold, K. H. Johansen, O. Mollestad, S. Forseth and N. Størkersen, J. Power Sources, 1999, 80, 254–260 CrossRef
- A. M. Cardenas-Valencia, J. Dlutowski, J. Bumgarner, L. Langebrake and W. Moreno, J. Micromech. Microeng., 2006, 16, 1511–1518 CrossRef CAS
- D. J. Brodrecht and J. J. Rusek, Appl. Energy, 2003, 74, 113–124 CrossRef CAS
- H. Zhang, Y. Yang, T. Liu and H. Chang, Energies, 2018, 11, 2316 CrossRef
- L. Sun, F. Wen, L. Shi and S. Li, J. Alloys Compd., 2020, 815, 152361 CrossRef CAS
- L. Sun, S. Li, W. He and F. Wen, Fuel Cells, 2019, 19, 748–754 CrossRef CAS
- F. L. Alfonso Moreno and D. E. Gómez Cervantes, Avances Investigación en Ingeniería, 2019, 16, 117–129 CrossRef
- D. Jia, F. Liu, D. Yang and W. Wang, Int. J. Electrochem. Sci., 2020, 15, 10584–10615 CrossRef
- C. J. Patrissi, R. R. Bessette, Y. K. Kim and C. R. Schumacher, J. Electrochem. Soc., 2008, 155, B558 CrossRef CAS
- C. Shu, E. Wang, L. Jiang, Q. Tang and G. Sun, J. Power Sources, 2012, 208, 159–164 CrossRef CAS
- F.-C. Wen, S. R. G. G. Li, Y. Chen, X.-H. Du, H. Tan, Y.-J. Song, M. Song and L.-M. Sun, Rare Met., 2022, 41, 2655–2663 CrossRef CAS
- L. Xu, J. Liu, P. Chen, Z. Wang, D. Tang, X. Liu, F. Meng and X. Wei, Cell Rep. Phys. Sci., 2020, 1, 100136 CrossRef
- C. Yue, N. Zhang, Z. Zhu, P. Chen, F. Meng, X. Liu, X. Wei and J. Liu, Small, 2022, 18, 2106532 CrossRef CAS PubMed
- Z. Zhu, L. Jin, M. Zhou, K. Fu, F. Meng, X. Wei and J. Liu, Chem. Commun., 2023, 59, 4356–4359 RSC
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC
- M. Zhou, K. Fu, Y. Xing, J. Liu, F. Meng, X. Wei and J. Liu, Sci. China Mater., 2024, 67, 2908–2914 CrossRef CAS
- S. Mehta, S. Kaur, K. Garg, M. Singh and T. C. Nagaiah, Angew. Chem., Int. Ed., 2025, 64, e202505593 CrossRef CAS PubMed
- X. Feng and L. Zhang, Appl. Catal., B, 2023, 337, 122955 CrossRef CAS
- H. Wei, J. Si, L. Zeng, S. Lyu, Z. Zhang, Y. Suo and Y. Hou, Chin. Chem. Lett., 2023, 34, 107144 CrossRef CAS
- Y. Tong, Z. Jiang, L. Yu, Y. Bai, L. Gu, P. Wang, Q. Chen, M. Li, Y. Cao, L. Song and L. Li, Inorg. Chem., 2025, 64, 9940–9952 CrossRef CAS PubMed
- F. Cheng, L. Wang, H. Wang, C. Lei, B. Yang, Z. Li, Q. Zhang, L. Lei, S. Wang and Y. Hou, Nano Energy, 2020, 71, 104621 CrossRef CAS
- P. Cai, Y. Li, G. Wang and Z. Wen, Angew. Chem., Int. Ed., 2018, 57, 3910–3915 CrossRef CAS PubMed
- H. Li, M. Zhang, L. Yi, Y. Liu, K. Chen, P. Shao and Z. Wen, Appl. Catal., B, 2021, 280, 119412 CrossRef CAS
- R. Tang, Y. Yang, Y. Zhou and X. Y. Yu, Adv. Funct. Mater., 2024, 34, 2301925 CrossRef CAS
- X. Wang, X. Xu, N. Liu, F.-N. Shi and G. Shi, ACS Sustainable Chem. Eng., 2019, 7, 10979–10985 CrossRef CAS
- L. Wang, Z. Li, K. Wang, Q. Dai, C. Lei, B. Yang, Q. Zhang, L. Lei, M. K. H. Leung and Y. Hou, Nano Energy, 2020, 74, 104850 CrossRef CAS
- Z. Lu, W. Sun, P. Cai, L. Fan, K. Chen, J. Gao, H. Zhang, J. Chen and Z. Wen, Energy Environ. Sci., 2025, 18, 2918–2930 RSC
- Ø. Hasvold, H. Henriksen, E. Melv, G. Citi, B. Ø. Johansen, T. Kjønigsen and R. Galetti, J. Power Sources, 1997, 65, 253–261 CrossRef
- R. Thimmappa, A. R. Kottaichamy, R. Bouchal, T. Marichelvam, M. O. Thotiyl and O. Fontaine, Hydrogen Exhaling Metal-H2O Battery, 2019, Available at SSRN 3318942.
- S. Paruvayakode, O. V. Athulya, K. A. Thomas and F. Fasmin, J. Power Sources, 2023, 578, 233175 CrossRef CAS
- Z. Le, W. Zhang, W. Li, J. Tan, R. Li, X. Wang, Y. V. Kaneti, X. Jiang, J. Chu, Y. Yamauchi and M. Hu, Matter, 2020, 3, 879–891 CrossRef
- G. Liu, H. Lu, Y. Xu, Q. Quan, H. Lv, X. Cui, J. Chen, L. Jiang and R. J. Behm, Chem. Eng. J., 2023, 455, 140875 CrossRef CAS
- A. Nadeema, G. Pandurang Kharabe, D. Prakash Biswal and S. Kurungot, ChemElectroChem, 2020, 7, 2582–2591 CrossRef CAS
- Q. Xia, Z. Li, D. Liu, N. Song, N. Zhang, S. Ma, Z. Wu and W. Yuan, Batteries Supercaps, 2024, 7, e202400307 CrossRef CAS
- F. Zhu, W. Guo and Y. Fu, Chem. Soc. Rev., 2023, 52, 8410–8446 RSC
- R. F. Service, Science, 2018, 362, 508–509 CrossRef CAS PubMed
- W. Li, W. Xu, Z. Sun, L. Tang, G. Xu, X. He, Y. Deng, W. Sun, B. Zhou, J. Song and W. Liu, Nat. Commun., 2025, 16, 4654 CrossRef CAS PubMed
- T. Ramachandran, R. Khan, A. Ghosh, M. Hussien, Y. A. Kumar, N. P. Reddy and M. Moniruzzaman, Biomass Bioenergy, 2025, 201, 107846 CrossRef
- Y. Zhao, T. Ramachandran, A. Ghosh, A. G. Al-Sehemi, Y. A. Kumar, S. S. Rao, A. K. Yadav and D. Mani, Biomass Bioenergy, 2025, 200, 108052 CrossRef CAS
- N. Roy, Y. Anil Kumar, T. Ramachandran, A. M. Fouda, H. H. Hegazy, M. Moniruzzaman and S. Woo Joo, J. Ind. Eng. Chem., 2025, 144, 228–254 CrossRef CAS
- C. Zhang, Z. Yuan and X. Li, ACS Energy Lett., 2024, 9, 3456–3473 CrossRef CAS
- C. Mu, T. Li, C. Zhan, Q. Fu, Y. Zhang, L. Zhang, F. Wang, Y. Zhang and X. Li, Angew. Chem., Int. Ed., 2025, e202508456 CAS
- Y. Hu, Z. Min, G. Zhu, Y. Zhang, Y. Pei, C. Chen, Y. Sun, G. Liang and H. M. Cheng, Nat. Commun., 2025, 16, 3255 CrossRef CAS PubMed
- Z. Wang, G. Lu, T. Wei, G. Meng, H. Cai, Y. Feng, K. Chu, J. Luo, G. Hu, D. Wang and X. Liu, Nat. Commun., 2025, 16, 2885 CrossRef CAS PubMed
- J. Lei, Y. Zhang, Y. Yao, Y. Shi, K. L. Leung, J. Fan and Y.-C. Lu, Nat. Energy, 2023, 8, 1355–1364 CrossRef CAS
- J. Ye, L. Xia, H. Li, F. P. G. de Arquer and H. Wang, Adv. Mater., 2024, 36, e2402090 CrossRef PubMed
- Z. Li, Y. Zhang, S. Zheng, H. Tan, Y. Deng and B. Fan, Chem. Eng. J., 2025, 516, 164200 CrossRef CAS
- G. Yang, Y. Zhu, Z. Hao, Y. Lu, Q. Zhao, K. Zhang and J. Chen, Adv. Mater., 2023, 35, e2301898 CrossRef PubMed
- K. Peng, G. Tang, C. Zhang, X. Yang, P. Zuo, Z. Xiang, Z. Yao, Z. Yang and T. Xu, J. Energy Chem., 2024, 96, 89–109 CrossRef CAS
- R. Feng, X. Zhang, V. Murugesan, A. Hollas, Y. Chen, Y. Shao, E. Walter, N. P. N. Wellala, L. Yan, K. M. Rosso and W. Wang, Science, 2021, 372, 836–840 CrossRef CAS PubMed
- D. Li, J. Lan, X. Ge, L. Cui, Y. Han, K. Li, L. Feng, L. Yang, H. Lin and J. J. Chen, Chem. – Asian J., 2025, 20, e202500020 CrossRef CAS PubMed
- J. Gao, L. Xia, M. Ou and Z. A. Tan, Batteries Supercaps, 2024, 7, e202400434 CrossRef CAS
- M. Pan, M. Shao and Z. Jin, SmartMat, 2023, 4, e1198 CrossRef CAS
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2014, 505, 195–198 CrossRef CAS PubMed
- M. Wu, Y. Jing, A. A. Wong, E. M. Fell, S. Jin, Z. Tang, R. G. Gordon and M. J. Aziz, Chem, 2020, 6, 1432–1442 CAS
- K. Lin, Q. Chen, M. R. Gerhardt, L. Tong, S. B. Kim, L. Eisenach, A. W. Valle, D. Hardee, R. G. Gordon, M. J. Aziz and M. P. Marshak, Science, 2015, 349, 1529–1532 CrossRef CAS PubMed
- Y. Jing, E. W. Zhao, M. A. Goulet, M. Bahari, E. M. Fell, S. Jin, A. Davoodi, E. Jonsson, M. Wu, C. P. Grey, R. G. Gordon and M. J. Aziz, Nat. Chem., 2022, 14, 1103–1109 CrossRef CAS PubMed
- T. Janoschka, N. Martin, U. Martin, C. Friebe, S. Morgenstern, H. Hiller, M. D. Hager and U. S. Schubert, Nature, 2015, 527, 78–81 CrossRef CAS PubMed
- J. Luo, B. Hu, C. Debruler and T. L. Liu, Angew. Chem., 2017, 130, 237–241 CrossRef
- J. Luo, B. Hu, M. Hu, W. Wu and T. L. Liu, Angew. Chem., Int. Ed., 2022, 61, e202204030 CrossRef CAS PubMed
- S. Hu, T. Li, M. Huang, J. Huang, W. Li, L. Wang, Z. Chen, Z. Fu, X. Li and Z. Liang, Adv. Mater., 2021, 33, e2005839 CrossRef PubMed
- K. Lin, Q. Chen, M. R. Gerhardt, L. Tong, S. B. Kim, L. Eisenach, A. W. Valle, D. Hardee, R. G. Gordon, M. J. Aziz and M. P. Marshak, Science, 2015, 349, 1529–1532 CrossRef CAS PubMed
- A. Hollas, X. Wei, V. Murugesan, Z. Nie, B. Li, D. Reed, J. Liu, V. Sprenkle and W. Wang, Nat. Energy, 2018, 3, 508–514 CrossRef CAS
- G. Tang, K. Peng, Y. Liu, J. Fang, W. Wu, Z. Yang and T. Xu, Angew. Chem., Int. Ed., 2025, 64, e202501458 CrossRef CAS PubMed
- Y. Liu, M.-A. Goulet, L. Tong, Y. Liu, Y. Ji, L. Wu, R. G. Gordon, M. J. Aziz, Z. Yang and T. Xu, Chem, 2019, 5, 1861–1870 CAS
- G. Tang, W. Wu, Y. Liu, K. Peng, P. Zuo, Z. Yang and T. Xu, Nat. Commun., 2025, 16, 47 CrossRef PubMed
- B. Hu, C. DeBruler, Z. Rhodes and T. L. Liu, J. Am. Chem. Soc., 2017, 139, 1207–1214 CrossRef CAS PubMed
- H. Lebel, D. Rochefort, C. Lai, T. Boulanger, A. Debiais, L. Hamlet, M. Maleki and M. A. Goulet, J. Am. Chem. Soc., 2025, 147, 17555–17560 CrossRef PubMed
- S. Pang, L. Li, Y. Ji and P. Wang, Angew. Chem., Int. Ed., 2024, 63, e202410226 CrossRef CAS PubMed
- G. Ge, F. Li, M. Yang, Z. Zhao, G. Hou, C. Zhang and X. Li, Adv. Mater., 2024, 36, e2412197 CrossRef PubMed
- G. Ge, C. Mu, Y. Wang, C. Zhang and X. Li, J. Am. Chem. Soc., 2025, 147, 4790–4799 CrossRef CAS PubMed
- B. Hu, H. Li, H. Fan and J. Song, Energy Storage Mater., 2023, 59, 102789 CrossRef
- Z. Zhao, T. Li, C. Zhang, M. Zhang, S. Li and X. Li, Nat. Sustainable, 2024, 7, 1273–1282 CrossRef
- P. Zhang, Y. Liu, J. Wei, Z. Wu, X. Song, G. Ding, H. Wang, J. Liang, Z. Tie and Z. Jin, Nat. Commun., 2025, 16, 4727 CrossRef CAS PubMed
- Y. Liu, P. Zhang, Z. Wu, J. Wei, G. Ding, X. Song, J. Ma, W. Wang and Z. Jin, J. Am. Chem. Soc., 2024, 146, 3293–3302 CrossRef CAS PubMed
- M. E. Carrington, K. Sokolowski, E. Jonsson, E. W. Zhao, A. M. Graf, I. Temprano, J. A. McCune, C. P. Grey and O. A. Scherman, Nature, 2023, 623, 949–955 CrossRef CAS PubMed
- X. Liu, X. Zhang, C. Bao, Z. Wang, H. Zhang, G. Li, N. Yan, M.-J. Li and G. He, CCS Chem., 2023, 5, 2334–2347 CrossRef CAS
- X. Liu, H. Zhang, C. Liu, Z. Wang, X. Zhang, H. Yu, Y. Zhao, M. J. Li, Y. Li, Y. L. He and G. He, Angew. Chem., Int. Ed., 2024, 63, e202405427 CrossRef CAS PubMed
- L. Li, E. Yao, Y. Ji and P. Wang, Angew. Chem., Int. Ed., 2025, e202423219 CAS
- C. Ye, R. Tan, A. Wang, J. Chen, B. Comesana Gandara, C. Breakwell, A. Alvarez-Fernandez, Z. Fan, J. Weng, C. G. Bezzu, S. Guldin, N. P. Brandon, A. R. Kucernak, K. E. Jelfs, N. B. McKeown and Q. Song, Angew. Chem., Int. Ed., 2022, 61, e202207580 CrossRef CAS PubMed
- C. Ye, A. Wang, C. Breakwell, R. Tan, C. Grazia Bezzu, E. Hunter-Sellars, D. R. Williams, N. P. Brandon, P. A. A. Klusener, A. R. Kucernak, K. E. Jelfs, N. B. McKeown and Q. Song, Nat. Commun., 2022, 13, 3184 CrossRef CAS PubMed
- A. Wang, R. Tan, D. Liu, J. Lu, X. Wei, A. Alvarez-Fernandez, C. Ye, C. Breakwell, S. Guldin, A. R. Kucernak, K. E. Jelfs, N. P. Brandon, N. B. McKeown and Q. Song, Adv. Mater., 2023, 35, e2210098 CrossRef PubMed
- P. Zuo, C. Ye, Z. Jiao, J. Luo, J. Fang, U. S. Schubert, N. B. McKeown, T. L. Liu, Z. Yang and T. Xu, Nature, 2023, 617, 299–305 CrossRef CAS PubMed
- J. Fang, G. Zhang, M. A. Goulet, P. Zuo, Y. Zhou, H. Li, J. Jiang, M. D. Guiver, Z. Yang and T. Xu, Nat. Commun., 2025, 16, 3282 CrossRef CAS PubMed
- K. Peng, C. Zhang, J. Fang, H. Cai, R. Ling, Y. Ma, G. Tang, P. Zuo, Z. Yang and T. Xu, Angew. Chem., Int. Ed., 2024, 63, e202407372 CrossRef CAS PubMed
- T. Wong, Y. Yang, R. Tan, A. Wang, Z. Zhou, Z. Yuan, J. Li, D. Liu, A. Alvarez-Fernandez, C. Ye, M. Sankey, D. Ainsworth, S. Guldin, F. Foglia, N. B. McKeown, K. E. Jelfs, X. Li and Q. Song, Joule, 2025, 9, 101795 CrossRef CAS
- Y. H. Zhu, Y. F. Cui, Z. L. Xie, Z. B. Zhuang, G. Huang and X. B. Zhang, Nat. Rev. Chem., 2022, 6, 505–517 CrossRef CAS PubMed
- D. Xi, A. M. Alfaraidi, J. Gao, T. Cochard, L. C. I. Faria, Z. Yang, T. Y. George, T. Wang, R. G. Gordon, R. Y. Liu and M. J. Aziz, Nat. Energy, 2024, 9, 479–490 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |