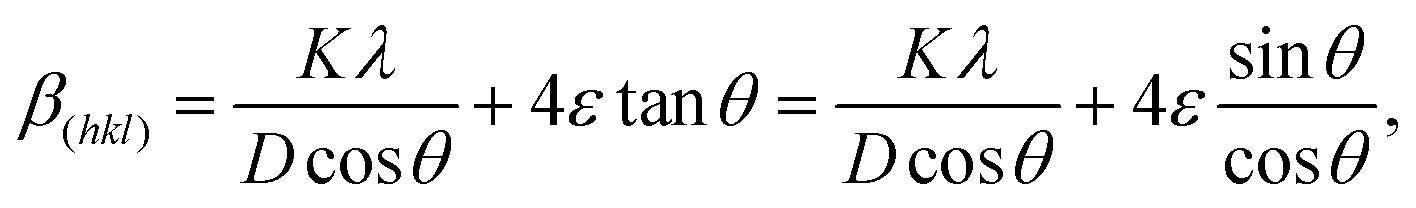DOI:
10.1039/D5CS00480B
(Review Article)
Chem. Soc. Rev., 2025,
54, 7870-7912
Lattice strain inhomogeneity in halide perovskite films: its origins and regulations
Received
1st May 2025
First published on 22nd July 2025
Abstract
Since perovskite solar cells reached an astonishing power conversion efficiency of 27%, the persistent stability issue has received significant attention, paving the way for a major step up in commercialization. Strain control has been proposed as a promising technology to address the stability issue because defects governing the structural stability are susceptible to lattice strain. Accordingly, various strategies have been developed and found to be effective in terms of device stability. Nevertheless, most of them are based on interface management by varying target molecules at the interface without understanding the underlying fundamentals. In this review article, we thoroughly investigate the origins of lattice strain in perovskite films, ranging from the atomic scale to the bulk scale, and introduced effective approaches to resolve the strain issue by listing relevant materials. Finally, the synergistic effect of strain regulation is discussed to recognize the feasible impact from the perspective of device performance.

Yun-Kyeong Hong
| Yun-Kyeong Hong is an integrated MS-PhD student under Prof. Hui-Seon Kim in the Department of Chemistry and Chemical Engineering, Inha University, Korea, where she also completed her undergraduate study in 2023. Her research focuses on perovskite solar cells (PSCs), with particular interest in strain engineering and the reliability assessment of perovskite solar modules. |

Yu-Na Lee
| Yu-Na Lee is an integrated MS-PhD student under the supervision of Prof. Hui-Seon Kim in the Department of Chemistry and Chemical Engineering, Inha University, Korea. She obtained her bachelor's degree from Gangneung-Wonju National University in 2023. Her research focuses on PSCs, particularly on controlling crystallographic orientation during crystal growth. |

Hui-Seon Kim
| Hui-Seon Kim is an Associate Professor at Inha University, Korea. Her research interest focus on energy materials and devices, including PSCs, since her first paper on solid-state PSCs in 2012. She received her BEng from the School of Chemical Engineering at Sungkyunkwan University (SKKU), Korea, in 2011. She then received her PhD from the Department of Energy Science at SKKU under the supervision of Prof. Nam-Gyu Park in 2016. She started her professional career as a post-doctoral researcher under Prof. Anders Hagfeldt at École Polytechnique Fédérale de Lausanne, Switzerland (2016–2019) and moved to the Department of Chemistry at Inha University as an Assistant Professor (2019–2023). |
1. Introduction
Since the development of all solid-state perovskite solar cells (PSCs) in 2012,1,2 a rapid transition has been observed in the research field of emerging photovoltaics.3,4 The certified power conversion efficiency (PCE) of PSCs recently reached 27.0% owing to intensive efforts from various perspectives, including compositional engineering, interface engineering and modulating crystal growth, along with in-depth fundamental understanding,5,6 which makes PSCs become one of the most competitive technologies in the photovoltaic market. In this regard, strong demand from industry is well reflected in a steep increase in the perovskite solar module efficiency, recently reaching a PCE of 21.1% over 843.5 cm2,7 by improving scalable technology for commercialization to reduce the gap between unit cell and module.8,9 The astonishing progress in the PCE of PSCs mainly relies on the intrinsic superb properties of the halide perovskite APbI3 (A+ = formamidinium (FA+), methylammonium (MA+), or Cs+),10 where its distinctive electronic band structure is favorable for photon absorption,11 low exciton binding energy,12 balanced long diffusion coefficient for electrons and holes,13,14 and suppressed charge recombination with high defect tolerance.15,16 Nevertheless, stability has been a chronic issue in PSCs, although significant progress has been certainly observed with satisfying stringent criteria for module test.17–19 Recently, the importance of residual lattice strain in the perovskite film has been highlighted not only because residual strain readily alters the optoelectronic properties by manipulating the electronic band structure20,21 but also because lattice stability is highly susceptible to residual strain.22 Despite controversial approaches to strain engineering,23,24 it is generally regarded that the in-plane tensile strain resides in the solution-processed halide perovskite film owing to a mismatch in thermal expansion coefficients between the soft perovskite film and the rigid inorganic substrate.25,26 As theoretically demonstrated, tensile strain is responsible for the increased bandgap and decreased hole mobility, while increased hole mobility is estimated under compressive strain.27,28 Therefore, compressive strain frequently induces favorable charge transport dynamics and thereby increases the PCE of PSCs.28,29 Furthermore, superb long-term stability of PSCs is generally enabled by alleviating in-plane tensile strain in the perovskite film.25 Similarly, many recent studies adopt strain engineering strategies toward achieving a strain-free film by fully relieving the in-plane tensile strain or even inducing in-plane compressive strain to benefit both photovoltaic performance and long-term stability.
Although structural inhomogeneity, being responsible for lattice strain, can be imposed by appreciable lattice tilting on an atomic scale,30,31 the local strain is further pronounced by the polycrystalline nature with randomly ordered crystals based on solution and low temperature process.27 In addition, advanced dopant engineering and annealing processes for crystallization lead to depth-dependent lattice strain in the perovskite film.32 Similarly, the lattice of halide perovskite unavoidably employs various multi-dimensional strains. Even though halide perovskite is widely known as soft lattice, which can adopt strains by lattice deformation,33 the lattice deformation modulates the band energy structure possibly in an undemanding way, and the accumulated strain can induce defects to release stress at a certain point,34 which is critical to performance reliability and structure stability in the long term. Therefore, understanding the origin and the effect of multi-dimensional lattice strain in perovskite film is extremely important in terms of deriving the maximum potential of optoelectronic properties as well as ensuring performance reliability and structural stability in various electronic devices. In this review paper, we discuss the origin of lattice strain from the nanoscale, spatially localized in a nanodomain, to the bulk scale (submicrometer). Characterization methods generally used to assess lattice strain is introduced, along with their validity and limitations. More importantly, we thoroughly investigate various strategies to regulate the residual lattice strain in bulk film by categorizing various approaches. This comprehensive review will not only provide an understanding of the origin-and-effect relationship but also provide a feasible insight to regulate the crystallization and design relevant interfaces based on the effective experimental strategies in terms of strain control. According to recent progress, FAPbI3 is of general interest as the composition of perovskite in this study although MAPbI3 and other compositions are also discussed in terms of fundamental strain studies.
2. Local strain on the atomic scale
It is apparent that FAPbI3 has taken the majority of the perovskite composition, particularly for PSCs with a single junction.22 FA+ cation plays a role in maintaining charge neutrality within the lattice by being located in the center of voids surrounded by eight PbI64− octahedral Conner-sharing cages.35,36 Moreover, FA+ stabilizes the lattice by nearly satisfying the ideal Goldschmidt tolerance factors (t = 1), where the geometrical steric effect is minimized.37 FA+ exhibits a larger size, compared to MA+,38 with hydrogen bonding sites at the opposite sides of the molecule,39 which contributes to a strong interaction with anionic inorganic frameworks.40 FAPbI3 is generally known as a cubic structure (α-phase with space group of Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) at room temperature with isotropic motion of FA+ freely rotating in the PbI64− inorganic network.41 As shown in Fig. 1(a), the high symmetry of a cubic structure undergoes phase transition to tetragonal (β-phase at 266 K) and orthorhombic (γ-phase at 153 K) with decreasing temperature,42 where the PbI64− octahedral tilting is enhanced, coupled with suppression of the dynamic motion of FA+.10 Cubic structure corresponds to a0a0a0 of Glazer notation, indicating high symmetry in terms of the average position of atoms. Although the high symmetry of a cubic structure would remind us of negligible strain in lattice based on neither octahedral rotation nor Pb2+ off-centering, appreciable PbI64− sublattice vibration and FA+ rotation is accompanied by thermal ellipsoids in a high frequency range (THz),30,31 opening up the local cage distortion and octahedral tilting in a high symmetry structure. It is further proposed that the lone pair orbitals of Pb2+ can encourage stereochemically off-center motions in the lattice,43,44 being distinguished from FA+ molecular dynamics mainly as the entropic thermal driving force.45,46 Such a static distribution of different low-symmetry local motif, enabled by different displacement of Pb2+, tilting angle, and rotation with respect to local octahedra in cubic structure, strongly indicates a polymorphous network,43 which is in accordance with extensive discussions that the experimentally emerging physical feature of the cubic structure reflects spatial-dependent inhomogeneity. However, macroscopically averaged (fictitious monomorphous) cubic unit cells are generally assumed to fit the X-ray diffraction pattern owing to a relatively long coherence length of X-ray diffraction44,47–49 although the property of the high symmetry (monomorphous) cubic structure would certainly differ from the properties of either the broken symmetry cubic structures (local structural motifs with low symmetry) or the macroscopically averaged polymorphous structures.43 To this end, space-resolved analysis techniques on the nanoscale are frequently used to examine the local structure. MAPbBr3 nanocrystals with a cubic structure, for example, exhibit 1–2 nm sized local domains with distinctly different orientations of MA+ from high-resolution transmission electron microscopy (HR-TEM).50 The local strain induced in the polymorphous network in a single formula unit can become more complicated in the mixed composition by doping. It is well known that an undesirable phase transition from cubic (α-phase) to non-perovskite hexagonal phase (δ-phase with space group of P63/mmc) occurs owing to the lower minimum in Gibbs free energy (G, ΔG = ΔH − TΔS, where H, T, and S denote enthalpy, temperature, and entropy, respectively) of δ-phase at ambient temperature (below 285 K51–350 K; as shown in bottom of Fig. 1(a)).52 The metastable α-phase can be kinetically trapped by an energy barrier (activation energy) between α- and -δ phases. Mixed-cation (MA+ and/or Cs+) and/or -anion (Br−) system with smaller radii compared to FA+ and I− is frequently adopted for FAPbI3 to enhance thermodynamic stability by lowering the minimum of G not only by increasing S based on an increased lattice disorder but also by decreasing H based on a unit cell contraction, which accordingly increases the energy barrier for α-to-δ transition and thus effectively retards the transition kinetics.10,53 Therefore, the doping strategy prevails in the FAPbI3 lattice,54,55 which inevitably alters the local lattice strain, as illustrated in Fig. 1(b) and (c). The breaking of local symmetry, mainly reflected in octahedral tilting, governs local strain. Larger amplitude octahedral tilts are generally responsible for larger strains in the crystal.56 Consequently, the local strain is inherent to the local chemical bonding environment along the inorganic framework, which enables the manipulation of the local electronic band structure and structural stability.
m) at room temperature with isotropic motion of FA+ freely rotating in the PbI64− inorganic network.41 As shown in Fig. 1(a), the high symmetry of a cubic structure undergoes phase transition to tetragonal (β-phase at 266 K) and orthorhombic (γ-phase at 153 K) with decreasing temperature,42 where the PbI64− octahedral tilting is enhanced, coupled with suppression of the dynamic motion of FA+.10 Cubic structure corresponds to a0a0a0 of Glazer notation, indicating high symmetry in terms of the average position of atoms. Although the high symmetry of a cubic structure would remind us of negligible strain in lattice based on neither octahedral rotation nor Pb2+ off-centering, appreciable PbI64− sublattice vibration and FA+ rotation is accompanied by thermal ellipsoids in a high frequency range (THz),30,31 opening up the local cage distortion and octahedral tilting in a high symmetry structure. It is further proposed that the lone pair orbitals of Pb2+ can encourage stereochemically off-center motions in the lattice,43,44 being distinguished from FA+ molecular dynamics mainly as the entropic thermal driving force.45,46 Such a static distribution of different low-symmetry local motif, enabled by different displacement of Pb2+, tilting angle, and rotation with respect to local octahedra in cubic structure, strongly indicates a polymorphous network,43 which is in accordance with extensive discussions that the experimentally emerging physical feature of the cubic structure reflects spatial-dependent inhomogeneity. However, macroscopically averaged (fictitious monomorphous) cubic unit cells are generally assumed to fit the X-ray diffraction pattern owing to a relatively long coherence length of X-ray diffraction44,47–49 although the property of the high symmetry (monomorphous) cubic structure would certainly differ from the properties of either the broken symmetry cubic structures (local structural motifs with low symmetry) or the macroscopically averaged polymorphous structures.43 To this end, space-resolved analysis techniques on the nanoscale are frequently used to examine the local structure. MAPbBr3 nanocrystals with a cubic structure, for example, exhibit 1–2 nm sized local domains with distinctly different orientations of MA+ from high-resolution transmission electron microscopy (HR-TEM).50 The local strain induced in the polymorphous network in a single formula unit can become more complicated in the mixed composition by doping. It is well known that an undesirable phase transition from cubic (α-phase) to non-perovskite hexagonal phase (δ-phase with space group of P63/mmc) occurs owing to the lower minimum in Gibbs free energy (G, ΔG = ΔH − TΔS, where H, T, and S denote enthalpy, temperature, and entropy, respectively) of δ-phase at ambient temperature (below 285 K51–350 K; as shown in bottom of Fig. 1(a)).52 The metastable α-phase can be kinetically trapped by an energy barrier (activation energy) between α- and -δ phases. Mixed-cation (MA+ and/or Cs+) and/or -anion (Br−) system with smaller radii compared to FA+ and I− is frequently adopted for FAPbI3 to enhance thermodynamic stability by lowering the minimum of G not only by increasing S based on an increased lattice disorder but also by decreasing H based on a unit cell contraction, which accordingly increases the energy barrier for α-to-δ transition and thus effectively retards the transition kinetics.10,53 Therefore, the doping strategy prevails in the FAPbI3 lattice,54,55 which inevitably alters the local lattice strain, as illustrated in Fig. 1(b) and (c). The breaking of local symmetry, mainly reflected in octahedral tilting, governs local strain. Larger amplitude octahedral tilts are generally responsible for larger strains in the crystal.56 Consequently, the local strain is inherent to the local chemical bonding environment along the inorganic framework, which enables the manipulation of the local electronic band structure and structural stability.
 |
| | Fig. 1 (a) Phase transition of FAPbI3 depending on temperature. Reproduced with permission from ref. 57. Copyright 2016, Wiley-VCH. (b) Schematic of local strain changes in FAPbI3 by employing MA+ and Br−. Reproduced with permission from ref. 58. Copyright 2021, American Chemical Society. (c) Schematic of local strain induced by incorporating methylenediammonium (MDA2+), Cs+, or MDA2+/Cs+ in FAPbI3 crystal. Reproduced with permission from ref. 59. Copyright 2020, American Association for the Advancement of Science. | |
3. Local strain on sub- and super-grain scales
Although the polymorphous network on the atomic scale would grant a macroscopically averaged structure, there still exists local strain on sub-grain and super-grain scales. Grains in perovskite film are usually identified by scanning electron microscopy (SEM) according to morphological criteria, which frequently and partially neglect crystallographic information. Nanodomains, according to structural criteria within a single grain, are specifically analyzed by the spatial crystal orientations or the spatial optoelectronic properties.60–63 Ultrasensitive electron backscatter diffraction (EBSD) is a useful technique for the acquisition of structural inhomogeneity by mapping the local crystal orientations of perovskite when minimizing the beam-induced damage.64Fig. 2(a) depicts the surface SEM image of MAPbI3 film, while Fig. 2(b) shows the inverse pole figure (IPF) map, obtained by EBSD, of the same region with the SEM image. The black arrow in the SEM image is aligned to cross a single grain boundary based on the morphological perspective (Fig. 2(a)), while the three distinct boundaries exist to separate four different crystal orientations according to the IPF map (Fig. 2(b)), where the separated domains are indexed by 1-4, matching the depicted orientations in Fig. 2(c). This implies that the so-called grains defined by SEM are likely to be composed of smaller domains in terms of structural orientation.60 Similarly, the IPF map depicted in Fig. 2(d) is converted to individual grains (Fig. 2(e)) determined by crystal orientation based on a threshold of 4° for crystal misorientation. The assigned grain is represented by a united color corresponding to its mean orientation. The color contrast with respect to grains accordingly implies the existence of appreciable local structural heterogeneity in the perovskite film. As depicted in Fig. 2(f), the grain orientation spread (GOS) is indicated by the average deviation in the crystal orientation between each point within a single grain and the mean orientation of the grain. The GOS ranges from 0° (perfectly ordered structure within a grain) to 4.3° (certain structural inhomogeneity within a single grain), strongly proposing a different degree of local strain residing in grain-to-grain. Therefore, when considering that the local variation in structural orientation determines local strain heterogeneity, a higher lattice strain is expected from the high GOS, while the minimized lattice strain is assumed on the grain scale from the 0° of the GOS.65
 |
| | Fig. 2 (a) SEM image and (b) corresponding IPF map of the MAPbI3 film generated by EBSD with a color code for facet orientation. (c) Depicted crystal orientation along the black arrows in (a) and (b). (d) Original IPF map, (e) conversion plot of grains identified from the IPF map with a threshold of 4° with an indication of mean orientation of the respective grains, and (f) conversion plot of GOS with an indication of average local misorientation. Scale bars in (a)–(f) represent 2 μm. Color cold for IPF mapping in (d)–(f) is the same as that in (b). Reproduced with permission from ref. 60. Copyright 2019, Cell Press. | |
The structural inhomogeneity at the sub-grain level is monitored within a single grain, defined by crystal orientation.60 Sub-grain boundaries, satisfying the equal threshold of 4°, exist as indicated by the solid line illustrated in Fig. 3(a). However, Fig. 3(b) depicts point-dependent misorientation with respect to the mean orientation of the corresponding grain, which demonstrates that a significant strain heterogeneity resides even in individual grains of MAPbI3 ranging from 0° to exceeding 6°. It is noted that the region closer to the grain boundary exhibits a higher misorientation angle, which indicates a higher degree of lattice deformation near the grain boundaries. Structural inhomogeneity becomes more exaggerated when estimating the misorientation angle between two extreme points in the same grain. Fig. 3(c) depicts the intra-grain misorientation angle by line mapping across the single grain (black arrow in the inset figure), where the misorientation angle reaches 9° between one end and the other end due to steadily varied crystal orientation. The local lattice imperfections induced by a sudden structural change in the ordering of the octahedral framework involve defects, which can act as non-radiative recombination centers,66 as evidenced by PL response,60 while the gradual strain change maintains lattice continuity with a spatially varied electronic band structure. Similarly, FAPbI3 exhibits atomic-level inhomogeneity in crystal orientation.67Fig. 3(d) shows a TEM image of FAPbI3, where the neighboring local regions indicated as 1-1, 1-2, and 1-3, corresponding to zoomed-in images in Fig. 3(e), represent the interplanar spacings of 0.29 nm along (210), 0.35 nm along (111), and 0.32 nm along (200), respectively. Furthermore, the local structure assigned as 1-4 presents 0.24 nm of interplanar spacing for (300) of PbI2, indicating a partial decomposition. The distinct variation in local crystal orientation, as illustrated in Fig. 3(f), consequently induces lattice strain and thus lowers the uniformity of lattice-level electronic properties.
 |
| | Fig. 3 (a) IPF of an individual grain (outer black line) in MAPbI3 with sub-grain boundaries (inner black lines). (b) Misorientation angle with respect to the mean orientation of a single grain. Scale bars in (a) and (b) represent 200 nm. (c) Plot of misorientation angle with respect to the first point (blue symbols) and its neighboring points (orange symbols) along the line mapping (the black arrow in the inset figure). Reproduced with permission from ref. 60. Copyright 2019, Cell Press. (d) HR-TEM image of FAPbI3 film. (e) High-magnification HR-TEM images of different spots (1-1 to 1-4 as indicated in (d)) of FAPbI3 film. The red lines and arrows in (e) indicate the (003) facet of PbI2 generated by perovskite decomposition. (f) Schematic of the crystalline film of FAPbI3. Reproduced with permission from ref. 61. Copyright 2025, Springer Nature. | |
In addition, structural coherency in super-grain is notably monitored from a single formula (almost phase-pure) of halide perovskite.62 Nano- and micro-XRD with corresponding spatial resolution are utilized to analyze residual strain stemming from the structural inhomogeneity in MAPbI3 polycrystalline film. Fig. 4(a) shows a quiver plot for the (110) orientation obtained by nanofocus XRD, where its azimuthal angle coordination is indicated by an arrow pointing direction with a corresponding color (inset color map) in each spatial diffraction spot. When considering the long-range cluster adjacent in both real and reciprocal space coordinates as super-grain, two super-grains are indicated with blue and red in Fig. 4(a). As expected from the earlier discussion on sub-grain strain inhomogeneity, local strain variation is similarly monitored within super-grains, as indicated by a structural inhomogeneity (q). Micro-XRD also reveals the long-range microstrain according to the Williamson–Hall equation, where complex local heterogeneity ranging from 0.1 to 0.2% is present (Fig. 4(b)). The different degrees of microstrain would grant different local strain environments along the octahedral framework, leading to an inhomogeneous photoluminescence response. Furthermore, the largest structural broadening by increasing local q (decreased interplanar spacing) from the correlation between microstrain and local q in Fig. 4(c) could imply compressive strain along the designated plane. Hence, a dimension of domain, which comprises a relatively uniform lattice configuration without a sudden breaking, can be widely varied from sub-grain level to super-grain level though the marginal local structural inhomogeneity is consistently inherent in the domain with different degrees.
 |
| | Fig. 4 (a) Quiver plots generated by nano-XRD measurements of the MAPbI3 film demonstrate two large super-grains along the (110) direction, highlighted in blue (left) and red (right), with local variations in the scattering vector q extracted from the respective super-grains. (b) Microstrain map for the (220) diffraction peak in the MAPbI3 film. (c) Histogram of the microstrain as a function of the scattering vector q for the (220) diffraction peak. The solid red line indicates a linear regression fit to a scatter plot of the data, confirming the correlation. Reproduced with permission from ref. 62. Copyright 2019, Wiley-VCH. | |
Structural coherency is also governed by crystallographic orientation. The atomic-scale high-resolution Cryo-TEM image depicted in Fig. 5(a) reveals a well-aligned grain boundary between (211)- and (001)-oriented grains,68 indicating a minimal accumulation of local strain at the (211)/(001) interface.69 The corresponding fast Fourier transform (FFT) analysis in Fig. 5(b) further confirms the coherency of this grain boundary. In contrast, the convergence of (001)-oriented grains tends to form incoherent grain boundaries, as shown in Fig. 5(c) and (d), with dangling bonds, dislocations, and lattice mismatch, which are responsible for interfacial local strain.70 Notably, the application of high Miller-index (211) facets to micron-thick FA-based perovskite films is effective in reducing dislocation density and local strain,68 which highlights the importance of crystallographic orientation at grain boundaries in regulating interfacial lattice strain and in encouraging the structural coherency between adjacent grains toward super-grain.
 |
| | Fig. 5 (a) Atomic-scale Cryo-TEM image of a coherent grain boundary, indicated in light orange, at the interface between (211)- and (001)-oriented perovskite grains. (b) FFT patterns corresponding to the coherent grain boundary along the (211) and (001) zone axes. (c) Cryo-TEM image of regular incoherent grain boundaries, indicated in light orange, and (d) the corresponding FFT patterns between three (001) perovskite grains. Reproduced with permission from ref. 68. Copyright 2025, Springer Nature. | |
4. Out-of-plane strain inhomogeneity across bulk film
4.1. Doping (mixed-composition effect)-induced strain inhomogeneity
Based on morphological identification, almost no horizontal grain boundaries in parallel with the substrate are evidenced by the cross-sectional SEM images of state-of-the-art high-performing perovskite films,71,72 indicating a single grain along the out-of-plane direction, while vertical grain boundaries separate grains along the in-plane direction from the cross-sectional view. Even though the cubic structure underlies a negligible difference between out-of-plane and in-plane directions, a distinctive structural inhomogeneity along the out-of-plane is observed within a single grain (sub-grain level), particularly in the mixed-composition system with a doping strategy to stabilize the α-phase of FAPbI3 or to achieve the wide bandgap material based on the mixed halide system (APbI3−xBrx).73,74 As depicted in Fig. 6(a) a perovskite film with mixed composition of FA0.85MA0.15Pb(I0.85Br0.15)3 represents a gradient of (004) interplanar distance along the vertical direction on sub-grain level by decreasing from 1.67 Å (local region III near bottom) to 1.64 Å (local region II at middle) and 1.60 Å (local region I near top surface), which is attributed to the depth-dependent gradient distribution of MA+, as evidenced by time-of-flight secondary ion mass spectrometry depth profiles,27 probably owing to the difference in coordination strength between FA+ and MA+ during the film growth. Similarly, FA1−xCsxPbI3 shows the out-of-plane cation inhomogeneity within a single grain, as illustrated in Fig. 6(b).74 Cs+-rich region is frequently observed near the bottom, resulting in 3.11 Å for (200) interplanar spacing, which is gradually decreased toward the top surface, leading to FA-rich composition with an enlarged (200) interplanar spacing of 3.20 Å (Fig. 6(c)). Although Cs+ is frequently incorporated to improve the phase stability of α-FAPbI3,75 the rapid crystallization in the presence of Cs+ inevitably results in an inhomogeneous distribution across the film.74 To this end, the incorporation of Rb+ is found to be highly effective on more uniform distribution across the film with a mixed-composition system based on quadruple cations (FA+, MA+, Cs+, and Rb+) and triple halides (Cl−, Br−, and I−). The improved distribution of Cs+ and the suppressed halide segregation are evidenced by employing Rb+,76,77 as confirmed by the relatively depth-independent halide distribution depicted in Fig. 6(d) and (e).76 The incorporation of Rb+ promotes more homogeneous distribution of Cs+ and halides throughout the perovskite film, which is definitely favorable for mitigating the lattice strain inhomogeneity along the out-of-plane direction, as evidenced by GIXRD results. Although an apparent peak shift in 2θ is observed in the absence of Rb+ from 28.66° to 28.56° with increasing ω from 0.3° to 4.0° (Fig. 6(f)), Rb+ induces a negligible shift in 2θ (Fig. 6(g)), indicating the enhanced lattice strain homogeneity along the out-of-plane direction.76 Similarly, the sub-grain level inhomogeneity of local strain in the doped-perovskite film should be carefully assessed by differentiating out-of-plane from in-plane owing to a possible gradient for cation and/or anion distribution along the out-of-plane direction.
 |
| | Fig. 6 (a) Cross-sectional TEM image and depth-dependent nano-beam electron diffraction patterns of the FA0.85MA0.15Pb(I0.85Br0.15)3-based device. Reproduced with permission from ref. 27. Copyright 2019, Springer Nature. (b) Illustration of the inhomogeneous phase distribution along the out-of-plane of the FA1−xCsxPbI3 film. (c) Cross-sectional high-angle annular dark-field TEM images (200 nm for the scale bar) and depth-dependent HR TEM images (7.3 × 7.3 nm) of FA1−xCsxPbI3. Reproduced with permission from ref. 74. Copyright 2023, Springer Nature. Depth profile XPS results of oxygen and halides in perovskite film (d) without and (e) with Rb+. GIXRD patterns with varying ω for the perovskite film (f) without and (g) with Rb+. Reproduced with permission from ref. 76. Copyright 2025, American Association for the Advancement of Science. | |
4.2. Processing (thermal effect)-induced strain inhomogeneity
The perovskite film is generally grown based on a solution process.78 Therefore, crystal growth is inevitably governed by solvent evaporation during the annealing process, where an anisotropic solvent evaporation and a difference in thermal expansion coefficient between rigid substrates and soft halide perovskite materials are considered the main reasons for strain inhomogeneity along the out-of-plane direction during the process.37,58,79 More specifically, the annealing process for the perovskite crystallization is frequently performed using a simple hot plate, which makes it difficult to guarantee a negligible temperature gradient across the loaded precursor between the bottom and the open top. A difficulty in controlling uniform solvent evaporation could induce different crystallization kinetics between the surface and bulk region, which imposes even more complexity when employing additives in the precursor solution. During the annealing process, the PbI64− inorganic framework near the bottom is prone to have a more expanded structure, while the lattice near the top surface has a relatively contracted volume owing to the temperature gradient, which increases the chance of larger cations being employed in the bottom.27 This can further complicate the aforementioned compositional inhomogeneity along the out-of-plane direction. Most importantly, a substantial difference in thermal coefficient by an order of magnitude (∼10−5 K−1 for rigid metal oxide substrates and ∼10−4 K−1 for soft organic–inorganic halide perovskite) consequently involves a substantial difference in lattice contraction when cooling from the higher annealing temperature for crystallization.25,80,81 During the crystallization at higher temperature (ca. 100–150 °C), the perovskite lattice is anchored to the underlying substrate, while the strong adhesion between the perovskite and the substrate constrains the perovskite from contracting during cooling to room temperature. The severe constraints of lattice contraction introduce the in-plane biaxial tensile strain, where the most pronounced in-plane tensile strain resides in the bottom region of the perovskite film, while the relatively reduced in-plane tensile strain is applied near the open top, being far away from the substrate.27 The different degrees of in-plane tensile strain along the out-of-plane direction leads to a vertical gradient in in-plane tensile strain, which is responsible for the strain inhomogeneity along the out-of-plane direction.25,27,78 It is noticeable that the out-of-plane strain inhomogeneity ranges across the whole bulk film, which is a collective strain on a bulk scale unlike various anisotropic local strains on the nanoscale.
5. Strain inhomogeneity by external stress
The lattice strain in perovskite films is also affected by external environmental factors, such as illumination, moisture, and aging effects. Under illumination, lattice expansion generally occurs in halide perovskite film, where the lattice strain is accordingly alleviated in the expanded unit cell.82 Nevertheless, the effect of halide phase segregation under illumination can further complicate the local lattice strain. The interaction between phonon and charged entities (usually photogenerated free carriers) induces the local strain by forming a polaron-to-screen charge in the lattice.83 The polaronic strain can markedly initiate the phase segregation for mixed I− and Br−, which can be rather restrained by composing a uniform and homogeneous distribution of local polaronic strain under high carrier density.84 The structural instability under illumination is suppressed in terms of a mechanical perspective. A graphene–polymer complex, coordinating on the perovskite surface, can mechanically limit structural deformation and thus effectively mitigate lattice distortion.85 In addition, halide perovskite films are forced to experience periodic strain alteration during diurnal cycles. Although the lattice strain is mitigated under continuous illumination, the lattice contraction under dark conditions induces a structural reversion to a more asymmetric structure with reinforced lattice strain.80,82,86 The repetitive strain dynamics can afford to facilitate the formation of deep trap states and potentially accelerate chemical degradation.87 The incorporation of additives, which can coordinate with Pb2+ on the surface, enables the interfacial anchoring at grain boundaries and effectively suppresses the lattice volume fluctuations.88 Furthermore, the decomposition of the perovskite film is frequently initiated by moisture ingress through the grain boundaries, which act as diffusion pathways for water molecules to penetrate the bulk of the film.89,90 The moisture subsequently propagates laterally along the crystal planes and leads to a local misorientation and the generation of localized lattice strain,60,89 suggesting a coupled relationship between lattice strain and moisture-induced degradation. Besides, the aging process of perovskite films often results in changes in the lattice strain.91 The addition of MACl to FAPbI3 films is generally adopted to ensure high film quality of α-FAPbI3, where the slow removal of Cl− from the perovskite film during the so-called ‘aging process’ is responsible for lattice strain relaxation, leading to an improved PCE after aging.91
6. Characterization of out-of-plane strain inhomogeneity across bulk films
Although the local strain and its inhomogeneity are generally assessed by various local mapping analysis techniques with high resolution focusing on the target region,92,93 strain measurements in terms of the collective bulk film scale have been most widely performed.94 It is meaningful because the collective strain across the whole bulk film can be more intuitively and directly linked to the device performance. The grazing incidence X-ray diffraction (GIXRD) can reflect the information of in-plane lattice distance, which is suitable for assessing the depth-dependent in-plane strain, while the micro strain, usually estimated by normal XRD, can indicate the deviation extent of interplanar spacing, assuming that the local structural randomness contributes to the strain inhomogeneity along the out-of-plane direction.95 GIXRD is the most routinely adopted method for assessing lattice strain because depth-dependent in-plane strain distinctly prevails in solution-processed halide perovskite films,96,97 which is often complemented by HR-TEM to evidence the real lattice distances depending on the depth from the cross-sectional device.98
6.1. GIXRD
Normal XRD can selectively recognize the crystal planes in parallel with the sample surface, implying the limited acquisition of crystal information on the planes perpendicular to the sample surface. Therefore, the structural in-plane information, in parallel with the sample surface (i.e., interplanar spacing along the in-plane direction), is effectively restricted in the normal XRD pattern. However, GIXRD enables the in-plane structural information to be reflected, coupled with an out-of-plane response.
6.1.1. Penetration depth.
Fig. 7 illustrates the geometric relationship between the X-ray beam and the sample with defined angles in normal XRD and GIXRD. Although the normal XRD is based on either θ–θ (left side of Fig. 7(a)) or θ–2θ (right side of Fig. 7(a)) scan modes, the GIXRD pattern is obtained by moving a detector to sweep 2θ with a fixed incidence angle (αi) of the X-ray beam (Fig. 7(b)).99 The penetration depth (z) of the X-ray beam from the surface is then determined by αi, where a small αi allows the shallow depth near the surface is then determined by XRD response while excluding the remaining deeper region below z,100 as illustrated in Fig. 7(c).
 |
| | Fig. 7 (a) Basic principle of θ/θ (left) and θ/2θ (right) goniometers of the normal XRD. Reproduced with permission from ref. 101. Copyright 2016, Elsevier. (b) Illustration of XRD measurement in grazing incidence geometry (GIXRD). Reproduced with permission from ref. 99. Copyright 2016, Royal Society of Chemistry. (c) Illustration of αi-dependent z (α1 < α2 < α3 < α4 and z1 < z2 < z3 < z4) in GIXRD characterization. Reproduced with permission from ref. 100. Copyright 2023, Springer Nature. | |
The diffracted intensity (Id) of the X-ray beam can be driven by the attenuation of the incidence intensity (I0) according to eqn (1), where μ is the attenuation coefficient (or linear absorption coefficient) of the material, L1 is the path length (L1 = z/sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) αi), L2 is the path length at the diffracted side (L2 = z/sin(2θ − αi)), and 2θ is the Bragg angle.99
αi), L2 is the path length at the diffracted side (L2 = z/sin(2θ − αi)), and 2θ is the Bragg angle.99
| |  | (1) |
The z is defined as a specific depth at which Id is reduced by e−1 (∼37%) from the original intensity, I0, under given parameters of μ by material (μ = ∼103 cm−1 for FAPbI3 with 1.54 Å of Cu Kα)102–104 and αi by setting input.105 In other words, z is a function of αi, as depicted in eqn (2), confirming a shallow z as αi decreases.99
| |  | (2) |
6.1.2. Residual in-plane lattice strain measurements.
Polycrystalline material, such as halide perovskite, consists of (hkl) planes oriented exactly parallel to the substrate and oriented in various directions. GIXRD enables the interplanar spacing (d) of randomly oriented (hkl) planes in polycrystalline thin films to be systematically measured by modulating the tilt angle, Ψ, between the crystalline plane normal (L3-axis; vertical to the (hkl) crystalline) and the surface normal (S3-axis; vertical to the film surface), while maintaining a small αi to ensure a limited z, as shown in Fig. 8(a) and (b).106 From the perspective of residual lattice strain, d of a specific (hkl) plane in a strained film can be simply compared with a reference d under the strain-free condition (d0). The acquisition of the d of the (hkl) plane at a given Ψ (dΨ) estimates the strain (ε) by a comparison with d0 according to eqn (3):107| |  | (3) |
 |
| | Fig. 8 (a) Iso-inclination method tilting the sample (tilting angle = Ψ) in the same direction with αi (χ = 0) along the axis for 2θ and αi variation. (b) Side-inclination method tilting the sample (tilting angle = Ψ = χ) along the axis perpendicular to the sample normal direction (S3) and diffraction vector (L3). Reproduced with permission from ref. 106. Copyright 2005, International Union of Crystallography. (c) Typical d–ω plot achieved from iso-inclination method-based GIXRD patterns with linear fittings. Reproduced with permission from ref. 108. Copyright 2025, American Chemical Society. (d) Typical 2θ–sin2Ψ plot achieved using side-inclination method-based GIXRD patterns with linear fittings. Reproduced with permission from ref. 110. Copyright 2024, Wiley-VCH. | |
There are two ways to make a difference in Ψ according to the tilting direction (either iso- or side-inclinations), which consequently vary z.106 As depicted in Fig. 8(a), the iso-inclination method applies the same direction of αi to Ψ, leading to the actual incidence angle (ω) as the sum of αi and Ψ (ω = αi + Ψ).106 Since ω becomes larger with increasing Ψ despite the fixed αi, a difference occurring in z makes the iso-inclination method suitable for measuring z-dependent ε. When iso-inclination is utilized for GIXRD measurements to analyze the residual strain of perovskite thin films, the plot is generally represented in d–ω, as shown in Fig. 8(c). As depicted in Fig. 8(c), the d of the perovskite film (particularly for the control sample with a large slope) expands as ω increases from 0.8° to 2.4°, implying that the d increases as it crosses the bulk perovskite film from the top surface to the bottom interface.108 The positive slope of the d–ω plot, therefore, indicates more pronounced tensile strain near the bottom interface compared to the top surface,94,108 which is in accordance with the strain tendency of the aforementioned processing-induced in-plane strain inhomogeneity along the out-of-plane direction. However, the marginal slope of the target film, which is close to zero, indicates a negligible depth-dependent tensile strain across the film. Fig. 7(b) illustrates the side-inclination method, where the Ψ is equivalent to the tilting of the Chi angle (χ).106 As χ increases, the X-ray beam enters the sample at a steeper inclination and accordingly increases its path length within the material.27 Therefore, a slight change in z is observed as χ varies.21,27 The GIXRD measurement with the side-inclination method can assume the residual strain (σ) according to eqn (4),109 suggesting a 2θ–sin2Ψ plot as a useful form to assume the strain, as shown in Fig. 8(d).
| |  | (4) |
where
E is the Young's modulus,
ν is the Poisson's ratio, and
θ0 is the diffraction peak position under the strain-free condition. Therefore, the slope of the 2
θ–sin
2Ψ plot implies the strain type with a negative slope (
σ > 0) for tensile strain and a positive slope (
σ < 0) for compressive strain within the film.
Fig. 8(d) is obtained by sweeping
Ψ from 15° to 60° by fixing
αi as 0.2°,
110 showing a decrease in 2
θ with increasing
Ψ (sin
2Ψ).
110 Since
θ is inversely proportional to
d according to Bragg's law and
Ψ is proportional to
z, the negative slopes imply enlarged
d near the bottom interface of the perovskite film (with deep penetration of the X-ray beam) compared to the top surface. This is further in accordance with the residual in-plane tensile strain, which originates from the routine annealing process of perovskite films. As depicted in
Fig. 8(d), the target film indicated in orange exhibits a relatively flat negative slope compared to that of the control film shown in blue, implying the released in-plane tensile strain with strain inhomogeneity to a lesser extent along the out-of-plane direction.
Many studies have exclusively relied on the GIXRD characterization to evaluate the residual lattice strain in halide perovskite films because the processing-induced strain generally originates from the interface and accordingly leads to the depth-dependent in-plane tensile strain, exhibiting a difference along the out-of-plane direction across the film. When considering that the variation in ω or χ enables the crystal information within different depths to be probed,101,111 it seems suitable for this purpose. However, one should be careful in interpreting GIXRD patterns owing to the acquisition of (hkl) along different orientations by varying ω or χ. More specifically, the response of a particular orientation, satisfying ω (αi), among randomly oriented (hkl) planes in a polycrystalline film is often referred to as ‘in-plane’ to indicate depth-dependency.27,102,112 Therefore, the assessment of strain inhomogeneity along the ‘out-of-plane’ direction by GIXRD is difficult, as a sample resembles a perfectly ordered single crystal.
6.2. Microstrain
Fig. 9(a) illustrates the change in the XRD peak depending on the lattice strain.104 Under the strain-free condition (no strain), the Bragg peak appears at 2θ0, corresponding to d0. In the second row, a uniform strain changes d, which results in a peak shift to satisfy Bragg's law (θ < θ0 for tensile strain and θ > θ0 for compressive strain).113 However, nonuniform strain induces multiple peaks from different d, where the overlap of multiple peaks at close 2θ generates a broad peak.79 The peak width is quantified using the full width at half maximum (FWHM, β), where not only lattice micro strain (ε) but also crystallite size (D) and the instrument effect serve as parameters determining β.114 Therefore, the adjusted FWHM (β(hkl)) can be estimated by removing the instrument effect, making β(hkl) only dependent on the sample (β(hkl) = βcrystallitesize + βstrain).115,116 Williamson–Hall method allows us to assess the effects of D and ε on β(hkl) using D = Kλ/(βcrystallitesizecosθ) and βstrain = 4εtanθ,114,117 where K is the Scherrer constant of the material (0.89 for cubic structure118) and λ is the wavelength of the X-ray beam source (1.54 Å for Cu Kα).104 Therefore, the plot of βcosθ–4sinθ is a useful form to assume ε, as illustrated in eqn (5) and (6), where the intercept is denoted by Kλ/D and the slope is denoted by ε. It is noted that ε values with positive and negative signs are reported for perovskite films, as shown in Fig. 9(b) and (c). The interpretation of the meaning of the sign is still controversial.104,114,119 Some authors suggest the dependence of the sign on the strain type (tensile/compressive),104,114 while others argue that a negative slope is physically meaningless.119 In addition to the ambiguity in interpreting the sign of strain, the microstrain is incapable of providing depth-dependent strain information along the out-of-plane direction.| |  | (5) |
| |  | (6) |
 |
| | Fig. 9 (a) Schematic of the correlation between d in the crystal lattice and the resultant pattern of the diffraction peak. Reproduced with permission from ref. 104. Copyright 2011, Elsevier. (b) βcosθ–4sinθ plot based on the Williamson–Hall method with ε obtained from the slope of the linear fitting. Reproduced with permission from ref. 120. Copyright 2024, Wiley-VCH. (c) βcosθ–4sinθ plot based on the Williamson–Hall method with ε as a percentage of strain. Reproduced with permission from ref. 121. Copyright 2024, Elsevier. | |
6.3. HR-TEM
In strain-related studies, TEM is frequently employed to directly verify d values in local spots.122,123 Specifically, cross-sectional HR-TEM imaging, combined with microscopic diffraction pattern analysis, enables the direct investigation of structural variations at different depths within the film, facilitating strain analysis along the out-of-plane direction.27,98,124 The acquisitions of d as a function of depth via cross-section HR-TEM analysis successfully complement the GIXRD analysis. Fig. 10(a) and (b) demonstrate the direct evidence of structural changes by calculating d values from TEM analysis. Fig. 10(a) illustrates three different spots across the perovskite bulk film, d values corresponding to (200) exhibit a gradual difference with d = 3.07 Å near the top surface (spot II), d = 3.11 Å in the middle (spot V), and d = 3.12 Å near the bottom interface (spot VIII),98 implying the strain inhomogeneity across the bulk film. However, Fig. 10(b) demonstrates consistent d values (d = 3.19 Å) for the (200) planes regardless of the depth, confirming a negligible strain variation.98 The depth-dependent TEM results are consistent with the corresponding GIXRD data presented in Fig. 10(c) and (d). As depicted in Fig. 10(c), the decrease in the Bragg angle 2θ with increasing Ψ results from increasing d at high z (toward bottom interface), being in accordance with d from TEM results (Fig. 10(a)), while the invariant 2θ in Fig. 10(d) confirms the consistent d values from TEM analysis (Fig. 10(b)).98 Cross-sectional TEM necessitates a sample preparation process using focused ion beam (FIB) milling, which typically utilizes high-energy ions (Ga+ ions) to trim the perovskite bulk film into lamella, followed by a cleaning process with low beam power to minimize beam-induced damage.125 It is revealed that conventional accelerating voltages (16–30 kV) and currents (>2.5 nA), which are commonly used for sampling metallic materials, can induce amorphization, chemical decomposition, and structural defects in organic samples.126,127 Although some studies have reported that the structural damage to perovskite materials is inevitable during FIB milling process, other studies suggest that the damage by FIB milling can be sufficiently avoided by controlling the milling condition. A negligible difference was evidenced from a comparison of TEM analysis results between the perovskite sample directly deposited onto the TEM grid and the FIB-milled perovskite sample by 30 kV Ga+ ion beam with a current of 28 nA to cut 0.5 μm-thick film from a bulk sample and 30 kV with 2.8 nA for further thinning the film.125 Therefore, a careful modulation of the beam power for the FIB milling can induce negligible damage to perovskite, confirming the reliability of the TEM results for analyzing the residual strain in the perovskite film. Despite the strong merit of TEM analysis in terms of strain evaluation, which can directly monitor d, TEM is basically a spot-dependent analysis,128 which demands statistical data analysis by measuring multiple spots to derive a reliable conclusion, particularly in halide perovskite film, as it is a polycrystalline material with a varied local structure. Furthermore, it is debatable that the validity of constant crystal structure (e.g., cubic for FAPbI3) despite the strain inhomogeneity along the out-of-plane direction because the residual in-plane tensile strain ideally induces the lattice contraction along the out-of-plane direction.129 More systematic in-depth study of the lattice distortion on the atomic scale will be significantly helpful for understanding the multiscale strain impact and thus interpreting depth-dependent TEM results.
 |
| | Fig. 10 Cross-sectional TEM images and HR-TEM analysis for obtaining the depth-dependent d of (a) pristine perovskite film and (b) target film with interface-formamidine active addition reaction (i-FAAR). GIXRD patterns of the (c) pristine and (d) i-FAAR-based perovskite films. GIXRD measurements are based on the side-inclination method with a probing depth of 50 nm determined by αi = 0.3°–0.4°. Reproduced with permission from ref. 98. Copyright 2024, Wiley-VCH. | |
7. Experimental strategies to control out-of-plane strain inhomogeneity
7.1. Strain control by regulating crystallization
7.1.1. Additive strategy.
Appreciable lattice strain with local structural inhomogeneity on sub- and super-grain levels is commonly and widely observed in halide perovskite films, which is apparently unfavorable for device applications in terms of both performance and reliability. It is regarded that less strain with less octahedral tilting increases the contribution of covalency nature to Pb–I bonding in the inorganic core framework by intensifying the orbital overlapping,37,56 which implies that the local strain with different degrees of octahedral tilting leads to different optoelectronic properties. The grain boundaries with a sudden lattice discontinuity are assumed to act similarly as boundaries for the electronic band structure.62 Therefore, the local strain, originating from the local structural inhomogeneity, can afford to induce spatial inhomogeneity in the energy band structure, increasing the sensitivity of material properties.27 Lattice matching between intra-grains as well as reducing the structural inhomogeneity on a sub-grain scale are highly important in terms of mitigating anisotropic behavior, including charge transport, recombination and stability. To this end, the formation of a uniform crystal orientation has been an experimental pursuit by lattice matching and strain engineering in the perovskite film. The additive strategy has been widely used to enhance the crystal orientation of the FAPbI3 film by utilizing thiocyanate (SCN−), formate (HCOO−), or acetate (CH3COO−),130–132 which is generally coupled with MACl or long alkyl chain-based chloride to form Cl/I hybrid octahedra,54,133 unavoidably leaving a small amount of MA, which can occupy the FAPbI3 lattice as a byproduct in the crystallized film,133 and mostly repels large components, being incapable of filling the lattice point due to a significant difference in ionic radii, to the grain boundaries. The additives play a synergistic role in regulating crystal growth for tuning lattice strain and in passivating defects at the interface.134,135 The additive engineering is also useful in resolving the inhomogeneous distribution of mixed cations along the out-of-plane composition, which otherwise induces detrimental effects by cation segregation.136,137
Small molecule-induced strain control.
FA1−xCsxPbI3 films tend to induce vertical inhomogeneity in composition between FA and Cs, as illustrated in Fig. 5(b) and (c). An organic molecule with a sulfone group (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO), such as 1-(phenylsulfonyl) pyrrole (PSP), is proposed to retard the segregation of cations in FA1−xCsxPbI3 films.74 A preferential crystallization with Cs+, possibly due to its soft base property or different solubility,138 can be kinetically hindered by accelerating crystallization rate with lowered energy barriers based on the devised interaction between the sulfone group of additive and Pb atoms in precursor solution (Fig. 11(a) and (b)). A negligible variation in interplanar distances along the out-of-plane direction (d(002) = 3.13 Å at the bottom, bulk, and open top) is depicted in Fig. 11(c), indicating a homogeneous distribution between FA and Cs with different ionic radii. The variation in normalized intensity over 3 nm illustrated in Fig. 11(d) confirms a consistent interplanar distance without depth dependency.
SO), such as 1-(phenylsulfonyl) pyrrole (PSP), is proposed to retard the segregation of cations in FA1−xCsxPbI3 films.74 A preferential crystallization with Cs+, possibly due to its soft base property or different solubility,138 can be kinetically hindered by accelerating crystallization rate with lowered energy barriers based on the devised interaction between the sulfone group of additive and Pb atoms in precursor solution (Fig. 11(a) and (b)). A negligible variation in interplanar distances along the out-of-plane direction (d(002) = 3.13 Å at the bottom, bulk, and open top) is depicted in Fig. 11(c), indicating a homogeneous distribution between FA and Cs with different ionic radii. The variation in normalized intensity over 3 nm illustrated in Fig. 11(d) confirms a consistent interplanar distance without depth dependency.
 |
| | Fig. 11 Schematics of computational results of free energy as a function of the reaction coordinate for (a) control and (b) target conditions with PSP. (c) Cross-sectional high-angle annular dark-field TEM images (200 nm for the scale bar) and depth-dependent HR TEM images (7.3 × 7.3 nm) of the crystal orientation-regulated FA1−xCsxPbI3 film. (d) Calculated intensity over 3 nm of the crystal orientation-regulated FA1−xCsxPbI3 film depending on the depth. Color code corresponds to surface, bulk, and bottom indicated in (c). Reproduced with permission from ref. 74. Copyright 2023, Springer Nature. | |
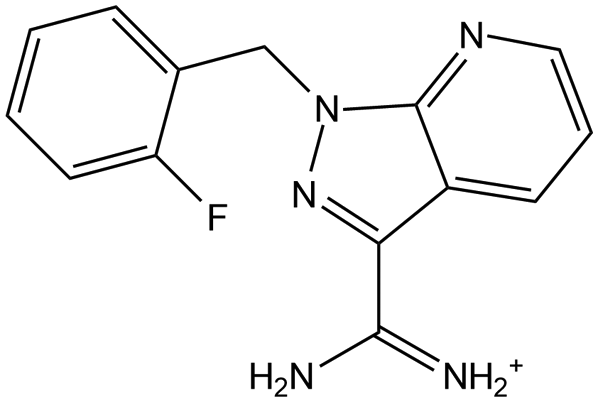
Furthermore, the gradient strain along the out-of-plane direction can be effectively alleviated by formulating the local structures with different orientations. A strong chemical coordination effect of the additive, 1-(2-fluorobenzyl)-1H-pyrazolo[3,4-b]pyridine-3-carboximidamide hydrochloride, to the inorganic framework results in a preferred interaction, particularly with the (111) facet and consequently lowers the surface energy of (111), which serves as a driving force for the (111)-oriented crystal growth on the surface.108Fig. 12(a)–(c) illustrates that the gradient in-plane tensile strain of the control film is mostly released by facet complementarity between (100) and (111), as illustrated in Fig. 12(d). Notably, the local regions with distinct crystal orientation are intentionally adopted to suppress the out-of-plane strain inhomogeneity on a relatively macro scale (bulk film), even though the microstrain is rather pronounced by aggravating crystal orientational mismatch at grain boundaries.108,139 When considering that the superior performance in both PCE and long-term stability is monitored from the target film with facet complementarity, the strategy for preferentially relieving the vertically gradient collective strain at the cost of bearing local strain at grain boundaries is effective. In other words, the collective strain inhomogeneity along the out-of-plane direction triggers a more detrimental impact than local structural mismatch mostly owing to its larger extent of effective strain range across the whole film, proposing that the depth-dependent biaxial in-plane strain, which is responsible for the out-of-plane strain inhomogeneity, should be resolved with a high priority in perovskite films.
 |
| | Fig. 12 Depth-dependent GIXRD patterns with varying ω of (a) control and (b) target perovskite films. (c) d(211)–ω plots converted from the GIXRD patterns. (d) Schematic of lattice orientation in the control (left) and target (right) films. Reproduced with permission from ref. 108. Copyright 2025, American Chemical Society. | |
Fig. 13(a) shows the chemical structure of N1,N4-bis(2,3,5,6-tetrafluoro-4-iodophenyl)terephthalamide (FIPh-A). The carbonyl (–CO), amino (–NH), and iodotetrafluorophenyl (–C6F4I) groups of FIPh-A facilitate the crystallization of perovskite and relieve strain as well.140 Notably, the crystallization of the FAPbI3 perovskite lattice in the presence of FIPh-A effectively increases the formation energy of I− vacancies from 4.86 eV to 5.19 eV (Fig. 13(b)),140 enhancing the stability of the inorganic framework. FIPh-A, as a multifunctional additive, is highly favorable for strongly interacting with various sites of the perovskite lattice (e.g., uncoordinated Pb2+ and I−), as illustrated in Fig. 13(c), which contributes to the stabilization of the octahedra inorganic framework in the perovskite lattice. The GIXRD results shown in Fig. 13(d) and (e) confirm that tensile strain is reduced upon the addition of FIPh-A.140 The synergistic effect of CO and –NH on stabilizing the inorganic framework while regulating lattice crystallization is also responsible for the residual strain alleviation in the lattice. As shown in Fig. 13(d), the control film suffers from intensive tensile strain by showing a gradual 2θ peak shift toward a lower angle as ω increases in the iso-inclination method of GIXRD. The use of FIPh-A encourages lattice crystallization bearing negligible depth-dependent strain difference, as confirmed by the little 2θ peak shift as ω varies in Fig. 13(e). The relieved tensile strain seems to be closely related to the increased crystalline quality with a preferred orientation.141
 |
| | Fig. 13 (a) Molecular structure of FIPh-A. (b) Formation energy of an I− vacancy calculated using density functional theory (DFT). (c) Illustration of the interaction mechanism between FIPh-A and the perovskite lattice at the grain boundary compared to the control perovskite film. GIXRD spectra of the (d) control film and (e) FIPh-A-employed film. ψ in (d) and (e) indicates ω in the iso-inclination method. Reproduced with permission from ref. 140. Copyright 2024, Wiley-VCH. | |
 |
| | Fig. 14 (a) Schematic of TMTA-CSRC treatment based on two stages (stages I and II) of perovskite formation. SEM images of (b) the control, (c) PTMTA-, and (d) TMTA-treated perovskite films at stage I. GIXRD spectra of (e) the control, (f) PTMTA-, and (g) TMTA (for SCRC)-treated perovskite films. Reproduced with permission from ref. 142. Copyright 2021, Wiley-VCH. | |
Similarly, acrylamide (Am) is introduced as a monomer that undergoes light-triggered polymerization and turns into polyacrylamide (PAm).145 When AM is coated after finishing the crystallization of the perovskite film (AAC treatment in Fig. 15(a)), Am is ruled out of the crystallization of perovskite and merely leads to a surface passivation effect. However, AM is treated on wet film with perovskite precursors before crystallization in the ABC treatment (Fig. 15(b)), which allows AM to participate in the nucleation and the growth of perovskite crystals and results in large and isolated grains, leaving PAm at grain boundaries and the top surface in the amorphous phase. It is worth noting that the liquidity of AM suppresses lattice distortion during crystal growth, thereby reducing lattice strain during the crystallization of the perovskite film, which balances the chronic in-plane tensile strain induced by different degrees of lattice thermal expansion/contraction with the underlying substrate. The GIXRD patterns of the (022) plane of the AAC-treated perovskite film exhibit the same tendency as the control film without AM inclusion, where the gradual 2θ peak shift toward a lower angle as Ψ increases in the side-inclination method indicates the residual tensile strain (Fig. 15(c) and (d)). Meanwhile, almost full release of the tensile strain is observed when the ABC strategy is applied, demonstrating a negligible peak shift regardless of varying Ψ (Fig. 15(e)).145 Similarly, the residual lattice strain can be effectively regulated by incorporating self-polymerizable additives into the perovskite wet film prior to crystallization.146–148 Self-polymerizing materials are prone to restrict the thermal expansion of the perovskite during the crystallization process at high temperatures, effectively mitigating the tensile strain in the perovskite film.146–148 Self-polymerizing n-methacrylamide (NMA) monomers interact with perovskite nuclei and constrain the perovskite lattice from thermally expanding during the annealing process, which reduces the residual tensile strain by approximately 34%.146 Furthermore, the inclusion of octafluoro-1,6-hexanediol diacrylate (OF-HDDA) induces coordination between the –CO of OF-HDDA and Pb2+ of perovskite during the annealing process and thus regulates crystallization. Consequently, the combined polymerization of OF-HDDA occurs with perovskite crystallization, effectively alleviating the residual tensile strain.147
 |
| | Fig. 15 Illustration of the perovskite lattice and schematic of perovskite crystallization using (a) AAC and (b) ABC treatments with corresponding grains and grain boundaries. GIXRD patterns for the (022) planes of (c) control, (d) AAC-treated, and (e) ABC-treated perovskite films. Reproduced with permission from ref. 145. Copyright 2024, Wiley-VCH. | |
7.1.2. Epitaxial growth strategy.
Epitaxial growth of three-dimensional (3D) perovskite film by utilizing two-dimensional (2D) perovskite as a substrate is also regarded as one of the effective ways to control the lattice strain by regulating crystallization. 2D perovskite (NAM)2PbI4 (NAM = nicotinamide) is devised to provide a highly matched heterointerface with FAPbI3. (NAM)2PbI4 has a weak hydrogen bonding between the organic molecule of NAM+ and the inorganic PbI64− framework, which loosens the octahedra tilting and allows for a large Pb–I–Pb angle (167.18°).122 The Pb–I–Pb angle of 2D perovskite is well matched with that of 3D FAPbI3 (176.85°), proposing the favorable heterointerface alignment between the (002) plane of (NAM)2PbI4 and the (100) plane of FAPbI3, as illustrated in Fig. 16(a). Although the 2D perovskite layer can be partially dissolved during the spin-coating of the 3D perovskite precursor solution, a portion remains at the bottom and serves as a substrate with a matched lattice for the heteroepitaxial growth of FAPbI3, while the dissolved part of 2D recrystallizes at the grain boundaries of FAPbI3, as shown in Fig. 16(b).122 As indicated by the 2θ shift toward lower angle with increasing Ψ, as depicted in Fig. 16(c), the in-plane tensile strain is inherent in the control perovskite film, while the heteroepitaxial growth by utilizing the lattice matching with underlying 2D perovskite enables the upper 3D perovskite to involve in-plane compressive strain near the bottom interface, showing 2θ shift toward higher angle (decreased d) with increasing Ψ (increased beam penetration from the surface) in Fig. 16(d). Therefore, in the 2θ–sin2Ψ plot (Fig. 16(e)), the negative slope for the control film is converted to the positive slope for the target film with (NAM)2PbI4.
 |
| | Fig. 16 (a) Illustration and Pb–I–Pb angles of (NAM)2PbI4 and FAPbI3. Free energy TOTEN refers to the total free energy calculated based on DFT. (b) Schematic mechanism for the crystallization of the perovskite film using (NAM)2PbI4. GIXRD patterns of (c) the control and (d) (NAM)2PbI4-assisted target films. (e) 2θ–sin2Ψ plots with linear fitting. Reproduced with permission from ref. 122. Copyright 2023, Wiley-VCH. | |
Moreover, 2D nanoflakes play a similar role in regulating lattice strain by providing a well-matched crystal lattice structure with the perovskite layer for heteroepitaxial growth.123,149,150Fig. 17(a)–(c) shows the HR-TEM images of WS2 nanoflakes. The 2.79 Å of d along the (100) plane in WS2 nanoflakes is twice that of the (100) plane in CsPbBr3, leading to favorable epitaxial growth of the CsPbBr3 layer on top of WS2 nanoflakes with lattice matching, as shown in Fig. 17(d) and (e). The van der Waals interactions between CsPbBr3 and WS2, along with the dangling bond-free surface of the WS2 (100) plane, facilitate the crystal growth of CsPbBr3 highly oriented along the out-of-plane direction layer with large grains and a low defect density.123 Furthermore, the weak interaction between 2D nanoflake WS2 and perovskite can act as a lubricant for lattice movements at the perovskite bottom interface during annealing and cooling (temperature changing) processes, which is beneficial in terms of residual strain.123 GIXRD analysis with iso-inclination method is carried out to define the residual strain, as depicted in Fig. 17(g)–(i). In the absence of WS2, no crystal growth regulation along the out-of-plane direction, 2θ peaks exhibit a noticeable shift toward lower angles as ω increases (Fig. 17(g)), while the introduction of WS2 at the bottom interface significantly reduces the diffraction peak shift, as shown in Fig. 17(h). Fig. 17(i) illustrates that the in-plane tensile strain at the bottom lattice, reflected in a steep slope, in the control film is mitigated by regulating crystallization by WS2, as noticed by a decreased slope of the d–ω plot.
 |
| | Fig. 17 (a) TEM, (b) HR-TEM, and (c) FFT images of WS2 nanoflakes. (d) Atomic arrangement of the WS2 (100) plane. (e) Cross-sectional atomic structure of the CsPbBr3/WS2 heterojunction. (f) Illustration of the van der Waals epitaxial growth of the CsPbBr3 film on the WS2-coated electron transport layer. Depth-dependent GIXRD patterns of the CSPbBr3 film (g) without and (h) with WS2 layer. (i) d(110)–ω plots depending on WS2 layer. Reproduced with permission from ref. 123. Copyright 2020, Wiley-VCH. | |
7.1.3. Solid conversion strategy.
More recently, noticeably oriented FAPbI3 was reported by solid-based conversion from two-dimensional (2D) to three-dimensional (3D) crystals. Although rapid nucleation by evaporating solvents in traditional methodologies (e.g., antisolvent) makes the control of crystal growth challenging,151 the solid-based conversion from 2D FAMAPbI4 perovskite (Imma space group) to 3D FAPbI3 lattice by the sublimation of MAI is found to be highly beneficial for regulating the crystal orientation, as illustrated in Fig. 18(a).61 The small ionic radius of MA+ facilitates the lower vaporization temperature, unlike longer alkyl chain-based ones, while the consistency in halide composition with I− helps reduce compositional entropy and align crystal orientation during the lattice reordering.61 Consequently, neighboring local spots in Fig. 18(b) demonstrate notably consistent structural ordering with analogous patterns and almost identical interplanar distance along (001), as confirmed in the high-magnification HR-TEM images (Fig. 18(c)). It is indeed worth noting that the lattice orientation in the single grain and the lattice matching between intra grains are obtained by the solid conversion (Fig. 18(d)), which is confirmed by the comparison of GIXRD patterns depicted in Fig. 18(e)–(g). The FAPbI3 film prepared by applying the control method exhibits a significant peak 2θ shift by varying the incidence angle of ω (Fig. 18(e)), implying the prominent in-plane tensile strain at the bottom, while the solid conversion methodology enables FAPbI3 film to bear insignificant in-plane tensile strain, namely negligible strain inhomogeneity along the out-of-plane direction by presenting a significantly lowered slope in the d–ω plot (Fig. 18(g)).
 |
| | Fig. 18 (a) Schematic of the phase transformation via solid conversion from FAMAPbI4 to FAPbI3. (b) HR-TEM image and (c) high-magnification HR-TEM images of different spots (1-1 to 1-4 as indicated in (b)) of the crystal orientation-regulated FAPbI3 film. (d) Schematic of the crystalline film of the crystal orientation-regulated FAPbI3 film. Depth-dependent GIXRD plots for the (100) plane of the (e) control and (f) target films based on the solid conversion. (g) Interplanar spacing values as a function of incidence angle. Reproduced with permission from ref. 61. Copyright 2025, Springer Nature. | |
The chemicals used for regulating the crystallization of the perovskite film in Section 7.1 are summarized in Table 1, involving related information on perovskite, strain and photovoltaic performance with long-term stability.
Table 1 Chemicals employed for regulating the crystallization of perovskite film to relieve the out-of-plane strain inhomogeneity
| Chemical type |
Chemical equation |
Chemical structure |
Interaction type |
Perovskite |
Other additives or passivation materials |
Characterization |
Strain type (control) |
Strain type (target) |
PCE (%) |
Long-term stability |
Long-term stress condition |
Ref. |
|
Value obtained from the 2θ–sin2Ψ plot (side-inclination method).
Value obtained from the d–ω plot (iso-inclination method).
Value obtained from the 2θ–sin2Ψ plot according to eqn (4) (side-inclination method).mpp: maximum power point tracking; +RH: relative humidity. |
| Small molecule |
Methylammonium iodide (MAI) |

|
|
FAPbI3 |
|
GIXRD |
Tensile |
Released tensile |
25.01 |
T
90.8 = 150 h |
mpp, one sun, ambient air, 30% RH |
61
|
| Small molecule |
1-(Phenylsulfonyl)pyrrole (PSP) |

|
Coordinated bond |
FA1−xCsxPbI3 |
MAPbBr3, PbI2, MACl |
TEM |
|
|
26.09 |
T
92 = 2500 h |
mpp, N2, RT, one sun |
74
|
| Small molecule |
1-(2-Fluorobenzyl)-1H-pyrazolo[3,4-b]pyridine-3-carboximidamide hydrochloride (FPPC) |

|
Coordinated bond/hydrogen bond |
FAPbI3 |
MACl |
GIXRD |
Tensile strain 0.0034b |
Released tensile 0.0002b |
25.63 |
T
90 = 500 h |
mpp, N2, one sun, 35 °C |
108
|
| 2D perovskites |
(NAM)2PbI4 |
|
|
FAPbI3 |
MACl |
GIXRD |
Tensile 61 Mpac |
Compressive 16 Mpac |
25.2 |
T
91 = 300 h |
mpp, one sun, N2 |
122
|
| 2D nanoflake |
WS2 |
|
|
CsPbBr3 |
|
GIXRD TEM |
Tensile |
Released tensile |
10.65 |
T
80 = 120 h |
Encapsulation, 80% RH |
123
|
| Small molecule |
N1,N4-Bis(2,3,5,6-tetrafluoro-4-iodophenyl)terephthalamide, FIPh-A |

|
Coordinated bond/hydrogen bond |
FA0.60MA0.36Cs0.04Pb(I0.97Br0.02Cl0.01)3 |
|
GIXRD |
Tensile |
Released tensile |
24.6 |
T
92 = 500 h |
One sun, aging track |
140
|
| Small molecule |
Trimethylolpropane triacrylate (TMTA) |

|
Coordinated bond |
(FAPbI3)0.95(MAPbBr3)0.05 |
|
GIXRD |
Tensile |
Compressive |
22.39 |
T
95 = 4000 h |
Stored in a dark, dry box, RT |
142
|
| Small molecule |
Acrylamide (Am) |

|
Coordinated bond/hydrogen bond |
(FAPbI3)0.95(MAPbBr3)0.05 |
|
GIXRD |
Tensile 36.5 MPac |
Released tensile 7.44 MPac |
24.45 |
T
80 = 1008 h |
One sun, 10% RH |
145
|
| Small molecule |
n-Methacrylamide (NMA) |

|
Coordinated bond/hydrogen bond |
FAxMA1−xPbClxI3−x |
|
GIXRD |
Tensile 85.0 ± 5.6 Mpac |
Released tensile 55.9 ± 5.2 Mpac |
22.0 |
T
91 = 1500 h |
Aging track, ambient air (25 °C ± 5 °C, 30 ± 5% RH), dark |
146
|
| Small molecule |
Octafluoro-1,6-hexanediol diacrylate (OF-HDDA) |

|
Coordinated bond/hydrogen bond |
Cs0.03FA0.95MA0.02Pb(I0.975Br0.025)3 |
|
GIXRD |
Tensile 0.613a |
Released tensile 0.117a |
24.76 |
T
92 = 1000 h |
N2, mpp, one sun, 35 °C |
147
|
| Small molecule |
N-Carbamoyl-2-propan-2-ylpent-4-enamide (apronal) |

|
Coordinated bond/hydrogen bond |
Cs0.05(FA0.85MA0.15)0.95Pb(I0.75Br0.25)3) |
|
GIXRD |
Tensile |
Compressive |
25.09 |
T
90.3 = 1404 h |
N2, mpp, encapsulation, one sun, 35 °C |
148
|
7.2. Strain control at the bottom interface
7.2.1. Small molecules as in-plane strain relievers.
Small molecules on the metal oxide selective contact.
As previously discussed, the solution-processed polycrystalline perovskite film generally exhibits residual in-plane tensile strain, which is the upmost pronounced at the bottom interface mainly owing to the low thermal coefficient of the rigid underlying substrate and gradually released across the film toward the top surface. The resultant strain inhomogeneity along the out-of-plane direction, which is attributed to the in-plane tensile strain with different degrees as a function of depth, is relieved by employing small molecules between the rigid substrate and the soft perovskite, as illustrated in Fig. 19(a). The control structure of the perovskite film, grown on SnO2, demonstrates a prominent in-plane tensile strain by shifting the 2θ toward a lower angle, indicating an increase in d, with increasing χ for deep penetration shown in Fig. 19(b).102 However, the depth-dependent strain inhomogeneity is greatly relieved by employing phosphorylethanolamine (PEA) as an interlayer to restrain the perovskite film from being directly anchored to rigid SnO2, which is evidenced by the negligible dependence of 2θ on χ, as illustrated in Fig. 19(c). The strain is quantified by converting the GIXRD results into the 2θ–sin2ψ plot, which reveals that a steep slope of −0.043 ± 0.007 from the control film is reduced to 0.009 ± 0.009 by employing PEA, suggesting the successful elimination of the in-plane tensile strain. PEA consists of phosphate (–OPO(OH)2) and amine groups (–NH2), which are linked by an alkyl chain, where the alkyl chain serves a pivotal role in relieving the in-plane strain of the perovskite film by rotating along the C–C bond,152 while the phosphate contributes to the passivation of oxygen vacancies of SnO2 by forming a strong interaction as Lewis base.153 Thus, –PO of the phosphate can form a strong coordinated bond with both uncoordinated Pb2+ of the perovskite film and Sn4+ of the SnO2 layer, while NH2 forms a hydrogen bond with I− of the perovskite framework.154,155 Therefore, various small molecules with the same functional groups of phosphate and amine have been widely utilized as an interlayer between the metal oxide substrate and the perovskite film.154–157 Methylphosphonic acid (MPA) with a phosphate group releases the residual in-plane tensile strain of the perovskite film, leading to the slope reduction in the 2θ–sin2Ψ plot from −0.043 ± 0.007 to −0.043 ± 0.007. The relatively weak effect of strain alleviation by MPA, compared to the slope of 0.009 ± 0.009 by PEA, could be attributed to the length of the alkyl chain. Longer alkyl chain linker connecting the bifunctional groups has a flexible molecular geometry and can accommodate the strain.102 Apart from phosphate, small molecules with other electronegative functional groups, such as –CO, –S–H, and pyridine, have been also utilized as an interlayer to release the residual strain by forming coordinated bonds with Pb2+ of upper perovskite film and Sn4+ of SnO2.120,154–159DL-Cysteine hydrochloride monohydrate (DLCH) is pre-employed in the SnO2 colloidal solution to suppress the spontaneous agglomeration of SnO2 nanoparticles using the coordinated bond between DLCH and Sn4+,156 which accordingly leads to smooth surface of SnO2 layer as surface roughness decreases from 2.43 nm to 1.48 nm (Fig. 19(d)). DLCH also comprises multi-functional groups of –COOH and –SH, serving as Lewis bases to passivate uncoordinated Pb2+ and Sn4+ defects,157,160–163 and –NH3+, forming a hydrogen bond with I−.156 Therefore, the formation of multiple bonding modes allows DLCH to perform bottom-up multifunctional modification, as illustrated in Fig. 19(e).164 In addition to the role of DLCH molecule in mitigating the in-plane tensile strain at the interface, the smoother surface of SnO2–DLCH is indicated as a parameter to reduce the strain by improving the bottom interface contact.165 Consequently, the in-plane tensile strain of the control film, with a prominent shift of 2θ toward lower angle with increasing ω (Fig. 19(f)), is effectively relieved by employing DLCH at the interface, showing a marginal shift in Fig. 19(g).
 |
| | Fig. 19 (a) Schematic of the regulation of in-plane tensile strain by surface modification of SnO2. GIXRD spectra of perovskite films on (b) bare SnO2 and (c) PEA-modified SnO2. Reproduced with permission from ref. 102. Copyright 2024, American Chemical Society. (d) Reduction in root-mean-square surface roughness after the incorporation of DLCH. (e) Schematic of bottom-up multilayer control by DLCH. GIXRD spectra of perovskite films deposited on (f) SnO2 and (g) DLCH-modified SnO2. Reproduced with permission from ref. 156. Copyright 2024, Wiley-VCH. | |
 |
| | Fig. 20 (a) Schematic of strain effect during cooling after the annealing of perovskite films for (I) ideal state and (II) real state. (III) Strain release by introducing a buffer layer using soft molecules with long alkyl chains, with the (IV) defect passivation effect. βcosθ–4sinθ plots of perovskite films deposited on (b) PTAA (control) and (c) DTAC-coated PTAA, with linear fitting to calculate strain by the slope. Reproduced with permission from ref. 169. Copyright 2022, Wiley-VCH. | |
 |
| | Fig. 21 (a) Schematic of SAM interaction between the substrate and perovskite. GIWAXS images of perovskite films on (b) Me-4PACz, (c) MeO-4PACz, and (d) GM-4PACz substrates. GIXRD spectra of perovskite films on (e) Me-4PACz, (f) MeO-4PACz, and (g) GM-4PACz substrates. Reproduced with permission from ref. 173. Copyright 2024, Wiley-VCH. | |
 |
| | Fig. 22 (a) Schematic of the lattice strain of perovskite grains deposited on MeO-2PACz and MeO-2PACz/PEACl substrates. GIXRD spectra of perovskite films on (b) MeO-2PACz and (c) MeO-2PACz/PEACl. Reproduced with permission from ref. 177. Copyright 2024, Wiley-VCH. | |
7.2.2. Polymer as an in-plane strain reliever.
Polymer on the metal oxide layer.
Polymer substrate can be preferred in terms of thermal expansion coefficient, which is closer to that of perovskite film compared to that of metal oxide.168,178,179 Therefore, various polymers are introduced between the soft perovskite film and rigid metal oxide substrate to reduce the extent of thermal contraction. A formation of a composite between polymer and SnO2 is proposed to control the residual strain. Polyacrylic acid (PAA) is mixed into SnO2 quantum dots (QDs) to prevent the agglomeration of QDs and to reduce oxygen vacancies caused by hydroxyl separation during the annealing process.180 The oxygenic group of PAA bonds with uncoordinated Sn4+ and fills the oxygen vacancies of both FTO and SnO2 QDs, forming a hard SnO2 film with few microcracks.181 Additionally, PAA serves as a buffer spring to release the residual strain at the interface and passivates two adjacent layers with functional groups responding to oxygen vacancies of SnO2 and the Pb dangling bonds.182 As shown in Fig. 23(a), PAA is present at the interface as an interlayer and diffuses into the perovskite bulk along the vertical grain boundaries owing to a relatively low volatility temperature of PAA (∼200 °C) compared to the high annealing temperature (250 °C) of the CsPbBr3 film according to the mechanism, as depicted in Fig. 23(b).183Fig. 23(c) and (d) show the analysis of GIXRD using the iso-inclination method, where the dependence of 2θ on ω (dashed guideline) greatly dwindles by employing PAA. The gentle slope in the converted d–ω plot depicted in Fig. 23(e) accordingly indicates the relaxation of the residual in-plane strain, that is the suppression of strain inhomogeneity along the depth of the perovskite film grown on PAA.183 Furthermore, the inverted structure employing NiOx as an HTM is similarly modulated with a polymer, poly(4-butylphenyldiphenylamine) (pTPD). Although a great difference in thermal coefficients of NiOx (14.0 × 10−6 K−1)178 and FAPbI3 (98.6–100 × 10−6 K−1)51 readily leads to the in-plane tensile strain at the bottom perovskite lattice, pTPD with a higher thermal expansion coefficient (1.0 × 10−3 K−1 above 60 °C and 1.2 × 10−4 K−1 below 60 °C)184 can even ideally induce the in-plane compressive strain, as illustrated in Fig. 23(f). The residual strain is characterized by applying the side-inclination method GIXRD, which clearly shows the distinct shift patterns of 2θ as Ψ increases depending on the pTPD interlayer (2θ shifts toward lower and higher angle without (upper figure) and with pTPD (lower figure), respectively, as depicted in Fig. 23(g)).185 The 2θ–sin2Ψ graph shown in Fig. 23(h) estimates σ of 0.0164 for NiOx substrate and −0.0079 for pTPD substrate, where positive and negative values indicate tensile and compressive strain, respectively.25 The effective regulation of the lattice in-plane strain from tensile to compressive using the polymer emphasizes the importance of considering the thermal expansion coefficient of the substrate for the solution-processed perovskite crystal growth but still would not guarantee the superior properties in terms of photovoltaic performance.
 |
| | Fig. 23 (a) Schematic of defect passivation by PAA at the bottom interface and grain boundaries of the perovskite film. (b) Schematic of the passivation and diffusion of PAA along the grain boundaries of the perovskite film. GIXRD spectra of the perovskite film on (c) SnO2 and (d) on PAA-modified SnO2. (e) d–ω plots with linear fitting depending on PAA. Reproduced with permission from ref. 183. Copyright 2023, Wiley-VCH. (f) Schematic of perovskite lattice strain development depending on the substrate. (g) GIXRD spectra of perovskite film on NiOx (top) and pTPD (bottom). (h) Linear fitting of 2θ–sin2Ψ depending on the substrate. Reproduced with permission from ref. 185. Copyright 2022, American Chemical Society. | |
 |
| | Fig. 24 SEM images of the buried interface of perovskite films grown on (a) PTAA, (b) CB-processed PTACz-PO, and (c) MeTHF-processed PTACz-PO. GlXRD spectra of perovskite films grown on (d) PTAA, (e) CB-processed PTACz-PO, and (f) MeTHF-processed PTACz-PO. Reproduced with permission from ref. 186. Copyright 2025, Royal Society of Chemistry. | |
7.2.3. Inorganic materials as in-plane strain relievers.
Although small molecules and polymers mostly serve as strain relievers with a flexible core structure forming an interlayer, inorganic materials tend to induce a selective lattice contraction at the bottom to release the in-plane tensile strain either by doping or by involving pre-compressive strain.
It reveals that doping of the perovskite lattice with alkali metal cations can alter the lattice strain.59,189 When rubidium dihydrogen phosphate (RbDP) is introduced into the SnO2, Rb+ is prone to diffuse into the perovskite lattice during the perovskite crystallization process, as illustrated in Fig. 25(a),121 owing to the lower adsorption energy of Rb+ on the SnO2 surface.190 X-ray photoelectron spectroscopy (XPS) spectra of the perovskite layer show signals of Rb and P, confirming the diffusion of RbDP, although the depth of diffusion remains unclear.121 Furthermore, XRD peak corresponding to the crystal planes of (110) and (220) exhibits a slight shift to a higher angle, indicating a decrease in d likely due to a partial replacement of FA+ (an ionic radius of 2.79 Å) with Rb+ (an ionic radius of 1.52 Å) for lattice contraction.121,191–193 As depicted in Fig. 25(b), the pristine perovskite film exhibits a gradual shift of 2θ toward a lower angle as Ψ increases, implying a larger d near the bottom interface of perovskite, compared to the upper lattice, which is in accordance with the routine in-plane residual strain. The incorporation of RbDP at the bottom interface of the perovskite film notably induces a slight shift in 2θ toward higher angle as Ψ increases, which confirms the effective role of RbDP in regulating the residual in-plane strain type in the perovskite lattice from tensile- to compressive-strain.121 The tensile strain is modulated by the synergistic effects of H2PO4− and Rb+, wherein H2PO4− interacts with SnO2 and uncoordinated Pb2+, while the diffusion-induced Rb+ doping substitutes FA+. The reduction in d occurs more prominently near the bottom interface, nullifying the formation of in-plane tensile strain. Similarly, the in-plane lattice strain of the perovskite film can be relieved by inducing selective lattice contraction near the bottom. For the case of CsPbBr3 film, the lattice contraction at the bottom film can be pursued by incorporating Cl− with a smaller ionic radius to replace Br−, as illustrated in Fig. 25(d).194 CsCl is employed in SnO2, where the increased interplanar spacing of SnO2 with CsCl suggests that Cs+ is well distributed within the SnO2 lattice.194 Owing to the smaller ionic radius of Cl− compared to Br−, a strong electronic coupling between Pb2+ and Cl− occurs and consequently induces the lattice contraction at the bottom.194 Time-of-flight secondary ion mass spectrometry (TOF-SIMS) confirms that Cl− components are selectively distributed at the interface of SnO2/CsPbBr3.194 The Cl−-induced lattice contraction at the bottom can facilitate uniform crystal growth and mitigate strain inhomogeneity along the out-of-plane direction by attenuating depth-dependent d mismatch.194 As illustrated in Fig. 25(e) and (f), the residual strains of perovskite grown on SnO2 and CsCl–SnO2 substrate are compared, where the pronounced shift in 2θ toward higher angle is slightly eased using the CsCl–SnO2 substrate, confirming the relieved in-plane tensile strain.194
 |
| | Fig. 25 (a) Illustration of RbDP diffusion into the perovskite layer. GIXRD patterns of (b) control and (c) RbDP-modified perovskite films. (d) Schematic of the bottom interface when CsCl is incorporated in SnO2. GIXRD patterns of perovskite films grown on (e) SnO2 and (f) CsCl–SnO2. Reproduced with permission from ref. 121. Copyright 2024, Elsevier and 194 Copyright 2024, Elsevier. | |
A strategy of pre-strain, which is intentionally introduced to the material prior to completion of the process, can be applied to the bottom interface of perovskite films to regulate the residual lattice strain.112 When the metastable material of Pb(CH3NH3)2Cl2 (PMC) is located at the interface between substrate and perovskite film, volatile MA+ is evaporated during the annealing process, forcing the bottom lattice of the perovskite film to contract (the left side of Fig. 26(a)).112 During the subsequent cooling process, the upper lattice of perovskite freely shrinks unlike the bottom lattice adhering to the substrate, which converts the in-plane compressive strain, in the form of pre-strain, into a strain-free state (the right side of Fig. 26(a)).112 This pre-strain concept is verified by grazing incidence wide-angle X-ray scattering (GIWAXS) along with TOF-SIMS. The conversion of PMC to PbCl2 at the bottom interface of the perovskite film during the annealing process is evidenced by GIWAXS,112 which is in accordance with the TOF-SIMS results, showing a gradual signal decrease for MA+ while maintaining Cl−.112Fig. 26(b)–(d) evidently illustrates the strain type conversion. As depicted in Fig. 26(b), the residual in-plane tensile strain is confirmed by a shift of the (100) diffraction peak toward lower q, indicating larger d as χ increases. The formation of the pre-strain is shown in Fig. 26(c), where the reverse shift toward higher q with increasing χ implies that the pre-strain is revealed as in-plane compressive strain, which turns into a strain-free condition after cooling, as shown in Fig. 26(d).112
 |
| | Fig. 26 (a) Illustration of the mechanism of the pre-strain strategy. GIWAXS of the (100) plane at different χ angles for (b) type 1, (c) type 2 and (d) type 3. Reproduced with permission from ref. 112. Copyright 2024, Royal Society of Chemistry. | |
The chemicals used to release the residual in-plane tensile strain at the bottom interface of the perovskite film in Section 7.2 are summarized in Table 2, having related information on perovskite, strain and photovoltaic performance with long-term stability.
Table 2 Chemicals employed at the bottom interface of the perovskite film to relieve out-of-plane strain inhomogeneity
| Chemical type |
Chemical equation |
Chemical structure |
Interaction type |
Perovskite |
Other additives or passivation materials |
Characterization |
Strain type (control) |
Strain type (target) |
PCE (%) |
Long-term stability |
Long-term stress condition |
Ref. |
|
Value obtained from 2θ–sin2Ψ plot (side-inclination method).
Value obtained from d–ω plot (iso-inclination method).
Value obtained from 2θ–sin2Ψ plot according to eqn (4) (side-inclination method).
Value obtained from the Williamson–Hall graph slope.
STEM (strain mapping).mpp: maximum power point tracking; RH: relative humidity; RT: room temperature. |
| Polymer |
Poly[9,9-bis(3′-(N,N-dimethyl)-nethylammoinium-propyl-2,7-fluorene)-alt-2,7-(9,9-dioctylfluorene)] dibromide (PFN-Br) |

|
Coordinated bond |
Cs0.05FA0.84MA0.11Pb(I0.86Br0.14)3 |
|
GIXRD, Williamson–Hall |
Compressive −112.2 MPac |
Released compressive −39.8 MPac |
21.93 |
T
97 = 240 h |
mpp, one sun, N2 |
23
|
| Small molecule |
Phosphorylethanolamine (PEA) |

|
|
(FA0.74MA0.26)PbI1−xBrx |
OABr |
GIXRD |
Tensile −0.043 ± 0.007a |
Strain-free 0.009 ± 0.009a |
24.35 |
T
90 = 423 h |
mpp, 0.95 sun, encapsulation, ∼40% RH |
102
|
| Small molecule |
1-(2-Fluorobenzyl)-1H-pyrazolo[3,4-b]pyridine-3-carboximidamide hydrochloride (FPPC) |

|
Coordinated bond/hydrogen bond |
FAPbI3 |
|
GIXRD |
Tensile 0.0034b |
Released tensile 0.0002b |
25.63 |
T
90 = 500 h |
mpp, one sun, N2 |
108
|
| Small molecule |
5-Aminopyridine-2-carboxylic acid (APC) |

|
Coordinated bond, hydrogen bond, π–π bond, π–π stacking |
Cs0.05FA0.85MA0.10Pb(I0.93Br0.07)3 |
RbCl, TEACl |
GIXRD, Williamson–Hall |
Tensile 61.57 ± 6.92 MPac |
Released tensile 27.54 ± 2.16 MPac |
24.87 |
T
97 = 1800 h |
mpp, one sun, N2 |
110
|
| Inorganic salt |
Pb(CH3NH2)2Cl2 (PMC) |
|
|
FAPbI3 |
MACl |
GIWAXS |
Tensile 26.77 MPac |
Strain-free −1.73 MPac |
25.36 |
T
90 = 1000 h |
mpp, one sun, N2, ∼40 °C |
112
|
| Small molecule |
4-Chloro-3-sulfamoylbenzoic acid (CSBA) |

|
Coordinated bond |
FA0.69MA0.25Rb0.06PbI3 |
MeO-PEAI |
GIXRD Williamson–Hall |
Tensile 0.09752b |
Released tensile 0.0063b |
25.32 |
T
85 = 1200 h |
One sun, N2 |
120
|
| Inorganic salt |
RbH2PO4 (RbDP) |

|
Coordinated bond |
FA1−xMAxPbI3 |
|
GIXRD Williamson–Hall |
Tensile −0.105a |
Compressive 0.009a |
24.52 |
T
90 = 150 days |
RT, RH ∼40% |
121
|
| Small molecule |
3-Amino-5-bromopicolinaldehyde (BD) |

|
Coordinated bond/hydrogen bond |
Cs0.05(FA0.95MA0.05)0.95Pb(I0.95Br0.05)3 |
|
GIXRD |
Tensile 48.8 MPac |
Compressive −4.1 MPac |
24.45 |
T
81 = 500 h |
One sun, N2 |
154
|
| Small molecule |
Bis(2,2,2-trifluoroethyl) (methoxycarbonylmethyl)phosphonate (BTP) |

|
Coordinated bond/hydrogen bond |
Rb0.02(FA0.95Cs0.05)0.98PbI2.91Br0.03Cl0.06 |
|
GIXRD |
Tensile |
Released tensile |
24.26 |
T
54.5 = 1728 h |
One sun, N2 |
155
|
| Small molecule |
DL-Cysteine hydrochloride monohydrate (DLCH) |

|
Coordinated bond/hydrogen bond |
(FA0.95Cs0.05)0.98Rb0.02Pb(Br0.03Cl0.06I2.91) |
|
GIXRD |
Tensile |
Released tensile |
24.01 |
T
93 = 1800 h |
Dark, N2, 65 °C |
156
|
| Small molecule |
Ergothioneine (EGT) |

|
Coordinated bond/hydrogen bond |
Rb0.02(FA0.95Cs0.05)0.98PbI2.91Br0.03Cl0.06 |
PEAI |
GIXRD |
Tensile |
Released tensile |
25.13 |
T
90.1 = 930 h |
mpp, one sun |
157
|
| Small molecule |
Cucurbit[5]uril (CB[5]) |
|
Coordinated bond |
Cs0.03FA0.95MA0.02Pb(I0.975Br0.025)3 |
|
GIXRD |
Tensile |
Released tensile |
24.83 |
T
87.5 = 1000 h |
mpp, under illumination, N2 |
158
|
| Small molecule |
1-Adamantaneacetic acid (ADAA) |

|
Coordinated bond |
Rb0.02(FA0.95Cs0.05)0.98PbI2.91Br0.03Cl0.06 |
|
GIXRD |
Tensile |
Released tensile |
22.83 |
T
92.4 = 1000 h |
N2, dark, 65 °C |
159
|
| Small molecule |
2-Butynoic acid (BA) |

|
Coordinated bond |
FA0.85Cs0.15PbI3 |
|
GIXRD |
Tensile −0.117a |
Released tensile −0.109a |
23.33 |
T
72 = 500 h |
mpp, one sun, RH (40 ± 10%) |
160
|
| Small molecule |
Hexadecyltrimethylammonium chloride (CTAC) |

|
Coordinated bond |
Rb0.05Cs0.05MA0.05FA0.85Pb(I0.95Br0.05)3 |
|
Williamson–Hall |
Tensile 7.0 × 10−4d |
Released tensile 3.5 × 10−4d |
22.04 |
T
73 = 1200 h |
Encapsulation, 1.0 cm2, one sun |
166
|
| Small molecule |
Piperazine dihydriodide (PDI2) |

|
Coordinated bond |
Cs0.05FA0.87MA0.08Pb(I0.97Br0.03)3 |
|
GIXRD |
Tensile −0.1035 ± 0.0132a |
Released tensile −0.0524 ± 0.0089a |
23.47 |
|
|
168
|
| Small molecule |
Dodecyltrimethylammonium chloride (DTAC) |

|
|
Cs0.05FA0.81MA0.14Pb(I0.86Br0.14)3 |
|
Williamson–Hall |
Tensile 7 × 10−4d |
Released tensile 5 × 10−4d |
19.7 |
T
84 = 6200 h |
∼65 °C |
169
|
| Small molecule |
[4-(3,6-Glycol monomethyl ether-9H-carbazol-9-yl)butyl]phosphonic acid (GM-4PACz) |

|
Coordinated bond, hydrogen bond |
(FA0.95MA0.05)0.95Cs0.05Pb(I0.95Br0.05)3 |
|
GIXRD |
Tensile |
Released tensile 23.31 MPac |
25.52 |
T
91 = 1000 h |
mpp, one sun, N2 |
173
|
| Me-4PACz: 35.88 MPac |
| MeO-4PACz: 51.83 MPac |
| Small molecule |
Phenylethylammonium chloride (PEACl) |

|
Coordinated bond |
Cs0.15FA0.85PbI3 |
|
GIXRD |
Tensile |
Released tensile |
24.11 |
T
97 = 1500 h |
Tracking under 0.9 V, one sun, N2 |
177
|
| Polymer |
Poly(acrylic acid) (PAA) |

|
Coordinated bond (with Sn) |
CsPbBr3 |
|
GIXRD |
Tensile |
Released tensile |
10.83 |
T = 100 h |
mpp, LED (100 mW cm−2) |
183
|
| Polymer |
Poly(4-butylphenyldiphenylamine) (pTPD) |
|
|
Cs0.05FA0.83MA0.12Pb(I0.88Br0.12)3 |
|
GIXRD |
Tensile −0.04235 ± 0.02084a |
Compressive 0.02035 ± 0.00749a |
|
T = 911 h |
Dry air (∼745 h) RH (35–45%) (745–911 h) |
185
|
| Polymer |
PTACz-PO |

|
|
Cs0.05(FA0.95MA0.05)0.95Pb(I0.95Br0.05)3 |
|
GIXRD |
Tensile 49.51 MPac |
Released tensile 7.70 MPac |
26.31 |
T
94 = 1450 h |
65 °C, 50% RH, one sun |
186
|
| Inorganic salt |
CsCl |
|
|
CsPbBr3 |
|
GIXRD |
Tensile |
Released tensile |
11.16 |
1000 h |
One sun, ambient air |
194
|
| Small molecule |
4-Sulfobenzoic acid monopotassium salt (K-SBA) |

|
Coordinated bond |
FA1−xMAxPbI3 |
|
GIXRD |
Tensile −0.16465a |
Released tensile −0.02928a |
24.56 |
T
90 = 500 h |
mpp, one sun, N2, 50–60 °C |
195
|
| 2D perovskite |
PEA2PbBr4 |
|
|
CsPbBr3 |
|
STEM (strain mapping) |
Strain ∼ 6%e |
Strain ∼ 2%e |
|
|
|
196
|
| Small molecule |
Rb2CO3 |

|
|
FA0.93Cs0.07PbI3 |
|
GIXRD |
Tensile 0.7 MPac |
Compressive −38 MPac |
22.7 |
T
95 = 2700 h |
mpp, one sun, N2 |
197
|
| Small molecule |
6FPPY |

|
|
FA0.95Cs0.05PbI3 |
|
GIXRD |
Tensile −0.0195a |
Released tensile 0.0044a |
24 |
T
90 = 200 h |
mpp, one sun, N2 |
198
|
| Small molecule |
p-Chlorobenzenesulfonic acid (CBSA) |

|
Covalent bond |
MAPbI3 |
|
GIXRD |
Tensile −0.097a |
Strain-free −0.013a |
20.7 |
T
84 = 3000 h |
N2 |
199
|
| Small molecule |
2-Thiopheneacetic acid (TPA) |

|
Coordinated bond |
CsPbBr3 |
|
GIXRD |
Tensile |
Released tensile |
11.23 |
T
84 = 120 h |
mpp, LED (100 mW cm−2) |
200
|
7.3. Strain control at the top interface
Materials deposited on top of the perovskite film can be similarly anchored to the top lattice and accordingly exert a strong interaction to formulate the residual lattice strain. Nevertheless, strain control at the top interface is not widely attempted because the fundamental origin of the residual in-plane lattice strain, causing the strain inhomogeneity along the out-of-plane direction, is inherent to the bottom interface. Therefore, the top interface approach is relatively limited compared to the bottom interface.
7.3.1. Small molecules as in-plane strain relievers.
The residual tensile strain of the perovskite film is modulated by engineering its top surface with 2-(methylthio)ethylamine hydrochloride (2MTEAC). 2MTEAC has both an electron-donating group (–S as Lewis base) and an electron-withdrawing group (–NH3+ as Lewis acid), which are favorable for coordinating with undercoordinated Pb2+ and I or N of the top perovskite lattice, respectively, as illustrated in Fig. 27(a).201 The effect of the top surface engineering on the residual strain of perovskite film is assessed by applying the side-inclination method, GIXRD, shown in Fig. 27(b)–(d). The target film with the surface treatment with 2MTEAC exhibits a relaxation of the scattering 2θ peak shift as Ψ increases (Fig. 27(c)) compared to the control film (Fig. 27(b)). The linear fitting shown in the 2θ–sin2Ψ graph estimates the slopes of the control and the target films as ε = 0.214 and 0.173, respectively (Fig. 27(d)).201 More specifically, an increased 2θ at higher sin2Ψ, indicating reduced d close to the bottom interface, is monitored from the target film, while d is even enlarged near the top interface of the target film by forming a strong interaction with the small molecules, confirming the relieved strain inhomogeneity along the out-of-plane direction for the 2MTEAC-treated target film. Furthermore, π-conjugated small molecules of aminobenzoic acid derivatives, meta-aminobenzoic acid (MABA) and para-aminobenzoic acid (PABA), are similarly introduced to the top surface of the perovskite film to passivate the defect and to release the residual in-plane tensile strain.202,203 The two molecules consist of two bioactive-site ligands with π-conjugated systems, –NH2 and –COOH, where the free movement of π-electrons occurs in the extended molecular orbital, and a lone pair electron can move from the strong electron donor –NH2 group to the weak electron withdrawing –COOH group owing to π–π intermolecular interactions.202 In addition, the presence of both electron-rich and electron-donor ligands allows MABA and PBBA to passivate both negatively and positively charged defects (e.g., uncoordinated Pb2+, Br− vacancy, and Cs+ vacancy) in the perovskite film, as depicted in Fig. 27(e).204 The iso-inclination method of GIXRD is used to confirm the effect of MABA- and PABA-treated perovskite films on the residual lattice strain. The control film in Fig. 27(f) exhibits an obvious peak shift of 2θ with increasing ω, implying the pronounced residual in-plane strain at the bottom region.205 The peak shift is gradually relieved by the top surface engineering of perovskite film with MABA (Fig. 27(g)) and PABA (Fig. 27(h)), suggesting relieved strain inhomogeneity along the out-of-plane direction. It is noteworthy that the top surface engineering of the perovskite film by coordinating pertinent small molecules affords to wield distinct influence over the residual in-plane tensile strain, which dominantly originates from the bottom interface of the perovskite film although their effects on the related mechanism are not thoroughly discussed in the relevant studies.
 |
| | Fig. 27 (a) Schematic of the interaction between 2MTEAC and perovskite. GIXRD spectra of (b) control and (c) 2MTEAC-treated perovskite films. (d) Linear fit of 2θ–sin2Ψ plots. Reproduced with permission from ref. 201. Copyright 2025, Elsevier. (e) Schematic of multifunctional passivation at different defect sites in CsPbBr3 by MABA and PABA. GIXRD spectra of (f) control, (g) MABA-treated, and (h) PABA-treated perovskite films. Reproduced with permission from ref. 205. Copyright 2024, American Chemical Society. | |
Ligands of quantum dots (QDs) can also act as SAM on the perovskite surface and play a similar role in regulating the residual lattice strain. CsPbI3 quantum dot (QD), capped with an optimal amount of ligand, is employed in toluene as an antisolvent, which is subsequently dropped on the spinning film with the FA0.95Cs0.05PbI3 perovskite precursor solution, as illustrated in Fig. 28(a).206 The ligand concentration is controlled by successive treatments with a mixed solvent of hexane and methyl acetate, where the strain degree varies with the ligand concentration. CsPbI3 QDs mainly function as nucleation sites and are responsible for enhanced film quality, while the ligands are detached from the QD surface during the film annealing process and transferred to the surface of perovskite film, such as SAM, as confirmed by the shifted CO stretching vibration of the oleic acid ligand from 1710 cm−1 in pure CsPbI3 QDs to 1750 cm−1 in the perovskite film. It is noteworthy that the self-assembled ligands on the perovskite surface play a critical role in determining the residual lattice strain.206Fig. 28(b) presents the GIXRD pattern of the control film with the (012) plane, which is frequently selected to acquire more grain information than orientation impact.142,206,207 As the Ψ angle increases, the peak shifts to a lower angle owing to the increased d of the (012) plane, suggesting in-plane tensile strain with increasing depth.206 However, the ligand content readily governs the residual lattice strain ranging from compressive strain to strain-free and even to tensile strain by decreasing the ligand concentration from QDs-I (Fig. 28(c)) to QDs-II (Fig. 28(d)) and to QDs-III (Fig. 28(e)), as observed by 2θ peak shift direction with increasing Ψ. As illustrated in Fig. 28(f), the slope of the 2θ–sin2Ψ plot indicates σ. When considering the negative slope of the control film as a reference point, the slope is widely varied from positive σ (compressive strain with QD-I) to negative σ (tensile strain with QD-III).
 |
| | Fig. 28 (a) Schematic of the fabrication process of perovskite films with CsPbI3 QDs, determining the number of surface ligands to modulate the lattice strain in the perovskite film. GIXRD patterns of (b) control film and films treated with (c) QD-I, (d) QDs-II, and (e) QDs-III. (f) Linear fit of 2θ–sin2Ψ depending on the employment of the QDs. Reproduced with permission from ref. 206. Copyright 2023, Royal Society of Chemistry. | |
The chemicals used to release the residual in-plane tensile strain dominantly on the top surface of the perovskite film in Section 7.3 are summarized in Table 3 with related information on perovskite, strain and photovoltaic performance with long-term stability.
Table 3 Chemicals employed at the top interface of the perovskite film to relieve out-of-plane strain inhomogeneity
| Chemical type |
Chemical equation |
Chemical structure |
Interaction type |
Perovskite |
Characterization |
Strain type (control) |
Strain type (target) |
PCE (%) |
Long-term stability |
Long-term stress conditions |
Ref. |
|
Value obtained from 2θ–sin2Ψ plot (side-inclination method).
Value obtained from 2θ–sin2Ψ plot according to eqn (4) (side-inclination method).mpp: maximum power point tracking; RH: relative humidity. |
| Small molecule |
2-(Methylthio)ethylamine hydrochloride (2MTEAC) |

|
Coordinated bond/hydrogen bond |
(FA0.98MA0.02)0.95Cs0.05Pb(I0.95Br0.05)3 |
GIXRD |
Tensile −0.214a |
Released tensile −0.173a |
24.52 |
T
90 = 1000 h |
N2, 85 °C |
201
|
| Small molecule |
para-Amino-benzoic acid (PABA) |

|
Coordinated bond |
CsPbBr3 |
GIXRD |
Tensile |
Released tensile |
11.02 |
T
89 = 50 days |
85% RH, 25 °C |
205
|
| Inorganic QD |
QD CsPbI3 |
|
|
FA0.95Cs0.05PbI3 |
GIXRD |
Tensile |
Strain-free |
23.3 |
T
90 = 360 h |
55% RH, ambient air |
206
|
| Small molecule |
1,1′-(Methylenedi-4,1-phenylene) bismaleimide, BMI |

|
Coordinated bond/hydrogen bond |
FA0.96Cs0.04PbI3 |
GIXRD |
Tensile 6.2 Mpab |
Compressive −7.4 Mpab |
22.7 |
T
91.8 = 1000 h |
mpp, one sun, N2 |
208
|
| Small molecule |
Organic polymer J71 |

|
Coordinated bond |
CsPbI2Br |
GIXRD TEM |
Tensile −0.26897 ± 0.0093a |
Released tensile −0.19753 ± 0.0075a |
16.15 |
T
85 = 826 h |
N2, 25% RH |
209
|
7.4. Strain control at grain boundaries
A flexible chain can bear strain and thus mitigate the strain in the system.210 Small molecules with long alkyl chains, which cannot fit in the perovskite lattice, are expelled from the lattice during crystallization but remain at the grain boundaries as spacers.99,104,106,211 The contribution of molecules sitting at grain boundaries to the lattice strain should be carefully assessed because the molecules unavoidably affect the lattice crystallization, thereby modulating the residual strain without accounting for the effect of molecules at the grain boundaries. This effect is closely related to the earlier-mentioned effect in subsection and Section ‘7.1. Strain control by regulating crystallization and 7.1.1. Additive strategy’, respectively. Here, we attempt to consider the predominant effect (either crystallization or a certain role at grain boundaries) on strain control to distinguish and categorize the effect although a single molecule/material can sufficiently introduce multiple synergistic effects, and it is difficult to decouple the contribution extent.
Alkylamines with different chain lengths, n-butylamine (BA), n-hexylamine (HA), and n-octylamine (OA), are introduced as dopants in the perovskite precursor solution to regulate the lattice strain.210 The electronegativity of HA is slightly higher than that of BA and OA.210 The perovskite film incorporating HA can induce the strongest coordination interaction between the amino group of HA and Pb2+ in the perovskite film, which is responsible for an obvious peak shift of Pb 4f to lower binding energy in XPS analysis, compared to the control film without any dopant. The GIXRD pattern of the control film reveals a significant 2θ shift toward a lower angle with increasing ω (Fig. 29(b)), confirming the residual in-plane tensile strain in the control film. The in-plane tensile strain in the control film is most effectively relieved by the HA dopant, where the ω-dependent 2θ peak shift becomes less prominent (Fig. 29(c)).
 |
| | Fig. 29 (a) Molecular structure of BA, HA and OA. Depth-dependent GIXRD patterns of (b) pristine perovskite film and (c) film with HA. Reproduced with permission from ref. 210. Copyright 2024, Royal Society of Chemistry. (d) Microstrain in perovskite films containing OAI, OAI:DAP, and DAP. Reproduced with permission from ref. 211. Copyright 2023, Wiley-VCH. (e) Molecular structure of 2-PyEAI. GIXRD patterns of (110) planes in the perovskite film with (f) 3D (control), (g) 2D@3D, and (h) 2D@3D/2D. Reproduced with permission from ref. 217. Copyright 2023, American Chemical Society. (i) Molecular structure of 3-PPA. GIXRD patterns of (j) control and (k) target (containing 3-PPA) perovskite films. (l) d–ω plots with linear fitting. Reproduced with permission from ref. 152. Copyright 2024, Wiley-VCH. | |
The perovskite lattice at the grain boundaries is generally coordinated with ammonium ligands by hydrogen bonds, while intermolecular interaction between ammonium ligands is based on van der Waals interactions, where the ligands function as spacers to define grain boundaries.212–214 Monoammonium ligands (n-octylamine iodide, OAI) and diammonium ligands (1,3-diaminopropane, DAP) with different alkyl chain lengths and different numbers of ammonium functional groups are also added in the perovskite precursor solution as additives. Monoammonium ligands with long alkyl chains (e.g., OAI) guarantee a wide grain boundary spacing capability but mostly rely on hydrogen bonding, coupled with weak van der Waals interactions.134 However, diammonium ligands with short alkyl chains (e.g., DAP) can induce a strong interaction based on hydrogen bond at the grain boundaries while maintaining a narrow spacing between grains, where a migration of ions across the grain boundary can be facilitated owing to the narrow spacing.215 Therefore, a mixed ammonium ligand passivation strategy, using OAI and DAP (OAI:DAP), is introduced to generate the synergistic effect of two ligands. Furthermore, OAI is prone to form a Ruddlesden–Popper (RP) 2D capping layer, which encourages the lattice stability while suppressing ion migration.216 Meanwhile, the Dion–Jacobson (DJ) 2D lattice exhibits an overall opposite effect to the RP phase, facilitating charge transport but with reduced lattice stability.212,213,216 Microstrain is estimated by the Williamson–Hall method, as shown in Fig. 29(d), where the strain of the control film is most effectively relieved by employing OAI with a long alkyl chain, while the DAP with a relatively short alkyl length is less effective in releasing the strain. Even though the microstrain has a limitation in evaluating the depth-dependent strain information, such as GIXRD, and simply possesses overall strain inhomogeneity, the relieved strain value certainly implies a more uniform strain distribution in the engineered film. Similarly, an extremely thin capping layer of the 2D perovskite is formed on 3D perovskite film (2D@3D) by incorporating 2-(2-pyridyl)ethylamine (2-PyEA), as shown in Fig. 29(e).217 The ammonium functional group (–NH3+) can occupy the vacancy at the FA+ site, while the N atom of pyridine interacts with the Pb2+ ions in the perovskite surface, resulting in the regulation of the surface termination and a partial relief of the residual tensile strain.217 Therefore, the significant 2θ peak shift toward lower angle as Ψ increases in the control 3D film (Fig. 29(f)) is slightly relieved in the 2D@3D film (Fig. 29(g)), which is further alleviated by the post-treatment of the 2D@3D film with 2-PyEAI (2D@3D/2D), as shown in Fig. 29(h). Fig. 29(i) shows the molecular structure of 3-phosphonopropanoic acid (3-PPA), which is also used as a dopant in the precursor solution and simultaneously influences the residual lattice strain. The bilateral head group of 3-PPA stabilizes the octahedral lattice, while the alkyl chain alleviates the residual stress through the rotation of carbon–carbon single bonds at the grain boundaries.152 The CO and PO bonds of 3-PPA coordinate with the Pb2+ ions of the perovskite, while the hydrogen bonds between 3-PPA and the N–H group of FA+ are formed.152 The GIXRD pattern of (001) planes in the control film shows a prominent 2θ shift toward a lower angle as ω increases (Fig. 29(j)), indicating the tensile strain. The strain inhomogeneity along the out-of-plane direction is considerably resolved in the target film (Fig. 29(k)), leading to a lower slope for the target film in the d–ω plot than that of the control film (Fig. 29(l)). Notably, relieved strain inhomogeneity is achieved by decreasing d at high ω (near the bottom interface) and by increasing d values near the top surface (at low ω) in the target film. The d values at low ω in the target film are even greater than those of the control film. In other words, 3-PPA enlarges the d of (001) particularly near the top interface, which readily contributes to the decrease in slope in the d–ω plots although the in-plane tensile strain is absolutely more involved at the top lattice of the target film.
The chemicals used to release the residual in-plane tensile strain dominantly at the grain boundaries of the perovskite film in Section 7.4. are summarized in Table 4, with related information on perovskite, strain and photovoltaic performance with long-term stability.
Table 4 Chemicals employed at grain boundaries of the perovskite film to release out-of-plane strain inhomogeneity
| Chemical type |
Chemical equation |
Chemical structure |
Interaction type |
Perovskite |
Characterization |
Strain type (control) |
Strain type (target) |
PCE (%) |
Long-term stability |
Long-term stress conditions |
Ref. |
|
Value obtained from the 2θ–sin2Ψ plot (side-inclination method).
Value obtained from the d–ω plot (iso-inclination method).
Value obtained from the 2θ–sin2Ψ plot according to eqn (4) (side-inclination method).
Value obtained from the Williamson–Hall graph slope.mpp: maximum power point tracking; RH: relative humidity; RT: room temperature. |
| Small molecule |
Pemirolast potassium, PP |

|
Coordinated bond |
FA0.88Cs0.12Pb(I0.88Br0.12)3 |
GIXRD |
Compressive 0.405a |
Released compressive 0.305a |
23.06 |
T
90 = 250 h |
85 °C, N2 |
24
|
| Small molecule |
3-Phosphonopropanoic acid (3-PPA) |

|
Coordinated bond/hydrogen bond |
Cs0.05FA0.95PbI3 |
GIXRD |
Tensile 0.045b |
Released tensile 0.016b |
24.05 |
T
86.4 = 1254 h |
Ambient air, 30–40% RH |
152
|
| Small molecule |
n-Hexylamine |

|
Coordinated bond |
CsPbIBr2 |
GIXRD |
Tensile |
Released tensile |
10.67 |
T
79 = 100 h |
One sun |
210
|
| Small molecule |
n-Octylammonium iodide, OAI/1,3-diaminopropane, DAP |

|
Hydrogen bond |
Cs0.175FA0.825Pb(I0.85Br0.15)3 |
Williamson–Hall |
Compressive −0.110d |
Compressive −0.142d |
21 |
T
94 = 1000 h |
Encapsulation, one sun |
211
|
| Small molecule |
2-(2-Pyridylthylamine (2-PyEA) |

|
Coordinated bond/hydrogen bond |
FA0.75MA0.25PbI3 |
GIXRD |
Tensile |
Released tensile |
23.2 |
T
92 = 3000 h |
RT, 30% RH, ambient air |
217
|
| Inorganic salt |
Lead formate, PbFa |

|
Coordinated bond |
(FAPbI3)0.95(MAPbBr3)0.05 |
GIXRD Williamson–Hall |
Tensile 38.5 MPac |
Released tensile 27.09 MPac |
24.32 |
T
90 = 300 h |
One sun, N2 |
218
|
| Small molecule |
2,2′-Diamino-[1,1′-biphenyl]-4,4′-dicarboxylic acid, DBDA |

|
Coordinated bond/hydrogen bond |
FA0.71FA0.29Pb(I0.88Br0.22Cl0.1)3 |
GIXRD |
Tensile 0.0628a |
Compressive −0.1814a |
24.57 |
T
92 = 1000 h |
mpp, one sun, N2 |
219
|
| Small molecule |
Methyl(methylsulfinyl)methyl sulfide, MMS |

|
Coordinated bond |
Cs0.05FA0.95PbI3 |
GIXRD |
Tensile 22.5 MPac |
Released tensile 9.4 MPac |
26.12 |
T
82 = 1650 h |
mpp, one sun, 30 ± 5% RH |
220
|
| Small molecule |
Methylamine formate, MAFa |

|
|
FAPbI3 |
GIXRD, Williamson–Hall |
Compressive −3.8 MPac |
More compressive −13.9 MPac |
24.08 |
T
90 = 1000 h |
30 ± 5% RH |
221
|
8. Synergistic effects of strain control
8.1. Improved optoelectronic properties and lattice stability
In terms of reducing the local strain inhomogeneity, the enhanced crystal orientation with a larger grain size is generally pursued by regulating crystallization to maintain structural homogeneity, where lattice deformation with octahedra tilting to afford the strain can be significantly suppressed. Consequently, long-range effective overlapping between Pb–I–Pb with less PbI64− octahedral tilting is enabled, which establishes a spatially independent uniform band structure. In the case of FAPbI3, the cubic-like structure with suppressed lattice deformation is favorable for a strong spin–orbit coupling effect to suppress the carrier recombination,222 as well as for light carrier effective masses to enhance charge transport properties.223,224 Therefore, the long-range lattice order with uniform orientation by minimizing the local strain inhomogeneity is preferred for deriving superior optoelectronic properties and for ensuring performance reliability. Similarly, the residual lattice strain even intensifies the lattice deformation and readily affects the Pb–I bonding nature in the inorganic framework; hence, its electronic band structure, particularly the valence band, is accordingly altered with modulating optoelectronic properties.60,62 It is noteworthy that even a small strain (about ±0.5%, positive value for the tensile strain and negative value for the compressive strain) can induce a noticeable physical change when considering a gradual change in the electronic band structure with varying strains.26,27 It is assumed that the experimentally feasible strain window is limited to ±4%,26–28,225 while the best performance is mostly observed from the strain ranging from 0% (strain-free) to −1.5% (slightly compressive strain).26–28 Generally, the residual in-plane tensile strain tends to form a dispersed curvature in the electronic valence band structure as a function of wave vector, while a large curvature results from the compressive strain. Therefore, in contrast with relatively constant electron effective mass, the hole effective mass significantly depends on the lattice strain, where the hole effective mass is gradually decreased by converting from tensile strain to strain-free, and to compressive strain.28 The hole mobility thus gradually increases by strengthening the compressive strain until the critical dislocation occurs to nullify the intense compressive strain.226 For the case of α-FAPbI3, −1.2% of compressive strain is suggested as a compromise point between a low hole effective mass and a high trap density owing to the dislocation.28 Besides, the residual strain is closely related to the defect dynamics,227 where the activation energies for defect formation and migration are governed by the lattice strain. The compressive strain generally increases the activation energies for defect and migration, while the tensile strain helps defects form and migrate by decreasing the activation energies.228 The high defect density under the in-plane tensile strain is well reflected in the higher ideality factor of 1.55 compared to that of 1.05 under the compressive strain,27 where the ideality factor varies from 1 for ideal radiative recombination to 2 for trap-assisted nonradiative recombination.229 The strain-dependent defect dynamics is critically important for FAPbI3 because the phase transition from α- to δ-phase should accompany atomic displacement in the soft lattice,10 where the activation energy highly depends on the defect density.230 Furthermore, the residual strain influences the unit cell volume in lattice, which can affect the lattice stability by H-driven change in G.22 Therefore, the lattice strain control toward improving homogeneity and relieving the tensile strain, possibly leading to a decrease in G by the volume reduction in the unit cell, simultaneously exhibits enhanced optoelectronic properties and lattice stability as synergistic effects.
8.2. Defect passivation at the interface and grain boundaries
The lattice strain control is dominantly triggered by additives during crystallization or interfacial management (bottom, top, and grain boundaries) with specific materials, as discussed earlier. The additives for regulating crystallization contain functional groups to induce a strong interaction with precursors of perovskite (e.g., Pb2+, I−, and organic cation) and are subsequently situated at grain boundaries after completing the crystallization.24,211 Similarly, the materials for interfacial management also consist of functional groups to interact with perovskite in common and optionally to interact with adjacent selective contact (electron or hole transport layer) depending on the target interface.110,160 Therefore, the strategies for suppressing the strain inhomogeneity and relieving the in-plane tensile strain usually accompany chemicals employing specific functional groups, which can act as Lewis bases or acids (e.g., –OPO(OH)2, –NH2, –NH3+, –CO, –SH, and –COOH) to efficiently coordinate with perovskite surface (e.g., Pb2+, I−, organic cation, and vacancies), resulting in surface passivation.205,208 Because the interfacial defect predominantly serves as a main route for facilitating nonradiative carrier recombination,231 the chemicals, which are utilized for modulating lattice strain, simultaneously suppress nonradiative recombination by coordinating with defect sites, as evidenced by increased photoluminescence lifetime and recombination resistance.201,209,218 The interfacial defect passivation is, therefore, responsible for the increased PCE by reducing the energy loss and the enhanced long-term stability by stabilizing defects.
8.3. Favorable band energy alignment by interface management
Although the residual lattice strain would manipulate the band structure of the perovskite bulk film, the interface energy level alignment is mainly governed by the chemicals used to modulate the residual lattice strain at the interface of the perovskite.120,159,195,218 The interfacial energy band alignment is generally estimated by Kelvin probe force microscopy for work function, ultraviolet photoelectron spectroscopy for valence band maximum, and UV-vis spectrometer for bandgap. The Fermi level of the selective contact is highly dominated by a change in the surface potential of the chemical-treated selective contact,120 which is accordingly reflected in the energy alignment at the interface with varying energy offsets. Notably, the modified interfacial energy alignment is frequently found to either be favorable for charge extraction or effectively suppress the charge recombination at the interface,120,159,195 contributing to improved photovoltaic performance.
9. Summary and perspective
Halide perovskite film exhibits a soft lattice, particularly for APbI3, which can accommodate strain by undergoing lattice deformation based on octahedra PbI6− tilting. The polycrystalline nature of the perovskite film imposes local variation in structural orientation and consequently increases local strain heterogeneity in the perovskite lattice. The local strain is directly reflected in the chemical bonding in the core framework, which determines the local electronic band structure. The reliability of device performance is closely related to the uniform electronic band structure by minimizing local dependency. Therefore, the control of strain inhomogeneity is highly important for achieving reliability and reproducibility from the perspective of device performance. In addition, the lattice strain plays an important role in determining defect dynamics in the lattice, which is closely related to its formation and migration. The lattice stability is, therefore, inherent to the lattice strain because the atomic displacement under phase conversion can be significantly facilitated by involving defects. To this end, various approaches for enhancing crystallinity with a long-range structural orientation effectively reduce the local strain inhomogeneity in the perovskite film. Apart from the local strain and its heterogeneity, originating from a polycrystalline soft lattice, depth-dependent in-plane tensile strain is generally entailed in the solution-processed perovskite film and regarded as a more detrimental factor to device performance. The strain inhomogeneity along the out-of-plane direction is found to be resolved by regulating crystallization and/or interface management. Although the control of crystallization is aimed principally at avoiding the formation of the in-plane tensile strain, interface management usually pursues relaxation of the residual strain by employing an interlayer based on a flexible alkyl chain to effectively dissipate the strain at the interface. Effective strategies to relieve the in-plane tensile strain consequently exhibit improved device stability with enhanced performance owing to the synergistic effect on interfacial defect passivation and interfacial energy band alignment, which is favorable for charge collection.
Strain control strategies have mostly focused on the problematic residual tensile strain along the in-plane direction and have accordingly been devised to relieve residual strain to improve the long-term stability of PSCs. However, regulating the lattice strain can be one of the fascinating methods for delicately manipulating the electronic band structure and lattice stability. In this regard, the development of advanced technology to control the residual lattice strain is in high demand to passively deal with the current issue and to actively derive the maximal benefits in optoelectronic properties and lattice stability, which would likely pave the way for opening new horizons by overcoming the persistent stability issue.
Conflicts of interest
The authors declare no conflict of interest.
Data availability
This is a review article and does not report any original data. All data discussed are from previously published sources, which are appropriately cited within the text.
Acknowledgements
This work was supported by the National Research Foundation (NRF) grant funded by the Korea government (Ministry of Science and ICT) under contract NRF-2021R1C1C1009686 and RS-2023-00233865.
References
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2, 591 CrossRef PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- A. Machín and F. Márquez, Materials, 2024, 17, 1165 CrossRef PubMed.
- P. Chen, Y. Xiao, S. Li, X. Jia, D. Luo, W. Zhang, H. J. Snaith, Q. Gong and R. Zhu, Chem. Rev., 2024, 124, 10623–10700 CrossRef CAS PubMed.
- H. Zhang, X. Ji, H. Yao, Q. Fan, B. Yu and J. Li, Sol. Energy, 2022, 233, 421–434 CrossRef CAS.
- National Renewable Energy Laboratory, Champion Photovoltaic Module Efficiency Chart, https://www.nrel.gov/pv/cell-efficiency, (accessed January 2025).
- National Renewable Energy Laboratory, Best Research-Module Efficiency Chart, https://www2.nrel.gov/pv/module-efficiency, (accessed December 2024).
- H.-S. Kim and N.-G. Park, Appl. Phys. Rev., 2025, 12, 021313 CAS.
- H.-J. Kim, H.-S. Kim and N.-G. Park, Adv. Energy Sustainability Res., 2021, 2, 2000051 CrossRef CAS.
- H.-S. Kim and N.-G. Park, Adv. Energy Mater., 2025, 15, 2400089 CrossRef CAS.
- E. M. Hutter, M. C. Gélvez-Rueda, A. Osherov, V. Bulović, F. C. Grozema, S. D. Stranks and T. J. Savenije, Nat. Mater., 2017, 16, 115–120 CrossRef CAS PubMed.
- K. Galkowski, A. Mitioglu, A. Miyata, P. Plochocka, O. Portugall, G. E. Eperon, J. T.-W. Wang, T. Stergiopoulos, S. D. Stranks, H. J. Snaith and R. J. Nicholas, Energy Environ. Sci., 2016, 9, 962–970 RSC.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- K. Handloser, N. Giesbrecht, T. Bein, P. Docampo, M. Handloser and A. Hartschuh, ACS Photonics, 2016, 3, 255–261 CrossRef CAS.
- J. M. Ball, M. M. Lee, A. Hey and H. J. Snaith, Energy Environ. Sci., 2013, 6, 1739 RSC.
- N.-Y. Jo, Y.-K. Hong, S. Yang and H.-S. Kim, Bull. Korean Chem. Soc., 2024, 45, 570–575 CrossRef CAS.
- J. Suo, B. Yang, E. Mosconi, D. Bogachuk, T. A. S. Doherty, K. Frohna, D. J. Kubicki, F. Fu, Y. Kim, O. Er-Raji, T. Zhang, L. Baldinelli, L. Wagner, A. N. Tiwari, F. Gao, A. Hinsch, S. D. Stranks, F. D. Angelis and A. Hagfeldt, Nat. Energy, 2024, 9, 172–183 CrossRef CAS PubMed.
- Y. S. Shin, J. Lee, D. G. Lee, J. W. Song, J. Seo, J. Roe, M. J. Sung, S. Park, G. Y. Shin, J. Yeop, D. Lee, C. H. Yoon, M. Kim, J. G. Son, G.-H. Kim, S. Cho, J. Y. Kim, T. K. Lee and D. S. Kim, Energy Environ. Sci., 2025, 18, 3269–3277 RSC.
- Z. Zheng, Z. Xue, K. Zhao, Y. Yang, X. Zhu, H. Li, S. Cheng, S. Li, N. Yan and Z. Wang, Sol. RRL, 2024, 8, 2301076 CrossRef CAS.
- G. Tang, P. Ghosez and J. Hong, J. Phys. Chem. Lett., 2021, 12, 4227–4239 CrossRef CAS PubMed.
- H.-S. Kim and N.-G. Park, NPG Asia Mater., 2020, 12, 78 CrossRef CAS.
- H.-S. Kim and N.-G. Park, Adv. Mater., 2023, 35, 2204807 CrossRef CAS PubMed.
- B. Hou, C. Shang, C. Wang, D. Qu, J. Qiao, X. Zhang, W. Zhao, R. Han, S. Dong, Y. Xue, Y. Ke, F. Ye, X. Yang, Y. Tu and W. Huang, Research, 2024, 7, 0309 CrossRef PubMed.
- J. Li, L. Xie, Z. Pu, C. Liu, M. Yang, Y. Meng, B. Han, S. Bu, Y. Wang, X. Zhang, T. Wang and Z. Ge, Adv. Funct. Mater., 2023, 33, 2301956 CrossRef CAS.
- D.-J. Xue, Y. Hou, S.-C. Liu, M. Wei, B. Chen, Z. Huang, Z. Li, B. Sun, A. H. Proppe, Y. Dong, M. I. Saidaminov, S. O. Kelley, J.-S. Hu and E. H. Sargent, Nat. Commun., 2020, 11, 1514 CrossRef CAS PubMed.
- J. Zhao, Y. Deng, H. Wei, X. Zheng, Z. Yu, Y. Shao, J. E. Shield and J. Huang, Sci. Adv., 2017, 3, eaao5616 CrossRef PubMed.
- C. Zhu, X. Niu, Y. Fu, N. Li, C. Hu, Y. Chen, X. He, G. Na, P. Liu, H. Zai, Y. Ge, Y. Lu, X. Ke, Y. Bai, S. Yang, P. Chen, Y. Li, M. Sui, L. Zhang, H. Zhou and Q. Chen, Nat. Commun., 2019, 10, 815 CrossRef CAS PubMed.
- Y. Chen, Y. Lei, Y. Li, Y. Yu, J. Cai, M.-H. Chiu, R. Rao, Y. Gu, C. Wang, W. Choi, H. Hu, C. Wang, Y. Li, J. Song, J. Zhang, B. Qi, M. Lin, Z. Zhang, A. E. Islam, B. Maruyama, S. Dayeh, L.-J. Li, K. Yang, Y.-H. Lo and S. Xu, Nature, 2020, 577, 209–215 CrossRef CAS PubMed.
- S. Cheng, Y. Yang, X. Zhu, Y. Li, H. Li, W. Xiong, Z. Zheng, S. Li, Y. Liu, X. Liu, Q. Lin, S. Yuan, E. Shi and Z. Wang, Energy Environ. Sci., 2025, 18, 2452–2461 RSC.
- A. Garrote-Márquez, L. Lodeiro, R. Suresh, N. C. Hernández, R. Grau-Crespo and E. Menéndez-Proupin, J. Phys. Chem. C, 2023, 127, 15901 CrossRef PubMed.
- X. Yue, C. Wang, B. Zhang, Z. Zhang, Z. Xiong, X. Zu, Z. Liu, Z. Hu, G. O. Odunmbaku, Y. Zheng, K. Sun and J. Du, Nat. Commun., 2023, 14, 917 CrossRef CAS PubMed.
- B. Jin, J. Cao, R. Yuan, B. Cai, C. Wu and X. Zheng, Adv. Energy Sustainability Res., 2023, 4, 2200143 CrossRef CAS.
- Y. Wang, L. Gao, Y. Yang, Y. Xiang, Z. Chen, Y. Dong, H. Zhou, Z. Cai and G.-C. Wang, Phys. Rev. Mater., 2018, 2, 076002 CrossRef CAS.
- M. I. Saidaminov, J. Kim, A. Jain, R. Quintero-Bermudez, H. Tan, G. Long, F. Tan, A. Johnston, Y. Zhao, O. Voznyy and E. H. Sargent, Nat. Energy, 2018, 3, 648–654 CrossRef CAS.
- I. Borriello, G. Cantele and D. Ninno, Phys. Rev. B:Condens. Matter Mater. Phys., 2008, 77, 235214 CrossRef.
- G. E. Eperon, S. D. Stranks, C. Menelaou, M. B. Johnston, L. M. Herz and H. J. Snaith, Energy Environ. Sci., 2014, 7, 982–988 RSC.
- R. Prasanna, A. Gold-Parker, T. Leijtens, B. Conings, A. Babayigit, H.-G. Boyen, M. F. Toney and M. D. McGehee, J. Am. Chem. Soc., 2017, 139, 11117–11124 CrossRef CAS PubMed.
- W. Travis, E. N. K. Glover, H. Bronstein, D. O. Scanlon and R. G. Palgrave, Chem. Sci., 2016, 7, 4548–4556 RSC.
- T.-Y. Wang, Y.-Y. Hao, M.-Z. Zhu, G.-R. Cao and Z.-M. Zhou, Chin. J. Chem., 2024, 42, 1284–1306 CrossRef CAS.
- M. Senno and S. Tinte, Phys. Chem. Chem. Phys., 2021, 23, 7376–7385 RSC.
- O. J. Weber, D. Ghosh, S. Gaines, P. F. Henry, A. B. Walker, M. S. Islam and M. T. Weller, Chem. Mater., 2018, 30, 3768–3778 CrossRef CAS.
- M. Šimėnas, S. Balčiūnas, M. Mączka and J. Banys, J. Mater. Chem., 2022, 10, 5210–5217 Search PubMed.
- X.-G. Zhao, G. M. Dalpian, Z. Wang and A. Zunger, Phys. Rev. B, 2020, 101, 155137 CrossRef CAS.
- G. Laurita, D. H. Fabini, C. C. Stoumpos, M. G. Kanatzidis and R. Seshadri, Chem. Sci., 2017, 8, 5628 RSC.
- A. M. A. Leguy, A. R. Goñi, J. M. Frost, J. Skelton, F. Brivio, X. Rodríguez-Martínez, O. J. Weber, A. Pallipurath, M. Isabel Alonso, M. Campoy-Quiles, M. T. Weller, J. Nelson, A. Walsh and P. R. F. Barnes, Phys. Chem. Chem. Phys., 2016, 18, 27051–27066 RSC.
- M. A. Carignano, Y. Saeed, S. A. Aravindh, I. S. Roqan, J. Even and C. Katan, Phys. Chem. Chem. Phys., 2016, 18, 27109–27118 RSC.
- C. Quarti, E. Mosconi, J. M. Ball, V. D’Innocenzo, C. Tao, S. Pathak, H. J. Snaith, A. Petrozza and F. D. Angelis, Energy Environ. Sci., 2016, 9, 155 RSC.
- J.-S. Park, S. Choi, Y. Yan, Y. Yang, J. M. Luther, S.-H. Wei, P. Parilla and K. Zhu, J. Phys. Chem. Lett., 2015, 6, 4304 CrossRef CAS PubMed.
- J. S. Bechtel, R. Seshadri and A. V. D. Ven, J. Phys. Chem. C, 2016, 120, 12403 CrossRef CAS.
- D. Zhang, Y. Zhu, L. Liu, X. Ying, C.-E. Hsiung, R. Sougrat, K. Li and Y. Han, Science, 2018, 359, 675–679 CrossRef CAS PubMed.
- D. H. Fabini, C. C. Stoumpos, G. Laurita, A. Kaltzoglou, A. G. Kontos, P. Falaras, M. G. Kanatzidis and R. Seshadri, Angew. Chem., Int. Ed., 2016, 55, 15392–15396 CrossRef CAS PubMed.
- T. Chen, B. J. Foley, C. Park, C. M. Brown, L. W. Harriger, J. Lee, J. Ruff, M. Yoon, J. J. Choi and S.-H. Lee, Sci. Adv., 2016, 2, e1601650 CrossRef PubMed.
- B. Charles, J. Dillon, O. J. Weber, M. S. Islam and M. T. Weller, J. Mater. Chem. A, 2017, 5, 22495–22499 RSC.
- M. Kim, G.-H. Kim, T. K. Lee, I. W. Choi, H. W. Choi, Y. Jo1, Y. J. Yoon, J. W. Kim, J. Lee, D. Huh, H. Lee, S. K. Kwak, J. Y. Kim and D. S. Kim, Joule, 2019, 3, 2179–2192 CrossRef CAS.
- F. Ye, J. Ma, C. Chen, H. Wang, Y. Xu, S. Zhang, T. Wang, C. Tao and G. Fang, Adv. Mater., 2021, 33, 2007126 CrossRef CAS PubMed.
- J. S. Bechtel and A. V. D. Ven, Phys. Rev. Mater., 2018, 2, 025401 CrossRef CAS.
-
R. J. Tilley, Perovskites: structure-property relationships, John Wiley & Sons, United Kingdom, 2016 Search PubMed.
- X. Zheng, C. Wu, S. K. Jha, Z. Li, K. Zhu and S. Priya, ACS Energy Lett., 2016, 1, 1014–1020 CrossRef CAS.
- G. Kim, H. Min, K. S. Lee, D. Y. Lee, S. M. Yoon and S. I. Seok, Science, 2020, 370, 108–112 CrossRef CAS PubMed.
- S. Jariwala, H. Sun, G. W. P. Adhyaksa, A. Lof, L. A. Muscarella, B. Ehrler, E. C. Garnett and D. S. Ginger, Joule, 2019, 3, 3048–3060 CrossRef CAS.
- Z. Liang, J. Cao, Z. Zhou, L. Ren, H. Wu, Z. Wang, C. Yi, D. Yang, K. Wang and C. Wu, Nat. Synth., 2025, 1–12 Search PubMed.
- T. W. Jones, A. Osherov, M. Alsari, M. Sponseller, B. C. Duck, Y.-K. Jung, C. Settens, F. Niroui, R. Brenes, C. V. Stan, Y. Li, M. Abdi-Jalebi, N. Tamura, J. E. Macdonald, M. Burghammer, R. H. Friend, V. Bulović, A. Walsh, G. J. Wilson, S. Lilliu and S. D. Stranks, Energy Environ. Sci., 2019, 12, 596–606 RSC.
- M. U. Rothmann, W. Li, J. Etheridge and Y.-B. Cheng, Adv. Energy Mater., 2017, 7, 1700912 CrossRef.
- G. W. P. Adhyaksa, S. Brittman, H. Āboliņš, A. Lof, X. Li, J. D. Keelor, Y. Luo, T. Duevski, R. M. A. Heeren, S. R. Ellis, D. P. Fenning and E. C. Garnett, Adv. Mater., 2018, 30, 1804792 CrossRef PubMed.
- Z. Guo, J. S. Manser, Y. Wan, P. V. Kamat and L. Huang, Nat. Commun., 2015, 6, 7471 CrossRef CAS PubMed.
- A. M. Stoneham, Rep. Prog. Phys., 1981, 44, 1251–1295 CrossRef.
- W. Li, S. K. Yadavalli, D. Lizarazo-Ferro, M. Chen, Y. Zhou, N. P. Padture and R. Zia, ACS Energy Lett., 2018, 3, 2669–2670 CrossRef CAS.
- S. Li, Y. Xiao, R. Su, W. Xu, D. Luo, P. Huang, L. Dai, P. Chen, P. Caprioglio, K. A. Elmestekawy, M. Dubajic, C. Chosy, J. Hu, I. Habib, A. Dasgupta, D. Guo, Y. Boeije, S. J. Zelewski, Z. Lu, T. Huang, Q. Li, J. Wang, H. Yan, H.-H. Chen, C. Li, B. A. I. Lewis, D. Wang, J. Wu, L. Zhao, B. Han, J. Wang, L. M. Herz, J. R. Durrant, K. S. Novoselov, Z.-H. Lu, Q. Gong, S. D. Stranks, H. J. Snaith and R. Zhu, Nature, 2024, 635, 874–881 CrossRef PubMed.
- S. Jariwala, H. Sun, G. W. P. Adhyaksa, A. Lof, L. A. Muscarella, B. Ehrler, E. C. Garnett and D. S. Ginger, Joule, 2019, 3, 3048–3060 CrossRef CAS.
- M.-J. Choi, J.-W. Lee and H. W. Jang, Adv. Mater., 2024, 36, 2308827 CrossRef CAS PubMed.
- J. Zhou, S. Fu, S. Zhou, L. Huang, C. Wang, H. Guan, D. Pu, H. Cui, C. Wang, T. Wang, W. Meng, G. Fang and W. Ke, Nat. Commun., 2024, 15, 2324 CrossRef CAS PubMed.
- Z. Qin, M. Chen, Z. Zhang, Y. Wang and L. Han, Energy Environ. Sci., 2025, 18, 2264–2272 RSC.
- M. Hao, J. Yang, W. Yu, B. J. Lawrie, P. Guo, X. Zhang, T. Duan, T. Xiao, L. Chen, Y. Xiang, P. Guo, M. Ahmadi and Y. Zhou, Nat. Nanotechnol., 2025, 20, 630–638 CrossRef CAS PubMed.
- Z. Liang, Y. Zhang, H. Xu, W. Chen, B. Liu, J. Zhang, H. Zhang, Z. Wang, D.-H. Kang, J. Zeng, X. Gao, Q. Wang, H. Hu, H. Zhou, X. Cai, X. Tian, P. Reiss, B. Xu, T. Kirchartz, Z. Xiao, S. Dai, N.-G. Park, J. Ye and X. Pan, Nature, 2023, 62, 557–563 CrossRef PubMed.
- T. Huang, F. Xu, J. Hu, J. Wu, S. Li, P. Chen, X. Jia, Q. Li, H. Yan, Y. Ji, D. Luo, D. Wang, J. Hu, H.-H. Chen, Z. Lu, H. Xu, L. Li, R. Sha, Q. Zhong, X. Bai, M. I. Dar, T. Song, Z. Li, X. Yang, L. Zhao, Z.-H. Lu, Q. Gong and R. Zhu, Energy Environ. Sci., 2024, 17, 5984–5992 RSC.
- L. Zheng, M. Wei, F. T. Eickemeyer, J. Gao, B. Huang, U. Gunes, P. Schouwink, D. W. Bi, V. Carnavali, M. Mensi, F. Biasoni, Y. Zhang, L. Agosta, V. Slama, N. Lempesis, M. A. Hope, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, L. Pfeifer, Y. Xuan and M. Grätzel, Science, 2025, 388, 88–95 CrossRef CAS PubMed.
- J. Xu, C. C. Boyd, Z. J. Yu, A. F. Palmstrom, D. J. Witter, B. W. Larson, R. M. France, J. Werner, S. P. Harvey, E. J. Wolf, W. Weigand, S. Manzoor, M. F. A. M. van Hest, J. J. Berry, J. M. Luther, Z. C. Holman and M. D. McGehee, Science, 2020, 367, 1097–1104 CrossRef CAS PubMed.
- P. Wang, Y. Wu, B. Cai, Q. Ma, X. Zheng and W.-H. Zhang, Adv. Funct. Mater., 2019, 29, 1807661 CrossRef CAS.
- D. Liu, D. Luo, A. N. Iqbal, K. W. P. Orr, T. A. S. Doherty, Z.-H. Lu, S. D. Stranks and W. Zhang, Nat. Mater., 2021, 20, 1337–1346 CrossRef CAS PubMed.
- M. Keshavarz, M. Ottesen, S. Wiedmann, M. Wharmby, R. Küchler, H. Yuan, E. Debroye, J. A. Steele, J. Martens, N. E. Hussey, M. Bremholm, M. B. J. Roeffaers and J. Hofkens, Adv. Mater., 2019, 31, 1900521 CrossRef PubMed.
- T. J. Jacobsson, L. J. Schwan, M. Ottosson, A. Hagfeldt and T. Edvinsson, Inorg. Chem., 2015, 54, 10678–10685 CrossRef CAS PubMed.
- H. Tsai, R. Asadpour, J.-C. Blancon, C. C. Stoumpos, O. Durand, J. W. Strzalka, B. Chen, R. Verduzco, P. M. Ajayan, S. Tretiak, J. Even, M. A. Alam, M. G. Kanatzidis, W. Nie and A. D. Mohite, Science, 2018, 360, 67–70 CrossRef CAS PubMed.
- H. Zhang and N.-G. Park, J. Phys. Energy, 2023, 5, 024002 CrossRef.
- W. Mao, C. R. Hall, S. Bernardi, Y.-B. Cheng, A. Widmer-Cooper, T. A. Smith and U. Bach, Nat. Mater., 2021, 20, 55 CrossRef CAS PubMed.
- Q. Li, Y. Zheng, H. Wang, X. Liu, M. Lin, X. Sui, X. Leng, D. Liu, Z. Wei, M. Song, D. Li, H. G. Yang, S. Yang and Y. Hou, Science, 2025, 387, 1069–1077 CrossRef CAS PubMed.
- N. Rolston, R. Bennett-Kennett, L. T. Schelhas, J. M. Luther, J. A. Christians, J. J. Berry and R. H. Dauskardt, Science, 2020, 368, eaay8691 CrossRef CAS PubMed.
- W. Meng, K. Zhang, A. Osvet, J. Zhang, W. Gruber, K. Forberich, B. Meyer, W. Heiss, T. Unruh, N. Li and C. J. Brabec, Joule, 2022, 6, 458–475 CrossRef CAS.
- Y. Shen, T. Zhang, G. Xu, J. A. Steele, X. Chen, W. Chen, G. Zheng, J. Li, B. Guo, H. Yang, Y. Wu, X. Lin, T. Alshahrani, W. Yin, J. Zhu, F. Wang, A. Amassian, X. Gao, X. Zhang, F. Gao, Y. Li and Y. Li, Nature, 2024, 635, 882–889 CrossRef CAS PubMed.
- Q. Wang, B. Chen, Y. Liu, Y. Deng, Y. Bai, Q. Dong and J. Huang, Energy Environ. Sci., 2017, 10, 516–522 RSC.
- J. S. Yun, J. Kim, T. Young, R. J. Patterson, D. Kim, J. Seidel, S. Lim, M. A. Green, S. Huang and A. Ho-Baillie, Adv. Funct. Mater., 2018, 28, 1705363 CrossRef.
- S. Shin, S.-H. Yang, S. Seo, H. Park, U. Amornkitbamrung, Y. In, C. Wang, T. Nakamura, A. Wakamiya, Y.-M. Kim, Y.-M. Kim, Y.-M. Kim and H. Shin, ACS Energy Lett., 2024, 9, 3618–3627 CrossRef CAS.
- C. Gammer, J. Kacher, C. Czarnik, O. L. Warren, J. Ciston and A. M. Minor, Appl. Phys. Lett., 2016, 109, 081906 CrossRef.
- D. Ivanov, S. Ivanov, S. Lomov and I. Verpoest, Opt. Lasers Eng., 2009, 47, 360–370 CrossRef.
- J. Wu, S.-C. Liu, Z. Li, S. Wang, D.-J. Xue, Y. Lin and J.-S. Hu, Natl. Sci. Rev., 2021, 8, nwab047 CrossRef CAS PubMed.
- C. Xing, Q. Bao, S. Liu, X. Chu, F. Lu, L. Zhang, L. Yu, T. Zhang and D. Wang, Sol. RRL, 2023, 7, 2300434 CrossRef CAS.
- G. Yuan, W. Xie, Q. Song, S. Ma, Y. Ma, C. Shi, M. Xiao, F. Pei, X. Niu, Y. Zhang, J. Dou, C. Zhu, Y. Bai, Y. Wu, H. Wang, Q. Fan and Q. Chen, Adv. Mater., 2023, 35, 2211257 CrossRef CAS PubMed.
- R. Wang, X. Li, J. Qi, C. Su, J. Yang, S. Yang, M. Yuan and T. He, Adv. Mater., 2023, 35, 2304149 CrossRef CAS PubMed.
- L. Ning, L. Song, Z. Yao, W.-H. Chen, P. Du, P.-C. Jiang and J. Xiong, Adv. Energy Mater., 2024, 14, 2401320 CrossRef CAS.
- M. Dimitrievska, A. Fairbrother, R. Gunder, G. Gurieva, H. Xie, E. Saucedo, A. Pérez-Rodríguez, V. Izquierdo-Roca and S. Schorr, Phys. Chem. Chem. Phys., 2016, 18, 8692–8700 RSC.
- C. Luo, G. Zheng, F. Gao, X. Wang, C. Zhan, X. Gao and Q. Zhao, Nat. Photonics, 2023, 17, 856–864 CrossRef CAS.
-
J. Epp, X-ray diffraction (XRD) techniques for materials characterization, Woodhead Publishing, 2016, pp. 81–124 Search PubMed.
- D.-A. Park, C. Zhang and N.-G. Park, ACS Energy Lett., 2024, 9, 2428–2435 CrossRef CAS.
-
M. J. Berger, J. H. Hubbell, S. M. Seltzer, J. Chang, J. S. Coursey, R. Sukumar, D. S. Zucker and K. Olsen, XCOM: photon cross sections database, https://www.nist.gov/pml/data/xcom/index.cfm, 1998 Search PubMed.
- A. K. Zak, W. H. A. Majid, M. E. Abrishami and R. Yousefi, Solid State Sci., 2011, 13, 251–256 CrossRef.
- J. Liu, R. E. Saw and Y.-H. Kiang, J. Pharm. Sci., 2010, 99, 3807–3814 CrossRef CAS PubMed.
- U. Welzel, J. Ligot, P. Lamparter, A. C. Vermeulenb and E. J. Mittemeijer, J. Appl. Crystallogr., 2005, 38, 1–29 CrossRef.
- C.-H. Ma, J.-H. Huang and H. Chen, Thin Solid Films, 2002, 418, 73–78 CrossRef CAS.
- T. Yang, Y. Yang, T. Niu, E. Zhao, N. Wu, S. Wang, X. Chen, S. Wang, Y. Wang, Y. Wu, Z. Zhang, C. Ma, Y. Gong, D. Liu and K. Zhao, ACS Energy Lett., 2024, 10, 427–438 CrossRef.
- C. Zhang, S. Wu, L. Tao, G. M. Arumugam, C. Liu, Z. Wang, S. Zhu, Y. Yang, J. Lin, X. Liu, R. E. I. Schropp and Y. Mai, Adv. Energy Mater., 2020, 10, 2002004 CrossRef CAS.
- X. Xu, Q. Du, H. Kang, X. Gu, C. Shan, J. Zeng, T. Dai, Q. Yang, X. Sun, G. Li, E. Zhou, G. Luo, B. Xu and A. K. K. Kyaw, Adv. Funct. Mater., 2024, 34, 2408512 CrossRef CAS.
- X. Wang and A. V. Riessen, Powder Diffr., 2017, 32, S9–S15 CrossRef.
- H. Xu, Y. Xiao, K. A. Elmestekawy, P. O. Caprioglio, Q. Li, Q. Zhong, Y. Ji, T. Huang, H. Yan, Y. Yang, L. M. Herz, Q. Gong, H. J. Snaith, R. Zhu and L. Zhao, Energy Environ. Sci., 2025, 18, 246–255 RSC.
- P. Bindu and S. Thomas, J. Theor. Appl. Phys., 2014, 8, 123–134 CrossRef.
- S. A. Hassanzadeh-Tabrizi, J. Alloys Compd., 2023, 968, 171914 CrossRef CAS.
- G. Srinet, S. Sharma, M. Kumar and A. Anshul, Phys. E, 2021, 125, 114381 CrossRef CAS.
- H. Bantikatla, L. D. N.S.M.P. and R. K. Bhogoju, Mater. Today: Proc., 2021, 47, 4891–4896 Search PubMed.
- A. M. Sami and K. H. Harbbi, AIP Conf. Proc., 2022, 2394, 090018 CrossRef.
- F. T. L. Muniz, M. A. R. Miranda, C. Morilla dos Santos and J. M. Sasake, Acta Crystallogr., 2016, 72, 385–390 CrossRef CAS PubMed.
- A. S. Abdel-Rahman and Y. A. Sabry, Int. J. Non-Linear Mech., 2024, 161, 104670 CrossRef.
- X. Wang, H. Huang, M. Wang, Z. Lan, P. Cui, S. Du, Y. Yang, L. Yan, Q. Zhang, S. Qu and M. Li, Adv. Mater., 2024, 36, 2310710 CrossRef CAS PubMed.
- P. Yang, J. Wu, W. Lin, X. Jiang, Y. Wang, W. Sun, Z. Lan and J. Lin, Chem. Eng., 2024, 488, 151128 CrossRef CAS.
- Y. Meng, Y. Wang, C. Liu, P. Yan, K. Sun, Y. Wang, R. Tian, R. Cao, J. Zhu, H. Do, J. Lu and Z. Ge, Adv. Mater., 2024, 36, 2309208 CrossRef CAS PubMed.
- Q. Zhou, J. Duan, X. Yang, Y. Duan and Q. Tang, Angew. Chem., Int. Ed., 2020, 132, 22181–22185 CrossRef.
- J. Kim, J.-S. Park, G. Y. Kim and W. Jo, Adv. Energy Mater., 2024, 14, 2402117 CrossRef CAS.
- Y. Zhou, A. L. Vasiliev, W. Wu, M. Yang, S. Pang, K. Zhu and N. P. Padture, J. Phys. Chem. Lett., 2015, 6, 2292–2297 CrossRef CAS PubMed.
- M. U. Rothmann, W. Li, Y. Zhu, A. Liu, Z. Ku, U. Bach, J. Etheridge and Y.-B. Cheng, Adv. Mater., 2018, 30, 1800629 CrossRef PubMed.
- N. D. Bassim, B. T. DE Gregorio, A. L. D. Kilcoyne, K. Scott, T. Chou, S. Wirick, G. Cody and R. M. Stroud, J. Microsc., 2012, 245, 288–301 CrossRef CAS.
- L. Hao and S. Cai, Microstructures, 2025, 5, 2025012 CrossRef CAS.
- Y. Miao, Y. Zhao, S. Zhang, R. Shi and T. Zhang, Adv. Mater., 2022, 34, 2200868 CrossRef CAS PubMed.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. Jin Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Grätzel and J. Y. Kim, Nature, 2021, 592, 381–385 CrossRef CAS PubMed.
- Q. Tai, P. You, H. Sang, Z. Liu, C. Hu, H. L. W. Chan and F. Yan, Nat. Commun., 2016, 7, 11105 CrossRef CAS PubMed.
- C. Liang, H. Gu, Y. Xia, Z. Wang, X. Liu, J. Xia, S. Zuo, Y. Hu, X. Gao, W. Hui, L. Chao, T. Niu, M. Fang, H. Lu, H. Dong, H. Yu, S. Chen, X. Ran, L. Song, B. Li, J. Zhang, Y. Peng, G. Shao, J. Wang, Y. Chen, G. Xing and W. Huang, Nat. Energy, 2021, 6, 38–45 CrossRef CAS.
- J. Park, J. Kim, H.-S. Yun, M. J. Paik, E. Noh, H. J. Mun, M. G. Kim, T. J. Shin and S. I. Seok, Nature, 2023, 616, 724–730 CrossRef CAS PubMed.
- A. Hassan, Z. Wang, Y. H. Ahn, M. Azam, A. A. Khan, U. Farooq, M. Zubair and Y. Cao, Nano Energy, 2022, 101, 107579 CrossRef CAS.
- X. Yuan, R. Li, Z. Xiong, P. Li, G. O. Odunmbaku, K. Sun, Y. Deng and S. Chen, Adv. Funct. Mater., 2023, 33, 2215096 CrossRef CAS.
- N. Li, Y. Luo, Z. Chen, X. Niu, X. Zhang, J. Lu, R. Kumar, J. Jiang, H. Liu, X. Guo, B. Lai, G. Brocks, Q. Chen, S. Tao, D. P. Fenning and H. Zhou, Joule, 2020, 4, 1743–1758 CrossRef CAS.
- Y. Bai, Z. Huang, X. Zhang, J. Lu, X. Niu, Z. He, C. Zhu, M. Xiao, Q. Song, X. Wei, C. Wang, Z. Cui, J. Dou, Y. Chen, F. Pei, H. Zai, W. Wang, T. Song, P. An, J. Zhang, J. Dong, Y. Li, J. Shi, H. Jin, P. Chen, Y. Sun, Y. Li, H. Chen, Z. Wei, H. Zhou and Q. Chen, Science, 2022, 378, 747–754 CrossRef CAS PubMed.
- Y. Zheng, X. Wu, J. Liang, Z. Zhang, J. Jiang, J. Wang, Y. Huang, C. Tian, L. Wang, Z. Chen and C.-C. Chen, Adv. Funct. Mater., 2022, 32, 2200431 CrossRef CAS.
- H. Min, S.-G. Ji and S. I. Seok, Joule, 2022, 6, 2175–2185 CrossRef CAS.
- S. Cai, J. Gao, Y. Wu, Y. Zou, J. Liang, Y. Li, X. He, Q. Cai, M. Wang, X. Huang, X. Wang, S. Sajid, D. Wei, R. Zhang, D. Song and Y. Wang, Adv. Funct. Mater., 2024, 34, 2411014 CrossRef CAS.
- H. Zhang, Z. Wang, H. Wang, X. Yao, F. Wang, S. Wang, S. Bai, J. Huang, X. Luo, S. Wu and X. Liu, J. Mater. Chem. A, 2023, 11, 15301–15310 RSC.
- H. Zhang, Z. Chen, M. Qin, Z. Ren, K. Liu, J. Huang, D. Shen, Z. Wu, Y. Zhang, J. Hao, C.-S. Lee, X. Lu, Z. Zheng, W. Yu and G. Li, Adv. Mater., 2021, 33, 2008487 CrossRef CAS PubMed.
- X. Li, W. Zhang, Y.-C. Wang, W. Zhang, H.-Q. Wang and J. Fang, Nat. Commun., 2018, 9, 3806 CrossRef PubMed.
- X. Li, S. Fu, S. Liu, Y. Wu, W. Zhang, W. Song and J. Fang, Nano Energy, 2019, 64, 103962 CrossRef CAS.
- H. Guo, G. W. Yoon, Z. J. Li, Y. Yun, S. Lee, Y.-H. Seo, N. J. Jeon, G. S. Han and H. S. Jung, Adv. Energy Mater., 2024, 14, 2302743 CrossRef CAS.
- F. Qiu, J. Sun and J. Qi, Chem. Eng. J., 2022, 430, 132869 CrossRef CAS.
- J. Zhang, Z. Li, F. Guo, H. Jiang, W. Yan, C. Peng, R. Liu, L. Wang, H. Gao, S. Pang and Z. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202305221 CrossRef PubMed.
- J. Zhang, X. Niu, C. Peng, H. Jiang, L. Yu, H. Zhou and Z. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202314106 CrossRef CAS PubMed.
- G. Tang, P. You, Q. Tai, A. Yang, J. Cao, F. Zheng, Z. Zhou, J. Zhao, P. K. L. Chan and F. Yan, Adv. Mater., 2019, 31, 1807689 CrossRef PubMed.
- M. B. Faheem, B. Khan, C. Feng, W. S. Subhani, S. Mabrouk, M. H. Sayyad, A. Yildiz, W.-H. Zhang and Q. Qiao, Adv. Mater. Interfaces, 2022, 9, 2200421 CrossRef CAS.
- Q. Gao, J. Qi, K. Chen, M. Xia, Y. Hu, A. Mei and H. Han, Adv. Mater., 2022, 34, 2200720 CrossRef CAS PubMed.
- K. Wang, Z. Xu, Z. Guo, H. Wang, S. M. H. Qaid, K. Yang and Z. Zang, Adv. Energy Mater., 2024, 14, 2402249 CrossRef CAS.
- B. Zhang, Y. Tian, J. X. Zhang and W. Cai, Appl. Phys. Lett., 2011, 98, 021906 CrossRef.
- F. Zeng, L. Xu, C. Hu, J. Xing, Y. Wu, X. Bai, B. Dong and H. Song, Adv. Funct. Mater., 2025, 35, 2415547 CrossRef CAS.
- B. Liu, Q. Zhou, Y. Li, Y. Chen, D. He, D. Ma, X. Han, R. Li, K. Yang, Y. Yang, S. Lu, X. Ren, Z. Zhang, L. Ding, J. Feng, J. Yi and J. Chen, Angew. Chem., Int. Ed., 2024, 63, e202317185 CrossRef CAS PubMed.
- Q. Zhou, B. Liu, Y. Chen, D. Ma, X. Han, D. He, Z. Zhang, H. Yang, L. Ding, J. Feng, J. Yi, C. Chen and J. Chen, Adv. Funct. Mater., 2024, 34, 2315064 CrossRef CAS.
- H. Yang, Z. Xu, H. Wang, S. M. H. Qaid, O. F. Mohammed and Z. Zang, Adv. Mater., 2024, 36, 2411721 CrossRef CAS PubMed.
- Z. Long, C. Peng, K. Dong, H. Jiang, M. Zhu, W. Yan, Y. Dong, W. Jiang, L. Wen, X. Jiang and Z. Zhou, Adv. Funct. Mater., 2024, 34, 2408818 CrossRef CAS.
- Q. Zhou, D. He, Q. Zhuang, B. Liu, R. Li, H. Li, Z. Zhang, H. Yang, P. Zhao, Y. He, Z. Zang and J. Chen, Adv. Funct. Mater., 2022, 32, 2205507 CrossRef CAS.
- J. Wang, S. Liu, X. Guan, K. Wang, S. Shen, C. Cong, C.-C. Chen and F. Xie, ACS Appl. Mater. Interfaces, 2024, 16, 35732–35739 CrossRef CAS PubMed.
- L. Xie, J. Chen, P. Vashishtha, X. Zhao, G. S. Shin, S. G. Mhaisalkar and N.-G. Park, ACS Energy Lett., 2019, 4, 2192–2200 CrossRef CAS.
- L. Zhu, X. Zhang, M. Li, X. Shang, K. Lei, B. Zhang, C. Chen, S. Zheng, H. Song and J. Chen, Adv. Energy Mater., 2021, 11, 2100529 CrossRef CAS.
- Z. Wu, M. Jiang, Z. Liu, A. Jamshaid, L. K. Ono and Y. Qi, Adv. Energy Mater., 2020, 10, 1903696 CrossRef CAS.
- T. Li, S. Wang, J. Yang, X. Pu, B. Gao, Z. He, Q. Cao, J. Han and X. Li, Nano Energy, 2021, 82, 105742 CrossRef CAS.
- H. Bi, B. Liu, D. He, L. Bai, W. Wang, Z. Zang and J. Chen, Chem. Eng. J., 2021, 418, 129375 CrossRef CAS.
- L. Wang, C. Wang, J. Li, C. Geng, Y. Mo, H. Li, T. Bu, J. Tong, Y.-B. Cheng and F. Huang, Sol. RRL, 2023, 7, 2300144 CrossRef CAS.
- Q. Song, H. Gong, F. Sun, M. Li, T. Zhu, C. Zhang, F. You, Z. He, D. Li and C. Liang, Small, 2023, 19, 2208260 CrossRef CAS PubMed.
- D. Music and L. Elalfy, J. Phys. Condens. Matter, 2019, 31, 125101 CrossRef PubMed.
- W. Lv, Z. Hu, W. Qiu, D. Yan, M. Li, A. Mei, L. Xu and R. Chen, Adv. Sci., 2022, 9, 2202028 CrossRef CAS PubMed.
- J. Fang, Z. Ding, X. Chang, J. Lu, T. Yang, J. Wen, Y. Fan, Y. Zhang, T. Luo, Y. Chen, S. (Frank) Liu and K. Zhao, J. Mater. Chem. A, 2021, 9, 13297–13305 RSC.
- J. Suo, B. Yang, D. Bogachuk, G. Boschloo and A. Hagfeldt, Adv. Energy Mater., 2025, 15, 2400205 CrossRef CAS.
- M. Azam, T. Du, Z. Wan, H. Zhao, H. Zeng, R. Wei, C. J. Brabec, J. Luo and C. Jia, Energy Environ. Sci., 2024, 17, 6974–7016 RSC.
- H. Zhou, W. Wang, Y. Duan, R. Sun, Y. Li, Z. Xie, D. Xu, M. Wu, Y. Wang, H. Li, Q. Fan, Y. Peng, Y. Yao, C. Liao, Q. Peng, S. Liu and Z. Liu, Angew. Chem., Int. Ed., 2024, 63, e202403068 CrossRef CAS PubMed.
- N. Rolston, K. A. Bush, A. D. Printz, A. Gold-Parker, Y. Ding, M. F. Toney, M. D. McGehee and R. H. Dauskardt, Adv. Energy Mater., 2018, 8, 1802139 CrossRef.
- S. Fu, N. Sun, H. Chen, Y. Li, Y. Li, X. Zhu, B. Feng, X. Guo, C. Yao, W. Zhang, X. Li and J. Fang, Energy Environ. Sci., 2025, 18, 3305–3312 RSC.
- Y. Zhang, R. Yu, M. Li, Z. He, Y. Dong, Z. Xu, R. Wang, Z. Ma and Z. Tan, Adv. Mater., 2024, 36, 2310203 CrossRef CAS PubMed.
- C. Zhang, C. Liang, F. Sun, T. Zhu, X. Huang, Y. Guo, X. Guo, K. Ge, D. Li, F. You and Z. He, Sci. China:Chem., 2025, 68, 163–173 CrossRef CAS.
- M. Mori, T. Yamamoto, H. Itoh, H. Inaba and H. Tagawa, J. Electrochem. Soc., 1998, 145, 1374 CrossRef CAS.
- E. C. Schueller, G. Laurita, D. H. Fabini, C. C. Stoumpos, M. G. Kanatzidis and R. Seshadri, Inorg. Chem., 2018, 57, 695–701 CrossRef CAS PubMed.
- Z. Xiong, L. Lan, Y. Wang, C. Lu, S. Qin, S. Chen, L. Zhou, C. Zhu, S. Li, L. Meng, K. Sun and Y. Li, ACS Energy Lett., 2021, 6, 3824–3830 CrossRef CAS.
- M. Kim, J. Jeong, H. Lu, T. K. Lee, F. T. Eickemeyer, Y. Liu, I. W. Choi, S. J. Choi, Y. Jo, H.-B. Kim, S.-I. Mo, Y.-K. Kim, H. Lee, N. G. An, S. Cho, W. R. Tress, S. M. Zakeeruddin, A. Hagfeldt, J. Y. Kim, M. Grätzel and D. S. Kim, Science, 2022, 375, 302–306 CrossRef CAS PubMed.
- B. Wang, J. Ma, Z. Li, G. Chen, Q. Gu, S. Chen, Y. Zhang, Y. Song, J. Chen, X. Pi, X. Yu and D. Yang, Nano Res., 2022, 15, 1069–1078 CrossRef CAS.
- Y. Zhao, L. Gao, Q. Wang, Q. Zhang, X. Yang, J. Zhu, H. Huang, J. Duan and Q. Tang, Carbon Energy, 2024, 6, e468 CrossRef CAS.
- P. Fenter, F. Schreiber, V. Bulović and S. R. Forrest, Chem. Phys. Lett., 1997, 277, 521–526 CrossRef CAS.
- S.-Y. Ju, W. I. Lee and H.-S. Kim, ACS Appl. Mater. Interfaces, 2022, 14, 39996–40004 CrossRef CAS PubMed.
- S. Yin, X. Luo, F. Tang, Z. Xiong, Y. Lin, W. Yang, Y. Shu, Y. Wang and L. Ying, Energy Environ. Sci., 2025, 18, 4153–4161 RSC.
- Y. Huang, T. Liu, D. Li, Q. Lian, Y. Wang, G. Wang, G. Mi, Y. Zhou, A. Amini, B. Xu, Z. Tang, C. Cheng and G. Xing, Small, 2022, 18, 2201694 CrossRef CAS PubMed.
- Y. Ma, J. Ge, A. K.-Y. Jen, J. You and S. F. Liu, Adv. Opt. Mater., 2024, 12, 2301623 CrossRef CAS.
- G. Kapil, T. Bessho, C. H. Ng, K. Hamada, M. Pandey, M. A. Kamarudin, D. Hirotani, T. Kinoshita, T. Minemoto, Q. Shen, T. Toyoda, T. N. Murakami, H. I. Segawa and S. Hayase, ACS Energy Lett., 2019, 4, 1991–1998 CrossRef CAS.
- J. Zhuang, P. Mao, Y. Luan, N. Chen, X. Cao, G. Niu, F. Jia, F. Wang, S. Cao and J. Wang, Adv. Funct. Mater., 2021, 31, 2010385 CrossRef CAS.
- X. Zhang, D. Zhang, Y. Zhou, Y. Du, J. Jin, Z. Zhu, Z. Wang, X. Cui, J. Li, S. Wu, J. Zhang and Q. Tai, Adv. Funct. Mater., 2022, 32, 2205478 CrossRef CAS.
- M. Saliba, T. Matsui, K. Domanski, J.-Y. Seo, A. Ummadisingu, S. M. Zakeeruddin, J.-P. Correa-Baena, W. R. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Science, 2016, 354, 206–209 CrossRef CAS PubMed.
- J.-W. Lee, D.-H. Kim, H.-S. Kim, S.-W. Seo, S. M. Cho and N.-G. Park, Adv. Energy Mater., 2015, 5, 1501310 CrossRef.
- L. Zhou, M. Sui, J. Zhang, K. Cao, H. Wang, H. Yuan, Z. Lin, J. Zhang, P. Li, Y. Hao and J. Chang, Chem. Eng. J., 2024, 496, 154043 CrossRef CAS.
- Q. Xiao, Y. Zhao, Z. Huang, Y. Liu, P. Chen, S. Wang, S. Zhang, Y. Zhang and Y. Song, Adv. Funct. Mater., 2024, 34, 2314472 CrossRef CAS.
- Z. Zhu, C. Zhu, L. Yang, Q. Chen, L. Zhang, J. Dai, J. Cao, S. Zeng, Z. Wang, Z. Wang, W. Zhang, J. Bao, L. Yang, Y. Yang, B. Chen, C. Yin, H. Chen, Y. Cao, H. Gu, J. Yan, N. Wang, G. Xing, H. Li, X. Wang, S. Li, Z. Liu, H. Zhang, L. Wang, X. Huang and W. Huang, Nat. Mater., 2022, 21, 1042–1049 CrossRef CAS PubMed.
- C. Zhu, X. Wang, H. Li, C. Wang, Z. Gao, P. Zhang, X. Niu, N. Li, Z. Xu, Z. Su, Y. Chen, H. Zai, H. Xie, Y. Zhao, N. Yang, G. Liu, X. Wang, H. Zhou, J. Hong, X. Gao, Y. Bai and Q. Chen, Interdiscip. Mater., 2023, 2, 348–359 CAS.
- H. Wang, W. Zhang, B. Wang, Z. Yan, C. Chen, Y. Hua, T. Wu, L. Wang, H. Xu and M. Cheng, Nano Energy, 2023, 111, 108363 CrossRef CAS.
- J. Zhang, J. Yang, R. Dai, W. P. Sheng, Y. Su, Y. Zhong, X. Li, L. Tan and Y. Chen, Adv. Energy Mater., 2022, 12, 2103674 CrossRef CAS.
- Q. Wang, J. Zhu, Y. Zhao, Y. Chang, N. Hao, Z. Xin, Q. Zhang, C. Chen, H. Huang and Q. Tang, Carbon Energy, 2024, 6, e566 CrossRef CAS.
- R. Wu, W. Miao, R. Yin, W. Sun, Y. Sun, K. Wang, T. You and P. Yin, Chem. Eng. J., 2025, 504, 158954 CrossRef CAS.
- Y. Lao, S. Yang, W. Yu, H. Guo, Y. Zou, Z. Chen and L. Xiao, Adv. Sci., 2022, 9, 2105307 CrossRef CAS PubMed.
- R. Guo, J. Xia, H. Gu, X. Chu, Y. Zhao, X. Meng, Z. Wu, J. Li, Y. Duan, Z. Li, Z. Wen, S. Chen, Y. Cai, C. Liang, Y. Shen, G. Xing, W. Zhang and G. Shao, J. Mater. Chem. A, 2023, 11, 408–418 RSC.
- F. H. Isikgor, F. Furlan, J. Liu, E. Ugur, M. K. Eswaran, A. S. Subbiah, E. Yengel, M. D. Bastiani, G. T. Harrison, S. Zhumagali, C. T. Howells, E. Aydin, M. Wang, N. Gasparini, T. G. Allen, A. Rehman, E. V. Kerschaver, D. Baran, I. McCulloch, T. D. Anthopoulos, U. Schwingenschlogl, F. Laquai and S. D. Wolf, Joule, 2021, 5, 1566–1586 CrossRef CAS.
- Z. Xin, Y. Zhao, L. Bian, Y. Cao, J. Duan, Q. Guo, J. Dou, Q. Zhang and Q. Tang, ACS Mater. Lett., 2024, 6, 3327–3334 CrossRef CAS.
- Y. Xu, Y. Ren, S. Cheng, L. Zhang, P. Niu, M. Lyu, H. Lu, M. Wang and J. Zhu, J. Mater. Chem. A, 2023, 11, 868–877 RSC.
- C.-C. Zhang, S. Yuan, Y.-H. Lou, Q.-W. Liu, M. Li, H. Okada and Z. K. Wang, Adv. Mater., 2020, 32, 2001479 CrossRef CAS PubMed.
- C. Shi, Q. Song, H. Wang, S. Ma, C. Wang, X. Zhang, J. Dou, T. Song, P. Chen, H. Zhou, Y. Chen, C. Zhu, Y. Bai and Q. Chen, Adv. Funct. Mater., 2022, 32, 2201193 CrossRef CAS.
- K. Zheng, C. Liu, K. Yu, Y. Meng, X. Yin, S. Bu, B. Han, C. Liu and Z. Ge, Chem. Eng. J., 2022, 446, 137307 CrossRef CAS.
- W. He, X. Duan, Q. Tang, J. Dou and J. Duan, Chem. Commun., 2024, 60, 4954–4957 RSC.
- H.-J. Lee, Y.-J. Kang, S.-N. Kwon, D.-H. Kim and S.-I. Na, Small Methods, 2024, 8, 2300948 CrossRef CAS PubMed.
- J. Yang, W. Sheng, X. Li, Y. Zhong, Y. Su, L. Tan and Y. Chen, Adv. Funct. Mater., 2023, 33, 2214984 CrossRef CAS.
- J. Gong, X. Fan, Z. Zong, M. Yang, Y. Sun and G. Zhao, RSC Adv., 2023, 13, 15531 RSC.
- O. V. Oyelade, O. K. Oyewole, D. O. Oyewole, S. A. Adeniji, R. Ichwani, D. M. Sanni and W. O. Soboyejo, Sci. Rep., 2020, 10, 7183 CrossRef CAS PubMed.
- W.-Q. Wu, Z. Yang, P. N. Rudd, Y. Shao, X. Dai, H. Wei, J. Zhao, Y. Fang, Q. Wang, Y. Liu, Y. Deng, X. Xiao, Y. Feng and J. Huang, Sci. Adv., 2019, 5, eaav8925 CrossRef CAS PubMed.
- B. Han, Y. Wang, C. Liu, K. Sun, M. Yang, L. Xie, S. Yang, Y. Meng, S. Lin, P. Xu, J. Li, Q. Qiu and Z. Ge, Angew. Chem., Int. Ed., 2023, 62, 202217526 CrossRef PubMed.
- G. Li, J. Song, J. Wu, Z. Song, X. Wang, W. Sun, L. Fan, J. Lin, M. Huang, Z. Lan and P. Gao, ACS Energy Lett., 2021, 6, 3614–3623 CrossRef CAS.
- Y. Sun, W. Miao, W. Sun, Z. Niu, R. Yin, X. Huo, K. Wang, T. You and P. Yin, Small, 2024, 20, 2404272 CrossRef CAS PubMed.
- Q. Wang, X. Jiang, C. Peng, J. Zhang, H. Jiang, H. Bu, G. Yang, H. Wang, Z. Zhou and X. Guo, Chem. Eng. J., 2024, 481, 148464 CrossRef CAS.
- J. Yang, Q. Wang, W. Hui, X. Chen, Y. Yao, W. Tang, W. Qiu, X. Xu, L. Song, Y. Wu and Q. Peng, Energy Environ. Sci., 2025, 18, 1732–1744 RSC.
- X. Jiang, X. Wang, X. Wu, S. Zhang, B. Liu, D. Zhang, B. Li, P. Xiao, F. Xu, H. Lu, T. Chen, A. K.-Y. Jen, S. Yang and Z. Zhu, Adv. Energy Mater., 2023, 13, 2300700 CrossRef CAS.
- F. Zheng, L. Z. Tan, S. Liu and A. M. Rapee, Nano Lett., 2015, 15, 7794–7800 CrossRef CAS PubMed.
- J.-H. Lee, N. C. Bristowe, J. H. Lee, S.-H. Lee, P. D. Bristowe, A. K. Cheetham and H. M. Jang, Chem. Mater., 2016, 28, 4259–4266 CrossRef CAS.
- A. Amat, E. Mosconi, E. Ronca, C. Quarti, P. Umari, M. K. Nazeeruddin, M. Grätzel and F. D. Angelis, Nano Lett., 2014, 14, 3608–3616 CrossRef CAS PubMed.
- L. Guo, G. Xu, G. Tang, D. Fang and J. Hong, Nanotechnology, 2020, 31, 225204 CrossRef CAS PubMed.
- G. Giorgi, J.-I. Fujisawa, H. Segawa and K. Yamashita, J. Phys. Chem. Lett., 2013, 4, 4213–4216 CrossRef CAS PubMed.
- G. Liu, M. Ghasemi, Q. Wei, B. Jia, Y. Yang and X. Wen, Adv. Energy Mater., 2025, 15, 2405239 CrossRef CAS.
- J. Wu, S.-C. Liu, Z. Li, S. Wang, D.-J. Xue, Y. Lin and J.-S. Hu, Sol. RRL, 2024, 8, 2400203 CrossRef.
- T. S. Sherkar, C. Momblona, L. Gil-Escrig, J. Ávila, M. Sessolo, H. J. Bolink and L. J. A. Koster, ACS Energy Lett., 2017, 2, 1214–1222 CrossRef CAS PubMed.
- S. Tan, I. Yavuz, M. H. Weber, T. Huang, C.-H. Chen, R. Wang, H.-C. Wang, J. H. Ko, S. Nuryyeva, J. Xue, Y. Zhao, K.-H. Wei, J.-W. Lee and Y. Yang, Joule, 2020, 4, 2426–2442 CrossRef CAS.
- E. Aydin, M. D. Bastiani and S. D. Wolf, Adv. Mater., 2019, 31, 1900428 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*