
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmerging inorganic–organic hybrid photocatalysts for solar-driven overall water splitting: progress and perspectives
De-Shan
Zhang
ac,
Lei
Wang
b,
Xiaodong
Zhang
 b,
Xu-Bing
Li
*ac,
Hangxun
Xu
*b,
Chen-Ho
Tung
ac and
Li-Zhu
Wu
*ac
b,
Xu-Bing
Li
*ac,
Hangxun
Xu
*b,
Chen-Ho
Tung
ac and
Li-Zhu
Wu
*ac
aNew Cornerstone Science Laboratory, Technical Institute of Physics and Chemistry, Chinese Academy of Science, Beijing 100190, P. R. China. E-mail: lixubing@mail.ipc.ac.cn; lzwu@mail.ipc.ac.cn
bSchool of Chemistry and Materials Science, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: hxu@ustc.edu.cn
cSchool of Future Technology, University of Chinese Academy of Science, Beijing 100049, P. R. China
First published on 24th September 2025
Abstract
The pursuit of sustainable energy technologies has long inspired the development of efficient photocatalysts capable of converting solar energy into hydrogen (H2) via overall water (H2O) splitting. While inorganic semiconductors, such as metal oxides, oxynitrides, and oxysulfides, have demonstrated reasonable activity and robustness, their intrinsic limitations in light harvesting and charge separation continue to hinder their photocatalytic performance. Conversely, organic semiconductors offer compelling advantages, including tunable electronic structures, visible-light absorption, and synthetic versatility. However, their application in overall H2O splitting remains constrained by short exciton diffusion lengths, low carrier mobility, and poor activity in multi-electron processes. Recently, integrating organic and inorganic materials into hybrid photocatalysts has emerged as a powerful strategy to overcome these bottlenecks. By synergistically combining the efficient charge transport of inorganic frameworks with the structural adaptability and optoelectronic tunability of organic materials, rationally designed hybrid systems have shown remarkable potential in enhancing light utilization, facilitating exciton dissociation, and suppressing recombination. These advances not only improve overall H2O splitting efficiency but also open new avenues for photocatalyst design. This review critically examines the fundamental principles, interfacial interactions, and photophysical mechanisms underpinning inorganic–organic hybrid photocatalysts for solar-driven overall H2O splitting. We highlight recent breakthroughs, analyse the remaining scientific and engineering challenges, and propose strategic directions for next-generation hybrid systems with improved scalability, efficiency, and durability. Our goal is to establish a forward-looking roadmap that defines the role of hybrid photocatalysts as a transformative platform in achieving a sustainable, carbon-neutral society.
 De-Shan Zhang | De-Shan Zhang received his BS degree from Shandong University in 2022. Currently, he is a PhD candidate at Technical Institute of Physics and Chemistry of Chinese Academy of Sciences (CAS) under the supervision of Prof. Li-Zhu Wu. His research interests mainly focus on artificial photosynthesis, including water splitting, carbon dioxide reduction and nitrogen fixation. |
 Lei Wang | Lei Wang obtained his PhD from the University of Science and Technology of China (USTC) in 2019 under the supervision of Professor Hangxun Xu. From 2020 to 2022, he served as a postdoctoral fellow at the Hefei National Laboratory for Physical Sciences at the Microscale. He is currently an associate researcher at USTC. His research interests focus on developing conjugated polymer photocatalysts for solar-to-chemical energy conversion. |
 Xiaodong Zhang | Xiaodong Zhang obtained his PhD degree from the Hefei National Research Center for Physical Sciences at the Microscale, University of Science and Technology of China (USTC) in 2013 under the tutelage of Prof. Yi Xie. After two years of Postdoctoral training in iChEM, he joined the Department of Applied Chemistry, USTC as an associate professor, and achieved promotion to a professor in 2019. His primary research interest is in the photoexcitation process study of low dimensional solids, and their applications in energy catalysis. |
 Xu-Bing Li | Xu-Bing Li obtained his BS degree from Wuhan University in 2010 and PhD degree from Chinese Academy of Sciences in 2015 under the supervision of Prof. Li-Zhu Wu and Chen-Ho Tung. After that, he continued his research career in the same group as an assistant researcher and was promoted to a professor in 2023. His research interests mainly focus on using semiconductor nanocrystals in photochemical transformations, including water splitting, carbon dioxide reduction, organic synthesis and mechanism study. |
 Hangxun Xu | Hangxun Xu obtained his PhD degree from the Department of Chemistry at the University of Illinois at Urbana-Champaign in 2011. After postdoctoral work with Professor John A. Rogers from 2011 and 2013, he returned to the University of Science and Technology of China as a professor. His primary research interest is the design and synthesis of functional/conjugated polymers for applications in energy, catalysis, and flexible electronics. |
 Chen-Ho Tung | Prof. Chen-Ho Tung graduated from the University of Science and Technology of China in 1963 and was awarded his PhD degree in 1983 from Columbia University in New York City under the supervision of Prof. Nicholas J. Turro. He was elected as an academician of CAS in 1999 and is currently a professor at Technical Institute of Physics and Chemistry, CAS. His research interests include photochemical reactions, photoinduced electron transfer and energy transfer in supramolecular systems, and photocatalytic water splitting. |
 Li-Zhu Wu | Li-Zhu Wu received her BS degree from Lanzhou University in 1990 and PhD from Institute of Photographic Chemistry of Chinese Academy of Sciences in 1995 under the supervision of Prof. Chen-Ho Tung. After a one-year post-doctoral stay at University of Hong Kong with Prof. Chi-Ming Che, she was appointed as a professor at Technical Institute of Physics and Chemistry, CAS. In 2019, she was elected as an academician of CAS. Her research interests include artificial photosynthesis, organic photoredox catalysis, and photoinduced electron transfer and energy transfer in supramolecular systems. |
1. Introduction
Hydrogen (H2), characterized by its high combustion enthalpy and zero-carbon emissions upon utilization, has long been recognized as an ideal secondary energy carrier for a sustainable future.1 However, over 90% of current H2 production is still based on conventional methods such as steam methane reforming (SMR) and coal gasification, both of which rely heavily on fossil fuels.2–5 These methods not only contribute to significant environmental concerns but also fail to align with the intensification strategies needed for sustainable energy development. In contrast, solar-driven H2 production represents a compelling alternative by directly converting solar energy into chemical energy stored in H2 molecules.6,7 This route not only offers a sustainable solution for solar energy storage but also enables clean H2 generation without fossil inputs. The process of photo-mediated catalysis, where photon energy drives the generation of electron–hole pairs that then participate in subsequent redox reactions, holds immense potential. Furthermore, when integrated with proton exchange membrane (PEM) fuel cells, solar-derived H2 can be converted back to electricity with high energy density, exceptional energy conversion efficiency, and zero carbon emissions, producing only water (H2O) as the by-product.8,9 This seamless coupling establishes a closed-loop solar-hydrogen-electricity pathway.10 Nevertheless, designing a stable and efficient photocatalytic system capable of driving H2 production through overall H2O splitting still remains one of the most formidable challenges in solar energy research. Overcoming this barrier, often referred to as the “holy grail” in the field, would mark a transformative step toward scalable and carbon–neutral H2 production.11–13Photocatalytic overall H2O splitting comprises two coupled half-reactions, which are H2O oxidation and proton reduction, necessitating the concerted action of photoexcited holes and electrons, respectively. Unlike proton reduction, H2O splitting involves more complex processes, including the cleavage of O–H bonds and the formation of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. This adds substantial complexity to the reaction, as it demands precise control over electron, proton, and bond dynamics.14–16 These complexities impose stringent requirements on the spatial and temporal control of charge carrier dynamics and reaction intermediates within the photocatalyst. Under typical illumination conditions, the absorption of photons by the light-harvesting material generates single excitons, but the resulting kinetic barriers and charge accumulation often impede multi-electron redox reactions.17,18 These challenges are exacerbated by competing dissipative pathways, including radiative and non-radiative transitions, energy transfer, and vibrational relaxation, all of which limit the efficiency of charge separation and transfer.19–22 Furthermore, the kinetics of proton transfer play a crucial role in optimizing communication between the catalytic centres and the substrates involved in the H2O splitting reaction. From a thermodynamic standpoint, solar-driven overall H2O splitting is a non-spontaneous process that must overcome a significant energy barrier. While the theoretical thermodynamic minimum for H2O splitting is about 1.23 eV, the practical systems typically experience overpotentials, often raising this requirement to over 1.7 eV.23–25 In 1972, Fujishima and Honda's pioneering work on TiO2-based photoanodes under UV light and applied bias voltage demonstrated the feasibility of photoelectrochemical cells, sparking numerous subsequent strategies aimed at improving solar-driven H2 and oxygen (O2) evolution from H2O during the past fifty years.26–29
O bonds. This adds substantial complexity to the reaction, as it demands precise control over electron, proton, and bond dynamics.14–16 These complexities impose stringent requirements on the spatial and temporal control of charge carrier dynamics and reaction intermediates within the photocatalyst. Under typical illumination conditions, the absorption of photons by the light-harvesting material generates single excitons, but the resulting kinetic barriers and charge accumulation often impede multi-electron redox reactions.17,18 These challenges are exacerbated by competing dissipative pathways, including radiative and non-radiative transitions, energy transfer, and vibrational relaxation, all of which limit the efficiency of charge separation and transfer.19–22 Furthermore, the kinetics of proton transfer play a crucial role in optimizing communication between the catalytic centres and the substrates involved in the H2O splitting reaction. From a thermodynamic standpoint, solar-driven overall H2O splitting is a non-spontaneous process that must overcome a significant energy barrier. While the theoretical thermodynamic minimum for H2O splitting is about 1.23 eV, the practical systems typically experience overpotentials, often raising this requirement to over 1.7 eV.23–25 In 1972, Fujishima and Honda's pioneering work on TiO2-based photoanodes under UV light and applied bias voltage demonstrated the feasibility of photoelectrochemical cells, sparking numerous subsequent strategies aimed at improving solar-driven H2 and oxygen (O2) evolution from H2O during the past fifty years.26–29
Inorganic photocatalysts, including metal oxides, nitrides, phosphides, sulfides, chlorides, oxynitrides, and oxyhalides, have long been at the forefront of solar-driven H2 production technologies, owing to their favorable light-response performances, chemical stability, and cost-effectiveness.30–33 These materials have been extensively studied for their potential in large-scale solar-driven H2 production. For example, Domen et al. successfully scaled up the aluminum-doped strontium titanate (SrTiO3:Al) photocatalyst from a 1.0 m2 panel reactor to a 100 m2 outdoor system, achieving stable, large-scale photocatalytic H2O splitting with integrated gas collection and separation modules.34 The system operated stably for months, with a solar-to-hydrogen (STH) conversion efficiency of 0.76%. By loading cocatalysts like Rh/Cr2O3 and CoOOH on the SrTiO3:Al surface, the anisotropic charge transport, facilitated by work function differences, suppresses recombination and enhances the system's efficiency. Notably, an external quantum efficiency of 96% was achieved in the 350–360 nm UV range.35 Despite these achievements, inorganic photocatalysts face limitations, such as narrow light absorption ranges and significant energy losses due to rapid recombination of photogenerated carriers. These factors contribute to a persistent gap between current performance and the benchmark STH efficiency of ≥5% required for economically viable solar H2 production. Bridging this gap calls for innovative strategies that extend light harvesting into the visible spectrum while enhancing charge transport and catalytic dynamics.
In contrast, organic semiconductors are gaining attention due to their synthetically tunable molecular structures, which allow for precise control over light absorption, energy levels, and charge transport properties.36,37 Their structural versatility offers unique opportunities for engineering charge migration pathways and enhancing exciton dissociation. For instance, Jiang and Scholes synthesized sp2 carbon-conjugated covalent organic frameworks (COFs) linked by CC bonds, which demonstrated efficient visible-light absorption and long-range exciton transport within 2D conjugated planes.38 The incorporation of cofacial pyrene moieties within these COFs facilitated exciton delocalization, leading to enhanced exciton mobility and extended diffusion lengths. Moreover, donor–acceptor conjugated COFs are shown to facilitate ultrafast charge separation, with phonon-assisted polaron pair generation being a key mechanism.39 Despite these advances, organic semiconductors face inherent challenges, including strong exciton binding energies, limited intrinsic carrier mobility, and relatively short carrier lifetimes, which collectively constrain their overall photocatalytic efficiency.
As depicted in Fig. 1, given the current state of research, the integration of inorganic and organic semiconductor components into hybrid systems presents a highly effective strategy to overcome persistent limitations in photocatalytic H2O splitting, such as suboptimal light absorption, inefficient charge carrier dynamics, and poor thermodynamic compatibility. A prime example is the hybridization of polyaniline with ZnO, which promotes directional charge transfer across the inorganic–organic interface, thereby improving both photocatalytic activity and stability.40 A growing body of literature has demonstrated that inorganic–organic hybrid photocatalysts can efficiently modulate energy levels, optimize charge transfer pathways, and extend the lifetime of photogenerated charge carriers.41 These improvements are critical for meeting the demands of multi-electron and multi-proton reactions required in the H2O splitting process. The rational design of such hybrid platforms thus offers a promising route toward achieving the benchmark STH conversion efficiency needed for scalable solar H2 production.
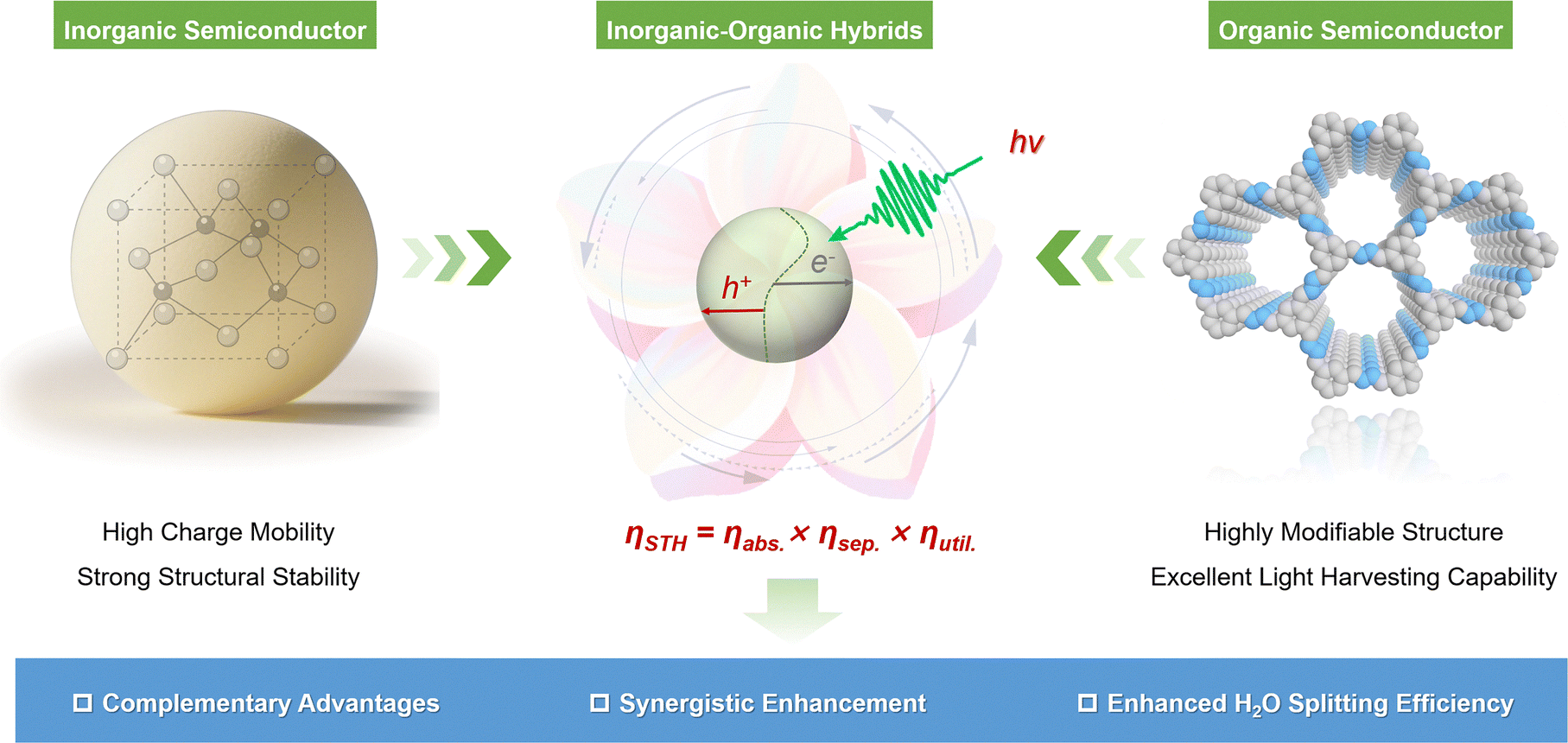 | ||
| Fig. 1 Harnessing the integrated advantages of inorganic semiconductors and organic counterparts for the enhanced overall H2O splitting toward H2 production. | ||
Fig. 2 clearly demonstrates that the development of integrated inorganic–organic hybrid platforms is of great significance for enhancing photocatalytic overall H2O splitting performance and for gaining deeper insights into the underlying mechanisms. In recent years, increasing attention has been directed toward inorganic–organic hybrid systems incorporating emerging materials such as amorphous conjugated polymers and crystalline covalent organic frameworks (COFs). However, despite important advancements, the literature remains fragmented, lacking a comprehensive and mechanistically focused overview of the design principles governing hybrid photocatalysts. Key aspects, such as the role of interfacial carrier dynamics, hybrid band structure tuning, and synergistic charge transport mechanisms, have not been systematically reviewed. This Review aims to fill these gaps by providing an in-depth discussion of hybridization strategies that enhance overall H2 production from H2O splitting. We emphasize the importance of charge generation, separation, and transport within hybrid systems, and classify representative inorganic–organic photocatalysts based on their electronic band structures. Finally, we outline the prospects and challenges facing the development of inorganic–organic hybrid photocatalysts, with the goal of inspiring future research to push the boundaries of this promising field.
 | ||
| Fig. 2 Schematic illustration of the advantages of inorganic–organic hybrid photocatalysts for overall H2O splitting to produce H2 and O2 (taking titanium dioxide and covalent organic frameworks as example). | ||
2. Principles of inorganic–organic hybridization to enhance photocatalytic H2O splitting efficiency
In a typical photocatalytic process, photocatalysts absorb photons with energy equal to or greater than its bandgap on the femtosecond (fs) timescale, promoting electrons from the valence band to the conduction band and generating electron–hole pairs. These charge carriers rapidly thermalize and migrate to surface active sites over tens to hundreds of picoseconds (ps). Interfacial charge transfer and reactions with adsorbed species then proceed on nanosecond (ns) to microsecond (ms) scales, in competition with radiative and nonradiative recombination.42 However, bulk and interfacial recombination processes proceed on picosecond–nanosecond timescales and frequently compete with (or exceed) the rates of productive interfacial charge transfer. Consequently, many photogenerated carriers recombine before accessing active sites, constraining the efficiency of overall photocatalytic H2O splitting. Thus, to enhance H2 production, it is essential to improve ultrafast charge dynamics, particularly at the nanoscale.43 Strategies such as crystal facet regulation, morphology design, and defect engineering have been shown to provide localized and directional control over charge migration in unitary semiconductor photocatalysts.44–46 Inorganic–organic hybridization further tailors carrier dynamics by forming intimate heterointerfaces with favorable band alignment and built-in electric fields, thereby improving charge-transfer efficiency, extending excited-state lifetimes, and suppressing recombination.47,48As shown in Fig. 3, the exciton dissociation pathways and interfacial electric field characteristics are systematically compared among representative systems, including inorganic semiconductors, organic semiconductors, inorganic–inorganic heterogeneous interface, organic–organic heterogeneous interface, and inorganic–organic heterogeneous interface. Inorganic–organic hybrid materials uniquely integrate the structural tunability of organic semiconductors with the superior charge transport properties of inorganic counterparts. The interfacial coupling between materials with drastically different dielectric constants and carrier concentrations is expected to generate strong internal electric fields (IEF), thereby enhancing interfacial charge separation and facilitating subsequent catalytic transformations.
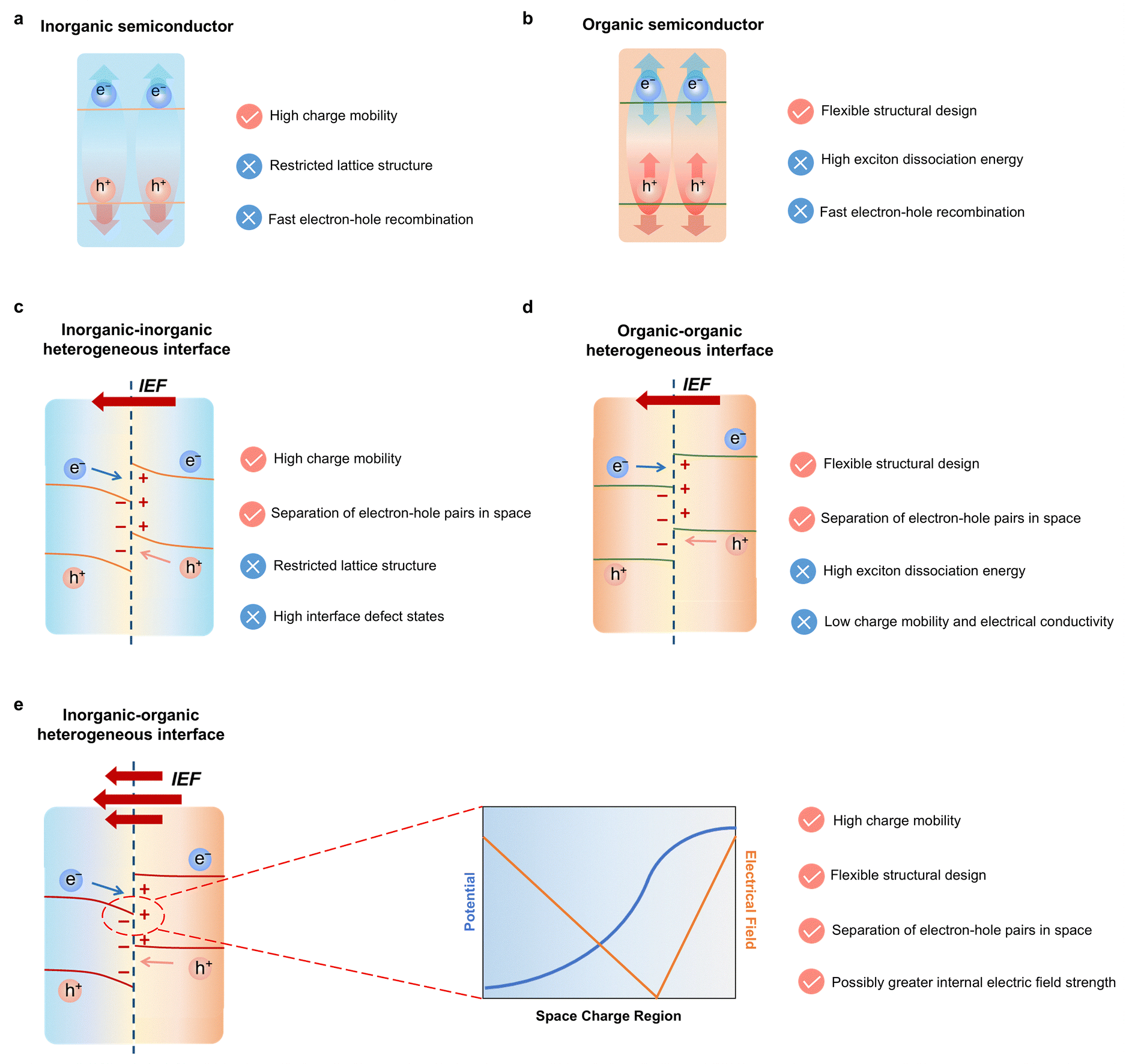 | ||
| Fig. 3 Diagram illustrating the charge transfer kinetics in (a) inorganic semiconductors and (b) organic semiconductors. Comparison of charge transfer at the (c) inorganic–inorganic heterojunction, (d) organic–organic heterojunction, and (e) inorganic–organic heterojunction (The left side represents inorganic semiconductors, while the right side represents organic semiconductors. The right figure illustrates the distribution of the IEF potential and intensity in the space charge region). | ||
To fully understand the benefits of inorganic–organic hybrid systems, it is crucial to first examine the exciton dynamics in semiconductor materials. The initial step in photocatalytic efficiency is light harvesting, which hinges on the balance between broad-spectrum absorption and suitable redox capability. The solar spectrum spans a wide range of wavelengths, but photocatalysts typically absorb photons that excite valence electrons from the valence band (VB) to the conduction band (CB), with the energy bandgap (Eg) determining the range of absorbed wavelengths. The relationship between the Eg and the shortest absorption wavelength follows eqn (1). Furthermore, much of the absorbed energy falls within the ultraviolet (UV) region, which is limited in terms of total energy and does not align with the need to improve solar energy conversion efficiency.49 By integrating inorganic and organic materials, hybrid systems can extend the absorption to lower energy photons by adjusting functional groups and energy levels, thus improving overall photocatalytic performance.50 In general, the VB and CB edge potentials of semiconductors can be approximately predicted from the Mulliken electronegativity theory.51 As shown in eqn (2) and (3), EVB and ECB denote the potentials of the valence and conduction bands; χ is the absolute electronegativity of the semiconductor; and Ee is the energy of a free electron on the hydrogen electrode scale.
 | (1) |
| EVB = χ − Ee − 0.5Eg | (2) |
| ECB = χ − Ee − 0.5Eg | (3) |
The differences in the dielectric properties between organic and inorganic semiconductors significantly influence their photogenerated charge carrier dynamics. When photons are absorbed by the semiconductor, ground state electrons in the valence band are excited to the conduction band, leading to the formation of bound excitons through Coulomb interactions between the excited electrons and the in situ generated holes.
In inorganic semiconductors, such as TiO2, Ta2O5, and CdS, the high dielectric constants (ε)—typically above 10, and sometimes exceeding 100—serve to screen the electric field between electron–hole pairs, reducing the exciton binding energy.52,53 In such a high-dielectric environment, the radius of the Wannier–Mott excitons often exceeds the lattice constant, and the binding energy is typically around 0.01 eV, which contributes to the easy dissociation of excitons into free carriers.54 In contrast, the molecular orbitals in organic semiconductors are highly localized, and the lower dielectric constants (ε < 5) result in higher exciton dissociation energies (eqn (4)). This limits the charge carrier lifetime (τ) and carrier separation efficiency (μ is the reduced exciton mass, Ry is the atomic Rydberg energy), both critical factors for efficient photocatalysis.55,56 The dissociation of Frenkel excitons in organic semiconductor materials often requires external electric fields or varying chemical potentials at interfaces to aid in carrier separation. The interaction of these excitons with the surrounding environment plays a key role in modulating the photophysical processes, including the lifetime and diffusion length of charge carriers, both of which are critical for enhancing photocatalytic redox efficiency. The relationship between the average exciton diffusion length (LD), diffusion coefficient (D), and exciton lifetime (τ) can be expressed by eqn (5). During the diffusion process, excitons tend to recombine and lose their energy either radiatively or non-radiatively.
The charge transfer efficiency (ηct) is determined by both the charge transfer rate constant (kct) and the excited-state lifetime (τ) (eqn (6)). Suppressing charge recombination and increasing kct both raise ηct. Within a Marcus-type framework, the kct depends on the electronic coupling between the donor and acceptor sites (HDA) and the reaction driving force at the semiconductor–solution interface. In addition, the HDA and reorganization energy (λi) involved in the transition from initial to final charge states are both significant dynamic parameters influencing recombination and relaxation processes. Thus, they are crucial parameters for optimizing charge dynamics (eqn (7)).57,58 Moreover, surface states must be managed judiciously. Shallow, catalytically productive (or co-catalyst-induced) states can transiently localize carriers, extend interfacial residence times, and facilitate charge transfer, whereas deep traps promote non-radiative recombination and should be minimized. While rational design approaches, such as crystal facet control, doping (including metal and non-metal atoms), and morphology optimization, have shown effectiveness in modulating charge trapping and improving photocatalytic activity, significant improvements in photocatalytic H2 production from H2O splitting are still required. Hybridization schemes combining inorganic and organic materials offer a promising strategy to address these challenges, optimizing the charge dynamics for efficient H2 production.
 | (4) |
 | (5) |
 | (6) |
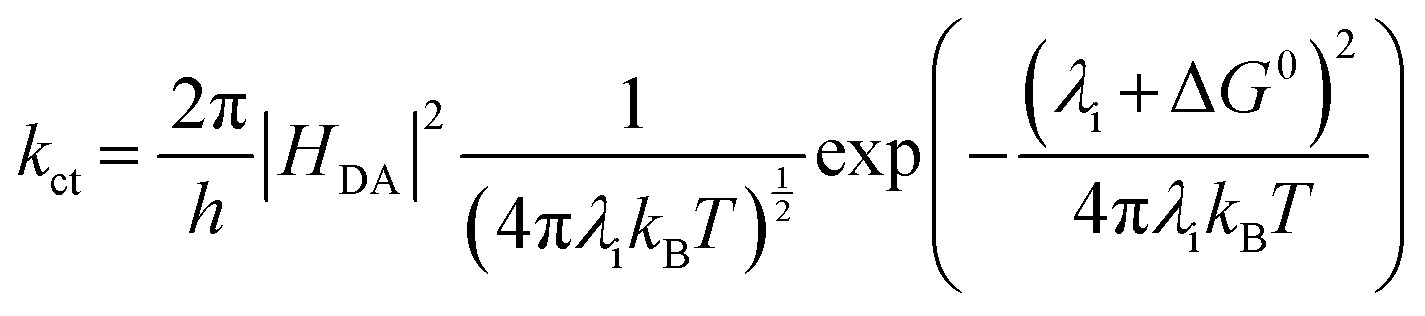 | (7) |
The hybridization strategy integrates distinct organic and inorganic materials, aligning surface potentials and energy levels to create favorable pathways for the dissociation and efficient transfer of excitons. From a carrier dynamics perspective, this enhanced interaction is primarily driven by the potential difference at the interface. When organic compounds or heterogeneous semiconductors come into close contact with a semiconductor photocatalyst, differences in molecular dipoles and work functions (WF) induce changes in the surface electric field, which in turn affects the distribution and movement of charge carriers. The WF represents the energy required for an electron to escape from the material, relative to the Fermi energy level (EF) (eqn (8)).59 The discontinuity in the EF at the hybrid interface causes electrons to drift from regions of higher EF to lower EF, in accordance with the thermodynamic potential difference. This results in the accumulation of charge at the interface, thus creating a dipole layer that forms an IEF across the surface of the inorganic–organic hybrid system. Moreover, the IEF can be quantified by the surface voltage (Vsurf) and surface charge density (ρ), which play a crucial role in enhancing interfacial charge interactions, further optimizing the charge separation and transfer efficiency essential for photocatalytic processes (eqn (9)).60
| W = −eφ − EF | (8) |
 | (9) |
 | (10) |
 | (11) |
As shown in Fig. 4, contact between the semiconductors induces carrier diffusion across the junction, establishing space-charge regions and an IEF. The IEF strengthens until it exactly balances diffusion (Fermi level align), at which point the net current vanishes and the band edges bend under the resulting built-in potential. The electric field is position-dependent and attains its maximum magnitude at the interface and decreases toward zero at the boundary of the space-charge region, where the band bending is most pronounced near the contact. The IEF at the junction of such heterostructures can be precisely measured using Kelvin Probe Force Microscopy.61 The derived eqn (10) and (11) of the Poisson equation describe how the surface potential (V(x)) evolves with interface coordinates, with D1 and D2 representing the depletion layer widths of the heterogeneous components (eqn (14) and (15)). The contact potential difference at the heterojunction is the sum of the electrical potential drops (VD1 and VD2) at the interface. These potential drops are strongly influenced by the ε and carrier concentrations (N) of the components, as shown in eqn (12) and (13). This IEF plays a critical role in enhancing charge separation and migration, significantly improving photocatalytic efficiency. Under illumination, non-equilibrium charges are generated, and their directional drift at the interface is influenced by the IEF. This non-equilibrium state closely resembles an applied voltage across the heterostructure, and the potential drop in the space charge region can still be described by eqn (13) (after accounting for the applied voltage). As photogenerated charges are transferred, the IEF strength decreases, and the space charge region narrows, eventually reaching a new equilibrium state.
| VD = VD1 + VD2 | (12) |
 | (13) |
 | (14) |
 | (15) |
| σ = nqμ | (16) |
 | ||
| Fig. 4 The charge transfer processes in heterojunction (i) before and (ii) after contact, (iii) under light irradiation and (iv) surface reactions. | ||
The unique properties of the inorganic–organic hybrid interface arise from the differences in the physical and photoelectric characteristics of the organic and inorganic components. These differences provide potential advantages in promoting exciton dissociation and migration compared to inorganic–inorganic or organic–organic interfaces. The ρ, N, and dielectric properties of different lattice phases play a critical role in enhancing the charge separation driven by the IEF. Directional carrier drift driven by the built-in IEF sweeps electrons and holes away from surface traps, suppressing nonradiative recombination. N-Doped organic semiconductors, owing to their high N-dopant density and favorable dielectric response, can further reinforce the IEF. However, their limited carrier mobility remains a bottleneck for further enhancing catalytic performance (eqn 16). These limitations can be partially mitigated by hybridizing inorganic and organic materials, whose complementary thermodynamic and charge transport characteristics promote more efficient charge separation and transfer. At the interface of inorganic–organic hybrid systems, the formation of an IEF–arising from differences in energy levels and work functions–serves as an internal driving force that facilitates directional carrier migration and suppresses recombination.62 Additionally, enhanced carrier mobility within the hybrid architecture extends carrier lifetimes, which is critical for sustaining multi-step photoredox sequences. Molecular- and atomic-scale interfaces further suppress trap-mediated losses, promoting more efficient interfacial charge transfer. These insights highlight the importance of rational hybrid design for engineering the IEF, is a key parameter to enhance the efficiency of overall photocatalytic H2O splitting.
The charge transfer processes between the organic and inorganic components in inorganic–organic hybrid photocatalysts can typically be classified into four primary modes: (1) the sensitization mechanism, (2) type I heterojunction, (3) type II heterojunction, and (4) Z-scheme heterojunction. Each of these modes involves distinct mechanisms for facilitating the transfer of photogenerated carriers across the interface, and their effectiveness is largely determined by the electronic structure, energy band alignment, and interface properties of the hybrid materials. The sensitization scheme involves the transfer of energy from the organic component to the inorganic semiconductor, promoting the generation of charge carriers in the latter. Type I and II heterojunctions, on the other hand, rely on the alignment of energy bands between the two materials to drive the separation of electrons and holes, with type I heterojunctions favoring electron transfer to the inorganic component, while type II heterojunctions facilitate more efficient separation and migration of both charge carriers across the interface.
The Z-scheme heterojunction, inspired by the natural photosynthesis process, allows for the simultaneous separation of electrons and holes at different components, thereby enhancing charge transfer and photocatalytic efficiency. Under two-photon excitation, electrons accumulate at the component with the more negative conduction band while holes concentrate at the one with the more positive valence band.63,64 This preserves strong reduction and oxidation potentials, relaxes the per-photon energy requirement compared with single-absorber schemes, and suppresses recombination by spatially isolating redox sites. When band positions appear similar, the direction of carrier flow is governed by the contact potential difference and the associated interfacial band bending established upon junction formation.65–68
The distinctive nature of charge transfer at inorganic–organic interfaces arise from the intrinsic composition and structure differences between inorganic and organic semiconductors. Inorganic semiconductors, such as metal oxides, are typically composed of atoms held together by strong covalent or ionic bonds, resulting in extended electronic delocalization and high ε. These features facilitate efficient light absorption and the generation of free charge carriers upon photoexcitation.69 In contrast, organic semiconductors are held together by weak intermolecular interactions (i.e., van der Waals forces, π–π stacking, hydrogen bonding), leading to the localized electronic states and lower ε than that of inorganic counterparts. These characteristics limit their charge separation efficiency, and the formation of carriers often requires external electric fields to assist in dissociation of the bound excitons. The four primary charge transfer modes outlined previously demonstrate that the chemical potential differences across the interfaces between inorganic and organic components provide an effective means for generating intrinsic photogenerated carriers and facilitating exciton dissociation. For example, the hierarchical ZnIn2S4/g-C3N4 inorganic–organic heterostructure photocatalyst developed by Huang et al. demonstrates enhanced photocatalytic performance by leveraging efficient interfacial charge transfer and reduced recombination loss.70 When compared to single semiconductors or conventional inorganic heterojunction photocatalysts, such as those materials based on Fe2O3 or CeO2, this inorganic–organic hybrid platform offers a more effective photo-induced charge transfer efficiency, ultimately leading to superior photocatalytic performance.71,72
In addition to optimizing charge transfer modes, inorganic–organic hybrid photocatalytic platforms offer the advantage of providing more active sites for surface reactions. The tailored integration of organic and inorganic components leads to changes in the surface lattice structure and functional group composition.73 Due to the inherent differences in crystal structure and bonding, hybrid interfaces expose a greater number of active sites, enhancing the dispersion of photocatalysts and promoting more efficient charge transfer. Compared to bulk semiconductors, where charge carriers tend to recombine before reaching the surface, the hybrid structure facilitates the migration of carriers to the surface, where they can more readily participate in subsequent reactions. For example, Wang et al. demonstrated the effectiveness of this optimization strategy by constructing a g-C3N4/polyaniline/ZnO ternary heterostructure, where carbon nitride nanosheets served as supports.74 The synergy between organic polymers and inorganic semiconductors increased the specific surface area of the photocatalytic system, enhanced charge separation efficiency, and broadened the light absorption range. As a result, the visible-light catalytic efficiency of the hybrid system was more than three times higher than that of the individual components. The flexibility in tuning the structure of organic components also accelerates surface reactions by adjusting functional groups and molecular arrangements. For instance, in the CuNi alloy nanoparticle/g-C3N4 hybrid system, the CO functional groups in the organic layer were used to modulate the work function and H2 adsorption free energy, resulting in a photocatalytic H2 production rate of 2.36 mmol g−1 h−1 under visible-light irradiation.75 Additionally, the introduction of functional groups such as carboxyl, amino, and hydroxyl groups can further enhance photocatalytic performance by improving substrate selectivity and expanding the width of the space charge region, thereby optimizing overall H2O splitting catalytic efficiency.76,77
3. Synthetic approaches toward inorganic–organic hybrid photocatalysts
Inorganic–organic hybrid systems, due to their unique hetero-interfacial properties, have gained significant attention for their potential to enhance carrier dynamics and address key challenges (e.g., mass transfer and stability) in photocatalytic H2 production via overall H2O splitting. These systems typically involve the coupling of organic molecules or semiconductors with inorganic counterparts, leveraging the complementary advantages of both material classes. By modulating the redox properties and the electrical conductivity of these hybrids, significant improvements in photocatalytic performance have been achieved.78–80 Notably, hybrid semiconductor architectures incorporating bandgap engineering provide more versatile charge modulation pathways than conventional sensitized or conductive systems, while maintaining strong redox capabilities. This versatility allows for greater flexibility in optimizing charge transport and separation processes, which are critical for enhancing photocatalytic performance of H2O splitting. In this context, we focus on hybrid systems composed of organic semiconductors and bandgap-engineered inorganic semiconductors. These systems can be further classified into two categories: amorphous organic polymers and crystalline COFs, based on the distinct structural characteristics of the organic semiconductor components.The synthesis strategies employed to construct such hybrid materials play a decisive role in determining their interfacial properties, structural integrity, and functional performance.81–85 Over the years, numerous reviews have extensively explored various approaches for constructing these hybrid catalysts. In this section, we provide a concise overview of the synthesis techniques most commonly employed for hybrid systems that integrate organic and inorganic semiconductors.
In situ synthesis is widely employed, either by growing inorganic phases from precursors on organic semiconductors or by polymerizing organic monomers directly on inorganic substrates.86,87 This method enables the formation of continuous, well-integrated hybrid networks with strong interfacial bonding and efficient electronic coupling.88–90 Techniques such as electrochemical deposition and solvothermal synthesis offer fine control over reaction parameters, including temperature, pH, and precursor concentration, allowing precise tuning of hybrid interface properties. This approach has been successfully applied to fabricate hybrid electrodes and powder photocatalysts with well-defined morphologies, enhanced stability, and improved charge separation efficiency.91,92 Nevertheless, achieving precise synthesis via this route remains highly sensitive to the substrate's surface chemistry and morphology, as well as to the specific processing conditions.
Physical mixing is a straightforward, versatile method that integrates organic and inorganic semiconductors by co-dispersing them in a common solvent, typically with ultra-sonication used to promote uniform distribution.93,94 Non-covalent interactions, such as hydrogen bonding and π–π stacking, play critical roles in stabilizing the hybrid system and defining the binding modes between the components.95 This method allows for the creation of highly ordered hybrid interfaces and offers the flexibility to tailor the interfacial structures by modulating the semiconductor properties. Such tailored interfaces are beneficial for facilitating efficient electron–hole separation and promoting directional charge transfer, which significantly enhances the catalytic performance of the hybrid materials. However, due to the relatively weak interfacial binding, challenges remain, including poor interfacial coupling and limited control over morphology and structure.
Mechanical ball milling offers an efficient, scalable route to integrate organic and inorganic components.96,97 High-energy impact and shear promote intimate, homogeneous mixing, suppress agglomeration, and can induce interfacial bonding. This approach often requires minimal or no solvent and utilizes mechanochemical conditions to drive the synthesis. The grinding process introduces defect sites into the hybrid products, which may result from uncontrollable reactions triggered by the applied high activation energy.98 Additionally, requirements regarding material viscosity and dispersibility can pose limitations on material development. Under the optimized conditions, ball milling can produce diverse hybrid interfaces with enhanced structural and functional properties, providing an effective means of material synthesis.
Surface functionalization provides an additional route for constructing hybrid interfaces via covalent bonding. Functional groups such as hydroxyl, amino, or carboxyl moieties can be introduced onto the surfaces of organic and inorganic semiconductors to facilitate chemical coupling.99,100 However, the limited reactivity of certain semiconductor surfaces may restrict the diversity of achievable hybrid structures. Furthermore, the inherent experimental complexity and associated costs pose challenges for large-scale fabrication. Despite these challenges, surface functionalization remains a powerful means to improve interfacial compatibility, accelerate charge transfer kinetics, and tailor the overall photocatalytic performance in hybrid systems.
Together, these synthesis methods offer a toolkit for tailoring the composition, morphology, and electronic properties of inorganic–organic hybrid photocatalysts. The careful selection and optimization of synthetic approaches are critical to achieving the structural precision and functional integration required for efficient, scalable solar-to-hydrogen conversion.
4. Typical inorganic–organic hybrid systems for photocatalytic overall H2O splitting
Organic semiconductors, primarily represented by amorphous organic polymers, have attracted extensive interest due to their facile synthesis, chemical stability, and amenability to molecular-level design. In hybrid systems, these materials can serve dual roles: harvesting light and facilitating charge transfer. A key advantage of organic semiconductors lies in their intrinsic ambipolar transport behavior, which enables the concurrent migration of electrons and holes. This property is particularly valuable in the construction of inorganic–organic hybrid photocatalysts, where efficient charge separation and transport are essential for high-performance solar-driven H2O splitting. Substantial progress has been made in integrating organic polymers with well-established inorganic semiconductors such as TiO2, CdS, and Fe2O3.101–103 The interfacial interaction between the organic and inorganic phases critically influences photocatalytic activity by modulating light absorption characteristics, exciton generation dynamics, and charge separation efficiency. These hybrid systems have shown considerable potential to meet both the thermodynamic and kinetic requirements of overall H2O splitting. The following sections will present case studies and detailed analysis of their working principles.4.1 Integrating inorganic semiconductors with 1D organic polymers for photocatalytic overall H2O splitting
Linear polymers are among the most well-developed organic semiconducting materials, consisting of hydrocarbon chains that are soluble in organic solvents (Fig. 5(a) and (b)).102 Their inherent stretchability makes them ideal for various applications. It has been demonstrated that the linear polymers can drive photocatalytic H2O splitting with the assistance of suitable cocatalysts (Fig. 5(c) and (e)).103 Moreover, their flexibility enhances their ability to meet interfacial contact area and compatibility requirements, which are essential for the development of efficient inorganic–organic hybrid photocatalysts. In this regard, Cooper and Sprick et al. demonstrated the coupling of the homopolymer of dibenzo[b,d]thiophene sulfone (P10) with BiVO4, which facilitated photocatalytic overall H2O splitting (Fig. 5(f) and (g)).104 The resulting inorganic–organic hybrid system was particularly effective in promoting a Z-scheme charge transfer mechanism, which enhanced the redox abilities of the photogenerated charge carriers, overcoming the thermodynamic challenges of H2O splitting. Conjugated linear polymers provide a dynamic platform for charge migration, improving charge transfer between the two phases. In this system, the linear polymer enriched photogenerated electrons to generate H2, while BiVO4 was responsible for oxidizing H2O to produce O2. Under broad-spectrum illumination (e.g., 300 W Xe light source), the hybrid photocatalyst achieved H2 and O2 production rates of 10.8 and 4.5 μmol h−1, respectively, thereby giving an STH conversion efficiency of ∼0.0014%. Zhang et al. synthesized Cu6Sn5/polyaniline composite via a combination of chemical reduction and hydrothermal methods and employed as photocatalyst for overall H2O splitting under sunlight irradiation.105 The hybrid photocatalyst exhibited H2 and O2 evolution rates of 121.3 and 58.6 μmol g−1 h−1, respectively. The enhanced photocatalytic performance was attributed to the synergistic effect of the bimetallic Cu6Sn5 component and polyaniline matrix. In this system, polyaniline served as an acceptor that effectively trapped plasmonic holes and promoted charge separation and utilization, thereby improving the efficiency of the light-driven overall H2O splitting. This successful integration of linear conjugated polymers with inorganic semiconductors paves the way for the use of other linear polymers, such as polydopamine and polypyrrole, in photocatalytic applications.106,107 | ||
| Fig. 5 (a) Structures of typical linear polymers for photocatalysis. (b) TD-B3LYP predicted potentials of the charge carriers and excitons in the linear polymer photocatalysts. Adapted with permission from ref. 102. Copyright 2020, Wiley-VCH. (c) Alignment of the P10 energy levels. (d) Photocatalytic H2O splitting performance of photocatalyst based on P10. (e) Dependence of gas evolution rates on the different co-catalyst loaded onto P10 under visible-light illumination. Adapted with permission from ref. 103. Copyright 2022, Wiley-VCH. (f) Time course of overall H2O splitting on P10 and BiVO4. (g) Wavelength dependence of the photocatalytic overall H2O splitting activity of P10 and BiVO4. Adapted with permission from ref. 104. Copyright 2020, Royal Society of Chemistry. | ||
4.2 Integrating inorganic semiconductors with 2D organic polymers for photocatalytic overall H2O splitting
While linear polymers offer flexibility and versatility, their relatively simplistic structure and limited stability under irradiation restrict their potential for further enhancement of photocatalytic efficiency. To overcome these limitations, extending the conjugated network in two dimensions or in a plane, thus expanding the delocalized system formed by overlapping π orbitals, presents a promising strategy to optimize their photoelectronic properties. Various amorphous polymers, based on different chemical bonds, have been explored for designing advanced hybrid systems. A notable example is the direct Z-scheme catalytic system formed by the photo-deposition of Fe2O3 on g-C3N4, which exhibits remarkable photocatalytic activity for H2O splitting.108 This hybrid system circumvents the need for redox couples, inducing band bending in both the organic and inorganic components, which facilitates the transfer of photogenerated electrons to the conduction band of g-C3N4 and holes to the valence band of Fe2O3. Charge tracking and reactive oxygen species (ROS) experiments further confirm the formation of the Z-scheme charge transfer mechanism. At 380 nm, the apparent quantum efficiency is enhanced by a factor of 3.5 compared to pristine g-C3N4.The efficiency of photocatalytic H2O splitting is highly dependent on the structural characteristics of both organic polymers and inorganic semiconductors. For example, Ajayan et al. demonstrated that loading α-Fe2O3 nanosheets onto ultrathin two-dimensional g-C3N4 significantly improved H2 production efficiency by reducing the migration distance of photogenerated carriers on the two-dimensional scale (Fig. 6(a)).109 The tightly integrated interface between the organic and inorganic components in this all-solid-state Z-scheme heterojunction structure resulted in H2 and O2 evolution rates of 38.2 and 19.1 μmol g−1 h−1, respectively, without the need for sacrificial donors. With the addition of TEOA as a sacrificial agent, the H2 production rate exceeded 30 mmol g−1 h−1. In addition, the external quantum efficiency of this system at 420 nm was significantly enhanced to 44.35%, surpassing that of other g-C3N4-based photocatalysts (Fig. 6(b) and (c)).
 | ||
| Fig. 6 (a) HRTEM image of α-Fe2O3/2D g-C3N4 hybrid photocatalyst. (b) The wavelength dependence of external quantum efficiency of α-Fe2O3/2D g-C3N4 hybrid photocatalyst. (c) Photocatalytic overall H2O splitting performance of the hybrid photocatalyst. Adapted with permission from ref. 109. Copyright 2017, Wiley-VCH. (d) Synthetic route of nitrogen-rich covalent organic polymers and TiO2@covalent organic polymers hybrids. (e) Transient photocurrent responses of TiO2 and hybrid photocatalyst. Adapted with permission from ref. 110. Copyright 2018, Elsevier. (f) Schematic for preparation process of Co9S8/CdS@PP12. (g) H2 and O2 evolution rates as a function of time. Adapted with permission from ref. 124. Copyright 2024, Wiley-VCH. | ||
Beyond conventional two-dimensional conjugated networks, nitrogen-rich covalent organic polymers can also be integrated with inorganic semiconductors to form photocatalysts with diverse structural characteristics.110 As shown in Fig. 6(d), in situ growth of covalent organic polymers on the TiO2 surface creates a core–shell structure, facilitating close contact between the organic and inorganic phases. Although pristine TiO2 typically responds only to ultraviolet light due to its wide bandgap, the hybridization with organic polymers with a narrow bandgap of 2.53 eV can enhance visible-light absorption. The graphene-like conjugated framework and nitrogen doping further improve the photocatalytic efficiency under visible light due to the optimized charge and mass transfer processes. The hybrid photocatalyst exhibited enhanced photoresponse performance (Fig. 6(e)), as confirmed by transient photocurrent measurements, and the H2 production rate reached 162.7 μmol h−1.
Liu et al. developed a one-step hydrothermal method to construct TiO2/g-C3N4 heterojunction, where the exposed (001) crystal face of anatase TiO2 facilitated the formation of a heterogeneous surface, thereby enhancing the separation of photogenerated carriers and improving photocatalytic H2 production efficiency.111 Incorporating β-Ni(OH)2 onto WO3 or BiVO4 improves the overall efficiency of H2O splitting for H2 and O2 generation in the presence of I−/IO3− or Fe2+/Fe3+ redox mediators. Under optimized conditions, the apparent quantum yield (QAY) of this Z-scheme hybrid system at 365 nm and 405 nm is ∼4.9% and ∼4.0%, respectively. The hybrid system composed of CoO and g-C3N4 demonstrates photocatalytic overall H2O splitting activity, with the type II charge transfer mechanism facilitating electron enrichment on CoO and hole accumulation on g-C3N4.112 The enhanced photocatalytic activity of the CoO/g-C3N4 heterojunction can be attributed to the synergistic effects at the junction and interface between CoO and g-C3N4. The H2 and O2 evolution rates reached 2.51 and 1.39 μmol h−1, respectively. Notably, 30 wt% CoO/g-C3N4 exhibited exceptional long-term photocatalytic stability, maintaining its stability for more than 15 cycles due to the large specific surface area and flexible two-dimensional structure of g-C3N4, which effectively prevents the aggregation-induced deactivation of CoO nanoparticles. The integration of CoO nanorods with C3N4 leads to the formation of a tightly coupled heterointerface, enabling the construction of a hybrid photocatalyst with an optimized H2 evolution rate of up to 92 μmol h−1. This enhanced photocatalytic performance is primarily attributed to the formation of a heterojunction at the CoO/C3N4 interface, which significantly facilitates the efficient separation and transfer of photogenerated charge carriers.113
Wang et al. designed and fabricated a Z-scheme composite photocatalyst consisting of g-C3N4/ITO/Co–BiVO4, in which g-C3N4 functions as the H2 evolution photocatalyst, Co-doped BiVO4 serves as the O2 evolution photocatalyst, and ITO nanoparticles act as a conductive electron mediator. Without the use of any sacrificial agents, the g-C3N4/ITO/Co–BiVO4 composite exhibited H2 and O2 evolution rates of 95.41 and 40.23 μmol g−1 h−1, respectively, under full arc irradiation, approximately four times higher than those of the g-C3N4/Co–BiVO4 system.114 Zou and co-workers designed the charge transfer pathway in a Z-scheme heterojunction by tuning the work function of semiconductors.115
A heterojunction composed of BiVO4 and polymeric carbon nitride (PCN) was constructed, in which the direction of band bending within the space charge region was reversed by controlling the crystal growth of BiVO4. Specifically, the oxygen vacancy concentration in BiVO4 was reduced from 8.9% to 3.8%, resulting in an increase in its work function from below to above that of PCN. As a consequence, the interfacial electric field was reoriented to promote more favorable charge transfer at the interface. A direct Z-scheme van der Waals heterojunction composed of ultrathin WO3·H2O and g-C3N4 nanosheets also demonstrated efficient overall H2O splitting without any sacrificial reagents.116 The WO3·H2O/g-C3N4 nanosheet hybrid achieved H2 and O2 evolution rates of 482 and 232 μmol g−1 h−1. Notably, this heterojunction exhibited a quantum efficiency of 6.2% at 420 nm.
In addition to conventional inorganic oxide semiconductors, a broader range of inorganic materials has been explored in combination with g-C3N4 for overall H2O splitting. Chen et al. developed a ternary CdS/Ni2P/g-C3N4 composite that exhibited overall H2O splitting activity under visible-light irradiation. The optimized composite containing 3 wt% Ni2P achieved H2 and O2 evolution rates of 15.56 and 7.75 μmol g−1 h−1, representing a ∼4-fold enhancement compared to the binary CdS/g-C3N4 system.117 The introduction of Ni2P acted as an efficient electron mediator, accelerating charge transfer from the conduction band of g-C3N4 to that of CdS. The Co3(PO4)3/g-C3N4 heterojunction, assembled via Coulombic electrostatic interaction, exhibited an expanded light absorption range and enhanced interfacial contact, which promoted effective charge separation and transfer across the interface. The 35% Co3(PO4)3/g-C3N4 composite demonstrated H2 and O2 evolution rates of 375.6 and 177.4 μmol g−1 h−1, respectively, along with an apparent quantum efficiency of 1.32% at 420 nm.118 Moreover, this hybrid system exhibited excellent stability and recyclability during prolonged photocatalytic operation. The simple coupling model composed of g-C3N4 nanosheets and CdS nanorods was developed, in which dual cocatalysts including 3 wt% Pt and 4 wt% MnOx were deposited in situ to enhance the photocatalytic performance.119 Under visible-light irradiation, the optimized system achieved a H2 evolution rate of 924.4 μmol g−1 h−1 and an O2 evolution rate of 460 μmol g−1 h−1. The apparent quantum efficiencies reached ∼3.4% at 400 nm and 1.7% at 420 nm. Wang et al. employed reduced graphene oxide (RGO) nanosheets as a solid-state electron mediator to construct an electron shuttle channel between the H2 evolution photocatalyst and the O2 evolution photocatalyst.120 By rationally tuning the interfacial contact in the Fe2O3/RGO/PCN composite, efficient charge transport between PCN and Fe2O3 was achieved, enabling overall H2O splitting with enhanced activity. The photocatalytic performance of overall H2O splitting can also be optimized by in situ growth of crystalline carbon nitride in LaOCl.121 The IEF at the hybrid interface facilitates spatial charge carrier separation, with crystalline carbon nitride promoting efficient photogenerated charge transfer across the heterogeneous interface. Compared with the polymer carbon nitride/LaOCl hybrid, the crystalline carbon nitride/LaOCl system significantly enhances H2 and O2 generation rates by a factor of 3 and 28, respectively.
In addition to the extensive development of carbon nitride-based materials, researchers have also pioneered alternative polymer-based hybrid photocatalysts for overall H2O splitting. Yang et al. made significant advancements by developing an inorganic–organic hybrid photocatalyst that integrates polymerized carbon–oxygen semiconductors (PCOS) with Ni2P.122 The strategic incorporation of Ni2P enhances hole accumulation, while the presence of NiS, supported on the surface of nickel phosphide, serves as an active site for H2 production. This Ni2P/NiS@PCOS catalyst promotes a two-step, two-electron reaction pathway, enabling selective H2O oxidation and efficient photocatalytic performance. The synergistic effects of PCOS and NiS modify the electron-rich hole states of Ni atoms, optimizing the dissociation thermodynamics of H2O and enhancing electron migration kinetics. As a result, this system yields photocatalytic H2 and O2 production rates of 150.7 and 70.2 μmol h−1via overall H2O splitting, respectively.
Perylene dimethylimide (PDI) is another commonly used organic component in the design of photocatalytic systems. Recent studies have demonstrated that overall H2O splitting can be efficiently achieved by anchoring ZnIn2S4 nanosheets onto the surface of highly crystalline PDI supramolecular nanorods.123 The unique hierarchical branching structure created by this inorganic–organic hybrid significantly increases the surface area and light-harvesting capabilities of the photocatalyst. Fourier-transform infrared spectroscopy (FT-IR) and X-ray photoelectron spectroscopy (XPS) confirm robust covalent bonding between HC-PDI and ZnIn2S4. This intimate connection, along with the IEF, facilitates rapid charge transfer and establishes a Z-scheme heterojunction. The optimized HC-PDI@ZnIn2S4 hybrid demonstrates remarkable visible-light-driven catalytic activity for overall H2O splitting, with H2 and O2 evolution rates reaching 275.4 and 138.4 μmol g−1 h−1, respectively, without the need for sacrificial agents. Moreover, this system exhibits excellent stability for over 40 hours of operation.
Organic polymers also serve as excellent supports in hybrid photocatalysts, stabilizing co-catalysts and promoting efficient interfacial charge transfer. Recently, a hydrothermal method was reported to in situ grow CdS and Co9S8 on porous polymer microreactor (PP12) as the light absorption component and co-catalyst,124 respectively (Fig. 6(f)). The interaction between PP12 and the inorganic components enhanced charge separation, while the bonding between Co9S8 and PP12 created abundant catalytic active sites. Without the use of sacrificial reagents, the hybrid photocatalyst achieved H2 and O2 production rates of ∼4.4 and ∼2.2 mmol g−1 h−1, respectively (Fig. 6(g)). This represents a significant enhancement in efficiency compared to conventional stirred tank reactors, which exhibit a much lower H2 rate of 0.004 mmol g−1 h−1.
The tunable structure and inherent stability of polymer-based photoelectrode coatings make them ideal for optimizing charge transport pathways in photocatalytic systems. For instance, Fonzo and Antognazza et al. demonstrated this by fabricating a hybrid organic/inorganic photocathode composed of FTO/CuI/P3HT:PCBM/TiO2/Pt layers through precise layer-by-layer deposition.125 The conjugated polymer P3HT, with its 1.9 eV bandgap, enhances the visible light response of the hybrid electrode. The energy band structure formed by the gradient arrangement of organic and inorganic materials matches the electrochemical potential needed for efficient H2 production, providing an inherent potential gradient. The inclusion of high-quality hole-selective layers (PCBM and CuI) effectively separates photogenerated electrons and holes, reducing recombination losses of free charge carriers. This hybrid system achieved a faradaic efficiency of 100% during the H2 evolution process and an incident photon-to-electron conversion efficiency (IPCE) exceeding 50%, demonstrating exceptional photocatalytic performance.
Optimizing the stability of organic components is essential for improving the efficiency of photocatalytic H2 production, as many organic polymers face challenges in terms of long-term durability under reaction conditions. Covalent triazine frameworks (CTFs), a chemically robust class of semiconducting polymers, offer an attractive solution due to their tunable electronic structures, which can be modified with lightweight functional groups to enhance performance. For example, we developed a durable photocathode by integrating triazine units with a bithiophene moiety,126 and further employed CTFs to modify the surface of Mo-doped BiVO4 to construct a high-performance photoanode (Fig. 7(a) and (b)). The well-matched band structure and alignment of the hybrid photoelectrochemical (PEC) H2O splitting system enable the modified CTF-BTh to form both p–n junctions with inorganic photocathodes and type II heterojunctions with photoanodes. The staggered energy levels and efficient charge transfer routes between the heterogeneous layers facilitate the sequential transfer of charges, optimizing the overall PEC performance (Fig. 7(c)). Moreover, the CTF-BTh film serves as an effective anti-corrosion layer, protecting the photoelectrodes from photo-corrosion and enhancing the stability of the system. Remarkably, the photocurrent density of both photoelectrodes coated with CTF-BTh only showed a marginal decrease of 10% after 150 hours of operation. As depicted in Fig. 7(d), the interfacial electron transfer and stability of the inorganic–organic hybrid photoelectrode enable a light energy conversion efficiency of 3.24%, even after continuous operation for 120 hours, which is in the top realm among related studies (Fig. 7(e)).
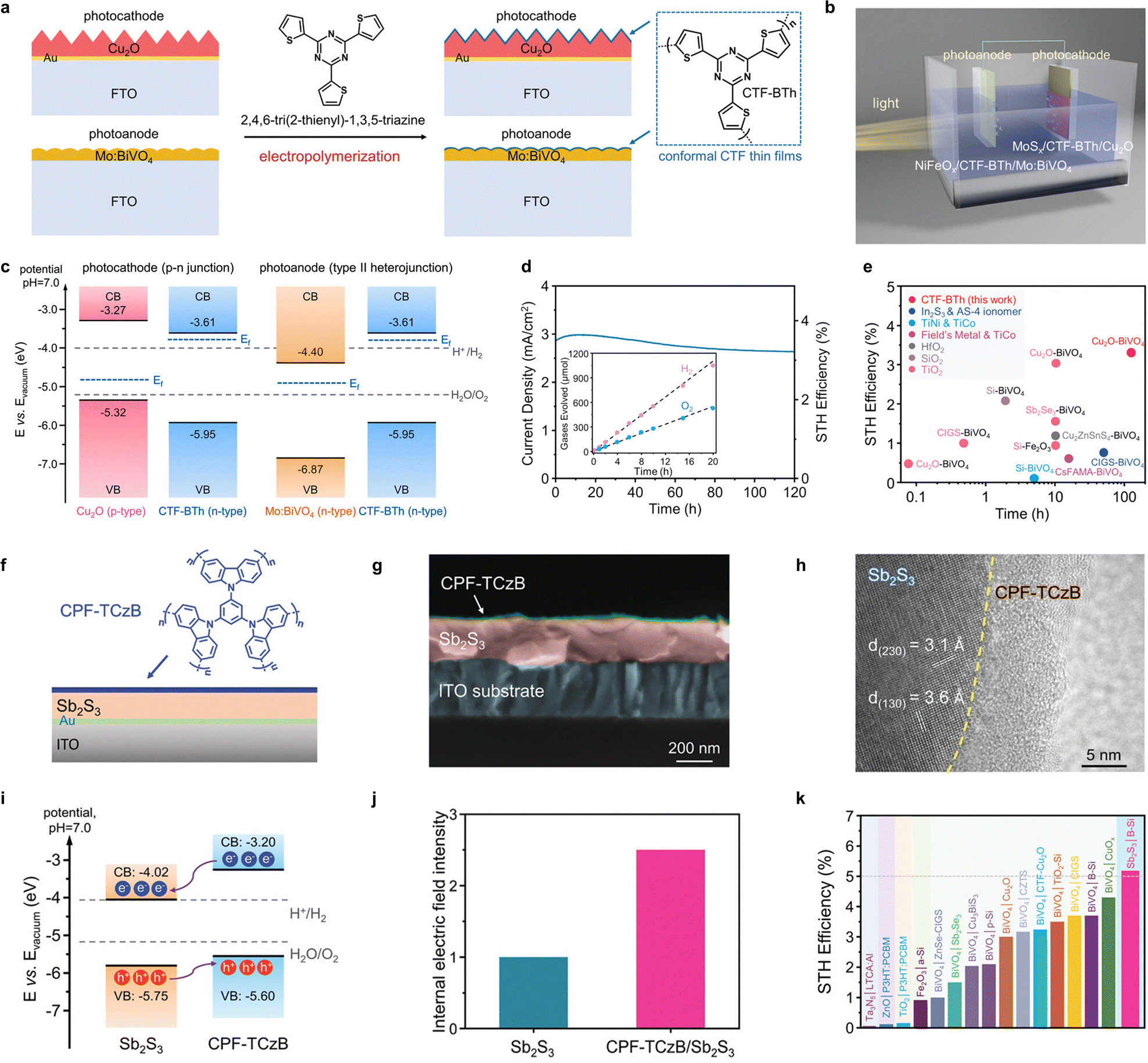 | ||
| Fig. 7 (a) Preparation process of the hybrid photoanode. (b) Schematic diagram of the photolysis H2O system. (c) Schematic representation of band-position alignments and heterojunction energy structure. (d) The long-term J–t curve of the tandem device with STH efficiencies. (e) STH efficiencies and operating durations summarized from reported tandem PEC H2O splitting devices. Adapted with permission from ref. 126. Copyright 2021, Wiley-VCH. (f) The illustration of the CPF-TCzB/Sb2S3 hybrid photoanode. (g) Cross-sectional SEM image of the CPF-TCzB/Sb2S3 photoanode. (h) HRTEM image showing the interface between Sb2S3 and CPF-TCzB. (i) Band structures of the Sb2S3 and CPF-TCzB. (j) Relative IEF intensity in hybrid interface. (k) Summarized STH efficiencies from reported unbiased PEC devices for overall H2O splitting. Adapted with permission from ref. 127. Copyright 2022, Wiley-VCH. | ||
In addition, coating conjugated polycarbazole frameworks (CPF-TCzB) on the Sb2S3 photoanode introduces a new pathway for charge transport (Fig. 7(f)), see the hybrid interface structure in Fig. 7(g) and (h).127 In the type II heterojunction configuration, the more negative conduction band energy level of the CPF-TCzB layer improves the stability of the tandem device. The appropriate band-edge energy shift of Sb2S3 induced by the heterojunction also enhances charge separation (Fig. 7(i)). As shown in Fig. 7(j), the enhanced IEF at the hybrid interface further facilitates the separation of photogenerated charges. The carbazole-based conjugated polymer, which is an effective hole-transport material with high mobility, contributes to a photocurrent density of 10.1 mA cm−2 at 1.23 V. This hybrid photoanode maintains stability for at least 12 hours under continuous illumination, producing H2 and O2 at a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The unbiased photoelectrochemical tandem device achieves a STH conversion efficiency of 5.21% (Fig. 7(k)), with a photocurrent density loss of less than 10% after 100 hours of continuous operation.
:1 ratio. The unbiased photoelectrochemical tandem device achieves a STH conversion efficiency of 5.21% (Fig. 7(k)), with a photocurrent density loss of less than 10% after 100 hours of continuous operation.
The chemically robust structure and diverse tunable properties of amorphous organic conjugated polymers have been extensively investigated for their use in corrosion-resistant coatings or as intermediate layers to modulate photophysical processes in photocatalysis.128,129 Given the advancements in polymer and semiconductor catalysis, there is significant potential for further exploration of a broader range of polymer materials combined with inorganic semiconductors. While current research is focused on a limited selection of conjugated organic polymers, expanding the scope to include materials with exceptional mechanical strength and optimizing the contact area and mass transfer performance between the organic and inorganic phases will be crucial for developing more efficient inorganic semiconductor–organic polymer hybrid systems. These innovations will provide new opportunities for enhancing the efficiency of photocatalytic H2O splitting for H2 production.
4.3 Integrating inorganic semiconductors with COFs for photocatalytic overall H2O splitting
COFs are crystalline porous polymers assembled via reversible bond-forming reactions, enabling the precise integration of molecular building blocks into highly ordered architectures.130,131 These materials exhibit low density, high specific surface area, well-defined pore channels, and tunable active sites, enabling them highly promising for a wide range of applications, including heterogeneous catalysis, gas adsorption, chemical sensing, and energy storage.132–134 Notably, COFs have emerged as promising crystalline organic semiconductors for photocatalysis due to their extended π-conjugation, tunable optoelectronic properties, and periodic porosity. They exhibit strong potential in H2 evolution, CO2 reduction, and pollutant degradation,135,136 and several COF-based photocatalysts have also demonstrated activity for overall H2O splitting.137–139 However, a fundamental challenge for COFs, as with many organic semiconductors, is the strong exciton effect and large exciton binding energy, which significantly hinder the generation of free carriers and impede efficient charge separation. These intrinsic properties limit their effectiveness in driving multi-electron processes required for photocatalytic overall H2O splitting. As discussed above, the integration of organic COF materials with inorganic semiconductors provides a viable strategy to overcome these inherent limitations and improve the photocatalytic performance of COFs.The light harvesting process is critical for the efficient operation of the interfacial photo-induced electron transfer process. The connectivity within COFs is primarily mediated by borate esters, imine, hydrazone, or triazine units, which exhibit high bond reversibility,140–143 but their chemical bonding modes are relatively constrained (Fig. 8(a)). Despite this, the structural flexibility and ease of modification inherent in the COF frameworks are invaluable in optimizing the light absorption performance of hybrid catalytic systems. In photocatalytic applications, organic dye molecules such as Eosin Y and Rhodamine B, as well as photosensitive metal complexes, are often incorporated to enhance the photoresponse performance.144–146 For flexible porous frameworks, chromophores like porphyrin and triphenylamine can be embedded into the COF framework via in situ chemical reactions to boost their light absorption properties.147,148 Due to these unique properties, COFs often function as self-sufficient photocatalytic platforms that obviate the need for additional photosensitizers.
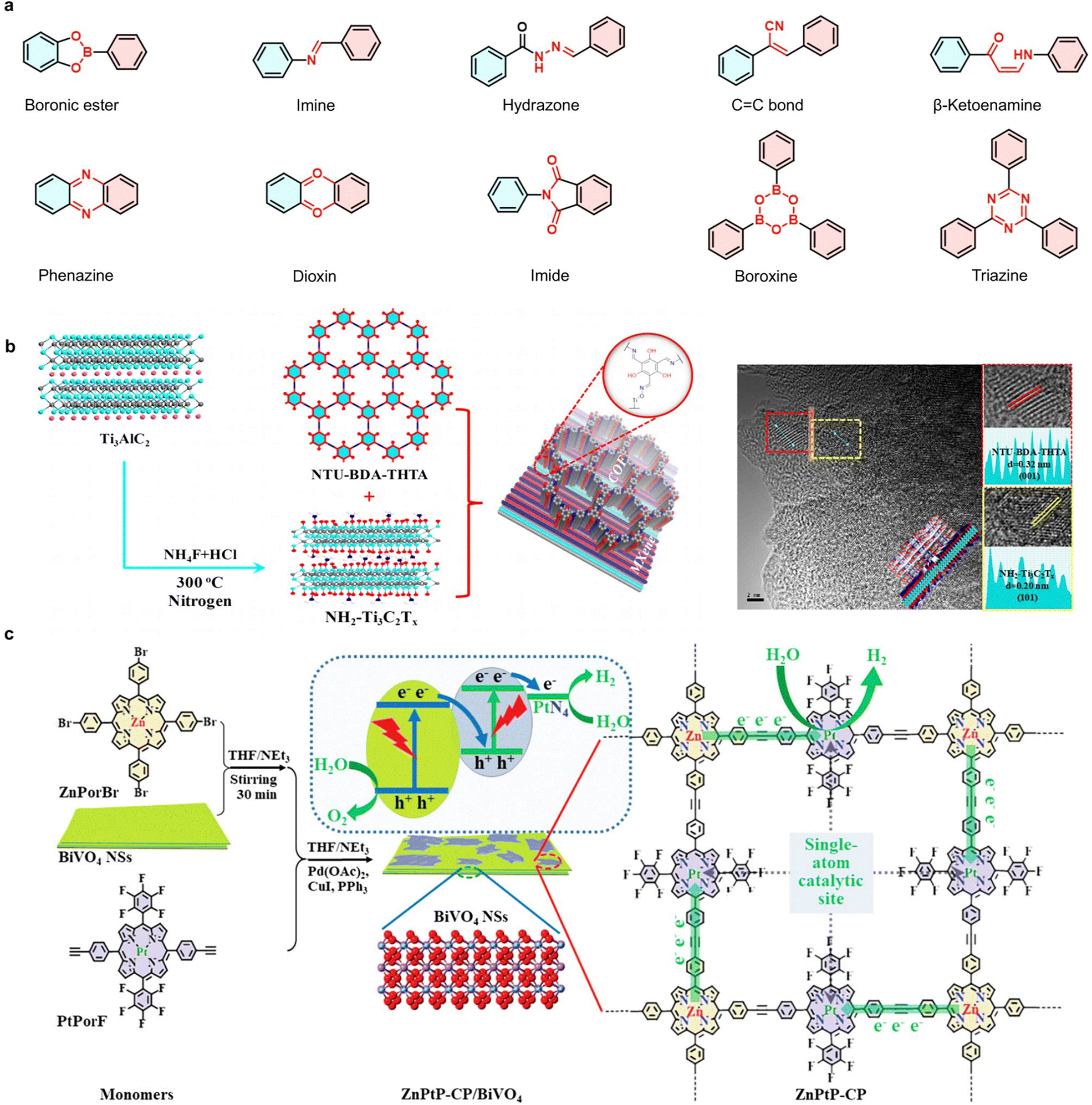 | ||
| Fig. 8 (a) Typical reversible covalent linkages in the reported COFs structures. (b) Synthesis of COFs and hybridization with NH2–Ti3C2Tx MXenes. Adapted with permission from ref. 150. Copyright 2020, American Chemical Society. (c) Schematic illustration of the synthesis, structure and energy band structure of the ZnPtP-CP/BiVO4 hybrid. Adapted with permission from ref. 151. Copyright 2021, Wiley-VCH. | ||
For example, Lan et al. utilized bipyridine ruthenium and zinc porphyrin as photosensitive moieties to construct a three-dimensional COF via Schiff-base reaction.149 Solid-state diffuse reflectance spectroscopy revealed that the resulting photocatalyst exhibited significantly broadened light absorption, resulting in enhanced H2 production efficiency. More recently, Zhao et al.150 synthesized a series of COFs with varied ratios of β-ketoenamine to imine moieties and evaluated the photocatalytic activity for H2 and O2 generation (Fig. 8(b)). The integration of COFs with inorganic NH2–Ti3C2Tx MXenes in a hybrid photocatalytic platform further enhances the lifetime of excited states and charge mobility. β-ketoenamine linkage in the backbone serves as a photosensitizer, optimizing the photoresponse performance of the hybrid system. The strong covalent coupling between these components endow the system with superior charge separation capabilities, enabling effective H2 and O2 production under visible light.
In addition, the ZnPtP-CP, a class of photosensitive COFs containing porphyrin groups,151 was connected to ultrathin BiVO4 through a Zn–O–V bridging bond by Li and Peng (Fig. 8(c)). This hybrid system forms a Z-scheme charge transfer mechanism between the inorganic and organic heterolayers, resulting in efficient electron enrichment on ZnPtP-CP and hole accumulation on BiVO4. The highly dispersed PtN4 centers within the grafted heterometallic porphyrins act as efficient single-atom catalytic sites for the reduction of H2O. The cascade charge transfer process, coupled with a two-step excitation mechanism, significantly enhances overall H2O splitting, producing H2 and O2 without requiring sacrificial reagents or external bias. The apparent quantum yield of the ZnPtP–CP/BiVO4 inorganic–organic hybrid photocatalyst at 400 nm reaches 9.85% for H2 evolution, demonstrating a remarkable improvement in photocatalytic efficiency.
In addition to the rational modification of the original framework, post-modification techniques, such as hybridizing with inorganic materials, provide an effective strategy to enhance the light absorption properties of COFs. For instance, the II–VI semiconductor CdS, with its size-dependent electronic characteristics and a suitable bandgap of 2.4 eV, is a promising candidate for photocatalytic H2O splitting. By integrating CdS with COFs to form a heterostructure photocatalyst, issues such as photocorrosion and low photocatalytic performance can be effectively addressed.152 The photocatalytic activity of the CdS–COF hybrid catalyst, formed via the photo-deposition method, is significantly improved due to the exposure of additional active sites. Time-resolved spectroscopy and electrochemical impedance spectroscopy confirm that the hybrid photocatalytic platform exhibits superior charge separation and transport efficiency. The hexagonal layered structure of SnS2,153 with exposed S atoms on its surface, facilitates the formation of hydrogen bonds with H2O molecules, enhancing the photocatalytic performance. To form the heterojunction, a ketoenamine-based COF (TpPa-1-COF) with well-matched energy band positions is selected, promoting the directional migration of photogenerated electrons and holes at the interface. UV-vis diffuse reflectance spectroscopy shows a red-shifted absorbance edge to 700 nm for the hybrid photocatalyst. Under 600 nm light irradiation, the hybrid system achieves an apparent quantum efficiency of 0.23% towards H2 evolution, which is 21.7 times higher than that of the original TpPa-1-COF. Unlike other organic semiconductors, this heterogeneous hybridization allows for the incorporation of inorganic materials within the pores of COFs, promoting uniform assembly and optimizing photocatalytic processes.
Due to their tunable electronic bandgap and high molar extinction coefficient,154 we have systematically investigated the use of semiconductor quantum dots (QDs) in artificial photosynthesis. For instance, by passivating the defect states on the surface,155 the turnover number of photocatalytic H2 evolution is greatly enhanced to (4.4 ± 0.3) × 105, ∼110-fold to that of unmodified CdSe QDs under identical conditions (Fig. 9(a)). Recently, a well-designed dot-on-rod nano-heterostructure is established to solve the problem of sluggish hole transfer and utilization,156 successfully coupling H2O oxidation with CO2 reduction under visible light (Fig. 9(b) and (c)). Moreover, the abundant surface sites and ultra-small size make them ideal candidates for encapsulation in COF channels through coordination or covalent interactions for advanced photochemical transformations.157 This encapsulation offers greater control over the size of the inorganic nanocrystals, enhances photocatalyst stability, and suppresses the aggregation-induced quenching effect commonly observed in QDs. Meanwhile, the ordered structure of COF channels improves QD dispersion, passivates defect states on QD surfaces, and significantly improves their stability in photocatalytic environments. This integration strategy also enhances the light-harvesting performance of the inorganic–organic hybrid photocatalyst. Recent studies have explored hybrid photocatalytic systems that incorporate other QDs with COFs, demonstrating the potential for significant improvements in efficiency.158
 | ||
| Fig. 9 (a) Quantum dots for photocatalytic H2 evolution without external cocatalysts. Adapted with permission from ref. 155. Copyright 2018, Wiley-VCH. (b) Scheme of charge transfer processes and radial distribution function. (c) Illustration of the overall reaction mechanism of CO2 photoreduction taking H2O as an electron donor. Adapted with permission from ref. 156. Copyright 2021, Wiley-VCH. (d) The schematic illustration of the BiFeO3@TpPa-1-COF synthetic processes and heterojunction structure. (e) Proposed photocatalytic pathway of H2 and O2 evolution. (f) The rate of H2 and O2 production of photocatalysis. (g) Comparison of photocatalytic overall H2O splitting rate of different systems based on COF and C3N4. Adapted with permission from ref. 159. Copyright 2022, Wiley-VCH. | ||
Despite the favorable carrier transport properties of single inorganic semiconductors, their photocatalytic performance often requires further enhancement in terms of suppressing carrier recombination, refining thermodynamics, and improving stability. In this context, COFs materials offer a stable platform with extensive conjugation and facilitate the formation of multiple electronic structures when coupled with inorganic semiconductors. The heterojunction formed by the energy level alignment between inorganic and organic components generates a robust IEF, which aids in directed charge transport. This internal field allows electrons and holes to migrate to energetically favorable positions for oxidation and reduction reactions, both thermodynamically and kinetically optimized.
For example, Lan et al. integrated TaPa-1-COF with piezoelectric BiFeO3 nanosheets through covalent bonds,159 resulting in the formation of highly efficient Z-scheme heterostructured photocatalysts for H2O splitting (Fig. 9(d)). The detailed analysis of the H2O splitting process driven by this hybrid photocatalyst (Fig. 9(e)) shows that the polarization potential generated by the IEF effectively separates charge carriers, thus leading to significant improvements in H2O splitting efficiency. TaPa-1-COF efficiently captures electrons, while BiFeO3 efficiently accumulates holes. Photoelectric tests show that the BiFeO3@TaPa-1-COF hybrid exhibits superior photocurrent density, resistance, and overpotential compared to control groups. Under ultrasonic and simulated sunlight irradiation, this photocatalytic system achieves H2 production at a rate of 1416.4 μmol g−1 h−1 and O2 at a rate of 708.2 μmol g−1 h−1. These results highlight the critical role of the ultrasound-driven polarization potential, which functions as an IEF, facilitating the separation and transfer of photogenerated carriers. Furthermore, the charge transport mode established by the COF-based hybrid platform significantly enhances the reaction thermodynamics for H2O splitting. The photocatalytic and piezoelectric photocatalytic efficiencies of BiFeO3@TpPa-1-COF surpass those of other COF or C3N4-based photocatalysts, demonstrating the outstanding performance of this hybrid system (Fig. 9(f) and (g)).
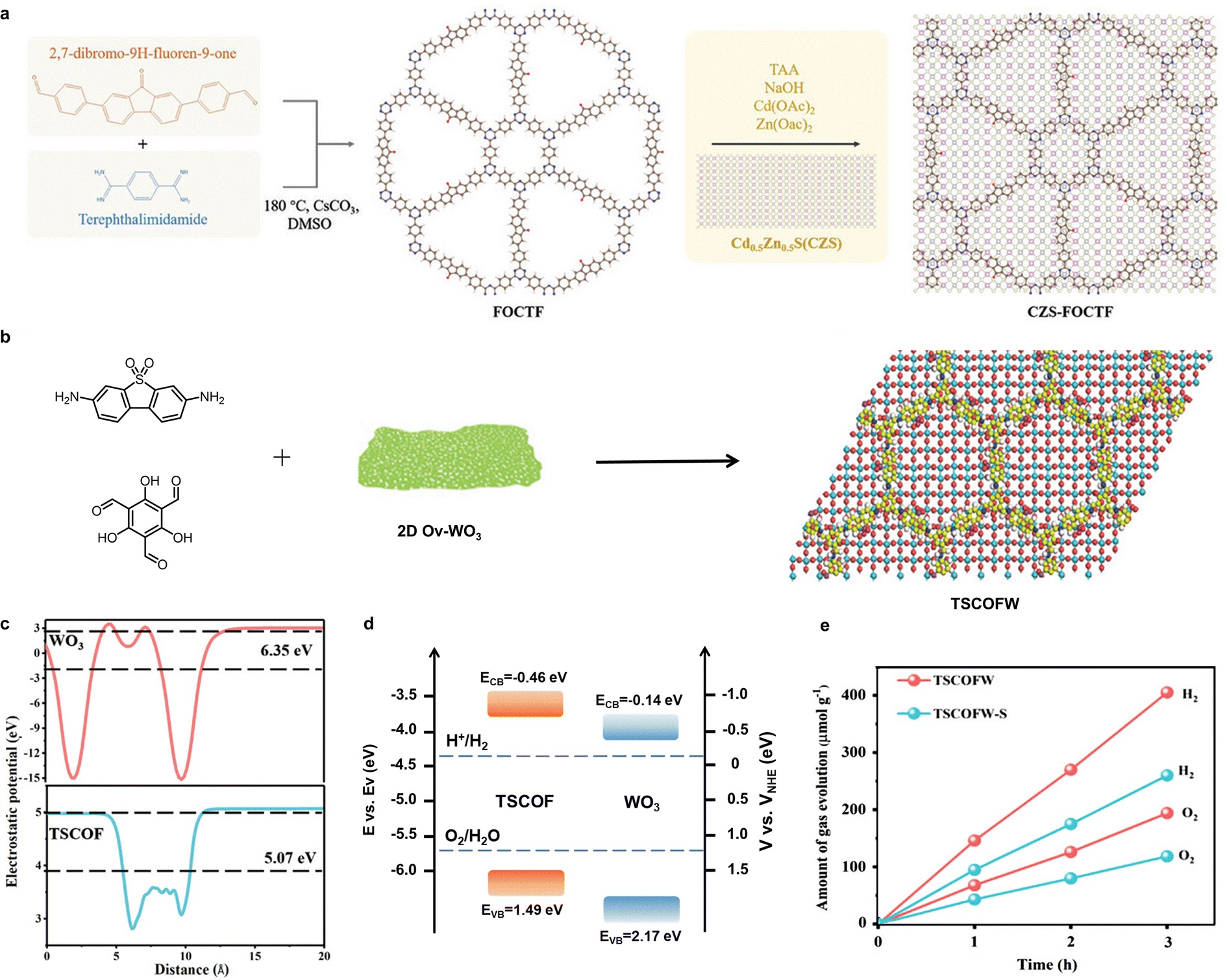
Li et al. reported a novel inorganic–organic hybrid S-scheme heterojunction photocatalyst for overall H2O splitting,160 constructed by integrating a fluorenone-based covalent triazine framework (FOCTF) with a Zn0.5Cd0.5S (CZS) solid solution (Fig. 10(a)). The in situ growth of CZS on the FOCTF surface enabled the formation of a well-defined S-scheme CZS-FOCTF heterojunction with enhanced charge separation and transfer efficiency. Both theoretical calculations and experimental analyses confirmed that the band alignment and work function disparity between FOCTF and CZS facilitated the generation of a strong IEF, which is critical for driving directional charge migration. Under illumination, photogenerated carriers with weak redox potentials underwent recombination via the S-scheme pathway, whereas those with strong redox abilities were preserved to participate in surface redox reactions. Consequently, the catalyst exhibited a H2 evolution rate of ∼247.6 mmol g−1 h−1, representing a 3.8-fold enhancement over pristine CZS.
 | ||
| Fig. 10 (a) Scheme of the CZS-FOCTF synthesis. Adapted with permission from ref. 160. Copyright 2024, Wiley-VCH. (b) Synthesis diagram of TSCOFW. (c) Total density of states and partial density of states of Ov-WO3 and TSCOF. (d) Energy band structure of WO3 and TSCOF. (e) Time profiles of photocatalytic H2 and O2 evolution. Adapted with permission from ref. 162. Copyright 2023, Wiley-VCH. | ||
In a separate study, Zhang et al. developed a noble-metal-free Cu2O/TpPa-2-COF photocatalyst exhibiting self-accelerating H2 evolution performance.161 Upon visible-light irradiation, the initial H2 evolution rate reached 4.41 mmol g−1 h−1 and continuously increased to 27.27 mmol g−1 h−1 after 25 h, indicating a 6.2-fold activity enhancement. The binary Cu2O/TpPa-2-COF system evolved into a ternary Cu-Cu2O/TpPa-2-COF heterojunction, which further improved charge carrier dynamics and catalytic efficiency. Density functional theory (DFT) calculations revealed that the Schottky barrier height at the Cu–Cu2O (111) interface was substantially lower than that at the Pt–Cu2O (111) interface. The tailored hybrid interface provides a robust and efficient platform for photogenerated charge separation, thereby boosting photocatalytic H2 production.
To overcome the challenges of charge recombination and poor H2O oxidation capability,162 Li et al. successfully in situ synthesized TSCOF on oxygen vacancy (O-vacancy) WO3 nanosheets through W–O–C chemical bonds (Fig. 10(b)). These interfacial covalent bonds not only enhance the IEF but also reduce carrier diffusion distance due to the layered structure of O-vacancy WO3 (Ov-WO3) and TSCOF, resulting in significant boost in the overall efficiency of photocatalytic H2O splitting. Combining ultraviolet photoelectron spectroscopy (UPS) and density functional theory (DFT) calculations, the authors determined that the work function difference between TSCOF and WO3 is approximately 1.3 eV (Fig. 10(c)). This difference in work function and Fermi level generates an IEF that facilitates the establishment of a Z-scheme charge transfer mechanism (Fig. 10(d)), promoting efficient charge separation and transfer. As a result, the light absorption performance in the long-wavelength region is notably enhanced, which improves the photocatalytic H2 evolution half-reaction rate of the TSCOF-WO3 composite to 593 mmol g−1 h−1. Furthermore, the composite demonstrates impressive photocatalytic activity with H2 and O2 evolution rates of 146 and 68 μmol g−1 h−1, respectively, under optimal conditions. These values surpass those of most previously reported hybrid photocatalysts for overall H2O splitting (Fig. 10(e)), highlighting the superior performance of this integrated system.
Hybrid materials composed of COFs and metal–organic frameworks (MOFs) have recently emerged as a promising class of photocatalysts for overall H2O splitting. As prototypical porous crystalline materials, MOFs are constructed by the coordination of metal ions or clusters with organic linkers, and their structural diversity and high designability have enabled broad applications in photocatalysis. Several representative MOF-based systems have demonstrated potential for overall H2O splitting through rational linker design to prolong charge carrier lifetimes, suppress radiative recombination, and the integration of appropriate cocatalysts.163 Building upon this, the incorporation of MOFs into COF platforms offers an opportunity to further regulate interfacial electric fields and promote directional charge transfer, which is critical for enhancing H2 evolution performance.
Recently, Lan et al. developed a novel MOF/COF hybrid photocatalyst by covalently integrating NH2-MIL-125(Ti)(DE-NM) with TpBpy-COF for overall H2O splitting,164 as shown in Fig. 11(a). By precisely controlling the exposed crystal facets, a strong IEF was established at the DE-NM/TpBpy-COF interface, which significantly accelerated the separation of photogenerated carriers. The resulting hybrid system exhibited excellent photocatalytic overall H2O splitting activity under visible light, achieving H2 and O2 evolution rates of 331.6 and 165.7 μmol g−1 h−1, respectively. In another study, Liu et al. reported a structurally analogous system by immobilizing a triangular prismatic metal–organic cage (MOC-Q3) onto a highly crystalline β-ketoenamine-linked COF (EA-COF), thus forming a Z-scheme piezo-photocatalytic system for H2O splitting (Fig. 11(b) and (c)).165 Obviously, these findings underscore the great potentials of MOF/COF hybrid systems for efficient solar-to-hydrogen energy conversion via H2O splitting.
 | ||
| Fig. 11 (a) Schematic illustration of the synthetic route and band structure of the NH2-MIL-125(Ti)/TpBpy-COF hybrid photocatalyst. Adapted with permission from ref. 164. Copyright 2025, Wiley-VCH. (b) Preparation of MOC-Q3 and EA-COF. (c) Working mechanism of the MOC-Q3/EA-COF system. Adapted with permission from ref. 165. Copyright 2024, American Chemical Society. | ||
Collectively, these examples highlight the potential of inorganic–organic hybrid materials in photocatalytic overall H2O splitting for solar H2 generation. Table 1 summarizes, for each system, the performance figures of merit, the operating parameters, and the stability metrics of representative inorganic–organic hybrid photocatalytic systems for overall H2O splitting.
| Photocatalyst | Light source | H2 production rate | O2 production rate | Stability | STH efficiency | Ref. |
|---|---|---|---|---|---|---|
| P10/BiVO4 | 300 W Xe lamp (λ > 420nm) |
10.8 μmol h−1 | 4.5 μmol h−1 | 70 h | 0.0014% | 104 |
| Cu6Sn5/PANI | 300 W Xe lamp | 121.3 μmol g−1 h−1 | 58.6 μmol g−1 h−1 | 20 h | — | 105 |
| α-Fe2O3/g-C3N4 | 300 W Xe lamp (λ > 400nm) |
38.2 μmol g−1 h−1 | 19.1 μmol g−1 h−1 | 5 h | — | 109 |
| CoO/C3N4 | 300 W Xe lamp | 2.51 μmol h−1 | 1.39 μmol h−1 | 25 h | — | 112 |
| CoO nanorod/C3N4 | 300 W Xe lamp (λ > 400 nm) | 92 μmol h−1 | — | — | — | 113 |
| g-C3N4/ITO/Co–BiVO4 | 300 W Xe lamp | 95.41 μmol g−1 h−1 | 40.23 μmol g−1 h−1 | 18 h | 0.028% | 114 |
| BiVO4/PCN | 300 W Xe lamp | 14 μmol h−1 | 6.8 μmol h−1 | 18 h | — | 115 |
| WO3·H2O/g-C3N4 | 300 W Xe lamp | 482 μmol g−1 h−1 | 232 μmol g−1 h−1 | 24 h | — | 116 |
| CdS/Ni2P/g-C3N4 | 300 W Xe lamp (λ > 420 nm) | 15.56 μmol g−1 h−1 | 7.75 μmol g−1 h−1 | 30 h | — | 117 |
| Co3(PO4)2/g-C3N4 | 300 W Xe lamp (λ > 400 nm) | 375.6 μmol g−1 h−1 | 177.4 μmol g−1 h−1 | 15 h | — | 118 |
| Pt-CdS/g-C3N4–MnOx | 300 W Xe lamp (λ > 400 nm) | 924.4 μmol g−1 h−1 | 460 μmol g−1 h−1 | 18 h | — | 119 |
| Fe2O3/RGO/PCN | 300 W Xe lamp | 43.6 μmol h−1 | 21.2 μmol h−1 | 24 h | — | 120 |
| CCN/LaOCl | 300 W Xe lamp | 60.6 μmol h−1 | 28.1 μmol h−1 | 20 h | — | 121 |
| Ni2P/NiS@PCOS | 300 W Xe lamp | 150.7 μmol h−1 | 70.2 μmol h−1 | 3 h | 0.91% | 122 |
| PDI@ZnIn2S4 | 300 W Xe lamp (λ > 400 nm) | 275.4 μmol g−1 h−1 | 138.4 μmol g−1 h−1 | 40 h | — | 123 |
| Co9S8/CdS@PP12 | 300 W Xe lamp (λ ≥ 420 nm) | 4.41 mmol g−1 h−1 | 2.20 mmol g−1 h−1 | 20 h | — | 124 |
| (MoSx/CTF-BTh/Cu2O): (NiFeOx/CTF-BTh/Mo:BiVO4) | 300 W Xe lamp | ∼50 μmol h−1 | ∼25 μmol h−1 | 120 h | 3.24% | 126 |
| (Pt/TiO2/Si):(NiCoFe–Bi/CPF-TCzB/Sb2S3) | 300 W Xe lamp | ∼0.067 mmol cm−2 h−1 | ∼0.033 mmol cm−2 h−1 | 12 h | 5.21% | 127 |
| ZnPtP–CP/BiVO4 | 300 W Xe lamp (λ > 400 nm) | 77.3 μmol h−1 | 39.5 μmol h−1 | 15 h | — | 151 |
| BiFeO3@TpPa-1-COF | 300 W Xe lamp (λ ≥ 420 nm) | 1416.4 μmol g−1 h−1 | 708.2 μmol g−1 h−1 | 2 h | — | 159 |
| Ov-WO3/TSCOF | 300 W Xe lamp (λ > 420nm) |
146 μmol g−1 h−1 | 68 μmol g−1 h−1 | 15 h | — | 162 |
| DI-NM/Pt@TpBpy-COF | 300 W Xe lamp | 331.6 μmol g−1 h−1 | 165.7 μmol g−1 h−1 | 25 h | 0.21% | 164 |
5. Conclusions and perspectives
Driven by the imperative of decarbonization and the pursuit of sustainable energy carriers, the global hydrogen economy is undergoing a pivotal transformation. As of 2023, global hydrogen demand has reached 97 million tonnes, the vast majority of which is still supplied by fossil-fuel-based processes such as steam methane reforming (SMR).166 This method continues to dominate due to its entrenched infrastructure and low production cost ($1–3 per kg H2) (Fig. 12(a)).167 However, the environmental cost is substantial as the so-called “gray H2” results in over 600 million tonnes of CO2 emissions annually. This highlights the urgent need to restructure H2 production pathways toward clean and sustainable alternatives.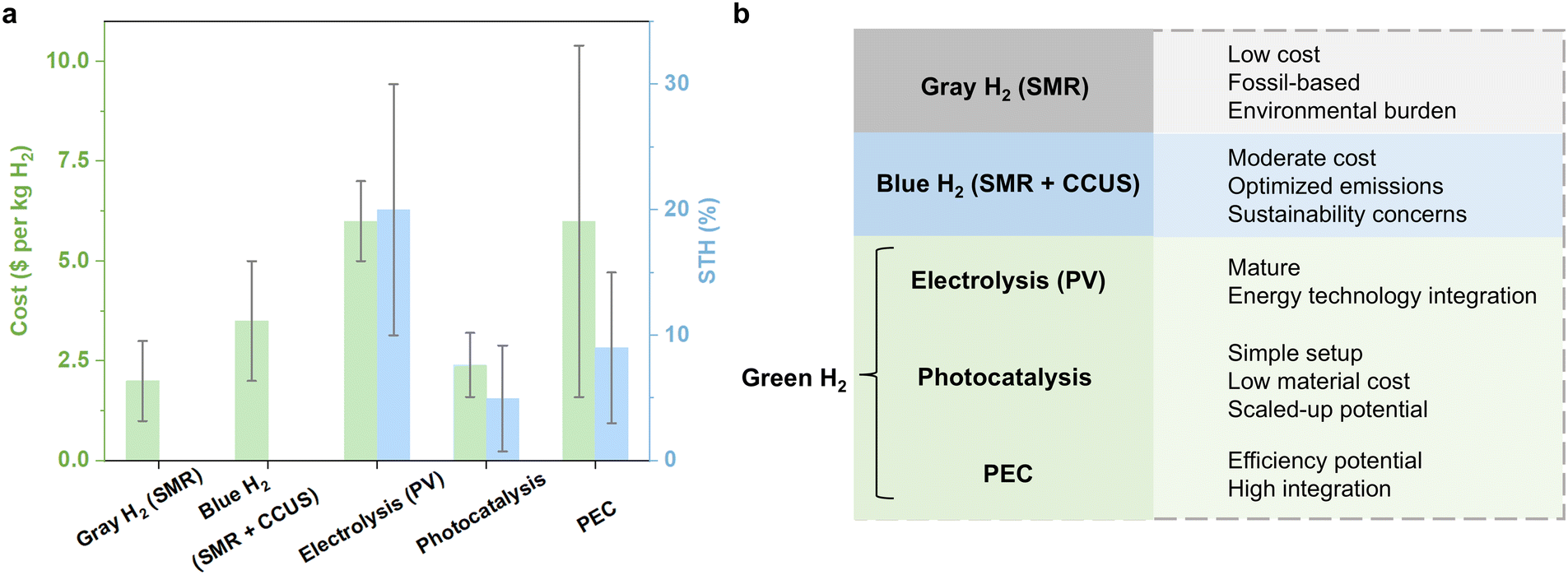 | ||
| Fig. 12 (a) Comparison of H2 production pathways in terms of cost ($ per kg H2, green bars) and STH efficiency (blue bars). Error bars represent the reported or estimated variability in cost and efficiency. (b) Summary of key characteristics for different H2 production technologies. | ||
To mitigate the carbon footprint associated with fossil-based H2, blue H2 has gained increasing attention. By integrating carbon capture, utilization, and storage (CCUS) technologies into conventional SMR processes, blue H2 can reduce CO2 emissions by more than 55%.168 However, this approach raises the production cost to approximately $2–5 per kg H2,167 depending on factors such as capture efficiency, energy losses, and the feasibility of geological storage (Fig. 12(a)). While large-scale blue H2 projects have been launched, concerns remain regarding methane leakage, the long-term stability of carbon storage, and the compatibility of blue H2 with long-term net-zero commitments.
In contrast, green H2 produced via H2O electrolysis powered by renewable energy—particularly photovoltaic (PV) electricity—offers a carbon–neutral solution. However, this pathway remains cost-intensive. Depending on electricity prices, electrolyser efficiency, and deployment scale, the average production cost of green H2 typically ranges from $5 to 7 per kg H2 (Fig. 12(a)).167 According to the International Energy Agency (IEA), the cost gap between fossil-derived and low-emission H2 is projected to narrow to $1–3 per kg by 2030. Recently, laboratory-scale photovoltaic (PV)-driven electrolyser has achieved STH efficiencies of up to 30%, and several pilot-scale facilities based on this technology have been established globally.169 Nevertheless, large-scale H2 production assisted by PV electricity is still anticipated to remain costlier than fossil-based H2 in the foreseeable future.
Among emerging technologies, photocatalytic H2O splitting, particularly particulate suspension systems, offers a structurally simplified and potentially scalable approach to solar H2 production. At STH efficiencies of ∼10%, particle-based systems may reach H2 production costs of $1.60–3.20 per kg H2 (Fig. 12(a)), rivalling both SMR and electrolysis, provided challenges in efficiency, gas separation, and catalyst durability are addressed.167 The compatibility of these systems with earth-abundant (Fig. 12(b)), low-cost materials and their suitability for decentralized deployment further strengthen their promise for distributed H2 infrastructure.170 PEC H2O splitting integrates solar harvesting and electrochemical conversion, offering system compactness but currently constrained by high materials costs, modest stability, and integration complexity. Technoeconomic assessments indicate that PEC-based centralized systems span a broad levelised cost of $1.60–10.40 per kg H2, with viability hinging on surpassing 10% STH threshold and ensuring long-term stability.171 While some perovskite-based PEC systems have demonstrated STH efficiencies of up to 15%, their scalability and long-term reliability remain critical challenges to overcome.172 We provide a detailed comparison of various H2 production technologies in Fig. 12, highlighting their technical characteristics and operational costs. Although photocatalytic approaches are still primarily at the laboratory research stage, they demonstrate significant potential in terms of sustainability and cost competitiveness.
Efficient overall H2O splitting through photo(electro)catalysis has long been a central goal in the field of renewable energy, which has been investigated for more than 50 years. Extensive research has focused on novel materials construction, including inorganic semiconductors, organic semiconductors, and their hybridized systems, to facilitate H2 and O2 production via photocatalytic H2O splitting. The inorganic–organic hybrids in Fig. 13(a) refer to the integration of organic and inorganic semiconductors with distinct bandgap structures. Although the overall efficiency of inorganic–organic hybrid materials remains relatively modest, there is significant potential for further improvement in this class of materials. Inorganic–organic hybrid photocatalysts combine heterogeneous organic and inorganic components, offering unique interfacial properties and superior carrier dynamics. Despite the demanding requirements for multiple charge and mass transfer processes in photocatalytic H2O splitting, these hybrid platforms present promising pathways for efficient H2 and O2 production through careful modulation of the hybrid interface.
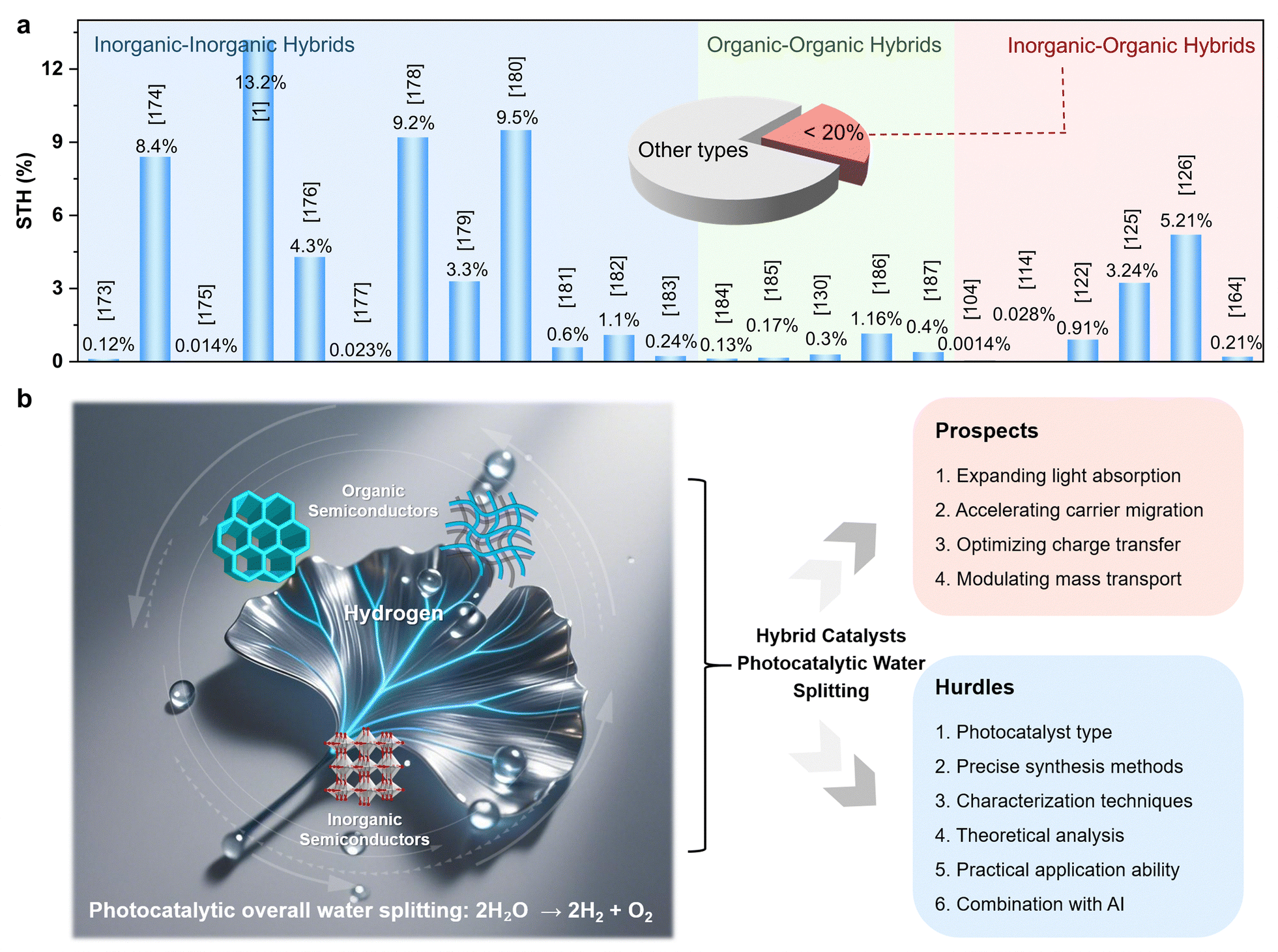 | ||
| Fig. 13 (a) The comparison of the STH conversion efficiencies for recently reported inorganic–inorganic hybrids, organic–organic hybrids, and inorganic–organic hybrids in photocatalytic and photo-electrocatalytic overall H2O splitting systems. The embedded statistical graph shows the proportion of inorganic–organic hybrids in photocatalytic and photo-electrocatalytic overall H2O splitting field. The data were adapted from ref. 1, 104, 114, 122, 125, 126, 130, 164, and 173–187. (b) Prospects and hurdles of photocatalytic H2O splitting employing inorganic–organic hybrid catalysts. | ||
The inorganic–organic hybrid strategy offers numerous advantages, positioning it as a highly promising platform for solar-driven H2O splitting and other photocatalytic processes. The diversity of hybrid catalyst structures and the design flexibility enable the incorporation of additional photosensitive units and the modulation of energy band structures, thus significantly enhancing the utilization of solar spectrum. The construction of large conjugated systems, combined with precise control over morphology, facilitates the directed migration of electrons and holes to specific active sites, optimizing photocatalytic performance. Furthermore, the IEF formed at the inorganic–organic hybrid interface plays a crucial role in promoting the directional movement of charges between the two phases, improving charge separation and transport efficiency. The interaction between the energy bands of organic and inorganic semiconductors fosters a broader range of charge transfer modes, such as Z-scheme heterojunctions, which are particularly beneficial for photocatalytic applications. Meanwhile, interface-mediated charge transfer mitigates the unfavorable charge recombination processes that often limit the efficiency of photocatalytic reactions. The multi-component nature of hybrid systems also enhances the exposure of surface active sites, accelerating kinetic processes such as mass transfer and the dissociation of reactants in photocatalytic reactions. More importantly, by employing rationally designed conjugated polymers, the common challenge of lattice mismatch in the fabrication of inorganic semiconductor-based heterojunctions can be effectively mitigated, leading to the formation of high-quality interfaces with improved compatibility and activity. This advantage in stability enhances the compatibility between the catalyst structure and reactive oxygen species during H2O splitting. Compared with conventional heterostructure, inorganic–organic hybrids offer suitable balance between production cost and photocatalytic performance. As highlighted in the previous sections, these integrated advantages are critical for significantly improving the efficiency and capability of photocatalytic H2O splitting systems.
Despite substantial progress in the development of inorganic–organic hybrid photocatalysts for overall H2O splitting, a significant gap remains between current performance levels and the energy efficiency thresholds required for practical, large-scale H2 production. As the field continues to mature, several critical challenges must be addressed to unlock the full potential of hybrid systems (Fig. 13(b)). Based on recent advances and our fundamental understanding of photocatalytic principles, future research should prioritize the following directions:
(1) The current hybrid photocatalytic platforms still face limitations in terms of efficiency, partly due to a restricted selection of inorganic and organic components. These limitations hinder the full potential of the hybrid systems. For instance, many COFs rely on a narrow set of linkages (e.g., imine, boronate ester, triazine), which restrict structural diversity and charge transport pathways. Expanding the chemical toolbox to include COFs with fully conjugated linkages (e.g., CC) and leveraging weak non-covalent interactions, such as hydrogen bonding, van der Waals interaction and π–π stacking, could provide enhanced flexibility in designing high-performance hybrids. Such interactions may facilitate better interfacial contact and charge delocalization, thereby improving catalytic efficiency.
(2) The structure of a photocatalyst plays a pivotal role in determining its physical and chemical properties, particularly in terms of exposing active sites for adsorption and catalysis. The controlled growth of specific crystal planes is critical for maximizing photocatalytic efficiency. However, achieving precise control over crystal facet orientation and surface morphology remains technically challenging. Future research should focus on advancing fabrication techniques that allow for fine-tuning of the catalyst's shape and surface properties, which can lead to more efficient exposure of active sites and improved photocatalytic performance.
(3) A deeper understanding of interfacial charge carrier dynamics is fundamental for performance enhancement. While techniques such as surface photovoltage spectroscopy and Kelvin probe force microscopy have shed light on IEF and surface potentials, they lack the temporal resolution to capture ultrafast charge transfer processes. Development and application of advanced time-resolved techniques, such as transient absorption spectroscopy, ultrafast photoluminescence, and operando scanning probe methods, will be crucial for visualizing charge migration pathways and identifying kinetic bottlenecks at relevant timescales.
(4) A more comprehensive understanding of catalytic mechanisms and charge dynamics at heterointerfaces is crucial for designing next-generation photocatalysts. As computational models and machine learning techniques continue to advance, their role in predicting and optimizing catalytic behavior is becoming increasingly important. Computational tools can assist in screening candidate materials, identifying optimal band alignments, and simulating charge dynamics at hybrid interfaces. Integrating theoretical insights with experimental work will be a valuable approach to optimizing hybrid systems and improving photocatalytic efficiency.
(5) Beyond performance metrics, the economic viability and long-term operational stability of hybrid photocatalysts are essential for real-world applications. The primary methods include two aspects: (i) enhancing interfacial bonding strength by constructing strong interfacial chemical bonds to replace weak physical interactions; (ii) developing more stable novel organic semiconductors, such as COFs and HOFs. The selection of earth-abundant elements, low-cost components and the mitigation of photocorrosion and interfacial degradation must be emphasized. Strategies to enhance stability include surface passivation, protective layer integration, and robust covalent bonding across interfaces.
(6) Artificial intelligence (AI) holds transformative potential for the rational design of inorganic–organic hybrid systems. Machine learning (ML) algorithms can extract structure–property relationships from high-throughput data, optimize interfacial energy alignments, and fine-tune parameters such as light absorption, carrier mobility, and synthesis conditions. AI can also facilitate in situ polymerization design and guide surface modification strategies, enabling faster, more cost-effective development of high-performance hybrid materials. This approach not only speeds up the development process but also reduces costs, facilitates in situ polymerization, and improves surface modifications, all of which are essential for advancing photocatalytic efficiency.
In summary, inorganic–organic hybrid photocatalysts represent a promising frontier in the pursuit of efficient solar-to-hydrogen energy conversion. While significant thermodynamic and kinetic barriers remain, continued progress in materials innovation, mechanistic understanding, and data-driven materials design is steadily closing the gap between laboratory research and practical implementation. With further interdisciplinary integration across chemistry, materials science, and computational modeling, inorganic–organic hybrid photocatalysts are promising to become an economically viable, low-cost strategy for addressing global energy demands and contributing to the future energy landscape.
Conflicts of interest
There are no conflicts to declare.Data availability
All data analyzed in this review are publicly available in the cited references and reported data. The datasets can be accessed through the corresponding links. No new data were generated for this study.Acknowledgements
We are grateful for financial support from the National Key R&D Program of China (2021YFA1500802 and 2022YFA1502900), the National Natural Science Foundation of China (22572205, 22088102, 22421005, and 52225307), the CAS Project for Young Scientists in Basic Research YSBR-004, and New Cornerstone Science Foundation.References
- S. Hwang, H. Gu, J. L. Young, M. A. Steiner, A. B. Laursen, R. A. Crichton, Y.-W. Yeh, P. E. Batson, L. C. Feldman, M. Li, K. Wyatt, A. Safari, T. G. Deutsch, E. Garfunkel and G. C. Dismukes, ACS Energy Lett., 2024, 9, 789–797 CrossRef CAS
.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K. Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed
- Y. Ling, H. Wang, M. Liu, B. Wang, S. Li, X. Zhu, Y. Shi, H. Xia, K. Guo, Y. Hao and H. Jin, Energy Environ. Sci., 2022, 15, 1861–1871 RSC
- S. Peng, F. Gong, L. Li, D. Yu, D. Ji, T. Zhang, Z. Hu, Z. Zhang, S. Chou, Y. Du and S. Ramakrishna, J. Am. Chem. Soc., 2018, 140, 13644–13653 CrossRef CAS PubMed
- L. Chen, Z. Qi, S. Zhang, J. Su and G. A. Somorjai, Catalysts, 2020, 10, 858 CrossRef CAS
- S. Ghosh, A. Nakada, M. A. Springer, T. Kawaguchi, K. Suzuki, H. Kaji, I. Baburin, A. Kuc, T. Heine, H. Suzuki, R. Abe and S. Seki, J. Am. Chem. Soc., 2020, 142, 9752–9762 CAS
- K. Li, Y.-Z. Lin, K. Wang, Y. Wang, Y. Zhang, Y. Zhang and F.-T. Liu, Appl. Catal., B, 2020, 268, 118402 CrossRef CAS
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC
- M. H. Sheikh-Mohseni and A. Nezamzadeh-Ejhieh, Electrochim. Acta, 2014, 147, 572–581 CrossRef CAS
- N. Zhang, H. Zheng, Y. Guo, J. Feng, Z. Li and Z. Zou, ACS Sustainable Chem. Eng., 2019, 7, 10509–10515 CrossRef CAS
- Y. Li, Y. Wang, C.-L. Dong, Y.-C. Huang, J. Chen, Z. Zhang, F. Meng, Q. Zhang, Y. Huangfu, D. Zhao, L. Gu and S. Shen, Chem. Sci., 2021, 12, 3633–3643 RSC
- C. Bie, L. Wang and J. Yu, Chem, 2022, 8, 1567–1574 CAS
- C. Kranz and M. Wachtler, Chem. Soc. Rev., 2021, 50, 1407–1437 RSC
- J. Low, S. Cao, J. Yu and S. Wageh, Chem. Commun., 2014, 50, 10768–10777 RSC
- X. B. Li, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed., 2019, 58, 10804–10811 CrossRef CAS PubMed
- X.-B. Li, C.-H. Tung and L.-Z. Wu, Nat. Rev. Chem., 2018, 2, 160–173 CrossRef CAS
- T. Xu, C. J. Yin, M. D. Wodrich, S. Mazza, K. M. Schultz, R. Scopelliti and X. Hu, J. Am. Chem. Soc., 2016, 138, 3270–3273 CrossRef CAS PubMed
- X. B. Li, Y. J. Gao, Y. Wang, F. Zhan, X. Y. Zhang, Q. Y. Kong, N. J. Zhao, Q. Guo, H. L. Wu, Z. J. Li, Y. Tao, J. P. Zhang, B. Chen, C. H. Tung and L. Z. Wu, J. Am. Chem. Soc., 2017, 139, 4789–4796 CrossRef CAS PubMed
- Y. Wang, Y. Ma, X.-Y. Gao, Z.-K. Xin, Y.-D. Wang, L. Qiao, L. Gao, C.-H. Tung, W.-Y. Wong, X.-B. Li and L.-Z. Wu, Adv. Mater., 2025, e02085 CrossRef PubMed
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed
- R. Martinez-Haya, J. Gomis, A. Arques, M. L. Marin, A. M. Amat and M. A. Miranda, Appl. Catal., B, 2017, 203, 381–388 CrossRef CAS
- F. Wang, Q. Li and D. Xu, Adv. Energy Mater., 2017, 7, 1700529 CrossRef
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC
- W. Hu, L. Lin, R. Zhang, C. Yang and J. Yang, J. Am. Chem. Soc., 2017, 139, 15429–15436 CrossRef CAS PubMed
- X. Niu, X. Bai, Z. Zhou and J. Wang, ACS Catal., 2020, 10, 1976–1983 CrossRef CAS
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed
- M. Shi, G. Li, J. Li, X. Jin, X. Tao, B. Zeng, E. A. Pidko, R. Li and C. Li, Angew. Chem., Int. Ed., 2020, 59, 6590–6595 CrossRef CAS PubMed
- X. Wang, R. Long, D. Liu, D. Yang, C. Wang and Y. Xiong, Nano Energy, 2016, 24, 87–93 CrossRef CAS
- H. Huang, B. Dai, W. Wang, C. Lu, J. Kou, Y. Ni, L. Wang and Z. Xu, Nano Lett., 2017, 17, 3803–3808 CrossRef CAS PubMed
- J. Zhang, Y. Liu, T. Dittrich, Z. Wang, P. Ji, M. Li, N. Ta, H. Zhang, C. Zhen, Y. Xu, D. Li, Z. Feng, Z. Li, Y. Luo, J. Cui, D. Su, Y. Weng, G. Liu, X. Wang, F. Fan and C. Li, Nat. Commun., 2025, 16, 1515 CrossRef CAS PubMed
- C. Wang, A. Li, C. Li, S. Zhang, H. Li, X. Zhou, L. Hu, Y. Feng, K. Wang, Z. Zhu, R. Shao, Y. Chen, P. Gao, S. Mao, J. Huang, Z. Zhang and X. Han, Adv. Mater., 2019, 31, 1903491 CrossRef CAS PubMed
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC
- Z. Zou, J. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414, 625–627 CrossRef CAS PubMed
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed
- Y. Wang, F. Silveri, M. K. Bayazit, Q. Ruan, Y. Li, J. Xie, C. R. A. Catlow and J. Tang, Adv. Energy Mater., 2018, 8, 1801084 CrossRef
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. Gonzalez Lopez, L. Zou, A. Wadsworth, W. Zhang, L. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Nat. Mater., 2020, 19, 559–565 CrossRef CAS PubMed
- X. Zhang, K. Geng, D. Jiang and G. D. Scholes, J. Am. Chem. Soc., 2022, 144, 16423–16432 CrossRef CAS PubMed
- T. W. Kim, S. Jun, Y. Ha, R. K. Yadav, A. Kumar, C. Y. Yoo, I. Oh, H. K. Lim, J. W. Shin, R. Ryoo, H. Kim, J. Kim, J. O. Baeg and H. Ihee, Nat. Commun., 2019, 10, 1873 CrossRef PubMed
- Z. Pei, L. Ding, M. Lu, Z. Fan, S. Weng, J. Hu and P. Liu, J. Phys. Chem. C, 2014, 118, 9570–9577 CrossRef CAS
- Y. Guo, H. Li, W. Ma, W. Shi, Y. Zhu and W. Choi, Carbon Energy, 2020, 2, 308–349 CrossRef CAS
- M. J. Munoz-Batista, M. M. Ballari, A. Kubacka, O. M. Alfano and M. Fernandez-Garcia, Chem. Soc. Rev., 2019, 48, 637–682 RSC
- T. Kosaka, Y. Teduka, T. Ogura, Y. Zhou, T. Hisatomi, H. Nishiyama, K. Domen, Y. Takahashi and H. Onishi, ACS Catal., 2020, 10, 13159–13164 CrossRef CAS
- M. Rezaei, A. Nezamzadeh-Ejhieh and A. R. Massah, Mater. Today Energy, 2025, 48, 101754 CrossRef CAS
- F. Qin, Y. Kang, X. San, Y.-L. Tang, J. Li, X. Zhang, K. Zhang and G. Liu, J. Am. Chem. Soc., 2025, 147, 12897–12907 CrossRef CAS PubMed
- H. Zou, Y. Qi, S. Du, Y. Bao, X. Xin, W. Fan, Y. Xiao, S. Jin, Z. Feng and F. Zhang, J. Am. Chem. Soc., 2024, 146, 28182–28189 CrossRef CAS PubMed
- Y. C. Zhang, N. Afzal, L. Pan, X. Zhang and J. J. Zou, Adv. Sci., 2019, 6, 1900053 CrossRef PubMed
- S. Kundu and A. Patra, Chem. Rev., 2017, 117, 712–757 CrossRef CAS PubMed
- A. H. Proppe, Y. C. Li, A. Aspuru-Guzik, C. P. Berlinguette, C. J. Chang, R. Cogdell, A. G. Doyle, J. Flick, N. M. Gabor, R. van Grondelle, S. Hammes-Schiffer, S. A. Jaffer, S. O. Kelley, M. Leclerc, K. Leo, T. E. Mallouk, P. Narang, G. S. Schlau-Cohen, G. D. Scholes, A. Vojvodic, V. W.-W. Yam, J. Y. Yang and E. H. Sargent, Nat. Rev. Mater., 2020, 5, 828–846 CrossRef CAS
- G. Liu, C. Zhen, Y. Kang, L. Wang and H. M. Cheng, Chem. Soc. Rev., 2018, 47, 6410–6444 RSC
- M. A. Butler and D. S. Ginley, J. Electrochem. Soc., 1978, 125, 228–232 CrossRef CAS
- N. Xu, L. Hu, Q. Zhang, X. Xiao, H. Yang and E. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 27373–27381 CrossRef CAS PubMed
- R. F. Cava, Jr., W. F. Peck and J. J. Krajewski, Nature, 1995, 377, 215–217 CrossRef
- B. Bernardo, D. Cheyns, B. Verreet, R. D. Schaller, B. P. Rand and N. C. Giebink, Nat. Commun., 2014, 5, 3245 CrossRef CAS PubMed
- B. T. Luppi, D. Majak, M. Gupta, E. Rivard and K. Shankar, J. Mater. Chem. A, 2019, 7, 2445–2463 RSC
- B. Siegmund, M. T. Sajjad, J. Widmer, D. Ray, C. Koerner, M. Riede, K. Leo, I. D. Samuel and K. Vandewal, Adv. Mater., 2017, 29, 1604424 CrossRef PubMed
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS
- M. Z. Rahman, T. Edvinsson and J. Gascon, Nat. Rev. Chem., 2022, 6, 243–258 CrossRef PubMed
- D. Chen, R. Lu, R. Yu, Y. Dai, H. Zhao, D. Wu, P. Wang, J. Zhu, Z. Pu, L. Chen, J. Yu and S. Mu, Angew. Chem., Int. Ed., 2022, 61, e202208642 CrossRef CAS PubMed
- Y. Guo, W. Shi and Y. Zhu, EcoMat., 2019, 1, e12007 CrossRef
- Y. Gao, J. Zhu, H. An, P. Yan, B. Huang, R. Chen, F. Fan and C. Li, J. Phys. Chem. Lett., 2017, 8, 1419–1423 CrossRef CAS PubMed
- L. Zhang, J. Zhang, H. Yu and J. Yu, Adv. Mater., 2022, 34, 2107668 CrossRef CAS PubMed
- N. Omrani and A. Nezamzadeh-Ejhieh, J. Mol. Liq., 2020, 315, 113701 CrossRef CAS
- Q. Wang, G. Zhang, W. Xing, Z. Pan, D. Zheng, S. Wang, Y. Hou and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202307930 CrossRef CAS PubMed
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef PubMed
- H. Gao, P. Zhang, J. Zhao, Y. Zhang, J. Hu and G. Shao, Appl. Catal., B, 2017, 210, 297–305 CrossRef CAS
- S. Zhang, S. Liu, Y. Sun, S. Li, J. Shi and Z. Jiang, Chem. Soc. Rev., 2021, 50, 13449–13466 RSC
- X. He, G. Zhu, J. Yang, H. Chang, Q. Meng, H. Zhao, X. Zhou, S. Yue, Z. Wang, J. Shi, L. Gu, D. Yan and Y. Weng, Sci. Rep., 2015, 5, 17076 CrossRef CAS PubMed
- L. Li, D. Ma, Q. Xu and S. Huang, Chem. Eng. J., 2022, 437, 135153 CrossRef CAS
- Z. Zhao, C. Shi, Q. Shen, W. Li, D. Men, B. Xu, Y. Sun and C. Li, CrystEngComm, 2020, 22, 8221–8227 RSC
- T. Hou, N. Luo, Y.-T. Cui, J. Lu, L. Li, K. E. MacArthur, M. Heggen, R. Chen, F. Fan, W. Tian, S. Jin and F. Wang, Appl. Catal., B, 2019, 245, 262–270 CrossRef CAS
- H. Yang, K. Dai, J. Zhang and G. Dawson, Chin. J. Catal., 2022, 43, 2111–2140 CrossRef CAS
- K. Pandiselvi, H. Fang, X. Huang, J. Wang, X. Xu and T. Li, J. Hazard. Mater., 2016, 314, 67–77 CrossRef CAS PubMed
- S. Chen, J. Liao, Z. Zhou, S. Yang, Q. Gao, X. Cai, F. Peng, Y. Fang and S. Zhang, Appl. Catal., B, 2021, 291, 120139 CrossRef CAS
- C. Jiang, H. An, G. Dong, J. Feng, M. Zhang, Y. Ren and J. Ma, Chem. Eng. J., 2022, 428, 131054 CrossRef CAS
- J. Gu, Y. Yu, S. Chen, W. Shi, Y. Wang, Y. Liao, H. Chen and F. Jiang, Chem. Eng. J., 2021, 424, 130539 CrossRef CAS
- Y. Zheng, Y. Chen, B. Gao, B. Lin and X. Wang, Adv. Funct. Mater., 2020, 30, 2002021 CrossRef CAS
- M. Ou, S. Wan, Q. Zhong, S. Zhang, Y. Song, L. Guo, W. Cai and Y. Xu, Appl. Catal., B, 2018, 221, 97–107 CrossRef CAS
- M. Liras, M. Barawi and V. A. de la Pena O’Shea, Chem. Soc. Rev., 2019, 48, 5454–5487 RSC
- H.-C. Yang, J. Hou, V. Chen and Z.-K. Xu, J. Mater. Chem. A, 2016, 4, 9716–9729 RSC
- R. Otero, A. L. Vázquez de Parga and J. M. Gallego, Surf. Sci. Rep., 2017, 72, 105–145 CrossRef CAS
- Y. Su, C. Chen, Y. Wang, M. Yao, R. Ma, W. Zhang, Q. Yuan and D. Hu, J. Mater. Chem. A, 2022, 10, 14187–14220 RSC
- Z. Q. Jiang, X. L. Chen, J. Lu, Y. F. Li, T. Wen and L. Zhang, Chem. Commun., 2019, 55, 6499–6502 RSC
- Y. Gao, X. Su, J. Zhang, H. Tan, J. Sun, J. Ouyang and N. Na, Small, 2021, 17, 2103773 CrossRef CAS PubMed
- M. Opanasenko, W. O. Parker, Jr., M. Shamzhy, E. Montanari, M. Bellettato, M. Mazur, R. Millini and J. Cejka, J. Am. Chem. Soc., 2014, 136, 2511–2519 CrossRef CAS PubMed
- H. Li, Y. Sun, Z. Y. Yuan, Y. P. Zhu and T. Y. Ma, Angew. Chem., Int. Ed., 2018, 57, 3222–3227 CrossRef CAS PubMed
- J.-F. Huang, J.-M. Liu, L.-M. Xiao, Y.-H. Zhong, L. Liu, S. Qin, J. Guo and C.-Y. Su, J. Mater. Chem. A, 2019, 7, 2993–2999 RSC
- D. Zhang, W. Zheng, R. Lin, Y. Li and F. Huang, Adv. Funct. Mater., 2019, 29, 1900935 CrossRef
- Z. Wei, F. Liang, Y. Liu, W. Luo, J. Wang, W. Yao and Y. Zhu, Appl. Catal., B, 2017, 201, 600–606 CrossRef CAS
- R. Chen, C. A. Tao, Z. Zhang, X. Chen, Z. Liu and J. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 43156–43165 CrossRef CAS PubMed
- X. Bai, L. Wang and Y. Zhu, ACS Catal., 2012, 2, 2769–2778 CrossRef CAS
- X. Ke, K. Dai, G. Zhu, J. Zhang and C. Liang, Appl. Surf. Sci., 2019, 481, 669–677 CrossRef CAS
- J. Yang, H. Miao, J. Jing, Y. Zhu and W. Choi, Appl. Catal., B, 2021, 281, 119547 CrossRef CAS
- L. Liu, L. Ding, Y. Liu, W. An, S. Lin, Y. Liang and W. Cui, Appl. Catal., B, 2017, 201, 92–104 CrossRef CAS
- S. E. Zhu, F. Li and G. W. Wang, Chem. Soc. Rev., 2013, 42, 7535–7570 RSC
- J. Zhou, M. Zhang and Y. Zhu, Phys. Chem. Chem. Phys., 2015, 17, 3647–3652 RSC
- H. Lv, X. Zhao, H. Niu, S. He, Z. Tang, F. Wu and J. P. Giesy, J. Hazard. Mater., 2019, 369, 494–502 CrossRef CAS PubMed
- D. Liu, M. Zhang, W. Xie, L. Sun, Y. Chen and W. Lei, Appl. Catal., B, 2017, 207, 72–78 CrossRef CAS
- X. Wang, X. Wang, T. Shi, Y. Yao, Y. Fang, A. Meng, Z. Li, L. Wang, S. Li, G. Li and J. Huang, Chem. Eng. J., 2022, 445, 136785 CrossRef CAS
- D.-I. Won, J.-S. Lee, J.-M. Ji, W.-J. Jung, H.-J. Son, C. Pac and S. O. Kang, J. Am. Chem. Soc., 2015, 137, 13679–13690 CrossRef CAS PubMed
- R. S. Sprick, Z. Chen, A. J. Cowan, Y. Bai, C. M. Aitchison, Y. Fang, M. A. Zwijnenburg, A. I. Cooper and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 18695–18700 CrossRef CAS PubMed
- Y. Bai, C. Li, L. Liu, Y. Yamaguchi, M. Bahri, H. Yang, A. Gardner, M. A. Zwijnenburg, N. D. Browning, A. J. Cowan, A. Kudo, A. I. Cooper and R. S. Sprick, Angew. Chem., Int. Ed., 2022, 61, 202201299 CrossRef PubMed
- Y. Bai, K. Nakagawa, A. J. Cowan, C. M. Aitchison, Y. Yamaguchi, M. A. Zwijnenburg, A. Kudo, R. S. Sprick and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 16283–16290 RSC
- P. Wei, P. Zhang, Y. Zhang and X. Li, J. Colloid Interface Sci., 2022, 609, 785–793 CrossRef CAS PubMed
- X. Zhu, Z. Song, Z. Wang, W. Liu, B. Hong, J. Bao, C. Gao and S. Sun, Appl. Catal., B, 2020, 274, 119010 CrossRef CAS
- C. Wang, L. Wang, J. Jin, J. Liu, Y. Li, M. Wu, L. Chen, B. Wang, X. Yang and B.-L. Su, Appl. Catal., B, 2016, 188, 351–359 CrossRef CAS
- W. Jiang, D. Qu, L. An, X. Gao, Y. Wen, X. Wang and Z. Sun, J. Mater. Chem. A, 2019, 7, 18348–18356 RSC
- X. She, J. Wu, H. Xu, J. Zhong, Y. Wang, Y. Song, K. Nie, Y. Liu, Y. Yang, M. T. F. Rodrigues, R. Vajtai, J. Lou, D. Du, H. Li and P. M. Ajayan, Adv. Energy Mater., 2017, 7, 1700025 CrossRef
- Z. Zhou, P. Peng and Z. Xiang, Sci. Bull., 2018, 63, 369–375 CrossRef CAS PubMed
- J. Yan, H. Wu, H. Chen, Y. Zhang, F. Zhang and S. F. Liu, Appl. Catal., B, 2016, 191, 130–137 CrossRef CAS
- F. Guo, W. Shi, C. Zhu, H. Li and Z. Kang, Appl. Catal., B, 2018, 226, 412–420 CrossRef CAS
- N. Wang and X. Li, Inorg. Chem. Commun., 2018, 92, 14–17 CrossRef CAS
- D. Dai, P. Wang, X. Bao, Y. Xu, Z. Wang, Y. Guo, Z. Wang, Z. Zheng, Y. Liu, H. Cheng and B. Huang, Chem. Eng. J., 2022, 433, 134476 CrossRef CAS
- S. Sun, R. Gao, X. Liu, L. Pan, C. Shi, Z. Jiang, X. Zhang and J. J. Zou, Sci. Bull., 2022, 67, 389–397 CrossRef CAS PubMed
- Y. Yang, M. Qiu, L. Li, Y. Pi, G. Yan and L. Yang, Sol. RRL, 2018, 2, 1800148 CrossRef
- H. He, J. Cao, M. Guo, H. Lin, J. Zhang, Y. Chen and S. Chen, Appl. Catal., B, 2019, 249, 246–256 CrossRef CAS
- W. Shi, M. Li, X. Huang, H. Ren, C. Yan and F. Guo, Chem. Eng. J., 2020, 382, 122960 CrossRef CAS
- X. Zhou, Y. Fang, X. Cai, S. Zhang, S. Yang, H. Wang, X. Zhong and Y. Fang, ACS Appl. Mater. Interfaces, 2020, 12, 20579–20588 CrossRef CAS PubMed
- Z. Pan, G. Zhang and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 7102–7106 CrossRef CAS PubMed
- Y. Lin, X. Wang, X. Fu and W. Su, J. Mater. Chem. A, 2022, 10, 8252–8257 RSC
- X. Yan, M. Xia, H. Liu, B. Zhang, C. Chang, L. Wang and G. Yang, Nat. Commun., 2023, 14, 1741 CrossRef CAS PubMed
- G. Zuo, W. Chen, Z. Yin, S. Ma, Y. Wang, Q. Ji, Q. Xian, S. Yang and H. He, Chem. Eng. J., 2023, 456, 141096 CrossRef CAS
- C. Duanmu, T. Wang, X. Y. Meng, J. J. Li, Y. N. Zhou and Y. X. Pan, Angew. Chem., Int. Ed., 2024, 64, 202412796 CrossRef PubMed
- H. C. Rojas, S. Bellani, F. Fumagalli, G. Tullii, S. Leonardi, M. T. Mayer, M. Schreier, M. Grätzel, G. Lanzani, F. Di Fonzo and M. R. Antognazza, Energy Environ. Sci., 2016, 9, 3710–3723 RSC
- Y. Zhang, H. Lv, Z. Zhang, L. Wang, X. Wu and H. Xu, Adv. Mater., 2021, 33, 2008264 CrossRef CAS PubMed
- L. Wang, W. Lian, B. Liu, H. Lv, Y. Zhang, X. Wu, T. Wang, J. Gong, T. Chen and H. Xu, Adv. Mater., 2022, 34, 2200723 CrossRef CAS PubMed
- X. Chen, J. Wang, Y. Chai, Z. Zhang and Y. Zhu, Adv. Mater., 2021, 33, 2007479 CrossRef CAS PubMed
- H. Zhang, R. Zong and Y. Zhu, J. Phys. Chem. C, 2009, 113, 4605–4611 CrossRef CAS
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O’Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed
- X. Liu, D. Huang, C. Lai, G. Zeng, L. Qin, H. Wang, H. Yi, B. Li, S. Liu, M. Zhang, R. Deng, Y. Fu, L. Li, W. Xue and S. Chen, Chem. Soc. Rev., 2019, 48, 5266–5302 RSC
- P. Pachfule, A. Acharjya, J. Roeser, T. Langenhahn, M. Schwarze, R. Schomäcker, A. Thomas and J. Schmidt, J. Am. Chem. Soc., 2018, 140, 1423–1427 CrossRef CAS PubMed
- C. Kang, Z. Zhang, S. Kusaka, K. Negita, A. K. Usadi, D. C. Calabro, L. S. Baugh, Y. Wang, X. Zou, Z. Huang, R. Matsuda and D. Zhao, Nat. Mater., 2023, 22, 636–643 CrossRef CAS PubMed
- M. Lu, J. Liu, Q. Li, M. Zhang, M. Liu, J. L. Wang, D. Q. Yuan and Y. Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 12392–12397 CrossRef CAS PubMed
- K. Sun, Y. Qian, D. Li and H. L. Jiang, Adv. Mater., 2024, 2411118 CrossRef PubMed
- J. Cheng, Y. Wu, W. Zhang, J. Zhang, L. Wang, M. Zhou, F. Fan, X. Wu and H. Xu, Adv. Mater., 2023, 36, 2305313 CrossRef PubMed
- B. B. Luan, X. Chu, Y. Wang, X. Qiao, Y. Jiang and F. M. Zhang, Adv. Mater., 2024, 36, 2412653 CrossRef CAS PubMed
- Y. Yang, X. Chu, H. Y. Zhang, R. Zhang, Y. H. Liu, F. M. Zhang, M. Lu, Z. D. Yang and Y. Q. Lan, Nat. Commun., 2023, 14, 593 CrossRef CAS PubMed
- N. Contreras-Pereda, S. Pané, J. Puigmartí-Luis and D. Ruiz-Molina, Coord. Chem. Rev., 2022, 460, 214459 CrossRef CAS
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC
- R. Sahoo, S. Mondal, S. C. Pal, D. Mukherjee and M. C. Das, Adv. Energy Mater., 2021, 11, 2102300 CrossRef CAS
- G. A. Leith, C. R. Martin, J. M. Mayers, P. Kittikhunnatham, R. W. Larsen and N. B. Shustova, Chem. Soc. Rev., 2021, 50, 4382–4410 RSC
- G. Meng, L. Zhen, S. Sun, J. Hai, Z. Zhang, D. Sun, Q. Liu and B. Wang, J. Mater. Chem. A, 2021, 9, 24365–24373 RSC
- A. López-Magano, A. E. Platero-Prats, S. Cabrera, R. Mas-Ballesté and J. Alemán, Appl. Catal., B, 2020, 272, 119027 CrossRef
- Z. Fu, X. Wang, A. M. Gardner, X. Wang, S. Y. Chong, G. Neri, A. J. Cowan, L. Liu, X. Li, A. Vogel, R. Clowes, M. Bilton, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 543–550 RSC
- X. Yu, Z. Chen and X. Li, Chem, 2022, 8, 2894–2897 Search PubMed
- M. Chen, H. Li, C. Liu, J. Liu, Y. Feng, A. G. H. Wee and B. Zhang, Coord. Chem. Rev., 2021, 435, 213778 CrossRef CAS
- M. Lu, S. B. Zhang, M. Y. Yang, Y. F. Liu, J. P. Liao, P. Huang, M. Zhang, S. L. Li, Z. M. Su and Y. Q. Lan, Angew. Chem., Int. Ed., 2023, 62, 202307632 CrossRef PubMed
- H. Wang, C. Qian, J. Liu, Y. Zeng, D. Wang, W. Zhou, L. Gu, H. Wu, G. Liu and Y. Zhao, J. Am. Chem. Soc., 2020, 142, 4862–4871 CrossRef CAS PubMed
- J. Wang, L. Xu, T. Wang, R. Li, Y. Zhang, J. Zhang and T. Peng, Adv. Energy Mater., 2021, 11, 2003575 CrossRef CAS
- D. Wang, H. Zeng, X. Xiong, M.-F. Wu, M. Xia, M. Xie, J.-P. Zou and S.-L. Luo, Sci. Bull., 2020, 65, 113–122 CrossRef CAS PubMed
- D. Shang, D. Li, B. Chen, B. Luo, Y. Huang and W. Shi, ACS Sustainable Chem. Eng., 2021, 9, 14238–14248 CrossRef CAS
- X. B. Li, Z. K. Xin, S. G. Xia, X. Y. Gao, C. H. Tung and L. Z. Wu, Chem. Soc. Rev., 2020, 49, 9028–9056 RSC
- Y.-J. Gao, X.-B. Li, H.-L. Wu, S.-L. Meng, X.-B. Fan, M.-Y. Huang, Q. Guo, C.-H. Tung and L.-Z. Wu, Adv. Funct. Mater., 2018, 28, 1801769 CrossRef
- Z. K. Xin, Y. J. Gao, Y. Gao, H. W. Song, J. Zhao, F. Fan, A. D. Xia, X. B. Li, C. H. Tung and L. Z. Wu, Adv. Mater., 2021, 34, e2106662 CrossRef PubMed
- H. Zhong, R. Sa, H. Lv, S. Yang, D. Yuan, X. Wang and R. Wang, Adv. Funct. Mater., 2020, 30, 2002654 CrossRef CAS
- S. Chen, T. Sun, M. Zheng and Z. Xie, Adv. Funct. Mater., 2020, 30, 2004680 CrossRef CAS
- M. L. Xu, M. Lu, G. Y. Qin, X. M. Wu, T. Yu, L. N. Zhang, K. Li, X. Cheng and Y. Q. Lan, Angew. Chem., Int. Ed., 2022, 61, 202210700 CrossRef PubMed
- H. Ding, R. Shen, K. Huang, C. Huang, G. Liang, P. Zhang and X. Li, Adv. Funct. Mater., 2024, 34, 2400065 CrossRef CAS
- Y. H. Liu, X. Chu, Y. Jiang, W. Han, Y. Wang, L. H. Shao, G. Zhang and F. M. Zhang, Adv. Funct. Mater., 2024, 34, 2316546 CrossRef CAS
- R. Shen, G. Liang, L. Hao, P. Zhang and X. Li, Adv. Mater., 2023, 35, 2303649 CrossRef CAS PubMed
- K. Sun, Y. Huang, F. Sun, Q. Wang, Y. Zhou, J. Wang, Q. Zhang, X. Zheng, F. Fan, Y. Luo, J. Jiang and H. L. Jiang, Nat. Chem., 2024, 16, 1638–1646 CrossRef CAS PubMed
- X. Chu, S. Liu, B. B. Luan, Y. Zhang, Y. Xi, L. H. Shao, F. M. Zhang and Y. Q. Lan, Angew. Chem., Int. Ed., 2025, 202422940 Search PubMed
- Z.-Z. Liang, X.-A. Li, Q.-Z. Chen, X.-L. Wang, P.-Y. Su, J.-F. Huang, Y. Zhou, L.-M. Xiao and J.-M. Liu, ACS Catal., 2024, 14, 10447–10461 CrossRef CAS
- M. J. Ginsberg, D. V. Esposito and V. M. Fthenakis, Cell Rep. Phys. Sci., 2023, 4, 101625 CrossRef CAS
- Global Hydrogen Review 2024, International Energy Agency, 2024.
- T. Hisatomi, Q. Wang, F. Zhang, S. Ardo, E. Reisner, H. Nishiyama, A. Kudo, T. Yamada and K. Domen, Front. Sci., 2024, 2, 101625 Search PubMed
- J. Jia, L. C. Seitz, J. D. Benck, Y. Huo, Y. Chen, J. W. D. Ng, T. Bilir, J. S. Harris and T. F. Jaramillo, Nat. Commun., 2016, 7, 13237 CrossRef CAS PubMed
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 RSC
- M. Daboczi, F. Eisner, J. Luke, S. W. Yuan, N. A. Lawati, M. Zhi, M. Yang, J. S. Muller, K. Stewart, J. S. Kim, J. Nelson and S. Eslava, Nat. Energy, 2025, 10, 581–591 CrossRef CAS PubMed
- Z. Song, C. Li, L. Chen, K. Dolia, S. Fu, N. Sun, Y. Li, K. Wyatt, J. L. Young, T. G. Deutsch and Y. Yan, ACS Energy Lett., 2023, 8, 2611–2619 CrossRef CAS
- C. Liu, J. Tang, H. M. Chen, B. Liu and P. Yang, Nano Lett., 2013, 13, 2989–2992 CrossRef CAS PubMed
- B. Liu, X. Wang, Y. Zhang, M. Zhu, C. Zhang, S. Li, Y. Ma, W. Huang and S. Wang, Nat. Commun., 2025, 16, 2792 CrossRef CAS PubMed
- Z. Wang, Y. Inoue, T. Hisatomi, R. Ishikawa, Q. Wang, T. Takata, S. Chen, N. Shibata, Y. Ikuhara and K. Domen, Nat. Catal., 2018, 1, 756–763 CrossRef CAS
- S. Ye, W. Shi, Y. Liu, D. Li, H. Yin, H. Chi, Y. Luo, N. Ta, F. Fan, X. Wang and C. Li, J. Am. Chem. Soc., 2021, 143, 12499–12508 CrossRef CAS PubMed
- K. Iwashina, A. Iwase, Y. H. Ng, R. Amal and A. Kudo, J. Am. Chem. Soc., 2015, 137, 604–607 CrossRef CAS PubMed
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhou, K. Sun and Z. Mi, Nature, 2023, 613, 66–70 CrossRef CAS PubMed
- F. A. Chowdhury, M. L. Trudeau, H. Guo and Z. Mi, Nat. Commun., 2018, 9, 1707 CrossRef PubMed
- M. Arunachalam, R. S. Kanase, K. Zhu and S. H. Kang, Nat. Commun., 2023, 14, 5429 CrossRef CAS PubMed
- Y. Qi, J. Zhang, Y. Kong, Y. Zhao, S. Chen, D. Li, W. Liu, Y. Chen, T. Xie, J. Cui, C. Li, K. Domen and F. Zhang, Nat. Commun., 2022, 13, 484 CrossRef CAS PubMed
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS PubMed
- Z. Wang, Y. Luo, T. Hisatomi, J. J. M. Vequizo, S. Suzuki, S. Chen, M. Nakabayashi, L. Lin, Z. Pan, N. Kariya, A. Yamakata, N. Shibata, T. Takata, K. Teshima and K. Domen, Nat. Commun., 2021, 12, 1005 CrossRef CAS PubMed
- X. Xu, L. Meng, J. Zhang, S. Yang, C. Sun, H. Li, J. Li and Y. Zhu, Angew. Chem., Int. Ed., 2024, 63, 202308597 CrossRef PubMed
- B. B. Luan, X. Chu, Y. Wang, X. Qiao, Y. Jiang and F. M. Zhang, Adv. Mater., 2024, 36, 2412653 CrossRef CAS PubMed
- D. Zhao, Y. Wang, C.-L. Dong, Y.-C. Huang, J. Chen, F. Xue, S. Shen and L. Guo, Nat. Energy, 2021, 6, 388–397 CrossRef CAS
- L. Wang, X. Zheng, L. Chen, Y. Xiong and H. Xu, Angew. Chem., Int. Ed., 2018, 57, 3454–3458 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |