
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScanning probe microscopy of metal–organic coordination systems: characterization of monolayers, single crystals, discrete architectures
Antonino
Cucinotta
 ,
Jack A.
Davies
,
Kunal S.
Mali
* and
Steven
De Feyter
*
,
Jack A.
Davies
,
Kunal S.
Mali
* and
Steven
De Feyter
*
Division of Molecular Imaging and Photonics, Department of Chemistry, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: kunal.mali@kuleuven.be; steven.defeyter@kuleuven.be
First published on 2nd October 2025
Abstract
Metal–organic coordination is omnipresent in a number of functional materials. Such systems are highly diverse in terms of their composition, complexity, and dimensionality. They include two-dimensional (2D) and three-dimensional (3D) structures ranging from monolayers of metal–organic coordination networks (MOCNs) physisorbed on solid surfaces, to crystalline metal–organic frameworks (MOFs), to discrete metallosupramolecular architectures (DMSAs). The use of these metal–organic materials in a wide variety of applications has been demonstrated, showing promise for their incorporation into emerging technologies. Several design strategies have been developed for the fabrication of MOCNs, MOFs, and DMSAs exhibiting a diverse array of structures, enabling precise control over their functional properties. As these strategies are designed at the molecular level, there has been considerable interest in the nanoscale resolution imaging of metal–organic coordination systems across different length scales. This review provides a glimpse of recent progress in the nanoscale characterization of metal–organic coordination systems using scanning probe microscopy (SPM). Systems ranging from surface-confined MOCN monolayers, to MOF thin films, surfaces of MOF single crystals, and DMSAs are discussed. Specifically, we discuss the contribution of scanning tunneling microscopy (STM), atomic force microscopy (AFM), and techniques that combine SPM with spectroscopic methods to obtain high-resolution chemical information, toward the nanoscale structural characterization of multinuclear metal–organic assemblies.
 Antonino Cucinotta | Antonino Cucinotta obtained his MSc in chemistry at the Università degli Studi di Messina (Italy) in 2020. In 2025, he obtained his PhD at KU Leuven (Belgium), under the supervision of Prof. Steven De Feyter and Dr. Kunal S. Mali. His doctoral research focused on the development of new strategies for the on-surface fabrication of metal–organic coordination networks and metal–organic frameworks and the investigation of their formation, growth, structure, and properties in two and three dimensions using scanning probe microscopy at the liquid–solid interface. He is currently a postdoctoral researcher in the De Feyter group. |
 Jack A. Davies | Jack A. Davies completed his PhD in 2022, at the University of Cambridge, UK. This doctoral work, under the supervision of Prof. Jonathan R. Nitschke, elucidated design principles underpinning the formation of discrete metal–organic architectures. Since joining the group of Prof. Steven De Feyter at KU Leuven in 2023, his research has focused on investigating the local structure in extended two-dimensional metal–organic coordination networks, primarily employing scanning probe microscopy to do so. |
 Kunal S. Mali | Kunal S. Mali obtained his PhD in 2008 from the University of Mumbai in India, while working at the Bhabha Atomic Research Centre, where he investigated polymer colloids and ionic liquids using time-resolved fluorescence spectroscopy. He then joined the group of Prof. Steven De Feyter at KU Leuven as a postdoctoral researcher, focusing on molecular self-assembly on surfaces studied by scanning tunneling microscopy. His current research interests span covalent and supramolecular chemistry at solid surfaces, explored using advanced scanning probe microscopy techniques with high spatial and chemical resolution. He is presently a research manager at KU Leuven. |
 Steven De Feyter | Steven De Feyter is a professor at KU Leuven, Belgium. He earned his PhD at KU Leuven in 1997 and subsequently pursued postdoctoral research at Caltech. His team explores both non-covalent, via self-assembly, and covalent modification of graphite and 2D materials, including graphene and transition metal dichalcogenides. A central focus of the work is the on-surface synthesis of covalent organic frameworks (COFs) and metal–organic frameworks (MOFs). At the core of the research lies high-resolution scanning probe microscopy, which enables real-space nanoscale characterization, with particular emphasis on processes occurring at the liquid–solid interface. |
1. Introduction
The coordination bond represents a highly versatile interaction, playing a pivotal role across various domains. The coordination of organic ligands to metal ions is ubiquitous in natural systems, with numerous proteins employing metal binding to perform diverse functions such as structural support, storage, electron transfer, dioxygen binding, and catalytic activity.1,2 However, metal ions are also prevalent pollutants in natural environments, and their complexation by organic ligands serves as an effective strategy for sequestration.3–5Over the past decades, the field of coordination chemistry has advanced significantly from the study of simple metal complexes, and coordination bonds have been incorporated into the design and synthesis of novel materials. Directionality is a key characteristic of coordination bonds, and the specific coordination geometries of d- and f-block metals enable the construction of scaffolds with well-defined architectures. By combining metal ions or clusters with a diverse array of organic ligands, a wide variety of structures can be formed with both extended and discrete structures. Examples of such structures include metal–organic frameworks (MOFs), surface-confined metal–organic coordination networks (MOCNs), and discrete metallosupramolecular architectures (indicated as DMSAs in this review).
MOFs are self-assembled polymeric coordination compounds consisting of metal ions or clusters connected by organic ligands, exhibiting exceptional porosity and tunable pore sizes.6–13 Although early reports on coordination polymers date back to the mid-20th century, the synthesis of the first crystalline MOFs in the late 1980s and 1990s marked a major breakthrough in coordination chemistry.12,13 This milestone laid the foundation for reticular chemistry, a field dedicated to the design of porous crystalline materials stabilized by strong bonds between molecular building blocks.14 Reticular chemistry also includes covalent organic frameworks (COFs), which are structurally analogous to MOFs but consist entirely of organic components linked by covalent bonds.15 Hydrogen-bonded organic frameworks (HOFs) represent porous reticular frameworks comprising ordered arrangements of organic linkers connected via hydrogen bonds, although they frequently derive additional stability from other complementary non-covalent interactions.16 In contrast to these designed reticular frameworks, zeolites are purely inorganic porous materials, which are characterized by relatively small pore sizes (<2 nm).17
A wide range of MOFs has been synthesized through the strategic combination of various metal centers and organic linkers.8 Notably, d-block transition metals offer predictable coordination geometries dictated by their electronic configuration, which result in a diverse array of framework geometries. This structural diversity is further expanded with the incorporation of f-block elements, particularly lanthanides, which typically form coordination complexes with high coordination numbers. These lanthanide-based complexes are generally labile, and their coordination chemistry is considered to be predominantly governed by electrostatic interactions.
In coordination polymers, the metal centers are typically referred to as primary building units (PBUs), which are linked by organic ligands to form the material's scaffold. Secondary building units (SBUs) are metal-containing clusters or coordination complexes that serve as nodes in the extended framework, connecting to organic linkers with a predictable geometry.18–20 The connectivity within the SBUs provides a stable scaffold, enhancing both the thermal and mechanical robustness of the overall network.19,20 Moreover, the use of SBUs in MOF design and synthesis enables the predictability of the final structure due to their well-defined geometry and connectivity.18,19
MOF classification may take into account several structural features, such as the dimensionality of the SBUs or of the crystals, among others.20,21 For the scope of this review, it is convenient to classify MOFs according to the connectivity of the building blocks. Therefore, MOFs which exhibit coordination bonds that extend in two dimensions are categorized as two-dimensional (2D) MOFs, while those with a periodic arrangement of coordination bonds throughout all three spatial dimensions are classified as three-dimensional (3D) MOFs (Scheme 1).8,10,22,23 MOFs can be fabricated as thin films with thicknesses ranging from several to a few hundred nanometers. Some 2D MOF thin films exhibit characteristic layered structures.22,24
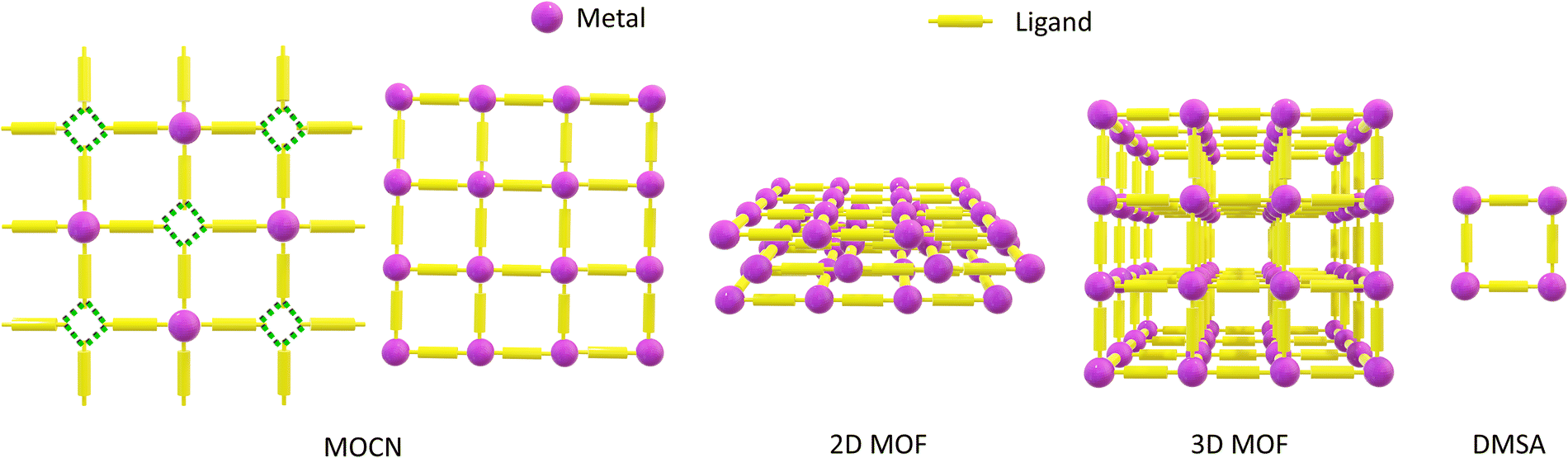 | ||
| Scheme 1 Schematic representation of the classification of metal–organic coordination systems discussed in this review. MOCNs: in this review, they include surface-supported 2D coordination networks stabilized by other directional supramolecular interactions, indicated by the green dashed lines, in combination with metal–ligand coordination. A porous MOCN featuring only coordination bonds within its structure can be conceptually regarded as a 2D MOF monolayer. 2D MOFs: MOFs exhibiting coordination bonds extending in two dimensions. 3D MOFs: MOFs exhibiting coordination bonds extending in three dimensions. Both 2D and 3D MOFs can be fabricated in the form of thin films, with the former presenting characteristic layered structures. DMSA: coordination compounds containing a specific number of metal ions and organic ligands that do not have extended structures. | ||
The uniform pore distribution, exceptional surface area, and incorporation of metal centers account for the extensive applications of MOFs in therapeutics and catalysis,25–27 as well as in gas storage,26,28,29 separation processes,26,30,31 sensing technologies,26 and charge transport and storage.26,32–34 When deposited onto solid surfaces, MOF thin films are more easily integrated into functional devices.35–37
MOFs are the porous subclass of coordination networks.6 However, in this review, the term MOCNs is used in reference to surface-confined metallosupramolecular monolayers extending in 2D (Scheme 1).38 We include in this definition 2D assemblies held together purely by metal–ligand coordination, as well as those held together by coordination bonds in combination with other directional interactions such as hydrogen bonding. MOCNs are examples of self-assembled molecular networks (SAMNs)—surface-confined supramolecular architectures consisting of individual molecules held together by various supramolecular interactions. While a variety of SAMNs containing coordination bonds in their scaffold has been reported, the term MOCN is mainly used specifically in reference to SAMNs featuring metal coordination in two dimensions.39 Surface-confined MOCNs featuring porous structures are indicated as porous MOCNs.
A distinctive feature of surface-confined MOCNs is their propensity toward planar coordination geometries, potentially resulting in coordination sites at metal centers that remain either unoccupied or occupied by labile ligands, such as solvent molecules.38 These open reactive sites can be utilized for catalytic or sensing applications, leveraging the unique electronic and magnetic properties of these materials.40–43 Additionally, given their crystalline nature, surface patterning with MOCNs presents a convenient method for templating the supramolecular organization of guest molecules in periodic arrangements.44
We distinguish between MOFs and MOCNs due to the structural differences arising from surface confinement. Crystals grown in 3D can be categorized into 230 3D space groups, whereas only 17 2D plane groups are possible for monolayered crystals.45 Additionally, substrate–adsorbate interactions can stabilize 2D structures that have no direct bulk counterparts, and analogous precursors can originate different structures from in-solution and on-surface processes. Furthermore, surface adsorption is associated with reduced symmetry in 2D crystals with consequences on the chirality of the assemblies, as will be discussed in the related sections. Therefore, surface-confined MOCNs are not necessarily the single-layered counterparts of 2D and 3D MOFs, and it is important to maintain a distinction between these two classes of materials.
Self-assembled materials that do not have extended structures can also be realized, including DMSAs (Scheme 1). Through the combination of carefully selected metal ions and ligands, discrete metal–organic architectures, consisting of a given number of metal ions and ligand units, can be selectively assembled, instead of extended frameworks.46–49
Given the inherent structural differences between MOFs, MOCNs, and DMSAs, these materials are studied using different techniques. The characterization of MOF morphology involves describing their crystal structure, size, shape, and porosity. To obtain a comprehensive understanding of MOF materials, a combination of complementary techniques is required.50–52 Due to their often crystalline nature, MOFs are typically characterized using X-ray diffraction techniques such as single-crystal X-ray diffraction (SC-XRD) and powder X-ray diffraction (PXRD). These techniques provide space- and time-averaged information about the unit cell of crystal samples with atomic resolution.53–55 Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) are valuable for investigating the shape of MOF particles. Additionally, the latter allows for nanometer or sub-nanometer resolution characterization on a particle-by-particle basis. This technique requires the samples to be typically under 200 nm thin and sufficiently resistant to the high-energy electron beam.56–58 The development of low-dose techniques and liquid cell TEM (LCTEM) holds promise for studying MOFs under dynamic conditions.59
Although the pore size can be calculated from the crystal structure determined by the aforementioned techniques, the accurate measurement of pore size distribution, surface area, and accessible volume is achieved by recording adsorption and desorption isotherms.60 Additionally, the hysteresis observed between adsorption and desorption isotherms can be an indirect indication of the occurrence of a reversible framework transformation upon guest adsorption, a characteristic feature of so-called third-generation MOFs.61,62
To elucidate the structure–function relationship in MOFs, examine their responsiveness to external stimuli, and directly visualize structural changes and phase transitions, utilizing local, spatially-resolved techniques is imperative. As previously discussed, LCTEM constitutes a promising methodological approach toward this objective. Ideally, minimally invasive analytical techniques that facilitate the characterization of material surfaces with molecular and sub-molecular resolution in real space would complement these TEM techniques.
As stated above, MOCNs are conceptually similar to MOFs. However, the aforementioned structural differences between the two classes of materials render structural characterization of MOCNs using analogous methods to those used for (bulk) MOFs challenging. Therefore, alternative techniques that enable molecular-resolution imaging of surface-confined monolayers are needed for investigating MOCNs.
It is common practice that the structures of DMSAs are elucidated using a combination of nuclear magnetic resonance (NMR) spectroscopy, mass spectrometry (MS), and SC-XRD.63 Solid-state structure determination by SC-XRD methods can become challenging when a metal–organic species exists as a mixture of distinct isomers. Techniques capable of structurally characterizing individual species in a mixture would thus be valuable in the thorough characterization of mixtures containing structurally different individual components.
As discussed, several challenges remain in the characterization of metal–organic systems, despite the wide range of techniques available. These include the need for nanoscale local characterization, also under dynamic experimental conditions and in the presence of external stimuli. Particularly in the case of MOCNs, the ability to image material surfaces with (sub)-molecular resolution is also required. To this end, scanning probe microscopy (SPM) offers a set of low-invasive, surface-sensitive characterization techniques that may provide complementary information to the insights obtained with the methods described above. SPM techniques also offer the capability to conduct imaging procedures in liquid environments and under dynamic experimental conditions.64,65
These techniques include scanning tunneling microscopy (STM) and atomic force microscopy (AFM). STM enables the analysis with (sub)-molecular resolution of single molecular species and extended single-layered (or few-layered) assemblies adsorbed on conductive surfaces.66,67 While AFM is also suitable for the study of atom-thick species on solid substrates, it is widely employed for the study of the surface of thicker samples, including thin films and nanoparticles.68–70 Both techniques can be operated under ultra-high vacuum (UHV) and ambient conditions (at the air–solid and liquid–solid interface), allowing for studies under dynamic conditions, as well as in the presence of external stimuli. In any instance, the firm immobilization of samples on solid substrates is essential for implementing SPM investigations.
Early works on the use of SPM for the characterization of metal–organic coordination systems date back to the early 2000s. Pivotal UHV-STM studies targeted the investigation of coordination complexes71,72 and MOCNs adsorbed on solid substrates,39,73 exploring ligand control strategies73 and designing chiral networks that incorporated coordination bonds.74 The fabrication of extended surface-confined MOCNs with large pore sizes demonstrated the stabilizing role exerted by solid substrates.75 Works targeting the STM characterization of coordination systems, including DMSAs, under ambient conditions emerged almost in parallel with UHV studies.76–78 However, MOCNs fabricated at the liquid–solid interface were reported 10 years later than UHV analogues,79 and only recently have in-depth investigations into the structural control and the design of chiral MOCNs at the liquid–solid interface been carried out.80,81 Initial AFM studies on MOF surfaces targeted the elucidation of nucleation and growth processes through the imaging of surface features such as defects and step heights.82,83 The use of functionalized substrates for MOF thin films and crystals growth was a significant contribution to the development of the field, as will be discussed in Section 4.84 In recent years, molecular-resolution AFM imaging of MOF surfaces was achieved,69 and the mechanism of complex surface dynamics was elucidated.68
This review examines the current status and methodological challenges associated with the characterization of MOCNs, MOFs, and DMSAs via SPM techniques, with particular emphasis on elucidating molecular structures, local features, and dynamic processes. Initially, the fundamental operating principles of the main SPM techniques will be briefly described (Section 2). Following this, a critical description of the characterization of MOCNs, MOFs, and DMSAs transversal to the length scale (monolayers, thin films, and crystal surfaces) performed using SPM will be provided. The experimental conditions used for materials preparation and the SPM techniques suitable for their characterization often vary based on the class of the material. In this review, categorization into three sections (Sections 3–5) is thus on the basis of material class. The characterization of MOCNs using STM in UHV and at the liquid–solid interface will be reviewed in Section 3. The characterization of MOF thin films and single crystals using AFM will then be discussed in Section 4. Finally, SPM characterization of self-assembled networks of DMSAs, and the use of SPM for the structural characterization of the metallosupramolecules themselves, will be addressed in Section 5. Throughout the review, the inherent challenges associated with achieving molecular-resolution imaging of MOCNs, MOFs, and DMSAs will be emphasized, and a perspective on the future developments in SPM characterization of these materials will be presented in the conclusions of this review.
2. SPM techniques
As discussed previously, MOFs, MOCNs, and DMSAs can be effectively characterized using SPM. STM and AFM have emerged as foundational SPM techniques for characterizing the aforementioned classes of materials. These techniques rely on the use of a physical probe that scans over the surface of the materials, providing information about their structure arising from the probe–sample interactions. One limitation of STM and AFM is their lack of chemical sensitivity, which can be overcome by combining them with spectroscopic techniques. In this regard, the combination of AFM with infrared spectroscopy (AFM-IR) has recently been applied to the study of MOFs.85 In this section, we present a concise overview of the key operational aspects of these analytical techniques that are relevant for the characterization of MOCNs, MOFs, and DMSAs. More detailed technical details can be found in the references contained in this section.2.1 Scanning tunneling microscopy
STM operates on the principle of quantum mechanical tunneling. In an STM setup (Scheme 2(a)), a sharp conducting tip is positioned very close (<1 nm) to a conducting substrate, and a bias voltage is applied between the two. Electrons tunnel through the gap, generating a measurable current. This tunneling current is exponentially dependent on the tip-sample distance, allowing for extraordinary vertical resolution down to picometer scales. The current is also proportional to the local density of states (LDOS) of the sample surface, enabling STM to provide information not only about the topography but also about the electronic structure of the sample.86–90 During STM measurements, the tip raster-scans the surface, as illustrated in Scheme 2(b), and a signal is recorded. In constant height mode, the tip-sample distance is maintained constant, and the variation in tunneling current is measured (Scheme 2(c)). In contrast, in constant current mode, the tip-sample distance varies to maintain the tunneling current constant (Scheme 2(d)). In scanning tunneling spectroscopy (STS), current–voltage measurements are performed to quantitatively study the electronic structure of the surface.91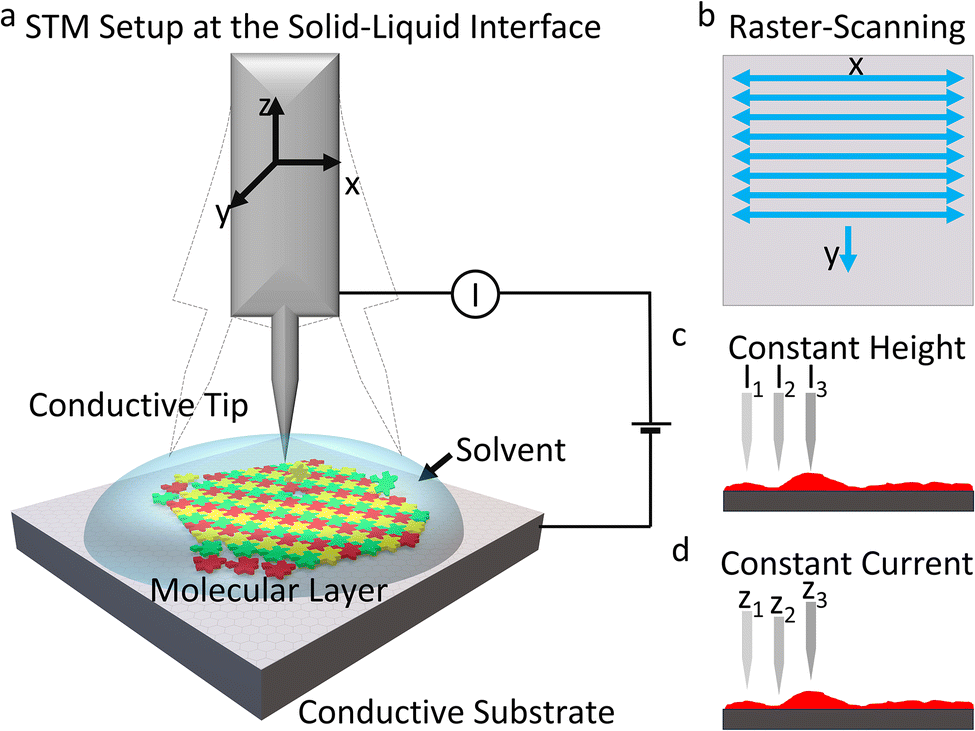 | ||
| Scheme 2 Representation of STM at the liquid–solid interface. (a) Machine setup. (b) Illustration of the tip movements. (c), (d) STM imaging modes. | ||
In STM performed at the liquid–solid interface, the solvent permittivity significantly influences the tunneling process. In polar solvents with high relative permittivity, such as water, an applied electric field induces molecular dipoles to orient parallel to the field, generating an induced electric field that opposes the external one. Furthermore, if the solvent is electrochemically active, the applied bias potential may trigger redox reactions that contribute to a background current, which can potentially dominate the tunneling current. Therefore, apolar solvents are selected, and low-volatile solvents are preferred to reduce solvent evaporation during imaging.92
Typical substrates employed in UHV processes include gold, copper, and silver surfaces, whereas gold and highly oriented pyrolytic graphite (HOPG) are preferred for studies conducted in air and at the liquid–solid interface due to their chemical inertness under ambient conditions.93
Nevertheless, MOFs are often fabricated using solution-based processes, in which the solvent is selected to enable control over crystal nucleation and growth, thereby determining the morphology of the products. MOCN fabrication and characterization at the liquid–solid interface offers the opportunity to gain molecular insights into the effect that different solvents play during the synthesis process directly in their growth solution. Moreover, as already stated, the liquid–solid interface is a dynamic environment, where in situ changes, including temperature-induced phase transitions and guest adsorption processes, can be monitored in real time for monolayers, thin films, and single crystals.
2.2 Atomic force microscopy
AFM overcomes STM's limitation of requiring conductive samples by measuring the forces between a sharp tip mounted on a cantilever and the sample surface. These interatomic forces, typically in the range of 10−8 to 10−9 N, follow the Lennard-Jones potential, and are attractive at longer distances due to van der Waals interactions and repulsive at shorter distances due to electrostatic repulsion and Pauli exclusion principle. As the tip raster-scans across the surface, cantilever deflections are detected by monitoring the reflection of a laser beam onto a position-sensitive photodiode (Scheme 3).88,89,94 | ||
| Scheme 3 Representation of the AFM setup operating in AM mode. Aexc represents the oscillation amplitude of the drive, whereas A is the oscillation amplitude of the cantilever. | ||
AFM offers multiple operational modes: contact mode, where the tip directly interacts with the surface in the repulsive regime; amplitude modulation (AM) mode, where the cantilever oscillation is driven at a constant excitation frequency near resonance and the amplitude changes are measured; and frequency modulation (FM) mode, which maintains the cantilever oscillation continuously at its resonance frequency through a phase-locked loop. Non-contact AFM (nc-AFM) is a special case of AFM that operates with sufficiently large amplitudes to avoid entering the repulsive regime.
AFM imaging at the liquid–solid interface enables in situ studies of dynamic processes, including crystallization, adsorption, and conformational changes in their native solution environments with molecular resolution.
2.3 Combining SPM with spectroscopy techniques
While not yet extensively employed, the use of AFM-IR in the field of metal–organic coordination systems is emerging. It represents a powerful integration of AFM with IR spectroscopy, overcoming the diffraction limit of conventional IR microscopy and the lack of chemical sensitivity of SPM. This technique often measures the local photothermal expansion caused by the absorption of mid-IR laser pulses, transduced by the AFM cantilever. When a wavelength-tunable IR laser pulse illuminates the sample region beneath the AFM tip, absorption leads to thermal expansion, causing the cantilever to oscillate with an amplitude proportional to the absorption coefficient (Scheme 4). Both “ringdown” detection (recording the cantilever oscillation after excitation by a single pulse) and resonance-enhanced detection (synchronizing the laser pulse repetition rate with a cantilever resonance frequency) are employed, with the latter dramatically enhancing sensitivity.95,96 Further operational details of AFM-IR, including discussion of the different available modes, have been recently reviewed.97 | ||
| Scheme 4 Illustration depicting AFM-IR. | ||
AFM-IR achieves chemical mapping with spatial resolution of 10–20 nm,95 far beyond the diffraction limit of conventional IR microscopy, enabling the nanoscale investigation of chemical composition, molecular conformation, and polymorphic states across diverse research fields including polymer science, biology, and materials science.
Another technique which can provide very high-resolution chemical information for metal–organic materials is synchrotron X-ray STM (SX-STM). In SX-STM, the STM tip acts as a detector for both photoejected electrons and the tunnelling current modulated by the absorption of X-rays.98
3. STM characterization of MOCNs
Surface-confined MOCN monolayers are typically obtained through the self-assembly of precursors in UHV conditions or at the liquid–solid interface.38,99 Several reviews have been published describing the various metal–organic architectures that can be fabricated on surfaces, detailing the variety of lattice geometries and electronic properties, investigated using mainly UHV-STM.38,100–105 Surfaces exert a significant influence on determining the outcome of the self-assembly process.75 In UHV, the formation of crystalline structures is governed by a balance between intermolecular and molecule–substrate interactions.Although rarer, MOCN synthesis and characterization at the liquid–solid interface have also been reported. Interactions between solvent molecules and the substrate, as well as between solvent and solute, actively modulate self-assembly processes at liquid–solid interfaces.93,106–110 The interrelated and occasionally competing intermolecular and molecule–substrate interactions, as well as interactions involving solvent or guest molecules, may facilitate the formation of distinct polymorphs on different substrates and/or when a different solvent is employed.107,108,111–113
In this section, we analyze the strategies for imparting structural control in MOCNs. Furthermore, investigation of substrate effect, phase transitions, stimuli-responsiveness, chirality, and network porosity will be discussed. While the selection of papers presented here does not aim to provide an exhaustive collection of works carried out on coordination systems on surfaces, it showcases how molecular-resolution imaging of local features achieved via STM characterization under UHV and at the liquid–solid interface provides unique insights into our understanding of MOCNs.
3.1 MOCNs fabricated and characterized in UHV
MOCN structures synthesized under UHV using different functional groups, including carboxylates, pyridyls, nitriles, and carbonyl-containing ligands, have been explored in the literature.114–118 Organic molecular beam deposition (OMBD) is a common technique used to fabricate monolayers containing small organic molecules. In this method, sublimated molecules either react with metal surface adatoms or with metal species generated by electron beam heating in a UHV chamber.119 This approach enables precise control over the deposition process, as both the molecular concentration and deposition rate can be finely tuned. However, some building blocks may have limited stability during sublimation.Early works relied on the use of UHV-STM to explore: novel supramolecular architectures obtained through metal coordination-induced phase transitions,120,121 the fabrication of cavities with tunable size,75,122 and the in situ examination of coordination dynamics.72 Following these foundational works, the field evolved to the study of surface-confined MOCNs aimed at elucidating their structure, the effect of experimental parameters, and the organizational properties of the assembly. Structural characterization enables structures to be distinguished based on the connectivity of the metal and ligand building blocks, and for their local chirality to be assigned. Experimental parameters, including the concentration, the temperature, and the choice of substrate, are shown to directly influence the outcome of the self-assembly process and the stability of the resulting networks. Leveraging on these insights, a high degree of control during the fabrication of surface-confined MOCNs is possible, and the high spatial resolution of STM enables different phases to be distinguished. Furthermore, quantitative information on the crystallinity of the networks can be obtained. Finally, the structural variability accessible in surface-confined MOCNs, and revealed by STM, is associated with functional variability. These aspects are discussed below.
The use of polytopic ligands carrying a different number of analogous coordinating groups also allows the synthesis of MOCNs with different crystal structures.126 Ligands with analogous core structures and different peripheral functional groups have been shown to form different architectures upon metal coordination on surfaces.127 Pre-organization of the ligands on the surface via supramolecular interaction is a useful strategy to readily access crystalline structures.128
Exotic phases, such as Kagome lattices, have also been fabricated using metal–organic coordination.115,129–132 Moreover, the use of f-block metals in MOCN synthesis expands the range of accessible coordination geometries, enabling the design of networks with higher coordination numbers at the metal nodes and distinct structural properties. Several examples have been reported that make use of lanthanides,104,105,127,133–135 whereas only discrete actinide complexes have been studied.136
A particularly striking example of structural control in MOCN fabrication is demonstrated by quasicrystalline (QC) coordination networks, which are ordered structures lacking translational symmetry. Through precise stoichiometric control of Eu centers and ditopic linear para-quaterphenyl-dicarbonitrile (QDC) molecular linkers (Fig. 1(a)) on Au(111) surfaces, 2D metal–organic quasicrystals exhibiting dodecagonal symmetry were successfully synthesized (Fig. 1).137 This breakthrough achievement utilized the large and flexible coordination sphere of lanthanide centers to generate three-coordinated, four-coordinated, five-coordinated, and six-coordinated vertices by controlling the Eu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :QDC stoichiometric ratio (Fig. 1(b)–(i)). Notably, a random square-triangle tessellation, featuring simultaneous four-coordinated, five-coordinated, and six-coordinated coordination geometries, with quasiperiodic order, was obtained at an Eu:QDC stoichiometric ratio of approximately 2:5, as shown in Fig. 1(d), (j) and (k). The formation of this dodecagonal quasicrystal (ddQC) structure was facilitated by the careful balance of adlayer–substrate interactions and entropic surface tessellation effects. UHV-STM characterization revealed a nanoporous structure with 12-fold fast Fourier transform (FFT) symmetry (Fig. 1(j)), representing a distinct manifestation of quasicrystallinity in surface-confined MOCNs. This work not only expands the structural diversity achievable through metal–organic coordination but also introduces f-block elements into QC architectures, opening pathways for functional surface nanoarchitectures that combine quasiperiodic order with the unique properties of lanthanide centers.
:QDC stoichiometric ratio (Fig. 1(b)–(i)). Notably, a random square-triangle tessellation, featuring simultaneous four-coordinated, five-coordinated, and six-coordinated coordination geometries, with quasiperiodic order, was obtained at an Eu:QDC stoichiometric ratio of approximately 2:5, as shown in Fig. 1(d), (j) and (k). The formation of this dodecagonal quasicrystal (ddQC) structure was facilitated by the careful balance of adlayer–substrate interactions and entropic surface tessellation effects. UHV-STM characterization revealed a nanoporous structure with 12-fold fast Fourier transform (FFT) symmetry (Fig. 1(j)), representing a distinct manifestation of quasicrystallinity in surface-confined MOCNs. This work not only expands the structural diversity achievable through metal–organic coordination but also introduces f-block elements into QC architectures, opening pathways for functional surface nanoarchitectures that combine quasiperiodic order with the unique properties of lanthanide centers.
 | ||
| Fig. 1 Random tiling in QC-MOCNs. (a) Molecular structure of ligand QDC. (b)–(e) High-resolution STM images of the distinct Eu-QDC MOCNs fabricated by controlling the Eu:QDC stoichiometric ratios and (f)–(i) molecular models of the corresponding coordination geometries. Scale bar: 5 nm. (j) Large-scale STM image of the ddQC structure observed at Eu:QDC 2:5 stoichiometric ratio. Inset: 2D-FFT depicting the 12-fold symmetry of the network. (k) Different orientations with respect of the surface of the square-triangle tessellation in the dodecagonal tiling. Adapted with permission from ref. 137. Copyright©2016, Springer Nature Limited. | ||
These examples demonstrate that spatially-localized imaging provided by STM enables visualization and distinction of such different geometries.
The study of chiral expression and amplification in 2D coordination networks has gained significant attention due to potential applications in chiral resolution and enantioselective catalysis. The sub-molecular resolution imaging power of STM enables differentiation between opposite orientations of the ligands in chiral complexes adsorbed on a surface. The earliest examples of chiral coordination compounds on metal surfaces are complexes of 1,3,5-benzenetricarboxylic acid (trimesic acid, BTC) and Fe metal centers.71,74,145Fe(BTC)4 complexes are prochiral, and two opposite enantiomers are distinguished on the surface. Their investigation on Cu(100) represents the first direct observation of a chiral metal–organic coordination complex on a surface. Annealing of such structures at higher temperatures facilitates the hierarchical formation of extended chiral MOCNs featuring chiral nanocavities.
Recent studies on the self-assembly of 4,4′-dihydroxybiphenyl (DHBP, Fig. 2(a)) on Ag(100) revealed that thermal annealing promotes phase transition in the hydrogen-bonded networks (Fig. 2(b)–(g)) and leads to coordination of the ligand to the surface adatoms at 490 K (Fig. 2(h) and (i)), resulting in the formation chiral windmill (DHBP)4Ag quartets (Fig. 2(j)).146 Enantiomeric quartets phase-separate into homochiral domains (Fig. 2(k)). Further annealing to 520 K for 10–30 minutes promotes C–C coupling between trans-positioned DHBP molecules in (DHBP)4Ag quartets, and the chirality of the windmill complexes is transferred to the assembly of C–C-coupled dimers (Fig. 2(l) and (m)). A true MOCN is not formed here, but rather a SAMN of metal complexes. Stepwise annealing and the formation of chiral (DHBP)4Ag metal complexes before the C–C coupling reaction are necessary to obtain an ordered arrangement of covalent dimers and the formation of chiral phases.
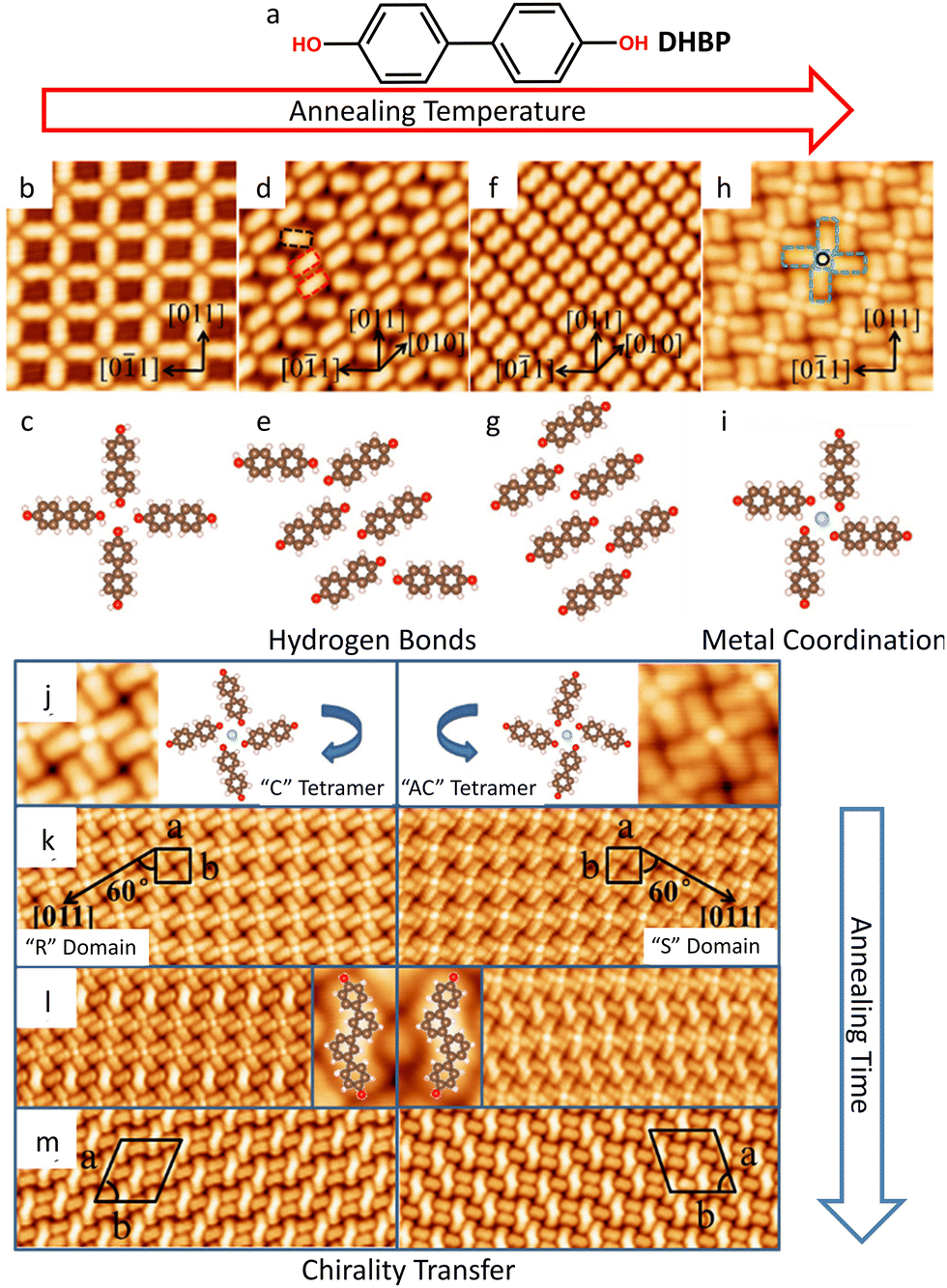 | ||
| Fig. 2 Chiral DHBP assemblies on Ag(100). (a) Molecular structure of DHBP. (b)–(i) Representative high-resolution STM images of the temperature-induced phase transition in DHBP self-assembly and corresponding molecular structures. Annealing temperatures: (b), (c) 200 K; (d), (e) 300 K; (f), (g) 370 K; (h), (i) 490 K. Metal-coordinated chiral quartets form at 490 K (h)–(j). (k) Homochiral domains of (DHBP)4Ag chiral quartets. (l) Mixed phase comprising of (DHBP)4Ag chiral quartets and C–C-coupled dimers, obtained after annealing at 520 K for 10 minutes. The molecular structures of the dimers are shown in the insets. (m) Enantiomorphous assemblies of C–C-coupled dimers obtained after annealing at 520 K for 30 minutes. The image sizes are: (b), (d), (f), (h) 7 nm × 7 nm; (k)–(m) 19 nm × 7 nm. Adapted with permission from ref. 146. Copyright©2019 American Chemical Society. | ||
These works underscore how sub-molecular resolution imaging via STM enables differentiation of enantiomers and distinct chiral phases of assembled coordination complexes through direct visualization of their local structures. However, several critical gaps remain in our understanding of chiral MOCNs compared to the extensive body of work on non-metal-containing self-assembled supramolecular networks. Despite decades of research on purely organic chiral networks where phenomena such as chiral amplification, spontaneous resolution, and sergeant-and-soldiers effects have been extensively documented, analogous systematic studies in metal-coordinated systems have not yet been reported. Detailed investigations into chiral selection processes and the influence of metal coordination strength on chiral expression in UHV-fabricated MOCNs have not been described. More critically, the functional implications of chirality in these metal–organic networks, such as their potential for enantioselective adsorption of chiral guests or chiral recognition in general, remain largely unexplored. The field would benefit from systematic comparative studies examining how metal coordination influences chiral amplification mechanisms and from functional demonstrations of these chiral MOCNs in applications that exploit their inherent asymmetry, especially given that fixed binding geometries within the cavities of porous MOCNs could enhance chiral discrimination.
Tuning the concentration of the precursors is an effective strategy to promote the formation of different phases starting from the same building blocks.125 A particularly instructive example of density-driven structural evolution is provided by the systematic study of TPB-Cu coordination networks on Au(111) as a function of molecular coverage.147 By incrementally increasing TPB sublimation while maintaining Cu excess, the authors observed a remarkable sequence of structural transformations driven by what they termed “intrinsic in-plane compression pressure”, as shown in Fig. 3. Starting from an open honeycomb network at low dosage (TPB concentration of 0.31 TPB per nm2, Fig. 3(b) and (c)), progressive density increases led to the sequential formation of pentagonal networks (0.34 TPB per nm2, Fig. 3(d) and (e)), rhombic structures (0.38 TPB per nm2, Fig. 3(f) and (g)), and finally two phases labelled as mixed honeycomb-rhombic and triangular phases (0.59 TPB per nm2, Fig. 3(h)–(k)). The corresponding molecular packing densities are indicated under the molecular models of each polymorph in Fig. 3. Most remarkably, this structural evolution was accompanied by a change in coordination chemistry: while at low TPB concentration only linear TPB2Cu complexes are observed (Fig. 3(b)–(g)), at the highest ligand concentration trigonal TPB3Cu complexes are also formed (Fig. 3(h)–(k)), demonstrating that compression can alter the preferred coordination geometry of metal nodes.
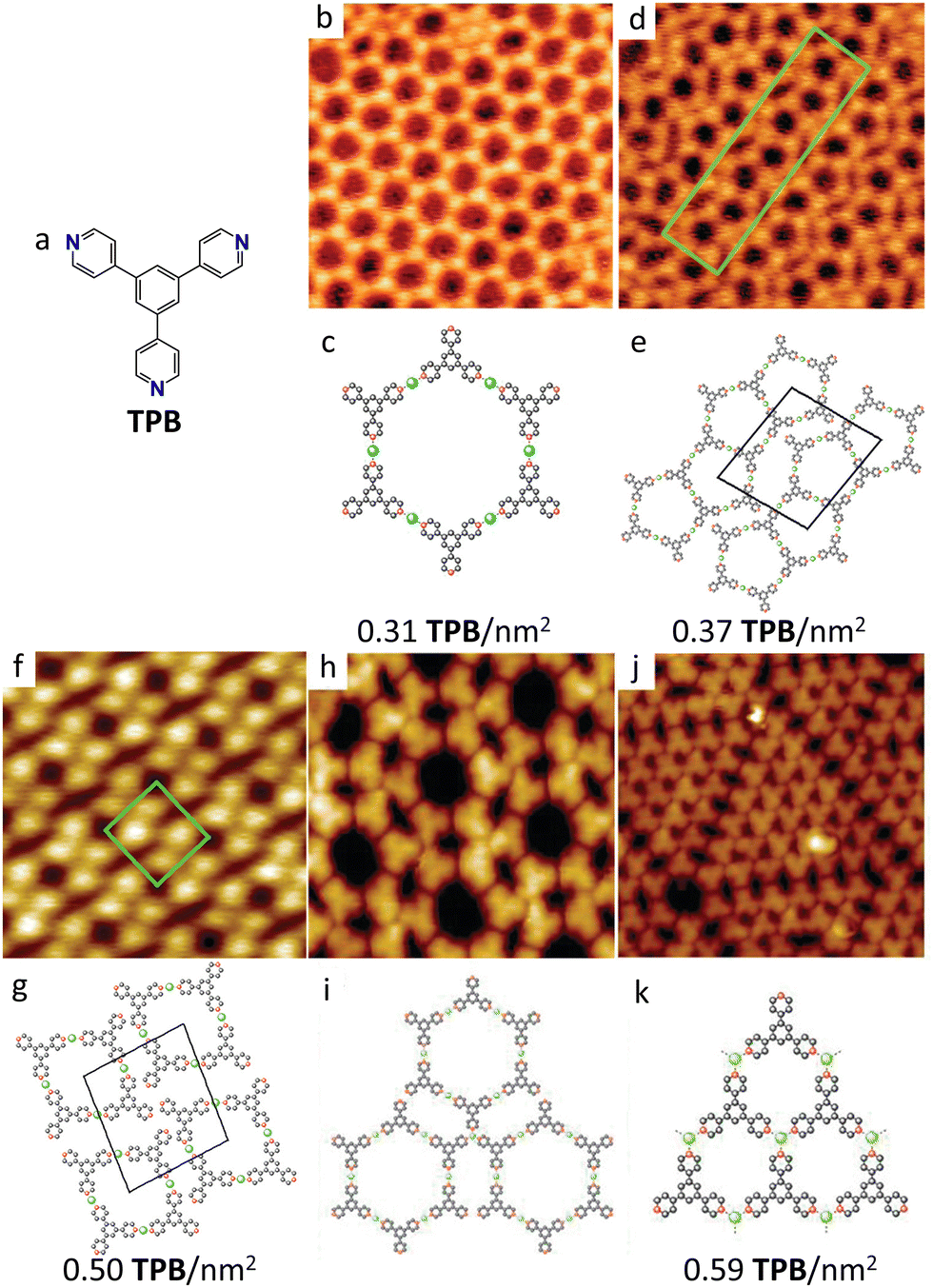 | ||
| Fig. 3 TPB-Cu polymorphs on Au(111). (a) Molecular structure of TPB. (b), (d), (f), (h), (j) high-resolution STM images and (c), (e), (g), (j), (k) corresponding molecular models of the different TPB-Cu MOCN polymorphs formed on Au(111) at different TPB densities, reported underneath the models. Models color code: grey: carbon, red: nitrogen, green: copper. The molecular packing densities of Cu and TPB are: (b) 0.46 Cu per nm2; 0.31 TPB per nm2. (d) 0.50 Cu per nm2; 0.37 TPB per nm2. (f) 0.50 Cu per nm2; 0.50 TPB per nm2. (j) 0.59 Cu per nm2; 0.59 TPB per nm2. The image sizes are: (b), (d), (f) 20 nm × 20 nm. (h) 10 nm × 10 nm. (j) 15 nm × 15 nm. Adapted with permission from ref. 147. Copyright©2011 American Chemical Society. | ||
This work provides valuable molecular-level insights into the role of ligand concentration as a control parameter in the synthesis of surface-supported MOCNs. The on-surface ligand concentration effects could be considered analogous to pressure effects in 3D MOF systems. The unique insights obtained by UHV-STM molecular resolution imaging may thus provide insights into bulk MOF-related dynamic phenomena.
 | ||
| Fig. 4 HATP and HITP supramolecular (metallo-)assemblies on Cu(111). (a) Molecular structure of HATP. (b), (c) STM images acquired after room-temperature deposition depicting incomplete and fused HATP hexamers stabilized by van der Waals interactions. (d) STM image captured after annealing at 375 K, showing Cu-coordinated cyclic triangular assemblies, van der Waals-stabilized hexamers, and interlinked dimers (indicated by the green arrows). (e), (f) STM images obtained after annealing at 475 K, showing the prevalence of aggregates consisting of Cu-coordinated trimers (highlighted in red), interlinked or terminated by single HATP molecules (red arrows). The inset in (f) is a molecular model of two linked trimers. (g)–(j) STM images of the HATP-Ni complexes on Cu(111). (g) STM image of the coordination oligomers formed by HATP and Ni on Cu(111) at room temperature. (h) STM image of the HATP + Ni system after annealing at 375 K showing partial Ni-coordination and pure HATP hexamers (yellow dashed oval). (i), (j) STM images of the HATP + Ni system after complete deprotonation at 475 K leading to Ni-coordinated hexamers (the red arrows indicate Ni atoms at the periphery of incomplete hexamers). Adapted with permission from ref. 148. Copyright©2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Upon Ni co-deposition, STM images show a dramatic shift in coordination behavior even at room temperature, with partial deprotonation and the emergence of bis(diimino)-Ni motifs (Fig. 4(g)–(j)). Despite the higher reactivity of Ni and the formation of local honeycomb-like features, extended networks were again not observed. This was attributed to the reduced reversibility of the HITP–Ni coordination bonds, which hinders network evolution by kinetically trapping the sub-units.
By correlating morphological evolution with stimuli such as temperature and metal identity, the authors not only provide an atomistic picture of coordination motifs but also reveal mechanistic insight into the reversibility and scalability of MOCNs under UHV. Moreover, they offer a critical perspective on how UHV conditions diverge from liquid-phase coordination environments, particularly by providing a less dynamic environment where reversibility is reduced and rearrangement processes are hindered.
It is important to note that such conclusions are relative to the specific experimental conditions used in this work. As a matter of fact, an extended crystalline MOCN formed by HITP–Ni complexes was obtained on Au(111),149 and a bimetallic HITP–Ni/Co network has recently been reported.150 Notably, in this latter work, the location of the different metals in the MOCN lattice was determined by differences in STM contrast at the metal nodes.
The ability to visualize dynamic processes in 2D networks may help elucidate how local coordination environments, metal–ligand binding preferences, and structural flexibility translate to bulk MOF properties. This may be particularly valuable for understanding how defects propagate, how different polymorphs form, and how structural transformations occur.
Coordination to different metals can also lead to different thermodynamic and kinetic stability, as demonstrated by the coordination of 1,3,5-tris(4-pyridyl)benzene (TPB, Fig. 3(a)) with Cu and Fe on Au(111).153 In the TPB-Cu MOCN, the ligands form linear complexes with the metal centers, whereas trigonal complexes are observed in the TPB-Fe MOCN. When the metals are deposited together and the ligand is added afterward, both networks are formed, with the TPB-Fe one to a larger extent. Annealing of this sample results in the TPB-Fe network dominating over the TPB-Cu one already at 400 K. Conversely, successive deposition of Fe onto a pre-formed TPB-Cu MOCN leaves the network almost unchanged, and the TPB-Fe network forms only after annealing, resulting in the complete disappearance of the TPB-Cu assembly at 500 K. Based on these observations, it was suggested that the TPB-Fe network is the thermodynamically most stable one and has a higher formation rate constant, whereas the TPB-Cu displays a low dissociation rate and is therefore kinetically stable up to higher temperatures if it forms first. These results highlight the significant influence of synthetic routes and kinetic vs. thermodynamic control on the MOCNs architecture.
The different affinities of ligand functional groups toward different metals may lead to different outcomes when ligands are deposited on different substrates. For instance, 1,3,5-tris(4-mercaptophenyl)benzene (TMB) was shown to coordinate to Cu adatoms on Cu(111) and form crystalline MOCNs upon annealing, whereas analogous experimental conditions on Ag(111) resulted in the formation of amorphous assemblies containing dimers connected through disulfide bridges.151
Interestingly, exceptions to the substrate effect may arise when coordination bonds are sufficiently strong to overcome possible orienting effects from the surface,155 whereas network flexibility may enable the formation of extended structures across surface defects.156
A comparative example is provided by the different coordination structures formed by 1,4,5,8,9,12-hexaazatriphenylene (HAT, Fig. 5(a)) in solution and on different substrates. In solution, HAT has been shown to form staircase-like coordination chains due to the steric hindrance that would result from the coordination of two bidentate sites around a metal center with square planar coordination geometry.157 In contrast, the deposition of HAT along with Ni, Fe, or Co atoms on Au(111) and Ag(111), followed by annealing at 450 K, leads to the formation of a porous hexagonal network via in-plane coordination geometries.41 Notably, the ligands are tilted out of the framework plane by up to 15° on Au(111), and consequently, the network has a buckled conformation, as indicated by the variation in contrast observed in the STM images and supported by DFT calculations. DFT calculations further indicated that the tilt angle reaches 23° in the gas phase to minimize the steric hindrance, and a flattening effect occurs upon surface absorption, highlighting the relevance of substrate–molecule interactions in controlling the morphology of adsorbed networks.
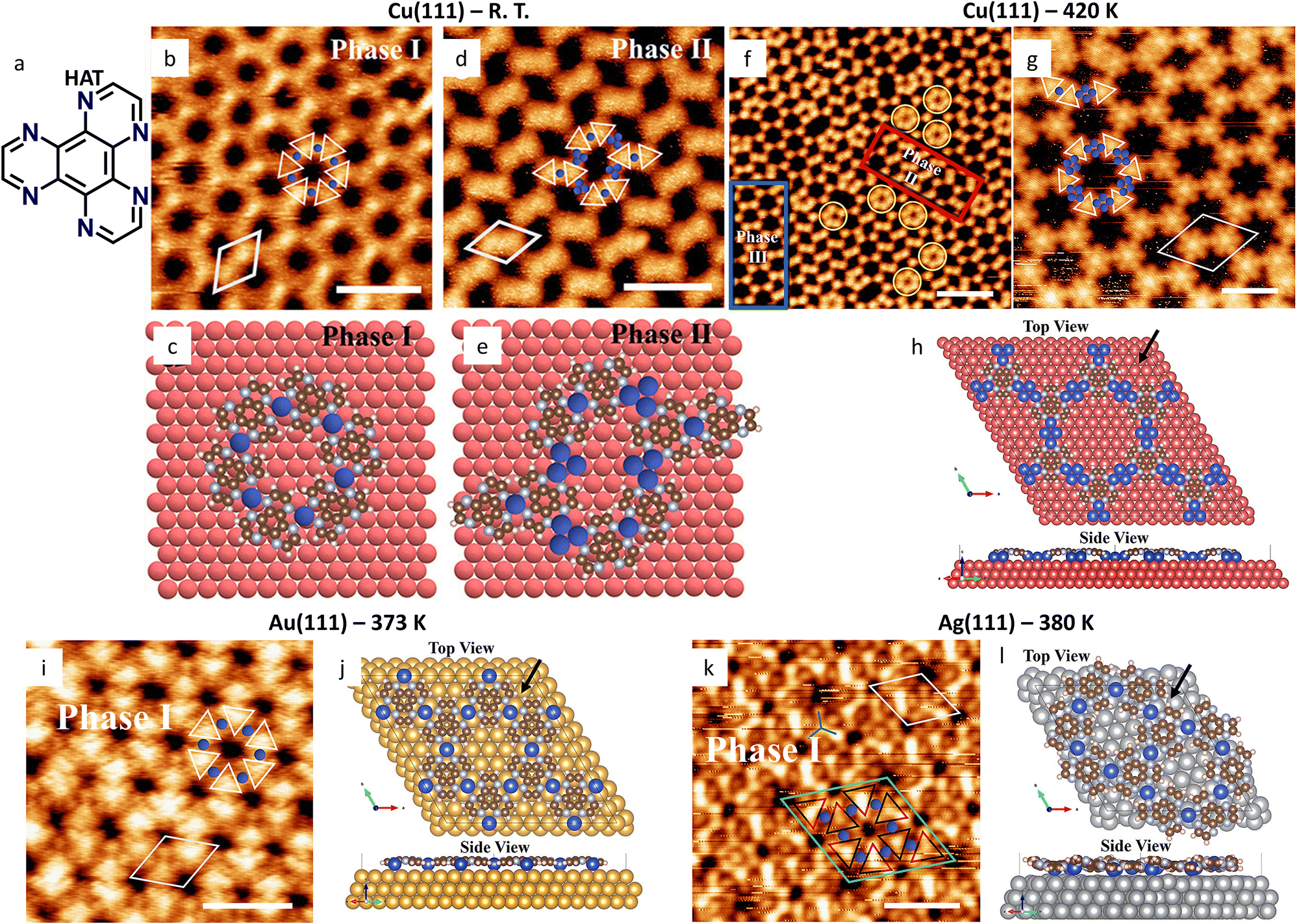 | ||
| Fig. 5 Substrate effect on the HAT-Cu MOCN revealed by UHV-STM. (a) Molecular structure of HAT. (b), (d) Small-scale STM images of the two polymorphs observed on Cu(111) without annealing treatment and (c), (e) corresponding molecular models. (f) Large-scale and (g) small-scale STM images of the Cu(111) sample after annealing at 420 K. The red and blue rectangles highlight phase II and phase III polymorphs, respectively. (h) DFT-optimized top and side views of phase III, depicting the (Cu4)3HAT2 stoichiometry. (i) Small-scale STM image of the Cu3HAT2 MOCN formed on Au(111) after annealing and (j) DFT-optimized top and side view of the corresponding molecular structure. (k) Small-scale STM images of the Cu3HAT2 MOCN formed on Ag(111) after annealing and (l) DFT-optimized top and side views of the corresponding molecular structure, depicting the buckled conformation. Scale bars (b) 2.4 nm; (d) 3 nm; (f) 5 nm; (g) 2 nm; (i) 2 nm (k) 2 nm. Adapted with permission from ref. 152. Copyright©2024 American Chemical Society. | ||
This substrate effect was explored in subsequent studies, where HAT was deposited on Cu(111) and, in combination with Cu, on Au(111), Ag(111), and MoS2 (Fig. 5).152 The same Cu3HAT2 stoichiometry was observed after depositing the precursors on Au(111), Ag(111), and MoS2 and annealing the samples (Fig. 5(i)–(l)). In contrast, two distinct polymorphs were observed on Cu(111) at room temperature, with the one denoted as phase I in Fig. 5(b) and (c) corresponding to the Cu3HAT2 stoichiometry (Fig. 5(b)–(e)). Annealing experiments at 420 K led to the disappearance of phase I and the formation of a new polymorph, denoted as phase III, featuring complexes with (Cu4)3HAT2 stoichiometry (Fig. 5(f)–(h)). This phase transition has been attributed to the different thermodynamic stability of the two phases. Moreover, the buckled network conformation was observed only on Ag(111) and MoS2, whereas higher adsorption energies on Cu(111) and Au(111) favor flatter conformations (Fig. 5(h), (j) and (l)).
In summary, HAT was demonstrated through UHV-STM to form extended MOCNs by coordinating metals on surfaces, in contrast with the 3D staircase structures observed in solution. Moreover, even when coordinating the same metals, adsorption on different substrates promotes different conformations and stoichiometries, underscoring the critical role of substrate-building blocks interactions in determining MOCN architecture.
This method was subsequently applied to quantitatively assess the crystallinity in an MOCN containing a mixture of two homologous ligands as a function of their stoichiometric ratio.159 The selected ligands are 2,4,6-tri(4-pyridinyl)-1,3,5-triazine (TPT, Fig. 6(a), top) and 4,4′-(5′-(4-(4-pyridyl)phenyl)-[1,1′:3′,1′′-terphenyl]-4,4′′-diyl)dipyridine (TPBT, Fig. 6(a), bottom), both of which form crystalline isoreticular hexagonal networks by coordinating Cu adatoms on Cu(111) over a temperature range between 300 K and 500 K (Fig. 6). Notably, 1:1 mixtures of the two ligands led to the formation of distinct structures depending on the annealing temperature: at lower temperatures, the ligands exhibited a tendency to segregate into separate crystalline phases, whereas annealing promoted the formation of a mixed, disordered phase, as illustrated in Fig. 6(b) and (c). Similarly, annealing samples prepared with TPT:TPBT stoichiometric ratios different from 1:1 results in amorphous phases with diverse morphologies, as shown in the series of images presented in Fig. 6(d). While no direct correlation between the PH score and other statistical parameters (Fig. 6(e)–(h)) could be established, PH remains a promising method for distinguishing between crystalline and amorphous phases. Furthermore, this study highlights the potential of STM imaging not only for visualizing different structural phases but also for identifying and quantifying amorphous domains with a high degree of precision.
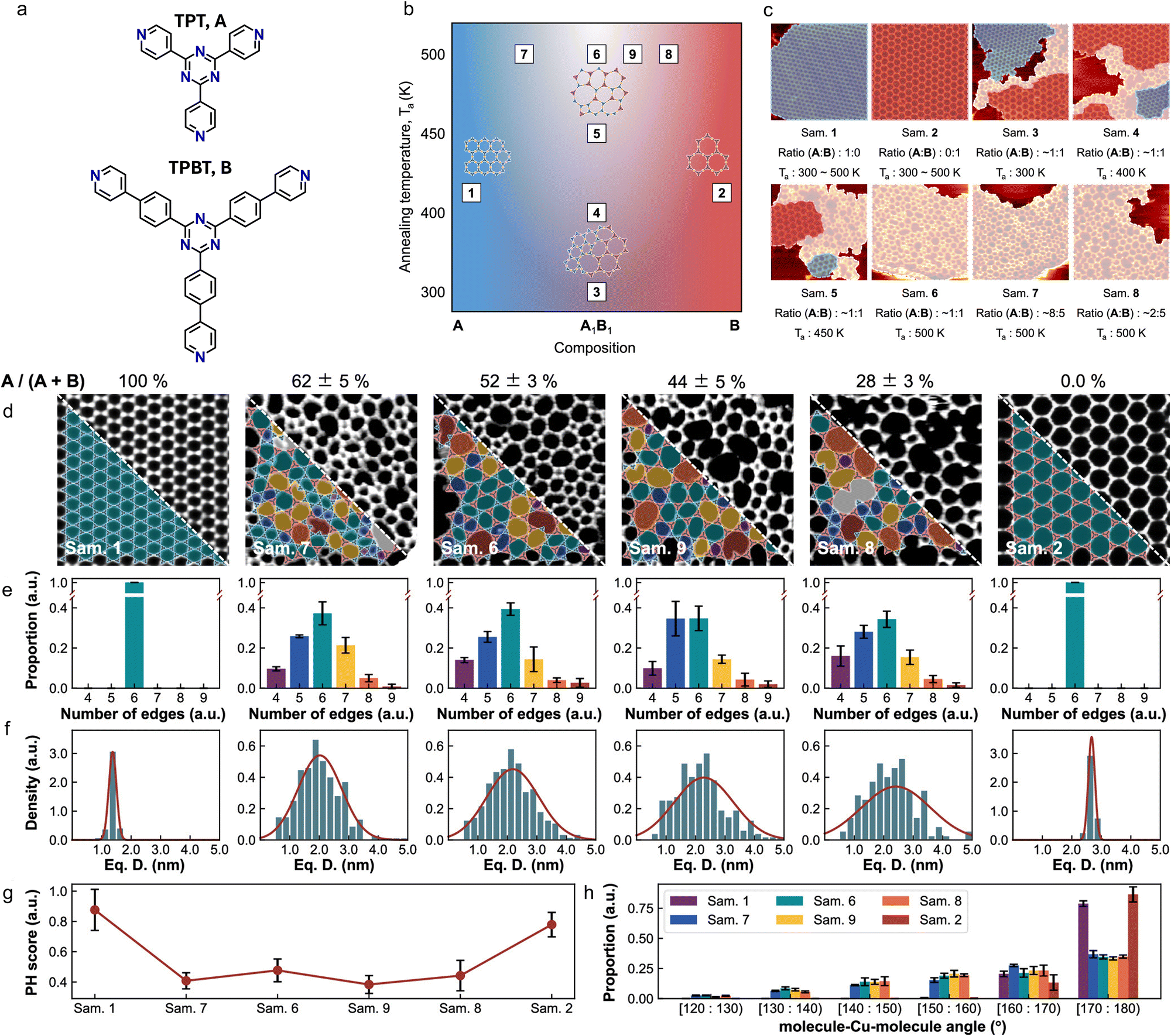 | ||
| Fig. 6 Amorphous and crystalline phases in a bi-ligand MOCN on Cu(111). (a) Molecular structures of TPT (A) and TPBT (B). (b) Composition diagram of the different phases observed as a function of the A:B stoichiometric ratio and of the annealing temperature. (c) Representative STM images of some of the samples marked in (b). The TPT crystalline phase is marked by the blue overlays; the TPBT crystalline phase is marked by the red overlays. (d)–(h) Statistical data for quality assessment. (d) Representative STM images of the samples marked in (b). The overlays indicate pores of various shapes: blue: tetragons, green: hexagons, yellow: heptagons, orange: octagons, red: higher order polygons. (e) Polygons distribution for each sample. On the x-axis, the number of edges of the polygon is reported. (f) Pore diameter distribution for each sample. The red line depicts a Gaussian distribution fit. (g) PH score values for each sample. (h) Molecule–Cu–molecule angle distribution for each sample. Adapted with permission from ref. 159. Copyright©2023 American Chemical Society. | ||
The above example concerns the formation of an MOCN formed from components that ordinarily form a 3D MOF. In the case of layered 2D MOFs, it would be intuitive that the single-layer surface-supported MOCN would be analogous to a single-layer of the stacked material. However, MOCNs with structures deviating from those matching single sheets of the layered 2D MOFs formed from the same building blocks have been reported. One such example is an MOCN obtained through the sequential deposition of 2,3,6,7,10,11-hexahydrocytriphenylene (H6HOTP) and Co on Au(111).163 A layered 2D MOF was previously fabricated using these precursors, consisting of an alternate stacking of two different layers. One layer is an extended honeycomb with Co3(HOTP)2(H2O)6 stoichiometry that features hexagonal pores, whereas the other layer contains discrete Co3(HOTP)(H2O)12 complexes.164 In contrast, surface confinement promotes a unique architecture in which each metal center is coordinated to three different HOTP ligands, as opposed to one or two in the layered material. STS measurements supported by DFT calculations showed that the HOTP-Co MOCN exhibits a smaller bandgap than the one that was previously reported for the layered 2D MOF thin film analog.165 The magnetic anisotropy displayed by the HOTP-Co MOCN, coupled with the evidence of antiferromagnetic interactions, opens up new opportunities for integrating such assemblies into spintronic applications.166
The use of organic radicals as ligands presents an effective strategy to promote strong magnetic coupling between metal nodes. In contrast to traditional diamagnetic linkers, radical ligands can serve as active spin carriers, enabling π–d interactions that enhance magnetic exchange and circumvent the requirement of using short linkers. A recent study demonstrates this approach through the on-surface synthesis of a large-scale, atomically precise MOCN using asymmetric radical ligands coordinated with Co centers (Fig. 7).167 This two-step strategy, involving initial on-surface formation and supramolecular organization of the radicals (Fig. 7(b)) followed by metalation, yields a high-quality magnetic network with long-range order (Fig. 7(c) and (d)). UHV-STM, STS, and nc-AFM techniques coupled with DFT calculations revealed two distinct magnetic couplings corresponding to different complexes. While no magnetic coupling is observed in the radical ligand SAMN (Fig. 7(f)), magnetic exchange is observed in the Co-coordinated structures, depending on how the ligands are connected within a coordination complex (Fig. 7(e), (g) and (h)). These results evidence strong π–d interactions and the coexistence of antiferromagnetic and ferromagnetic coupling modes. Notably, this methodology maintains porosity while achieving a complex spin texture, expanding the design space for 2D magnets. The radical-as-ligand approach thus offers a powerful pathway to engineer multifunctional MOCNs with tunable electronic and magnetic properties, holding promise for integration in quantum materials and spintronic architectures.
 | ||
| Fig. 7 Magnetic coupling in a radical-based MOCN. (a) Molecular structure of the radical ligand. The arms on the ligand are numbered 1-3. (b) Structure of the hydrogen-bonded SAMN formed by the self-assembly of the radical ligand. (c) Structure of a Co-coordinated trimer of radical ligands in the MOCN formed by addition of Co on the radical ligand SAMN. (d) Constant-current STM image of the radical ligand-Co MOCN (scale bar = 28 nm; lattice parameters: a1 = a2 = 6.4 nm, α = 60°). Inset: nc-AFM of two adjacent Co-coordinated molecular ligand trimers. (e) Long-range differential conductance spectra recorded at different locations, marked in the inset of (d). (f)–(h) Differential conductance spectra in the absence of Co and in the case of Co-coordinated radical ligand dimers connected at the arms 3-3 and 1-2, respectively. Adapted from ref. 167. Copyright©2025 The Authors. Published by American Chemical Society. | ||
Isoreticular chemistry represents a well-established approach in MOF design that enables the fabrication of frameworks with similar topologies but varying pore dimensions through the systematic use of ligands with analogous shapes and functional groups and different molecular lengths. While this strategy has been extensively employed in 3D MOF synthesis, its application to surface-confined MOCN fabrication remains relatively underexplored.114,133,159
A compelling demonstration of this approach is the synthesis of a Dy-based MOCNs on Cu(111) using two isoreticular carboxylate linkers: p-terphenyl-4,4′-dicarboxylic acid (TDA) and BDC.133 These ligands bring analogous carboxylate groups while having different molecular lengths. Each of the two oxygen atoms of the carboxylates occupies a coordination site around the Dy centers, and the large radius of the lanthanide allows for the accommodation of four ligands around the cation, resulting in an eightfold coordination geometry, regardless of the ligand length. As a result, isoreticular porous MOCNs are obtained with an internodal distance of 11.8(5) Å in the Dy-BDC network and 20.5(5) Å in the Dy-TDA network, as revealed by UHV-STM measurements. Despite the significant difference in pore dimensions between the two networks, the magnetic properties show only subtle variations. Both systems exhibit similar enhancement of magnetic anisotropy compared to isolated Dy atoms, with the only difference seemingly being a slightly smaller tilting angle of the anisotropy axis for the Dy-BDC MOCN. While the authors conclude that magnetic properties result from a delicate balance between oxygen–Dy3+ interactions and crystal field geometry, the experimental evidence suggests this balance is largely insensitive to the internodal distance variations achieved through isoreticular design.
A similar investigation of the structure–function relationship in isoreticular MOCNs was presented on the networks based on dicyanobiphenyl (DCBP) and dicyanoanthracene (DCA) ligands and Co, sequentially deposited on epitaxial graphene grown on Ir(111).168 Low-temperature UHV-STM and nc-AFM studies revealed that both ligands formed extended honeycomb MOCNs, containing Ligand3Co complexes. DCBP4Co complexes were also observed, whereas DCA formed exclusively DCA3Co complexes due to steric hindrance. STS measurements supported by DFT calculations demonstrated stronger coupling between the molecular building blocks of DCA3Co2 MOCN than in the DCBP3Co2 isoreticular analog, resulting in a significant bandwidth in the former network, which could be exploited for engineering new electronic materials.
3.2 MOCNs fabricated and characterized at the liquid–solid interface
In liquid-phase synthesis, a solution of precursors is drop-cast onto a flat surface under ambient atmospheric pressure. This method is fast and cost-effective; however, exposure to ambient conditions increases the risk of contamination.106,107The disparity between the number of reports on MOCNs fabricated in UHV and the ones fabricated at the liquid–solid interface arises from several interconnected experimental challenges. First, the sensitivity of certain metal surfaces to atmospheric conditions can lead to oxidation and surface contamination, compromising both the substrate quality and the fidelity of metal–organic coordination. Second, solubility limitations present a significant obstacle: many metal salts exhibit poor solubility in organic solvents suitable for STM imaging, while conversely, solvents that effectively dissolve metal precursors may be incompatible with ambient STM operation due to high volatility, conductivity, or chemical reactivity with the tip. Third, organic ligands and metal adatoms are constrained to the surface in UHV conditions, guiding the formation of planar complexes more readily than at liquid–solid interfaces. In the latter scenario, three-dimensional complexes may preferentially form in solution, subsequently competing with or impeding the formation of extended 2D monolayers.
Furthermore, ambient-STM studies of MOCNs demand precise control over multiple experimental parameters simultaneously, including deposition protocols, concentration ratios, solution chemistry, and environmental conditions. The complexity of optimizing these interdependent variables often makes liquid–solid interface studies more technically demanding than their UHV counterparts, where individual parameters can be controlled more independently.
Despite these challenges, the liquid-phase environment offers unique advantages that justify the additional experimental complexity. The presence of solvent introduces an additional parameter that can be tuned to direct self-assembly outcomes, through specific solvation effects and solvent–substrate interactions. Moreover, the liquid medium facilitates dynamic processes through continuous adsorption–desorption equilibria, as illustrated in Fig. 8, enabling error correction mechanisms and access to thermodynamically favored structures that may be kinetically inaccessible under vacuum conditions. This dynamic environment also permits real-time observation of assembly evolution and reorganization processes.
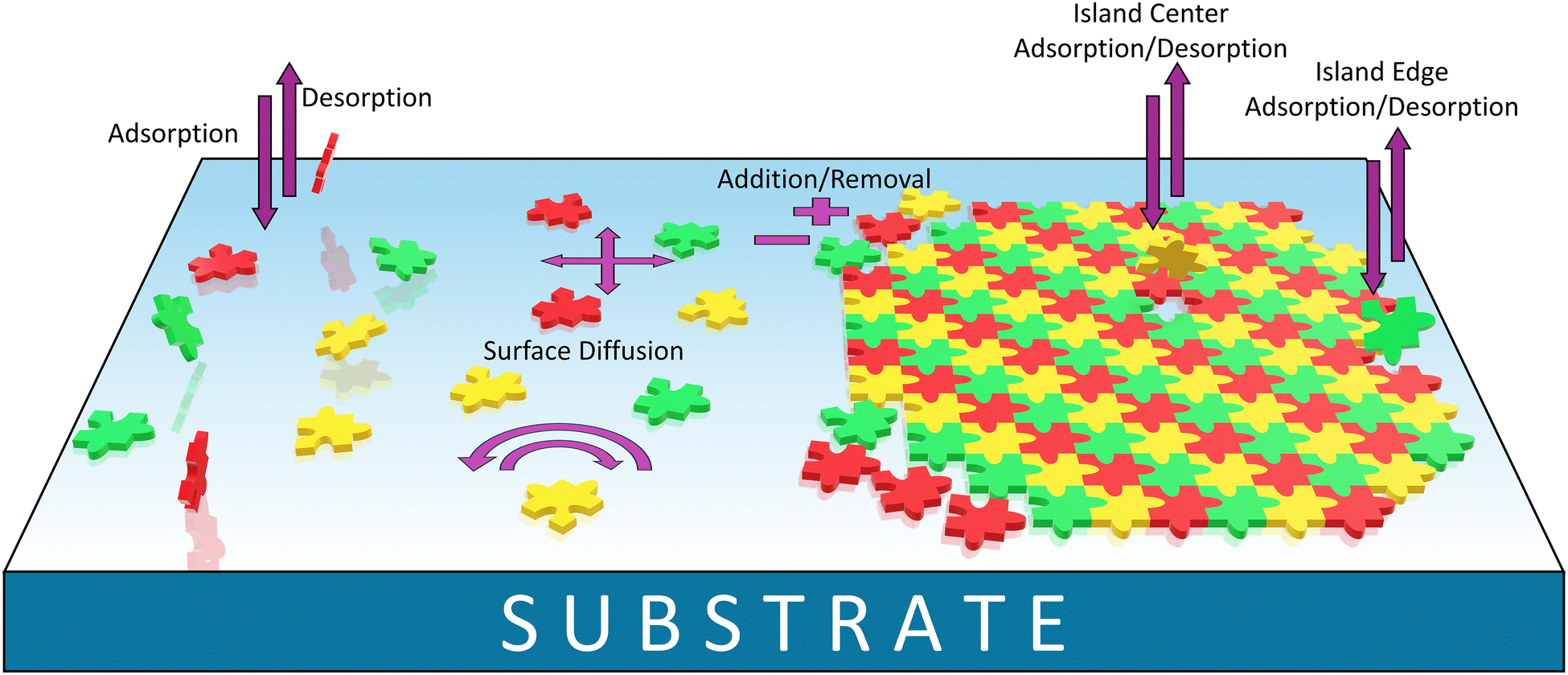 | ||
| Fig. 8 Cartoon depicting typical interface equilibria during the formation of supramolecular networks, such as MOCNs, at the liquid–solid interface. Puzzle pieces of different colors indicate different building blocks. For example, a three-component equilibrium may involve metal ions, organic ligands, and guest molecules. | ||
Finally, as already noted, MOF thin films and crystal powders are often synthesized using wet chemistry approaches. Consequently, while technically more challenging, liquid–solid interface studies offer complementary information essential for understanding the full scope of MOCN formation mechanisms, their potential for practical applications under ambient conditions, and their correlation with analogues of higher dimensionality.
The introduction of metal ions into SAMNs of coordinating molecules has been shown to induce dramatic structural transformations and promote the formation of new phases templated by metal coordination geometry.169,170 In addition to these examples, the majority of reported STM studies investigating metal coordination in supramolecular assemblies at the liquid–solid interface have focused on SAMNs exhibiting coordination bonds along a single direction (one-dimensional, 1D).171–177 These studies have nonetheless provided valuable insights into the fundamental principles governing the formation of metal-coordinated supramolecular structures at liquid–solid interfaces. Key findings have elucidated the role of metal coordination in forming new architectures at the liquid–solid interface, the critical influence of mixing procedures (ex situ versus in situ metalation) on polymorph selection, and the effects of metal-to-ligand concentration ratios on assembly outcomes.
This section will first examine these crucial aspects that govern metal–organic assembly at liquid–solid interfaces. Subsequently, we will review reports on more complex 2D MOCNs fabricated under ambient conditions, with particular emphasis on how the liquid environment enables unique structural control compared to UHV conditions. Throughout this discussion, we will highlight the intrinsic mechanistic differences between liquid-phase and vacuum-based MOCN formation, including the role of solvation effects, dynamic equilibria, and the influence of substrate–solvent interactions on final network architectures.
The ability to achieve equilibrium structures through dynamic processes suggests that liquid-phase assembly may provide access to thermodynamically favored coordination geometries and packing arrangements that are otherwise inaccessible through vacuum-based preparation methods. Furthermore, the successful interplay between metal coordination (providing directional bonding) and van der Waals interactions (enabling lateral organization) demonstrates a hierarchical assembly strategy that could be extended to more complex multi-component systems requiring precise structural control across multiple length scales.
These observations reveal substantial differences between in-solution and on-surface coordination-mediated self-assembly processes. As a possible reason for the different outcomes of the two procedures, the authors suggested that when metalation occurs ex situ, domains of dimers cannot evolve into columnar structures due to depletion of free Cu(II) ions in solution. Conversely, when metalation proceeds on the surface, sufficient unreacted precursors remain available to facilitate continued oligomer formation. These longer oligomers exhibit enhanced surface affinity due to their increased number of alkyl chains, providing stronger van der Waals interactions with the substrate. Additionally, surface-assisted growth may promote the incorporation of residual monomers into pre-adsorbed oligomers through a templating effect.
Notably, in situ addition of excess Cu(II) to samples prepared by pre-mixing did not induce structural changes in the on-surface assemblies, suggesting that the pathway-dependent polymorphs may represent kinetically trapped states rather than equilibrium structures. This observation raises important mechanistic questions about the role of surface confinement in determining coordination stoichiometry and assembly pathways. A compelling follow-up experiment would involve thermal annealing of the pre-mixed polymorphs to determine whether they constitute metastable states that can be converted to the thermodynamically favored columnar structure. Such studies would elucidate whether in situ mixing facilitates the formation of the true thermodynamic product or merely provides kinetic access to alternative assembly pathways. Furthermore, systematic variation of metal-to-ligand ratios and coordination kinetics by exploring different metal ions could provide deeper insights into the competition between solution-phase complexation and surface-mediated assembly processes.
In the absence of metal ions, TATB spontaneously forms a porous honeycomb network stabilized by intermolecular hydrogen bonds between carboxylic acid groups. The introduction of Cu(II) ions considerably alters this assembly behavior, and the final architectures depend critically on the Cu(II)/TATB molar ratio. At a Cu(II)/TATB molar ratio of 1:2, TATB–Cu(II)–TATB dimers form, with these coordination units serving as building blocks for extended structures where adjacent dimers interact through residual hydrogen bonding.
Increasing the relative concentration of Cu(II) ions with respect to the ligand to a Cu(II)/TATB 1:1 molar ratio produces a markedly different architecture featuring alternating TATB and Cu(II) rows, where hydrogen-bonded TATB chains are interconnected through Cu(II) coordination bridges.
The structural templating effect becomes even more pronounced with Fe(III) ions. At a 1:1 Fe(III)/TATB stoichiometry, the system self-assembles into discrete trigonal trimers, where Fe(III) centers template the formation of hydrogen-bonded TATB trimers. Further increasing the Fe(III) relative concentration to a 3:2 Fe(III)/TATB molar ratio triggers a structural transition, yielding chain architectures where Fe(III) ions intercalate between hydrogen-bonded TATB rows. These observations underscore how both metal identity and metal-to-ligand ratio serve as powerful tools for directing supramolecular assembly, enabling access to structurally diverse networks from a single organic building block through judicious selection of self-assembly conditions.
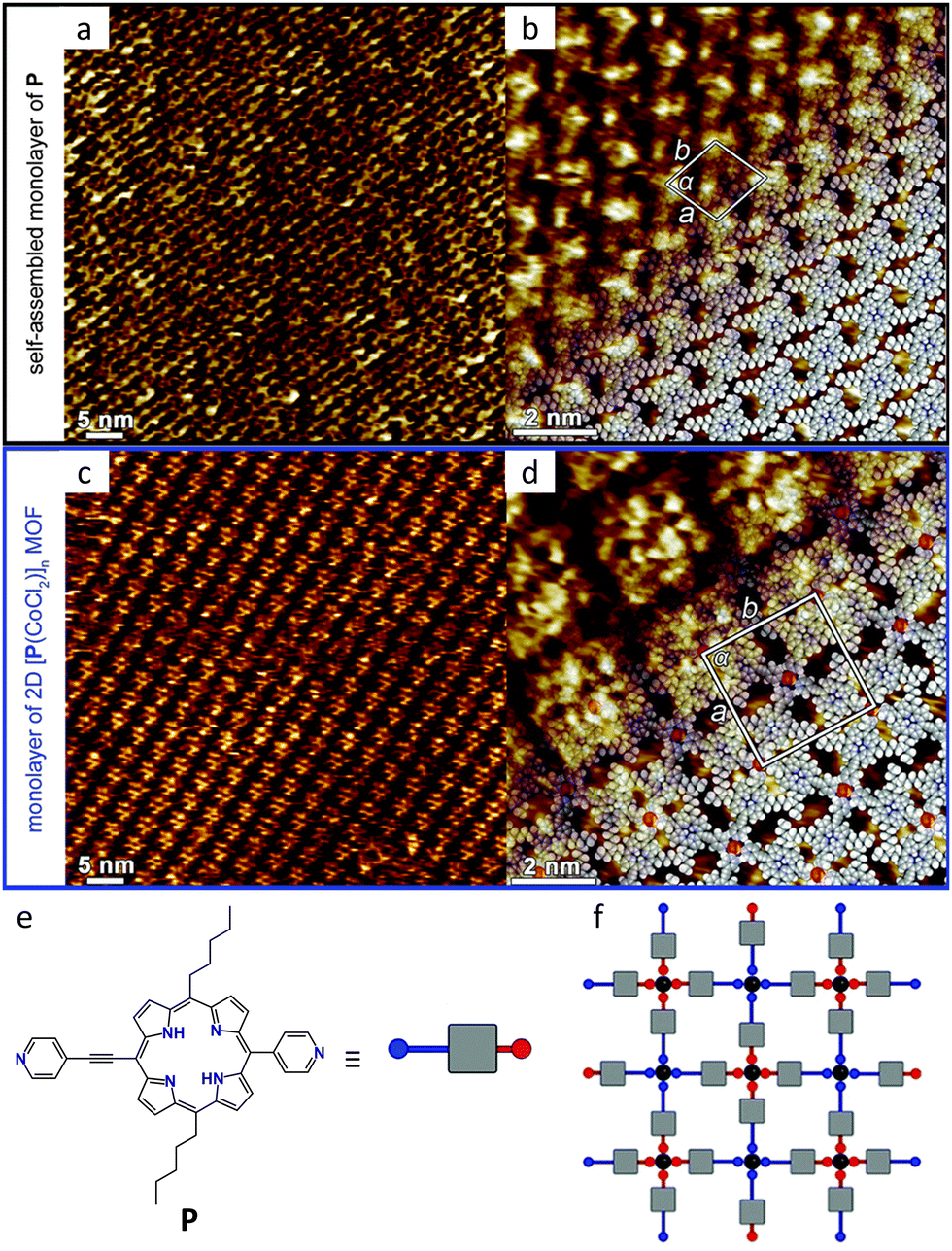 | ||
| Fig. 9 Porphyrin MOCNs at the 1-phenyloctane/HOPG interface. (a), (b) STM current image of a monolayer of free “tecton” P. The proposed molecular model is shown in (b). (c), (d) STM current image of a monolayer of the 2D P-Co(II) MOCN. The proposed molecular model is shown in (d). (e) Molecular structure of P. (f) Schematic representation of the MOCN with the smallest unit cell that may be formed upon combining the neutral acentric tecton P with CoCl2 (black sphere). In (e), (f), the grey square represents the porphyrin core, whereas the red and blue segments correspond to the pyridyl unit directly connected to the porphyrin backbone and the ethynyl pyridyl moiety, respectively. Chloride anions occupying the two apical positions on the octahedron around the Co(II) cation are not presented for clarity. Adapted with permission from ref. 79. Copyright©2014 The Royal Society of Chemistry. | ||
Notably, the preparation method proved critical for achieving structural order. While ex situ premixing produced the ordered 2D network, in situ addition of a CoCl2 solution onto the pre-formed SAMN of the ligand resulted in disordered structures on the substrate. The authors attributed this difference to the strong interfacial interactions between the substrate and the building block P, which likely prevent efficient reorganization when metal coordination occurs after initial surface adsorption. This observation highlights a crucial distinction between liquid–solid interface assembly and UHV-based approaches: the pre-existing molecular organization on the surface can kinetically trap disordered states when subsequent metal addition disrupts established intermolecular contacts. Conversely, ex situ coordination allows the system to explore its full configurational space in solution before surface deposition, enabling access to the thermodynamically favored ordered structure. This work thus establishes important guidelines for synthetic strategies targeting 2D MOCNs at liquid–solid interfaces. In a similar way, mellitic acid (MA) was pre-mixed with PdCl2 or ZnCl2 in methanol before drop-casting onto HOPG.178 STM results revealed distinct network structures due to the differing coordination properties of the two metal ions.
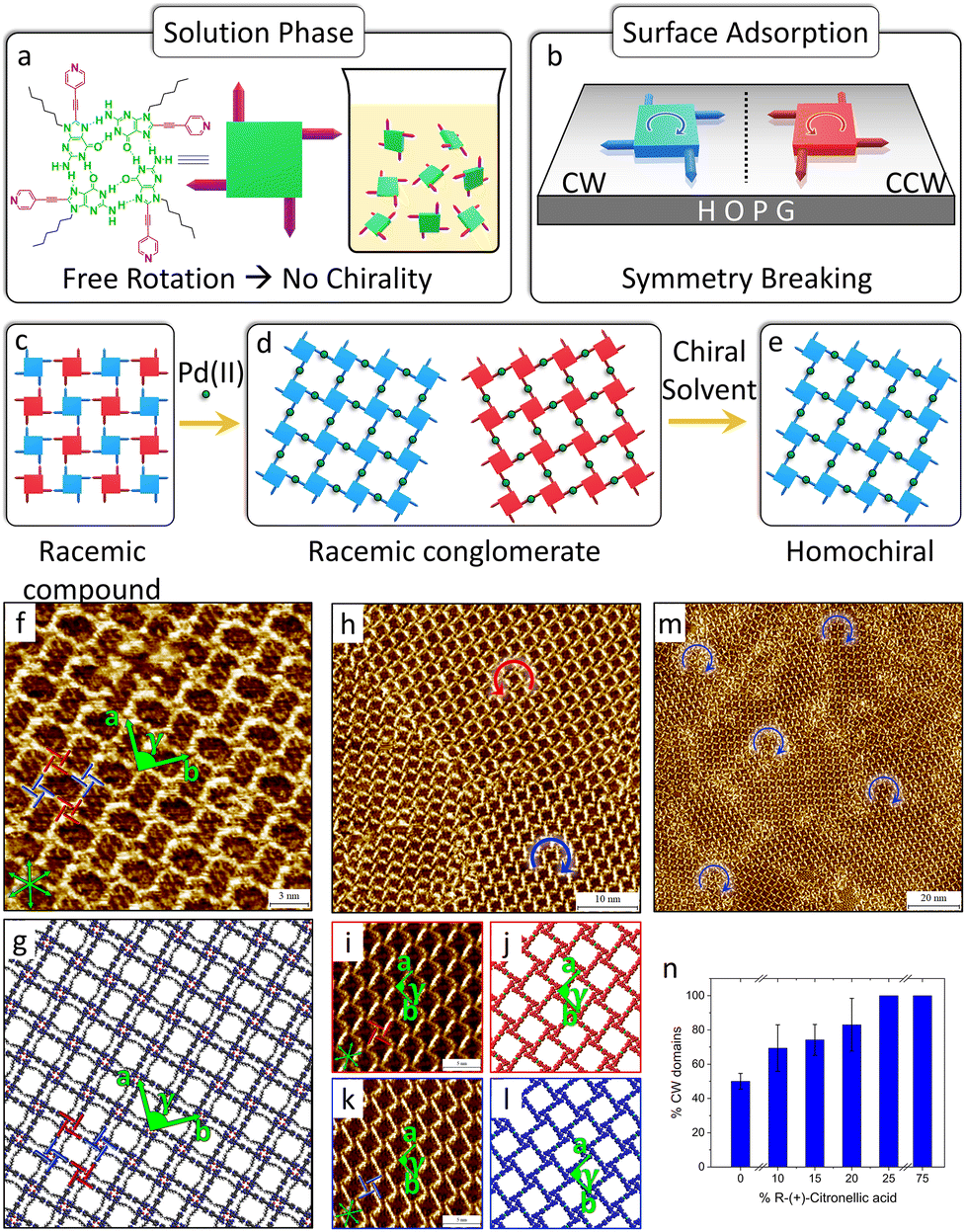 | ||
| Fig. 10 Enantiomers segregation and solvent induced chiral selection in the C6G4Py-Pd(II) MOCN. (a) Molecular structure of the C6G4Py hydrogen-bonded quartets. In the solution phase, they are free to rotate and are therefore prochiral. (b) Symmetry breaking in C6G4Py quartets upon surface confinement. The opposite enantiomers are indicated as clockwise (CW, blue) and counterclockwise (CCW, red). (c)–(e) Successive chiral transitions observed starting from the C6G4Py SAMN, following Pd(II) addition, and the use of an enantiopure solvent. (f) Representative high-resolution STM image of the racemic compound of C6G4Py quartets and (g) corresponding molecular model. Lattice parameters: a = 3.9 ± 0.1 nm, b = 3.9 ± 0.1 nm, γ = 90 ± 1° (p4gm plane group). (h) Representative large-scale STM image of the racemic conglomerate formed by mixing C6G4Py and Pd(II). (i), (k) High-resolution STM images of CCW and CW domains of the C6G4Py-Pd(II) MOCN, respectively, and (j), (l) corresponding molecular models (the alkyl chains and the counterions are omitted for clarity). (m) Large-scale STM image showing the formation of homochiral domains in (R)-(+)-citronellic acid. (n) Variation in the surface coverage of CW G-quartets as a function of volume % of (R)-(+)-citronellic acid in a heptanoic acid/(R)-(+)-citronellic acid solution mixture. Adapted with permission from ref. 80. Copyright©2022 American Chemical Society. | ||
In the absence of added metal ions, C6G4Py forms a SAMN where C6G4Py quartets adsorbed with opposite handedness co-crystallize and distribute periodically within the same phase, creating a racemic compound on HOPG, as depicted in Fig. 10(c), (f) and (g). When Pd(II) is introduced into the system, each Pd(II) connects two C6G4Py quartets, and spontaneous chiral resolution occurs upon this metal coordination. Enantiomorphous domains, forming racemic conglomerates, are characterized at the HOPG/heptanoic acid interface, as shown in Fig. 10(d), (h)–(l). The resulting chiral MOCN comprises homochiral hydrogen-bonded C6G4Py quartets bridged by Pd(II) centers that coordinate to the pyridyl moieties. Remarkably, chiral resolution was also observed following in situ addition of Pd(II) salt solution to the pre-formed C6G4Py SAMN.
Furthermore, complete chiral selection was achieved using enantiopure (R)-(+)-citronellic acid at low concentration (25% v/v, Fig. 10(e), (m) and (n)), while the opposite handedness resulted from using the S enantiomer. The possibility of hosting guest molecules inside the pores of the network was also confirmed.
In analogy with the works performed in UHV, STM at the liquid–solid interface offers the opportunity to identify and differentiate domain-specific chirality and even the opposite handedness of enantiomeric building blocks within one domain. Additionally, this work demonstrates the distinct advantages of liquid–solid interface studies: the ability to employ chiral solvents for controlling network handedness and achieving complete chiral amplification through solvent-mediated chirality transfer, which is fundamentally unavailable in UHV environments and provide an inexpensive, generic pathway toward homochiral coordination networks.
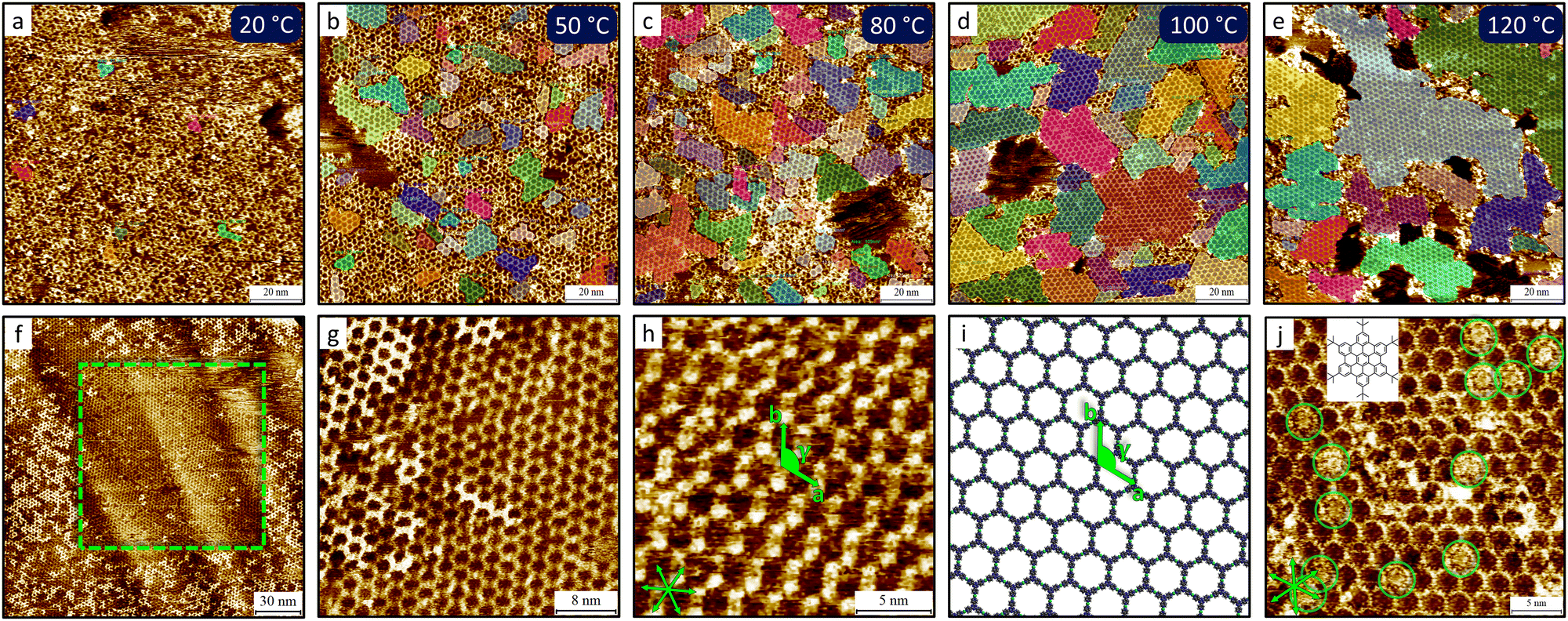 | ||
| Fig. 11 Molecular insights into the morphological changes of the TPT-Pd(II) porous MOCN. (a)–(e) Representative large-scale STM images showing the increase in crystallinity as a function of the annealing temperatures of samples prepared using the sequential protocol. The crystalline domains are marked by the coloured overlays. The images were acquired at the HOPG/1-phenyloctane interface. (f) Large-scale image showing the removal of the top layer of the TPT-Pd(II) MOCN prepared with the one-pot protocol within the nanoshaved area, marked by the dashed square; the top layer in the surrounding areas is intact. (g) High-resolution image of the bilayer, revealing the A–A stacking. (h) High-resolution image of the TPT-Pd(II) MOCN and (i) corresponding molecular model. The images were obtained at the HOPG/nonanoic acid interface. Lattice parameters: a = 2.6 ± 0.1 nm, b = 2.6 ± 0.1 nm, γ = 120 ± 1°. (j) High-resolution STM image of the TPT-Pd(II) MOCN hosting 6tBu-HBC molecules in its pores, as highlighted by the green circles. 6tBu-HBC molecular structure is shown in the inset. Imaging was carried out at the HOPG/1-phenyloctane interface. Adapted with permission from ref. 81. Copyright©2025 The Authors. Published by American Chemical Society. | ||
The sequential protocol involved separate drop-casting of TPT followed by Pd(II), with annealing at temperatures ranging from 20 to 120 °C. STM characterization at the HOPG/1-phenyloctane interface revealed that increasing the annealing temperature progressively enhanced network ordering, as illustrated by the selection of STM images shown in Fig. 11(a)–(e). Quantitative analysis demonstrated improved crystallinity through measurements of surface coverage, domain number and size distribution, and PH score, introduced in Section 3.1.
In contrast, the one-pot protocol involved premixing TPT and Pd(II) in a 2:3 ratio at 100 °C in nonanoic acid prior to deposition. Annealing was essential to obtain the TPT-Pd(II) MOCN. Compared to the sequential protocol, premixing yielded networks with different morphological characteristics, including vacancy defects and bilayer features (Fig. 11(f) and (g)). Apart from these differences, the porous honeycomb network featuring hexagonal cavities shown in Fig. 11(h) and (i) was obtained through both procedures.
The morphological differences arising from the two fabrication protocols suggest that the two synthetic routes likely operate through distinct growth pathways. The sequential approach appears to follow a chain-growth-like mechanism, wherein crystallization initiates at specific nucleation sites with subsequent binding of neighboring building blocks until grain boundary formation. Conversely, the one-pot synthesis in nonanoic acid likely proceeds through a step-growth-like mechanism, where oligomers are preformed in solution during annealing and subsequently assembled, and connected, on the surface during solvent evaporation.
Crucially, the study demonstrates the presence of accessible pores through STM imaging of guest molecule adsorption. Addition of hexa-tert-butyl-hexa-peri-hexabenzocoronene (6tBu-HBC) as a molecular guest on the pre-formed TPT-Pd(II) MOCN confirmed pore accessibility within the honeycomb structure (Fig. 11(j)). Notably, guest adsorption was observed exclusively in networks synthesized via the sequential protocol, highlighting notable differences between the two fabrication routes and their impact on pore accessibility.
This comprehensive investigation thus establishes key structure–property relationships governing MOCN synthesis at liquid–solid interfaces while demonstrating the critical role of STM characterization in understanding both underlying assembly mechanisms and functional properties that are potentially relevant to catalysis and molecular recognition applications.
The molecular-level insights provided by STM hold great potential for advancing research on metal–organic coordination systems. The emerging capability to fabricate porous MOCNs at the liquid–solid interface and characterize them under ambient conditions represents a significant methodological advancement, effectively bridging the research domains of MOCNs and MOFs. This methodological convergence enables unprecedented comparative investigations, allowing researchers to explore critical material properties across different dimensions. Specifically, STM emerges as a pivotal technique for systematic exploration of:
– Network stability: investigating how structural dimensionality influences material stability.
– Local features: for example, the identification of chiral centers and their differentiation.
– Guest adsorption mechanisms: examining molecular interactions and adsorption dynamics.
– Stimuli responsiveness: analyzing structural and functional changes under various external stimuli (light, chemical).
4. AFM characterization of MOFs
AFM is a powerful technique for characterizing materials, including MOFs, beyond layers that are only one (or a few) atoms thick. Beyond providing structural insights and probing electrical properties, AFM enables the investigation of dynamic phenomena such as crystal growth, phase transitions, and solvent-induced delamination.68,179–187 The local chemical composition can be analyzed by AFM-IR, offering a complementary method for characterizing MOFs.95This section analyzes recent advances in AFM and AFM-IR techniques for MOF characterization, with particular emphasis on liquid–solid interface studies. A distinction will be made between studies performed on MOF thin films vs. single crystals, revealing both similarities and differences between these two material classes.
Many studies are performed on HKUST-1, which represents one of the foundational and most extensively studied MOFs, establishing critical benchmarks for the field through its unique combination of chemical tunability and thermal stability up to 240 °C.188HKUST-1 can be synthesized as either a crystalline powder or a thin film, typically using copper acetate (CuOAc2) and BTC precursors. Its structure consists of paddlewheel dicopper(II) tetracarboxylate metal nodes interconnected by BTC linkers. The framework can be synthesized under a range of different conditions, and it has a well-defined structure, thus positioning HKUST-1 as an ideal MOF for exploring and advancing unconventional characterization methodologies in MOF research.189
The application of AFM to MOF characterization marked a pivotal advancement in understanding crystalline defects and enabled unprecedented real-time monitoring of growth pathways in HKUST-1 crystals.82,83 This breakthrough was significantly enhanced by the development of in situ growth protocols, which demonstrated substantial advantages over traditional ex situ preparation methods.190 Direct on-substrate growth not only yielded superior structural quality but also facilitated systematic optimization of synthesis procedures and growth conditions, representing a methodological leap forward in MOF fabrication control. Moreover, comparative studies conducted under UHV vs. ambient conditions revealed surface erosion phenomena upon air exposure.190 This finding established the critical importance of environmental control in accurate structural characterization, setting new standards for experimental protocols in the field.
4.1 Epitaxially-grown MOFs
Different synthetic procedures can yield MOFs in the form of thin films.43,191–196 As mentioned earlier, sample immobilization is a critical requirement to perform AFM characterization. Liquid-phase epitaxy (LPE) has been demonstrated as an efficient strategy for growing MOF thin films on solid substrates. These surface-mounted MOFs (SURMOFs) are synthesized using the layer-by-layer (LbL) technique on Au(111) substrates functionalized with self-assembled monolayers (SAMs) bearing functional groups that can coordinate with the metal nodes in the MOF.197 As illustrated in Scheme 5, this process involves sequential immersion of the substrates in two discrete solutions: one containing metal nodes and the other comprising organic ligands. A rinsing step is frequently incorporated after each immersion to eliminate unreacted precursors. Alternative approaches employ spray coating or spin coating techniques to deposit the solutions on the substrate. SURMOFs fabricated via LPE exhibit specific orientations, and their thickness can be precisely modulated by varying the number of immersion cycles. Furthermore, LPE helps to overcome the interpenetration issue encountered when targeting MOFs with large pore sizes.197,198 Additionally, as-synthesized SURMOFs are inherently amenable to AFM characterization since they are immobilized on solid substrates. | ||
| Scheme 5 LPE method for the growth of SURMOFs on SAM-functionalized Au(111). | ||
 | ||
| Fig. 12 Selective HKUST-1 growth with the LbL approach on patterned surfaces. (a) AFM topographic and (b) friction images for the SAM patterned substrate before SURMOF growth. (c) AFM topography for a patterned sample after 45 immersion cycles. (d) Series of topographic AFM images for different samples corresponding to n = 10, 20, 23, 30 and 45 immersion cycles, from left to right, respectively. The total color scale (total height range) is 110 nm for all the images. Due to the low topography of the 10 cycles sample, the inset shows the same image with the vertical scale magnified by a factor of two. (e) Film thickness as a function of the number of immersion cycles. The red dashed line corresponds to the proposed “half-layer” growth whereas the grey one would correspond to a one-unit cell or complete layer growth. (f) Root mean square (rms) surface roughness as a function of the number of immersion cycles calculated for different scan sizes (see inset). The black horizontal line corresponds to the rms of the starting substrate while the blue dashed line has been drawn just as a visual aid. Error bars are the standard deviation values. Adapted with permission from ref. 84. Copyright©2008 The Royal Society of Chemistry. | ||
HKUST-1 selectively grows on MHDA regions, as confirmed by topography AFM images, where the MOF thin films appear as bright squares, as shown in Fig. 12(c). This approach enabled film thickness quantification as a function of LbL cycles by measuring the height of the bright (MOF-covered) squares compared with the dark (MOF-free) areas. Notably, this measurement is impossible for extended films. Systematic thickness analysis across 10, 20, 23, 30, and 45 immersion cycles (Fig. 12(e)) revealed the mechanism underlying the growth process: after 20 cycles, thickness increase follows a strictly linear trend, establishing the existence of a “critical thickness” threshold beyond which a steady LbL growth regime dominates. The measured thickness increase of 1.317 nm per complete cycle, corresponding to half the HKUST-1 unit cell length (2.634 nm), provided direct experimental validation of the expected growth behavior based on the known crystal structure. This finding confirms the epitaxial relationship between substrate and MOF structure, where every second BTC layer is shifted relative to the first, requiring two LbL cycles to complete one crystallographic unit cell. AFM measurements also revealed that surface roughness reaches a steady value for high cycle numbers (Fig. 12(f)), providing direct evidence for LbL completion mechanisms rather than island growth modes.
Building upon this patterning approach, nanografting techniques offered an alternative strategy for in situ surface modification through AFM-mediated molecular replacement.199 This method involves removing matrix molecules in selected areas with an AFM tip while the sample remains immersed in COOH-terminated thiol solutions, enabling precise replacement of alkyl thiols with carboxyl-terminated alternatives. MHDA was grafted within a 1-octadecanethiol (ODT) matrix on Au(111), with subsequent HKUST-1 growth on these functionalized surfaces.
The MHDA/ODT combination revealed critical limitations, producing SURMOFs with elevated surface roughness (28.9 ± 3.9 nm) and detecting small crystals of comparable height on the ODT layer, indicating uncontrolled ODT displacement by MHDA molecules. This observation established that thiol selection represents a crucial parameter governing SURMOF quality, with molecular compatibility between matrix and patterned regions proving essential for maintaining pattern fidelity. These investigations demonstrated the broader potential of spatially controlled MOF growth while defining critical design principles for multi-component surface functionalization strategies.
Temperature effects proved particularly striking: synthesis conducted at an elevated temperature (50 °C) dramatically increased nucleation density, resulting in enhanced surface coverage and reduced overall roughness compared to ambient temperature conditions. Most significantly, temperature-dependent morphological control was demonstrated through distinct crystallite geometries: trigonal prismatic shapes dominated at 50 °C, while square pyramidal morphologies were characteristic of growth at 25 °C. This discovery challenged the prevailing paradigm that crystal morphology derives exclusively from substrate functionalization, revealing instead that thermal conditions exert equally critical influence over the final crystal architecture. These findings established temperature as a primary control parameter for MOF morphology engineering, expanding the toolkit for tailoring SURMOF properties beyond substrate-based approaches.
Thermal stability investigations yielded unexpected results that challenged established assumptions about HKUST-1 thermal properties. Material degradation occurred at surprisingly low temperatures (40–60 °C), despite reported bulk thermal stability. Critical methodological insights emerged from comparing in situ versus ex situ characterization: while in situ AFM revealed ∼65% reduction in grain features, ex situ measurements showed only ∼10% decrease. This discrepancy was attributed to scanning artifacts arising from enhanced thin film mobility at elevated temperatures, causing AFM tip-induced removal of surface features. It must be noted that the films used in this study were grown for just 5 LbL cycles and were intentionally made inhomogeneous through incomplete washing steps, which likely made them less stable than fully grown crystals. Nonetheless, these investigations demonstrated in situ AFM to be a potentially important tool for understanding MOF stability under realistic operating conditions while revealing the critical importance of experimental methodology in stability assessments.
The chemical sensitivity of AFM-IR enabled detection of (partially) unexchanged [Cu2(OAc)4] paddlewheels within HKUST-1 films. These paddlewheels, likely physisorbed onto the crystal even after washing and drying, contribute to defects in the SURMOF. To address this issue, the study suggests using Cu(NO3)2 as the metal precursor instead of Cu(OAc)2, as the former exhibits lower steric hindrance and weaker surface affinity, reducing defect formation.
Building upon these mechanistic insights, AFM-IR enabled sophisticated investigations of guest–host interactions through the fabrication of bifunctional surfaces containing both hydrophilic and hydrophobic MOF domains.204 Patterned Au(111) surfaces functionalized with 4-(mercaptomethyl)-benzoic acid (MMBA) and 4-mercaptopyridine (MPyr) SAMs, separated by 1-undecanethiol (UDT) SAM regions, were fabricated to epitaxially grow two different MOFs: the hydrophilic HKUST-1 and the hydrophobic ZIF-8, the latter composed of 2-methylimidazolate ligands and Zn2+ cations (Fig. 13(a)). The distinct morphologies of these MOFs allowed for differentiation in AFM topography, with HKUST-1 forming triangular {100} surfaces and ZIF-8 presenting octahedral crystals (indicated by the yellow and red markers in Fig. 13(c), respectively). AFM-IR provided definitive chemical confirmation of these assignments and revealed framework-specific responses to environmental changes.
 | ||
| Fig. 13 Top: Outline of the mixed MOF approach. (a) Different thiol compositions were used for Au substrate functionalization prior to LbL synthesis of SURMOFs. HKUST-1 (green) selectively grows on carboxylic acid-terminating SAMs, whereas ZIF-8 (blue) growth is promoted on pyridine-terminating SAMs. Methyl-thiols were used to suppress growth. (b) Left: In situ AFM-IR was carried out on these SURMOFs to deduce their interaction with D2O at the nanoscale. Right: Amphiphilic nature of the mixed HKUST-1/ZIF-8 surface revealed by in situ AFM-IR. (c) AFM image of 10 layers of HKUST-1/ZIF-8 grown on MPyr/MHDA. Point spectra were taken before and after vacuum/N2 purge cycles at the locations marked yellow for HKUST-1 and red for ZIF-8. (d) IR spectra showing the removal of water from HKUST-1 and the water-independent IR spectra of ZIF-8. (e) IR spectra of HKUST-1 and ZIF-8 after the introduction of 1000 ppm D2O. (f) Zoom-in IR spectra of the two areas marked in (e) highlighting the appearance of new peaks in the HKUST-1 spectrum. Adapted from ref. 204.©2020 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH. | ||
Water adsorption studies demonstrated the power of AFM-IR for probing guest–host dynamics: HKUST-1 exhibited spectral changes upon N2 purging attributed to water desorption, while ZIF-8 domains remained spectroscopically unchanged (Fig. 13(d)). D2O backfilling experiments provided molecular-level insights into hydration mechanisms, with new bands at 890 cm−1 (adsorbed D2O) and 1725 cm−1 (deuterated carboxylic acid groups) confirming water adsorption in HKUST-1 while ZIF-8 remained unaffected (Fig. 13(f)). These findings provide direct experimental validation of a previously proposed water adsorption mechanism, which can occur either at the axial or equatorial positions at Cu(II) centers. The latter instance promotes D2O dissociative adsorption, which eventually leads to the cleavage of metal–carboxylate bonds, contributing to the hydrolytic degradation of the framework. In summary, this work presents an efficient method to fabricate a bifunctional surface using epitaxial MOFs and provides new insights into the molecular processes occurring in MOF-based materials.
The integration of external stimuli with AFM-IR opened up new frontiers in understanding dynamic MOF behavior under operational conditions. Investigations of rotor dynamics in Cu2(F-BDC)2(DABCO) (F-BDC = 2-fluorobenzene-1,4-dicarboxylate and DABCO = 1,4-diazabicyclo[2.2.2]octane) thin films under external electric fields demonstrated the technique's versatility for probing stimuli-responsive behavior.205 Thin films of Cu2(F-BDC)2(DABCO) were epitaxially grown on gold wafers functionalized with MHDA as well as SrTiO3 substrates. By connecting both the substrate and AFM probe to an external power supply, force modulation through AFM setpoint adjustment enabled precise control over measurement conditions. AFM-IR measurements conducted under an applied electric field revealed inhibition of rotational motions parallel to the field direction, with this effect tunable through mechanical force exerted by the AFM probe. This work established direct correlations between external stimuli and molecular rotor dynamics, demonstrating AFM-IR as a powerful tool for understanding responsive MOF behavior under realistic operating conditions.
4.2 MOF nanoparticles
3D MOFs are commonly synthesized via solution-based methods at temperatures ranging from 25 to 220 °C, often using solvothermal conditions. The precursors are typically mixed in a reaction vessel or an autoclave for several hours or even days until crystals are formed. Alternative synthesis methods, such as microwave-assisted, mechanochemical, electrochemical, or ultrasound techniques, offer more efficient and environmentally friendly options by avoiding direct heating of the precursor mixture. These methods enable reactions to occur at lower temperatures, offering more economical and sustainable alternatives to solvothermal approaches.206,207The investigation of MOF crystal surfaces provides critical insights into material structure and structure–function relationships, as surface transformations may be indicative of related bulk processes occurring within the framework. Characterizing MOF nanoparticles with SPM techniques presents unique challenges, in particular, the immobilization of small crystals on substrates for high-resolution imaging. This limitation has driven the development of innovative approaches, including the use of SAM-functionalized substrates for direct crystal growth, enabling unprecedented real-time observations of nucleation and growth dynamics. In contrast with the LbL approach described in the previous section, growth of 3D MOFs on functionalized substrates occurs by immersing the substrates in a solution of both the metal and ligand precursors.
Building upon these observations, the authors employed SAM-functionalized gold substrates to gain deeper insights into the growth mechanism for HKUST-1.208 The key discovery was that crystal growth can initiate from single nucleation points rather than multiple simultaneous sites. The authors attributed this difference to the smaller variations in crystal size and lower defect concentration in the present study, which, in contrast, were both larger in the earlier studies. Furthermore, the discrepancy may stem from differences in the solvent system: the initial experiments used an N,N-dimethylformamide (DMF) solution, while the later investigations employed a water–ethanol mixture, highlighting the importance of the solvent as a critical parameter for controlling growth pathway selection.
The influence of temperature on MOF nucleation was systematically elucidated through AFM-IR imaging of HKUST-1 growth on MHDA SAMs across three distinct temperature regimes—13, 25, and 50 °C.209 This comprehensive study established that crystallization follows the Volmer–Weber model, producing 3D islands rather than uniform films. The temperature-dependent behavior revealed three distinct growth regimes: continuous nucleation and growth at 25 °C, creating both large grains and background “carpet” layers, incomplete growth at 13 °C due to kinetic limitations, and premature nucleation termination at 50 °C caused by SAM desorption.
From this work emerges an important limitation of SAM functionalization for anchoring MOF crystals to substrates, which is the thermal stability of the SAM. This moderate thermal stability limits the possibility of exploring various experimental conditions, including performing temperature-dependent studies.
MOF-5 was observed to follow a similar growth process.211 Additionally, it was found that the ligand-to-metal stoichiometry of the growth solution plays a crucial role in determining the morphology of the resulting crystals through differential directional growth rates. When the metal-to-ligand ratio is greater than 1, the growth rate along the [111] direction is slower than along the [110] direction, and large square terraces are formed. When the metal-to-ligand ratio is approximately 1, the growth rate along the [111] direction remains slower than along the [110] direction, and the resulting morphology consists of rhombus-shaped terraces; however, these are not squares. This indicates that subtle variations in the precursor ratio can significantly influence the final crystal structure and surface topography of MOF-5, opening up possibilities for MOF crystal shape engineering.
The controlled SBU approach (CSA) for MOF synthesis was validated through systematic liquid AFM studies of surface nucleation of MOF-5.212 This approach consists of the use of monotopic ligands and metal ions to form pre-assembled coordination complexes. Such pre-organization results in faster reaction times during MOF growth. To elucidate the mechanism of this process, nucleation seeds were grown on glass substrates, and 2D surface nucleation was investigated using liquid AFM in diluted MOF-5 growth solution. This study revealed that the CSA does not proceed through direct substitution of surface dangling BDC ligands by [ZnO4(O2CC6H5)6] SBUs as initially hypothesized. Instead, topographical measurements revealed that partial SBU dissociation precedes framework incorporation, substantially revising the understanding of how pre-assembled building blocks participate in MOF construction.
The study established that supersaturation conditions play a decisive role in directing this unconventional growth mode, providing quantitative parameters for interfacial engineering in MOF heterostructures. Moreover, these results show that isostructural MOFs exhibit nearly identical growth mechanisms.
By controlling lattice mismatch, it becomes possible to engineer specific interfacial orientations and properties in core–shell architectures. This mechanistic insight bridges the gap between single-phase MOF growth and complex multi-component systems, establishing a framework for the design of MOF heterostructures with tailored interfacial properties.
 | ||
| Fig. 14 Real-time in situ imaging of guest-induced flexing mechanism in Ga-MIL-53. (a)–(h) Error signal AFM images of a {011} facet during pore expansion induced by EtOH adsorption. Image size: 6.0 × 6.0 μm2. The white dashed lines in (a)–(f) indicate the wavefronts and the white arrows their propagation directions. The green dashed line in (a) indicates a defect. Inset in (h) is an optical microscope image of a Ga-MIL-53 immerged in DMF prior to transformation. (i) Schematic representation of the expansion process. (j)–(q) Error signal AFM images of a {011} facet during pore contraction induced by DMF. Transforming parts of the crystals are indicated by white arrows in (k) and (m). Inset in (q) is an optical microscope image of a Ga-MIL-53 immerged in EtOH prior to transformation. (r) Schematic representation of the contraction process. Adapted from ref. 68. Copyright©2025 The Authors. Published by American Chemical Society. | ||
The expansion transformation from Ga-MIL-53·0.96DMF to Ga-MIL-53·xEtOH revealed wavefronts traveling at constant velocities of approximately 0.9 μm min−1 across {011} facets, shown in the image sequence in Fig. 14(a)–(h). This image sequence indicated Case II non-Fickian diffusion behavior, where transformation kinetics are controlled by framework expansion rather than guest diffusion. Quantitative analysis demonstrated that individual wavefronts involve at least 76 unit cells in depth and 1429 unit cells laterally, establishing the cooperative scale of structural transformations. The observation that consecutive rows of unit cells parallel to the (011) plane transform cooperatively, followed by adjacent row transformation in the [100] direction (Fig. 14(i)), provided direct evidence for the row-by-row shear mechanism underlying framework flexibility.
The reverse contraction transformation from Ga-MIL-53·xEtOH to Ga-MIL-53·0.96DMF proceeded through a crystallographically distinct mechanism involving shearing parallel to the (01![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) at 0.6 μm min−1 (Fig. 14(r)). The image sequence of the transformation is presented in Fig. 14(j)–(q). The measured interface angle of 162.1° ± 1.1° between coexisting phases showed excellent agreement with crystallographically predicted values (160.9°), validating the coexistence of phases with different expansion degrees within single crystals. This phase coexistence represents a paradigm shift in understanding MOF flexibility, demonstrating that transformation gradients rather than uniform phase changes characterize flexible framework behavior.
) at 0.6 μm min−1 (Fig. 14(r)). The image sequence of the transformation is presented in Fig. 14(j)–(q). The measured interface angle of 162.1° ± 1.1° between coexisting phases showed excellent agreement with crystallographically predicted values (160.9°), validating the coexistence of phases with different expansion degrees within single crystals. This phase coexistence represents a paradigm shift in understanding MOF flexibility, demonstrating that transformation gradients rather than uniform phase changes characterize flexible framework behavior.
The study revealed that structural transformations create characteristic surface textures, including ridges formed by opposing shears on adjoining unit cell layers parallel to (011) planes. These features serve as stress relief mechanisms accommodating volume changes during transformation and provide diagnostic signatures of flexibility mechanisms.
The ability demonstrated in this work to visualize phase boundaries, measure transformation kinetics, and quantify structural parameters during dynamic processes establishes in situ AFM as a valuable technique for understanding framework flexibility. These insights have profound implications for designing flexible MOFs with controlled transformation properties and predicting their behavior in applications requiring reversible structural changes.
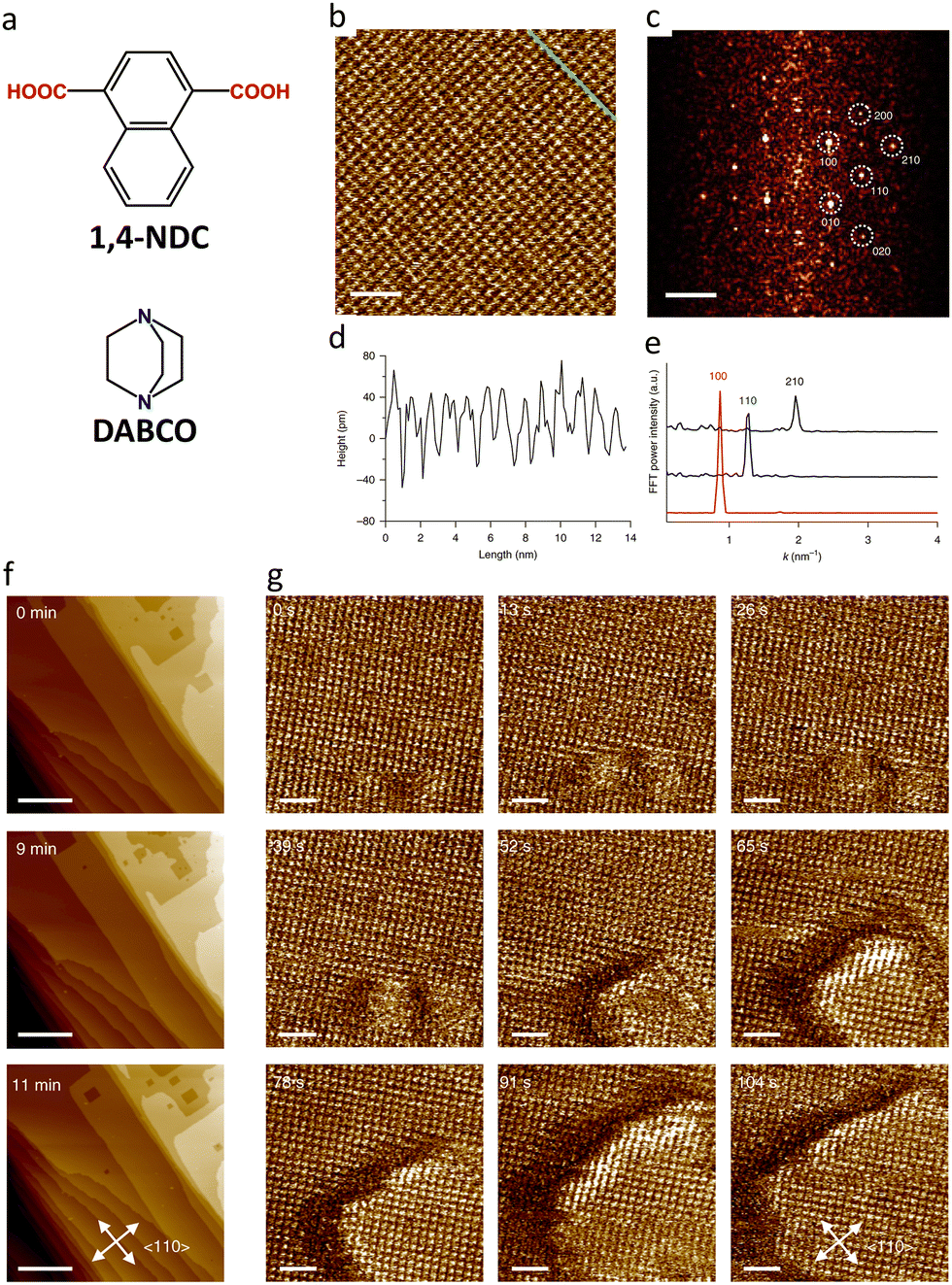 | ||
| Fig. 15 In Situ molecular resolution AFM imaging of Zn2(1,4-NDC)2(DABCO). (a) Molecular structures of DABCO and 1,4-NDC (b) High-resolution AFM topography of Zn2(1,4-NDC)2(DABCO) imaged in DMF (scale bar: 5 nm) and (c) corresponding 2D FFT spectrum (scale bar: 1 nm−1). (d) Height profile along the green line in (b). (e) 1D FFT power spectra. (f) Large scale AFM topography images captured 0, 9, and 11 minutes after the injection of a 200 mM BPY solution (scale bars: 250 nm). (g) High-resolution AFM phase images of the Zn2(1,4-NDC)2(DABCO) surface under the same conditions as in (g) collected 13 seconds apart, depicting the BPY-induced layer-by-layer exfoliation at the molecular level (scale bar: 5 nm). Adapted with permission from ref. 69. Copyright©2018, The Author(s), under exclusive license to Springer Nature Limited. | ||
In this work, a MOF crystal was glued on a steel disk and imaged in DMF. A representative high-resolution AFM image is shown in Fig. 15(b), while Fig. 15(c) is the corresponding 2D fast Fourier transform (2D FFT). Subsequently, a solution of biphenyl was injected into the AFM liquid cell. During real-time imaging, it was found that biphenyl guest injection induced reversible unit cell transformations from tetragonal (α = 89.6°) to rhombohedral (α ≈ 87°) structures exclusively at the crystal surface, while powder XRD analysis confirmed that bulk crystals remained unchanged even after extended exposure. This surface-selective responsiveness demonstrates that the reactivity of framework surfaces is different from bulk phases, experiencing dynamic coordination equilibria and enhanced structural flexibility due to reduced bonding connectivity at interfaces.
These observations suggest that surface structural changes possibly follow stochastic rather than cooperative mechanisms, contrasting sharply with the cooperative LbL shear transformations observed in bulk flexible frameworks. Deformation events occurred within approximately 10 minutes of guest concentration changes, with statistical analysis indicating that individual, competing adsorption/desorption of guest and solvent molecules seemingly trigger localized lattice modifications. Perhaps most remarkably, the introduction of competing ligands (4,4′-bipyridine (BPY) and 1,2-di(4-pyridyl)ethylene (DPYE)) enabled real-time visualization of LbL delamination processes along specific crystallographic directions. The delamination process is evident from the large-scale AFM topography images shown in Fig. 15(f), where pits with regular edges form and grow with time. The phase images presented in Fig. 15(g) show the formation of a delamination pit with molecular resolution. The directional preference for 〈110〉 delamination was rationalized through surface energy considerations based on sub-building unit saturation numbers, where {110} planes with two unsaturated coordination sites exhibit higher surface energies than {100} planes with single unsaturated sites.
This work provides a new perspective on our understanding of MOF surface behavior, demonstrating that surfaces represent distinct chemical environments with unique responsiveness and transformation mechanisms that complement but differ from the cooperative bulk flexibility mechanisms observed in framework transformations.
From the works reviewed in this section, it emerges that AFM characterization of MOF thin films and nanoparticles is well-established. This technique enables not only the determination of crystal morphological features—such as roughness and height—but also the elucidation of dynamic processes essential to MOF research, including crystal growth, guest adsorption, phase transitions, solvent-induced degradation, and delamination. In most studies, epitaxial growth and MOF immobilization have been performed on gold substrates functionalized with SAMs. Despite experimental evidence highlighting the influence of the substrate on MOF morphology, the exploration of alternative substrates and anchoring strategies remains limited. Moreover, manually gluing a single crystal onto a sample disk is only feasible when relatively large crystals can be obtained.
The potential for molecular-resolution imaging of MOF surfaces remains largely unexplored, likely due to the technical challenges associated with the approach. Achieving molecular resolution in liquid AFM requires firm immobilization of the crystals on the substrate in such a way that the crystal surface remains parallel to the scan direction.
5. Discrete metallosupramolecular architectures
Like for extended MOCNs, DMSAs have been investigated by SPM. In this section, studies will be presented that can be broadly categorized into two groups. Firstly, the use of ambient STM to investigate the on-surface self-assembly of preformed metal–organic architectures that have already been structurally characterized. Secondly, seminal work primarily demonstrating how UHV-STM can be employed to structurally characterize metal–organic architectures will be discussed.5.1 Self-assembled networks of DMSAs
Pre-formed metal–organic architectures, structurally characterized in solution, the gas phase, and the solid-state by techniques such as NMR spectroscopy, MS, and SC-XRD, respectively, have been demonstrated to self-assemble into networks at the liquid–solid interface, which can be characterized by STM under ambient conditions.76–78,214–218 On-surface assemblies characterized by STM (performed in solution or in air) include those consisting of 2D—rhombic (Fig. 16(a))76–78 and rectangular (Fig. 16(b))78,215—metallosupramolecular species and also 3D—trigonal bipyramidal (Fig. 16(c))78 and trigonal prismatic218—architectures. | ||
| Fig. 16 X-ray crystal structures of discrete metallosupramolecular species assembled into SAMNs on surfaces and studied by ambient STM. (a) Square.219 (b) Rectangle.220 (c) Trigonal bipyramidal cage.221 Solvent, counterions, disorder and hydrogen atoms are omitted for clarity. Pt(II): silver, C: grey, N: blue, P: orange, O: red. | ||
In work investigating the on-surface organization of metal–organic squares, the opening of these squares to form short oligomer chains under acidic conditions was revealed by STM and further supported by NMR studies.76 The high resolution of the STM images made the inspection of individual metal–organic species possible, and thus each species could be assigned as either the closed metal–organic square or oligomer chain.
5.2 Structural characterization of DMSAs
In the above examples, the metal–organic architectures had been structurally characterized prior to their assembly on the surface; it is the on-surface assembly of these species that is then characterized by STM. SPM techniques have, however, also been used to structurally characterize discrete metal–organic species, which in some cases have highly complex structures.:2.225 Two of the DLL1 ligand arms form part of the outer ring of the double-layered macrocycle, and the remaining two arms form part of the inner ring. When six DLL1 ligands and twelve Zn(II) cations combine to form the double-layered macrocycle DLM1, four distinct isomers (Fig. 17(b)–(i)) arise from two conformational isomers of ligand DLL1 (Fig. 17(a)). Individual ligand units are in the same conformation (conformer 1 in Fig. 17(a)) within three of the isomers (Fig. 17(b), (d) and (f)); their difference comes from the relative orientation, and thus connectivity, of the six DLL1 units that form the double-layered hexagon. In the fourth DLM1 isomer (Fig. 17(h)), the ligand units adopt a different conformation (conformer 2 in Fig. 17(a)) compared with the conformations in the first three DLM1 isomers.
 | ||
| Fig. 17 Isomer assignment of individual metal–organic double-layered hexagons. (a) Ligand DLL1 can exist in two distinct two-dimensional conformations. Ligand DLL1 reacts with Zn(NO3)2·6H2O to form Zn12DLL16 double-layered macrocycles (DLM1). (b), (d), (f), (h) Schematic representations of the four isomers of the metallosupramolecular complex DLM1. (c), (e), (g), (i) STM images containing the double-layered macrocycle DLM1 isomers displayed in b, d, f and h, respectively. The red and blue dashed lines are used to guide isomer assignment. Scale bars: 5 nm. Adapted with permission from ref. 225. Copyright©2021 American Chemical Society. | ||
UHV-LT-STM studies enabled the determination of the isomeric configuration of individual double-layered macrocycles (Fig. 17(c), (e), (g) and (i)), which was not possible using NMR spectroscopy and MS techniques. The authors assigned the isomeric configuration of a given DLM1 on the basis of the angles between the straight lines connecting the two nearest neighbor “inner” and “outer” TPY-Zn(II)-TPY (where, TPY = 2,2′:6′,2′′-terpyridine moiety) junctions (red dashed lines in Fig. 17), which appear as bright spots in the STM images, and the lines describing a polygon by connecting the “outer” Zn(II) cations (blue dashed lines in Fig. 17).
Electrospray ionization-MS (ESI-MS) studies indicated that a hexamer:heptamer:octamer mixture formed when ligand DLL1 was replaced by a ligand in which all three arms of the central core (colored black in the structures in Fig. 17(a)) were shortened. STM studies confirmed the presence of all three of these Zn2xLx double-layered macrocycles. The formation of metal–organic structures, including the species with higher nuclearity, containing the ligand in conformation 2 only was attributed to the reduction in unfavorable steric clashes between TPY-Zn(II)-TPY junctions on the outer and inner rings.
 | ||
| Fig. 18 Metal–organic hexagonal grid HG1; the relative positions of Ru(II) and Fe(II) cations, and thus the isomeric configuration, can be assigned for individual hexagonal grids.226 (a) Molecular structures of the organic ligands used to assemble metal–organic intermediate I1 and thus grid HG1. (b) TPY-Ru(II)-TPY and TPY-Fe(II)-TPY junctions. (c) Schematic representation of metal–organic intermediate I1. (d) Schematic representation of one of the several possible isomeric configurations of metal–organic grid HG1. | ||
When six intermediate I1 units are combined to form a hexagonal grid HG1, distinct isomers form based upon the relative position of the six Fe(II) cations in the aforementioned site type. A schematic representation of one isomer of hexagonal grid HG1 is displayed in Fig. 18(d). The presence of multiple isomers was hypothesized to broaden the signals in the 1H NMR spectrum of HG1, due to the overlapping of signals for the different isomers. Distinct sets of signals for each isomer could not be identified in the 1H NMR spectrum. UHV-LT-STM studies indicated the presence of hexagonal grid structures and the identification of the isomeric configuration adopted by a given HG1 complex was achieved by combining STM with STS. STS was used to determine the energy gap between the HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) at each metal center, which allowed each to be assigned as either Fe(II) or Ru(II), in turn allowing the assignment of the isomeric configuration of the complex being investigated. In a separate study, distinct isomers of hexagonal grids containing three different metal cations were similarly distinguished by using a combination of STM and STS data.227 Hexameric units, with a similar core structure to intermediate I1 before metalation with Fe(II), were used to form (metallo-)polymers.228 These units were appended with two azide functional groups and underwent a copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) “click” reaction with a pentaethylene glycol derivative, terminated at each end by alkyne groups. Combined STM and STS studies similar to those conducted in the above work provided insights into the metal–organic species formed after the addition of Fe(II) cations to the (metallo-)polymers. The study demonstrated that species formed in which the Fe(II) cation was chelated by TPY units in the same hexameric unit to close the hexagon. It also occurred that hexameric units in a linear conformation were instead joined together.
The combination of synchrotron X-rays with STM (SX-STM) can provide detailed information about a single atom/ion.229 The elemental identities of a Fe(II) cation chelated by two TPY units in intermediate I1, and Tb(III) coordinated by three brominated pyridine-2,6-dicarboxamide ligands, were confirmed using STM-X-ray absorption spectroscopy (STM-XAS). Furthermore, the chemical states of Fe(II) and Tb(III) were confirmed by STM-near-edge X-ray absorption fine structure (STM-NEXAFS) spectra.
This section has demonstrated the power of SPM, in particular STM and STS, for the structural characterization of discrete metal–organic assemblies. The work discussed above indicates that UHV-LT-STM can be employed as a complementary technique to solution and gas-phase characterization techniques, namely NMR spectroscopy and MS, respectively. In several cases, it appears that the structures present in solution are the same as those observed on the surface, following their deposition.
6. Conclusions
This review has examined the current state and methodological advances in SPM techniques for the characterization of MOCNs, MOFs at various length scales, and DMSAs, summarized in Table 1. The analysis reveals that SPM methodologies have emerged as indispensable tools for elucidating structure–function relationships in these materials, complementary to X-ray and electron diffraction techniques. They offer unparalleled molecular-resolution insights into material surfaces that bridge the gap between 2D surface-supported networks and 3D bulk frameworks.| Technique | Material | Experimental conditions | Information | Examples | Ref. |
|---|---|---|---|---|---|
| STM | MOCNs | UHV | Connectivity of the building blocks | Imaging of distinct crystalline and amorphous phases, coordination geometries, Kagome lattices, and quasicrystalline networks. | 104, 105, 115, 123–127 |
| Chirality | Distinction of single enantiomers and enantiomorphous phases | 71, 74, 145, 146 | |||
| Concentration and temperature | Exploit temperature and concentration as tunable parameters to fabricate distinct surface-supported MOCNs | 125, 147–150 | |||
| Stability | Competition between kinetic and thermodynamic factors | 151–153 | |||
| Substrate effect | Formation of new phases favored by substrate-building block interactions | 114–157 | |||
| Quantification of crystallinity | Distinction and quantification of crystalline and amorphous phases in multicomponent systems | 159 | |||
| Structure–function relationship | Magnetic anisotropy and magnetic coupling; tuning the magnetic properties using the isoreticular approach. | 96, 119, 124–126, 128, 132, 133 | |||
| Liquid–solid interface | Structural control | Effect of synthesis protocol on the self-assembly outcome, control in the formation and quantification of amorphous and crystalline phases. | 79–81 | ||
| Chirality | Metal-induced enantiomers separation and fabrication of enantiopure MOCNs aided by solvent selection. | 80 | |||
| Porosity | Adsorption of guest molecules in the MOCN free pores | 80, 81 | |||
| DMSAs | UHV | Structural characterization of DMSAs | Structure-type characterization | 222–224 | |
| Isomers assignment | 225–227 | ||||
| Liquid–solid and air–solid interface | Self-assembled networks of DMSAs | Impact of assembly conditions on SAMNs of DMSAs | 76, 215 | ||
| Co-adsorption of different types of discrete architectures | 78, 217 | ||||
| AFM | MOF thin films | Liquid–solid interface | Epitaxial growth | Use of patterned substrates | 84, 199 |
| Nanoscale insights into nucleation and growth mechanisms | Temperature dependence of the LPE outcome | 200,201 | |||
| Stability | High-temperature and high-pressure tests | 202 | |||
| Alternative synthesis routes | Vapor-assisted epitaxial growth of MOF thin films | 203 | |||
| MOF nanoparticles | Observation of nucleation and growth | Distinction between nucleation and growth processes | 83 | ||
| Relationship between nucleation and growth processes and experimental conditions | 83, 208 | ||||
| Elucidating the nucleation and growth mechanisms | Molecular insights into the nucleation and growth mechanisms and comparative studies across different MOFs | 210–213 | |||
| Real-time observation of surface processes | Framework flexibility | 68 | |||
| Molecular-resolution imaging of surface dynamics | 69 | ||||
| AFM-IR | MOF thin films | Liquid–solid interface | Nanoscale Insights into nucleation and growth mechanisms | Elucidation of the LPE mechanism | 85 |
| Chemical information | Water adsorption in samples patterned with different MOFs | 85, 204 | |||
| MOF nanoparticles | Stimuli responsiveness | Characterization of molecular motion under an applied electric field | 201 | ||
| Mechanistic insights | Temperature dependence of nucleation and growth processes | 209 |
The integration of STM and AFM techniques has considerably transformed our understanding of metal–organic coordination systems at the nanoscale. STM characterization under UHV conditions has demonstrated exceptional capabilities in providing molecular-level structural information for MOCNs, enabling the identification of local features and polymorphs, chirality differentiation, and the characterization of complex architectures, including quasicrystalline and amorphous phases. Similarly, UHV-STM has been demonstrated to be a valuable tool for the characterization of DMSAs, including complex mixtures of isomers. Notably, the extension of STM investigations of MOCNs to the liquid–solid interface represents a significant methodological advancement, allowing for real-time monitoring of solvent effects on network formation and guest adsorption and providing the capability to employ synthetic conditions that align with conventional MOF synthesis approaches.
AFM has proven to be valuable for characterizing MOF thin films and single crystals, offering unique capabilities for investigating dynamic processes such as crystal growth, guest adsorption, phase transitions, and structural degradation. The AFM-IR combination has further expanded the SPM analytical toolkit, enabling simultaneous topographical and chemical characterization of surface-supported MOF thin films and crystal surfaces with nanoscale spatial resolution.
Despite these remarkable advances, several challenges remain that limit the full potential of SPM characterization of metal–organic systems. The limited number of successful demonstrations of porous MOCN fabrication and STM characterization at the liquid–solid interface underscores the need for systematic methodological development in this area, which include targeting the control of molecular dynamics at the interface and achieving reproducible network formation in the presence of competing solvation and surface interactions. Such insights would be beneficial because MOCNs fabricated at the liquid–solid interface are more closely related to MOFs and in-depth studies on the structures and dynamics of the former can be relevant to advance our understanding of the latter.
An additional challenge relates to the statistical analysis using such local, small-scale techniques. The field currently lacks standardized methodologies for quantitatively assessing structural order and crystallinity in surface-supported monolayers, including MOCNs. This limitation hinders reproducible comparisons across different studies and impedes the development of structure–property correlations that are essential for rational material design.
The immobilization of MOF samples on solid substrates remains the primary technical barrier to achieving consistent molecular-resolution imaging of MOF particles. Current anchoring strategies, predominantly relying on SAMs on gold substrates, lack the versatility required for comprehensive investigations across diverse MOF systems. Furthermore, the substrate influence on MOF morphology and properties, while documented, remains inadequately understood and it would be beneficial for it to be explored in future works.
The convergence of experimental SPM techniques with computational modelling and machine learning approaches presents promising perspectives for advancing MOF characterization. Computational methods will be crucial for providing mechanistic insights into observed structural phenomena and for interpreting complex experimental data. Machine learning integration holds potential for the development of automated quantification algorithms and establishment of standardized analytical protocols for objective structural assessments of both extended networks and discrete supramolecules.
The continued refinement of SPM methodologies, coupled with the integration of complementary analytical techniques and computational approaches, will undoubtedly unlock new frontiers in our understanding of metal–organic systems, ultimately contributing to the design of materials with unprecedented functionality and performance in applications.
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Acknowledgements
This work was in part supported by Research Foundation – Flanders (FWO) (G0H2122N) and FNRS under the Excellence of Science (EOS) program (40007495). Additional support by FWO (G0A4120N, G0A5U24N, I006922N, G0K9822N) is acknowledged. A. C. acknowledges financial support through a FWO Fellowship (1159922N), J. A. D. acknowledges funding by the European Union under Horizon Europe, grant agreement ID: 101155157 (SURF-2DHYB). Financial support from KU Leuven-Internal Funds (C14/23/093) is acknowledged. S. D. F. acknowledges funding by the European Union under the Horizon Europe grant 101046231 (FantastiCOF) and M-ERA.NET 2021 (SuperSuper, FWO G0K9822N).Notes and references
- J. Anastassopoulou and T. Theophanides, Bioinorg. Chem., 1995, 209–218 CrossRef CAS.
- J. A. Cotruvo, ACS Cent. Sci., 2019, 5, 1496–1506 CrossRef CAS.
- G. Rando, S. Sfameni and M. R. Plutino, Gels, 2022, 9, 9 CrossRef.
- D. Pathania, A. K. Srivastava and A. Sharma, Curr. Res. Green Sustainable Chem., 2021, 4, 100073 CrossRef CAS.
- J. Wang, T. Feng, J. Chen, V. Ramalingam, Z. Li, D. M. Kabtamu, J. H. He and X. Fang, Nano Energy, 2021, 86, 106088 CrossRef CAS.
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O’Keeffe, M. Paik Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef CAS.
- E. Miguel-Casañ, G. R. F. Orton, D. E. Schier and N. R. Champness, Adv. Mater., 2025, 2414509 CrossRef.
- S. L. Griffin and N. R. Champness, Coord. Chem. Rev., 2020, 414, 213295 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490–2509 CrossRef CAS.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- O. M. Yaghi, H. Li, C. Davis, D. Richardson and T. L. Groy, Acc. Chem. Res., 1998, 31, 474–484 CrossRef CAS.
- S. Kitagawa and M. Kondo, Bull. Chem. Soc. Jpn., 1998, 71, 1739–1753 CrossRef CAS.
- R. Freund, S. Canossa, S. M. Cohen, W. Yan, H. Deng, V. Guillerm, M. Eddaoudi, D. G. Madden, D. Fairen-Jimenez, H. Lyu, L. K. Macreadie, Z. Ji, Y. Zhang, B. Wang, F. Haase, C. Wöll, O. Zaremba, J. Andreo, S. Wuttke and C. S. Diercks, Angew. Chem., Int. Ed., 2021, 60, 23946–23974 CrossRef CAS.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- R. B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC.
- L. H. Chen, M. H. Sun, Z. Wang, W. Yang, Z. Xie and B. L. Su, Chem. Rev., 2020, 120, 11194–11294 CrossRef CAS.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O’Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS.
- J. Ha, J. H. Lee and H. R. Moon, Inorg. Chem. Front., 2020, 7, 12–27 RSC.
- F. M. Amombo Noa, M. Abrahamsson, E. Ahlberg, O. Cheung, C. R. Göb, C. J. McKenzie and L. Öhrström, Chem, 2021, 7, 2491–2512 CAS.
- Y. Luo, M. Ahmad, A. Schug and M. Tsotsalas, Adv. Mater., 2019, 31, 1901744 CrossRef.
- G. Chakraborty, I.-H. Park, R. Medishetty and J. J. Vittal, Chem. Rev., 2021, 121, 3751–3891 CrossRef CAS.
- P. Payamyar, B. T. King, H. C. Öttinger and A. D. Schlüter, Chem. Commun., 2016, 52, 18–34 RSC.
- O. Shekhah, J. Liu, R. A. Fischer and C. Wöll, Chem. Soc. Rev., 2011, 40, 1081–1106 RSC.
- C. Simms, A. Mullaliu, S. Swinnen, F. de Azambuja and T. N. Parac-Vogt, Mol. Syst. Des. Eng., 2023, 8, 270–288 RSC.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef CAS PubMed.
- H. G. T. Ly, G. Fu, A. Kondinski, B. Bueken, D. De Vos and T. N. Parac-Vogt, J. Am. Chem. Soc., 2018, 140, 6325–6335 CrossRef CAS.
- P. Wu, Y. Li, J. J. Zheng, N. Hosono, K. ichi Otake, J. Wang, Y. Liu, L. Xia, M. Jiang, S. Sakaki and S. Kitagawa, Nat. Commun., 2019, 10, 1–8 CrossRef.
- L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC.
- Y. Gu, J. Zheng, K. Otake, M. Shivanna, S. Sakaki, H. Yoshino, M. Ohba, S. Kawaguchi, Y. Wang, F. Li and S. Kitagawa, Angew. Chem., Int. Ed., 2021, 60, 11688–11694 CrossRef CAS.
- J. Navarro-Sánchez, A. I. Argente-García, Y. Moliner-Martínez, D. Roca-Sanjuán, D. Antypov, P. Campíns-Falcó, M. J. Rosseinsky and C. Martí-Gastaldo, J. Am. Chem. Soc., 2017, 139, 4294–4297 CrossRef PubMed.
- V. Rubio-Giménez, S. Tatay and C. Martí-Gastaldo, Chem. Soc. Rev., 2020, 49, 5601–5638 RSC.
- D. Sheberla, J. C. Bachman, J. S. Elias, C. J. Sun, Y. Shao-Horn and M. Dinca, Nat. Mater., 2017, 16, 220–224 CrossRef CAS.
- L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS.
- Y.-H. Xiao, Z.-G. Gu and J. Zhang, Nanoscale, 2020, 12, 12712–12730 RSC.
- G. Genesio, J. Maynadié, M. Carboni and D. Meyer, New J. Chem., 2018, 42, 2351–2363 RSC.
- P. De Luna, W. Liang, A. Mallick, O. Shekhah, F. P. García De Arquer, A. H. Proppe, P. Todorović, S. O. Kelley, E. H. Sargent and M. Eddaoudi, ACS Appl. Mater. Interfaces, 2018, 10, 31225–31232 CrossRef CAS.
- L. Dong, Z. A. Gao and N. Lin, Prog. Surf. Sci., 2016, 91, 101–135 CrossRef CAS.
- A. Dmitriev, H. Spillmann, N. Lin, J. V. Barth and K. Kern, Angew. Chem., Int. Ed., 2003, 42, 2670–2673 CrossRef CAS PubMed.
- X. B. Wang, B. Xia, C. K. Lyu, D. Kim, E. Li, S. Q. Fu, J. Y. Chen, P. N. Liu, F. Liu and N. Lin, Commun. Chem., 2023, 6, 73 CrossRef CAS.
- C. Lyu, Y. Gao, Z. Gao, S. Mo, M. Hua, E. Li, S. Fu, J. Chen, P. Liu, L. Huang and N. Lin, Angew. Chem., Int. Ed., 2022, 134, 1–5 Search PubMed.
- W. Pang, B. Shao, X. Q. Tan, C. Tang, Z. Zhang and J. Huang, Nanoscale, 2020, 12, 3623–3629 RSC.
- R. Dong, M. Pfeffermann, H. Liang, Z. Zheng, X. Zhu, J. Zhang and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 12058–12063 CrossRef CAS PubMed.
- S. Stepanow, M. Lingenfelder, A. Dmitriev, H. Spillmann, E. Delvigne, N. Lin, X. Deng, C. Cai, J. V. Barth and K. Kern, Nat. Mater., 2004, 3, 229–233 CrossRef CAS PubMed.
- K. E. Plass, A. L. Grzesiak and A. J. Matzger, Acc. Chem. Res., 2007, 40, 287–293 CrossRef CAS.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- H. Wang, Y. Li, N. Li, A. Filosa and X. Li, Nat. Rev. Mater., 2020, 6, 145–167 CrossRef.
- B. S. Pilgrim and N. R. Champness, ChemPlusChem, 2020, 85, 1842–1856 CrossRef CAS PubMed.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- C. Gropp, S. Canossa, S. Wuttke, F. Gándara, Q. Li, L. Gagliardi and O. M. Yaghi, ACS Cent. Sci., 2020, 6, 1255–1273 CrossRef CAS PubMed.
- E. Ploetz, H. Engelke, U. Lächelt and S. Wuttke, Adv. Funct. Mater., 2020, 30, 1909062 CrossRef CAS.
- M. M. Modena, B. Rühle, T. P. Burg and S. Wuttke, Adv. Mater., 2019, 31, 1–26 Search PubMed.
- D. Y. Kang, J. S. Lee and L. C. Lin, Langmuir, 2022, 38, 9441–9453 CrossRef CAS PubMed.
- M. A. Soldatov, A. Martini, A. L. Bugaev, I. Pankin, P. V. Medvedev, A. A. Guda, A. M. Aboraia, Y. S. Podkovyrina, A. P. Budnyk, A. A. Soldatov and C. Lamberti, Polyhedron, 2018, 155, 232–253 CrossRef CAS.
- S. Øien-ØDegaard, G. C. Shearer, D. S. Wragg and K. P. Lillerud, Chem. Soc. Rev., 2017, 46, 4867–4876 RSC.
- X. Gong, K. Gnanasekaran, Z. Chen, L. Robison, M. C. Wasson, K. C. Bentz, S. M. Cohen, O. K. Farha and N. C. Gianneschi, J. Am. Chem. Soc., 2020, 142, 17224–17235 CrossRef CAS PubMed.
- Y. Zhou, X. Xu, A. Carlsson, S. Lazar, Z. Pan, Y. Ma, O. Terasaki and H. Deng, Chem. Mater., 2020, 32, 4966–4972 CrossRef CAS.
- X. Gong, H. Noh, N. C. Gianneschi and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 6146–6151 CrossRef CAS.
- S. Pu, C. Gong and A. W. Robertson, R. Soc. Open Sci., 2020, 7, 191204 CrossRef CAS.
- J. W. M. Osterrieth, J. Rampersad, D. Madden, N. Rampal, L. Skoric, B. Connolly, M. D. Allendorf, V. Stavila, J. L. Snider, R. Ameloot, J. Marreiros, C. Ania, D. Azevedo, E. Vilarrasa-Garcia, B. F. Santos, X. Bu, Z. Chang, H. Bunzen, N. R. Champness, S. L. Griffin, B. Chen, R. Lin, B. Coasne, S. Cohen, J. C. Moreton, Y. J. Colón, L. Chen, R. Clowes, F. Coudert, Y. Cui, B. Hou, D. M. D’Alessandro, P. W. Doheny, M. Dincă, C. Sun, C. Doonan, M. T. Huxley, J. D. Evans, P. Falcaro, R. Ricco, O. Farha, K. B. Idrees, T. Islamoglu, P. Feng, H. Yang, R. S. Forgan, D. Bara, S. Furukawa, E. Sanchez, J. Gascon, S. Telalović, S. K. Ghosh, S. Mukherjee, M. R. Hill, M. M. Sadiq, P. Horcajada, P. Salcedo-Abraira, K. Kaneko, R. Kukobat, J. Kenvin, S. Keskin, S. Kitagawa, K. Otake, R. P. Lively, S. J. A. DeWitt, P. Llewellyn, B. V. Lotsch, S. T. Emmerling, A. M. Pütz, C. Martí-Gastaldo, N. M. Padial, J. García-Martínez, N. Linares, D. Maspoch, J. A. Suárez del Pino, P. Moghadam, R. Oktavian, R. E. Morris, P. S. Wheatley, J. Navarro, C. Petit, D. Danaci, M. J. Rosseinsky, A. P. Katsoulidis, M. Schröder, X. Han, S. Yang, C. Serre, G. Mouchaham, D. S. Sholl, R. Thyagarajan, D. Siderius, R. Q. Snurr, R. B. Goncalves, S. Telfer, S. J. Lee, V. P. Ting, J. L. Rowlandson, T. Uemura, T. Iiyuka, M. A. van der Veen, D. Rega, V. Van Speybroeck, S. M. J. Rogge, A. Lamaire, K. S. Walton, L. W. Bingel, S. Wuttke, J. Andreo, O. Yaghi, B. Zhang, C. T. Yavuz, T. S. Nguyen, F. Zamora, C. Montoro, H. Zhou, A. Kirchon and D. Fairen-Jimenez, Adv. Mater., 2022, 34, 2201502 CrossRef CAS PubMed.
- S. Krause, N. Hosono and S. Kitagawa, Angew. Chem., Int. Ed., 2020, 59, 15325–15341 CrossRef CAS PubMed.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- A. J. McConnell, Chem. Soc. Rev., 2022, 51, 2957–2971 RSC.
- K. Bian, C. Gerber, A. J. Heinrich, D. J. Müller, S. Scheuring and Y. Jiang, Nat. Rev. Methods Primers, 2021, 1, 36 CrossRef CAS.
- D. Wang, T. Haposan, J. Fan, A. Arramel and A. T. S. Wee, ACS Nano, 2024, 18, 30919–30942 CrossRef CAS PubMed.
- G. Zhan, Z. F. Cai, K. Strutyński, L. Yu, N. Herrmann, M. Martínez-Abadía, M. Melle-Franco, A. Mateo-Alonso and S. De Feyter, Nature, 2022, 603, 835–840 CrossRef CAS PubMed.
- K. S. Mali, J. Adisoejoso, E. Ghijsens, I. De Cat and S. De Feyter, Acc. Chem. Res., 2012, 45, 1309–1320 CrossRef CAS PubMed.
- M. Trueman, R. J. S. Pooley, A. R. B. J. Lutton-Gething, A. Hasija, G. F. S. Whitehead, S. J. O’Shea, M. W. Anderson and M. P. Attfield, J. Am. Chem. Soc., 2025, 147, 17201–17208 CrossRef CAS PubMed.
- N. Hosono, A. Terashima, S. Kusaka, R. Matsuda and S. Kitagawa, Nat. Chem., 2019, 11, 109–116 CrossRef CAS PubMed.
- T. Ando, T. Uchihashi and S. Scheuring, Chem. Rev., 2014, 114, 3120–3188 CrossRef CAS PubMed.
- P. Messina, A. Dmitriev, N. Lin, H. Spillmann, M. Abel, J. V. Barth and K. Kern, J. Am. Chem. Soc., 2002, 124, 14000–14001 CrossRef CAS PubMed.
- N. Lin, A. Dmitriev, J. Weckesser, J. V. Barth and K. Kern, Angew. Chem., Int. Ed., 2002, 41, 4779–4783 CrossRef CAS PubMed.
- M. A. Lingenfelder, H. Spillmann, A. Dmitriev, S. Stepanow, N. Lin, J. V. Barth and K. Kern, Chem. – Eur. J., 2004, 10, 1913–1919 CrossRef CAS PubMed.
- A. Dmitriev, H. Spillmann, M. Lingenfelder, N. Lin, J. V. Barth and K. Kern, Langmuir, 2004, 20, 4799–4801 CrossRef CAS PubMed.
- D. Kühne, F. Klappenberger, R. Decker, U. Schlickum, H. Brune, S. Klyatskaya, M. Ruben and J. V. Barth, J. Am. Chem. Soc., 2009, 131, 3881–3883 CrossRef PubMed.
- C. Safarowsky, L. Merz, A. Rang, P. Broekmann, B. A. Hermann and C. A. Schalley, Angew. Chem., Int. Ed., 2004, 43, 1291–1294 CrossRef CAS PubMed.
- K. S. Jeong, S. Y. Kim, U.-S. Shin, M. Kogej, N. T. M. Hai, P. Broekmann, N. Jeong, B. Kirchner, M. Reiher and C. A. Schalley, J. Am. Chem. Soc., 2005, 127, 17672–17685 CrossRef CAS PubMed.
- Q.-H. Yuan, L.-J. Wan, H. Jude and P. J. Stang, J. Am. Chem. Soc., 2005, 127, 16279–16286 CrossRef CAS PubMed.
- M. El Garah, A. Ciesielski, N. Marets, V. Bulach, M. W. Hosseini and P. Samorì, Chem. Commun., 2014, 50, 12250–12253 RSC.
- A. Cucinotta, C. Kahlfuss, A. Minoia, S. Eyley, K. Zwaenepoel, G. Velpula, W. Thielemans, R. Lazzaroni, V. Bulach, M. W. Hosseini, K. S. Mali and S. De Feyter, J. Am. Chem. Soc., 2023, 145, 1194–1205 CrossRef CAS PubMed.
- A. Cucinotta, S. Eyley, J. A. Davies, W. Thielemans, K. S. Mali and S. De Feyter, J. Am. Chem. Soc., 2025, 147, 7682–7693 CrossRef CAS PubMed.
- M. Shöâeè, J. R. Agger, M. W. Anderson and M. P. Attfield, CrystEngComm, 2008, 10, 646–648 RSC.
- M. Shoaee, M. W. Anderson and M. P. Attfield, Angew. Chem., Int. Ed., 2008, 47, 8525–8528 CrossRef CAS PubMed.
- C. Munuera, O. Shekhah, H. Wang, C. Wöll and C. Ocal, Phys. Chem. Chem. Phys., 2008, 10, 7257–7261 RSC.
- G. Delen, Z. Ristanović, L. D. B. Mandemaker and B. M. Weckhuysen, Chem. – Eur. J., 2018, 24, 187–195 CrossRef CAS PubMed.
- G. Binnig, H. Rohrer, C. Gerber and E. Weibel, Appl. Phys. Lett., 1982, 40, 178–180 CrossRef CAS.
- G. Binnig and H. Rohrer, Rev. Mod. Phys., 1987, 59, 615–625 CrossRef CAS.
- E. Meyer, R. Bennewitz and H. J. Hug, Scanning Probe Microscopy, Springer, Cham, 2021 Search PubMed.
- B. Voigtländer, Non-Destructive Material Characterization Methods, Springer, 2015 Search PubMed.
- J. Atkins and P. W. de Paula, Atkins’ Physical Chemistry, Oxford University Press, 9th edn, 2010 Search PubMed.
- H. J. W. Zandvliet and A. van Houselt, Annu. Rev. Anal. Chem., 2009, 2, 37–55 CrossRef CAS PubMed.
- W. Mamdouh, H. Uji-I, J. S. Ladislaw, A. E. Dulcey, V. Percec, F. C. De Schryver and S. De Feyter, J. Am. Chem. Soc., 2006, 128, 317–325 CrossRef CAS PubMed.
- K. S. Mali, N. Pearce, S. De Feyter and N. R. Champness, Chem. Soc. Rev., 2017, 46, 2520–2542 RSC.
- G. Binnig, C. F. Quate and C. Gerber, Phys. Rev. Lett., 1986, 56, 930–933 CrossRef PubMed.
- J. J. Schwartz, D. S. Jakob and A. Centrone, Chem. Soc. Rev., 2022, 51, 5248–5267 RSC.
- A. Dazzi and C. B. Prater, Chem. Rev., 2017, 117, 5146–5173 CrossRef CAS PubMed.
- Q. Xie and X. G. Xu, Langmuir, 2023, 39, 17593–17599 CrossRef CAS PubMed.
- V. Rose, K. Wang, T. Chien, J. Hiller, D. Rosenmann, J. W. Freeland, C. Preissner and S. Hla, Adv. Funct. Mater., 2013, 23, 2646–2652 CrossRef CAS.
- L. Sosa-Vargas, E. Kim and A.-J. Attias, Mater. Horiz., 2017, 4, 570–583 RSC.
- X. Zhao and X. Miao, iScience, 2024, 27, 109392 CrossRef CAS PubMed.
- Y. Wang, X. Li, Q. Yang, Q. Shen, Y. He, Y. Zhang and Y. Wang, Fundam. Res., 2025, 5, 1524–1537 CrossRef CAS PubMed.
- J. Liu and N. Lin, ChemPlusChem, 2023, 88, e202200359 CrossRef CAS PubMed.
- H. Liang, Y. He, Y. Ye, X. Xu, F. Cheng, W. Sun, X. Shao, Y. Wang, J. Li and K. Wu, Coord. Chem. Rev., 2009, 253, 2959–2979 CrossRef CAS.
- S. O. Parreiras, J. M. Gallego and D. Écija, Chem. Commun., 2023, 59, 8878–8893 RSC.
- D. Écija, J. I. Urgel, A. P. Seitsonen, W. Auwärter and J. V. Barth, Acc. Chem. Res., 2018, 51, 365–375 CrossRef PubMed.
- A. Ciesielski, C. Palma, M. Bonini and P. Samorì, Adv. Mater., 2010, 22, 3506–3520 CrossRef CAS PubMed.
- D. P. Goronzy, M. Ebrahimi, F. Rosei, A. Arramel, Y. Fang, S. De Feyter, S. L. Tait, C. Wang, P. H. Beton, A. T. S. Wee, P. S. Weiss and D. F. Perepichka, ACS Nano, 2018, 12, 7445–7481 CrossRef CAS PubMed.
- K. G. Nath, O. Ivasenko, J. M. MacLeod, J. A. Miwa, J. D. Wuest, A. Nanci, D. F. Perepichka and F. Rosei, J. Phys. Chem. C, 2007, 111, 16996–17007 CrossRef CAS.
- U. Mazur and K. W. Hipps, Chem. Commun., 2015, 51, 4737–4749 RSC.
- R. Gutzler, L. Cardenas and F. Rosei, Chem. Sci., 2011, 2, 2290–2300 RSC.
- O. Ochs, M. Hocke, S. Spitzer, W. M. Heckl, N. Martsinovich and M. Lackinger, Chem. Mater., 2020, 32, 5057–5065 CrossRef CAS.
- I. Destoop, A. Minoia, O. Ivasenko, A. Noguchi, K. Tahara, Y. Tobe, R. Lazzaroni and S. De Feyter, Faraday Discuss., 2017, 204, 215–231 RSC.
- M. Lackinger, S. Griessl, W. M. Heckl, M. Hietschold and G. W. Flynn, Langmuir, 2005, 21, 4984–4988 CrossRef CAS PubMed.
- S. Stepanow, N. Lin, J. V. Barth and K. Kern, J. Phys. Chem. B, 2006, 110, 23472–23477 CrossRef CAS PubMed.
- Z. Shi and N. Lin, J. Am. Chem. Soc., 2009, 131, 5376–5377 CrossRef CAS PubMed.
- S. Vijayaraghavan, D. Ecija, W. Auwärter, S. Joshi, K. Seufert, M. Drach, D. Nieckarz, P. Szabelski, C. Aurisicchio, D. Bonifazi and J. V. Barth, Chem. – Eur. J., 2013, 19, 14143–14150 CrossRef CAS PubMed.
- J. I. Urgel, D. Ecija, W. Auwärter, D. Stassen, D. Bonifazi and J. V. Barth, Angew. Chem., Int. Ed., 2015, 54, 6163–6167 CrossRef CAS PubMed.
- J. F. Schultz, B. Yang and N. Jiang, J. Chem. Phys., 2021, 154, 1–10 CrossRef PubMed.
- S. Kowarik, A. Gerlach and F. Schreiber, J. Phys.: Condens. Matter, 2008, 20, 184005 CrossRef.
- A. Dmitriev, N. Lin, J. Weckesser, J. V. Barth and K. Kern, J. Phys. Chem. B, 2002, 106, 6907–6912 CrossRef CAS.
- T. Classen, M. Lingenfelder, Y. Wang, R. Chopra, C. Virojanadara, U. Starke, G. Costantini, G. Fratesi, S. Fabris, S. de Gironcoli, S. Baroni, S. Haq, R. Raval and K. Kern, J. Phys. Chem. A, 2007, 111, 12589–12603 CrossRef CAS PubMed.
- U. Schlickum, R. Decker, F. Klappenberger, G. Zoppellaro, S. Klyatskaya, M. Ruben, I. Silanes, A. Arnau, K. Kern, H. Brune and J. V. Barth, Nano Lett., 2007, 7, 3813–3817 CrossRef CAS PubMed.
- F. Bischoff, Y. He, K. Seufert, D. Stassen, D. Bonifazi, J. V. Barth and W. Auwärter, Chem. – Eur. J., 2016, 22, 15298–15306 CrossRef CAS PubMed.
- N. Lin, S. Stepanow, F. Vidal, J. V. Barth and K. Kern, Chem. Commun., 2005, 1681–1683 RSC.
- U. Schlickum, F. Klappenberger, R. Decker, G. Zoppellaro, S. Klyatskaya, M. Ruben, K. Kern, H. Brune and J. V. Barth, J. Phys. Chem. C, 2010, 114, 15602–15606 CrossRef CAS.
- Z. Shi and N. Lin, J. Am. Chem. Soc., 2010, 132, 10756–10761 CrossRef CAS PubMed.
- X. F. Mao, T. Lin, J. Adisoejoso, Z. Shi, X. S. Shang, P. N. Liu and N. Lin, Phys. Chem. Chem. Phys., 2013, 15, 12447 RSC.
- D.-D. Zhou, J. Wang, P. Chen, Y. He, J.-X. Wu, S. Gao, Z. Zhong, Y. Du, D. Zhong and J.-P. Zhang, Chem. Sci., 2021, 12, 1272–1277 RSC.
- D. Moreno, S. O. Parreiras, S. K. Mathialagan, M. Tenorio, K. Lauwaet, J. I. Urgel, J. M. Gallego and D. Écija, Chem. – Eur. J., 2025, 31, 2–7 CrossRef PubMed.
- L.-X. Kang, B.-X. Wang, X.-Y. Zhang, Y.-C. Zhu, D.-Y. Li and P.-N. Liu, J. Phys. Chem. Lett., 2024, 15, 6108–6114 CrossRef CAS PubMed.
- R. Adhikari, J. Kuliga, M. Ruppel, N. Jux, H. Marbach and H.-P. Steinrück, J. Phys. Chem. C, 2021, 125, 7204–7212 CrossRef CAS.
- D. Kumar, J. Hellerstedt, B. Field, B. Lowe, Y. Yin, N. V. Medhekar and A. Schiffrin, Adv. Funct. Mater., 2021, 31, 1–9 Search PubMed.
- S. O. Parreiras, D. Moreno, B. Cirera, M. A. Valbuena, J. I. Urgel, M. Paradinas, M. Panighel, F. Ajejas, M. A. Niño, J. M. Gallego, M. Valvidares, P. Gargiani, W. Kuch, J. I. Martínez, A. Mugarza, J. Camarero, R. Miranda, P. Perna and D. Écija, Small, 2021, 17, e2102753 CrossRef PubMed.
- J. I. Urgel, B. Cirera, Y. Wang, W. Auwärter, R. Otero, J. M. Gallego, M. Alcamí, S. Klyatskaya, M. Ruben, F. Martín, R. Miranda, D. Ecija and J. V. Barth, Small, 2015, 11, 6358–6364 CrossRef CAS PubMed.
- D. Écija, J. I. Urgel, A. C. Papageorgiou, S. Joshi, W. Auwärter, A. P. Seitsonen, S. Klyatskaya, M. Ruben, S. Fischer, S. Vijayaraghavan, J. Reichert and J. V. Barth, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6678–6681 CrossRef PubMed.
- E. Rheinfrank, M. Pörtner, M. del C. Nuñez Beyerle, F. Haag, P. S. Deimel, F. Allegretti, K. Seufert, J. V. Barth, M. L. Bocquet, P. Feulner and W. Auwärter, J. Am. Chem. Soc., 2021, 143, 14581–14591 CrossRef CAS PubMed.
- J. I. Urgel, D. Écija, G. Lyu, R. Zhang, C.-A. Palma, W. Auwärter, N. Lin and J. V. Barth, Nat. Chem., 2016, 8, 657–662 CrossRef CAS PubMed.
- S. M. Barlow and R. Raval, Surf. Sci. Rep., 2003, 50, 201–341 CrossRef CAS.
- J. A. A. W. Elemans, I. De Cat, H. Xu and S. De Feyter, Chem. Soc. Rev., 2009, 38, 722–736 RSC.
- F. Vidal, E. Delvigne, S. Stepanow, N. Lin, J. V. Barth and K. Kern, J. Am. Chem. Soc., 2005, 127, 10101–10106 CrossRef CAS PubMed.
- T. G. Gopakumar, F. Matino, B. Schwager, A. Bannwarth, F. Tuczek and R. Berndt, J. Phys. Chem. C, 2010, 114, 18247–18251 CrossRef CAS.
- I. Destoop, E. Ghijsens, K. Katayama, K. Tahara, K. S. Mali, Y. Tobe and S. De Feyter, J. Am. Chem. Soc., 2012, 134, 19568–19571 CrossRef CAS PubMed.
- M. Schöck, R. Otero, S. Stojkovic, F. Hümmelink, A. Gourdon, E. Lægsgaard, I. Stensgaard, C. Joachim and F. Besenbacher, J. Phys. Chem. B, 2006, 110, 12835–12838 CrossRef PubMed.
- J. Seibel, L. Verstraete, B. E. Hirsch, A. M. Bragança and S. De Feyter, J. Am. Chem. Soc., 2018, 140, 11565–11568 CrossRef CAS PubMed.
- H. Spillmann, A. Dmitriev, N. Lin, P. Messina, J. V. Barth and K. Kern, J. Am. Chem. Soc., 2003, 125, 10725–10728 CrossRef CAS PubMed.
- B. Yang, N. Cao, H. Ju, H. Lin, Y. Li, H. Ding, J. Ding, J. Zhang, C. Peng, H. Zhang, J. Zhu, Q. Li and L. Chi, J. Am. Chem. Soc., 2019, 141, 168–174 CrossRef CAS PubMed.
- J. Liu, T. Lin, Z. Shi, F. Xia, L. Dong, P. N. Liu and N. Lin, J. Am. Chem. Soc., 2011, 133, 18760–18766 CrossRef CAS PubMed.
- M. Lischka, R. Dong, M. Wang, N. Martsinovich, M. Fritton, L. Grossmann, W. M. Heckl, X. Feng and M. Lackinger, Chem. – Eur. J., 2019, 25, 1975–1983 CrossRef CAS PubMed.
- Z. Gao, C.-H. Hsu, J. Liu, F.-C. Chuang, R. Zhang, B. Xia, H. Xu, L. Huang, Q. Jin, P. N. Liu and N. Lin, Nanoscale, 2019, 11, 878–881 RSC.
- F. Gu, S. Shu, C. E. Patrick and M. R. Castell, J. Phys. Chem. C, 2024, 128, 17797–17805 CrossRef CAS.
- H. Walch, J. Dienstmaier, G. Eder, R. Gutzler, S. Schlögl, T. Sirtl, K. Das, M. Schmittel and M. Lackinger, J. Am. Chem. Soc., 2011, 133, 7909–7915 CrossRef CAS PubMed.
- C. Lyu, Y. Gao, K. Zhou, M. Hua, Z. Shi, P.-N. Liu, L. Huang and N. Lin, ACS Nano, 2024, 18, 19793–19801 CAS.
- Z. Shi, J. Liu, T. Lin, F. Xia, P. N. Liu and N. Lin, J. Am. Chem. Soc., 2011, 133, 6150–6153 CrossRef CAS PubMed.
- S. Stepanow, N. Lin, D. Payer, U. Schlickum, F. Klappenberger, G. Zoppellaro, M. Ruben, H. Brune, J. V. Barth and K. Kern, Angew. Chem., Int. Ed., 2007, 46, 710–713 CrossRef CAS PubMed.
- S. L. Tait, A. Langner, N. Lin, R. Chandrasekar, O. Fuhr, M. Ruben and K. Kern, ChemPhysChem, 2008, 9, 2495–2499 CrossRef CAS PubMed.
- C. S. Kley, J. Čechal, T. Kumagai, F. Schramm, M. Ruben, S. Stepanow and K. Kern, J. Am. Chem. Soc., 2012, 134, 6072–6075 CrossRef CAS PubMed.
- H. Grove, J. Sletten, M. Julve and F. Lloret, J. Chem. Soc., Dalton Trans., 2001, 1029–1034 RSC.
- A. Gutierrez, M. Buchet and S. Clair, ChemPhysChem, 2019, 20, 2286–2291 CrossRef CAS PubMed.
- J. Lu, D. Nieckarz, H. Jiang, Z. Zhu, Y. Yan, F. Zheng, W. Rżysko, J. Lisiecki, P. Szabelski and Q. Sun, ACS Nano, 2023, 17, 20194–20202 CrossRef CAS PubMed.
- S. L. Tait, Y. Wang, G. Costantini, N. Lin, A. Baraldi, F. Esch, L. Petaccia, S. Lizzit and K. Kern, J. Am. Chem. Soc., 2008, 130, 2108–2113 CrossRef CAS PubMed.
- P. Gambardella, S. Stepanow, A. Dmitriev, J. Honolka, F. M. F. De Groot, M. Lingenfelder, S. Sen Gupta, D. D. Sarma, P. Bencok, S. Stanescu, S. Clair, S. Pons, N. Lin, A. P. Seitsonen, H. Brune, J. V. Barth and K. Kern, Nat. Mater., 2009, 8, 189–193 CrossRef CAS PubMed.
- K. M. L. Taylor-Pashow, J. Della Rocca, Z. Xie, S. Tran and W. Lin, J. Am. Chem. Soc., 2009, 131, 14261–14263 CrossRef CAS PubMed.
- C. Martín-Fuentes, S. O. Parreiras, J. I. Urgel, V. Rubio-Giménez, B. Muñiz Cano, D. Moreno, K. Lauwaet, M. Valvidares, M. A. Valbuena, P. Gargiani, W. Kuch, J. Camarero, J. M. Gallego, R. Miranda, J. I. Martínez, C. Martí-Gastaldo and D. Écija, J. Am. Chem. Soc., 2022, 144, 16034–16041 CrossRef PubMed.
- M. Hmadeh, Z. Lu, Z. Liu, F. Gándara, H. Furukawa, S. Wan, V. Augustyn, R. Chang, L. Liao, F. Zhou, E. Perre, V. Ozolins, K. Suenaga, X. Duan, B. Dunn, Y. Yamamto, O. Terasaki and O. M. Yaghi, Chem. Mater., 2012, 24, 3511–3513 CrossRef CAS.
- A. Mahringer, A. C. Jakowetz, J. M. Rotter, B. J. Bohn, J. K. Stolarczyk, J. Feldmann, T. Bein and D. D. Medina, ACS Nano, 2019, 13, 6711–6719 CrossRef PubMed.
- V. Baltz, A. Manchon, M. Tsoi, T. Moriyama, T. Ono and Y. Tserkovnyak, Rev. Mod. Phys., 2018, 90, 15005 CrossRef CAS.
- F. Frezza, M. Kumar, A. Sánchez-Grande, D. Soler-Polo, M. Carrera, O. Stetsovych, P. Mutombo, D. Curiel and P. Jelínek, J. Am. Chem. Soc., 2025, 147, 19575–19582 CrossRef CAS PubMed.
- A. Kumar, K. Banerjee, A. S. Foster and P. Liljeroth, Nano Lett., 2018, 18, 5596–5602 CrossRef CAS PubMed.
- S. Yoshimoto, Y. Ono, K. Nishiyama and I. Taniguchi, Phys. Chem. Chem. Phys., 2010, 12, 14442–14444 RSC.
- S. Li, C. Gong, Y. Zhang, S. Fu, Z. Wang, Y. Lu, S. Gu, X. Liu and L. Wang, RSC Adv., 2022, 12, 3892–3896 RSC.
- D. J. Díaz, G. D. Storrier, S. Bernhard, K. Takada and H. D. Abruña, Langmuir, 1999, 15, 7351–7354 CrossRef.
- H. M. Zhang, W. Zhao, Z. X. Xie, L. S. Long, B. W. Mao, X. Xu and L. S. Zheng, J. Phys. Chem. C, 2007, 111, 7570–7573 CrossRef CAS.
- Y. Kikkawa, E. Koyama, S. Tsuzuki, K. Fujiwara, K. Miyake, H. Tokuhisa and M. Kanesato, Surf. Sci., 2007, 601, 2520–2524 CrossRef CAS.
- M. Surin, P. Samorì, A. Jouaiti, N. Kyritsakas and M. W. Hosseini, Angew. Chem., Int. Ed., 2007, 46, 245–249 CrossRef CAS PubMed.
- M. A. Carvalho, H. Dekkiche, M. Nagasaki, Y. Kikkawa and R. Ruppert, J. Am. Chem. Soc., 2019, 141, 10137–10141 CrossRef CAS PubMed.
- Y. Kikkawa, M. Nagasaki, S. Tsuzuki, T. N. J. Fouquet, S. Nakamura, Y. Takenaka, Y. Norikane and K. Hiratani, Chem. Commun., 2022, 58, 1752–1755 RSC.
- M. El Garah, N. Marets, M. Mauro, A. Aliprandi, S. Bonacchi, L. De Cola, A. Ciesielski, V. Bulach, M. W. Hosseini and P. Samorì, J. Am. Chem. Soc., 2015, 137, 8450–8459 CrossRef CAS PubMed.
- V. Mishra, S. H. Mir, J. K. Singh and T. G. Gopakumar, ACS Appl. Mater. Interfaces, 2020, 12, 51122–51132 CrossRef CAS PubMed.
- J. Liu, M. Paradinas, L. Heinke, M. Buck, C. Ocal, V. Mugnaini and C. Wöll, ChemElectroChem, 2016, 3, 713–718 CrossRef CAS.
- S. Goswami, M. Rimoldi, R. Anderson, C. Lee, X. Li, A. Li, P. Deria, L. X. Chen, R. D. Schaller, D. A. Gómez-Gualdrón, O. K. Farha and J. T. Hupp, Chem. Mater., 2022, 34, 9446–9454 CrossRef CAS.
- A. J. Brandt, D. M. Shakya, K. Metavarayuth, E. Dolgopolova, L. Hensley, A. S. Duke, S. Farzandh, M. Stefik, N. B. Shustova and D. A. Chen, Langmuir, 2020, 36, 9900–9908 CrossRef CAS PubMed.
- O. Lugier, U. Pokharel and S. Castellanos, Cryst. Growth Des., 2020, 20, 5302–5309 CrossRef CAS.
- L. G. S. Albano, T. P. Vello, D. H. S. de Camargo, R. M. L. da Silva, A. C. M. Padilha, A. Fazzio and C. C. B. Bufon, Nano Lett., 2020, 20, 1080–1088 CrossRef CAS PubMed.
- A. J. Cruz, I. Stassen, M. Krishtab, K. Marcoen, T. Stassin, S. Rodríguez-Hermida, J. Teyssandier, S. Pletincx, R. Verbeke, V. Rubio-Giménez, S. Tatay, C. Martí-Gastaldo, J. Meersschaut, P. M. Vereecken, S. De Feyter, T. Hauffman and R. Ameloot, Chem. Mater., 2019, 31, 9462–9471 CrossRef CAS.
- L. Pan, G. Liu, W. Shi, J. Shang, W. R. Leow, Y. Liu, Y. Jiang, S. Li, X. Chen and R.-W. Li, Nat. Commun., 2018, 9, 3813 CrossRef PubMed.
- S. Rana, R. Rajendra, B. Dhara, P. K. Jha and N. Ballav, Adv. Mater. Interfaces, 2016, 3, 1–8 Search PubMed.
- I. Christodoulou, T. Bourguignon, X. Li, G. Patriarche, C. Serre, C. Marlière and R. Gref, Nanomaterials, 2021, 11, 722 CrossRef CAS PubMed.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- T. Wang, H. Zhu, Q. Zeng and D. Liu, Adv. Mater. Interfaces, 2019, 6, 1–27 Search PubMed.
- K. Szelagowska-Kunstman, P. Cyganik, M. Goryl, D. Zacher, Z. Puterova, R. A. Fischer and M. Szymonski, J. Am. Chem. Soc., 2008, 130, 14446–14447 CrossRef CAS PubMed.
- C. Crivello, S. Sevim, O. Graniel, C. Franco, S. Pane, J. Puigmarti-Luis and D. Munoz-Rojas, Mater. Horiz., 2021, 8, 168–178 RSC.
- Y. Zhang and C.-H. Chang, Processes, 2020, 8, 377 CrossRef CAS.
- V. Chernikova, O. Shekhah and M. Eddaoudi, ACS Appl. Mater. Interfaces, 2016, 8, 20459–20464 CrossRef CAS PubMed.
- P. Su, M. Tu, R. Ameloot and W. Li, Acc. Chem. Res., 2022, 55, 186–196 CrossRef CAS PubMed.
- R. Ameloot, F. Vermoortele, W. Vanhove, M. B. J. Roeffaers, B. F. Sels and D. E. De Vos, Nat. Chem., 2011, 3, 382–387 CrossRef CAS PubMed.
- V. Rubio-Giménez, S. Tatay, F. Volatron, F. J. Martínez-Casado, C. Martí-Gastaldo and E. Coronado, J. Am. Chem. Soc., 2016, 138, 2576–2584 CrossRef PubMed.
- L. Heinke and C. Wöll, Adv. Mater., 2019, 31, 1806324 CrossRef PubMed.
- J. Liu and C. Wöll, Chem. Soc. Rev., 2017, 46, 5730–5770 RSC.
- T. Ladnorg, A. Welle, S. Heißler, C. Wöll and H. Gliemann, Beilstein J. Nanotechnol., 2013, 4, 638–648 CrossRef PubMed.
- M. L. Ohnsorg, C. K. Beaudoin and M. E. Anderson, Langmuir, 2015, 31, 6114–6121 CrossRef CAS PubMed.
- E. Biemmi, C. Scherb and T. Bein, J. Am. Chem. Soc., 2007, 129, 8054–8055 CrossRef CAS PubMed.
- R. P. Brand, L. D. B. Mandemaker, G. Delen, N. Rijnveld and B. M. Weckhuysen, ChemPhysChem, 2018, 19, 2397–2404 CrossRef CAS PubMed.
- K. Sakata, S. Kashiyama, G. Matsuo, S. Uemura, N. Kimizuka and M. Kunitake, ChemNanoMat, 2015, 1, 259–263 CrossRef CAS.
- G. Delen, M. Monai, F. Meirer and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2021, 60, 1620–1624 CrossRef CAS PubMed.
- D. Shu, R. Fan, S. Yao, H. Jiang, X. Zhang and Y. Zheng, J. Phys. Chem. C, 2024, 128, 12551–12557 CrossRef CAS.
- D. Sud and G. Kaur, Polyhedron, 2021, 193, 114897 CrossRef CAS.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- N. S. John, C. Scherb, M. Shöâeè, M. W. Anderson, M. P. Attfield and T. Bein, Chem. Commun., 2009, 6294–6296 RSC.
- L. D. B. Mandemaker, M. Filez, G. Delen, H. Tan, X. Zhang, D. Lohse and B. M. Weckhuysen, J. Phys. Chem. Lett., 2018, 9, 1838–1844 CrossRef CAS PubMed.
- P. Y. Moh, P. Cubillas, M. W. Anderson and M. P. Attfield, J. Am. Chem. Soc., 2011, 133, 13304–13307 CrossRef CAS PubMed.
- P. Cubillas, M. W. Anderson and M. P. Attfield, Chem. – Eur. J., 2012, 18, 15406–15415 CrossRef CAS PubMed.
- P. Cubillas, K. Etherington, M. W. Anderson and M. P. Attfield, CrystEngComm, 2014, 16, 9834–9841 RSC.
- F. I. Pambudi, M. W. Anderson and M. P. Attfield, Faraday Discuss., 2021, 231, 112–126 RSC.
- S. S. Li, B. H. Northrop, Q.-H. Yuan, L.-J. Wan and P. J. Stang, Acc. Chem. Res., 2009, 42, 249–259 CrossRef CAS PubMed.
- J.-R. Gong, L.-J. Wan, Q.-H. Yuan, C.-L. Bai, H. Jude and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 971–974 CrossRef CAS PubMed.
- T. Chen, G.-B. Pan, H. Wettach, M. Fritzsche, S. Höger, L.-J. Wan, H.-B. Yang, B. H. Northrop and P. J. Stang, J. Am. Chem. Soc., 2010, 132, 1328–1333 CrossRef CAS PubMed.
- S.-S. Li, H.-J. Yan, L.-J. Wan, H.-B. Yang, B. H. Northrop and P. J. Stang, J. Am. Chem. Soc., 2007, 129, 9268–9269 CrossRef CAS PubMed.
- Q.-H. Yuan, C.-J. Yan, H.-J. Yan, L.-J. Wan, B. H. Northrop, H. Jude and P. J. Stang, J. Am. Chem. Soc., 2008, 130, 8878–8879 CrossRef CAS PubMed.
- P. J. Stang, D. H. Cao, S. Saito and A. M. Arif, J. Am. Chem. Soc., 1995, 117, 6273–6283 CrossRef CAS.
- C. J. Kuehl, C. L. Mayne, A. M. Arif and P. J. Stang, Org. Lett., 2000, 2, 3727–3729 CrossRef CAS PubMed.
- C. J. Kuehl, Y. K. Kryschenko, U. Radhakrishnan, S. R. Seidel, S. D. Huang and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4932–4936 CrossRef CAS PubMed.
- P. Su, B. Wei, C. Guo, Y. Hu, R. Tang, S. Zhang, C. He, J. Lin, X. Yu, Z. Chen, H. Li, H. Wang and X. Li, J. Am. Chem. Soc., 2023, 145, 3131–3145 CrossRef CAS PubMed.
- Y. Li, G.-F. Huo, B. Liu, B. Song, Y. Zhang, X. Qian, H. Wang, G.-Q. Yin, A. Filosa, W. Sun, S. W. Hla, H.-B. Yang and X. Li, J. Am. Chem. Soc., 2020, 142, 14638–14648 CrossRef CAS PubMed.
- F. Su, S. Zhang, Z. Chen, Z. Zhang, Z. Li, S. Lu, M. Zhang, F. Fang, S. Kang, C. Guo, C. Su, X. Yu, H. Wang and X. Li, J. Am. Chem. Soc., 2022, 144, 16559–16571 CrossRef CAS PubMed.
- J. Shi, Y. Li, X. Jiang, H. Yu, J. Li, H. Zhang, D. J. Trainer, S. W. Hla, H. Wang, M. Wang and X. Li, J. Am. Chem. Soc., 2021, 143, 1224–1234 CrossRef CAS PubMed.
- Z. Zhang, Y. Li, B. Song, Y. Zhang, X. Jiang, M. Wang, R. Tumbleson, C. Liu, P. Wang, X.-Q. Hao, T. Rojas, A. T. Ngo, J. L. Sessler, G. R. Newkome, S. W. Hla and X. Li, Nat. Chem., 2020, 12, 468–474 CrossRef CAS PubMed.
- L. Wang, B. Song, Y. Li, L. Gong, X. Jiang, M. Wang, S. Lu, X.-Q. Hao, Z. Xia, Y. Zhang, S. W. Hla and X. Li, J. Am. Chem. Soc., 2020, 142, 9809–9817 CAS.
- Z. Li, Y. Li, Y. Zhao, H. Wang, Y. Zhang, B. Song, X. Li, S. Lu, X.-Q. Hao, S.-W. Hla, Y. Tu and X. Li, J. Am. Chem. Soc., 2020, 142, 6196–6205 CrossRef CAS PubMed.
- T. M. Ajayi, N. Shirato, T. Rojas, S. Wieghold, X. Cheng, K. Z. Latt, D. J. Trainer, N. K. Dandu, Y. Li, S. Premarathna, S. Sarkar, D. Rosenmann, Y. Liu, N. Kyritsakas, S. Wang, E. Masson, V. Rose, X. Li, A. T. Ngo and S.-W. Hla, Nature, 2023, 618, 69–73 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |