
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Top-down engineering of zeolite porosity†
Wieslaw J.
Roth
 *,
Barbara
Gil
,
Karolina A.
Tarach
and
Kinga
Góra-Marek
*
*,
Barbara
Gil
,
Karolina A.
Tarach
and
Kinga
Góra-Marek
*
Jagiellonian University, Faculty of Chemistry, Gronostajowa 2, Kraków, Poland. E-mail: wieslaw.roth@uj.edu.pl; kinga.gora-marek@uj.edu.pl
First published on 8th July 2025
Abstract
Synthetic zeolites present a near-ideal environment for catalytic conversion and separation of molecules. They have framework structures with discrete uniform micropores allowing selective processing and sorption based on the size and shape of molecules. Zeolites have been deployed in numerous industrial applications motivating continuous innovation efforts to synthesise new structures and pore systems. The conventional zeolite syntheses are carried out directly as hydrothermal bottom-up assemblies from molecular or amorphous precursors, producing robust hard-to-modify structures/crystals. To diversify zeolite structures, especially to alleviate diffusional limitations and active site accessibility, top-down modification approaches have been initiated. This article presents an overview of the various top-down methods for modifying already synthesised zeolite crystals or precursors to engineer additional porosity and functionality. They include: demetallation focused on Si and Al but also Ge and Ti, formation of micro/mesoporous hybrids by recrystallisation, mechanochemical methods, pore engineering with low-dimensional zeolite forms, especially 2D and nanozeolites, and treatments by microwaves, ultrasounds, plasma and lasers. The discussion presents illustrative examples of relevant properties, such as textural, acidic and catalytic, of materials obtained by the applied treatments. The methods and descriptors used to characterise changes in porosity are described in detail. An extended compilation of reported materials with textural properties is provided.
 Wieslaw J. Roth | Wieslaw J. Roth is a professor at the Faculty of Chemistry of the Jagiellonian University in Krakow, Poland. He joined the JU Faculty in 2012 after working as a researcher at Mobil R&D and ExxonMobil R&E in New Jersey, USA, from 1988 till 2009, specializing in zeolite synthesis and commercialisation. He received the D. W. Breck Award of the International Zeolite Association in 1994 for the (co)discovery of ordered mesoporous materials, Thomas Alva Edison Award in 2008 for the patent and commercialisation of mesoporous materials, and the A. F. Cronsted Award of the Federation of European Zeolite Associations in 2020, jointly with J. Cejka, for the contributions to the development of layered zeolites. |
 Barbara Gil | Prof. Barbara Gil is a full Professor and the Head of Zeolite Chemistry Group in the Faculty of Chemistry of the Jagiellonian University. She has specialized in the synthesis and characterisation of micro- and mesoporous materials. Her main areas of interest are classical 3D zeolites and the new class of lamellar 2D zeolites, MOFs and ordered silica mesoporous materials. Her research combines IR spectroscopy with catalysis and drug delivery. She is an expert in quantitative characterisation of acidic and redox centres in general and in environmentally important reactions. She is a co-author of over 150 scientific papers. |
 Karolina A. Tarach | Karolina A. Tarach is an associate professor in the Zeolite Chemistry Group within the Faculty of Chemistry at Jagiellonian University in Kraków, Poland. She earned her PhD from Jagiellonian University in 2014. In 2019, she completed a one-year postdoctoral fellowship at the Instituto de Tecnología Química in Valencia, Spain. Her primary research areas involve advanced in situ and operando FT-IR and UV-vis spectroscopic investigations of the catalytic properties of zeolites, utilizing two-dimensional correlation spectroscopy (2D COS) and multivariate curve resolution-alternating least squares (MCR-ALS) analyses. She is currently focusing on applying modulation excitation-phase sensitive detection (ME-PSD) for in situ and operando characterisation of heterogeneous catalysts. |
 Kinga Góra-Marek | Kinga Góra-Marek has been a Professor at the Faculty of Chemistry, Jagiellonian University (JU) since 2020. She holds an MSc in chemistry (2000) and a PhD in chemistry from JU (2005). In 2005–2006, she completed a one-year postdoctoral fellowship at the Instituto de Tecnología Química in Valencia, Spain. She is the Head of the Department of Inorganic Chemistry at the Faculty of Chemistry, Jagiellonian University. Her research focuses on characterising porous catalysts and investigating reaction mechanisms in heterogeneous catalysts through advanced spectroscopic studies, including rapid scan IR, in situ & operando FT-IR and UV-vis techniques, supported by 2D correlation (2D COS) and Multivariate Curve Resolution (MCR) analysis. She has co-authored over 150 scientific publications. |
Introduction
Porous solids have special practical significance because they provide internal void spaces, pores, and extended surface areas that can selectively adsorb guest molecules from the outside enabling separation and facilitation of catalytic conversions.1–3 Initially available porous solids such as amorphous (alumino)silicas and carbons, clays, etc., were characterised largely by polydisperse and/or flexible, non-discrete pore sizes and shapes.4 A notable transforming change occurred in the 1950s due to aluminosilicate zeolites, which presented pores and channels with uniform, fixed, well-defined sizes and shapes combined with high catalytic activity.5,6 This commenced the practical development of molecular sieves as materials with fixed structures and well-defined porosity. The above mentioned polydisperse/flexible porous materials continue to be widely used and have many valuable applications. Zeolites are microporous crystalline solids with framework structures that contain discrete pores and channels optionally incorporating isolated active centres due to heteroatoms such as Al, Ti, Fe and other metals. This enables selective separations and catalytic transformations based on the size, shape and chemical properties of the guest molecules. Zeolites have additional valuable qualities like potential high acid activity, stability, chemical resistance, easy manufacture and others. As a result, numerous zeolite-based processes have been implemented on an industrial scale as superior to other alternatives. There are continuous efforts to further expand the applications and uses of zeolites, especially because of significant environmental benefits. Zeolite frameworks are composed of oxygen-sharing tetrahedra, denoted TO4, with a central atom in the middle, originally Si and Al, but also can include other elements, e.g. as aluminophosphates and related compositions. Formally, these structures should be 4-connected, i.e. each TO4 should have 4 TO4 neighbours.7 The practical functional definition emphasises fixed internal porosity enabling sorption of guest molecules. Consequently, some materials with tetrahedral frameworks lacking porosity are not considered to be zeolites.8 On the other hand, materials with zeolite-like porosity but incomplete 4-connectivity have been included among zeolites.9The discrete nature and rigidity of zeolite frameworks demand tailored pore systems/structures for given (types of) molecules. This motivated continuous research and development to discover new zeolites to diversify available pore sizes, shapes and other properties, and ultimately, to control preparation by design of desired pore systems. The goal has been to enable the tailoring of pores and activity for processing of particular compounds and refinery fractions or to maximise yields of desired products. There are two basic approaches to the preparation of different pores systems: bottom-up synthesis from basic reagents according to the common procedures of obtaining zeolites or by top-down modification (engineering) of already synthesised available materials. The obstacle to the latter approach is the intrinsic stability of zeolite frameworks. They are rigid and immutable due to covalent bonding in 3D, which makes them not amenable to massive or regular structure adjustment or engineering, except through decomposition. This has favoured the direct synthesis of different zeolites, i.e. the bottom-up approach. It has been very successful in providing various new zeolite structures and forms. The basic limitation of the bottom-up syntheses is that new frameworks cannot be prepared at will or designed but rather rely on trial-and-error approaches, namely screening of the composition space and finding what comes out. A particularly successful and dominant approach involved syntheses with the addition of organic compounds, especially nitrogen-containing ones, which acted as and were named templates or organic structure directing agents (OSDAs). This allowed the synthesis of numerous novel frameworks with diverse pore sizes and channel systems. There is no well-established correlation between the nature of an OSDA and the zeolite structure it produces, although recently some progress in individual cases has been reported. The trial-and-error character of the bottom-up syntheses of new pore systems motivated an interest in the exploration of top-down options. Most, possibly all currently practised zeolite-based technologies employ directly synthesised zeolites, i.e. obtained by bottom-up preparations, proving the successes of this approach and the excellence of the frameworks. Nevertheless, post-synthesis tailoring of zeolite porosity has been gaining interest and can be justified by the following specific factors:
1. impossibility of targeting the synthesis of particular structures with pre-selected pore systems,
2. diminishing returns from discoveries – few if any new zeolite frameworks discovered after 1990 have been advanced to commercial readiness,
3. traditional frameworks, mostly with pores below 2 nm, were believed to limit access of larger molecules, were not open enough, and restricted diffusion of reactants and products.
One of the leading top-down approaches to modifying pore systems of existing zeolites has been based on selective degradation by desilication or dealumination. Hence, one of the main topics in this review focuses on the impact of the ring size opening of 8-, 10-, and 12-membered rings of dealuminated and desilicated zeolites, examining its effect on the characteristics of the resultant materials. In another development, the bottom-up synthesis efforts to obtain new zeolites produced an unexpected fundamental breakthrough – the discovery of layered zeolite precursors, consisting of nanosheets that could condense topotactically to produce the complete framework. Unlike the rigid 3D frameworks their corresponding 2D congeners could be modified, e.g. expanded without degradation. This enabled a different type of top-down engineering based on well-known methods of modifying and characterisation of 2D solids. There is now a documented case of a 3D zeolite formation from a 1D (chain) material, vide supra (Section 5.8). This allows referring to these materials as lower-dimensional zeolites, although for now, it is almost exclusively the domain of 2D forms. Various other methods have been reported as alternatives for zeolite pore adjustment.
The review is focused on the methods of creating and characterisation of new zeolite-based structures. It is to be understood as synonymous with pore engineering, specifically as top-down from pre-existing materials.
1 Modification of zeolite porosity by dealumination and desilication
The subject of large-pore and hierarchical zeolite preparation by bottom-up approaches has been presented in various reviews, mini-reviews, and perspectives covering particular synthetic methods.10–22 Post-synthesis treatments, including dealumination and desilication, referred to as demetallation, are top-down techniques wherein the starting substrates are already formed crystalline zeolites, which are then modified with the maintenance of the original form/shape or are rebuilt into new structures.13,23 The challenges in improving catalyst effectiveness by these demetallation strategies depend on the particular structure and chemical composition: ring sizes, Si/Al ratio, structural defects, and the distribution of aluminium atoms within the zeolite structure.Dealumination and desilication can affect the acid properties of zeolites, both the acid site concentration and their strength. The two types of acidic centres in zeolites are Brønsted acid sites (BAS) and Lewis acid sites (LAS). The former are typically identified as bridging hydroxyl groups, represented as Si(OH)Al. The BAS strength reflects its proton dissociation energy. The Lewis acid sites (LAS) may consist of charge-compensating extraframework metal cations, coordinatively unsaturated framework sites, or extraframework species (charged or neutral). The LAS strength refers to its tendency to remain in the form of an adduct with electron donors.
1.1 Dealumination and mechanistic insight
The Si/Al atomic ratio of the zeolite framework plays a critical role in determining ion exchange capacity, thermal and hydrothermal stability, hydrophobicity, and the concentration and strength of Brønsted acid sites, all of which influence catalytic behaviour. While zeolites with low Si/Al ratios are advantageous for ion exchange and adsorption processes, those with higher Si/Al ratios – lower aluminium content – are often preferred for catalytic applications. Brønsted acid sites, which form around aluminium atoms in zeolites, provide protonic catalytic activity; however, an excess of these sites can lead to undesirable side reactions that result in catalytic deactivation. Additionally, zeolites with low Si/Al ratios are more prone to degradation. The need for zeolites with an optimised Si/Al ratio has driven research into dealumination processes. ‘Dealumination’ refers to the removal of aluminium from zeolite frameworks via chemical processes that cause partial hydrolysis of the Al–O and Si–O bonds, leading to the formation of framework defects. Over time, post-synthesis dealumination has expanded to include various inorganic and organic acids. Mineral acids are more effective at removing aluminium atoms from the framework than organic acids, but excessive use may compromise structural integrity. Thermal dehydroxylation – in the case of hydrogen forms of zeolites – is also a process that increases the Si/Al ratio of zeolite structures by removing aluminium from the zeolite framework, resulting in the formation of defects. Steam treatment is employed to create mesopores by inducing partial hydrolysis of the framework, resulting in the breaking of Al–O and Si–O bonds. Ultrastabilisation of zeolites involves intense treatments, such as steaming at temperatures above 600 °C, followed by acid leaching to remove the extra-framework species that partially block the micro- and mesopores. In the latter two processes, in addition to the extraction of aluminium from the zeolite framework, there is an additional step. Both silicon and oxygen atoms fill in vacancies under high temperature and pressure. A T-jump mechanism is proposed for the healing of defects, involving the migration of framework vacancies to the crystal surface as a result of neighbouring Si atoms refilling the latter. The aforementioned dealumination methods, whether used individually or in combination, enable precise adjustments of the final zeolite properties like acidity and hydrothermal stability and improve the diffusivity of reagents.As early as 1958, it was reported24 that the structure of the faujasite-type zeolites collapsed completely when treated with strong mineral acids. Rare-earth exchanged zeolite Y (FAU) was identified as ‘superactive, superselective cracking catalyst’ marking the breakthrough of zeolites in catalysis.25 In 1968, McDaniel and Maher26 described a process of increasing the thermal resistance of Y zeolites. This ‘ultrastabilisation’ methodology quickly garnered substantial attention due to the technical significance of zeolite Y as a catalyst. The fundamental features of the stabilisation mechanism were postulated by Kerr.27,28 He suggested the stabilisation mechanism comprising hydrolytic cleavage of –O–Al–O– bonds by ‘self-steaming’, i.e. by the contact with gaseous water filling the pores and subsequent extraction of hydroxyaluminium species from the framework, then filling of cationic positions by positively charged aluminium species. Similarly, ultrastabilised materials were produced through direct steaming,29,30i.e. the thermal treatment of protonic zeolites with externally provided water steam under moderate partial pressures (up to 1 bar). Furthermore, it has been demonstrated that ultrastabilisation is linked to incorporating silica, derived from other crystal regions, into the framework vacancies created by dealumination. This process is a crucial step in the stabilisation procedure. It was first documented by IR spectroscopy31 and later supported by the X-ray study of Y zeolite dealuminated with H4EDTA.32 When Al–O–Si bonds are broken, aluminium is removed, which leads to the generation of defects in the zeolite structure. The migration of less stable extraframework silicon species to previously occupied aluminium sites leads to the development of silanol-rich domains. The outlined process can partially heal the defected structure, and as a result, numerous mesopores are generated. The evidence for releasing the framework aluminium and refilling the framework vacancies by the framework silicon atoms in the hydrothermally treated Y zeolite was also derived from the very early 27Al and 29Si MAS NMR studies.33–35
Theoretical investigations by Swang et al.36,37 compared two possible reaction routes for the interaction of zeolites with water steam. In theory, steaming can lead to the elimination of both Al and Si atoms; however, dealumination occurs just by interaction with water vapour, while a base addition must aid desilication. The authors concluded that dealumination has lower activation energy (by 40–50 kJ mol−1), making it more favourable than desilication. They considered the stepwise addition of four water molecules. The first hydration step (addition of the first water molecule) resulted in the formation of a vicinal silanol defect. Subsequently, one of the OH groups reoriented and moved to the opposite side of the framework Al atom. Next, adding the second water resulted in two OH groups bound to the Si and two to the Al. Sorption of the third H2O gave partially bonded Al(OH)3, and the fourth H2O molecule led to detachment of the final Al(OH)3(H2O) EFAl (extraframework aluminium) species. This mechanism accounts for the migration of the extraframework species because the Al atom extracted from the framework is reinserted into the neighbouring silanol nest. In these studies, the first Al–O bond breaking was predicted to have a high activation energy, which was inconsistent with the experimental observation of the dealumination of various zeolites at moderate temperatures.38 Silaghi and co-workers proposed a more favourable reaction pathway involving non-dissociative water adsorption on Al in the anti-position to the Si(OH)Al group for CHA, FAU, MOR, and MFI zeolites.39 They identified a universal mechanism based on water adsorption on the aluminium atom in the anti-position to the Brønsted acid site, resulting in a pentahedral or distorted tetrahedral aluminium atom coordination. Then, Al–O hydrolysis occurs via a water molecule dissociation. This allows successive Al–O bond hydrolysis until the framework Al is dislodged to a non-framework position and forms LAS, Al(OH)3H2O. The authors also quantified the confinement effect responsible for stabilisation of the EFAl species inside the zeolite cavities. The confinement effect was identified as a thermodynamic driving force for the aluminium extraction. The free energy barrier of Al–O hydrolysis, calculated based on this mechanism, was much lower than previously reported by Swang36,37 and discussed above. This barrier was effectively reduced by the introduction of more water molecules and accounted for collective effects among them, as discovered by Nielsen et al.40,41 This cooperative role was noted in the hydrolysis of the first Al–O bond in ZSM-5 with two water molecules present.42
The hydrolysis mechanism in the cation-exchanged zeolites is less understood, especially in the case of I and II group metals. The presence of La3+ in zeolite Y increases the activation energies of the dealumination pathways, indicating a stabilising impact of rare earth ions.43 Sun et al.,44 by applying periodic density functional theory (DFT) calculations, have demonstrated that the cationic forms (Na+, K+, and Ca2+) of LTA zeolites are more kinetically stable than the protonic ones, similarly as was found for La–Y,43 demonstrating higher reaction barriers for dealumination. The DFT analysis identified the mechanism for H2O dissociation, including proton transfers and rotations within protonic and cationic zeolites, further disrupting the Al–O bond. For cationic-LTA, the second transfer of the proton was observed, and the Al–O bonds on the side of the hexagonal prism did not break concurrently with the dissociation of H2O, which resulted in considerably higher energy barriers. From the two factors examined, the decreased Si/Al ratio promoted hydrolysis, whereas the type of the extraframework cation had only a marginal influence. The correlation between the Al–O bond length in the initial water-adsorbed states and the reaction activation energies has been identified.
1.2 Dealumination and its implications
Dealumination occurs by partial or complete hydrolysis of aluminium in the framework, resulting in the cleavage of Al–O bonds. The affected Al atoms can condense to form extraframework Al (EFAl) and Si–OH as defect sites referred to as ‘hydroxyl nests’.45,46 The rate-limiting step is hydrolysis rather than condensation, as initial dealumination rates have been documented to be second-order reactions with respect to the water vapour pressure.38,46 Numerous reports indicate that dealumination may not be only zeolite-specific but also site-specific.47–49 Spectroscopic NMR and FT-IR analysis of steamed H-mordenite (H-MOR) demonstrated the preferential dealumination of the T3 and T4 sites in 4-MR (4-membered rings).47,48 For the *BEA zeolite, distinct sequential stages in the framework dealumination process were identified. While the aluminium atoms in positions T1 and T2 exhibit resistance to dealumination and do not adopt the octahedral oxygen coordination, the T3 to T9 positions transform into at least two distinct types of octahedral aluminium atoms integrated into the framework (the T-sites for MOR and *BEA framework are visualised in Fig. 1). | ||
| Fig. 1 Labelling T positions in the MOR and *BEA frameworks. | ||
A combination of factors influences the behaviour of zeolites towards dealumination, with the Si/Al ratio being one of the most important ones (Fig. 2). When the structure, size of the crystals, and Si/Al ratio in the framework are the same, the behaviour towards dealumination depends on the number of defect sites in the framework, which is governed by the synthesis conditions and subsequent calcination to burn off the OSDA. Based on 1H MAS NMR, Müller et al.47 defined two kinds of protons – Brønsted protons interacting with the framework with a characteristic wide peak at 5.1–5.9 ppm, and ‘free’ protons (3.8 and 4.2 ppm). The defect sites (interrupted framework) resulting from synthesis conditions or template removal account for the increase in the flexibility of the zeolite framework, facilitating interaction with Brønsted protons with the framework oxygen atoms.
 | ||
| Fig. 2 Schematic representation of the co-dependence between applied treatments (detemplation, dealumination, desilication with NaOH, and with NaOH & Pore Directing Agent, PDA) and zeolite properties (Si/Al ratio), emphasising the type of generated porosity. The direction of the arrows represents changes in Si/Al. The spheres show occluded mesoporosity, while cylindrical pores visualise open mesoporosity. | ||
The Si/Al ratio and the number of defect sites formed by dealumination in the framework of zeolites (beta (*BEA), ferrierite (FER), mordenite (MOR), and ZSM-5 (MFI)) were investigated employing 1H, 29Si, and 27Al MAS NMR spectroscopy.47 Dealumination was performed through thermal treatment, complexation by oxalic acid, and direct aluminium replacement with silicon using gaseous silicon tetrachloride.
The number of Al-atoms in 4-MR was found to influence the stability towards dealumination because the tension in the smaller rings is larger. The more aluminium atoms there are in an environment with tension, the easier it is to dealuminate the zeolite sample. Therefore, the degree of dealumination diminished in the following sequence based on the number of Al-sites in 4-MR in the zeolites: beta (*BEA) > mordenite (MOR) > ZSM-5 (MFI) > ferrierite (FER). Additionally, the synthesis protocol led to important differences in the dealumination process even among the zeolites of the identical framework types. 1H MAS NMR results displayed that the enhanced dealumination was found for the zeolites with higher number of Brønsted acid sites, whereas 29Si MAS NMR revealed a concurrent increase in the number of defect sites within the zeolites. Depending on the zeolite synthesis conditions, the frameworks are disrupted to varying degrees, leading to increased flexibility and facilitating interaction between Brønsted protons and the oxygen atoms within the framework. The arrangement and size of the pores affected the accessibility of aluminium atoms within the framework, as well as their extraction and diffusion from the crystals, which is consistent with the findings in other studies. As a result, ferrierite containing both 10- and 8-MR channels demonstrated the highest stability against dealumination. The small size of 8-MR channels limits the transfer of the extracted species, and the fact that all Al-sites are bonded to 5-MR and none to 4-MR enhances the stability of ferrierite. As a general trend, the post-synthesis dealumination through high-temperature calcination, steaming, and treatment with HCl, oxalic acid, (NH4)2SiF6, or SiCl4 did not significantly alter the porosity of ferrierite,47,50,51 in contrast to the noticeable effect on other zeolite structures. However, sequential treatments, including dealumination and the subsequent FER zeolite desilication, facilitated hierarchical mesoporosity development, achieving nearly 110 m2 g−1 of mesopore surface area.52
The dealumination with gaseous SiCl4 offers the benefit of allowing silicon atoms to immediately substitute the removed aluminium atoms, in contrast to other dealumination techniques. This prevents the formation of new hydroxyl nests and may possibly facilitate the healing of existing defects.
Nonetheless, hydrochloric acid is generated during the SiCl4 treatment, which may lead to dealumination of the materials. Furthermore, washing the dealuminated samples with water leads to the hydrolysis of the aluminium trichloride generated in this process, resulting in aluminium hydroxide, which is poorly soluble. Consequently, EFAl species remain in the pore system post-washing.
Defect sites vacated by the aluminium atoms can be refilled by amorphous debris, which shows high migration tendencies. The defects can continue to grow and form mesoporous holes. The growth of spherical mesoporous holes may create larger mesoporous channels or cavities in regions with intensified dealumination or high density of defects. Nonetheless, the pores produced through dealumination primarily consist of mesoporous cavities within a zeolite linked to the external surface through the micropores. Such mesopores do not interconnect effectively with the zeolite external surface.53 Mild treatments result in many cavities inside zeolite crystals and frequently cause undesirable material deposition within the micro- and mesopores.53 More severe treatments further decrease crystallinity and reduce the micropore volume.53
The extraframework aluminium (EFAl) and extraframework silicon (EFSi) species within zeolite cavities have been extensively studied.38,48,54–59 The EFAl octahedral Al species, which are well-dispersed and highly charged, strongly interact with the zeolite framework. The interaction of the BAS protons attached to the oxygens bridging the framework Si and Al atoms becomes weaker with increasing amounts of the EFAl in H-USY zeolite. This weakening is caused by the polarisation induced by the EFAl, which increases the Brønsted acid strength of these centres. Solid-state NMR studies, supported by the density functional theory (DFT) calculations, demonstrated that the synergy between Brønsted and Lewis acids significantly increased the Brønsted acid strength of dealuminated HY zeolite. The extraframework Al(OH)3 and Al(OH)2+ species located in the supercages, along with the Al(OH)2+ species in the sodalite cages, were identified as the preferred Lewis acid sites. Moreover, the EFAl species coordination with the oxygen atom nearest the aluminium framework leads to enhanced HY zeolite acidity. However, no direct interactions, such as hydrogen bonding, exist between the EFAl species and the Brønsted acid sites. Van Bokhoven60 used 27Al 3Q MAS NMR, a quantitative 27Al MAS NMR, and 29Si MAS NMR to show that increased Brønsted acid strength is a result of the gradual increase in average Si–O–Al and Si–O–Si angles throughout the zeolite framework, induced by the EFAl species. Schroeder et al. showed,61 using zeolite Y as an example, the existence of two different pairs of Brønsted acid sites (located in the supercages and the sodalite cages), which were transformed into Brønsted–Lewis acid pairs upon ultrastabilisation. Using 1H DQ MAS NMR spectroscopy, they could assign the specific 1H NMR resonances to the zeolite Brønsted and Lewis acid sites, which allowed them to prove that both species become spatial neighbours after mild post-synthetic modification via ultrastabilisation. In turn, the changes in the polarisation transfer for BAS–BAS pairs and BAS–LAS pairs were validated and identified as the factors contributing to the increased acid strength of BAS and, as a consequence, higher catalytic activity.60,61
The role of the Lewis acid sites in acid-catalysed reactions is widely discussed in the literature. Extraframework Al species (EFAl), containing coordinatively unsaturated Al atoms, are themselves regarded as strong Lewis acid sites, facilitating the formation of carbenium ions by abstracting hydride ions from saturated hydrocarbons.62 Their role, however, is not so simple. Lewis sites can be produced when the zeolite framework is damaged either during the burning of OSDA or, in much larger quantities, during steaming and/or dealumination. Early works of Sohn et al.63 showed that cracking activity increased for mildly steamed faujasites and then decreased with more severe treatment, as exemplified by a 10-fold increase of n-hexane cracking over dealuminated Y zeolite (FAU).64 It was also found that steamed faujasites are much more active than dealuminated (by chemical treatment) ones with the same framework Al content.65 Wang et al.66 investigating a series of steamed Y zeolites (FAU) claimed that ‘superacid sites’, formed by the inductive influence of LAS on the neighbouring zeolitic protons are responsible for the activity enhancement. They also found that activity in monomolecular processes (carbocation formation) depended both on the number of framework and extraframework Al sites, while the bimolecular reactions (hydride transfer, coking) depended mostly on the proximity of acid sites. To enhance the catalytic activity of the catalyst, the proximity of the Brønsted site and extraframework Al is required. The Hensen group67 compared the activity of commercial ultrastabilised Y zeolite with the zeolite in which extraframework Al species were introduced by incipient wetness impregnation and ion exchange. They found that although commercial Y contained less EFAl, they were mostly cationic. The rate of propane cracking showed a strong correlation with the number of Brønsted acid sites influenced by cationic EFAl. A few years later, they confirmed, using H–D exchange followed by in situ1H MAS NMR that the protons, located inside sodalite cages, interact with the cationic EFAl species; thus, the enhancement of the acidity is very specific, and concerns BAS sites, located inside the faujasite cages.68 In the real FCC (fluid catalytic cracking) catalyst, Lewis acid centres may be located at clay (filler) or silica–alumina (binder) surfaces, or zeolite–clay and zeolite–alumina interfaces, further complicating the picture. The role of different additives in the FCC catalyst is reviewed in a well-known paper by Voght and Weckhuysen, revealing some of the secrets of ‘the grand old lady of zeolite catalysis’.62
1.3 Resistance to water
Zeolites maintain stability at ambient conditions when in contact with water or water vapour. Nonetheless, their crystalline structures may be compromised at extreme conditions, including elevated temperature, high water vapour pressure, or high acidity/basicity.69 The structural changes induced by water are proposed to start from the hydrolysis of terminal Si–O–Si–OH groups,70 forming a defect propagating inward throughout the framework, as shown by the examples of MCM-41 aluminosilicate.71 Indeed, the stabilising effect of Al in the zeolite framework may be dominated by the concentration of defect sites, as was shown by the example of the *BEA structure studied with Al K-edge extended X-ray absorption fine structure, and 27Al MAS NMR spectroscopies in combination with DFT calculations.72 The observed zeolite degradation by the treatment in hot liquid water was related to selective hydrolysis of the T1- and T2-sites that form the 4-MR in the framework (see Fig. 1). Furthermore, as the hydrolytic disintegration of the zeolite framework started at Si–OH groups, the structural stability of the zeolite crystal domains strongly depended on the defect site concentration. The local strain and the specific substitution significantly impact the hydrothermal stability of the zeolite structures. For these reasons, enhancing the quality of materials (absence of the defective Si–OH groups), their surface modification to eliminate Si–OH groups, and mitigating local stress are hypothesised to be the keys to future, more stable zeolite materials. On the contrary, Maag et al.73 suggested that treatment with liquid water promotes the dealumination of ZSM-5 (MFI), functioning through a mechanism akin to that seen during steaming: it begins with Al–O hydrolysis, followed by aluminium species migrating to the surface, and culminates in the deposition of EFAl or possible full dissolution under specific conditions. Dealumination occurs most strongly at temperatures ranging from 300 to 400 °C. Multiple models were assessed to address the non-Arrhenius temperature effect on amorphization and dealumination. The most effective one employed temperature-dependent values of the water auto-ionisation constant. Also, the stability of ZSM-5 in liquid water was claimed to be influenced by the concentration of the framework Al.74 The slope of the non-Arrhenius temperature dependence upon amorphization of ZSM-5 zeolite with Al was more pronounced than that of the siliceous silicalite-1. Moreover, the crystal tip oriented in the (001) direction functioned as a more effective dissolution centre. In contrast, except for the edge, such a centre was absent on the (010) face. The elevated content of Al in HY-zeolite led to enhanced stability in hot (150 °C) water.75 H-USY zeolites with high concentration of the framework and extraframework aluminium, demonstrated increased resistance to hot liquid water.76 Furthermore, the nature and the concentration of the extraframework Al species (i.e. EFAl and charge-compensating cations) have been documented to influence the stability of zeolites in water.77 It has been hypothesised that EFAl species engage with terminal Si–OH and Si–O–Si groups near the surface, protecting them against the attack by OH− anions.78 Ravenelle et al.75 noted the emergence of an amorphous aluminium phase, which maintained tetrahedral coordination even upon removal from the framework. It was proposed that the Si–O–Si groups within the zeolite framework could be cleaved selectively depending on the treatment conditions, such that only a few, but not all, pores collapse.75 The authors concluded that Al stabilises the framework against hydrolysis. Although this statement may be true, additional factors could influence the stability of zeolites in water, like the concentration of the framework defects in samples. The alterations caused by water vapour significantly impact the stability of the zeolite structure at high temperatures.1.4 Dealumination via steaming treatment
Among known zeolite structures, 51 are categorised as small-pore zeolites (International Zeolite Association: Structure Commission, Database of the zeolite structures).9 These zeolites, e.g. CHA, AEI, AFX, ERI, and RHO, are promising catalysts for the selective catalytic reduction of NOx with NH3 (NH3-SCR),79,80 selective oxidation of ammonia81 and methanol-to-olefin conversions.82 Adjusting the Si/Al ratio has become increasingly important for these structures. Additionally, the step of special importance is transporting the generated extraframework Al species having the kinetic diameter of ca. 0.5 nm83 to the liquid phase through the narrow 8-ring channels (0.3–0.4 nm) for removal from inside the crystals, as represented in Fig. 3. The dealumination via steaming of the small-pore zeolites, described by Davis et al.84,85 allowed preservation of crystallinity at 500 °C to 600 °C. However, the EFAl species blocked the pores, suppressing catalytic performance. In this case, approximately 85% of the total aluminium atoms remain in the zeolite structure of the CHA zeolite. After removing aluminium atoms through acid washing, the relative crystallinity of the zeolite was reduced. Steaming at higher temperatures (>700 °C) degraded the crystal structure of narrow pore zeolites. | ||
| Fig. 3 Dependence of factors influencing applied treatments (detemplation, dealumination, desilication with NaOH, and NaOH & Pore Directing Agent) and zeolite channel sizes. | ||
The organic additives used as the structure- or pore-directing agents (OSDA or PDA) during crystallisation, occupy the pores within the structure and stabilise the frameworks.86 These organic molecules are typically removed in the calcination procedure prior to subsequent post-synthetic treatment or application in catalysis and adsorption.
Organic cations have been documented to serve as stabilising agents in extracting aluminium from as-synthesised beta zeolites.10,87 The zeolite that still contained the template (OSDA) enabled obtaining mesostructured beta zeolites with the final Si/Al ratios exceeding 1000 in a single step without notable loss of crystallinity and pore volume, while maintaining thermal stability. Recently, a liquid-mediated treatment using cooperatively fluoride anions, hydroxide anions, and pore-filler cations was reported to stabilise the high-silica *BEA-, MFI-, and MOR-type topologies.88 These modified zeolites can endure exceptionally high-temperature steaming conditions (900–1150 °C). The stabilised zeolites maintained crystallinity and micropore volume, while the parent commercial zeolites underwent complete degradation. Defect sites in the treated zeolites are markedly diminished through a self-defect-healing mechanism that entails the movement of silicate species.
Agostini et al.38 reported that dealumination of the steamed Y zeolite occurs at milder temperatures (180–230 °C) rather than at the high ones, where the initial water molecules begin repopulating the pores. It has been pointed out that most of the aluminium is dislodged from the zeolitic framework, and there is a remarkable structural collapse due to steaming that does not occur at elevated temperatures. They also indicate the presence of 30–35% of the total aluminium within the sodalite cage. In addition, water can re-enter the pores at lower temperatures, resulting in defect formation and substantial migration of the framework Al3+ to extraframework positions.
1.5 Dealumination via acid treatment
The debris formed in the steaming process may deposit on the surface of zeolite particles or within the micropores, resulting in their blockage. Therefore, steaming is typically used alongside acid treatment to remove the debris produced. Chemical degradation in acids can also lead to remarkable meso- and macroporosity since it affects the zeolite structure and forces its reorganisation. Removing Al atoms from the framework positions significantly alters zeolite thermal stability and acidic properties. The severity of the dealumination process, which is influenced by factors such as acid type, concentration, temperature of the treatment, and zeolite crystallite size, can result in the removal of up to 100% of the aluminium atoms. The thermal stability of the final micro–mesoporous zeolite materials diminish progressively with the increase in the dealumination degree, as this process correlates with a partial reduction in crystallinity or amorphization of the zeolite structure.89–91Yoshioka et al.92 documented the compositional tuning of small-pore zeolites through leaching in an acidic solution based on the framework stabilisation using pore-filling organic molecules (Fig. 3). A liquid-mediated treatment in the presence of ammonium fluoride and TEAOH (tetraethylammonium hydroxide) healed the structural defects produced by the dealumination. The number of silanol groups resulting from structural defects significantly diminished during the defect-healing process, while the overall crystallinity and composition remained intact.
The literature reports concerning acid dealumination of zeolite ZSM-5 discuss a variety of results. Weckhuysen et al.93 demonstrated that sinusoidal channels in ZSM-5 zeolite exhibit greater susceptibility to dealumination and mesopore production following steam treatment than the straight channels. Kooyman et al.94 reported that dealumination did not occur using aqueous HBr or H2SO4 and was hardly noticeable in the presence of 1 M HCl even at temperatures as high as 160 °C. This high stability towards dealumination was attributed to the absence of structural defects, which resulted from the extended synthesis duration. The temperature and the duration of the acid treatment did not affect the degree of dealumination. The steaming process demonstrated greater efficacy in the dealumination of ZSM-5 zeolites, and many of the different extraframework Al species were produced. As this had a detrimental effect on the catalytic and transport properties45,46 it is usually followed by acid leaching. The defects formed during steaming can lead to undesirable effects, specifically to the occurrence of further dealumination of HZSM-5 zeolites if mineral acids, such as HCl, are employed in the leaching treatments. The acidity of HZSM-5 zeolites was adjusted through steaming and citric acid treatments, leading to an increase in the amount of the framework Al exclusively. This increase was attributed to reinserting extraframework Al into the defective sites of the steamed HZSM-5 framework. The reintegration of Al atoms into the framework increased Brønsted acidity, especially the strong Brønsted acidity, and nearly restored the pore structure to that of the original HZSM-5.95 Noteworthy differences were observed in the susceptibility of zeolite to dealumination, which arises from the varying numbers of defects created depending on the synthesis conditions for each type of zeolite.47 Zeolite synthesised via the fluoride method showed superior stability due to a minimal number of defect sites and the reduced fraction of Brønsted acid sites interacting with the surrounding framework. The number of defect sites in the framework correlated with the amount of Brønsted protons that interacted with oxygens and, simultaneously, with a decreasing stability towards dealumination. Zholobenko et al.96 observed a reduction in the number of strong acid sites and a concurrent increase in weaker acid sites during the dealumination of ZSM-5 samples. This phenomenon can be attributed to the formation of defect sites that facilitate the interaction of the remaining Brønsted protons with the framework while reducing acidity. Loeffler et al.97 proposed that the ease of dealumination in ZSM-5 depends on the presence of aluminium atoms that are not fully integrated into the zeolite framework, thereby classifying them as defect sites. In summary, the dealumination of ZSM-5 zeolites often results in poor mesopore formation. At the same time, it reduces the aluminium content, resulting in decreased acidity of the zeolite. Therefore, a follow-up desilication treatment is applied for Al-rich MFI zeolites to enhance their mesoporosity.98
It was noted that applying AlCl3 vapour at elevated temperatures integrated Al atoms into the structure of high-silica ZSM-5 zeolite, modifying its acidity and activity.99–101 Furthermore, the treatment with mineral acid (e.g. HCl) successfully facilitated the reinsertion of non-framework aluminium into the framework of dealuminated HZSM-5 zeolites.102 This is opposite to the established dealumination impact of mineral acids on aluminium-rich zeolites. In contrast, Omegna et al.103 observed no evidence of realumination, and instead, dealumination occurred following the mineral acid treatment of dealuminated HZSM-5 zeolites. Xie et al.104 reported the realumination effect of a single citric acid treatment on beta zeolite. Prior to the citric acid treatment, steaming was also essential for the realumination of HZSM-5 zeolites to occur, indicating that framework defect sites produced upon steaming were required for the citric acid treatment to be effective.95 The realumination due to the citric acid treatment on the steamed HZSM-5 zeolite could nearly recover the pore structure of the steamed zeolite to that of the parent. Solid AlF3 treated as a realuminating agent,105 was used in a simple and efficient method for dealuminating and realuminating H-beta zeolite. The structure, texture properties, and acidity of the final hierarchical beta zeolite were adjusted by varying the amount of AlF3 employed.
Lee and Rees106 have estimated the maximum amount of HClaq (10 mmol of HCl per 1 g Na–Y) which allows for preservation of the Y zeolite structure, while 56% of the framework aluminium atoms are extracted. Janssen et al.53 compared the shape of mesopores in the series of Y zeolites modified through different post-synthesis treatments, showing that acid treatment is pivotal for proper mesoporosity development. The distinction between cylindrical mesopores and mesoporous cavities connected to the external surface via micropores was shown. Many mesoporous cavities were observed after steaming and acid leaching (Fig. 2). Most of the mesopore volume is in the cylindrical mesopores. Only a special hydrothermal treatment involving strong dealumination by steaming and acid leaching allowed the formation of zeolite Y crystals with very high mesopore volumes. Even though some cavities remained, the resulting mesopore system consisted of strongly interconnected cylindrical mesopores. Such a system of interconnected cylindrical mesopores is expected to enhance diffusion much more than the mesoporous cavities inside the crystals. Dealumination of zeolite Y exemplifies an important process, yielding the ultra-stable (US) zeolite Y discussed above, a crucial component in contemporary cracking and hydrocracking catalysts.107 Reducing acid site density minimises the extent of proton transfer reactions, thereby enhancing the olefin yield.
Müller et al.47 investigated the stability of the framework aluminium in connection with the acid-assisted dealumination of various zeolites. The number of Al-atoms in 4-MR is a significant structural determinant, given that the tension in these rings exceeds that of 5-MR. Consequently, the dealumination of zeolite beta, characterised by 75% of Al-atoms present in 4-MR, proved to be more straightforward than that of ZSM-5, which contains only 17% of Al-atoms in similar configurations. In the context of the Al-atoms arrangement, the susceptibility of the CHA framework to desilication was also evaluated.108 Each Al-atom is integrated into three 4-MR in the chabazite structure, diminishing the framework aluminium stability in H-SSZ-13. This reduces the protective framework effect of aluminium, leading to the desilication of the H-SSZ-13 samples even with the low initial Si/Al ratio.
1.6 Desilication
Desilication is the second oldest method employed to create mesopores in zeolites, commonly by contacting with a base. It is typically defined as the process in which silicon atoms are selectively removed from the zeolite framework through the preferential cleavage of Si–O–Si bonds.109 Unlike dealumination, which creates closed mesopores inside the microporous zeolite framework, desilication leads to auxiliary mesoporous structures that reduce diffusion path while enhancing the accessibility to pristine microporosity.23 The hierarchical interconnections of pores in multicomponent industrial catalysts influence the catalyst activity and selectivity, presenting a significant challenge. The significance of the hierarchical pore interconnectivity in the multi-component zeolite-based industrial catalysts is exemplified in fluid catalytic cracking and hydroisomerization processes. Contrary to dealumination, desilication produces a more efficiently interconnected mesopore network that is directly linked to the external surface of zeolites. By evaluating different parameters of the desilication approach, specifically its duration and concentration of NaOH, it became apparent that both silicon and aluminium are extracted, albeit the latter to a markedly lesser extent.110 The quantity of dissolved silicon increased with the duration of desilication; however, the analysis of aluminium was not carried out. The tetrahedral Al centres are considered highly resistant to hydroxide attack owing to their negative charges, which impede the hydrolysis of the Si–O–Al bond.74,111–116 Consequently, the alkaline treatment is categorised as a ‘desilication’ procedure, in contrast to ‘dealumination’, which focuses on the selective extraction of Al atoms during acid leaching.The number of aluminium atoms in the zeolite framework governs the desilication process, analogous to dealumination. A comprehensive study conducted by Groen et al.98,115,119–121 on 10-MR zeolite ZSM-5 subjected to alkaline treatments showed the potential of mesoporous zeolites. An improvement of two orders of magnitude in the average characteristic diffusion time of neopentane over desilicated ZSM-5 crystals was observed, with a homogeneous distribution of the framework aluminium being a significant feature for the mesopore growth.122 These studies demonstrated that mesoporosity is distinctly influenced by the parent zeolite Si/Al molar ratio, with an optimum ratio ranging from 25 to 50 (Fig. 3). At the lower Si/Al ratio (<25, higher Al content), less mesoporosity was produced due to re-insertion (realumination) of the extraframework Al species alongside desilication.115,123 Therefore, subsequent acid washing, which eliminated Al-rich amorphous residues from meso- and micropores, was added to the procedures for desilication of Al-rich zeolites (Si/Al < 25). The elevated Si/Al ratio (>50, high Si concentration) results in more macropores due to uncontrolled Si extraction. The framework aluminium atoms are classified as ‘pore-directing agents’ (PDAs) because they influence mesopore development. The ‘realumination’ process was provisionally proposed to elucidate reintegration of Al onto the external zeolite surface, thereby inhibiting additional surface degradation (Fig. 4). This impact was attributed to the reduced Si/Al ratio range for desilication, a constraint of the alkaline treatment method. Finally, the Si/Al ratio range (Si/Al > 50) of parent zeolites susceptible to mesopores generation has been broadened to cover all Si/Al ratio ranges up to 1000 (Fig. 2)23 using metal salts or intentionally introduced PDAs, such as Al(OH)4− or tetrapropylammonium (TPA+) to inhibit excessive Si dissolution. Tetraalkylammonium salts are classified as pore-directing agents, as they preserve the zeolite during desilication by preferentially engaging with the zeolite crystal surface, thus inhibiting the attack of OH− ions on the zeolite.124–127 The significance of the role of tetraalkylammonium ions as pore-directing agents was demonstrated in neopentane sorption, supported by FT-IR analyses and using the Crank solution.128,129
 | ||
| Fig. 4 Schematic representation of desilication and co-occurring realumination followed by acid-wash aimed to remove Al-debris. | ||
 | ||
| Fig. 5 Representation of the most optimised hierarchical zeolites with the highest increase of Smeso (ΔSmeso) obtained via dealumination and desilication methods (based on Table S1, ESI†). | ||
Similarly to the CHA topology zeolites, the micropores of ZSM-58 (DDR) exhibit narrow pore sizes of 0.45 nm × 0.36 nm, facilitating the requisite shape selectivity for light olefins in the MTO process.133
The structural properties of ZSM-58 cause diffusion constraints of the formed hydrocarbons that are distant from the reactive acid site and outside the crystalline pore system, resulting in fast coking. Hierarchical porosity in zeolite ZSM-58 was attained through desilication using 0.1–0.5 M NaOH per 1 gram of zeolite at 95 °C. Applying desilication and re-assembly of dissolved silica species with added cetyltrimethylammonium bromide (CTAB) (0.05 M) produced hierarchical ZSM-58 materials. It should be noted that the mechanistic significance of PDA cations strongly depends on their concentration in the desilicating solution. They act as isolated cations attached to the zeolite surface at low concentrations, protecting the material from significant dissolution, as mentioned in Section 1.6.1. At high concentrations, surfactants form micelles that contribute to the formation of mesopores, as exemplified by mesoporous zeolites synthesised with CTAB during alkaline treatment.134 Surfactant micelles assisted redeposition of dissolved zeolite fragments onto parent zeolite crystals without the need for hydrothermal treatment. The synthesis of mesoporous zeolite by a combination of dissolution (e.g. desilication)135 and re-assembly processes under hydrothermal conditions was adapted from the principle of pseudomorphic transformations and is discussed in more detail in Section 3. The application of pure NaOH solutions to zeolite ZSM-58, initially with Si/Al = 50, yielded macroporous zeolite ZSM-58 exhibiting a wide pore size distribution, with pore volume dependent on the concentration of sodium hydroxide. No secondary porosity was formed due to high Si/Al. Incorporating CTAB into the desilication solution produced hierarchical ZSM-58 materials characterised by additional narrow pores measuring 3–4 nm, attributed to an amorphous shell on the outer surface of crystals. This layer exhibited long-range order at moderate NaOH concentrations, irrespective of the initial Si/Al ratio employed. The total specific surface area was significantly improved to 868 m2 g−1, with high external surface area. At the same time, the desilication yield (measured as weight loss) increased due to re-assembly of dissolved silica species. Consequently, elevated hierarchy factor values and desilication efficiency may be attained. The concept of hierarchy factor (HF) is described in the Section 9. In brief, it quantifies increase of the mesopore surface area/volume at the expense of the micropore area/volume. An increase in HF results in the formation of additional mesopores without a reduction in micropore volume. The quantity of strong acid sites in the desilicated samples above diminished with rising NaOH concentration, as established by temperature-programmed desorption of ammonia. The formation of silanol groups and pentahedral and octahedral coordinated additional framework aluminium was identified as the cause of reduced acidity in the hierarchical samples, as verified by 27Al and 29Si MAS NMR spectroscopy.
A hierarchical zeolite ZSM-58 architecture was generated using post-synthetic desilication with sodium hydroxide solutions of different concentrations and tetraethylammonium ions (TEA+) as a pore growth moderator. The latter was employed to enhance the size control of generated mesopores during desilication, as detailed by Pérez-Ramírez et al.124,136 Pore growth moderators are cations that engage with the negatively charged zeolite surface, hence offering a degree of protection to the zeolite structure against excessive leaching. Thus, a base leaching in the presence of a pore growth moderator allowed the introduction of intracrystalline mesopores with a volume of up to 0.50 cm3 g−1. All desilicated ZSM-58 samples had extended catalytic lifetimes and increased product yields in the methanol-to-olefins (MTO) reaction, attributable to improved adsorption/desorption kinetics, compared to simply microporous ZSM-58. Optimal catalytic performance was reached using ZSM-58 desilicated with 0.3 M NaOH and 0.05 M TEABr. The hierarchical ZSM-58 attains a methanol conversion rate over twelve times greater than a purely microporous zeolite.
Sequential leaching with nitric acid (HNO3) and sodium hydroxide (NaOH) to regulate the extraction of silicon and aluminium atoms from an erionite framework (Si/Al = 3.5) was identified as the most efficacious method for generating intracrystalline porosity in hierarchical zeolites with 8-MR137 (Fig. 5). Adding mesoporosity in erionite using a top-down technique (149 m2 g−1) is equally effective as the bottom-up design of the structurally analogous nanosized zeolite UZM-12 (178 m2 g−1). Despite the rod-like shape of erionite zeolites, they exhibited significant susceptibility to mesopore formation. The demetallation process generates mesopores in the zeolite and enhances the accessibility of its acid sites. The improved accessibility of protonic sites in hierarchical erionite, characterised by optimally developed microporosity and nanosized UZM-12, greatly influenced the conversion of 1,3,5-triisopropylbenzene dealkylation. UZM-12, a bottom-up synthesised zeolite, exhibited nearly identical exterior acidity and surface area to its top-down modified equivalent, demonstrating comparable catalytic efficiency.
Consecutive post-synthesis modifications, namely alkaline and acid treatments, can precisely refine zeolite characteristics. Modified mesoporous zeolites exhibit a favourable impact on reactions where the external surface is crucial, since the accessibility to their pore openings is remarkably enhanced. Subsequent dealumination and desilication processes were identified as effective techniques for generating a secondary system of mesopores in ferrierite (Si/Al = 9 and 23) and maintaining its microporous properties.52,138 Dealumination was performed using HNO3, while LiOH was employed for desilication. This modification facilitated the formation of mesopores (Smeso = 108 m2 g−1, Vmeso = 0.24 cm3 g−1) within the ferrierite structure while preserving crystallinity (86%). The increased quantity of Al sites accessed by pyridine indicated an enhancement in the accessibility of acid sites attributable to the formation of supplementary mesoporosity. The catalytic performance of hierarchical ferrierites is greatly influenced by acidity and textural characteristics during ethanol dehydration. The research showed that hierarchical zeolites exhibiting well-preserved microporous properties, including a substantial micropore area and inherent acidity, show enhanced catalytic activity and selectivity for ethylene production.52
Recent reports indicate that hierarchically structured micro-mesoporous H-form FER (Si/Al molar ratio of 27) typically require two sequential post-treatment procedures: a partial dissolution step in NaOH solution to generate nanocrystals, followed by a hydrothermal recrystallisation step using cetyltrimethylammonium bromide (CTAB) as a cationic surfactant. This mitigates in situ structural degradation of FER frameworks, including irregular and excessive structure collapse, as shown in prior studies.22,139–144 A one-step method, including desilication with NaOH solution and concurrent recrystallisation with CTAB, was documented for the selective production of mesoporous ferrierite.145 One-step recrystallisation method of commercial FER and home-made seed-derived FER were separately used to prepare mesoporous FER through one-step desilication method. The impact of desilication duration and mesopore architectures based on the kinds of FER used was assessed for catalytic performance and structural stability, focusing on surface acidity and coke deposition during the gas-phase carbonylation of dimethyl ether to methyl acetate. The newly developed mesoporous structures, measuring 5–40 nm, on the pristine seed-derived FER with a Si/Al molar ratio of 10.4 were substantially altered by the duration of desilication/recrystallisation. The mesopores markedly enhanced the surface acidic sites while maintaining a comparable level of crystallinity, even at the reduced Si/Al ratio of 6.7–8.6. The enhanced strong acidic sites associated with Brønsted acid sites, following an optimal desilication lasting approximately three hours, primarily contributed to the boosted DME carbonylation activity, with reduced formation of coke precursors, attributed to efficient mass transport through its larger mesopores.
Verboekend et al.146 indicated that low desilication effectiveness, and consequently restricted development of mesopore surface area (maximum approximately 100 m2 g−1), is attributable to the crystal shape. The efficacy of desilication significantly diminishes with the reduction in the crystal diameter. The desilication of zeolites with needle-like crystals (ZSM-22) or platelet (ferrierite) is less advantageous, as the major portion of the resulting mesopore surface originates from intercrystalline mesoporosity. The incorporation of mesoporosity was complex due to the unique characteristics of ZSM-22, specifically its rod-like crystal morphology, irregular aluminium distribution, and unidimensional structure of the elliptical micropores.146 Due to the predominant presence of Al on the external surface of the nanorods, a significant re-deposition of extracted Al during alkaline treatment resulted in the clogging of up to 80% of the micropore volume. Subsequent acid treatment in aqueous HCl effectively recovered up to 90% of the micropore volume and enhanced the mesopore surface area (from 95 m2 g−1 to 114 m2 g−1). A comparison with other alkaline-treated zeolites highlights the low effectiveness of desilication in ZSM-22, linking its origin to the morphology of the ZSM-22 nanorods. Undesirable crystal morphologies are particularly common in zeolites with micropore networks of limited dimensionality, leading to comparatively constrained pore openings.23 Moreover, in these instances, the micropore channels typically form along the longest axes of the crystal, signifying a diminished number of pore openings. The subsequent ‘less efficient’ introduction of the mesoporous surface via alkaline treatment is still expected to improve access to the micropores.
The desilication process has been extensively investigated for producing zeolites with hierarchical porosity and has become a standard procedure predominantly employed on a laboratory scale. A noteworthy contribution to advancing desilication originated from the Perez-Ramirez group.23,98,121,147 However, Ogura and co-workers carried out the first desilication process with increased mesopore formation applied to zeolite ZSM-5.148 The treatment of ZSM-5 in an alkaline solution significantly altered the shape of zeolite crystals. This created mesopores of nearly homogeneous sizes without significantly deteriorating the microporous structure. The alkaline environment permitted the preservation of the BET surface areas (316 and 320 m2 g−1 for pristine and alkali-treated ZSM-5, respectively), while concurrently slightly decreasing the micropore volume from 0.17 to 0.13 cm3 g−1. Moreover, the alkali treatment significantly enhanced mesopore volume from 0.07 to 0.28 cm3 g−1 (Fig. 5). Ogura et al.149 postulated that mesopores develop along the borders of ZSM-5 crystallite twinning, which are believed to exhibit low resistance to alkaline environments. This type of disorder, characterised by linear, two-dimensional, and macro-defects, has been extensively examined through different methodologies. Structural disorder and stacking faults may disrupt the three-dimensional pore system of zeolites (Fig. 6). Consequently, particular parts of the porous system may become inaccessible and inefficient in catalytic processes. For instance, the diffusion rates of toluene in polycrystalline ZSM-5 were three orders of magnitude lower than in a single crystal.150 Therefore, significant implications for the catalysis of boundary intergrowths in zeolites have been thoroughly investigated.151–153
 | ||
| Fig. 6 SEM and TEM images of zeolites with different pore sizes subjected to desilication. | ||
Traditionally, defects such as stacking faults and the intergrowth of various crystal forms in zeolites have been examined using electron microscopy and atomic force microscopy.154–156 The intergrowth of two distinct zeolite structures and the intergrowth of structural subunits can be recognised by these techniques. Weckhuysen, Roeffaers and their co-workers developed a new methodology of confocal fluorescence microscopy systems that allow the examination of intergrowth structures and the mapping of catalytic activity within individual zeolite crystals (discussed in Section 9).157–161 For an intergrown silicalite-1, the dissolution occurs predominantly along the interfaces of intergrown regions, leading to pronounced cracks. Conversely, the pores do not manifest in certain areas of defect-free crystals synthesised in the fluoride media. Notwithstanding the variability in pore creation, several similar characteristics are also evident.162 Li et al.162 corroborated the protective function of aluminium in desilication by applying integrated microscopic methods and ptychographic X-ray computed tomography (PXCT). The NaOH leaching of ZSM-5 crystals, with aluminium predominantly situated in the rim zone, resulted in the development of hollow crystals due to selective extraction of the core region. Alkaline-leached ZSM-5 with uniform aluminium distribution did not develop a hollow structure. Svelle et al.163 demonstrated that mesopores are generated by two mechanisms: one resulting from the Al-directed dissolution of siliceous regions and the other from selective dissolution or etching along boundaries, intergrowths, and defects throughout each particle. The authors determined an optimal grain morphology for the effective desilication of ZSM-5. Zeolite grains built from smaller fused subparticles appeared to be very susceptible to desilication-induced mesopore generation, regardless of their size. Carbon templating generally resulted in a mesoporous material with a higher defect density than desilication. Other high-silica 10-MR zeolites that could replace ZSM-5 in some petrochemical processes were also subjected to desilication. Hierarchical IM-5 and TNU-9 zeolites were successfully prepared by alkaline treatment, with values of mesopore surface areas higher than 120 m2 g−1. The hierarchical IM-5 and TNU-9 zeolites performed better than their pristine analogues in toluene alkylation, and n-alkane and polypropylene cracking.164–166
A carefully controlled desilication process, using NaOH and TBAOH (tetrabutylammonium hydroxide) as alkaline agents, was implemented to synthesise a hierarchical zeolite beta that integrated mesoporosity and zeolitic microporosity while preserving the intrinsic acidity of the original zeolite.168 A nearly 12-fold increase in mesopore surface area was observed for the desilicated samples (468–510 m2 g−1) relative to the parent zeolite (44 m2 g−1) (Fig. 5). The study of the associated isotherms indicated that the micropore volume of the NaOH-treated zeolite beta diminished, consisting with the XRD data demonstrating the loss of crystallinity in NaOH-desilicated zeolite compared to the parent material. Nonetheless, the zeolite treated with NaOH and TBAOH maintained its micropore volume (Vmicro) following the alkaline treatment. This suggested that treating beta zeolites with a combination of NaOH and TBAOH has less impact on their structure. An optimised catalyst beta exhibiting exceptionally high LCO (light cycle oil) and propylene selectivity during the gas–oil cracking reaction has been obtained. The catalytic cracking of low-density polyethylene using *BEA zeolites demonstrated the interrelation between the speciation of acidic sites and their accessibility towards reactants, which are either intrinsic to the microporous structure or accessible via the newly formed mesoporous surface.169
Zeolite beta shows relatively low stability of aluminium in the framework upon desilication compared to MFI, MOR, and MTW-type zeolites, and it has undesirable impact on the effectiveness of alkaline treatments. As mentioned above, this lower framework stability has been attributed to the high density of 4-MR in the *BEA framework.47,170 This precludes high framework stability for zeolite beta, so aluminium will not function as a silicon extraction moderator. This differs from MOR and MFI, where the formation of mesopores through silicon extraction is often beneficial. Thus, enhancing mass transfer in beta zeolites for future catalytic applications may require alternative methods, such as templating,171–176 the preparation of zeolite nano-composites177–179 or partial detemplation of zeolites followed by desilication.180 As discussed above, the latter method is an effective and advanced option for creating hierarchical zeolites, as the template-containing framework exhibits significantly higher resistance to silicon leaching when subjected to treatments in aqueous NaOH solutions. It is especially appealing for zeolites exhibiting limited aluminium stability or having high silicon-to-aluminium ratio. Partial elimination of the structure-directing agent generates areas inside the crystal vulnerable to mesopore formation by desilication, while other areas, still holding the template, are shielded from silicon extraction. The change of calcination temperature between 230–550 °C influences the quantity of template elimination and facilitates the regulation of mesopore development in beta zeolite (Smeso = 20–230 m2 g−1) following alkaline treatment (Fig. 2). Samples of MFI, MTW, and MOR containing various template contents were treated in 0.2 M NaOH at 65 °C for 30 minutes, which represents typical conditions for desilication of calcined zeolites toward mesopore formation. The findings indicated that an elevated level of detemplation during the initial calcination phase results in outstanding enhancement of mesopore surface area following NaOH treatment (by a factor of 10 to 20). This was accompanied by a reduction in micropore volume due to silicon extraction from the template-free areas of the zeolite, thereby facilitating intracrystalline mesoporosity. The resulting improved effectiveness in the catalytic pyrolysis of low-density polyethylene (LDPE) illustrated the utility of this newly incorporated mesoporosity inside the hierarchical beta crystals. The LDPE conversion, adjusted for the aluminium level in each catalyst, shows that the turnover frequency is improved due to greater active site utilisation.
Desilication of mordenite, having a unidimensional channel system, remarkably improves catalytic activity compared to structures with two- or three-dimensional channel systems (Fig. 5). The preference of alkaline solutions to remove silicon allows preservation of the acid Si(OH)Al sites, but the quantity of Lewis acid centres increases, and Brønsted acidity remains constant. Mordenite was subjected to alkaline treatment using aqueous NaOH solutions with concentrations ranging from 0.05 to 2.5 M between room temperature and 90 °C for 10 minutes to 2 hours.181–183 Groen et al.183 demonstrated that desilication of high-silica mordenite in an alkaline environment produces significant intracrystalline mesoporosity while preserving its microporous and, crucially, acidic properties. In addition to maintaining the inherent zeolitic characteristics, the mesopore surface area generated in mesoporous mordenite surpasses that reached through dealumination. The mesoporous mordenite had improved catalytic performance in the liquid-phase alkylation of benzene with ethylene. Coke formation was inhibited during ethylene oligomerization, and in the above alkylation, which enhanced productivity and selectivity for ethylbenzene. The higher catalytic performance was ascribed to improved molecular transport in the effectively reduced length of unidimensional micropores and maintained acidity. Van Laak et al.184 investigated the effectiveness of mordenite in a comparable reaction, namely the alkylation of benzene with propene to produce cumene. The authors evaluated the outcomes of post-synthetic NaOH treatment for H-MOR and Na-MOR. Both zeolites exhibited enhanced mesoporosity while maintaining their crystallinity. To achieve equivalent hierarchical materials, the Na-form of mordenite requires double the treatment duration compared to the H-form. Consequently, it was determined that the conversion to the H-form before the treatment could be bypassed if the duration for post-synthetic treatment is extended. Lin et al.185 examined acid-treated, alkali-treated, and metal-loaded mordenite in the alkylation of benzene with α-olefin as environmentally sustainable and industrially effective catalysts. Hierarchically desilicated mordenites exhibited superior catalytic activity compared to the samples modified by the alternative treatments (HCl treatment and loading of Pt, Pd, and Zn). Upon precise selection of the treatment conditions and parent samples, the NaOH-modified catalysts exhibited steady dodecene conversions of around 100%, with linear alkylbenzene selectivity approaching 98%. Furthermore, a correlation was established between elevated concentrations of NaOH employed in the treatment of mordenite and the enhanced diffusion characteristics of the catalysts, accompanied by a reduction in the reaction temperature during the selective synthesis of ethylenediamine (EDA) through the condensation amination of monoethanolamine.186
The effect of enhanced accessibility of acid sites in micro/mesoporous mordenite zeolites was also reported for the hydroisomerization of n-hexane.187 Secondary mesopores were generated by three different sequential treatments of a commercial mordenite: NaOH alkali followed by acid, acid–alkali–acid and fluorination with NH4F–alkali–acid. The alkaline treatment caused desilication, predominantly along crystal defects, forming a secondary mesoporous structure defined by 5–20 nm cavities. The appearance of extraframework species and terminal Si–OH groups facilitated the development of mesopores. The extraframework species generated by the hydrolysis of perturbed or displaced framework aluminium constricted part of the mordenite unidimensional channel structure. Subsequent extraction of the extraframework species through mild acid leaching or concurrent elimination of Si and Al atoms via desilication, possible for a fluorinated mordenite, yielded a micro/mesoporous structure with numerous unrestricted channel openings, significantly improving the accessibility of the Si(OH)Al groups for n-hexane. Successive leaching treatments facilitate the development of active acid sites within unconstrained microporous channels, simultaneously improving the accessibility of these sites and the transport of molecules. The micro/mesoporous structure, with high density of accessible Brønsted sites, facilitated hydroisomerization, yielding increased amounts of branched isomers. Additionally, reducing the primary 12-MR channels and increasing the number of channel openings enhanced selectivity, thereby minimising nonselective cracking reactions.
Biomass has been drawing increasing attention as an alternative to petrochemical supplies. α-Pinene, a by-product of the softwood industry, may be readily converted through isomerization into chemicals such as limonene, camphene, and terpinolene, which have diverse applications. Liu et al.188 investigated the impact of alkaline treatment combined with microwave radiation to produce catalysts with a homogeneous mesopore distribution. The application of a microwave oven in the post-synthesis treatment of mordenite resulted in an enhanced conversion of α-pinene to 94.7%, attributable to catalyst improvement due to selective extraction of silicon atoms while preserving the aluminium atoms responsible for Brønsted acidity within the 12-MR channels. Another study examined an alkaline treatment of two varieties of mordenite samples: the first containing the synthesis template (organic molecule) and the second subjected to high-temperature calcination.189 The catalytic efficiency of both samples in dimethyl ether carbonylation remained highly consistent with the amount of Brønsted acid sites preserved inside the mordenite after desilication. It was observed that strong acid sites were retained in the 8-MR channels in mordenite with an organic template, inhibiting side reactions and coke accumulation. The pre-calcined samples had lower crystallinity due to partial dissolution of the zeolite in alkaline medium. The removal of strong acid sites led to a significant drop in carbonylation reaction.
Ghosh and Kydd190 examined the activity of mordenite after treatment with hydrofluoric acid. The treatment attempted substituting O atoms or surface OH groups with F−. The enhanced electronegativity of fluorine compared to the groups it replaces leads to an increase in framework polarity, subsequently elevating the acidity and reactivity of the surface. Consequently, they observed a notable (3-fold) reduction of the zeolite surface area and a slight alteration in the Si/Al ratio following treatment with a more concentrated solution. Conversely, a reduction in the solution concentration resulted in the observed removal of aluminium atoms. Nevertheless, the samples exhibited enhanced catalytic activity for cumene cracking. In the case of reduced active site content, accelerated deactivation was observed due to coke production.
Zeolite Y with FAU topology is the primary active phase in cracking catalysts, which dominate the industrial usage of zeolites. Its improvements through hierarchization have been studied with particular interest. The hierarchical zeolites Y prepared through intense alkaline treatments (368 K, 0.5–1.3 M NaOH) had good crystallinity, indicating that the NaOH treatment had a negligible damaging effect on the framework of pristine zeolite Y (Si/Al = 4.1, Smeso = 18 m2 g−1). The resulting increase of mesoporosity was moderate (Smeso = 61 m2 g−1).191 Successive desilication and dealumination with ammonium hexafluorosilicate (AHFS) resulted in zeolites with a homogeneous aluminium–silicon distribution. The framework defects induced by desilication significantly enhanced the intracrystalline transport of reactant molecules during the subsequent dealumination process. This enhanced the uniformity of dealumination and prevented surface silicon deposition in the final zeolites. The framework defects can coalesce and develop further into well-accessible mesoporous structures. Upon desilication under mild conditions (298 K, 0.05–0.1 M NaOH) applied to steamed and acid-leached mesoporous zeolite USY (CBV760, Si/Al = 28.4, Smeso = 213 m2 g−1) additional mesoporosity was generated manifested by a significant external surface development (Smeso = 443 m2 g−1).192 The NaOH-leached samples exhibited a distinct bimodal mesoporosity as a result of the enhancement of larger mesopores but, more importantly, the considerable rise of small mesopores. Specifically, the volume of small mesopores was tripled while the volume of large mesopores only increased by about 50%. However, this significant increase in mesoporosity came at the cost of significant amorphization of the USY zeolite, which was confirmed by a reduction in the micropore volume from 0.21 cm3 g−1 to 0.07 cm3 g−1. These studies clearly demonstrated that zeolite Y and its ultrastabilised counterpart USY behave very differently in alkaline environments. Therefore, the combinations of post-synthetic modifications involving acid (H4EDTA, Na2H2EDTA, citric acid, and HCl) and base solutions (NaOH and TPAOH) were further applied to facilitate the design of a diverse array of hierarchical Y and USY zeolites (FAU topology), irrespective of the Si/Al ratio (Fig. 5).193 By selecting different starting zeolites – such as pristine (Y, Si/Al = 2.4), steamed (USY, Si/Al = 2.6), and steamed and dealuminated (USY, Si/Al = 15 and 30) – and applying a wide range of dealuminating and desilicating treatments and their combinations, the relationship between the fragile FAU framework and its high Al content was better understood. The zeolites treated with acids and a base resulted in samples with mesoporous surfaces of up to 500 m2 g−1 and maintained intrinsic zeolite characteristics. The alkaline treatment of zeolites with high aluminium content (Si/Al = 4–6) resulted in a significant accumulation of extraframework Al-rich residue, which could be eliminated with a subsequent mild acid wash. The zeolites with Si/Al > 4 required mild dealumination for effective subsequent desilication. On the other hand, extensively steamed and dealuminated Si-rich USY zeolites (Si/Al = 15 and 30) exhibited remarkable sensitivity to alkaline solutions, which resulted in rapid dissolution and significant amorphization.194 In the case of ultrastable Y zeolites, including TPA+ or TBA+ in the alkaline solution preserved the zeolite structures during mesoporosity introduction via desilication, maintaining crystallinity and micropore volume. The advantage of hierarchical Y and USY was verified by the gravimetric adsorption of toluene, the liquid-phase alkylation of benzyl alcohol with toluene, and the pyrolysis of polyethylene. To alleviate diffusion constraints and enhance accessibility of active sites, and to improve selectivity for desired products, Manrique et al.195 synthesised Y zeolites with the 100–200 nm size range with a low Si/Al (∼2.4). Then, the impact of sequential desilication–dealumination (NaOH treatment and steaming) and the reverse process of sequential dealumination–desilication was examined.195 The process of desilication followed by dealumination resulted in structurally more stable zeolites, featuring a more ordered and controlled dealumination. This approach helped retain the number of Brønsted acid sites. Additionally, it was observed that the structural aluminium was protected during the initial desilication step before the dealumination. This method effectively generated both intraparticle and interparticle mesoporosity concurrently. Such post-synthesis techniques facilitate the adjustment of porosity and acidity in microporous Y zeolites, resulting in hierarchical zeolites with the preservation of Brønsted acid sites.
A highly crystalline hollow Y zeolite was obtained through post-synthesis modification of common NaY crystals.196 The applied top-down transformation included unusual dealumination of the zeolite framework using SiCl4, followed by acid leaching and, ultimately, selective dissolution of the crystal core with present protective aluminium species. The development of interior cavities depends on the degree of dealumination of the zeolite and the effectiveness of the extraframework Al species removal prior to realumination. Hollow Y zeolite crystals exhibited relatively uniform internal cavities of 0.8 μm in diameter while maintaining exterior dimensions and morphology comparable to the original NaY crystals. The obtained hierarchical Y zeolites were characterised by significantly elevated Si/Al ratios, enhanced crystallinities, and unchanged microporous shells of 0.12 μm in thickness. In conclusion, the family of hollow faujasites has been expanded considerably to include more siliceous and less defective materials. Their development is likely attributed to an Al-rich shell produced by NaAlO2 species during the last step of the process, which shields the outside regions of the grains from desilication.
2 Acidity of desilicated and dealuminated zeolites
Demetallation is important for creating mesoporosity, but it also influences the acid sites and their properties in hierarchical zeolites. It causes significant disordering of the crystalline structure, compromising symmetry and generating defect sites. Hierarchical zeolites that integrate interconnected micro-, meso-, and macro-porosities consistently exhibit defects to varying degrees.197 Isomorphous substitution of Al3+ for Si4+ in the zeolite framework generates a negative charge that necessitates compensation by an extraframework positive charge. When the compensating entity is a proton, Brønsted acid sites, referred to as bridge hydroxyls and represented by Si(OH)Al, are obtained. During demetallation with bases, silicon and aluminium atoms are removed from the framework sites. However, higher solubility of silicates relative to aluminate species results in considerably greater desilication and significantly limited dealumination. This extraction of Al and Si atoms from the zeolite structure significantly alters the type and number of acidic sites and their accessibility. The resulting, substantially improved, accessibility of acid sites and markedly less impeded diffusion of bulky molecules within the mesopore channels can contribute to improved catalytic activity. In numerous cases, the sole advantage observed was improved textural features without catalytic benefits.198 Steaming-induced dealumination affected the crystal structure, leading to its degradation and disintegration. This resulted from the transformation of framework aluminium into extraframework species. Consequently, the Brønsted acid sites attributed to Si(OH)Al groups were converted into Lewis acid sites, leading to significant decrease in protonic acidity.54 The proximity of positively charged extraframework aluminium species to the Si(OH)Al groups markedly enhances the acid strength of the Si(OH)Al hydroxyls. It is crucial to highlight that modification of acidic properties governed by dealumination is generally unpredictable and challenging to control.The dealumination mechanism was suggested to explain the reintegration of Al onto the outer zeolite surface during demetallation to prevent additional surface degradation. The aluminium removed from the framework was primarily retained on the zeolite, leading to Lewis acidity; an extra acid treatment was necessary to remove this extraframework aluminium, which frequently hindered reactant diffusion in zeolites with unidimensional channels, due to obstruction in the natural microporosity. The degree of silicon extraction was shown to rely upon the spatial distribution of aluminium on the surface or within the interior of the zeolite crystals.199,200 Higher Al concentration has been recognised to hinder OH− ions attack on the surface; consequently, aluminium-enriched surfaces demonstrated diminished susceptibility to silicon extraction.120,201,202 Desilication of zeolite frameworks is considered a silicon-selective process; however, it is widely acknowledged that some extraction of aluminium species also occurs. The elimination of Al, its role during desilication, its function as a pore-directing agent (PDA), and the reintroduction of Al are the subject of extensive studies.115,147,203 Reintroduction of Al atoms may lead to the formation of novel acid sites. Groen et al.115 thoroughly investigated the mechanism of zeolite desilication. The changes noted post-desilication led to the conclusion that removed aluminium species were reintegrated, predominantly on the external surfaces. Desilication with a low concentration of NaOH201 influenced the Si/Al ratio on both the external surfaces (Si/Alsurf) and in the bulk of the zeolite (Si/Albulk); yet, the Si/Alsurf/Si/Albulk ratio remained unchanged. Two factors can substantiate this phenomenon: (i) uniform extraction of silicon atoms from the surface and bulk of the zeolite grains and (ii) realumination. Ultimately, integration of Al atoms derived from the zeolite into the mesopore walls has been proposed.201,204 Differentiation between intact (originally located in the framework) and altered (re-aluminated) aluminium in a complex hierarchical porous system was validated by 27Al MAS NMR studies conducted by Verboekend and Pérez-Ramírez.203 The authors showed the formation of both Brønsted and Lewis acid sites in the silicalite-1 framework due to the partial integration of Al species, previously introduced into the desilicating mixture. The FT-IR studies of pyridine sorption demonstrated the presence of minor amounts of both Lewis and Brønsted acid sites, the latter correlating with the presence of aluminium atoms in tetrahedral coordination. The freshly produced Brønsted acid sites do not originate from the framework aluminium, i.e. the Si(OH)Al structural hydroxyls, as their IR spectra lack the distinctive band. The formation of new acid sites in silicalite-1 via the reinsertion of aluminium from an external source was convincingly confirmed.
The concept of ‘defects’ is essential to the unique properties of zeolites in acid-catalysed reactions, as these sites and the reactants are confined within a precisely specified molecular-scale environment.197 Silanol groups (Si–OH) are regarded as defects within the micropore architecture or on the exterior surface of zeolites. The external silanols arise due to the termination of the zeolite crystal growth. In contrast, the internal silanols originate from unbalanced charges inside the zeolitic structure. Internal silanol defect sites in zeolites can occur as hydrogen-bonded pairs. These sites are moderately acidic hydroxyls, which are less acidic than Al-associated bridging Si(OH)Al hydroxyls, but considerably more acidic than the external silanols. Geminal, vicinal, and bridged silanols, along with clusters of silanols, referred to as ‘silanol nests’, are identified as defect sites resulting from the absence of one or more T–O–T bonds. Understanding defects in zeolitic structures and exploring alternate passivation approaches to managing defect sites is crucial. In situ FT-IR analyses addressed numerous questions on the different kinds of silanols involved in the desilication process. It was observed that the number of silanol nests diminished in the parent MFI after desilication, whereas the quantity of isolated silanols increased.205 This suggests that desilication was initiated at the crystal boundaries and/or silanol sites, leading to the development of mesoporosity where isolated silanols were essential for terminating the crystals. Alternative possibilities suggest that the lack of silanol nests may correlate with the healing effect due to the presence of aluminium atoms. A significant silanols population growth in ZSM-5 was concluded from the increased intensity of the representative IR band, which was directly related with the expansion of the mesopore system. This was confirmed by the linear dependence between the area of the Si(OH) bands and the mesopore surface area.206
Acid leaching of zeolites represents selective demetallation because aluminium is preferentially extracted from the framework and predominantly eliminated, increasing the overall and framework Si/Al ratios.93,94 During dealumination of zeolites with acids, the generated silanol nests enabled subsequent incorporation of transition metal cations into the framework locations, as evidenced by 29Si MAS NMR, FT-IR, and UV-vis analyses.207 An increase in the number of external silanols was seen as a function of steaming time. Upon steaming, the structure was reconfigured to heal these defects effectively.208 Additional acidity produced by silanols is frequently regarded as a contributing factor to coke production and catalyst deactivation. In situ FT-IR analysis established a link, indicating that coke generation during xylene isomerization decreased bands associated with silanol groups.209 The existence of internal silanols in the zeolite modified by caustic treatment was identified as the cause of rapid catalyst degradation. UV-Raman examination indicated that the deactivation of the zeolite synthesised in a basic solution was associated with the coke formation within the internal structural defects. The deposition of coke in the interior microporosity obstructed reagent diffusion, hence diminishing catalytic effectiveness.210 The existence of mesopores, which enhanced the diffusion of coke precursor molecules, decreased coke deposition within the ZSM-5 structure during n-heptane aromatization.211 The formation of mesopores and the healing of silanol defects were both efficient in diminishing the catalyst deactivation rate. Despite the significant influence of mesopores, annealing of silanols was far more beneficial in reducing coke formation. These findings confirmed that the inner silanols also produced coke precursors.
In desilicated zeolites with an extensive mesopore system, smaller micropore fragments are retained between mesopores, reducing the average micropore length. It enables the transport of large molecules, making the active sites within micropores accessible.176,212–214 The accessibility of Lewis acid sites in desilicated zeolites surpasses that of Brønsted sites. Considering that in desilicated zeolites most Lewis acid sites arise from the dehydroxylation of Si(OH)Al groups, which are generated by the reinsertion of aluminium previously removed from the zeolite during alkaline treatment, the high accessibility of LAS seems reasonable. The newly created Lewis sites are located on the surface of mesopores, enhancing accessibility for bulky compounds.
3 Formation of micro/mesoporous hybrids by recrystallisation
Composite micro/mesoporous materials integrate the advantages of zeolites, which offer high acid site concentrations and catalytic activity, with those of mesoporous materials, which reduce diffusion constraints and facilitate high reaction rates, at least in theory. Typically, the mesoporous materials have amorphous walls prompting attempts at the synthesis of mesoporous materials exhibiting SBA or mesoporous MCM-41-type pore ordering but with crystalline walls. This has proven extremely challenging, usually resulting in the formation of separate phases,215 despite being approached in many ways.216 Zeolitization of porous materials or the production of mesoporous phases in zeolites may be classified as a top-down process only if the secondary porosity is created at the expense of the parent material, with an option of providing only small amount of external silicon or aluminium.142,216 The in situ dual-templating processes are considered beyond the scope of this review, as they represent a modification of the classic bottom-up methodology. Fig. 7 provides a mind-map, representing possible methodologies leading to the formation of micro/mesoporous composites, which are discussed in this chapter. | ||
| Fig. 7 Schematic representation of possible top-down methodologies leading to the formation of micro/mesoporous composites. | ||
3.1 Production of zeolitic phases through the consumption of mesoporous materials
Crystallisation of the amorphous walls of mesoporous materials seems to be the most direct technique for obtaining materials that combine the best features of the microporous and mesoporous solids. However, in this case, it is extremely hard to form material with interconnectivity between zeolitic micropores and primary mesopores, since zeolite crystals obtained from mesoporous solids often formed separate phases.217 The main advantage of direct zeolitization is that it circumvents the issue of direct incorporation of aluminium into the mesoporous materials to create stable active BAS. Disregarding the problem of micropore and mesopore connectivity, the criterion of success is catalytic activity and durability exceeding those of each component separately. Additionally, the composites should possess the capacity to facilitate reactions that are unattainable by microporous zeolites alone, which have restricted access to their active sites. | ||
| Fig. 8 Scheme of direct zeolitization of the mesoporous material (exemplified by MCM-41 ordered mesoporous silica) with the amorphous walls. | ||
A wormhole-type mesoporous material with Si/Al = 100 was synthesised in the presence of the triblock polymer (EO20PO70EO20, Pluronic P-123) and then impregnated with TPAOH, which acted as the OSDA for the transformation of amorphous walls into ZSM-5, reaching 42% crystallinity after 5 days of crystallisation at 130 °C.221 The micropore volume increased from 0.020 to 0.158 cm3 g−1, with a negligible change in the micropore diameter (about 5.7 Å). Both the wide-angle XRD and the FT-IR analyses confirmed the presence of the ZSM-5 crystals.
Analogous synthesis was proposed by Campos et al.222,223 who impregnated SBA-15 with a solution of TPAOH and sodium aluminate and further modified this procedure by adding glycerol in water. In all preparations separate particles of SBA-15 and ZSM-5 were obtained; therefore, meso- and micropores were not connected. However, generation of the zeolite phase led to increased catalytic activity in the isomerization of m-xylene (up to 7% conversion), dependent on the content of crystalline ZSM-5. Additionally, alternative combinations were employed: SBA-15 treatment at 180 °C with fluoride anions and water resulted in the formation of large MFI crystals, whereas in the absence of water, nanocrystalline MFI was produced.224 As in the earlier studies, zeolite and mesoporous materials constituted distinct phases. To ensure higher degree of zeolitization and to preserve the structure of the mesoporous precursor, microwave heating was used to decrease the crystallisation time.225 The composites with a discrete zeolitic phase were formed, with a surface area of 300–350 m2 g−1, total pore volume 0.60–0.70 cm3 g−1, but quite negligible micropore content of 6–7%. The incorporation of zeolite in the SBA-15 mesopores network was demonstrated using 129Xe NMR showing the presence of micropores, but without proving the connectivity between both pore systems. The concentration of acid sites was the sum of ZSM-5 and Al-SBA-15, as a result of the formation of zeolite crystals on the mesopores surfaces.
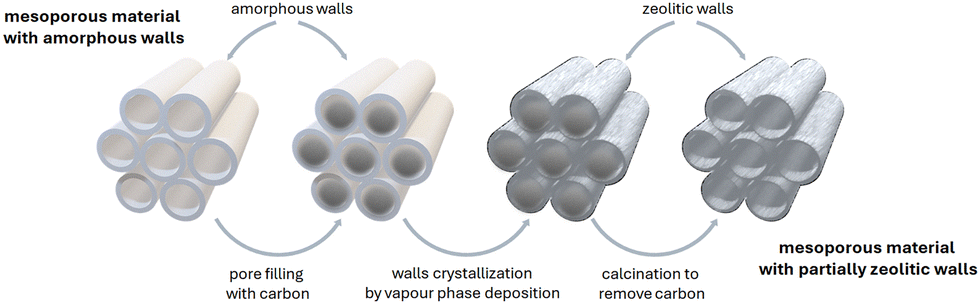 | ||
| Fig. 9 Graphic representation of the MCM-41 mesopore recrystallisation via vapour phase deposition with mesoporosity protection by filling the pores with carbon. | ||
Prokesova et al.227,228 studied a series of micro/mesoporous composites synthesized by mixing zeolite *BEA seeds with the precursor solution of MCM-48 or MCM-41. Both types of composites contained micropores, mesopores, and a high interparticle porosity so the total mesopore volume was higher than that for pure mesoporous components. Longer crystallisation times resulted in increased zeolite concentration but decreased micropore volume. For MCM-48, the size of mesopores was nearly independent of crystallisation time.
The formation of a steam-stable AlMCM-41 was achieved by surfactant assisted assembly of FAU zeolite seeds upon lowering the pH of the seeds solution to ca. 9 and introduction of HDTMABr (hexadecyltrimethylammonium bromide, also called cetyltrimethylammonium bromide) and sulphuric acid after the initial ageing period.229 The important part was subsequent heating with ammonia solution, ensuring stability of the structure. The resultant Al-MSU-S with a Si/Al = 9 was active in cumene cracking and had a large BET area of 713 m2 g−1 and a total pore volume of 0.56 cm3 g−1. After steaming at 800 °C, the BET area and pore volume decreased to 652 m2 g−1 and 0.42 cm3 g−1, respectively. This approach was then continued by the same authors with the use of zeolite ZSM-5 (MFI) and beta (*BEA) seeds.230 In both cases, the final mesostructured materials had MCM-41 XRD characteristics. The obtained high hydrothermal stability (upon steaming at 600 and 800 °C) was attributed to the presence of zeolite-like subunits, comprised of AlO4 and SiO4 tetrahedra in the mesoporous walls. In all three cases, the evidence of the presence of zeolitic fragments was provided by the 27Al NMR and IR spectroscopy. The porosity and acidity were not examined in detail. On and Kaliaguine prepared zeolite-coated aluminosilicate MSU foams, using similar approach with preformed MSU as a carrier.231 Clear gels, containing zeolite seeds, FAU or MFI, were used as zeolites precursors. In the resultant material the authors observed slight deformation of the spherical MSU cells and decrease of their pore sizes from 31.5 nm to 17.5 and 15.5 nm, for the MFI-coated and FAU-coated foam, respectively. The samples showed 129Xe NMR signal at ∼174 ppm proving microporosity, while qualitative pyridine adsorption revealed enhanced acidity (increased intensity of the 1545 cm−1 IR band, indicative of the pyridinium PyH+ ion).
Zhang et al.232 prepared a micro/mesoporous material denoted MAS-5 with ordered hexagonal structure. The authors used preformed aluminosilicate precursors and HDTMABr surfactant as the template. The resultant MAS-5 had BET area of 1170 m2 g−1, total pore volume of 1.17 cm3 g−1, mesopores with the diameter of 2.7 nm, and micropores with the diameter 0.58 nm. The acidity, measured by ammonia TPD was comparable to Beta zeolite (only qualitative comparison was given).
In all presented approaches, the formation of mesoporous matrix was evident and the zeolitic phase was ‘scattered’ over its mesopore surface. Zeolite crystals were XRD-invisible; thus, it was assumed that only ultra-small crystals were present. Due to low zeolite content, the acidity was low or moderate and the connectivity between zeolitic micropores and the pores of mesoporous material (MCM-41, SBA-15 or MSU) was limited. The main advantage was increased (hydro)thermal stability of the resultant mesoporous materials.
Zhou et al.233 introduced a variation of the above-mentioned process, by recrystallising preformed micro-mesoporous MFI/binder monoliths using controlled steaming in the presence n-butyl amine vapour as the OSDA. As a result, the binder was transformed into pure zeolitic phase, yielding monoliths with high mechanical strength. During shaping of the monoliths, the mesoporous binder plugged some of the micropores. The amine treatment resulted in total binder conversion and the introduction of intracrystalline mesopores, opening the microporous system. Catalytic performance of the shaped zeolites was evaluated using methanol-to-hydrocarbons and olefin catalytic cracking processes. The hierarchical zeolitic monolith outperformed commercial zeolite catalysts in terms of activity; the reaction rate for both reactions increased by ca. 40% and the cycle time increased from 780 to 2030 h during the MTH process. Deactivation was also delayed due to reduced coke formation, and the yield of the target product, propylene, was improved.
3.2 Co-development of micro and mesoporous phases
An interesting material, build of nanosheets of silicalite-1 with the MFI structure and bimodal sub-nano- and mesoscale porosity was proposed by Messinger et al.234 together with with the original mechanism of non-topotactic co-development of crystalline and mesoscopic phases. The material was synthesised using the C22-6-6Br2 bifunctional surfactant designed by Ryoo et al.235 to produce >3.5 nm thick nanosheets of the MFI zeolite. It has been demonstrated that hydrothermal synthesis of siliceous zeolite MFI in the presence of these structure-directing di-quaternary ammonium surfactant species underwent atomic and mesoscopic structural changes in a specific order. The process started with mesostructured amorphous silica and progressed via an intermediate nanolayered silicate phase (hexagonal-to-lamellar mesophase transition) to the final crystalline zeolite MFI nanosheets (Fig. 10). The 2D 29Si correlation NMR spectra confirmed the transition, showing interactions between pairs of dipole–dipole-coupled 29Si framework sites during this phase transformation, i.e. for signals corresponding to the transforming nanolayered silicates and the developing Q4 29Si signals from the crystalline MFI.236 In the XRD patterns the scattering intensities were attributed to zeolite MFI frameworks with lamellar mesophase ordering and d-spacing of 6.3 nm. The porosity or catalytic data are not available. The proposed above mechanism of zeolite crystallisation in which both structures, the mesoporous and microporous, co-exist in sub-nanometre-scale proximities was later questioned by Goesten et al.,237 who noted that for zeolites, the majority, if not all, such transitions are realised via dissolution and recrystallisation and pointed out apparent inconsistencies in NMR and TEM analysis, which in their opinion were contradictory. In conclusion, they stated that layered phases ‘work as an artefact rather than a species possibly playing a significant role in MFI nanosheet formation’. This critique was not unanswered; the authors of the original work defended and strengthened their conclusions, noting that the state-of-the-art characterisation techniques allow the observation of phenomena that previously went unnoticed due to lower spatial or temporal resolutions.238 They specifically mentioned the ‘high-spectral resolution and unprecedented sensitivity provided by newly available DNP-enhanced 2D J-mediated 29Si–29Si NMR’, which was employed in both the original and the follow-up papers. | ||
| Fig. 10 TEM images of the products obtained after 1 day (a) and 8.5 days (b) during hydrothermal synthesis of mesostructured zeolite MFI nanosheets in the presence of structure-directing di-quaternary ammonium surfactant, C22-6-6Br2. (c) Solid-state 2D dipolar-mediated 29Si{29Si} correlation NMR spectrum of the intermediate product of crystallising zeolite MFI nanosheets after 10 days of hydrothermal synthesis, (d) TEM image illustrating a zone where intermediate nanolayered silicates are converting into zeolite MFI nanosheets.234 Reproduced from ref. 234 with permission Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2015. | ||
Despite extensive efforts to facilitate the direct transformation of mesoporous materials into microporous counterparts, the characteristics of the meso/microporous hybrids remain somewhat unsatisfactory; however, this experience has significantly enhanced our understanding of specific complications impeding this transformation.
Ultimately, it is important to acknowledge that the materials that were intended to be produced via top-down approaches were instead synthesised bottom-up using a bolaform structure-directing agent, with a central biphenyl group connected by C10 alkyl chains to quinuclidinium end groups (BCPh10Qui-1,1′-(([1,1′-biphenyl]-4,4′-diylbis(oxy))bis(decane-10,1-diyl))bis(quinuclidin-1-ium)bromide).239 The obtained product was identified as single-walled zeolite nanotubes with a mesoporous core channel of 3 nm in width and zeolitic walls composed of linked fragments that resemble the *BEA and MFI frameworks (Fig. 11). The inner wall surface contains 10-MR micropores akin to zeolite MFI, whereas the outside wall surface features 12-MR micropores resembling zeolite beta. The material is acidic, with acid sites concentration 479 μmol g−1 measured by NH3-TPD and 151 μmol g−1 by pyridine adsorption. These single-walled zeolitic nanotubes were recently used as supports for poly(ethylenimine) and applied as sorbents for CO2 sorption from simulated flue gas or air.240
 | ||
| Fig. 11 ADF-STEM imaging of an individual (A) and fused (B) single-walled zeolitic nanotubes perpendicular (A) and (B) and along (C) and (D) the channel axis.239 Reproduced from ref. 239 with permission from the American Association for the Advancement of Science, copyright 2022. | ||
The bottom-up strategy for the formation of mesoporous zeolite utilising a phosphonium-based (Tri-Cy-dC8) bolaform template has recently progressed, resulting in the synthesis of zeolite ZMQ-1 (Zeolitic Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, no. 1). This zeolite features an intersecting intrinsic meso-microporous channel system characterised by a 28 × 10 × 10-MR framework, with the 28-MR exhibiting a free diameter of 2.276 × 1.183 nm.241 The results from Ar adsorption show record-high BET area of 1447 m2 g−1, and total pore volume of 0.47 cm3 g−1. The material was tested in catalytic cracking of VGO and showed conversion rate comparable to USY and beta zeolites. These initial materials exhibit considerable promise, and the field merits further examination.
3.3 Production of mesoporous phases through partial consumption of a zeolitic phase
The formation of mesoporous phases may be realised via the partial dissolution of a zeolite when zeolite fragments are extracted and consequently reassembled into a mesoporous phase. Sachse et al.242 defined two terms describing the procedures used: ‘zeolite recrystallisation’ and ‘zeolite surfactant-templating’. The former involves subjecting a zeolite to extremely basic conditions before adding the surfactant, the latter – treating it with a milder basic solution that already contains the surfactant. Again, the process can be considered top-down only if zeolite is formed at the expense of the mesoporous phase, so no external source of Al or Si is added. The degree of zeolite dissolution determines whether the mesoporous phase covers the zeolite surface, forms composites or completely absorbs the residual zeolite crystals.The same method was applied to FAU of lower Si/Al = 2.55 (CBV300, Zeolyst), which had been pretreated with citric acid to increase the Si/Al ratio prior to base treatment. The authors proposed that controlled, regular mesoporosity occurs via ‘surfactant-assisted crystal rearrangement’. In the proposed mechanism, mesopores are formed around the micelles, akin to the formation of surfactant-templated mesoporous silicas, but in the confined space of zeolite crystals (Fig. 12). It is worth mentioning that no separate mesoporous phase was detected and that a new mesopore system replaced irregular mesopores present in the parent material. FCC catalysts built from mesostructured zeolite Y showed significantly higher selectivity to gasoline and LCO and much lower to gaseous or uncracked products, together with suppressed coke formation.
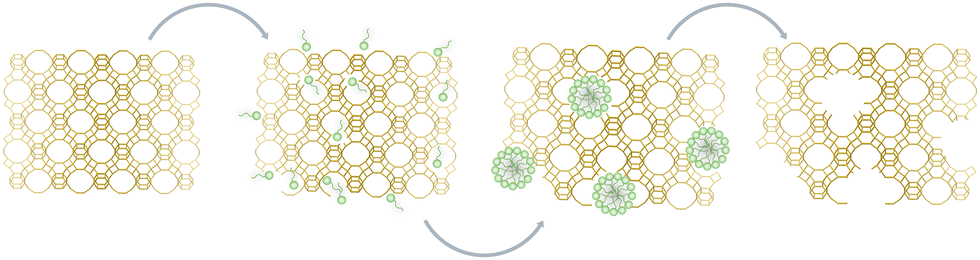 | ||
| Fig. 12 Illustration of the zeolite surfactant-templating process. Green shapes represent surfactant molecules. | ||
Kasyanov et al.246 presented a more complex mechanism (Fig. 13) of the zeolite surfactant-templating process, examining the stepwise formation of mesopores.
 | ||
| Fig. 13 Schematic depiction of the key stages of the zeolite recrystallisation process. Blue spheres represent sodium cations, green shapes represent surfactant molecules. | ||
The mechanism was explained as follows. (1) The addition of a base (desilication) breaks the Si–O–Si bonds, first on the crystal surfaces and later inside the crystallites. Simultaneously, zeolite protons from Si–OH–Al and Si–OH groups are ion-exchanged for sodium cations. (2) The addition of a surfactant removes water from the pores and replaces it with bulkier surfactant molecules; sodium cations are replaced by surfactant cations, which are attracted to negatively charged defect sites. This ion exchange was reported to be the driving force for the formation of micelles, resulting in the nucleation of the mesoporous phase inside the zeolitic mesopores and on the crystallites exterior surfaces. (3) Hydrothermal treatment causes condensation of the siliceous species (removed from the zeolite) around preformed micelles, resulting in the formation of a mesoporous phase: inter- and intracrystalline pores are enveloped by disordered mesoporous phase. (4) The ordered mesoporous phase is formed after the reaction mixture is acidified to pH of around 8. The authors emphasise that an ordered mesoporous phase can only be formed with a high degree of zeolite dissolution, i.e. when a large amount of siliceous species is available to form a mesoporous phase. The zeolite surfactant-templating has been expanded and refined using diverse surfactants,229,247,248 incorporation of porogens,249,250 or co-solvents,251 but the overall process illustrated in Fig. 13 remained largely unchanged.
Overall, the self-assembly process is hindered by relatively weak interactions between the zeolite surface and surfactants, as well as the size disparity between zeolite domains and the micelles, particularly when micelle self-organisation is to occur within zeolite crystals.
Lv et al.252 using this approach synthesized ZSM-5/SBA-15 composites with varying morphologies – from thin platelets to elongated rods. The former morphology, upon modification with Co and Mo, was the most suitable for hydro-upgrading of FCC gasoline. Despite morphology differences, the pore diameters and surface areas were similar, 7.7–9.6 nm and 648 to 660 m2 g−1. The acid site concentrations and Co and Mo amounts were similar, which pointed to the importance of morphology of the mesoporous support.
Over the years, different zeolites were introduced into TUD-1 material, a hydrothermally stable foam-like amorphous mesoporous silica, reported by Jansen et al. in 2001.257 TUD-1 contains well-defined pores that can be adjusted in a wide range from 2.5 to 25 nm, surface area of around 1000 m2 g−1, and three-dimensional pore connectivity. As the first example, well-defined, pre-formed *BEA nanocrystals were ‘blended’ with the TUD-1 synthesis mixture,178 followed by MFI (ZSM-5) crystals,178 ITQ-2258 and FAU (Y zeolite).259 In all of these samples, zeolites and TUD-1 were separate phases, with zeolite concentrations ranging widely, however at higher zeolite content, the zeolite formed agglomerates, which reduced the activity of the catalyst.
Commercial zeolites were also used to form composites. *BEA (CP811E, Zeolyst) and MOR (CBV90A, Zeolyst) were added to Al-SBA-15 crystallisation gel and then impregnated with Pt nanoparticles. Their catalytic properties were compared to the respective physical mixtures.260 The corresponding XRD patterns confirmed the presence of both zeolitic and mesoporous phases in the composites. The addition of zeolites to the synthesis gel resulted in the formation of secondary porosity not only as uniform mesoporosity inside the newly formed SBA-15 phase but also within the zeolite crystals, as seen in Fig. 14. The authors calculated the contribution of secondary mesopores generated in the presence of *BEA and MOR. The predicted mesoporous surface for the SBA-15 in the composites was 518 (*BEA) and 509 m2 g−1 (MOR). The experimental mesoporous surface calculated from N2 sorption was higher in both cases, at 613 and 584 m2 g−1, including both types of mesopores (interzeolite and secondary). Even after accounting for the presence of the intercrystalline mesopores (formed at zeolite-SBA-15 junctions), the rise in zeolite mesoporosity was noticeable.
 | ||
| Fig. 14 Microscopic images of Pt/SBA-15_BEA.260 (a) TEM image of BEA with visible mesopores (some are indicated by blue arrows for easy identification), (b) TEM image of AlSBA-15 phase. Reproduced from ref. 260 with permission from Elsevier B.V., copyright 2019. | ||
To evaluate benefits from the presence of both pore systems, the composites and physical mixtures were used as supports for metal nanoparticles (Pt in this case) for the reaction requiring both acidic and redox functions, such as olefin hydroisomerization. The authors assumed that including zeolite would improve thermal stability and acidity (both acid site concentration and acid strength) of the catalyst, while mesoporous SBA-15 would reduce the contact time between the catalyst and reagents, preventing coke formation. The test reaction, hydroisomerization of n-hexadecane, did not show that composites outperformed physical mixtures, most likely because the reaction was carried out on a catalyst mixed with a binder (which changed the porosity of the final catalyst) and because intracrystalline mesopores were used sparingly, being too small for such long hydrocarbon molecules. The composite catalysts performed significantly better than the pure Pt/SBA-15, with the Pt/SBA-15_BEA composite being the most active, affording high yield of isomers (together with the highest contribution of monobranched ones) and the lowest yield of the cracking products. However, the differences between physical mixtures and composites were not so significant. At 320 °C, the conversion of n-hexadecane was 96.6 vs. 96.4% (for composite and mixture of SBA-15/*BEA, respectively) and 90.9 vs. 92.2% (for composite and mixture of SBA-15/MOR, respectively).
ZSM-5/MCM-48 composites were synthesised using MFI seeds at various stages of crystallisation and assembled into a mesostructured MCM-48 type material.261 The synthesis process consisted of two steps: preparation of colloidal zeolite ZSM-5 seeds and their assembly into a mesoporous MCM-48 in the presence of CTAB at pH = 12. The authors produced a series of materials as a function of the ageing time of ZSM-5 seeds. These materials ranged from entirely XRD-amorphous MCM-48 (ageing time 2–3 h) to pure microporous ZSM-5 after 8 h of seed ageing. The materials obtained from zeolite seeds aged 2–3 hours had no microporosity. For materials matured for 8 hours, showing a considerable amount of the zeolitic phase, the micropore contribution was 46% (surface area) and 25% (pore volume). The authors claimed that progressive transformation of seed-containing MCM-48 into microporous ZSM-5 zeolite was responsible for the changes in the textural properties. It is worth noting that the concentration of acid sites (derived from the TPD of cyclohexylamine) decreased as the ageing time increased. The acid site concentration for the shortest ageing time was 0.47 mmol g−1. In comparison, the product with a 6-hour ageing period had the concentration of only 0.31 mmol g−1. This was explained by the fact that the solid products incorporated a greater amount of silica as the ageing process progressed.
The obtained low degree of zeolitization discussed in this section is typically linked to the low temperature (usually 100–150 °C) used to avoid surfactant degradation, essential for the mesophase formation.262 Vu et al.263,264 increased the zeolitization temperature to 200 °C and increased pH to 3.5 to obtain ZSM-5/SBA-15 composites, using ZSM-5 seeds and nanocrystals. Even after using zeolite nanocrystals, the BAS concentration was only 2–3 times higher than in the parent AlSBA-15 (30% of BAS concentration in ZSM-5). The composite was hydrothermally stable – its pore size and pore volume did not change after steaming at 800 °C (pore diameter 7.5 nm, pore volume 0.84 cm3 g−1) while SBA-15 lost ca. 60% of porosity. The catalytic activity was tested in gas-phase cracking of cumene and 1,3,5-tri-isopropylbenzene (TIPB). The best composite sample showed 86.3% of cumene conversion (compared to 13.2% for Al-SBA-15 and 94.4% for ZSM-5). This demonstrates that, despite much lower acidity of the composite, the catalytic activity was comparable to that of pure zeolite, highlighting the importance of zeolitic phase dispersion as well as the effectiveness of increasing the crystallisation temperature and using larger zeolite precursor crystals.
3.4 Reorganisation and recrystallisation of zeolitic domains
Section 3.3 discussed the formation of micro/mesoporous composites using mesopore-forming templates; this section presents methods of reorganisation/recrystallisation without the use of a secondary template and preparations in which the primary zeolite crystals/nanocrystals/seeds are modified before reorganisation to form mesoporous materials.One of the approaches relies on self-organisation of small zeolite crystals or seeds. The secondary mesoporosity in this case is formed between the nanocrystals. One of the early examples is mesosil, a material constructed using *BEA (beta) and MFI (silicalite-1) seeds.265,266 During synthesis, a clear gel, containing zeolite seeds was acidified and hydrothermally treated, which resulted in the formation of a large pore mesoporous material with intercrystalline mesoporosity controlled by the crystallisation time, temperature and pH. Despite large surface area of 600 m2 g−1 and pore volume of 1.35 cm3 g−1 for the best sample, the micropore volume was close to zero.
Micro/mesoporous composites were synthesised using silanization, which involves the attachment of organosilyl groups to the zeolite surface, resulting in conversion from silanol-terminated to alkylsiloxy-terminated surface. This impedes particle growth during crystallisation due to the presence of surface organosilanes, promoting the development of organic–inorganic composites. The secondary mesoporosity is formed upon calcination of the organosilanes, therefore the mesopore volume depends on the space occupied by the organic species. Serrano et al.267 identified three hierarchisation methods based on silanization: (1) crystallisation of silanized protozeolitic units, (2) crystallisation using amphiphilic organosilanes, and (3) crystallisation using silylated polymers.
In the first method, crystallisation of silanized protozeolitic units, the authors investigated the nature of such units prior to the silanization treatment. These units were X-ray amorphous but in their IR spectra a clear shoulder appeared in the region of the zeolitic framework vibrations, 500–600 cm−1, depending on the zeolite type, confirming short-range ordering.268,269 The exterior surfaces of XRD-amorphous protozeolitic units (*BEA and MFI), obtained during precrystallisation of the zeolite synthesis gel were functionalised with an organosilane (phenylaminopropyltrimethoxysilane). It inhibited further growth of the units during subsequent hydrothermal treatment which completed their crystallisation. Finally, calcination eliminated both the zeolite-forming OSDAs and organosilane. The samples were highly crystalline, with high micropore volume (0.117 cm3 g−1 for ZSM-5 and 0.258 cm3 g−1 for beta) and high external surface area (314 m2 g−1 for ZSM-5 and 120 m2 g−1 for beta). The authors noted that neighbouring nanocrystals exhibited the same crystalline orientations, suggesting significant degree of intergrowth. When the units were allowed to grow without the organosilane, the external surfaces were much smaller, 94 and 22 m2 g−1 for ZSM-5 and beta, respectively. Both samples exhibited superior conversion in the test reaction of polypropylene cracking (100% vs. 27.3% for ZSM-5 and 19.7% vs. 3.8% for beta), which was apparently influenced by the increased (large) external surface areas and secondary porosity in the zeolites derived from the silylated units. These enhancements reduce steric and diffusional barriers, especially for medium-pore ZSM-5.
In the second approach, crystallisation using amphiphilic organosilanes, the following compounds were tested: 3-aminopropyltrimethoxysilane (APTMS), isobutyltriethoxysilane (IBTES), phenylaminopropyltrimethoxysilane (PHAPTMS), and octadecyltrimethoxysilane (ODTMS).270 The attachment of the ODTMS to zeolite seeds was very limited (only 1.3%), and the resulting material had characteristics close to the reference nanocrystalline ZSM-5 with crystallite sizes 20–80 nm, SBET = 450 m2 g−1, Sext = 100 m2 g−1. Silanization by APTMS or IBTES resulted in ZSM-5 samples with higher surface areas, SBET around 500 m2 g−1 and Sextca. 200 m2 g−1, with wide 8 nm mesopores. The seed silanization by PHAPTMS significantly increased surface area to SBET = 573 m2 g−1 with much more developed outer surface (Sext = 225 m2 g−1) of nanounits (5–10 nm) within aggregates. The mesopores obtained after removal of PHAPTMS were in the range 2.0 to 3.0 nm.
The third technique, crystallisation with silylated polymers, employed silylated polypropylene oxide diamine polymers as the organosilane.271 The resultant material, hierarchical ZSM-5 had larger mesopores (4–20 nm) in higher proportion than the hierarchical ZSM-5 synthesised using smaller organosilanes, PHAPTMS, mentioned above. The catalytic performance (cracking of low-density polyethylene, LDPE) was comparable to the ZSM-5 synthesized with PHAPTMS, despite lower concentration and acid strength of the active sites. These results indicated that the increased accessibility of acid sites resulting from larger mesopores compensated for the reduced quantity and strength of its acid sites.
This methodology was recently extended to an amphiphilic organosilane (dimethyloctadecyl[3-(trimethoxysilyl)propyl]ammonium chloride, TPOAC) to functionalise zeolitic seeds, leading to a branched aggregative growth and formation of dendritic MFI (ZSM-5) crystals.272 The secondary porosity was formed by interconnected network of radially-oriented mesopores (3–10 nm) and large cavities (20–80 nm). The BET surface areas ranged from 476 to 630 m2 g−1, surpassing those of conventional ZSM-5, typically around 400 m2 g−1. The mesopore volume (0.390–0.809 cm3 g−1) depended on the crystallisation temperature and time. The total pore volume for the best sample was 0.945 cm3 g−1. Dendritic ZSM-5 samples had lower BAS concentration relative to the reference materials, and nano- and hierarchical ZSM-5 (0.12 mmol g−1vs. 0.21 and 0.18 mmol g−1, respectively), whereas all samples had comparable LAS concentration (0.10–0.12 mmol g−1). As a result, the dendritic ZSM-5 had significantly improved catalytic performance, such as enhanced capacity for VOCs (toluene) adsorption, increased activity in LDPE catalytic cracking, and superior activity and deactivation resistance in hydrogen production by methane decomposition.
In 2016, Valtchev et al.273 proposed novel type of zeolite preparations, resulting in the formation of so-called ‘embryonic zeolites’. The synthesis time was shortened, and the resulting structures were XRD-amorphous but due to more open micropores embryonic zeolites provided shorter and easier diffusion pathways. The synthesis used TEOS as the silica source producing clear gels of general composition 4.5(TPA)2O:yAl2O3:25SiO2:430H2O:100EtOH, where y = 0.0625 to 0.50. After completion of TEOS hydrolysis, carried out at ambient temperature, the mixtures were freeze-dried and calcined in air at 550 °C to obtain the H-form. Micropores of different sizes (1–5 nm) were present in all samples. Following the burning off of the OSDA (TPAOH), most of the aluminium stays tetrahedral, with only a little quantity of the octahedral species. In the following work,274 to synthesize zeolites with bigger micropores, a variety of tetraalkylammonium cations with sizes ranging from 6 to 12 Å were used, namely tetramethylammonium (TMA+), tetrapropylammonium (TPA+), trimethyladamantylammonium (TMAd+), tetrabutylammonium (TBA+), and tetrahexylammonium (THA+). The resultant materials were microporous with pore sizes of 1–2 nm, high specific surface areas (up to 1095 m2 g−1) and micropore volumes (up to 0.47 cm3 g−1), much higher than conventional zeolites made from the same templates. Despite relatively low number of active sites and their moderate acid strength, they had considerable activity in catalysis of bulky molecules, in this case, 1,3,5-triisopropylbenzene dealkylation.
Embryonic zeolites may be also supported, either by nanocasting of preformed zeolite embryos or in situ formation under hydrothermal conditions, to prepare composite catalysts. As an example, the embryos of ZSM-5 were deposited on silica-doped alumina matrix (Siralox 30). Both composites contained pores larger than 0.83 nm, including a fraction of pores with sizes 1.42 and 2.25 nm.
An interesting example was presented by Parsapur et al.,275 who built mesoporous materials with 2D-hexagonal and 3D-cubic mesopore systems using nanosized, microporous FAU-type fragments. The parent, commercially available FAU zeolite crystals were fragmented by desilication using in situ generated urea to obtain zeolitic fragments. Subsequently, due to their interaction with a surfactant (organosilane-dimethyloctadecyl-(3-trimethoxy-silyl-propyl)-ammonium chloride), they were reassembled to form ordered mesoporous structures by topotactic condensations (Fig. 15). The mechanism is somehow related to ADOR,276 because the parent material, cut into unit-cell size fragments, is subsequently reorganised around micelles and re-crystallised in the new form – not different zeolite, but as a mesostructured zeolite of the same framework topology. The BAS acidity of the parent zeolite was 0.28 mmol g−1, the new material contained 0.25 mmol g−1 BAS and 0.1 mmol g−1 LAS. The observed slight reduction in acidity the authors attributed to enhanced porosity, resulting in decreased material density. This material was tested in the low-pressure hydrocracking of the VGO-recycle stream and had enhanced naphtha and middle distillate yields by 10.5% and decreased internal coke deposition by 42–57% compared to the parent material (FAU, CBV 720) and is currently at the stage of pilot-scale catalyst formulation.
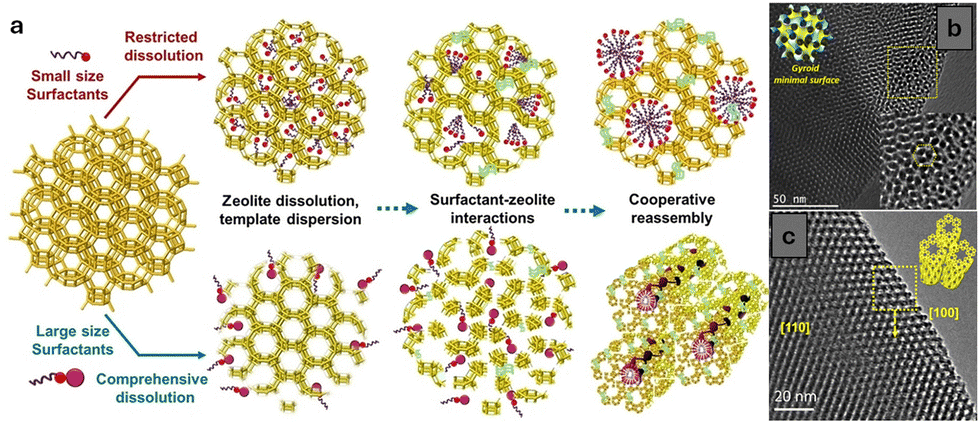 | ||
| Fig. 15 (a) Hierarchical ordering of zeolite fragments using small and large size surfactants, (b) and (c) SEM images of ZAK-1 and ZAK-2 materials,275 Reproduced from ref. 275 with permission from Wiley-VCH GmbH, copyright 2023. | ||
Another approach for reorganising zeolite crystals involves the production of thin films using pulsed laser ablation (see Section 6.5).
Devos et al.281 categorised three forms of IZC based on the origin of Si and Al during the transition. The ‘true IZC’ employs zeolitic precursors as their sole source, ‘partial IZC’ incorporates supplementary Si or Al sources alongside the parent zeolite, and ‘mixed IZC’ refers to zeolite formulations that integrate two or more zeolitic source materials. Chengeng et al.282 proposed the categorisation of IZCs depending whether they occur with or without using OSDA molecules. The interzeolite transformations from layered precursors are reviewed in Section 5.9.
The application of IZC started in the 1990s, when Zones investigated the use of zeolite P (GIS)283 as the aluminosilicate source to produce SSZ-13 (CHA). Later it was shown that P can be replaced by FAU zeolites,284,285 which also enhanced the synthesis rate of the daughter zeolites, SSZ-13 (CHA)283 and Nu-3 (LEV).286 The SSZ-13 could be also synthesised by IZC of beta (*BEA) and L (LTL) zeolites.287
Recently, Peng et al.288 modified this method by shortening the induction and extending the crystallisation period, by 40% and 100%, respectively, to obtain SSZ-13 zeolite crystals on hollow fibre substrates for application as the membrane for high-pressure CO2/CH4 separation. The authors used FAU-CHA seeds which were hydrothermally synthesized using commercial FAU zeolite and N,N,N-trimethyl-1-adamantammonium hydroxide (TMAdaOH) as the OSDA. After secondary growth exceeded the crucial time of 24 hours, the membrane thickness remained steady at around 3 μm. The intercrystalline pores were self-healed when synthesis time was extended. The obtained batch-synthesized membranes had CO2 permeance of 5.2 × 10−7 mol m−2 s−1 Pa−1 and CO2/CH4 selectivity of 143 ± 7 at 0.2 MPa and up to 126 at 6.1 MPa, both at 30 °C. These values exceeded industry requirements. The SSZ-13 zeolite can be also obtained using 1-(1-methylpropyl)-4-aza-1-azoniabicyclo[2.2.2]octane hydroxide, to obtain CHA with Si/Al ratio up to 7.2.289
Xu et al.290 modified IZC using internally confined organic structure directing agents and called it the ‘OSDA-confined’ strategy. Nanosized high-silica ZSM-5 zeolite was obtained in high yield using *BEA zeolite as the Si and Al source (73.8% solid yield after 3 h). The innovation of the approach lies in the mixing of the parent zeolite with the OSDA solution used for the synthesis of ZSM-5 (tetrapropylammonium hydroxide, TPAOH) before its dissolution in NaOH. A high concentration of organics inhibited zeolite degradation and facilitated the organisation of zeolite particles into MFI (ZSM-5).
In the reaction of benzyl alcohol etherification, the obtained ZSM-5 showed TOF seven times higher than a commercial MFI zeolite.
An example of IZC, allowing to expand Si/Al composition range and OSDA-free synthesis was provided by Lozinska et al.291 who synthesized high silica zeolite ZK-5 (KFI, Si/Al = 4.8) by interzeolite conversion from a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture of two ultrastable Y zeolites (CBV712, Si/Al = 6.2 and CBV720, Si/Al = 16.2), using alkali metal cations (K+ and Cs+) and nitrate anions considered the sole structure directing agents. A conversion mechanism was proposed, in which the transition is directed by both inorganic cations and inorganic anions. K+ (and Cs+) cations occupy all three available extraframework cation sites in 8-MR with high occupancy, therefore the charge-balancing nitrate anions are directed to the remaining space inside the pores. The maximum Si/Al was attained due to a balance between the K+ required to maintain the assembly of nitrate ions, compensating for the negative charges generated by Al substitution, and Cs+ ions, favouring ZK-5 crystallisation over erionite, (K+ + Cs+)/Al > 2.6. The templating effect was only observed for K+ and nitrate ions; when these ions were replaced by others, such as SO42− and PO43−, or Na+ for K+, ZK-5 did not form.
:50 mixture of two ultrastable Y zeolites (CBV712, Si/Al = 6.2 and CBV720, Si/Al = 16.2), using alkali metal cations (K+ and Cs+) and nitrate anions considered the sole structure directing agents. A conversion mechanism was proposed, in which the transition is directed by both inorganic cations and inorganic anions. K+ (and Cs+) cations occupy all three available extraframework cation sites in 8-MR with high occupancy, therefore the charge-balancing nitrate anions are directed to the remaining space inside the pores. The maximum Si/Al was attained due to a balance between the K+ required to maintain the assembly of nitrate ions, compensating for the negative charges generated by Al substitution, and Cs+ ions, favouring ZK-5 crystallisation over erionite, (K+ + Cs+)/Al > 2.6. The templating effect was only observed for K+ and nitrate ions; when these ions were replaced by others, such as SO42− and PO43−, or Na+ for K+, ZK-5 did not form.
An alternative approach for synthesizing novel zeolite frameworks from pre-existing structures involves a 3D–3D topotactic transformation.292 This process initiates with breaking of relatively weak aluminium-oxygen bonds. The authors specified that the neighbouring, 5-coordinated AlO4(OH) or 6-coordinated AlO4(OH)2 units are the most vulnerable. Such bond cleavage facilitates structural reorganisation at the atomic level, enabling the transformation from one zeolite topology to another while maintaining a coherent framework continuity.
Such bonds can be viewed as ‘built-in weakness’, by analogy with the Ge–O bonds in germanozeolites. They are common in AlPOs but can also appear as defects in zeolites. The mechanism was studied during transformation of AlPO4 PST-5 to PST-6 by changing their dcc chains to narsarsukite (nsc) chain. Upon calcination, the OH groups were removed from the units containing pentacoordinated Al atoms. That resulted in the breakage of the links connecting this unit with the neighbouring PO4 tetrahedron. Both AlO4 and PO4 inverted their orientations and re-established connections with the adjacent units. This led to a domino-like modification of the tetrahedral configuration. As a result, the 1D channel system of PST-5 was transformed to the 2D channel system in PST-6 (Fig. 16).
 | ||
| Fig. 16 Zeolite and AlPO frameworks (a1)–(a10) transferred into (b1)–(b10) frameworks by changing dcc to narsarsukite (nsc) chains.292 Magenta: dcc; cyan: nsc; yellow: double four-ring (d4r). Reproduced from ref. 292 under CC-BY-4.0 licence https://creativecommons.org/licenses/by/4.0/. | ||
The authors identified 12 framework types containing infinite dcc chains and generated a series of novel hypothetical nsc-containing zeolite structures (Fig. 16), demonstrating that the 3D–3D topotactic transition may be regarded as supplementary to current methodologies for synthesising new zeolite structures. IZC has also been carried out using thermally controlled grinding of commercial zeolite Y, which was transformed into five different zeolite phases with minimal added water,293 as discussed in Section 4.1. Mendoza-Castro et al.294 showed a modification of the IZC leading to the production of a series of micro/mesoporous composites. The interzeolite transformation intermediates (ITI), were obtained by interruption of the conversion of FAU into *BEA and addition of OSDA. The ITI intermediate showed increased active sites accessibility due to the development of mesoporosity. Strong acidity was caused by the presence of ultrasmall zeolite crystals. Three different approaches were considered.
In the first method, CBV720 zeolite (FAU) was hydrothermally processed in the presence of TEAOH. The second procedure involved treating an uncalcined surfactant-templated CBV720 with TEAOH. Finally, CBV720 was treated with TEAOH and HDTMABr. The products were denoted ITI1 through ITI3. A prolonged treatment, regardless of the system, resulted in the development of *BEA zeolite. The authors found that the presence of quaternary amines inhibits desilication while the presence of surfactant allows to obtain materials with narrow pore size distribution (average pore size ca. 4 nm). The ITIs materials were studied in two test reactions involving bulky products: Friedel–Crafts alkylation of indole with benzohydrol and Claisen-Schmidt condensation of benzaldehyde with hydroxyacetophenon. In both cases, the ITIs showed comparable TOF increase over the parent FAU, 6-fold and 3-fold, respectively.
IZC may be used as a method to synthesize hybrid zeolites, composed of building units from different frameworks, because at some point during IZC process, building units from both the parent and the final zeolite coexist, not necessarily in close proximity. García-Martínez et al.295 used the specific OSDA, cetyltripropylammonium bromide, to drive the interconversion of FAU into MFI. This molecule has both the OSDA function (tripropylammonium as a polar head) and the surfactant activity (hexadecyl/cetyl as a long aliphatic chain), which allowed to synthesize a series of micro-mesoporous hybrids. The combination of well-defined mesoporosity and the presence of zeolitic building blocks in their structure resulted in enhanced formation of desired products (diisopropylbenzenes, DiPBz) in catalytic cracking.
The primary challenge in studying interzeolite transition chemistry is identifying which, if any, parent zeolite fragments contribute to nucleation or crystal development, as they cannot be directly detected experimentally, due to low spatial resolution of contemporary experimental methods.
Structural resemblance between the initial zeolite and the resultant one was initially believed to be the necessary condition for interzeolite conversion.296 From that time three other significant hypotheses were proposed. The first one is thermodynamics-based, readily accounting for the transition from a less dense to denser zeolite phases but it fails in the opposite direction.297 The second hypothesis was based on the kinetics; the increased crystallisation rate was attributed to the presence of common secondary building units (SBU) both in the dissolving parent zeolite and the precursors of the newly formed zeolitic phase. Transformation of LEV into CHA (D6R – double six ring)298 or FAU into *BEA and MAZ (common chains of 4-MR) may be examples.299 The third hypothesis, called the ring building units (RBU) theory, was proposed for transformation between zeolites not having any common SBUs, such as FAU to MOR or HEU.300 The smallest common units, such as 4-MR and 6-MR, were recommended as the units facilitating the transition. The formation of 5-MR in the daughter zeolite, not present in the parent zeolite (FAU), was explained by the fact that zeolites dissolve incongruently, i.e. silica fragments are more readily dissolved than Al-containing ones. According to the authors, the 5-MR usually not contain any Al atoms, therefore they should form in Al-free environment at the beginning of dissolution. Schwalbe-Koda et al.301 determined that 65% of the analysed parent-daughter interconversions lack common units (including SBUs or RBUs) undermining this explanation for the undelaying mechanism which clearly shows that the conversion process requires more investigation. They used a graph theory to create a supercell-invariant metric allowing comparison between crystal structures. They showed that diffusionless, i.e. topotactic and reconstructive modifications occur exclusively between graph-similar pairs. Experimentally, Mlekodaj et al.302 showed that during IZC the parent zeolite is decomposed into linear or branching (Si–Al)n chains. Among them, a large fraction (about 45%) was relocated between parent-daughter structures in the form of noncyclic sequences including a Si–Al chain with a length of at least five T-atoms. The authors obtained zeolite SSZ-13 (CHA) from zeolites X (FAU), A (LTA), and ZSM-5 (MFI), having no mutual structural building units, such as hexagonal prisms (D6R) or 6-MR. Similar results were obtained by Liu et al.303 who investigated transformation of a series of dealuminated FAU into AEI zeolite under various conditions. They also concluded that the parent zeolite is partially dissolved into the liquid phase, containing small (alumino)silicate oligomers. There is a dynamic interaction with the residual crystals and when conditions favour the crystal growth of the daughter zeolite the parent zeolite material is gradually consumed.
Calculations using a coarse-grained reactive model were recently employed to examine the acceleration of a daughter zeolite synthesis at the expense of the parent zeolite.304 The author studied the system in which the parent zeolite, CHA, had neither common plane nor any crystal building units shared with the daughter zeolite, AFI. The simulations showed that interzeolite transformation can be explained by cross-nucleation of the daughter zeolite on the parent seed if both zeolites had domain matching on a crystal plane. These shared fragments allow formation of a stable interfacial transition layer, lowering the energy barrier for cross-nucleation. The cross-nucleation is defined as the nucleation of a new crystal structure on the surface of a pre-existing surface of an other crystal, usually a polymorph of the same substance.305,306 For that reason, zeolite pairs with intergrowths were proposed for cross-nucleation. These zeolites share a common crystal plane, so they do not need to form a separate interfacial layer to connect different zeolite types. This shared plane makes it easier to grow the daughter structure.
As mentioned above, the IZC process may produce different locations of Al atoms inside the daughter zeolite. Devos et al.281,307 studied internal distribution of Al and acid sites in ZSM-5 (MFI) and SSZ-13 (CHA) synthesized from FAU zeolites. They also proposed a ‘quasigeneric’ model for the IZC process where Al-dense environment promotes fast nucleation, thus zeolite frameworks which cannot accommodate high concentrations of Al (such as MFI) should be substantially slower in assembly. In the structures that are able to accommodate large number of Al (such as CHA), the ‘Al-dense’ regions are observed. Their presence may be explained by the preservation of the parent, Al-rich zeolite fragments during fast nucleation, not giving enough time for Al-redistribution (possible for much slower crystallisation for Al-poor structures). According to the authors, the distribution of Al in the final zeolite structure may also be influenced by the framework charge mobility, allowing rearrangement of cations and acid sites (thus T-atoms) during maturation stages.
3.5 Formation of hollow zeolites
The introduction of secondary mesoporosity may be achieved by forming hollow structures or building zeolite-on-zeolite assemblies.With the attention-catching statement ‘No core required! Why keep unused catalyst components?’ an enhanced effectiveness factor (see Section 9, the ratio of the reaction rate with pore diffusion to the reaction rate at the particle surface) from 0.63 to 0.97 of catalysts based on hollow zeolite crystals was shown for hollow NaY zeolite (FAU) containing Pt nanoparticles in the test reaction of cyclohexene hydrogenation.308 Hollow NaY was produced in three steps: NaY crystals (Si/Al = 2.5) were first dealuminated with silicon tetrachloride, then leached with hydrochloric acid, and finally desilicated (to final Si/Al = 33.8). For Pt@Y hollow, the appearance of a large H2-type hysteresis loop closing at p/p0 = 0.47 implied that the large internal cavity was accessible only through relatively smaller entrances, i.e. microporous shell built of NaY zeolite. The observed reaction rate increased from 8.1 (for Pt@NaY bulk) to 19.7 mmol kg−1 s−1 (for Pt@NaY hollow) and was higher than for commercially used catalyst Pt/Al2O3 (11.5 mmol kg−1 s−1).
Van Bokhoven et al.309 conducted systematic studies to determine the effect of parent MFI-type zeolite (ZSM-5 and silicalite-1) synthesis conditions and leaching conditions on hollow zeolite characteristics. If, during synthesis, part of the TPAOH was replaced by TPABr, the zeolite became less stable and could form hollow structures more easily. Increased water content in the synthesis gel resulted in larger hollow crystals following leaching. The authors found that the optimal conditions to obtain hollow ZSM-5 crystals with a large void and unbroken shell was leaching in a 0.2 M NaOH solution. At lower NaOH concentrations (0.1 M) the inner mesopores appeared, while higher NaOH concentrations (0.4 M) introduced mesopores into the crystal shells. For silicalte-1 both the inner and external parts of the crystals were damaged. Hollow crystals with double shells were also obtained by a two-step crystallisation process followed by base leaching.
Controlled desilication of Al-zoned MFI crystals is a fairly simple way to obtain ‘hollow’ zeolites.120 ZSM-5 zeolites with small and large crystals and Si/Al ratios of 25–50 were desilicated using NaOH, which led to the formation of ‘hollow’ particles with a well-preserved Al-rich exterior. The effect varied between small and large crystals due to differences in Al-zoning. The zeolite crystals did not contain regular voids; a broad range of mesopore diameters was characteristic of desilication, as mentioned in Section 1.6. This different behaviour of small and large ZSM-5 crystals was also presented by Fodor et al.310 The base leaching of small (100 nm) MFI crystals resulted in the formation of hollow interiors, whereas in the larger crystals (micrometre range), irregular mesopores were formed.
The same methodology, namely subsequent treatment with SiCl4, HCl and NaOH of the Al-zoned material was used to prepare hollow Y zeolite with a 200 nm shell and a hierarchy factor of 0.18, which is more than with HF for steamed USY zeolite (0.08).311
Similar but somehow ‘inverted’ materials were recently referred to as ‘core–shell-like zeolites’.312 They were prepared by diffusion-controlled chemical etching of commercially available FAU zeolite with a mixture of oxalic acid and ammonium fluoride. The resultant crystals contained a rough, mesoporous, Si-rich surface with about 100 nm thickness and a microporous interior, with a smooth transition between them, ensuring interconnectivity between microporous and mesoporous zones.
Following the same thread, HZSM-5 microboxes with regular cuboid shapes were produced by dealumination with 5 M HCl to remove non-framework aluminium followed by calcination in air to remove the TPAOH template, and finally desilication with 0.6 M Na2CO3. The crystalline shell of 15–25 nm thickness was preserved, with a total crystallinity loss of around 16%. The Si/Al ratio decreased from 72 to 49, whereas the acidity increase was not so pronounced, from 0.27 to 0.34 mmol g−1. Total pore volume increased from 0.30 to 1.55 cm3 g−1 (66% of which was macropore volume). Two test reactions, α-pinene isomerization in liquid phase and gas-phase cumene cracking, were used to evaluate the catalytic activity. Hollow crystals were more efficient than bulk ones. Still, these differences became less pronounced as the reaction temperature increased, particularly for cumene cracking (at 250 °C, the conversion was about 30% for bulk and 45% for hollow ZSM-5, and at 350 °C it was about 75% and 80%, respectively). The hollow catalysts performed far better in the liquid phase process, with a conversion difference of over 100% showing clear advantage in diffusion-controlled processes.
Liu et al.313 proposed the synthesis of hollow ZSM-48 zeolite crystals (*MRE) via alumination-recrystallisation method, using NaOH as the desilication agent and Al2(SO4)3 as the external aluminium source. This allowed adjustment of the Si/Al ratio between 130 and 45. The authors postulated a mechanism of synthesis based on the equilibrium between desilication, leading to the development of both mesopores and the hollow interior, and OSDA-aided healing of the outer shell. The presence of initially formed mesopores allowed Al species to diffuse into the internal part of crystals, where they were re-incorporated into the framework. The hollow ZSM-48 demonstrated good catalytic stability and high conversion (50% vs. 5% for standard ZSM-48) in m-xylene isomerization. Interestingly, the initially low selectivity towards p-xylene increased over time on stream, finally reaching approximately 65%. Despite excellent shape selectivity, the conversion of xylene is extremely low, owing to high diffusional limitations and high Si/Al ratio, both mitigated through the synthesis of hollow structures.
The preparation of hollow zeolite was also linked to the Ti for Al exchange.314 Silicalite-1 was employed as the parent zeolite, (NH4)2SO4 as the dual-function crystallisation-mediating agent, and tetrabutylorthotitanate as a titanium source. The resultant crystals had a regular shape, which the author called ‘nanobox’. The nanobox wall thickness was ca. 30 nm, with a Si/Ti ratio of around 40. The authors found that when the molar ratio of (NH4)2SO4/TEOS reached 0.25, the dissolution rate of silicalite matched the crystallisation rate of titanosilicate, resulting in a perfect hollow structure. The concentration of (NH4)2SO4 was critical in achieving equilibrium between the rate of dissolution and crystallisation. The catalytic activity of synthesized hollow titanosilicate was assessed using 1-hexene epoxidation. The conversion of 1-hexene was significantly higher than that of hollow TS-1 synthesized using the usual approach.
Pre-formed mesoporous materials of a certain shape may be used to produce hollow zeolites. The conventional approach for preparing hollow zeolites involves template-assisted synthesis;315 however, top-down methods, which use mesoporous materials as a feed during zeolite formation, are also developed. This technology offers the added advantage of enabling functionalisation of the developing zeolite by encapsulating the required species already within the mesoporous material.
Hollow and core/shell ZSM-5 spheres were produced via in situ transformation of mesoporous silica spheres (MSS) into MFI zeolite, utilizing isopropylamine as an OSDA.316 Isopropylamine initiated recrystallisation of MSS into small, highly crystalline ZSM-5 crystals, following the addition of Al source. The synthesized ZSM-5 spheres preserved the morphology of the original MSS, while displaying a hollow core/shell architecture. The authors named their synthesis methodology as ‘templating and surface to core’ crystallisation. Their methodology was used by Rutkowska et al.317 to synthesise spherical ZSM-5 for the dehydration of methanol to dimethyl ether. The spherical ZSM-5 sample was stable in the long-term catalytic test, the methanol conversion of about 90% remained unchanged for about 15 h.
The synthesis proposed by Dong et al.318 involved coating mesoporous silica (MS) spheres with PDDA (poly(diallyldimethyl-ammonium)) to form a layer of PDDA, allowing deposition of negatively charged silicalite-1 seeds. The dried seeded MS spheres were subsequently treated with a mixture of 3 ml of triethylamine, 0.1 ml of ethylenediamine, and 0.5 ml of H2O via vapour-phase transfer (VPT). The seeds on the surfaces of the spheres grew by assimilating the silica cores. The resultant material was predominantly zeolitic. Alternatively, silica spheres containing Fe2O3 were produced by wet impregnation and subsequently subjected to the aforementioned treatment to form hollow material with functionalised interior. The authors obtained well-intergrown zeolite crystals decorated with clusters of Fe2O3 nanoparticles approximately 13 nm in size, comparable to the pore size of the MS spheres. This represents simplification of the proposed earlier strategy,319 in which mesoporous silica spheres (diameter 1.3 μm) were initially modified by tri-layered polyelectrolyte films PDDA/PSS/PDDA, with PDDA and PSS denoting poly(sodium 4-styrene sulfonate), to formulate a positively charged outer surface on the spheres. Subsequently, electrostatic attraction deposited uniform, negatively charged silicalite-1 seeds (size ca. 80 nm) on the polyelectrolyte-modified MS spheres. The seeds subsequently crystallised, consuming silica from the spheres and TEOS from a diluted solution, which also contained TPAOH template. The functionalisation of the hollow zeolite interior with silver and palladium oxide nanoparticles or polymers was also previously reported by the same authors.320
Pt-incorporated microporous CIT-6 zincosilicate, which has the same framework structure as *BEA, was employed as a scaffold and partial feed to construct hollow beta zeolite.321 In this synthesis, CIT-6 accounts for just 10% of the overall silica content (Al and Si sources are added during synthesis). The presence of the template inside CIT-6 permits beta zeolite to crystallise on the zincosilicate scaffold before it is completely dissolved. Even though Pt nanoparticles were only present inside the thin layer of *BEA (90–190 nm), the catalytic characteristics of the final catalyst were not significantly different from the simple Pt/SiO2 in test reactions of toluene and mesitylene hydrogenation.
Hollow crystals can also be obtained for Ti-MFI, TS-1.322 In this particular case, the distribution of Ti species in the resulting material changed due to Ti reincorporation into the zeolite structure. The authors proposed that a controlled treatment may be used to eliminate undesired extraframework Ti species and improve catalytic performance of zeolites. The calcined TS-1 was treated with a TPAOH solution and then heated at 170 °C for 24 hours. As a result, the recrystallised TS-1 had large voids of varying widths, visible in TEM images.
4 Top-down mechanochemical approaches
Mechanical post-synthetic treatment is proposed as an environmentally friendly method to adjust the properties of zeolites and to improve catalytic performance at both laboratory and industrial scale.323 Mechanochemistry can facilitate insertion of heteroatoms (as exchangeable cations or isomorphous substitution, depending on supplied energy), thereby circumventing wet chemistry and enhancing atom efficiency of the reactions. Rainer and Morris324 recently reviewed mechanochemistry of zeolites, presenting a compilation of the benefits associated with mechanochemical techniques for top-down modifications.4.1 Milling techniques
Mechanical treatment allows modification of the textural properties, including particle size reduction, deagglomeration, and secondary pore development. Milling, followed by recrystallisation, facilitates regulation of crystal sizes, which is challenging in bottom-up synthesis. Simple grinding/milling of a zeolite after synthesis reduces particle size but causes significant amorphization.325 Consecutive recrystallisation may result in undesirable particle growth, compromising the results.324 Additional treatment is thus needed to stop unwanted crystal growth processes or cause appropriate spatial ordering of crystals. Most mechanochemical modifications are at least two-step processes, but their simplicity and environmental sustainability make them attractive despite the loss of previously formed crystalline components. The presented examples show that milling produces mixed or moderate improvements and should be applied judiciously.Breaking zeolite crystals requires considerable energy, and the extent of both wanted and unwanted damage is proportional to the force used. Bead milling, known to exert less damage than conventional milling, was used to reduce the crystal size of zeolites LTA, FAU, and MFI. In all cases the agglomerates of the size of ca. 3.5 μm composed of individual particles with sizes 50–200 were micronized to nanocrystals with average sizes ca. 100 nm.326–328 All these zeolites can be synthesised as nanocrystals using bottom-up synthesis. However, it is expensive and ecologically unfavourable due to the use of OSDAs and low synthesis yields. The mechanochemical technique was applied for homogenising zeolite suspension in water or ethanol using sonication, then milling for 120 and 360 minutes with 300 and 100 μm zirconia beads, respectively. This process allowed the recovery of almost the whole powder, which may then be recrystallised in a diluted solution with the composition that is approximately in equilibrium with the zeolite. The structure of milled zeolites was investigated using XRD and high-energy X-ray diffraction (HEXRD).329 The standard XRD measurement showed almost 90% loss of crystallinity, but the pair distribution function analysis showed that close-range ordering was intact, with only minor changes in the distances between the T-atom (T = Si or Al) and the second oxygen (the nearest T–O), which for X zeolite (FAU) was attributed to ring structure distortion or partial destruction. Recrystallisation did not restore crystallinity, as evidenced by the analysis of the XRD patterns; additionally, in the TEM images, both well-faceted and poorly faceted particles were visible for MFI, whereas for LTA of much higher crystallinity (98%) the nanocrystals showed sharp planes and edges. The authors also noted that large ZSM-5 crystals (above 1 μm) can be obtained by recrystallising totally amorphized solids formed through extended milling treatment under the same conditions.
Vapour–solid interzeolite transformation (IZC), facilitated by mechanochemistry (milling), was demonstrated in the conversion of FAU zeolite into CHA, using only Na-type FAU zeolite and KOH as the starting materials.330 The authors showed that the transformation occurred by local rearrangement of Si and Al species without macroscopic dissolution of the parent zeolite, provided that KOH was appropriately loaded into the FAU cages. The resulting CHA zeolite retained the Si/Al ratio of the initial FAU zeolite. This technology was further enhanced by combining the milling and heating steps and employing temperature-controlled milling.293
The mechanochemical method offered advantages in the passivation of zeolite external surfaces. Some processes (such as toluene disproportionation to p-xylene, xylene isomerization, or naphtha cracking) suffer from the presence of acid sites on the external crystal surfaces, which can catalyse the same reaction but not selectively, thus decreasing yields of desired products. Commercially available ZSM-5 zeolite (840NHA, Si/Al = 19.7, Tosoh Co.) was bead-milled and tested in the 1,3,5-triisopropylbenzene (TIPB) cracking reaction, which can occur only at the external surface of the ZSM-5 crystals, and toluene alkylation with methanol to xylene, catalysed by both internal and external acid sites.331 Relatively short milling time (up to 30 min) did not cause significant changes in the crystal morphology, acidity or textural properties (BET, external surface area and micropore volume were almost unchanged; 423 m2 g−1 for both, 40 vs. 46 m2 g−1 and 0.173 vs. 0.163 cm3 g−1, respectively). The catalysts were tested in a pulse reactor. The TIPB conversion decreased after milling to 28% versus 68% for the parent material. While the parent material gradually lost activity, the milled one was inactive already after the second pulse due to the complete deactivation of the remaining active sites at the surface. The para-selectivity in the alkylation reaction increased after milling from 22.4 to 41.9% at practically the same conversion level (28.4 vs. 25.5%). Lima et al.332 also confirmed that the mechanochemical treatment caused significant modifications of the external surface of ball-milled ZSM-5 zeolite crystals (20 mm alumina balls). The milled zeolite was used as a catalyst in the liquid-phase glycerol ketalization with acetone. The authors observed that when ZSM-5 was milled for 8 hours (the longest milling time), the contact angle of glycerol decreased (from 38.3 to 18.9°), which could lead to the collection of a denser glycerol phase at the bottom of the reactor (closer to zeolite) and thus impede contact with acetone, reducing conversion from approximately 20 to ca. 10%.
4.2 Formation of patterned zeolites films
Electronics, optics, mechanics, and healthcare are all being transformed by micro- and nanoscale technologies such as smart implants, environmental sensors, and microchips.333,334 Making or moulding zeolites into useful micro- and nano-scale devices remains the primary barrier to employing zeolites for these applications. Recent advances in exfoliating zeolites nanosheets described in Section 5.7 enable manipulation of zeolite layers at microscale but it is worth mentioning some of the other efforts to date.Photolithography is a common technique for semiconductor microfabrication. It uses light to project patterns onto a substrate using photosensitive materials, undergoing a chemical change in reaction to light, called resist (or photoresist). The layer of photoresist is coated onto the substrate and exposed to UV light through a mask, that way transferring a desired pattern. The resist can be developed and selectively dissolved to expose the desired parts of the underlying substrate. This technique can be employed to produce zeolite thin films, exemplified by the fabrication of highly oriented thin film of silicalite-1 through seeding (Fig. 17).335
 | ||
| Fig. 17 Diagram for photolithographic fabrication of zeolite-based micropatterns on a silicon substrate. Zeolite seeds are placed on polished Si surface (the other side is covered with Teflon tape) and then allowing to grow in the contact with the synthesis gel. Then, zeolite-silicon composite is rinsed and dried. On the zeolite layer, the photoresist is spin-coated. The micropattern etched on the photomask is transferred to the polymer resist layer via contact printing following exposure to UV light. The sample is heated to harden the photoresist and improve its adhesion. The pattern is etched using BOE (buffered oxide etching solution). At the final step the photoresist is stripped from the surface using acetone. | ||
A zeolite–silicon composite was prepared by growing oriented polycrystalline zeolite film onto a silicon wafer. The design pattern was subsequently transferred and etched onto the zeolite-silicon composite with standard photolithography. The film thickness and crystal orientation were directly manipulated by regulating the seed population, synthesis mixture, and hydrothermal treatment conditions. The crystal growth along the c-axis was facilitated by seeding the substrate and keeping regular nutrient concentration gradient above the developing zeolite layer. Only full coverage of the silicon layer with the seeds guaranteed the formation of c-oriented crystals. The films were further etched to produce, complex catalyst arrays with features as small as 3 μm. They were then incorporated in microchemical devices, such as zeolite-based microreactors, membrane microseparators and microelectrochemical cells.335,336
The lithography technique was also applied to TS-1 and ZSM-5.337 To create enclosed channel structures that might be used as microreactors or membrane microseparators, Chau and Yeung used patterned sacrificial layers on flat substrate surfaces on which zeolite layers were grown.338 Microtunnels were formed as channels enclosed by a zeolite film on top. They were formed by underetching (removing photoresist on silicon substrate), while microchannels were formed by etching the silicon substrate penetrating through the zeolite film.
Pellejero et al.339 examined different etching methods of silicalite-1 films to obtain micropatterns. Dry etching technologies, including ion milling (bombarding with argon ions) and reactive ion etching (using a commercial reactive ion etching system) provided effective control over etching rates, particularly for c-oriented silicalite films. The etching rates differed between calcined and uncalcined films. Underetching, undesirable for supported membranes, was used to release free-standing structures. The wet etching, using HF or commercial buffered etching mixtures (such as AF875-125 semiconductor grade from Riedel-de Haen) caused the formation of both intra- and intercrystalline defects (etching of the crystal walls).
An original method of spatial organisation of zeolite crystals was proposed by Huang et al.340 who combined micromoulding with a self-assembly of zeolite nanoparticles (Fig. 18).
 | ||
| Fig. 18 SEM image of calcined micro-patterned silicalite film composed of zeolite nanocrystals.340 Reproduced from ref. 340 with permission from the American Chemical Society, copyright 2000. | ||
The ‘stamping and moulding’ process involved multiple steps, including (1) applying an ethanol dispersion of silicalite nanocrystals to a flat surface, (2) pressing a patterned polydimethylsilane (PDMS) stamp and evaporating ethanol for at least 12 hours, and (3) calcining. Ethanol was chosen because it wets both hydrophilic and hydrophobic surfaces; also, the authors anticipated that ethyl groups might be partially grafted onto the crystal surfaces during stamping and minimise early aggregation during self-assembly. The latter process is driven by capillary forces, which move nanoparticles closer together and finally allow for the formation of hydrogen bonding, ‘locking’ the positions of individual crystals.
5 Pore engineering with low-dimensional zeolites
Low-dimensional solids, especially 2D layered ones, had been used to generate novel porous materials by expansion and permanent separation of the layers.5 This supplemented zeolites with larger pore materials which they did not provide themselves. For example, layers comprising clay minerals were expanded and supported by solid props, called pillars, providing so-called pillared layered clays, PILCs, with pores greater than available with zeolites.341 One of the goals of clay pillaring was application for catalytic refining of heavier fractions but compared to zeolites PILCs were less active and hydrothermally unstable.342 This suggested zeolite-like layers as the possible solution, which seemed like a pipe-dream because zeolites were known and developed as exclusively 3D frameworks. More than that, their basic properties that made them exceptional, especially the fixed porosity, but also strong acid sites and stability, seemed to be inextricably linked to 3D structures. In a surprising twist, in the early 1990s, layered zeolite forms called precursors were discovered. They could condense topotactically to form 3D frameworks with a particular topology but were also susceptible to modification and generation of interlayer porosity. It enabled generation by design of new expanded architectures providing novel pore systems and modulated catalytic activities. Layered zeolites precursors represented a fundamental expansion of the basic zeolite paradigm and have not affected the development of the traditional 3D zeolites. They added a new direction for advancement and the fundamental concept of zeolites as exclusively 3D solids had to be amended. The discovery itself was unexpected so it can be helpful to review the circumstances and background. It is relevant beyond a historical account as an introduction to novel practices and methodologies of synthesis and characterisation that had to be developed to address challenges peculiar to these new forms of zeolites. These challenges and attendant complexities are not always appreciated and properly addressed. This have resulted in sometimes inadequate or even erroneous reporting and interpretations.5.1 The origins of 2D and lower-dimensional zeolites
One of the consequences of the 3D character of zeolites was preservation of the structure upon heat treatment to 500 °C and higher, as usually required for activation as a catalyst and for example to check if a new unknown material could be a zeolite. Comparing the powder X-ray diffraction pattern (XRD) before and after calcination was the most convenient test. If the pattern showed unchanged Bragg peak positions, indicating retention of the crystallographic unit cell and the structure in general, the unknown material was assumed to have a 3D covalent structure and provisionally regarded and tested as a zeolite (structure solution from powder XRDs could take a long time then). In contrast, for lower-dimensional solids like compositionally related 2D clay minerals, the XRD positions of some peaks change upon heating and other treatments resulting from inherent flexibility due absence of the 3D covalent connectivity.343,344 The assumption of invariable 3D structure was implicit for zeolites. Before this was recognized as false, several reported (alumino)silicates, designated NU-6(2),345 EU-19346 and ilerite,347 were noted for showing differences in XRDs before and after calcination. They were later confirmed to be layered zeolite precursors. The first was in fact called a ‘zeolite’ by the inventors, but this was not established until after the idea of 2D zeolites was accepted and validated with other materials.The mismatch between XRD before and after calcination was noticed for the zeolite MCM-22 (assigned the framework type code MWW) and led to the recognition of layered zeolite forms.348,349 The calcined material was confirmed to be a 4-connected framework, i.e. a zeolite, by structure solution.349 The XRD pattern of the as-synthesized form, denoted MCM-22P, i.e. precursor, showed visible discrepancies between peak position and appearance in comparison to the calcined zeolite form. Additionally, the XRD of MCM-22P contained an uncharacteristic combination of sharp and broad peaks preventing crystallographic solution and refinement.350,351 This XRD was rationalized as resulting from a 2D structure – 2.5 nm layers with the MWW topology, separated by 0.15–0.2 nm. Despite this separation the layers were stacked with largely preserved vertical alignment indicated by clearly identifiable interlayer reflections at lower angles (below 10° 2θ). Other zeolites, later recognized as isomorphous with MCM-22 have been patented earlier but were not recognized explicitly for their structural and XRD anomalies nor exploited for swelling and pillaring until much later (see IZA Structure Database MWW tab ‘Materials – MWW all materials’).9 As an accompanying development, the complete 3D MWW zeolite (MCM-49) was obtained directly via one-pot synthesis.351 This confirmed the possibility of 2 pathways to zeolite framework formation: direct assembly in 3D, and indirect via a layered precursor. MWW was a new zeolite so its 3D/2D duality could be viewed as an exception or even a rare anomaly. However, subsequent recognition of the 2D ferrierite form,352,353 corresponding to the well-known 3D framework material found in nature and one of 5 major zeolites in catalysis, validated this as possibly a common, maybe ubiquitous, phenomenon. To date, layered 2D forms have been identified for close to 20 frameworks.354–358 It was postulated that 3D/2D pairs may be ubiquitous and exist for all zeolites because any periodic 3D structure can be hypothetically cut into identical topotactically congruent slices.359 There may be synthetic obstacles or other fundamental restrictions to this but, for now, no such barriers have been identified yet. So far, most findings of new layered zeolite representatives have been accidental. Zeolite MFI, one of the most valuable and profitable 3D framework representatives, has been obtained in a layered form by design using a bifunctional template,235 exemplifying a viable route to further new layered frameworks.360 Mechanistic explanation for the formation of zeolite layers assumes termination of a propagating (crystallising) framework with OH groups instead of continuous attachment of TO4 units, which represents growth/expansion in the 3rd dimension. The key lies in finding suitable synthetic pathways conducive to such termination as in most situations (conditions used to date) the propagation leading to 3D frameworks seems to be preferred. The role of framework heteroatoms, especially Al, remains obscure but may be significant, as exemplified by the formation of isolated monolayers of MWW-MCM-56361 and bifer362 in high Al systems, as elaborated below. Surface Al was also postulated to assist the preparation of MWW nanosheets with enhanced activity due to added surfactant in the synthesis gel.363
The existence of complementary 3D and 2D forms represented by zeolite is quite rare among solid materials.343,364 Zeolites are quite unique, which in itself is a noteworthy distinguishing feature, probably allowed by the flexibility of their basic (T–O)3TO building units to attach either H or T at the apex to form either 2D or 3D structures. Recent results indicate the possibility of stepping further down the dimensionality ladder, i.e. to 1D.365 It can be envisioned as further cutting of periodic structures like 2D layers into chains but, aside from paucity of currently known examples, it will be more challenging to synthesize by design as well as to control and characterise upon modifications.366 Although the title of this discussion refers to low-dimensional zeolites in general, it will focus almost exclusively on the layered forms, due to the aforementioned scarcity of 1D zeolites. As demonstrated by extensively developed 2D materials, especially clays,367 layered zeolites enabled non-destructive pore and structure engineering. It is possible by relatively mild and moderate treatments, since no covalent bonds have to be broken. Besides identification by XRD, the existence of 2D zeolites needed practical confirmation by swelling with surfactants and intercalation, which are intrinsic features of 2D solids in general, stemming from their definition.368,369 Subsequent extensive efforts produced initially 4 basic classes of derivatives representing pore engineering of 2D zeolites: swollen, pillared, delaminated, and interlayer expanded (IEZ).355,356,370 In recent advances, layered zeolites were completely exfoliated as nanosheets in solution371,372 and, going in the opposite direction, i.e. 2D to 3D, zeolite layers have been used to produce new frameworks via a process called ADOR.276,373,374 One of the highlights of ADOR is the possibility to synthesize frameworks considered unfeasible for a bottom-up preparation by the conventional hydrothermal synthesis based on thermodynamic considerations.375 The various layered forms of layered zeolites are presented in Fig. 19, which highlights their basic post-synthetic transformations.
 | ||
| Fig. 19 Various as-synthesized forms of zeolites, exemplified by MWW and MFI as models for other topologies, and their transformations. | ||
5.2 Overview of known layered zeolites
Although it is possible that most, maybe all, zeolite frameworks can crystallise as modifiable layered forms, the practical reality is that till now the discoveries of new layered zeolites have been predominantly accidental.356,357 Layered MFI is an exception and has been synthesized by design using bifunctional templates consisting of a core, templating the framework, and long hydrocarbon chains at the ends, preventing structure propagation beyond thin nanosheets. Other zeolites have been synthesized by the same strategy but have not been studied for post-synthesis modification.360 To date, approximately 20 out of over 250 frameworks are represented by well-defined layered forms.355,359 They are listed in Table 1 as candidates for pore engineering. The majority are precursors with known IZA approved structure. The name precursor refers to the ability to condense topotactically to form a complete 3D framework.351| Precursor condensing to 3D | Layers with imbedded OSDA | Single layers | |
|---|---|---|---|
| Al containing layers are in bold. ‘hor’ and ‘vert’ designate in-plane (horizontal) and perpendicular channels. MR pore sizes after topotactic condensation are given after framework codes or in front for dense layers. | |||
| Layers with internal pores | MWW (MCM-22P) | MWW (UJM-1) | MWW (MCM-56) |
| hor-10-MR, 12-MR surface pockets | |||
| MFI; hor-10-MR, vert-10-MR | |||
| bifer (unknown) | |||
| hor-10 and/or 8-MR | |||
| Layers with perpendicular 8-MR | RTH(8 × 8) | ||
| SAS (ITQ-8;8) | |||
| Layers forming 2 or more frameworks | FER(10 × 8)-CDO(8 × 8) | ||
| CAS(8)-NSI(8) | |||
| RRO(10 × 8)-HEU(10 × 8) | |||
| PCR(10 × 8), IPC-9(10 × 7) | |||
| Dense layers | 6-MR: SOD, AST | ||
| 8-MR: MTF, RWR | |||
| 10-MR: AFO | |||
| 10 × 8-MR: STI | |||
| 12 × 8-MR: SFO | |||
| Chain silicate zeolite precursor (1D) | ZEO-2 | ||
The second most important zeolite MFI was already mentioned as represented by nanosheets with embedded templates and lined with extended surfactant tails, designed to produce the MFI layers, which cannot form otherwise, at least not so far. This has consequences for post-synthesis modifications, which must deal with the presence of long hydrocarbon tails lining the surface and protecting against external reagents. The removal of this coating requires conditions that avoid undesired condensation or crosslinking of the layers with formation of Si–O–Si bridges as this would obstruct layer separation.376,377 MFI consisting of thin layers can be synthesized by simple OSDAs, e.g. TBA+, as a self-pillared material with layers intergrown at right angles.378 It is remarkable in its own right but is not structurally modifiable and not amenable to pore engineering via layer manipulation. Layers in the precursor that can produce zeolite ferrierite, e.g. ZSM-55, are approximately 1 nm thick and are called ferrierite layers.357 Another layered material designated bifer has similar planar unit cell dimensions but its layer thickness is doubled, i.e. approximately 2 nm. Bifer is obtained when aluminium is added to the synthesis gel to make ZMS-55, which is siliceous or with added boron. The structure of bifer is unknown but may be composed of two fused ferrierite layers as a monolayered nanosheet.362 It does not have a 3D counterpart yet, just as HUS-2, which is also not recognized officially as a zeolite.379 Recently reported material ECNU-28 was identified to have layers with the SZR topology.380 It is almost certain that it is isomorphous with bifer so the SZR structure is still questionable and requires further validation.372,380 The majority of layered zeolites are siliceous and therefore may be less attractive for acid catalysis, especially in processes demanding high acidity. Those available with Al in the framework are marked in red in Table 1 to highlight potential usefulness in acid-catalysed reactions. Siliceous 2D zeolites can be functionalised by heteroatoms like Ti, Sn, etc. and be used for redox activity.381 Some zeolite layers, lacking internal horizontal mirror symmetry, can produce 2 different zeolite topologies upon stacking by translation or mirror reflection symmetry. The MWW layer has lateral intralayer mirror plane producing the same structure by translation and mirror reflection. In some cases, like FER/CDO382,383 and PCR/IPC-9,375 the corresponding structures are further related by a lateral translation due to symmetry, so they can be interconverted experimentally by inducing lateral layer shift. The layer PCR, discussed in a separate section (Section 5.9), can theoretically produce 4 different structures by such lateral translation.
In most cases, the layers in as-synthesized zeolite precursors are separated by OSDA molecules, which may be held tight and obstruct transformations like swelling with surfactants or interlayer silylation (IEZ formation). It is a common practice to remove these original templates, typically by contacting with an acid in organic solvents like methanol.384,385 Such acidic environment can cause dealumination resulting in decreasing acid site concentration leading to lowering of catalytic activity. The original template may also determine the course of the layer condensation upon calcination to either ordered 3D framework or a disordered one.386 Sometimes replacing the original OSDA with another intercalant can help in better ordering of the layers upon condensation resulting in a zeolite framework without defects.387 Multi-layered zeolite MWW precursor (MCM-22P) prepared with HMI can be usually swollen without prior template extraction.
5.3 Producing different structures and materials from zeolite layers
The basic types of layered zeolite structures that have been obtained to date are shown in Fig. 19. They comprise 15 different types of materials that have been identified for the frameworks MWW and MFI. They represent various arrangements of rigid nanosheets and might be expected to be possible for all zeolites available as layers. So far, other zeolites have much fewer representatives. SSZ-70 with regularly offset layers388 and self-pillared MFI378 shown in Fig. 19 may be exceptions due to peculiarities of the MWW and MFI topologies, respectively. Currently, zeolite MWW is represented by the largest number, approximately 14, of distinct layered forms by direct synthesis and post-synthesis modifications.355 Particularly striking is the variety and diversity of its layered structures obtained by direct synthesis numbering 7.389 The MWW zeolite family is unmatched by any other zeolite and clearly dominates this area. It is uncertain if this is permanent due to particular chemistry of the MWW topology or it can be more or less mimicked by other frameworks. Since MWW is dominant in the literature on 2D zeolites, it is unavoidable that it will also be most extensively covered in this discussion. It is possible that MWW is indeed special and particularly prone to produce various architecture due to its low density of surface TOH groups and the corresponding low maximum possible charge (see Table 2).| MWW | MFI | FER | PCR | NSI | RWR | SOD | |
|---|---|---|---|---|---|---|---|
| Rub-18 | Rub-15 | ||||||
| Layer thickness, nm | 2.5 | 3.0 | 0.9 | 0.9 | 0.9 | 0.74 | 0.77 |
| Maximum layer charge, q/nm2 | 1.14 | 1.49 | 1.89 | 2.35 | 2.69 | 3.7 | 4.1 |
| d-spacing swollen, nm | >5.0368 | 6.0 | 3.7–3.8400 | 3.4–3.8401 | 3.7–3.7402 | 2.74403 | 2.85 to 31–32.5404 |
| d-spacing pillared-calcined | 5.2–4.9361 | 3.1405 | |||||
| Swelling agent | HDTMA-OH | surfactant OSDA | HDTMA-OH | HDTMA-OH | HDTMA-OH | HDTMA-Br; Ion exchange | HDTMA-Br; Ion exchange |
As silanols are weakly acidic and their density can influence both stacking during synthesis and the ability to separate, e.g. upon swelling (see Tables 2 and 3). The dominance of MWW in the development of 2D zeolites and its present status are reflected in several respects. The MWW zeolite appears to show a particular ease of forming independent layers and different forms by direct synthesis. It is illustrated by crystallisation of 3 different forms with one template HMI by changing alkalinity, Si/Al and the HMI content: the multilayered ordered (MCM-22P),348,349 the standard 3D material (MCM-49)351 and monolayered disordered (MCM-56).390,391 MCM-56 is unique, now matched by bifer,362,392 as a delaminated form obtained by direct synthesis with high Al content.391,393 Its interesting and valuable properties as a benchmark are discussed further in the section concerning delaminated zeolites (Section 5.6). Another feature of MWW is crystallisation of layered forms in its entire range of Si/Al from 10 to infinity.394 All other layered zeolites except MWW and bifer are typically purely siliceous or with Al content much below the maximum known for a given topology. This has implications for catalytic uses. Zeolite MWW provided the first examples of almost all classes of derivative materials produced post-synthesis (swollen,368 pillared,368 delaminated,395 IEZ,396 exfoliated (monolayers in solution)371,397 and monolayers (delaminated) obtained by direct synthesis).390 MFI was the only zeolite producing some unprecedented forms (by design with bifunctional template and self-pillared) before MWW. Coincidentally the XRD pattern of zeolite MWW and its derivatives is exceptionally convenient to interpret and informative. It may seem secondary but is extremely helpful for identification that, fortuitously, all critical reflection and features allowing recognition of MWW layers (100 peak), interlayer spacing (001 and 002) and order/disorder (101 and 102) are located below 10° 2θ and are usually well-resolved allowing even visual recognition of various structures. The basic principles of identifying various structures are the same for all layered materials (zeolites) but seem not as convenient and unequivocal to apply as with MWW, especially with regard to identifying order/disorder as highlighted in Fig. 20. In the range 8 to 10° 2θ 3D ordered MWW shows 2 distinct 101 and 102 reflections, which appear as a broad band as the layers become vertically disordered. Furthermore, partial order and its extent can be estimated based on a valley in the middle of this band. No other layered zeolite has been found to show such feature. This is important because many transformations of layered materials involve transition from order to disorder, e.g. swelling, delamination, and its easy identification and quantification by XRD are critical for proving success/completeness of the attempted modification. The reality is that many reported materials are not transformed completely into the intended products. In the case of MWW derivatives the 8–10° band provides an immediate visual check of the extent (yield of transformation) and allows distinguishing between ordered and disordered layer arrangements and their mixtures. This is particularly useful for instant identification of single-layered species393,397–399 and for proving successful swelling, pillaring and delamination as discussed in the corresponding sections.
| MCM-22P multilayered385 | MCM-22P multilayered361,368 | MCM-56 monolayered361,420 | |
|---|---|---|---|
| Abbreviations: HDTMA – hexadecyltrimethylammonium cations as representative surfactants, X – halide anion, usually Cl or Br, TPAOH – tetrapropylammonium hydroxide. | |||
| Si/Al | >20 | ∼12 | 10–11 |
| HDTMA-OH | Yes | Yes | Yes |
| HDTMAX/TPAOH | Yes | Yes | Yes |
| HDTMAX-NaOH | No | No | Yes |
| Temperature | Ambient-high | High | Ambient |
| Used for | Pillaring (pore size 20 ± 7 nm); delamination (with high temp swelling) | Pillaring (pore size 32 ± 5 nm) | Pillaring, direct exfoliation w/o swelling (zeolite monolayers in solution) |
 | ||
| Fig. 20 Selected XRD patterns of MWW materials highlighting the detection of layer order/disorder based on the 8–10 deg. features. Note low angle line in the swollen MWW and unique pattern of the c-oriented disc. | ||
Layered MFI is potentially of comparable or greater interest than MWW as a more versatile catalyst and because of vertical pores across the layers allowing additional path of molecular diffusion.397,406,407 MFI ranks among the 2 most important zeolites for catalysis and shows versatile utility, evidenced by diverse industrial applications.408 It is unique with regard to layer formation by allowing preparation of nanosheets with different thicknesses and as intergrown house-of-cards assemblies.409–411 Compared to MWW its known layered forms are much less numerous and not as diverse but there is a unique one, self-pillared (SPP) with intergrown thin nanosheets at right angles (see Fig. 19). It is not amenable to structure alteration but is interesting for basic investigation of MFI nanosheets.378
The 3rd most studied zeolite precursors are composed of ferrierite layers, which can produce 2 different frameworks FER and CDO with unidimensional 10-MR channels separated by 6-MR (FER) and 8-MR ones (CDO). Synthesis conditions and the type of OSDA determine which configuration, CDO or FER, is produced. They can be interconverted post-synthesis upon suitable in-plane layer shift. Upon swelling the original organisation is disturbed and the CDO/FER distinction disappears. FER is the preferred structure if the swollen layers are condensed, e.g. upon calcination.382 Deswelling in the presence of appropriate organic compounds and high pH can produce CDO instead of FER.383 Ferrierite layers synthesized in alkaline media often have low Al content, which has consequences for catalytic applications. Lower Si/Al, e.g. 15 CDO and >20 FER and higher, have been obtained in the presence of fluoride ions.353,412 Increasing Al content in the synthesis of the CDO precursor ZSM-55 templated by choline produces unexpected outcome. It results in mixtures of ZSM-55 composed of ferrierite layers (0.9 nm thick) and layers of bifer with planar unit cell similar to FER/CDO and thickness doubled to ∼2 nm. The amount of bifer increases with added amount of Al and the content of ZSM-55 decreases. Similar unit cells suggest that bifer could be FER/CDO with doubled layer thickness but its structure has not been confirmed. ZSM-55 can be swollen but not exfoliated (as single layers into solution) but bifer exfoliates readily (vide infra).362 XRD patterns for various derivatives of zeolite ZSM-55 with ferrierite layers are shown in Fig. 21. Zeolite RWR (ilerite) is notable as the first synthesized layered zeolite precursor, initially viewed only as a layered silicate.347,413 It is also the only precursor synthesized without organic OSDA and is also distinguished by swelling through ion exchange with cationic surfactants, not by the typical high pH treatment.403 Lastly, it was shown to form nematic phases as a demonstration of easy exfoliation into single layers upon intercalation of meglumine.414 The above discussion of individual zeolites does not diminish the significance of the other layered precursors, which can play important and diverse roles in the top-down engineering of zeolites. To illustrate typical textural and acidic properties of MWW materials obtained directly or by modification a table is provided in the ESI,† Section with selected literature data (Table S3, ESI†).
 | ||
| Fig. 21 Selected XRD patterns for layered zeolite precursor ZSM-55 and derivative structures. | ||
5.4 Surfactant-expanded layered zeolites
Swelling of zeolite precursors, i.e. expansion of interlayer galleries by intercalation of surfactant-like molecules, represented practical validation of their 2D character and demonstrated the possibility of exploitation to engineer new porous structures akin to PILCs368 and delaminated materials.395,397 Various methods had been known for the expansion of clays and other diverse layered solids.344,415,416 They proved ineffective when applied with the first identified layered zeolite MCM-22P. It is instructive to discuss the circumstances and details of the successful swelling as it shows the underlying complexities, how they motivated/impacted subsequent efforts and how to confirm positive outcome. Swelling was achieved by increasing pH and temperature but only with surfactant hydroxide, hexadecyltrimethyl-ammonium-OH (HDTMA-OH), obtained by ion exchange of the commercial solutions of halides. Equivalent solutions combining surfactant salts and hydroxides of sodium or small tetraalkylammonium (TAA) cations were not effective for swelling with high yield.417 Cationic surfactants induce interlayer expansion by interacting with SiO− centres on the surface generated due to high pH. The smaller cations seemed to interact more favourably with these sites and obstruct swelling by excluding surfactant cations. This rationale was validated by successful swelling with mixtures of surfactant salt with tetrapropylammonium hydroxide (TPA-OH), see Table 3. In this case the larger size of the TPA cations made it less favoured for interaction with the SiO− groups than the surfactant molecules with smaller head groups. This had practical implications because both ingredients are industrial commercial reagents while the surfactant hydroxide had to be custom made. Nonetheless, the use of pure surfactant hydroxide appears to be the most reliable method of swelling, especially with high Al MWW precursors. Successful swelling of MCM-22P was confirmed by XRD, which exhibited all essential and expected features, present and absent in comparison to the starting parent. Specifically, the pattern contained prominent 001 and 003 reflections at ca. 5.0 and 1.7 nm corresponding to approximately 5 nm basal spacing, 2.5 nm expansion between 2.5 nm thick layers. It also displayed invariant intralayer reflections 100, 200, 220 and 310 proving preserved MWW structure. Lastly, a new broad band in the range 8–10° 2θ replaced the 101 and 102 reflections observed for a 3D ordered MWW material, as discussed above. This broad band without a valley proved to represent essential evidence of successful swelling of MWW and is related to the loss of vertical layer alignment. Final validation was obtained upon treatment-pillaring with TEOS (tetraethylorthosilicate) which produced permanently expanded silica pillared material denoted MCM-36 with expected enhanced textural properties confirmed by detailed characterisation.The initially applied solutions with high pH required for efficient swelling of MCM-22P raised concerns about potential layer damage due to desilication and motivated studies on alternative methods aimed at layer preservation. One of the modifications was swelling at room temperature. It has been adopted later on in many reported preparations of swollen MCM-22P but mainly with zeolites having lower Al content, typically Si/Al > 20. Pillaring resulted in smaller pores than with high temperature swollen MCM-22P, which also appear not to delaminate upon sonication (see Table 3). On the other hand, the aforementioned monolayered MCM-56 with high Al content (Si/Al 10–11) can be swollen at room temperature and even with a surfactant salt/NaOH combination, which does not swell the multilayered MCM-22P. The Al content plays a crucial role in catalysis as the acid activity of zeolites tends to rise with increasing Al concentration.418 It may be affecting the stability or be offset by other factors but in high severity processes maximizing Al content is often desired. Thus, it is essential to know the properties of zeolites and layered precursors at their high Al end and how it impacts their modification. A particular example demonstrating that MCM-22P with higher Al content affords more active pillared MCM-36,361,419 is discussed in Section 5.10. This study also showed that MCM-36 materials prepared from the same precursor via swelling at room temperature with layer preservation and at higher temperatures with partial degradation (indicated by Si/Al dropping from 45 to 24 due to desilication) showed comparable catalytic activity with some selectivity advantages for the latter.419 It is evident that swelling of layered zeolites has many variables, which must be balanced for optimal outcome. It is highlighted by the summary in Table 2 for the zeolite MWW showing basic approaches to achieve high yield swelling.
The MWW zeolite precursors can be swollen directly in as-synthesized form but other 2D zeolites usually require removal of the OSDA imbedded between layers, which is referred to as detemplation.384 Effective treatment can involve extraction with acids in alcohol, optionally at elevated temperature. The obtained protonated H-zeolites are easier to swell by contacting with surfactant hydroxide solutions. Table 3 presents selected examples of swollen zeolite precursors with basic structural characteristics. Layered precursors of the zeolites RWR and SOD did not require high pH and could be expanded by direct ion exchange with surfactant halide solutions. A possible reason for such undemanding swelling can be high layer charge density compensated by small cations Na+ and tetramethylammonium (TMA), respectively.
Swelling is typically carried out as an intermediate step for subsequent transformations like pillaring and delamination discussed in the subsequent sections. A recent study analysed calcination of swollen MCM-22P as a method of delamination without additional post-synthesis treatments.399 Details are presented in the section on delamination (Section 5.6).
Swelling of zeolite precursors was also used to generate highly stable small metal clusters below 2 nm in size and single atoms (Pt) inside the MWW structure. Siliceous zeolite MWW (ITQ-1) was contacted with basic surfactant solution (pH ∼ 12.5) containing Pt clusters in DMF. The swollen product was calcined resulting in condensation to well-ordered MWW zeolite.421 The obtained Pt-MWW materials were characterised by TEM, HAADF-STEM, EXAFS, and XANES confirming small size and uniform distribution of Pt clusters. Catalytic hydrogenation of propylene was more than 2 times faster in comparison to isobutylene. This was attributed to the accessibility of the propylene to the clusters located inside the zeolite. The clusters showed high stability upon heating to 650 °C in air followed by hydrogen reduction. In four cycles the size of Pt clusters remained below 2 nm and showed only minor coarsening.
A follow up study showed that the size of nanoparticles formed between the layers increased with the length of the alkyl chain in the alkyltrimethylammonium hydroxide used to swell MCM-22P (Si/Al = 50).422 The swelling with alkyl chains containing 12, 14, 16 and 18 carbon atoms resulted in the final average size of Pt nanoparticles equal to 0.85, 1.0, 1.55 and 2.08 nm, respectively. The content of Pt was equal to approximately 0.25% in all samples. Measured micropore volumes indicated a trend of diminishing with increasing hydrocarbon chain length but the differences were small and could be due to experimental fluctuation. The study demonstrated the ability to control the size of metal particles by adjusting the size of swelling surfactants.
HDTMA-swollen layered ferrierite (RUB-36) was used to encapsulate Pd by ion exchange (deswelling) with diethylenediamine-Pd acetate complex in ethanol followed by calcination which resulted in topotactic formation of a 3D FER zeolite (Pd@FER).423 The Pd nanoparticles located inside had uniform diameter distribution of about 1.4 nm and showed size selectivity in catalysis different from the catalyst Pd/RUB-37 obtained by wet impregnation.
Palladium containing MCM-22 (0.79% Pd Si/Al = 60) was obtained by swelling in the presence diethylenediamine-Pd acetate complex and calcination.424 The average Pd(0) particle size was 1.9 nm. Catalytic testing confirmed shape-selectivity in the hydrogenation reaction of various nitroarenes to anilines. Pd@MCM-22 catalysed rapid conversion of nitrotoluene in contrast to negligible reactivity towards nitronaphthalene. This can be explained by exclusion of the latter from inside zeolite pores where the metal particles were located. Commercial Pd/C catalysed hydrogenation of nitronaphthalene at slightly lower rate than catalytic hydrogenation of nitrotoluene by Pd@MCM-22.
Layered precursor of zeolite PCR (IPC-1P) described below produced 2 different 3D structures containing nanoparticles.425 The precursor IPC-1P is available through disassembly of zeolite UTL (14 × 12-MR channels) and can be reassembled into PCR and OKO zeolites with 10 × 8-MR and 12 × 10-MR channels, respectively. OKO has extra S4R units as (SiO)4 bridging PCR layers that can form through insertion of silica present in the surroundings. The larger pore OKO with encapsulated nanoparticles (Pt@OKO) was obtained by intercalation of IPC-1P with platinum(0)-2,4,6,8-tetramethyl-2,4,6,8 tetravinyl-cyclotetra siloxane. The Pt@PCR zeolite was prepared by combining IPC-1P, dodecyltrimethylammonium hydroxide as the swelling agent and Pt(acac)2 in dimethylformamide (DMF), stirring and calcining the isolated solid. The nature and quality of the products was confirmed by XRD, nitrogen sorption, and electron microscopy (SEM and STEM). The content of platinum and the average size of platinum nanoparticles were comparable in both materials: 0.34% and 0.32%, and 0.98 nm and 0.96 nm for the OKO and PCR structures, respectively. The former exhibited broader distribution of Pt sizes, which can be ascribed to larger pores of its zeolite channels.
The above studies providing metal particles of small size and exceptional stability inside zeolites represent pore engineering with regard to content rather than size and shape. Their preparation via layered precursors can be used to tailor selectivity in catalytic reactions.
5.5 Pillared zeolites
Pillaring refers to introducing oxide precursors between expanded layers to enable formation of permanent supports after heating to high temperature, which removes water and organic components, producing porous structure. Organic molecules have been also introduced as pillars, but they are sensitive to degradation and collapse at higher temperatures. Liquid TEOS (tetraethylorthosilicate, Si(OC2H5)4) stands out as the most efficient pillaring reagent for layered oxides and zeolites.416 The treatment of swollen materials with TEOS, optional hydrolysis in water, and calcination converts the alkoxide into amorphous silica between layers producing pore systems with distinct pore size maxima in the low mesopore range >2 nm. Pillared MWW zeolite is denoted MCM-36.368 Pillars do not appear as discrete features in TEM because of their amorphous nature. The presence of pillars is evidenced indirectly in TEMs of calcined particles with edge-on orientation, which show equally spaced parallel layers separated by 2–3 nm region with no distinguishable features.368,426 The downside of using TEOS is catalytic inertness of silica meaning the pillars reduce density of active sites. Still, pillared zeolites often outperform the parent despite a two-fold or greater reduction of the concentration of acid site. One of the remedies has been the addition of heteroatoms like Al or pillaring with other oxides.427–429 This had mixed effects on quality and porosity of the pillared structures which were often lowered or inferior in comparison to the silica pillared counterparts. Sometimes this could be evidenced by XRDs showing for MWW the 8–10° band but with a valley in the middle indicating partial (3D) ordering. Pillaring with other metal oxides introduced control over acid–base properties of the products with implications for catalysis.428 Incorporation of Ti into swollen MWW zeolites was achieved by using tetrabutylorthotitanate in combination with TEOS in ratios 0–120. It was possible to obtain Ti-MCM-36 with high quality XRD and textural characteristics, e.g. BET up to 800 m2 g−1. The products obtained with Si:Ti 40:1 were investigated in detail including the conditions of post-pillaring hydrolysis at different pH between 5–9.430 The basic pH provided by NH3 but not NaOH was most advantageous for porosity. The pillared products contained mesopores in the range of 2 nm which proved beneficial for accessibility of bulky molecules in comparison to model microporous titanates Ti-MWW, Ti-MWW-IEZ and TS-1.431 MCM-36 with Ti was catalytically active for epoxidation of cyclo-hexene with t-butylhydroperoxide (TBHP). The presence of acid sites due to Al in the framework thwarted this oxidation reaction so the samples were acid extracted which had a multiple effect: removal of Al and octahedral Ti and increasing hydrophobicity.432 This made the samples active and was most pronounced with boron-MWW, denoted ERB-1, which was Al free from the start. MCM-22 pillared with pure Ti oxide was reported and used for CO2 capture after loading with polyamines. The reported XRD and low BET surface area indicate incomplete pillaring.433
Pillaring of zeolite MFI has been studied using the multilayered nanosheets obtained with bifunctional surfactants first reported by Ryoo et al.235,434 The as-synthesized layered materials are already swollen with OSDA cations partially embedded in the pores and lining the surface creating 3–4 nm interlayer separation. This allows direct pillaring of as-synthesized precursors, which was carried out by the standard TEOS treatment followed by hydrolysis with water and calcination. The products have basal spacing around 6 nm with ca. 3 nm interlayer spacing (layers are 1.5-unit cell thick, 3 nm). The Si/Al increased from 49 to 57 due to added silica. The typical pore size determined by nitrogen sorption is ca. 4.0 nm while BET is below 700 m2 g−1 (up from ca. 500 m2 g−1 for the calcined layered MFI). Pillaring of MFI was also carried out by a vapor-phase method.435 The sample was exposed to TEOS vapours heated to 150 °C and optionally hydrolysed with water. The ratio TEOS to the sample was varied and the best ratio was determined. It was equal to 0.5 and did not require the hydrolysis step. The textural properties were similar to the conventionally pillared product: ca. 4 nm pore size and BET ∼ 650 m2 g−1. Catalytic testing was carried out in comparison to the calcined parent zeolite and commercial MFI. In the reaction between mesitylene and benzyl alcohol (see Section 5.10), which reflects acid site accessibility, the conversion increased in the order: commercial MFI < lamellar MFI calcined < pillared MFI.
It was postulated that pillaring of MFI may produce silica pillars as ‘crystalline microporous structure corresponding with that of MFI zeolite’.434 They could be formed due to free OSDA or organics present between layers as fillers. The evidence included TEM images showing MFI-like domains between layers. The typical silica pillars are amorphous, hydrothermally unstable and catalytically inert. Crystalline zeolite pillars can show enhanced stability and activity, which if observed would validate their zeolitic nature and result in improved catalysts.
A typical zeolite layer is covered with a regular array of silanol groups which are capable to deprotonate but also to condense with suitable organic groups such as alkoxysilanes.436 This has been exploited to produce organic–inorganic hybrids with organic pillars based on prior studies of layered silicates including ilerite-octosilicate.437,438 Since the latter is now recognized as a precursor to zeolite RWR these studies represent the first examples of organic-pillared zeolites. In this case, zeolites can provide additional functionality due to having Brønsted and Lewis acid centres. Layered zeolites are usually intercalated or swollen before condensation with organic molecules. Ilerite in the acid form was kept in contact with n-hexylamine and then reacted with alkoxysilanes of 4 4′-biphenylene containing 3, 2 and 1 ethoxysilanes complemented with methyl groups: 0, 1 and 2 respectively.438 The products were extracted with an acid to remove the intercalant and dried at 50 °C, after optional washing with EtOH, to afford porous materials. The obtained interlayer space values were 0.87, 1.65 and 0.60 nm. The 1st product contained standing molecules bridging both layers. In the second the molecules were standing but only with one end attached to the layer showing 40% higher organic content (35 vs. 25%). In the 3rd product the rings were horizontal to the layers. Nitrogen adsorption/desorption carried out after degassing at 150 °C gave BET surface areas equal to 616, 669 and 241 m2 g−1. They declined to 508, 578 and 35 m2 g−1 upon prolonged storage (300 days).
MCM-22 (Si/Al = 50) was used to prepare a multifunctional hybrid organic–inorganic material by pillaring with 1,4-bis(triethoxysilyl)benzene (BTEB).439 The precursor was swollen with HDTMA-OH and reacted with the above organic reagent in dioxane followed by extraction with an acid. The benzene ring was also functionalised with amine. The products contained micropores in the layers and mesopores in between. The BET was increased to ∼550 m2 g−1. The amine-containing product demonstrated a 2-step chemical process in one-step: benzaldehyde dimethylacetal was hydrolysed by the acid sites to produce benzaldehyde which reacted with malononitrile on basic sites to give benzylidene malononitrile.
Lamellar MFI was used to prepare a hybrid similar to the above MWW one. Here the initial challenge was removal of the template embedded in and lining the layers without calcination which would cause layer condensation.377 The applied treatment involved contacting with an acid in alcohol at temperatures above 75 °C, twice, followed by a solution containing H2O2 and UV irradiation. This eliminated organic from both outside and inside the layers. Catalytic testing was carried out like for the MWW hybrid and showed similar type of activity validating the one-pot 2-step approach. The MWW hybrid was more active than the MFI one.
Layered zeolite precursor PCR (IPC-1P) which is obtained from zeolite UTL by disassembly was extensively studied to produce organic–inorganic hybrids with tuneable textural properties.440,441 Precursor samples swollen with HDTMA/TPAOH mixtures were contacted in chloroform with various silsesquioxanes and polyhedral oligomeric siloxanes of different types forming pillars between crystalline zeolite-derived layers. The variables studied included the presence of S and N in the hydrocarbon chain length and rigidity of the backbone, nature and types of substituents and density of functional groups. Thorough characterisation of the products allowed formulation of rules for choosing molecules to produce high sorption properties.
5.6 Delaminated zeolites
Delamination refers to splitting materials into layers. It can be spontaneous with 2D solids contacted with the right medium and is widely exploited in practice.364 Technically speaking swelling is already a form of delamination but for 2D zeolites this term acquired a particular meaning. The goal of delamination was to separate precursors down to single layers to maximize access to ‘zeolite-type catalytic sites (…) contained within thin readily accessible sheets’.395 It is important to distinguish two scenarios. One, based on the original approach, initially involving surfactants or pre-swollen layered precursors, is carried out in heterogeneous systems. The treatments include disorganisation of layers in solution, e.g. by sonication, acidification and calcination. It can be called heterogenous delamination as it is carried out in a liquid with constant presence of a solid phase and affords solid as the final product. It is unknown if and how the layers become separated into individual nanosheets. The second scenario is concerned precisely with ensuring complete separation into independent nanosheets in solution. After such complete dispersion and formation of a homogeneous liquid phase the layers can be reassembled into many different materials including composites with various other components, films etc.372 The dispersion can be achieved directly in one step but usually requires purification from larger aggregates by centrifugation. To differentiate these homogeneous layer dispersions in solution from heterogeneously delaminated zeolites the former have been called exfoliated following the definition adopted by the clay science community and recommended in the Glossary of Clay Science.372 This allows maintaining the original name and concept of delaminated zeolites for those not exfoliated explicitly (via a liquid phase) and characterised as solids. The problem with the proposed distinction is that delamination and exfoliation have been used as equivalent terms for zeolites and sometimes other materials, often interchangeably, sometimes in the same publications356,395 This was not a serious problem before the zeolites exfoliated into a liquid were obtained. The recommendation going forward is a strict and consistent differentiation between delamination and exfoliation as defined above in accordance with the Glossary and making sure these terms are not used as synonyms. As there is no certainty for compliance, readers are advised to be aware of this possible ambiguity. Both classes will be discussed separately in this review. As mentioned above and demonstrated in practice, the exfoliated zeolites are more versatile with few limits for production of diverse materials, e.g. with other compositions and particles to combine different functionalities. They are effectively liquid solutions of zeolite nanosheets as giant molecules that can be mixed with any compound, particle etc., which is rather limited in the heterogeneous delamination processes. Exfoliated zeolites in solution are also valuable fundamentally because they represent a clearly defined stage where all layers are definitely separated and independent, so it is easier for monitoring and design of basic studies. In contrast, delaminations are carried out in complex heterogenous systems where it is difficult to discern and observe intermediate elementary stages and to control events.Delaminated zeolites via the heterogeneous process attracted a lot of attention for catalysis due to increased mesopore volume and BET surface area, which had potential for enhancing activity especially towards larger molecules.445 Frequently they were used as carriers for active species, especially metals (see Section 5.10). Considerable efforts have been made to improve/simplify the procedure and to gain knowledge about parameters influencing delamination and properties of the products. One of the concerns was amorphization caused by silica dissolution due to high pH of the swelling mixture and its effect during sonication at elevated temperatures.363,446,447 The amorphization had potential for reducing effective zeolite content and catalytic activity. An additional identified side-effect was possible formation of mesoporous MCM-41 or related surfactant templated phases if conditions became too severe. It was also established that ‘the delamination process is favoured by the decreasing aluminium concentration of parent materials’,446 in other words delamination is less effective with increasing Al content. As already noted, this has implications for catalytic activity, which is controlled by the Al content and for activity-demanding processes must often be high. The above limitations of delamination and its multi-step procedure motivated attempts to find less severe methods and with fewer processing steps. One of the methods applied a treatment of MCM-22P with a mixture of hexadecyltrimethylammonium bromide, tetrabutylammonium fluoride and tetrabutylammonium chloride at 80 °C with addition of tetrapropylammonium hydroxide (TPAOH) to rise the pH to 9.447 A similar approach in DMF without TPAOH was applied to the ferrierite precursor PREFER.448 Later on, one-step delamination without surfactants of boron-containing MWW zeolite was reported upon reaction with aluminium nitrate at temperatures from 100 to 170 °C.449 In the last case the notable feature was contraction of the apparent basal spacing to 2.353 nm, which is below the nominal thickness of the MWW layer (2.476 nm based on directly calcined 3D structure). Similar behaviour was noted previously with NSI and FER zeolites. Because their basal spacing was below the apparent layer thickness these forms were called sub-zeolites.402,450 Their formation is rationalized by the fact that zeolite surfaces are not smooth but have indentations, which in the standard 3D form are opposite each other and generate interlayer pores. When the layers are offset it can result in heights falling into troughs (pores) causing contraction below the apparent formal crystallographic thickness.
Direct calcination of a swollen MCM-22P (Si/Al = 50) was proposed as a simple method to obtain delaminate zeolite layers.399 The effects of ambient (RT) vs. elevated (80 °C) temperature swelling were compared. The latter was subjected to sonication for 1 h and acidification to pH 2 in the swelling mother liquor. Its XRD showed significant decrease in crystallinity and substantial content of amorphous silica, which could be due to both high pH/high temperature swelling conditions and sonication also at high pH. The RT swollen product was isolated without sonication and calcined. It was regarded as delaminated based on estimated average layer thickness. The comparison of its XRD with DIFFaX simulated plots gave approximate layer thickness equal to 1.3 unit cells (UC), consistent with slight but visible valley in the 8–10° 2θ band used to evaluated disorder of MWW layers. Their RT swollen form showed no increase in overall BET surface area, equal to ∼570 m2 g−1, but the external surface area increased over 3 times. The HT treated and sonicated product exhibited nitrogen isotherm with capillary condensation (inflection at p/p0 ≈ 0.3) similar to surfactant-templated mesoporous silicas. Despite profound structural difference, both RT and HT treated MWW materials exhibited similar acid site concentrations both overall and external, determined by adsorption of pyridine and 2,6-di-tert-butylpyridine, respectively. Catalytic activity of the RT delaminated MCM-22 was tested in methanol-to-hydrocarbon (MTH) and toluene methylation processes. Both can take place in micropores. No improvement in performance was observed in comparison to the starting zeolite but the delaminated derivative showed long-term stability in the MTH. The authors also quantified average layer thickness based on XRD calibration lines of half-width at half-maximum of the 101 reflection and ratio of 10-MR volume to entire micropore volume plotted against (#unit cells)−1. These plots gave values of 0.98 and 0.87, respectively, corresponding to 1.04 and 1.14 unit-cell average thickness. This study is illustrative of the characterisation complexities associated with determining the extent of delamination when modifying layered structures. They involve many variables, and it is hard to correlate textural and acid properties with activity.
More recent efforts to obtain delaminated or related materials focused on designing simplified synthesis procedures, especially by direct preparations. They are formally bottom-up and therefore beyond the scope of this article but because of direct interest for the top-down delamination they are relevant to the present discussion. Directly synthesized delaminated zeolite with MWW topology consisting of single layers was already reported in 1995 and designated MCM-56.390,391 It is worth presenting in greater details because of its unusual properties, commercial potential and facile top-down modification capacity (top-down engineering) superior to the multi-layered MCM-22P. MCM-56 is frequently benchmarked against delaminated and other catalysts, so it is important to understand its unique features, especially as an intermediate during synthesis. The XRD of MCM-56 exemplifies fully disordered MWW showing a band between 8–10° 2θ instead of 101 and 102 peaks (at 8 and 10°) observed with the ordered 3D MCM-22 and MCM-49. It shows no depression in the middle and no pointed maxima at the ends, which when observed suggest contamination with ordered MCM-22/49 domains. This is critical because MCM-56 is an intermediate converting to the 3D structure, MCM-49, if its synthesis is continued. It must be stopped at the right time (based on XRD) to ensure obtaining an optimal representative, e.g. for catalytic benchmarking. The XRD evidence in some reported studies indicates that MCM-56 used as the benchmark was over-crystallised (containing MCM-49, e.g. 70:30)451 and therefore not truly representing an optimal product for comparison. MCM-56 has high Al content with Si/Al 10–11 and is catalytically very active. Industrially formulated and tested MCM-56 catalyst outperformed MCM-22 and MCM-49 in small olefin aromatic alkylation.452 This proves commercial potential of MCM-56 because the MWW zeolites are prominent commercial catalysts used in this process.452,453 The formation of MCM-56 at the low Si/Al ratio 10–11 while most layered zeolite are synthesized at higher Si/Al is interesting fundamentally, especially since the product is a single-layered precursor.361 Exfoliable zeolite bifer is another such example, which suggests that high Al content may play a role in producing delaminated zeolites directly. Last but not least, MCM-56 can be engineered top-down, e.g. swollen under much less demanding conditions and exfoliated directly, more conveniently than the multilayered precursors MCM-22P as shown in Table 3.361 BET and pore volumes of MCM-56 are comparable to the multilayered MCM-22 and MCM-49 so it was proposed that it indicates face-to-face layer packing.395 There is a nominally related material referred to as MCM-56 analogue. It is obtained by acid treatment of MCM-22P resulting in XRD resembling MCM-56. It is not known whether its chemistry (exfoliation and facile swelling) matches MCM-56. It has higher Si/Al so it may be not equivalent to MCM-56.454
Other efforts to synthesize directly delaminated MWW zeolites adopted dual temple strategies, especially by combining the typical MWW OSDA, hexamethyleneimine (HMI), with long chain surfactant-like molecules. Directly synthesized DS-ITQ-2 was obtained with HMI and N-hexadecyl-N′-methyl-DABCO from a gel with Si/Al = 15 after synthesis for 7 days at 150 °C.455 The crystals exhibited external BET of 304 m2 g−1 (545 m2 g−1 total). Ar physisorption isotherm indicated significant reduction in the inflection corresponding to 12-MR in MCM-22. The estimation of layer thickness of crystallites gave 35%, 35% and 20% of mono-, double- and triple-layers. Catalytic activity of DS-ITQ-2 was evaluated in comparison to ITQ-2, MCM-22 and MCM-56 in the liquid-phase alkylation of benzene with propene to cumene at weight hourly space velocity (WHSV) = 25, 50 or 100 h−1, T = 125 °C, P = 3.5 MPa. MCM-22 performance was usually lower but the other three were comparable, except for WHSV = 100 h−1. At these conditions the estimated propene conversions were 60, 58 and 55 for ITQ-2, DS-ITQ-2 and MCM-56. The DS-ITQ-2 showed much slower deactivation rate as the main difference between the tested materials. It should be noted that the MCM-56 sample appeared not to be optimal. Its XRD exhibited distinct peaks at 8 and 10° 2θ corresponding to the 101 and 102 reflections indicating significant amounts of ordered MWW zeolites.
In another study, carefully adjusted amount of hexadecyl-trimethylammonium (8% w/w) was added to the MCM-22 synthesis mixture producing MWW zeolite with XRD similar to MCM-56 and enhanced textual properties.363 The average layer thickness was estimated by statistical analysis of TEM images and the value 3.5 nm (∼1.5 unit cells) was obtained. Catalytic activity was evaluated in a model reaction of benzene alkylation with benzyl alcohol (at 80 °C for a fixed catalyst loading – 30 mg of catalyst per 8.86 g of reaction mixture; benzene/BA = 196 mol mol−1). The compared catalysts were: standard MCM-22 (Si/Al = 11, BET 634 m2 g−1), 5.5% surf-MCM-22 (13558 m2 g−1), 8% surf-MCM-22 (13557 m2 g−1) and ITQ-2 (20662 m2 g−1). The approximate conversions ratios after 9 h reaction were equal to: 6:4.5:9:4. The turnover ratios had similar proportions.
Delamination by mechanochemical approach was applied to MCM-22. It involved ball-milling producing changes in crystal size and increased mesopore volume.456 Catalytic benefits were evaluated for 4-propylphenol cracking as a model reaction for lignin-derived aromatics. The parent MCM-22 was initially more active, but the ball-milled product showed reduced deactivation and became more active in the long run.
Quantitative evaluation of delamination typically relies on gas adsorption measurements, which provide information such BET surface area, pore size distribution and micro vs. mesopore volume. The results are referenced to the starting material. Typical BET values for 3D MWW are 400–600 m2 g−1 and for MFI 400–500 m2 g−1. Upon delamination BET has been reported to be increased up to 1000 m2 g−1 but sometimes the increases are more moderate, e.g. in the range 600–700 m2 g−1, especially with samples with higher Al content.395,446 The differences can be observed in the split between micro- (internal) and meso- (external) surface areas. The former can drop down to near zero while the latter increases significantly. This is reflected in the pore volume – decreasing for micropores and increasing for mesopores. Zeolite MWW is particularly convenient for investigation because of its intralayer 10-MR pores and 12-MR cups on the surface of layers. Delamination and increasing disorder can be monitored by Ar physisorption which can show qualitative data about the amount of each type of micropores.455 Typically, the population of 12-MR pores can diminish down to zero while the 10-MR pores can remain.
The enhanced porosity translates into increased access of active sites to larger molecules. It is quantified by base titration or adsorption of probe molecules and measurement in IR (amines) or NMR.
Delaminated zeolites showed advantage in various catalytic processes especially those involving bulky molecules.460 Examples of processes highlighted for delaminated zeolites included epoxidation of olefins, Beckman rearrangements of bulky ketone-oximes, acetalization of alcohols, and as catalysts for the synthesis of diamino diphenyl methane (DADPM, a monomer for polyurethane) for possible use in an industrial process.460 Extended surface areas with defined structures461 and increased mesopore volumes make expanded 2D zeolites attractive as supports, e.g. for metal and oxide loading in processes like Fischer–Tropsch,462,463 SCR,464 N2O reduction,464 carbon dioxide reforming by Ni,465 renewable diesel with Ni clusters,466 phosphorylation of dodecanol with CaO,467 dibutyl sulphide oxidation with Nb, Zr and Mo,468,469 oxidation and oxidative dehydrogenation with V/vandia,470–472 electrocatalytic oxygen reduction with metalloporphyrins,473 epoxidation by Ti.474
5.7 Exfoliated zeolite monolayers in solution
These are potentially the most effective, versatile, and least constrained 2D systems for producing new zeolite-layered architectures and composite materials. They are effectively homogeneous solutions of zeolite nanosheets presenting no physical obstacles to reassembly and combination with any other component(s). This can generate structural and functional hybrids, including structured materials like films and membranes. The initial problem encountered with these systems was the resistance of layered zeolites to exfoliate into solutions despite expectation grounded in the known behaviour of other 2D solids and fundamental principles. Even layered precursors showing expansion by swelling,475 formation of different frameworks by layer shifts375,382 and heterogeneous delamination442 did not show signs of sufficient direct exfoliation. In the end, layered zeolites did reveal spontaneous exfoliation into solution as single monolayers, as described below, but with suitable samples/preparations. Before that, the first successful approach used surfactant-swollen layered zeolites MWW and MFI, the latter from direct synthesis, which were dispersed in a polymer by melt blending in a co-rotating twin screw extruder at elevated temperature under N2 atmosphere. The obtained polymer composites were sonicated in toluene and after centrifugation to remove larger particles, solutions containing approximately 1.25% w/w polymer and 0.01% w/w nanosheets were obtained. Characterisation by TEM and AFM confirmed the presence of MWW and b-oriented MFI monolayers with 1- and 1.5-unit cell thicknesses, respectively. The dispersed nanosheets were deposited as coatings on anodized alumina membranes and α-alumina discs to evaluate potential for membrane fabrication. The organic content was burned off by calcination at 540–580 °C. These films showed no selectivity for separation of xylenes. TEM revealed nanometre-size defects, which were subsequently repaired by additional, secondary growth of zeolite on the original film. This generated selectivity for para- and ortho-xylenes giving separation factor between 40 to 70 and p-xylene permeance equal to 3 × 10−7 mol m−2 s−1 Pa−1 at 150 °C. The secondary growth was also applied to the zeolite MWW, which has 6-MR pores perpendicular to the layer. It showed increased selectivities for He/H2 and He/N2, up to 3 and 17, respectively. Similar procedure was carried out with the SOD precursor RUB-15 having 6-MR openings in the layer.405 The changes in procedure versus the earlier methodology included swelling with a surfactant bromide at neutral pH, purification of the toluene extract by gradient centrifugation and dispersing nanosheets in ethanol, ion-exchange of HDTMA with H+ in a 0.2 M solution of sulphuric acid in ethanol at 80 °C for 16 hours. Membranes were obtained by filtration of the solutions through commercial aluminium supports. Calcination produced defects. They were cured by filtration through of an additional nanosheets solution, which plugged the gaps in the nanosheet coverage. This increased selectivity of H2/CO2 separation 10 times to values above 25 and reaching up to 100.The above procedures are multi-step and require swelling as the first step, which results in layers coated with surfactants. In contrast, a direct one-step exfoliation of layered zeolites has been demonstrated recently.371 It was achieved with zeolites MWW (MCM-56),371 MFI,376 bifer with layers of unknown structure possibly related to ferrierite (same synthesis mixture, similar unit cell)362 and ilerite (zeolite RWR).414 High pH TBAOH (tetrabutylammonium hydroxide) solutions were used with the first three zeolites mimicking its common usage for exfoliation of negatively charged layered oxides.476 Ilerite was exfoliated to produce nematic suspension of monolayers ‘by forceless spontaneous delamination via repulsive osmotic swelling’. The ilerite was repeatedly contacted with a 1 M solution of meglumine in water with pH adjusted to 9 to achieve ion exchange of all Na+ with the organic intercalant. Other organocations including TBAOH proved ineffective for osmotic swelling of ilerite. The suggested reasons included too low hydration enthalpy and too small equivalent areas to exceed the apparent threshold for separation. The ilerite charge density (area per unit charge) at pH 9 is 0.7 nm2 compared to 0.77 nm2 of meglumine, which results in remaining charge that generates separation pressure. The efficiency of exfoliation with TBAOH, i.e. the percent of solid that became dispersed into solution, varied depending on zeolite framework and sample type. Multi-layered zeolites like MCM-22P and the ferrierite precursor (ZSM-55) showed negligible exfoliation. MCM-56 with the same topology as MCM-22P shows substantial yield variation from 0 to 70%. This is tentatively postulated due to layer intergrowth but remains to be further investigated. As an illustration of possible uses, the obtained monolayer liquid dispersions demonstrated potential for producing zeolites with encapsulated metal atoms or particles, mixed zeolite layers, house-of-cards with nanoparticles, oriented films and discs (see Fig. 22). These exfoliated zeolite systems have enormous synthetic potential but there is a practical constraint impeding larger scale usage. So far, the dispersions can be produced only at high dilution, 1–2% in a liquid, to minimize layer aggregation. Since the preparation involves high speed centrifugation this makes these systems impractical for traditional applications as catalysts in bulk, which typically requires large volumes of catalysts. However, they can be attractive for high-value high-cost uses, e.g. as membranes, films and other still to be identified applications. Exfoliated layers can be readily deposited as oriented films that can be useful for gas separations.477
 | ||
| Fig. 22 Modified materials obtained from exfoliated nanosheets in solution. | ||
The above exfoliations of MCM-56, MFI and bifer have been carried out using TBAOH solutions but other tetralkyl hydroxides can be also effective. A portion of the solid remains unexfoliated and is separated by high-speed centrifugation. It could not be assumed a priori that the solid dispersed in solution was comprised only of monolayers. It had to be proven, and no single characterisation method could be considered conclusive. This led to a protocol of 5–6 techniques to prove the predominace of monolayers in solution and characterisation of the structure. The first method was AFM, which determined thickness of the layers deposited from the solution. The layers were then characterised by in-plane XRD to determine the planar unit cell and in situ XRD confirming agreement of experimental profile with the calculated one for dispersions of single layers. Electron microscopy (ED, TEM) further confirmed the structure. A good indication of zeolite monolayers in solution is a reaction with surfactants like HDTMA which typically precipitates layered composites. They can be characterised by XRD. These products are equivalent to swollen derivates but often show XRD with higher intensity, which may be indicative of higher content of surfactant-separated layers, but preferred crystal orientation effects can also contribute to this.
High crystallinity of the exfoliated layers is confirmed by in-plane XRD and TEM images. Testing of quality included catalytic alkylation of benzyl alcohol with mesitylene and comparing conversion vs. time to that of the starting zeolite like MCM-56. The exfoliated and recovered MWW layers were as active as the starting material indicating good retention of activity even despite potentially damaging condition of exfoliation and purification. There is also evidence that hybrids/composites could have better catalytic characteristics than that the sum of its parts. For example, the unprecedented mixture of zeolite layers, MWW and bifer, was more active in the above test reaction than normalized contribution from each component.362
5.8 Interlayer expanded zeolites
The methods of modifying structures/pores of layered zeolites described above can be viewed as extensions of the approaches already known and developed with other 2D materials. The present one, affording IEZ materials with discrete interlayer expansion and ‘pillars’, is completely novel. It is possible because of terminal Si–OH groups on the layer. Typically, the silanols on opposite layers can condense topotactically according to the reaction 2Si–OH → Si–O–Si + H2O, to produce the regular zeolite framework with the interlayer pore systems as part of the overall structure. In the early 2000 it was discovered that Si(OR)2, R = alkyl or H, bridging moieties can be inserted between these silanols to form expanded O–Si(OR)2–O interlayer connections instead of just oxygen atoms.396,478 The interlayer pores are effectively expanded by two SiO links, e.g. from 8-MR to 10-MR. These new products were denoted interlayer expanded zeolites or IEZ, e.g. IEZ-MWW. Sometimes the term ‘pillared’ is also applied. There are caveats concerning their nature and pore openings. First, the new SiO2(OR)2 interlayer bridges are not 4-connected (to other TO4 tetrahedra) so formally these new products are not zeolites, except when 4 such units can condense restoring full 4-connectivity (see below).373 Second, the OR and OH groups attached to the Si-link occupy some volume within a pore so the aperture is constricted with lower effective cross-section than the nominal MR size after enlargement. In the XRD the IEZ zeolites produced interesting effects contrasting the topotactic transformation of as-synthesized 2D precursors to 3D zeolites. The latter process results in contraction of the interlayer distance producing XRD peak shifts, especially noticeable for the basal reflections, to lower d-spacing. The formation of IEZ resulted in preservation of the peak positions in XRD upon calcination. For this reason IEZ products e.g. of MWW, were initially referred to as stabilized precursors.396 Typical syntheses were carried out in acid solutions with added silica sources like SiR2(OR′)2 or even detritus silica on zeolites at relatively high temperatures, up to 170 °C. This treatment can be also called silylation for brevity. The efficiency was also dependent on framework Si/Al and was usually lower with increasing Al.479 The postulated reason was strong interaction between the intercalated template molecules like HMI with MWW layers. MWW zeolites synthesized with piperidine and Si/Al = 10 and higher proved to be more amenable to form the IEZ-MWW derivatives.479 On the other hand, the Al-MWW precursor with piperidine was not synthesized directly. First, the boron-containing MWW was prepared, then deboronated in 6 M HNO3 and afterwards used as a silica source to prepare Al-MWW precursors with Si/Al = >10 templated with piperidine. The obtained precursors could be readily silylated providing Al-containing IEZ-MWWs that were catalytically active. They were tested in alkylation of anisole with benzyl alcohol and acylation of anisole with acetic anhydride, which showed promising activity in comparison to zeolites USY, MOR, *BEA, MFI, MWW and IEZ-MWW. Porosity was characterised by cyclohexane adsorption at 25 °C for several zeolites. It increased in the order MWW < MOR < IEZ-MWW < *BEA < USY. The acidic environment, 1 M acid and higher, in which the silylation of zeolite precursors is carried out to produce the IEZ derivatives is conducive to dealumination resulting in lowering of the acid site concentration and reduced potential acid activity. The methods adopted to alleviate this problem were vapour phase silylation with Si(OR)2Cl2 and a two-step process: first using a dilute acid like 0.1 M followed by 1 M ammonia solution. The final product showed high activity as a catalyst for the acylation of anisole with acetic anhydride.480,481Other elements, especially metals have been used to provide linkers in the IEZ-zeolites. Fe-COE-3 (Fe IEZ-CDO) was investigated by powder XRD and Rietveld refinement. Fe atoms in the linker positions were confirmed. Elemental analysis indicated about 50/50 population of Fe and Si in the linkers. Isostructural interlayer expanded materials were prepared with Ti, Sn, Zn, Eu, and Al validating this method as a general approach for introducing active centres in the IEZ structures.482
Detailed characterisation of pore size distribution in IEZ-MWW in comparison to the parent zeolite MWW, both containing Al, was reported and based on Ar adsorption and Hovarth–Kawazoe calculations.481 Zeolite MWW showed a maximum at 0.6 nm and a shoulder peak at 0.7 nm assigned to intralayer 10-MR pores and interlayer entrances. The IEZ-MWW exhibited two distinct peaks of almost equal heights at 0.6 and 0.8 nm with the latter reflecting interlayer expansion through silylation. This expansion enhanced adsorption of cyclohexane from 1.5 cm3 g−1 (STP at p/p0 = 1) in MWW to 22 cm3 g−1 for IEZ-MWW and reflected increased accessibility of interlayer porosity including expanded supercages. In contrast, the difference in nitrogen sorption was small 0.16 cm3 g−1vs. 0.18 cm3 g−1 because such small molecules can penetrate equally easy both pore systems in both materials.
The silicon interlayer linkers in IEZ materials have two terminal alkyl groups, usually methyl groups in the as-synthesized form. They become converted to geminal hydroxyls upon calcination. These methyl groups give characteristic signals in both IR and NMR.478 They showed bands at 2970 and 850 cm−1 assigned to asymmetric stretching and rocking of CH3 groups attached to Si. 13C MAS NMR contained a new resonance at −2.0 ppm. 29Si MAS NMR spectra of the precursor contained Si Q3 and Q4 signals near −100 ppm. Upon silylation the Q3 diminished significantly, and a new signal appeared at −15 ppm. It was assigned to Si(CH3)2 and it disappeared after calcination when OH groups replaced the methyl groups. FT-IR of the calcined material showed a new signal at 3730 cm−1 assigned to geminal OH with internal H-bonding.
The O–Si(OR)2–O interlayer connectors in IEZ zeolite derivatives can be isolated (MWW), condensed in pairs by forming –O– connection between Si atoms (FER)483 or condensed in four to produce horizontal (parallel with the layers) S4R units (zeolite OKO) resulting in fully 4-connected framework, i.e. a zeolite.401 A vertical (perpendicular to the layers) S4R was inserted into Al-free ferrierite precursor PREFER by reaction with 1,3,5,7-tetramethylcyclotetrasiloxane in HNO3 solution at 150 °C for 24 h.484 The methyl groups initially attached to Si atoms showed IR bands at 1280 cm−1 and 780 cm−1 associated with Si–CH3 stretching and wagging modes, respectively, and 13C MAS NMR signal at −5.6 ppm. They remained intact up to 450 °C but were oxidized to OH at 550 °C. According to 29Si MAS NMR to amount of Si–OH (Q3) in the last product was approximately 30%. The silylated IEZ product had formally 14 × 12-MR channels. Its Ar determined average pore diameter was 0.6 nm vs. 0.39 nm in 3D ferrierite.
Catalytic benefits of the interlayer expansion have been observed with reactions catalysed by metal atoms inside zeolite frameworks.485 Both IEZ-MWW and IEZ-CDO containing Ti showed increased conversion of cyclo-hexene in the oxidation reaction compared to the regular Ti form. IEZ-MWW adsorbed more Ce3+ than both MCM-22 (calcined) and MCM-56. This induced the capability for spontaneous room temperature conversion of adsorbed CO to CO2, which did not occur with MCM-22 and was lower with MCM-56.486
It is fitting to conclude this section with the series of zeolites obtained via 1D to 3D condensation. Its first member is the precursor denoted ZEO-2 obtained with tricyclohexylmethylphosphonium cation as the OSDA. It consists of chains analogous to units in zeolite beta. Calcination leading to condensation of ZEO-2 to complete a 4-connected framework affords a zeolite with 16 × 14 × 14-MR channels.365 Silylation with dimethyldichlorosilane (DCDMS with one Si per molecule) or 2,4,6,8-tetramethylcyclotetrasiloxane (TMCTS; with a S4R ring of Si–O units) produced silylated derivatives similar to as-synthesized IEZ forms with dangling –CH3 groups. Upon calcination complete framework was obtained with 20 × 16 × 16-MR channels.366 Among its remarkable features is a triple S4R unit (designated TSR). The pore sizes increased to 1.43 × 1.35 nm windows on one side and 1.07 × 1.07 nm on the other. The structure has the lowest framework density (11.07 Si atoms per 1 nm3), low density of 1.10 g cm−3 and BET surface area 1533 and 1832 m2 g−1 for materials obtained with the different silylating agents. ZEO-5 is fully siliceous but has been functionalised with Ti by contacting with TiCl4. The product showed advantage in the epoxidation of cyclooctene with tert-butylhydroperoxide in comparison to Ti beta, attributed to larger pores. Designated Ti-ZEO-5 is promising for the commercial CHP-PO process (epoxidation of propylene with cumene hydroperoxide).
5.9 New zeolites produced by ordered reassembly of layers
So far, we presented two methods for producing zeolite layers: by direct synthesis using small OSDAs and with designed surfactant bifunctional templates which become embedded inside and line the layers. There is a third method involving degradation of 3D frameworks that have built-in weakness such as Ge in the structure especially in cubic D4R units connecting the layers.487,488 The first such case was zeolite UTL with intersecting 14 and 12-MR pores created by D4R bridges.489,490 Acid hydrolysis resulted in structure collapse due to degradation of the D4R units producing layers with ca. 0.9 nm thickness. The layers designated IPC-1P were swollen, pillared and silylated similarly to the traditional zeolite precursors.487,491 Subsequently it was found that intercalation of organic molecules like octylamine and calcination resulted in organisation of the layers and reassembly (condensation) into a new zeolite framework topology designated PCR (IPC-4) with 10- and 8-MR pores between layers. The entire process has been formulated as ADOR (Assembly of a 3D zeolite, Disassembly into layers, Organisation and Reassembly into a zeolite). As is illustrated in Fig. 23 there can be many additional transformations associated with the ADOR producing new materials and pore systems. A notable case is the silylated IEZ-PCR, which is a zeolite too (OKO) because its interlayer (–O)2Si(OH)2 linkers are close enough to condense producing fully 4-connected S4R bridge between the PCR layers. There is also a direct top-down pathway to OKO from UTL via inverse sigma transformation.492 Another new framework that was obtained from the IPC-1P (PCR) layers by lateral shift and alternative interconnection is denoted IPC-9. It has been designated ‘unfeasible’ because its formation via the traditional bottom-up hydrothermal synthesis is deemed impossible on thermodynamic grounds.375 The IPC-9 precursor can be also silylated before calcination yielding IEZ-IPC-9, designated IPC-10 as another novel zeolite framework.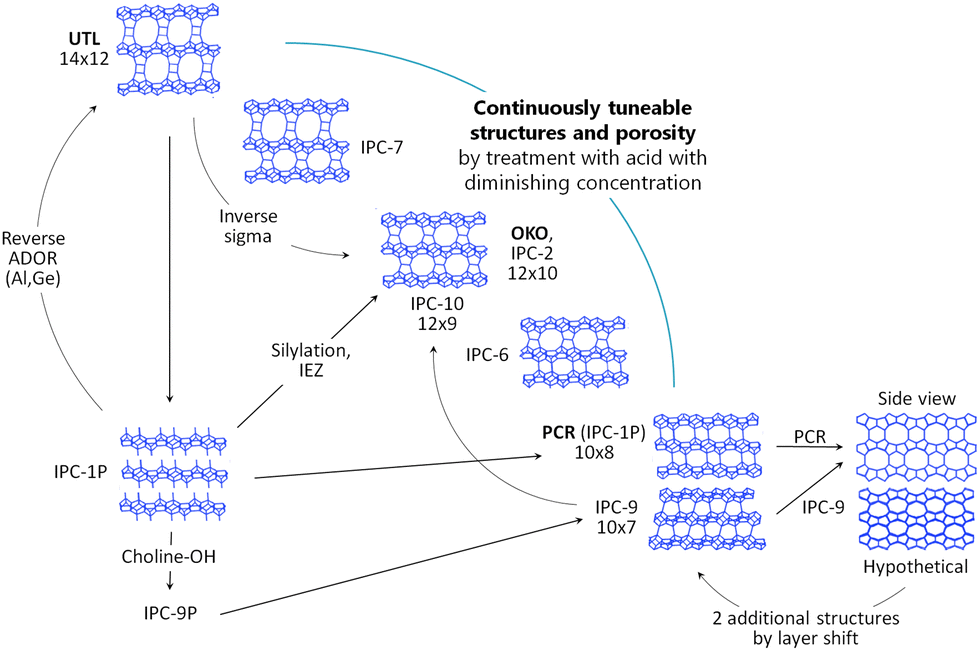 | ||
| Fig. 23 Transformations of zeolite UTL as a model for the ADOR strategy. | ||
There are several other zeolites with D4R units between layers that were transformed to new zeolite derivatives with SiO–S4R, equivalent to OKO, and simply O connections (PCR equivalent) between layers.373 Their chemistry is expected to mimic UTL but so far has not been as elaborate. Most of these zeolites were reviewed recently373 and there is also an extensive family of CIT materials based on the layer found in the zeolite CIF.370,493,494 In this case the zeolite with D4R bridges (*CTH) can be transformed into S4R connected materials (CIT-14/IPC-16) or CIF having a continuous bridge formed by fused S4R links (like a ribbon). The latter process can be included as another new type of transformation within ADOR. So far, the formation of independent layers like IPC-1P precursor, i.e. swellable etc., is scarce among the ADOR type materials reported to date. Besides UTL producing PCR separated layers *CTH and UOV were disassembled to layers from which O and S4R connected zeolites (for *CTH) and O-connected (UOV – IPC-12) were produced.495,496 Swelling, pillaring and delamination have not been reported yet with these and other layers related to ADOR (except PCR). Table 4 summarizes zeolites and materials prepared by the ADOR strategy.
| Layer or structure | Interlayer bridge | |||
|---|---|---|---|---|
| D4R | S4R | O | Other | |
| IPC-15P, SAZ-1P | *CTH | CIT-14 | CIT-15 | |
| CIT-13, SAZ-1 | IPC-16 | IPC-15 | ||
| CIF (continuous S4R) | ECNU-21 (EWO) | |||
| UOV | UOV | IPC-12 | ||
| IWW | IWW | IPC-18 | ||
| PCR layer | UTL | OKO | PCR (IPC-4) | IPC-7, IPC-6 – mixed D4R, S4R and O bridges |
| PCR layer shifted | IPC-10 | IPC-9 | +2 hypothetical | |
| IWV | IWV | IPC-20 | ||
| IWR | IWR | IPC-17 | ||
Zeolites with Ge containing D4R units that are potentially ADORable but so far have not been transformed to corresponding derivatives are ITR and ITH.
Zeolites of the ADOR family are in principle microporous with discrete large to small pore sizes depending on the type of bridging units between layers. Zeolite UTL with 14 × 12-MR pores between layers can undergo continuous removal of single and double four rings in acid solutions with decreasing concentration from 12 M to 0.1 M.497 This allows controllable tuning of the structure and porosity: basal spacing decreased from 2.4 nm to 1.8 nm, BET from 550 to 150 m2 g−1 and micropore volume from 0.22 to 0.05 cm3 g−1 as the molarity changed from 3 to 0.1. The layered precursor IPC-1P showed the possibility of producing odd-membered rings (7- and 9-MR, zeolite IPC-9, IPC-10), which are rare but can provide unique catalytic activities.
The preparations based on ADOR involve degradation by exploiting the presence of Ge, which usually leads to highly siliceous products with low acid activity. They can be functionalised for oxidation by introduction of Ti, Sn and other elements.498 Brønsted acid sites can be generated by insertion of Al in the lattice. It is exemplified by the process called Reverse ADOR in which UTL framework was recreated from the IPC-1P layers initially produced from Ge-rich UTL material (see Fig. 23).499 IPC-1P was intercalated with TBA+ and reacted with solutions containing HCl, diethoxydimethylsilane, ammonium fluoride and germanium methoxide or aluminium nitrate. The mixture was heated at 170 °C for 20 hours. The product was identified as UTL based on XRD. It showed higher activity than zeolite beta in the tetrahydropyranylation of 1-decanol. In another case, isoreticular series of catalysts with tuneable pores from 14-MR to 10-MR were obtained from zeolites UTL or Al-UTL, and contacted with Al(NO3)3 at 60–90 °C. The obtained materials: IPC-7, IPC-2 (OKO), IPC-6, and IPC-4 (PCR) had comparable Si/Al ratios between 20–26, total concentration of acid sites ∼0.4 mmol g−1 with LAS slightly higher (>0.2 mmol g−1) than BAS. Catalytic activity was tested in a liquid-phase tetrahydropyranylation of 1-decanol increasing with average pore size from IPC-4 to IPC-7.500 Another test involved gas-phase isomerization of m-xylene.501 The highest conversion and p-xylene yields were observed with the middle representative (IPC-2 with 12 × 10-MR pores). The smaller zeolites showed slowed conversions, while selectivity to p-xylene was reduced with the larger-pore materials due to xylene disproportionation.
5.10 The effects of post-synthesis modification on acidity and catalytic properties of 2D zeolites
The self-evident purpose of post-synthesis modifications of zeolites is usually to improve or impart novel catalytic activity. Modified sorption characteristics can be also of interest and subsequently exploited but catalytic benefits are the dominant objectives. There are many reports demonstrating better or new performances of modified layered zeolites in comparison to the parent 3D frameworks. Some were already mentioned above when discussing particular examples of modification and additional examples are mentioned below.Top-down modification of 2D zeolites assumes preservation of the integrity and properties of zeolite layers but in practice partial deterioration is often unavoidable. The treatments often require basic or acidic pH, elevated temperature or mechanical force, which can create defects, fragmentation or deterioration of the structure. The possible undesired effects include amorphization but even when the structure is preserved other adverse phenomena like dealumination, loss of Brønsted acid sites and increased silanol population are commonly observed. Mitigating actions, some already mentioned, include lower temperature (ambient) treatments and finding less severe reagents. On the other hand, harsher condition may be necessary for maximizing efficiency of swelling or delamination, as already emphasized for materials with higher Al content (e.g. see Table S3, ESI†). There are few systematic studies of these issues so we will focus on selected illustrative cases with general implications.
One of such studies concerned pillared MCM-36 obtained from MCM-22P precursors with different Al content, swollen at ambient (RT) and higher (HT, 80 °C). The RT treatment represented layer preservation while HT caused significant desilication tantamount to some degradation (Si/Al changing from 45 to 24). The product were compared in various catalytic reactions that are usually enhanced by higher zeolite acidity: decane and vacuum oil cracking, m-xylene isomerization, and alkylation of benzene and toluene with iso-propanol.419 The first conclusion that could be drawn is that higher Al content in the starting MCM-22P led to higher activity in all reactions. Second, degradation due to swelling at HT did not diminish activity in comparison to the same precursor swollen at RT with layer preservation.361 In some instances the HT treatment gave more desired outcomes, e.g. better selectivities of C3 and C4 olefin/alkane and lower C1–C4 gas vs. conversion. In detail, the studied materials were pillared MCM-22P with starting Si/Al (in the gel) equal to 20 and 45. The pillared MCM-36 products had Si/Al equal to 23 (gel 20, swollen RT), 54 (gel 45, swollen RT) and 24 (gel 45, swollen HT). The sample swollen at HT had more than doubled Al content compared to its RT swollen counterpart (Si/Al 24 vs. 54) due to desilication. This was undoubtedly associated with serious defect formation but had hardly any impact on the overall catalytic activity (conversion) and selectivities. The results and trends were similar for both catalysts derived from the same MCM-22P. The HT form with less preserved layer had not only comparable activity but some selectivities were better. These results emphasize high Al content as the primary factor determining activity of layered zeolite materials and that layer preservation may play a minor role or even be slightly inferior. In other words, in this instance desilication leading to Si/Al reduction does not change substantially the effective acidity, which remains equivalent to the parent (non-desilicated form). The emerging working hypothesis is that for acid catalysed reaction it is the priority to select high Al layered precursors and worry less about harsher conditions to transform into derivative structure.
The primary acid parameters of zeolites are acid site concentration and acid strength, while for 2D zeolite materials the accessibility is frequently invoked as the acquired quality. Detailed data and review of the acid characteristics of 2D zeolite derivatives has been published recently.356 We will focus on highlighting selected important topics and cases worth closer attention. The first important parameter is the acid site concentration. It is studied primarily by 27Al MAS NMR, TPD and FT-IR. The last technique is often based on determining quantitative adsorption of probe molecules. It allows distinguishing Brønsted and Lewis acid sites, denoted BAS and LAS, respectively. Accessibility of acid sites is evaluated by adsorption of larger molecules followed by FT-IR or 31P NMR of phosphine oxides of different sizes.235,363 The typical results are that accessibility, which is related to the concentration of acid sites on external surfaces, is increased sometimes up to 100% of all sites. FT-IR of pyridine adsorbed at 150 ± 20 °C can provide a very good measure of the overall concentration of acid sites. One of the striking feature of reported results for zeolite MWW which has been most often investigated is that many values of acid site concentration from different studies appear too low.356,502 Even for the parent MCM-22 and other zeolites the values can be 5 times lower than expected based on the reported Si/Al. These discrepancies are hard to explain, so the corresponding data cannot be used to reliably assess the effects of modifications on acid characteristics. Even comparison on relative basis raises doubts about correctness of the conclusions. More realistic, acceptable values are exemplified by Brønsted acid site concentrations approaching and exceeding 1 mmol g−1 for MWW with Si/Al near 10.361,502 The effect of swelling is illustrated by MCM-56 with determined BAS 0.94 mmol g−1. First, we note that a frequent result of swelling at high pH is desilication showed by Si/Al going down, especially for materials with low and medium Al content. Al atoms are treated as largely retained because of greatly reduced solubility in the presence of dissolved silica (in Al,Si-oxide systems at high pH). After swelling of MCM-56 the surfactant was removed by calcination or by de-swelling with ammonium nitrate and calcined. The BAS values were close, 0.635 and 0.649 mmol g−1, respectively, indicating about 30% loss. At the same time the values of LAS, which are typically in the range 0.1–0.2 mmol g−1, did not increase beyond 0.2 mmol g−1. Pillaring introduced silica as a neutral component resulting in dilution of BAS by 1/3 to approximately 0.4–0.5 mmol g−1. The LAS remain in the range 0.1–0.2 mmol g−1, which means relative increase when factoring the dilution by pillars. This suggests that the LAS concentration increases probably by some dealumination (of BAS).502 The observed 30% loss of BAS is not ‘transferred’ to increase the LAS and is not easy to account for. 27Al MAS NMR results indicate the presence of some octahedral Al at 0 ppm, but it does not appear to account for all ‘missing’ BAS. This suggests the possibility of invisible pentacoordinate Al. The discussed case represents a high Al MWW, which must be converted to the ammonium form and calcined for the FT-IR examination. Calcination is conducive to the formation of (undesired) additional octahedral Al, which may be absent in the as-synthesized form.
The evaluation of acid sites in MWW with lower Al content was carried out for the swelling at room temperature discussed above. It results in better structure preservation, i.e. smaller desilication, but as emphasized it may not be efficient for zeolites with high Al content. The study used MCM-22P with Si/Al = 46.7.385 The swelling with HDTMACl/TPAOH was carried out at room temperature and at 80 °C, which resulted in lowering of Si/Al (desilication) to 43.2 and 11.8, respectively. The latter clearly involved significant desilication/degradation. The products were studied by 27Al MAS NMR, which showed only tetrahedral Al and the octahedral Al signal at 0 was absent. One of the Al signals, at 50 ppm assigned to T sites inside the layer not on the surface was changed upon swelling, more for the HT process. The RT swollen spectrum was judged to more closely resemble the staring material indicative of smaller degradation. The provided data do not allow full assessment of the impact of the treatments because it would require quantitative comparison of calcined products, especially how much octahedral Al was formed upon calcination. The study of MCM-22P delamination showed that octahedral Al appeared in the 27Al MAS NMR only after calcination and was absent in the parent, swollen and delaminated products prior to the thermal treatment.446 The determined T/O ratios were 65/35 and 71/29 for delaminated materials (after calcination) with Si/Al = 20 and 50 (in the synthesis gel). Similar results were observed in 27Al MAS NMR of as-synthesized MWW zeolite (Si/Al ∼ 50) delaminated at mild conditions.447 As they were also reported without the data for calcined samples the benefits of milder conditions cannot be fully appraised. Since performance in catalysis is the main goal, the possible detrimental effects on physical/chemical characteristics may not be crucial as illustrated by the catalytic studies of MCM-36 described in the 3rd paragraph of this Section. They indicated that the primary determinant of activity is the Si/Al ratio of the parent zeolite and the extent of destruction upon modification could have lesser or little impact, thus better correlations between performance and properties are needed.
Given the definite non-negligible effect of post-synthesis transformation on the concentration of Al in 2D zeolite derivative it was a natural question to consider the strength of the resultant acid sites. It is reasonable to take into account that increased accessibility of Al sites may cause weakening of their strength, e.g. as a result of lost micropore environment. As an example, the study of MCM-22 and ITQ-2 with a range of Si/Al ratios from 12 to ∼50 concluded that delamination caused weakening of the acid strengths.503 Model catalytic reactions in the liquid state, namely isomerization of α-pinene and alkylation of hydroquinone indicated predominant influence of the accessibility over the apparent reduction of the acid strength. In a study discussed below the etherification of benzyl alcohol by layered MFI is said to be influenced by different strengths of the acid sites in mesopores.504 Overall, this issue is not settled, but in general, it seems that variations in already high Brønsted acid strength in zeolites with Si/Al greater than 10 are not particularly significant for catalysis. Recent results based on theoretical calculation with partial support of experimental data suggest preservation of the acid strength despite acquired external exposure. The conclusion was that the strength of BAS is the same for internal and external locations.505 Another study involving zeolites MWW, MFI and PCR applied both calculation and FT-IR measurements to evaluate acidity descriptors upon interaction with CO as the probe molecule.506 It concluded that ‘BAS strength of 2D forms is the same or slightly lower than that of the 3D form’.
Improved catalysis is the main goal and criterion for evaluation of the benefits of engineering of 2D zeolites. All above modifications, except ADOR focused to a large extent on structure expansion leading to increased accessibility of active sites and enhanced diffusion. These effects have been confirmed first by physical and chemical characterisation and eventually in catalytic tests employing various processes in which zeolites are used. Catalytic results for 2D zeolites have been reviewed in dedicated articles.445,507,508 They touch on all major catalytic processes and often employ model reactions but there are few leads that present commercial potential. Despite notable advances these derivative materials face, in most cases, hard to overcome competition from regular zeolites, if not in performance than economics and logistics (novelty, extra labour and cost). For this reason, it is helpful to focus on highlighting some fundamental issues that emerged from the catalytic studies.
Modified layered zeolite provide catalysts that contain both micro and mesopores with strong acid sites. This allows overcoming the limitations of microporous zeolites leading to the possibility of processing larger molecules for producing fine chemical, biomass conversion and heavy fractions in oil refining. Open architectures also allow incorporating other active elements like Ti, Sn and even enzymes.460,509 One of the first studies showing advantage of expanded layered zeolites over the conventional 3D materials concerned alkylation presented in the Newsbrief section that is not available online now, in an article by Schweitzer and van den Oosterkamp entitled ‘Overview of paraffin alkylation over solid acid catalysts’510 summarized in a review.511 The process itself requires strongly acidic media and is typically carried out in a liquid phase in hydrofluoric or sulphuric acid. High aluminium zeolites FAU, EMT and beta showed promise in replacing these highly efficient but inconvenient and harmful catalysts. Zeolite MCM-22 showed rather poor activity and performance but its pillared version with Si/Al > 15 proved much better and not much behind the above conventional zeolites. It required higher operation temperature and produced less favourable isomer ratios but excelled in one category: the stability, expressed in grams of product per grams of catalyst, was twice as much as for beta and over 5 times higher than FAU/EMT. This outcome was attributed to reduced coking resulting from a more open structure. Similar coke effect was observed with methanol to olefin process with layered MFI.235 It was explained by fast diffusion of coke precursors from the layers and resulting coke formation mainly on the external surface of the layers. The opposite occurred with Mo-layered MFI in the methane aromatization, which exhibited strong coke formation. Here, the explanation invoked higher amount of external active sites facilitating faster coke deposition and blockage of micropore entrances.512 Analogous enhancement of coking in pillared MCM-22P was reported for Aldol condensation of furfural and acetone.513 These rationalizations seem plausible but are made in hindsight, so more detailed studies may be needed to allow predictability and understanding of the coking effects.
To establish the fundamental properties and behaviour of modified 2D zeolites, model reactions of bulky molecules have been used. Alkylation of aromatic molecules such as benzene, toluene and mesitylene with benzyl alcohol have become popular model reactions.514,515 Benzylation of mesitylene is particularly valuable for evaluating activity in larger pores and studying the mechanism of reactions.378 Catalytic alkylation of mesitylene occurs only on external acid sites because of its size exclusion from micropores. Benzyl alcohol molecules can react with each other to produce the ether both inside micropores, if present and outside. The ether formed can be consumed to produce more of the alkylated product. In the literature the applied conditions vary (amounts of reactants and catalyst, temperature). A particularly instructive study was carried out for MWW, 3D and pillared, and MFI: 3D crystals of various sizes, zeolite prepared by confined growth within 3D carbon (3DOm-i), pillared and self-pillared.515 Pillared MWW exhibited much faster conversion of benzyl alcohol (100% in under 4 h) than the 3D MWW (95% 8 h). It was also more active than pillared MFI and self-pillared MFI (90% conversion in 7 h) which were in turn more active than 3D MFI samples (below 60% conversion after 7 h). Both MWW catalysts produced more benzylated mesitylene than ether (initially 60 and 40%, respectively) and the ratio increased with conversion. For MFI, the ether was produced with yields of 60% and higher and in most cases this selectivity changed little with conversion. These results underline the benefits of interlamellar pore expansion to enhance reactions of larger molecules and in this case MWW showed advantage over MFI. The study also evaluated the activity and selectivity on external Brønsted acid sites. The fraction of these sites in MWW was 70% in pillared vs. 8% in 3D MWW. For MFI the fractions of external acid sites were 38, 29, 14 and below 3.2% for the self-pillared, 3DOm-1, and the 3D crystals of different sizes, respectively. The difference between pore systems in pillared MWW and MFI (including self-pillared) is that the former has separated micro- and mesopores in and between layers, respectively. In MFI both types are interconnected. For MWW it was shown that both alkylation and etherification occurred only in the mesopores, based on the poisoning of the external acid sites with 2,6-di-tert-butylpyridine (DTBP). Both pillared MFIs catalysed alkylation only in the mesopores while etherification took place in both micro- and mesopores.
Further investigation of the parallel alkylation and etherification reaction was reported for hybrids of layered and bulk MFI with tuneable micro- to mesopore ratio from approximately 2.5:1 to 1:2.504 These materials were prepared by including in the synthesis mixture various amounts of the bifunctional C22-6-6 template promoting MFI layers. The alkylation on external acid sites remained unaffected but etherification was influenced due to different strengths of the acid sites in mesopores. Changing Si/Al of the lamellar MFI from Si/Al 144 to 25 also had an effect on ether formation (lowering TOF and effectiveness) but not on alkylation (TOF).516 Quantification of acid sites in different locations, e.g. internal and external, has been evaluated by selective poisoning with molecules of suitable size/accessibility. The situation can be more complex as illustrated by a recent study showing further differentiation including pore mouth as the third loci for catalysis. This was done by combining dimethyl ether (DME) titration and methanol dehydration in the presence of 2,6-di-tert-butylpyridine (DTBP) or triphenylphosphine (TPP), respectively.517 These molecules can access all sites (DME), external and pore mouth (DTBP) and external only (TPP). The studied zeolites were pillared MWW and pillared MFI. The former showed the pore mouth and external sites to constitute 1/3 each of the population. For pillared MFI the amounts were 6% and ∼32%, respectively. This is consistent with previous results showing greater activity of pillared MWW than pillared MFI.
6 Other techniques
There are additional noteworthy top-down treatment methods, including demetallation that involves the removal of atoms other than silicon or aluminium, such as titanium and germanium, albeit in a context distinct from the creation of layered zeolites as in Section 5.9. Degermanation is typically employed to enhance the stability of germanosilicates or to introduce Lewis acidity by the insertion of Sn or Zr. Conversely, detitanation is infrequently employed, serving to introduce secondary porosity while maintaining the initial redox functionality of the titanium framework species.Aside from mechanochemistry, zeolite treatments with microwaves, ultrasound or plasma are becoming increasingly popular. These methods often serve as alternative sources of energy, because they accelerate reaction rates and can speed up structural modifications; in general, they are seen as environmentally friendly and energy-efficient alternatives to the traditional thermal or chemical modification operations.
Laser ablation is the technique of removing material from a solid substance by using various types of lasers to evaporate a target material, which subsequently condenses into nanoparticles inside a liquid medium. Depending on the detached fragments, they may be deposited as thin films on various substrates, achieving uniform and controllable film thickness and orientation or, in the case of metal nanoparticles, become inserted into zeolites.
6.1 Degermanation
Ge in zeolite synthesis mixtures promotes the formation of frameworks with small 3- and 4-MR, as well as D4Rs, which can generate extra-large channels.11,518–520 Ge–O bonds are sensitive to hydrolytic breaking, which often reduces the hydrothermal stability of zeolites.521 Ge has higher tendency to leave tetrahedral coordination than Al, which is facilitated by the straining of Ge–O–Ge bonds in small rings. Subsequent coordination of water molecules can cause rearrangements of both extraframework species and the framework itself, promote defect development, and even amorphization. This instability may be helpful, as demonstrated in the case of generating novel zeolite structures via ADOR401 or inverse sigma transformation,492 (reviewed in Section 5.9). However, Ge is frequently removed to increase the stability of frameworks.Structural degradation of germanozeolites may already occur during the calcination carried out to remove the organic template. It was demonstrated by two Ge-AST-type fully connected germanozeolites that degradation is caused by the change of the Ge oxidation state.522 Under non-oxidising conditions (N2), template-bearing Ge-AST can be converted to amorphous GeO2 but metallic Ge may also appear. The authors also observed the sublimation of GeO. Valtchev et al.523 addressed this issue by demonstrating that one-pot simultaneous OSDA removal, Ge extraction, and Al inclusion stabilise the resulting zeolite beta-type material. Subsequent alumination produced extraframework Al, so the sample was again acid-leached. The initial ITQ-17 zeolite (BEC-type, beta polymorph C) with a Si/Ge ratio of 3.6 was converted to a material with the following characteristics (sample calcined at 600 °C): Si/Al = 30.8, Si/Ge = 6.7, Ge/Al = 4.6, high BET surface area of 609 m2 g−1, and micropore volume of 0.174 cm3 g−1.
Burel et al.524 showed that similar treatment for ITQ-22 zeolite was unsuccessful. Thus, the as-made ITQ-22 was treated with a concentrated HCl solution (37%). The authors demonstrated that crystallinity of the zeolite was preserved, and neither amorphization nor formation of a lamellar phase was observed. After the initial treatment, the starting Si/Ge = 5.6 was increased to 100 and then to 185 after repeated treatment. Degermanation resulted in the formation of mesopores (mesoporous volumes of up to 0.135 cm3 g−1), which were visible in TEM images.
Recently synthesised HPM-16, a germanozeolite with a multidimensional mixed medium-large pore system containing supercages, is unstable in the presence of water.525 Its degermanation was carried out after the OSDA was removed by ozone treatment in an alcoholic acidic solution. Following the treatment, the zeolite showed stability up to 800 °C; the authors noted a minor reduction in crystallinity following a 24-hour exposure to water at 200 °C.
Wu et al.526 found that for germanozeolites with high Si/Ge ratio (4.7), the substitution Si for Ge may be achieved with an acid treatment at elevated temperatures; however, for those with lower Si/Ge ratios, an extra silicon source and much gentler acid treatment is required. This adaptable (depending on Si/Ge ratio) Si for Ge substitution technique produced hydrothermally stable siliceous zeolites from germanosilicates with UTL (IM-12), UWY (IM-20), BEC (ITQ-17), and IWR (ITQ-24) topologies. The calcined forms of the parent Ge-zeolites were unstable in liquid water, regardless of structure. Stabilising Ge zeolites with isomorphous Si substitution resulted in highly stable structures that kept crystallinity and porosity even after treatment with concentrated (65%) HNO3 at 150 °C and calcination at 550 °C.
Post-synthesis stabilisation of OSDA-containing germanozeolites ITH (Si/Ge = 2–13), IWW (Si/Ge = 3–7), and UTL (Si/Ge = 4–6) by isomorphous substitution of Al for Ge was proposed by Shamzhy et al.527 It was found that this alumination treatment depends on the framework topology and germanium content. In Ge-poor IWW (Si/Ge = 7) and ITH (Si/Ge = 13), 70–78% of the Ge was replaced by Al atoms, while in Ge-rich IWW (Si/Ge = 3), only 30% was replaced. Alumination produced more acid sites in large-pore IWW (711–757 μmol g−1) than medium-pore ITH (211–330 μmol g−1), suggesting diffusion control of the alumination process.
Recently, Sn and Zr were incorporated into extra-large pore zeolites UTL and *CTH, with Si/Sn = 40–51 and Si/Zr = 14–15, through a four-step approach: synthesis of parent germanosilicate zeolites, followed by post-synthesis stabilisation, degermanation, and metal incorporation.528 Stabilisation was achieved by incorporation of Si atoms (via acid leaching and TEOS hydrolysis), while degermanation (treatment with distilled water) led to the formation of silanol defects, subsequently healed by metal incorporation. The resultant materials were Lewis acids with LAS concentrations 210 μmol g−1 for Sn/UTL, 586 μmol g−1 for Zr/UTL and 183 μmol g−1 for Sn/CTH, 474 μmol g−1 for Zr/CTH. The incorporation of Zn was much easier than Sn, independently of the structure. All materials were active in the test reaction, reduction of furfural to furfuryl alcohol under batch and flow conditions.
The removal of germanium may lead to zeolite-to-zeolite transformation, such as the transition of Ge-CIT-13 (*CTH) to Ge-CIT-5 (CFI) in the presence of water (mentioned in Section 5.9).493 Partial Ge leaching rearranges germanium-rich D4R in CIT-13 to form double-zigzag chains in CIT-5. Ge-CIT-5 can be converted into CIT-15 (isostructural to IPC-15 and ECNU-21) using ammonium hydroxide solution as a delaminating agent or aluminated (by acid treatment and Al(NO3)3) to produce high-silica CFI-type aluminogermanosilicate with Si/Al = 14–230.
6.2 Detitanation
The incorporation of titanium into a zeolite structure affects its catalytic activity, making it appropriate for various industrial and environmental applications, especially in the field of selective oxidation processes.15,529,530 The opposite treatment, purposeful removal of Ti atoms, is rarely practiced.Classic demetallation methods could be used to obtain hierarchical TS-1 zeolite, but it is not focused on the removal of Ti alone. In most cases hierarchization is achieved due to removal of silica (by base etching)531–533 or Si and Ti species together by NH4F leaching.534
In contrast to that, Pavel et al.535 documented the development of supermicropores (0.76 × 1.98 nm), 5–20 nm mesopores and surface macropores in the ETS-10 structure through post-synthesis microwave assisted H2O2 treatment. The mesopore size and distribution was controlled by changing H2O2 concentration, contact time, and temperature. Systematic studies indicated that mesoporosity started to develop at 100 °C even with low H2O2 concentration. In ETS-10 the Ti–O–Ti chains are perpendicular to one another in two dimensions, and their removal produces pores orientated primarily along these chains.
6.3 Microwave and ultrasound treatment
Microwave (MW) irradiation is recognised as a cost-effective alternative to existing heating technologies. The efficacy of microwave heating surpasses that of conventional thermal methods due to the direct interaction of microwaves with the medium.536 This interaction effectively addresses heat and mass transfer limitations, allowing homogeneous and rapid heating of irradiated materials, thereby reducing heating times from hours to minutes.537 However, despite the promising results from lab-scale microwave applications, the translation of these findings into industrial-scale microwave processes remains minimal,538 and this is also true for zeolite technology.Almost 40 years ago, with the development of microwave technology and commercially available microwave ovens, Komarneni and Roy reported ‘anomalous microwave melting of zeolites’ and demonstrated that some zeolites can be melted in a few seconds using a kitchen-type microwave oven (at 2.45 GHz).539 Although it could hardly be called a top-down ‘modification’ because it resulted in the total destruction of the material, it demonstrated that ceramic materials absorb microwaves and may be processed using this radiation. The strongest effect observed for sodium forms of FAU (Linde 13X) and LTA was attributed to ionic ‘rattling’ in rigid cages. These studies were continued during following years, with a focus on the interaction of zeolitic water and zeolitic cations with MW radiation. It was found that initially, the major absorbing medium is zeolitic water, while a zeolite begins to absorb microwaves directly around 300–400 °C, when it is completely dehydrated, and the absorption efficiency increases with increasing temperature.540
Nowadays, for modification of the zeolite properties MW treatment is not used as a ‘stand alone’ methodology, but rather as a method of providing extra energy during standard zeolite treatments, usually during synthesis, but there are examples of MW application during ion-exchange or demetallation, shortly described below. The ‘microwave effect’ not only shortens the operation timeline, but it can also cause changes in the characteristics of the MW-treated materials.
The effect of MW treatment during desilication with NaOH was investigated for two ZSM-5 zeolites, synthesized with different Si/Al ratios, 200 and 15.541 The effect of 10 minutes MW treatment (at 700 W) was compared to conventional heating at 80 °C during 2 h. Unfortunately, the Si/Al ratio for both zeolites were outside of the optimal values for desilication, therefore the effects of MW treatment were not beneficial for the catalytic properties.123 For the Si-rich ZSM-5, the microporosity was almost completely destroyed during desilication (micropore volume decreased from 0.110 to 0.041 and 0.034 cm3 g−1, for MW and thermal treatment). In contrast, for Al-rich ZSM-5 the effect on microporosity was negligible (0.122 vs. 0.120 and 0.101 cm3 g−1). The changes extended to external surfaces, which increased 12–13 times for the ZSM-5 with Si/Al = 200 and only 1.4–3 times for ZSM-5 with Si/Al = 15. In n-heptane cracking, the MW-modified zeolite samples, independently of Si/Al, showed only slightly higher (and similar for MW and thermal desilication) catalytic cracking efficiency than the original MFI zeolites. The obtained results show that the ‘microwave effect’ did not change the mechanism of desilication.
The effect of MW treatment during ion-exchange was investigated by Kuroda et al.,542 who compared the effectiveness of room-temperature N2 sorption in Cu-ZSM-5. The authors showed that the number of monovalent Cu able to irreversibly chemisorb dinitrogen at RT increased 4 times in the MW-exchanged sample, and that the total amount of chemisorbed N2 increased from 0.60 to 1.66 cm3 g−1.
Microwave-assisted chelation (MWAC) was developed as a versatile approach of post-synthesis zeolite modification that includes controlled dealumination and surface hydrophobization. It was proposed by Zhang et al.,543 who modified commercial Y zeolite (FAU, CBV 300, Si/Al = 2.6) by a combination of the microwave-assisted dealumination and hydrothermal alkaline treatment. The authors investigated the effects of the type of dealuminating agent (mineral acids versus organic carboxylic acids) on the characteristics of the modified zeolites. Their findings indicated that the mineral acid (HCl) caused hydrolysis of the zeolite, while carboxylic acids, through their chelating function, were effective in extracting framework aluminium and generating mesopores. Depending on the acid used, Si/Al increased to 3.8 and 6.3, the external surface areas increased from 9 to 148 and 214 m2 g−1, for oxalic and diethylenetriaminepentaacetic acid (EDTA), respectively. The effectiveness of the formation of mesopores depended on the number of coordination sites present in the carboxylic acid. In the following work the mechanism of dealumination and mesopore formation was studied.544 The authors concluded that under MWAC conditions, EDTA directly complexed framework aluminium, and that the hydrolysis of framework Al–O bonds and the production of silanol groups was facilitated by zeolitic protons rather than by water molecules.
Abdulridha et al.545 investigated the acid and catalytic properties of MWAC-treated zeolite Y, called mesostructured zeolitic materials (MZMs) with mesopore sizes ranging from 2 to 10 nm. The obtained materials had high mesopore area (260 m2 g−1) and volume (0.37 cm3 g−1). The material was tested in two reaction involving bulky reactants, 1,3,5-triisopropylbenzene dealkylation and benzaldehyde condensation with 1-heptanal. In both reactions the performance of the MZM material was superior, which was attributed to the presence of mesopores (despite moderate acidity, 0.6 mmol g−1).
The MWAC treatment was also used to dealuminate zeolite beta and increase its hydrophobicity for applications in organics removal from aqueous media.546 The MWAC-treated zeolite beta had enhanced surface hydrophobicity (contact angle increased from 16.2 to 35.2°), which the authors attributed to reduced framework polarity resulting from the elimination of polar OH groups and the healing of silanol nests through calcination. The benzene adsorption capacity increased by ca. 31% in comparison to the parent zeolite beta.
The MW treatment was also used to functionalise beta (*BEA) and SBA-15 with sulfonic groups.547 During sulfonation new silanol groups were formed, which in turn reacted with the organosulfonating agent, chlorosulfonylphenylethyltrimethoxysilane (CSPTMS), resulting in the incorporation of the (organo)sulfonic acid groups. MW-sulfonated catalysts showed high selectivity towards glycerol di- and triethers (83–91%) in the test reaction of glycerol etherification with isobutene, compared to the conventionally sulfonated materials (61% for SBA-15 and 51% for beta).
Sonochemistry uses ultrasonic waves at frequencies ranging from 20 kHz to 10 MHz, with high enough energy to accelerate chemical processes and trigger specific reactions.548 Ultrasounds produce alternating expanding and compressive acoustic waves, resulting in oscillating microbubbles (cavitation phenomena). Their collapse generates localised hot spots with temperatures of approximately 4500 °C and pressures of around 1000 bar.549
In the zeolite science sonication is most frequently used to accelerate the crystallisation process. Ultrasound treatments enable adjustments of the properties of zeolites because the cavitation effect generated by ultrasound can lead to better dispersion of reactants and improved molecular interactions.550 The application of ultrasonication in synthesis and modification of layered zeolites was mentioned in Section 5.6.
Sonication is also frequently used for modification of the properties of extraframework species, such as exchangeable cations, metal oxides or nanoparticles. In such cases sonication usually does not change the morphology, porosity or framework composition of the treated zeolites.551–553
A sequential treatment – hydrothermal dealumination (combined with chelation by EDTA) followed by ultrasound-assisted alkaline treatment was investigated for FAU with Si/Al = 2.6.554 The resulting mesoporous zeolite Y had specific external surface area of 160 m2 g−1 and mesopore volume of 0.22 cm3 g−1, slightly higher than subjected to classic desilication (128 m2 g−1 and 0.19 cm3 g−1, respectively). The acidic properties and catalytic activity (n-octane cracking) of mesoporous Y zeolites produced by the two methods were comparable. Sonication was advantageous for shortening the reaction time.
Secondary mesoporosity was generated in EMT zeolite using an NH4F treatment with addition of the HDTMABr surfactant at low temperature with ultrasound irradiation. The results indicated that this is an effective technique for obtaining hierarchical EMT, although prolonging the sonication time or increasing the temperature (from ambient to 65 °C) of this treatment caused amorphization of the zeolite. The catalytic activity was tested by glycerol dehydration reaction showing 80% selectivity to acrolein.
Kulak et al.555 employed water droplets dispersed in toluene as templates for assembling zeolite nanocrystals into microspherulites. As model nonspherical nanoparticles (with sizes of ca. 150 nm) the nanocrystals of zeolite X (FAU, octahedral), LTA (cubic) and LTL (cylindrical) were used. The authors proposed that water droplets, dispersed in toluene, acted as the templates, attracting zeolite crystals and the subsequent sonication caused formation of siloxane bridges via dehydroxylation of the adjacent silanols. When the surfactant (sodium dodecylsulfate) was added to water, previously randomly distributed crystals became ordered, up to two layers from the surface. The procedure worked well with FAU and LTA, but not for the LTL zeolite, probably due to elongated shape of the crystals. When intermediate sonication times were used, perforated spherules were formed, and the presence of surfactant aided in the hole formation.
Ultrasonication allows organisation of zeolite crystals on carrier surfaces. Kim et al.556 used sonication to selectively deposit plate-like Si-CHA particles (minor product), co-synthesized with near-cubic ones (major product), onto an α-Al2O3 disc, resulting in a membrane with high potential for CO2 separation in the presence of H2O. Two major variables contributed to the successful formation of the oriented layer. Sonication allowed homogeneous deposition of calcined, dry Si-CHA crystals on the disc. Cover glasses sandwiching the disc prevented larger near-cubic particles from reaching the disc surface, which was thus covered with smaller platelike crystals. The authors noted that physical interactions between the platelike CHA and the disc were sufficient to form a homogeneous layer, eliminating the necessity for chemical attachment between the zeolite and the support.
An interesting combination of ultrasound and microwave treatments was used by Schmidt et al.557 who, by post-synthetic modification of twinned MFI prisms obtained single-crystal segments of MFI (silicalite-1 and ZSM-5). The microwave irradiation of the parent crystallites in an alkaline solution with hydrogen peroxide, followed by ultrasonic treatment, resulted in disintegration of the twinned zeolite crystals. The resultant wedge-shaped segments had exceptional crystallinity as evidenced by optical and X-ray analyses. The integrity of the crystallite interior was validated using N2 and Ar sorption. It was attributed to the protective role of the template, which prevented desilication inside micropores. The authors examined internal and external structures of the obtained pyramidal segments of ZSM-5, silicalite-1, and calcined ZSM-5, and found that the straight pores were aligned parallel to the ridge of the roof at the blunt end (Fig. 24). This altered the quantity of pore openings exposed on the external surfaces of the MFI crystals and affected the diffusivity of reactant molecules. In the following work it was shown that the sorption kinetics of pure isobutane in silicalite-1 crystal fragments was primarily governed by intracrystalline diffusion.153 Nonetheless, even traces of water significantly altered the scenario, with sorption becoming governed by resistances on the crystal surface, signifying development of surface barriers.558
 | ||
| Fig. 24 SEM images of wedge shaped (pyramidal) ZSM-5 crystals, cut perpendicular to the c-axis. The inset shows typical morphology of the pyramidal segment.557 Reproduced from ref. 557 with permission from the American Chemical Society, copyright 2007. | ||
6.4 Plasma treatment
Plasma technologies are employed for green synthesis because of their rapidity, diminished reliance on hazardous chemicals, and reduced generation of harmful waste. Presently, there are three primary applications of plasma, all of which may be applied to zeolites: (1) the synthesis of ultrafine particles, (2) plasma-assisted deposition of catalytically active chemicals onto carriers, and (3) plasma-enhanced fabrication or modification of catalysts.559 Most of these procedures are bottom-up approaches, but a few reports of the top-down methods can be also found.Batur et al.560 subjected seeds of three zeolites (MFI, LTA, and LTL) to argon cold atmospheric-pressure plasma jet (Ar-CAPPJ) treatment for up to 30 min during the aging period. Three, coexisting mechanisms of plasma influence were proposed. In the first, the species formed by plasma jests were attached to the surface of the seeds, thus modifying their chemistry. In the second, plasma enhanced the production of soluble silicates and increased Al concentration in the supernatant. The third mechanism assumed that the shape of the crystals was modified by ‘sculpting’ the precursors. The surfaces of the plasma-treated seeds were more rounded and noticeably larger than those of the control (100–130 nm vs. ca. 90 nm) by the end of the aging period.
El-Roz et al.561 used low-temperature SiCl4 and TiCl4 plasma treatment of as-synthesized Ge-silicates to achieve structural stabilisation of a BEC-type germanosilicate. The plasma treatment resulted in simultaneous removal of the template, partial extraction of Ge from the framework, and incorporation of Si and Ti into the micropores. The plasma-treated material was subsequently calcined, which completed the incorporation of the cations into the zeolite framework.
Plasma treatment was also employed to modify the surface of mesoporous SBA-15.562 During the plasma treatment in an O2 atmosphere, the surface grafting rate of amine was significantly accelerated and the reaction time shortened from 18 h to 2 h. The authors observed that the number of silanol groups increased, and zeolites were ‘activated’, most likely due to the production of free radicals in the reactant solution, allowing the grafting of a large number of amine groups (2.56 mmol APTS per g of sample). The positioning of in situ coordinated Ag nanoparticles allowed easy detection of the highly scattered amine groups within the channels. Furthermore, the amine-modified SBA-15 via plasma treatment had a higher CO2 adsorption capacity (1.26 mmol g−1) than the typical amine-modified SBA-15.
The same methodology was used to activate silanol groups in MCF (mesostructured cellular foam) silica. The introduced silver nanoparticles had smaller average size, higher loading, and better dispersion, which resulted in improved catalytic activity in the test reaction of the reduction of 4-nitrophenol. MCF-100-Ag-0.01, containing 2.6 wt% of immobilised Ag nanoparticles (average size of 6.0 nm), displayed the highest catalytic activity, quantified as very high TOF (8.97 × 1018 molecules g−1 s−1).
A DBD (dielectric-barrier discharge) plasma method was used to accelerate the synthesis of core–shell zeolite composites made from 13X zeolite (FAU, core) and NaA zeolite (LTA, shell).563 The 13X powders, or a mixture of NaA precursor gel and 13X particles, were treated with oxygen plasma which resulted in increased concentration of surface silanols (13X zeolite) and hydroxyl radicals (NaA gel) and consequently, rapid formation of LTA shell (under 30 minutes). The thickness of the LTA shell and pore size of the resulting FAU@LTA composites were regulated by potassium cation exchange, which reduced the pore size of the A zeolite, impeding the N2 sorption. The composites exhibited high selectivity of CO2 over N2 sorption, 149–380, while maintaining good sorption capacity of 1.84–3.41 mmol g−1.
6.5 Laser ablation
Laser ablation employs high-energy laser pulses to remove material from solid surfaces, enabling precise modifications while eliminating the need for chemical etching. This method can be used to obtain nanozeolites (LTA).564 Laser fragmentation is a popular top-down approach for the generation of metallic nanoparticles or their implantation on various carriers. The process may be carried out in both liquid and solid states. It was also used to prepare metal nanoparticles inside zeolites.565–567Laser ablation demonstrates potential as an effective technique for the formation of zeolite-based mesoporous structures and zeolite films. This technique was used to prepare oriented, or partially oriented membranes (MOR, FAU – X type, UTD, UTD/ZSM-48 or AlPO4) by seeding on the support.568–573
In this method, the zeolitic material is ‘torn’ from a pressed zeolite pellet by a high-intensity excimer laser beam. The fragments are deposited on a temperature-controlled substrate and undergo a hydrothermal treatment which restructures the laser-deposited zeolite film.568–570 In the case of UTD-1 zeolite, a laser-deposited film was X-ray amorphous, and served as a seed layer or a nucleation site for film recrystallisation.569 The author proposed that the oriented growth along the b-axis resulted from the dense packing of the nanocrystals, which served as an autogenous rigid template, permitting crystal growth exclusively perpendicular to the support. The production of thin films using pulsed laser ablation presents numerous advantages compared to direct synthesis techniques, such as the ability to produce well-adhered continuous and (in some cases) oriented films, with quite precise control over film thickness ranging from a few hundred nanometres to several microns, depending upon the experimental settings. For comparison, see zeolite film fabrication from exfoliated nanosheets in Section 5.7.
7 Top-down engineering of nanozeolites
One approach for improving the contact between an active centre and reactant molecules is to reduce the size of crystals, which shortens the passage of the reagent through the micropores. What are nanozeolites, then? There is no clear distinction between a standard zeolite crystal and a nanozeolite, so the change in properties should be used as the criterion. A nanozeolite should exhibit properties different from those of a classical zeolite crystal with micrometre dimensions. For that reason, Mintova et al.574 recommended restricting the crystal size of the materials known as ‘nanozeolites’ to the range of 5–500 nm. Nanosized zeolites can be fabricated in several forms, including colloidal suspensions, powders, self-supported pellets, self-supported or on-support thin films, membranes, and optical-quality films.575 Nanozeolites may be produced either by bottom-up574–576 (in situ synthesis) or a top-down strategy. In contrast to numerous bottom-up methods applied for the synthesis from simple components (zeolite growth into enclosed spaces, microwave and ultrasound irradiation, synthesis from the clear solutions, and dry gel conversion),577 the top-down strategies are to date mostly limited to desilication or mechanochemistry accompanied by recrystallisation.Some examples of desilication, which were also applied to nanocrystals, was reviewed in Sections 1.6, 3.4.1 and 3.5.
Okubo et al.578 reported ultra-fast crystallisation of industrially important materials: zeolite SSZ-13 and aluminophosphate AlPO4 using continuous flow reactor and seed-assisted synthesis. The synthesis lasted only a few minutes and gave large, micron-sized crystals. Ultrafast synthesis, by extension, required ultrafast reduction of the crystal sizes, leading to the development of a technology wherein zeolites are bead milled and recrystallised in a tube reactor using the supernatant liquid obtained from the synthesis of conventional zeolites, which would otherwise be discarded.579 The optimal recrystallisation duration was 10 min and the recrystallised SSZ-13 maintained cubic morphology of the original material. The intensities of the XRD reflections were restored, the porosity was only slightly affected; the mesopore volume decreased from 0.26 to 0.24 cm3 g−1, and the BET surface area decreased from 710 to 685 m2 g−1.
A mechanochemical top-down strategy to form nanozeolites proposed by Wakihara et al.327 involves two stages: bead milling of micrometre-sized ZSM-5 zeolite crystals (840NHA Tosoh Co., Japan) to generate nanoparticles, followed by recrystallisation of the damaged outer layers in a diluted solution containing only amorphous silica and sodium hydroxide. The synthesised nano ZSM-5, approximately 60 nm in size, was evaluated in the cumene cracking process to produce benzene and propylene. Regardless of comparable Si/Al ratios and Brønsted acid site concentrations (20.9 vs. 19.7; 497 vs. 443 mol g−1 for the initial and nano ZSM-5, respectively), the authors noted superior benzene yield (96.5 vs. 70.2%), attributed to augmented external surface area of the milled and recrystallised nanozeolite (69 m2 g−1), since all examined materials exhibited identical acid strength. The observed extended catalyst lifespan was attributed to inhibited coke accumulation although the coke was not examined. The procedure was also applied to a commercial X zeolite (FAU).328
Pure-phase nano-MOR (crystal sizes 20–160 nm) was produced by high-energy wet ball milling of a natural mordenite composed of MOR, HEU (clinoptilolite), and quartz, followed by recrystallisation in sodium silicate solution.580 The impurities amorphized faster than the mordenite, resulting in decreased crystal size and impurity elimination; recrystallisation to pure mordenite was seen after just two hours of hydrothermal treatment, and it stayed as the pure phase until six hours, when quartz emerged as a secondary phase.
A sequential mechanochemical treatment and recrystallisation with HDTMA surfactant as a mesopore-forming template was shown to be effective in the production of nanosized ZSM-5 aggregates with intercrystalline mesopores and enhanced external crystal surfaces. As a result, the indexed hierarchy factor (IHF, depicting the enhancement of the external surface and mesopore volume) increased from 0.18 to 0.32.581 HDTMA inhibited excessive coalescence and crystal growth; in its absence, only micron-sized ZSM-5 crystals were produced. The hierarchical nano-ZSM-5 had higher activity in LDPE pyrolysis; the T50 value (temperature of 50% conversion) decreased from 461 to 395 °C with respective change in the activation energy from 341 to 132 kJ mol−1.
More complex structures, such as hollow nanozeolites or yolk–shell composites, were also produced, using the same approaches as for micrometre-size zeolites, i.e. either dissolution–recrystallisation or mechanochemistry followed by recrystallisation. The yolk–shell composite catalysts were formed using solvent-free mechanochemical grinding of nanoscale silicalite-1 crystals in the presence of NH4F and tetrapropylammonium bromide (TPABr), followed by hydrothermal treatment.582 Platinum and cobalt nanoparticles were added either before (Co) or after (Pt) the milling-heating process. The TPA+ cations established protective coating on the external crystal surfaces, allowing F− ions to diffuse into the crystal interior and form hollow structures. The dissolution started at structural defects to generate smaller mesopores, which were subsequently merged to form macropores and, finally, void volume in the centre of the crystals. Hollow nanozeolites with double shells were produced using the dissolution–recrystallisation method, starting with regular ZSM-5 ‘nanoboxes’ of sizes 120 nm × 180 nm.583 In the first step, hollow ZSM-5 was prepared and subsequently added to silicalite-1 synthesis gel to obtain hollow ZSM-5@Silicalite-1 or to ZSM-5 synthesis gel to obtain ZSM-5@void@ZSM-5. In turn, hollow ZSM-5@Silicalite-1 crystals were used to obtain even more complex hollow ZSM-5@Silicalite-1@ZSM-5. The synthesis scheme and resultant ZSM-5@void@ZSM-5 crystal are presented in Fig. 25.
 | ||
| Fig. 25 The scheme of construction of hollow ZSM-5 with double shells (a), TEM images of hollow ZSM-5@void@ZSM-5 (b) and (c).583 Reproduced with permission from ref. 583 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2015. | ||
8 Hierarchical zeolites in acid-catalysed reactions
Hierarchical zeolites are designed for acid-catalysed conversion of reactants that have large kinetic diameters. Conventional zeolites tend to deactivate quickly due to significant adsorption of heavy products and the build-up of carbon deposits within their relatively small micropores. The extended lifetime of hierarchical catalysts is attributed to the presence of open mesopores with a well-connected intracrystalline hierarchical pore network, as opposed to mesopores with poor connectivity. Hierarchical zeolites with open pore structures enhance a catalytic activity in processes such as Fischer–Tropsch synthesis by providing improved accessibility of acid sites, combined with expanded external surface area, improving metal dispersion. The balance between acid and redox functions is crucial in designing new bifunctional catalysts for hydrocracking, hydroisomerization, and hydrodesulphurization. In these catalysts, the acidic components facilitate cracking and isomerization, while the supported metal sites support hydrogenation functions. Introducing mesopores into zeolites or reducing the zeolite crystal size significantly accelerates mass transfer to and from the catalytic sites, thus limiting the possibility of secondary reactions promoting coke formation and catalyst deactivation. The effect of mesoporosity on the activity of the methanol-to-hydrocarbons (MTH) process is well-documented. The reason for prolonged lifetime of the MTH catalyst was the improved diffusion of coke precursors from the micropores to external surfaces. Coke build-up leads to significant catalyst deactivation, since it covers the acidic sites and blocks micropores, even at low levels of coking. The overall connectivity of the intracrystalline pore systems is a crucial factor in extending the lifespan of zeolite catalysts involved in the transformation of organic molecules. In the fluid catalytic cracking process (FCC), such open hierarchical porosity reduces the undesirable secondary reactions, like the overcracking of the valuable gasoline fraction, interconversion of olefins, and, particularly, coking. Hierarchically porous zeolites demonstrate significant potential for addressing the environmental issue of plastic waste. During the cracking process, large polyolefin molecules cannot enter the micropores. The catalytic activity of these catalysts is highly dependent on their external surface area; therefore, the presence of mesopores decreases the unused volume of the catalyst. Other notable examples, where hierarchical zeolites demonstrate greater activity and selectivity compared to conventional zeolites are industrially important Friedel–Crafts alkylation and acylation reactions.The appropriate strategy for top-down modification must be customized for each specific catalytic reaction. Therefore, a crucial aspect of designing targeted catalysts is to gain a thorough understanding of how each method influences factors such as the type and distribution of acid sites, pore structure, and structural stability under operational conditions. Each modification method offers a compromise between pore diameter enlargement, and maintaining structural stability and zeolite acidity. Dealumination, while relatively straightforward to implement, may lead to undesirable acid site density reduction if not optimised. Excessive dealumination can reduce the total density of acid sites, which impairs catalytic activity or even changes reaction mechanism (favouring bimolecular catalysis) or promotes unwanted side reactions. The same issue arises with desilication; excessive framework degradation can lead to significant reduction in Brønsted acid sites concentration and loss of zeolite stability. Recrystallisation, a more controlled process, allows the restoration or preservation of the ordered zeolitic framework while generating hierarchical porosity. This technique is highly effective in retaining Brønsted acidity; however, it is limited by industrial scalability. Mechanochemical approaches, such as milling and patterning, offer rapid and scalable alternatives to traditional chemical methods. These processes can induce nanoscale restructuring, which enhances external surface area and pore architecture. However, the intense mechanical forces can also damage the zeolite framework, potentially resulting in a loss of acid sites or decreased thermal stability. Low-dimensional and layered zeolites can enhance accessibility by undergoing delamination, pillaring, or surfactant-assisted expansion. These processes increase the external surface area and interconnectivity of the pores. As a result, these materials often exhibit superior performance in reactions involving large, bulky molecules due to their shorter diffusion paths. However, their stability under typical reaction conditions, especially in the presence of moisture or at elevated temperatures, can be an issue limiting their long-term effectiveness as catalysts.
9 Methods and descriptors used to characterise changes in porosity
Reactions in porous catalysts require many steps; in addition to the reaction taking place on specific active sites, the reactants must adsorb and diffuse towards and away from the active centres.Van Donk et al.584 clearly showed how important is characterisation of the porosity and how limited diffusivity in micropores reduces accessibility of the active sites. The authors determined that for mordenite, crystallised in the form of elongated crystals, the accessible length of the micropores was as low as 30% of the value expected from the crystal size (Fig. 26).
 | ||
| Fig. 26 SEM image of HMOR (Si/Al = 5.5). The white bars indicate the accessible pore length. Reproduced from ref. 584 with permission from WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2005. | ||
The formation of secondary porosity in zeolites, regardless of the process, should produce materials with hierarchical structure of pores, usually called ‘hierarchical zeolites’. The broadest definition of hierarchical zeolites is that they are a category of porous materials featuring both micropores (diameter less than 2 nm) and mesopores (2–50 nm). They are designed to improve mass transfer characteristics and catalytic activity by integrating the advantages of conventional microporous zeolites with supplementary mesoporosity. Schwieger et al.585 emphasise that introduction of the secondary porosity is advantageous only if the system is ‘truly hierarchical’, meaning that various pore systems are coupled appropriately, i.e. the larger pores lead to smaller ones. The following two types of hierarchy are distinguished: type I, in which each level of bigger pores subdivides into several levels of narrower pores at the subsequent level, and type II, where the larger pores intersect with the smaller pore system.
Better micro- and mesopore interconnectivity ensures fast desorption and diffusion of coke precursors generated on the active sites and helps to increase catalyst lifetime.586,587 The advantageous effect of secondary porosity on the speciation and accessibility of acidic sites, and consequently on catalytic activity and selectivity, has been demonstrated for numerous hierarchical zeolites.212,213,588 Examples include improved selectivity for xylenes during toluene disproportionation and branched hydrocarbons in the hydrocracking process, while minimizing the formation of undesired secondary products. These changes can be attributed to decreased contact time between the reactants and active sites.198,589,590 This can be exemplified by the improved performance of the hierarchical ZSM-5 zeolites (MFI) in the methanol-to-hydrocarbon (MTH) reaction. Enhanced diffusion of aromatic intermediates inhibits the aromatic-based catalytic cycle in favour of the olefin-based cycle, which leads to the formation of light olefins instead of aromatics.591 In bi-functional hydrocracking catalysts, the principle that ‘the closer, the better’ regarding the proximity of metal and acidic active sites is considered the golden rule. Nevertheless, some studies on bi-functional hierarchical zeolitic catalysts, providing reduced diffusion pathway, contradicted the aforementioned assumptions.592–594 On the other hand, secondary porosity can negatively impact zeolite shape selectivity, particularly in processes like xylene isomerization, where the selectivity for p-xylene diminishes in hierarchical zeolites compared to purely microporous counterparts.192,595 The other example where mesoporosity may be counterproductive, is modification of wide-pore (12-MR) zeolites, because the decreased acidity (acid sites concentration) is not balanced by a substantial number of readily accessible sites.169 Therefore, the connectivity between porous structures must find a balance between enhancing mass transport and maintaining the intrinsic acidity, shape and transition state selectivity of zeolites.
The optimisation of pore systems necessitates precise descriptors. There are several proposed to date, such as bio-inspired Murray’s Law, the hierarchy factor based on the porosity measurement, or the effectiveness factor defined as the ratio of the reaction rate with pore diffusion to the reaction rate at the particle surface. The so-called ‘generalised Murray’s Law’ is based on the rule that the optimal design of the pores should provide least diffusion resistance while maintaining the lowest feasible volume of the pores.596 This law applies to materials with pore diameters that drop throughout several scales and eventually terminate in size-invariant units, with well-defined regular pores. Such organisation is uncommon in materials obtained by most top-down approaches and hence will not be covered here.
Hierarchy factor (HF) is an important descriptor that directly correlates with the porosity changes.124 It is calculated as the product of relative micropore volume and mesopore surface area (eqn (1))
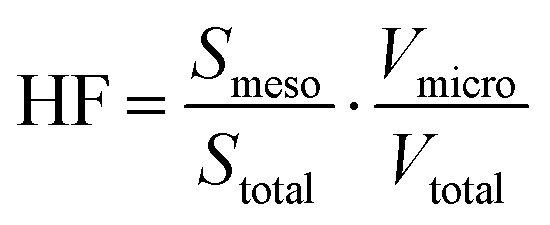 | (1) |
 | ||
| Fig. 27 A contour plot of hierarchy factor HF as a function of the relative mesoporous surface area and relative microporous volume based on the data from Tables S1 and S2 (ESI†) when sufficient details were available in the publications. The points are marked in the following way: catalysts discussed in Section 1: rectangles for desilication, diamonds for dealumination followed by desilication (full symbols for parent materials, empty for modified), black for 8-MR, red for 10-MR, magenta for 12-MR, orange for 12 × 8-MR, grey for 10 × 8-MR; Section 3 (recrystallisation): full circles; Section 5 (low-dimensional zeolites): full stars; Section 6 (other): full down triangles; Section 7 (nanozeolites): full up triangles. | ||
Aside from porosity, the most significant metric for modified material is acidity, specifically the concentration of acid sites available for a given reactant. As a result, another descriptor is useful: the accessibility index (ACI), which standardises acid site accessibility.213,597 The ACI is determined by comparing the number of acid sites able to react with substituted, bulky pyridines to all acid sites present in the zeolite. The effectiveness factor (η, eqn (2)) was introduced as the first quantitative descriptor for estimating the effect of diffusion limitation on catalytic reactions.598 It was originally introduced for macroscopic spherical catalyst pellets, but is applicable to the majority of irreversible processes.599 It is calculated as the ratio of observed (apparent) and intrinsic reaction rates (eqn (2)) and depends on the Thiele modulus (ϕ, eqn (3)), a dimensionless number characterising the ratio of the reaction rate to the diffusion rate.600 For low Thiele modulus (ϕ close to 0), the effectiveness factor η is close to 1. This means that the intrinsic and apparent reaction rates are nearly the same, and the entire catalyst surface participates in the reaction.
 | (2) |
 | (3) |
Thiele modulus and the effectiveness factor provide a universal criterion for the region of pronounced diffusion limitation (Fig. 28). A low Thiele modulus indicates high mass transport efficiency, attainable via shortening the diffusion path or enhancing effective diffusivity within the zeolite pores.198,308
 | ||
| Fig. 28 Effectiveness factor η as a function of Thiele modulus ϕ. | ||
Among all above descriptors, only the hierarchy factor can be extracted from the literature data, but as shown in Tables S1 and S2 (ESI†), not all essential statistics are published. For example, pore volumes can often be replaced by corresponding surface areas, but recalculation is not always straightforward.
Although the hierarchy factor accounts for the presence (and relative content) of micro- and mesopores, determining pore network connection is still problematic.
Some of the indices mentioned above are calculated on the basis of porosity measurement, which is a bulk technique and does not account for pore connectivity. Porosity is determined based on physisorption of gases at subcritical temperatures: N2 at 77 K, Ar at 87 K, CO2 at 273 K, but also organic vapours and supercritical gases.601–603 Currently, the standard is the application of non-local density functional theory (NLDFT) to interpret adsorption isotherms, yielding detailed pore size distributions that are crucial for analysis of hierarchical porosity.604,605 NLDFT offers the most accurate modelling of pore structure and the ability to account for confinement effects within nanoporous materials. It also takes into account the effects of pore blockage or cavitation.
To gain detailed information on pore network connectivity, sophisticated adsorption tests must be integrated with DFT and molecular simulation methods.606,607 One example is reducing the Ar adsorption temperature by 10–15 K, which brings it below the hysteresis critical temperature even for very small pores. As a result, the hysteresis loop shape changes. In the reported case of mesostructured zeolite Y, the new hysteresis shape, having two plateaux on the desorption branch, demonstrated that the majority of the mesopores were accessible without any constriction, while the remainder could only be accessed through the micropores.608
Insights into pore connectivity may also be provided by the technique of hysteresis scanning curves derived from N2 or Ar adsorption/desorption isotherms.609,610 Upon adsorption, a fraction of the porous network is initially filled, and desorption is investigated just for this contribution. Partial or full filling does not affect the desorption scan if pores are independent, i.e. directly accessible from the external surface. If large pores are interconnected with small pores the desorption occurs through pore blocking or cavitation. This allows the study of the accessibility of pores, pore mouths, and potential diffusion limitations.
Adsorption on zeolites can be studied not only experimentally,609,611 but also via computational methods, that were recently thoroughly reviewed by Pérez-Botella, Valencia, and Rey.612
Several techniques for determining accessibility in porous materials exist, each with a different spatial or time resolution. Very short overview of the techniques providing information on the local scale is presented below. This is by no means a comprehensive review of methods, but rather a snapshot of currently used technologies and literature reports as an introduction to further exploration of the topic. Vreeswijk and Weckhuysen613 recently discussed new developments and future directions in zeolite material characterisation, particularly in working environments.
9.1 Pulsed field gradient NMR diffusion measurement
Pulse field gradient (PFG) NMR offers unique ability to determine the rate of molecular transport as a function of distance travelled by the molecule, which can be between hundreds of nanometres to tens of micrometers.614,615 PFG NMR is also known as NMR diffusometry. The principle is based on the detection of the interaction of the spin of a particular molecule with superimposed inhomogeneous magnetic field and a steady one over brief time intervals (the field gradient pulses).616 Molecules may be followed on their diffusion path through individual crystals, powders, or even shaped materials, thus the method can be used to determine the intra and interparticle transport.Recently, Mantle et al.617 proposed a simple 1H PFG NMR method to determine intracrystalline molecular self-diffusivities for weakly adsorbing hydrocarbon gases (methane and ethane). The method used T1/T2 relaxation measurements from two different locations of a sample: the pure gas phase region well above the zeolite and the zeolite crystallite bed, to determine intracrystalline diffusion coefficients. The authors emphasised that the measurements may be performed with a single, flame-sealed sample. The method is independent of the microporous material crystallite size, and can be routinely used on small, benchtop NMR systems.
9.2 2D T2–T2 relaxation exchange NMR (REXSY)
The ‘classic’ NMR measurement is realized in the frequency domain; the chemical shift, i.e. the position of an NMR signal relative to a standard reflects the local electronic environment of the nucleus in the molecule. The exchange NMR is specifically designed to study dynamic processes, i.e. exchange between different environments or states over time. Relaxation times, denoted as T1 (longitudinal relaxation time or spin–lattice relaxation) and T2 (transverse relaxation time or spin–spin relaxation), are fundamental parameters, describing how nuclear spins return to equilibrium and lose coherence following excitation. Molecules confined in the porous materials collide with pore surfaces, which influences the interaction of their nuclei spin with the external magnetic field. Collisions with pore walls accelerate dephasing, leading to shortened T2 for molecules in smaller pores that encounter more frequent collisions. To evaluate pore connectivity, it is crucial to determine whether molecular transport occurs between distinct pores and, if so, at what velocity. This information can be acquired by measuring temporal fluctuations in T2, or even better, measuring two separate T2 relaxation times for a given spin system, under different conditions or using paired pulse sequences.618–620 The results are combined to construct 2D T2–T2 correlation maps with cross-peaks, indicating exchange or interaction between different relaxation domains. Diagonal peaks represent homogeneous or non-exchanging environments while off-diagonal peaks suggest exchange or interaction between environments.6219.3 Frequency response (FR) diffusion
Another method, or rather a set of methods, suited to study how molecules or ions diffuse within a porous material, is frequency response diffusion.622–626 In FR methods, periodic (oscillating, alternating) perturbation produces response with lower amplitude and phase shift (lag). The data across the frequency spectrum offers information on various diffusion processes, whether fast or slow, restricted or free. They can be used to characterise pore connectivity and size, because different pore environments respond differently at various frequencies. They are also used to determine kinetic barriers, hopping rates, and interactions within porous or complex structures.627 Yasuda et al.628 used the FR method (sinusoidal perturbation of reactant partial pressure in a steady-flow reactor) for investigation of the kinetics of CO2 formation by CO catalytic oxidation.9.4 Positron annihilation spectroscopy
The positron annihilation spectroscopy (PAS, also called PALS – positron annihilation lifetime spectroscopy)629 was proposed as a method for distinguishing complex pore geometries in zeolites.630 The principle of PAS is based on the fact that positrons placed inside a material interact with its electrons to generate positronium (an electron–positron bound state) of different spin states (para and ortho). The para-positronium has a short lifetime (0.125 ns), but the long-living ortho-positronium diffuses within the pore network, with a lifespan dependent on pore size and shape (average lifetime 2–10 ns in micropores, 10–80 ns in mesopores, above 80 ns in interparticle space or vacuum). PAS was applied to a series of zeolites with different pore connectivity (1D, 2D, and 3D), such as *BEA, FAU, FER, MFI, MOR, and UTL, and was then extended to all known zeolitic structures. PALS was then used to demonstrate the interconnectivity of pores of different dimensions by calculating the fraction of o-positronium escaping into vacuum,586 as positrons are more likely to reach zeolite crystals if they diffuse from micropores to mesopores (than the other way around), and then to the surface. Positrons from closed or restricted pores would have to diffuse from larger pores through micropores, where they would be annihilated rather than reach the surface (Fig. 29). To prove their assertions, the authors used differently treated ZSM-5 zeolites with well-characterised pores, including micropores, as well as occluded, constricted, and open mesopores. PALS is a very useful technique that can differentiate pores of various sizes as well as their interconnectivity; however, its utility is limited by the measuring setup, which requires access to a slow positron beam. | ||
| Fig. 29 Positron decay scheme. Positrons implanted in a sample can annihilate immediately into two gamma rays or bind with electrons to generate positronium. para-Positronium (p-Ps) decays quickly, whereas ortho-Positronium (o-Ps) can permeate through porous material, escape into vacuum or decay with a lifetime proportional to the pore size. Based on ref. 586. | ||
9.5 129Xe NMR spectroscopy
The 129Xe NMR spectroscopy was first proposed by Ito and Fraissard as a method for characterising porous substances with FAU zeolites as the example.631 There are several parameters, that can be derived from the spectra, such as the chemical shift and its anisotropy, the line width, and the longitudinal relaxation time T1. They provide information about pore size, surface chemistry, symmetry, connectivity, and dynamic behaviour of Xe inside zeolitic structures. Comprehensive information is available in the illustrative papers632–635 and reviews636,637 on this subject, which are condensed below to provide a brief overview of the data obtainable via spectral analysis.(1) Chemical shift. Larger chemical shifts usually suggest the presence of narrower channels or smaller cavities due to enhanced contact between Xe and pore walls. Variations in chemical shift might also indicate the presence of various functional groups on the pore walls.
(2) Chemical shift anisotropy. High anisotropy is characteristic of asymmetric pores, while low anisotropy indicates symmetric and generally more isotropic environments.
(3) Line width. Narrow lines are characteristic of uniform pore sizes and types; broader lines point to more dynamic environments, i.e. broader pore size distribution and/or higher diffusion rates. The broadening of signals can be used to detect pore blockage, for example, due to coke deposition.
(4) The T1 relaxation time. This parameter is the most useful in the investigation of pore connectivity. Longer T1 times suggest isolated or poorly linked pores, where Xe have fewer collisions and longer residence times. Shorter T1 indicates more space for the movement of Xe atoms, thus improved connection which allows easier Xe exchange with neighbouring pores. Another factor influencing the T1 relaxation time is the composition of the material, especially the presence of paramagnetic centres.
Telkki et al.638 developed a technique known as 129Xe porometry. The principle is based on the observation that Xe can be ‘dissolved’ in the liquid phase but is excluded from the solid phase. In the experiment, a xenon-loaded liquid (such as acetonitrile or cyclohexane)639 is sorbed in the porous material, after which the entire system is frozen. The 129Xe NMR signal vanishes as the confined medium melts. The melting transition temperature depends on surface curvature, thus the pore size.640 Therefore, the temperature at which a specific xenon signal vanishes enables the calculation of pore size. 129Xe NMR spectra collected in a wide temperature range can be used to calculate pore size distribution.
Laser-hyperpolarized 129Xe NMR and two-dimensional exchange spectroscopy (EXSY) allow observation of the exchange of Xe atoms in different types of pores.641,642 In a 2D spectrum, the peaks of Xe in different pores are located along the diagonal. Off-diagonal cross peaks appear if Xe can be transferred between these pores, indicating whether the pores are interconnected. Cross-peak intensities correlate with the quantity of exchanged xenon, allowing the calculation of exchange rates.
In summary, 129Xe NMR spectroscopy enables investigation of the inner architecture of porous materials.
9.6 Quasielastic neutron scattering (QENS)
QENS is a common neutron scattering technique for studying dynamics in porous materials.643 It detects small broadening around elastic peaks caused by diffusive motions of molecules within the pores. The width and shape of quasielastic signals allow to determine diffusion rates of guest molecules, mobility and rotational motions of adsorbed species as well as flexibility and vibrational dynamics of the zeolite framework itself.In principle, QENS measures the movement of guest molecules (e.g. water, reactants) within the hosting pore network. Changes in molecular mobility at different temperatures or conditions can indicate whether molecules move seamlessly in the pore system or are restricted to specific domains. This helps to confirm the presence of connected hierarchies.644
QENS can be used to follow the molecular motion over wide length scales from Angstroms to nanometers and equally wide time scales, ranging from a few femtoseconds to over a hundred nanoseconds, using time-of-flight (TOF) measurement, neutron backscattering (BS), and neutron spin-echo (NSE) methodologies.645 QENS results are usually accompanied by molecular dynamics (MD) simulations, a method perfectly suited to predict the movement and status of molecules in the confined, well-defined environment of zeolite channels.646,647
9.7 Confocal fluorescence microscopy
Confocal fluorescence microscopy cannot directly visualise pores; nevertheless, it can detect the fluorescence of molecules located within the channel system. Through judicious selection of molecules and confocal imaging, three-dimensional spatial distribution data (pore size distribution, accessibility, and interconnectivity) may be obtained.157,648The spatial resolution of confocal fluorescence microscopy is generally around 200–300 nm laterally and approximately 500–700 nm axially, limited by two factors: diffraction of light and brightness limitations at high magnifications.649 Some super-resolution techniques have extended these boundaries further, achieving resolutions below 100 nm.650
Weckhuysen et al.651 used DAMPI (4-(4-diethylaminostyryl)-N-methylpyridinium-iodide) fluorescent dye to visualize external entrances to the MFI (ZSM-5) straight channels. The aromatic part of DAMPI can enter the straight channels of ZSM-5 (MFI), but its terminal diethylamino-group, like the open canopy of an umbrella, blocks its further diffusion. The authors successfully monitored the desilication process within an individual ZSM-5 crystal, because the generated mesopores facilitated the entry of DAMPI into the micropores through openings on the surfaces of mesopores. This showed direct connectivity between micropores (straight channels) and mesopores. The changes of porosity upon dealumination were also investigated using fluorescent 4-fluorostyrene oligomerization, which allowed mapping of the surface distribution of active sites.93
9.8 Direct visualisation methods
Electron microscopy, whether scanning electron microscopy (SEM) or transmission electron microscopy (TEM), is unquestionably a key tool for materials characterisation. The high-resolution SEM and TEM allow direct observation of internal structure of materials, including their porosity, but they only provide 2D information by projecting through a 3D structure.652Because of their many advantages when imaging electron beam-sensitive materials and light elements, scanning transmission electron microscopy (STEM) imaging techniques like 4D-STEM ptychography and integrated differential phase contrast/optimum bright-field STEM (iDPC/OBF-STEM) have demonstrated huge potential for atomic resolution characterisation of zeolites.653
Three-dimensional images can be acquired by electron tomography. In this method a sequence of pictures is captured from a sample as a function of tilt angle. Combining these images enables a 3D reconstruction of the structure.53,654 Time-resolved cryo-TEM analysis coupled with electron tomography illustrated the pore evolution during zeolite leaching by a base eventually leading to the formation of hollow ZSM-5 crystals.655 Apeleo Zubiri et al.656 coupled electron tomography and lab-based nano X-ray computed tomography (nano-CT) enabling 3D analyses of the same particle by both methods. The authors followed the formation mechanism of porous materials with the example of steam-assisted crystallisation of macroporous zeolite particles.
Very recently a perspective paper on the new characterisation technique, multiscale X-ray tomographic imaging was published, focusing on its applicability to the characterisation of membranes.657 The authors pointed out important advantage of X-rays over visible photons or electrons, which is their longer penetrating length. They emphasised that high resolution imaging, particularly with synchrotron sources, can be used to study nanoscale phenomena such as scaling, membrane fouling, and pore blocking, or to characterise the inner structure of membranes in terms of pore size distribution, tortuosity, or chemical robustness.
The integration of machine learning algorithms with spectroscopic and microscopic data facilitates automatic and more precise analysis of complex structures, representing the future of direct visualisation methods. These methodological improvements are expected to aid in the design and optimisation of zeolite-based catalysts and adsorbents by offering more comprehensive and detailed structural information.
Summary and outlook
Zeolites and related molecular sieve materials with uniform micropores made an enormous impact in catalysis and separation technologies. This motivated research to diversify pore characteristics and sizes of zeolites to facilitate the processing of larger molecules. The primary effort focused on the bottom-up discovery of new materials and structures. An alternative approach adopted top-down strategies of modifying bottom-up synthesised zeolites to generate new or additional porosity to improve catalysis or sorption characteristics. Crystalline molecular sieves are robust and not amenable to easy modification of the structure and porosity. Various methods were explored to overcome this problem and are overviewed in this article. The discussed results include data about porosity enhancement and its impact on catalytic properties and stability of the products.A frequent theme in developing top-down engineering of zeolites has been the generation of mixed micro/mesoporous systems. The goal was to enhance the diffusion of molecules and increase accessibility of active sites in zeolites by the presence of mesopores. One of the convenient approaches entailed removing atoms from the frameworks through dealumination and desilication, both methods often referred to as demetallation.
The relative simplicity of demetallation contrasts to its underlying complexity. The mechanisms behind mesopore development and the accompanying processes, including the formation of defect sites and extraframework species, are important and still not fully understood. Some issues regarding precise characterisation of molecular-scale modifications and an accurate description of the reconstructed zeolitic framework and amorphous phase (extraframework species) remain unresolved. The competition between partial dissolution of the framework and reincorporation of dissolved species back into zeolite framework determines the character of the final mesoporosity, the acidic properties, and catalytic performance. In order to understand reaction mechanisms for mesopore formation, the use of theoretical tools, such as density functional theory (DFT) and molecular dynamics computations are of paramount importance. There is still ample room for research integrating advanced theoretical methodologies with in situ experimental techniques to enable targeted design of mesoporosity.
Due to the progress made in the characterisation techniques, more precise but still far from complete description of the extraframework species and defect sites associated with mesopore production was achieved. The important issue, currently gaining a lot of attention, are experimental mechanistic data obtained by operando techniques, complementing the reaction profile, including, for example, the dynamics of water molecules and their joint involvement in hydrolysis or formation of Al–O/Si–O bond in different zeolite structures.
The regulatory effect of framework Al on mesopore formation have been elaborated, allowing better control over demetallation processes and the transition of demetallated zeolites from the laboratory to the industrial scale. Only a few, however important, examples of industrial use of porosity-engineered zeolites exist. The limitations lay on the practical side, i.e. multistep preparation methods, the necessity of using organic surfactants, and additional waste streams related to generating secondary porosity. The same reasoning also brings economic aspects that are challenging for the wider application of hierarchically ordered zeolites. The most straightforward example of top-down hierarchically porous zeolite employed in the industry is dealuminated ultrastabilised micro/mesoporous Y zeolite (USY, FAU structure). USY is the most extensively utilised catalyst in the process of fluid catalytic cracking (FCC) of crude oil. The newly developed secondary mesoporosity enabled the effective cracking of bulky feed molecules, i.e. vacuum gas oil, by increasing the accessibility of active sites. Other examples include severely dealuminated mordenite-based catalysts to optimise cumene production. It has been claimed that a newly developed 3-dimensional porous structure allowed molecular traffic control of benzene and propylene during alkylation.
The early methods of generating micro/mesoporous hybrids had a bottom-up character of combining conventional zeolite synthesis mixtures with surfactants as mesopore generating agents. This had limited success as phase separation was the common outcome, resulting in poor connectivity between micro- and mesopores.
Methodology adjustment enabled the most successful technology to date, referred to as zeolite surfactant-templating. It entails a hydrothermal treatment of zeolite in moderately basic cationic surfactant solutions. This method introduces mesoporosity while preserving the primary characteristics of the original zeolite, its crystallinity, microporosity and, most importantly, acidity. The surfactant-templated zeolites are reported as commercially implemented, with annual production at thousands of tons. Therefore, it is feasible to implement hierarchically ordered zeolites in large-scale industry, but under certain conditions, i.e. affordable and recyclable surfactants, limited steps of optimised modification methods, also the use of existing infrastructures.
There are other strategies of the top-down development of micro/mesoporous materials, which are still at the laboratory scale. One such strategy relies on self-organisation of nanosized zeolite crystals or zeolite seeds, where secondary mesoporosity is generated between the nanocrystals. Such composites can be synthesized through silanization, a process that entails the attachment of organosilyl groups to the seeds surface. The presence of surface organosilanes hinders particle growth during crystallisation, facilitating the formation of organic–inorganic composites. Recently, a unique type of zeolite preparation has led to the formation of ‘embryonic zeolites.’ These materials are XRD-amorphous, yet the increased openness of the micropores facilitates shorter diffusion paths. Despite a relatively small number of active sites and moderate acid strength, embryonic zeolites exhibited significant catalytic activity. The interzeolite conversion (IZC) facilitates the synthesis of novel zeolitic phases, as well as the broadening of Si/Al composition ranges, or the modification of Al siting, relative to zeolites synthesised by the bottom-up methodology. IZC can be employed as a technique for synthesising mixed zeolite phases, consisting of building units from different frameworks. Very specific form of the introduction of secondary mesoporosity is formation of hollow structures or the construction of zeolite-on-zeolite assemblies. This is often accomplished through desilication of aluminium-zoned or OSDA-containing zeolite crystals. The hollow structures can be also obtained by zeolitization of hollow amorphous silica spheres, referred to as ‘templating and surface to core’ crystallisation. The resulting all-zeolite material retains the original shape of the mesoporous sacrificial matrix.
In addition to traditional wet chemistry, mechanical post-synthetic treatments are suggested as eco-friendly approaches to modifying zeolite characteristics. Mechanochemistry can enable not only the bottom-up synthesis of zeolites but also the incorporation of heteroatoms (as exchangeable cations or through isomorphous substitution); improving the atom efficiency of the reactions and reducing waste production, in line with the requirements of the green (or at least greener) chemistry. Milling, followed by recrystallisation, enables the adjustment of crystal sizes, which is difficult in bottom-up synthesis. In recent years, advancements in photolithography, originally developed for semiconductor microfabrication, have been transferred to zeolite chemistry, aiding the production of zeolite thin films, including the option for highly oriented thin films on substrates. An example of the novel techniques, adapted from the production of semiconductors, is dry etching, encompassing ion milling (bombarding with argon ions) and reactive ion etching to fabricate c-oriented silicalite films. In addition to mechanochemistry, the treatment of zeolites using microwaves, ultrasound, or plasma is investigated to modify the properties. These technologies are frequently used simply to provide alternative energy sources. Generally, they are regarded as environmentally sustainable and energy-efficient substitutes for conventional thermal or chemical modification processes.
The possibilities for structure and pore engineering have expanded enormously with the discovery that zeolites can produce not only extended 3D forms but also 2D precursors consisting of layers with thicknesses of a few nanometers or less, depending on the structure. These precursors could condense to produce a complete 3D zeolite framework but also be modified post-synthesis to generate additional porosity in a controlled way. Based on methodologies developed with other 2D solids, zeolite layers were expanded by swelling with surfactants, pillared, delaminated or otherwise disorganised, and eventually exfoliated as monolayers dispersed in liquid media. The last method is most versatile for producing new structures and composites as the layers are effectively independent gigantic molecules that can be combined with any other substance of choice (not always yielding meaningful products). The regular arrays of silanols on the surface of zeolite layers allow production of unique materials, called interlayer expanded zeolites with interlayer pores expanded by two MR units (SiO links). These pores are constricted with additional groups, e.g. OH, attached to complete the 4-coordination of the bridging Si. Zeolite MWW, with 2 independent 10-MR systems and surface cavities, which are favourable for monoalkylation of aromatics with small molecules, is the most prolific representative of 2D zeolites, so far affording ∼15 different structural forms and derivatives, seven by direct bottom-up synthesis. The MWW zeolite produces modifiable layered forms within the entire range of Si/Al from 10 to infinity. The second most important/profitable zeolite MFI/ZSM-5 has also been obtained in layered forms with bifunctional templates containing surfactant tails. Its pillared and exfoliated forms have also been obtained. The special feature of the MFI layers are perpendicular 10-MR pores across, allowing additional molecular diffusion and possible usefulness for assembly into permeable films and membranes. About 20 out of >250 zeolite topologies have already been obtained and demonstrated some of the standard modifications mentioned above. A crucial parameter in zeolite modification may be the maximum charge density (silanol density per nm2). It is the lowest for MWW, equal to 1.14 charge nm−2. The dependence on charge density may be manifest in the swelling with surfactants, which with MWW requires high pH and often pure surfactant hydroxide (no other cations). On the other hand, zeolites with charge density near 4 units nm−2 are expanded by surfactant salts in a neutral solution (RWR, SOD). Most of the precursors have been obtained as siliceous layers or with low Al content. The layered material designated bifer, obtained in a system related to ferrierite, and MWW produce exfoliable disorganised assemblies of monolayers in Al-rich syntheses. High Al content drives synthesis of the other layered zeolite preparations to 3D materials. Understanding the role of Al in these systems may be important to the synthesis of more exfoliable zeolite monolayers. Other hetero-atoms, especially Ge, have also been found to produce new results related to zeolite layers. Ge-rich zeolite UTL with 14 and 12-MR between layers supported by double 4-MR units, can be degraded by hydrolysis and produce layers, from which another zeolite, previously unknown, PCR with 10 and 8-MR, can be obtained. These layers can be shifted in the plane to produce theoretically 3 more zeolite structures, one of which, denoted IPC-9 has already been obtained experimentally. Silylation of the PCR precursor layers affords the IEZ from with 12 and 10-MR. It becomes a 4-connected zeolite, OKO, due to 4 interlayer IEZ bridges condensing into a square of SiO units. These elaborate transformations of UTL have been named the ADOR process (Assembly of a zeolite, Disassembly, Organisation, Reassembly). The ADOR transformations of UTL have been partially replicated with other zeolites with Ge-rich D4R units. Recent synthesis showed potential for producing zeolites via condensation of 1D units exemplified by ZEO-2, producing a 16 × 14 × 14-MR framework upon calcination. The silylation produced an IEZ-related derivative with a 20 × 16 × 16-MR pore system. This illustrates the potential for pore engineering to lower-dimensional structures with 1D chains as the building units.
Many of the transformations of 2D zeolites affording larger or modified porosity have been followed by catalytic testing as evidence of meaningful improvement over the parent. The results often confirmed increased active sites accessibility and enhanced conversion of larger molecules. With a few exceptions (delaminated NSI, ITQ-18), there have been few systematic efforts to build on the promising results and prove conclusively meaningful advantages over industrial state-of-the-art competitors. One of the emerging trends is to focus on the direct synthesis of layered forms, as they have a better chance of competing against already practised technologies. The typical modifications of 2D zeolites increase the number of processing steps, increasing cost and labour, which can be more favourable with direct syntheses of delaminated and other types of products. In terms of possible practical applications, the exfoliated layered zeolite in solution can be considered promising for development of structured materials and nanodevices. So far, such applications have been tried for zeolites by bottom-up growth or grouting.
Author contributions
KGM and KAT authored Sections 1, 2 and 8, BG authored Sections 3, 4, 6, 7 and 9, WJR authored Section 5.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review. The editable version of Fig. 27 with data is included in ESI.†Acknowledgements
Financial support from National Science Centre is acknowledged, grant no. 2023/51/I/ST4/00658 (BG), 2020/37/B/ST5/01258 (WJR), 2020/37/B/ST4/01215 (KAT), 2021/43/B/ST4/00307 (KGM). The open-access publication of this article has been supported by a grant from the Faculty of Chemistry under the Strategic Programme Excellence Initiative at Jagiellonian University.References
- J. Rouquerol, F. Rouquerol, P. Llewellyn, G. Maurin and K. S. W. Sing, Adsorption by Powders and Porous Solids: Principles, Methodology and Applications, Elsevier Science, 2013 Search PubMed.
- P. Van Der Voort, K. Leus and E. De Canck, Introduction to Porous Materials, Wiley, 2019 Search PubMed.
- B. Lindström and L. J. Pettersson, CATTECH, 2003, 7, 130–138 CrossRef.
- F. Schüth, K. S. W. Sing and J. Weitkamp, Handbook of Porous Solids, Wiley, 2002 Search PubMed.
- R. M. Barrer, Pure Appl. Chem., 1986, 58, 1317–1322 CrossRef CAS.
- A. F. Masters and T. Maschmeyer, Microporous Mesoporous Mater., 2011, 142, 423–438 CrossRef CAS.
- J. V. Smith, Chem. Rev., 1988, 88, 149–182 CrossRef CAS.
- R. M. Barrer, Hydrothermal chemistry of zeolites, Academic Press, 1982 Search PubMed.
- C. Baerlocher and L. B. McCusker, International Zeolite Association, Database of Zeolite Structures, (https://www.iza-structure.org/databases/).
- C. Martinez and A. Corma, Coord. Chem. Rev., 2011, 255, 1558–1580 CrossRef CAS.
- J. Y. Li, A. Corma and J. H. Yu, Chem. Soc. Rev., 2015, 44, 7112–7127 RSC.
- A. Corma, C. Li and M. Moliner, Angew. Chem., Int. Ed., 2018 Search PubMed.
- X. Jia, W. Khan, Z. Wu, J. Choi and A. C. K. Yip, Adv. Powder Technol., 2019, 30, 467–484 CrossRef CAS.
- J. Cejka, R. Millini, M. Opanasenko, D. P. Serrano and W. J. Roth, Catal. Today, 2020, 345, 2–13 CrossRef CAS.
- T. Pang, X. Yang, C. Yuan, A. A. Elzatahry, A. Alghamdi, X. He, X. Cheng and Y. Deng, Chin. Chem. Lett., 2021, 32, 328–338 CrossRef CAS.
- I. El Bojaddayni, M. Emin Küçük, Y. El Ouardi, I. Jilal, S. El Barkany, K. Moradi, E. Repo, K. Laatikainen and A. Ouammou, Miner. Eng., 2023, 198, 108086 CrossRef CAS.
- C. Cundy and P. Cox, Microporous Mesoporous Mater., 2005, 82, 1–78 CrossRef CAS.
- Q. Wu, X. Meng, X. Gao and F. Xiao, Acc. Chem. Res., 2018, 51, 1396–1403 CrossRef CAS PubMed.
- R. Morris and S. Weigel, Chem. Soc. Rev., 1997, 26, 309–317 RSC.
- M. Moliner, Y. Román-Leshkov and A. Corma, Acc. Chem. Res., 2019, 52, 2971–2980 CrossRef CAS PubMed.
- J. Shin, D. Jo and S. Hong, Acc. Chem. Res., 2019, 52, 1419–1427 CrossRef CAS PubMed.
- R. Chal, C. Gérardin, M. Bulut and S. van Donk, ChemCatChem, 2011, 3, 67–81 CrossRef CAS.
- D. Verboekend and J. Pérez-Ramírez, Catal. Sci. Technol., 2011, 1, 879–890 RSC.
- R. H. Peters, Proc. Chem. Soc., 1958, 93–124 Search PubMed.
- C. J. Plank, E. J. Rosinski and W. P. Hawthorne, I&EC Product Res. Dev., 1964, 3, 165–169 Search PubMed.
- C. McDaniel and P. Maher, in Molecular Sieves, ed. R. M. Barrer, Society of Chemical Industry, London, 1968, pp. 186–195 Search PubMed.
- G. T. Kerr, J. Phys. Chem., 1967, 71, 4155 CrossRef CAS.
- G. T. Kerr, J. Catal., 1969, 15, 200–204 CrossRef CAS.
- R. C. Hansford, US Pat., 3354077, 1967, Union Oil Co Search PubMed.
- J. W. Ward, J. Catal., 1970, 18, 348–351 CrossRef CAS.
- J. Scherzer, J. Catal., 1973, 28, 101–115 CrossRef CAS.
- P. Gallezot, R. Beaumont and D. Barthomeuf, J. Phys. Chem., 1974, 78, 1550–1553 CrossRef CAS.
- J. Klinowski, J. M. Thomas, C. A. Fyfe and G. C. Gobbi, Nature, 1982, 296, 533–536 CrossRef CAS.
- J. Stabenow, L. Marosi and M. Schwarzmann, Ger. Pat., 2510740, 1976, BASF AG Search PubMed.
- G. Engelhardt, U. Lohse, A. Samoson, M. Mägi, M. Tarmak and É. Lippmaa, Zeolites, 1982, 2, 59–62 CrossRef CAS.
- T. Fjermestad, S. Svelle and O. Swang, J. Phys. Chem. C, 2013, 117, 13442–13451 CrossRef CAS.
- S. Malola, S. Svelle, F. L. Bleken and O. Swang, Angew. Chem., Int. Ed., 2012, 51, 652–655 CrossRef CAS PubMed.
- G. Agostini, C. Lamberti, L. Palin, M. Milanesio, N. Danilina, B. Xu, M. Janousch and J. A. van Bokhoven, J. Am. Chem. Soc., 2010, 132, 667–678 CrossRef CAS PubMed.
- M.-C. Silaghi, C. Chizallet, J. Sauer and P. Raybaud, J. Catal., 2016, 339, 242–255 CrossRef CAS.
- M. Nielsen, A. Hafreager, R. Y. Brogaard, K. De Wispelaere, H. Falsig, P. Beato, V. Van Speybroeck and S. Svelle, Catal. Sci. Technol., 2019, 9, 3721–3725 RSC.
- M. Nielsen, R. Y. Brogaard, H. Falsig, P. Beato, O. Swang and S. Svelle, ACS Catal., 2015, 5, 7131–7139 CrossRef CAS.
- K. Stanciakova, B. Ensing, F. Göltl, R. E. Bulo and B. M. Weckhuysen, ACS Catal., 2019, 9, 5119–5135 CrossRef CAS.
- J. N. Louwen, S. Simko, K. Stanciakova, R. E. Bulo, B. M. Weckhuysen and E. T. C. Vogt, J. Phys. Chem. C, 2020, 124, 4626–4636 CrossRef CAS.
- J. Sun, H. Fang, P. I. Ravikovitch and D. S. Sholl, J. Phys. Chem. C, 2020, 124, 668–676 CrossRef CAS.
- S. M. Campbell, D. M. Bibby, J. M. Coddington, R. F. Howe and R. H. Meinhold, J. Catal., 1996, 161, 338–349 CrossRef CAS.
- T. Masuda, Y. Fujikata, S. R. Mukai and K. Hashimoto, Appl. Catal., A, 1998, 172, 73–83 CrossRef CAS.
- M. Müller, G. Harvey and R. Prins, Microporous Mesoporous Mater., 2000, 34, 135–147 CrossRef.
- P. C. Van Geem, K. F. M. G. J. Scholle, G. P. M. Van der Velden and W. S. Veeman, J. Phys. Chem., 1988, 92, 1585–1589 CrossRef CAS.
- J. A. van Bokhoven, D. C. Koningsberger, P. Kunkeler, H. van Bekkum and A. P. M. Kentgens, J. Am. Chem. Soc., 2000, 122, 12842–12847 CrossRef CAS.
- D. P. B. Peixoto, S. M. Cabral de Menezes and M. I. Pais da Silva, Mater. Lett., 2003, 57, 3933–3942 CrossRef CAS.
- G. Onyestyák, J. Valyon, G. Pál-Borbély and L. V. C. Rees, Appl. Surf. Sci., 2002, 196, 401–407 CrossRef.
- K. Brylewska, K. A. Tarach, W. Mozgawa, Z. Olejniczak, U. Filek and K. Gora-Marek, J. Mol. Struct., 2016, 1126, 147–153 CrossRef CAS.
- A. H. Janssen, A. J. Koster and K. P. de Jong, J. Phys. Chem. B, 2002, 106, 11905–11909 CrossRef CAS.
- R. A. Beyerlein, C. Choi-Feng, J. B. Hall, B. J. Huggins and G. J. Ray, Top. Catal., 1997, 4, 27–42 CrossRef CAS.
- S. van Donk, A. H. Janssen, J. H. Bitter and K. P. de Jong, Catal. Rev., 2003, 45, 297–319 CrossRef CAS.
- B. H. Wouters, T. H. Chen and P. J. Grobet, J. Am. Chem. Soc., 1998, 120, 11419–11425 CrossRef CAS.
- L. Benco, T. Demuth, J. Hafner, F. Hutschka and H. Toulhoat, J. Catal., 2002, 209, 480–488 CrossRef CAS.
- D. L. Bhering, A. Ramírez-Solís and C. J. A. Mota, J. Phys. Chem. B, 2003, 107, 4342–4347 CrossRef CAS.
- O. Lisboa, M. Sánchez and F. Ruette, J. Mol. Catal. A: Chem., 2008, 294, 93–101 CrossRef CAS.
- J. A. v Bokhoven, A. L. Roest, D. C. Koningsberger, J. T. Miller, G. H. Nachtegaal and A. P. M. Kentgens, J. Phys. Chem. B, 2000, 104, 6743–6754 CrossRef.
- C. Schroeder, M. R. Hansen and H. Koller, Angew. Chem., Int. Ed., 2018, 57, 14281–14285 CrossRef CAS PubMed.
- E. T. C. Vogt and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7342–7370 RSC.
- J. Sohn, S. Decanio, P. Fritz and J. Lunsford, J. Phys. Chem., 1986, 90, 4847–4851 CrossRef CAS.
- A. I. Biaglow, D. J. Parrillo, G. T. Kokotailo and R. J. Gorte, J. Catal., 1994, 148, 213–223 CrossRef CAS.
- F. Lónyi and J. H. Lunsford, J. Catal., 1992, 136, 566–577 CrossRef.
- Q. L. Wang, G. Giannetto and M. Guisnet, J. Catal., 1991, 130, 471–482 CrossRef CAS.
- S. M. T. Almutairi, B. Mezari, G. A. Filonenko, P. C. M. M. Magusin, M. S. Rigutto, E. A. Pidko and E. J. M. Hensen, ChemCatChem, 2013, 5, 452–466 CrossRef CAS.
- B. Mezari, P. C. M. M. Magusin, S. M. T. Almutairi, E. A. Pidko and E. J. M. Hensen, J. Phys. Chem. C, 2021, 125, 9050–9059 CrossRef CAS PubMed.
- C. J. Heard, L. Grajciar, F. Uhlík, M. Shamzhy, M. Opanasenko, J. Čejka and P. Nachtigall, Adv. Mater., 2020, 32, 2003264 CrossRef CAS PubMed.
- S. C. Shen and S. Kawi, J. Phys. Chem. B, 1999, 103, 8870 CrossRef CAS.
- R. Mokaya, J. Phys. Chem. B, 2000, 104, 8279–8286 CrossRef CAS.
- A. Vjunov, J. L. Fulton, D. M. Camaioni, J. Z. Hu, S. D. Burton, I. Arslan and J. A. Lercher, Chem. Mater., 2015, 27, 3533–3545 CrossRef CAS.
- A. R. Maag, G. A. Tompsett, J. Tam, C. A. Ang, G. Azimi, A. D. Carl, X. Huang, L. J. Smith, R. L. Grimm, J. Q. Bond and M. T. Timko, Phys. Chem. Chem. Phys., 2019, 21, 17880–17892 RSC.
- T. Sano, Y. Nakajima, Z. B. Wang, Y. Kawakami, K. Soga and A. Iwasaki, Microporous Mater., 1997, 12, 71–77 CrossRef CAS.
- R. M. Ravenelle, F. Schüβler, A. D’Amico, N. Danilina, J. A. van Bokhoven, J. A. Lercher, C. W. Jones and C. Sievers, J. Phys. Chem. C, 2010, 114, 19582–19595 CrossRef CAS.
- T. Ennaert, J. Geboers, E. Gobechiya, C. M. Courtin, M. Kurttepeli, K. Houthoofd, C. E. A. Kirschhock, P. C. M. M. Magusin, S. Bals, P. A. Jacobs and B. F. Sels, ACS Catal., 2015, 5, 754–768 CrossRef CAS.
- A. Guzman, I. Zuazo, A. Feller, R. Olindo, C. Sievers and J. A. Lercher, Microporous Mesoporous Mater., 2005, 83, 309–318 CrossRef CAS.
- W. Lutz, W. Geßner, R. Bertram, I. Pitsch and R. Fricke, Microporous Mater., 1997, 12, 131–139 CrossRef CAS.
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. F. Peden and J. Szanyi, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC.
- N. Martín, C. Paris, P. N. R. Vennestrøm, J. R. Thøgersen, M. Moliner and A. Corma, Appl. Catal., B, 2017, 217, 125–136 CrossRef.
- C. W. Lopes, J. Martinez-Ortigosa, K. Góra-Marek, K. Tarach, J. A. Vidal-Moya, A. E. Palomares, G. Agostini, T. Blasco and F. Rey, J. Mater. Chem. A, 2021, 9, 27448–27458 RSC.
- T. Nishitoba, N. Yoshida, J. N. Kondo and T. Yokoi, Ind. Eng. Chem. Res., 2018, 57, 3914–3922 CrossRef CAS.
- D. W. Fickel, E. D’Addio, J. A. Lauterbach and R. F. Lobo, Appl. Catal., B, 2011, 102, 441–448 CrossRef CAS.
- Y. Ji, M. A. Deimund, Y. Bhawe and M. E. Davis, ACS Catal., 2015, 5, 4456–4465 CrossRef CAS.
- Y. Ji, J. Birmingham, M. A. Deimund, S. K. Brand and M. E. Davis, Microporous Mesoporous Mater., 2016, 232, 126–137 CrossRef CAS.
- A. Corma, F. Rey, J. Rius, M. J. Sabater and S. Valencia, Nature, 2004, 431, 287–290 CrossRef CAS PubMed.
- É. Bourgeat-Lami, F. Fajula, D. Anglerot and T. D. Courières, Microporous Mater., 1993, 1, 237–245 Search PubMed.
- K. Iyoki, K. Kikumasa, T. Onishi, Y. Yonezawa, A. Chokkalingam, Y. Yanaba, T. Matsumoto, R. Osuga, S. P. Elangovan, J. N. Kondo, A. Endo, T. Okubo and T. Wakihara, J. Am. Chem. Soc., 2020, 142, 3931–3938 CrossRef CAS PubMed.
- P. Peng, X. H. Gao, Z. F. Yan and S. Mintova, Natl. Sci. Rev., 2020, 7, 1726–1742 CrossRef PubMed.
- K. A. Tarach, J. Tekla, U. Filek, A. Szymocha, I. Tarach and K. Gora-Marek, Microporous Mesoporous Mater., 2017, 241, 132–144 CrossRef CAS.
- D. Verboekend, T. C. Keller, M. Milina, R. Hauert and J. Pérez-Ramírez, Chem. Mater., 2013, 25, 1947–1959 CrossRef CAS.
- T. Yoshioka, K. Iyoki, Y. Hotta, Y. Kamimura, H. Yamada, Q. Han, T. Kato, C. A. J. Fisher, Z. Liu, R. Ohnishi, Y. Yanaba, K. Ohara, Y. Sasaki, A. Endo, T. Takewaki, T. Sano, T. Okubo and T. Wakihara, Sci. Adv., 2022, 8, eabo3093 CrossRef CAS PubMed.
- L. R. Aramburo, L. Karwacki, P. Cubillas, S. Asahina, D. A. M. de Winter, M. R. Drury, I. L. C. Buurmans, E. Stavitski, D. Mores, M. Daturi, P. Bazin, P. Dumas, F. Thibault-Starzyk, J. A. Post, M. W. Anderson, O. Terasaki and B. M. Weckhuysen, Chem. – Eur. J., 2011, 17, 13773–13781 CrossRef CAS PubMed.
- P. J. Kooyman, P. van der Waal and H. van Bekkum, Zeolites, 1997, 18, 50–53 CrossRef CAS.
- Y. Fan, X. Bao, X. Lin, G. Shi and H. Liu, J. Phys. Chem. B, 2006, 110, 15411–15416 CrossRef CAS PubMed.
- V. L. Zholobenko, L. M. Kustov, V. B. Kazansky, E. Loeffler, U. Lohser, C. Peuker and G. Oehlmann, Zeolites, 1990, 10, 304–306 CrossRef CAS.
- E. Loeffler, U. Lohse, C. Peuker, G. Oehlmann, L. M. Kustov, V. L. Zholobenko and V. B. Kazansky, Zeolites, 1990, 10, 266–271 CrossRef CAS.
- J. C. Groen, J. A. Moulijn and J. Pérez-Ramírez, J. Mater. Chem., 2006, 16, 2121 RSC.
- C. D. Chang, C. T. W. Chu, J. N. Miale, R. F. Bridger and R. B. Calvert, J. Am. Chem. Soc., 1984, 106, 8143–8146 CrossRef CAS.
- R. M. Dessau and G. T. Kerr, Zeolites, 1984, 4, 315–318 Search PubMed.
- K. Yamagishi, S. Namba and T. Yashima, J. Catal., 1990, 121, 47–55 CrossRef CAS.
- T. Sano, Y. Uno, Z. B. Wang, C. H. Ahn and K. Soga, Microporous Mesoporous Mater., 1999, 31, 89–95 CrossRef CAS.
- A. Omegna, M. Haouas, A. Kogelbauer and R. Prins, Microporous Mesoporous Mater., 2001, 46, 177–184 Search PubMed.
- X. Zaiku, C. Qingling, Z. Chengfang, B. Jiaqing and C. Yuhua, J. Phys. Chem. B, 2000, 104, 2853–2859 CrossRef.
- J. Li, H. Liu, F. Li, T. An and X. Bao, Ind. Eng. Chem. Res., 2018, 57, 10876–10882 CrossRef CAS.
- E. F. T. Lee and L. V. C. Rees, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 1531–1537 RSC.
- L. Zhang, A. N. C. van Laak, P. E. de Jongh and K. P. de Jong, Zeolites and Catalysis, 2010, pp. 237–282 Search PubMed.
- L. Sommer, D. Mores, S. Svelle, M. Stöcker, B. M. Weckhuysen and U. Olsbye, Microporous Mesoporous Mater., 2010, 132, 384–394 CrossRef CAS.
- D. Zhai, L. Zhao, Y. Liu, J. Xu, B. Shen and J. Gao, Chem. Mater., 2015, 27, 67–74 CrossRef CAS.
- M. Ogura, S.-y Shinomiya, J. Tateno, Y. Nara, M. Nomura, E. Kikuchi and M. Matsukata, Appl. Catal., A, 2001, 219, 33–43 CrossRef CAS.
- J. Wiegmann, Acta Polym., 1980, 31, 406 CrossRef.
- R. M. Dessau, E. W. Valyocsik and N. H. Goeke, Zeolites, 1992, 12, 776–779 Search PubMed.
- A. Čimek, B. Subotić, I. Šmit, A. Tonejc, R. Aiello, F. Crea and A. Nastro, Microporous Mater., 1997, 8, 159–169 CrossRef.
- A. Čižmek, B. Subotić, R. Aiello, F. Crea, A. Nastro and C. Tuoto, Microporous Mater., 1995, 4, 159–168 Search PubMed.
- J. C. Groen, L. A. A. Peffer, J. A. Moulijn and J. Pérez-Ramírez, Chem. – Eur. J., 2005, 11, 4983–4994 CrossRef CAS PubMed.
- H. Lechert and H. Kacirek, Zeolites, 1991, 11, 720–728 CrossRef CAS.
- M. Jin, M. Liu, P. Nachtigall, L. Grajciar and C. J. Heard, Chem. Mater., 2021, 33, 9202–9212 CrossRef CAS.
- M. S. Holm, S. Svelle, F. Joensen, P. Beato, C. H. Christensen, S. Bordiga and M. Bjørgen, Appl. Catal., A, 2009, 356, 23–30 CrossRef CAS.
- J. C. Groen, L. A. A. Peffer and J. Pérez-Ramírez, Microporous Mesoporous Mater., 2003, 60, 1–17 CrossRef CAS.
- J. C. Groen, T. Bach, U. Ziese, A. M. Paulaime-van Donk, K. P. de Jong, J. A. Moulijn and J. Pérez-Ramírez, J. Am. Chem. Soc., 2005, 127, 10792–10793 CrossRef CAS PubMed.
- D. Verboekend, S. Mitchell, M. Milina, J. C. Groen and J. Pérez-Ramírez, J. Phys. Chem. C, 2011, 115, 14193–14203 CrossRef CAS.
- J. C. Groen, W. Zhu, S. Brouwer, S. J. Huynink, F. Kapteijn, J. A. Moulijn and J. Pérez-Ramírez, J. Am. Chem. Soc., 2007, 129, 355–360 CrossRef CAS PubMed.
- J. C. Groen, J. C. Jansen, J. A. Moulijn and J. Pérez-Ramírez, J. Phys. Chem. B, 2004, 108, 13062–13065 CrossRef CAS.
- J. Pérez-Ramírez, D. Verboekend, A. Bonilla and S. Abelló, Adv. Funct. Mater., 2009, 19, 3972–3979 CrossRef.
- D. Verboekend, J. C. Groen and J. Pérez-Ramírez, Adv. Funct. Mater., 2010, 20, 1441–1450 CrossRef CAS.
- K. Gora-Marek, K. Tarach, J. Tekla, Z. Olejniczak, P. Kustrowski, L. Liu, J. Martinez-Triguero and F. Rey, J. Phys. Chem. C, 2014, 118, 28043–28054 CrossRef CAS.
- M. Gackowski, K. Tarach, Ł. Kuterasiński, J. Podobiński, B. Sulikowski and J. Datka, Microporous Mesoporous Mater., 2019, 281, 134–141 CrossRef CAS.
- K. A. Tarach, G. Jajko, M. Palomino, F. Rey and K. Góra-Marek, Langmuir, 2024, 40, 6918–6932 CrossRef CAS PubMed.
- M. Strzempek, K. Tarach, K. Góra-Marek, F. Rey, M. Palomino, S. Valencia and W. Piskorz, Phys. Chem. Chem. Phys., 2021, 23, 2981–2990 RSC.
- T. Zhang, F. Qiu and J. Li, Appl. Catal., B, 2016, 195, 48–58 CrossRef CAS.
- R. Oord, I. C. ten Have, J. M. Arends, F. C. Hendriks, J. Schmidt, I. Lezcano-Gonzalez and B. M. Weckhuysen, Catal. Sci. Technol., 2017, 7, 3851–3862 RSC.
- M. K. Wardani, G. T. M. Kadja, A. T. N. Fajar, Subagjo, I. G. B. N. Makertihartha, M. L. Gunawan, V. Suendo and R. R. Mukti, RSC Adv., 2019, 9, 77–86 RSC.
- Y. Kumita, J. Gascon, E. Stavitski, J. A. Moulijn and F. Kapteijn, Appl. Catal., A, 2011, 391, 234–243 CrossRef CAS.
- F. Schmidt, M. R. Lohe, B. Büchner, F. Giordanino, F. Bonino and S. Kaskel, Microporous Mesoporous Mater., 2013, 165, 148–157 CrossRef CAS.
- W. C. Yoo, X. Zhang, M. Tsapatsis and A. Stein, Microporous Mesoporous Mater., 2012, 149, 147–157 CrossRef CAS.
- D. Verboekend, M. Milina, S. Mitchell and J. Pérez-Ramírez, Cryst. Growth Des., 2013, 13, 5025–5035 CrossRef CAS.
- J. Tekla, L. Lakiss, V. Valchev, K. A. Tarach, M. Jabłońska, V. Girman, A. Szymocha, A. Kowalczyk, K. Góra-Marek and J.-P. Gilson, Microporous Mesoporous Mater., 2020, 299, 110088 CrossRef CAS.
- A. Bonilla, D. Baudouin and J. Pérez-Ramírez, J. Catal., 2009, 265, 170–180 CrossRef CAS.
- X. Cheng, T. Cacciaguerra, D. Minoux, J.-P. Dath, F. Fajula and C. Gérardin, Microporous Mesoporous Mater., 2018, 260, 132–145 CrossRef CAS.
- Y. P. Khitev, Y. G. Kolyagin, I. I. Ivanova, O. A. Ponomareva, F. Thibault-Starzyk, J. P. Gilson, C. Fernandez and F. Fajula, Microporous Mesoporous Mater., 2011, 146, 201–207 CrossRef CAS.
- Y. P. Khitev, I. I. Ivanova, Y. G. Kolyagin and O. A. Ponomareva, Appl. Catal., A, 2012, 441–442, 124–135 CrossRef CAS.
- I. I. Ivanova and E. E. Knyazeva, Chem. Soc. Rev., 2013, 42, 3671–3688 RSC.
- J. García-Martínez, M. Johnson, J. Valla, K. Li and J. Y. Ying, Catal. Sci. Technol., 2012, 2, 987–994 RSC.
- R. Chal, T. Cacciaguerra, S. van Donk and C. Gérardin, Chem. Commun., 2010, 46, 7840–7842 RSC.
- T. X. Nguyen, J. W. Moon, H. S. Jung, G. Y. Han and J. W. Bae, Korean J. Chem. Eng., 2021, 38, 1231–1239 CrossRef CAS.
- D. Verboekend, A. M. Chabaneix, K. Thomas, J.-P. Gilson and J. Pérez-Ramírez, CrystEngComm, 2011, 13, 3408–3416 RSC.
- J. C. Groen, G. M. Hamminga, J. A. Moulijn and J. Perez-Ramirez, Phys. Chem. Chem. Phys., 2007, 9, 4822–4830 RSC.
- M. Ogura, S. Y. Shinomiya, J. Tateno, Y. Nara, E. Kikuchi and M. Matsukata, Chem. Lett., 2000, 882 CrossRef CAS.
- M. Ogura, E. Kikuchi and M. Matsukata, in Studies in Surface Science and Catalysis, ed. A. Galarneau, F. Fajula, F. Di Renzo and J. Vedrine, Elsevier, 2001, vol. 135, p. 216 Search PubMed.
- G. Muller, T. Narbeshuber, G. Mirth and J. A. Lercher, J. Phys. Chem., 1994, 98, 7436–7439 CrossRef CAS.
- P. Kortunov, S. Vasenkov, C. Chmelik, J. Kärger, D. M. Ruthven and J. Wloch, Chem. Mater., 2004, 16, 3552–3558 CrossRef CAS.
- O. Geier, S. Vasenkov, E. Lehmann, J. Kärger, U. Schemmert, R. A. Rakoczy and J. Weitkamp, J. Phys. Chem. B, 2001, 105, 10217–10222 CrossRef CAS.
- D. Tzoulaki, L. Heinke, W. Schmidt, U. Wilczok and J. Kärger, Angew. Chem., Int. Ed., 2008, 47, 3954–3957 CrossRef CAS PubMed.
- M. W. Anderson, K. S. Pachis, F. Prébin, S. W. Carr, O. Terasaki, T. Ohsuna and V. Alfreddson, J. Chem. Soc., Chem. Commun., 1991, 1660–1664 RSC.
- G. González, W. Stracke, Z. Lopez, U. Keller, A. Ricker and R. Reichelt, Microsc. Microanal., 2004, 10, 224–235 CrossRef PubMed.
- M. W. Anderson, J. R. Agger, L. I. Meza, C. B. Chong and C. S. Cundy, Faraday Discuss., 2007, 136, 143–156 RSC.
- L. Karwacki, E. Stavitski, M. H. F. Kox, J. Kornatowski and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2007, 46, 7228–7231 CrossRef CAS PubMed.
- E. Stavitski, M. H. F. Kox and B. M. Weckhuysen, Chem. – Eur. J., 2007, 13, 7057–7065 CrossRef CAS PubMed.
- M. H. F. Kox, E. Stavitski and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2007, 46, 3652–3655 CrossRef PubMed.
- M. H. F. Kox, E. Stavitski, J. C. Groen, J. Pérez-Ramírez, F. Kapteijn and B. M. Weckhuysen, Chem. – Eur. J., 2008, 14, 1718–1725 CrossRef CAS PubMed.
- E. Stavitski, M. H. F. Kox, I. Swart, F. M. F. de Groot and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2008, 47, 3543–3547 CrossRef CAS PubMed.
- T. Li, J. Ihli, Z. Ma, F. Krumeich and J. A. van Bokhoven, J. Phys. Chem. C, 2019, 123, 8793–8801 CrossRef CAS.
- S. Svelle, L. Sommer, K. Barbera, P. N. R. Vennestrøm, U. Olsbye, K. P. Lillerud, S. Bordiga, Y.-H. Pan and P. Beato, Catal. Today, 2011, 168, 38–47 CrossRef CAS.
- K. A. Tarach, J. Martinez-Triguero, S. Valencia, K. Wojciechowska, F. Rey and K. Góra-Marek, Appl. Catal., B, 2023, 338, 123066 CrossRef CAS.
- M. Kubů, N. Žilková and J. Čejka, Catal. Today, 2011, 168, 63–70 CrossRef.
- Q. Yu, H. Sun, H. Sun, L. Li, X. Zhu, S. Ren, Q. Guo and B. Shen, Microporous Mesoporous Mater., 2019, 273, 297–306 CrossRef CAS.
- J. C. Groen, S. Abelló, L. A. Villaescusa and J. Pérez-Ramírez, Microporous Mesoporous Mater., 2008, 114, 93–102 CrossRef CAS.
- K. Tarach, K. Gora-Marek, J. Tekla, K. Brylewska, J. Datka, K. Mlekodaj, W. Makowski, M. C. Igualada Lopez, J. Martinez Triguero and F. Rey, J. Catal., 2014, 312, 46–57 CrossRef CAS.
- K. Pyra, K. A. Tarach, D. Majda and K. Góra-Marek, Catal. Sci. Technol., 2019, 9, 1794–1801 RSC.
- J. Pérez-Ramírez, J. C. Groen, A. Brückner, M. S. Kumar, U. Bentrup, M. N. Debbagh and L. A. Villaescusa, J. Catal., 2005, 232, 318–334 CrossRef.
- C. H. Christensen, K. Johannsen, I. Schmidt and C. H. Christensen, J. Am. Chem. Soc., 2003, 125, 13370–13371 CrossRef CAS PubMed.
- Y. Tao, H. Kanoh and K. Kaneko, Langmuir, 2005, 21, 504–507 CrossRef CAS PubMed.
- M. Y. Kustova, P. Hasselriis and C. H. Christensen, Catal. Lett., 2004, 96, 205–211 CrossRef CAS.
- Y. Tao, H. Kanoh and K. Kaneko, J. Phys. Chem. B, 2003, 107, 10974–10976 CrossRef CAS.
- K. Egeblad, M. Kustova, S. K. Klitgaard, K. Zhu and C. H. Christensen, Microporous Mesoporous Mater., 2007, 101, 214–223 CrossRef CAS.
- Y. Wei, T. E. Parmentier, K. P. de Jong and J. Zecevic, Chem. Soc. Rev., 2015, 44, 7234–7261 Search PubMed.
- K. Möller, B. Yilmaz, U. Müller and T. Bein, Chem. – Eur. J., 2012, 18, 7671–7674 CrossRef PubMed.
- P. Waller, Z. Shan, L. Marchese, G. Tartaglione, W. Zhou, J. C. Jansen and T. Maschmeyer, Chem. – Eur. J., 2004, 10, 4970–4976 CrossRef CAS PubMed.
- S. A. Bagshaw, N. I. Baxter, D. R. M. Brew, C. F. Hosie, N. Yuntong, S. Jaenicke and C. G. Khuan, J. Mater. Chem., 2006, 16, 2235–2244 RSC.
- J. Perez-Ramirez, S. Abello, A. Bonilla and J. C. Groen, Adv. Funct. Mater., 2009, 19, 164–172 CrossRef CAS.
- C. Bertrand-Drira, X.-W. Cheng, T. Cacciaguerra, P. Trens, G. Melinte, O. Ersen, D. Minoux, A. Finiels, F. Fajula and C. Gerardin, Microporous Mesoporous Mater., 2015, 213, 142–149 CrossRef CAS.
- N. Chaouati, A. Soualah, I. Hussein, J. D. Comparot and L. Pinard, Appl. Catal., A, 2016, 526, 95–104 CrossRef CAS.
- J. C. Groen, T. Sano, J. A. Moulijn and J. Pérez-Ramírez, J. Catal., 2007, 251, 21–27 CrossRef CAS.
- A. N. C. van Laak, R. W. Gosselink, S. L. Sagala, J. D. Meeldijk, P. E. de Jongh and K. P. de Jong, Appl. Catal., A, 2010, 382, 65–72 CrossRef CAS.
- J.-S. Lin, J.-J. Wang, J. Wang, I. Wang, R. J. Balasamy, A. Aitani, S. Al-Khattaf and T.-C. Tsai, J. Catal., 2013, 300, 81–90 Search PubMed.
- F.-W. Zhao, Q. Zhang, F. Hui, J. Yuan, S.-N. Mei, Q.-W. Yu, J.-M. Yang, W. Mao, Z.-W. Liu, Z.-T. Liu and J. Lu, Catalysts, 2020, 10, 10040386 Search PubMed.
- J. Pastvova, D. Kaucky, J. Moravkova, J. Rathousky, S. Sklenak, M. Vorokhta, L. Brabec, R. Pilar, I. Jakubec, E. Tabor, P. Klein and P. Sazama, ACS Catal., 2017, 7, 5781–5795 Search PubMed.
- Y. Liu, D. Zheng, B. Li, Y. Lyu, X. Wang, X. Liu, L. Li, S. Yu, X. Liu and Z. Yan, Microporous Mesoporous Mater., 2020, 299, 110117 CrossRef CAS.
- X. Wang, R. Li, C. Yu, Y. Liu, L. Zhang, C. Xu and H. Zhou, Fuel, 2019, 239, 794–803 CrossRef CAS.
- A. K. Ghosh and R. A. Kydd, J. Catal., 1987, 103, 399–406 Search PubMed.
- Z. Qin, B. Shen, X. Gao, F. Lin, B. Wang and C. Xu, J. Catal., 2011, 278, 266–275 CrossRef CAS.
- K. P. de Jong, J. Zečević, H. Friedrich, P. E. de Jongh, M. Bulut, S. van Donk, R. Kenmogne, A. Finiels, V. Hulea and F. Fajula, Angew. Chem., Int. Ed., 2010, 49, 10074–10078 CrossRef CAS PubMed.
- D. Verboekend, G. Vilé and J. Pérez-Ramírez, Adv. Funct. Mater., 2012, 22, 916–928 CrossRef CAS.
- M. Gackowski, K. Tarach, L. Kuterasinski, J. Podobinski, S. Jarczewski, P. Kustrowski and J. Datka, Microporous Mesoporous Mater., 2018, 263, 282–288 CrossRef CAS.
- C. Manrique, R. Solano, C. Mendoza, S. Amaya and A. Echavarría, New J. Chem., 2024, 48, 6188–6200 RSC.
- C. Pagis, A. R. Morgado Prates, N. Bats, A. Tuel and D. Farrusseng, CrystEngComm, 2018, 20, 1564–1572 Search PubMed.
- I. C. Medeiros-Costa, E. Dib, N. Nesterenko, J.-P. Dath, J.-P. Gilson and S. Mintova, Chem. Soc. Rev., 2021, 50, 11156–11179 RSC.
- M. S. Holm, E. Taarning, K. Egeblad and C. H. Christensen, Catal. Today, 2011, 168, 3–16 CrossRef CAS.
- R. von Ballmoos and W. M. Meier, Nature, 1981, 289, 782–783 Search PubMed.
- R. Althoff, B. Schulz-Dobrick, F. Schüth and K. Unger, Microporous Mater., 1993, 1, 207–218 CrossRef CAS.
- K. Sadowska, A. Wach, Z. Olejniczak, P. Kustrowski and J. Datka, Microporous Mesoporous Mater., 2013, 167, 82–88 CrossRef CAS.
- B. Gil, Ł. Mokrzycki, B. Sulikowski, Z. Olejniczak and S. Walas, Catal. Today, 2010, 152, 24–32 CrossRef CAS.
- D. Verboekend and J. Pérez-Ramírez, Chem. – Eur. J., 2011, 17, 1137–1147 CrossRef CAS PubMed.
- K. Sadowska, K. Gora-Marek, M. Drozdek, P. Kustrowski, J. Datka, J. Martinez Triguero and F. Rey, Microporous Mesoporous Mater., 2013, 168, 195–205 CrossRef CAS.
- K. Sadowska, K. Gora-Marek and J. Datka, Vib. Spectrosc., 2012, 63, 418–425 CrossRef CAS.
- K. A. Tarach, K. Góra-Marek, J. Martinez-Triguero and I. Melián-Cabrera, Catal. Sci. Technol., 2017, 7, 858–873 RSC.
- K. Gora-Marek, J. Datka, S. Dzwigaj and M. Che, J. Phys. Chem. B, 2006, 110, 6763–6767 CrossRef CAS PubMed.
- L. H. Ong, M. Dömök, R. Olindo, A. C. van Veen and J. A. Lercher, Microporous Mesoporous Mater., 2012, 164, 9–20 Search PubMed.
- F. Thibault-Starzyk, A. Vimont and J.-P. Gilson, Catal. Today, 2001, 70, 227–241 CrossRef CAS.
- M. Grahn, A. Faisal, O. G. W. Öhrman, M. Zhou, M. Signorile, V. Crocellà, M. S. Nabavi and J. Hedlund, Catal. Today, 2020, 345, 136–146 CrossRef CAS.
- K. Lee, S. Lee, Y. Jun and M. Choi, J. Catal., 2017, 347, 222–230 CrossRef CAS.
- K. Sadowska, K. Gora-Marek and J. Datka, J. Phys. Chem. C, 2013, 117, 9237–9244 CrossRef CAS.
- F. Thibault-Starzyk, I. Stan, S. Abelló, A. Bonilla, K. Thomas, C. Fernandez, J.-P. Gilson and J. Pérez-Ramírez, J. Catal., 2009, 264, 11–14 CrossRef CAS.
- K. A. Tarach, K. Pyra and K. Góra-Marek, Molecules, 2020, 25, 2878 CrossRef CAS PubMed.
- J. S. Beck, J. C. Vartuli, G. J. Kennedy, C. T. Kresge, W. J. Roth and S. E. Schramm, Chem. Mater., 1994, 6, 1816–1821 CrossRef CAS.
- J. Cejka and S. Mintova, Catal. Rev.: Sci. Eng., 2007, 49, 457–509 CrossRef CAS.
- M. Hartmann, M. Thommes and W. Schwieger, Adv. Mater. Interfaces, 2021, 8, 2001841 CrossRef.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- K. Richard Kloetstra and J. C. Jansen, Chem. Commun., 1997, 2281–2282 RSC.
- M. J. Verhoef, P. J. Kooyman, J. C. Van Der Waal, M. S. Rigutto, J. A. Peters and H. Van Bekkum, Chem. Mater., 2001, 13, 683–687 CrossRef CAS.
- D. Trong On and S. Kaliaguine, Angew. Chem., Int. Ed., 2001, 40, 3248–3251 CrossRef PubMed.
- A. A. Campos, C. R. Silva, M. Wallau, L. D. Dimitrov and E. A. Urquieta-González, in Studies in Surface Science and Catalysis, ed. J. Čejka, N. Žilková and P. Nachtigall, Elsevier, 2005, vol. 158, pp. 573–580 Search PubMed.
- A. A. Campos, C. R. Silva, L. D. Dimitrov, M. Wallau and E. A. Urquieta-Gonzalez, in Studies in Surface Science and Catalysis, ed. E. M. Gaigneaux, M. Devillers, D. E. De Vos, S. Hermans, P. A. Jacobs, J. A. Martens and P. Ruiz, Elsevier, 2006, vol. 162, pp. 347–354 Search PubMed.
- A. A. Campos, L. Martins, L. L. de Oliveira, C. R. da Silva, M. Wallau and E. A. Urquieta-González, Catal. Today, 2005, 107–108, 759–767 CrossRef CAS.
- S. Habib, F. Launay, H. E. Zakhem, M. Mazaj, F. Guenneau, P. Beaunier, D. Brouri, N. N. Tušar, V. Kaučič and A. Gédéon, Mater. Res. Bull., 2013, 48, 1288–1295 CrossRef CAS.
- Y. Zhang, T. Okubo and M. Ogura, Chem. Commun., 2005, 2719 RSC.
- P. Prokesova, S. Mintova, J. Cejka and T. Bein, Mater. Sci. Eng., C, 2003, 23, 1001–1005 CrossRef.
- P. Prokešová-Fojtíková, S. Mintova, J. Čejka, N. Žilková and A. Zukal, Microporous Mesoporous Mater., 2006, 92, 154–160 CrossRef.
- Y. Liu, W. Z. Zhang and T. J. Pinnavaia, J. Am. Chem. Soc., 2000, 122, 8791–8792 CrossRef CAS.
- Y. Liu, W. Z. Zhang and T. J. Pinnavaia, Angew. Chem., Int. Ed., 2001, 40, 1255–1258 CrossRef CAS PubMed.
- D. Trong On and S. Kaliaguine, J. Am. Chem. Soc., 2003, 125, 618–619 CrossRef PubMed.
- Z. Zhang, Y. Han, F.-S. Xiao, S. Qiu, L. Zhu, R. Wang, Y. Yu, Z. Zhang, B. Zou, Y. Wang, H. Sun, D. Zhao and Y. Wei, J. Am. Chem. Soc., 2001, 123, 5014–5021 CrossRef CAS PubMed.
- J. Zhou, J. Teng, L. Ren, Y. Wang, Z. Liu, W. Liu, W. Yang and Z. Xie, J. Catal., 2016, 340, 166–176 CrossRef CAS.
- R. J. Messinger, K. Na, Y. Seo, R. Ryoo and B. F. Chmelka, Angew. Chem., Int. Ed., 2015, 54, 927–931 CrossRef CAS PubMed.
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo, Nature, 2009, 461, 246–249 CrossRef CAS PubMed.
- N. Ni, X. Gao, E. Xing, Y. Zhu, J. Liu, M. Xin, Y. Ouyang, Y. Luo and X. Shu, Microporous Mesoporous Mater., 2023, 360, 112706 CrossRef CAS.
- M. G. Goesten, X. Zhu, B. Mezari and E. J. M. Hensen, Angew. Chem., Int. Ed., 2017, 56, 5160–5163 CrossRef CAS PubMed.
- Z. J. Berkson, R. J. Messinger, K. Na, Y. Seo, R. Ryoo and B. F. Chmelka, Angew. Chem., Int. Ed., 2017, 56, 5164–5169 CrossRef CAS PubMed.
- A. Korde, B. Min, E. Kapaca, O. Knio, I. Nezam, Z. Wang, J. Leisen, X. Yin, X. Zhang, D. S. Sholl, X. Zou, T. Willhammar, C. W. Jones and S. Nair, Science, 2022, 375, 62–66 CrossRef CAS PubMed.
- G. N. Short, E. Burentugs, L. Proaño, H. J. Moon, G. Rim, I. Nezam, A. Korde, S. Nair and C. W. Jones, JACS Au, 2023, 3, 62–69 CrossRef CAS PubMed.
- P. Lu, J. Xu, Y. Sun, R. Guillet-Nicolas, T. Willhammar, M. Fahda, E. Dib, B. Wang, Z. Qin, H. Xu, J. Cho, Z. Liu, H. Yu, X. Yang, Q. Lang, S. Mintova, X. Zou and V. Valtchev, Nature, 2024, 636, 368–373 CrossRef CAS PubMed.
- A. Sachse and J. Garcia-Martinez, Chem. Mater., 2017, 29, 3827–3853 CrossRef CAS.
- D. Wang, L. Xu and P. Wu, J. Mater. Chem. A, 2014, 2, 15535–15545 RSC.
- J. Ying and J. Martinez, US Pat., 7584041, 2004 Search PubMed.
- M. Reisch, C&EN Global Enterprise, 2019, 97, 16 Search PubMed.
- I. A. Kasyanov, A. A. Maerle, I. I. Ivanova and V. I. Zaikovskii, J. Mater. Chem. A, 2014, 2, 16978–16988 RSC.
- F. S. Xiao, L. Wang, C. Yin, K. Lin, Y. Di, J. Li, R. Xu, D. S. Su, R. Schlögl, T. Yokoi and T. Tatsumi, Angew. Chem., Int. Ed., 2006, 45, 3090–3093 CrossRef CAS PubMed.
- J. Song, L. Ren, C. Yin, Y. Ji, Z. Wu, J. Li and F.-S. Xiao, J. Phys. Chem. C, 2008, 112, 8609–8613 CrossRef CAS.
- N. Nikolopoulos, L. A. Parker, A. Wickramasinghe, O. Van Veenhuizen, G. Whiting and B. M. Weckhuysen, Chem. Biomed. Imaging, 2023, 1, 40–48 CrossRef CAS PubMed.
- R. Sabarish and G. Unnikrishnan, SN Appl. Sci., 2019, 1, 989 CrossRef CAS.
- F. N. Gu, F. Wei, J. Y. Yang, N. Lin, W. G. Lin, Y. Wang and J. H. Zhu, Chem. Mater., 2010, 22, 2442–2450 CrossRef CAS.
- Y. Lv, X. Wang, D. Gao, X. Ma, S. Li, Y. Wang, G. Song, A. Duan and G. Chen, Ind. Eng. Chem. Res., 2018, 57, 14031–14043 CrossRef CAS.
- V. S. P. Ganjala, C. K. P. Neeli, C. V. Pramod, M. Khagga, K. S. R. Rao and D. R. Burri, Catal. Commun., 2014, 49, 82–86 CrossRef CAS.
- M. Liu, L.-A. Hou, S. Yu, B. Xi, Y. Zhao and X. Xia, Chem. Eng. J., 2013, 223, 678–687 CrossRef CAS PubMed.
- L. Gao, Z. Shi, U. J. Etim, P. Wu, D. Han, W. Xing, S. Mintova, P. Bai and Z. Yan, Microporous Mesoporous Mater., 2019, 277, 17–28 CrossRef CAS.
- Z. Wang and S. Yu, Adv. Chem. Eng. Sci., 2016, 06, 305–315 CrossRef CAS.
- J. C. Jansen, Z. Shan, T. Maschmeyer, L. Marchese, W. Zhou and N. V. D. Puil, Chem. Commun., 2001, 713–714 RSC.
- C. C. Aquino, H. O. Pastore, A. F. Masters and T. Maschmeyer, ChemCatChem, 2011, 3, 1759–1762 CrossRef CAS.
- M. S. Hamdy and G. Mul, ChemCatChem, 2013, 5, 3156–3163 CrossRef CAS.
- K. Jaroszewska, M. Fedyna and J. Trawczyński, Appl. Catal., B, 2019, 255, 117756 CrossRef.
- Y. Xia and R. Mokaya, J. Mater. Chem., 2004, 14, 863 RSC.
- X. Vu, U. Armbruster and A. Martin, Catalysts, 2016, 6, 183 CrossRef.
- X. H. Vu, U. Bentrup, M. Hunger, R. Kraehnert, U. Armbruster and A. Martin, J. Mater. Sci., 2014, 49, 5676–5689 CrossRef CAS.
- X. H. Vu, N. Steinfeldt, U. Armbruster and A. Martin, Microporous Mesoporous Mater., 2012, 164, 120–126 CrossRef CAS.
- C. Van Oers, W. Stevens, E. Bruijn, M. Mertens, O. Lebedev, G. Van Tendeloo, V. Meynen and P. Cool, Microporous Mesoporous Mater., 2009, 120, 29–34 CrossRef CAS.
- W. Stevens, V. Meynen, E. Bruijn, O. Lebedev, G. Van Tendeloo, P. Cool and E. Vansant, Microporous Mesoporous Mater., 2008, 110, 77–85 CrossRef CAS.
- D. P. Serrano, J. M. Escola and P. Pizarro, Chem. Soc. Rev., 2013, 42, 4004–4035 RSC.
- D. P. Serrano, J. Aguado, J. M. Escola, J. M. Rodriguez and A. Peral, Chem. Mater., 2006, 18, 2462–2464 CrossRef CAS.
- D. P. Serrano, R. A. Garcia, M. Linares and B. Gil, Catal. Today, 2012, 179, 91–101 CrossRef CAS.
- D. P. Serrano, J. Aguado, J. M. Escola, J. M. Rodriguez and A. Peral, J. Mater. Chem., 2008, 18, 4210 RSC.
- D. P. Serrano, T. J. Pinnavaia, J. Aguado, J. M. Escola, A. Peral and L. Villalba, Catal. Today, 2014, 227, 15–25 CrossRef CAS.
- M. del Mar Alonso-Doncel, C. Ochoa-Hernández, G. Gómez-Pozuelo, A. Oliveira, J. González-Aguilar, Á. Peral, R. Sanz and D. P. Serrano, J. Energy Chem., 2023, 80, 77–88 CrossRef CAS.
- K.-G. Haw, J.-M. Goupil, J.-P. Gilson, N. Nesterenko, D. Minoux, J.-P. Dath and V. Valtchev, New J. Chem., 2016, 40, 4307–4313 RSC.
- M. Akouche, J.-P. Gilson, N. Nesterenko, S. Moldovan, D. Chateigner, H. E. Siblani, D. Minoux, J.-P. Dath and V. Valtchev, Chem. Mater., 2020, 32, 2123–2132 CrossRef CAS.
- R. Kumar Parsapur, A. M. Hengne, G. Melinte, O. Refa Koseoglu, R. P. Hodgkins, A. Bendjeriou-Sedjerari, Z. Lai and K. W. Huang, Angew. Chem., Int. Ed., 2024, 63, e202314217 CrossRef CAS PubMed.
- P. Eliášová, M. Opanasenko, P. S. Wheatley, M. Shamzhy, M. Mazur, P. Nachtigall, W. J. Roth, R. E. Morris and J. Čejka, Chem. Soc. Rev., 2015, 44, 7177–7206 RSC.
- T. Sonoda, T. Maruo, Y. Yamasaki, N. Tsunoji, Y. Takamitsu, M. Sadakane and T. Sano, J. Mater. Chem. A, 2015, 3, 857–865 RSC.
- N. Martín, C. R. Boruntea, M. Moliner and A. Corma, Chem. Commun., 2015, 51, 11030–11033 RSC.
- S. Inagaki, Y. Tsuboi, Y. Nishita, T. Syahylah, T. Wakihara and Y. Kubota, Chem. – Eur. J., 2013, 19, 7780–7786 CrossRef CAS PubMed.
- D. Bruter, V. Pavlov and I. Ivanova, Microporous Mesoporous Mater., 2025, 381, 113364 CrossRef CAS.
- J. Devos, M. A. Shah and M. Dusselier, RSC Adv., 2021, 11, 26188–26210 RSC.
- C. Li, M. Moliner and A. Corma, Angew. Chem., Int. Ed., 2018, 57, 15330–15353 CrossRef CAS PubMed.
- S. I. Zones, J. Chem. Soc., Faraday Trans., 1990, 86, 3467 RSC.
- S. Zones, J. Chem. Soc., Faraday Trans., 1991, 87, 3709–3716 RSC.
- S. I. Zones, C. M. Lew, D. Xie, T. M. Davis, J. E. Schmidt and R. J. Saxton, Microporous Mesoporous Mater., 2020, 300, 110162 CrossRef CAS.
- I. Y. Chan and S. I. Zones, Zeolites, 1989, 9, 3–11 CrossRef CAS.
- L. Tang, K.-G. Haw, Y. Zhang, Q. Fang, S. Qiu and V. Valtchev, Microporous Mesoporous Mater., 2019, 280, 306–314 CrossRef CAS.
- X. Peng, L. Chen, L. You, Y. Jin, C. Zhang, S. Ren, F. Kapteijn, X. Wang and X. Gu, Angew. Chem., 2024, 63, e202405969 CrossRef CAS PubMed.
- Z. Wu, H. Sun, Q. Deng, L. Han, P. Lv, H. Chen, H. Duan and K. Zhu, ChemCatChem, 2024, 16, e202401010 CrossRef CAS.
- L. Xu, Y. Yuan, Q. Han, L. Dong, L. Chen, X. Zhang and L. Xu, Catal. Sci. Technol., 2020, 10, 7904–7913 RSC.
- M. M. Lozinska, E. L. Bruce, J. Mattock, R. G. Chitac, P. A. Cox, A. Turrina and P. A. Wright, Chem. – Eur. J., 2022, 28, e202201689 CrossRef CAS PubMed.
- Z. Huang, S. Seo, J. Shin, B. Wang, R. G. Bell, S. B. Hong and X. Zou, Nat. Commun., 2020, 11, 3762 CrossRef PubMed.
- N. Jakupec, K. J. Ardila-Fierro, V. Martinez, I. Halasz, J. Volavšek, G. Algara-Siller, M. Etter, V. Valtchev, K. Užarević and A. Palčić, ACS Sustainable Chem. Eng., 2024, 12, 5220–5228 CrossRef CAS.
- M. J. Mendoza-Castro, E. De Oliveira-Jardim, N.-T. Ramírez-Marquez, C.-A. Trujillo, N. Linares and J. García-Martínez, J. Am. Chem. Soc., 2022, 144, 5163–5171 CrossRef CAS PubMed.
- M. J. Mendoza-Castro, Z. Qie, X. Fan, N. Linares and J. García-Martínez, Nat. Commun., 2023, 14, 1256 CrossRef CAS PubMed.
- T. Sano, M. Itakura and M. Sadakane, J. Jpn. Pet. Inst., 2013, 56, 183–197 CrossRef CAS.
- S. Goel, S. I. Zones and E. Iglesia, Chem. Mater., 2015, 27, 2056–2066 CrossRef CAS.
- I. Goto, M. Itakura, S. Shibata, K. Honda, Y. Ide, M. Sadakane and T. Sano, Microporous Mesoporous Mater., 2012, 158, 117–122 CrossRef CAS.
- K. Honda, M. Itakura, Y. Matsuura, A. Onda, Y. Ide, M. Sadakane and T. Sano, J. Nanosci. Nanotechnol., 2013, 13, 3020–3026 CrossRef CAS PubMed.
- D. Suhendar, Buchari, R. R. Mukti and Ismunandar, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 349, 012016 CrossRef.
- D. Schwalbe-Koda, Z. Jensen, E. Olivetti and R. Gómez-Bombarelli, Nat. Mater., 2019, 18, 1177–1181 CrossRef CAS PubMed.
- K. Mlekodaj, M. Bernauer, J. E. Olszowka, P. Klein, V. Pashkova and J. Dedecek, Chem. Mater., 2021, 33, 1781–1788 CrossRef CAS.
- Z. Liu, A. Chokkalingam, S. Miyagi, M. Yoshioka, T. Ishikawa, H. Yamada, K. Ohara, N. Tsunoji, Y. Naraki, T. Sano, T. Okubo and T. Wakihara, Phys. Chem. Chem. Phys., 2022, 24, 4136–4146 RSC.
- C. Chu-Jon, E. Martinez, A. A. Bertolazzo, S. Banik, J. D. Rimer, S. K. R. S. Sankaranarayanan and V. Molinero, J. Am. Chem. Soc., 2024, 146, 33204–33213 CrossRef CAS PubMed.
- S. F. S. P. Looijmans, D. Cavallo, L. Yu and G. W. M. Peters, Cryst. Growth Des., 2018, 18, 3921–3926 CrossRef CAS PubMed.
- D. Cavallo, F. Galli, L. Yu and G. C. Alfonso, Cryst. Growth Des., 2017, 17, 2639–2645 CrossRef CAS.
- J. Devos, S. Robijns, C. Van Goethem, I. Khalil and M. Dusselier, Chem. Mater., 2021, 33, 2516–2531 CrossRef CAS.
- C. Pagis, F. Meunier, Y. Schuurman, A. Tuel, M. Dodin, R. Martinez-Franco and D. Farrusseng, ChemCatChem, 2018, 10, 4525–4529 CrossRef CAS.
- T. Li, Z. Ma, F. Krumeich, A. J. Knorpp, A. B. Pinar and J. A. Van Bokhoven, ChemNanoMat, 2018, 4, 992–999 CrossRef CAS.
- D. Fodor, F. Krumeich, R. Hauert and J. A. Van Bokhoven, Chem. – Eur. J., 2015, 21, 6272–6277 CrossRef CAS PubMed.
- D. Yuan, C. Kang, W. Wang, H. Li, X. Zhu, Y. Wang, X. Gao, B. Wang, H. Zhao, C. Liu and B. Shen, Catal. Sci. Technol., 2016, 6, 8364–8374 RSC.
- Y. Shen, M. Xu, J. Li, Z. Qin, C. Wang, S. Mintova and X. Liu, Inorg. Chem. Front., 2021, 8, 2144–2152 RSC.
- W. Liu, J. Li, Q. Yu, H. Chen, W. Liu, Z. Yang, X. Liu, Z. Xu, S. Xu, X. Zhu and X. Li, ACS Appl. Mater. Interfaces, 2022, 14, 52025–52034 CrossRef CAS PubMed.
- M. Li, Y. Zhai, X. Zhang, F. Wang, G. Lv, A. Rosine, M. Li, Q. Zhang and Y. Liu, Microporous Mesoporous Mater., 2022, 331, 111655 CrossRef CAS.
- C. Dai, A. Zhang, C. Song and X. Guo, in Advances in Catalysis, ed. C. Song, Academic Press, 2018, vol. 63, pp. 75–115 Search PubMed.
- Z. Wang, Y. Liu, J.-G. Jiang, M. He and P. Wu, J. Mater. Chem., 2010, 20, 10193–10199 RSC.
- M. Rutkowska, W. Dubiel, A. Kowalczyk, A. Jankowska, Z. Piwowarska, K. Maćkosz, J. Kawałko, B. Gil and L. Chmielarz, J. Phys. Chem. Solids, 2025, 201, 112645 CrossRef CAS.
- A. Dong, Y. Wang, Y. Tang, N. Ren, Y. Zhang and Z. Gao, Chem. Mater., 2002, 14, 3217–3219 CrossRef CAS.
- A. Dong, Y. Wang, Y. Tang, Y. Zhang, N. Ren and Z. Gao, Adv. Mater., 2002, 14, 1506–1510 CrossRef CAS.
- A. Dong, N. Ren, W. Yang, Y. Wang, Y. Zhang, D. Wang, J. Hu, Z. Gao and Y. Tang, Adv. Funct. Mater., 2003, 13, 943–948 CrossRef CAS.
- A. R. Morgado Prates, C. Pagis, F. C. Meunier, L. Burel, T. Epicier, L. Roiban, S. Koneti, N. Bats, D. Farrusseng and A. Tuel, Cryst. Growth Des., 2018, 18, 592–596 CrossRef CAS.
- Y. Wang, M. Lin and A. Tuel, Microporous Mesoporous Mater., 2007, 102, 80–85 CrossRef CAS.
- L. F. Acevedo-Córdoba, O. J. Vargas-Montañez, E. A. Velasco-Rozo, I. D. Mora-Vergara, J. N. Díaz de León, D. Pérez-Martínez, E. M. Morales-Valencia and V. G. Baldovino-Medrano, Catal. Today, 2024, 429, 114474 CrossRef.
- D. N. Rainer and R. E. Morris, Dalton Trans., 2021, 50, 8995–9009 RSC.
- P. A. Zielinski, A. Van Neste, D. B. Akolekar and S. Kaliaguine, Microporous Mater., 1995, 5, 123–133 CrossRef CAS.
- T. Wakihara, R. Ichikawa, J. Tatami, A. Endo, K. Yoshida, Y. Sasaki, K. Komeya and T. Meguro, Cryst. Growth Des., 2011, 11, 955–958 CrossRef CAS.
- T. Wakihara, A. Ihara, S. Inagaki, J. Tatami, K. Sato, K. Komeya, T. Meguro, Y. Kubota and A. Nakahira, Cryst. Growth Des., 2011, 11, 5153–5158 CrossRef CAS.
- T. Wakihara, K. Sato, K. Sato, J. Tatami, S. Kohara, K. Komeya and T. Meguro, J. Ceram. Soc. Jpn., 2012, 120, 341–343 CrossRef CAS.
- T. Wakihara and J. Tatami, J. Jpn. Pet. Inst., 2013, 56, 206–213 CrossRef CAS.
- S. Miyagawa, K. Miyake, Y. Hirota, N. Nishiyama, M. Miyamoto, Y. Oumi and S. Tanaka, Microporous Mesoporous Mater., 2019, 278, 219–224 CrossRef CAS.
- S. Inagaki, K. Sato, S. Hayashi, J. Tatami, Y. Kubota and T. Wakihara, ACS Appl. Mater. Interfaces, 2015, 7, 4488–4493 CrossRef CAS PubMed.
- D. S. D. Lima, I. W. Zapelini, L. L. Silva, S. Mintova and L. Martins, Catal. Today, 2024, 441, 114842 CrossRef CAS.
- M. M. Alvarez, J. Aizenberg, M. Analoui, A. M. Andrews, G. Bisker, E. S. Boyden, R. D. Kamm, J. M. Karp, D. J. Mooney, R. Oklu, D. Peer, M. Stolzoff, M. S. Strano, G. Trujillo-De Santiago, T. J. Webster, P. S. Weiss and A. Khademhosseini, ACS Nano, 2017, 11, 5195–5214 CrossRef CAS PubMed.
- F. J. Tovar-Lopez, Sensors, 2023, 23, 5406 CrossRef CAS PubMed.
- J. L. H. Chau, Y. S. S. Wan, A. Gavriilidis and K. L. Yeung, Chem. Eng. J., 2002, 88, 187–200 CrossRef CAS.
- Y. S. S. Wan, J. L. H. Chau, A. Gavriilidis and K. L. Yeung, Microreaction Technology, Springer Berlin Heidelberg, 2001, pp. 94–102 Search PubMed.
- J. L. H. Chau, A. Y. L. Leung and K. L. Yeung, Lab Chip, 2003, 3, 53–55 RSC.
- J. L. H. Chau and K. L. Yeung, Chem. Commun., 2002, 960–961 RSC.
- I. Pellejero, M. Urbiztondo, M. Villarroya, J. Sesé, M. P. Pina and J. Santamaría, Microporous Mesoporous Mater., 2008, 114, 110–120 CrossRef CAS.
- L. Huang, Z. Wang, J. Sun, L. Miao, Q. Li, Y. Yan and D. Zhao, J. Am. Chem. Soc., 2000, 122, 3530–3531 CrossRef CAS.
- D. E. W. Vaughan, Catal. Today, 1988, 2, 187–198 CrossRef CAS.
- A. Clearfield, H. P. Perry and K. J. Gagnon, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk, K. Poeppelmeier, Elsevier, Amsterdam, 2013, pp. 169–211 Search PubMed.
- S. M. Auerbach, K. A. Carrado and P. K. Dutta, Handbook of Layered Materials, Marcel Dekker, New York, 2004 Search PubMed.
- G. Lagaly and K. Beneke, Colloid Polym. Sci., 1991, 269, 1198–1211 CrossRef CAS.
- T. V. Whittam, US Pat., 4397825, 1983.
- A. J. Blake, K. R. Franklin and B. M. Lowe, J. Chem. Soc., Dalton Trans., 1988, 2513–2517 RSC.
- U. Brenn, H. Ernst, D. Freude, R. Herrmann, R. Jähnig, H. G. Karge, J. Kärger, T. König, B. Mädler, U. T. Pingel, D. Prochnow and W. Schwieger, Microporous Mesoporous Mater., 2000, 40, 43–52 CrossRef CAS.
- M. K. Rubin and P. Chu, US Pat., 4954325, 1990.
- M. E. Leonowicz, J. A. Lawton, S. L. Lawton and M. K. Rubin, Science, 1994, 264, 1910–1913 CrossRef CAS PubMed.
- J. M. Bennett, C. D. Chang, S. L. Lawton, M. E. Leonowicz, D. N. Lissy and M. K. Rubin, US Pat., 5236575, 1993 Search PubMed.
- S. L. Lawton, A. S. Fung, G. J. Kennedy, L. B. Alemany, C. D. Chang, G. H. Hatzikos, D. N. Lissy, M. K. Rubin, H. K. C. Timken, S. Steuernagel and D. E. Woessner, J. Phys. Chem., 1996, 100, 3788–3798 CrossRef CAS.
- L. Schreyeck, P. Caullet, J.-C. Mougenel, J.-L. Guth and B. Marler, J. Chem. Soc., Chem. Commun., 1995, 2187–2188 RSC.
- L. Schreyeck, P. Caullet, J. C. Mougenel, J. L. Guth and B. Marler, Microporous Mater., 1996, 6, 259–271 CrossRef CAS.
- W. J. Roth, P. Nachtigall, R. E. Morris and J. Čejka, Chem. Rev., 2014, 114, 4807–4837 CrossRef CAS PubMed.
- M. Shamzhy, B. Gil, M. Opanasenko, W. J. Roth and J. Čejka, ACS Catal., 2021, 11, 2366–2396 CrossRef CAS.
- E. Schulman, W. Wu and D. X. Liu, Materials, 2020, 13, 1822 CrossRef CAS PubMed.
- B. Marler and H. Gies, Eur. J. Mineral., 2012, 24, 405–428 CrossRef CAS.
- F. S. O. Ramos, M. K. de Pietre and H. O. Pastore, RSC Adv., 2013, 3, 2084–2111 RSC.
- W. J. Roth, B. Gil, W. Makowski, B. Marszalek and P. Eliasova, Chem. Soc. Rev., 2016, 45, 3400–3438 RSC.
- K. Na, C. Jo, J. Kim, K. Cho, J. Jung, Y. Seo, R. J. Messinger, B. F. Chmelka and R. Ryoo, Science, 2011, 333, 328–332 CrossRef CAS PubMed.
- W. J. Roth, J. Čejka, R. Millini, E. Montanari, B. Gil and M. Kubu, Chem. Mater., 2015, 27, 4620–4629 CrossRef CAS.
- W. J. Roth, T. Sasaki, K. Wolski, Y. Ebina, D. M. Tang, Y. Michiue, N. Sakai, R. Z. Ma, O. Cretu, J. Kikkawa, K. Kimoto, K. Kalahurska, B. Gil, M. Mazur, S. Zapotoczny, J. Cejka, J. Grzybek and A. Kowalczyk, J. Am. Chem. Soc., 2021, 143, 11052–11062 CrossRef CAS PubMed.
- Y. Zhou, Y. Mu, M.-F. Hsieh, B. Kabius, C. Pacheco, C. Bator, R. M. Rioux and J. D. Rimer, J. Am. Chem. Soc., 2020, 142, 8211–8222 CrossRef CAS PubMed.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 1–18 CrossRef.
- J. Li, Z. R. Gao, Q.-F. Lin, C. Liu, F. Gao, C. Lin, S. Zhang, H. Deng, A. Mayoral, W. Fan, S. Luo, X. Chen, H. He, M. A. Camblor, F.-J. Chen and J. Yu, Science, 2023, 379, 283–287 CrossRef CAS PubMed.
- Z. R. Gao, H. Yu, F.-J. Chen, A. Mayoral, Z. Niu, Z. Niu, X. Li, H. Deng, C. Márquez-Álvarez, H. He, S. Xu, Y. Zhou, J. Xu, H. Xu, W. Fan, S. R. G. Balestra, C. Ma, J. Hao, J. Li, P. Wu, J. Yu and M. A. Camblor, Nature, 2024, 628, 99–103 CrossRef CAS PubMed.
- F. Bergaya and G. Lagaly, in Developments in Clay Science, ed. F. Bergaya and G. Lagaly, Elsevier, 2013, vol. 5, pp. 1–19 Search PubMed.
- W. J. Roth, C. T. Kresge, J. C. Vartuli, M. E. Leonowicz, A. S. Fung and S. B. McCullen, Catal. Microporous Mater., 1995, 94, 301–308 CAS.
- R. Millini, G. Perego, W. O. Parker Jr, G. Bellussi and L. Carluccio, Microporous Mater., 1995, 4, 221–230 CrossRef CAS.
- H. Xu and P. Wu, Nat. Sci. Rev., 2022, 9, nwac045 CrossRef CAS PubMed.
- W. J. Roth, T. Sasaki, K. Wolski, Y. Song, D. M. Tang, Y. Ebina, R. Z. Ma, J. Grzybek, K. Kalahurska, B. Gil, M. Mazur, S. Zapotoczny and J. Cejka, Sci. Adv., 2020, 6, eaay8163 CrossRef CAS PubMed.
- W. J. Roth, M. Opanasenko, M. Mazur, B. Gil, J. Cejka and T. Sasaki, Adv. Mater., 2024, 36, 2307341 CrossRef CAS PubMed.
- M. Opanasenko, M. Shamzhy, Y. Wang, W. Yan, P. Nachtigall and J. Čejka, Angew. Chem., Int. Ed., 2020, 59, 19380–19389 CrossRef CAS PubMed.
- Q. Yue, V. Kasneryk, M. Mazur, S. Abdi, Y. Zhou, P. S. Wheatley, R. E. Morris, J. Čejka, M. Shamzhy and M. Opanasenko, J. Mater. Chem. A, 2024, 12, 802–812 RSC.
- M. Mazur, P. S. Wheatley, M. Navarro, W. J. Roth, M. Položij, A. Mayoral, P. Eliášová, P. Nachtigall, J. Čejka and R. E. Morris, Nat. Chem., 2016, 8, 58–62 CrossRef CAS PubMed.
- W. J. Roth, T. Sasaki, K. Wolski, B. Gil, S. Zapotoczny, J. Cejka, M. Kubu, M. Mazur, Y. Ebina, N. Sakai, D. M. Tang and R. Z. Ma, Inorg. Chem. Front., 2023, 10, 1511–1521 RSC.
- B. Y. Liu, C. Wattanaprayoon, S. C. Oh, L. Emdadi and D. X. Liu, Chem. Mater., 2015, 27, 1479–1487 CrossRef CAS.
- X. Y. Zhang, D. X. Liu, D. D. Xu, S. Asahina, K. A. Cychosz, K. V. Agrawal, Y. Al Wahedi, A. Bhan, S. Al Hashimi, O. Terasaki, M. Thommes and M. Tsapatsis, Science, 2012, 336, 1684–1687 CrossRef CAS PubMed.
- N. Tsunoji, T. Ikeda, Y. Ide, M. Sadakane and T. Sano, J. Mater. Chem., 2012, 22, 13682–13690 RSC.
- J. L. Wang, Y. Q. Fan, J. A. Jiang, Z. Wan, S. Y. Pang, Y. J. Guan, H. Xu, X. He, Y. H. Ma, A. S. Huang and P. Wu, Angew. Chem., Int. Ed., 2023, 62, e202304734 CrossRef CAS PubMed.
- A. Corma, U. Diaz, M. E. Domine and V. Fornés, Angew. Chem., Int. Ed., 2000, 39, 1499–1501 CrossRef CAS PubMed.
- Z. Zhao, W. Zhang, P. Ren, X. Han, U. Mueller, B. Yilmaz, M. Feyen, H. Gies, F.-S. Xiao, D. De Vos, T. Tatsumi and X. Bao, Chem. Mater., 2013, 25, 840–847 CrossRef CAS.
- W. J. Roth, B. Gil, W. Makowski, A. Sławek, J. Grzybek, M. Kubu and J. Čejka, Chem. Mater., 2016, 28, 3616–3619 CrossRef CAS.
- A. Burton, R. J. Accardi, R. F. Lobo, M. Falcioni and M. W. Deem, Chem. Mater., 2000, 12, 2936–2942 CrossRef CAS.
- S. Maheshwari, E. Jordan, S. Kumar, F. S. Bates, R. L. Penn, D. F. Shantz and M. Tsapatsis, J. Am. Chem. Soc., 2008, 130, 1507–1516 CrossRef CAS PubMed.
- B. Marler, Y. Wang, J. Song and H. Gies, Dalton Trans., 2014, 43, 10396–10416 RSC.
- T. Moteki, W. Chaikittisilp, A. Shimojima and T. Okubo, J. Am. Chem. Soc., 2008, 130, 15780–15781 CrossRef CAS PubMed.
- S. Smeets, Z. J. Berkson, D. Xie, S. I. Zones, W. Wan, X. Zou, M.-F. Hsieh, B. F. Chmelka, L. B. McCusker and C. Baerlocher, J. Am. Chem. Soc., 2017, 139, 16803–16812 CrossRef CAS PubMed.
- J. Grzybek, W. J. Roth, B. Gil, A. Korzeniowska, M. Mazur, J. Čejka and R. E. Morris, J. Mater. Chem. A, 2019, 7, 7701–7709 RSC.
- A. S. Fung, S. L. Lawton and W. J. Roth, US Pat., 5362697, 1994 Search PubMed.
- W. J. Roth, Catal. Microporous Mater., 2005, 158A and B, 19–26 Search PubMed.
- J. Dewing, M. S. Spencer and T. V. Whittam, Catal. Rev.: Sci. Eng., 1985, 27, 461–514 CrossRef.
- G. G. Juttu and R. F. Lobo, Microporous Mesoporous Mater., 2000, 40, 9–23 CrossRef CAS.
- M. A. Camblor, A. Corma, M.-J. Díaz-Cabañas and C. Baerlocher, J. Phys. Chem. B, 1998, 102, 44–51 CrossRef CAS.
- A. Corma, V. Fornes, S. B. Pergher, T. L. M. Maesen and J. G. Buglass, Nature, 1998, 396, 353–356 CrossRef CAS.
- W. B. Fan, P. Wu, S. Namba and T. Tatsumi, Angew. Chem., Int. Ed., 2004, 43, 236–240 CrossRef CAS PubMed.
- K. Varoon, X. Y. Zhang, B. Elyassi, D. D. Brewer, M. Gettel, S. Kumar, J. A. Lee, S. Maheshwari, A. Mittal, C. Y. Sung, M. Cococcioni, L. F. Francis, A. V. McCormick, K. A. Mkhoyan and M. Tsapatsis, Science, 2011, 334, 72–75 CrossRef CAS PubMed.
- H. Y. Luo, V. K. Michaelis, S. Hodges, R. G. Griffin and Y. Román-Leshkov, Chem. Sci., 2015, 6, 6320–6324 RSC.
- G. H. Lee, E. Jang, T. H. Lee, Y. Jeong, H. Kim, S. L. Lee, Y. G. Chung, K. S. Ha, H. Baik, H. G. Jang, S. J. Cho and J. Choi, Catal. Today, 2023, 411–412, 113856 CrossRef CAS.
- W. J. Roth, B. Gil, A. Mayoral, J. Grzybek, A. Korzeniowska, M. Kubu, W. Makowski, J. Cejka, Z. Olejniczak and M. Mazur, Dalton Trans., 2018, 47, 3029–3037 RSC.
- W. J. Roth, P. Nachtigall, R. E. Morris, P. S. Wheatley, V. R. Seymour, S. E. Ashbrook, P. Chlubná, L. Grajciar, M. Položij, A. Zukal, O. Shvets and J. Čejka, Nat. Chem., 2013, 5, 628–633 CrossRef CAS PubMed.
- W. J. Roth and C. T. Kresge, Microporous Mesoporous Mater., 2011, 144, 158–161 CrossRef CAS.
- M. Ogawa and D. Iwata, Cryst. Growth Des., 2010, 10, 2068–2072 CrossRef CAS.
- A. Kawai, T. Ikeda, T. Kodaira, A. Endo and F. Mizukami, J. Nanosci. Nanotechnol., 2013, 13, 2864–2870 CrossRef CAS PubMed.
- M. Dakhchoune, L. F. Villalobos, R. Semino, L. M. Liu, M. Rezaei, P. Schouwink, C. E. Avalos, P. Baade, V. Wood, Y. Han, M. Ceriotti and K. V. Agrawal, Nat. Mater., 2021, 20, 362–369 CrossRef CAS PubMed.
- H. Zhang, Q. Xiao, X. Guo, N. Li, P. Kumar, N. Rangnekar, M. Y. Jeon, S. Al-Thabaiti, K. Narasimharao, S. N. Basahel, B. Topuz, F. J. Onorato, C. W. Macosko, K. A. Mkhoyan and M. Tsapatsis, Angew. Chem., Int. Ed., 2016, 55, 7184–7187 CrossRef CAS PubMed.
- Y. C. Li, G. F. Zhu, Y. Wang, Y. M. Chai and C. G. Liu, Microporous Mesoporous Mater., 2021, 312, 110790 CrossRef CAS.
- T. F. Degnan, G. K. Chitnis and P. H. Schipper, Microporous Mesoporous Mater., 2000, 35–36, 245–252 CrossRef CAS.
- K. Na, W. Park, Y. Seo and R. Ryoo, Chem. Mater., 2011, 23, 1273–1279 CrossRef CAS.
- W. Park, D. Yu, K. Na, K. E. Jelfs, B. Slater, Y. Sakamoto and R. Ryoo, Chem. Mater., 2011, 23, 5131–5137 CrossRef CAS.
- J. Jung, C. Jo, K. Cho and R. Ryoo, J. Mater. Chem., 2012, 22, 4637–4640 RSC.
- R. Martínez-Franco, C. Paris, J. Martínez-Triguero, M. Moliner and A. Corma, Microporous Mesoporous Mater., 2017, 246, 147–157 CrossRef.
- B. Marler, N. Stroter and H. Gies, Microporous Mesoporous Mater., 2005, 83, 201–211 CrossRef CAS.
- P. Loch, D. Schuchardt, G. Algara-Siller, P. Markus, K. Ottermann, S. Rosenfeldt, T. Lunkenbein, W. Schwieger, G. Papastavrou and J. Breu, Sci. Adv., 2022, 8, eabn9084 CrossRef CAS PubMed.
- G. Lagaly, Adv. Colloid Interface Sci., 1979, 11, 105–148 CrossRef CAS.
- M. E. Landis, B. A. Aufdembrink, P. Chu, I. D. Johnson, G. W. Kirker and M. K. Rubin, J. Am. Chem. Soc., 1991, 113, 3189–3190 CrossRef CAS.
- W. J. Roth and J. C. Vartuli, Catal. Microporous Mater., 2002, 141, 273–279 CAS.
- W. O. Haag, R. M. Lago and P. B. Weisz, Nature, 1984, 309, 589–591 CrossRef CAS.
- S. Maheshwari, C. Martínez, M. Teresa Portilla, F. J. Llopis, A. Corma and M. Tsapatsis, J. Catal., 2010, 272, 298–308 CrossRef CAS.
- Z. Zhang, W. Zhu, S. Zai, M. Jia, W. Zhang and Z. Wang, J. Porous Mater., 2013, 20, 531–538 CrossRef CAS.
- L. C. Liu, U. Diaz, R. Arenal, G. Agostini, P. Concepcion and A. Corma, Nat. Mater., 2017, 16, 132–138 CrossRef CAS PubMed.
- Y. Zhang, M. Kubů, M. Mazur and J. Čejka, Catal. Today, 2019, 324, 135–143 CrossRef CAS.
- Z. Zhao, Y. Li, M. Feyen, R. McGuire, U. Müller and W. Zhang, ChemCatChem, 2018, 10, 2254–2259 CrossRef CAS.
- Y. Zhang, K. Fulajtárová, M. Kubu, M. Mazur, M. Shamzhy, M. Hronec and J. Cejka, Mater. Today Nano, 2019, 8, 100056 CrossRef.
- Y. Zhang, M. Kubů, M. Mazur and J. Čejka, Microporous Mesoporous Mater., 2019, 279, 364–370 CrossRef CAS.
- A. G. Machoke, I. Y. Knoke, S. Lopez-Orozco, M. Schmiele, T. Selvam, V. R. R. Marthala, E. Spiecker, T. Unruh, M. Hartmann and W. Schwieger, Microporous Mesoporous Mater., 2014, 190, 324–333 CrossRef CAS.
- J. O. Barth, J. Kornatowski and J. A. Lercher, J. Mater. Chem., 2002, 12, 369–373 RSC.
- J. O. Barth, A. Jentys, J. Kornatowski and J. A. Lercher, Chem. Mater., 2004, 16, 724–730 CrossRef CAS.
- J. Kornatowski, J. O. Barth, K. Erdmann and M. Rozwadowski, Microporous Mesoporous Mater., 2006, 90, 251–258 CrossRef.
- F. Jin, S. Y. Chen, L. Y. Jang, J. F. Lee and S. F. Cheng, J. Catal., 2014, 319, 247–257 CrossRef CAS.
- F. Jin, C. C. Chang, C. W. Yang, J. F. Lee, L. Y. Jang and S. Cheng, J. Mater. Chem. A, 2015, 3, 8715–8724 RSC.
- F. Jin, S. J. Huang, S. F. Cheng, Y. X. Wu, C. C. Chang and Y. W. Huang, Catal. Sci. Technol., 2015, 5, 3007–3016 RSC.
- C. F. Cogswell, T. P. Nigl, A. Stavola, A. Wolek, Y. C. Wang, J. Zummo, Y. Lin, L. Dukaye, R. Chinn and S. Choi, Microporous Mesoporous Mater., 2019, 280, 151–156 CrossRef CAS.
- K. Na, M. Choi, W. Park, Y. Sakamoto, O. Terasaki and R. Ryoo, J. Am. Chem. Soc., 2010, 132, 4169–4177 CrossRef CAS PubMed.
- L. Wei, K. C. Song, W. Wu, S. Holdren, G. H. Zhu, E. Shulman, W. J. Shang, H. Y. Chen, M. R. Zachariah and D. X. Liu, J. Am. Chem. Soc., 2019, 141, 8712–8716 CrossRef CAS PubMed.
- R. Millini and G. Bellussi, Catal. Sci. Technol., 2016, 6, 2502–2527 RSC.
- D. Mochizuki, A. Shimojima, T. Imagawa and K. Kuroda, J. Am. Chem. Soc., 2005, 127, 7183–7191 CrossRef CAS PubMed.
- R. Ishii, T. Ikeda, T. Itoh, T. Ebina, T. Yokoyama, T. Hanaoka and F. Mizukami, J. Mater. Chem., 2006, 16, 4035–4043 RSC.
- A. Corma, U. Díaz, T. García, G. Sastre and A. Velty, J. Am. Chem. Soc., 2010, 132, 15011–15021 CrossRef CAS PubMed.
- M. Opanasenko, W. O. Parker, M. Shamzhy, E. Montanari, M. Bellettato, M. Mazur, R. Millini and J. Cejka, J. Am. Chem. Soc., 2014, 136, 2511–2519 CrossRef CAS PubMed.
- M. Opanasenko, M. Shamzhy, F. Yu, W. Zhou, R. E. Morris and J. Čejka, Chem. Sci., 2016, 7, 3589–3601 RSC.
- A. Corma, V. Fornes, J. Martinez-Triguero and S. B. Pergher, J. Catal., 1999, 186, 57–63 CrossRef CAS.
- A. Corma, U. Diaz, M. E. Domine and V. Fornes, J. Am. Chem. Soc., 2000, 122, 2804–2809 CrossRef CAS.
- A. Corma, V. Fornes and U. Diaz, Chem. Commun., 2001, 2642–2643 RSC.
- U. Díaz and A. Corma, Dalton Trans., 2014, 43, 10292–10316 RSC.
- P. Frontera, F. Testa, R. Aiello, S. Candamano and J. B. Nagy, Microporous Mesoporous Mater., 2007, 106, 107–114 CrossRef CAS.
- I. Ogino, M. M. Nigra, S. J. Hwang, J. M. Ha, T. Rea, S. I. Zones and A. Katz, J. Am. Chem. Soc., 2011, 133, 3288–3291 CrossRef CAS PubMed.
- I. Ogino, E. A. Eilertsen, S. J. Hwang, T. Rea, D. Xie, X. Y. Ouyang, S. I. Zones and A. Katz, Chem. Mater., 2013, 25, 1502–1509 CrossRef CAS.
- X. Y. Ouyang, S. J. Hwang, R. C. Runnebaum, D. Xie, Y. J. Wanglee, T. Rea, S. I. Zones and A. Katz, J. Am. Chem. Soc., 2014, 136, 1449–1461 CrossRef CAS PubMed.
- B. Yang, J.-g Jiang, H. Xu, P. Ji and P. Wu, Microporous Mesoporous Mater., 2015, 203, 54–62 CrossRef CAS.
- M. Položij, H. V. Thang, M. Rubeš, P. Eliášová, J. Čejka and P. Nachtigall, Dalton Trans., 2014, 43, 10443–10450 RSC.
- D. J. Klocke, S. L. Lawton, D. N. Lissy, W. J. Roth, C. M. Smith and D. E. Walsh, US Pat., 5453554, 1995 Search PubMed.
- T. F. Degnan, Jr., C. M. Smith and C. R. Venkat, Appl. Catal., A, 2001, 221, 283–294 CrossRef.
- L. L. Wang, Y. M. Liu, W. Xie, H. H. Wu, Y. W. Jiang, M. Y. He and P. Wu, Catal. Microporous Mater., 2007, 170, 635–640 Search PubMed.
- V. J. Margarit, M. E. Martinez-Armero, M. T. Navarro, C. Martinez and A. Corma, Angew. Chem., Int. Ed., 2015, 54, 13724–13728 CrossRef CAS PubMed.
- L. L. Silva, M. J. Stellato, M. V. Rodrigues, B. J. Hare, J. C. Kenvin, A. S. Bommarius, L. Martins and C. Sievers, J. Catal., 2022, 411, 187–192 CrossRef CAS.
- W. Dubiel, M. Kobielusz, K. Mróz, M. Mazur, L. Ang, L. Chmielarz, W. Macyk, W. J. Roth, J. Čejka and B. Gil, Appl. Mater. Today, 2024, 41, 102473 CrossRef.
- S. Lawton, M. E. Leonowicz, R. Partridge, P. Chu and M. K. Rubin, Microporous Mesoporous Mater., 1998, 23, 109–117 CrossRef CAS.
- K. Kalahurska, P. P. Ziemianski, W. J. Roth and B. Gil, Molecules, 2021, 26, 2076 CrossRef CAS PubMed.
- P. Miró, J. Lázaro, M. Pérez Pascual, J. Frontela, F. Rey and J. Pérez Pariente, Microporous Mesoporous Mater., 2023, 358, 112403 CrossRef.
- N. A. Grosso-Giordano, C. Schroeder, A. Olkrut, A. Solovyoy, C. Schöttle, W. Chassé, N. Marinkoyic, H. Koller, S. I. Zones and A. Katz, J. Am. Chem. Soc., 2018, 140, 4956–4960 CrossRef CAS PubMed.
- Q. Q. Hao, C. Y. Lei, Y. H. Song, Z. T. Liu and Z. W. Liu, Catal. Today, 2016, 274, 109–115 CrossRef CAS.
- G. Prieto, A. Martínez, P. Concepción and R. Moreno-Tost, J. Catal., 2009, 266, 129–144 CrossRef CAS.
- A. Swies, A. Kowalczyk, M. Rutkowska, U. Díaz, A. E. Palomares and L. Chmielarz, Catalysts, 2020, 10, 734 CrossRef CAS.
- S. Kweon, Y. W. Kim, J. Bae, E. J. Kim, M. B. Park and H. K. Min, J. CO2 Util., 2022, 58, 101921 CrossRef CAS.
- H. Pang, G. J. Yang, L. Li and J. H. Yu, NPG Asia Mater., 2023, 15, 24 CrossRef CAS.
- J. H. Liang, X. Hu, J. G. Wang, X. Q. Ren, X. R. Yang and J. L. Zhu, Catal. Commun., 2015, 69, 174–178 CrossRef CAS.
- A. Wojtaszek-Gurdak, M. Trejda, D. Kryszak and M. Ziolek, Microporous Mesoporous Mater., 2014, 197, 185–193 CrossRef CAS.
- A. Wojtaszek-Gurdak and M. Ziolek, RSC Adv., 2015, 5, 22326–22333 RSC.
- M. K. de Pietre, F. A. Bonk, C. Rettori, F. A. Garcia and H. O. Pastore, Microporous Mesoporous Mater., 2012, 156, 244–256 CrossRef CAS.
- M. K. de Pietre, F. A. Bonk, C. Rettori, F. A. Garcia and H. O. Pastore, Microporous Mesoporous Mater., 2011, 145, 108–117 CrossRef CAS.
- B. Solsona, J. M. L. Nieto and U. Díaz, Microporous Mesoporous Mater., 2006, 94, 339–347 CrossRef CAS.
- A. Fuerte, A. Corma, M. Iglesias, E. Morales and F. Sánchez, Catal. Lett., 2005, 101, 99–103 CrossRef CAS.
- D. Nuntasri, P. Wu and T. Tatsumi, Chem. Lett., 2003, 32, 326–327 CrossRef CAS.
- T. Maluangnont, Y. Yamauchi, T. Sasaki, W. J. Roth, J. Cejka and M. Kubu, Chem. Commun., 2014, 50, 7378–7381 RSC.
- T. Taniguchi, L. Nurdiwijayanto, R. Ma and T. Sasaki, Appl. Phys. Rev., 2022, 9, 021313 CAS.
- K. V. Agrawal, Chimia, 2018, 72, 313–321 CrossRef CAS PubMed.
- P. Wu, J. Ruan, L. Wang, L. Wu, Y. Wang, Y. Liu, W. Fan, M. He, O. Terasaki and T. Tatsumi, J. Am. Chem. Soc., 2008, 130, 8178–8187 CrossRef CAS PubMed.
- W. Fan, S. Wei, T. Yokoi, S. Inagaki, J. Li, J. Wang, J. N. Kondo and T. Tatsumi, J. Catal., 2009, 266, 268–278 CrossRef CAS.
- T. Yokoi, S. Mizuno, H. Imai and T. Tatsumi, Dalton Trans., 2014, 43, 10584–10592 RSC.
- S. Inagaki, H. Imai, S. Tsujiuchi, H. Yakushiji, T. Yokoi and T. Tatsumi, Microporous Mesoporous Mater., 2011, 142, 354–362 CrossRef CAS.
- H. Gies, M. Feyen, T. De Baerdemaeker, D. E. De Vos, B. Yilmaz, U. Mueller, X. J. Meng, F. S. Xiao, W. P. Zhang, T. Yokoi, T. Tatsumi and X. H. Sao, Microporous Mesoporous Mater., 2016, 222, 235–240 CrossRef CAS.
- J. Ruan, P. Wu, B. Slater, Z. Zhao, L. Wu and O. Terasaki, Chem. Mater., 2009, 21, 2904–2911 CrossRef CAS.
- S. Q. Li, L. Han, Z. L. Zhao, H. Xu, J. G. Jiang and P. Wu, Inorg. Chem. Front., 2021, 8, 5277–5285 RSC.
- C. Bian, Y. Yang, X. Luo, W. Zhang, J. Zhang, L. Zhu and J. Qiu, Molecules, 2021, 26, 5916 CrossRef CAS PubMed.
- W. J. Roth, W. Makowski, B. Marszalek, P. Michorczyk, W. Skuza and B. Gil, J. Mater. Chem. A, 2014, 2, 15722–15725 RSC.
- W. J. Roth, O. V. Shvets, M. Shamzhy, P. Chlubna, M. Kubu, P. Nachtigall and J. Cejka, J. Am. Chem. Soc., 2011, 133, 6130–6133 CrossRef CAS PubMed.
- R. E. Morris and J. Cejka, Nat. Chem., 2015, 7, 381–388 CrossRef CAS PubMed.
- A. Corma, M. J. Diaz-Cabañas, F. Rey, S. Nicolooulas and K. Boulahya, Chem. Commun., 2004, 1356–1357 RSC.
- J. L. Paillaud, B. Harbuzaru, J. Patarin and N. Bats, Science, 2004, 304, 990–992 CrossRef CAS PubMed.
- P. Chlubna, W. J. Roth, H. F. Greer, W. Z. Zhou, O. Shvets, A. Zukal, J. Cejka and R. E. Morris, Chem. Mater., 2013, 25, 542–547 CrossRef CAS.
- E. Verheyen, L. Joos, K. Van Havenbergh, E. Breynaert, N. Kasian, E. Gobechiya, K. Houthoofd, C. Martineau, M. Hinterstein, F. Taulelle, V. Van Speybroeck, M. Waroquier, S. Bals, G. Van Tendeloo, C. E. A. Kirschhock and J. A. Martens, Nat. Mater., 2012, 11, 1059–1064 CrossRef CAS PubMed.
- J. H. Kang, D. Xie, S. I. Zones and M. E. Davis, Chem. Mater., 2019, 31, 9777–9787 CrossRef CAS.
- J. H. Kang, D. Xie, S. I. Zones and M. E. Davis, Chem. Mater., 2020, 32, 2014–2024 CrossRef CAS.
- D. S. Firth, S. A. Morris, P. S. Wheatley, S. E. Russell, A. M. Z. Slawin, D. M. Dawson, A. Mayoral, M. Opanasenko, M. Položij, J. Čejka, P. Nachtigall and R. E. Morris, Chem. Mater., 2017, 29, 5605–5611 CrossRef CAS.
- V. Kasneryk, M. Shamzhy, M. Opanasenko, P. S. Wheatley, S. A. Morris, S. E. Russell, A. Mayoral, M. Trachta, J. Čejka and R. E. Morris, Angew. Chem., Int. Ed., 2017, 56, 4324–4327 CrossRef CAS PubMed.
- P. S. Wheatley, P. Chlubna-Eliasova, H. Greer, W. Z. Zhou, V. R. Seymour, D. M. Dawson, S. E. Ashbrook, A. B. Pinar, L. B. McCusker, M. Opanasenko, J. Cejka and R. E. Morris, Angew. Chem., Int. Ed., 2014, 53, 13210–13214 CrossRef CAS PubMed.
- M. Peng, Y. Zhao, H. Xu, J. Jiang and P. Wu, Chem. – Eur. J., 2024, 30, e202303657 CrossRef CAS PubMed.
- O. Vesely, M. Mazur, J. Prech and J. Cejka, Inorg. Chem. Front., 2022, 9, 5444–5453 RSC.
- Y. Zhou, S. A. Kadam, M. Shamzhy, J. Čejka and M. Opanasenko, ACS Catal., 2019, 9, 5136–5146 CrossRef CAS.
- N. Remperová, J. Prech, M. Kubu, K. Golabek, J. F. F. Minambres, M. F. Hsieh, A. Turrina and M. Mazur, Catal. Today, 2022, 390, 78–91 CrossRef.
- B. Gil, W. Makowski, B. Marszalek, W. J. Roth, M. Kubu, J. Čejka and Z. Olejniczak, Dalton Trans., 2014, 43, 10501–10511 RSC.
- J. Wang, X. Tu, W. Hua, Y. Yue and Z. Gao, Microporous Mesoporous Mater., 2011, 142, 82–90 CrossRef CAS.
- L. Emdadi, S. C. Oh, Y. Q. Wu, S. N. Oliaee, Y. X. Diao, G. H. Zhu and D. X. Liu, J. Catal., 2016, 335, 165–174 CrossRef CAS.
- H. Balcom, A. J. Hoffman, H. Locht and D. Hibbitts, ACS Catal., 2023, 13, 4470–4487 CrossRef CAS.
- H. V. Thang, J. Vaculik, J. Prech, M. Kubu, J. Cejka, P. Nachtigall, R. Bulanek and L. Grajciar, Microporous Mesoporous Mater., 2019, 282, 121–132 CrossRef CAS.
- G. T. M. Kadja, N. J. Azhari, S. Mardiana, N. T. U. Culsum and A. Maghfirah, Results Eng., 2023, 17, 100910 CrossRef CAS.
- H. Xu and P. Wu, Catal. Rev., 2021, 63, 234–301 CrossRef CAS.
- H. X. Zhang, Z. B. Jiang, Q. H. Xia and D. Zhou, Biochem. Eng. J., 2021, 172, 108033 CrossRef CAS.
- E. J. A. Schweitzer and P. F. van den Oosterkamp, Microporous Mesoporous Mater., 1998, 20, 397–401 Search PubMed.
- W. J. Roth and J. Čejka, Catal. Sci. Technol., 2011, 1, 43–53 RSC.
- N. Kosinov, E. A. Uslamin, L. Meng, A. Parastaev, Y. Liu and E. J. M. Hensen, Angew. Chem., Int. Ed., 2019, 58, 7068–7072 CrossRef CAS PubMed.
- O. Kikhtyanin, P. Chlubna, T. Jindrova and D. Kubicka, Dalton Trans., 2014, 43, 10628–10641 RSC.
- N. Narender, K. K. Mohan, S. J. Kulkarni and I. A. K. Reddy, Catal. Commun., 2006, 7, 583–588 CrossRef CAS.
- D. X. Liu, X. Y. Zhang, A. Bhan and M. Tsapatsis, Microporous Mesoporous Mater., 2014, 200, 287–290 CrossRef CAS.
- A. Korde, B. Min, Q. Almas, Y. D. Chiang, S. Nair and C. W. Jones, ChemCatChem, 2019, 11, 4548–4557 CrossRef CAS.
- Y. Q. Wu, L. Emdadi, D. C. Qin, J. Y. Zhang and D. X. Liu, Microporous Mesoporous Mater., 2017, 241, 43–51 CrossRef CAS.
- J. Jiang, Y. Xu, P. Cheng, Q. Sun, J. Yu, A. Corma and R. Xu, Chem. Mater., 2011, 23, 4709–4715 CrossRef CAS.
- M. Moliner, C. Martinez and A. Corma, Angew. Chem., Int. Ed., 2015, 54, 3560–3579 CrossRef CAS PubMed.
- G. Sastre and A. Corma, J. Phys. Chem. C, 2010, 114, 1667–1673 CrossRef CAS.
- S. E. Henkelis, M. Mazur, C. M. Rice, P. S. Wheatley, S. E. Ashbrook and R. E. Morris, J. Am. Chem. Soc., 2019, 141, 4453–4459 CrossRef CAS PubMed.
- L. A. Villaescusa and M. A. Camblor, Chem. Mater., 2016, 28, 7544–7550 CrossRef CAS.
- F. Gao, M. Jaber, K. Bozhilov, A. Vicente, C. Fernandez and V. Valtchev, J. Am. Chem. Soc., 2009, 131, 16580–16586 CrossRef CAS PubMed.
- L. Burel, N. Kasian and A. Tuel, Angew. Chem., Int. Ed., 2014, 53, 1360–1363 CrossRef CAS PubMed.
- Z. R. Gao, S. R. G. Balestra, J. Li and M. A. Camblor, Angew. Chem., Int. Ed., 2021, 60, 20249–20252 CrossRef CAS PubMed.
- H. Xu, J. G. Jiang, B. Yang, L. Zhang, M. He and P. Wu, Angew. Chem., Int. Ed., 2014, 53, 1355–1359 CrossRef CAS PubMed.
- M. V. Shamzhy, P. Eliašová, D. Vitvarová, M. V. Opanasenko, D. S. Firth and R. E. Morris, Chem. – Eur. J., 2016, 22, 17377–17386 CrossRef CAS PubMed.
- J. Zhang, T. Zakeri, Q. Yue, M. Kubů, R. Barakov, J. Přech, M. Opanasenko and M. Shamzhy, Catal. Today, 2024, 440, 114825 CrossRef CAS.
- G. Yang, X. Lan, J. Zhuang, D. Ma, L. Zhou, X. Liu, X. Han and X. Bao, Appl. Catal., A, 2008, 337, 58–65 CrossRef CAS.
- C. Perego, A. Carati, P. Ingallina, M. A. Mantegazza and G. Bellussi, Appl. Catal., A, 2001, 221, 63–72 CrossRef CAS.
- Y. Xue, Y. Xie, H. Wei, Y. Wen, X. Wang and B. Li, New J. Chem., 2014, 38, 4229 RSC.
- S.-T. Tsai, P.-Y. Chao, T.-C. Tsai, I. Wang, X. Liu and X.-W. Guo, Catal. Today, 2009, 148, 174–178 CrossRef CAS.
- Y. Jiao, A.-L. Adedigba, Q. He, P. Miedziak, G. Brett, N. F. Dummer, M. Perdjon, J. Liu and G. J. Hutchings, Catal. Sci. Technol., 2018, 8, 2211–2217 RSC.
- S. Du, X. Chen, Q. Sun, N. Wang, M. Jia, V. Valtchev and J. Yu, Chem. Commun., 2016, 52, 3580–3583 RSC.
- C. C. Pavel and W. Schmidt, Chem. Commun., 2006, 882 RSC.
- D. A. Jones, T. P. Lelyveld, S. D. Mavrofidis, S. W. Kingman and N. J. Miles, Resour., Conserv. Recycl., 2002, 34, 75–90 CrossRef.
- D. E. Clark, D. C. Folz and J. K. West, Mater. Sci. Eng., A, 2000, 287, 153–158 CrossRef.
- P. P. Falciglia, P. Roccaro, L. Bonanno, G. De Guidi, F. G. A. Vagliasindi and S. Romano, Renewable Sustainable Energy Rev., 2018, 95, 147–170 CrossRef CAS.
- S. Komarneni and R. Roy, Mater. Lett., 1986, 4, 107–110 CrossRef CAS.
- T. Ohgushi, S. Komarneni and A. S. Bhalla, J. Porous Mater., 2001, 8, 23–35 CrossRef CAS.
- Q. G. Ho, L. T. Bui, P. T. T. Nguyen, D. T. H. Ngo and L. Q. Nguyen, Chem. Eng. Trans., 2023, 106, 907–912 Search PubMed.
- Y. Kuroda, T. Okamoto, R. Kumashiro, Y. Yoshikawa and M. Nagao, Chem. Commun., 2002, 1758–1759 RSC.
- R. Zhang, D. Raja, Y. Zhang, Y. Yan, A. A. Garforth, Y. Jiao and X. Fan, Top. Catal., 2020, 63, 340–350 CrossRef CAS.
- R. Zhang, R. Zou, W. Li, Y. Chang and X. Fan, Microporous Mesoporous Mater., 2022, 333, 111736 CrossRef CAS.
- S. Abdulridha, Y. Jiao, S. Xu, R. Zhang, A. A. Garforth and X. Fan, Front. Chem., 2020, 8, 482 CrossRef CAS PubMed.
- Z. Qie, Z. Ji, H. Xiang, X. Zhang, A. Alhelali, L. Lan, H. Alhassawi, G. Zhao, X. Ou and X. Fan, Sep. Purif. Technol., 2023, 320, 124148 CrossRef CAS.
- M. González, Y. Cesteros, J. Llorca and P. Salagre, J. Catal., 2012, 290, 202–209 CrossRef.
- A. Rosales Pérez and K. Esquivel Escalante, ChemPlusChem, 2024, 89, e202300660 CrossRef PubMed.
- S. V. Ley and C. M. R. Low, Reactivity and Structure Concepts in Organic Chemistry, Springer Berlin Heidelberg, 1989, pp. 2–28 Search PubMed.
- L. Zhou, M. Wang, S. Yang, W. Guo, X. Pu, Y. He, J. Zhu, B. Wang, M. Zheng, S. Liu and Y. Zhang, Ultrason. Sonochem., 2022, 88, 106098 CrossRef CAS PubMed.
- V. A. Reyes Villegas, J. I. De León Ramirez, L. Pérez-Cabrera, S. Pérez-Sicairos, R. I. Yocupicio-Gaxiola, J. R. Chávez-Méndez, L. Huerta-Arcos and V. Petranovskii, Mater. Chem. Phys., 2025, 331, 130199 CrossRef CAS.
- D. Chlebda, P. Stachurska, R. Jędrzejczyk, Ł. Kuterasiński, A. Dziedzicka, S. Górecka, L. Chmielarz, J. Łojewska, M. Sitarz and P. Jodłowski, Nanomaterials, 2018, 8, 21 CrossRef PubMed.
- N. Sobuś, B. Michorczyk, M. Piotrowski, Ł. Kuterasiński, D. K. Chlebda, J. Łojewska, R. J. Jędrzejczyk, P. Jodłowski, P. Kuśtrowski and I. Czekaj, Catal. Lett., 2019, 149, 3349–3360 CrossRef.
- R. Zhang, P. Zhong, H. Arandiyan, Y. Guan, J. Liu, N. Wang, Y. Jiao and X. Fan, Front. Chem. Sci. Eng., 2020, 14, 275–287 CrossRef CAS.
- A. Kulak, Y. J. Lee, Y. S. Park, H. S. Kim, G. S. Lee and K. B. Yoon, Adv. Mater., 2002, 14, 526–529 CrossRef CAS.
- E. Kim, W. Cai, H. Baik and J. Choi, Angew. Chem., Int. Ed., 2013, 52, 5280–5284 CrossRef CAS PubMed.
- W. Schmidt, U. Wilczok, C. Weidenthaler, O. Medenbach, R. Goddard, G. Buth and A. Cepak, J. Phys. Chem. B, 2007, 111, 13538–13543 CrossRef CAS PubMed.
- D. Tzoulaki, W. Schmidt, U. Wilczok and J. Kärger, Microporous Mesoporous Mater., 2008, 110, 72–76 CrossRef CAS.
- C.-J. Liu, G. P. Vissokov and B. W. L. Jang, Catal. Today, 2002, 72, 173–184 CrossRef CAS.
- J. Batur, Z. Duan, M. Jiang, R. Li, Y. Xie, X.-F. Yu and J.-R. Li, Chem. Mater., 2023, 35, 3867–3879 CrossRef CAS.
- M. El-Roz, L. Lakiss, A. Vicente, K. N. Bozhilov, F. Thibault-Starzyk and V. Valtchev, Chem. Sci., 2014, 5, 68–80 RSC.
- J. Gao, X. Zhu, Z. Bian, T. Jin, J. Hu and H. Liu, Microporous Mesoporous Mater., 2015, 202, 16–21 CrossRef CAS.
- J. Huang, J. Hu, W. Du, H. Liu, F. Qian and M. Wang, J. Mater. Chem. A, 2017, 5, 18801–18807 RSC.
- W. Nichols, T. Kodaira, Y. Sasaki, Y. Shimizu, T. Sasaki and N. Koshizaki, J. Phys. Chem. B, 2006, 110, 83–89 CrossRef CAS PubMed.
- S. Hashimoto, T. Uwada, H. Masuhara and T. Asahi, J. Phys. Chem. C, 2008, 112, 15089–15093 CrossRef CAS.
- Y. Takeda, N. Toyama and K. Egashira, Eur. Phys. J.: Appl. Phys., 2023, 98, 63 CrossRef CAS.
- Y. Takeda, N. Toyama and K. Egashira, Surf. Sci., 2023, 728, 122199 CrossRef CAS.
- T. Muñoz and K. J. Balkus, J. Am. Chem. Soc., 1999, 121, 139–146 CrossRef.
- K. J. Balkus, T. Muñoz and M. E. Gimon-Kinsel, Chem. Mater., 1998, 10, 464–466 CrossRef.
- K. J. Balkus, L. J. Sottile, S. J. Riley and B. E. Gnade, Thin Solid Films, 1995, 260, 4–9 CrossRef CAS.
- D. Coutinho and K. Balkus, Microporous Mesoporous Mater., 2002, 52, 79–91 CrossRef CAS.
- T. Pisklak and K. Balkus, J. Porous Mater., 2004, 11, 191–209 CrossRef CAS.
- T. Pisklak and K. Balkus, Microporous Mesoporous Mater., 2005, 81, 125–134 CrossRef CAS.
- S. Mintova, M. Jaber and V. Valtchev, Chem. Soc. Rev., 2015, 44, 7207–7233 RSC.
- S. Mintova, J.-P. Gilson and V. Valtchev, Nanoscale, 2013, 5, 6693 RSC.
- L. Tosheva and V. P. Valtchev, Chem. Mater., 2005, 17, 2494–2513 CrossRef CAS.
- Y. Tao, H. Kanoh, L. Abrams and K. Kaneko, Chem. Rev., 2006, 106, 896–910 CrossRef CAS PubMed.
- Z. Liu, T. Wakihara, D. Nishioka, K. Oshima, T. Takewaki and T. Okubo, Chem. Mater., 2014, 26, 2327–2331 CrossRef CAS.
- Z. Liu, N. Nomura, D. Nishioka, Y. Hotta, T. Matsuo, K. Oshima, Y. Yanaba, T. Yoshikawa, K. Ohara, S. Kohara, T. Takewaki, T. Okubo and T. Wakihara, Chem. Commun., 2015, 51, 12567–12570 RSC.
- T. Kurniawan, O. Muraza, A. S. Hakeem and A. M. Al-Amer, Cryst. Growth Des., 2017, 17, 3313–3320 CrossRef CAS.
- G. T. M. Kadja, T. R. Suprianti, M. M. Ilmi, M. Khalil, R. R. Mukti and Subagjo, Microporous Mesoporous Mater., 2020, 308, 110550 CrossRef CAS.
- J. Huang, Y. Fan, G. Zhang and Y. Ma, RSC Adv., 2020, 10, 13583–13590 RSC.
- C. Dai, A. Zhang, M. Liu, X. Guo and C. Song, Adv. Funct. Mater., 2015, 25, 7479–7487 CrossRef.
- S. Van Donk, J. H. Bitter, A. Verberckmoes, M. Versluijs-Helder, A. Broersma and K. P. De Jong, Angew. Chem., Int. Ed., 2005, 44, 1360–1363 CrossRef CAS PubMed.
- W. Schwieger, A. G. Machoke, T. Weissenberger, A. Inayat, T. Selvam and M. Klumpp, Chem. Soc. Rev., 2016, 45, 3353–3376 RSC.
- M. Milina, S. Mitchell, D. Cooke, P. Crivelli and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 1591–1594 CrossRef CAS PubMed.
- F. L. Bleken, K. Barbera, F. Bonino, U. Olsbye, K. P. Lillerud, S. Bordiga, P. Beato, T. V. W. Janssens and S. Svelle, J. Catal., 2013, 307, 62–73 CrossRef CAS.
- N. S. Nesterenko, F. Thibault-Starzyk, V. Montouillout, V. V. Yuschenko, C. Fernandéz, J. P. Gilson, F. Fajula and I. I. Ivanova, Microporous Mesoporous Mater., 2004, 71, 157–166 CrossRef CAS.
- C. Fernandez, I. Stan, J.-P. Gilson, K. Thomas, A. Vicente, A. Bonilla and J. Pérez-Ramírez, Chem. – Eur. J., 2010, 16, 6224–6233 CrossRef CAS PubMed.
- Z. Musilová, N. Žilková, S.-E. Park and J. Čejka, Top. Catal., 2010, 53, 1457–1469 CrossRef.
- R. Khare, D. Millar and A. Bhan, J. Catal., 2015, 321, 23–31 CrossRef CAS.
- J. Zecevic, G. Vanbutsele, K. P. de Jong and J. A. Martens, Nature, 2015, 528, 245–248 CrossRef CAS PubMed.
- M. Y. Kim, K. Lee and M. Choi, J. Catal., 2014, 319, 232–238 CrossRef CAS.
- P. B. Weisz, in Advances in Catalysis, ed. D. D. Eley, P. W. Selwood, P. B. Weisz, A. A. Balandin, J. H. De Boer, P. J. Debye, P. H. Emmett, J. Horiuti, W. Jost, G. Natta, E. K. Rideal and H. S. Taylor, Academic Press, 1962, vol. 13, pp. 137–190 Search PubMed.
- M. Y. Kim, J.-K. Kim, M.-E. Lee, S. Lee and M. Choi, ACS Catal., 2017, 7, 6256–6267 Search PubMed.
- X. Zheng, G. Shen, C. Wang, Y. Li, D. Dunphy, T. Hasan, C. J. Brinker and B.-L. Su, Nat. Commun., 2017, 8, 14921 CrossRef CAS PubMed.
- D. Verboekend, L. A. Villaescusa, K. Thomas, I. Stan and J. Perez-Ramirez, Catal. Today, 2010, 152, 11–16 CrossRef CAS.
- R. Baur and R. Krishna, Catal. Today, 2005, 105, 173–179 Search PubMed.
- R. Aris, Ind. Eng. Chem. Fundam., 1965, 4, 227–229 CrossRef CAS.
- D. M. Ruthven, in Studies in Surface Science and Catalysis, ed. J. Čejka, H. van Bekkum, A. Corma and F. Schüth, Elsevier, 2007, vol. 168, pp. 737–785 Search PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- M. Thommes and K. A. Cychosz, Adsorption, 2014, 20, 233–250 CrossRef CAS.
- K. A. Cychosz and M. Thommes, Engineering, 2018, 4, 559–566 CrossRef CAS.
- J. Jagiello and J. Kenvin, J. Colloid Interface Sci., 2022, 625, 178–186 CrossRef CAS PubMed.
- S. Mitchell, A. B. Pinar, J. Kenvin, P. Crivelli, J. Kaerger and J. Perez-Ramirez, Nat. Commun., 2015, 6, 8633 Search PubMed.
- K. Morishige, M. Tateishi, F. Hirose and K. Aramaki, Langmuir, 2006, 22, 9220–9224 CrossRef CAS PubMed.
- Y. Zhu, T. Qiu, T. Liu, C. Li, Y. Huai and W. Ge, Ind. Eng. Chem. Res., 2024, 63, 17402–17412 Search PubMed.
- M. Thommes and C. Schlumberger, Annu. Rev. Chem. Biomol. Eng., 2021, 12, 137–162 CrossRef CAS PubMed.
- K. A. Cychosz, R. Guillet-Nicolas, J. García-Martínez and M. Thommes, Chem. Soc. Rev., 2017, 46, 389–414 Search PubMed.
- R. Cimino, K. A. Cychosz, M. Thommes and A. V. Neimark, Colloids Surf., A, 2013, 437, 76–89 CrossRef CAS.
- W. Kellouai, P. Judeinstein, M. Plazanet, S. Baudoin, M. Drobek, A. Julbe and B. Coasne, Langmuir, 2022, 38, 5428–5438 Search PubMed.
- E. Pérez-Botella, S. Valencia and F. Rey, Chem. Rev., 2022, 122, 17647–17695 CrossRef PubMed.
- S. H. van Vreeswijk and B. M. Weckhuysen, Nat. Sci. Rev., 2022, 9, nwac047 CrossRef CAS PubMed.
- E. Weiland, M.-A. Springuel-Huet, A. Nossov, F. Guenneau, A.-A. Quoineaud and A. Gédéon, New J. Chem., 2016, 40, 4447–4454 RSC.
- E. Thompson, K. Graf, E. Brendlé, H. Schreyer, T. De Baerdemaeker, A. Parvulescu and M. Kellermeier, ChemCatChem, 2024, 16, e202400434 CrossRef CAS.
- J. Kärger, M. Avramovska, D. Freude, J. Haase, S. Hwang and R. Valiullin, Adsorption, 2021, 27, 453–484 CrossRef.
- M. D. Mantle, M. Ainte, A. P. E. York, M. Bentley and L. F. Gladden, Catal. Today, 2024, 431, 114561 CrossRef CAS.
- J. Lee, C. Labadie, C. Springer and G. Harbison, J. Am. Chem. Soc., 1993, 115, 7761–7764 Search PubMed.
- M. Van Landeghem, A. Haber, J. B. D'Espinose De Lacaillerie and B. Blümich, Concepts Magn. Reson., Part A, 2010, 36A, 153–169 CrossRef CAS.
- M. N. d’Eurydice, E. T. Montrazi, C. A. Fortulan and T. J. Bonagamba, J. Chem. Phys., 2016, 144, 204201 CrossRef PubMed.
- M. S. Ullah, O. Mankinen, V. V. Zhivonitko and V.-V. Telkki, Phys. Chem. Chem. Phys., 2022, 24, 22109–22114 RSC.
- L. Song and L. Rees, Microporous Mesoporous Mater., 2000, 35–36, 301–314 CrossRef CAS.
- D. Shen and L. Rees, Zeolites, 1991, 11, 666–671 CrossRef CAS.
- L. Song, Z. Sun and L. Rees, Microporous Mesoporous Mater., 2002, 55, 31–49 CrossRef CAS.
- N. Vandenbegin, L. Rees, J. Caro and M. Bulow, Zeolites, 1989, 9, 287–292 CrossRef CAS.
- J. Valyon, G. Onyestyák and L. V. C. Rees, Langmuir, 2000, 16, 1331–1336 CrossRef CAS.
- G. Onyestyták, J. Valyon, A. Bóta and L. Rees, Helv. Chim. Acta, 2002, 85, 2463–2468 CrossRef.
- Y. Yasuda, H. Mizusawa and T. Kamimura, J. Phys. Chem. B, 2002, 106, 6706–6712 CrossRef CAS.
- A. Wagner, W. Anwand, M. Butterling, F. Fiedler, F. Fritz, M. Kempe and T. E. Cowan, J. Phys.: Conf. Ser., 2014, 505, 012034 CrossRef CAS.
- A. Zubiaga, R. Warringham, S. Mitchell, L. Gerchow, D. Cooke, P. Crivelli and J. Pérez-Ramírez, ChemPhysChem, 2017, 18, 470–479 CrossRef CAS PubMed.
- T. Ito and J. Fraissard, J. Chem. Phys., 1982, 76, 5225–5229 CrossRef CAS.
- H. C. Hoffmann, B. Assfour, F. Epperlein, N. Klein, S. Paasch, I. Senkovska, S. Kaskel, G. Seifert and E. Brunner, J. Am. Chem. Soc., 2011, 133, 8681–8690 CrossRef CAS PubMed.
- M. J. Annen, M. E. Davis and B. E. Hanson, Catal. Lett., 1990, 6, 331–339 CrossRef CAS.
- M. Li, K. Ideta, K. Hata, H. Kil, K. Kuroda, X. Zhai, K. Nakabayashi, S. Yoon and J. Miyawaki, J. Am. Chem. Soc., 2024, 146, 34401–34412 CrossRef CAS PubMed.
- M. Gackowski, A. Selent, I. Ainasoja, M. Mazur, M. Hunger, J. Datka and V. Telkki, Microporous Mesoporous Mater., 2023, 359, 112626 CrossRef CAS.
- D. Wisser and M. Hartmann, Adv. Mater. Interfaces, 2021, 8, 2001266 CrossRef.
- J. Bonardet, J. Fraissard, A. Gédéon and M. Springuel-Huet, Catal. Rev.: Sci. Eng., 1999, 41, 115–225 CrossRef CAS.
- V.-V. Telkki, J. Lounila and J. Jokisaari, in Hyperpolarized Xenon-129 Magnetic Resonance: Concepts, Production, Techniques and Applications, ed. T. Meersmann and E. Brunner, The Royal Society of Chemistry, 2015 Search PubMed.
- V.-V. Telkki, J. Lounila and J. Jokisaari, J. Phys. Chem. B, 2005, 109, 24343–24351 CrossRef CAS PubMed.
- J. H. Strange, M. Rahman and E. G. Smith, Phys. Rev. Lett., 1993, 71, 3589–3591 CrossRef CAS PubMed.
- Y. Liu, W. Zhang, Z. Liu, S. Xu, Y. Wang, Z. Xie, X. Han and X. Bao, J. Phys. Chem. C, 2008, 112, 15375–15381 CrossRef CAS.
- Y. Liu, W. Zhang, S. Xie, L. Xu, X. Han and X. Bao, J. Phys. Chem. B, 2008, 112, 1226–1231 CrossRef CAS PubMed.
- T. Springer, in Springer Tracts in Modern Physics, ed. T. Springer, Springer Berlin Heidelberg, Berlin, Heidelberg, 1972, vol. 64, pp. 1–100 Search PubMed.
- H. Jobic, J. Kärger and M. Bée, Phys. Rev. Lett., 1999, 82, 4260–4263 CrossRef CAS.
- J. Kärger, R. Valiullin, S. Brandani, J. Caro, C. Chmelik, B. Chmelka, M. Coppens, S. Farooq, D. Freude, H. Jobic, M. Kruteva, E. Mangano, R. Pini, W. Price, A. Rajendran, P. Ravikovitch, G. Sastre, R. Snurr, A. Stepanov, S. Vasenkov, Y. Wang and B. Weckhuysen, Pure Appl. Chem., 2025, 97, 1–89 CrossRef.
- S. Rives, H. Jobic, A. Beale and G. Maurin, J. Phys. Chem. C, 2013, 117, 13530–13539 CrossRef CAS.
- H. Jobic and D. Theodorou, Microporous Mesoporous Mater., 2007, 102, 21–50 CrossRef CAS.
- D. Mores, E. Stavitski, M. H. F. Kox, J. Kornatowski, U. Olsbye and B. M. Weckhuysen, Chem. – Eur. J., 2008, 14, 11320–11327 CrossRef CAS PubMed.
- B. Huang, M. Bates and X. Zhuang, Annu. Rev. Biochem., 2009, 78, 993–1016 CrossRef CAS PubMed.
- S. Basak, A. Chizhik, J. I. Gallea, I. Gligonov, I. Gregor, O. Nevskyi, N. Radmacher, R. Tsukanov and J. Enderlein, Nat. Photonics, 2025, 19, 229–237 CrossRef CAS.
- M. Filez, M. Vesely, I. Garcia-Torregrosa, M. Gambino, Ö. Attila, F. Meirer, E. A. Katrukha, M. B. J. Roeffaers, J. Garrevoet, L. C. Kapitein and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2021, 60, 13803–13806 CrossRef CAS PubMed.
- M. Weyland, Top. Catal., 2002, 21, 175–183 CrossRef CAS.
- G. Wang, H. Xiong, F. Wei and X. Chen, Chem. Res. Chin. Univ., 2025, 41, 196–210 CrossRef CAS.
- A. H. Janssen, A. J. Koster and K. P. De Jong, Angew. Chem., Int. Ed., 2001, 40, 1102–1104 CrossRef CAS PubMed.
- T. Li, H. Wu, J. Ihli, Z. Ma, F. Krumeich, P. H. H. Bomans, N. A. J. M. Sommerdijk, H. Friedrich, J. P. Patterson and J. A. Van Bokhoven, J. Mater. Chem. A, 2019, 7, 1442–1446 RSC.
- B. Apeleo Zubiri, J. Wirth, D. Drobek, S. Englisch, T. Przybilla, T. Weissenberger, W. Schwieger and E. Spiecker, Adv. Mater. Interfaces, 2021, 8, 2001154 CrossRef CAS.
- G. Rudolph-Schöpping, E. Larsson, T. N. Pingel, M. Guizar-Sicairos, P. Villanueva-Perez, S. Hall and F. Lipnizki, J. Membr. Sci., 2024, 690, 122245 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cs00319a |
| This journal is © The Royal Society of Chemistry 2025 |