
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress towards catalytic asymmetric construction of inherently chiral scaffolds
Kai
Zhu
a,
David R.
Spring
 *b,
Bing-Feng
Shi
*c and
Fengzhi
Zhang
*d
*b,
Bing-Feng
Shi
*c and
Fengzhi
Zhang
*d
aSchool of Medicine, Huanghuai University, Zhumadian 463000, Henan, P. R. China
bDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: spring@ch.cam.ac.uk
cDepartment of Chemistry, Zhejiang University, Hangzhou 310027, Zhejiang, P. R. China. E-mail: bfshi@zju.edu.cn
dSchool of Pharmacy, Hangzhou Medical College, Hangzhou 310014, Zhejiang, P. R. China. E-mail: zhangfengzhi@hmc.edu.cn
First published on 20th October 2025
Abstract
Inherently chiral scaffolds are a unique type of structurally fascinating building blocks, which have found increasing applications in the field of material chemistry, medicinal chemistry, asymmetric synthesis, molecular recognition and assembly. Owing to the steady demand for these chiral entities, numerous efforts have been made on their enantioselective synthesis. Among the plethora of accomplishments reported, the catalytic asymmetric strategy is emerging as one of the most efficient and sustainable approaches. This protocol provides a powerful platform to achieve enantiomerically pure inherently chiral architectures with structural diversity. In this review article, we aim to generalize the booming and remarkable advancements in asymmetric synthesis of inherently chiral calix[n]arenes, pillar[n]arenes, saddle-shaped scaffolds, mechanically interlocked molecules, and prism-like cages under catalyst control, which would offer valuable insights for future research on the rational design of conceptually novel and streamlined asymmetric synthetic systems, thereby expanding the scope/chemical space and improving the added value of inherently chiral molecules.
 Kai Zhu | Kai Zhu received his PhD (2020) from Zhejiang University of Technology under Professor Fengzhi Zhang. After postdoctoral work with Professor Fengzhi Zhang (2020–2022) at the same university, he took up a faculty position at Huanghuai University. His research interests include the discovery of novel catalytic enantioselective reactions and their synthetic applications. |
 David R. Spring | David Spring is currently Professor of Chemistry and Chemical Biology at the University of Cambridge within the Chemistry Department. He received his DPhil (1998) from Oxford University under Sir Jack Baldwin. He then worked as a Wellcome Trust Postdoctoral Fellow at Harvard University with Stuart Schreiber (1999–2001), after which he joined the faculty at the University of Cambridge. His research programme is focused on the use of chemistry to explore biology. |
 Bing-Feng Shi | Bing-Feng Shi received his BS degree from Nankai University in 2001 and a PhD degree from the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences under the guidance of Professor Biao Yu in 2006. Following time as a postdoctoral fellow at the University of California, San Diego (2006–2007), he moved to The Scripps Research Institute working with Professor Jin-Quan Yu as a research associate. In 2010, he joined the Department of Chemistry at Zhejiang University as a professor. His research focus is on transition metal-catalyzed C–H functionalization and its application in the synthesis of biologically important small molecules. |
 Fengzhi Zhang | Fengzhi Zhang received his MSc (2003) from Lanzhou University with Prof. Yulin Li and PhD (2008) from the University of Nottingham (UK) with Prof. Nigel Simpkins. He then worked with Profs. John Moses (2008–2009, Nottingham), Mike Greaney (2009–2011, Edinburgh), Matthew Gaunt (2011–2013, Cambridge) and David Spring (2013–2014, Cambridge) before he took up a faculty position at Zhejiang University of Technology (2014, China). Currently, he is a full professor at Hangzhou Medical College, and his research interests include the asymmetric synthesis, electrochemical synthesis, green chemistry and medicinal chemistry. |
Key learning points(1) The definition of inherent chirality and its distinction from classical stereogenic elements.(2) The key factors influencing the configurational stability of inherently chiral scaffolds. (3) Recent advancements in catalytic asymmetric synthesis of inherently chiral scaffolds. (4) The reaction mechanism and how enantiocontrol is achieved in catalytic asymmetric processes. (5) The utility and promising applications of inherently chiral molecules. |
1. Introduction
Inherent chirality is a manifestation of molecular chirality that arises when the stereogenic unit is the entire molecular scaffold itself, rather than localized stereogenic elements (centers, axes, or planes), and it represents a compelling frontier in modern stereochemistry. Building upon the foundational principles of chirality established by Kurt Mislow and others,1–4 Böhmer and colleagues introduced the term “inherent chirality” in 1994 to characterize calixarenes that exhibit no symmetry elements except for a C1 asymmetry axis, thereby expanding the theoretical framework of stereochemistry.5 To provide a rigorous theoretical basis, Schiaffino and Mandolini established the first scientific definition in 2004, characterizing inherent chirality as arising from “the introduction of curvature into an ideal planar structure that is devoid of symmetry axes in its two-dimensional representation”.6 This definition was later refined by Szumna in 2010, replacing “devoid of symmetry axes” with “devoid of perpendicular symmetry planes”.7 According to this criterion, calix[n]arenes,8–11 pillar[n]arenes,12–15 rigid saddle-shaped scaffolds,16,17 mechanically interlocked molecules,18–20 and prism-like cages21–23 are capable of displaying this form of molecular chirality (Fig. 1).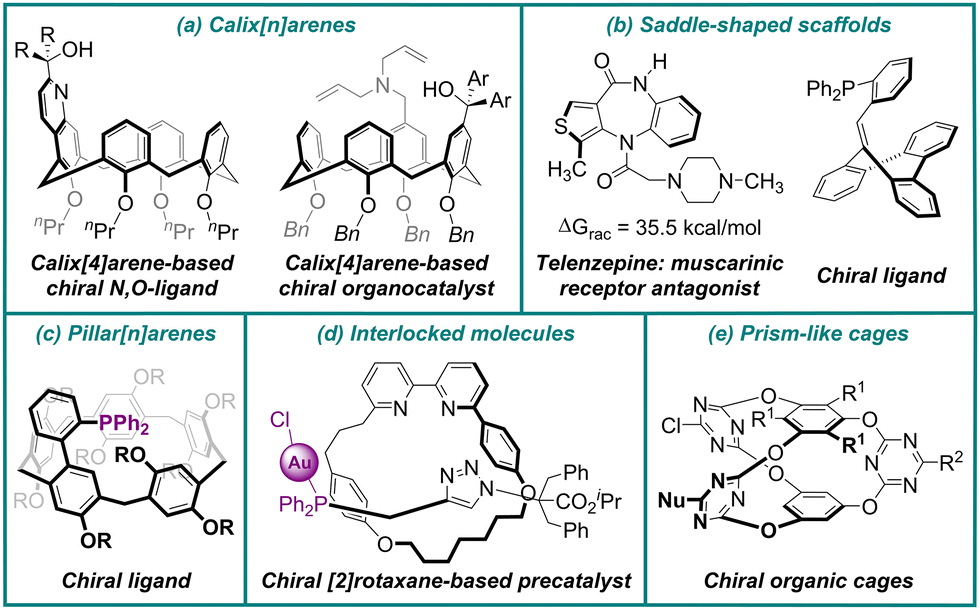 | ||
| Fig. 1 Selected examples of inherently chiral scaffolds. | ||
At present, these stereogenic units are widely employed in enantioselective molecular recognition and assembly and serve as essential building blocks in functional materials, exerting a substantial impact on the development of asymmetric synthesis and material chemistry. For instance, chiral calixarenes, in particular, the tunable three-dimensional calix[4]arenes, have been extensively investigated as potential artificial hosts for chiral recognition24 and as efficient ligands and organocatalysts in catalytic systems like Suzuki–Miyaura cross-coupling, Tsuji–Trost allylic substitution, Henry reaction and Michael addition, etc. (Fig. 1a).25,26 Furthermore, the seven-membered saddle-shaped heterocycle telenzepine was found to exhibit significant antimuscarinic activity, where the (+)-enantiomer was 500 times more potent than its (−)-enantiomer, which demonstrated the importance of their enantioselective synthesis (Fig. 1b).27,28
Owing to their intriguing structural properties and diverse applications, significant efforts have been dedicated to the enantioselective construction of these chiral architectures. Nevertheless, access to these enantiopure entities still predominantly relies on inefficient and tedious optical resolutions of racemic samples by means of chiral chromatographic techniques or with stoichiometric amounts of chiral auxiliaries. It is worth mentioning that, over the past few years, among the plethora of accomplishments reported, the catalytic asymmetric strategy has been emerging as one of the most efficient and sustainable approaches. This protocol provides a powerful platform to generate the inherently chiral scaffolds with structural diversity and enantiomeric purity, thus enabling the expansion of their chemical space and potential applications.11,15,17,20,29
In 2023, Yang and co-workers provided a timely summary of advancements in the catalytic asymmetric synthesis of inherently chiral scaffolds.29 Since then, numerous elegant works related to the catalytic asymmetric construction of such scaffolds have emerged. Additionally, several recent reviews have organized prior research and specialized perspectives according to specific compound classes.11,15,17,20
However, a comprehensive review that systematically summarizes and discusses all advancements in this rapidly evolving field remains lacking. Therefore, a comprehensive overview of this burgeoning research area is highly desirable. In this review article, we aim to systematically generalize the booming and remarkable advancements in the catalytic enantioselective synthesis of inherently chiral scaffolds, including calix[n]arenes, pillar[n]arenes, saddle-shaped scaffolds, mechanically interlocked molecules, and prism-like cages, in which the molecular chirality results when the stereogenic unit is the entire molecular scaffold itself, rather than localized stereogenic elements. We anticipate that this tutorial review will offer valuable insights for future research on the rational design of conceptually novel and streamlined asymmetric synthetic systems, thereby expanding the chemical space of inherently chiral scaffolds and improving the added value of these molecules.
2. Enantioselective construction of calixarenes
Inherently chiral calixarenes, a highly attractive type of privileged macrocyclic structures, exhibit broad applications in chiral recognition and sensing, circularly polarized luminescence, asymmetric catalysis, and separation science.8–11,24–26 In general, inherent chirality can be introduced into the calixarene platform in a sophisticated manner by incorporation of achiral substituents into the bridging units, or upper- and/or lower-rim. In most cases, approaches for stereocontrolled preparation of these scaffolds are primarily limited to chiral auxiliary-based resolutions and couplings, often suffering from poor atom economy and low efficiency. Catalytic enantioselective synthesis, which can be considered to be a powerful and versatile synthetic method for accessing enantioenriched calixarenes, remains a longstanding challenge and is still in its infancy.112.1. Biocatalytic approach
To meet the demands of both economic value and high efficiency, earliest attempts toward the catalytic pathways were focused on the enzymatic reactions. In 1998, McKervey and colleagues discovered that the acylation of achiral triethyl alcohol monophenols 1 with acylating agent vinyl acetate 2 can be promoted by the cross-linked enzyme crystals (CLECs) of Aspergillus niger lipase to derive optically pure monoacetylated calix[4]arenes 3 with high to excellent enantioselectivities, albeit in poor yields (Scheme 1).30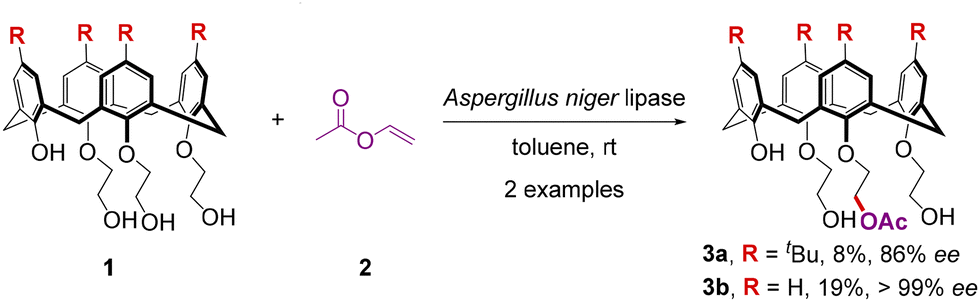 | ||
| Scheme 1 Biocatalytic synthesis of enantiopure calix[4]arenes via enantioselective desymmetrization. | ||
These results demonstrate the importance of enzymatic catalysis as a means to effect asymmetric synthesis of large, inherently chiral calixarenes. After McKervey and co-workers disclosed the biocatalytic access to enantiopure calix[4]arenes via an enantioselective desymmetrization reaction, it took more than a decade to see another catalytic method leading to the inherently chiral azacalix[4]arene.
2.2. Transition-metal-catalyzed methods
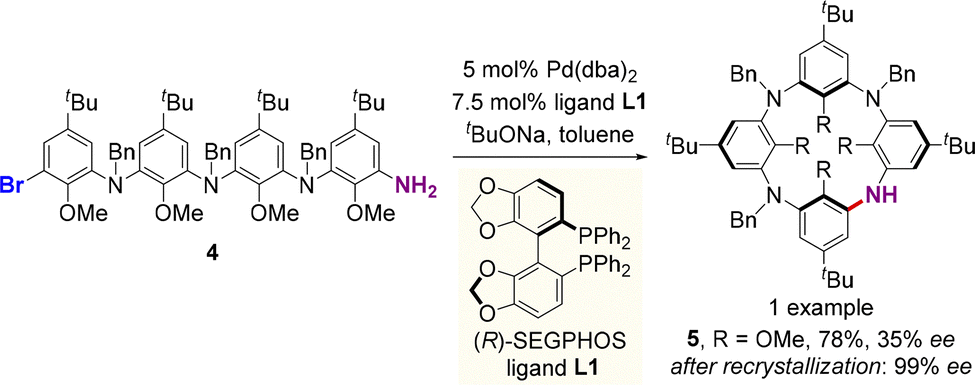 | ||
| Scheme 2 Palladium-catalyzed enantioselective synthesis of azacalix[4]arene via intramolecular Buchwald–Hartwig cyclization. | ||
In 2020, Wang, Tong and co-workers used a similar strategy for the catalytic enantioselective intramolecular C–N coupling of ABCD-type linear tetramer 6 to produce inherently chiral heteracalix[4]aromatics 7 (Scheme 3).34
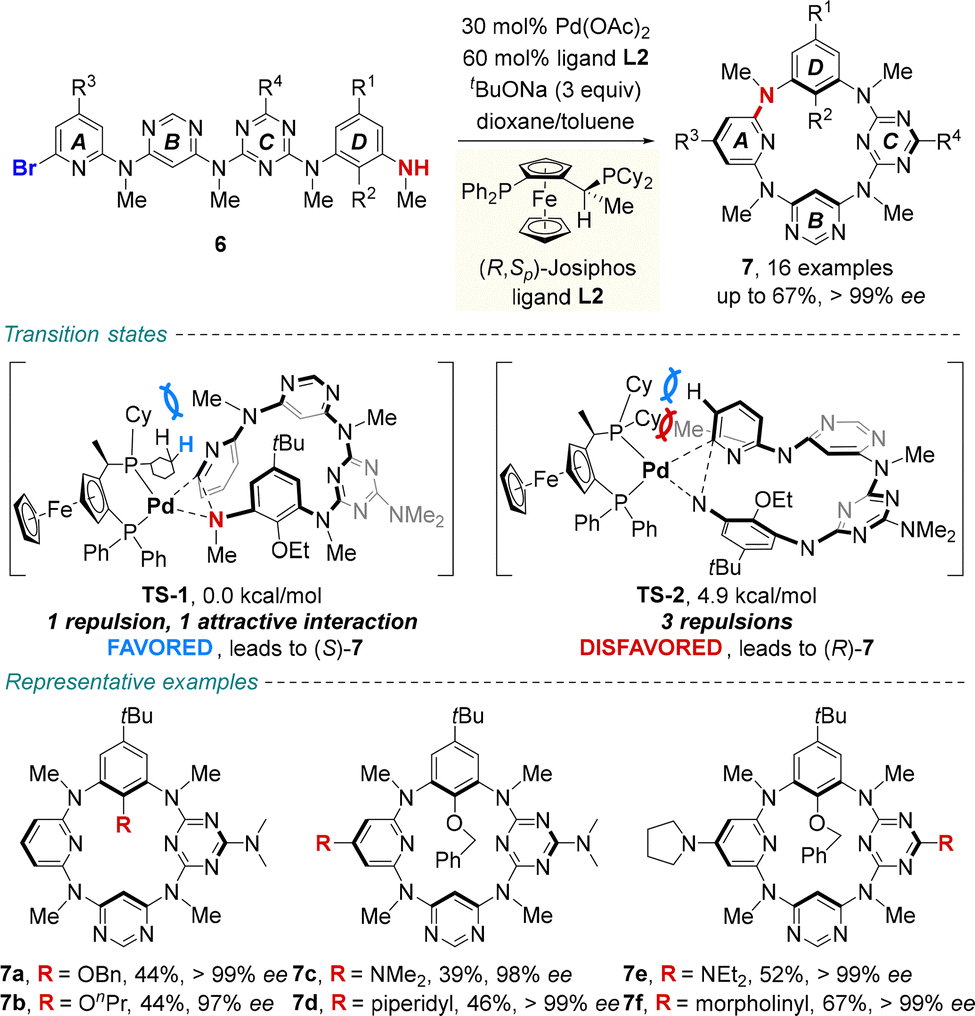 | ||
| Scheme 3 Palladium-catalyzed enantioselective synthesis of ABCD-type inherently chiral tetraazacalix[4]aromatics. | ||
The asymmetric macrocyclization promoted by a palladium/(R,Sp)-JOSIPHOS complex afforded ABCD-type inherently chiral tetraazacalix[4]aromatics 7 in moderate to good yields (up to 67%) and generally excellent enantiomeric excesses (up to >99% ee). Density functional theory (DFT) calculations revealed that the activation free energy of transition state TS-1 is lower than that of TS-2. In the (S)-pathway, the favorable C–H⋯π interaction, along with weaker steric repulsion between cyclohexyl substituents on the ligand and the N-methyl group of substrate 6, is thought to efficiently stabilize the unique transition state geometry, thereby governing the enantiocontrol. Remarkably, the acquired enantiomerically enriched macrocycles showed excellent, pH-triggered switchable electronic circular dichroism (ECD) and circularly polarized luminescence (CPL) properties. These inherently chiral macrocycles are poised to serve as guest-responsive chiroptical systems by leveraging the versatile molecular recognition capabilities of heteracalix[4]aromatics.
Palladium-catalyzed asymmetric Suzuki–Miyaura couplings have traditionally been employed to generate axial chirality through the stereoselective construction of aryl–aryl bonds.35 Recently, Tong and colleagues applied this approach for the preparation of inherently chiral macrocycles (Scheme 4).36
 | ||
| Scheme 4 Pd-catalyzed enantioselective synthesis of inherently chiral nor-heteracalixarenes via intramolecular Suzuki–Miyaura coupling. | ||
With the linear achiral precursor as the substrate, represented as 8 in Scheme 4, in the presence of the chiral phosphoramidite ligand (R)-SIPHOS (L3), the intramolecular Pd-catalyzed Suzuki–Miyaura cross-coupling macrocyclization reaction afforded the corresponding 15-membered inherently chiral nor-heteracalixarenes 9 in good to high yields and enantioselectivities. Racemization studies revealed that the restricted rotation of the aromatic ring A and the steric effect of the C2 substituents determine the configurational stability of the inherent chirality. Moreover, the intriguing chiroptical properties of these rigid molecules, including high fluorescence quantum yields and CPL brightness values (BCPL, up to 0.65), made them a promising platform for fabricating CPL emitters.
 | ||
| Scheme 5 Palladium-catalyzed enantioselective synthesis of inherently chiral calix[4]arenes via intramolecular C–H arylations. | ||
Concurrently, from the same starting materials 10, Tong and colleagues reported an alternative palladium-catalyzed sequential intramolecular transannular dehydrogenative coupling reaction (Scheme 6).38 With this protocol, a wide range of 9H-fluorene-embedded inherently chiral calixarenes 12 were obtained in good yields with high enantioselectivities using PdBr2 as the catalyst and chiral phosphoramidite L5 as the ligand. The absolute configuration of 12a was confirmed by X-ray crystallography analysis. Mechanistically, the reaction might initiate from the oxidative addition of Pd/L5 with 10, followed by asymmetric C–H activation/1,5-palladium migration to form int-1, which would undergo a second C–H activation/reductive elimination to deliver the final meta–meta bridged calix[4]arenes 12. These highly rigid structures exhibit unique chiroptical properties.
 | ||
| Scheme 6 Palladium-catalyzed enantioselective synthesis of inherently chiral calixarenes via transannular dehydrogenative coupling. | ||
Very recently, Zhou, Cheng and co-workers inventively designed an efficient palladium/chiral norbornene cooperative catalyzed enantioselective Catellani-type cascade reaction for the synthesis of enantiomerically enriched calix[4]arenes 15 featuring both axial and inherent chirality with excellent diastereo- and enantioselectivities (Scheme 7).39
 | ||
| Scheme 7 Palladium/chiral norbornene cooperative catalyzed enantioselective method for the synthesis of calix[4]arenes featuring both axial and inherent chirality. | ||
The suggested reaction pathway involves the formation of axially chiral PdII complex int-2, which undergoes an asymmetric intramolecular C–H activation process and realizes the axial-to-inherent chirality conversion. This class of phosphine oxide-substituted calix[4]arenes 15 with axial and inherent chirality could be facilely transformed into a novel chiral phosphine ligand, which exhibits high catalytic activity in the silver-catalyzed enantioselective [3+2] cyclization reaction.
Regarding the key importance of cavitands as privileged scaffolds in supramolecular chemistry, their enantioselective preparation has attracted considerable attention.40,41 More recently, Zhu, Gandon and co-workers achieved the synthesis of inherently chiral resorcinarene cavitands 17 in modest to good yields and high enantiomeric excess by engineering an ionic chiral palladium-catalyzed cross-coupling of the corresponding prochiral cavitands 16 (Scheme 8).42 Using Pd2(dba)3 and amino acid-derived ligand L6 as the catalyst system, the authors developed conditions for stereoselective Suzuki–Miyaura coupling, Buchwald–Hartwig amination, and Sonogashira coupling reactions with a variety of coupling partners. Mechanistic studies revealed that synergistic electrostatic steering, along with electrostatic catalysis by the interactions between the ionic catalyst and the substrate, could be responsible for the selectivity control observed. Additionally, based on these chiral catalytic systems, a hierarchical heterofunctionalization protocol was designed for the enantioselective construction of functionalized inherently chiral ABCD-type cavitands, which bear programmable substitution patterns, through sequential cross-coupling reactions. Using the products thus synthesized, a chiral hemicarcerand was designed that exhibits enantioselective binding toward a BINOL-derived guest through specific host–guest interactions. This system demonstrates efficient discrimination of nanometer-sized enantiomers, underscoring its potential as an artificial chiral receptor.
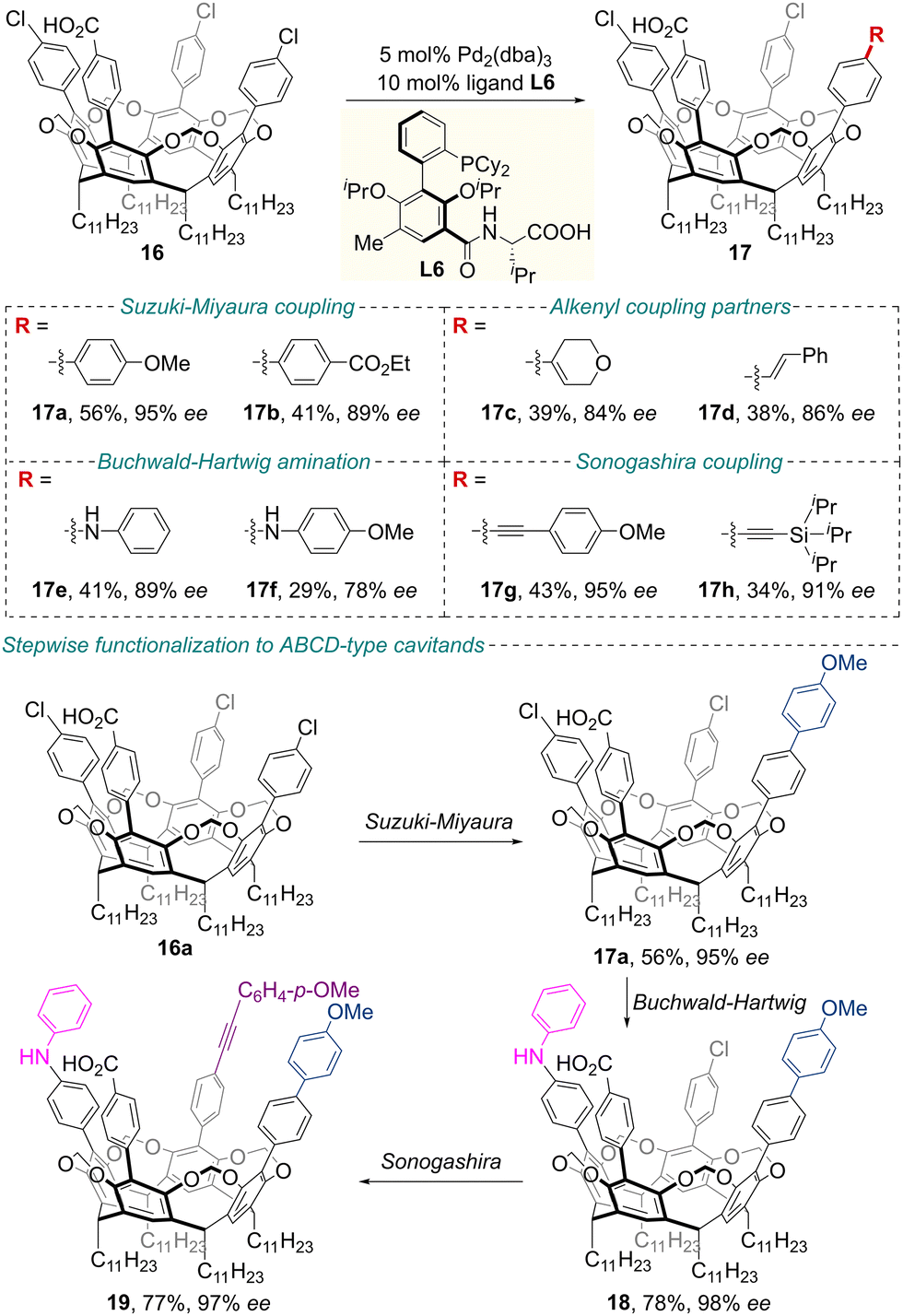 | ||
| Scheme 8 Ionic chiral palladium-catalyzed desymmetrizing cross-coupling to inherently chiral resorcinarene cavitands. | ||
In 2023, Cera, Secchi and co-workers reported the synthesis of inherently chiral calix[4]arenes 21via a step- and atom-economical, gold(I)-catalyzed intramolecular hydroarylation of alkynes 20 (Scheme 9).43 Nevertheless, the corresponding catalytic enantioselective version of this method was not fruitful, and only a single example was presented with 98% yield and 28% ee.
 | ||
| Scheme 9 Gold-catalyzed asymmetric synthesis of inherently chiral calix[4]arenes via intramolecular hydroarylation of alkynes. | ||
Due to its less toxic and cost-efficient features, the cobalt-catalyzed asymmetric C–H activation strategy has garnered considerable attention for the synthesis of a panel of highly enantioenriched inherently chiral calix[4]arenes.44
Very recently, Niu, Yang and colleagues unraveled that the utilization of a chiral salicyloxazoline (Salox) ligand L7 for the cobalt-catalyzed enantioselective intermolecular C–H activation/annulation of benzamide tethered calix[4]arenes 22 with exogenous alkynes 23 provided the inherently chiral products 24, with multiple C–N axial chirality, in good to high yields (up to 94%) with excellent diastereoselectivity (all >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) and high enantioselectivity (up to 96% ee) (Scheme 10).45 The utility of this protocol was highlighted by the gram-scale reaction, synthetic transformations and catalytic application. Additionally, compound 24a effectively functions as a host molecule for the enantioselective recognition of C2 symmetric chiral binaphthols, demonstrating its potential for applications in chiral discrimination. The proposed catalytic cycle initiates with the coordination of chiral ligand L7 to Co(II) and undergoes oxidation with O2 to generate the active Co(III) species. Subsequent coordination with substrate 22 is followed by chelation of the N-oxide group to the cobalt center. This key interaction helps organize a rigid chiral environment through synergy with ligand L7, thereby controlling the enantioselectivity during C–H activation and alkyne 23 migratory insertion. Reductive elimination then affords the product as well as the Co(I) species, which is reoxidized to the active Co(III) catalyst by O2 to complete the catalytic cycle.
:1 dr) and high enantioselectivity (up to 96% ee) (Scheme 10).45 The utility of this protocol was highlighted by the gram-scale reaction, synthetic transformations and catalytic application. Additionally, compound 24a effectively functions as a host molecule for the enantioselective recognition of C2 symmetric chiral binaphthols, demonstrating its potential for applications in chiral discrimination. The proposed catalytic cycle initiates with the coordination of chiral ligand L7 to Co(II) and undergoes oxidation with O2 to generate the active Co(III) species. Subsequent coordination with substrate 22 is followed by chelation of the N-oxide group to the cobalt center. This key interaction helps organize a rigid chiral environment through synergy with ligand L7, thereby controlling the enantioselectivity during C–H activation and alkyne 23 migratory insertion. Reductive elimination then affords the product as well as the Co(I) species, which is reoxidized to the active Co(III) catalyst by O2 to complete the catalytic cycle.
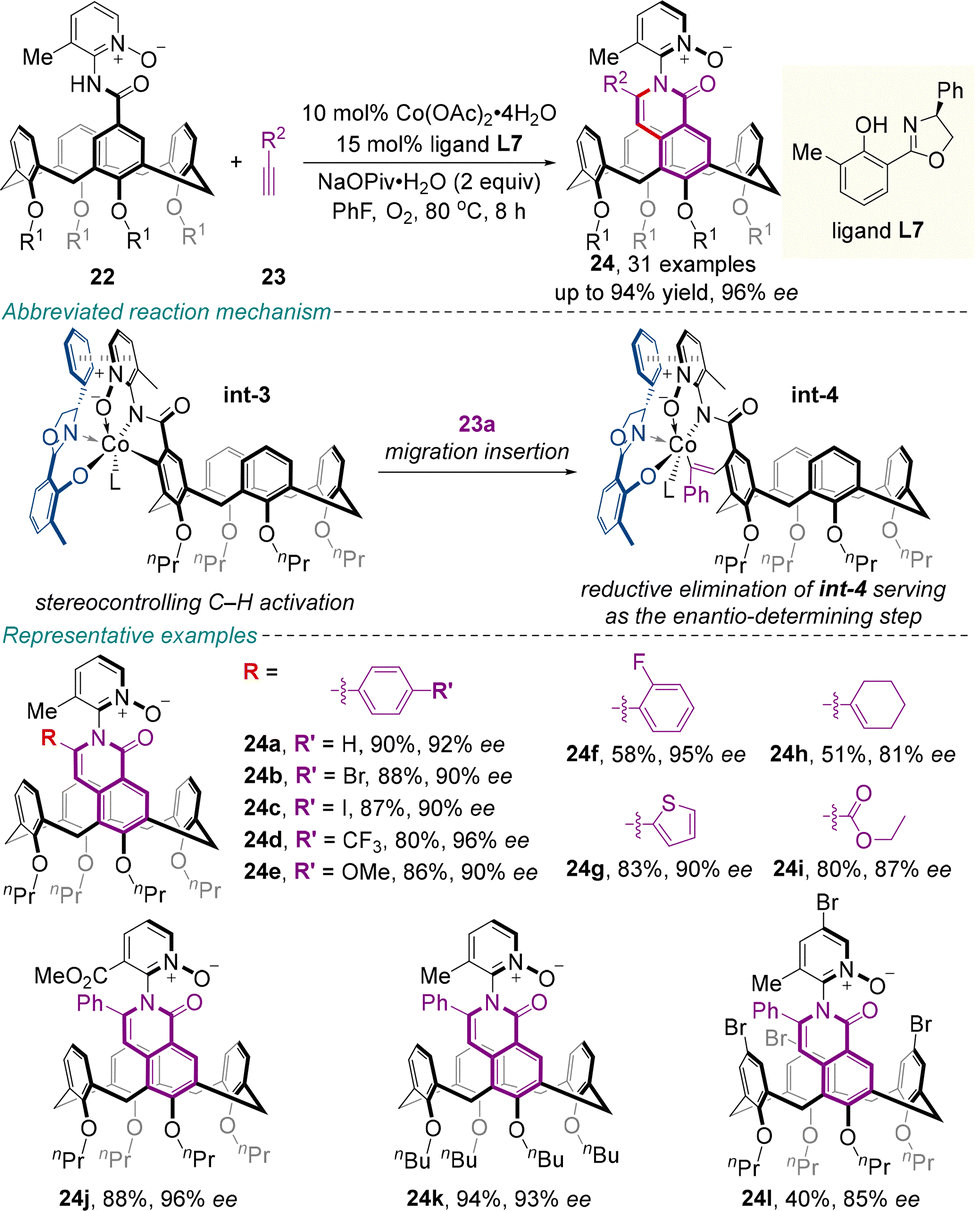 | ||
| Scheme 10 Cobalt-catalyzed asymmetric C–H activation/annulation strategy for the synthesis of calix[4]arenes featuring both inherent and axial chirality elements. | ||
Almost simultaneously, a similar transformation was independently accomplished by Shi and co-workers using different chiral Salox ligands (L8, L9) and directing groups (8-aminoquinoline derivatives) (Scheme 11).46 In terms of the substrate scope, acetylene gas, ethylene gas, substituted aryl/alkyl alkynes, and allene were all well tolerated, resulting in the formation of a remarkably broad range of inherently or both inherently and axially chiral calix[4]arenes 27 in high to excellent yields (up to 99%) with excellent enantioselectivities (up to >99% ee). Confirmed by the single-crystal X-ray diffraction of cobaltacyle and previous investigations,44 the π–π interaction between quinolyl in the substrate and phenyl in the Salox ligand was characterized as a dominant factor for outstanding stereocontrol. Moreover, the measured photophysical properties of these products indicate their potential applications in organic optoelectronic materials science.
 | ||
| Scheme 11 Shi's cobalt-catalyzed enantioselective intermolecular C–H annulation strategy for the synthesis of calix[4]arenes with inherent or both inherent and axial chirality. | ||
In recent years, the renaissance of organic electrosynthesis has given new impetus to catalytic enantioselective synthesis.47–49 In the same report, Shi further implemented an electrochemical asymmetric synthetic version of this catalytic system and delivered the desired products 27 with high yields (74–91%) and enantioselectivities (87–93% ee), which avoided the drawbacks posed by the employment of stoichiometric sacrificial chemical oxidant Mn(OAc)3·2H2O (Scheme 12).
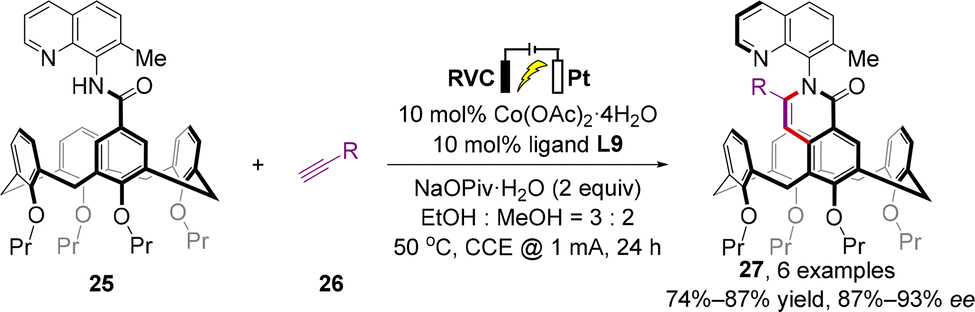 | ||
| Scheme 12 Electrochemically driven cobalt-catalyzed enantioselective intermolecular C–H annulation strategy. | ||
Meanwhile, Niu, Dou and colleagues demonstrated an elegant example of electrochemically driven cobalt-catalyzed asymmetric aryl C–H acyloxylation of prochiral calix[4]arenes 28, enabling the generation of various enantioenriched inherently chiral acyloxylated products 30 with good to high yields (up to 94%) and excellent enantioselectivities (up to >99% ee) (Scheme 13).50 The success of this protocol was attributed to the combination of the directing group pyridine N-oxide with chiral Salox ligand L10, as well as the perfect match of electrochemistry with chiral cobalt catalysts.
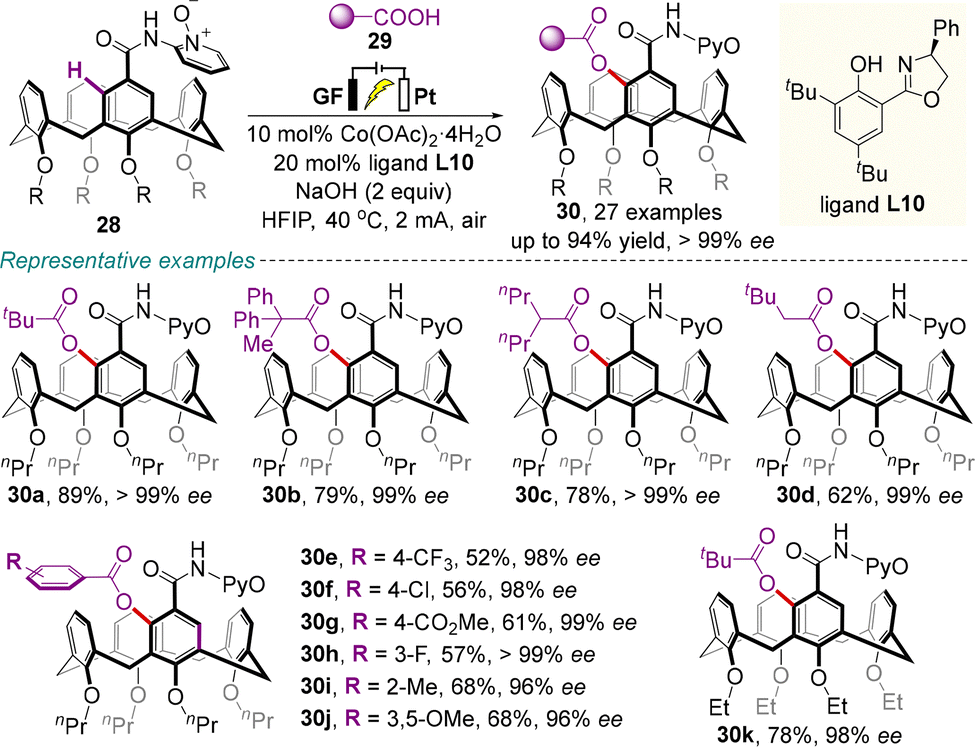 | ||
| Scheme 13 Electrochemically driven cobalt-catalyzed asymmetric C–H acyloxylation for the synthesis of inherently chiral calix[4]arenes. | ||
 | ||
| Scheme 14 Copper-catalyzed asymmetric synthesis of inherently chiral zigzag-type molecular belt via KR and subsequent cyclization. | ||
2.3. Organocatalytic strategy
 | ||
| Scheme 15 CPA-catalyzed enantioselective synthesis of inherently chiral N3,O-calix[2]arene[2]triazines via an intramolecular SNAr reaction. | ||
This strategy has been further extended to assemble inherently chiral tetraazacalix[4]aromatics using CPA2 as the catalyst by Wang, Cheng and colleagues recently (Scheme 16).56 It should be pointed out that the inherent chirality of the resulting tetraazacalix[1]arene[1]pyridine[2]triazines 36 originated from the variation of a single substituent at the bridging nitrogen units.
 | ||
| Scheme 16 CPA-catalyzed asymmetric synthesis of inherently chiral tetraazacalix[4]aromatics via an intramolecular SNAr reaction. | ||
Furthermore, Tong and colleagues demonstrated that the SNAr reaction could be applied in an intermolecular macrocyclic condensation process (Scheme 17).57 Applying cinchonine-derived chiral phase-transfer catalyst PTC1, the asymmetric one-pot cross-cyclotetramerization of 2,6-dichloro-3-nitropyridine 37 with resorcinol derivatives 38 delivered inherently chiral tetraoxacalix[2]arene[2]pyridines 39 with moderate to good enantioselectivities (up to 77% ee). However, poor yields were obtained, potentially due to the complexity of the multiple competing reaction pathways.
 | ||
| Scheme 17 PTC-catalyzed enantioselective synthesis of inherently chiral tetraoxacalix[2]arene[2]pyridines via cross-cyclotetramerization. | ||
Recently, by employing this strategy, Liu's group and Yang's group independently realized the enantioselective synthesis of inherently chiral calix[4]arenes, through an effective central-to-inherent chirality conversion paradigm (Scheme 18).61,62 Under different CPA (CPA3 or CPA4) catalysis, amino group-substituted calix[4]arenes 40, (di)enamides 41 and aldehydes 42 underwent an asymmetric Povarov-type annulation to generate the key optically enriched cycloadduct with two well-defined carbon stereocenters, which was oxidized with DDQ to obtain a diverse array of novel inherently chiral 4-amido-quinoline-containing calix[4]arenes 43 or 44 with excellent enantioselectivities (up to 99% ee). Mechanistic studies assumed that the high selectivity arises from the steric repulsions between the vertical plane of calix[4]arenes 40 and the bulky aryl substituents on the CPA catalyst.
 | ||
| Scheme 18 CPA-catalyzed asymmetric synthesis of chiral 4-amido-quinoline-containing calix[4]arenes via the Povarov reaction. | ||
Very recently, Yang and co-workers successfully accomplished the enantioselective synthesis of inherently chiral calix[4]arenes 47 through CPA-catalyzed asymmetric electrophilic aromatic aminations of phenol-containing prochiral calix[4]arenes 45 with high to excellent yields and enantioselectivities (Scheme 19).63
 | ||
| Scheme 19 CPA-catalyzed asymmetric synthesis of inherently chiral calix[4]arenes via electrophilic aromatic aminations. | ||
This transformation evolves through CPA-controlled stereoselective addition to produce a stereoenriched dearomatized intermediate with both central and inherent chirality. Rapid keto–enol tautomerism follows with central-to-inherent chirality conversion to render the ortho-C–H aminated phenol products 47 stereochemically defined. In analogy with the previous research on the origin of enantioselectivity, the parallel alignment of the substrates relative to one of the catalyst's anthracenyl groups, along with a series of C–H⋯π interactions between the reactant and the anthracenyl group, is probably responsible for the enantiocontrol. Moreover, the chiral meta-amino-substituted calix[4]arene 47a was readily converted into the primary amine catalyst 48. This novel catalyst efficiently promoted the asymmetric α-amination of aldehyde 49 with azodicarboxylate 50, highlighting its significant potential for the development of useful chiral organocatalysts.
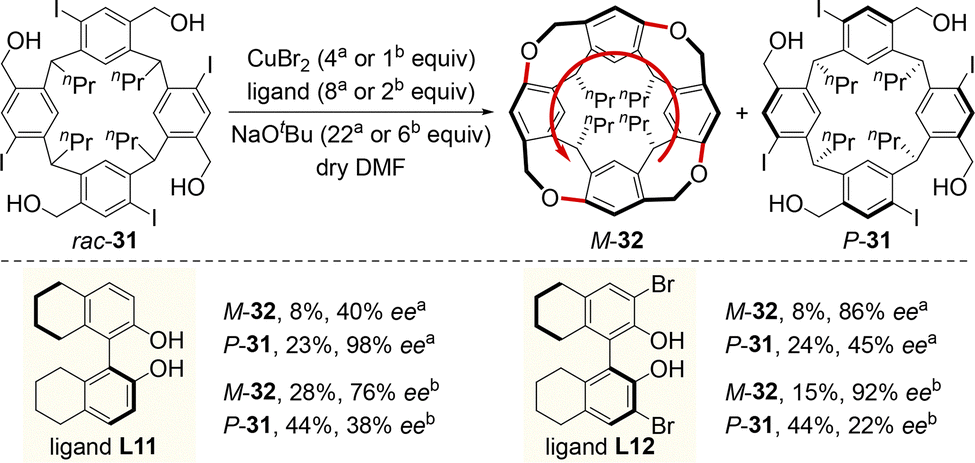
In 2024, Chen and colleagues developed an efficient chiral Lewis base-catalyzed desymmetrization approach for the enantioselective electrophilic sulfenylation of various prochiral calix[4]arenes 52 (Scheme 20).64 Upon treatment with sulfenylating reagent 53, 10 mol% of sulfide catalyst LB1, and hexafluoroisopropanol (HFIP), the reaction proceeded smoothly to give the enantioenriched inherently chiral sulfur-containing calix[4]arenes 54 in moderate to excellent yields (most ≥80%) with high enantioselectivities (most ≥90% ee). Mechanistically, the thermodynamically favored C-SAr product 54 is gradually formed from the rapidly formed kinetically favored N-SAr product 55. In addition, the combination of the chiral Lewis base catalyst LB1 and HFIP has a dramatic effect on both enantioselectivity and reactivity, since the hydrogen bonds between the p-toluenesulfonic acid (pTSA) anion/HFIP species (A−) and the N–H moiety of substrate 52 may decrease the energy barrier of this transformation and contribute to stabilize the favorable transition state int-9.
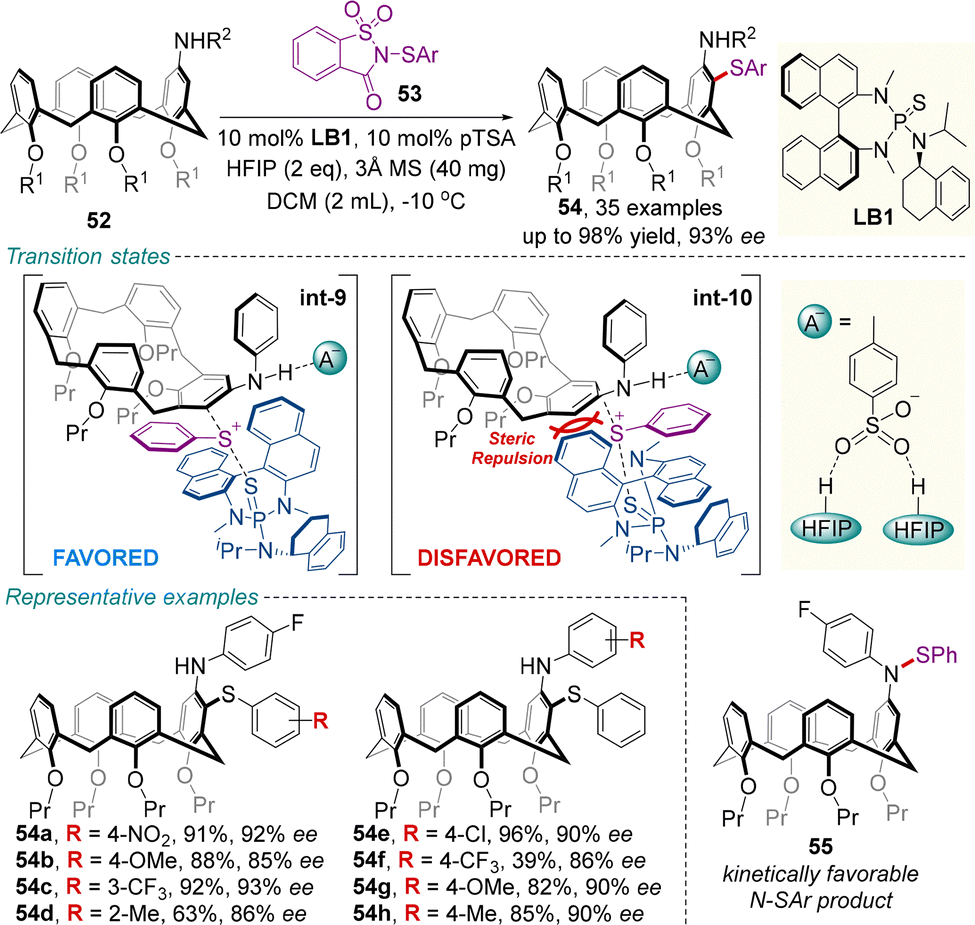 | ||
| Scheme 20 Chiral sulfide catalyzed desymmetrizing electrophilic sulfenylation of racemic calix[4]arenes. | ||
Chiral NHC-catalyzed desymmetrization of prochiral diformyl derivatives stands out as a promising platform for asymmetric transformations. Recently, this chemistry was further popularized to access inherently chiral ABCC-type calix[4]arenes by Veselý, Dočekal and colleagues (Scheme 21).65 Under oxidative conditions (tetra-tert-butyldiphenyl-quinone, DQ), the reaction of prochiral diformylcalix[4]arenes 56 with bifunctional chiral NHC1 effectively generates an NHC-bound acylazolium intermediate int-11, which then reacts with aromatic alcohols 57 to complete the esterification process. Mechanistic investigations indicated that desymmetrization is the enantio-determining step. Remarkably, this method has been applied for the late-stage modification of several natural products and bioactive compounds, including Umbelliferone, Mecarbinate, Carvacrol and Capsaicin, which furnished the desired drug-like molecules (58k–58n) with high yields (61–88%) and ee values (71–95% ee), showing good substrate tolerance.
 | ||
| Scheme 21 Chiral NHC-catalyzed enantioselective desymmetric esterification of prochiral diformylcalix[4]arenes. | ||
More recently, Tong and co-workers reported that achiral resorcin[4]arenes 59 underwent a 4-fold Mannich/cyclization reaction to generate C4-symmetric chiral resorcin[4]arenes 61 in the presence of chiral N,N′-dioxide L3-PiMe3 derived from (S)-piperidine-2-carboxamide (Scheme 22).66
 | ||
| Scheme 22 N,N′-Dioxide catalyzed enantioselective desymmetrization of resorcin[4]arenes via the Mannich/cyclization reaction. | ||
The desymmetrization of symmetric resorcin[4]arene 59 through the first Mannich/cyclization reaction enantioselectively formed the 1,3-oxazinane-fused resorcin[4]arene intermediate int-12 of C1 symmetry. This intermediate adopts a conformation stabilized by intramolecular hydrogen bonding interaction, enabling exclusive regioselectivity during the subsequent transformation to afford C4-symmetric products 61. Furthermore, such chiral resorcin[4]arenes could be transformed into novel chiral catalysts for the asymmetric addition of diethylzinc to benzaldehyde. This system successfully demonstrated the effective transfer of inherent chirality from the macrocyclic scaffold to the product's central chirality, underscoring its potential in enantioselective catalysis.
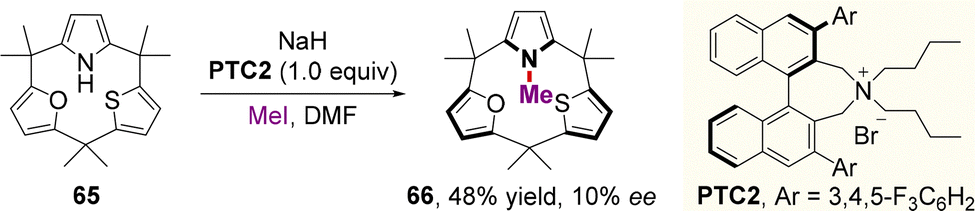 | ||
| Scheme 23 Dynamic kinetic resolution (DKR) by enantioselective N-methylation of the pyrrole moiety. | ||
In 2024, the dynamic kinetic resolution protocol was utilized by Tong and colleagues to access inherently chiral calix[4](het)arenes (Scheme 24).68 The reaction of diazadioxacalix[2]arene[2]quinazolines 67 with bromomethylarenes 68 as alkylation partners and cinchonine-derived PTC3 as the catalyst delivered highly enantioenriched 69 in excellent yields. Guided by DFT studies and observed stereochemical outcomes, the excellent enantiocontrol could be attributed to the host–guest-like interaction between the substrate and the catalyst.
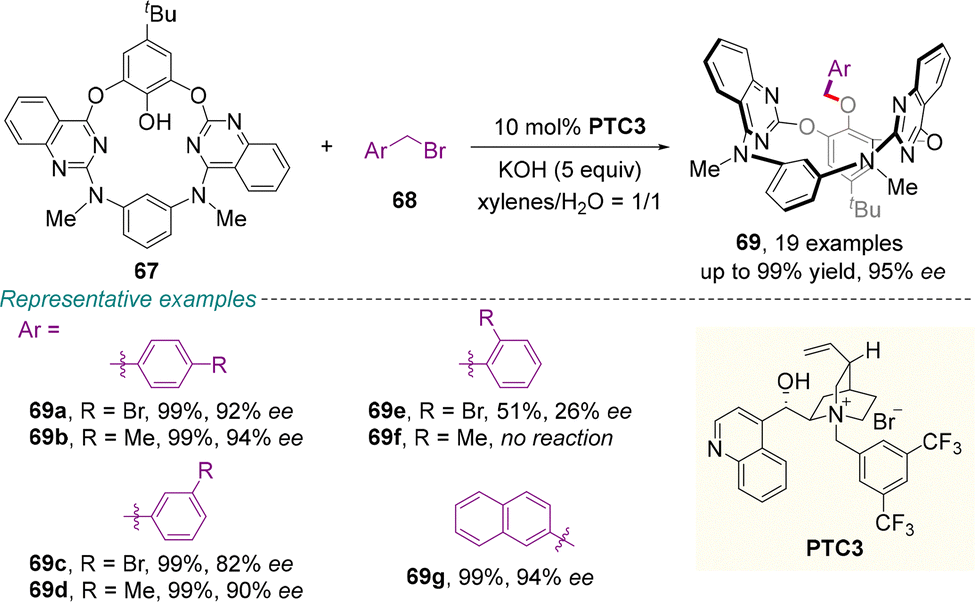 | ||
| Scheme 24 Chiral PTC-catalyzed asymmetric synthesis of inherently chiral heteracalixarenes via dynamic kinetic resolution. | ||
After the success of DKR in preparing inherently chiral macrocycles, Wang and co-workers implemented an NHC-catalyzed DKR protocol to forge inherently chiral heteracalixarenes 72via esterification of diazadioxacalix[2]arene[2]quinazolines 70 (Scheme 25).69 DFT analysis revealed that int-13 exhibits a 9.2 kcal mol−1 lower energy barrier for hydroxyl addition to acyl azolium compared to int-14, kinetically favoring S-configured products. Furthermore, employing the same catalytic system, the kinetic resolution of racemic rac-73 provided access to enantioenriched 74 with good to high enantioselectivities. Additionally, optically active ent-73 was recovered in acceptable yields with high to excellent enantioselectivities (Scheme 26).
 | ||
| Scheme 25 Chiral NHC-catalyzed asymmetric synthesis of inherently chiral calix[4](het)arenes via dynamic kinetic resolution. | ||
 | ||
| Scheme 26 Chiral NHC-catalyzed asymmetric synthesis of inherently chiral calix[4](het)arenes via kinetic resolution. | ||
3. Enantioselective construction of pillar[n]arenes
Among various chiral macrocycles, pillar[n]arenes have garnered significant attention due to their unique topological structures and broad utility in materials science,70,71 host–guest chemistry,72,73 and molecular machinery.74,75 In particular, inherently chiral pillar[5]arenes, composed of five repeating paraphenylene methylene units, stand out for their synthetic accessibility and versatile functionality. However, free rotation of these repeating units leads to the coexistence of up to eight conformers in solution, presenting a major obstacle to obtaining configurationally stable enantiomers.12–15 This dynamic behavior can be restrained through the introduction of sterically hindering substituents or the construction of mechanically interlocked and self-locking architectures, thereby enabling the isolation of stable enantiopure forms. Nevertheless, to date, the preparation of enantiopure pillar[5]arenes still predominantly relies on chiral HPLC resolution or diastereomeric crystallization, approaches that suffer from limited efficiency and scalability. Catalytic enantioselective synthesis offers an attractive alternative to overcome these drawbacks.3.1. Enantioselective Sonogashira coupling
Recently, a breakthrough strategy for the enantioselective synthesis of inherently chiral pillar[5]arenes has been developed by Wang and co-workers via a Pd-catalyzed Sonogashira coupling reaction (Scheme 27).76 This method enables efficient access to a diverse range of homo- and hetero-rimmed pillar[5]arenes 77 in good to high yields with high to excellent enantioselectivities (up to 97% ee). The absolute configuration of product 77a was determined as a pS conformer by single-crystal X-ray diffraction. Notably, once the stereochemical configuration of one repeating unit in the pillar[5]arene framework is fixed as pS through the introduction of two sterically bulky alkynes, the remaining four units adopt the same chiral conformation through conformational induction, resulting in the all-pS conformer as the thermodynamically most stable enantiomer. Mechanistic studies revealed that the second Sonogashira coupling step is enantio-determining in this sequential double coupling process. Importantly, IRI (interaction region indicator) analysis indicated that reduced steric repulsion between the Pd center of catalyst Pd/L13 and the phenyl group of the pillar[5]arene in transition state int-15 leads to a lower activation energy barrier, which is crucial for achieving high enantiocontrol. Furthermore, product 77b was successfully employed as a chiral wheel component for the construction of chiral rotaxane 82, which exhibits remarkable CPL with glum reaching up to −0.005 (Scheme 28). | ||
| Scheme 27 Palladium-catalyzed asymmetric synthesis of inherently chiral pillar[5]arenes by DKR via Sonogashira coupling. | ||
 | ||
| Scheme 28 Application of inherently chiral pillar[5]arene 77b in the synthesis of chiral rotaxanes. | ||
3.2. Enantioselective Suzuki–Miyaura coupling
Very recently, Liu, Xi and colleagues disclosed a DKR based on the enantioselective Pd-catalyzed Suzuki–Miyaura coupling of pillar[5]arene-based bifunctional triflate 83 and boronic acids 84 with the use of chiral Sadphos ligand L14 (Scheme 29a).77 The significant steric hindrance and electronic properties of the aryl group in L14 were reasoned to control the high reaction enantioselectivity (Scheme 30a). | ||
| Scheme 29 Palladium-catalyzed asymmetric synthesis of inherently chiral pillar[5]arenes by DKR via Suzuki–Miyaura coupling. | ||
 | ||
| Scheme 30 Diastereomeric transition states in the Pd-catalyzed Suzuki–Miyaura coupling. | ||
Concurrently, a catalytic system comprising Pd(TFA)2 and sterically chiral AKphos ligand L15 was reported by Mazet, Leforestier and colleagues (Scheme 29b).78 As with the previous approach, a high enantiomeric ratio was achieved with a broad spectrum of boronic acids. Stereodifferentiation of this transformation originates from both stabilizing secondary interactions and repulsive steric factors, as established computationally and experimentally (Scheme 30b).
Soon after, a Pd(II)/chiral phosphoramidite L16 catalytic system featuring higher enantioselectivity (95–99% ee) was provided by the Tu, Ma and Zhang's group (Scheme 29c).79 By employing bulkier boronic acids, this protocol was successfully extended to the synthesis of configurationally stable, inherently chiral pillar[6]arenes. Mechanistic studies indicated that axial steric hindrance governs the conformational chirality-locking process in pillar[n]arenes. It is also noteworthy that in all three reactions above, the origin of stereoselectivity consistently identifies the second coupling as the enantio-determining step in the sequential double Suzuki–Miyaura coupling process.
3.3. Enantioselective copper-catalyzed alkyne–azide cycloaddition
Catalytic asymmetric Cu-catalyzed azide–alkyne cycloaddition (CuAAC) offers an efficient way for constructing structurally diverse centrally chiral triazoles and heterobiaryl atropisomers.80,81 However, this type of click reaction remained unexplored in producing inherently chiral scaffolds until recently. Based on the extended side-arm strategy, success with enantioselective CuAAC was achieved by Liu and co-workers using Cu(I) catalysis with chiral oxazoline-containing ligand L17, starting from 2-ethynyl-pillar[5]arene 86 (Scheme 31).82 | ||
| Scheme 31 Copper-catalyzed asymmetric synthesis of inherently chiral pillar[5]arenes by DKR via alkyne–azide cycloaddition. | ||
The introduction of sterically hindered triazolyl groups afforded a diverse array of inherently chiral triazolyl-pillar[5]arenes 88 in high yields with exceptional enantioselectivities. Ligand control studies confirmed that chiral induction occurs during the second catalytic step. The resulting products demonstrate high configurational stability; 88a, for example, exhibits no racemization at 140 °C in xylene. Moreover, these enantiopure products enable the construction of chiral pillar[5]arene-based rotaxanes.
4. Enantioselective construction of saddle-shaped scaffolds
Beyond inherently chiral calix(het)arenes and pillar[n]arenes, rigid saddle-shaped medium-sized scaffolds, exemplified by tetraphenylene and its derivatives, represent another privileged class of persistent inherently chiral architectures. This unique chirality arises from their conformationally constrained structures, which impose high energy barriers to racemization. Such topologically defined frameworks underpin significant applications, ranging from chiral functional materials to bioactive molecules (e.g., telenzepine).27,28 However, the enantioselective synthesis of these saddle-shaped targets still remains a fundamental challenge. Modern catalytic asymmetric strategies now offer powerful and potentially transformative solutions to this long-standing problem.17 This section surveys significant recent advances in this rapidly evolving field, organized by transition-metal catalysis and organocatalysis.4.1. Transition-metal-catalyzed methods
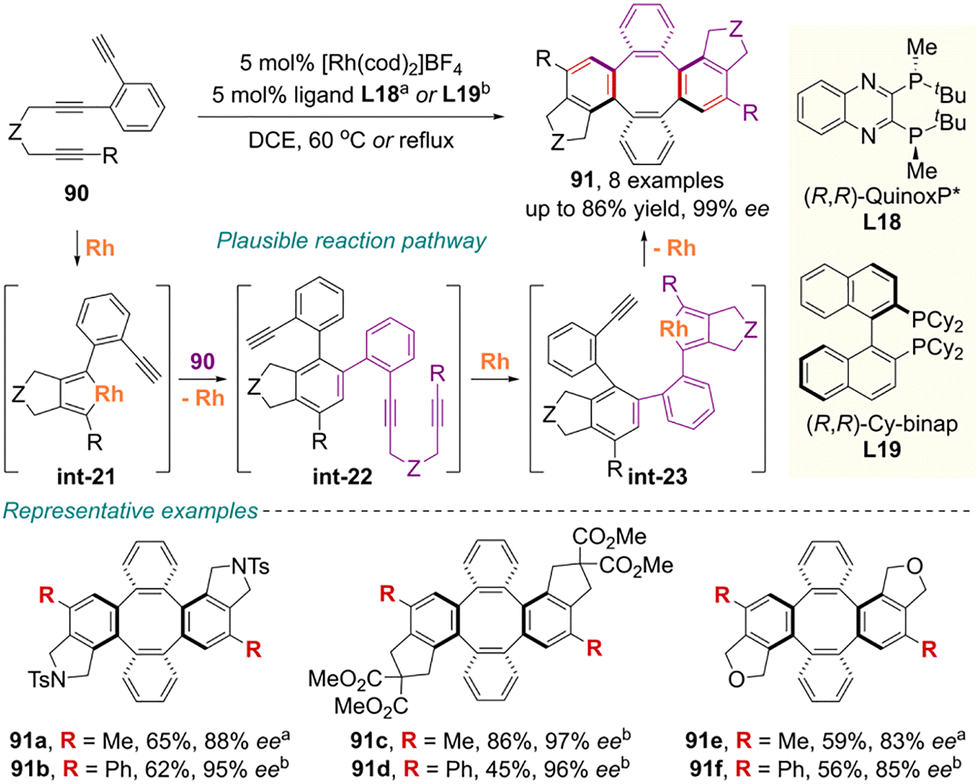 | ||
| Scheme 32 Rhodium-catalyzed enantioselective synthesis of inherently chiral tetraphenylenes via consecutive cycloadditions. | ||
This synthetic operation commences with the formation of rhodacyclopentadiene intermediate int-21 from the 1,6-diyne moiety of the first triyne. Subsequently, the terminal alkyne moiety of the second triyne undergoes selective insertion into this intermediate to generate the primary cycloadduct int-22 with axial chirality. Following this, oxidative coupling of the 1,6-diyne moiety in the second triyne forms the metallacycle int-23, and intramolecular coupling of the remaining terminal alkyne from the first triyne ultimately yields the eight-membered cycloadduct 91.84
In 2016, the same group advanced the chemistry of enantioselective arene forming [2+2+2] cycloaddition further to derive enantioenriched saddle-shaped tribenzothiepins (Scheme 33).85 This powerful and atom-economical approach provides efficient access for the synthesis of inherently chiral multisubstituted tribenzothiepins 94 and 96 from readily available diphenyl-sulfidetethered diynes 92 or 2-phenyl sulfanylbenzene-tethered diynes 95 using a rhodium catalyst in the presence of a chiral bidentate phosphorus ligand, respectively. Racemization measurements on optically pure tribenzothiepin 94a revealed a racemization energy barrier of 29.1 kcal per mole, and the corresponding half-life of racemization at 20 °C was estimated to be 9.2 years.
 | ||
| Scheme 33 Rhodium-catalyzed enantioselective synthesis of chiral saddle-shaped tribenzoheteropins via intermolecular cycloadditions. | ||
As an extension of this strategy, Shibata and colleagues achieved the synthesis of the configurationally stable saddle-shaped cyclic polyarylenes 99 through the catalytic asymmetric intermolecular [2+2+2] cycloaddition of 1,8-naphthylene-based 1,10-diynes 97 with alkynoates 98 or aryl cyanides (Scheme 34).86 The use of [Rh(cod)2]NTf2/(S)-BINAP afforded the corresponding inherently chiral cycloadducts 99 with good to high yields and enantioselectivities.
 | ||
| Scheme 34 Rhodium-catalyzed enantioselective synthesis of saddle-shaped cyclic polyarylenes via [2+2+2] cycloaddition. | ||
More recently, rhodium-catalyzed asymmetric synthesis of seven-membered saddle-shaped tribenzogermepins featuring inherent chirality has been demonstrated by Xiao and co-workers (Scheme 35).87 In the presence of [Rh(CH2CH2)2Cl]2 with a chiral JosiPhos-based diphosphine ligand L23, a wide range of tribenzogermepins 101 were conveniently constructed in good to high yields with impressive enantioselectivities via dehydrogenative C(sp2)–H germylation. These products are configurationally stable; for example, the theoretical half-life for racemization (t1/2) of 101d was determined to be 503 years at 25 °C.
 | ||
| Scheme 35 Rhodium-catalyzed enantioselective synthesis of inherently chiral seven-membered tribenzogermepins via C–H germylation. | ||
A prominent recent report by Zhu, Luo and colleagues documented the highly enantioselective three-component coupling reaction of various 2,2′-diisocyano-1,1′-biphenyl 102, aryl iodide 103, and carboxylate, promoted by a catalytic system comprising palladium(0) complex and Josiphos ligand L24 (Scheme 36).88 This reaction involves a sequence of oxidative addition of phenyl iodide 103, migratory insertion of the first isocyano group of 102, coordination of the second isocyano group to the Pd center, a second migratory insertion of the isocyano moiety and reductive elimination before migration of the Piv group to the N-atom of intermediate int-27 to reveal the final saddle-shaped aza analog of tetraphenylene 104. Stereochemical stability of this aza-bridged framework was typified by 104a, for which the half-life was estimated to be 16 h at 100 °C. In addition, atropisomerically enriched compound 104a could serve as a recyclable chiral acylating reagent for primary amines. This approach provides a sustainable route to enantiomerically enriched amides, with promising applications in asymmetric synthesis and pharmaceutical chemistry.
 | ||
| Scheme 36 Palladium-catalyzed enantioselective synthesis of a chiral saddle-shaped aza analog of tetraphenylene. | ||
In 2024, the same authors developed a palladium-catalyzed enantioselective carbene-based cross-coupling protocol using N-arylsulfonylhydrazone derivatives 108 and benzyl bromides 109 in the presence of spiro-phosphoramidite ligand L25, enabling the efficient construction of inherently chiral saddle-shaped tribenzoannulene derivatives 110 in high yields with exceptional enantioselectivities (Scheme 37, condition a).89
 | ||
| Scheme 37 Pd-catalyzed enantioselective synthesis of saddle-shaped tribenzoannulene derivatives via carbene-based cross-coupling. | ||
The proposed mechanism begins with oxidative addition of benzyl bromide to the Pd(0) catalyst to form Pd(II) complex int-28, which reacts with in situ generated diazo species to afford the palladium carbene intermediate int-30. Subsequent carbene migratory insertion, followed by β-hydride elimination, yields the insertion alkene products 110. The β-hydride elimination step was identified as the stereocontrolling step of the entire reaction. The stability of inherent chirality of saddle-shaped 110 was substantial (110a, 31.7 kcal mol−1 at 140 °C). Notably, phosphine ligand 113, derived from product 110e, exhibited excellent catalytic performance in enantioselective Rh-catalyzed 1,4-addition and Pd-catalyzed Tsuji–Trost reactions, demonstrating its potential as a versatile platform for chiral phosphine ligand design. Concurrently, utilizing Pd2(dba)3 with chiral Feringa ligand L26, Liu and colleagues independently accomplished the same transformation with high yields and excellent enantioselectivities (Scheme 37, condition b).90
 | ||
| Scheme 38 Copper-catalyzed enantioselective synthesis of inherently chiral aryl oxime ethers via dynamic kinetic resolution. | ||
4.2. Organocatalytic strategy
 | ||
| Scheme 39 CPA-catalyzed enantioselective synthesis of inherently chiral tribenzocycloheptene oxime ethers via a condensation reaction. | ||
In 2023, catalytic enantioselective cyclocondensation of easily available [1,1′-biphenyl]-2,2′-diamines 123 and substituted benzils 124 to construct saddle-shaped 6,7-diphenyldibenzo[e,g][1,4]diazocines 125 portraying inherent chirality was contrived by Zhu, Luo and colleagues using H8-BINOL-based chiral phosphoric acid catalysts (Scheme 40).94 Precipitation and phase separation steps help increase the enantiomeric excess of the product. By changing the CPA catalyst CPA7 to CPA8, an inversed product configuration was intriguingly acquired, although the two catalysts had the same configuration. The enantioselectivity was found to originate from the CPA-catalyzed dehydration step through non-covalent interactions, as elucidated by DFT calculations. Moreover, the configurationally stable diphenol derivative 126 served as a versatile synthetic platform, enabling the efficient construction of architecturally novel phosphoramidite ligands 127 and diphosphine ligands 128 while fully preserving enantiomeric excess. Preliminary catalytic tests showed that both ligands were effective in Pd- or Rh-catalyzed asymmetric reactions, providing enantioselectivities comparable to those from privileged scaffolds like BINOL and BINAP, highlighting the practical value of this methodology.
 | ||
| Scheme 40 CPA-catalyzed asymmetric synthesis of inherently chiral 6,7-diphenyldibenzo[e,g][1,4]diazocines via cyclocondensation. | ||
In related work, Yang and co-workers obtained good yields and an excellent enantioselective gateway to inherently chiral saddle-shaped dibenzo[b,f][1,5]diazocines 130 from asymmetric dimerization–cyclization of 2-acylbenzoisocyanates 129 enabled by chiral phosphoric acid CPA10 (Scheme 41).95 Notably, the incorporation of 2-acylaniline 131 as a co-catalyst enhanced the reaction efficiency, and the phase separation process improved enantioselectivity. The reaction initiates with the nucleophilic addition of 131 to int-38, followed by CO2 release, generating the imine-type intermediate int-40. Following this, two plausible pathways were identified. The first pathway involves iterative addition, expulsion of CO2, cyclization of int-41 and elimination of aniline 131 with central-to-inherent chirality conversion of intermediate int-42 to reveal the final product. Alternatively, the second pathway proceeds through CPA-guided stereoselective intramolecular condensation of int-40, followed by dehydration to generate the inherently chiral scaffold 130.
 | ||
| Scheme 41 CPA-catalyzed asymmetric synthesis of inherently chiral dibenzo[b,f][1,5]diazocines via dimerization of 2-acylbenzoisocyanates. | ||
In the same year, the same research group reported the CPA-catalyzed enantioselective synthesis of saddle-shaped inherently chiral 9,10-dihydrotribenzoazocines through two alternative methods: KR and DKR (Schemes 42 and 43).96 In the KR pathway, using BINOL-derived catalyst CPA5, the intermolecular electrophilic amination proceeds with excellent selectivities (s-factors up to >1000). Racemization studies on optically pure (–)-132a revealed a racemization energy barrier of 27.33 kcal mol−1, which suggested decent stereochemical stability of this scaffold. In the process of DKR, using SPINOL-derived CPA11 as the catalyst in combination with Hantzsch ester 136 as the hydrogen source, asymmetric transfer hydrogenation of configurationally labile imine-type azaheterocycles 135 delivered 9,10-dihydrotribenzoazocines (+)-132a with good yields and high enantioselectivities.
 | ||
| Scheme 42 CPA-catalyzed asymmetric synthesis of inherently chiral 9,10-dihydrotribenzoazocines via kinetic resolution. | ||
 | ||
| Scheme 43 CPA-catalyzed asymmetric synthesis of inherently chiral 9,10-dihydrotribenzoazocines via dynamic kinetic resolution. | ||
 | ||
| Scheme 44 Quinine-catalyzed enantioselective synthesis of chiral tub-shaped dianthranilides via dynamic kinetic resolution. | ||
 | ||
| Scheme 45 Quinidine-catalyzed enantioselective synthesis of chiral tub-shaped dianthranilides via kinetic resolution. | ||
Recently, Yan, Liu and co-workers approached the highly chemo-, diastereo-, and enantioselective synthesis of novel rigid eight-membered O-heterocycles 144 and 146 possessing inherent chirality through a cross-[4+4] cycloaddition reaction, by harnessing the synthetic versatility of quinone methides (QMs) (Scheme 46).98 Mechanistically, the quinidine-derived catalyst cat3 enantioselectively processes the tautomerization of 2-alkynylnaphthol 142 to give axially chiral vinylidene ortho-quinone methide (VQM) intermediate int-50 on which nucleophilic addition of ortho-quinone methide (o-QM) 143 proceeds to give 144. Critically, the inherent chirality of the product was relayed from the enantioenriched VQM intermediate int-50. Apart from o-QMs 143, ortho-hydroxyphenyl substituted para-quinone methides (p-QMs) 145 also react well for this transformation. DFT calculations confirmed the high stereochemical stability of these O-heterocycles (ΔG‡rac = 48.2 kcal mol−1 for compound 144a), with rigidity primarily arising from the bulky alkenes.
 | ||
| Scheme 46 Quinine-catalyzed enantioselective synthesis of chiral eight-membered O-heterocycles via cross-[4+4] cycloaddition. | ||
 | ||
| Scheme 47 Bifunctional iminophosphorane-catalyzed enantioselective synthesis of chiral seven- and eight-membered rings. | ||
The identical catalyst cat4 facilitated the formation of seven- and eight-membered rings via two distinct mechanistic paradigms. The rotational barrier of model products 148a and 150a was determined to be >36 kcal mol−1 (t1/2 > 900000 years at 25 °C) and 27.5 kcal mol−1 (t1/2 ≈ 200 days at 25 °C), respectively, confirming their configurational stability. Computational studies demonstrated that hydrogen-bonding interactions and minimization of substrate–catalyst dipole moments govern transition state assembly, whereas attractive dispersion forces substantially dictate the observed enantioselectivity and enantiodivergent behavior.
 | ||
| Scheme 48 NHC-catalyzed enantioselective synthesis of saddle-shaped eight-membered lactones via [5+3] annulation. | ||
Recently, Yang, Zhang, Chi, and colleagues reported an efficient NHC-catalyzed, base-controlled enantiodivergent construction of inherently chiral, saddle-shaped 8-membered lactones from triaryl aldehydes 154via intramolecular cyclization (Scheme 49).101 Employing NHC catalysts sharing an identical chiral scaffold (NHC3 or NHC5), but with different achiral bases, afforded both enantiomers of the lactones in high to excellent yields (up to 99%) with good to excellent enantioselectivities (up to 99:1 er for 155 and 1:99 er for ent-155). DFT studies revealed that this enantiodivergence stems from base-dependent noncovalent interactions governing the enantiodetermining transition state of the key acyl azolium intermediate. Notably, several enantiopure lactones exhibited potent antibacterial activity against plant pathogens, underscoring their potential for agricultural applications.
 | ||
| Scheme 49 NHC-catalyzed base-controlled enantiodivergent synthesis of 8-membered saddle-shaped lactones via intramolecular cyclization. | ||
5. Enantioselective construction of rotaxanes
Mechanically interlocked molecules (MIMs), such as chiral rotaxanes and catenanes, can exhibit mechanical chirality (also referred to as inherent chirality) even when all covalent subcomponents are achiral.18–20 This chirality originates from the relative spatial orientation enforced by the mechanical bond between the interlocked components, such as a ring and an axle, each possessing structural dissymmetry. In rotaxanes, mechanical chirality can arise from the symmetry breaking of achiral subunits due to conformational restrictions imposed by the mechanical bond, or through the introduction of steric hindrance to limit relative motion between the axle and the macrocycle. In catenanes, chirality can emerge when the two rings are traversed in a specific directional sequence, or when the macrocyclic faces are distinguishable (Fig. 2).102–106 Despite holding considerable promise for applications spanning chiral sensing, supramolecular catalysis, and molecular machinery, the asymmetric synthesis of such architectures faces substantial challenges stemming from their conformational flexibility and noncovalent nature of mechanical bonds.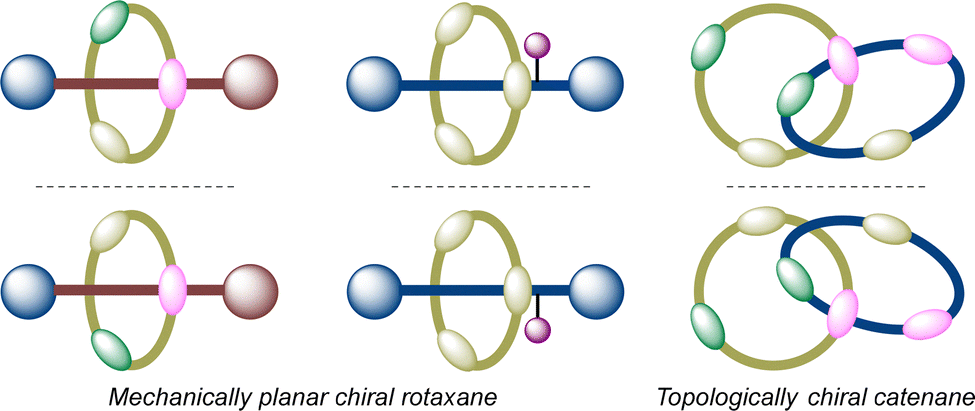 | ||
| Fig. 2 Schematic presentation of a mechanically planar chiral rotaxane and topologically chiral catenane. | ||
5.1. Dynamic kinetic resolution
In 2007, Takata and colleagues pioneered the catalytic enantioselective synthesis of inherently chiral rotaxanes 159via a trialkylphosphane-catalyzed O-acylation of secondary ammonium salt 157 with bulky benzoic anhydride 158 (Scheme 50).107 The enantioselectivity was governed by hydrogen bonding interactions between the phosphane groups in catalyst cat5 and the amide moiety on the crown ether wheel 156, with the rather low ee value (4.4%) suggesting that these interactions were insufficient to achieve effective facial discrimination of the crown ether. | ||
| Scheme 50 Chiral trialkylphosphane-catalyzed asymmetric synthesis of inherently chiral rotaxanes via O-acylation. | ||
In 2008, Leigh and colleagues reported the catalytic asymmetric synthesis of configurationally stable chiral benzyl ester rotaxanes 162via DKR of enantiomeric hydroxyl-rotaxanes 160 (Scheme 51).108 The two hydroxyl-rotaxane enantiomers, featuring four-point hydrogen bonding between a single fumaramide moiety on the thread and the benzylic amide macrocycle, exist in equilibrium due to macrocycle shuttling. Employing a bulky chiral DMAP-derived catalyst (S)-cat6 (2.0 equivalent) and benzoic anhydride 161, asymmetric benzoylation installs a sterically demanding benzoyl group on the thread. This modification kinetically traps the macrocycle on one side, inhibiting shuttling and affording enantioenriched (S)-162 with 34% ee.
 | ||
| Scheme 51 Chiral DMAP derivative-catalyzed asymmetric synthesis of inherently chiral rotaxanes via dynamic kinetic resolution. | ||
5.2. Kinetic resolution
In 2021, Kawabata and colleagues reported the catalytic synthesis of enantiomerically enriched rotaxane 163 through kinetic resolution of the corresponding racemate (Scheme 52).109 Treatment of racemic 163 with 0.8 equivalent amount of acetic anhydride 164 in the presence of 1.5 equivalent amount of catalyst cat7 resulted in acylated rotaxane 165 after 37 h with 71% conversion. Analysis of the unreacted starting material ent-163 revealed an enantiomeric excess >99.9% and a selectivity factor (s) > 14.5. Control experiments and ONIOM (our own N-layered integrated molecular orbital and molecular mechanics) calculations identified hydrogen-bonding interactions between the amide carbonyl group of catalyst cat7 and the NsN–H moiety, along with general base catalysis by the proximal carboxylate anion adjacent to the reacting hydroxy group, as key factors enabling the effective remote asymmetric acylation. | ||
| Scheme 52 Chiral 4-pyrrolidinopyridine-catalyzed enantioselective synthesis of mechanically planar chiral rotaxane via kinetic resolution. | ||
5.3. Desymmetrization
In a significant advance, Zhu, Tian and co-workers developed a transformative catalyst-controlled desymmetrization strategy for the enantioselective synthesis of mechanically planar chiral (MPC) rotaxanes (Scheme 53).110 This approach employs a Pd-catalyzed Suzuki–Miyaura coupling of prochiral, symmetrical bis(chloroaryl)macrocycle rotaxanes 166 with arylboronic acids 167, utilizing the novel anionic chiral phosphine ligand L28 to achieve high enantioselectivity (up to 93% ee). Of note, distal electrostatic interactions between the rotaxane and catalyst, mediated by carboxylate anions, predominantly govern precise stereocontrol, outweighing contributions from other types of non-covalent interactions. This strategy decouples chirality induction from mechanical bond formation, providing modular post-assembly access to MPC architectures and establishing the ionic ligand design as a robust paradigm for stereochemical manipulation in mechanically interlocked molecules. | ||
| Scheme 53 Pd-catalyzed enantioselective synthesis of mechanically planar chiral rotaxane via desymmetrization. | ||
6. Enantioselective construction of prism-like cages
In recent years, chiral peptide-mimic phosphonium salt (PPS) catalysis has emerged as a powerful and versatile strategy for accessing diverse enantiomerically enriched molecules.111 In this realm, the first phosphonium-containing foldamer-catalyzed asymmetric synthesis of inherently chiral prism-like cages was realized by Wang and colleagues in 2024 via SNAr desymmetrization (Scheme 54).112 Employing a mild cationic foldamer catalysis system, a wide range of stereochemically rich, high-value prism-like cages 171 were prepared in good-to-high yields with high-to-excellent enantioselectivities across various aryl alcohol and thiol nucleophiles. From a mechanistic standpoint, the dual phosphonium centers of catalyst PPS-1 would bind naphtholate anions through ion pairing, while N–H and benzyl C–H hydrogen-bonding sites within the peptide core would provide complementary non-covalent interactions. This synergistic ion-pairing/hydrogen-bonding network generates a chiral pocket that directs the stereoselective nucleophilic substitution of the chloride group in prochiral cage 169 by naphthol (via key intermediate int-64), enabling highly effective stereocontrol. Additionally, an enantiomer-selective (M-selective) kinetic resolution cycle involving intermediate int-65 further enhanced the enantioselectivity. Notably, the versatility of these stereogenic-at-cage building blocks was further highlighted by their application in the late-stage diversification of drug molecule skeletons. | ||
| Scheme 54 Cationic foldamer-catalyzed enantioselective synthesis of inherently chiral cages via desymmetrization. | ||
7. Summary and outlook
Inherently chiral scaffolds constitute a structurally unique class of building blocks that are increasingly encountered in modern functional materials, medicinal chemistry, supramolecular catalysts, molecular recognition and self-assembly. Driven by persistent demand, significant efforts have been focused on the development of novel enantioselective synthetic strategies. In line with advances regarding enantioselective catalysis, methodologies have evolved from chiral separation techniques and stoichiometric chiral auxiliary-based approaches toward efficient catalytic protocols. Consequently, the catalytic asymmetric synthesis of inherently chiral scaffolds has undergone remarkable development, emerging as one of the rapidly evolving frontiers in synthetic chemistry.Numerous efficient catalytic strategies, stemming from biocatalysis, transition-metal catalysis and organocatalysis, have been established, enabling access to diverse, value-added, and previously inaccessible enantiomerically pure inherently chiral architectures. These include heteracalix[4](het)arenes, rigid saddle-shaped medium-sized rings, and prism-like cages. Such progress offers valuable insights into designing novel catalytic asymmetric systems, expanding structural diversity and functional utility. Nevertheless, this field remains in its nascent stage. Moving forward, innovative research focusing on the following suggested directions holds considerable promise for accelerating the exploration of inherently chiral scaffolds in both academia and industry.
Firstly, practical access to potentially useful inherently chiral frameworks is often hampered by the limited substrate scope of contemporary synthetic methods. The vast majority of transformations rely on highly reactive, specifically designed precursors, constraining the accessible range of functionalized chiral scaffolds. Furthermore, catalytic asymmetric syntheses have been successfully developed for only a select few classes of inherently chiral systems to date, notably calix[4]arenes, seven- and eight-membered saddle-shaped scaffolds, mechanically interlocked rotaxanes, and prism-like cages. In contrast, significant synthetic challenges remain for other important classes, including calix[n]arenes (n = 3, 5, and 6),67,113 additional rigid saddle-shaped medium-sized rings, mechanically interlocked catenanes and knots,19,114,115 and other shape-persistent molecular cages. Expanding into these underrepresented architectures is imperative for exploring uncharted chemical space. Secondly, while inherent chirality arises from the overall spatial arrangement and intrinsic geometry of the entire molecular framework, it can often be generated through the enantioselective transformation of specific local regions. Consequently, established general catalytic enantioselective strategies that have been successful toward controlling classical chiral elements (central, axial,116–118 planar,119,120 and helical chirality121) may provide a valuable foundation for designing novel catalytic asymmetric syntheses of complex inherently chiral scaffolds. Building upon these proven methodologies will not only facilitate access to such unique architectures but may also reveal novel stereocontrol mechanisms. Finally, leveraging the power of computational and data-driven tools, such as artificial intelligence,122 machine learning,123 and advanced computational modeling, allows us to identify key factors governing conformational stability and characterizing rotational energy barriers in inherently chiral scaffolds. Such efforts may enable quantitative mapping of structure–chirality relationships, thus providing conceptual frameworks to guide the rational design of novel chemical transformations.
By systematically summarizing and discussing the advancements in this rapidly evolving field, this overview aims to inspire the development of conceptually novel catalytic strategies. The methodologies highlighted herein thus provide a fertile foundation for future innovation. Looking ahead, the pursuit of more efficient and truly versatile enantioselective catalytic methods is promising. By addressing current limitations, the field is poised to unlock access to hitherto unseen inherently chiral scaffolds. The development of such novel architectures and chemical space holds significant potential for applications in areas including medicinal chemistry, advanced functional materials, asymmetric synthesis, and molecular recognition and assembly.
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Acknowledgements
The authors gratefully acknowledge the financial support provided by the Key Science and Technology Project of Henan Province [Grant No. 252102310415], the Cultivation Fund of Huanghuai University for National Scientific Research Project (XKPY-2023012), and the Seeking Truth Talent Project of Hangzhou Medical College.Notes and references
- K. Mislow, Introduction to stereochemistry, W. A. Benjamin Publisher, New York, 1965 Search PubMed.
- K. Mislow and M. Raban, Top. Stereochem., 1967, 1, 1–38 CrossRef CAS.
- K. Mislow and J. Siegel, J. Am. Chem. Soc., 1984, 106, 3319–3328 CrossRef CAS.
- K. Mislow, Croat. Chem. Acta, 1996, 69, 485–511 CAS.
- V. Böhmer, D. Kraft and M. Tabatabai, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 19, 17–39 CrossRef.
- A. Dalla Cort, L. Mandolini, C. Pasquini and L. Schiaffino, New J. Chem., 2004, 28, 1198–1199 RSC.
- A. Szumna, Chem. Soc. Rev., 2010, 39, 4274–4285 RSC.
- Y. Zheng and J. Luo, J. Incl. Phenom. Macro., 2011, 71, 35–56 CrossRef CAS.
- G. E. Arnott, Chem. – Eur. J., 2018, 24, 1744–1754 CrossRef CAS PubMed.
- P. Lhoták, RSC Adv., 2024, 14, 23303–23321 RSC.
- W. Qin and G. Cera, Chem. Rec., 2025, e202400237 CrossRef CAS.
- P. J. Cragg and K. Sharma, Chem. Soc. Rev., 2012, 41, 597–607 RSC.
- K. Kato, S. Fa and T. Ogoshi, Angew. Chem., Int. Ed., 2023, 62, e202308316 CrossRef CAS PubMed.
- T. Ogoshi, T. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed.
- X. Zhou and W. Wang, Chem. – Eur. J., 2025, 31, e202501185 CrossRef CAS PubMed.
- J. Han, J. Chen, X. Li, X. Peng and H. N. C. Wong, Synlett, 2013, 2188–2198 CrossRef CAS.
- Y. Luo, S. Luo and Q. Zhu, J. Org. Chem., 2025, 90, 5307–5322 CrossRef CAS PubMed.
- E. M. G. Jamieson, F. Modicom and S. M. Goldup, Chem. Soc. Rev., 2018, 47, 5266–5311 RSC.
- S. M. Goldup, Acc. Chem. Res., 2024, 57, 1696–1708 CrossRef CAS PubMed.
- H. Yang, H. Feng, J. Chen and L. Zhou, Chem. – Eur. J., 2025, 31, e202500898 CrossRef CAS PubMed.
- K. E. Jelfs, X. Wu, M. Schmidtmann, J. T. A. Jones, J. E. Warren, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2011, 50, 10653–10656 CrossRef CAS.
- Y. Chen, G. Wu, B. Chen, H. Qu, T. Jiao, Y. Li, C. Ge, C. Zhang, L. Liang, X. Zeng, X. Cao, Q. Wang and H. Li, Angew. Chem., Int. Ed., 2021, 60, 18815–18820 CrossRef CAS PubMed.
- T. M. Fukunaga, T. Katoa, K. Ikemoto and H. Isobe, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2120160119 CrossRef CAS.
- C. V. Maftei, E. Fodor, P. G. Jones, M. H. Franz, C. M. Davidescu and I. Neda, Pure Appl. Chem., 2015, 87, 415–439 CrossRef CAS.
- G. Sachdeva, D. Vaya, C. M. Srivastava, A. Kumar, V. Rawat, M. Singh, M. Verma, P. Rawat and G. K. Rao, Coord. Chem. Rev., 2022, 472, 214791 CrossRef CAS.
- O. Santoro and C. Redshaw, Coord. Chem. Rev., 2021, 448, 214173 CrossRef CAS.
- P. Eveleigh, E. C. Hulme, C. Schudt and N. J. Birdsall, Mol. Pharmacol., 1989, 35, 477–483 CrossRef CAS PubMed.
- H. R. Chobanian, Y. Guo, P. Liu, T. J. Lanza, M. Chioda, L. Chang, T. M. Kelly, Y. Kan, O. Palyha, X. Guan, D. J. Marsh, J. M. Metzger, K. Raustad, S. Wang, A. M. Strack, J. N. Gorski, R. Miller, J. Pang, K. Lyons, J. Dragovic, J. G. Ning, W. A. Schafer, C. J. Welch, X. Gong, Y. Gao, V. Hornak, M. L. Reitman, R. P. Nargund and L. S. Lin, Biorg. Med. Chem., 2012, 20, 2845–2849 CrossRef CAS PubMed.
- M. Tang and X. Yang, Eur. J. Org. Chem., 2023, e202300738 CrossRef CAS.
- J. K. Browne, M. A. McKervey, M. Pitarch, J. A. Russell and J. S. Millership, Tetrahedron Lett., 1998, 39, 1787–1790 CrossRef CAS.
- C. Lu, Q. Xu, J. Feng and R. Liu, Angew. Chem., Int. Ed., 2023, 62, e202216863 CrossRef CAS PubMed.
- J. Feng, L. Xi, C. Lu and R. Liu, Chem. Soc. Rev., 2024, 53, 9560–9581 RSC.
- K. Ishibashi, H. Tsue, H. Takahashi and R. Tamura, Tetrahedron: Asymmetry, 2009, 20, 375–380 CrossRef CAS.
- S. Tong, J. T. Li, D. D. Liang, Y. E. Zhang, Q. Y. Feng, X. Zhang, J. Zhu and M. X. Wang, J. Am. Chem. Soc., 2020, 142, 14432–14436 CrossRef CAS.
- G. Hedouin, F. Hazra, S. Gallou and S. Handa, ACS Catal., 2022, 12, 4918–4937 CrossRef CAS.
- Y. Jiang, S. Tong, J. Zhu and M. Wang, Chem. Sci., 2024, 15, 12517–12522 RSC.
- Y. Z. Zhang, M. M. Xu, X. G. Si, J. L. Hou and Q. Cai, J. Am. Chem. Soc., 2022, 144, 22858–22864 CrossRef CAS PubMed.
- X. Zhang, S. Tong, J. P. Zhu and M. X. Wang, Chem. Sci., 2023, 14, 827–832 RSC.
- Y. You, H. Cheng, X. Huang, P. Wang, H. Cong, H. Cheng and Q. Zhou, ChemRxiv, 2024, preprint DOI:10.26434/chemrxiv-2024-d2zh7-v2.
- Y. Yu, J. M. Yang and J. Rebek, Chem., 2020, 6, 1265–1274 CAS.
- I. Martín-Torres, G. Ogalla, J. Yang, A. Rinaldi and A. M. Echavarren, Angew. Chem., Int. Ed., 2021, 60, 9339–9344 CrossRef PubMed.
- M. Li, C. K. S. Ho, I. K. W. On, V. Gandon and Y. Zhu, Chem, 2024, 10, 3323–3341 CAS.
- G. Giovanardi, G. Scarica, V. Pirovano, A. Secchi and G. Cera, Org. Biomol. Chem., 2023, 21, 4072–4083 RSC.
- Q. Yao and B. Shi, Acc. Chem. Res., 2025, 58, 971–990 CrossRef CAS PubMed.
- T. Li, Y. Zhang, C. Du, D. Yang, M. Song and J. Niu, Nat. Commun., 2024, 15, 7673 CrossRef CAS PubMed.
- P. Qian, G. Zhou, J. Hu, B. Wang, A. Jiang, T. Zhou, W. Yuan, Q. Yao, J. Chen, K. Kong and B. Shi, Angew. Chem., Int. Ed., 2024, 63, e202412459 CrossRef CAS PubMed.
- X. Chang, Q. Zhang and C. Guo, Angew. Chem., Int. Ed., 2020, 59, 12612–12622 CrossRef CAS PubMed.
- C. Ma, J. Guo, S. Xu and T. Mei, Acc. Chem. Res., 2025, 58, 399–414 CrossRef CAS PubMed.
- X. Jiang, C. Zou, W. Zhuang, R. Li, Y. Yang, C. Yang, X. Xu, L. Zhang, X. He, Y. Yao, X. Sun and W. (Walter) Hu, Green Chem., 2025, 27, 915–945 RSC.
- L. Zhang, C. Yang, X. Wang, T. Yang, D. Yang, Y. Dou and J. Niu, Green Chem., 2024, 26, 10232–10239 RSC.
- Y. Mao, Y. Zhang, S. Tong, J. Zhu and M. Wang, Org. Lett., 2023, 25, 3936–3940 CrossRef CAS PubMed.
- R. Maji, S. C. Mallojjala and S. E. Wheeler, Chem. Soc. Rev., 2018, 47, 1142–1158 RSC.
- E. I. Jiménez, Org. Biomol. Chem., 2023, 21, 3477–3502 RSC.
- I. O. Betinol, Y. Kuang, B. P. Mulley and J. P. Reid, Chem. Rev., 2025, 125, 4184–4286 CrossRef CAS PubMed.
- X. Li, Y. Cheng, X. Wang, S. Tong and M. Wang, Chem. Sci., 2024, 15, 3610–3615 RSC.
- X. Li, Y. Cheng, R. Jian, S. Tong, Y. Xia and M. Wang, Org. Lett., 2025, 27, 4603–4608 CrossRef CAS PubMed.
- R. Ibragimova, Q. Lu, M. Wang, J. Zhu and S. Tong, Chem. – Asian J., 2025, 20, e202401621 CrossRef CAS PubMed.
- J. Clerigué, M. T. Ramos and J. C. Menéndez, Org. Biomol. Chem., 2022, 20, 1550–1581 RSC.
- B. C. Lemos, E. V. Filho, R. G. Fiorot, F. Medici, S. J. Greco and M. Benaglia, Eur. J. Org. Chem., 2022, e202101171 CrossRef CAS.
- W. Xie, J. Zhou, W. Liu, T. Qin and X. Yang, Cell Rep. Phy. Sci., 2024, 5, 101993 CrossRef CAS.
- Y. Jiang, Y. Tian, J. Feng, H. Zhang, L. Wang, W. Yang, X. Xu and R. Liu, Angew. Chem., Int. Ed., 2024, 63, e202407752 CrossRef CAS PubMed.
- S. Yu, M. Yuan, W. Xie, Z. Ye, T. Qin, N. Yu and X. Yang, Angew. Chem., Int. Ed., 2024, 63, e202410628 CrossRef CAS PubMed.
- M. Yuan, W. Xie, S. Yu, T. Liu and X. Yang, Nat. Commun., 2025, 16, 3943 CrossRef CAS PubMed.
- X. Zhang, D. Zhu, R. Cao, Y. Huo, T. Ding and Z. Chen, Nat. Commun., 2024, 15, 9929 CrossRef CAS PubMed.
- V. Dočekal, L. Lóška, A. Kurčina, I. Císařová and J. Veselý, Nat. Commun., 2025, 16, 4443 CrossRef PubMed.
- H. Han, X. Wang, S. Tong, J. Zhu and M. Wang, ACS Catal., 2025, 15, 6018–6024 CrossRef CAS.
- Y. Inaba, J. Yang, Y. Kakibayashi, T. Yoneda, Y. Ide, Y. Hijikata, J. Pirillo, R. Saha, J. L. Sessler and Y. Inokuma, Angew. Chem., Int. Ed., 2023, 62, e202301460 CrossRef CAS PubMed.
- Q. Lu, X. Wang, S. Tong, J. Zhu and M. Wang, ACS Catal., 2024, 14, 5140–5146 CrossRef CAS.
- Z. Li, J. Zhang, W. Zhu, T. Wang, Y. Tang and J. Wang, Chem. Sci., 2025, 16, 11021–11026 RSC.
- H. Zhu, Q. Li, B. Shi, H. Xing, Y. Sun and S. Lu, J. Am. Chem. Soc., 2020, 142, 17340–17345 CrossRef CAS PubMed.
- J. F. Chen, Q. X. Gao, L. Liu, P. Chen and T. B. Wei, Chem. Sci., 2023, 14, 987–993 RSC.
- H. Zhu, L. Chen, B. Sun, M. Wang, H. Li, J. F. Stoddart and F. Huang, Nat. Rev. Chem., 2023, 7, 768–782 CrossRef CAS PubMed.
- K. Wada and T. Ogoshi, Mater. Chem. Front., 2024, 8, 1212–1229 RSC.
- H. Liang, B. Hua, F. Xu, L. S. Gan, L. Shao and F. Huang, J. Am. Chem. Soc., 2020, 142, 19772–19778 CrossRef CAS PubMed.
- Y. Sun, L. Liu, L. Jiang, Y. Chen, H. Zhang, X. Xu and Y. Liu, J. Am. Chem. Soc., 2023, 145, 16711–16717 CrossRef CAS PubMed.
- X. Zhou, X. Zhang, Y. Song, X. Li, L. Bao, W. Xu, X. Wang, H. Yang and W. Wang, Angew. Chem., Int. Ed., 2025, 64, e202415190 CrossRef CAS PubMed.
- T. Luan, C. Sun, Y. Tian, Y. Jiang, L. Xi and R. Liu, Nat. Commun., 2025, 16, 2370 CrossRef CAS PubMed.
- A. Konter, J. Rostoll-Berenguer, C. Besnard, B. Leforestier and C. Mazet, ACS Catal., 2025, 15, 2607–2619 CrossRef CAS.
- Y. Zhang, Y. Jiang, Y. Zhang, H. Wang, Z. Wu, R. Li, Y. Ma and T. Tu, Chin. Chem. Lett., 2025, 36, 111201 CrossRef CAS.
- W. D. G. Brittain, B. R. Buckley and J. S. Fossey, ACS Catal., 2016, 6, 3629–3636 CrossRef CAS.
- C. Qin, C. Zhao, G. Chen and Y. Liu, ACS Catal., 2023, 13, 6301–6311 CrossRef CAS.
- W. Zhou, L. Xi, M. Zhang, H. Wang, M. An, J. Li and R. Liu, Angew. Chem., Int. Ed., 2025, 64, e202502381 CrossRef CAS PubMed.
- T. Shibata, T. Chiba, H. Hirashima, Y. Ueno and K. Endo, Angew. Chem., Int. Ed., 2009, 48, 8066–8069 CrossRef CAS PubMed.
- T. Shibata, T. Uchiyama, H. Hirashima and K. Endo, Pure Appl. Chem., 2011, 83, 597–605 CAS.
- Y. Tahara, R. Matsubara, A. Mitake, T. Sato, K. S. Kanyiva and T. Shibata, Angew. Chem., Int. Ed., 2016, 55, 4552–4556 CrossRef CAS PubMed.
- T. Shibata, M. Nakada, K. Senda and M. Ito, Chem. – Eur. J., 2025, 31, e202501193 CrossRef CAS PubMed.
- C. Qian, H. Yan, J. Li, Z. Zhang, Z. An and B. Xiao, Org. Lett., 2025, 27, 4118–4123 CrossRef CAS PubMed.
- Y. Luo, S. Cheng, Y. Peng, X. Wang, J. Li, C. Gan, S. Luo and Q. Zhu, CCS Chem., 2022, 4, 2897–2905 CrossRef CAS.
- X. Wang, C. Wang, Y. Luo, J. Li, C. Gan, S. Luo and Q. Zhu, Chem. Catal., 2024, 4, 100904 CAS.
- H. Zhang, C. Lu, G. Cai, L. Xi, J. Feng and R. Liu, Nat. Commun., 2024, 15, 3353 CrossRef CAS PubMed.
- P. R. Singh, A. Banerjee and A. K. Simlandy, ACS Catal., 2025, 15, 3096–3115 CrossRef CAS.
- M. Zhang, H. Wang, H. Shan, L. Xi, C. Lu, X. Du, C. Sun, L. Xu and R. Liu, Nat. Commun., 2025, 16, 2505 CrossRef CAS PubMed.
- J. Li, X. Li, J. Feng, W. Yao, H. Zhang, C. Lu and R. Liu, Angew. Chem., Int. Ed., 2024, 63, e202319289 CrossRef CAS PubMed.
- Y. Luo, X. Wang, W. Hu, Y. Peng, C. Wang, T. Yu, S. Cheng, J. Li, Y. He, C. Gan, S. Luo and Q. Zhu, CCS Chem., 2023, 5, 982–993 CrossRef CAS.
- J. Zhou, M. Tang and X. Yang, Chin. J. Chem., 2024, 42, 1953–1959 CrossRef CAS.
- D. Zhang, J. Zhou, T. Qin and X. Yang, Chem. Catal., 2024, 4, 100827 CAS.
- C. Guan, S. Zou, C. Luo, Z. Li, M. Huang, L. Huang, X. Xiao, D. Wei, M. Wang and G. Mei, Nat. Commun., 2024, 15, 4580 CrossRef CAS PubMed.
- D. Xu, G. Zhou, B. Liu, S. Jia, Y. Liu and H. Yan, Angew. Chem., Int. Ed., 2025, 64, e202416873 CrossRef CAS PubMed.
- N. Tampellini, B. Q. Mercado and S. J. Miller, J. Am. Chem. Soc., 2025, 147, 4624–4630 CrossRef CAS PubMed.
- S. Shi, C. Cui, L. Xu, J. Zhang, W. Hao, J. Wang and B. Jiang, Nat. Commun., 2024, 15, 8474 CrossRef CAS PubMed.
- L. Wei, Y. Chen, Q. Zhou, Z. Wei, T. Tu, S. Ren, Y. R. Chi, X. Zhang and X. Yang, J. Am. Chem. Soc., 2025, 147, 30747–30756 CrossRef CAS PubMed.
- A. W. Heard and S. M. Goldup, ACS Cent. Sci., 2020, 6, 117–128 CrossRef CAS PubMed.
- J. R. J. Maynard and S. M. Goldup, Chem., 2020, 6, 1914–1932 CAS.
- J. P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080–11093 CrossRef CAS PubMed.
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed.
- D. A. Leigh, Angew. Chem., Int. Ed., 2016, 55, 14506–14508 CrossRef CAS PubMed.
- Y. Makita, N. Kihara, N. Nakakoji, T. Takata, S. Inagaki, C. Yamamoto and Y. Okamoto, Chem. Lett., 2007, 36, 162–163 CrossRef CAS.
- M. Alvarez-Pérez, S. M. Goldup, D. A. Leigh and A. M. Z. Slawin, J. Am. Chem. Soc., 2008, 130, 1836–1838 CrossRef PubMed.
- A. Imayoshi, B. V. Lakshmi, Y. Ueda, T. Yoshimura, A. Matayoshi, T. Furuta and T. Kawabata, Nat. Commun., 2021, 12, 404 CrossRef CAS PubMed.
- M. Li, X. L. Chia, C. Tian and Y. Zhu, Chem, 2022, 8, 2843–2855 CAS.
- S. Fang, Z. Liu and T. Wang, Angew. Chem., Int. Ed., 2023, 62, e202307258 CrossRef CAS PubMed.
- S. Fang, Z. Bao, Z. Liu, Z. Wu, J. Tan, X. Wei, B. Li and T. Wang, Angew. Chem., Int. Ed., 2024, 63, e202411889 CrossRef CAS.
- G. Sachdeva, D. Vaya, C. M. Srivastava, A. Kumar, V. Rawat, M. Singh, M. Verma, P. Rawat and G. K. Rao, Coord. Chem. Rev., 2022, 472, 214791 CrossRef CAS.
- R. Jamagne, M. J. Power, Z. Zhang, G. Zango, B. Gibbera and D. A. Leigh, Chem. Soc. Rev., 2024, 53, 10216–10252 RSC.
- L. Wu, M. Tang, L. Jiang, Y. Chen, L. Bian, J. Liu, S. Wang, Y. Liang and Z. Liu, Nat. Synth., 2023, 2, 17–25 CrossRef CAS.
- G. Liao and B. Shi, Acc. Chem. Res., 2025, 58, 1562–1579 CrossRef CAS PubMed.
- T. A. Schmidt, V. Hutskalova and C. Sparr, Nat. Rev. Chem., 2024, 8, 497–517 CrossRef PubMed.
- S. Xiang, W. Ding, Y. Wang and B. Tan, Nat. Catal., 2024, 7, 483–498 CrossRef CAS.
- G. Yang and J. Wang, Angew. Chem., Int. Ed., 2024, 63, e202412805 CAS.
- K. Zhu, L. Yang, Y. Yang, Y. Wu and F. Zhang, Chin. Chem. Lett., 2025, 36, 110678 CrossRef CAS.
- Y. Wang, Z. Wu and F. Shi, Chem. Catal., 2022, 2, 3077–3111 CAS.
- A. M. Mroz, A. R. Basford, F. Hastedt, I. S. Jayasekera, I. Mosquera-Lois, R. Sedgwick, P. J. Ballester, J. D. Bocarsly, E. A. R. Chanona, M. L. Evans, J. M. Frost, A. M. Ganose, R. L. Greenaway, K. K. (Mimi) Hii, Y. Li, R. Misener, A. Walsh, D. Zhang and K. E. Jelfs, Chem. Soc. Rev., 2025, 54, 5433–5469 RSC.
- J. Xia, Y. Zhang and B. Jiang, Chem. Soc. Rev., 2025, 54, 4790–4821 RSC.
| This journal is © The Royal Society of Chemistry 2025 |