
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadical retrosynthesis: a powerful strategy for modern assembly of terpenoid natural products
Yong
Zhang†
ab,
Yanbo
Zhang†
 b and
Chao
Li
*bc
b and
Chao
Li
*bc
aAcademy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, China
bNational Institute of Biological Sciences, Beijing, 102206, China. E-mail: lichao@nibs.ac.cn
cTsinghua Institute of Multidisciplinary Biomedical Research, Tsinghua University, Beijing, 100084, China
First published on 7th October 2025
Abstract
Radical retrosynthesis has emerged as a powerful strategy for the modern assembly of complex natural products, offering a one-electron logic that complements traditional polar disconnections. This review highlights recent advances—particularly developed over the past decade—in radical methodologies and their strategic applications in the total synthesis of terpenoid natural products. Emphasis is placed on how various radical precursors, including alkenes, carboxylic acids, halides, alcohols, carbonyl compounds, and alkanes, have been transformed through innovative processes such as photoredox catalysis, metal–hydride hydrogen atom transfer (MHAT), redox-active ester (RAE) chemistry, cross-electrophile couplings (XEC), and HAT-based C–H functionalization. By organizing the discussion around precursor classes and corresponding reaction modes, we illustrate how radical-based synthetic strategies enable efficient construction of quaternary stereocenters and modular assembly of complex polycyclic scaffolds.
 Yong Zhang | Yong Zhang earned his BSc in Pharmacy from Hebei University of Science and Technology in 2013 and his Master's degree under Prof. Yunfei Du from Tianjin University in 2018. He is currently a PhD candidate at the National Institute of Biological Sciences, Beijing (NIBS) and Peking University, supervised by Prof. Chao Li. His research focuses on 3d metal-catalyzed cross-coupling reactions and radical retrosynthesis strategies for natural product synthesis. |
 Yanbo Zhang | Yanbo Zhang graduated from The High School Affiliated to Renmin University of China in 2025 and has recently begun his undergraduate studies in the Weinberg College of Arts and Sciences at Northwestern University. He is also enrolled in the Integrated Science Program and is majoring in chemistry. Prior to starting his undergraduate studies, he conducted research as an intern in Dr. Chao Li's laboratory, where he focused on organic synthesis and methodology development. His academic interests center on the design and development of novel synthetic strategies in organic chemistry. |
 Chao Li | Chao Li received his BS from Qingdao University of Science and Technology in 2008 and earned his PhD in Organic Synthesis in 2013 under the supervision of Prof. Xiaoguang Lei at Tianjin University and the National Institute of Biological Sciences (NIBS), Beijing. He then carried out postdoctoral research with Prof. Phil S. Baran at Scripps Research. In late 2017, he joined NIBS as an Assistant Investigator and was promoted to Associate Investigator in 2023. His research focuses on the total synthesis of complex natural products and the development of novel, efficient synthetic methods. |
1. Introduction
Retrosynthetic analysis, since its formalization by E. J. Corey,1,2 has traditionally rested on polar disconnections as the foundation of synthetic planning. These intuitive disconnections rely on charged synthons, often requiring prefunctionalization and relatively harsh conditions to generate ionic intermediates (Scheme 1(A)). Radical retrosynthesis—articulated by Baran and co-workers in 2018—offers a complementary one-electron framework that exploits innate radical reactivity.3 Radical intermediates can frequently be accessed under milder, more functional-group-tolerant conditions, enabling bond formations such as radical additions, metal-catalyzed cross-couplings, and cascade sequences. Recent advances—including MHAT,4–6 RAE chemistry,7,8 metallaphotoredox catalysis,9 cross-electrophile couplings (XEC),10 and HAT-based C–H functionalization11,12—have expanded the radical synthesis toolbox and begun to address longstanding selectivity challenges.13 By providing alternative disconnections and harnessing the unique reactivity of radicals, this approach is reshaping synthetic logic and opening new avenues for the efficient assembly of complex natural products.14,15 | ||
| Scheme 1 (A) Polar and radical disconnection strategies. (B) Representative terpenoid-based drugs. (C) Modern radical precursors employed in the total synthesis of terpenoids. | ||
Terpenoid natural products represent one of the most structurally and functionally diverse classes of secondary metabolites.16 Biosynthetically derived from simple isoprene units, terpenoids give rise to an astonishing array of carbon skeletons, often characterized by densely functionalized frameworks, multiple contiguous stereocenters, and complex polycyclic scaffolds. Representative examples such as taxol and artemisinin not only exemplify the architectural complexity of terpenoids but also underscore their potent biological activities (Scheme 1(B)). Beyond their inherent diversity, terpenoids also serve as precursors to structurally elaborate natural products, including terpenoid-alkaloids,17 meroterpenoids,18 and steroids,19 further expanding their structural and functional landscape.
The remarkable structural complexity and diversity of terpenoids, combined with their potent biological activities,20 have positioned them as enduring targets in synthetic chemistry.21–24 Over the years, several reviews have summarized strategies for the chemical synthesis of terpenoids, focusing on bioinspired logic,25 convergent assembly tactics,26 radical cascade processes,14 or chiral pool-based approaches.27,28 In contrast, retrosynthetic strategies that systematically incorporate radical logic remain largely underrepresented in this context. While the concept of radical retrosynthesis was articulated in Baran's seminal 2018 Acc. Chem. Res. article,3 that perspective was not specific to terpenoid synthesis and primarily laid the conceptual foundation for broader applications of radical disconnections. In the years since, however, radical chemistry has undergone remarkable methodological advances, and its utility in the total synthesis of complex terpenoids has begun to emerge more prominently. At this juncture, a comprehensive review of radical retrosynthesis as a strategic platform for assembling terpenoid natural products is timely and warranted.
From the perspective of radical retrosynthesis, a given chemical bond can be strategically disconnected into radical fragments, thereby simplifying complex molecular architectures. The success of this tactic hinges on the identification of suitable and stable radical precursors—readily available building blocks that can be converted into the corresponding radical species under controlled conditions. In the forward direction, appropriate radical generation methods and coupling paradigms enable the re-formation of the target bond through a variety of radical transformations. Guided by this logic, the present review is organized around the classes of radical precursors commonly employed in terpenoid synthesis (Scheme 1(C)), including alkenes, carboxylic acids, halides, alcohols, aldehydes, ketones, and alkanes. Within each category, representative examples are discussed according to the types of radical transformations utilized, such as Giese-type radical additions, metal-catalyzed cross-coupling reactions, and radical-polar crossover processes.
By articulating this framework, we aim to highlight the synthetic flexibility and strategic versatility that radical retrosynthetic analysis offers in assembling terpenoid skeletons. While numerous studies have demonstrated the growing impact of radical chemistry in total synthesis,15,29–34 this review focuses specifically on advances in radical chemistry made over the past decade—summarizing representative methodologies that have enabled the efficient assembly of complex terpenoid scaffolds. We hope this perspective offers a constructive framework for appreciating the potential of radical strategies in natural product synthesis, and encourages both experienced practitioners and newcomers to further explore and develop this promising area.
2. Alkenes as radical precursors
Alkenes, long revered for their ubiquity and chemical versatility, have ascended as privileged radical precursors in modern synthetic design, offering a dynamic platform to forge intricate molecular architectures. Their innate electronic polarization—paired with exceptional stability under diverse reaction regimes—renders them uniquely adept at generating carbon-centered radicals. Over the past decade, innovations such as MHAT have catalyzed a paradigm shift, redefining alkenes from passive structural units into strategic linchpins for radical-mediated transformations. This section delineates two recently emerged strategies—MHAT and dealkenylative fragmentation—that exploit alkenes as radical precursors, each offering distinct solutions to key synthetic challenges in terpenoid assembly.2.1. MHAT reactions
The MHAT reaction has revolutionized the functionalization of unactivated alkenes, converting C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds into reactive carbon-centered radicals under remarkably mild conditions.4–6,35 By employing abundant transition metals (e.g., Fe, Co, Mn) to deliver hydrogen atoms to alkenes via metal–hydride intermediates,36,37 MHAT circumvents traditional ionic mechanisms reliant on carbocations or carbanions, thereby obviating harsh acidic/basic conditions and prefunctionalization requirements. Pioneered by Tabushi in 1979 for alkene hydration and refined by Mukaiyama in 1989 using silicon hydrides,38–41 this radical-based paradigm was systematically expanded by researchers including Carreira,42–48 Boger,49–52 Norton,53–57 Baran,58–62 Shenvi,63–69 Herzon,70–73 and Pronin,74–76 among many others,77–81 who demonstrated its versatility in forging diverse bonds (C–O, C–N, C–C, C–H) through radical intermediates, even within polyfunctional molecular landscapes.
C bonds into reactive carbon-centered radicals under remarkably mild conditions.4–6,35 By employing abundant transition metals (e.g., Fe, Co, Mn) to deliver hydrogen atoms to alkenes via metal–hydride intermediates,36,37 MHAT circumvents traditional ionic mechanisms reliant on carbocations or carbanions, thereby obviating harsh acidic/basic conditions and prefunctionalization requirements. Pioneered by Tabushi in 1979 for alkene hydration and refined by Mukaiyama in 1989 using silicon hydrides,38–41 this radical-based paradigm was systematically expanded by researchers including Carreira,42–48 Boger,49–52 Norton,53–57 Baran,58–62 Shenvi,63–69 Herzon,70–73 and Pronin,74–76 among many others,77–81 who demonstrated its versatility in forging diverse bonds (C–O, C–N, C–C, C–H) through radical intermediates, even within polyfunctional molecular landscapes.
A transformative leap occurred in 2014 with Baran's reductive olefin coupling (BROC),59,60 which enabled the direct coupling of unactivated alkenes with electron-deficient partners (e.g., Michael acceptors) using Fe(acac)3 as the catalyst and PhSiH3 as the hydrogen source. This method allowed for the modular construction of sterically congested motifs—such as quaternary centers, bicyclic systems, and cyclopropanes—with high chemoselectivity. In the years that followed, MHAT chemistry emerged as a powerful strategy for assembling complex terpenoid architectures. Its ability to engage unactivated alkenes under mild conditions has proven particularly valuable in constructing polycyclic frameworks with high efficiency and step economy.80 Today, MHAT-based transformations play an increasingly central role in radical-mediated approaches to terpenoid synthesis.
From the lens of radical retrosynthesis, sterically congested motifs such as quaternary carbons—long considered synthetic bottlenecks—can be strategically deconstructed into two modular fragments: a bulky carbon radical and a tailored radical coupling partner. The former is readily generated via MHAT activation of alkenes, while the latter encompasses a diverse array of electrophilic or nucleophilic partners that dictate bond-forming selectivity (Scheme 2). In terpenoid synthesis, key radical coupling partners include electron-deficient olefins (e.g., α,β-unsaturated carbonyls), nitriles, alkenes (as linchpins for radical cascades), alkenylsilanes, carbonyls (ketones/aldehydes), electron-deficient or electron-rich arenes (depending on radical polarity), benzyliron (TPP) (for SH2 homolytic substitution), and quinone monoacetals (for C–O bond formation)—each enabling distinct bond-forming events. The following case studies illustrate how these partnerships, orchestrated via MHAT, overcome steric and electronic challenges in terpenoid assembly. Note that, given the prolific application of MHAT in terpenoid skeleton construction over the past decade, we categorize MHAT-enabled reactions into four classes based on mechanistic parallels: MHAT-radical additions, MHAT-radical-polar crossover cascades, MHAT-SH2 homolytic substitution, and MHAT-fragmentations cascades.
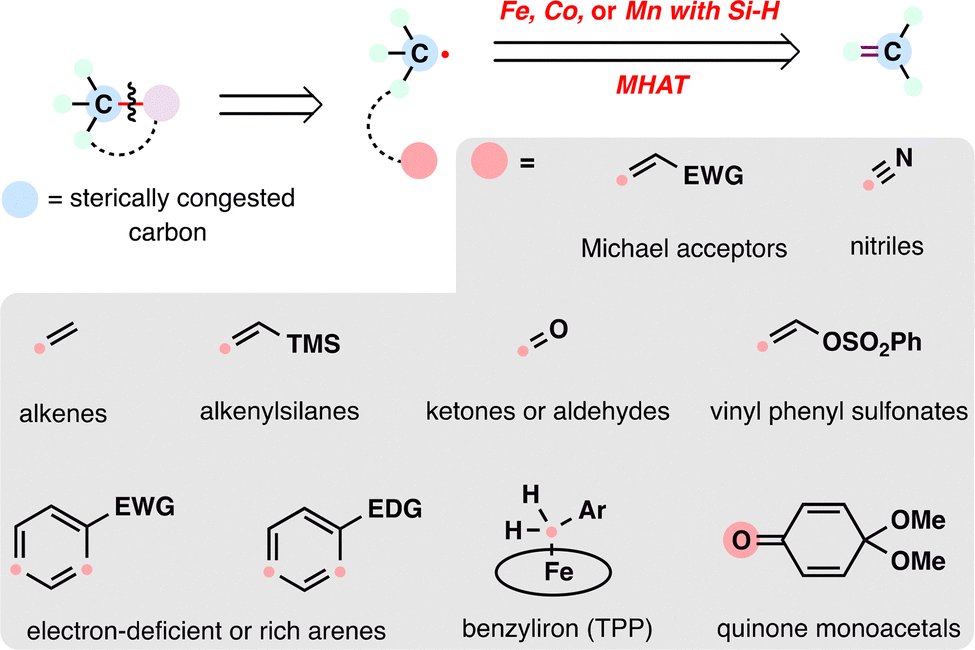 | ||
| Scheme 2 Contemporary radical retrosynthesis of sterically congested carbon employing the MHAT reaction. | ||
 | ||
| Scheme 3 Total synthesis of (±)-emindole SB (1) by Pronin and co-workers. | ||
Taking this reaction as an illustrative case, the mechanism of BROC, although intricate and influenced by solvent effects, can be outlined as follows:58 Fe(acac)3 first reacts with a silane in an alcoholic solvent to generate an Fe(III)–H intermediate. This species engages the donor olefin to produce a tertiary carbon radical along with an Fe(II) complex. The resulting radical then undergoes an intramolecular Giese-type addition to form an α-aldehyde radical, which is subsequently reduced by the Fe(II) species and protonated to afford the desired product. Treatment of 6 with KHMDS induced an intramolecular aldol reaction to deliver pentacycle 7, which was advanced to (±)-emindole SB (1) in a few further steps.
Hispidanin A (8) is a dimeric diterpenoid isolated from the rhizomes of Isodon hispida, notable for its polycyclic structure with eleven stereocenters.82 One of the monomeric units features a polycyclic structure that can be efficiently assembled via an MHAT-enabled radical cascade.83,84 As illustrated in Scheme 4, B. Liu and co-workers initiated the synthesis with an epoxide opening of 9, followed by TBS protection of the resulting propargyl alcohol to furnish alkynyl ether 10. Au(I)-catalyzed hydration afforded ester 11, which was then formylated and tosylated to yield enol tosylate 12. Kumada cross-coupling of 12 with furanyl Grignard reagent 13 efficiently delivered Z-alkene 14. The furan moiety subsequently underwent oxidative elaboration followed by acid-mediated rearrangement to generate lactone 15. A BROC radical cyclization, initiated at the gem-disubstituted terminal alkene bearing the least steric hindrance and greatest electron density, then constructed the tricyclic core 16. This radical cascade forged two carbocyclic rings and installed four contiguous stereocenters in a single operation, efficiently assembling the labdane scaffold with high stereocontrol. Further manipulations of 16 provided compound 17 bearing a diene fragment, which engaged in a Diels–Alder cycloaddition with totarane-type dienophile 18, furnishing the complex polycyclic framework of hispidanin A (8) through a concise, strategically orchestrated sequence featuring high endo-selectivity and mild conditions.
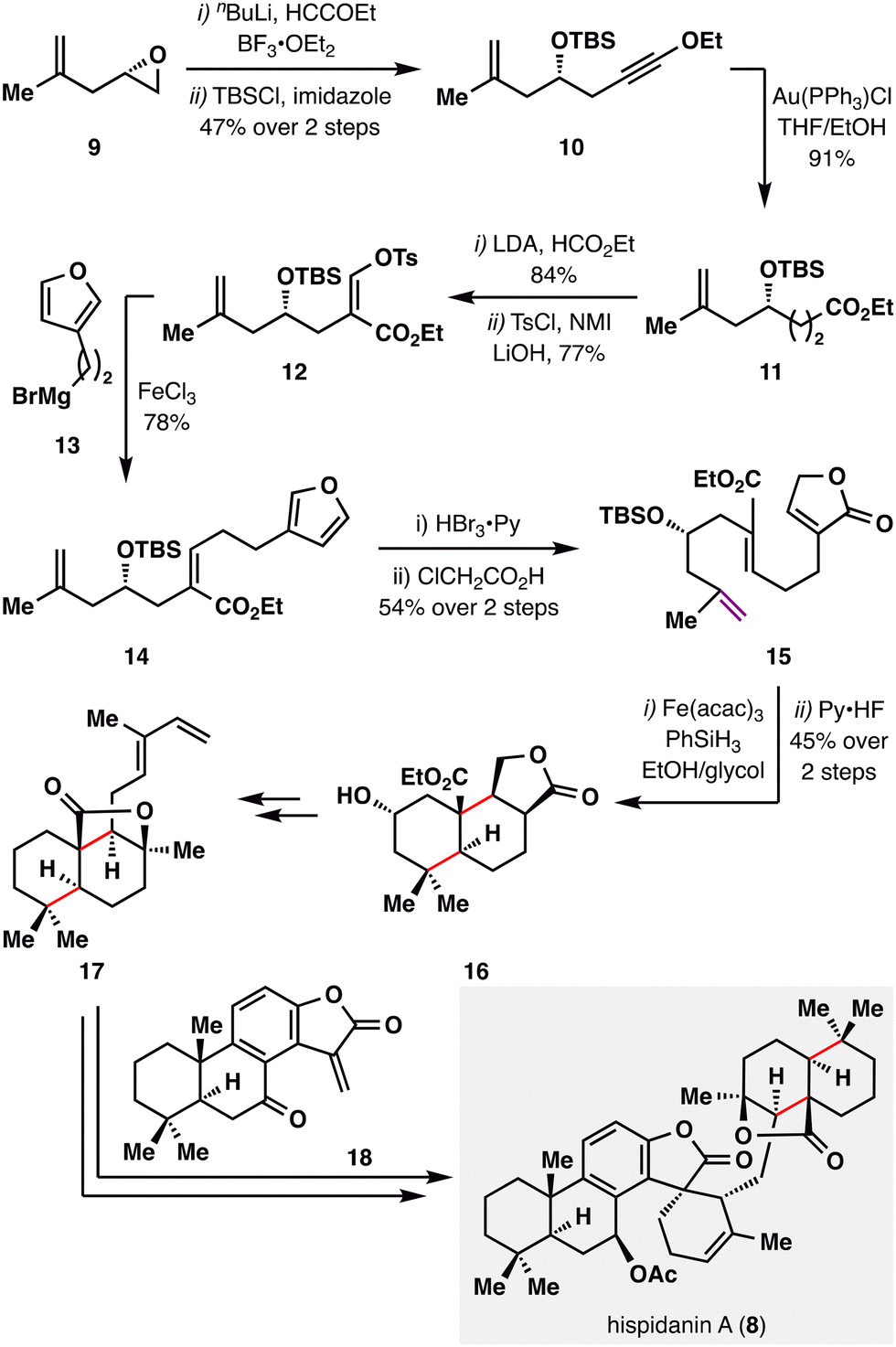 | ||
| Scheme 4 Liu's synthesis of (−)-hispidanin A (8). | ||
Another noteworthy example of employing BROC radical cyclization to construct complex polycyclic frameworks is the total synthesis of aplysiasecosterol A (19) reported by A. Li and co-workers (Scheme 5),85 in which three contiguous stereocenters—including a quaternary carbon center—were forged in a single step. The synthesis adopted a convergent strategy, involving the preparation of two polyoxygenated fragments 24 and 29, each bearing multiple stereocenters. The left-hand fragment 24 was constructed from compound 20 through a sequence involving key desymmetrizing reduction, asymmetric 2-bromoallylation, ketalization, and IBX dehydrogenation to furnish intermediate 23, which then underwent intramolecular radical cyclization, bromination, and ozonolysis. The right-hand fragment 29 was derived from (+)-citronellol, with an Aggarwal lithiation–borylation serving as the key transformation to install the C17 stereocenter.86 Coupling of fragments 24 and 29via a radical-initiated Reformatsky-type reaction furnished the adduct,87 which was subjected to Burgess dehydration to give compound 30. Subsequent treatment of 30 with a modified BROC cyclization protocol [Fe(dpm)3 and PhSiH2(OiPr)] provided tricyclic product 31 in 56% yield with high diastereoselectivity. Final deprotections of 31 delivered aplysiasecosterol A (19) in high yield.
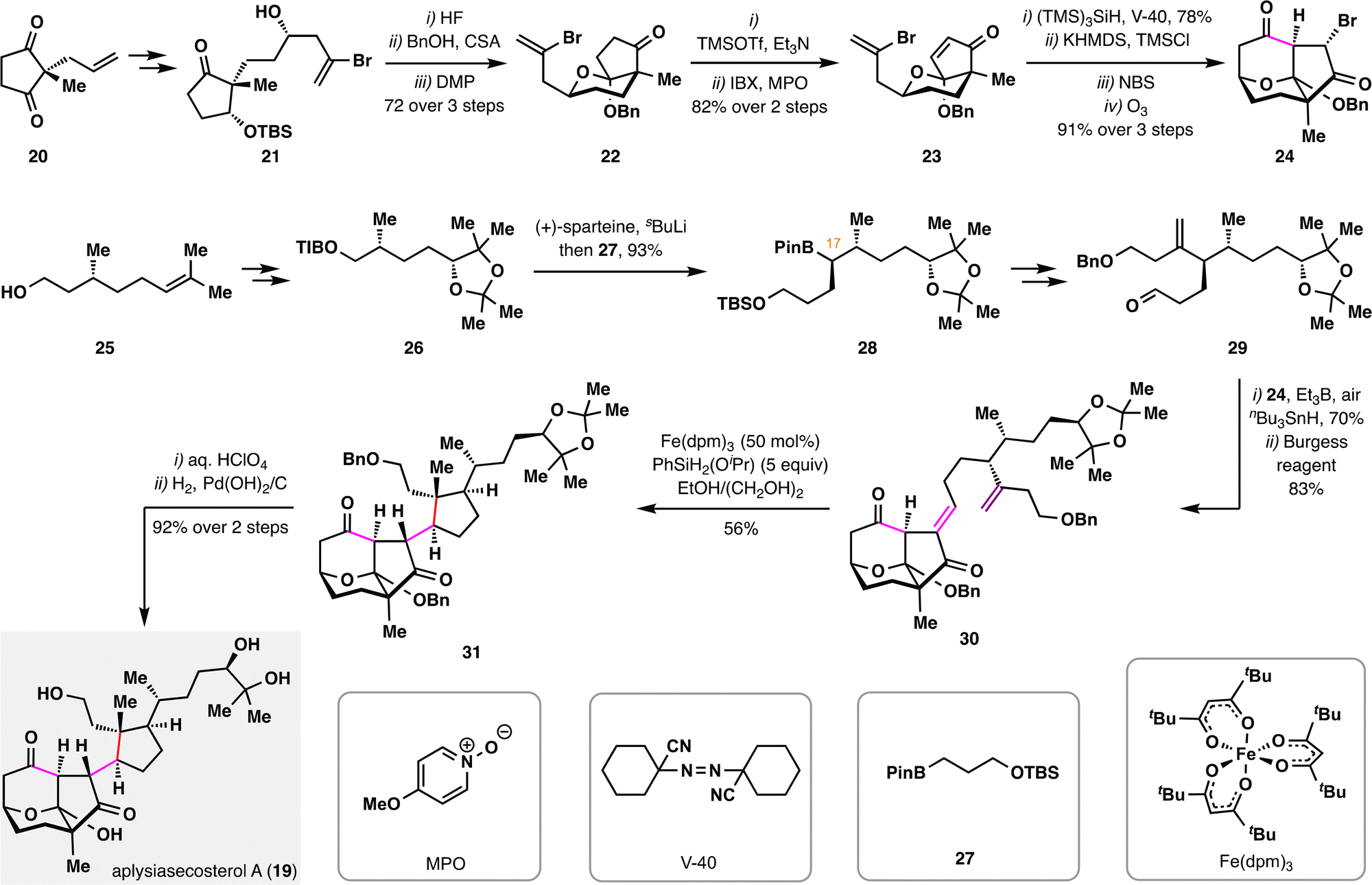 | ||
| Scheme 5 Total synthesis of aplysiasecosterol A (19) by A. Li and co-workers. | ||
The stereospecific MHAT radical addition directed by a quaternary center has been effectively explored by Snyder and co-workers in their elegant construction of the conidiogenone family (32–34).88 As illustrated in Scheme 6, enone 35 underwent a BROC radical cyclization to afford bicyclic ketone 36. Due to the steric bias imparted by the adjacent quaternary center, the tertiary radical generated under Fe(acac)3/PhSiH3 conditions approached exclusively from the bottom face, furnishing 36 as a single diastereomer in 83% yield. Subsequent functional group manipulations converted 36 into enol triflate 37, bearing a terminal alkene, which underwent a Pd-catalyzed ring closure to construct the tricyclic scaffold 38. This intermediate was then elaborated to alkenyl iodide 40, which participated in a reductive Heck coupling to generate 42—a versatile branching point for the divergent synthesis of three structurally related natural products: conidiogenone B (32), conidiogenone (33), and conidiogenol (34).
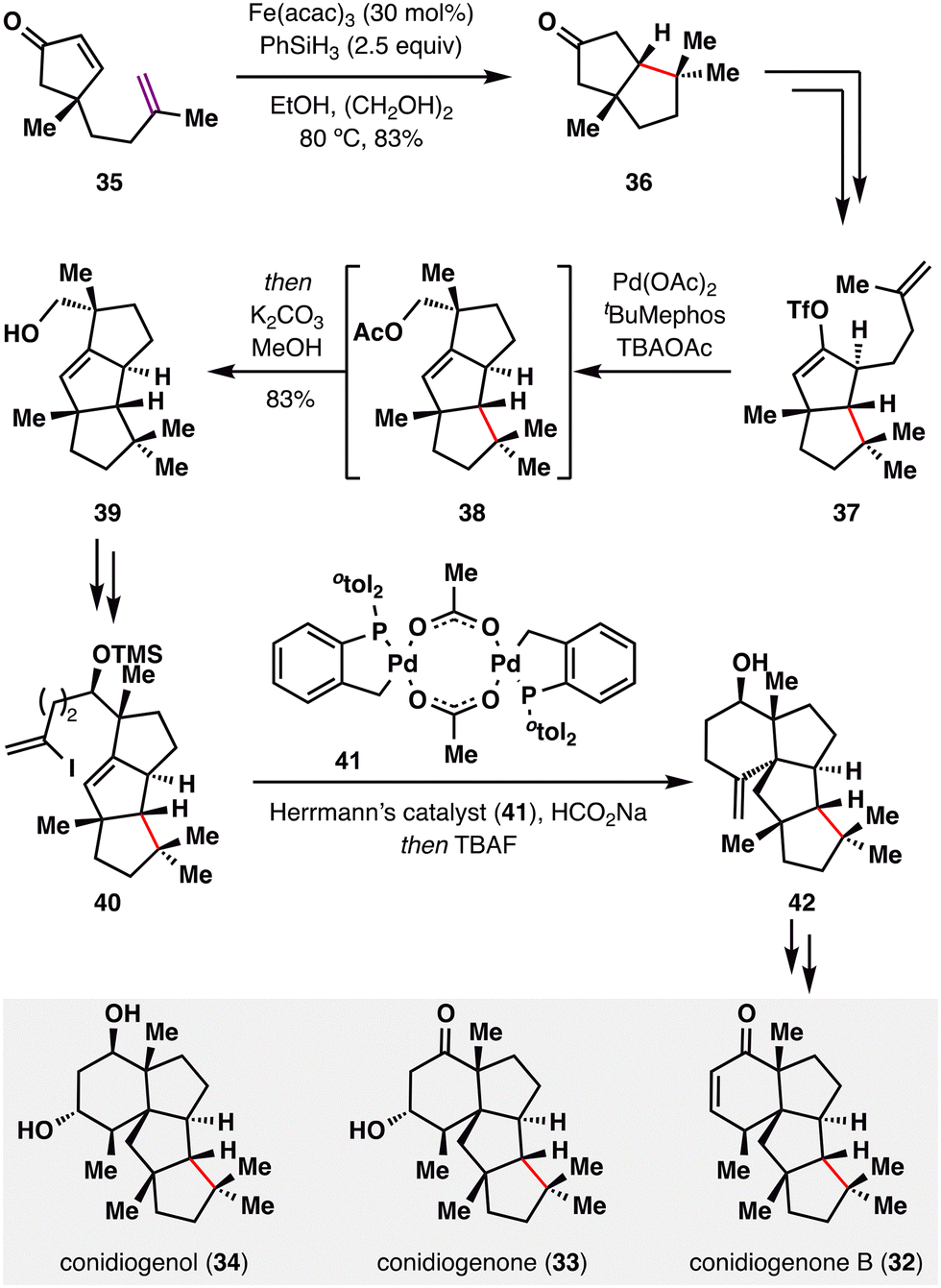 | ||
| Scheme 6 Quaternary-center-guided synthesis of conidiogenones (32–34) by Snyder and co-workers. | ||
Meroterpenoids represent a vast and structurally diverse class of hybrid terpene natural products, many of which exhibit notable biological activities.89 Among them, taondiol (43) and chevalone A (44) share a characteristic C3-oxidized ent-isocopalane core. Recently, Renata and co-workers employed an intramolecular BROC to stereoselectively construct the synthetically challenging adjacent C8 and C14 stereocenters (Scheme 7).90 Starting from sclareol (45), a readily available diterpene with an isocopalane-like framework, a concise three-step sequence transformed it into lactone 46, thereby establishing the structural platform for a BROC radical cyclization. Under classical conditions, the resulting tertiary radical underwent a highly diastereoselective top-face addition to the enone, furnishing compound 47 as a single diastereomer in 95% yield. This selectivity was attributed to rapid conformational reorganization of the tertiary radical intermediate and conformational control imposed by the rigid trans-decalin and butyrolactone motifs. Subsequent lactone opening via elimination yielded carboxylic acid 48, completing the construction of the ent-isocopalane core. Site-selective C3 oxidation of 48 was achieved using the engineered biocatalyst MERO1 L75A, delivering compound 49 in 62% yield. Further functionalization furnished iodinated intermediate 50, which served as a common precursor for the synthesis of both taondiol (43) and chevalone A (44) via cross-electrophile coupling (XEC), as outlined in Scheme 73.
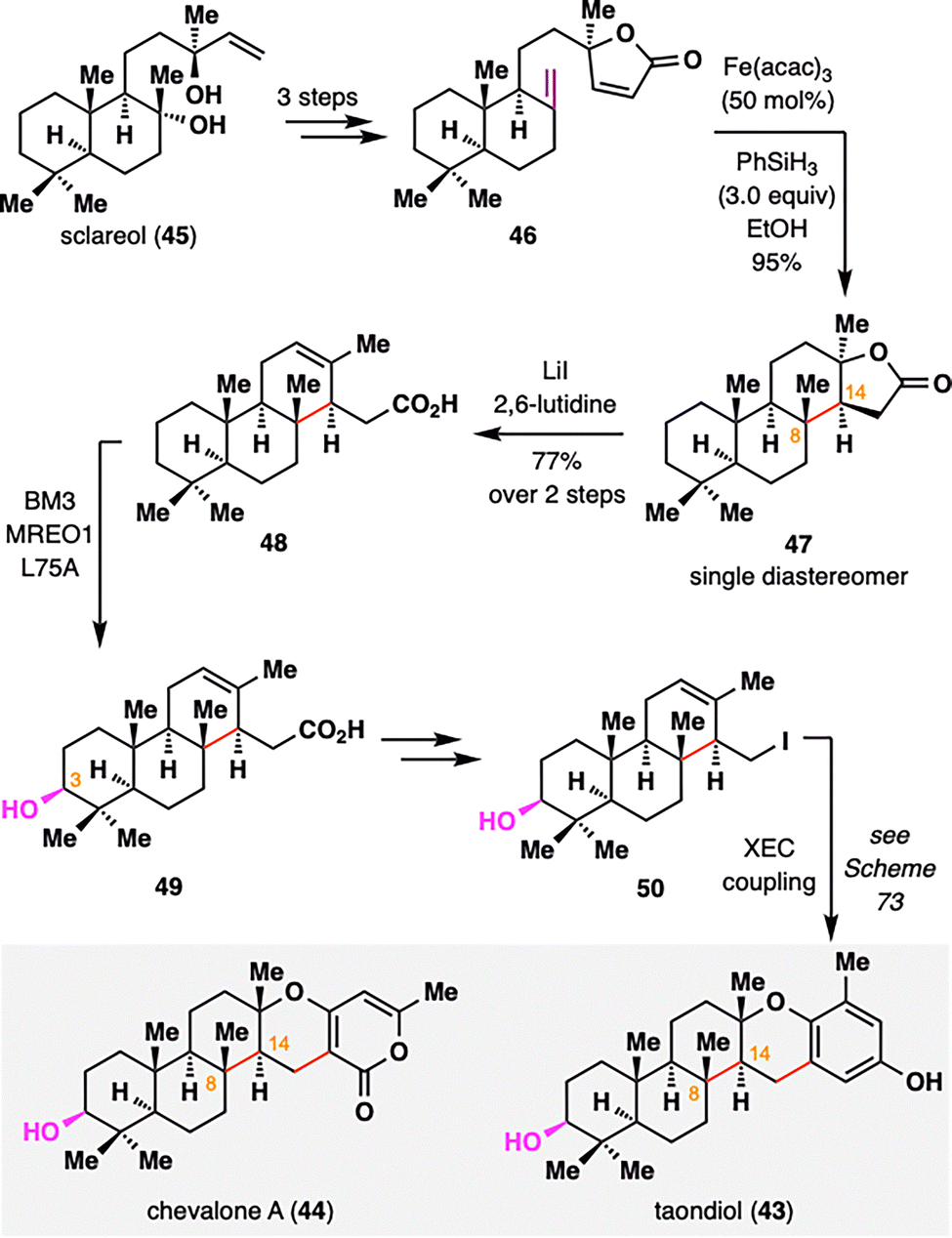 | ||
| Scheme 7 Renata's synthesis of taondiol (43) and chevalone A (44). | ||
Daphnezomines A (51) and B (52), first isolated by Kobayashi and co-workers from the leaves of Daphniphyllum humile, feature a rare and synthetically formidable aza-adamantane core (Scheme 8).91 Our group addressed this challenge through a late-stage construction of the topologically strategic C8–C9 bond via BROC radical cyclization.92 The synthesis commenced from commercially available chiral pool material (S)-(+)-carvone (53). A sequence involving hydrogenation, Sharpless allylic amination,93,94 and palladium-catalyzed oxidative cyclization furnished azabicyclo[3.3.1]nonane scaffold 55. The bridgehead ketone was subsequently transformed into an olefin, and an enone moiety was installed to set the stage for the BROC radical cyclization. Notably, the use of low concentrations of Fe(acac)3 and Ph(iPrO)SiH2 proved optimal, delivering compound 57 in moderate yield. This transformation simultaneously established two stereocenters at C8–C9 and completed the construction of the tetracyclic skeleton in a single operation. Final treatment of 57 with trifluoroacetic acid in methanol furnished the natural products daphnezomine A (51) and daphnezomine B (52) in 34% and 54% yields, respectively.
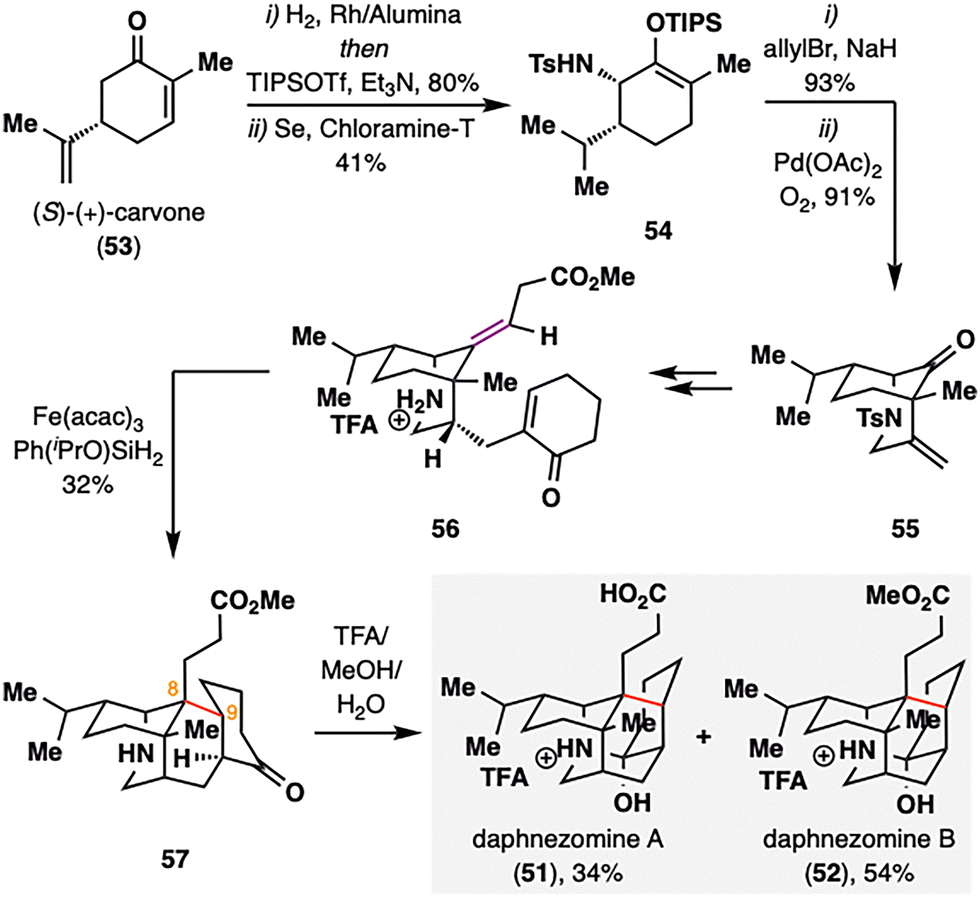 | ||
| Scheme 8 C. Li's total synthesis of daphnezomines A (51) and B (52). | ||
Gedunin (58), a member of the tetranortriterpenoid family, features a highly oxidized and structurally rearranged limonoid scaffold that poses a significant synthetic challenge. Renata and co-workers achieved the first total synthesis of gedunin (58),95 through a combination of BROC radical cyclization and enzyme-catalyzed C–H oxidation. As outlined in Scheme 9, the synthesis commenced from bicyclic enone 59, readily prepared from commercially available sclareolide. A bioengineered enzyme-catalyzed C3 hydroxylation of 59 furnished alcohol 60, which was coupled with lactone 61via Luche's protocol to deliver ketone 62 as a single diastereomer in 93% yield.96 Methylenation of the ketone followed by a second C3 oxidation provided intermediate 63. Subsequent C7 allylic oxidation, followed by treatment of the resulting allylic alcohol with Fe(acac)3 and PhSiH3, successfully established the crucial quaternary stereocenter and the sterically congested C/D-ring junction with excellent diastereoselectivity and high efficiency. Final double desaturation of the A and D rings, followed by diastereo- and chemoselective epoxidation, delivered the natural product gedunin (58) in a highly streamlined and efficient sequence.
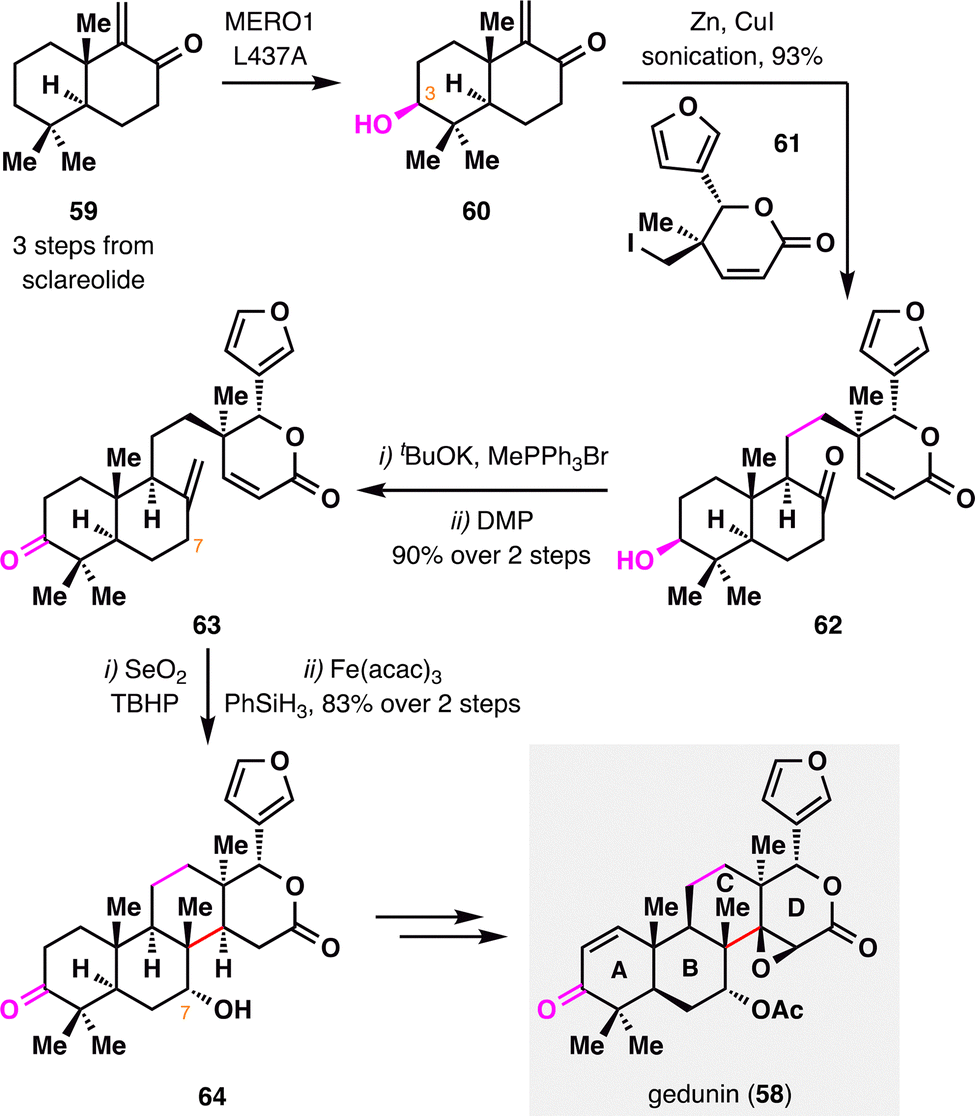 | ||
| Scheme 9 Chemoenzymatic synthesis of gedunin (58) by Renata and co-workers. | ||
MHAT-radical addition also plays a central role in the synthesis of longiborneol sesquiterpenoids (65–73) via a “functionalized camphor” strategy, as reported by Sarpong and co-workers (Scheme 10).97,98 Notably, this work revealed vinyl sulfonates as viable radical acceptors, thereby expanding the scope of MHAT chemistry to include polarity-reversed enol alkylation. The synthesis commenced with epoxidation of (S)-carvone (53), followed by a Ti(III)-mediated intramolecular reductive cyclization of the resulting epoxy-carvone to yield cyclobutanol 76.99 A Brønsted acid-promoted semipinacol rearrangement of 76 furnished 8-hydroxycamphor (77), which was further elaborated to compound 78, bearing a skipped diene and an enol vinyl phenyl sulfonate. Exposure of 78 to BROC radical cyclization conditions delivered compound 80 in excellent yield. This transformation proceeded via HAT from an Fe(III)–H species to the terminal position of the 1,1-disubstituted alkene, generating a tertiary radical that underwent intramolecular addition into the vinyl sulfonate. Subsequent homolytic cleavage of the S–O bond furnished the corresponding ketone. The successful application of MHAT in this “functionalized camphor” strategy highlights its versatility in enabling the total synthesis of structurally diverse longiborneol sesquiterpenoids (65–73) bearing the bicyclo[2.2.1]bornane core.
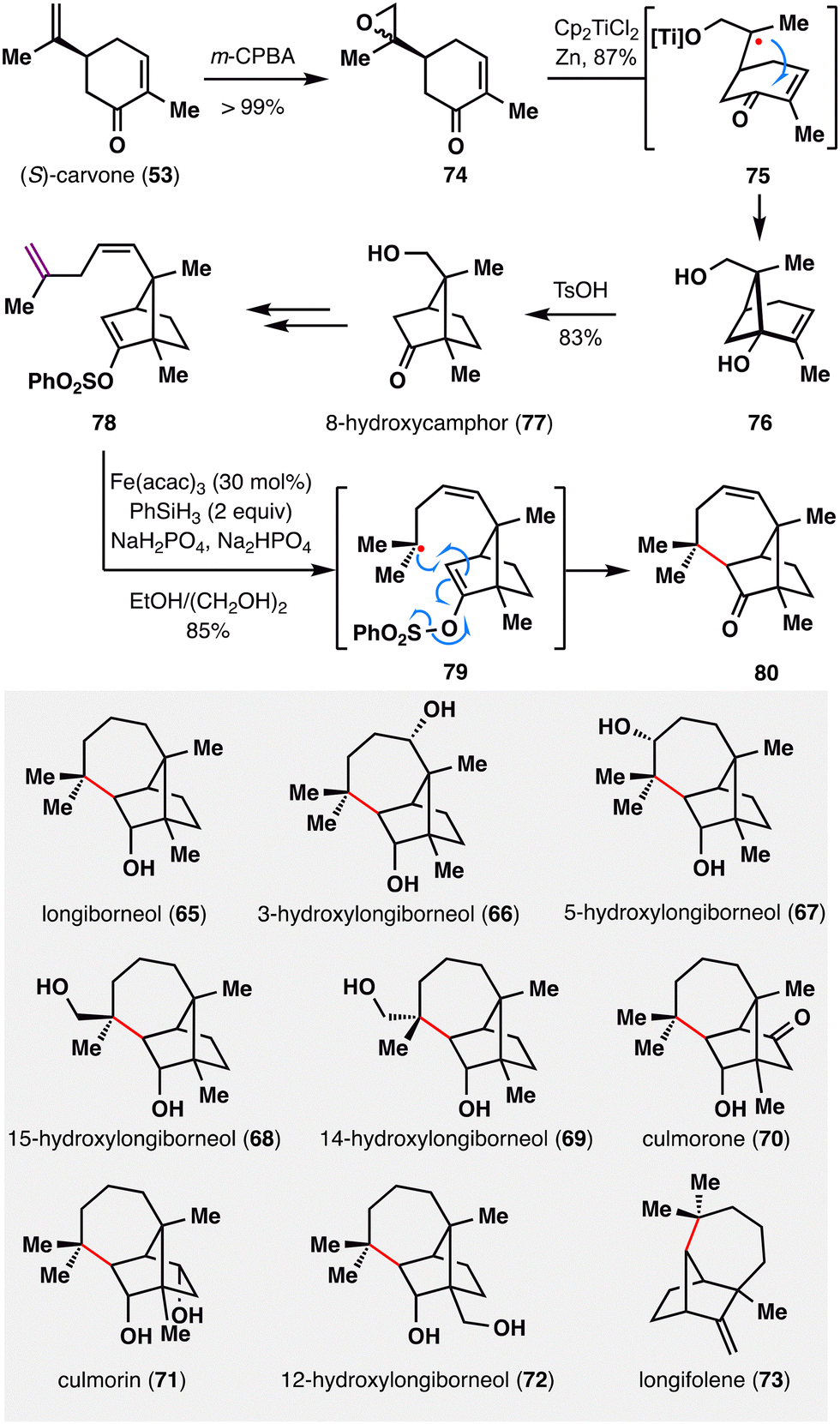 | ||
| Scheme 10 Collective synthesis of longiborneol sesquiterpenoids (65–73) by Sarpong and co-workers. | ||
Pleuromutilin (81), a diterpenoid natural product isolated from Clitopilus species, is well known for its antibacterial activity.100 Pronin and co-workers achieved rapid access to its core perhydroindanone framework through a sequence featuring an intermolecular Diels–Alder cycloaddition followed by a key BROC radical cyclization.101 As outlined in Scheme 11, the Diels–Alder reaction between diene 82 and dienophile 83 afforded bicyclic adduct 84 in 41% yield with moderate diastereoselectivity (exo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) : endo = 2:1). Oxidation of the resulting enol ether yielded enone 85, bearing both a terminal alkene and an enone moiety poised for BROC cyclization. Treatment of 85 with Fe(acac)3/PhSiH2(OiPr) delivered tricycle 86 in 44% yield and excellent diastereoselectivity (d.r. > 20:1). This radical cyclization proceeded via formation of a secondary radical at the terminal alkene, which added to a sterically hindered β-disubstituted enone—demonstrating the power of MHAT in challenging radical additions. From 86, formation of a strained four-membered ring in 87 enabled a photoredox-catalyzed oxidative decarboxylation (air/TEMPO/Ir cat. 88), generating a cyclobutyl radical intermediate that was trapped by TEMPO or oxygen. The resulting intermediate underwent Grob-type fragmentation to furnish functionalized cyclooctane 89. Final elaboration of 89 completed the total synthesis of pleuromutilin (81) in three additional steps.
: endo = 2:1). Oxidation of the resulting enol ether yielded enone 85, bearing both a terminal alkene and an enone moiety poised for BROC cyclization. Treatment of 85 with Fe(acac)3/PhSiH2(OiPr) delivered tricycle 86 in 44% yield and excellent diastereoselectivity (d.r. > 20:1). This radical cyclization proceeded via formation of a secondary radical at the terminal alkene, which added to a sterically hindered β-disubstituted enone—demonstrating the power of MHAT in challenging radical additions. From 86, formation of a strained four-membered ring in 87 enabled a photoredox-catalyzed oxidative decarboxylation (air/TEMPO/Ir cat. 88), generating a cyclobutyl radical intermediate that was trapped by TEMPO or oxygen. The resulting intermediate underwent Grob-type fragmentation to furnish functionalized cyclooctane 89. Final elaboration of 89 completed the total synthesis of pleuromutilin (81) in three additional steps.
 | ||
| Scheme 11 Pronin's synthesis of pleuromutilin (81). | ||
B. Liu and co-workers reported the total synthesis of sculponin U (90),102 an ent-kaurane-type diterpenoid characterized by a distinctive 7,20-lactone–hemiketal bridge. The synthetic strategy prominently features a radical cascade initiated by photoinduced electron transfer (PET) and a subsequent BROC radical cyclization as key transformations. As outlined in Scheme 12, silyl enol ether formation from intermediate 91, followed by a Diels–Alder reaction with acrolein, furnished adduct 92. The core bicyclo[3.2.1]octane scaffold 94 was then constructed via a PET-mediated radical cyclization developed by the Mattay group,103 employing 9,10-dicyanoanthracene (93) as photosensitizer under 430 nm irradiation. Remarkably, the transformation proceeded exclusively through a 6-endo-trig cyclization followed by a 5-exo-dig closure. Subsequent steps included dehydrogenation at C5–C6 and installation of a terminal alkene at C10, thereby setting the stage for BROC radical cyclization. Treatment of 95 with Fe(dpm)3/PhSiH3, followed by desilylation at C17, furnished compound 96 in excellent yield. Notably, the steric hindrance from the bulky TMS group at C17 effectively suppressed undesired hydrogenation of the C16C17 double bond under HAT conditions, thus ensuring the chemoselectivity of the radical cyclization. Final elaboration, including conversion of the cis- to the trans-A/B ring fusion, completed the total synthesis of sculponin U (90).
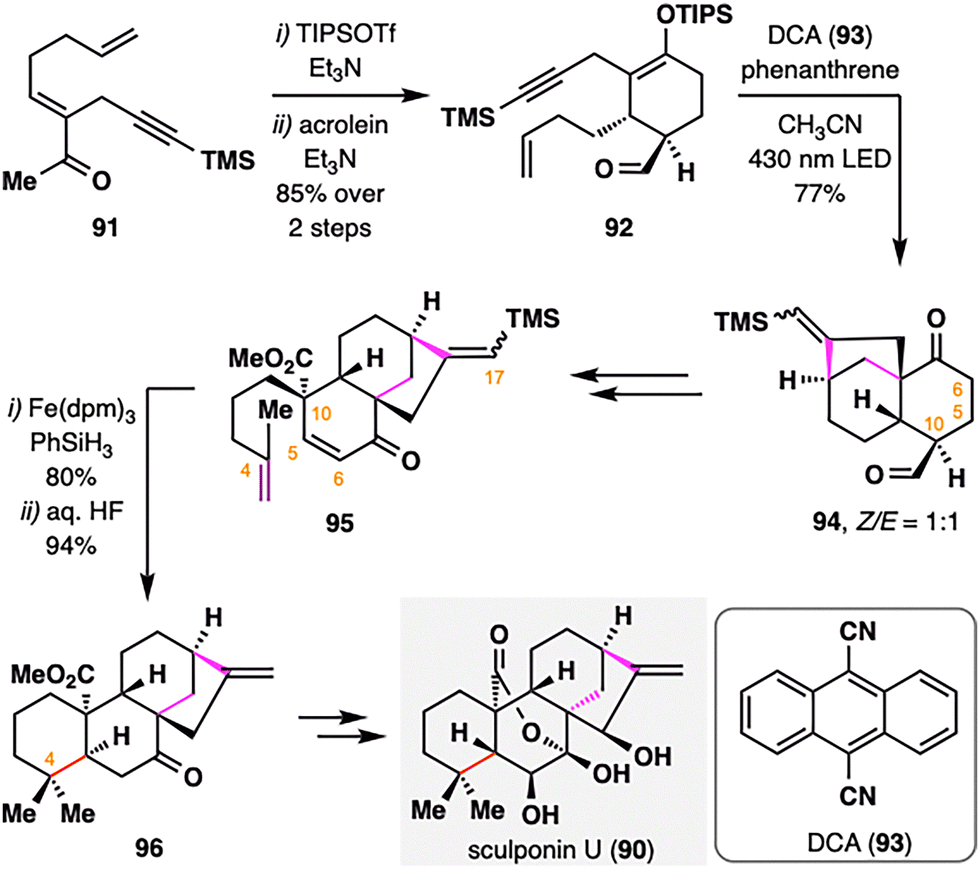 | ||
| Scheme 12 Total Synthesis of sculponin U (90) by Liu, Fu, and co-workers. | ||
Beyond the construction of the C4 quaternary center in the aforementioned synthesis of sculponin U (90), BROC radical cyclization also demonstrates powerful utility in forging the C10 quaternary carbon in other tetracyclic diterpenoids. In 2023, C. Fan and co-workers developed an asymmetric strategy for the divergent synthesis of nine C8-ethano-bridged tetracyclic diterpenoids, including natural products 97–101, with MHAT-mediated BROC radical cyclization as a key step.104 As illustrated in Scheme 13, the bicyclo[3.2.1]octane scaffold 103 was efficiently assembled from readily accessible cyclohexenone 102via a Au(I)-catalyzed Conia-ene cyclization. Subsequent functional group manipulation of 103 furnished intermediate 104, which underwent intermolecular reductive coupling with fragment 105 to generate a mixture of C7 diastereomers, 106a and 106b. Strategic application of BROC radical cyclization on 106a and 106b delivered the corresponding tetracyclic products 107 and 108, each bearing a C10 quaternary center, in good yields and with high diastereoselectivity. Notably, the optimized protocol—employing Fe(dbm)3 and Ph(iPrO)SiH2 in the presence of Na2HPO4—proved crucial in suppressing both the formation of undesired diastereomers at C10 and over-reduction of the C16C17 olefin, thereby ensuring excellent regio- and stereocontrol. Tetracycles 107 and 108 subsequently served as versatile intermediates for the downstream collective synthesis of C8-ethano-bridged diterpenoids.
 | ||
| Scheme 13 C. Fan's divergent synthesis of ent-kaurane-, ent-atisane-, ent-beyerane-, ent-trachylobane-, and ent-gibberellane-type diterpenoids (97–101). | ||
(−)-Retigeranic acid A (109), isolated from Himalayan lichens,105 features a unique pentacyclic architecture comprising an angular triquinane subunit and eight stereocenters—rendering it an exceptionally challenging and attractive synthetic target. Recently, the groups of S.-H. Wang and X. Chen collaboratively achieved its total synthesis,106 highlighted by a late-stage 5-endo-trig BROC radical cyclization—an anti-Baldwin closure that is topologically disfavored (Scheme 14). The synthesis commenced from ester 110, which underwent α-substitution, Krapcho decarboxylation, Wittig olefination, and a Conia–ene cyclization to furnish the [5.5]-fused D/E ring system 111. In parallel, aldehyde 112 was elaborated into the A/B ring fragment 113via a sequence comprising Horner–Wadsworth–Emmons (HWE) homologation, Prins cyclization, hydrogenation, and Appel chlorination. Intramolecular substitution followed by triflation with PhNTf2 afforded vinyl triflate 114, which underwent Nozaki–Hiyama–Kishi (NHK) coupling with fragment 111. Subsequent DMP oxidation furnished ketone 115. The pivotal BROC radical cyclization was then applied to forge the central C ring in 116via a topologically disfavored 5-endo-trig pathway, proceeding in an impressive 91% yield and showcasing the unique capability of this radical approach in accessing otherwise inaccessible ring systems. Although the resulting stereochemistry at C2 was epimeric relative to the natural product, a subsequent IBX dehydrogenation installed enone 117, enabling downstream stereochemical correction. With the core skeleton fully constructed, a final four-step sequence completed the synthesis of (−)-retigeranic acid A (109).
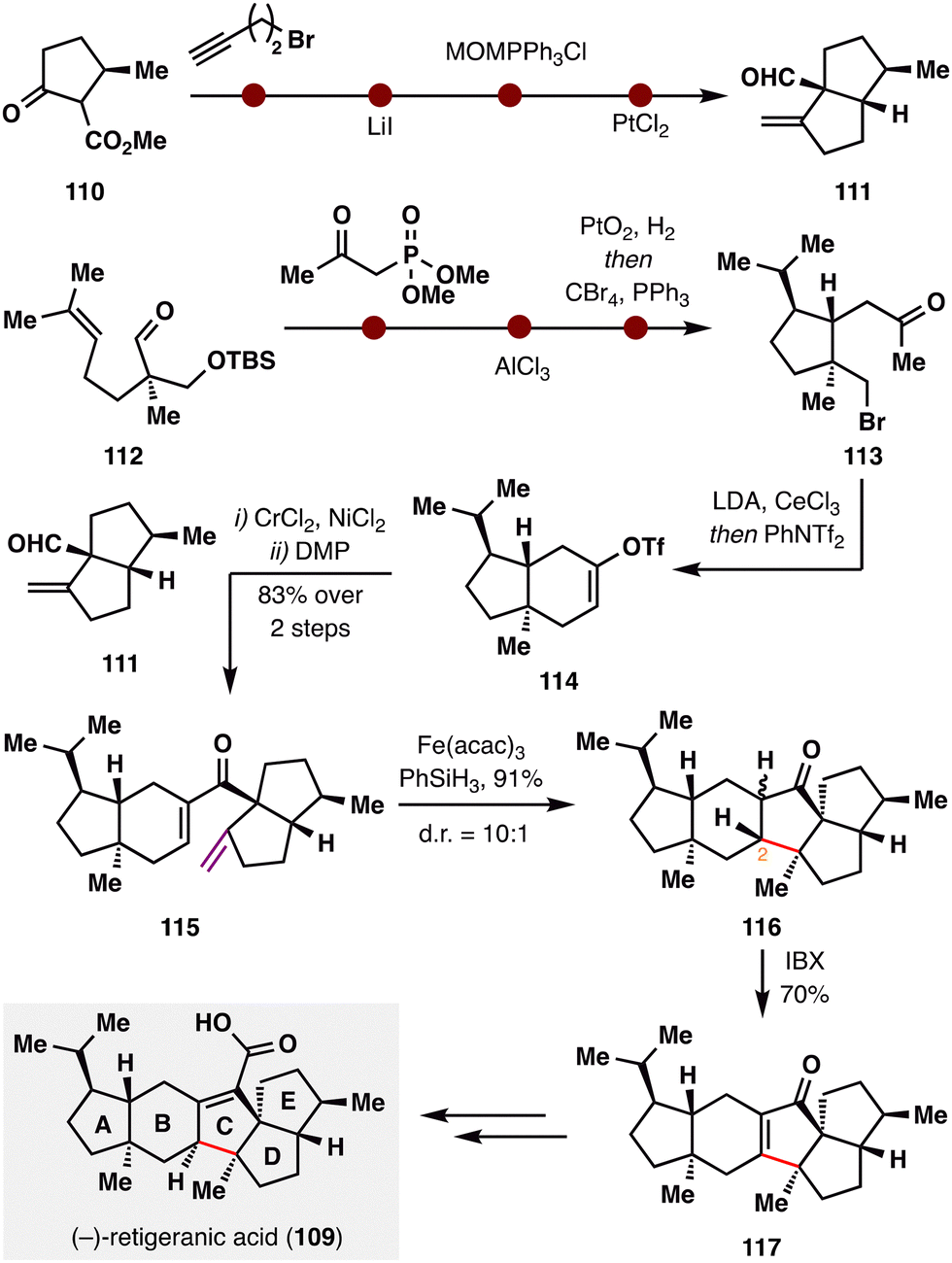 | ||
| Scheme 14 Total synthesis of (−)-retigeranic acid (109) by X. Chen, S.-H. Wang, and co-workers. | ||
Cycloaurenones and dysiherbols represent a class of sesquiterpene quinones/quinols characterized by a fused 6/6/5/6 tetracyclic framework that incorporates either a cis- or trans-decalin motif and four contiguous stereocenters.107 Recently, the Lu group reported the first enantioselective and divergent total synthesis of these natural products,108 featuring a desymmetrizing BROC radical cyclization and a newly developed copper-catalyzed enantioselective conjugate addition. In their synthesis of cycloaurenones (Scheme 15(A)), the route commenced with the installation of two nonadjacent 1,4-stereocenters via a desymmetrizing enantioselective hydrogenation of bis-α,β-unsaturated Weinreb amide 121, delivering 122 in d.r. > 99:1 and 99% e.e. Subsequent transformations converted 122 into cyclohexadienone 124, thereby setting the stage for the key BROC radical cyclization. Treatment of 124 with Fe(acac)3 and PhSiH3 induced an intramolecular cyclization to furnish fused tricyclic intermediate 125 in 85% yield. The high stereoselectivity of this desymmetrizing radical cyclization was attributed to pseudo-1,3-diaxial interactions between the methyl groups at C5 and C9, which directed the formation of the stereogenic quaternary center at C9. Further elaboration of 125, including SeO2-mediated allylic oxidation and Pd-catalyzed reductive Heck coupling, yielded the tetracyclic intermediate 126, which was advanced to cycloaurenones (118–120). In the synthesis of dysiherbols (Scheme 15(B)), a Cu-catalyzed desymmetrizing 1,4-conjugate methylation to 124 furnished 132, which upon exposure to the same BROC radical cyclization conditions delivered fused decalin 133 in 78% yield as a single diastereomer. Subsequent IBX dehydrogenation followed by an intramolecular conjugate addition furnished common intermediate 135, which was further functionalized to afford dysiherbols (127–131). In both sequences, BROC radical cyclization proved pivotal in constructing the polycyclic cores and establishing multiple stereocenters in a single transformation, thereby enabling a concise and modular approach to these architecturally complex sesquiterpenoids.
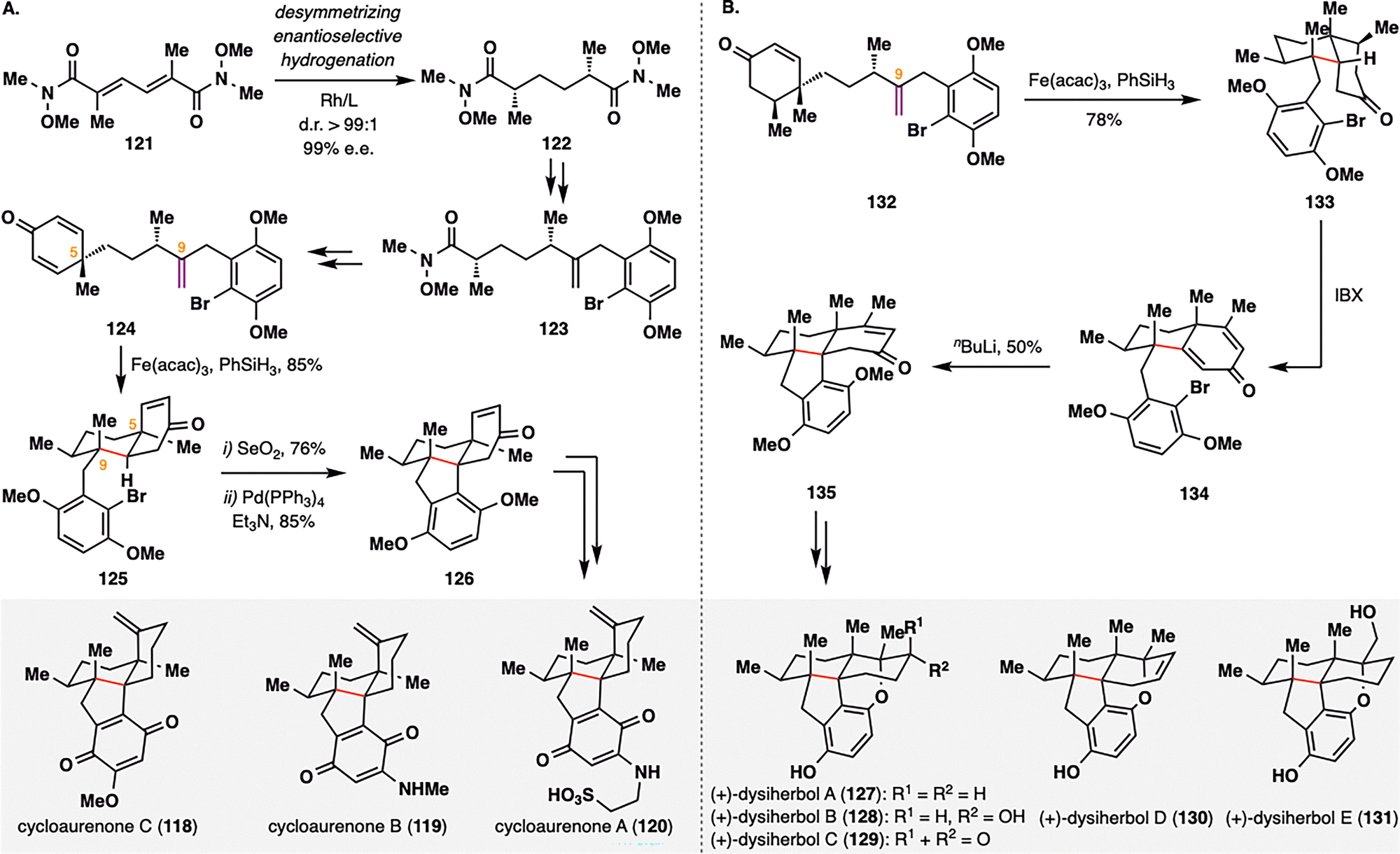 | ||
| Scheme 15 Lu's collective total synthesis of cycloaurenones (118–120) and dysiherbols (127–131). | ||
Building upon these elegant applications of MHAT chemistry in assembling complex polycyclic terpenoid frameworks, Dai and co-workers further demonstrated its versatility in addressing one of the most formidable topological challenges in diterpenoid synthesis: the stereoselective construction of a sterically congested cyclopropane ring (Scheme 16).109 (−)-Peyssonnoside A (137), a sulfated diterpene glycoside isolated from the red alga Peyssonnelia sp.,110 features an unprecedented 5/6/3/6 tetracyclic carbon skeleton incorporating a highly hindered pentasubstituted cyclopropane moiety—posing a significant synthetic challenge. Dai and co-workers addressed this complexity by leveraging a counterintuitive BROC radical cyclization to forge the cyclopropane ring with remarkable stereocontrol. The synthesis commenced from 2-methylcyclopentenone (138), which underwent asymmetric conjugate addition of iPrMgCl in the presence of Cu(OTf)2/chiral N-heterocyclic carbene,111 alkylation with alkyne 139, and Pd-catalyzed syn-hydroarylation of the alkyne with aryl boronic acid 140 to furnish alkene 141.112 Subsequent Ir-catalyzed asymmetric hydrogenation delivered intermediate 142 with exceptional diastereoselectivity (d.r. = 51:1). The MHAT precursor 144 was assembled in three steps from 142, involving vinyl triflation, demethylation, and a Pd(II)/RuPhos-catalyzed dearomative cyclization.113 Upon treatment with BROC radcial cyclization conditions, a tertiary radical was generated that underwent an intramolecular Giese-type addition to the adjacent enone, constructing the tetracyclic product 145 in 65% yield as a single diastereomer. The success of this unusual transformation was attributed to rapid single-electron reduction of the radical intermediate to a stabilized carbanion, thereby preventing fragmentation of the newly formed cyclopropane ring. A final six-step sequence completed the total synthesis of (−)-peyssonnoside A (137) in just 13 steps from 138.
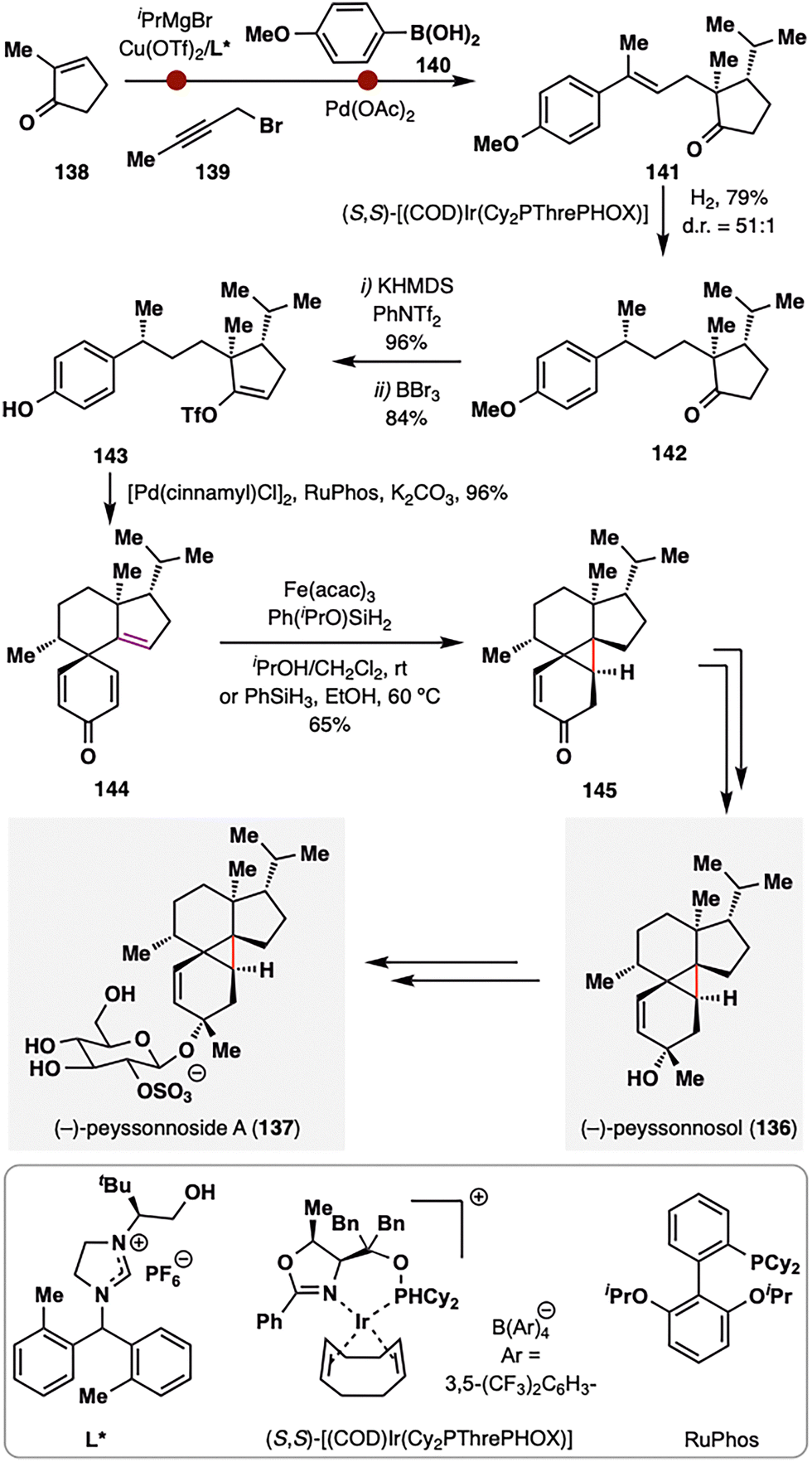 | ||
| Scheme 16 Dai's total synthesis of (−)-peyssonnoside A (137). | ||
As illustrated in the previous example, the BROC radical cyclization has proven particularly effective for constructing small-ring cycloalkanes with substantial ring strain. Building on this strategy, Carreira and co-workers achieved the first total synthesis of (+)-pedrolide (146, Scheme 17),114 a densely substituted carane-containing diterpenoid isolated from Euphorbia pedroi.115 The carane scaffold, frequently encountered in spurge-family natural products, has long been regarded as a formidable challenge for synthetic chemists due to its strain and stereochemical complexity. The synthesis began with the efficient construction of ketone 152 from intermediate 147 through a sequence involving oxidative dearomatization, enantioselective 1,4-conjugate methyl addition, Rubottom oxidation, a diastereoselective Grignard addition, and TES protection of the resulting diol. Treatment of 152 with Fe(acac)3 and PhSiH3 under BROC radical cyclization conditions enabled a MHAT-driven radical addition into the adjacent enone, furnishing the highly strained cyclopropyl ketone 153 in an impressive 98% yield. This transformation underscores the utility of MHAT-based radical chemistry in assembling highly functionalized carane skeletons with exceptional efficiency. A late-stage, strategically designed Diels–Alder cascade was then employed to elaborate intermediate 156, ultimately culminating in the concise total synthesis of (+)-pedrolide (146).
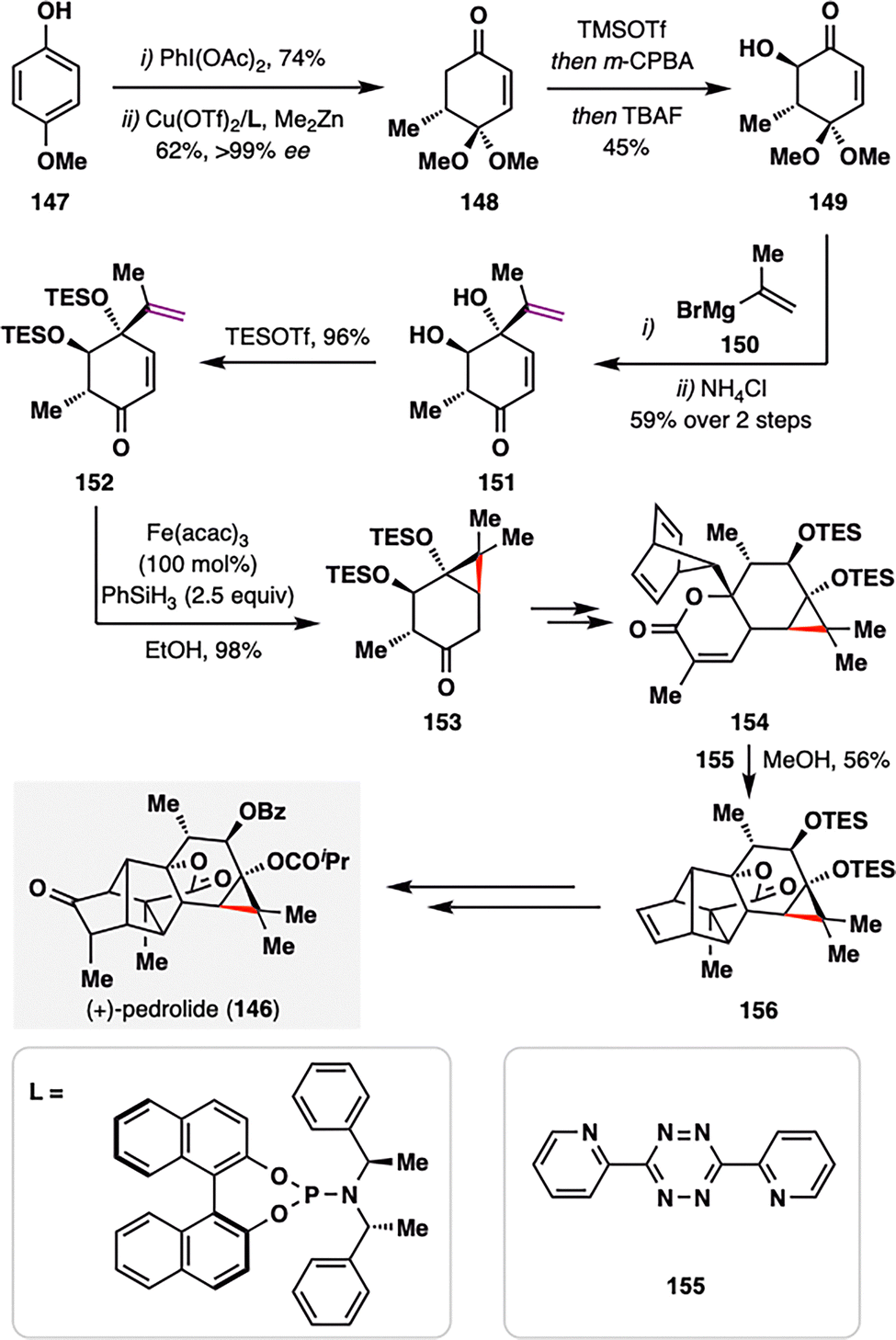 | ||
| Scheme 17 Carreira's total synthesis of (+)-pedrolide (146). | ||
Recently, Ma and co-workers completed the total synthesis of ent-polytrichastrene B (157), a structurally complex natural product bearing a highly strained and sterically congested tetrasubstituted cyclopropane motif, featuring MHAT-based key transformations (Scheme 18).116 The installation and stereochemical control of this feature posed significant synthetic challenges, which were elegantly addressed through the BROC radical cyclization similar to previous examples. The synthesis commenced with the asymmetric construction of cyclohexenone-aldehyde 161, achieved via a redox-relay Heck alkenylation between enone triflate 158 and a homoallylic alcohol 159 under Sigman's conditions.117 Following a series of elaborations, the core framework of ent-polytrichastrene B was efficiently assembled. The pivotal diastereoselective reduction of a highly hindered, unactivated tetrasubstituted olefin of 162 was accomplished using MHAT-initiated hydrogenation, enabling stereocontrolled access to the desired intermediate 163. Subsequent seven-step manipulations led to the formation of an unsaturated ester 164, which underwent a successful 3-exo-trig radical cyclization under BROC conditions, furnishing tetracyclic intermediate 165 as a single diastereomer. This intermediate, featuring the target cyclopropane motif, was converted to ent-polytrichastrene B (157) in two additional steps.
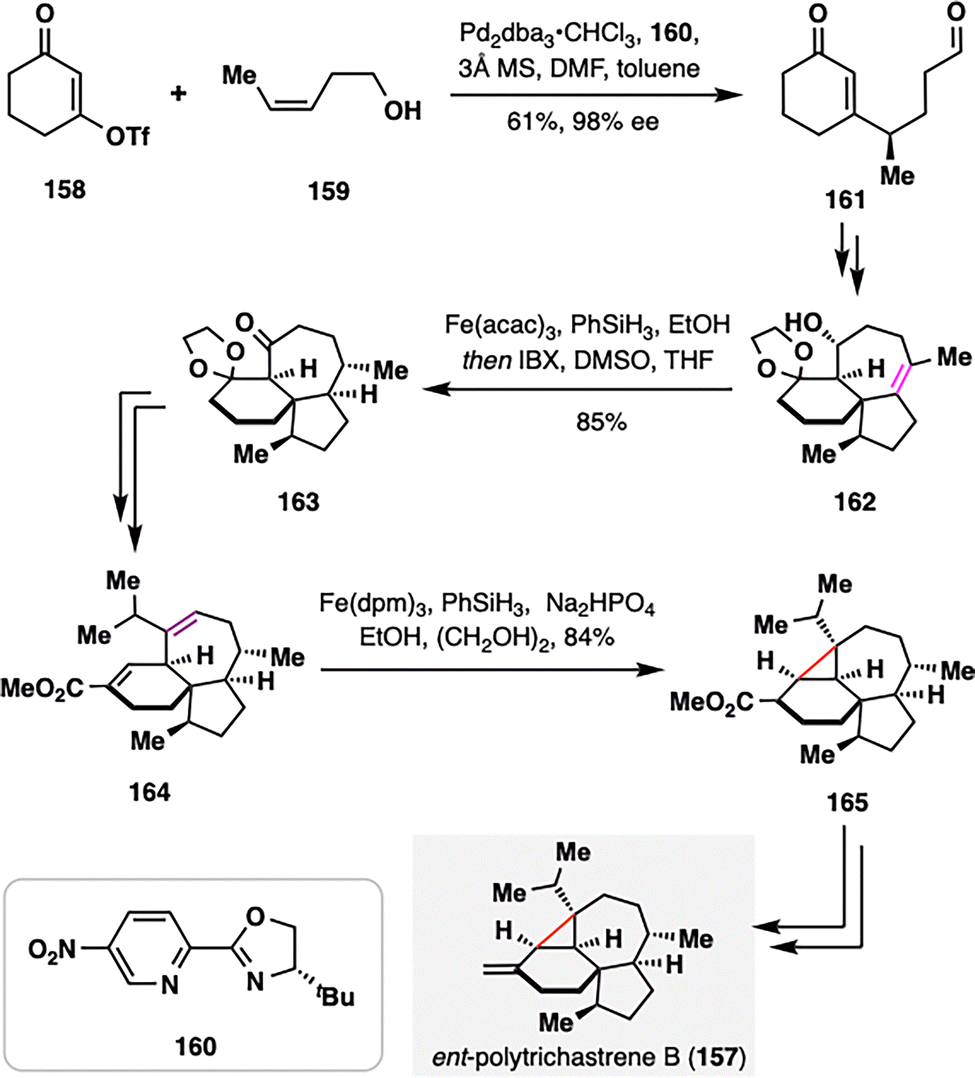 | ||
| Scheme 18 Z. Ma's asymmetric synthesis of ent-polytrichastrene B (157). | ||
Building on their prior success in strained cyclopropane construction, Dai and co-workers recently extended this strategy to forge a cyclobutane motif in their total synthesis of psathyrin A (166),118 a diterpene natural product featuring an unique 6/4/5/5 tetracyclic carbon skeleton isolated from the fermentation broth of Psathyrella candolleana.119 As outlined in Scheme 19, the synthesis began with a copper-catalyzed enantioselective conjugate addition of an isopropyl Grignard reagent to cyclopentenone 167, followed by propargylation with 1-bromo-2-butyne to afford 169. Subsequent elaboration provided intermediate 170, which underwent a gold(I)-catalyzed Conia-ene cyclization to furnish 171. Birch reduction of the aromatic ring in 171, accompanied by alkene migration, gave 172 and established the requisite alkene–enone pairing for the forthcoming cyclization. Subjecting 172 to BROC conditions successfully delivered the strained cyclobutene 173, overcoming the intrinsic ring strain of this scaffold. From this intermediate, further elaboration completed the synthesis of psathyrin A (166). This achievement underscores the power of BROC cyclization for constructing highly strained carbocycles in complex diterpene architectures.
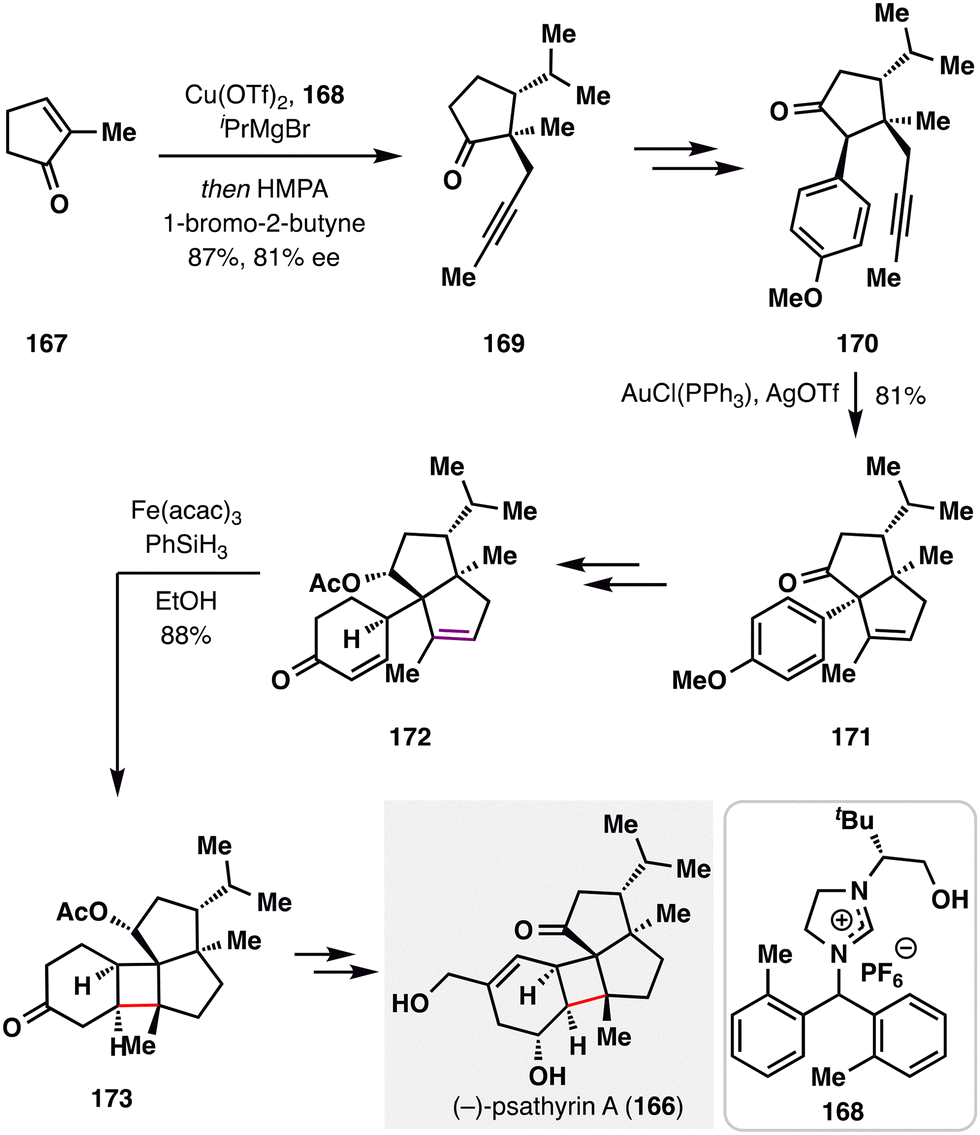 | ||
| Scheme 19 Dai's asymmetric total synthesis of psathyrin A (166). | ||
MHAT radical cyclization can extend beyond traditional alkene–enone systems to encompass alkene–nitrile couplings, as exemplified in the total synthesis of cyclopianes (32–34) by Zhai and co-workers (Scheme 20).120 These architecturally complex natural products—previously addressed in Snyder's synthesis (Scheme 6)—pose significant synthetic challenges due to their dense, angular polycyclic frameworks. Zhai's synthesis began with the asymmetric Corey–Bakshi–Shibata (CBS) reduction of readily available trimethylcyclopentenone (174), followed by a tandem vinylation/Claisen rearrangement and van Leusen homologation to furnish nitrile 177, thereby establishing the crucial alkene–nitrile coupling motif. Subjecting 177 to BROC radical cyclization conditions [Fe(acac)3, PhSiH3] effected an intramolecular radical cyclization followed by acid-mediated hydrolysis to deliver the cis-biquinane scaffold 178 in 94% yield as a single diastereomer, establishing a new quaternary center with excellent stereocontrol. Subsequent elaboration of 178 yielded 179, featuring an alkene–alkyne system primed for a Co-mediated Nicholas/Pauson–Khand reaction, which efficiently assembled the triquinane core 180. A final Danheiser annulation with cumulene 181 forged the highly strained angular triquinane skeleton 182 in 89% yield and with good diastereoselectivity (d.r. > 4:1), thus completing the construction of the core cyclopiane framework. Final functional group manipulations converted 182 into natural products 32–34.
 | ||
| Scheme 20 Zhai's total synthesis of cyclopianes (32–34). | ||
As the scope of MHAT chemistry continues to expand beyond classical radical acceptors, its compatibility with unconventional motifs—such as alkenylsilanes—further underscores its versatility in complex molecule construction. (−)-Habiterpenol (183), a microbial G2 checkpoint inhibitor isolated from Phytohabitans sp.,121 was synthesized by Nagamitsu, Ohtawa, and co-workers via a key MHAT-initiated redox radical cyclization involving an atypical alkene–alkenylsilane pairing (Scheme 21).122 The synthesis commenced with the coupling of readily available aldehyde 185 and allylsilane 186, followed by DMP oxidation to furnish alkenylsilane 187 with moderate diastereoselectivity at C3 (d.r. = 1.2:1). Subjecting 187 to BROC radical cyclization conditions [Fe(acac)3, PhSiH3] triggered a redox radical cyclization that efficiently forged the central six-membered ring and simultaneously established two stereocenters with excellent diastereocontrol (single diastereomer), affording pentacyclic intermediate 188 in 73% yield. Final deoxygenation steps completed the total synthesis of (−)-habiterpenol (183). Beyond enabling this total synthesis, the success of this transformation highlights the potential of MHAT chemistry to engage previously unreactive functional groups, thereby opening new avenues for radical-based bond constructions.
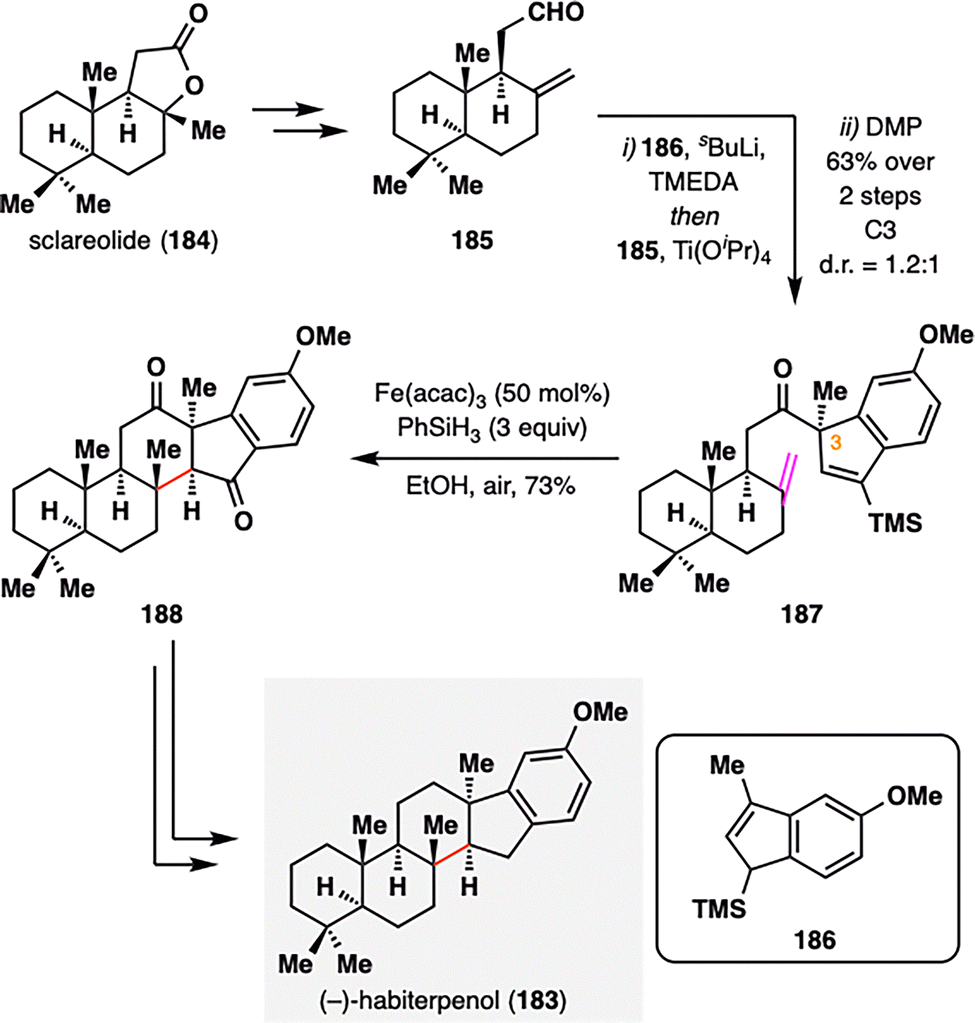 | ||
| Scheme 21 Synthesis of (−)-habiterpenol (183) by Nagamitsu, Ohtawa, and co-workers. | ||
While prior examples of MHAT-based radical cyclization largely relied on 1,1-disubstituted or monosubstituted alkenes as radical donors, Gui and co-workers strategically employed a rare diene moiety as a radical donor in the total synthesis of swinhoeisterols A–C (189–191),123 complex marine-derived steroids isolated from the Xisha sponge Theonella swinhoei,124,125 thereby broadening the synthetic utility of MHAT chemistry (Scheme 22). These natural products feature a synthetic challenging seven-membered ring and three contiguous stereocenters at C13, C17, and C20. To address this, Gui's team employed a BROC radical cyclization, treating 193 with Fe(dpm)3 and Ph(iPrO)SiH2, which furnished the desired intramolecular cyclized intermediate as a mixture of diastereomers with a ratio of 2.3:1 at C17 and a ratio of 1:1.5 at C20. Upon treatment with tBuOK, base-induced epimerization at C20 selectively furnished the desired (S)-configured isomer 194 with high diastereoselectivity (7.7:1). Interestingly, the E/Z geometry of 193 had minimal effect on stereochemical outcome. This strategy enabled construction of the key 6/6/5/7 tetracyclic core in 44% yield over two steps. Subsequent functional group manipulations from this common scaffold allowed for the divergent synthesis of swinhoeisterols A–C (189–191).
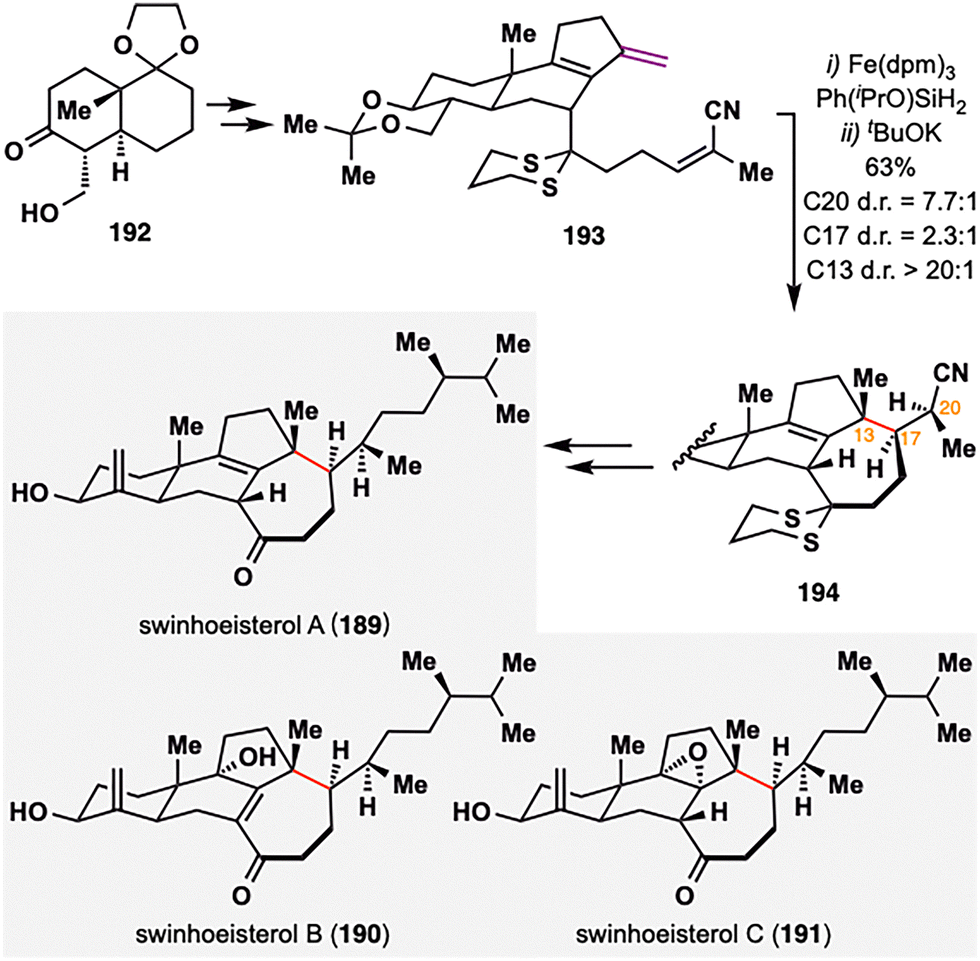 | ||
| Scheme 22 Gui's total synthesis of swinhoeisterols A–C (189–191). | ||
The expanding scope of MHAT-based radical additions—from classical alkene–enone systems to more unconventional partners such as nitriles, alkenylsilanes, and vinyl phenyl sulfonates—has enabled new retrosynthetic strategies for assembling structurally diverse natural products. A striking example of this versatility is the application of MHAT chemistry to alkene–ketone pairs, as demonstrated by Bradshaw and co-workers in their concise total synthesis of (−)-4-epi-presilphiperfolan-8α-ol (195),126 a fungal-derived sesquiterpenoid featuring a fused 6/5/5 tricyclic core (Scheme 23). The synthesis commenced from (R)-pulegone (196), which was transformed into diene 197via the installation of two requisite alkene units. A ring-closing olefin cross-metathesis using the Grubbs II catalyst efficiently forged the eight-membered bridged ring, affording the key keto–alkene intermediate 198 in 54% yield. Exposure of 198 to BROC radical cyclization conditions [Fe(acac)3, PhSiH3] generated a tertiary alkyl radical, which underwent a transannular cyclization onto the ketone to form alkoxy radical intermediate 199. Subsequent reduction delivered (−)-4-epi-presilphiperfolan-8α-ol (195) as a single diastereomer in 66% yield. Notably, this transannular radical cyclization proceeded with excellent efficiency and stereocontrol, further exemplifying the potential of MHAT-based strategies in constructing complex and strained polycyclic architectures.
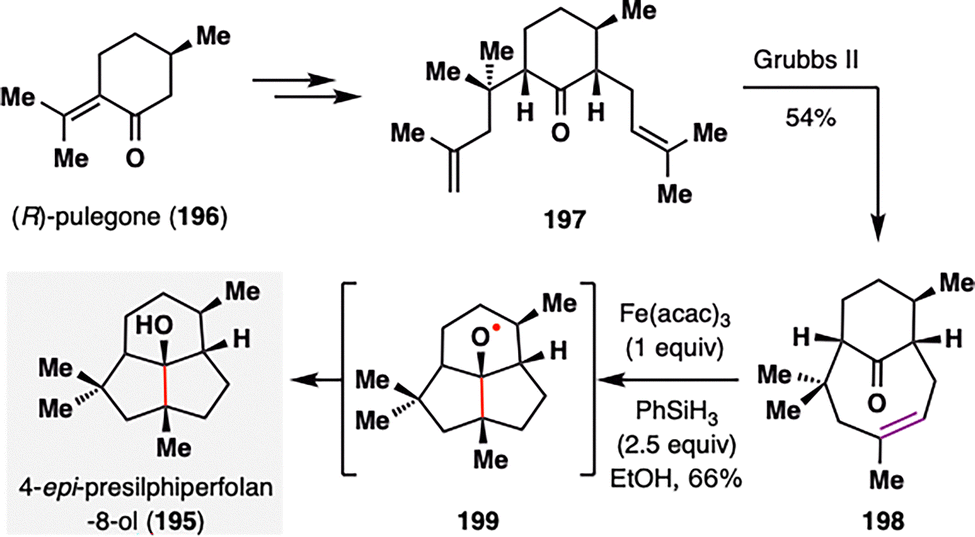 | ||
| Scheme 23 Bradshaw's synthesis of (−)-4-epi-presilphiperfolan-8α-ol (195). | ||
Beyond classical thermal conditions, photochemical activation has recently emerged as a powerful strategy for modulating MHAT reactivity—enabling new catalytic manifolds and improving functional group compatibility. In this context, Luo and co-workers employed a visible-light-promoted, Co(TPP)-catalyzed MHAT radical addition in their total synthesis of (−)-triptonide (200),127 offering valuable mechanistic and synthetic insights into the optimization of MHAT-initiated coupling reactions (Scheme 24). The synthesis began with the preparation of enone 206 from ketone 201via a sequence involving vinylMgBr addition, bromination, allylation, and acetal deprotection. Initial attempts at MHAT-mediated radical cyclization using conventional BROC radical cyclizayion conditions [Fe(acac)3, PhSiH3] furnished tricyclic product 208 in only 41% yield. To improve the synthetic efficiency, the authors investigated a cobalt-catalyzed alternative. Remarkably, the combination of Co(TPP), PhSiH3, and visible light enabled the cyclization to proceed efficiently, affording 208 in 68% yield as a single diastereomer. This enhanced performance was attributed to photoexcitation-assisted homolysis of the Co–C bond, which facilitated effective radical generation and efficient turnover of the active catalyst. Final transformations—including acid-mediated deprotection, an intramolecular Friedel–Crafts cyclization, and global oxidation—completed the concise synthesis of (−)-triptonide (200).
 | ||
| Scheme 24 Luo's total synthesis of (−)-triptonide (200). | ||
Building upon the expanding versatility of MHAT chemistry, its application has recently been extended to the synthesis of aromatic steroids—a structural class traditionally considered less amenable to radical-based strategies. Viridin (209),128 isolated from Gliocladium virens, and its biogenetic congener viridiol (210) feature a highly oxygenated steroidal framework incorporating a fused furan ring.129 In a pioneering study, Gao and co-workers reported the first strategic use of MHAT radical cyclization in the synthesis of aromatic steroids, providing a new platform for the rapid construction of terpenoid architectures such as aromatic abietane diterpenoids (Scheme 25).130 The synthesis began with an intramolecular [3+2] cycloaddition of readily accessible precursor 211, furnishing compound 212 bearing a densely substituted six-membered ring. Subsequent functional group manipulations delivered 213, which underwent coupling with dihydroindenol fragment 214 to yield ketone 215—the substrate for the pivotal MHAT step. Subjecting 215 to Co-catalyzed MHAT cyclization promoted intramolecular radical addition between the terminal alkene and the aromatic ring, forging a new six-membered ring and establishing an all-carbon quaternary center. Notably: (i) the transformation proceeded with excellent substrate-controlled stereoselectivity via a twisted boat-like transition state that minimized 1,3-diaxial interactions between the OTBS group and the nascent methyl substituent; (ii) only catalytic amounts of Co-catalyst and PhSiH3 were required, as the aromatization-driven process released a hydrogen atom, thus regenerating Co–H. This highly efficient sequence furnished tetracyclic intermediate 216 in 79% overall yield across the MHAT cyclization, deprotection, and oxidation steps. Final elaboration, including furan ring installation and oxidation-state adjustments, completed the total synthesis of viridin (209) and viridiol (210), showcasing a powerful new strategy for constructing oxygenated steroid frameworks using radical-based methodologies.
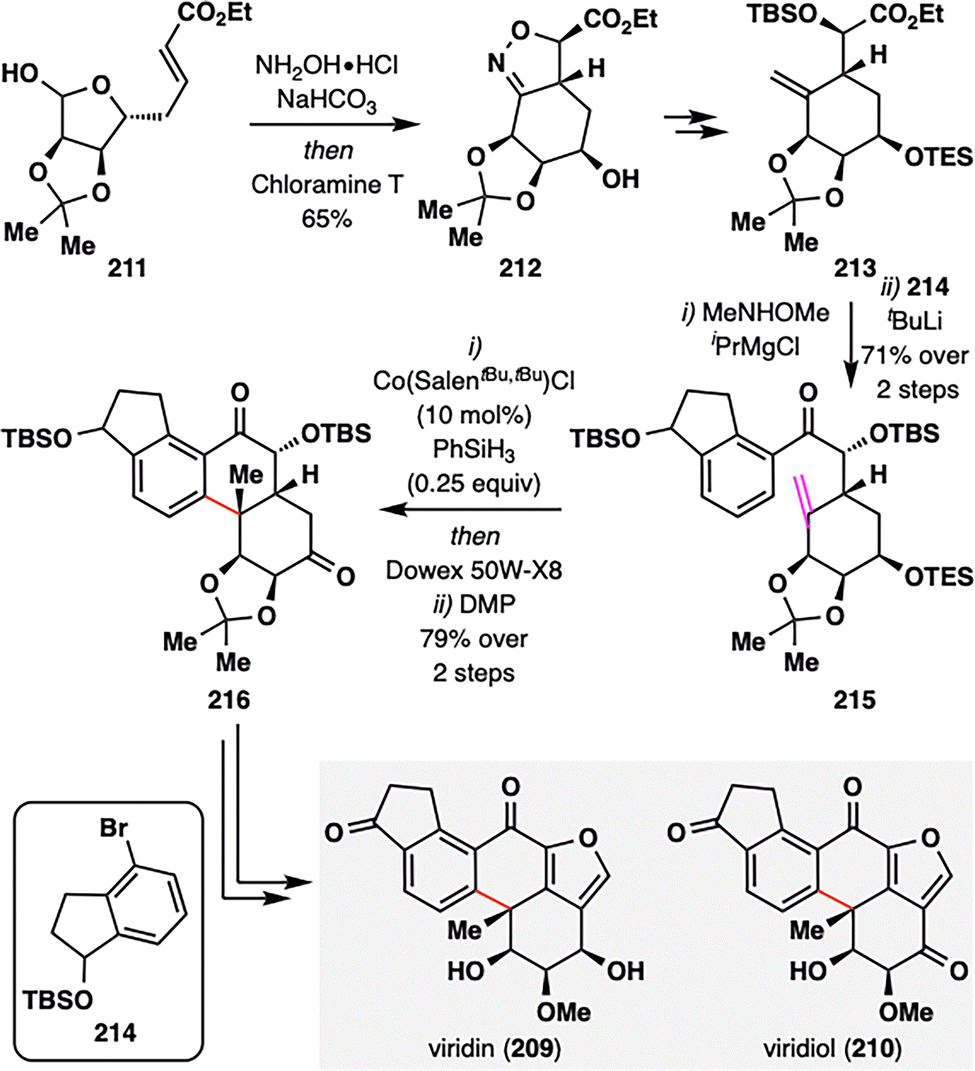 | ||
| Scheme 25 Gao's total synthesis of viridin (209) and viridiol (210). | ||
Dankasterones A (217) and B (218), and periconiastone A (219), isolated from marine sponges,131–133 share an angularly fused tricyclic framework featuring two adjacent quaternary stereocenters—structural elements that present formidable challenges for chemical synthesis. Z. Ma and co-workers reported an enantioselective total synthesis of these complex terpenoids, employing a key MHAT-mediated radical cyclization to forge the cis-decalin core embedded within a 6/6/6/5 tetracyclic architecture (Scheme 26).134 The requisite MHAT precursor 222 was assembled via a convergent strategy from aldehyde 220 and bromide 221, followed by DMP oxidation. Upon exposure to an in situ-generated CoIII–H complex, a tertiary radical was generated at the terminal alkene, which then underwent intramolecular addition to the enone moiety. Subsequent PhI(OAc)2-mediated oxidation furnished enone 224. Remarkably, the MHAT cyclization proceeded with high diastereoselectivity, selectively delivering the desired cis-fused decalin framework. From the isomerized enone 225, a radical rearrangement was employed: allylic oxidation generated a hydroperoxide intermediate,135 which underwent Fe(II)-mediated reductive rearrangement to furnish spirocyclic intermediate 226. Final functional group manipulations enabled divergent elaboration to the target natural products—dankasterones A and B (217–218) and periconiastone A (219).
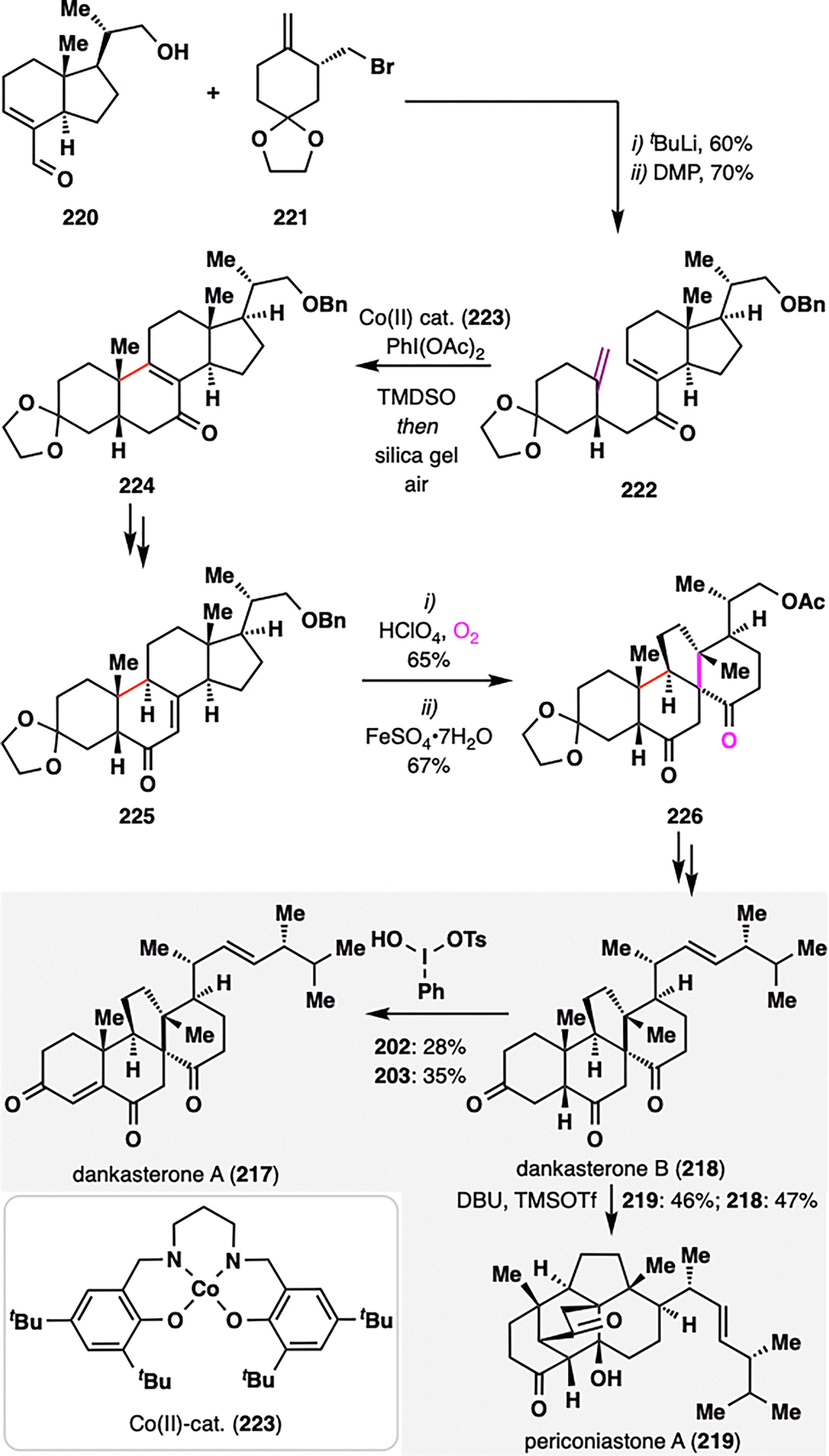 | ||
| Scheme 26 Z. Ma's asymmetric total synthesis of dankasterones A and B (217–218), and periconiastone A (219). | ||
Ding and co-workers accomplished the first asymmetric total synthesis of (+)-davisinol (227),136 a C11-oxygenated hetisine-type alkaloid isolated from Delphinium davisii Munz (Scheme 27).137 A pivotal feature of this synthesis is a HAT-initiated transannular redox radical addition, in which a tertiary radical—generated from an electron-deficient enone—undergoes intramolecular coupling with an electron-rich alkene, followed by oxygen trapping. The synthesis began with the construction of diketone 233, accessed via a highly diastereoselective (d.r. > 32:1) oxidative dearomatization/intramolecular Diels–Alder (IMDA) cascade from intermediate 232, which was in turn derived from 229 through a sequence of transformations, including a key acylative kinetic resolution.138 Crucially, The MHAT-mediated transannular redox cyclization was made possible by the favorable spatial orientation of the olefinic carbons at C9 and C10 in 233. Under optimized conditions, the desired polycyclic product 235 was obtained in 83% yield as a single diastereomer through radical transformation shown in 234. Notably, fine-tuning the oxidation state at specific positions proved essential for downstream diversification toward other hetisine alkaloids, underscoring the broad utility of MHAT-based redox cyclizations in accessing this architecturally complex family. Final oxidation-state adjustments completed the total synthesis of the natural product (+)-davisinol (227).
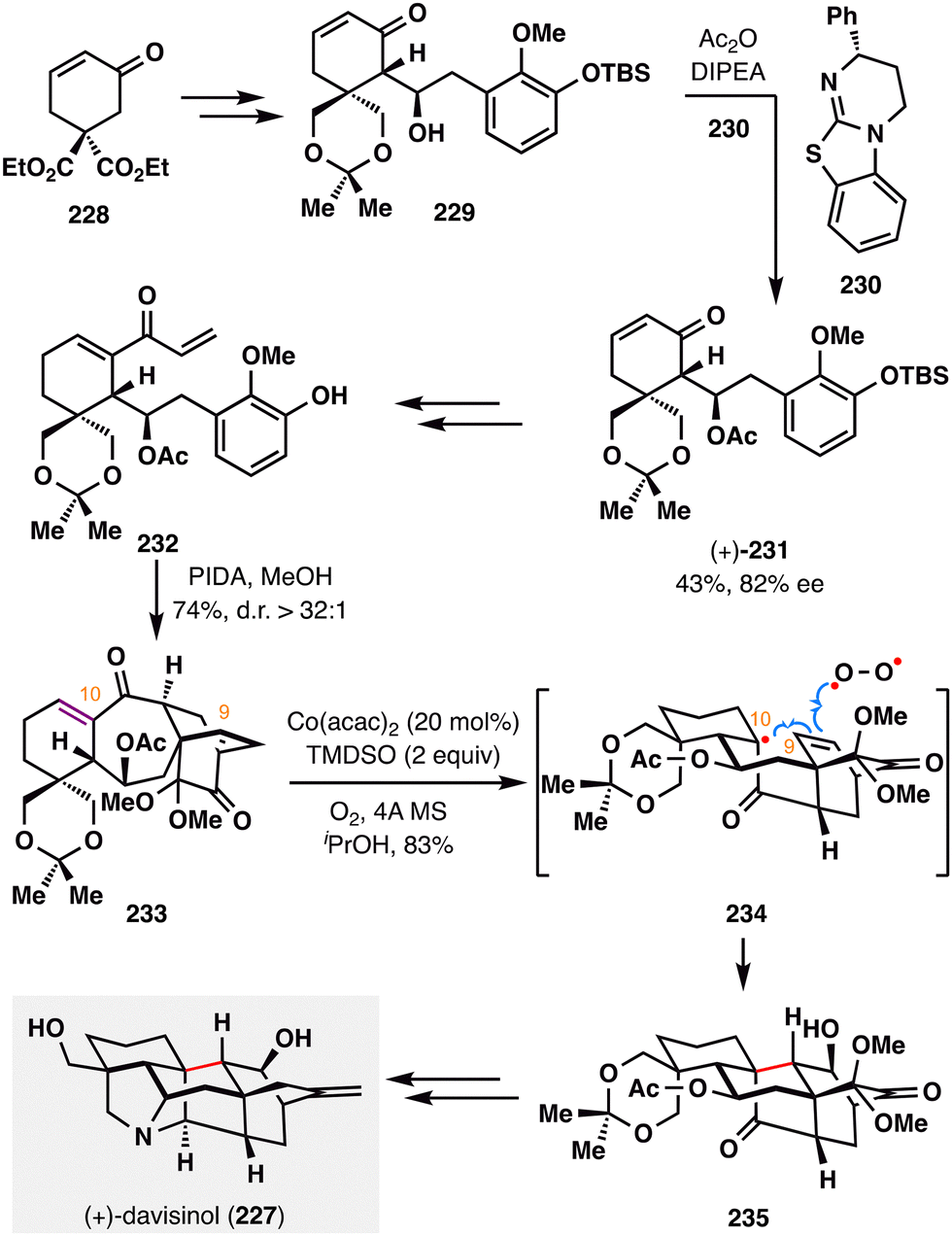 | ||
| Scheme 27 Ding's total synthesis of (+)-davisinol (227). | ||
Navirine C (236), a hetidine-type alkaloid isolated from Aconitum naviculare,139 was synthesized by D. Ma and co-workers through a concise and efficient route that leveraged a Mn-catalyzed alkene–nitrile MHAT radical cyclization to establish the core framework for subsequent nitrogen heterocycle construction (Scheme 28).140 The synthesis began with the preparation of phenol 239, in which a chelation-controlled conjugate addition between nitrile 237 and Grignard reagent 238 was employed—marking the first use of this strategy in natural product synthesis.141 Subsequent elaboration of 239, including oxidative dearomatization and an intramolecular Diels–Alder cascade, furnished tetracyclic intermediate 240. This was further converted to dinitrile 241, strategically positioned to serve as the radical acceptor in the key MHAT cyclization. Upon treatment with Shenvi's MHAT conditions [Mn(dpm)3, PhSiH3, TBHP],142 an intramolecular alkene–nitrile coupling occurred wherein the alkyl radical, derived from the terminal olefin, added to the cyano group to form an imine, thereby forging the complex pentacyclic framework 242 in 68% yield. The resulting imine intermediate was then subjected to Pd/C-catalyzed hydrogenation, followed by a one-pot Mannich reaction, delivering tertiary amine 243. Final functional group manipulations completed the synthesis of navirine C (236), highlighting the power of alkene–nitrile MHAT radical cyclization as a strategic tool for efficiently assembling nitrogen-containing alkaloid scaffolds.
 | ||
| Scheme 28 Total synthesis of the proposed structure of navirine C (236) by D. Ma and co-workers. | ||
Despite the broad utility of MHAT cyclizations, achieving efficient radical closure onto trans-fused 6/5 ring systems remains a formidable synthetic challenge. Liu and co-workers addressed this issue in their asymmetric total synthesis of euphane triterpenoids (245–247),143 a structurally diverse family of natural products characterized by a 6/6/6/5 tetracyclic core derived from six isoprene units (Scheme 29).144 Their approach explored how substrate architecture and catalytic conditions govern MHAT reactivity and selectivity in the formation of such complex frameworks. The synthesis began with the preparation of key MHAT precursors 251, constructed via a Liebeskind stannane–thioester coupling between organotin reagent 248 and thioester derivative 249,145 followed by DIBAL-H reduction to afford alcohol 250 as a single detectable diastereomer. Acetylation of the hydroxyl groups furnished the fully protected MHAT substrate 251. Under Mn-catalyzed MHAT conditions (Shenvi's protocol),142251 underwent smooth radical cyclization, delivering pentacycle 252 in 78% yield with excellent diastereoselectivity (d.r. = 10:1). This Mn-mediated cyclization represents a previously unreported transformation in the synthesis of this structural motif, as similar outcomes had only been achieved under cobalt catalysis. In contrast, modifying the protecting groups to pivaloyl esters and switching the catalyst system to Fe(III)/Fe(II)/PhSiH3 conditions triggered a mechanistically distinct cyclization-elimination pathway, furnishing the product 254 in an impressive 93% brsm yield. Subsequent functional group manipulations of core intermediates 252 and 254 enabled access to the natural products 245–247, showcasing how fine-tuning catalytic systems and protecting-group strategies can expand the synthetic reach of MHAT cyclizations to encompass even topologically challenging trans-fused ring systems.
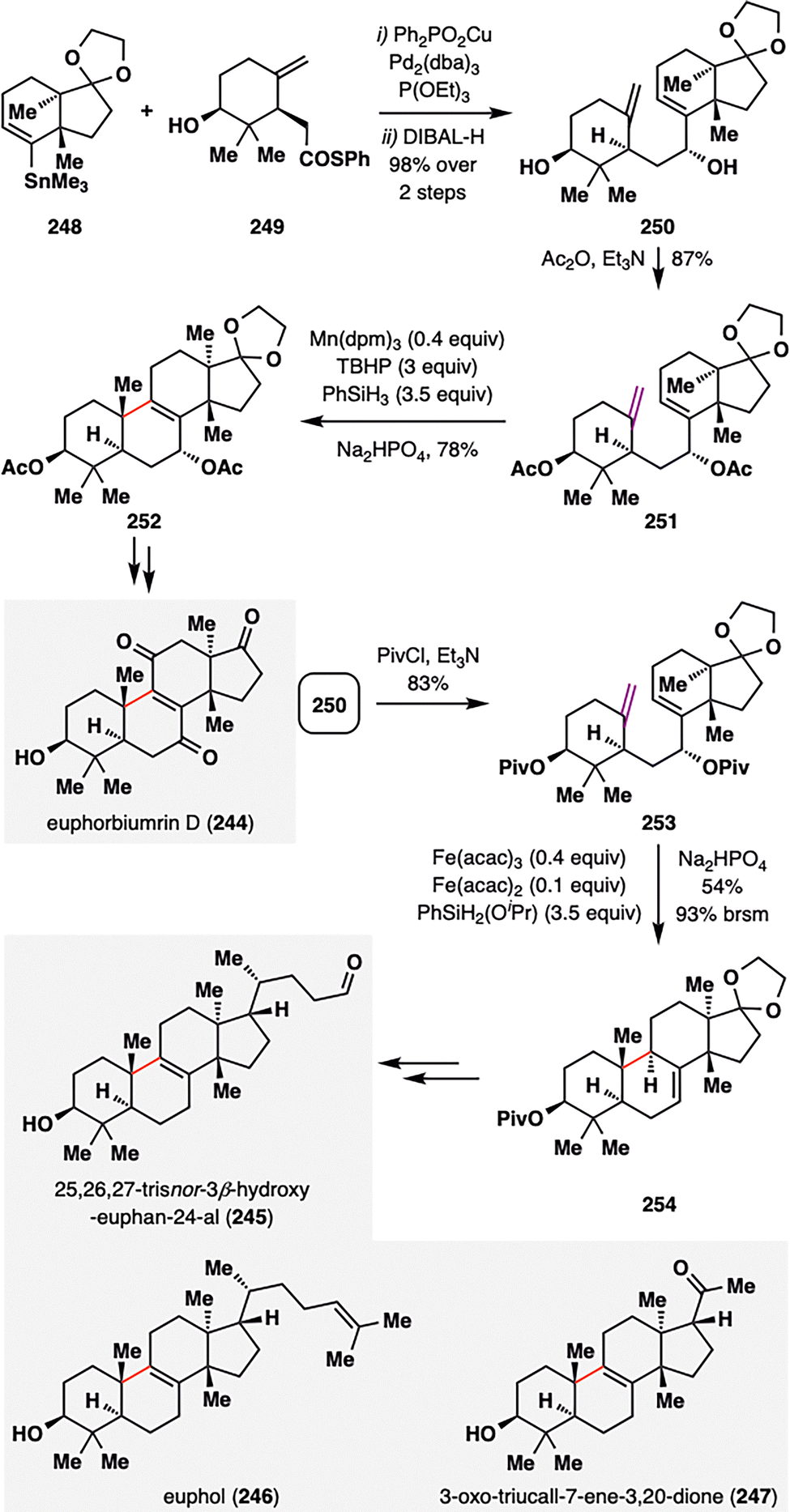 | ||
| Scheme 29 Total synthesis of euphane triterpenoids (245–247) by B. Liu and co-workers. | ||
Meroterpenes derived from dimethylorsellinic acid (DMOA) and farnesyl pyrophosphate (FPP) have long captivated biosynthetic chemists due to their intricate polycyclic frameworks and intriguing biosynthetic origins.89 Only recently, however, have these structures been synthetically realized.150–155 Maimone, Newhouse, and co-workers accomplished the first bio-inspired total synthesis of andrastin- and terretonin-type meroterpenes (255–258) by leveraging a MHAT-initiated radical cyclization/radical fragmentation/oxidative RPCO cascade, which closely mimics the proposed biosynthetic transformations (Scheme 30).156 Their synthesis began with the protoaustinoid-derived bicyclo[3.3.1]nonane core 259, which, under modified Shigehisa conditions [Co cat. (266), PhSiH3, and N-fluoropyridinium salt (F+)],157 underwent a radical cascade to furnish the trisubstituted alkene 260. Interestingly, substituting F+ with TsCl led instead to the formation of the less substituted alkene 261, a thermodynamically less favored isomer. Both divergent pathways proceeded through a common tertiary radical intermediate 262, generated via HAT from a Co(III)–H species to the terminal alkene. This radical rapidly engaged in a 3-exo-trig cyclization onto the adjacent enone, followed by β-fragmentation to yield 264. In the oxidative pathway (F+), the resulting radical was oxidized to a carbocation that underwent β-elimination to furnish 260. In contrast, under non-oxidative conditions (TsCl), radical quenching via HAT occurred preferentially at the less hindered methyl site, leading to 261. This oxidation-state-dependent bifurcation elegantly illustrates how the MHAT-RPCO cascade can emulate biosynthetic logic and selectively guide divergent molecular outcomes from a common intermediate. Final elaboration of both intermediates enabled the convergent total synthesis of the meroterpenoid natural products andrastins and terretonins (255–258), highlighting the strategic power of RPCO logic in complex terpene synthesis.
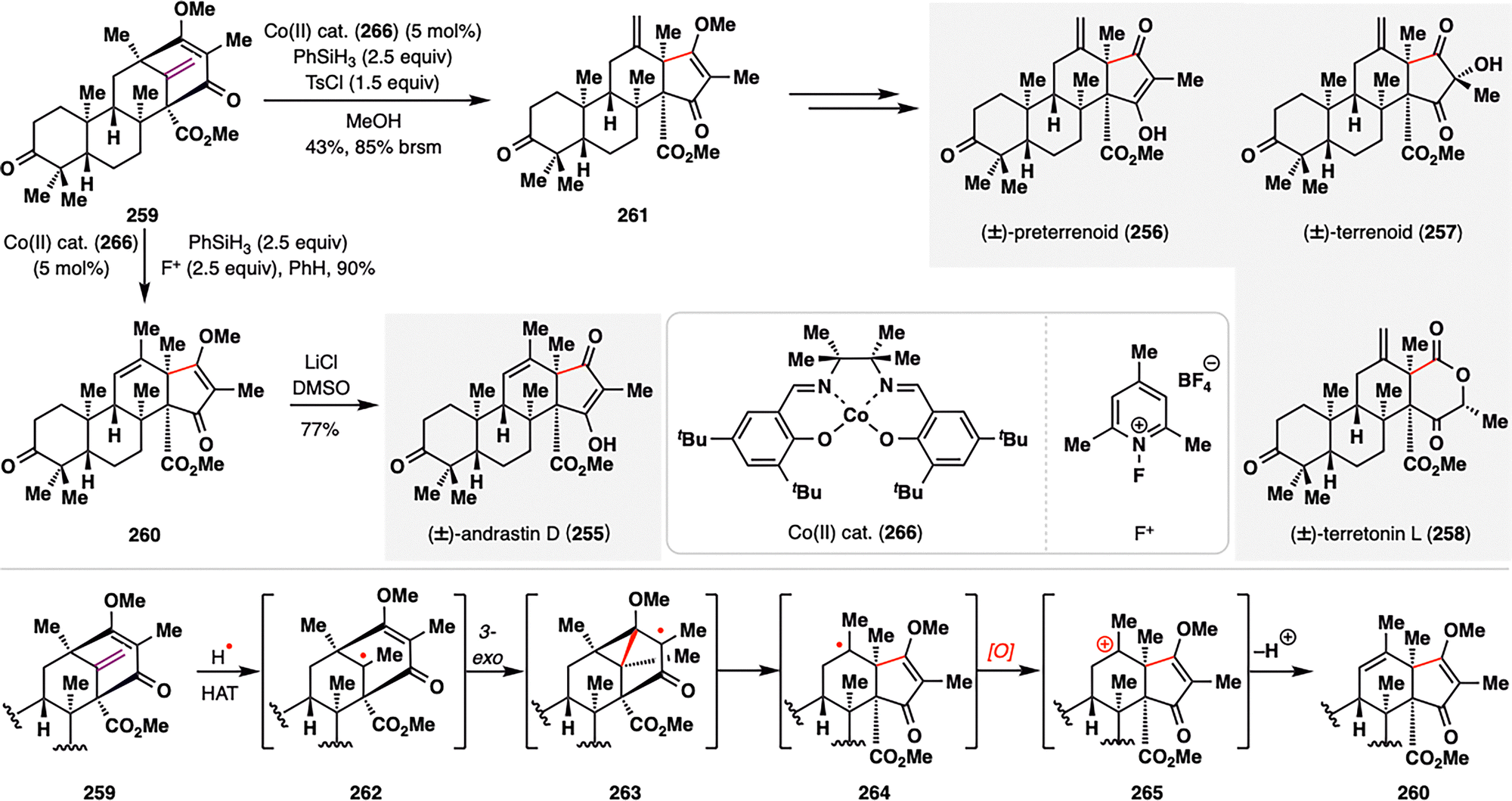 | ||
| Scheme 30 Total synthesis of andrastin and terretonin meroterpenes (255–258) by Newhouse, Maimone, and co-workers. | ||
Building upon the success of bioinspired MHAT-RPCO cascades in meroterpene synthesis, Vanderwal and co-workers extended this strategy to address a long-standing challenge: achieving polycyclization in highly oxidized contexts, a hallmark of abietane diterpenoids.158–160 The poor compatibility of traditional acid-catalyzed carbocationic cyclizations with oxygen-rich substrates has historically impeded synthetic access to such densely functionalized frameworks. To overcome this limitation, Vanderwal and co-workers devised an RPCO-based approach that exhibited exceptional tolerance to multiple oxidation states and delivered high stereocontrol—even on substrates functionalized at nearly every position. As outlined in Scheme 31, a family of abietane diterpenoids (267–270) was accessed by tuning the oxidation patterns on the radical precursors. The cyclization sequence began with a Co(III)–H-mediated HAT to the terminal alkene of diene 271, generating a tertiary radical that cyclized onto a proximal electron-deficient olefin. The resulting α-cyano radical then engaged in a second radical addition to the adjacent aromatic ring, forming a transient cyclohexadienyl σ-complex radical 272. Upon oxidation and rearomatization, the tricyclic product 274 was furnished, forming two carbon–carbon bonds and three stereocenters, including two sterically demanding quaternary centers, in a single transformation. This elegant example underscores the strategic potential of MHAT-RPCO chemistry to invert the traditional sequence of cyclization followed by oxidation, enabling direct cyclization of highly oxidized substrates—a capability rarely achievable by conventional ionic approaches. It thus expands the synthetic logic for constructing complex polyoxygenated diterpenoid architectures.
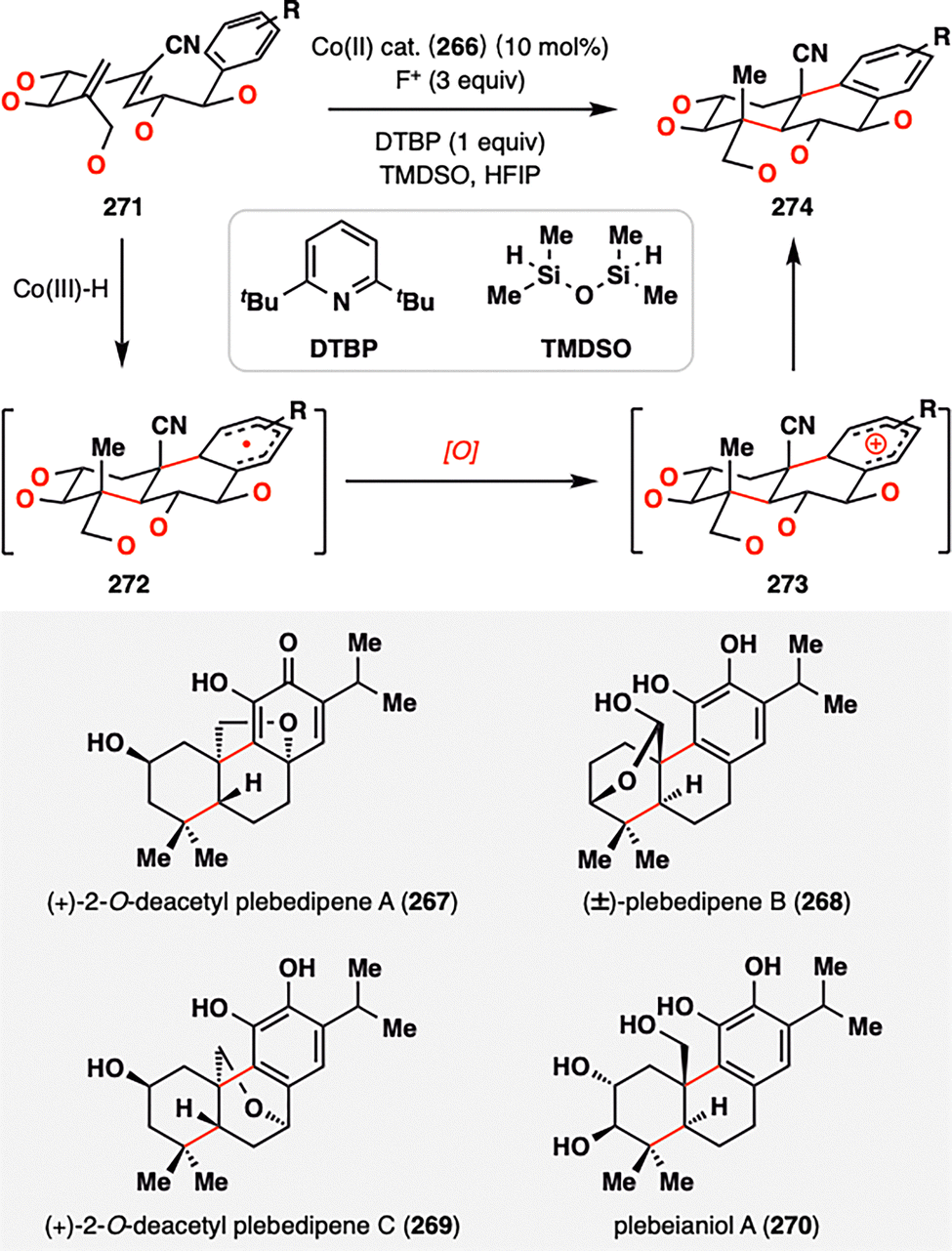 | ||
| Scheme 31 Total synthesis of abietane diterpenoids (267–270) by Vanderwal and co-workers. | ||
Ding and co-workers further disclosed an elegant HAT-initiated Dowd–Beckwith rearrangement/radical-polar crossover cascade in the collective total synthesis of (−)-crinipellins (275–282).161 As shown in Scheme 32, the key rearrangement precursor, ketone 286, was efficiently assembled through a sequence involving an oxidative dearomatization-induced (ODI) [5+2] cycloaddition followed by a pinacol rearrangement from substrate 283. To enable the critical rearrangement, the authors extensively screened conditions and identified two optimal Co(II)-catalyzed systems—one employing TBHP, and the other using N-fluoropyridinium salt (F+) as oxidant—both affording the desired rearranged tetracycle 289 in excellent yields (98% and 90%, respectively). Interestingly, while both conditions proceeded via similar MHAT-initiated radical intermediates, they diverged mechanistically in the termination step: the TBHP pathway favored β-hydrogen elimination to regenerate the Co(III)–H catalyst, whereas the F+ system facilitated a RPCO event, oxidizing the intermediate radical to a carbocation. This MHAT-enabled Dowd–Beckwith rearrangement not only provided an elegant solution for assembling the compact tetraquinane core but also established a platform for modular late-stage diversification. Through further functional group elaboration, the team successfully accessed the full series of (−)-crinipellins (275–282), underscoring the rearrangement's strategic utility in natural product synthesis.
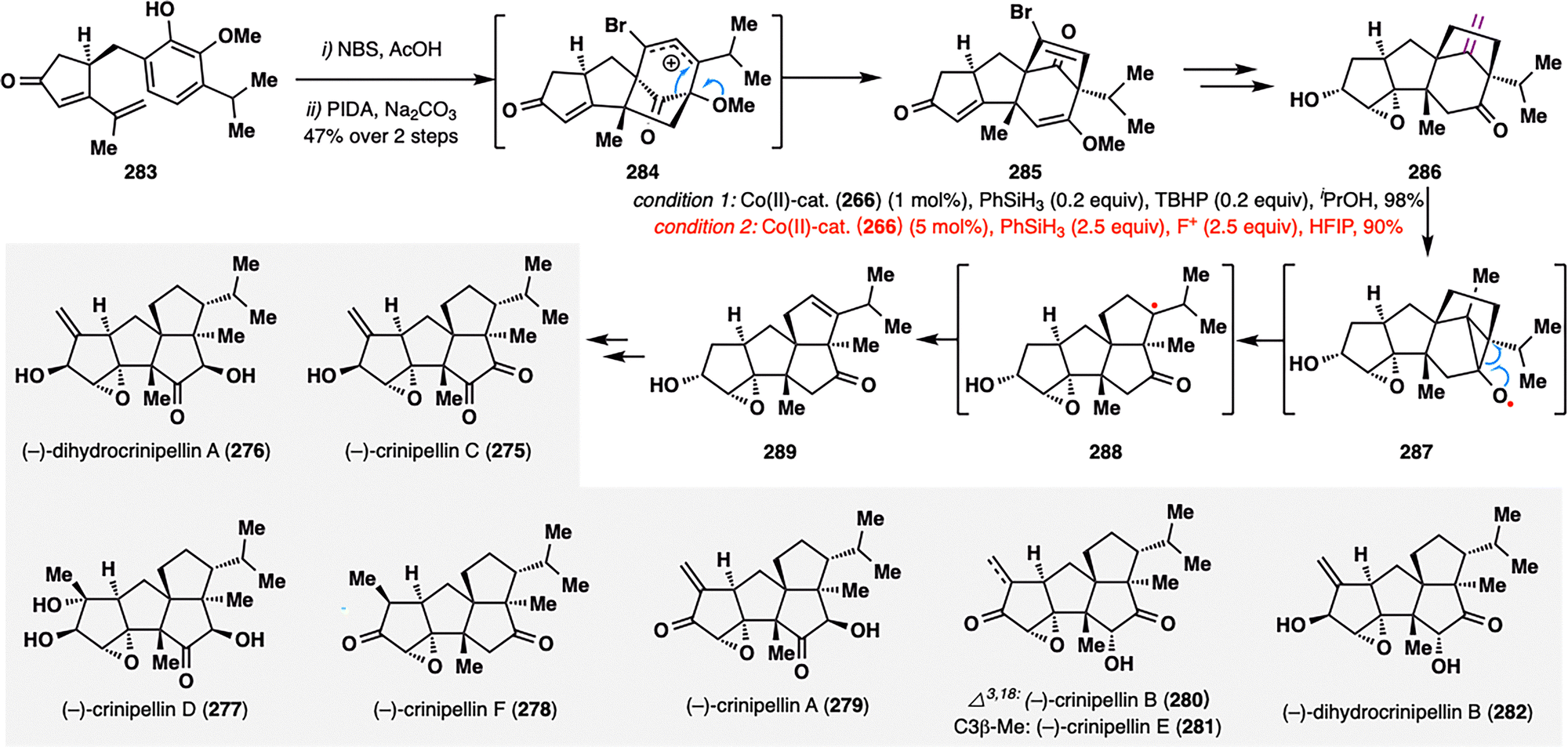 | ||
| Scheme 32 Divergent total synthesis of (−)-crinipellins (275–282) by Ding, Xie, and co-workers. | ||
In Vincent's total synthesis of dactyloquinone A (290), a pivotal transformation involved an MHAT-initiated RPCO event proceeding via an intramolecular redox process—remarkably achieved without the use of external oxidants (Scheme 33).162 The radical coupling partners were constructed through a convergent sequence from 291 and 292 comprising intermolecular substitution and oxidative dearomatization, ultimately affording tricyclic quinone 295. Upon treatment with Fe(acac)3 and PhSiH3, a tertiary radical was generated at the trisubstituted olefin. Rather than undergoing conventional carbon–oxygen radical coupling, the radical was more likely to engage in intramolecular single-electron oxidation via the pathway illustrated in 296, forming a zwitterionic intermediate 297. This process effectively completed the RPCO event, furnishing the polycyclic intermediate 290 in a synthetically satisfying 50% yield from 295. A subsequent Wittig olefination, followed by ceric ammonium nitrate (CAN)-mediated oxidation, delivered dactyloquinone A (290) in 71% yield, thus completing the total synthesis in a concise and mechanistically elegant fashion.
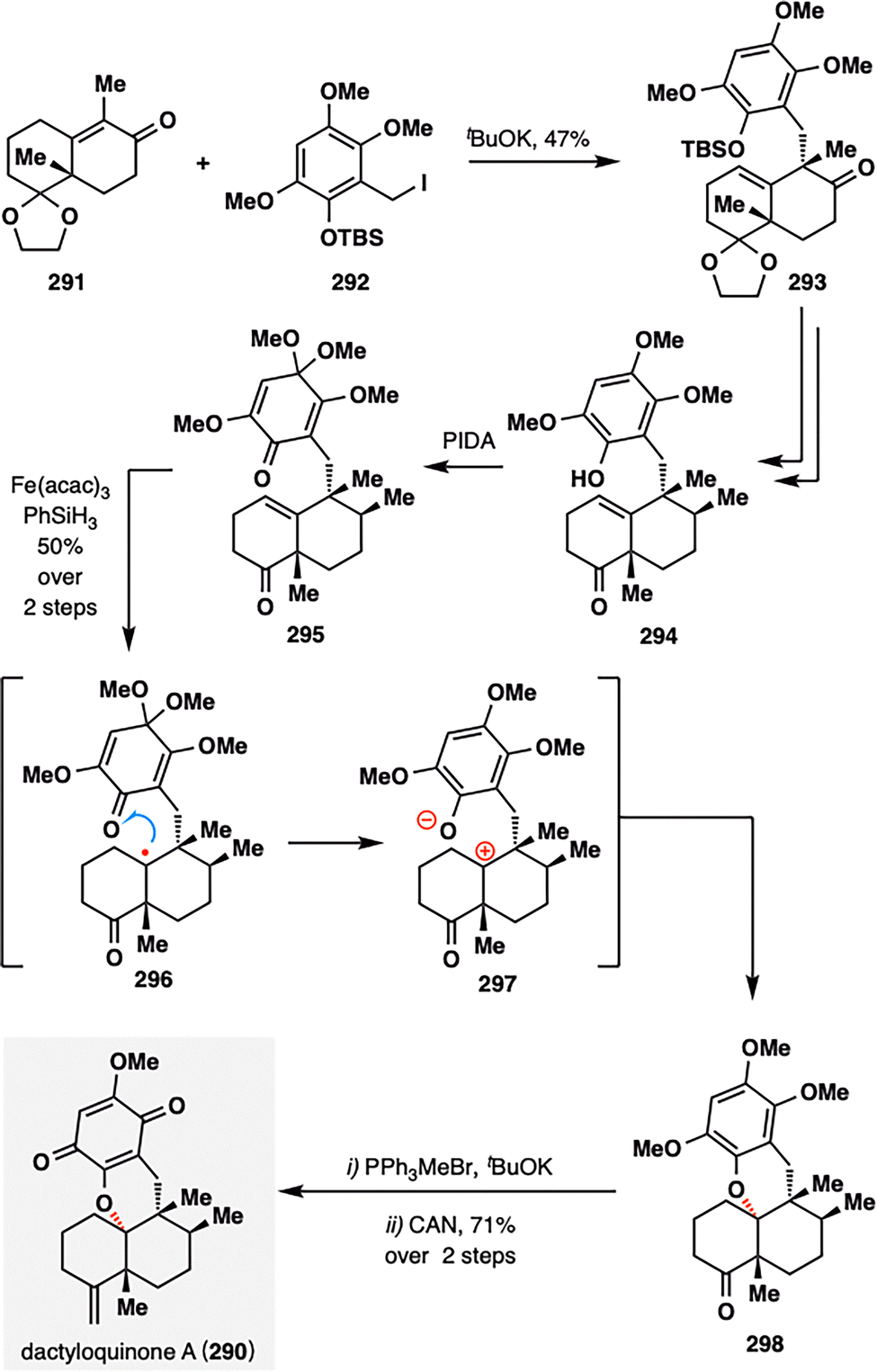 | ||
| Scheme 33 Total synthesis of dactyloquinone A (290) by Vincent and co-workers. | ||
In contrast to the previously described oxidative MHAT-RPCO transformations, the Pronin group pioneered a reductive variant of the MHAT-RPCO cascade for the rapid construction of complex terpenoid architectures.23,163 Their strategy centered on MHAT-initiated BROC additions followed by intramolecular aldol condensations, offering a direct and convergent approach for forming multiple carbon–carbon bonds and stereocenters under mild conditions. In their total synthesis of (±)-forskolin (299) and (+)-quassin (305) (Schemes 34(A) and (B)),164,165 the MHAT-RPCO cascade was strategically employed to unite two advanced fragments while concurrently forging ring systems, efficiently delivering polycyclic intermediates 303 and 308, respectively. These transformations proceeded via Fe(III)–H-initiated intramolecular radical addition to an enone, generating a stabilized radical intermediate that was subsequently reduced to an enolate anion. The resulting anion underwent intramolecular aldol condensation, forming two new C–C bonds and establishing three to four contiguous stereocenters in a single synthetic operation—dramatically enhancing molecular complexity.
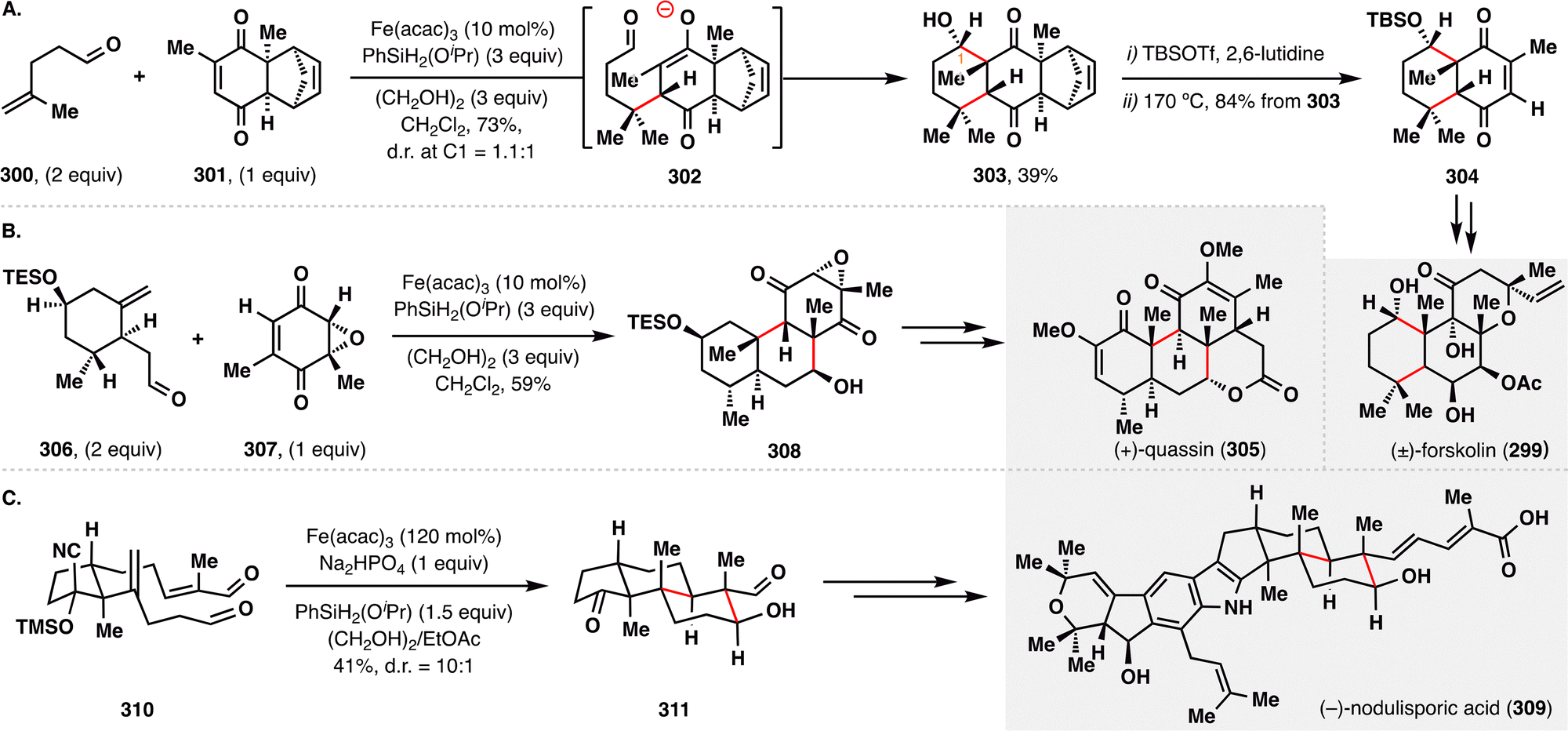 | ||
| Scheme 34 Pronin's reductive radical-polar crossover in the total synthesis of forskolin (299), quassin (305), and nodulisporic acid (309). | ||
In contrast, the total synthesis of (−)-nodulisporic acid (309) featured an intramolecular application of the MHAT-RPCO cascade (Scheme 34(C)).166 Here, dialdehyde 310 underwent efficient cyclization to afford fused bicyclic intermediate 311 with excellent diastereoselectivity (d.r. = 10:1), guided by a stereogenic methyl substituent that favored trans-decalin formation. With the core polycyclic scaffolds efficiently assembled via these elegant reductive cascades, subsequent functional group elaborations enabled streamlined access to the complex natural product nodulisporic acid (309).
The power of reductive MHAT-RPCO chemistry was further exemplified by Liu, Fu, and co-workers in their total synthesis of rumphellclovane E (312),167 a clovane-type sesquiterpenoid isolated from the gorgonian coral Rumphella antipathies.168 As illustrated in Scheme 35, the authors employed a MHAT-mediated intramolecular reductive aldol strategy to construct the tricyclic core of the molecule. Following Fe(III)-mediated MHAT activation, a tertiary radical intermediate 314 was generated, which underwent a RPCO to form the corresponding enolate 315. This enolate then participated in a highly diastereoselective intramolecular aldol cyclization, furnishing tricyclic product 316. This concise cascade not only efficiently assembled the core ring system but also established two contiguous stereocenters—including a challenging all-carbon quaternary center—with excellent stereocontrol.
 | ||
| Scheme 35 Total synthesis of rumphellclovane E (312) by Liu, Fu, and co-workers. | ||
In a pioneering study, Shenvi and co-workers applied MHAT-SH2 chemistry in the total synthesis of (−)-eugenial C (317),169 a densely functionalized meroterpenoid isolated from the fruit of Eugenia umbelliflora (Scheme 36).170 The synthesis culminated in an iron-catalyzed hydrobenzylation between alkene 321 and benzyl bromide 322, forging the quaternary center with high diastereocontrol (d.r. = 10:1) and delivering the coupled product 324 in 50% yield. The alkene fragment 321 was constructed from 318via a sequence involving MHAT-mediated hydration, acylation, site-selective C–H oxidation, and elimination of a tertiary alcohol. Under the optimized Fe(acac)3/PhSiH3 conditions, MHAT generated a tertiary radical from 321, which then reacted with a benzyl–Fe(TPP) species via homolytic SH2 substitution (illustrated in 323), stereoselectively installing the quaternary center. The final elaboration of the coupling product 324 delivered (−)-eugenial C (317) in just four steps.
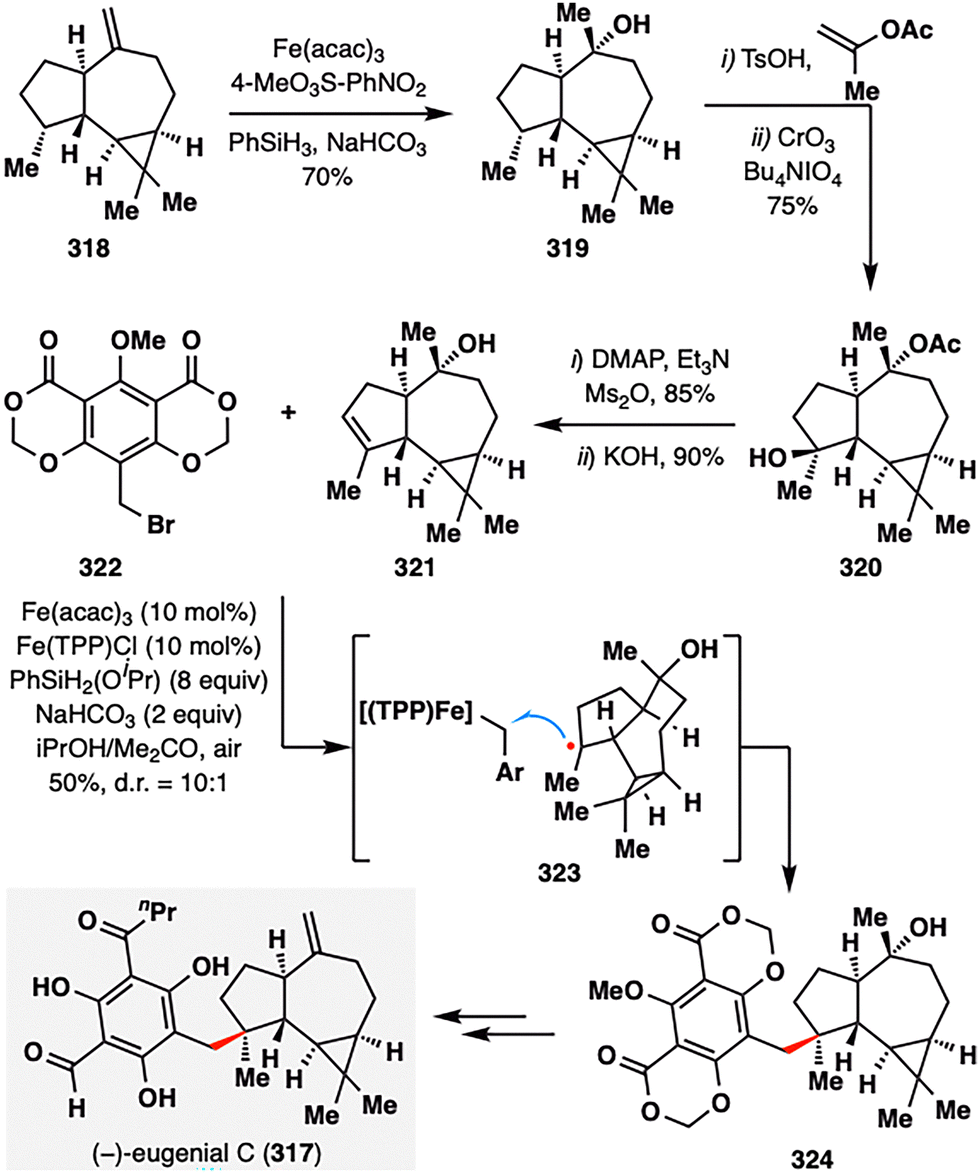 | ||
| Scheme 36 Shenvi's total synthesis of (−)-eugenial C (317). | ||
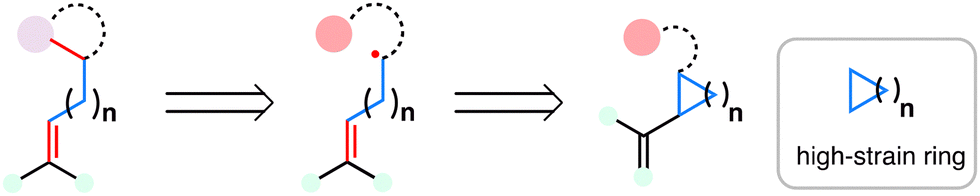 | ||
| Scheme 37 Radical retrosynthesis employing the MHAT-fragmentation cascade. | ||
Extending the scope of MHAT chemistry beyond bond formation, Shenvi and co-workers demonstrated its unique potential for skeletal rearrangement through a MHAT-fragmentation cascade—a transformation first disclosed in 2014 during their investigation of catalytic, chemoselective olefin isomerization.171 In this foundational study, catalytic amounts of Co(SaltBu,tBu)Cl in the presence of catalytic amounts organosilanes were found not only to mediate positional isomerization of alkenes, but also to enable cycloisomerization of dienes and retrocycloisomerization of strained ring systems under mild conditions. This work highlighted the versatility of MHAT catalysis in promoting radical-induced skeletal editing processes.
Building on these insights, Shenvi's group remained at the forefront of leveraging MHAT-fragmentation logic in terpenoid synthesis. A notable example is the transformation of (−)-caryophyllene oxide (326) into (−)-humulene oxide II (325), a naturally occurring sesquiterpene commonly found in essential oils. As illustrated in Scheme 38, a single-step MHAT-induced retrocycloisomerization efficiently remodeled the bicyclic framework of 326 into the uncommon eleven-membered macro ring system in an impressive 95% yield. Mechanistically, the reaction was initiated by Co(III)–H-mediated HAT at the terminal alkene, generating tertiary radical intermediate 327. This species underwent β-scission via cleavage of the adjacent cyclobutane ring to yield intermediate 328, which then underwent β-hydrogen elimination to complete the ring expansion. This pioneering application of MHAT-fragmentation not only underscores the synthetic power of radical-based skeletal editing, but also continues to inspire novel strategies for terpenoid diversification and complexity generation.
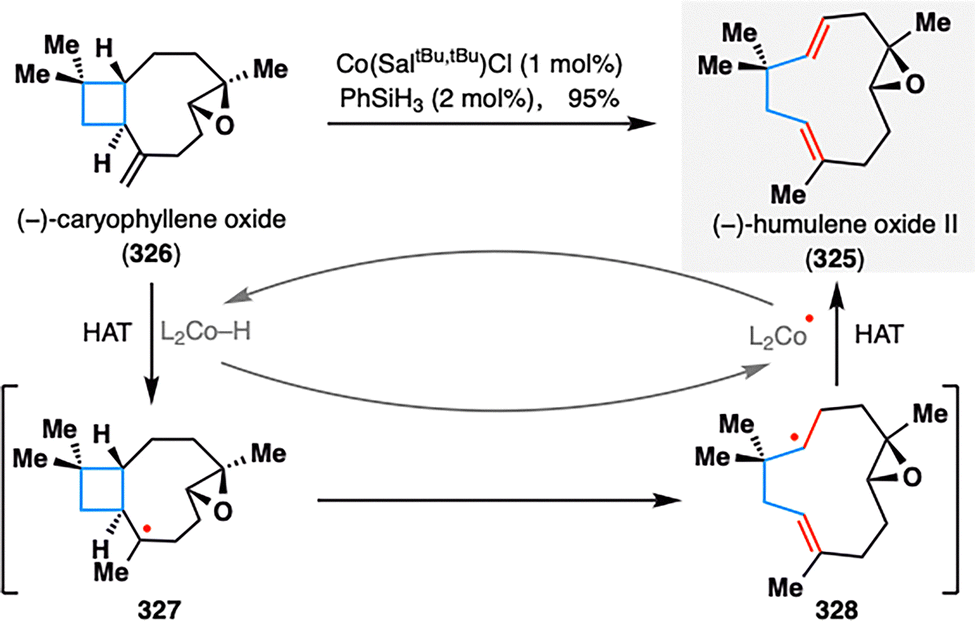 | ||
| Scheme 38 Shenvi's synthesis of (−)-humulene oxide II (325) by HAT isomerization. | ||
Inspired by Shenvi's pioneering work on HAT-induced retrocycloisomerization, Sarlah and co-workers adopted a similar strategy in the synthesis of monobistropolones (329–331)—natural products isolated from Phoma sp. cultures that share a characteristic 11-membered humulene-derived core bearing pendent tropolone motifs (Scheme 39).172,173 The fragmentation precursor 333 was assembled from readily available chiral pool material (−)-kobusone (332). Upon treatment of 333 with Shenvi's retrocycloisomerization conditions, the hydroxylated caryophyllene oxide derivative 333 underwent efficient ring-opening rearrangement, delivering the desired eleven-membered macrocyclic framework 334. Subsequent epoxide reduction and TBS deprotection furnished (−)-10-hydroxyhumulene (329) in a concise and efficient sequence, setting the stage for further elaboration into the target monobistropolone natural products 330 and 331.
 | ||
| Scheme 39 Total synthesis of monobistropolones (329–331) by Sarlah and co-workers. | ||
To further expand the skeletal editing capabilities of MHAT-initiated retrocycloisomerization, Gaich and co-workers strategically employed an Fe(III)/PhSiH3-catalyzed system within the context of the complex taxane scaffold (Scheme 40).174 This transformation enabled the conversion of cyclotaxane 337 into the canonical taxane core 336, thereby significantly broadening synthetic access to this pharmaceutically important diterpene family. Remarkably, the fragmentation proceeded only in the presence of an enol ether moiety, which enforced a stereoelectronic alignment between the exocyclic methylene and enol ether π-systems with the σ-bond undergoing cleavage (illustrated in 340). This alignment, together with the formation of a stabilized allylic radical, provided the necessary thermodynamic driving force for the rearrangement. Following MHAT-triggered retrocycloisomerization, chemoselective oxidation of the enol ether with singlet oxygen delivered taxinine A (336), illustrating the utility of MHAT-radical fragmentation for late-stage skeletal remodeling of complex terpenoid frameworks. Notably, irradiation of 336 at 254 nm led to the formation of a [2+2] photocycloaddition product 338 after, along with taxinine K (337), proposed to arise via a 1,5-HAT process.
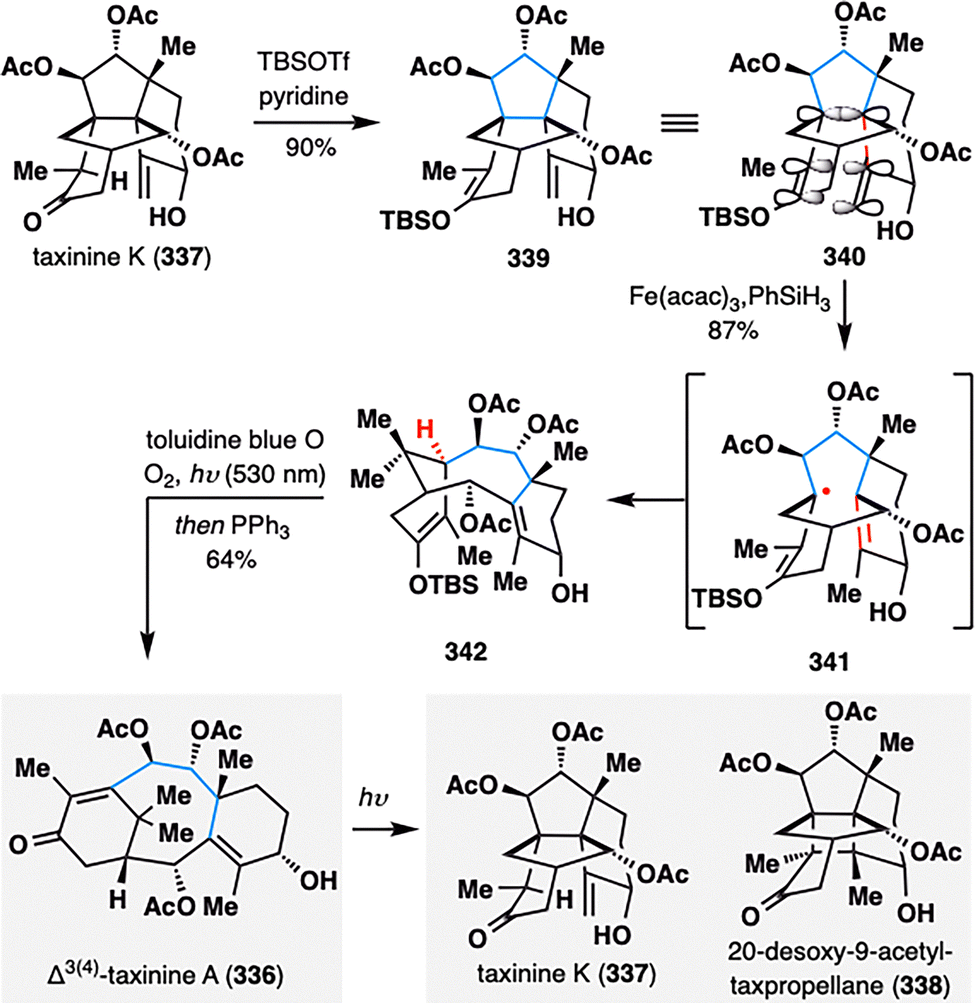 | ||
| Scheme 40 Collective total synthesis of taxane diterpenes (336–338) by Gaich, Pan, and co-workers. | ||
Analogous to MHAT-mediated radical fragmentations, Yang and co-workers developed a notable photocatalytic thiyl radical-initiated fragmentation–intramolecular annulation cascade.175,176 As illustrated in Scheme 41, this strategy was harnessed to construct the intricate tetracyclic core of (−)-pavidolide B (343), a bioactive metabolite isolated from the marine soft coral Sinularia pavida.177 The radical precursor 346 was obtained in 74% yield via a Mitsunobu coupling between enone 344 and vinylcyclopropane (VCP) 345. Upon irradiation with blue LEDs in the presence of an Ir-based photocatalyst (88), a phenylthiyl radical was generated and selectively added to the electron-rich alkene, triggering β-scission of the adjacent cyclopropane ring. The resulting α-ester radical 347 underwent a 5-exo-trig cyclization onto the enone from the top face, yielding α-ketone radical 348, which subsequently cyclized onto the allyl phenyl thioether moiety. Elimination of the thiyl radical then delivered tricyclic intermediate 349. Notably, the regio- and stereoselectivity of the entire cascade were governed by the tethered ester group, which directed the facial selectivity of cyclization and relayed chirality from C14 to both the newly formed stereocenter at C13 and the challenging quaternary center at C4. This highly orchestrated radical sequence established all key stereocenters of the C-ring in a single step, enabling a streamlined completion of (−)-pavidolide B (343) through minimal downstream elaboration.
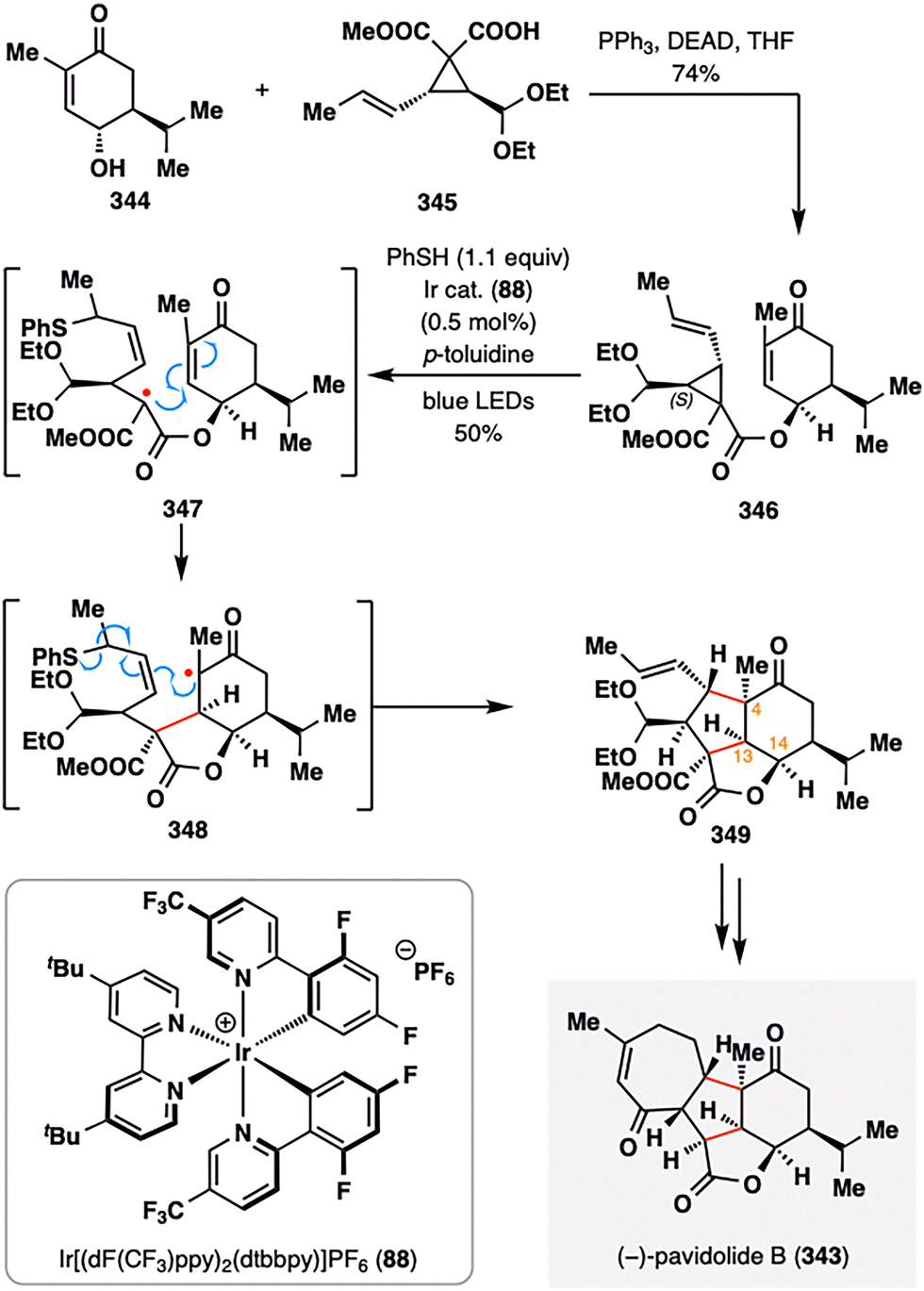 | ||
| Scheme 41 Total synthesis of (−)-pavidolide B (343) by Yang and co-workers. | ||
By marrying MHAT's chemofidelity with programmable coupling partners, modern synthesis achieves unprecedented brevity in constructing terpenoid skeletons—a testament to the synergy of radical reactivity and strategic disconnection.
2.2. Dealkenylative fragmentation
Dealkenative fragmentation has emerged as a powerful tool in synthetic chemistry for the selective remodeling of molecular frameworks through radical-mediated cleavage of alkenes. Pioneering work by Kwon and co-workers has substantially expanded the scope of this strategy,178,179 enabling its integration with a wide array of transformations—including hydrogenation,180 thiylation,181 hydroxylation,182,183 alkenylation,184 and alkynylation.185 In recent years, dealkenative fragmentation, particularly the hydrodealkenylation, has gained increasing traction in the total synthesis of complex natural products, providing access to frameworks that are otherwise challenging to construct using classical disconnections. Mechanistically, the hydrodealkenylation process is initiated by ozonolysis (Scheme 42), which results in the elimination of formaldehyde and the formation of a hydroperoxide intermediate. Upon reduction by Fe(II), the resulting alkoxy radical undergoes β-scission to cleave the adjacent C–C bond, generating an alkyl radical. This radical subsequently undergoes HAT with a suitable hydrogen atom donor (HAD) to afford the hydrodealkenylated product.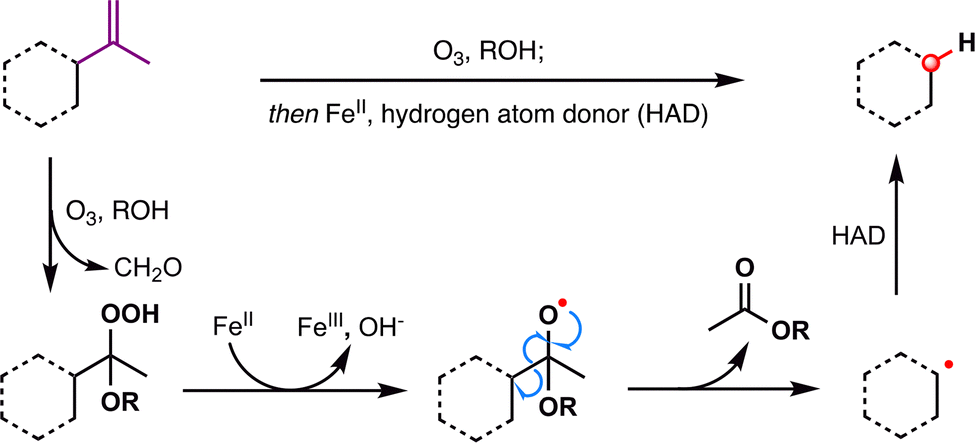 | ||
| Scheme 42 General mechanism of hydrodealkenylation. | ||
From a retrosynthetic standpoint, Kwon's dealkenylative alkylation represents a compelling strategic disconnection by enabling selective cleavage of C(sp3)–C(sp2) bonds in alkenes to reveal synthetically valuable alkyl radical fragments. This approach inverts conventional logic, shifting from forward alkene functionalization to a deconstructive paradigm in which complexity is introduced through fragmentation. In retrosynthesis, complex alkyl-substituted motifs can be traced back to terminal or internal alkenes, which function as latent radical precursors (Scheme 43). The strategic power of this method lies in its ability to cleave unactivated C–C bonds, a longstanding challenge in traditional polar disconnection-based strategies.
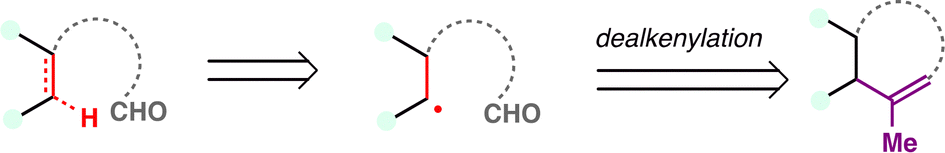 | ||
| Scheme 43 Radical retrosynthesis employing the hydrodealkenylation. | ||
Notably, hydrodealkenylation was first applied in total synthesis by Newhouse and co-workers in their construction of the complex indole diterpenoid shearilicine (350), a natural product featuring a highly oxidized right-hand ring (Scheme 44).186 The synthesis began with the chiral pool compound (R)-carvone (351), enabling diastereoselective conjugate addition to yield 352, which then underwent CsF-mediated desilylation and hydrolysis to afford 353, followed by a Pauson–Khand reaction to furnish tricyclic intermediate 354 with excellent stereocontrol. While the isopropylidene motif in carvone proved invaluable for asymmetric induction, it is absent in the native natural product. To resolve this structural incongruity, the authors employed a reductive dealkenylation strategy: ozonolysis of 354 followed by treatment with Fe(II) as a reductant and benzenethiol as a hydrogen atom donor delivered the dealkenylated product 355 in 53% yield.180 This transformation not only capitalized on the stereochemical utility of the carvone scaffold in early-stage synthesis but also enabled its strategic removal to match the substitution pattern of shearilicine. A final late-stage convergent coupling with the indole fragment completed the total synthesis of shearilicine (350).
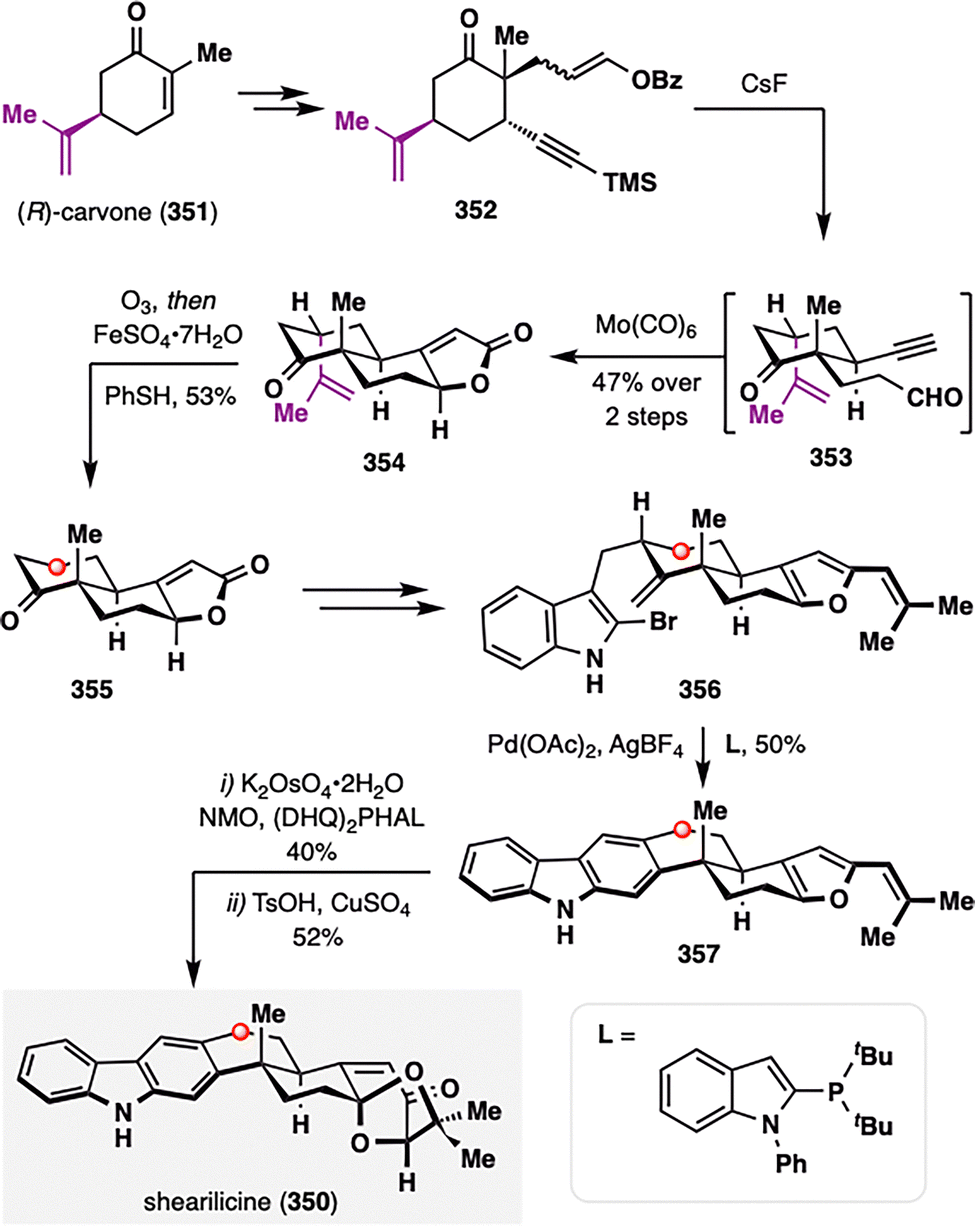 | ||
| Scheme 44 Newhouse's total synthesis of (+)-shearilicine (350). | ||
Building upon the strategic precedent established by Newhouse, Ito and co-workers similarly leveraged the asymmetric potential of (S)-carvone (53) as a native chiral pool in their total synthesis of (−)-lemnalemnane A (358) and (+)-lemnardosinane A (363)—two sesquiterpenoids isolated from marine soft corals of the Lemnalia genus (Scheme 45).187–189 The isopropylidene group in carvone served as a highly effective stereocontrol element, enabling the diastereoselective installation of two stereocenters in intermediate 360 from (S)-carvone (53) in 99% yield with excellent diastereoselectivity (d.r. = 30:1). To remove this now-superfluous functionality, Kwon's hydrodealkenylation protocol was applied to 360, delivering the dealkenylated product 361 in 71% yield. In a variant of this strategy used en route to (+)-lemnardosinane A (358), the same isopropylidene moiety was excised from a related intermediate to furnish enone 365. In this case, the radical intermediate was intercepted by Cu(II) and underwent β-elimination,190 directly installing the oxidized enone motif. Through subsequent elaborations of these intermediates, both (−)-lemnalemnane A (358) and (+)-lemnardosinane A (363) were accessed in asymmetric fashion, further highlighting the strategic utility of chiral-pool-based hydrodealkenylation in natural product synthesis.
 | ||
| Scheme 45 Ito's asymmetric synthesis of (−)-lemnalemnane A (358) and (+)-lemnardosinane (363). | ||
Unlike previous studies that focused on terminal alkene cleavage to achieve traceless stereocontrol, Qin and co-workers uniquely applied the hydrodealkenylation strategy to a trisubstituted internal alkene in their total synthesis of viloraconitine (367),191 a C19-diterpenoid alkaloid isolated from medicinal plants of the Aconitum genus.192 This natural product features a highly congested heptacyclic framework, including a rigid cyclopropane unit. Notably, this strategy allowed the resulting fragment to remain embedded within the molecular framework, facilitating not only precise control over N-heterocycle construction but also streamlined oxidation-state management in downstream transformations. As shown in Scheme 46, phenol derivative 368 underwent an IMDA cascade to furnish tricyclic scaffold 369 bearing an extraneous alkene moiety on the bridged ring system. To address this, hydrodealkenylative fragmentation was applied to oxidatively cleave the alkene in 370, generating an aldehyde group in 371, thereby enabling a subsequent Mannich-type annulation to construct the N-heterocycle 372en route to the intricate viloraconitine skeleton. Following a series of elaborations—including a Robinson annulation and a second IMDA reaction—the desired natural product viloraconitine (367), featuring a rigid cyclopropane motif, was successfully assembled.
 | ||
| Scheme 46 Total synthesis of vilmoraconitine (367) by Qin, Liu, and co-workers. | ||
Dai and co-workers showcased the utility of hydrodealkenylation at the outset of their total synthesis of neocucurbol C (375),193 a structurally intricate natural product isolated from Neocucurbitaria unguis-hominis FS685,194 thereby demonstrating that this transformation can be performed on large scale. As depicted in Scheme 47, the synthesis commenced with Kwon's hydrodealkenylative fragmentation of commercially available (+)-nootkatone (376), delivering intermediate 377 on a gram scale and underscoring the expanded utility of chiral pool terpenoids in complex molecule synthesis. Subsequent site-selective C–H hydroxylation of 377 and TMSN3-promoted α-iodination of 378 furnished intermediate 379. Benzyl deprotection of 380 then unveiled a phenol primed for dearomatization. Owing to its inherent instability—attributed to para-quinone methide formation—this phenol was directly subjected to oxidative dearomatization with PIDA in HFIP to give cyclized intermediate 381. Treatment of 381 with Fe(acac)3 and PhSiH3 initiated a pivotal BROC radical cyclization, delivering 382 in 75% yield. This sequential oxidative dearomatization/BROC radical cyclization forged the challenging C9–C10 bond, thereby completing the unprecedented 6/6/5/5/6 fused polycyclic skeleton while simultaneously establishing two contiguous stereocenters at C9 and C10. With the neocucurbol core in hand, subsequent functional group manipulations enabled completion of the first total synthesis of neocucurbol C (375) in 24 steps.
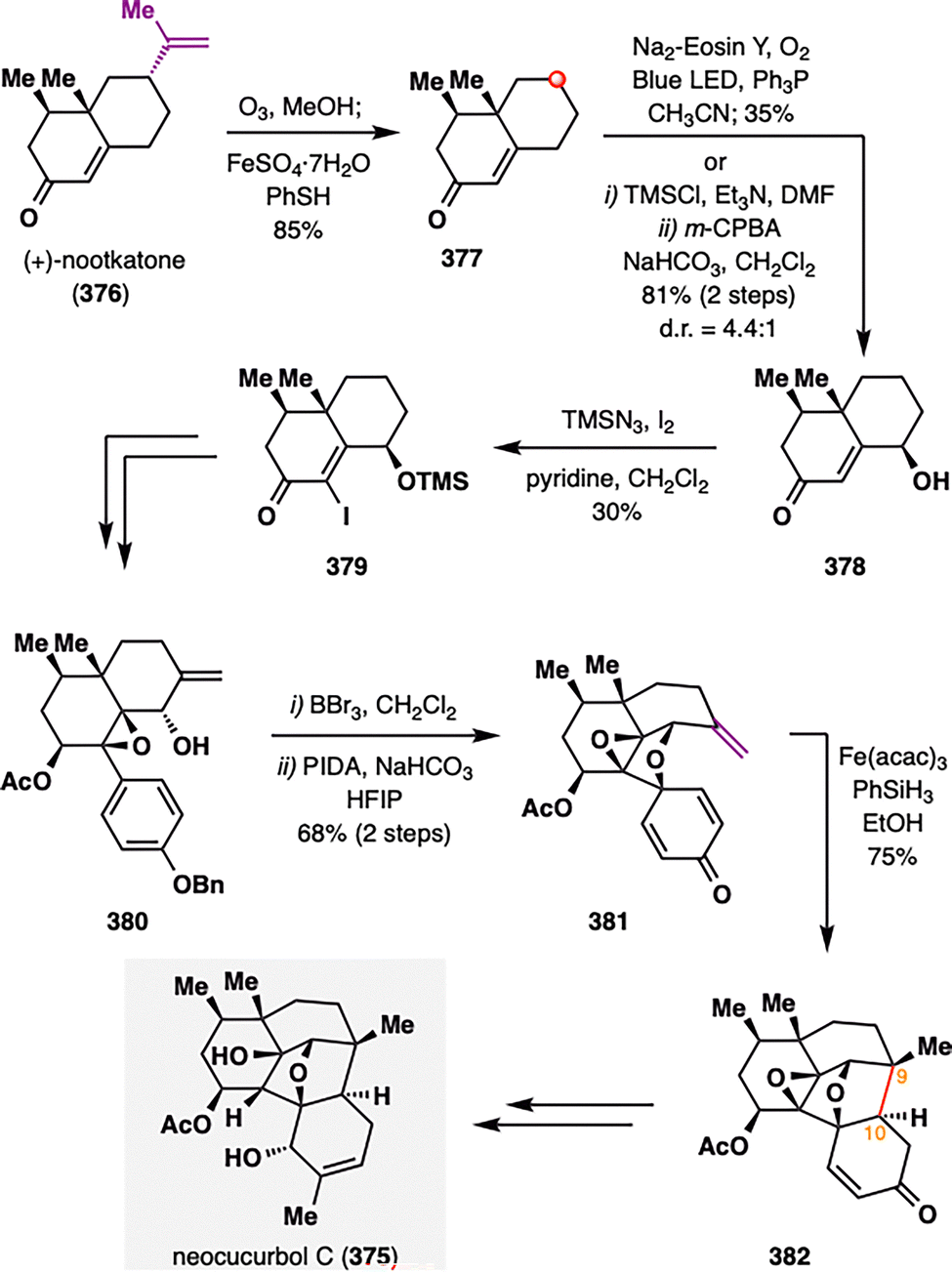 | ||
| Scheme 47 Dai's total synthesis of neocucurbol C (375). | ||
3. Carboxylic acids as radical precursors: versatile handles for terpenoid assembly
The strategic utilization of carboxylic acids as radical precursors has evolved from classical Barton decarboxylation into a cornerstone of modern terpenoid synthesis. Over the past decade, innovations such as photoredox-mediated decarboxylative couplings,9,195 Et3B-driven decarbonylation of α-alkoxyacyl tellurides,196,197 and reductive decarboxylation of redox-active esters [RAEs, e.g., N-(acyloxy)phthalimide esters] have revolutionized the field.198 These methods transform abundant carboxylic acids into modular radical sources under mild conditions. By enabling direct C–C and C–heteroatom bond formations—critical for constructing quaternary carbons, polycyclic cores, and oxygenated side chains—decarboxylative strategies now offer unparalleled efficiency in assembling sterically congested terpenoid architectures. From taxane diterpenes to meroterpenoids, these advances exemplify how radical reactivity bridges the gap between feedstock simplicity and molecular complexity, redefining ideality in natural product synthesis.From a radical retrosynthetic perspective, the disconnection of key bonds in terpenoid skeletons into carbon radicals and their coupling partners has been revolutionized over the past decade by three principal carboxylate-derived precursors (Scheme 48): (1) RAEs, which undergo single-electron reduction to liberate alkyl radicals;198–200 (2) carboxylate anions, which undergo single-electron oxidation followed by decarboxylation (e.g., photoredox-mediated processes);195 and (3) α-alkoxyacyl tellurides, activated via Et3B-mediated decarbonylation to generate stabilized alkoxy radicals.197 Each precursor class offers distinct advantages in chemoselectivity and functional group tolerance, with RAEs emerging as the most versatile due to their mild reaction conditions. The coupling partners for these radicals—ranging from electron-deficient olefins and alkenyl/aryl zinc reagents (Ni/Fe-catalyzed cross-couplings) to vinyl halides and arylboronic acids—form a versatile toolkit for forging critical bonds in terpenoid synthesis. While these partners have been widely employed over the past decade, emerging strategies such as alkynyl reagents for C(sp)–C(sp3) bond formation remain underexplored in terpenoid contexts.201,202
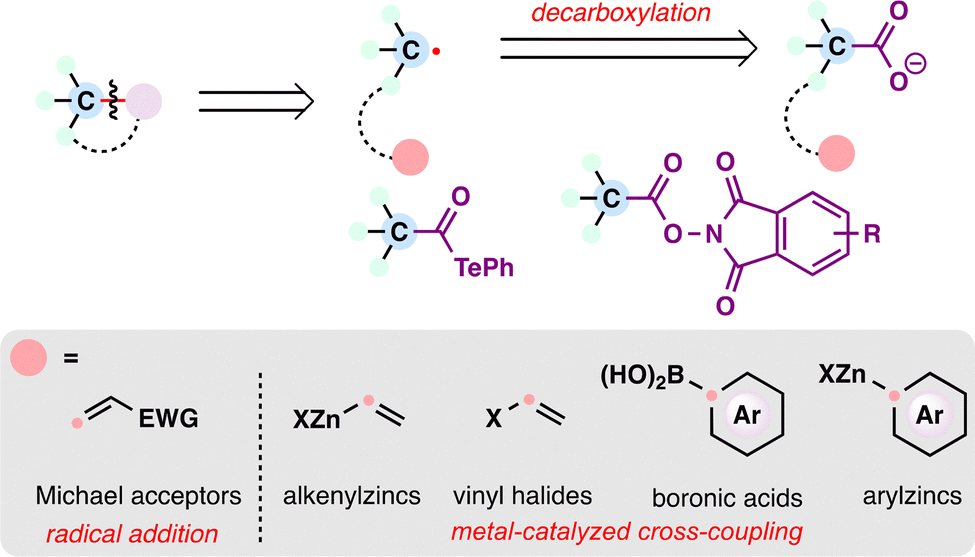 | ||
| Scheme 48 Advanced radical retrosynthesis of C–C bonds employing the decarboxylative reactions. | ||
The choice of carboxylate precursor and coupling partner is dictated by the target bond type [e.g., C(sp3)–C(sp2) and C(sp3)–C(sp3)]. In the following sections, we analyze the application of decarboxylative radical chemistry in terpenoid synthesis through the lens of bond formation types, including decarboxylative C(sp3)–C(sp3) bond formation and decarboxylative C(sp2)–C(sp3) bond formation.
3.1. Decarboxylative C(sp3)–C(sp3) bond formation
Over the past decade, decarboxylative radical addition to electron-deficient alkenes has appeared as a transformative strategy for stereoselective construction of C(sp3)–C(sp3) bonds.203 This approach leverages carboxylate-derived precursors to generate carbon radicals, which engage in conjugate additions to α,β-unsaturated carbonyl systems or related electrophilic partners. By bypassing classical ionic pathways prone to steric hindrance and regiochemical limitations, these radical-mediated reactions enable efficient access to sterically congested architectures. Given the extensive application of decarboxylative C(sp3)–C(sp3) bond formation in terpenoid synthesis, we herein categorize advancements in this field based on three pivotal carboxylate precursors: (1) RAEs,204,205 (2) carboxylate anions,206 and (3) α-alkoxyacyl tellurides—each offering distinct practical advantages.197As shown in Scheme 49(A), the quaternary stereocenter at C8 of (−)-aplyviolene (383)—a rearranged spongian diterpenoid bearing a cis-perhydroazulene core and a fused 6-acetoxy-2,7-dioxabicyclo[3.2.1]octan-3-one fragment—was constructed via a photoredox-catalyzed decarboxylative coupling. The requisite RAE 387 was prepared from inexpensive (+)-fenchone (385) via a Beckmann fragmentation to yield tertiary nitrile 386, followed by formation of a seven-membered ring and subsequent functional group interconversions. Irradiation of a mixture of 387, enone 388, Ru(bpy)3(BF4)2, Hantzsch ester, and DIPEA in CH2Cl2 provided the coupled product 389 in 61% yield as a single diastereomer. This transformation replaced the two-electron Michael addition previously used to construct the same bond in their earlier synthesis,209 thereby enabling completion of (−)-aplyviolene (383) by following the established downstream sequence. This revision not only streamlined the construction of the C8–C14 quaternary–tertiary junction but also reduced the number of isolated intermediates by five relative to the polar pathway, highlighting the strategic efficiency of radical disconnections in complex settings.
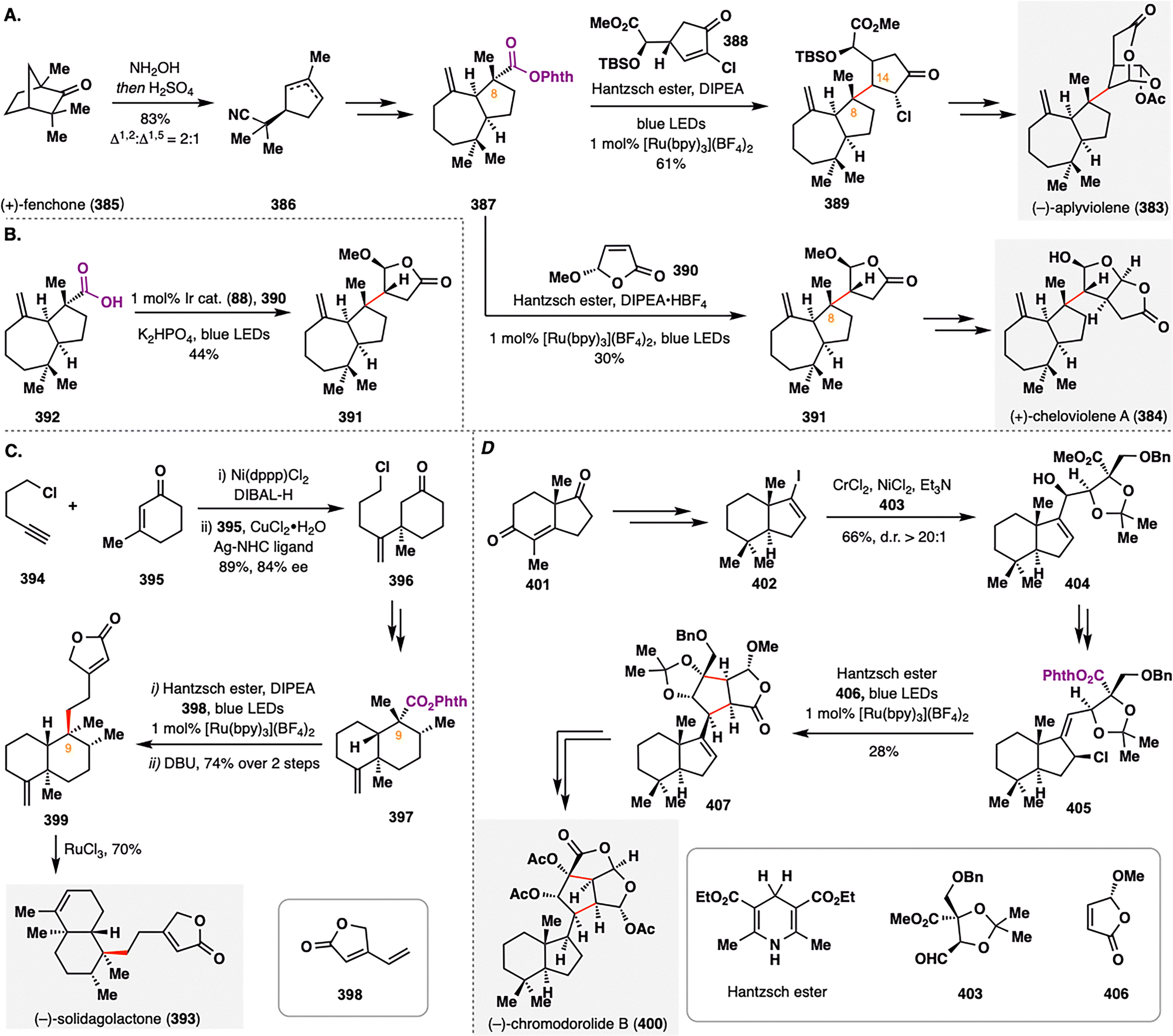 | ||
| Scheme 49 Overman's total synthesis of spongian (383–384), clerodane (393), chromodorolide (400) diterpenoids. | ||
Building on this success, Overman and co-workers extended their radical-based approach to the synthesis of cheloviolene A (384),210 another rearranged spongian diterpenoid. The same RAE 387 was employed in a decarboxylative coupling with enone 390. Interestingly, the same product 391 could also be accessed using MacMillan's Ir(III)-catalyzed oxidative photoredox conditions via direct coupling of carboxylic acid 392 with enone 390 (Scheme 49(B)).210 This alternative approach improved the coupling efficiency, raising the yield from 30% to 44%, and further underscored the utility and adaptability of photoredox-enabled decarboxylative strategies.
In 2015, Overman's group reported a concise and enantioselective synthesis of trans-clerodane diterpenoids using a similar radical strategy (Scheme 49(C)).211 The trans-decalin RAE 397 was constructed from chloroalkyne 394via a nickel-catalyzed regioselective hydroalumination, followed by an enantioselective conjugate addition to 3-methylcyclohex-2-en-1-one (395) catalyzed by a silver–NHC complex developed by Hoveyda.212 The pivotal 1,6-addition of a tertiary radical derived from RAE 397 to β-vinylbutenolide 399 stereoselectively forged the key C9 quaternary center. This was the first demonstration of tertiary radical 1,6-addition to electron-deficient dienes, broadening the reactivity landscape of decarboxylative radical chemistry.
The power of this platform was further illustrated in the total synthesis of (−)-chromodorolide B (400),213 a highly complex member of the rearranged spongian diterpenoid family (Scheme 49(D)). The tertiary RAE 405 was synthesized from methyl ester 404, which was itself prepared via a modified NHK coupling between iodide 402 and aldehyde 403.214 The final radical cascade—an intermolecular decarboxylative addition/cyclization/fragmentation (ACF) sequence—was initiated under photoredox conditions with a Hantzsch ester in the absence of DIPEA, which improved the reaction efficiency. This cascade formed two C–C bonds and established four contiguous stereocenters, furnishing tetracyclic intermediate 407 in a single operation. This work showcased the unique ability of radical cascades to rapidly generate complexity.
Building on the conceptual framework established by Overman, Yoshimitsu and co-workers developed a decarboxylation-initiated radical addition/cyclization/oxidative aromatization cascade to construct the tetralin motif of (±)-hamigeran B (408),215 a tricyclic natural product isolated from the marine sponge Hamigera tarangaensis (Scheme 50).216 In this transformation, a novel Zn(II)–porphyrin photocatalyst (411) was employed in place of conventional Ru(II)-based systems, significantly improving the yield of tetralin product 413 from 52% to 82%. Upon irradiation, photoexcited 411 facilitated single-electron reduction of RAE 409 in the presence of DIPEA, generating a tertiary carbon radical that underwent addition to an acrylate acceptor 410, forming an α-ester radical. This intermediate then cyclized onto the aromatic ring, and the resulting dearomatized species was oxidized via SET by the photocatalyst to regenerate aromaticity—completing the radical annulation sequence. Notably, the reduced by-product 412 could be converted to 413 in 93% yield via DDQ-mediated oxidation, further highlighting the efficiency and robustness of this decarboxylative cascade. Subsequent elaboration of 413 provided access to (±)-hamigeran B (408) in a concise fashion.
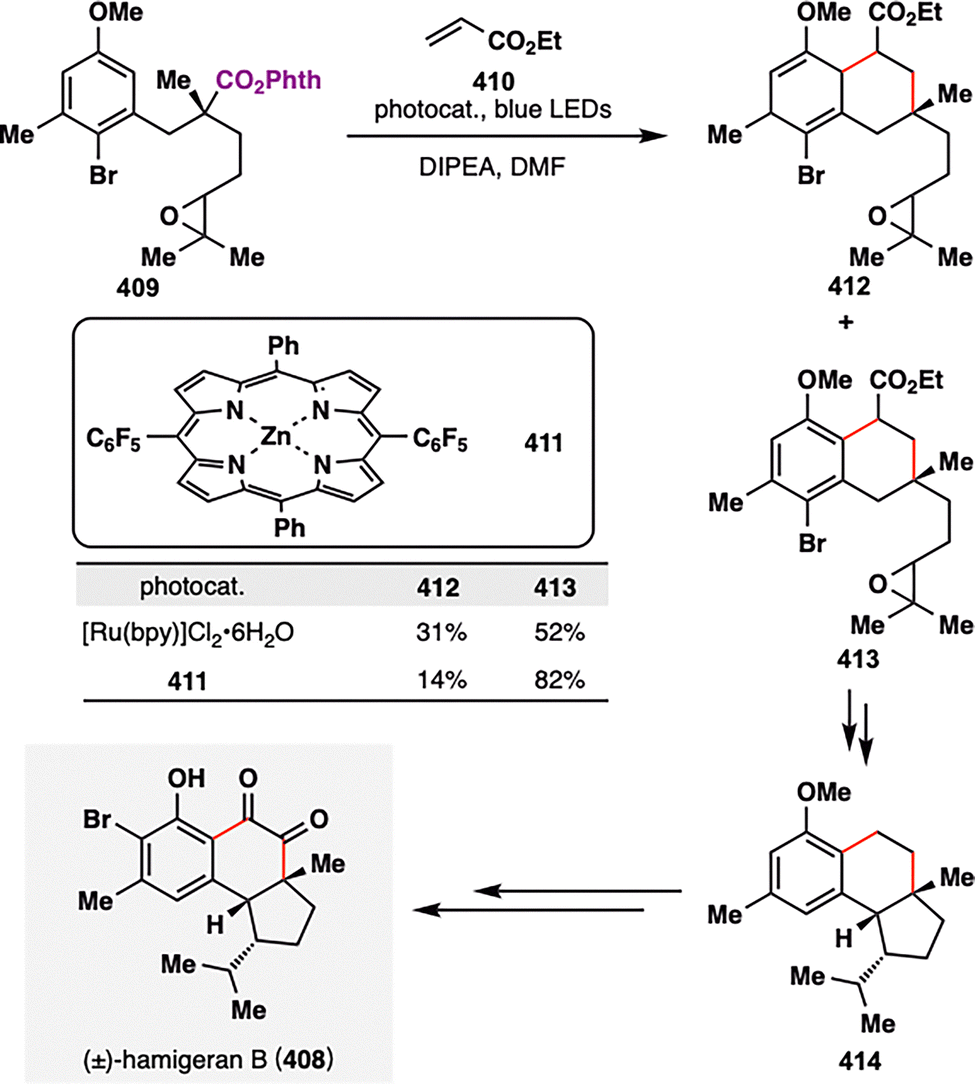 | ||
| Scheme 50 Yoshimitsu's formal synthesis of hamigeran B (408). | ||
While prior studies have predominantly employed photocatalysts to mediate decarboxylation, Wu and co-workers developed a photocatalyst-free, photoinduced decarboxylative allylation based on a donor–acceptor (EDA) complex strategy to achieve side-chain elaboration in the total synthesis of (25S)-Δ4-dafachronic acids (415–417),217 a class of steroidal hormones identified in nematodes (Scheme 51). In the key transformation, the EDA complex 420—formed between the Hantzsch ester (donor) and the RAE 419—underwent visible-light excitation to enable single-electron transfer, generating an alkyl radical. This radical engaged allyl sulfone 421 in a radical addition followed by β-scission, furnishing ester 422 in 69% yield under optimized conditions. Subsequent transformations enabled a divergent route to (25S)-Δ4-dafachronic acid (415). Notably, the same EDA-mediated strategy was also applied to the synthesis of (25S)-Δ7-dafachronic acid (416) and desulfated boophiline (417),218 further demonstrating the generality and utility of this mild, catalyst-free radical methodology in complex steroidal frameworks.
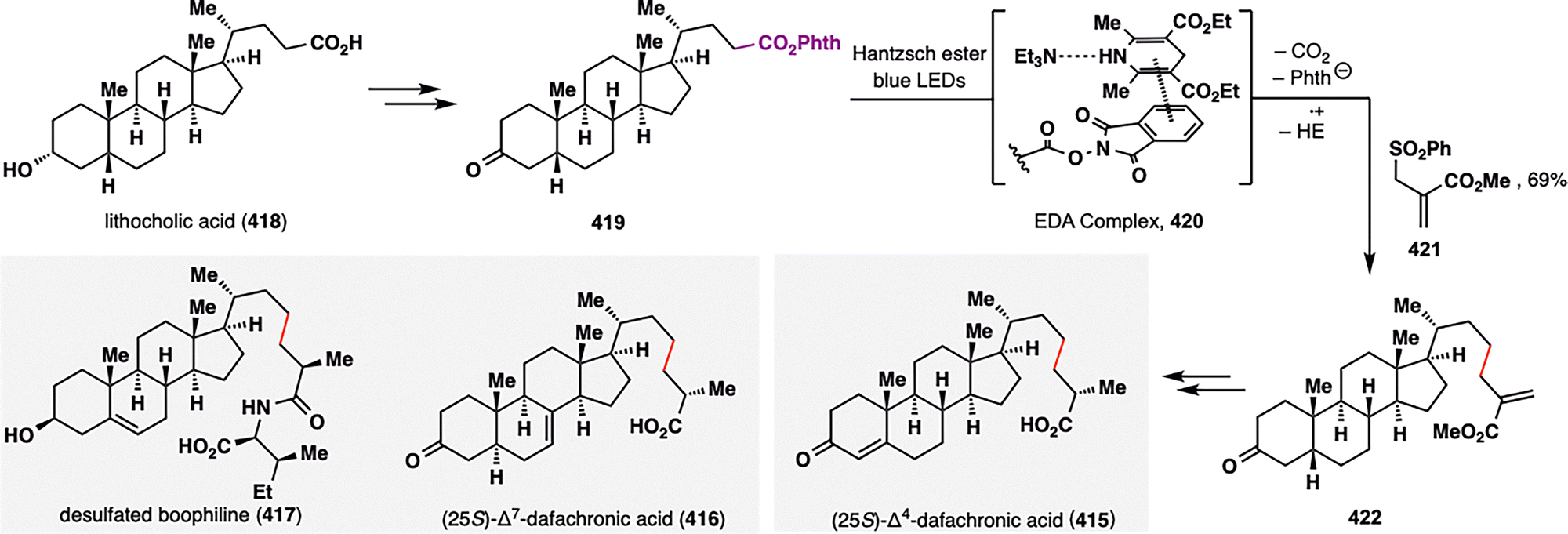 | ||
| Scheme 51 Wu's synthesis of (25S)-Δ4-dafachronic acid (415), (25S)-Δ7-dafachronic acid (416), and desulfated boophiline (417). | ||
Recently, our group demonstrated the synthetic utility of reductive, metal-mediated decarboxylative coupling in the enantioselective total synthesis of (−)-macrocalyxoformins A (423) and B (424), as well as (−)-ludongnin C (425)—three highly oxygenated enmein-type ent-kauranoids (Scheme 52).219 The first key transformation involved the construction of tetrahydrofuran fragment 428via a cascade comprising reductive decarboxylative cyclization, RPCO, and subsequent carboxylation. Initiated by Ni(II)/Zn-mediated reduction of a RAE embedded in enone 426, the resulting radical underwent intramolecular conjugate addition to form radical intermediate 429. This intermediate was then reduced to enolate 430, which intercepted CO2—either from the decarboxylation event or released during the following esterification step with Boc2O—to afford carboxylate 431. In the presence of Boc2O, 431 was converted into the tert-butyl ester 428. Notably, the dual role of CO2—as both a byproduct and a reactive species—was confirmed by isotopic labeling experiments conducted by our team. After installation of a hydroxymethyl substituent at C10, a Suárez-type radical cyclization furnished the second tetrahydrofuran ring, yielding intermediate 433. This was then converted into RAE 434, which underwent a second Ni(II)/Zn-mediated decarboxylative coupling—this time with 2,2,2-trifluoroethyl acrylate—stereoselectively constructing the C10 all-carbon quaternary center in 435.205 Final elaboration of the C/D-ring system ultimately delivered (−)-macrocalyxoformins A (423) and B (424), as well as (−)-ludongnin C (425), underscoring the strategic value of sequential decarboxylative radical disconnections in the synthesis of architecturally complex natural products.
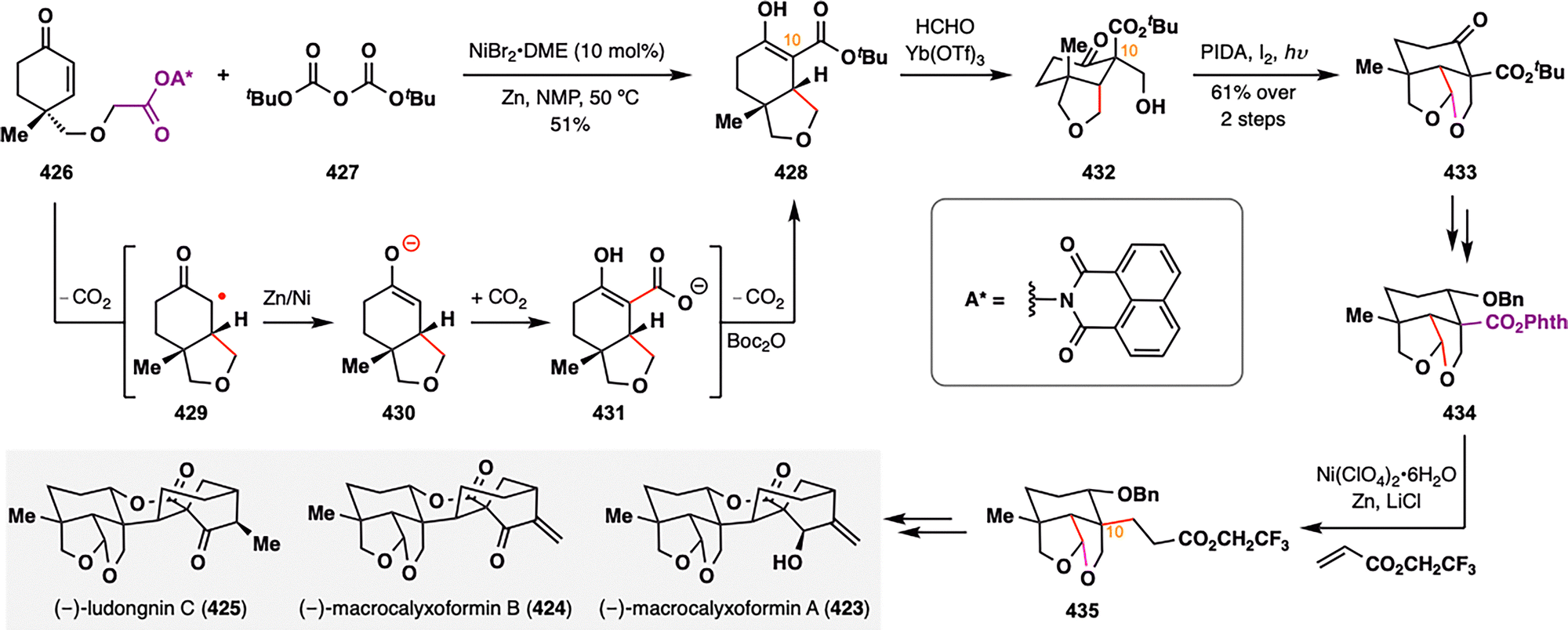 | ||
| Scheme 52 Li's total synthesis of (−)-macrocalyxoformins A (423) and B (424), and (−)-ludongnin C (425). | ||
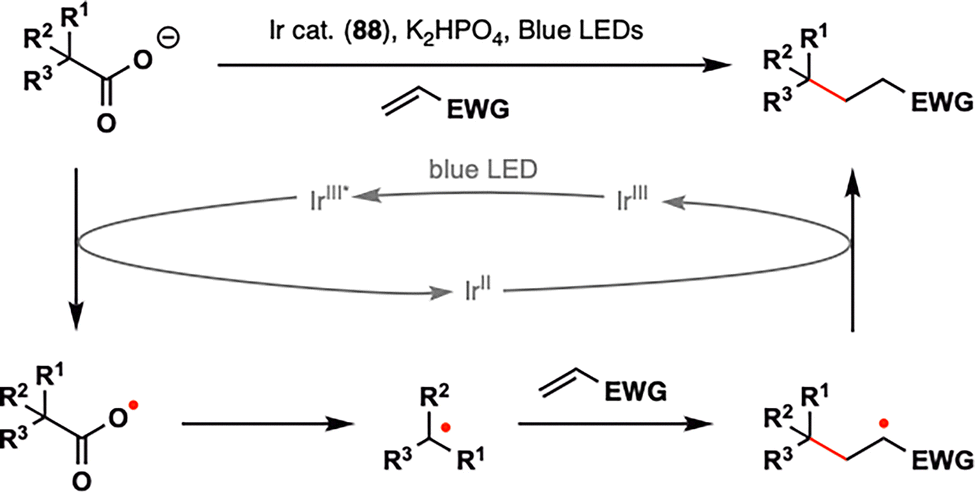 | ||
| Scheme 53 Macmillan's pioneered study on direct decarboxylative Giese reaction. | ||
Reisman and co-workers achieved the total synthesis of the cyclobutane-containing meroterpenoid (+)-rumphellaone A (436) by employing enantioenriched cyclobutene derivatives as chiral starting materials, leveraging C–H arylation and oxidative decarboxylative radical addition as key steps (Scheme 54).221 A directed C–H arylation of 437 with 438, facilitated by an 8-aminoquinolinamide auxiliary (8AQ), furnished cis-arylated cyclobutene 439 in excellent yield (90%). Hydrolysis of 439 triggered epimerization at the carboxyl center, delivering the thermodynamically favored trans-diastereomer 440. Subsequent oxidation, methylation, and hydrogenation of 440 delivered lactone 441, which was then subjected to oxidative decarboxylative radical addition. In the presence of K2HPO4, photoexcited Ir(III)* catalyzed a direct decarboxylative Giese reaction with methyl vinyl ketone (MVK), providing (+)-rumphellaone A (436) in good yield. This concise sequence underscores the strategic value of oxidative radical disconnections for the stereocontrolled assembly of highly substituted cyclobutane frameworks.
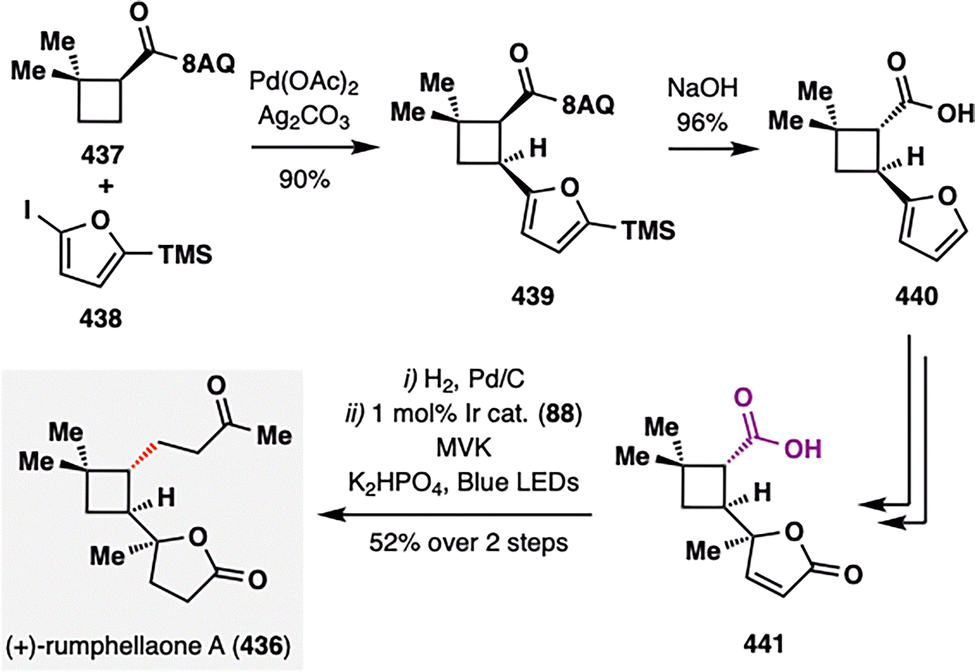 | ||
| Scheme 54 Reisman's total synthesis of meroterpenoid (+)-rumphellaone A (436). | ||
Inoue and co-workers strategically employed an oxidative decarboxylative radical cyclization to achieve a pivotal 7-endo-trig ring closure in their total synthesis of resiniferatoxin (442),222 a highly oxygenated daphnane diterpenoid isolated from Euphorbia resinifera (Scheme 55).223 The key cyclopentanone intermediate 446 was constructed through a Barton dehydroxylative allylation of 443 followed by a Trost-type cross-coupling between fragments 444 and 445. Upon hydrolysis and base-promoted deprotonation, the resulting carboxylate underwent single-electron oxidation by photoexcited Ir(III)*, generating a conformationally rigid bridgehead radical via CO2 extrusion.206 This radical then engaged in a stereocontrolled intramolecular 7-endo-trig addition. Crucially, a pre-installed OTBS group served as a steric control element, enforcing radical approach from the opposite face; simultaneously, β-elimination of the OTBS group delivered tricyclic scaffold 448. Subsequent side-chain elaboration furnished resiniferatoxin (442), highlighting the utility of radical-based ring closures for constructing densely functionalized diterpenoid frameworks.
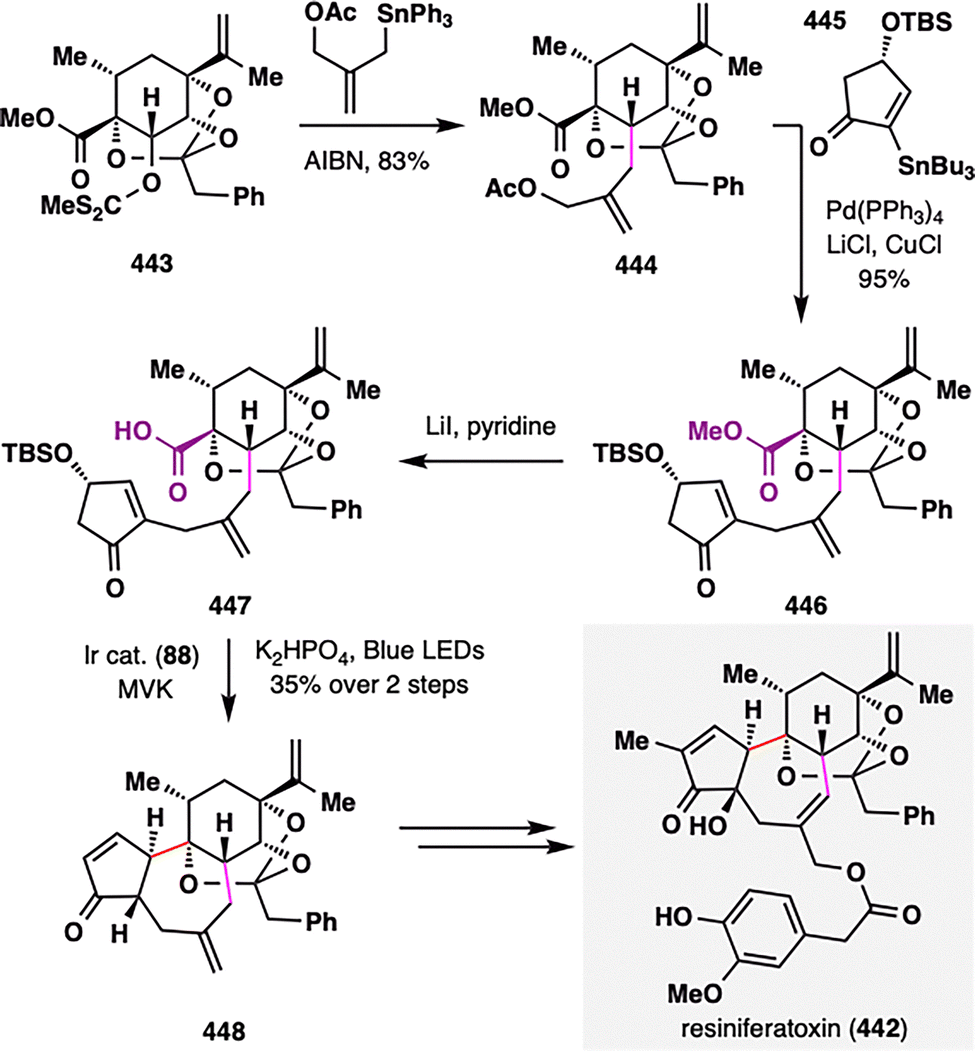 | ||
| Scheme 55 Inoue's total synthesis of resiniferatoxin (442). | ||
While most previous examples of oxidative decarboxylation rely on base-mediated deprotonation to generate carboxylate anions, Inoue and co-workers developed a base-free decarboxylative coupling protocol in their total synthesis of taxol (449)—a structurally intricate taxane diterpenoid with well-established anticancer activity—thereby substantially broadening the substrate scope and functional group compatibility of this strategy (Scheme 56).224 The key carboxylic acid precursor 451 was prepared enantioselectively from 2,2-dimethylcyclohexane-1,3-dione (450). The decarboxylative coupling was accomplished under neutral conditions using a Pt-doped TiO2 photocatalyst (MPT-623) and violet LED irradiation in ethyl acetate,225 effecting a direct union of acid 451 with enone 452. A subsequent one-pot DDQ oxidation furnished enone 453 in 45% overall yield, with full preservation of stereochemical integrity. Unlike conventional Ir(III)-based photoredox methods—which typically require basic conditions to generate the carboxylate—this neutral protocol avoids the use of external bases, rendering it particularly advantageous for substrates bearing base-sensitive functional groups. The mild conditions significantly enhance chemoselectivity and broaden the functional group tolerance. With this complex enone 453 in hand, a series of intramolecular cyclizations and late-stage elaborations ultimately enabled completion of the total synthesis of taxol (449).
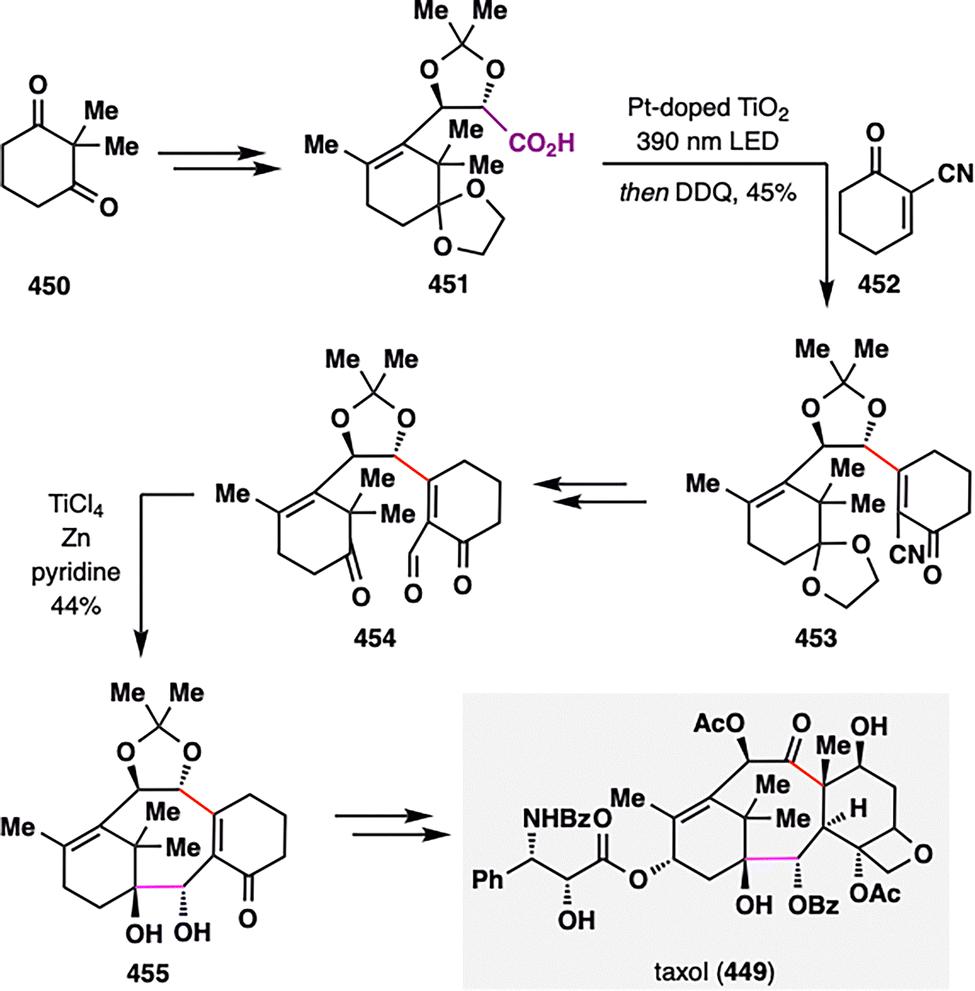 | ||
| Scheme 56 Inoue's total synthesis of taxol (449). | ||
Following their successful application of base-free decarboxylative cross-coupling in the total synthesis of taxol (449), Inoue and co-workers extended this strategy to achieve the first total synthesis of euphorbialoid A (456),226 a highly oxygenated member of the rare premyrsinane diterpenoid family characterized by a 5/7/6/3-membered tetracyclic framework (Scheme 57). Initial attempts employing Ir(III)-based photocatalysis under basic conditions resulted in substantial substrate decomposition. To circumvent this limitation, they turned again to their previously developed Pt-doped TiO2 (MPT-623) photocatalyst,225 which enabled efficient coupling of carboxylic acid 458 with enone 460 under neutral conditions. Remarkably, the decarboxylative radical addition proceeded with high stereoselectivity, delivering a single diastereomer and forging a sterically congested quaternary center. The diastereoselectivity was rationalized by the steric shielding imparted by the TMS-alkyne on the bottom face of fragment 458 and the dimethylcyclopropane moiety on the top face of fragment 460, directing the radical addition to the least hindered trajectory. This transformation efficiently installed two contiguous stereocenters and markedly increased molecular complexity in a single operation. A late-stage Pauson–Khand reaction then forged the 5/7/6/3 fused carbocyclic core 463, setting the stage for final oxidation-state adjustments to furnish euphorbialoid A (456).
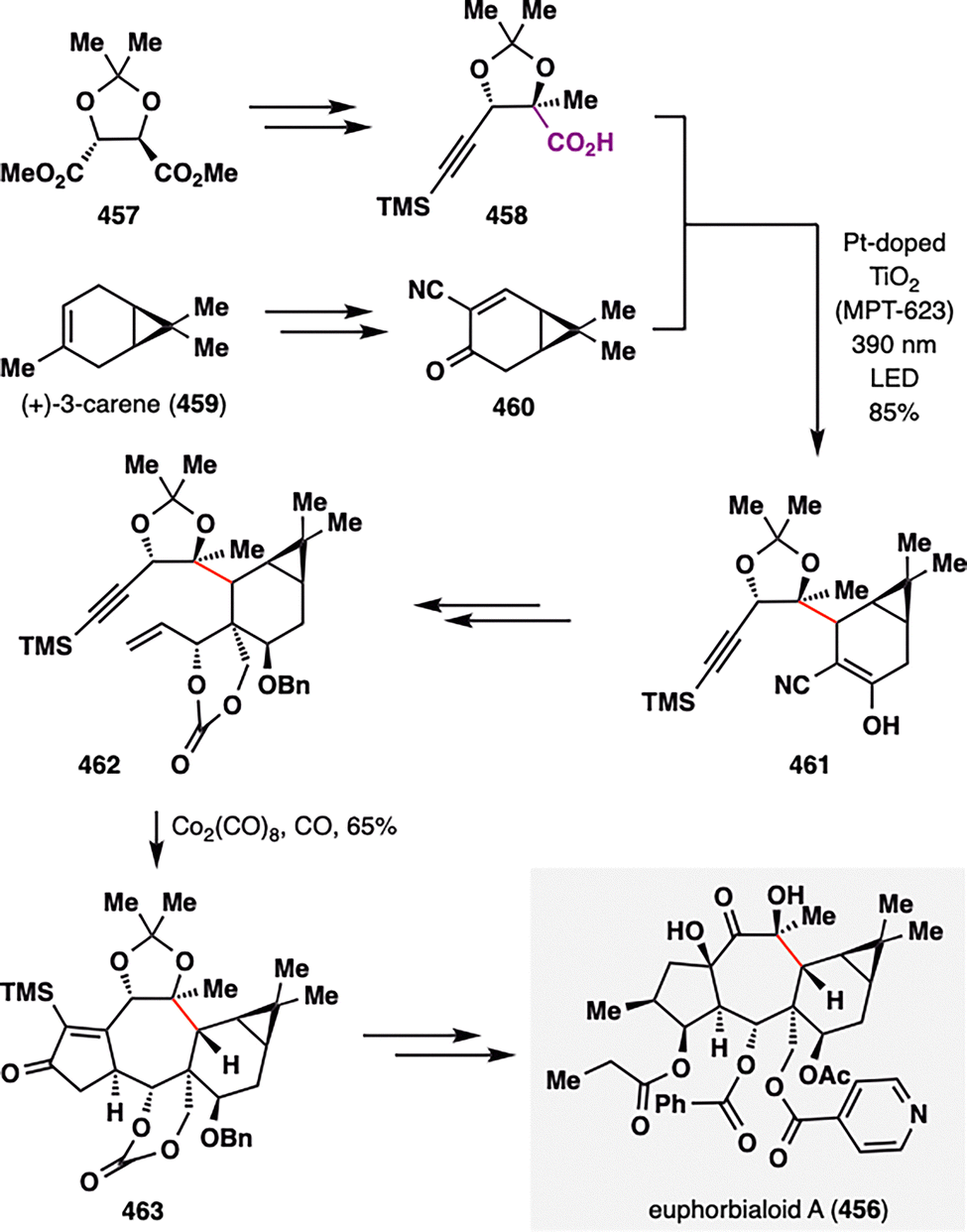 | ||
| Scheme 57 Inoue's total synthesis of euphorbialoid A (456). | ||
Inoue and co-workers resourcefully employed their developed Et3B/O2-mediated decarbonylation of α-alkoxyacyl tellurides as a key transformation in their total synthesis of 1-hydroxytaxinine (464),227 particularly for constructing sterically hindered C–C bonds within highly oxygenated frameworks (Scheme 58). This strategy effectively demonstrated excellent functional group compatibility under mild radical conditions. Mechanistically, homolytic cleavage of the C–Te bond, initiated by Et3B/O2, generated a ketyl radical that underwent spontaneous decarbonylation to afford α-alkoxy radical 467. Importantly, while this step erased the original stereochemistry at the radical center, the subsequent radical addition to an enone 452 re-established stereocontrol, as the coupling partner selectively approached from the face opposite to the bulky C4 substituent. The resulting radical intermediate was then trapped by Et3B to generate a boron enolate 468, which upon oxidation furnished enone 469 as a single diastereomer in 65% yield. Inoue further demonstrated the synthetic utility of this radical-based approach in their synthesis of taxol (449),224 using it as an alternative to MPT-623 photocatalysis for stereocontrolled decarboxylative coupling—thereby underscoring the versatility of radical-mediated strategies in the construction of complex taxane architectures.
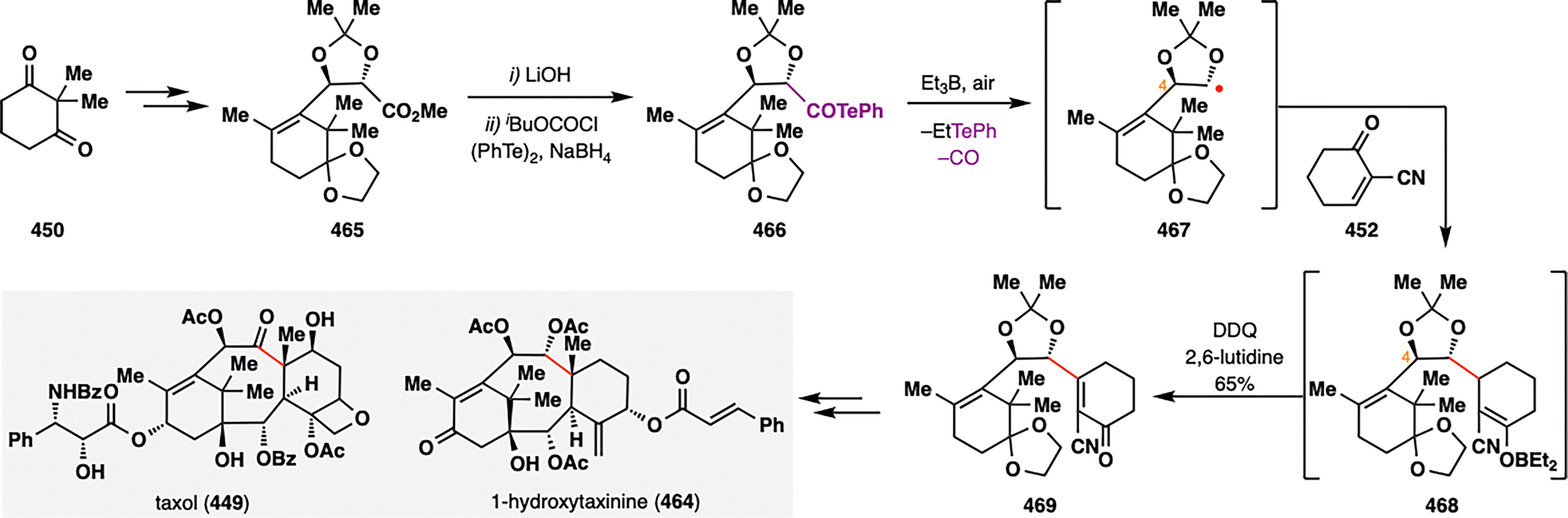 | ||
| Scheme 58 Inoue's total synthesis of 1-hydroxytaxinine (464) and taxol (449). | ||
Building on the success of their earlier taxane synthesis, Inoue and co-workers further refined their approach in 2023 by unveiling a streamlined route to taxol (449) that shortened the overall sequence by six steps while preserving the key Et3B/O2-mediated decarbonylation of α-alkoxyacyl tellurides as a pivotal transformation (Scheme 59).228 Specifically, guided by theoretical calculations, they envisioned that bicycle 475's 6–H/Br/CN would all deliver C3-stereoselectivity, and the radical accepting propensities was expected to be 6–CN > 6–Br > 6–H, based on their respective LUMO energies. Upon experimental validation, 6–Br emerged as the optimal coupling partner, serving as electron-acceptor with α-alkoxyketyl radical through radical addition. The observed stereoselectivity was attributed to the steric shielding imposed by the proximal dimethyl group in intermediate 470 and the ether bridge within 475, which collectively blocked the bottom face and directed radical addition to the less-hindered top face. Following the coupling event, the bromide was reductively removed under Zn/AcOH conditions, enabling downstream elaboration toward the complex taxol (449) with minimal functional group manipulations.
 | ||
| Scheme 59 Inoue's total synthesis of taxol (449). | ||
Continuing their pioneering application of α-alkoxyacyl telluride couplings, Inoue and co-workers accomplished a formal total synthesis of batrachotoxin (477),229,230 a potent steroidal alkaloid originally isolated from a Colombian poison-dart frog,231 featuring a densely functionalized 6/6/6/5-membered carbocyclic framework (Scheme 60). A central element of this strategy was the Et3B/O2-mediated decarbonylation of an α-alkoxyacyl telluride, which enabled the union of the A/B ring system 478 and the D-ring fragment 479 with stereospecific establishment of a quaternary center. Notably, the decarbonylative transformation proceeded with high chemoselectivity: selective homolytic cleavage of the C–Te bond in 478 occurred without perturbing the adjacent C–Br bond, a distinction attributed to the activating influence of the neighboring oxygen substituent. This process generated a bridgehead α-alkoxy radical with fixed stereochemistry, which underwent a radical addition to the electron-deficient alkene of 479. While the radical coupling afforded two diastereomers (480 and 481) at C11 without significant stereoselectivity, both intermediates could be funneled toward the desired diastereomer through subsequent elaborations.
 | ||
| Scheme 60 Inoue's formal total synthesis of batrachotoxin (477). | ||
3.2. Decarboxylative C(sp2)–C(sp3) bond formation
The construction of C(sp2)–C(sp3) bonds, particularly through alkenylation, is indispensable in constructing terpenoid frameworks. Building on Baran's pioneering work in 2016, which established the viability of Ni-catalyzed cross-coupling of RAEs with aryl zinc reagents,232 subsequent advancements have expanded this paradigm to alkenyl systems—encompassing alkenylzinc reagents,233 vinyl halides,234,235 and related electrophiles.236,237 These methodological innovations have profoundly streamlined terpenoid synthesis, offering unparalleled efficiency in forging their complex architectures. Notably, all reported decarboxylative C(sp2)–C(sp3) bond formations in terpenoid synthesis exclusively utilize RAEs as radical precursors, underscoring their unique combination of practicality, stability, and chemoselectivity. The defining feature of RAEs lies in their ability to undergo single-electron reduction under transition metal catalysis (e.g., Ni, Ir, Cu), generating alkyl radicals that engage alkenyl or aryl coupling partners. This radical-mediated pathway circumvents the electronic limitations inherent to classical ionic mechanisms, thereby enabling the convergent assembly of sterically congested terpenoid skeletons with precision.Following Baran's pioneering 2016 work on RAEs,232 the strength of this methodology was swiftly illustrated in Baran's synthesis of (+)-2-oxo-yahazunone (482) using a decarboxylative arylation.238 As detailed in Scheme 61, electrochemical C–H oxidation of sclareolide (184) at a 50 g scale delivered ketone 483, which was subsequently converted to RAE 484 and coupled via Ni-catalyzed decarboxylative cross-coupling to form the critical C–C bond,232 culminating in the efficient synthesis of 486. Followed by CAN-mediated oxidative dearomatization, (+)-2-oxo-yahazunone (482) was obtained in high efficiency. This seamless integration of electrochemical oxidation and RAE-based decarboxylative coupling exemplifies the transformative potential of these methodologies in streamlining complex natural product assembly.
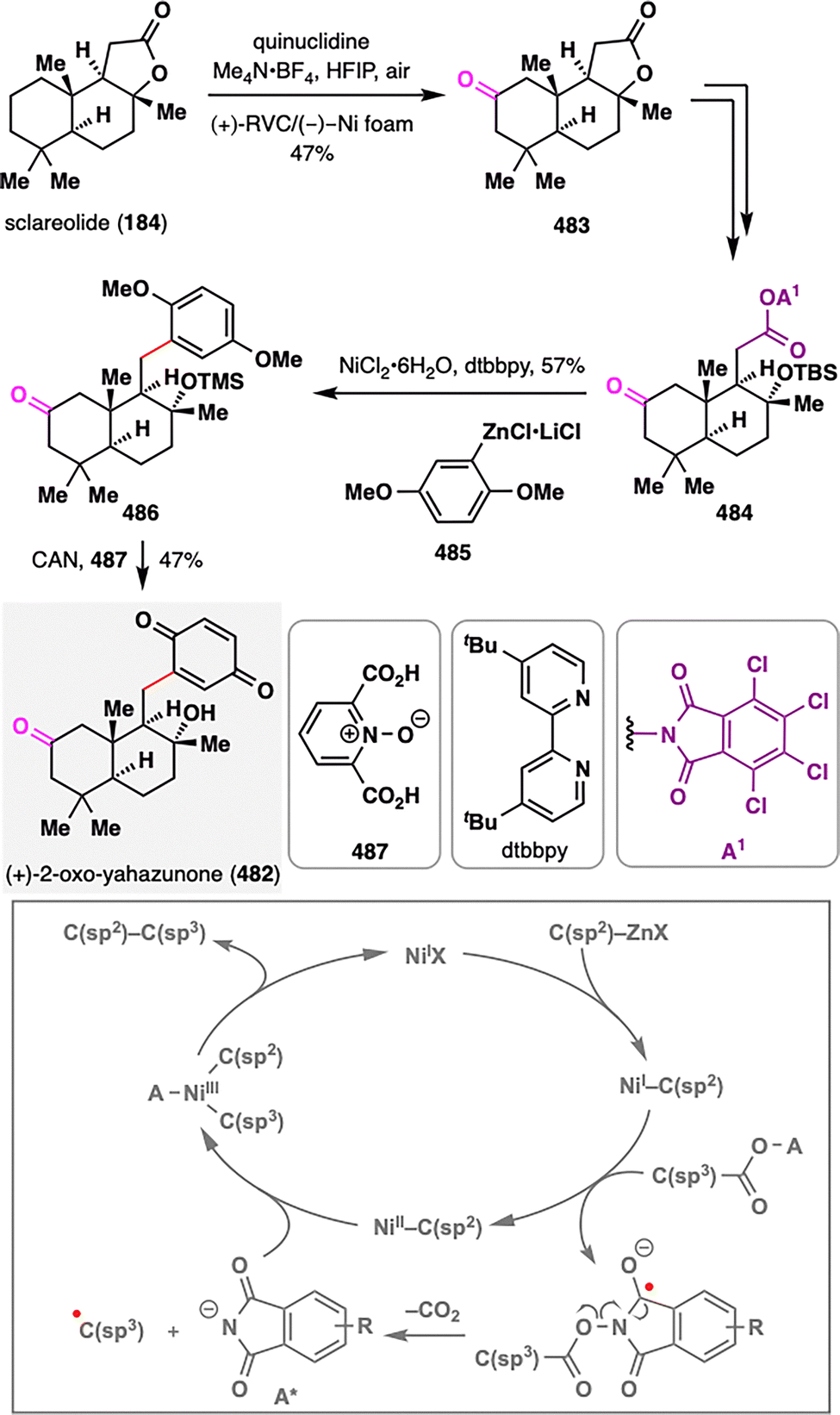 | ||
| Scheme 61 Baran's synthesis of (+)-2-oxo-yahazunone (482). | ||
Regarding the Ni-catalyzed cross-coupling of RAEs with aryl zinc reagents, a plausible mechanism is outlined in Scheme 61.232 The catalytic cycle is proposed to initiate with transmetalation between a NiI complex and the aryl zinc reagent, furnishing an aryl–NiI intermediate. This species then undergoes single-electron reduction of the NHPI ester, generating an alkyl radical—following decarboxylation—and an aryl–Ni(II) complex. Subsequent radical capture by the aryl–Ni(II) species delivers a key aryl–Ni(III)–alkyl intermediate, which undergoes reductive elimination to provide the desired C(sp2)–C(sp3) cross-coupled product. Notably, the same mechanistic paradigm can be extended to the following decarboxylative alkenylation reactions.
Building upon their earlier work on decarboxylative arylation in 2016,232 Baran and co-workers subsequently disclosed an efficient decarboxylative alkenylation strategy,233 enabling the construction of C(sp2)–C(sp3) bonds across a broad substrate scope—including mono- to fully substituted olefins and a wide range of carboxylic acids, from primary to tertiary (Scheme 62(A)). The decarboxylative alkenylation, which proceeded between RAEs and organozinc reagents in the presence of inexpensive Ni(II) catalysts, had proven exceptionally efficient for the streamlined synthesis of diterpenes and steroids, as illustrated in Scheme 62(B).233 Utilizing carboxylic acid 495 as a common intermediate, Baran and co-workers demonstrated a divergent approach to access three clerodane diterpenes (492–494) via the above alkenylation sequence, significantly reducing the total synthesis of (−)-kolavenol (492) from 21 steps to just 9 steps with excellent stereoselectivity (E:Z > 20:1), in particular. Similarly, employing carboxylic acid 497 as the decarboxylative precursor, α-tocotrienol (496) was efficiently assembled in only two steps—substantially improving upon the previously reported seven-step sequence.
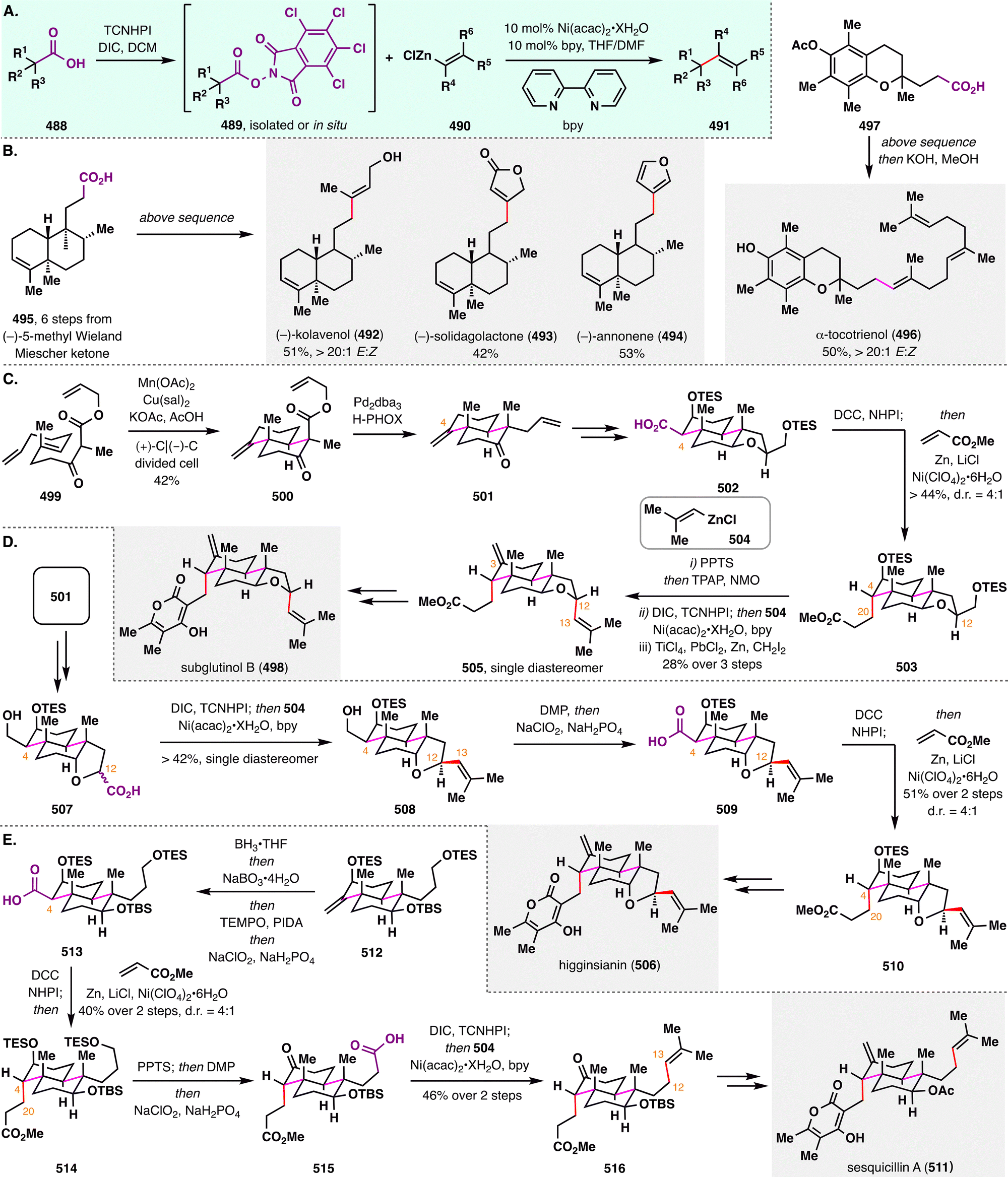 | ||
| Scheme 62 Baran's decarboxylative alkenylation and their applications in terpenoid synthesis. | ||
As shown in Scheme 62(C)–(E), Baran and co-workers further expanded the scope of their decarboxylative alkenylation platform by applying it to the divergent total synthesis of several structurally distinct pyrone diterpenoids,239 all derived from a common trans-decalin intermediate (501). As outlined in Scheme 62(C), the trans-decalin scaffold was efficiently assembled via an electrochemically assisted, Mn-catalyzed oxidative radical polycyclization of 499 to furnish intermediate 500, followed by a Tsuji allylation to deliver 501. Conversion of the C4 exo-methylene into a carboxylic acid, along with construction of the right-hand tetrahydrofuran ring, afforded carboxylic acid 502. Under the group's previously developed reductive decarboxylative radical addition conditions,205502 underwent coupling with methyl acrylate to furnish ester 503. Oxidation of the C12 hydroxymethyl group then generated a carboxylic acid, which was subjected to nickel-catalyzed decarboxylative alkenylation to install an isopropenyl substituent at C12 with high diastereoselectivity. Notably, this radical-based approach was particularly advantageous in this context, as traditional α-oxy radical precursors such as halides are inherently unstable at this position. Subsequent olefination at C3 yielded olefin 505, a versatile intermediate that could be converted to subglutinol B (498) following Hong's reported protocol.240
Leveraging insights from the synthesis of subglutinol B (498), the team next achieved the total synthesis of higginsianin A (506)—another architecturally complex member of the pyrone diterpenoid family—by reversing the order of the decarboxylative transformations (Scheme 62(D)). Specifically, decarboxylative radical addition at C4 was carried out after the nickel-catalyzed alkenylation at C12. This sequence inversion preserved high stereoselectivity at both sites but led to an opposite diastereoselectivity at C12, thereby enabling access to the stereochemical configuration required for higginsianin A (506). The resulting intermediate 510 was further elaborated to complete the first total synthesis of higginsianin A (506). Remarkably, the decarboxylative alkenylation proceeded smoothly even in the presence of an unprotected primary alcohol, underscoring the functional group tolerance of the method. The same modular strategy was extended to the synthesis of sesquicillin A (511) (Scheme 62(E)), where the radical-based disconnections effectively addressed stereochemical challenges that had previously defied solution using traditional two-electron disconnections. These studies collectively highlight the synthetic power of radical cross-coupling in constructing highly oxidized and stereochemically complex terpenoid architectures.
Advancing their decarboxylative alkenylation platform, Baran and co-workers developed an electrochemical reductive decarboxylative C(sp2)–C(sp3) cross-coupling strategy between RAEs and alkenyl halides (Scheme 63(A)).235 This method provided a powerful and modular approach to terpene synthesis (Scheme 63(B)–(F)), obviating the need for preformed alkenylzinc reagents and minimizing functional group manipulations, protecting group interconversions, and redox fluctuations. A key mechanistic insight emerged from their studies: although pure silver electrodes were ineffective, silver ions proved indispensable. Under electrochemical conditions, Ag-based nanoparticles formed in situ on the cathode surface, playing a crucial role in promoting the cross-coupling.
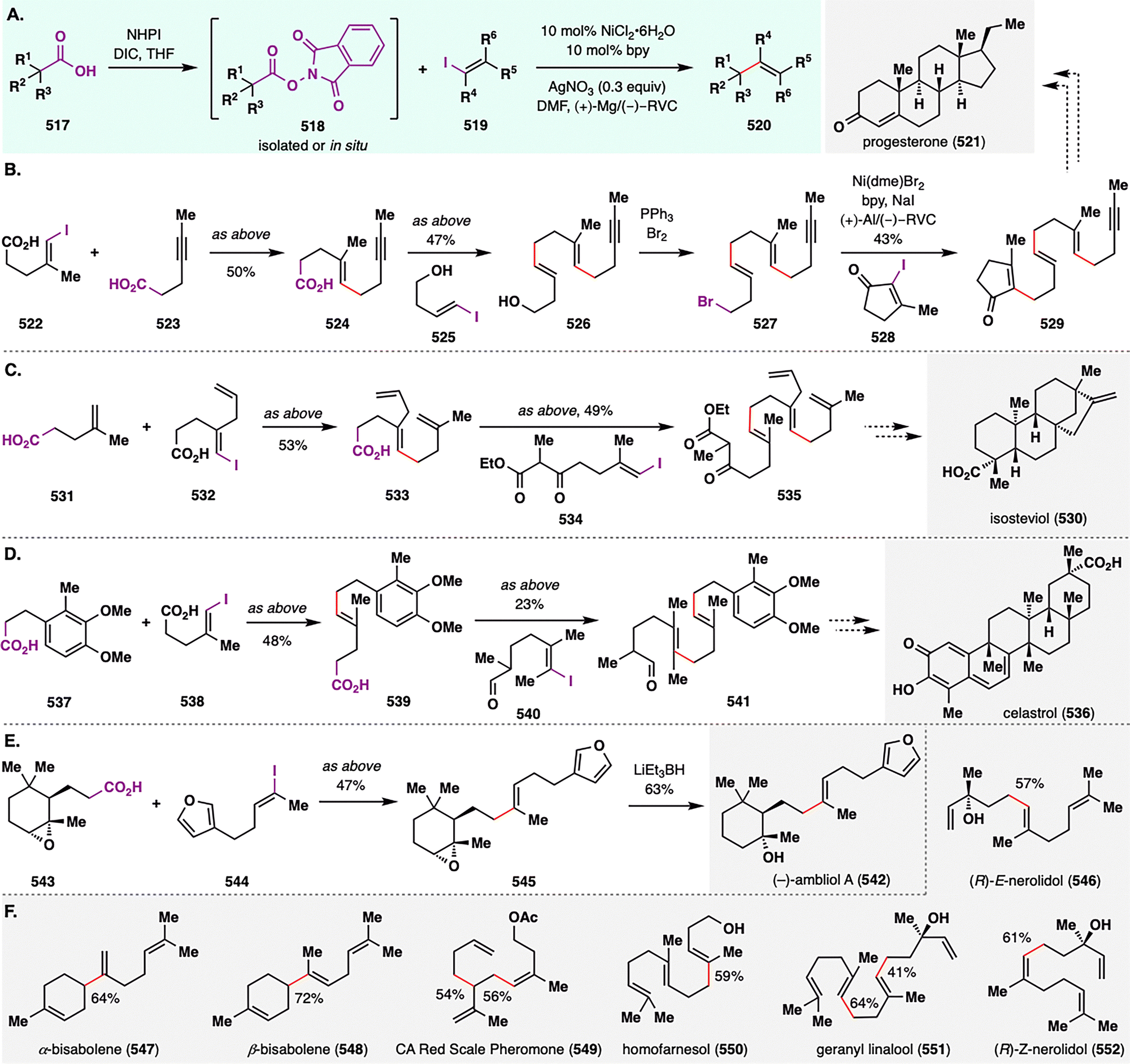 | ||
| Scheme 63 Baran's modular terpene synthesis. | ||
To demonstrate the utility of this approach in complex molecule synthesis, the group streamlined the preparation of a polyene cyclization precursor en route to progesterone (521) (Scheme 63(B)). Conventional retrosynthetic disconnections toward this intermediate were both non-intuitive and inefficient. In contrast, their electrocatalytic decarboxylative strategy enabled direct forging of a pivotal C(sp2)–C(sp3) bond between well-defined building blocks, thereby simplifying the overall synthetic route. Through sequential electrocatalytic couplings of geometrically defined fragments, they accessed the key polyene intermediate in a modular and convergent fashion, ultimately enabling an efficient total synthesis of progesterone (521).
Applying a similar strategy, Baran's group efficiently synthesized two additional polyene precursors en route to structurally complex terpenoids: isosteviol (530) and celastrol (536) (Scheme 63(C) and (D)), using their electrochemical decarboxylative alkenylation twice in each case. These examples further highlight the robustness of the method, which exhibited broad functional group tolerance—including epoxides, alkynes, unprotected alcohols, and carboxylic acids.
To assess the generality of this strategy, the group extended its application to a wider range of terpenoid targets. In particular, they accomplished the first chemical synthesis of ambliol A (542), a natural product originally isolated from the sponge Dysidea amblia, by cross-coupling an epoxy acid 543 with alkenyl iodide 544, followed by reductive epoxide opening (Scheme 63(E)). Further examples—including the synthesis of polyene terpenoids 546–552 (Scheme 63(F))—demonstrate the remarkable flexibility of this electrocatalytic decarboxylative platform for the modular construction of complex terpenoid scaffolds.
Building on the success of their electrocatalytic decarboxylative platform for modular terpene synthesis, Baran and co-workers expanded its application to a more synthetically demanding target—(+)-calcipotriol (553),241 a clinically relevant secosteroid and structural analog of vitamin D. This class of molecules, widely recognized for their broad-spectrum biological activities, has long served as a privileged framework in therapeutic development.242 Guided by their radical-based retrosynthetic logic, the team designed a route that strategically combined electrochemical reductive alkenylation with an intramolecular BROC radical cyclization. As illustrated in Scheme 64, the key carboxylic acid 555 was synthesized from readily available cyclohexenone (554) and subjected to their electrocatalytic decarboxylative alkenylation with alkenyl iodide 556.235 This transformation efficiently forged a critical C(sp2)–C(sp3) bond, installing an α,β-unsaturated ester motif and furnishing intermediate 557 in 63% yield—while simultaneously embedding the radical handle for the forthcoming BROC radical cyclization.
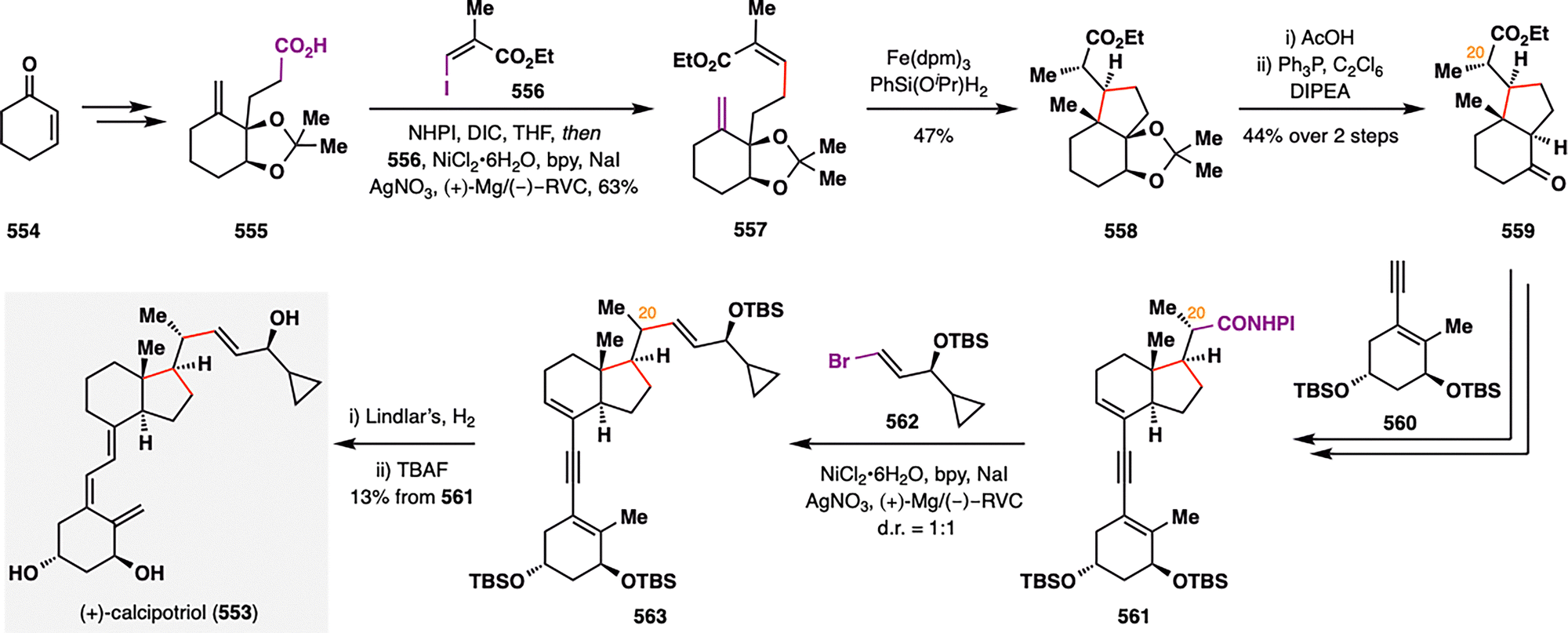 | ||
| Scheme 64 Baran's total synthesis of (+)-calcipotriol (553). | ||
The thermodynamically disfavored trans-6/5 ring system 559 was then constructed through a BROC reaction followed by a semipinacol rearrangement. Coupling 559 with alkyne 560 and converting the C20 ester into a RAE set the stage for a second decarboxylative alkenylation with 562 to afford 563. Although this transformation exhibited modest diastereoselectivity (d.r. = 1:1), subsequent hydrogenation of the alkyne and global deprotection of the TBS group successfully delivered (+)-calcipotriol (553). This synthesis exemplifies the power of integrating sequential decarboxylative radical couplings with innovative radical cyclization strategies to streamline access to architecturally complex and pharmacologically relevant secosteroids.
Building upon the demonstrated versatility of their electrocatalytic reductive decarboxylative platform, Baran and co-workers further highlighted its synthetic utility in the concise total synthesis of the marine diterpenoid (−)-bipinnatin J (564),243 employing sequential decarboxylative couplings using succinate as a linchpin for modular C–C bond formation (Scheme 65). A central feature of this synthesis was the strategic use of a succinate unit as a two-carbon ethylene bridge to unite sp2-hybridized fragments with high efficiency. The sequence began with commercially available monomethyl succinate (565), which underwent electrochemical decarboxylative alkenylation with alkenyl iodide 566. Subsequent hydrolysis delivered carboxylic acid 567. A second decarboxylative coupling—this time with alkenyl bromide 568—furnished butanolide intermediate 569, effectively stitching together the two alkenyl fragments through the succinate-derived ethylene linker. Finally, a Ni(II)/Zn-mediated 1,6-conjugate addition using J. Zhou's protocol introduced the furan unit in a convergent fashion,244 enabling late-stage elaboration to the natural product (−)-bipinnatin J (564).
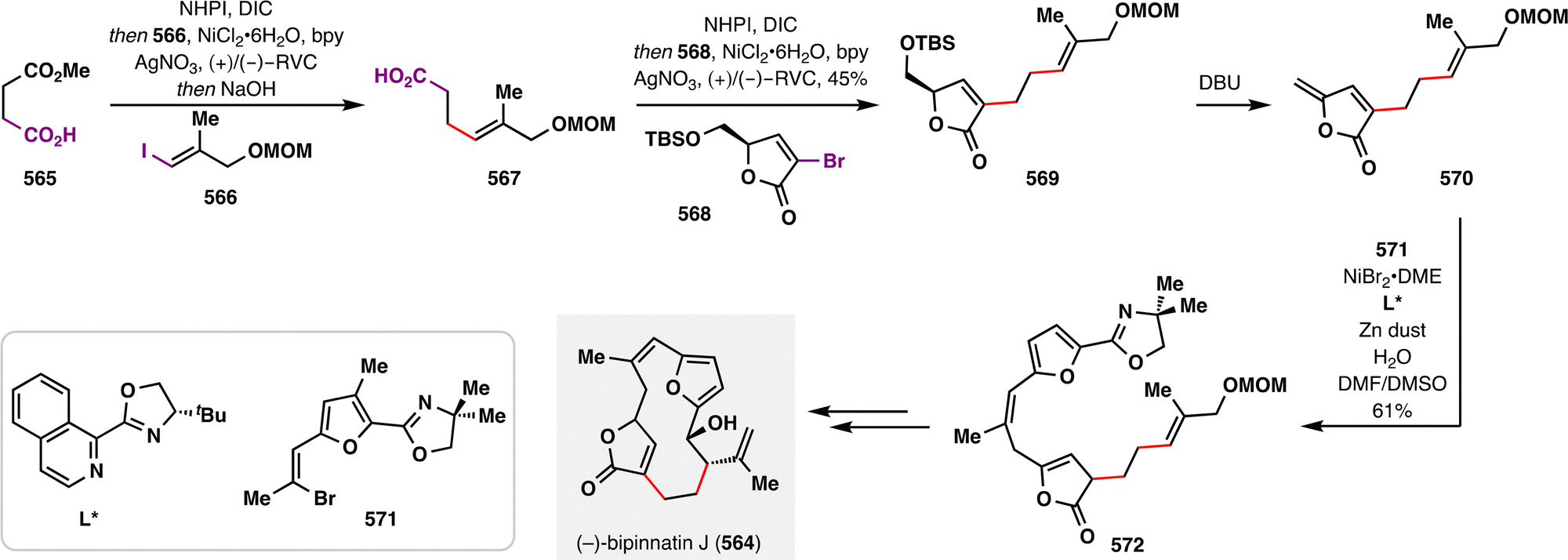 | ||
| Scheme 65 Baran's total synthesis of (−)-bipinnatin J (564). | ||
Recently, the Tomanik group accomplished a convergent and stereoselective total synthesis of the nominal structure of (+)-talaromyolide D (Scheme 66).245 Their strategy pivoted on a stereoretentive, nickel-catalyzed electrochemical decarboxylative cross-coupling between sp2 and sp3 carbons, complemented by strategic C–H functionalizations. The synthesis commenced with the preparation of the cis-substituted cyclobutane building block 575 from (−)-verbenone in three steps: oxidative cleavage of the alkene followed by Baeyer–Villiger oxidation with m-CPBA furnished the key intermediate, whose derived carboxylic acid was subsequently converted to the redox-active NHPI ester 575. A pivotal decarboxylative cross-coupling between 575 and vinyl iodide 576, under Baran's nickel–electrocatalytic conditions using silver nanoparticle-modified electrodes, afforded the 577 in 85% yield with 11:1 dr. An aromatic moiety was then installed at C14 via an oxime-directed C(sp3)–H arylation; subsequent removal of the directing group and Wittig olefination delivered alkene 578. This alkene underwent diastereoselective iron-catalyzed Mukaiyama-type hydration, and the resulting tertiary alcohol participated in an intramolecular SNAr reaction with a pendant aryl fluoride to construct the C-ring, yielding 579 in 80% over two steps. Finally, two late-stage carboxylic-acid-directed C(sp2)–H oxidations installed the isocoumarin lactone, thereby completing the synthesis of talaromyolide D.
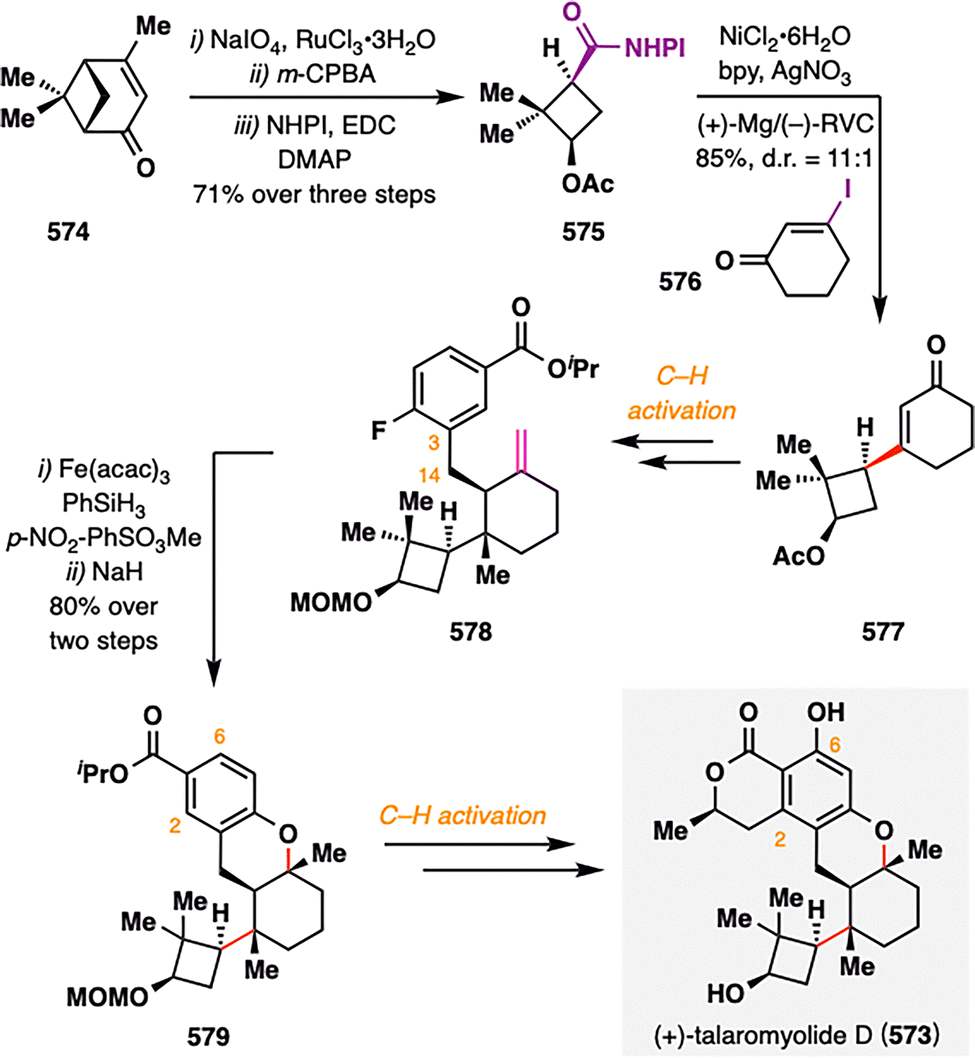 | ||
| Scheme 66 Tomanik's total synthesis of talaromyolide D (573). | ||
While previous C(sp2)–C(sp3) decarboxylative coupling strategies have primarily relied on organozinc reagents or alkenyl halides as coupling partners, Gui and co-workers expanded the scope of this methodology by employing organoboron reagents—originally developed for such transformations by the Baran group—in the first total synthesis of myrmenaphthol A (580),246,247 a structurally unique phenolic steroid featuring a naphthyl-fused A/B ring system (Scheme 67). The synthesis began with sitolactone (581), which was converted into the corresponding carboxylic acid via a one-pot lactone hydrolysis followed by Jones oxidation. Subsequent condensation with N-hydroxytetrachlorophthalimide furnished RAE 582, setting the stage for a Baran's Ni-catalyzed decarboxylative cross-coupling with boronic acid 583. This strategy again demonstrated its efficacy for constructing C(sp2)–C(sp3) bonds, affording tricyclic intermediate 584 in 60% yield. A sequence of downstream elaborations—including a key Friedel–Crafts cyclization/olefin isomerization/oxidative aromatization cascade—culminated in a concise 10-step total synthesis of myrmenaphthol A (580).
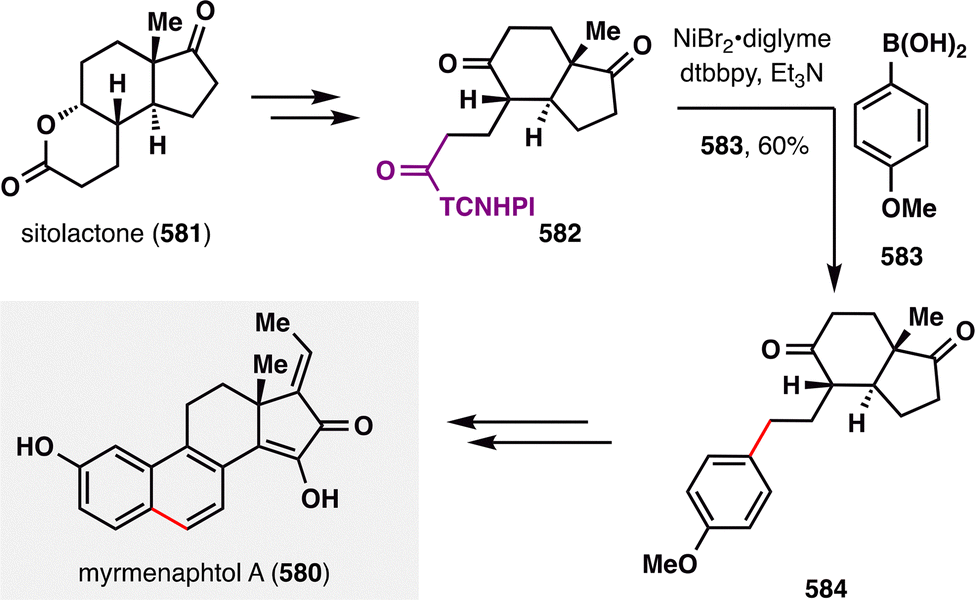 | ||
| Scheme 67 Gui's total synthesis of myrmenaphthol A (580). | ||
Inspired by Baran's pioneering work on decarboxylative alkenylation, our group developed a tandem decarboxylative cyclization/alkenylation strategy that enabled the divergent total synthesis of (+)-effusin (585), (+)-longirabdolactone (586), and (+)-longirabdiol (587), three highly oxygenated spiro-lactone type ent-kauranoids (Scheme 68).248 Notably, we employed a modified decarboxylative cross-coupling protocol originally developed by Weix and Reisman,249,250 utilizing RAEs and C(sp2)-haloalkenes as coupling partners. In contrast to Baran's approach, which employed alkenylzinc reagents and often led to undesired direct alkenylation at C6 due to the high reactivity of the organozinc species,233 our protocol minimized such side reactions and improved site-selectivity. Key to the success of this transformation was the use of CuCl2 as the catalyst, Davephos as ligand, and 2-bromopropionic acid as an additive, which significantly enhanced the coupling efficiency. Under these conditions, the desired vicinal alkene 590 was obtained as a diastereomeric mixture (d.r. = 4.2:1) in 60% yield. Subsequent elaboration furnished monocyclic carboxylic acid 591, which then underwent a reductive decarboxylative Giese-type radical addition with an acrylate, installing the all-carbon quaternary stereocenter at C10. This transformation set the stage for divergent synthesis of the spiro-lactone ent-kauranoid core, culminating in the successful preparation of (+)-effusin (585), (+)-longirabdolactone (586), and (+)-longirabdiol (587).
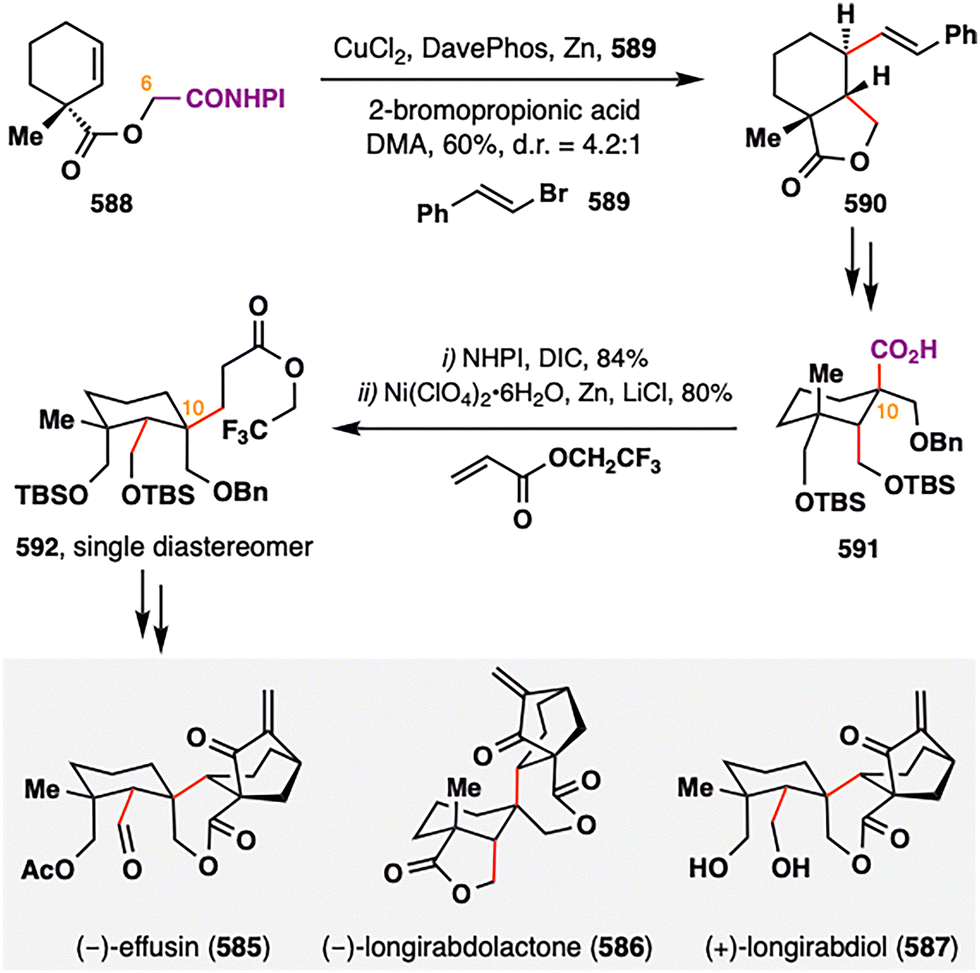 | ||
| Scheme 68 C. Li's total synthesis of (+)-effusin (585), (+)-longirabdolactone (586), and (+)-longirabdiol (587). | ||
In contrast to the previously discussed Ni-catalyzed decarboxylative cross-coupling strategies involving RAEs and alkenylzinc, alkenyl halide, or boronic acid coupling partners, Magauer and co-workers developed a tandem photochemical decarboxylation/Friedel–Crafts cyclization that elegantly incorporates a RPCO event for the efficient construction of the tetralone scaffold in lingzhiol (593)—a meroterpenoid natural product isolated from Ganoderma fungi, distinguished by its 1,2,4-trisubstituted aromatic ring and polycyclic terpenoid core (Scheme 69).251,252 In this transformation, RAE 596 underwent a photoinduced single-electron reduction by an excited-state Ir(III)* complex, generating stabilized tertiary radical 598 after extruding CO2. Subsequent single-electron oxidation by Ir(IV) produced the corresponding tertiary carbocation 599—thereby completing the RPCO sequence. This carbocation was then rapidly trapped by a tethered electron-rich arene via an intramolecular Friedel–Crafts cyclization, furnishing tetralone intermediate 600. A critical aspect of this strategy involved strategic protection of phenolic hydroxyl groups to prevent undesired overoxidation of the electron-rich aromatic ring during the RPCO step. Final global deprotection of 600 delivered lingzhiol (593) in a concise and convergent fashion, highlighting the utility of RPCO-based radical strategies in the efficient assembly of complex meroterpenoid architectures.
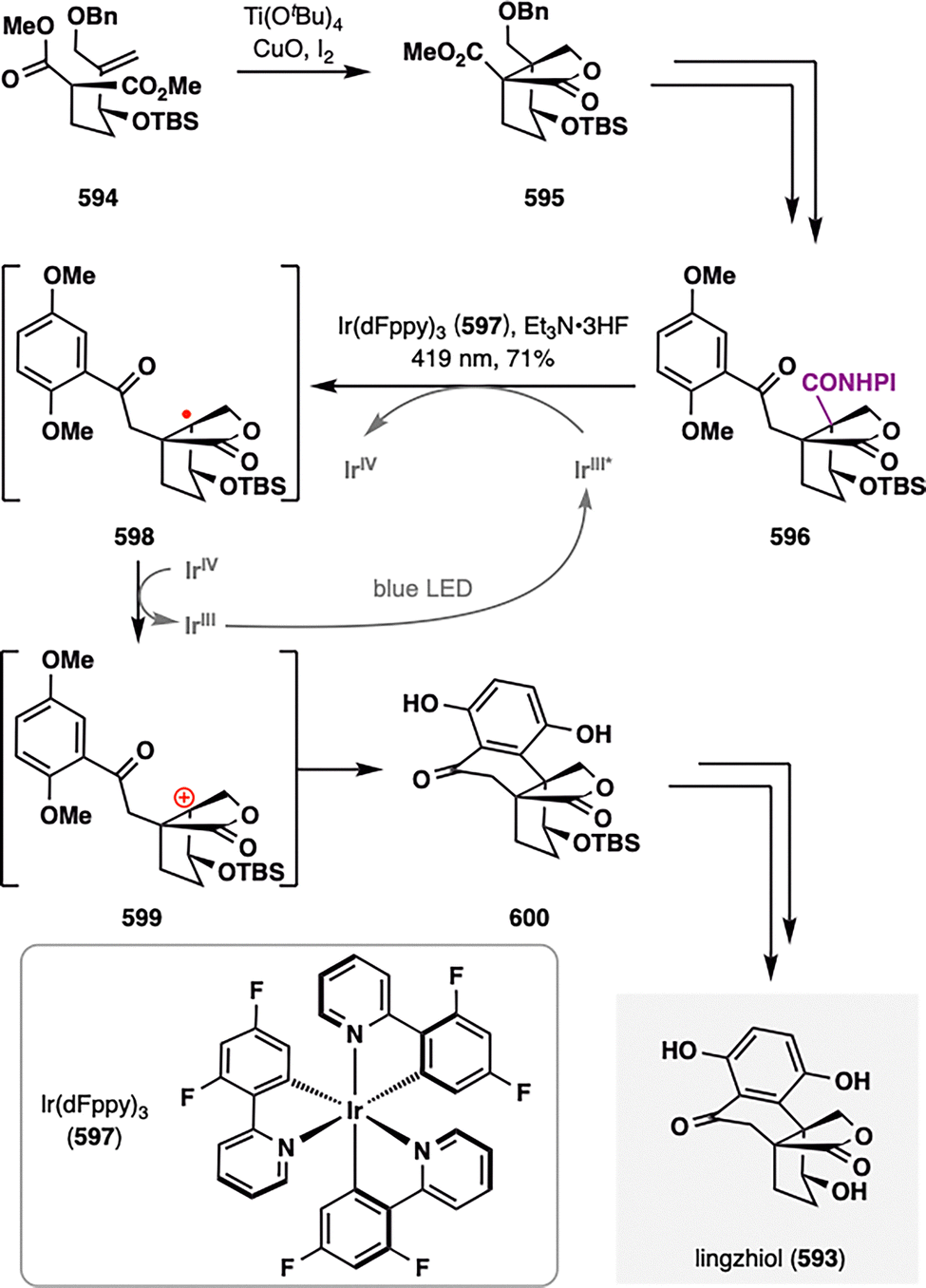 | ||
| Scheme 69 Magauer's total synthesis of lingzhiol (593). | ||
4. Organohalides as radical precursors: versatile handles for terpenoid assembly
The utilization of organohalides as radical precursors embodies a classically rooted yet dynamically evolving strategy in synthetic organic chemistry. The homolytic cleavage of carbon–halogen bonds, a foundational mechanism for generating carbon-centered radicals, has historically underpinned diverse bond-forming reactions through its operational simplicity and broad applicability. Over the past decade, methodological breakthroughs—spanning metal-catalyzed systems,253 photoredox platforms,9,254 and catalyst-free protocols255—have substantially augmented the functional group compatibility, regioselectivity, and environmental sustainability of dehalogenative transformations. These advances have reinvigorated the strategic value of organohalide-derived radicals in addressing synthetic challenges posed by structurally intricate molecules. Within the framework of terpenoid natural product synthesis,three organohalide-enabled radical strategies have garnered significant attention in contemporary methodologies: dehalogenative XEC,10,256 visible-light-induced dehalogenative radical addition,9 and dehalogenative cross-coupling.257From a retrosynthetic perspective (Scheme 70), the construction of a C(sp3)–C(sp2) bond can be traced to its precursors via dehalogenative XEC reactions, disconnecting to an alkyl halide and an aryl or vinyl halide. Alternatively, this bond formation may be retrosynthetically envisioned through dehalogenative cross-coupling, derived from an alkyl halide (serving as the radical precursor) and an organometallic coupling partner (such as organotin reagents). Similarly, the formation of a C(sp3)–C(sp2) bond can be retrosynthetically analyzed through radical addition pathways, retracing to an alkyl halide and an alkene (typically an electron-deficient olefin). Note that this retrosynthetic analysis is grounded in the current application of organohalides as radical precursors within the context of reported terpenoid natural product synthesis. We anticipate that the recent emerging novel synthetic methodologies leveraging organohalides as radical sources, particularly the asymmetric dehalogenative cross-couping,257 will find increasingly fruitful applications in the efficient construction of complex terpenoid architectures.
 | ||
| Scheme 70 Modern radical retrosynthesis of C–C bonds from organohalides. | ||
4.1. Dehalogenative cross-electrophile coupling (XEC)
Dehalogenative XEC—a strategy enabling the direct union of two electrophiles through single electron transfer (SET) pathways—has redefined C(sp2)–C(sp3) bond construction in complex molecule synthesis.10 Pioneered by Périchon in the 1990s via electrochemical reductive cross-couplings,258 this methodology was later advanced by Weix,259 Gong,260 Reisman261et al. through transition-metal catalysis (e.g., Ni, Co), which bypasses the need for preparing organometallic reagents. XEC facilitates the direct coupling of two electrophilic components via SET pathways, exhibiting unparalleled tolerance toward diverse functional groups, including alcohols and carbonyls—features that are indispensable for manipulating multifunctional terpenoid intermediates. By leveraging the innate reactivity of organohalides and avoiding stoichiometric metal reagents, XEC achieves streamlined synthetic workflows and enhanced step economy, making it particularly suited for forging polycyclic terpenoid frameworks. In current total synthesis of terpenoid natural products, XEC has been predominantly employed to forge C(sp2)–C(sp3) bonds with exceptional functional group tolerance.10Christmann and co-workers exemplified the synthetic power of XEC by achieving the first protecting-group-free total synthesis of the cytotoxic diterpenoid alkaloids vitepyrroloids A (601, IC50 = 8.7 μM) and B (602),262 isolated from the dried leaves of Vitex trifolia L.263 As shown in Scheme 71, the key transformation was a Ni(II)/Zn-mediated dehalogenative XEC between alkyl iodide 603—readily prepared from sclareolide (184)—and pyrrole bromide 604. Mechanistically, the process is proposed initiated by the reduction of Ni(II) to low-valent Ni(0) by zinc. The alkyl iodide 603 undergoes SET with Ni(0) to generate a carbon-centered radical, while the aryl halide 604 undergoes oxidative addition to Ni(0), forming an aryl–Ni(II) complex. Radical capture by this aryl–Ni(II) species forms a high-valent Ni(III) intermediate, which undergoes reductive elimination to forge the C(sp3)–C(sp2) bond, thereby delivering vitepyrroloid A (601). Importantly, the inclusion of NaI was found to effectively suppress undesired homocoupling side products by modulating the halide speciation. It is worth noting that multiple mechanistic pathways have been proposed for XEC reactions, and the exact mechanism may vary depending on the nature of the substrates and ligands involved.10 From this intermediate, vitepyrroloid B (602) was obtained in 79% yield via a straightforward SN2 displacement with a brominated ester, completing the efficient and concise synthesis of both natural products.
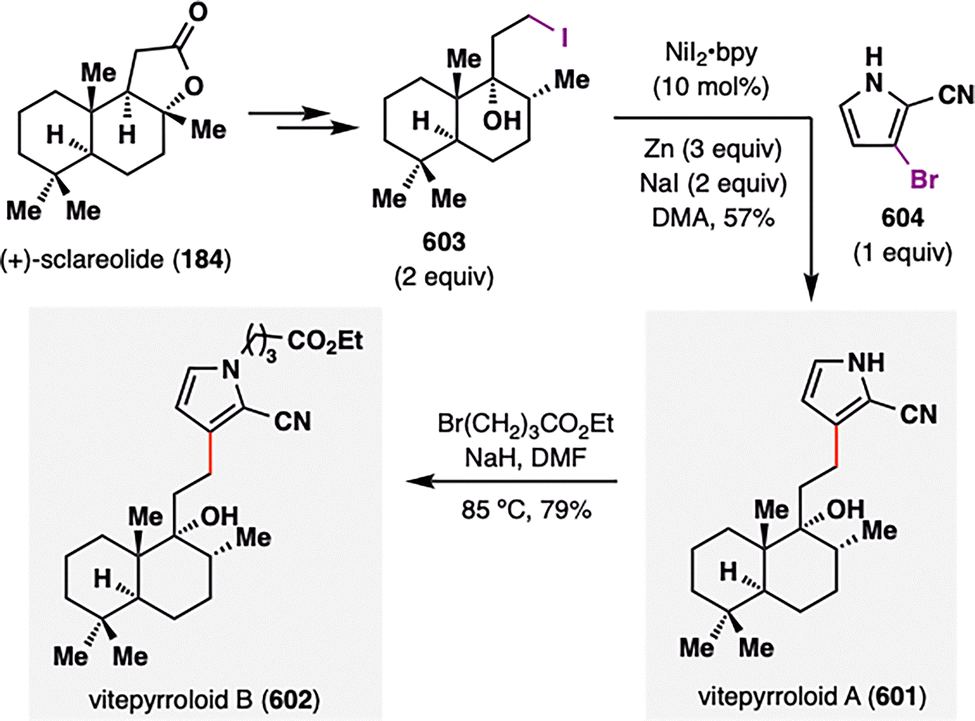 | ||
| Scheme 71 Christmann's synthesis of (+)-vitepyrroloids A (601) and B (602). | ||
Similarly, Burtoloso and co-workers harnessed a dehalogenative XEC strategy to achieve concise total synthesis of the icetaxane diterpenoids (±)-brussonol (605) and (±)-komaroviquinone (606),264 featuring a densely functionalized fused 6/7/6 tricyclic scaffold, in only six and eight steps, respectively (Scheme 72). A key aspect of their approach was the in situ generation of alkyl iodide 609 through NaI-mediated epoxide opening of 607, enabling the subsequent XEC with arylbromide 608. This approach exhibited excellent functional group tolerance—including compatibility with sensitive aldehyde moieties—and enabled the convergent union of two complex fragments to forge bycyclic 610, setting stage for further manipulations to furnish (±)-brussonol (605) and (±)-komaroviquinone (606).
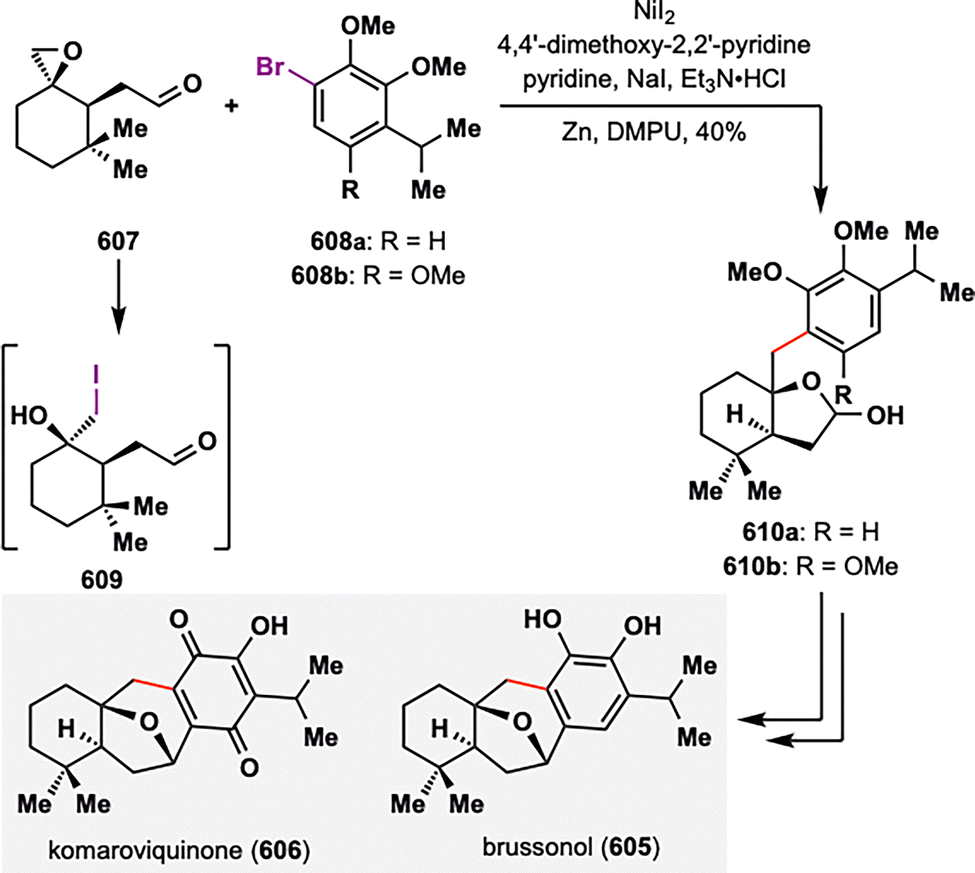 | ||
| Scheme 72 Burtoloso's total synthesis of (±)-brussonol (605) and (±)-komaroviquinone (606). | ||
As previously discussed in Section 2.1.1 (Scheme 7), Renata and co-workers employed a strategically designed sequence combining chemoenzymatic oxidation with BROC to construct the key tricyclic intermediate 50, which served as a versatile branching point en route to taondiol (43), and chevalone A (44).90 As shown in Scheme 73, the team subsequently adopted a nickel-catalyzed dehalogenative XEC to forge the critical C(sp2)–C(sp3) bonds. This strategic pivot was driven by the failure of decarboxylative couplings between C11 RAE and either alkenyl iodide 611 or aryl iodide 613, prompting the use of the corresponding C11 iodide 50 as a more reliable radical precursor. Under Weix's Ni-catalyzed conditions,259 the cross-couplings proceeded efficiently, delivering the coupled products in excellent yields of 85% and 76%, respectively. Subsequent acid-catalyzed cyclizations completed the formation of the dihydropyran rings, culminating in the total synthesis of taondiol (43) and chevalone A (44).
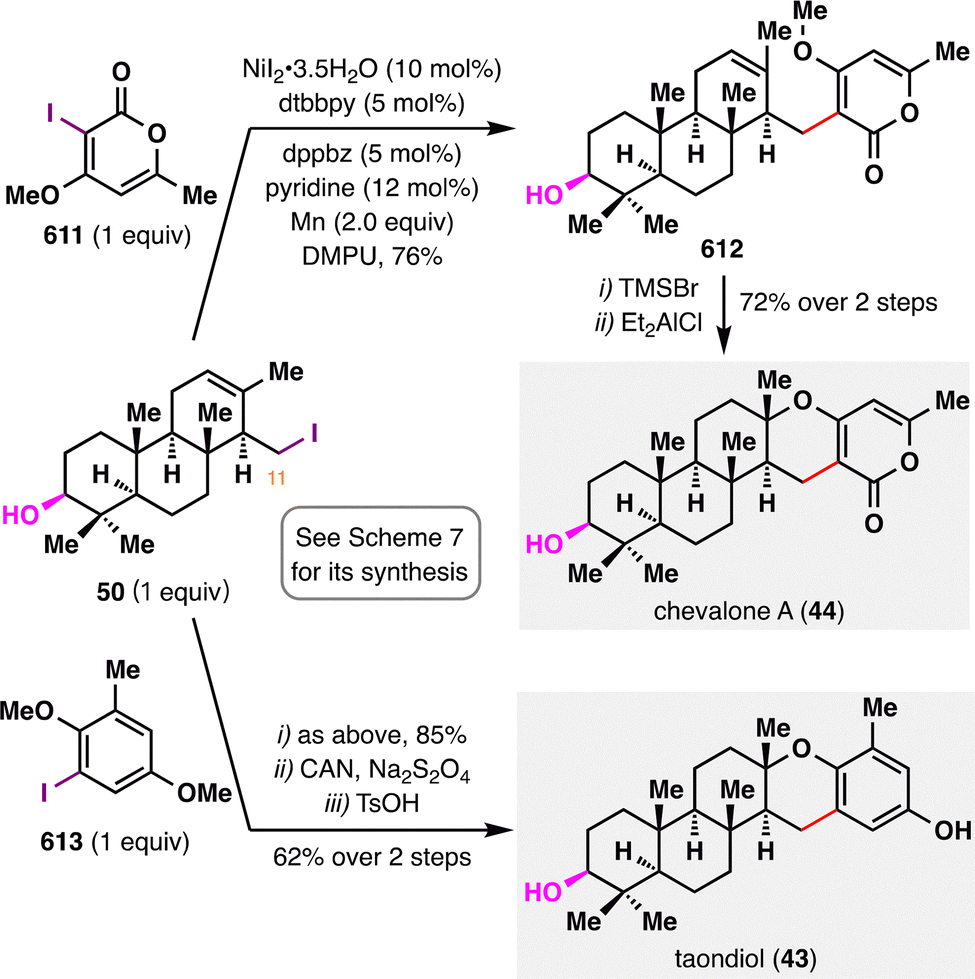 | ||
| Scheme 73 Renata's synthesis of oxidized meroterpenoids taondiol (43) and chevalone A (44). | ||
Li, Puno, and co-workers strategically employed a protecting-group-free dehalogenative XEC to install the side chain in their concise total synthesis of (−)-isoscopariusin A (614),265 an enantioenriched immunosuppressive meroditerpenoid characterized by a sterically congested 6/6/4 tricyclic carbon framework bearing seven contiguous stereocenters. As shown in Scheme 74, under Ni-catalyzed XEC conditions, functionalized alkyl bromide 615 coupled efficiently with (E)-3-bromobut-2-en-1-ol (616) under Weix's protocol,266 enabling the regio- and stereoselective installation of the (E)-crotyl alcohol side chain in 68% yield. Notably, this XEC transformation exhibited excellent functional group tolerance, accommodating both free allylic alcohols and carboxylic acids without the need for protecting groups. Moreover, the reaction was readily scalable to gram quantities. Following this key cross-coupling event, a streamlined sequence of elaborations culminated in the total synthesis of (−)-isoscopariusin A (614) in just 12 steps from commercially available (+)-sclareolide (184).
 | ||
| Scheme 74 Total synthesis of (−)-isoscopariusin (614) A by Li, Puno and co-workers. | ||
M. Yang and co-workers further showcased the remarkable utility of dehalogenative XEC in their terpenoid synthesis of trans-clerodanes and sesquiterpene (hydro)quinones to install different side chain with sterically hindered alkyl halides.267 As illustrated in Scheme 75(A), a NiBr2/Mn-catalyzed XEC under Shu's conditions successfully coupled sterically congested alkyl bromide 620 with oxalate ester 621 to forge a key C(sp3)–C(sp3) bond,268 yielding annonene (618). This terpenoid then underwent [4+2] addition with the singlet oxygen, furnishing PL3 (619) in 78% yield. In a related transformation (Scheme 75(B)), a XEC enabled the formation of a C(sp2)–C(sp3) bond via coupling of alkyl iodide 624 with aryl bromide 625, affording phenolic intermediates that were further oxidized to yield avarone-type natural products (622–623). The same dehalogenative arylation strategy was also employed in the synthesis of dysidavarones A–D (627–629, 634), further showcasing the broad applicability of XEC for assembling diverse terpenoid and quinonoid frameworks under mild, functional-group-tolerant conditions.
 | ||
| Scheme 75 Modular total synthesis of trans-clerodanes and sesquiterpene (hydro)quinones by Yang and co-workers. | ||
Recently, Jia and co-workers stratagically combined the Ni(II)/Mn catalyzed dehalogenative XEC and Mn(III)-mediated radical tandem cyclization to achieve a protecting-group-free total synthesis of the diterpenoid (+)-aberrarone (637, Scheme 76),269 a cyclohexane-angularly-fused triquinane natural product isolated from the West Indian gorgonian octocoral Pseudopterogorgia elisabethae.270 Specifically, Ni-catalyzed XEC between alkyl iodide 638 and alkenyl iodide 639 proceeded efficiently in the presence of an unprotected hydroxyl group to deliver compound 640 in 68% yield. Oxidation of 640 with IBX, followed by ketone acylation, furnished enyne 641. Then, a Mn(OAc)3-mediated radical cascade cyclization forged three rings in a single operation,271 delivering triquinane 642 in 40% yield. Further elaboration of this intermediate ultimately led to the completion of (+)-aberrarone (637).
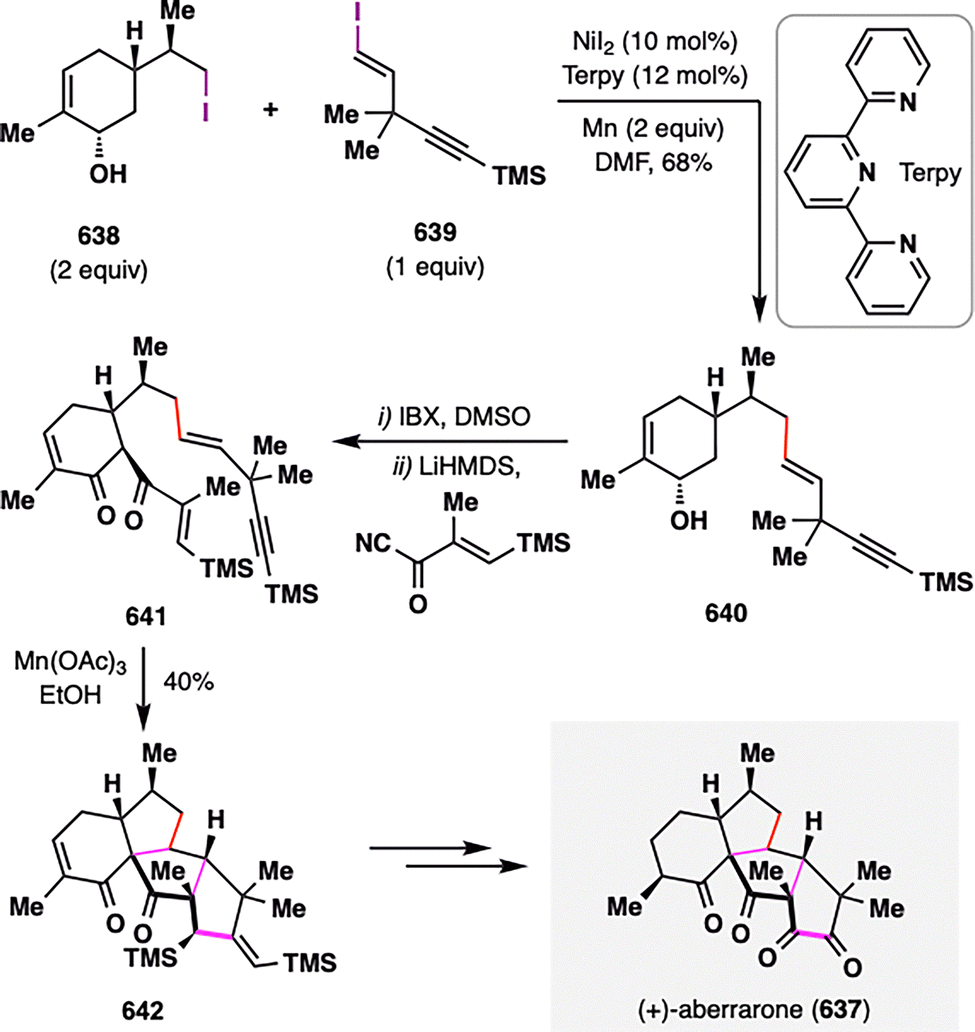 | ||
| Scheme 76 Jia's total synthesis of (+)-aberrarone (637). | ||
Together, these studies highlight the growing impact of XEC in complex terpenoid synthesis, offering a robust and modular platform for constructing highly congested carbon frameworks under mild and operationally simple conditions, with broad compatibility for sensitive functional groups.
4.2. Visible-light-induced dehalogenative radical addition
Visible-light-induced dehalogenative radical addition represents a paradigm shift in radical-mediated bond formation, harnessing photoredox catalysis to generate carbon-centered radicals under spatiotemporally controlled and environmentally benign conditions.9,254,272,273 Unlike traditional dehalogenative radical additions reliant on toxic initiators (e.g., AIBN, nBu3SnH), Modern photoredox systems employ tunable visible-light irradiation to drive SET events, selectively reducing organohalides to radicals while bypassing hazardous byproducts.From a retrosynthetic perspective, the bond disconnection logic for visible-light-induced dehalogenative radical addition aligns with classical dehalogenative radical addition strategies: target C–C bonds are traced to halogenated precursors and olefins. However, the photoredox platform sometimes exhibits superior efficiency, as demonstrated in Yang's total synthesis of haperforin G (643) (Scheme 77).274,275 Traditional methods employing azobis(isobutyronitrile) (AIBN)/nBu3SnH-mediated coupling of enone 644 and alkyl iodide 645 generated the desired product in only 21–40% yield. By contrast, a photoredox system employing a catalytic iridium-based photocatalyst ([Ir(ppy)2(dtbbpy)]PF6 (88), 2.5 mol%) under blue LED irradiation, with Hantzsch ester as a mild reductant, enabled efficient activation of the unactivated C(sp3)-hybridized iodide 645. The generated alkyl radical underwent intermolecular stereoselective addition to enone 644, delivering the adduct 646 as a single diastereoisomer in 64% yield. Subsequent intramolecular aldol condensation and dehydration then furnished haperforin G (643).
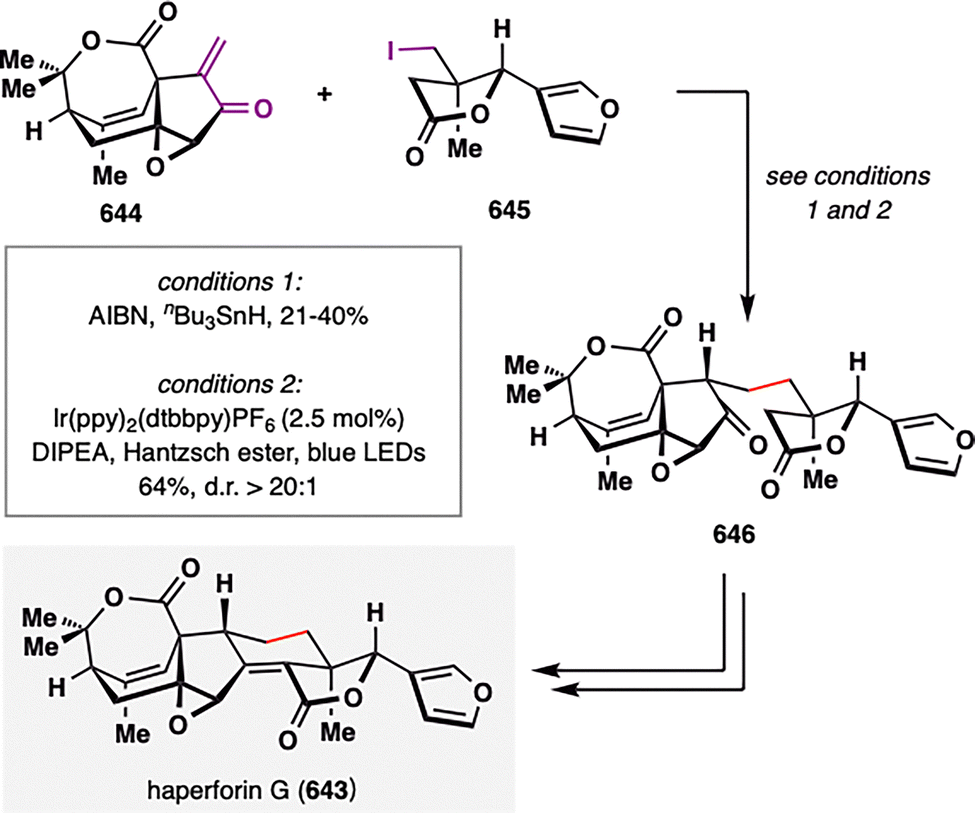 | ||
| Scheme 77 Yang's total synthesis of (+)-haperforin G (643). | ||
Notably, the redox-ambiphilic nature of photoredox catalysts enables unique mechanistic bifurcations. Following the initial single-electron reduction (SET) of the organohalide, the resulting carbon-centered radical may undergo a secondary SET oxidation by the now-oxidized photocatalyst—generating a carbocation and thereby opening pathways to radical-polar crossover (RPCO) transformations or tandem cationic cyclizations. This versatility significantly broadens the reactivity landscape of halogenated synthons, enabling sequential bond constructions within a single photocatalytic cycle—a powerful strategy for streamlining the assembly of structurally complex terpenoids.
As exemplified in the synthesis of norascyronone A (647) (Scheme 78),276 Lou, Cheng, and co-workers developed a photoredox-mediated aryl radical addition/cyclization/oxidative RPCO/Friedel–Crafts cascade that efficiently constructs densely functionalized polycyclic frameworks using diaryliodonium salts as aryl radical precursors. Mechanistically, photoexcited Ru(bpy)3Cl2 reduced diaryliodonium salt 649 to generate an aryl radical, which underwent intermolecular addition to enone 648, forming tertiary radical intermediate 650. This intermediate cyclized intramolecularly onto the alkenyl side chain to form alkyl radical 651, which was then oxidized by Ru(III) to generate carbocation 652. A subsequent intramolecular Friedel–Crafts-type reaction with a proximal arene furnished tetracyclic scaffold 653 in an optimized 84% yield. Final benzylic oxidation afforded the natural product norascyronone A (647).
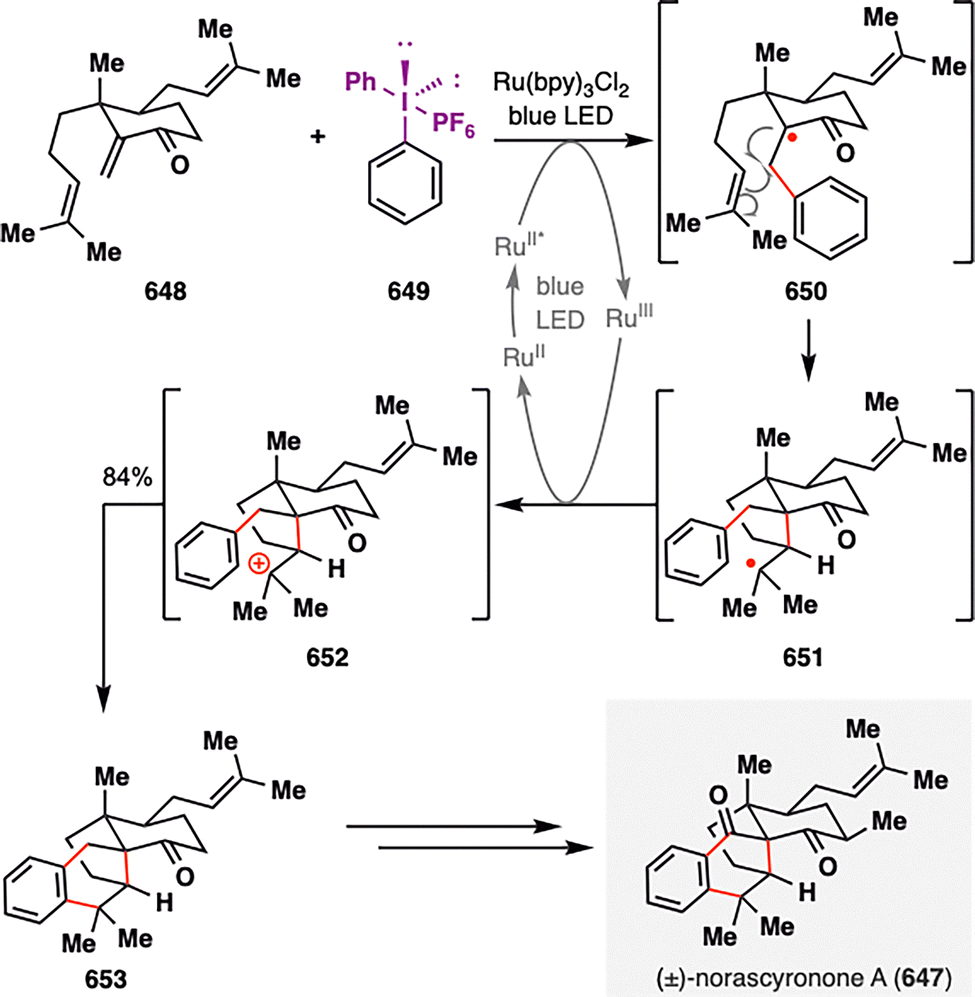 | ||
| Scheme 78 Total synthesis of norascyronone A (647) by Lou, Cheng, and co-workers. | ||
While visible-light-induced dehalogenative radical additions are typically redox-neutral, Peng and co-workers developed a photocatalytic reductive radical cascade—employing DIPEA as a sacrificial electron donor—that features an unusual 5-exo-trig/6-endo-trig cyclization sequence (Scheme 79).277 This transformation enabled the efficient and divergent construction of tricyclic diterpenoid frameworks, providing the first synthetic access to 10-epi-epoxyhinoliol (654), a structurally distinct abietane diterpenoid bearing a rare dihydrobenzofuran motif. The cascade was initiated by SET from Ir(II) to aryl iodide 655, triggering C–I bond homolysis and generating aryl radical 656. Note that Ir(II) was generated from the reduction of photoexcited Ir(III)* by DIPEA. This intermediate underwent sequential 5-exo-trig and 6-endo-trig cyclizations to form benzyl radical 657, which was subsequently quenched by HAT from cationic radical of DIPEA. Deprotection and regioselective bromination of the aromatic ring yielded aryl bromide 659, which then underwent a photoredox/nickel dual-catalyzed dehalogenative XEC with isopropyl bromide—using conditions developed by the MacMillan group—to deliver 10-epi-epoxyhinoliol (654) in 60% yield.
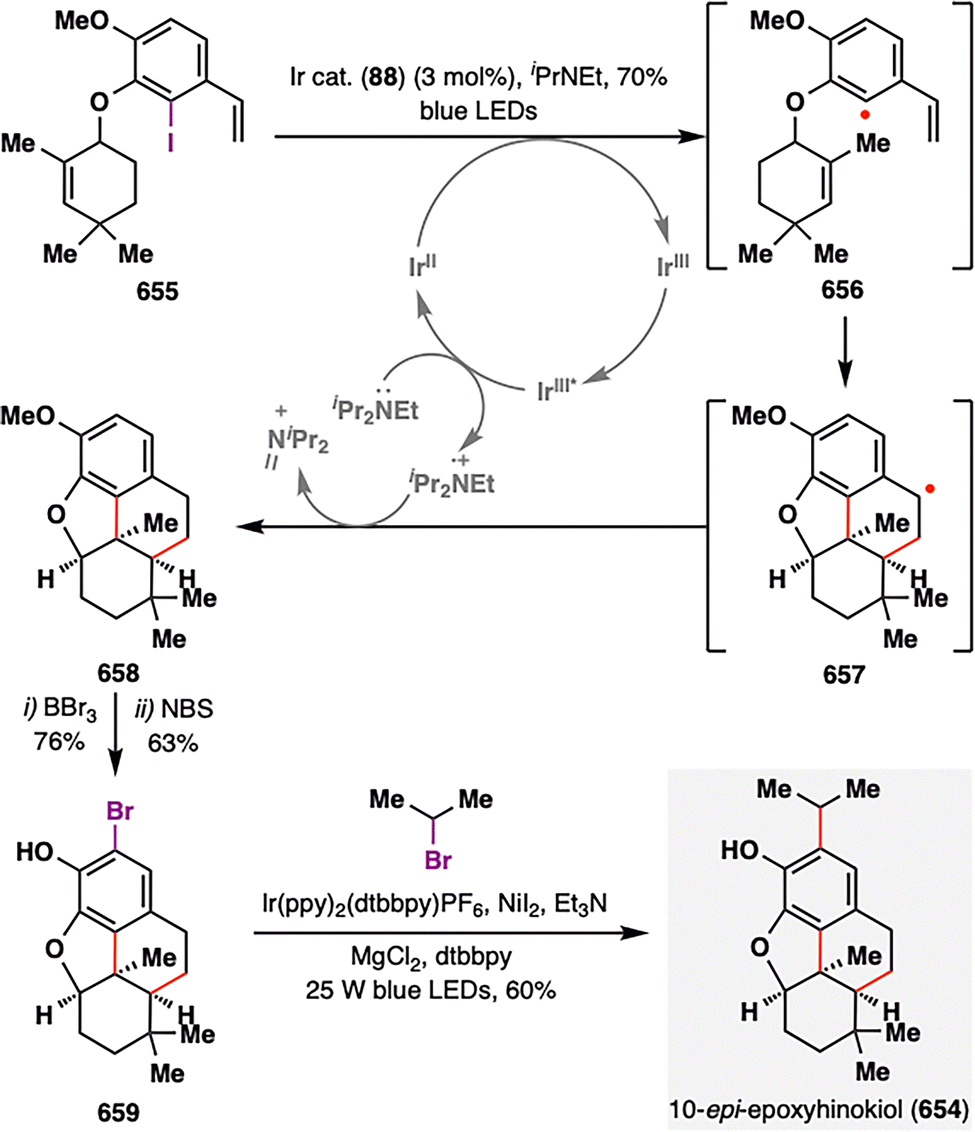 | ||
| Scheme 79 Total synthesis of 10-epi-epoxyhinoliol (654) by Peng and co-workers. | ||
The emerging strategy of visible-light-induced dehalogenative radical addition not only overcomes long-standing challenges associated with C–C bond formation between two electrophilic partners but also enables the orchestration of cascade processes under mild and redox-economical conditions. Its growing integration into terpenoid synthesis underscores its potential as a versatile platform for forging densely functionalized carbon architectures.
4.3. Dehalogenative cross-coupling
While dehalogenative cross-coupling has yet to gain broad traction in terpenoid assembly, recent methodological advances in radical involved transition-metal-catalyzed nucleophilic substitutions—particularly those achieving enantioselective C–C bond formation via chiral ligand design—signal its growing relevance.257 For instance, Ni-catalyzed asymmetric construction of quaternary carbons now enable the programmed installation of chiral motifs in congested environments.278–281 These developments, when adapted to terpenoid systems, will unlock novel disconnections for stereochemically dense terpenoids.To date, the most illustrative application of dehalogenative cross-coupling is found in P. Tang's landmark total synthesis of schilancidilactones A (660) and B (661), 20-epi-schilancitrilactone (664), and schilancitrilactone A (665), as illustrated in Scheme 80.282 At the final step, a Ni-catalyzed cross-coupling between alkyl bromide 662 and vinyl stannane 663 was employed to forge the sterically encumbered C(sp2)–C(sp3) bond in these schisandraceae triterpenoid architectures (660–661, 664–665).283 Notably, conventional radical conditions (AIBN and nBu3SnH) exclusively yielded hydrodebromination byproducts, underscoring the inefficacy of classical initiators in such demanding contexts. In contrast, the nickel-mediated system achieved good efficiency and exquisite functional group tolerance toward hydroxyl group and proximal acetal group, delivering the desired coupling products. This success underscores the strategy's unique capacity to access sterically hindered junctions in polyoxygenated terpenoid frameworks where traditional polar reactivity falters.
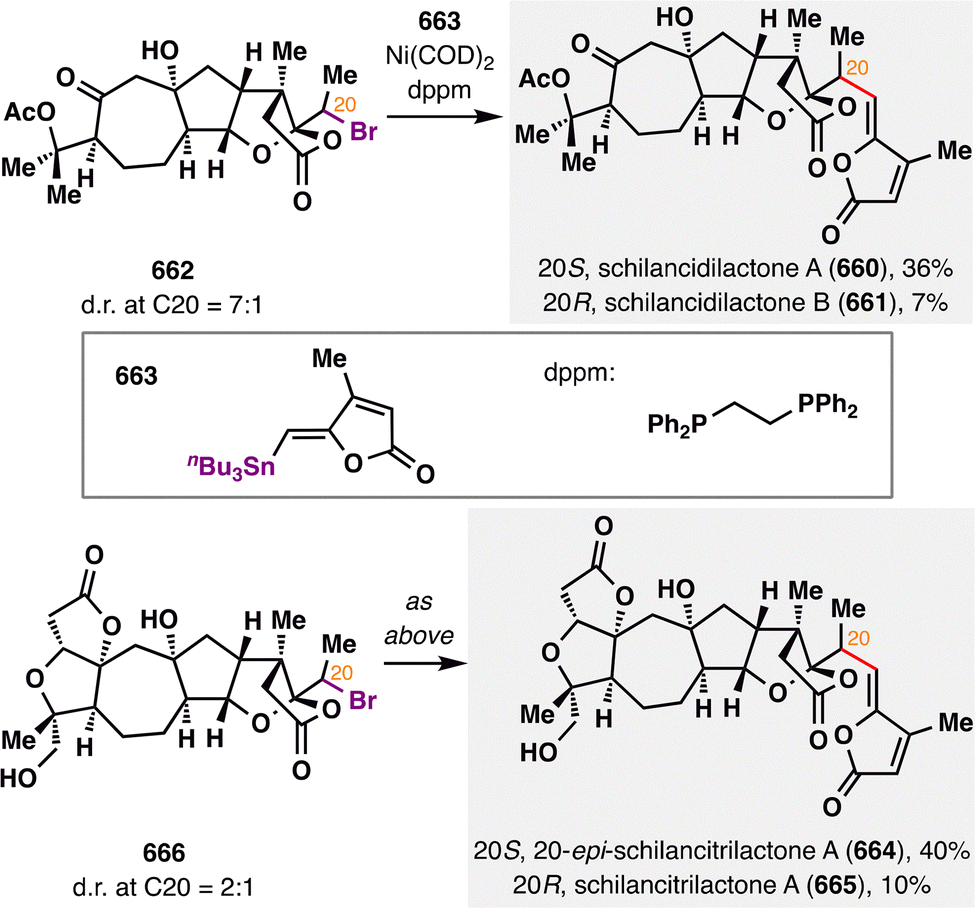 | ||
| Scheme 80 Tang's total synthesis of schilancidilactones A and B (660–661), schilancitrilactone A (664), and 20-epi-schilancitrilactone (665). | ||
Transition-metal-catalyzed dehalogenative cross-coupling has witnessed significant advances in forging C–C bonds. Nonetheless, the development of milder, more economical, and transition-metal-free alternatives to achieve carbon–heteroatom bond constructions remains highly appealing. A striking example of such an approach emerged in our total synthesis of berkeleyacetal D (667)—a highly oxidized member of the DMOA-derived meroterpenoid family (Scheme 81).284 Specifically, treatment of intermediate 670 with tBuOLi in DMSO directly furnished the key 2,3-dihydrofuran moiety of 667 in 40% yield, effectively bypassing multiple anticipated steps. Mechanistic studies propose that the transformation proceeds via an intramolecular SET from an in situ–generated trienolate 671 to the adjacent C–Br bond (highlighted in purple), a hypothesis supported by the observation of enhanced yields under photoirradiation. The resulting diradical intermediate 672 is proposed to undergo rapid intramolecular radical recombination, forging the strained dihydrofuran ring embedded within the berkeleyacetal D (667) framework.
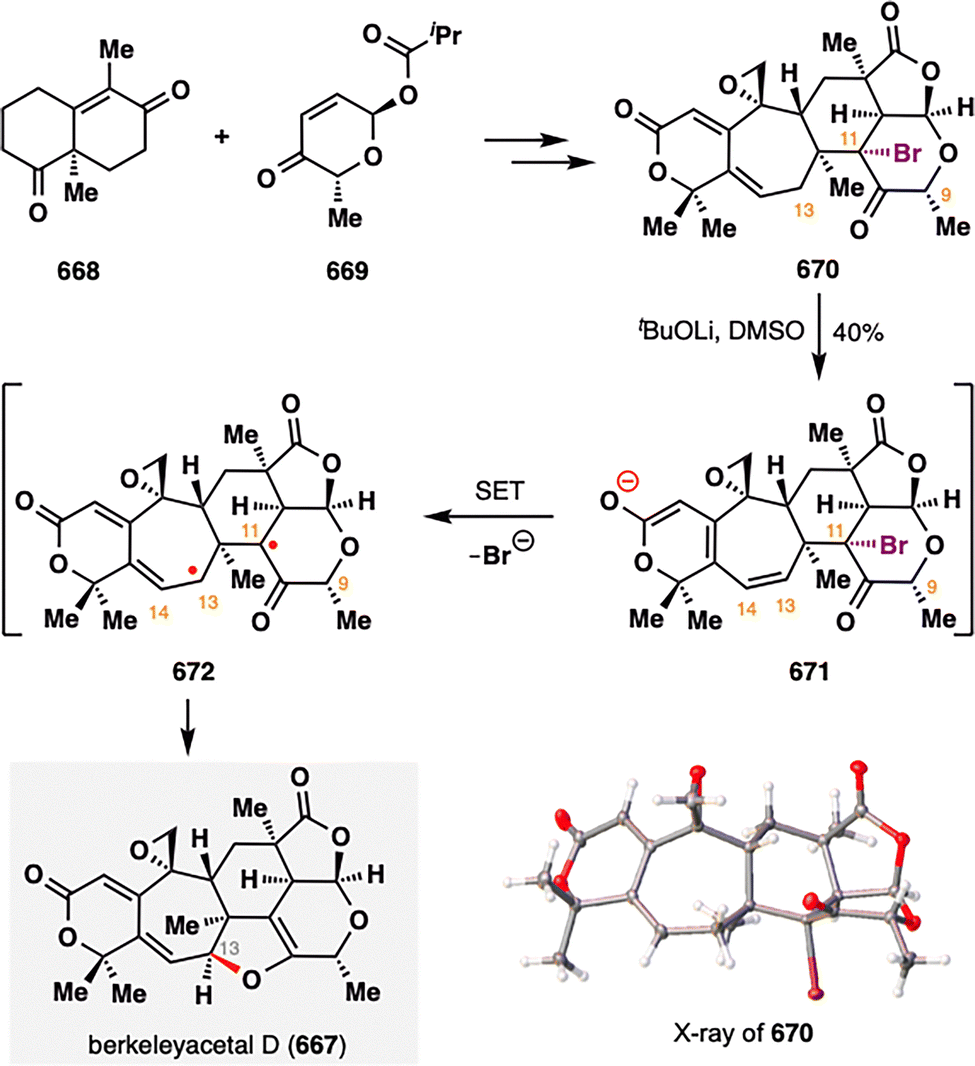 | ||
| Scheme 81 C. Li's total synthesis of berkeleyacetal D (667). | ||
5. Alcohol derivatives as radical precursors
The pervasive presence of hydroxyl functionalities across diverse molecular scaffolds makes alcohols as intrinsically appealing candidates for generating C(sp3) radicals. However, the inherent stability conferred by the high bond dissociation energy (BDE) of the C–O bond, coupled with the poor leaving group propensity of the hydroxide (OH−), typically precludes their direct utilization as alkylating agents in cross-coupling manifolds.285,286 Consequently, strategic derivatization of the alcohol moiety is essential to convert this ubiquitous functional group into a viable radical precursor amenable to SET processes. Common activating groups enabling this transformation include tosylates,287–290 mesylates,291 oxalates,292 and halides (this category having been discussed in the preceding section).Among these activated species, oxalate-based derivatives have emerged as particularly prominent radical precursors in contemporary terpenoid total synthesis (Scheme 82). Their prominence arises from two distinct activation pathways utilizing different types of oxalate derivatives: (i) SET reduction of methyl oxalate esters (e.g., R-OC(O)C(O)OCH3) to generate nucleophilic alkyl radicals;268,292 (ii) SET oxidation of oxalate salts (e.g., cesium/potassium alkyl oxalates, R-OC(O)COO−Cs+/K+) to yield electrophilic alkyl radicals.293 The carbon-centered radicals generated via these pathways are efficiently captured in pivotal C–C bond-forming reactions, most notably Giese-type radical additions to electron-deficient alkenes and Keck-type radical allylations.
 | ||
| Scheme 82 Emerging radical retrosynthesis of C–C bonds employing the dehydroxylative reactions. | ||
Critically, both reaction manifolds exhibit exceptional efficiency for constructing challenging quaternary carbon centers—a common structural motif in complex terpenoids. This synthetic prowess translates into powerful retrosynthetic disconnections: a quaternary carbon center within a target molecule can often be traced retrosynthetically to a tertiary alcohol precursor, transformed into its corresponding oxalate derivative (methyl ester or cesium/potassium salt, Scheme 82), and an appropriate coupling partner (e.g., an electron-deficient alkene for the Giese pathway or an allylic acceptor for the Keck pathway). The strategic power of this approach lies in the accessibility of the tertiary alcohol precursor. Such alcohols are readily synthesized via nucleophilic addition to readily available ketones, significantly simplifying the retrosynthetic tree. By leveraging oxalate activation of tertiary alcohols and radical-based C–C bond formation, synthetic chemists gain a streamlined, convergent route to architecturally demanding terpenoid frameworks containing all-carbon quaternary stereocenters.
Soon after Overman, MacMillan, and co-workers introduced a redox-neutral dehydroxylative coupling strategy using alkyl oxalates as radical precursors in 2015,293 Overman and co-workers extended this methodology to the synthesis of a diverse array of structurally complex diterpenoids (384, 673, 677, 682, and 683 in Scheme 84). By developing stable tert-alkyl oxalate salts as radical progenitors, they addressed a longstanding challenge in radical chemistry—the effective utilization of tertiary alcohols in C–C bond formation—via a net redox-neutral process. Mechanistically, photoexcited Ir(III)* oxidizes the tert-alkyl oxalate salt, generating an alkyl oxalate radical (Scheme 83). This intermediate undergoes sequential extrusion of two molecules of CO2 to furnish a stabilized tertiary carbon-centered radical. The resulting radical then engages in a Giese-type addition to an electron-deficient alkene, forming a new C–C bond. The resulting adduct radical is subsequently reduced by Ir(II), completing the catalytic cycle in a redox-neutral manner.
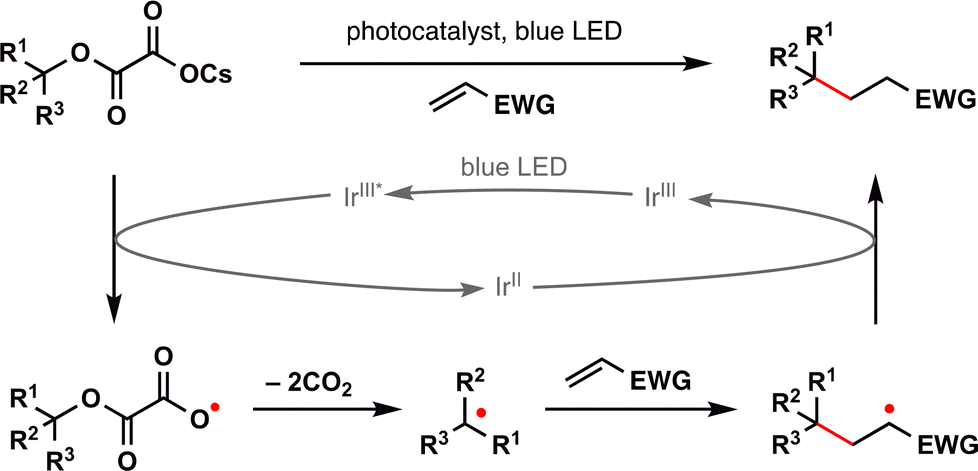 | ||
| Scheme 83 Dehydroxylative Giese reaction through SET-oxidation of oxalate salts. | ||
This methodological innovation enabled an enantioselective synthesis of trans-clerodane (673) via a highly diastereoselective photoredox-catalyzed coupling between a tertiary cesium alkyl oxalate (675) and an electron-deficient butenolide (676), simultaneously constructing the quaternary stereocenter with exceptional selectivity (d.r. > 20:1) (Scheme 84(A)).293 The generality of the approach was further showcased in concise synthesis of (−)-macfarlandin C (677) and cheloviolenes A, B, and C (384, 682, and 683, Scheme 84(B) and (C)),210,294,295 underscoring how the oxalate-based strategy, when paired with visible-light photocatalysis, offers a unified and efficient platform for accessing complex diterpenoid frameworks with high yield and stereocontrol—outcomes that were previously difficult to achieve using conventional methods.
 | ||
| Scheme 84 Overman's total synthesis of trans-clerodane (673), macfarlandin C (677), and cheloviolenes (384, 682, 683). | ||
During the peer-review process of our manuscript, Gui and co-workers reported a striking demonstration of radical retrosynthetic logic in their total synthesis of harziandione (689), a harziane diterpenoid isolated from Trichoderma species featuring a highly caged 6/5/7/4 tetracyclic framework and four contiguous stereocenters.296,297 As illustrated in Scheme 85, the synthesis combined a BROC radical cyclization, a photocatalytic dehydroxylative Giese reaction, and an acyl radical cyclization to rapidly assemble the bridged [3.2.1] ring system and three of the four contiguous stereocenters. The sequence commenced with formation of the C13–C14 bond in 692via an Fe(acac)3/Ph(iPrO)SiH2-mediated BROC reaction, simultaneously establishing a prochiral quaternary center at C13 and a tertiary center at C14 in 83% yield. Installation of an oxalate group in 693 then set the stage for a photocatalytic dehydroxylative Giese reaction, which—through substrate-controlled stereocontrol (694)—furnished the C6 quaternary center in 695 as a single diastereomer in 54% yield. A subsequent desymmetrizing intramolecular aldol reaction under tBuOK established the remaining stereochemistry at C13 while constructing the strained bicyclo[5.2.0]nonene motif in 696. This concise sequence enabled access to harziandione (689) in only 12 steps from commercially available intermediates 690/691, underscoring the potential of radical retrosynthetic strategies for assembling complex bridged architectures.
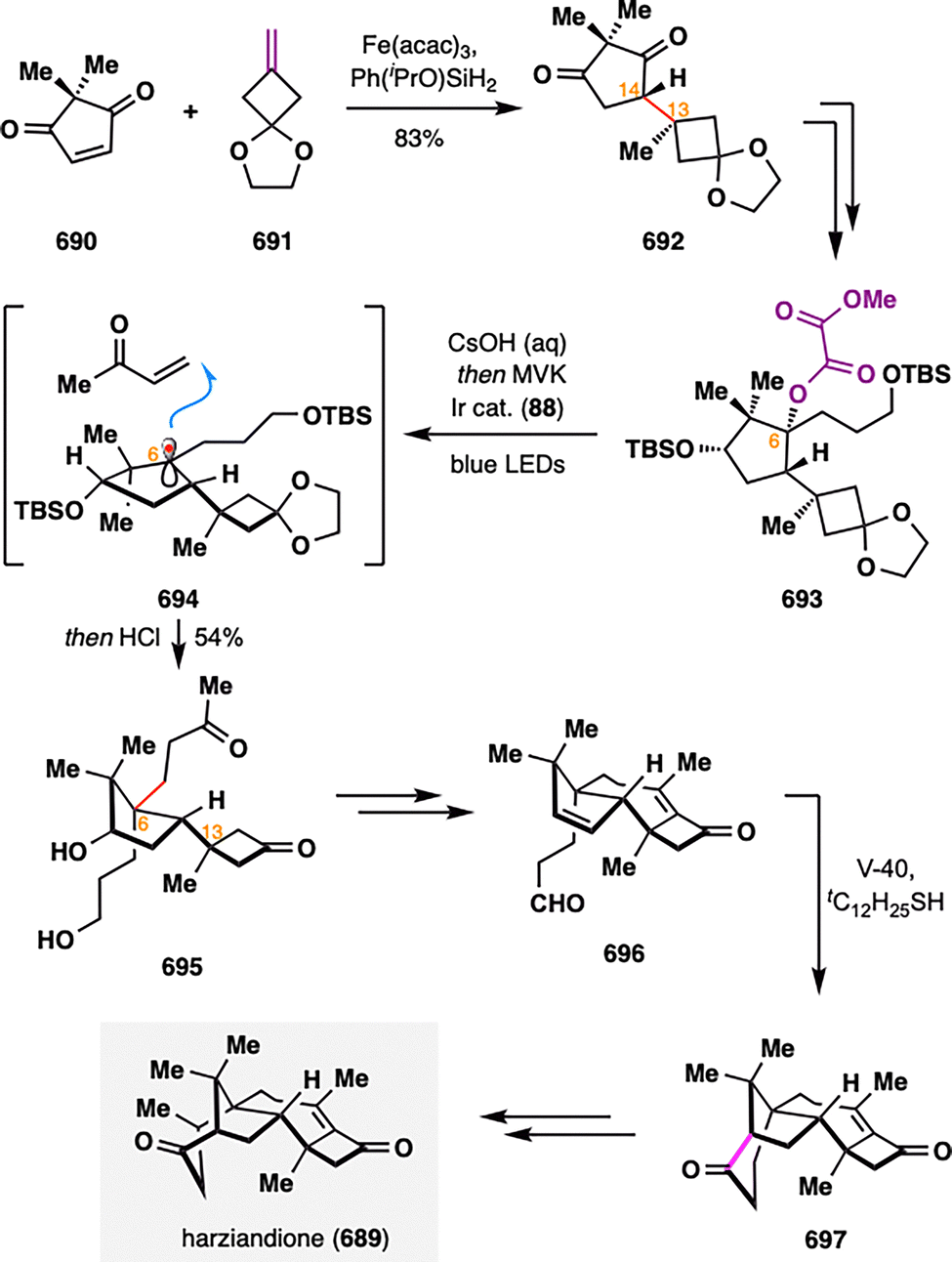 | ||
| Scheme 85 Gui's total synthesis of harziandione (689). | ||
Inspired by Gong's pioneering work on the single electron reduction of methyl oxalates,292 our group employed a tertiary alkyl oxalate as a radical precursor to achieve the total synthesis of a series of ent-kaurane and beyerane-type diterpenoids (698–707),298 featuring a strained and bridged bicyclo[3.2.1]octane core, via a dehydroxylative radical addition/annulation protocol (Scheme 86). This radical-based [3+2] annulation strategy enabled the efficient conversion of precursor 708 to the key intermediate 710 in synthetic useful yield. The reaction was facilitated by a MgCl2/PBI/Zn reductive system, with TEMPO serving as a crucial additive; the in situ generation of TEMPO–H or Zn(TEMPO)2 can selectively quench the secondary radical intermediate through a HAT or SH2 process, thereby preventing uncontrolled radical addition of 713 to the acrylate. Because of the steric hindrance, the reaction of tertiary radical 712 with TEMPO–H or Zn(TEMPO)2 is not favorable. With rapid access to the core skeleton secured, further elaborations enabled the divergent synthesis of both ent-kaurane and beyerane-type diterpenoids (698–707).
 | ||
| Scheme 86 C. Li's collective synthesis of tetracyclic ent-kauranoids (698–707) via [3+2] radical annulation. | ||
While previous studies have demonstrated the utility of tandem dehydroxylative Giese-type radical addition reactions, Baudoin and co-workers pursued an alternative strategy by employing a dehydroxylative Keck-type allylation to accomplish the total synthesis of (+)-randainin D (716) and (+)-barekoxide (719)—two architecturally complex diterpenoids bearing a trans-hydroazulenone core and a butenolide moiety (Scheme 87).299 In their initial attempts, dehydroxylative radical addition of methyl oxalate 717 to electron-deficient olefins proved ineffective, leading primarily to undesired C8-hydrogenated by-products due to inefficient radical trapping.
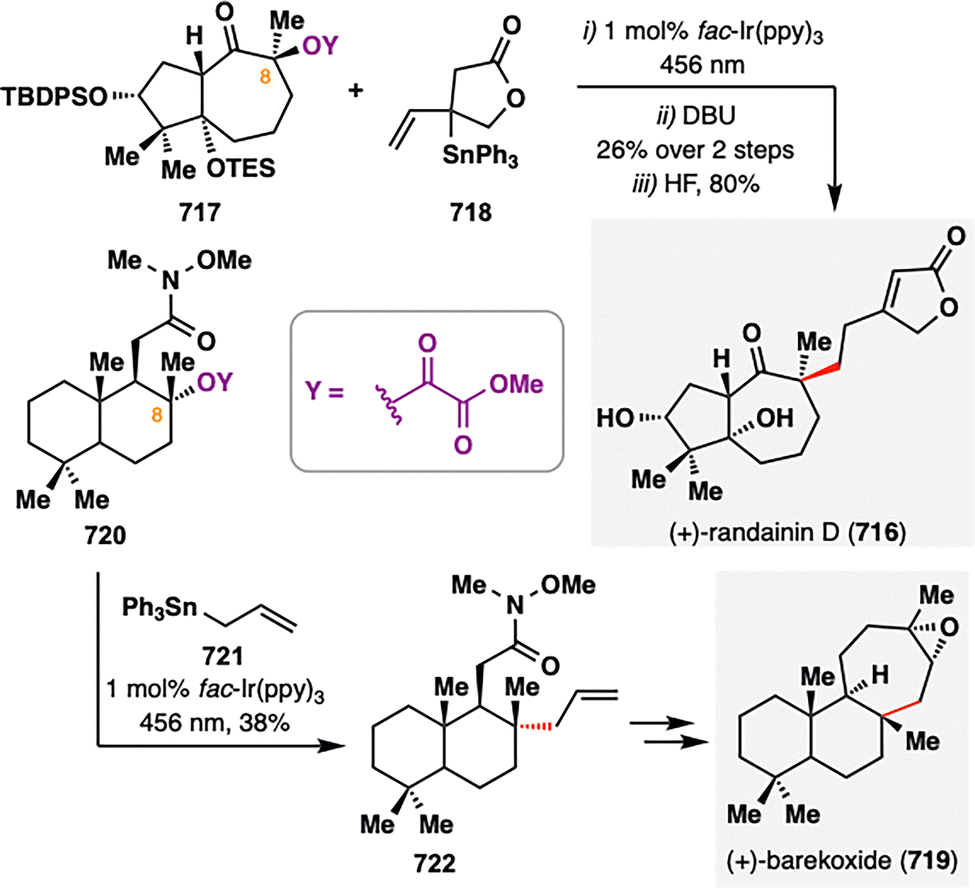 | ||
| Scheme 87 Baudoin's total synthesis of (+)-randainin D (716) and (+)-barekoxide (719). | ||
To circumvent this limitation, the team adopted a Keck-type radical allylation using allylstannane 718 as the radical acceptor.300,301 Upon single-electron reduction of 717 by photoexcited Ir(III)*, an α-ketone radical was generated. This radical was successfully intercepted by 718, forging the sterically hindered C8 all-carbon quaternary center with high efficiency. Notably, the Ir(IV) species generated in the reduction step was reduced back to Ir(III) by the leaving group (a methyl oxalate salt), thereby completing the redox-neutral photocatalytic cycle. Following the radical allylation, DBU-mediated alkene isomerization and global deprotection with HF furnished (+)-randainin D (716). Encouraged by this success, the same photoredox-mediated allylation strategy was applied to install the side chain in the synthesis of (+)-barekoxide (719), further demonstrating the efficacy of organostannane-mediated radical allylation in constructing sterically congested quaternary stereocenters.
6. Aldehyde derivatives as radical precursors
The aldehyde functional group occupies a privileged position as a multifaceted radical precursor, enabling innovative and streamlined disconnections in complex molecule assembly. While the classical generation of nucleophilic ketyl radicals via single-electron reduction (most notably employing SmI2) has found extensive historical application in total synthesis.302–306 A major modern paradigm leverages the relatively weak aldehyde C(O)–H bond (BDE ≈ 88 kcal mol−1) for direct generation of synthetically valuable ketyl radicals through efficient HAT using photoredox catalysis (e.g., tetrabutylammonium decatungstate).307,308 The resulting ketyl radicals can be engaged in radical additions or be harnessed in metal-catalyzed cross-coupling reactions, enabling the formation of C(sp2)–C(sp3) or C(sp2)–C(sp2) bonds.Complementing this approach, the strategic SOMO-activation of aldehydes represents another powerful modern tactic.309–312 This method involves the condensation of aldehydes with secondary amines to form electron-rich enamines, which subsequently undergo single-electron oxidation—facilitated by chemical oxidants like CAN or photoredox catalysis—to generate electrophilic α-amino radical cations (SOMOphiles). These highly reactive species excel in intramolecular radical cyclizations with tethered alkenes. This reactivity provides efficient and often stereoselective routes to access challenging ring systems ubiquitous in polycyclic terpenoid frameworks. The development of enantioselective variants employing chiral catalysts further enhances the synthetic utility of this method for establishing critical stereocenters.
From a retrosynthetic perspective, the modern radical methodologies involving aldehydes enable powerful disconnections in terpenoid planning (Scheme 88). A ketone functionality within the target molecule can be strategically traced back to its precursors through an ketyl radical intermediate. This disconnection implies the convergent coupling of an aldehyde (serving as the ketyl radical precursor) with either an alkene (for radical addition) or an organohalide (for transition metal-catalyzed cross-coupling), effectively forging the new C–C bond via the pathways previously described. Furthermore, when the carbon atom α to an aldehyde group resides within a ring system—a common feature in polycyclic terpenoids—the SOMO-activation strategy offers a unique disconnection opportunity. Here, the critical insight is to cleave the C–C bond between the α and β carbons (Cα–Cβ) relative to the aldehyde. This retrosynthetic step effectively deconstructs the cyclic framework, revealing a synthetically accessible, linear or acyclic aldehyde precursor bearing a tethered alkene. The superiority of this approach depends on its power to utilize the aldehyde both as the radical precursor (via enamine formation and oxidation) and as the anchor point for the intramolecular radical cyclization that rebuilds the ring during the forward synthesis. This represents a particularly elegant application of radical chemistry for simplifying the synthesis of complex carbocyclic structures prevalent in terpenoid natural products.
 | ||
| Scheme 88 Modern radical retrosynthesis of C–C bonds from aldehydes. | ||
Cross-dehydrogenative coupling (CDC) reactions have proven to be powerful tools for constructing C–C bonds directly from C–H bonds.313 However, intermolecular CDC reactions between aldehydes and electron-deficient alkenes remain relatively underexplored—particularly in the context of complex terpene synthesis. Addressing this gap, Park, Han, and co-workers employed their self-developed Cu-catalyzed CDC platform to forge key fragment couplings in the total synthesis of elodeoidins A (723) and B (724) (Scheme 89).314 The first CDC transformation utilized tetrabutylammonium decatungstate (TBADT) as a photocatalyst under 370 nm irradiation to generate conjugated enol intermediate 727via direct coupling of enedione 725 and isobutyraldehyde 726,315,316 followed by a selenylation/oxidation sequence to yield enetrione 728 in 68% over two steps. Mechanistically, photoexcited TBADT initiated HAT from aldehyde 726, generating a ketyl radical that underwent Giese-type radical addition to enedione 725. Subsequent back-HAT from TBADTH to the resulting radical intermediate enabled catalytic turnover under mild, redox-neutral conditions—well suited for complex molecular settings. Besides, the second CDC event featured a Cu(dap)2PF6-mediated acylation that formed a key C–C bond in 49% yield. This transformation proceed via photoexcitation of [Cu(I)(dap)2]*, which reduces TBPB to form a tert-butoxyl radical. The latter abstracts a hydrogen atom from acetaldehyde 729, generating a ketyl radical that undergoes a Giese-type addition to enetrione 728. A subsequent concerted deprotonation furnishes intermediate 730 and regenerates the Cu(i) catalyst. Final treatment with AcCl/MeOH furnished elodeoidins A (723) and B (724).
 | ||
| Scheme 89 Total synthesis of (−)-elodeoidins A and B (723–724) by Park, Han and co-workers. | ||
A similar strategy involving ketyl radical generation from aldehydes was strategically employed by Chen, Wang, and co-workers in their inaugural total synthesis of the complex sesterterpenoid (−)-calidoustene (731) (Scheme 90).317 The key challenge—construction of the sterically congested C-ring—was addressed through a dual-strategy approach. Initially, a traditional NHK coupling of 733 with 735 followed by IBX oxidation of the resulting alcohol provided the dienone intermediate 736 in 79% yield over two steps. However, this route suffered from competitive dimerization side reactions. To overcome this limitation, the team first utilized Reisman's bromination protocol mediated by LiBr/Ni catalysis to afford the corresponding alkenyl bromide in 90% yield.318 Next, using a metallaphotoredox method developed by MacMillan was implemented, giving the same product 736 with improved selectivity (52% isolated yield, 68% brsm).319 Mechanistically, photoexcited Ir(III)* oxidize quinuclidine to generate a N-radical cation, which selectively abstract the formyl hydrogen from aldehyde 735via HAT, generating a ketyl radical. This radical is intercepted by a Ni(II) complex formed via oxidative addition of Ni(0) to alkenyl bromide 734, yielding an alkenyl–Ni(III) intermediate that undergo reductive elimination to furnish the desired coupling product 736 in 58% yield. Subsequent transformations, including an anti-Baldwin BROC cyclization, completed the ring closure and finalized the total synthesis of (−)-calidoustene (731).
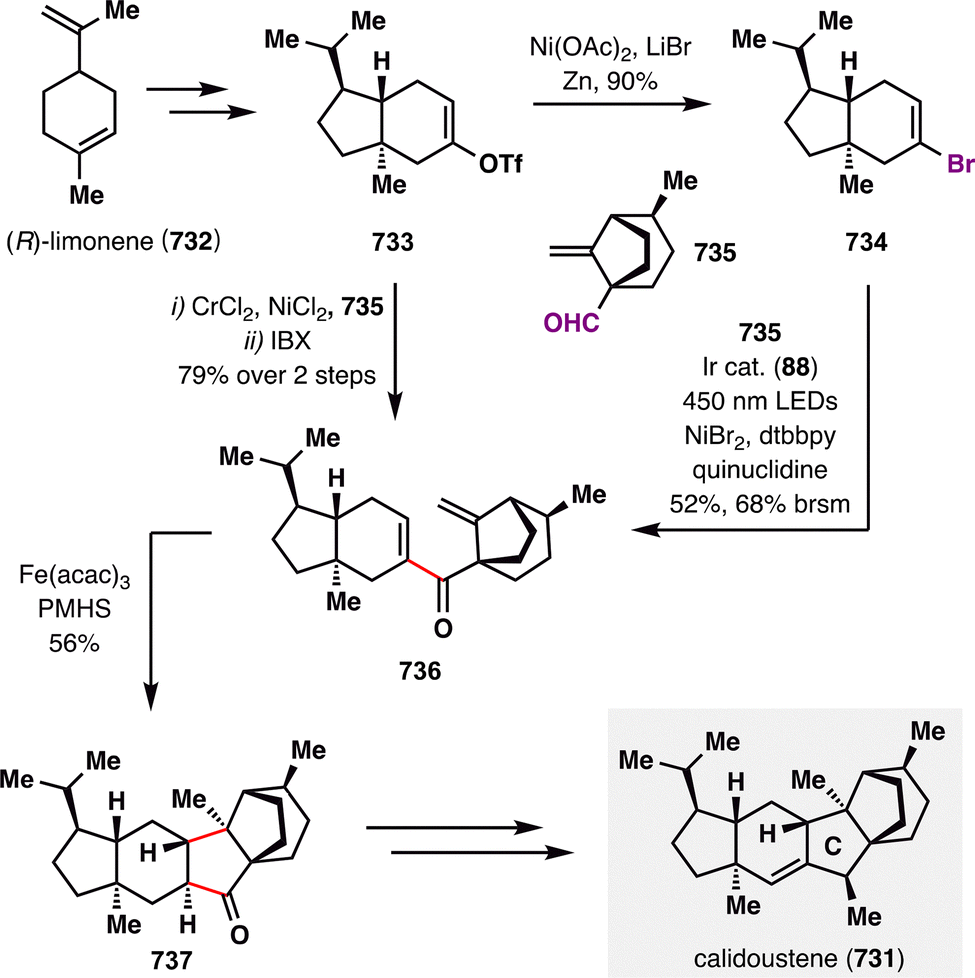 | ||
| Scheme 90 Total synthesis of (−)-calidoustene (731) by Chen, Wang and co-workers. | ||
Another milestone in aldehyde activation was MacMillan's development of SOMO catalysis using imidazolidinone organocatalysts. Building on this concept, Ma and co-workers accomplished the first total synthesis of leucosceptroids A (738) and B (739) through a strategically designed route featuring an enantioselective ring closure enabled by SOMO catalysis (Scheme 91).320 Specifically, an (S)-citronellol-derived aldehyde 741 underwent an efficient intramolecular SOMO-catalyzed allylation, delivering the cyclized product 742 in 89% yield on gram scale with excellent diastereoselectivity (d.r. = 33:2:1). This enantioselective aldehyde–ene cyclization proceeded via formation of a chiral enamine intermediate, followed by single-electron oxidation to generate an electrophilic enamine radical cation. Re-face selective radical addition to the tethered olefin furnished an alkyl radical, which was further oxidized to a carbocation via a RPCO process. Subsequent desilylation and hydrolysis of the imidazolidinone yielded the cyclization product 742. Further elaboration involved a highly diastereoselective aldol reaction between chiral aldehyde 743 and functionalized dihydrofuranone 744 (78% yield), followed by a pivotal SmI2-mediated intramolecular ketyl–olefin radical cyclization efficiently constructed the complex polycyclic core 746 in 89% yield with complete stereocontrol, thereby setting the stage for late-stage modifications en route to leucosceptroids A (738) and B (739). In a parallel advance, S. Li and co-workers also employed an enantioselective SOMO-catalyzed cyclization of commercially available (S)-citronellal (748) with cat. 749 to afford 750, thereby setting the stage for the total synthesis of the sesterterpenoid gracilisoid A (747) (Scheme 92),321 further highlighting the utility of SOMO catalysis in assembling architecturally complex terpenoids.
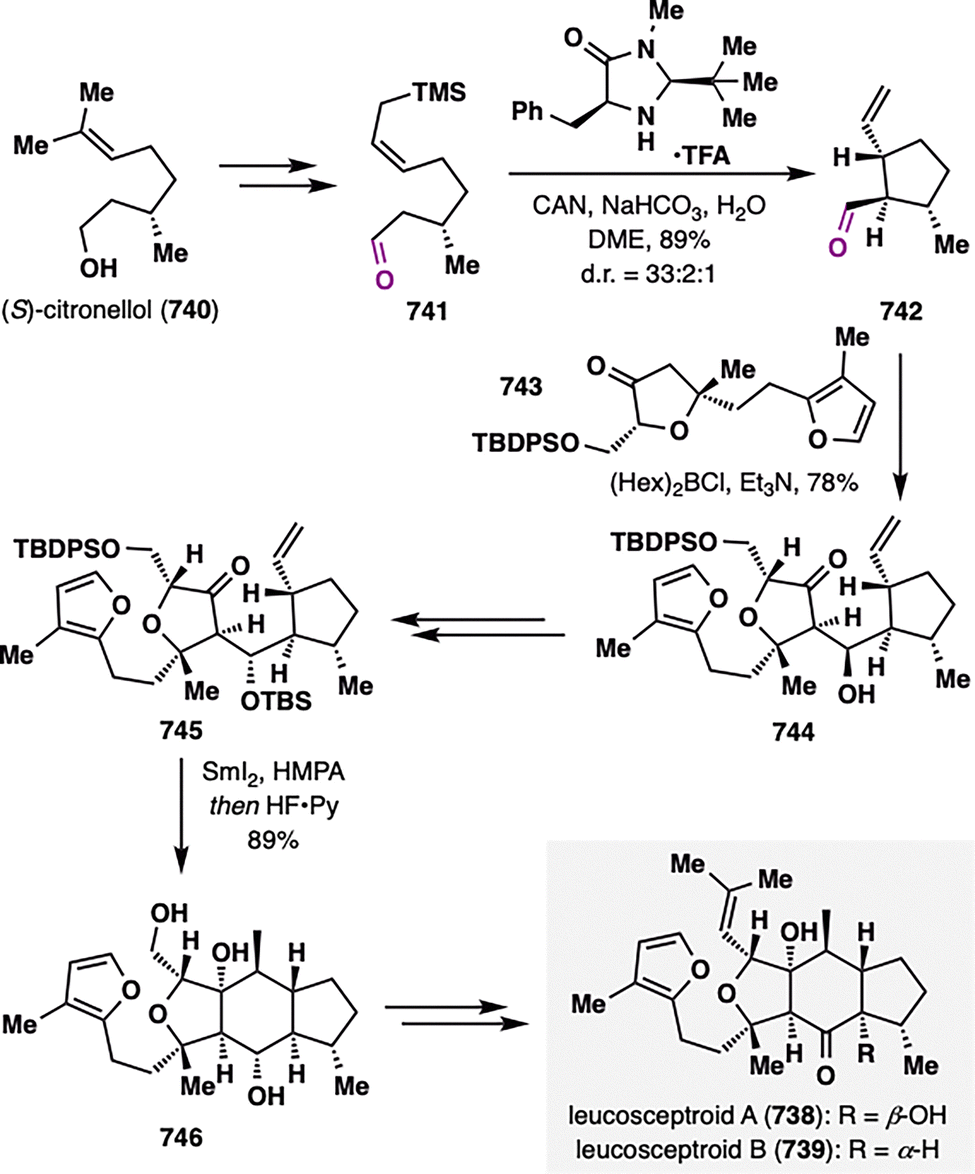 | ||
| Scheme 91 D. Ma's total synthesis of leucosceptroids A and B (738–739). | ||
 | ||
| Scheme 92 Total synthesis of gracilisoid A by S. Li and co-workers (747). | ||
Lei and co-workers achieved the total synthesis of (+)-ent-kauradienone (751) and (−)-jungermannenone C (752) by strategically employing a tandem SOMO-catalyzed aldehyde polycyclization (Scheme 93).322 Treatment of precursor 753 with imidazolidinone organocatalyst ent-749 and Cu(II) enabled efficient enantioselective cyclization, furnishing tricyclic aldehyde 754 in 72% yield with 85% ee (further upgraded to 96% ee via recrystallization). The ent-kaurane core was then constructed via a highly selective tin-mediated radical 1,6-dienyne cyclization of 755, which selectively formed the bicyclo[3.2.1]octene scaffold 756. 756 could be further elaborated to (+)-ent-kauradienone (751). Luche reduction of (+)-ent-kauradienone (751) afforded intermediate 757, which underwent UV-induced (254 nm) radical-mediated rearrangement through 758a and 758b to deliver (−)-jungermannenone C (752) in 58% yield. Notably, this transformation was shown to be thermodynamically reversible: irradiation at 365 nm enabled the back-conversion of 752 to 757 in 21% yield, underscoring the equilibrium nature of this structural interconversion.
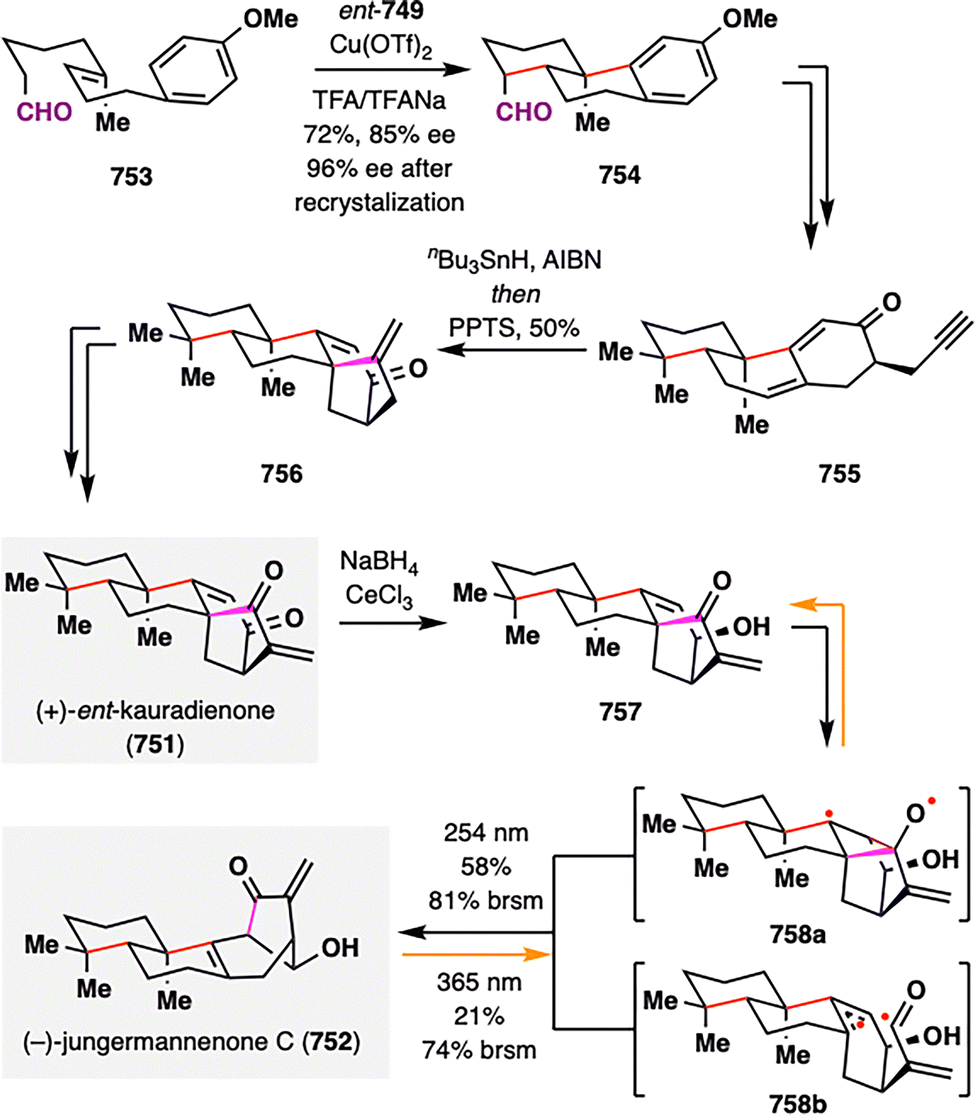 | ||
| Scheme 93 Total synthesis of ent-kauradienone (751) and jungermannenone C (752) by Lei, Li, and co-workers. | ||
Inspired by MacMillan's pioneering work on aldehyde organo-SOMO catalysis, Tu, Zhang, and co-workers developed a tandem enantioselective aldehyde α-alkylation/semipinacol rearrangement strategy that enabled the efficient construction of synthetically challenging α-quaternary 1,5-dicarbonyl scaffolds.323 This approach was elegantly demonstrated in their concise total synthesis of (+)-cerapicol (759) (Scheme 94). The synthesis commenced with α-allylation of isobutyraldehyde using (E)-1,3-dibromobut-2-ene (760), followed by a sequence of Wittig homologation, nucleophilic addition, acetal deprotection, and regioselective hydrogenation to furnish aldehyde 761. The key transformation employed imidazolidinone organocatalyst ent-749 to promote a gram-scale, enantioselective tandem α-alkylation/oxidative RPCO/semipinacol rearrangement of allylic alcohol 761, delivering the pivotal intermediate 764 in 67% yield with excellent enantioselectivity (96% ee). This cascade strategy not only provided an efficient solution to constructing all-carbon quaternary stereocenters but also enabled the streamlined synthesis of (+)-cerapicol (759), highlighting the versatility and synthetic utility of SOMO-catalyzed transformations in the context of complex molecule assembly.
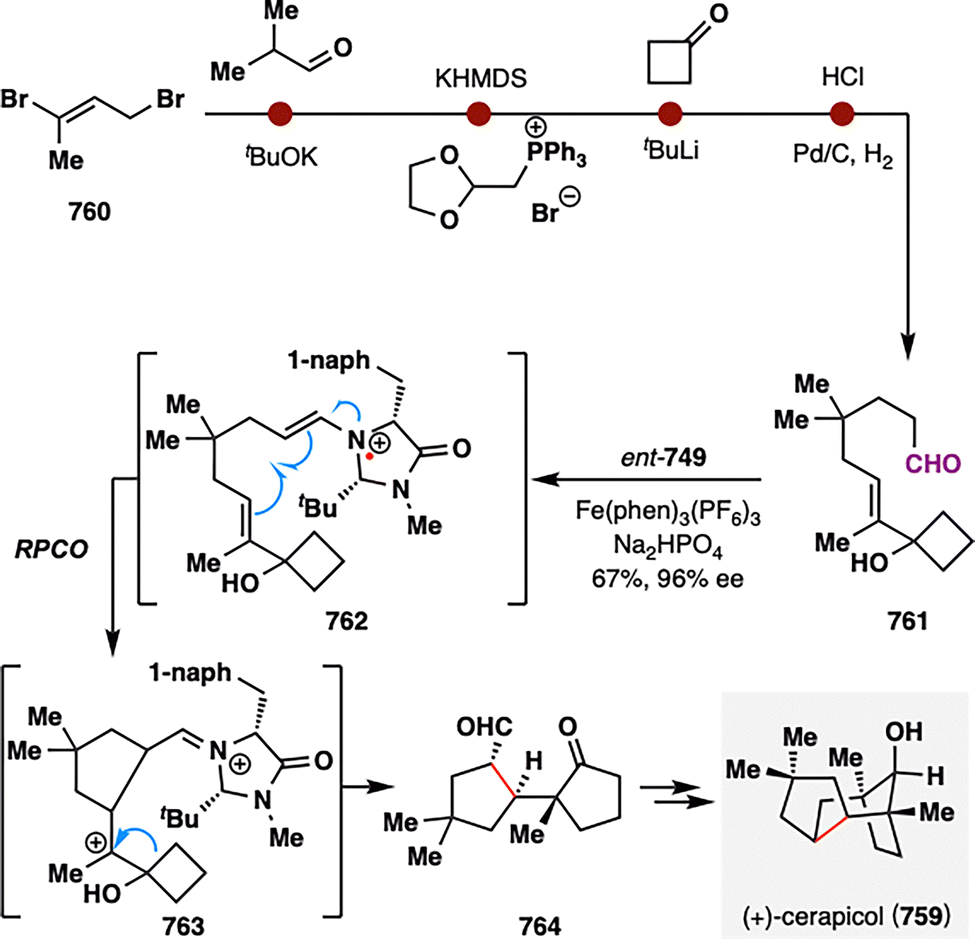 | ||
| Scheme 94 Total synthesis of (+)-cerapicol (759) by Zhang and Tu. | ||
While previous approaches to SOMO activation typically relied on stoichiometric metal oxidants, Xuan, Ding, and co-workers applied a recent method developed by Macmillan that combined photoredox catalysis and organocatalysis to achieve the enantioselective α-alkylation of aldehydes without the requirement of stoichiometric oxidants.309 This strategy was applied in their asymmetric total synthesis of three pleurotin natural products (765–767),324 originally isolated from the fungus Pleurotus griseus,325 and featuring several innovative transformations that address key stereochemical challenges (Scheme 95). The synthesis began with a regioselective ozonolysis of a trisubstituted alkene 768, followed by intramolecular aldol condensation, and reduction to furnish allylic alcohol 769. A particularly noteworthy Johnson–Claisen rearrangement of 769 proceeded with excellent diastereoselectivity (d.r. > 12:1) and high yield (80%, gram scale), delivering diene 770. Subsequent hydroboration–oxidation furnished diol 771, which was further elaborated to aldehyde 772. Application of MacMillan's dual photoredox–organocatalytic system to 772 enabled stereocontrolled α-alkylation, affording 773 as a single diastereomer in 52% yield, which set the stage for downstream elaboration to complete the total synthesis of pleurotin congeners 765–767.
 | ||
| Scheme 95 Total synthesis of pleurotins (765–767) by Ding and Xuan. | ||
Mechanistically, the α-alkylation is initiated by condensation of the pyrrolidine catalyst with the aldehyde substrate to generate an enamine (Scheme 95). Upon photoexcitation, the IrIII* catalyst undergoes single-electron transfer (SET) from the enamine, affording an enaminyl radical and reduced IrII. The resulting radical undergoes a 6-exo-trig cyclization, forging a new C–C bond and simultaneously establishing a stereocenter while generating a benzylic radical. HAT from ArSH then furnishes an iminium ion, which hydrolyzes to release the pyrrolidine catalyst and the α-alkylated aldehyde. Finally, the thiyl radical formed in the HAT step is reduced by IrII and protonated to regenerate both the ground-state IrIII photocatalyst and the thiol catalyst.
Collectively, these modern strategies utilizing aldehyde—direct C–H functionalization to ketyl radicals and SOMO-enabled α-radical generation—significantly expand the synthetic chemist's arsenal for terpenoid construction. They circumvent the need for stoichiometric organometallic reagents, unlock novel retrosynthetic disconnections, and provide robust solutions for assembling the intricate and stereochemically demanding architectures characteristic of bioactive terpenoid natural products.
7. Ketone derivatives as radical precursors
Alcohol functionalities, particularly tertiary alcohols, represent foundational handles for radical-based disconnections of to ketones in complex molecule assembly (Scheme 96). Like aldehydes, ketones primarily generate nucleophilic ketyl radicals via single-electron reduction, classically employing samarium diiodide (SmI2). The diverse reactivity of ketyl radicals has been extensively cataloged in contemporary reviews.302–306 Notably, Procter's comprehensive recent tutorial reviews provide authoritative survey of modern generation methods (e.g., photoredox catalysis) and their evolving synthetic applications.326–331 This section highlights a complementary, strategically powerful approach: the ketyl radical-mediated skeletal reorganization pioneered by Takatori et al.332 This method efficiently converts fused bicyclo[4.2.0] frameworks into synthetically challenging bicyclo[3.2.1] architectures through a SmI2-induced 1,2-rearrangement with ring expansion.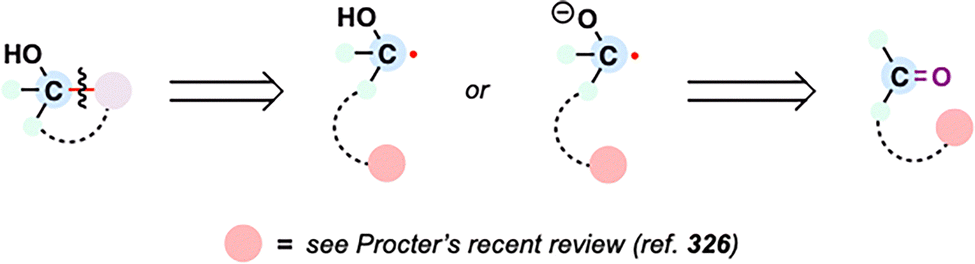 | ||
| Scheme 96 Radical retrosynthesis of C–C bonds from ketone based on recent studies. | ||
Its significant recent adoption in terpenoid synthesis—demonstrating robust efficiency in forging key polycyclic intermediates—motivates focused discussion here, with mechanistic principles exemplified by Dai's synthesis of (−)-GA18 methyl ester (774),333 a member of the gibberellin (GA) family of plant hormones that regulate growth and development (Scheme 97).334 Given the importance of GAs in agricultural and biochemical research, synthetic access to these compounds is of considerable value. In this work, Dai and co-workers reported a concise nine-step total synthesis of (−)-GA18 methyl ester (774), starting from the inexpensive and readily available diterpenoid andrographolide (775). Their strategy merged photochemical [2+2] cyclization with a SmI2-mediated radical addition/fragmentation cascade to construct the methylenebicyclo[3.2.1]octanol motif—a central feature of the gibberellin scaffold—in 70% yield from intermediate 777. Mechanistically, SET from SmI2 to the carbonyl group of 777 generated an α-alkoxy radical, which underwent a 3-exo-trig cyclization onto the adjacent alkene to form intermediate 778. A subsequent radical ring expansion of 778 relieved ring strain, furnishing the methylenebicyclo[3.2.1]octanol framework. This transformation effectively set the stage for final manipulations, ultimately delivering (−)-GA18 methyl ester (774).
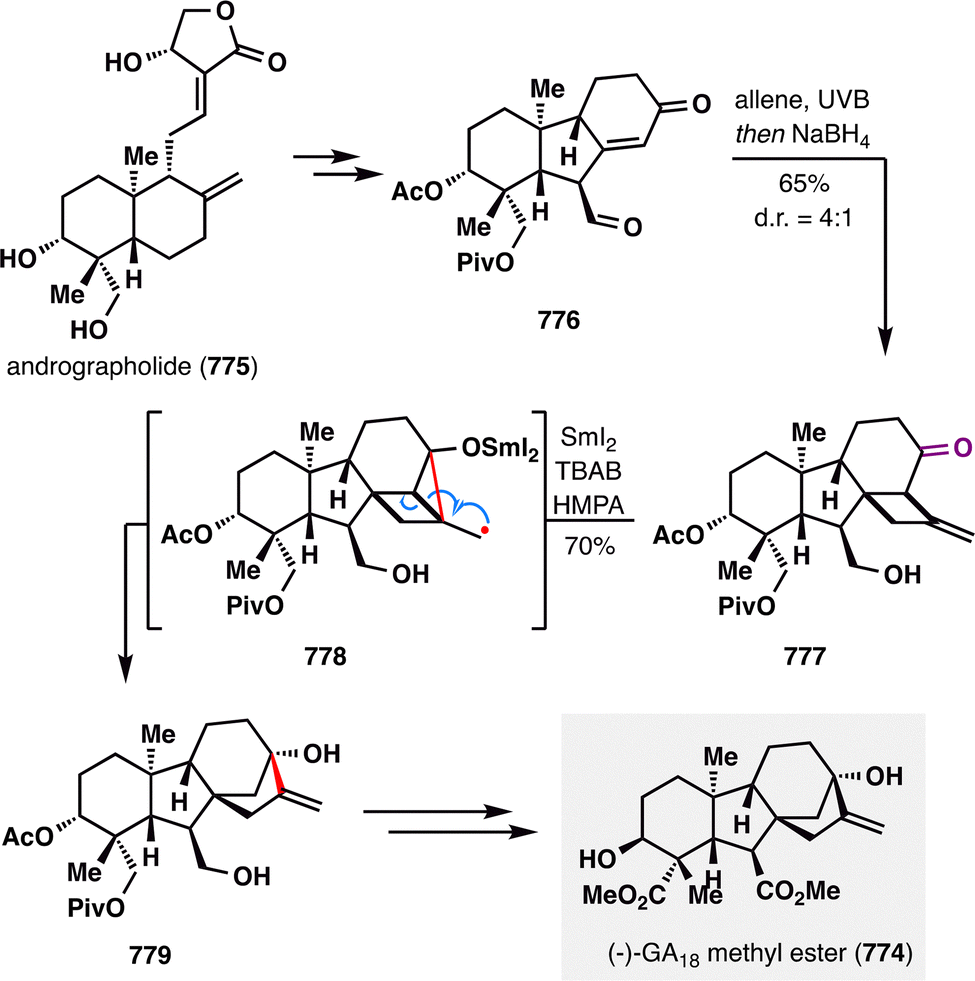 | ||
| Scheme 97 Dai's total synthesis of (−)-GA18 methyl ester (774). | ||
Notably, Wang and co-workers employed a related radical-based skeletal rearrangement strategy to construct the methylenebicyclo[3.2.1]octanol core in their total synthesis of (−)-erythroxylisin A (780) (Scheme 98).335 The enone precursor 784 was synthesized from 783, which in turn was assembled via a Au-catalyzed asymmetric polyene cyclization of alkyne 781—a key transformation establishing the tricyclic framework. Subsequent exposure of enone 784 to allene under photoirradiation conditions furnished strained cyclobutene 785, which then underwent a SmI2-mediated radical addition/fragmentation cascade. Final oxidation with DMP delivered the target natural product (−)-erythroxylisin A (780).
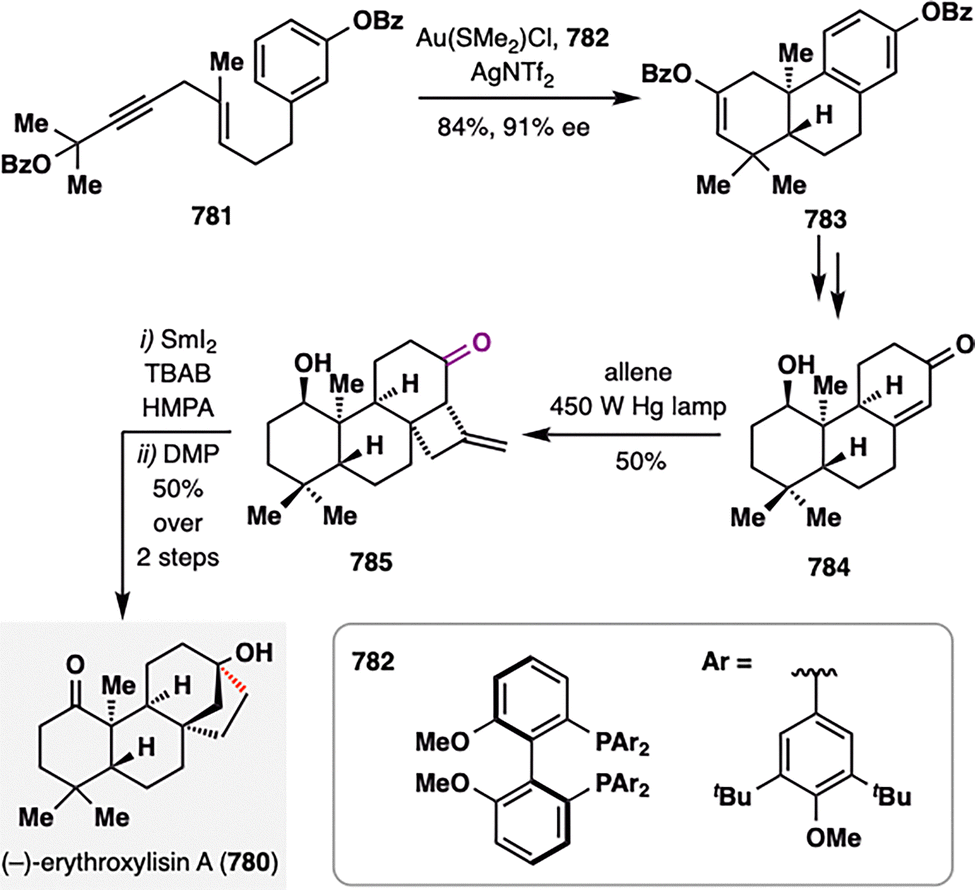 | ||
| Scheme 98 Total synthesis of (−)-erythroxylisin A (780) by Wang and co-workers. | ||
Similarly, Kalesse and co-workers applied a comparable sequence, combining photoinduced [2+2] cycloaddition with SmI2-mediated radical addition/fragmentation cascade to access methylenebicyclo[3.2.1]octanol intermediate 789, as shown in Scheme 99.336 This species subsequently underwent diazo formation followed by a photoinduced Wolff rearrangement to achieve a ring contraction, thereby forging the bicyclo[3.1.1]heptane core in 791 and setting the stage for final elaborations en route to acanthodoral (786).
 | ||
| Scheme 99 Kalesse's total synthesis of acanthodoral (786). | ||
Notably, in all three total synthesis described above, the SmI2-triggered radical addition/fragmentation was strategically employed at a late stage, underscoring its exceptional chemoselectivity, mildness, and compatibility with complex, functionally dense molecular architectures.
8. Alkanes as radical precursors
The direct functionalization of aliphatic C–H bonds has become a powerful strategy for late-stage diversification and improved step- and atom-economy in complex molecule synthesis.12,337–339 Among methods for C–H activation, HAT has gained prominence by generating carbon-centered radicals under mild, tunable conditions. Unlike classical two-electron pathways, HAT can achieve C–H functionalization without requiring prefunctionalization, enabling unique modifications of complex molecular architectures.Recent advances in photocatalysis, electrochemistry, and biocatalysis have dramatically expanded the HAT toolbox. Photocatalytic HAT enables selective activation of aliphatic C–H bonds under visible light,11,340 and electrochemical strategies offer scalable, redox-economical radical generation.238,341–344 Engineered enzymes can perform highly site- and stereoselective HAT-mediated oxidations.345–348 Collectively, these developments have elevated HAT from a mechanistic curiosity to a powerful platform for constructing complex targets, including terpenoid frameworks.
In retrosynthetic analysis, leveraging HAT of alkanes to formally disconnect a C–X bond into a C(sp3)–H bond presents both conceptual opportunities and practical challenges. The ubiquity of aliphatic C–H bonds demands precise control of selectivity—dictated by factors such as bond dissociation energies (BDEs), steric and electronic environments, and the presence of directing groups that facilitate intramolecular HAT. Consequently, no structural pattern can be generalized to use HAT disconnections; instead, effective retrosynthetic design requires detailed molecular analysis and careful reaction condition optimization. In terpenoid synthesis (Scheme 100), key HAT-initiated transformations include C–H oxidation (e.g., hydroxylation, carboxylation),238,349 C(sp3)–C(sp3) desaturation,350 stereocenter epimerization,351 alkene isomerization,352 radical addition,353 and radical-mediated skeletal rearrangements.354
 | ||
| Scheme 100 Modern radical retrosynthesis using alkane as radical precursors. | ||
An alkane C–H functionalization strategy enabled by amidyl radicals was strategically employed by Sarpong and co-workers in their unified approach to functionalized camphor derivatives (85, 792, and 793)—a class of versatile chiral building blocks long valued in organic synthesis (Scheme 101).353 The C8-functionalization transformation hinged on a 1,5-HAT process initiated from an amidyl radical, which was generated from camphor-derived aminonitrile 795via photoexcited Ir(III)*-catalyzed oxidation. The resulting C8-centered primary radical then underwent a Giese-type conjugate addition to MVK, affording adduct 798, which enabled streamlined access to several structural intricate sesquiterpenoids, including copacamphor (792), longicamphor (793), and longiborneol (85).
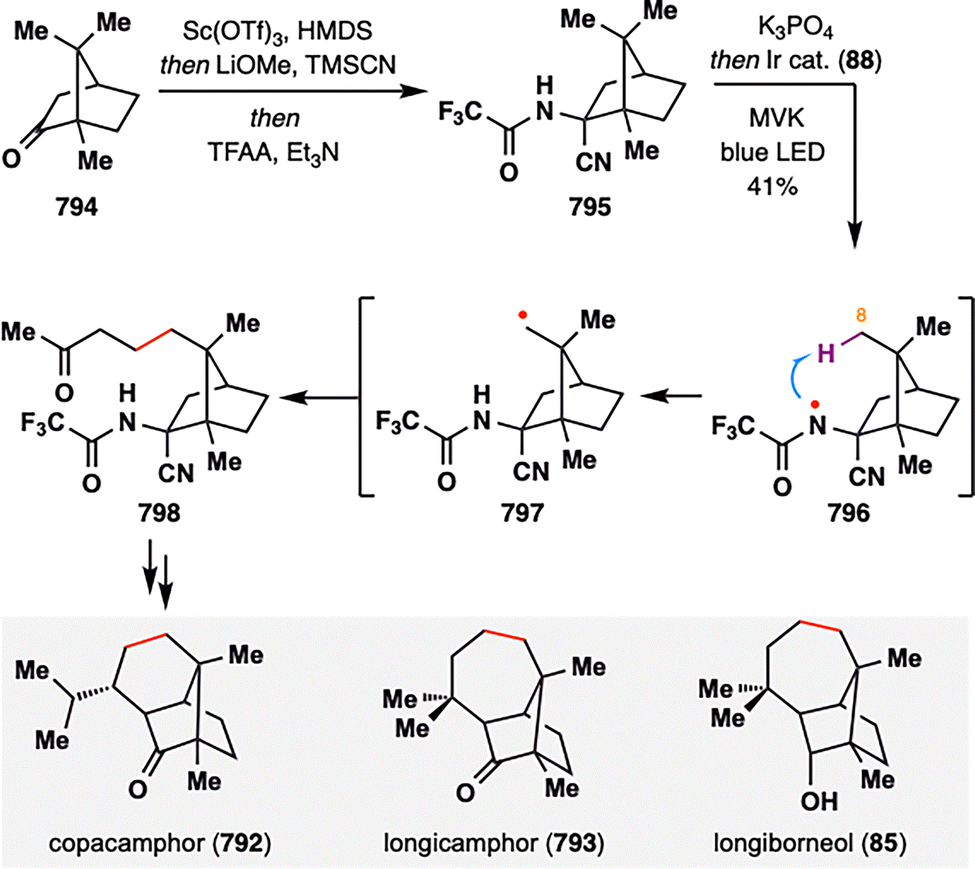 | ||
| Scheme 101 Sarpong's total synthesis of copacamphor (792), longicamphor (793), and longiborneol (85). | ||
While previous efforts in C–H activation have predominantly relied on heteroatom-initiated intramolecular 1,5-HAT processes, Luo and co-workers achieved a photo-induced direct C–H activation to trigger a Dowd–Beckwith rearrangement, serving as a pivotal step in the total synthesis of (+)-kobusine (799) and (+)-spirasine IX (800)—two polycyclic C20-diterpenoid alkaloids renowned for their intricate cage-like architectures (Scheme 102).354 Specifically, upon irradiation, the photoexcited TBADT undervent HAT reversibly with C12–H bond of precursor 802, generating a carbon-centered radical intermediate 803. This radical underwent a 3-exo-trig cyclization to furnish intermediate 804, which then engaged in a Dowd–Beckwith rearrangement to forge the characteristic hetisine skeleton 805 efficiently. This strategically executed skeletal rearrangement laid the foundation for a concise total synthesis of both natural products 799 and 800 through subsequent functional group manipulations, showcasing the synthetic power of HAT-driven transformations in constructing highly complex diterpenoid frameworks.
 | ||
| Scheme 102 Luo's total synthesis of (+)-kobusine (799) and (+)-spirasine IX (800). | ||
9. Conclusion and outlook
Radical retrosynthesis has emerged as a powerful and conceptually distinct platform for the synthesis of complex terpenoid natural products. By reconceiving bond disconnections in terms of radical fragments, this strategy enables synthetic solutions that circumvent the limitations of traditional polar approaches—facilitating late-stage functionalization, efficient quaternary center construction, and modular assembly of polycyclic scaffolds. In this review, we have summarized the recent applications of diverse classes of radical precursors—including alkenes, carboxylic acids, organohalides, alcohol derivatives, aldehydes, ketones, and alkanes—in the context of terpenoid synthesis. Particular emphasis has been placed on how each precursor class has been transformed through newly developed radical processes to enable bond-forming reactions that are well-suited to the densely functionalized and stereochemically rich nature of terpenoid frameworks.These advances have been driven by a wave of methodological innovation over the past decade. Developments in photoredox catalysis, electrochemical activation, MHAT, redox-active ester chemistry, and HAT based C–H functionalization have significantly expanded the repertoire of radical generation and reactivity under synthetically useful conditions. In terpenoid contexts, such tools have enabled installation of quaternary stereocenters, ring constructions, and skeletal rearrangements with improved efficiency and convergence. By organizing the discussion around radical precursor classes and mapping them to representative reaction modes, this review provides a conceptual and practical framework for integrating radical retrosynthetic logic into the total synthesis of architecturally intricate terpenoid natural products.
Looking forward, the broader integration of radical retrosynthesis into complex molecule synthesis will benefit from continued advances on multiple fronts. Asymmetric radical transformations, though rapidly developing, remain underutilized in total synthesis and demand expansion beyond substrate-controlled systems. The rational design of radical cascade reactions also holds great promise for enhancing step economy, particularly in the assembly of polycyclic terpenoid frameworks. In addition, while radical additions to alkenes provide a valuable strategy for constructing quaternary carbon centers, further development of alternative approaches—such as SH2-based couplings and other non-Giese-type mechanisms—will be critical for expanding structural diversity and stereochemical control. Finally, achieving higher levels of regio- and chemoselectivity in C–H functionalization remains a key goal, one that will enable more precise and programmable bond disconnections. Continued innovation in these areas will further establish radical retrosynthesis as a central and enabling logic in the synthesis of architecturally complex natural products. We hope that the ideas outlined in this review will inspire practitioners to think beyond conventional retrosynthetic paradigms and to embrace the unique reactivity of radicals as a foundation for creative and efficient synthesis.
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
We are grateful to all members of our laboratory for their support in gathering and surveying the pertinent literature. Financial support was provided by the National Natural Science Foundation of China (grant no. 22171025), MOST of China, and Tsinghua Institute of Multidisciplinary Biomedical Research, Tsinghua University.References
- E. J. Corey, Chem. Soc. Rev., 1988, 17, 111–133 RSC.
- E. J. Corey and X.-M. Cheng, Logic of chemical synthesis, Wiley, New York, New ed., 1995 Search PubMed.
- J. M. Smith, S. J. Harwood and P. S. Baran, Acc. Chem. Res., 2018, 51, 1807–1817 CrossRef CAS.
- S. L. Shevick, C. V. Wilson, S. Kotesova, D. Kim, P. L. Holland and R. A. Shenvi, Chem. Sci., 2020, 11, 12401–12422 RSC.
- S. A. Green, S. W. M. Crossley, J. L. M. Matos, S. Vásquez-Céspedes, S. L. Shevick and R. A. Shenvi, Acc. Chem. Res., 2018, 51, 2628–2640 CrossRef CAS PubMed.
- S. W. M. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Chem. Rev., 2016, 116, 8912–9000 CrossRef CAS.
- S. Murarka, Adv. Synth. Catal., 2018, 360, 1735–1753 CrossRef CAS.
- S. Karmakar, A. Silamkoti, N. A. Meanwell, A. Mathur and A. K. Gupta, Adv. Synth. Catal., 2021, 363, 3693–3736 CrossRef CAS.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- L. E. Ehehalt, O. M. Beleh, I. C. Priest, J. M. Mouat, A. K. Olszewski, B. N. Ahern, A. R. Cruz, B. K. Chi, A. J. Castro, K. Kang, J. Wang and D. J. Weix, Chem. Rev., 2024, 124, 13397–13569 CrossRef CAS PubMed.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS.
- S. Sarkar, K. P. S. Cheung and V. Gevorgyan, Chem. Sci., 2020, 11, 12974–12993 RSC.
- S. Mondal, F. Dumur, D. Gigmes, M. P. Sibi, M. P. Bertrand and M. Nechab, Chem. Rev., 2022, 122, 5842–5976 CrossRef CAS.
- K. Hung, X. Hu and T. J. Maimone, Nat. Prod. Rep., 2018, 35, 174–202 RSC.
- K. J. Romero, M. S. Galliher, D. A. Pratt and C. R. J. Stephenson, Chem. Soc. Rev., 2018, 47, 7851–7866 RSC.
- A. Frank and M. Groll, Chem. Rev., 2017, 117, 5675–5703 CrossRef CAS.
- E. C. Cherney and P. S. Baran, Isr. J. Chem., 2011, 51, 391–405 CrossRef CAS.
- E. Goyer, C. Lavaud and G. Massiot, Nat. Prod. Rep., 2023, 40, 1071–1077 RSC.
- R. B. Clayton, Q. Rev. Chem. Soc., 1965, 19, 168–200 RSC.
- J. Gershenzon and N. Dudareva, Nat. Chem. Biol., 2007, 3, 408–414 CrossRef CAS PubMed.
- Y. Wang and J. Gui, Chem. Soc. Rev., 2025, 54, 6807–6831 RSC.
- A. Kanwal, M. Bilal, N. Rasool, M. Zubair, S. A. A. Shah and Z. A. Zakaria, Pharmaceuticals, 2022, 15, 1392 CrossRef CAS.
- W. P. Thomas and S. V. Pronin, Acc. Chem. Res., 2021, 54, 1347–1359 CrossRef CAS PubMed.
- Y. Chen, J. Zhao, S. Li and J. Xu, Nat. Prod. Rep., 2019, 36, 263–288 RSC.
- J. Justicia, L. Álvarez De Cienfuegos, A. G. Campaña, D. Miguel, V. Jakoby, A. Gansäuer and J. M. Cuerva, Chem. Soc. Rev., 2011, 40, 3525 RSC.
- D. Urabe, T. Asaba and M. Inoue, Chem. Rev., 2015, 115, 9207–9231 CrossRef CAS PubMed.
- C. N. Stout and H. Renata, Acc. Chem. Res., 2021, 54, 1143–1156 CrossRef CAS.
- Z. G. Brill, M. L. Condakes, C. P. Ting and T. J. Maimone, Chem. Rev., 2017, 117, 11753–11795 CrossRef CAS PubMed.
- S. P. Pitre, N. A. Weires and L. E. Overman, J. Am. Chem. Soc., 2019, 141, 2800–2813 CrossRef CAS.
- C. P. Jasperse, D. P. Curran and T. L. Fevig, Chem. Rev., 1991, 91, 1237–1286 CrossRef CAS.
- G. J. Rowlands, Tetrahedron, 2009, 65, 8603–8655 CrossRef CAS.
- G. J. Rowlands, Tetrahedron, 2010, 66, 1593–1636 CrossRef CAS.
- T. Yoshimitsu, Chem. Rec., 2014, 14, 268–279 CrossRef CAS.
- M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed.
- R. W. Hoffmann, Chem. Soc. Rev., 2016, 45, 577–583 RSC.
- Y. Li, X. Lu and Y. Fu, CCS Chem., 2024, 6, 1130–1156 CrossRef CAS.
- S. H. Kyne, G. Lefèvre, C. Ollivier, M. Petit, V.-A. Ramis Cladera and L. Fensterbank, Chem. Soc. Rev., 2020, 49, 8501–8542 RSC.
- S. Isayama and T. Mukaiyama, Chem. Lett, 1989, 18, 569–572 CrossRef.
- I. Tabushi and N. Koga, J. Am. Chem. Soc., 1979, 101, 6456–6458 CrossRef CAS.
- S. Inoki, K. Kato, T. Takai, S. Isayama, T. Yamada and T. Mukaiyama, Chem. Lett, 1989, 18, 515–518 CrossRef.
- T. Mukaiyama, S. Isayama, S. Inoki, K. Kato, T. Yamada and T. Takai, Chem. Lett., 1989, 449–452 CrossRef CAS.
- B. Gaspar and E. M. Carreira, J. Am. Chem. Soc., 2009, 131, 13214–13215 CrossRef CAS.
- J. Waser and E. M. Carreira, Angew. Chem., Int. Ed., 2004, 43, 4099–4102 CrossRef CAS PubMed.
- J. Waser and E. M. Carreira, J. Am. Chem. Soc., 2004, 126, 5676–5677 CrossRef CAS PubMed.
- J. Waser, H. Nambu and E. M. Carreira, J. Am. Chem. Soc., 2005, 127, 8294–8295 CrossRef CAS.
- J. Waser, B. Gaspar, H. Nambu and E. M. Carreira, J. Am. Chem. Soc., 2006, 128, 11693–11712 CrossRef CAS.
- B. Gaspar and E. M. Carreira, Angew. Chem., Int. Ed., 2007, 46, 4519–4522 CrossRef CAS.
- B. Gaspar and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 5758–5760 CrossRef CAS PubMed.
- J. E. Sears and D. L. Boger, Acc. Chem. Res., 2015, 48, 653–662 CrossRef CAS PubMed.
- H. Ishikawa, D. A. Colby and D. L. Boger, J. Am. Chem. Soc., 2008, 130, 420–421 CrossRef CAS PubMed.
- H. Ishikawa, D. A. Colby, S. Seto, P. Va, A. Tam, H. Kakei, T. J. Rayl, I. Hwang and D. L. Boger, J. Am. Chem. Soc., 2009, 131, 4904–4916 CrossRef CAS PubMed.
- E. K. Leggans, T. J. Barker, K. K. Duncan and D. L. Boger, Org. Lett., 2012, 14, 1428–1431 CrossRef CAS.
- D. C. Eisenberg and J. R. Norton, Isr. J. Chem., 1991, 31, 55–66 CrossRef CAS.
- Y. Hu, A. P. Shaw, D. P. Estes and J. R. Norton, Chem. Rev., 2016, 116, 8427–8462 CrossRef CAS PubMed.
- L. Tang, E. T. Papish, G. P. Abramo, J. R. Norton, M.-H. Baik, R. A. Friesner and A. Rappé, J. Am. Chem. Soc., 2003, 125, 10093–10102 CrossRef CAS.
- J. Choi, L. Tang and J. R. Norton, J. Am. Chem. Soc., 2006, 129, 234–240 CrossRef PubMed.
- J. L. Kuo, J. Hartung, A. Han and J. R. Norton, J. Am. Chem. Soc., 2015, 137, 1036–1039 CrossRef CAS.
- J. C. Lo, D. Kim, C.-M. Pan, J. T. Edwards, Y. Yabe, J. Gui, T. Qin, S. Gutiérrez, J. Giacoboni, M. W. Smith, P. L. Holland and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 2484–2503 CrossRef CAS PubMed.
- J. C. Lo, Y. Yabe and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 1304–1307 CrossRef CAS.
- J. C. Lo, J. Gui, Y. Yabe, C.-M. Pan and P. S. Baran, Nature, 2014, 516, 343–348 CrossRef CAS.
- H. T. Dao, C. Li, Q. Michaudel, B. D. Maxwell and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 8046–8049 CrossRef CAS.
- J. Gui, C.-M. Pan, Y. Jin, T. Qin, J. C. Lo, B. J. Lee, S. H. Spergel, M. E. Mertzman, W. J. Pitts, T. E. La Cruz, M. A. Schmidt, N. Darvatkar, S. R. Natarajan and P. S. Baran, Science, 2015, 348, 886–891 CrossRef CAS PubMed.
- L. Kong, X. Gan, V. A. Van Der Puyl Lovett and R. A. Shenvi, J. Am. Chem. Soc., 2024, 146, 2351–2357 CrossRef CAS PubMed.
- S. A. Green, J. L. M. Matos, A. Yagi and R. A. Shenvi, J. Am. Chem. Soc., 2016, 138, 12779–12782 CrossRef CAS PubMed.
- S. A. Green, S. Vásquez-Céspedes and R. A. Shenvi, J. Am. Chem. Soc., 2018, 140, 11317–11324 CrossRef CAS PubMed.
- J. L. M. Matos, S. Vásquez-Céspedes, J. Gu, T. Oguma and R. A. Shenvi, J. Am. Chem. Soc., 2018, 140, 16976–16981 CrossRef CAS PubMed.
- S. L. Shevick, C. Obradors and R. A. Shenvi, J. Am. Chem. Soc., 2018, 140, 12056–12068 CrossRef CAS.
- S. A. Green, T. R. Huffman, R. O. McCourt, V. van der Puyl and R. A. Shenvi, J. Am. Chem. Soc., 2019, 141, 7709–7714 CrossRef CAS PubMed.
- X. Gan, B. Zhang, N. Dao, C. Bi, M. Pokle, L. Kan, M. R. Collins, C. C. Tyrol, P. N. Bolduc, M. Nicastri, Y. Kawamata, P. S. Baran and R. Shenvi, Science, 2024, 384, 113–118 CrossRef CAS PubMed.
- X. Ma and S. B. Herzon, Beilstein J. Org. Chem., 2018, 14, 2259–2265 CrossRef CAS PubMed.
- S. M. King, X. Ma and S. B. Herzon, J. Am. Chem. Soc., 2014, 136, 6884–6887 CrossRef CAS PubMed.
- X. Ma and S. B. Herzon, Chem. Sci., 2015, 6, 6250–6255 RSC.
- X. Ma, H. Dang, J. A. Rose, P. Rablen and S. B. Herzon, J. Am. Chem. Soc., 2017, 139, 5998–6007 CrossRef CAS PubMed.
- D. E. Essayan, M. J. Schubach, J. M. Smoot, T. Puri and S. V. Pronin, J. Am. Chem. Soc., 2024, 146, 18224–18229 CrossRef CAS.
- E. E. Touney, N. J. Foy and S. V. Pronin, J. Am. Chem. Soc., 2018, 140, 16982–16987 CrossRef CAS PubMed.
- C. A. Discolo, E. E. Touney and S. V. Pronin, J. Am. Chem. Soc., 2019, 141, 17527–17532 CrossRef CAS.
- L.-J. Li, Y. He, Y. Yang, J. Guo, Z. Lu, C. Wang, S. Zhu and S.-F. Zhu, CCS Chem., 2024, 6, 537–584 CrossRef CAS.
- Y. Wang, Y. He and S. Zhu, Acc. Chem. Res., 2022, 55, 3519–3536 CrossRef CAS PubMed.
- G. Zhang and Q. Zhang, Chem. Catal., 2023, 3, 100526 CAS.
- J. Wu and Z. Ma, Org. Chem. Front., 2021, 8, 7050–7076 RSC.
- D. T. George, E. J. Kuenstner and S. V. Pronin, J. Am. Chem. Soc., 2015, 137, 15410–15413 CrossRef CAS.
- B. Huang, C.-J. Xiao, Z.-Y. Huang, X.-Y. Tian, X. Cheng, X. Dong and B. Jiang, Org. Lett., 2014, 16, 3552–3555 CrossRef CAS.
- W. Cao, H. Deng, Y. Sun, B. Liu and S. Qin, Chem. – Eur. J., 2018, 24, 9120–9129 CrossRef CAS.
- H. Deng, W. Cao, R. Liu, Y. Zhang and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 5849–5852 CrossRef CAS.
- Z. Lu, X. Zhang, Z. Guo, Y. Chen, T. Mu and A. Li, J. Am. Chem. Soc., 2018, 140, 9211–9218 CrossRef CAS.
- D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174–3183 CrossRef CAS.
- K. Nozaki, K. Oshima and K. Utimoto, Tetrahedron Lett., 1988, 29, 1041–1044 CrossRef CAS.
- P. Hu, H. M. Chi, K. C. DeBacker, X. Gong, J. H. Keim, I. T. Hsu and S. A. Snyder, Nature, 2019, 569, 703–707 CrossRef CAS.
- Y. Matsuda and I. Abe, Nat. Prod. Rep., 2016, 33, 26–53 RSC.
- J. Li, F. Li, E. King-Smith and H. Renata, Nat. Chem., 2020, 12, 173–179 CrossRef CAS PubMed.
- J. Kobayashi and T. Kubota, Nat. Prod. Rep., 2009, 26, 936 RSC.
- G. Xu, J. Wu, L. Li, Y. Lu and C. Li, J. Am. Chem. Soc., 2020, 142, 15240–15245 CrossRef CAS.
- P. Magnus, J. Lacour, I. Coldham, B. Mugrage and W. B. Bauta, Tetrahedron, 1995, 51, 11087–11110 CrossRef CAS.
- K. B. Sharpless, T. Hori, L. K. Truesdale and C. O. Dietrich, J. Am. Chem. Soc., 1976, 98, 269–271 CrossRef CAS.
- J. Li, F. Chen and H. Renata, J. Am. Chem. Soc., 2022, 144, 19238–19242 CrossRef CAS PubMed.
- C. Petrier, C. Dupuy and J. L. Luche, Tetrahedron Lett., 1986, 27, 3149–3152 CrossRef CAS.
- R. F. Lusi, G. Sennari and R. Sarpong, J. Am. Chem. Soc., 2022, 144, 17277–17294 CrossRef CAS.
- R. F. Lusi, G. Sennari and R. Sarpong, Nat. Chem., 2022, 14, 450–456 CrossRef CAS PubMed.
- F. A. Bermejo, A. Fernández Mateos, A. Marcos Escribano, R. Martín Lago, L. Mateos Burón, M. Rodríguez López and R. Rubio González, Tetrahedron, 2006, 62, 8933–8942 CrossRef CAS.
- O. Goethe, A. Heuer, X. Ma, Z. Wang and S. B. Herzon, Nat. Prod. Rep., 2019, 36, 220–247 RSC.
- N. J. Foy and S. V. Pronin, J. Am. Chem. Soc., 2022, 144, 10174–10179 CrossRef CAS PubMed.
- W. Cao, Z. Wang, Y. Hao, T. Wang, S. Fu and B. Liu, Angew. Chem., Int. Ed., 2023, 62, e202305516 CrossRef CAS PubMed.
- A. Heidbreder and J. Mattay, Tetrahedron Lett., 1992, 33, 1973–1976 CrossRef CAS.
- X.-H. Zhao, L.-L. Meng, X.-T. Liu, P.-F. Shu, C. Yuan, X.-T. An, T.-X. Jia, Q.-Q. Yang, X. Zhen and C.-A. Fan, J. Am. Chem. Soc., 2023, 145, 311–321 CrossRef CAS PubMed.
- P. S. Rao, K. G. Sarma and T. R. Seshadri, Curr. Sci., 1965, 34, 9–11 CAS.
- X. Chen, W. Yao, H. Zheng, H. Wang, P.-P. Zhou, D.-Y. Zhu and S.-H. Wang, J. Am. Chem. Soc., 2023, 145, 13549–13555 CrossRef CAS.
- X.-H. Tian, L.-L. Hong, W.-H. Jiao and H.-W. Lin, Nat. Prod. Rep., 2023, 40, 718–749 RSC.
- Y. Huang, Q. Gu, Q. Chao, H. Xiao and H. Lu, Angew. Chem., Int. Ed., 2025, 64, e202507638 CrossRef CAS PubMed.
- B. Xu, C. Liu and M. Dai, J. Am. Chem. Soc., 2022, 144, 19700–19703 CrossRef CAS PubMed.
- B. Khatri Chhetri, S. Lavoie, A. M. Sweeney-Jones, N. Mojib, V. Raghavan, K. Gagaring, B. Dale, C. W. McNamara, K. Soapi, C. L. Quave, P. L. Polavarapu and J. Kubanek, J. Org. Chem., 2019, 84, 8531–8541 CrossRef CAS.
- N. Germain and A. Alexakis, Chem. – Eur. J., 2015, 21, 8597–8606 CrossRef CAS.
- C. H. Oh, H. H. Jung, K. S. Kim and N. Kim, Angew. Chem., Int. Ed., 2003, 42, 805–808 CrossRef CAS.
- K. Du, P. Guo, Y. Chen, Z. Cao, Z. Wang and W. Tang, Angew. Chem., Int. Ed., 2015, 54, 3033–3037 CrossRef CAS PubMed.
- M. Fadel and E. M. Carreira, J. Am. Chem. Soc., 2023, 145, 8332–8337 CrossRef CAS.
- R. J. Ferreira, G. Spengler, A. Orthaber, D. J. V. A. Dos Santos and M.-J. U. Ferreira, Org. Lett., 2021, 23, 274–278 CrossRef CAS.
- H. Wang, L. Wei, J. Huang, L. Wang, H. Peng, H. Jiang, J. Wu and Z. Ma, Angew. Chem., Int. Ed., 2025, 64, e202507961 CrossRef CAS PubMed.
- H. H. Patel and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 3462–3465 CrossRef CAS.
- W. Zhao, R. Al-Ahmad and M. Dai, J. Am. Chem. Soc., 2025, 147, 32365–32369 CrossRef CAS PubMed.
- Y.-P. Liu, Q. Dai, W.-X. Wang, J. He, Z.-H. Li, T. Feng and J.-K. Liu, J. Nat. Prod., 2020, 83, 1725–1729 CrossRef CAS.
- B. Xu, W. Xun, S. Su and H. Zhai, Angew. Chem., Int. Ed., 2020, 59, 16475–16479 CrossRef CAS PubMed.
- R. Uchida, S. Yokota, D. Matsuda, A. Matsumoto, S. Iwamoto, H. Onodera, Y. Takahashi and H. Tomoda, J. Antibiot., 2014, 67, 777–781 CrossRef CAS.
- H. Taguchi, M. Kawaguchi, T. Nagamitsu and M. Ohtawa, Org. Biomol. Chem., 2023, 21, 6129–6133 RSC.
- G. Huang, X. Zhang, Y.-C. Gu and J. Gui, J. Am. Chem. Soc., 2025, 147, 20239–20245 CrossRef PubMed.
- J. Li, H. Tang, T. Kurtán, A. Mándi, C.-L. Zhuang, L. Su, G.-L. Zheng and W. Zhang, J. Nat. Prod., 2018, 81, 1645–1650 CrossRef CAS.
- J. Gong, P. Sun, N. Jiang, R. Riccio, G. Lauro, G. Bifulco, T.-J. Li, W. H. Gerwick and W. Zhang, Org. Lett., 2014, 16, 2224–2227 CrossRef CAS.
- M. Saladrigas, E. Gómez-Bengoa, J. Bonjoch and B. Bradshaw, Chem. – Eur. J., 2023, 29, e202203286 CrossRef CAS.
- X. Fang, N. Zhang, S.-C. Chen and T. Luo, J. Am. Chem. Soc., 2022, 144, 2292–2300 CrossRef CAS.
- P. W. Brian and J. G. Mcgowan, Nature, 1945, 156, 144–145 CrossRef CAS.
- J. S. Moffatt, J. D. Bu’Lock and T. H. Yuen, J. Chem. Soc. D, 1969, 839a RSC.
- Y. Ji, Z. Xin, H. He and S. Gao, J. Am. Chem. Soc., 2019, 141, 16208–16212 CrossRef CAS.
- W. Gao, C. Chai, Y. He, F. Li, X. Hao, F. Cao, L. Gu, J. Liu, Z. Hu and Y. Zhang, Org. Lett., 2019, 21, 8469–8472 CrossRef CAS.
- T. Amagata, M. Doi, M. Tohgo, K. Minoura and A. Numata, Chem. Commun., 1999, 1321–1322 RSC.
- T. Amagata, M. Tanaka, T. Yamada, M. Doi, K. Minoura, H. Ohishi, T. Yamori and A. Numata, J. Nat. Prod., 2007, 70, 1731–1740 CrossRef CAS PubMed.
- P. Chen, C. Wang, R. Yang, H. Xu, J. Wu, H. Jiang, K. Chen and Z. Ma, Angew. Chem., Int. Ed., 2021, 60, 5512–5518 CrossRef CAS PubMed.
- L. Canonica, B. Danieli, G. Lesma and G. Palmisano, J. Chem. Soc., Chem. Commun., 1985, 1321–1322 RSC.
- K. Yu, F. Yao, Q. Zeng, H. Xie and H. Ding, J. Am. Chem. Soc., 2021, 143, 10576–10581 CrossRef CAS PubMed.
- A. Ulubelen, H. K. Desai, S. K. Srivastava, B. P. Hart, J.-C. Park, B. S. Joshi, S. W. Pelletier, A. H. Meriçli, F. Meriçli and R. Ilarslan, J. Nat. Prod., 1996, 59, 360–366 CrossRef CAS.
- V. B. Birman, E. W. Uffman, H. Jiang, X. Li and C. J. Kilbane, J. Am. Chem. Soc., 2004, 126, 12226–12227 CrossRef CAS PubMed.
- S. Dall’Acqua, B. B. Shrestha, M. B. Gewali, P. K. Jha, M. Carrara and G. Innocenti, Nat. Prod. Commun., 2008, 3, 1985–1989 Search PubMed.
- J. Liu and D. Ma, Angew. Chem., Int. Ed., 2018, 57, 6676–6680 CrossRef CAS.
- F. F. Fleming, J. Guo, Q. Wang and D. Weaver, J. Org. Chem., 1999, 64, 8568–8575 CrossRef CAS.
- K. Iwasaki, K. K. Wan, A. Oppedisano, S. W. M. Crossley and R. A. Shenvi, J. Am. Chem. Soc., 2014, 136, 1300–1303 CrossRef CAS.
- C. Zhou, W. Qin, C. Tu, Y. Chen, S. Fu and B. Liu, Angew. Chem., Int. Ed., 2025, 64, e202503943 CrossRef CAS.
- H. Hussain, J. Xiao, A. Ali, I. R. Green and B. Westermann, Nat. Prod. Rep., 2023, 40, 412–451 RSC.
- R. Wittenberg, J. Srogl, M. Egi and L. S. Liebeskind, Org. Lett., 2003, 5, 3033–3035 CrossRef CAS PubMed.
- R. J. Wiles and G. A. Molander, Isr. J. Chem., 2020, 60, 281–293 CrossRef CAS.
- N. Müller, T. Magauer and O. Kováč, J. Org. Chem., 2025, 90, 5083–5092 CrossRef.
- L. Pitzer, J. L. Schwarz and F. Glorius, Chem. Sci., 2019, 10, 8285–8291 RSC.
- T. Long, S. He and C. Li, Chin. J. Org. Chem., 2025, 45, 748–763 CrossRef CAS.
- X. Li, Z. Chang, S. Duan and Z. Xie, Angew. Chem., Int. Ed., 2025, 64, e202416211 CrossRef CAS.
- F. Tang, Z.-C. Zhang, Z.-L. Song, Y.-H. Li, Z.-H. Zhou, J.-J. Chen and Z. Yang, J. Am. Chem. Soc., 2025, 147, 4731–4735 CrossRef CAS PubMed.
- C. P. Ting, G. Xu, X. Zeng and T. J. Maimone, J. Am. Chem. Soc., 2016, 138, 14868–14871 CrossRef CAS.
- M. Elkin, S. M. Szewczyk, A. C. Scruse and T. R. Newhouse, J. Am. Chem. Soc., 2017, 139, 1790–1793 CrossRef CAS PubMed.
- F. Yang and J. A. Porco, J. Am. Chem. Soc., 2022, 144, 12970–12978 CrossRef CAS.
- Y. Zhang, Y. Ji, I. Franzoni, C. Guo, H. Jia, B. Hong and H. Li, Angew. Chem., Int. Ed., 2021, 60, 14869–14874 CrossRef CAS PubMed.
- G. Xu, M. Elkin, D. J. Tantillo, T. R. Newhouse and T. J. Maimone, Angew. Chem., Int. Ed., 2017, 56, 12498–12502 CrossRef CAS PubMed.
- H. Shigehisa, T. Aoki, S. Yamaguchi, N. Shimizu and K. Hiroya, J. Am. Chem. Soc., 2013, 135, 10306–10309 CrossRef CAS.
- L. K. Johnson, S. W. Niman, D. Vrubliauskas and C. D. Vanderwal, Org. Lett., 2021, 23, 9569–9573 CrossRef CAS PubMed.
- D. Vrubliauskas, B. M. Gross and C. D. Vanderwal, J. Am. Chem. Soc., 2021, 143, 2944–2952 CrossRef CAS PubMed.
- D. Vrubliauskas and C. D. Vanderwal, Angew. Chem., Int. Ed., 2020, 59, 6115–6121 CrossRef CAS PubMed.
- Y. Zhao, J. Hu, R. Chen, F. Xiong, H. Xie and H. Ding, J. Am. Chem. Soc., 2022, 144, 2495–2500 CrossRef CAS PubMed.
- G. Schoenn, C. Kouklovsky, R. Guillot, T. Magauer and G. Vincent, Angew. Chem., Int. Ed., 2025, 64, e202505270 CrossRef CAS.
- D. J. Schatz, E. J. Kuenstner, D. T. George and S. V. Pronin, Nat. Prod. Rep., 2022, 39, 946–968 RSC.
- W. P. Thomas and S. V. Pronin, J. Am. Chem. Soc., 2022, 144, 118–122 CrossRef CAS PubMed.
- W. P. Thomas, D. J. Schatz, D. T. George and S. V. Pronin, J. Am. Chem. Soc., 2019, 141, 12246–12250 CrossRef CAS PubMed.
- N. A. Godfrey, D. J. Schatz and S. V. Pronin, J. Am. Chem. Soc., 2018, 140, 12770–12774 CrossRef CAS PubMed.
- G. Liu, Z. Zhang, S. Fu and B. Liu, Org. Lett., 2021, 23, 290–295 CrossRef CAS.
- H.-M. Chung, J.-H. Su, T.-L. Hwang, J.-J. Li, J.-J. Chen, Y.-H. Chen, Y.-C. Chang, Y.-D. Su, Y.-H. Chen, L.-S. Fang, J.-H. Sheu, W.-H. Wang and P.-J. Sung, Tetrahedron, 2013, 69, 2740–2744 CrossRef CAS.
- X. Gan, S. Kotesova, A. Castanedo, S. A. Green, S. L. B. Møller and R. A. Shenvi, J. Am. Chem. Soc., 2023, 145, 15714–15720 CrossRef CAS PubMed.
- L. G. Faqueti, I. V. Farias, E. C. Sabedot, F. Delle Monache, A. San Feliciano, I. T. A. Schuquel, V. Cechinel-Filho, A. Bella Cruz and C. Meyre-Silva, J. Agric. Food Chem., 2015, 63, 8151–8155 CrossRef CAS PubMed.
- S. W. M. Crossley, F. Barabé and R. A. Shenvi, J. Am. Chem. Soc., 2014, 136, 16788–16791 CrossRef CAS.
- G. H. Harris, K. Hoogsteen, K. C. Silverman, S. L. Raghoobar, G. F. Bills, R. B. Lingham, J. L. Smith, H. W. Dougherty, C. Cascales and F. Peláez, Tetrahedron, 1993, 49, 2139–2144 CrossRef CAS.
- C. Y. Bemis, C. N. Ungarean, A. S. Shved, C. S. Jamieson, T. Hwang, K. S. Lee, K. N. Houk and D. Sarlah, J. Am. Chem. Soc., 2021, 143, 6006–6017 CrossRef CAS PubMed.
- L. Pan, F. Schneider, M. Ottenbruch, R. Wiechert, T. List, P. Schoch, B. Mertes and T. Gaich, Nature, 2024, 632, 543–549 CrossRef PubMed.
- P. Zhang, Y. Li, Z. Yan, J. Gong and Z. Yang, J. Org. Chem., 2019, 84, 15958–15971 CrossRef CAS PubMed.
- P.-P. Zhang, Z.-M. Yan, Y.-H. Li, J.-X. Gong and Z. Yang, J. Am. Chem. Soc., 2017, 139, 13989–13992 CrossRef CAS.
- S. Shen, H. Zhu, D. Chen, D. Liu, L. V. Ofwegen, P. Proksch and W. Lin, Tetrahedron Lett., 2012, 53, 5759–5762 CrossRef CAS.
- J. H. Dworkin, B. W. Dehnert and O. Kwon, Trends in Chem., 2023, 5, 174–200 CrossRef CAS.
- M. Šimek, J. H. Dworkin and O. Kwon, Acc. Chem. Res., 2025, 58, 1547–1561 CrossRef.
- A. J. Smaligo, M. Swain, J. C. Quintana, M. F. Tan, D. A. Kim and O. Kwon, Science, 2019, 364, 681–685 CrossRef CAS.
- A. J. Smaligo and O. Kwon, Org. Lett., 2019, 21, 8592–8597 CrossRef CAS PubMed.
- A. J. Smaligo, J. Wu, N. R. Burton, A. S. Hacker, A. C. Shaikh, J. C. Quintana, R. Wang, C. Xie and O. Kwon, Angew. Chem., Int. Ed., 2020, 59, 1211–1215 CrossRef CAS PubMed.
- B. W. Dehnert, J. H. Dworkin and O. Kwon, Synthesis, 2024, 71–86 CAS.
- M. Swain, G. Sadykhov, R. Wang and O. Kwon, Angew. Chem., Int. Ed., 2020, 59, 17565–17571 CrossRef CAS PubMed.
- M. Swain, T. B. Bunnell, J. Kim and O. Kwon, J. Am. Chem. Soc., 2022, 144, 14828–14837 CrossRef CAS PubMed.
- D. E. Kim, Y. Zhu, S. Harada, I. Aguilar, A. E. Cuomo, M. Wang and T. R. Newhouse, J. Am. Chem. Soc., 2023, 145, 4394–4399 CrossRef CAS.
- M. Liu, P. Li, X. Tang, X. Luo, K. Liu, Y. Zhang, Q. Wang and G. Li, J. Org. Chem., 2021, 86, 970–979 CrossRef CAS PubMed.
- T. Kobayashi, R. Sugitate, K. Uchida, Y. Kawamoto and H. Ito, Org. Lett., 2024, 26, 1803–1806 CrossRef CAS PubMed.
- T. Kobayashi, F. Hayano, A. Kamiya, Y. Kawamoto and H. Ito, Org. Lett., 2024, 26, 5105–5109 CrossRef CAS PubMed.
- S. L. Schreiber, J. Am. Chem. Soc., 1980, 102, 6163–6165 CrossRef CAS.
- J. Ji, J. Chen, S. Qin, W. Li, J. Zhao, G. Li, H. Song, X.-Y. Liu and Y. Qin, J. Am. Chem. Soc., 2023, 145, 3903–3908 CrossRef CAS.
- F.-P. Wang and Q.-H. Chen, The Alkaloids: Chem. Bio., Elsevier, 2010, vol. 69, pp. 1–577 Search PubMed.
- L.-P. Zhong, C. Gudeman, J. Zhen, O. A. Wanasinghe, J. Hellmig, M. J. E. Collins, J. Bacsa, A. Adibekian and M. Dai, J. Am. Chem. Soc., 2025, 147, 28589–28594 CrossRef PubMed.
- J. Hu, Z.-X. Zou, Y. Chen, S. Li, X. Gao, Z. Liu, Y. Wang, H. Liu and W. Zhang, J. Nat. Prod., 2022, 85, 1967–1975 CrossRef CAS.
- S. B. Beil, T. Q. Chen, N. E. Intermaggio and D. W. C. MacMillan, Acc. Chem. Res., 2022, 55, 3481–3494 CrossRef CAS PubMed.
- T. Kato, K. Hagiwara and M. Inoue, Chem. Pharm. Bull., 2024, 72, 767–771 CrossRef CAS PubMed.
- M. Nagatomo, D. Kamimura, Y. Matsui, K. Masuda and M. Inoue, Chem. Sci., 2015, 6, 2765–2769 RSC.
- G. Laudadio, M. D. Palkowitz, T. El-Hayek Ewing and P. S. Baran, ACS Med. Chem. Lett., 2022, 13, 1413–1420 CrossRef CAS PubMed.
- C. R. Jamison and L. E. Overman, Acc. Chem. Res., 2016, 49, 1578–1586 CrossRef CAS.
- S. P. Pitre and L. E. Overman, Chem. Rev., 2022, 122, 1717–1751 CrossRef CAS PubMed.
- L. Huang, A. M. Olivares and D. J. Weix, Angew. Chem., Int. Ed., 2017, 56, 11901–11905 CrossRef CAS PubMed.
- J. M. Smith, T. Qin, R. R. Merchant, J. T. Edwards, L. R. Malins, Z. Liu, G. Che, Z. Shen, S. A. Shaw, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 11906–11910 CrossRef CAS.
- D. M. Kitcatt, S. Nicolle and A.-L. Lee, Chem. Soc. Rev., 2022, 51, 1415–1453 RSC.
- G. Pratsch, G. L. Lackner and L. E. Overman, J. Org. Chem., 2015, 80, 6025–6036 CrossRef CAS.
- T. Qin, L. R. Malins, J. T. Edwards, R. R. Merchant, A. J. E. Novak, J. Z. Zhong, R. B. Mills, M. Yan, C. Yuan, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 260–265 CrossRef CAS.
- L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS PubMed.
- M. J. Schnermann and L. E. Overman, Angew. Chem., Int. Ed., 2012, 51, 9576–9580 CrossRef CAS.
- K. Okada, K. Okamoto, N. Morita, K. Okubo and M. Oda, J. Am. Chem. Soc., 1991, 113, 9401–9402 CrossRef CAS.
- M. J. Schnermann and L. E. Overman, J. Am. Chem. Soc., 2011, 133, 16425–16427 CrossRef CAS.
- M. R. Garnsey, Y. Slutskyy, C. R. Jamison, P. Zhao, J. Lee, Y. H. Rhee and L. E. Overman, J. Org. Chem., 2018, 83, 6958–6976 CrossRef CAS.
- D. S. Müller, N. L. Untiedt, A. P. Dieskau, G. L. Lackner and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 660–663 CrossRef PubMed.
- F. Gao and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 10961–10963 CrossRef CAS PubMed.
- D. J. Tao, Y. Slutskyy and L. E. Overman, J. Am. Chem. Soc., 2016, 138, 2186–2189 CrossRef CAS.
- Z.-K. Wan, H. Choi, F.-A. Kang, K. Nakajima, D. Demeke and Y. Kishi, Org. Lett., 2002, 4, 4431–4434 CrossRef CAS PubMed.
- Y. Okanishi, T. Ishikawa, T. Jinnouchi, S. Hayashi, T. Takanami, H. Aoyama and T. Yoshimitsu, J. Org. Chem., 2023, 88, 1085–1092 CrossRef CAS.
- K. D. Wellington, R. C. Cambie, P. S. Rutledge and P. R. Bergquist, J. Nat. Prod., 2000, 63, 79–85 CrossRef CAS PubMed.
- X. Li, Y. Zhang, Z. Zhang and J. Wu, Org. Chem. Front., 2024, 11, 3939–3945 RSC.
- X. Li, Z. Zhang and J. Wu, Angew. Chem., Int. Ed., 2025, 64, e202500341 CrossRef CAS.
- Z. Cao, W. Sun, J. Zhang, J. Zhuo, S. Yang, X. Song, Y. Ma, P. Lu, T. Han and C. Li, Nat. Commun., 2024, 15, 6052 CrossRef CAS.
- L. Li, Y. Yao and N. Fu, Eur. J. Org. Chem., 2023, 26, e202300166 CrossRef CAS.
- J. C. Beck, C. R. Lacker, L. M. Chapman and S. E. Reisman, Chem. Sci., 2019, 10, 2315–2319 RSC.
- Y. Hikone, T. Kato, M. Nagatomo and M. Inoue, Org. Lett., 2022, 24, 929–933 CrossRef CAS.
- M. Hergenhahn, W. Adolf and E. Hecker, Tetrahedron Lett., 1975, 16, 1595–1598 Search PubMed.
- Y. Imamura, K. Takaoka, Y. Komori, M. Nagatomo and M. Inoue, Angew. Chem., Int. Ed., 2023, 62, e202219114 CrossRef CAS PubMed.
- D. Kuwana, Y. Komori, M. Nagatomo and M. Inoue, J. Org. Chem., 2022, 87, 730–736 CrossRef CAS PubMed.
- J. Taguchi, S. Fukaya, H. Fujino and M. Inoue, J. Am. Chem. Soc., 2024, 146, 34221–34230 CrossRef CAS PubMed.
- Y. Imamura, S. Yoshioka, M. Nagatomo and M. Inoue, Angew. Chem., Int. Ed., 2019, 58, 12159–12163 CrossRef CAS.
- T. Watanabe, K. Oga, H. Matoba, M. Nagatomo and M. Inoue, J. Am. Chem. Soc., 2023, 145, 25894–25902 CrossRef CAS.
- M. M. Logan, T. Toma, R. Thomas-Tran and J. Du Bois, Science, 2016, 354, 865–869 CrossRef CAS.
- Y. Watanabe, K. Sakata, D. Urabe, K. Hagiwara and M. Inoue, J. Org. Chem., 2023, 88, 17479–17484 CrossRef CAS.
- T. Tokuyama, J. Daly, B. Witkop, I. L. Karle and J. Karle, J. Am. Chem. Soc., 1968, 90, 1917–1918 CrossRef CAS PubMed.
- J. Cornella, J. T. Edwards, T. Qin, S. Kawamura, J. Wang, C.-M. Pan, R. Gianatassio, M. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 2174–2177 CrossRef CAS.
- J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D.-H. Bao, F.-L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213–218 CrossRef CAS PubMed.
- J. Lu, Y. Yao, L. Li and N. Fu, J. Am. Chem. Soc., 2023, 145, 26774–26782 CrossRef CAS PubMed.
- S. J. Harwood, M. D. Palkowitz, C. N. Gannett, P. Perez, Z. Yao, L. Sun, H. D. Abruña, S. L. Anderson and P. S. Baran, Science, 2022, 375, 745–752 CrossRef CAS.
- H. Chen, S. Sun and X. Liao, Org. Lett., 2019, 21, 3625–3630 CrossRef CAS.
- S. K. Pedersen, S. Clementson, K. El-Chami, J. L. Kristensen and M. Jessing, Chem. – Eur. J., 2023, 29, e202300265 CrossRef CAS.
- Y. Kawamata, M. Yan, Z. Liu, D.-H. Bao, J. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448–7451 CrossRef CAS.
- R. R. Merchant, K. M. Oberg, Y. Lin, A. J. E. Novak, J. Felding and P. S. Baran, J. Am. Chem. Soc., 2018, 140, 7462–7465 CrossRef CAS.
- H. Kim, J. B. Baker, S.-U. Lee, Y. Park, K. L. Bolduc, H.-B. Park, M. G. Dickens, D.-S. Lee, Y. Kim, S. H. Kim and J. Hong, J. Am. Chem. Soc., 2009, 131, 3192–3194 CrossRef CAS PubMed.
- J. Gu, K. Rodriguez, Y. Kanda, S. Yang, M. Ociepa, H. Wilke, A. Abrishami, L. Joergensen, T. Skak-Nielsen, J. Chen and P. Baran, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2200814119 CrossRef CAS.
- D. D. Bikle, Chem. Bio., 2014, 21, 319–329 CrossRef CAS PubMed.
- A. J. Rodriguez, M. S. Pokle, G. L. Barnes and P. S. Baran, J. Am. Chem. Soc., 2025, 147, 16781–16785 CrossRef CAS PubMed.
- L. Zhang, M. Zhao, M. Pu, Z. Ma, J. Zhou, C. Chen, Y.-D. Wu, Y. R. Chi and J. S. Zhou, J. Am. Chem. Soc., 2022, 144, 20249–20257 CrossRef CAS PubMed.
- B. Qin, A. Szyperek and M. Tomanik, J. Am. Chem. Soc., 2025, 147, 31221–31227 CrossRef CAS PubMed.
- J. Wang, T. Qin, T. Chen, L. Wimmer, J. T. Edwards, J. Cornella, B. Vokits, S. A. Shaw and P. S. Baran, Angew. Chem., Int. Ed., 2016, 55, 9676–9679 CrossRef CAS PubMed.
- M. Zhang, H. Fan, Y. Wang and J. Gui, Org. Lett., 2022, 24, 7383–7387 CrossRef CAS.
- J. Zhang, Z. Li, J. Zhuo, Y. Cui, T. Han and C. Li, J. Am. Chem. Soc., 2019, 141, 8372–8380 CrossRef CAS PubMed.
- K. M. M. Huihui, J. A. Caputo, Z. Melchor, A. M. Olivares, A. M. Spiewak, K. A. Johnson, T. A. DiBenedetto, S. Kim, L. K. G. Ackerman and D. J. Weix, J. Am. Chem. Soc., 2016, 138, 5016–5019 CrossRef CAS PubMed.
- N. Suzuki, J. L. Hofstra, K. E. Poremba and S. E. Reisman, Org. Lett., 2017, 19, 2150–2153 CrossRef CAS PubMed.
- Y.-M. Yan, J. Ai, L. Zhou, A. C. K. Chung, R. Li, J. Nie, P. Fang, X.-L. Wang, J. Luo, Q. Hu, F.-F. Hou and Y.-X. Cheng, Org. Lett., 2013, 15, 5488–5491 CrossRef CAS.
- A. Rode, N. Müller, O. Kováč, K. Wurst and T. Magauer, Org. Lett., 2024, 26, 9017–9021 CrossRef CAS.
- J. Choi and G. C. Fu, Science, 2017, 356, eaaf7230 CrossRef PubMed.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219–9280 CrossRef CAS.
- W. Xue, X. Jia, X. Wang, X. Tao, Z. Yin and H. Gong, Chem. Soc. Rev., 2021, 50, 4162–4184 RSC.
- G. C. Fu, ACS Cent. Sci., 2017, 3, 692–700 CrossRef CAS.
- M. Durandetti, J.-Y. Nédélec and J. Périchon, J. Org. Chem., 1996, 61, 1748–1755 CrossRef CAS.
- D. A. Everson, R. Shrestha and D. J. Weix, J. Am. Chem. Soc., 2010, 132, 920–921 CrossRef CAS.
- X. Yu, T. Yang, S. Wang, H. Xu and H. Gong, Org. Lett., 2011, 13, 2138–2141 CrossRef CAS.
- A. H. Cherney, N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 7442–7445 CrossRef CAS PubMed.
- M. Menger, D. Lentz and M. Christmann, J. Org. Chem., 2018, 83, 6793–6797 CrossRef CAS.
- P. Luo, W. Xia, S. L. Morris-Natschke, K.-H. Lee, Y. Zhao, Q. Gu and J. Xu, J. Nat. Prod., 2017, 80, 1679–1683 CrossRef CAS PubMed.
- A. Ahmad and A. C. B. Burtoloso, Org. Lett., 2019, 21, 6079–6083 CrossRef CAS.
- B. Yan, M. Zhou, J. Li, X. Li, S. He, J. Zuo, H. Sun, A. Li and P. Puno, Angew. Chem., Int. Ed., 2021, 133, 12969–12977 Search PubMed.
- K. A. Johnson, S. Biswas and D. J. Weix, Chem. – Eur. J., 2016, 22, 7399–7402 Search PubMed.
- W. Zhu, Q. Yin, Z. Lou and M. Yang, Nat. Commun., 2022, 13, 6633 CrossRef CAS.
- X.-B. Yan, C.-L. Li, W.-J. Jin, P. Guo and X.-Z. Shu, Chem. Sci., 2018, 9, 4529–4534 RSC.
- Y. Wang, Y. Su and Y. Jia, J. Am. Chem. Soc., 2023, 145, 9459–9463 CrossRef CAS.
- I. I. Rodríguez, A. D. Rodríguez and H. Zhao, J. Org. Chem., 2009, 74, 7581–7584 CrossRef PubMed.
- B. B. Snider, J. E. Merritt, M. A. Dombroski and B. O. Buckman, J. Org. Chem., 1991, 56, 5544–5553 CrossRef CAS.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- D. Staveness, I. Bosque and C. R. J. Stephenson, Acc. Chem. Res., 2016, 49, 2295–2306 CrossRef CAS.
- Z. Zhang, W. Zhang, J. Tang, J. Che, Z. Zhang, J. Chen and Z. Yang, J. Org. Chem., 2023, 88, 10539–10554 CrossRef CAS PubMed.
- W. Zhang, Z. Zhang, J.-C. Tang, J.-T. Che, H.-Y. Zhang, J.-H. Chen and Z. Yang, J. Am. Chem. Soc., 2020, 142, 19487–19492 CrossRef CAS PubMed.
- Z.-J. Xu, X.-Y. Liu, M.-Z. Zhu, Y.-L. Xu, Y. Yu, H.-R. Xu, A.-X. Cheng and H.-X. Lou, Org. Lett., 2021, 23, 9073–9077 CrossRef CAS.
- J. Xiao, H. Wu, J.-R. Liang, P. Wu, C. Guo, Y.-W. Wang, Z.-Y. Wang and Y. Peng, Org. Lett., 2024, 26, 3481–3486 CrossRef CAS PubMed.
- G. V. Ramakrishna, L. P. Pop, Z. Latif, H. K. V. Suryadevara, L. Santo and F. Romiti, J. Am. Chem. Soc., 2023, 145, 20062–20072 CrossRef CAS PubMed.
- Z. Wang, H. Yin and G. C. Fu, Nature, 2018, 563, 379–383 CrossRef CAS.
- Z. Wang, Z.-P. Yang and G. C. Fu, Nat. Chem., 2021, 13, 236–242 CrossRef CAS.
- G. V. Ramakrishna, Z. Latif and F. Romiti, J. Am. Chem. Soc., 2025, 147, 4613–4623 CrossRef CAS PubMed.
- H. Wang, X. Zhang and P. Tang, Chem. Sci., 2017, 8, 7246–7250 Search PubMed.
- D. A. Powell, T. Maki and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 510–511 CrossRef CAS PubMed.
- J. Zhang, X. Luo, J. Zhang and C. Li, J. Am. Chem. Soc., 2025, 147, 5933–5942 CrossRef CAS.
- Z. Dong and D. W. C. MacMillan, Nature, 2021, 598, 451–456 CrossRef CAS.
- Z. Li, W. Sun, X. Wang, L. Li, Y. Zhang and C. Li, J. Am. Chem. Soc., 2021, 143, 3536–3543 CrossRef CAS PubMed.
- J. Wang, J. Zhao and H. Gong, Chem. Commun., 2017, 53, 10180–10183 RSC.
- K. Komeyama, R. Ohata, S. Kiguchi and I. Osaka, Chem. Commun., 2017, 53, 6401–6404 RSC.
- J. Liu, C. Yang, X. Lu, Z. Zhang, L. Xu, M. Cui, X. Lu, B. Xiao, Y. Fu and L. Liu, Chem. – Eur. J., 2014, 20, 15334–15338 CrossRef CAS PubMed.
- G. A. Molander, K. M. Traister and B. T. O’Neill, J. Org. Chem., 2015, 80, 2907–2911 CrossRef CAS PubMed.
- L. K. G. Ackerman, L. L. Anka-Lufford, M. Naodovic and D. J. Weix, Chem. Sci., 2015, 6, 1115–1119 RSC.
- Y. Ye, H. Chen, J. L. Sessler and H. Gong, J. Am. Chem. Soc., 2019, 141, 820–824 CrossRef CAS PubMed.
- C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 11270–11273 CrossRef CAS PubMed.
- T. K. Allred, A. P. Dieskau, P. Zhao, G. L. Lackner and L. E. Overman, Angew. Chem., Int. Ed., 2020, 59, 6268–6272 CrossRef CAS PubMed.
- T. K. Allred, A. P. Dieskau, P. Zhao, G. L. Lackner and L. E. Overman, J. Org. Chem., 2020, 85, 15532–15551 CrossRef CAS PubMed.
- J. Wu, J. Bao, J. Deng, H. Tian and J. Gui, J. Am. Chem. Soc., 2025, 147, 30599–30605 CrossRef CAS PubMed.
- Y.-P. Song and N.-Y. Ji, Nat. Prod. Bioprospect., 2024, 14, 14 CrossRef CAS PubMed.
- J. Zhuo, C. Zhu, J. Wu, Z. Li and C. Li, J. Am. Chem. Soc., 2022, 144, 99–105 CrossRef CAS PubMed.
- O. Vyhivskyi and O. Baudoin, J. Am. Chem. Soc., 2024, 146, 11486–11492 CAS.
- M. Kosugi, K. Kurino, K. Takayama and T. Migita, J. Organomet. Chem., 1973, 56, C11–C13 CrossRef CAS.
- G. E. Keck and J. B. Yates, J. Am. Chem. Soc., 1982, 104, 5829–5831 CrossRef CAS.
- M. M. Heravi and A. Nazari, RSC Adv., 2022, 12, 9944–9994 RSC.
- D. J. Edmonds, D. Johnston and D. J. Procter, Chem. Rev., 2004, 104, 3371–3404 CrossRef CAS PubMed.
- K. C. Nicolaou, S. P. Ellery and J. S. Chen, Angew. Chem., Int. Ed., 2009, 48, 7140–7165 CrossRef CAS PubMed.
- T. Nakata, Chem. Soc. Rev., 2010, 39, 1955–1972 RSC.
- Y. Gao and D. Ma, Nat. Synth., 2022, 1, 275–288 CrossRef CAS.
- B.-C. Hong and R. R. Indurmuddam, Org. Biomol. Chem., 2024, 22, 3799–3842 RSC.
- S. Le, J. Li, J. Feng, Z. Zhang, Y. Bai, Z. Yuan and G. Zhu, Nat. Commun., 2022, 13, 4734 CrossRef CAS PubMed.
- A. G. Capacci, J. T. Malinowski, N. J. McAlpine, J. Kuhne and D. W. C. MacMillan, Nat. Chem., 2017, 9, 1073–1077 CrossRef CAS PubMed.
- S. Rendler and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 5027–5029 CrossRef CAS PubMed.
- P. V. Pham, K. Ashton and D. W. C. MacMillan, Chem. Sci., 2011, 2, 1470–1473 RSC.
- R. J. Comito, F. G. Finelli and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 9358–9361 CrossRef CAS PubMed.
- C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS.
- C. Lee, G. Kang, J. You, T. Kim, H.-S. Lee, Y. Park and S. Han, JACS Au, 2025, 5, 1096–1103 CrossRef CAS PubMed.
- S. Esposti, D. Dondi, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2007, 46, 2531–2534 CrossRef CAS.
- D. Gorbachev, E. Smith, S. P. Argent, G. N. Newton and H. W. Lam, Chem. – Eur. J., 2022, 28, e202201478 CrossRef CAS PubMed.
- W. Yao, Z. Liu, H. Ling, H. Wang, H. Zheng, S.-H. Wang, D.-Y. Zhu, S.-Y. Zhang and X. Chen, J. Am. Chem. Soc., 2025, 147, 15963–15969 CrossRef CAS PubMed.
- J. L. Hofstra, K. E. Poremba, A. M. Shimozono and S. E. Reisman, Angew. Chem., Int. Ed., 2019, 58, 14901–14905 CrossRef CAS.
- X. Zhang and D. W. C. MacMillan, J. Am. Chem. Soc., 2017, 139, 11353–11356 CrossRef CAS PubMed.
- S. Guo, J. Liu and D. Ma, Angew. Chem., Int. Ed., 2015, 54, 1298–1301 CrossRef CAS PubMed.
- Y. Zheng, L. Teng, T. Zhou, Z. Liu, K. Guo, H. Li, T. Li, L. Wang, Y. Liu and S. Li, Angew. Chem., Int. Ed., 2025, 64, e202421497 CrossRef CAS.
- B. Hong, W. Liu, J. Wang, J. Wu, Y. Kadonaga, P.-J. Cai, H.-X. Lou, Z.-X. Yu, H. Li and X. Lei, Chem, 2019, 5, 1671–1681 CAS.
- J. Yang, X. Zhang, F. Zhang, S. Wang, Y. Tu, Z. Li, X. Wang and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 8471–8475 CrossRef CAS PubMed.
- Y. Gao, Q. Xia, A. Zhu, W. Mao, Y. Mo, H. Ding and J. Xuan, J. Am. Chem. Soc., 2024, 146, 18230–18235 CrossRef CAS PubMed.
- W. J. Robbins, F. Kavanagh and A. Hervey, Proc. Natl. Acad. Sci. U. S. A., 1947, 33, 171–176 CrossRef CAS PubMed.
- Á. Péter, S. Agasti, O. Knowles, E. Pye and D. J. Procter, Chem. Soc. Rev., 2021, 50, 5349–5365 RSC.
- M. Szostak, M. Spain and D. J. Procter, Chem. Soc. Rev., 2013, 42, 9155–9183 RSC.
- M. Szostak, N. J. Fazakerley, D. Parmar and D. J. Procter, Chem. Rev., 2014, 114, 5959–6039 CrossRef CAS PubMed.
- X. Just-Baringo and D. J. Procter, Acc. Chem. Res., 2015, 48, 1263–1275 CrossRef CAS PubMed.
- M. P. Plesniak, H.-M. Huang and D. J. Procter, Nat. Rev. Chem., 2017, 1, 0077 CrossRef.
- H.-M. Huang, M. H. Garduño-Castro, C. Morrill and D. J. Procter, Chem. Soc. Rev., 2019, 48, 4626–4638 RSC.
- K. Takatori, S. Ota, K. Tendo, K. Matsunaga, K. Nagasawa, S. Watanabe, A. Kishida, H. Kogen and H. Nagaoka, Org. Lett., 2017, 19, 3763–3766 CrossRef CAS.
- L. Li, W. Liang, M. E. Rivera, Y.-C. Wang and M. Dai, J. Am. Chem. Soc., 2023, 145, 53–57 CrossRef CAS PubMed.
- J. R. Annand, A. R. Henderson, K. S. Cole, A. J. Maurais, J. Becerra, Y. Liu, E. Weerapana, A. N. Koehler, A. K. Mapp and C. S. Schindler, ACS Med. Chem. Lett., 2020, 11, 1913–1918 CrossRef CAS PubMed.
- T.-L. Zheng, C.-Y. Huo, W. Bao, X.-T. Xu, W.-H. Dai, F. Cheng, D.-S. Duan, L.-L. Yang, X.-M. Zhang, D.-Y. Zhu and S.-H. Wang, Org. Lett., 2023, 25, 7476–7480 CrossRef CAS PubMed.
- A. Eggert, K. T. Schuppe, H. L. S. Fuchs, M. Brönstrup and M. Kalesse, Org. Lett., 2024, 26, 2893–2896 CrossRef CAS PubMed.
- D. L. Golden, S.-E. Suh and S. S. Stahl, Nat. Rev. Chem., 2022, 6, 405–427 CrossRef CAS PubMed.
- H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS.
- L. Stateman, K. Nakafuku and D. Nagib, Synthesis, 2018, 50, 1569–1586 CrossRef CAS PubMed.
- L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 2056–2071 CrossRef CAS.
- Y. Wang, S. Dana, H. Long, Y. Xu, Y. Li, N. Kaplaneris and L. Ackermann, Chem. Rev., 2023, 123, 11269–11335 CrossRef CAS PubMed.
- M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC.
- C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179–7189 CrossRef CAS.
- M. Munda, S. Niyogi, K. Shaw, S. Kundu, R. Nandi and A. Bisai, Org. Biomol. Chem., 2022, 20, 727–748 RSC.
- S. Chakrabarty, Y. Wang, J. C. Perkins and A. R. H. Narayan, Chem. Soc. Rev., 2020, 49, 8137–8155 RSC.
- J. C. Lewis, P. S. Coelho and F. H. Arnold, Chem. Soc. Rev., 2011, 40, 2003–2021 RSC.
- F. Li, X. Zhang and H. Renata, Curr. Opin. Chem. Biol., 2019, 49, 25–32 CrossRef CAS PubMed.
- Y. Nakano, K. F. Biegasiewicz and T. K. Hyster, Curr. Opin. Chem. Biol., 2019, 49, 16–24 CrossRef CAS PubMed.
- I. Bakanas, R. F. Lusi, S. Wiesler, J. Hayward Cooke and R. Sarpong, Nat. Rev. Chem., 2023, 7, 783–799 CrossRef CAS PubMed.
- Y. Shen, L. Li, X. Xiao, S. Yang, Y. Hua, Y. Wang, Y. Zhang and Y. Zhang, J. Am. Chem. Soc., 2021, 143, 3256–3263 CrossRef CAS PubMed.
- J. F. Hoskin and E. J. Sorensen, J. Am. Chem. Soc., 2022, 144, 14042–14046 CrossRef CAS.
- B. Xu, Z. Zhang and M. Dai, J. Am. Chem. Soc., 2025, 147, 17592–17597 CrossRef CAS PubMed.
- G. Sennari, H. Yamagishi and R. Sarpong, J. Am. Chem. Soc., 2024, 146, 7850–7857 CrossRef CAS PubMed.
- M. Deng, F. Wu, T. Liu, Z. Jiang and T. Luo, J. Am. Chem. Soc., 2025, 147, 8132–8137 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |