
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembled nanotubes from the supramolecular polymerization of discrete cyclic entities
Marina
González-Sánchez
 a,
Jorge S.
Valera
a,
Jacobo
Veiga-Herrero
a,
Paula B.
Chamorro
a,
Fátima
Aparicio
*ab and
David
González-Rodríguez
*ab
a,
Jorge S.
Valera
a,
Jacobo
Veiga-Herrero
a,
Paula B.
Chamorro
a,
Fátima
Aparicio
*ab and
David
González-Rodríguez
*ab
aNanostructured Molecular Systems and Materials Group, Organic Chemistry Department, Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: fatima.aparicio@uam.es; david.gonzalez.rodriguez@uam.es
bInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
First published on 7th May 2025
Abstract
Inspired by the extraordinary attributes displayed by nanotubes in Nature, the creation of self-assembled nano-sized hollow tubes is an area of significant and growing interest given its potential application in transmembrane ion channels, ion sensing or catalysis, among others. One of the most utilized strategies employed to build these supramolecular entities implies the stacking of discrete cyclic units. Given the intrinsic dynamicity of the forces that drive the self-assembly processes, this approach offers substantial advantages when compared to inorganic or covalent approaches, ranging from tunable pore designs to error correction, to name a few. Herein we focus on the different approaches explored to design discrete cyclic entities as building blocks for the construction of self-assembled nanotubes, as well as the analytical tools used to elucidate the resulting structures. Attending to the nature of the bond involved in the formation of the cycle, we have distinguised three main categories: covalent, non-novalent and dynamic-covalent cycles. This review thus constitutes a roadmap to build self-assembled nanotubes based on soft matter and paves the way to expand their current applications.
 Marina González-Sánchez | Marina González-Sánchez obtained her Chemistry degree in 2017 from the Universidad de Extremadura (UEx) and completed her Master's degree in Organic Chemistry at the Universidad Autónoma de Madrid (UAM). In April 2019, she joined Prof. David González's research group (MSMn) to work on the non-covalent synthesis of self-assembled nanotubes. In February 2025, she successfully defended her doctoral thesis on the study of self-assembled architectures based on nucleobase pairing. |
 Jorge S. Valera | Jorge S. Valera obtained his PhD in 2018 from Universidad Complutense de Madrid (Spain), working under the supervision of Prof. Luis Sánchez and Prof. Rafael Gómez. Afterwards he conducted postdoctoral research in the group of Prof. Thomas Hermans’, firstly in the Institut de Sciences et d’Ingénierie Supramoléculaires (ISIS) at the Université de Strasbourg (France) and then at IMDEA Nanociencia. Since April 2024 he has worked in Prof. David González's group at Universidad Autónoma de Madrid thanks to a MSCA Individual Fellowship. His research interests include complex supramolecular polymerization processes, systems chemistry and light driven self-assembly. |
 Jacobo Veiga-Herrero | Jacobo Veiga-Herrero obtained his Degree in Chemistry in 2021 from the Universidad Autónoma de Madrid (UAM). He did his master's degree in Organic Chemistry, working on self-assembled nanotubes able to host DNA at Prof. David González's group (MSMn). In 2022, he began his PhD studies within the same group focused on the formation of water-soluble self-assembled nanotubes from non-covalent cycles. |
 Fátima Aparicio | Fátima Aparicio obtained her PhD degree in 2014 from the Universidad Complutense de Madrid working on Chirality in Supramolecular Polymers. She carried out a predoctoral stay in Prof. Carsten Schmuck's research group (Universität Duisburg-Essen, Germany) and a postdoctoral stay in Prof. Marc Sallé's research group (Université d'Angers, France). In 2016, she joined Prof. David González's research group at the Universidad Autónoma de Madrid where she works on the formation of self-assembled nanotubes, funded by a MSCA-Cofund and a MSCA-Individual Fellowships, and a project as main researcher from the Spanish MICINN. She is now a senior Ramon y Cajal researcher. |
 David González-Rodríguez | David González-Rodríguez obtained his PhD degree in 2003 in the group of Prof. T. Torres at the Universidad Autonoma de Madrid. Between 2005 and 2008, he was a Marie Curie fellow in the laboratories of Prof. E. W. Meijer at the Eindhoven University of Technology. He is Full Professor at the Universidad Autónoma de Madrid, where he leads from 2012 the Nanostructured Molecular Systems and Materials (MSMn) Group. The group's research interests focus on the development of versatile strategies to improve or create novel functions in organic materials by rationally ordering molecules at the nanoscale using the tools of self-assembly. |
1. Introduction
The natural world is full of diverse tubular forms that play crucial roles in numerous biological and geological activities. For example, nanotubes (NTs) serve as pathways for chemical signals, similar to transmembrane ion channels,1 and provide enclosed spaces for reactions, as demonstrated by protein-folding chaperonins2,3 and protein-degradation enzymes.4,5 Some of the most relevant tubular systems in nature are microtubules, which are essential elements of the cytoskeleton in eukaryotic cells.6 Microtubules are supramolecular polymers of the protein tubulin and are fundamental for the upholding of cellular structure,7,8 enabling transport within the cell, and ensuring the separation of chromosomes during cell division.9 These functions are critical for the normal operation and reproduction of cells, highlighting the significance of nanotubular structures in essential biological processes.Motivated by the extraordinary roles that tubular structures play in biological systems, many researchers have focused on developing functional artificial NTs, mostly exploiting inorganic motifs10,11 as well as carbon nanotubes (CNTs)12,13 to yield the hollow unidimensional structure. Another less explored approach involves the use of self-assembled nano-sized tubular systems based on individual organic molecules bound through dynamic supramolecular interactions.14–18 Illustrative examples of applications of these materials involve the implementation of artificial NTs in transfection for short interfering RNA,19 ion channels,20 drug delivery,21 asymmetric catalysts,22 antimicrobial materials,23 charge carriers,24 networks,25 fiber mats,26 ion sensors,27 and organic yarns,28 to cite a few.
Compared to inorganic and CNTs, self-assembled NTs offer substantial advantages including synthetic convergence, integrated error correction, tunable pore designs, higher biocompatibility and notably, the ability to self-organize. Different approaches for creating self-assembled NTs making use of weak and reversible non-covalent interactions, i.e., H-bonds, hydrophobic effects, π–π stacking, and/or metal–ligand coordination, among others, can be found in the literature. Some examples of these methods comprise: (a) the helical folding of oligomers,29 (b) the bundling of rod-like structures,30–32 (c) the rolling up of sheets made of amphiphilic molecules,33 and (d) the stacking of discrete cyclic units.27,34–36
Typical internal diameters reached by NTs assembled from classical amphiphilic molecules (method c) exceed 10 nm, which makes them perfect environments for integrating meso-scale entities such as (bio)macromolecules or nanomaterials, but are exceedingly large for conventional molecules.37 An interest in producing tubular structures with smaller pores, of a few nanometers, and thus compatible with molecular dimensions, has emerged in the scientific community as novel unconventional chemical processes could be explored in confined 1D nanospaces. While many advances have been reported towards that goal regarding (a) and (b) methods,33,38 this review is focused on the contributions made by diverse authors to the preparation of tubular structures from the stacking of discrete cyclic entities (strategy d), which can give access to pores with dimensions ranging from a few nanometers to the subnanometer scale (Fig. 1).
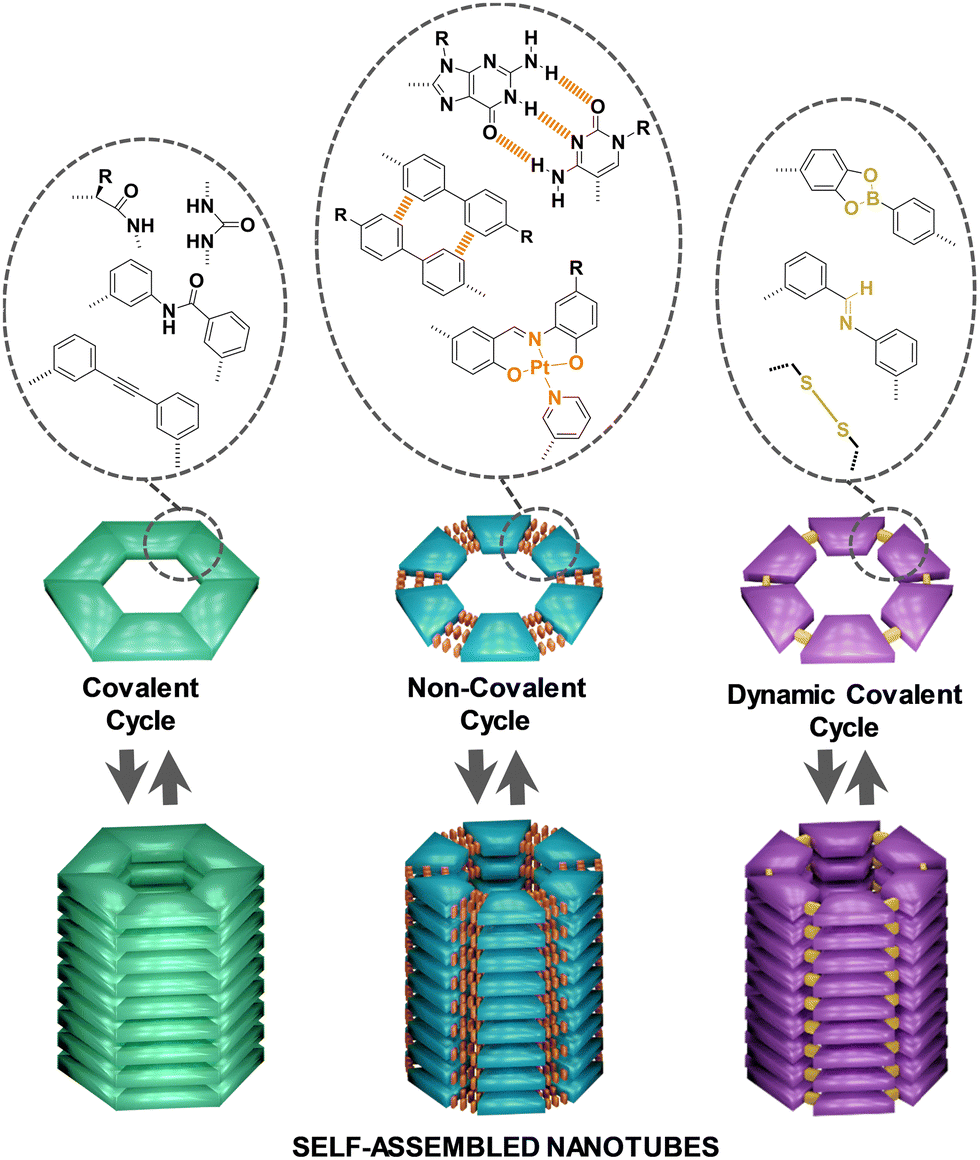 | ||
| Fig. 1 Classification of discrete cyclic entities into covalent, non-covalent and dynamic covalent cycles and schematic representation of their stacking into self-assembled NTs. | ||
The classification of the sections of this review takes into account the nature of the forces joining the individual units to form the corresponding cycles. Thus, the traditional and more developed approach consists in making use of organic reactions to form covalent bonds between small individual fragments to generate the corresponding covalent cycles, affording robust structures that cannot be disconnected back to their primary units. However, the advent in the last decades of supramolecular chemistry has introduced alternatives to connect in a reversible manner the small fragments that constitute the cycle, leading to non-covalent cycles that can be disassembled and assembled again by means of different stimuli. More recently, the incorporation of the new tools developed in the field of dynamic covalent chemistry has permitted the application of these versatile methodologies to the construction of dynamic covalent cycles of reversible nature but with higher stability compared to non-covalent cycles, as the interactions used to fuse the units are more robust. As self-assembled nanostructures, the vertical piling of all these kinds of cycles considered in this review into supramolecular polymers is achieved through the participation of additional supramolecular interactions, mainly H-bonds, π–π stacking and/or solvophobic interactions. Since the monomers are cyclic in nature, the resulting polymer's shape is tubular, generating a lumen in the inner part of the supramolecular structure. Although host–guest chemistry has been successfully employed for the construction of tubular structures,39–42 only examples of cycles that stack by themselves into tubular architectures, without the assistance of any other chemical species as template, have been contemplated in this review article.
Thus, the review starts with a general view of the well-known and consolidated strategies for the formation of covalent cycles, and progresses to describe in more detail the most recent approaches towards non-covalent cycles. It finishes with the introduction to the less explored use of dynamic covalent tools for the construction of cycles with the ability to form tubular structures. For the sections that have been explored in detail in other reviews, the description of the principles of the model and the most relevant examples developed so far are provided here, and only some interesting recent works are explained in depth in order to reach an overview in the context of the rest of the strategies.
2. Covalent cycles
The use of covalent cycles as scaffolds for creating NTs has been extensively used in the literature.43 In this review we will cover the main examples in which the flat shape of discrete cycles allows their vertical stacking to create tubular structures (Fig. 2). In the structures compiled in this section, H-bonds, π–π stacking and/or solvophobic forces constitute the main driving forces for the stacking of the cycles towards NTs. Examples of covalent cyclic structures that are intrinsically tubular and not planar, such as, cyclodextrins,30,44,45 cucurbiturils,31,46 or chaperonins,32,47,48 among others, and that can assemble on top of each other to generate higher order tubular architectures are not considered in this work. | ||
| Fig. 2 Schematic representation of the non-reversible organic synthesis of covalent macrocycles from individual precursors and subsequent reversible stacking of cyclic entities into self-assembled NTs. | ||
A key benefit of making use of covalent cycles is the ability to regulate the diameter of the NTs, which is precisely determined by the size of the cycle as the fundamental unit involved in the self-assembly process.
2.1. Cyclic peptides
Ghadiri's group was the pioneer in the exploitation of this assembly design. Their first structure consisted in the octapeptide of α-amino acids 1: cyclo[-(D-Ala-L-Glu-D-Ala-L-Gln)2-].50 The alternation of an even number of D- and L-amino acids led to a flat conformation in which the amide groups were perpendicular to the plane of the cyclic structure and the side chains pointed outwards. Thus, rings could stack with each other through intermolecular hydrogen bonds (H-bonds) in a β-sheet fashion (Fig. 3a). Based on this first chemical structure, many investigations have focused on expanding this approach to different α-cyclic peptides, with successful applications as transmembrane ion channels,51 antibacterial agents52 or proton conductivity in films,53 among others. Moreover, taking advantage of the intrinsic chirality of peptides, stereochemical control of the co-assembly process of enantiomers by imposing steric restrictions upon the H-bonding pattern has also been achieved.54 Regarding the potential use of these systems for biological applications, it is important to remark that the interchanged L- and D-amino acids in the cycle can have implications in the biological stability of the systems as it is known that the behaviour of D-amino acids can largely alter biological signalling processes.55–57
 | ||
| Fig. 3 (a) Chemical structure of an α-cyclic peptide (top) and schematic representation of the NT (bottom) formed by the self-assembly of 1. Structure of cyclic peptides containing (b) β-, (c) γ- and (d) α- and γ-amino acids, and illustration of the NT formation in (d) by the self-assembly of α,γ-cyclic peptides. Adapted with permission from ref. 20 and 58. Copyright 2016-Elsevier and 2013-American Chemical Society, respectively. | ||
This strategy to create NTs has also been extended to other kinds of amino acids or combinations of those. However, there are not many examples of cyclic structures entirely formed by β-amino acids in the literature. In contrast to other peptides integrated by other type of amino acids, those formed by β-amino acids are usually very insoluble and difficult to handle. For that reason, different functional groups have been often combined with amino acids in the skeleton of the cycle to facilitate their aggregation studies (see Heterocyclic peptide cycles section below). Seebach et al. reported the synthesis and characterization of the three different cyclic-β-peptides 2a, 2b and 2c, with no additional groups in their cyclic structure (Fig. 3b).59 Due to the solubility limitations mentioned before, they could not obtain single crystals of their compounds. However, the combination of powder X-ray diffraction (XRD) measurements and computer analysis enabled them to determine the solid-state structure which resulted in tubular aggregates in a similar fashion than those already observed for certain cyclic-α-peptides.
An attractive example in which the cyclic peptide contained exclusively γ-amino acids was reported by Deng and co-workers (Fig. 3c).60 This example is highly remarkable because it is one of the scarce cyclic peptides formed by this kind of amino acids and because, despite not having a planar conformation, compounds 3a and 3b were able to give rise to NTs through H-bond-mediated parallel stacking.
Granja's research group incorporated a novelty in the structure of cyclic peptides combining both α- and γ-amino acids. The introduction of γ-amino acids in compounds 4a, 4b and 4c (Fig. 3d) allowed the functionalization in the interior of the ensemble, for example, to create an hydrophobic pore, without distorting the required flatness of the cycle described by Ghadiri for α-peptides.61 In contrast, in NTs formed solely by α- or β-cyclic peptides, the amino acid side chains lie on the exterior of the ensemble, hampering the tuning of pore properties.
Based on the aforementioned Ghadiri's model, numerous research groups have studied the formation of NTs from different peptidic structures. Most of the examples have been collected in many general reviews about tubular supramolecular structures14,15,36 and also in more specific reviews where the use of cyclic peptides is described in detail.15,27,58,62–64 For that reason, we have mainly focused in this Cyclic peptides section on briefly describing the principles of the model and the most relevant examples developed so far, while some interesting recent works have been explained here in depth.
 | ||
| Fig. 4 (a) Compound 5 and its crystal structure. (b) Structure of cyclic peptide 6 (top) and TEM image of the NTs formed in acetonitrile (bottom left) and AFM image of the individual NTs of PEG-conjugated to 6 in DMF (bottom right). (c) Compounds 7a and 7b and illustrations of the arrangement of their corresponding assemblies. (d) Structure of compounds 8a and 8b. Adapted with permission from ref. 58, 68 and 69. Copyright 2019-American Chemical Society, 2016-Elsevier and 2006-American Chemical Society, respectively. | ||
A very recent article published by Xu and co-workers showed an example in which a benzimidazole group was incorporated in a cyclic peptide formed by α-amino acids (Fig. 4b).68 The presence of the imidazole moiety in the interior of cycle 6 provided pH-responsive properties to the resulting NTs. When the pH was decreased below its lower pKa, the NTs disassembled due to ionic repulsion from the positively charged benzimidazole groups. This was confirmed by UV-vis spectroscopy, unravelling different absorption bands at low and moderate pHs. Circular dichroism (CD) measurements corroborated the loss of the β-sheet structure at low pHs through the cancellation of the CD signal at 220 nm in these conditions. The organization of the macrocycle in the form of NTs in acetonitrile solutions was visualized by transmission electron microscopy (TEM), displaying a measured persistent length of 1.5 μm (Fig. 4b, bottom left). Finally, and with the future aim of dispersing the nanostructures and integrating them into polymer films, poly(ethylene glycol) (PEG) was conjugated to the outer lysine handles. Its deposition from DMF solutions resulted in the observance of individual NTs by atomic force microscopy (AFM) (Fig. 4b, bottom right).
A variety of modifications have been also carried out on β-peptides. Among others, Ghorai's research group has also utilized the triazole unit as an analogue of amides in terms of planarity and polarity, as well as H-bond donating and accepting ability (Fig. 4c).70 This kind of macrocycles exhibits two different rotameric forms that experience both parallel and antiparallel H-bonding between their backbones in solution. The formation of well-defined tubular nanostructures from these two macrocycles occurred through a comparable parallel homo-stacking through H-bonding between the amide NH and the carbonyl oxygen, even though the orientations of their functional groups differ (Fig. 4c).
Other heterocycles have also been incorporated in β-peptides, like saccharides in Kimura's and Chandrasekhar's works.69,71,72 The investigation of the ensembles of cyclic peptides in solution turned out difficult to perform due to the low solubility of these structures. The introduction of pyranose rings as a side chain of the platform of compounds 8a and 8b improved the solubility of the ensembles, thus overcoming this problem (Fig. 4d).69
In an example of the first strategy, Kimura's research group included tetrathiafulvalene (TTF) moieties at the side chains of cyclic β-peptides.73 With this modification, it was possible to alter the electronic properties of the final tubular structure. Another interesting example was introduced by Schmuck and co-workers. In their work, a non-natural arginine analogue was incorporated in the structure of a cyclic α-peptide.74 The presence of the weakly basic guanidiniocarbonyl-pyrrole moiety allowed the formation of positively charged cyclic peptide NTs under physiological conditions. They also demonstrated the usefulness of these aggregates in gene transfection through a non-endocytic cellular uptake pathway.
In the last years, Granja's research group has emerged as one of most active groups in the study of the self-assembly of cyclic peptides and they have reported numerous examples of functionalized systems,49,75–80 with successful applications as transmembrane transporters.20,81 They have exploited different combinations of α- and γ-amino acids to control the diameter of the nanostructures.82,83 Likewise, they have also described the combination of other amino acids, like the hybrids α-δ-cyclic peptides in which the inclusion of the δ-amino acid 4-aminocyclohexanecarboxylic acid in the chemical structure of the cycle, enabled the formation of NTs with large diameters and hydrophobic pores (compound 9 in Fig. 5a).84 Moreover, the presence of glutamic acid and lysine as side chains improved the solubility of the system and provided control on the stacking of cyclic peptides by salt bridge formation. In addition, the incorporation of histidine in the design of the peptide, afforded control on the self-assembly through pH modifications. The use of thioflavin, which is a well-known fluorescent probe that shows fluorescence when it binds to β-sheet structures, allowed to confirm the formation of NTs. Thus, addition of increasing amounts of compound 9 to an aqueous acidic solution of thioflavin, did not exhibit any fluorescence response. In contrast, similar additions to a slightly basic solution of thioflavin produced a rise in dye emission because of the binding between thioflavin and cyclic peptides interacting in a β-sheet fashion in the NT structure. Taking advantage of the hydrophobic properties and the large dimensions of the pore, the authors managed to encapsulate efficiently C60 fullerene molecules in the interior of the NT.
 | ||
| Fig. 5 (a) Structure of cyclic peptide 9 and side and top views of a computer assisted model of C60 encapsulated in the inner cavity of the NTs formed. (b) Structures of cyclic peptides 10a, 10b, 10a–10b and 10c and model of the proposed tubular structure formed by the assembly of 10a–10b and 10c. (c) Structure and model of pH-triggered self-assembly of cyclic peptide 11. Schematic illustration of drops containing compound 11 in different assembly degrees and locations, and confocal 3D projections of the droplets (bottom). Adapted with permission from ref. 84–86. Copyright 2018 and 2016-Royal Society of Chemistry and 2020-Wiley-VCH GmbH, respectively. | ||
The formation of one-dimensional structures by stacking of individual units has the limitation of the control of NT length. Granja and co-workers tackled this problem by using a method in which two cyclic peptides with different self-assembling properties were joint covalently. This led to the interaction between cyclic peptides and the formation of nanostructures with a cavity that resembled the shape of Venturi tubes.85 The two cycles that formed the covalently linked dimer were a combination of the small cyclic peptide 10a, with a high tendency to dimerize, and the large cyclic peptide 10b, which is able to homodimerize and to form heterodimers with the other unit described in the work: 10c (Fig. 5b). The homodimerization of 10a and 10b, the heterodimerization between 10b and 10c and the formation of supramolecular assemblies by the covalently linked dimer 10a–10b were studied by NMR separately, and the corresponding NMR signals for each assembled structure were identified. These results were compared to those obtained by 10c and by mixing a solution of the covalent dimer 10a–10b, in conditions in which they constitute NT structures with no defined length and confirmed the formation of the target NTs. Considering that the interacting distance between stacking cyclic peptides is established as 4.85 Å, the final NT would have a fixed length of approximately 30 Å.
Recently, Montenegro, Granja et al. have published several articles where the supramolecular polymerization process involved in the formation of NTs is adjusted using microfluidics and droplet encapsulation (Fig. 5c).86–88 The protonation of histidine and lysine residues in the cyclic peptide at acidic pH circumvents the assembly of supramolecular structures due to electrostatic repulsions.86 In contrast, deprotonation (alkalinisation) or counterion shielding (ionic strength) attenuates these repulsive forces and favours the stacking of the cycles to form the corresponding NTs. Moreover, the formation of bundles of NTs is assisted by the interaction of pyrene units, which can be monitored by the quenching and the bathochromic shift of the pyrene fluorescence emission, due to excimer formation. Taking into account these principles and regulating mixing regimes and trigger signals in microfluidics, it is feasible to control the pathways in the supramolecular polymerization process and to tune the packing degree, as well as the chemical nature and the spatial positioning of the resulting supramolecular peptide fibers. Experiments performed with pure water in one channel and an acidic aqueous solution of cyclic peptide 11 in another channel, thus allowing the final encapsulation in droplets once the channels were merged, disclosed emission properties that were characteristic of equally distributed dispersed cyclic peptides and monomeric pyrene units. However, when an alkaline solution was instead injected, a red-shifted green, fluorescent emission was detected at the edges of the droplet, and the related micrographs showed that the supramolecular peptide NTs tend to be adsorbed at the boundary of the water droplet interface. In contrast, when a solution of high ionic strength (CaCl2, 1 M) was used, the NTs were localized at the droplet core.
Granja et al. have also created a novel type of cyclic peptide hybrids with the ability to stack together in a parallel manner, so as to create NTs that can flex when exposed to light.89 In this scenario, the anthracene attachment on each cyclic element undergoes photodimerization, leading to alterations in the NT's dimensions and potentially prompting structural modifications in the way the NTs are packed, causing them to curve.
Regarding the second strategy, in which polymeric entities are covalently attached to cyclic peptides, the work of Biesalski and co-workers results essential, since they described the first example of a peptide–polymer hybrid structure.90 To this end, they conjugated a polymer initiator group to an α-peptide (compound 12 in Fig. 6a). In their approach, the polymer was linked to the surface of the structure after the formation of the ensemble by embedding the peptide NT in a polymeric shell. The morphology of the cyclic peptide NTs and the peptide–polymer NTs was compared by AFM (Fig. 6b and c). Single NTs and 2D aggregates with an average height of 1.7 ± 0.2 nm were observed for the first ones, whilst the peptide–polymer NTs were present as distinct rod-shaped structures that were 12 ± 3 nm in height.
 | ||
| Fig. 6 (a) Structure of cyclic peptide 12 and schematic outline of the synthesis of its corresponding peptide–polymer hybrid NT. AFM image of NTs and height distribution from the statistical analysis of adsorbed NTs from the assembly of (b) cyclic-peptides and (c) peptide–polymer hybrids. Adapted with permission from ref. 90. Copyright 2005-Wiley-VCH GmbH. | ||
Additionally, the preparation of hybrids of cyclic peptides and polymers can also be performed before NT formation, as firstly demonstrated by Börner's group.91 More recently, Perrier and Jollife's group has shown many examples using this convergent methodology.92–94 Many of these systems have unravelled possible potential applications derived from their aggregation behaviour and activity inside cells,95,96 and their membrane permeabilization abilities.97 In one of their most representative works, they prepared NTs with dual functionality by the connection of two different polymer chains to the peptide structure (Fig. 7a).98 The combination of immiscible polymers and a cyclic peptide led to the creation of Janus NTs, which assembled into artificial pores comprised of bundles of these tubes through a phase segregation process. Polystyrene (PS) and poly(n-butyl acrylate) were chosen as non-miscible polymers in compound 13a. With the aim to compare it with the Janus structures, the hybrid cyclic peptide–polymer conjugate 13b, bearing two miscible polymers: PS and poly(cyclohexyl acrylate), which should form a single microphase corona, was also prepared. The formation of NTs was confirmed by FTIR, dynamic light scattering (DLS) and small angle scattering experiments (SAXS). Additional nuclear Overhauser enhancement spectroscopY (NOESY) NMR experiments enabled to distinguish the Janus versus mixed nanostructures by analysing the absence or presence of contacts between different polymeric chains, respectively. Moreover, differential scanning calorimetry (DSC) analyses corroborated the microphase separation in Janus NTs by the observance of two distinct glass transition temperatures (Tg). In contrast, the same analysis performed in the mixed NTs led to a single Tg, because of the miscibility of the polymeric chains.
 | ||
| Fig. 7 (a) Structure of cyclic polymer–peptides 13a and 13b and illustration of the Janus vs. mixed assemblies. (b) Structure of amphiphilic cyclic peptide-diblock polymer conjugate 14 (top), schematic of stable non-exchanging (middle right) and dynamically exchanging mixed (middle left) cyclic polymer–peptide NTs, and schematic and stochastic optical reconstruction microscopy (STORM) of a co-injected cyclic peptide–polymer–dye conjugates (bottom). (c) Chemical structures of fluorophore-cyclic peptide–polymer conjugates 15 (left), cartoon illustration of the artificial light-harvesting system (right) and photograph showing different emission colours at different conjugates ratios (bottom). Adapted with permission from ref. 84, 98 and 99. Copyright 2013 and 2019-Springer Nature, and 2021-American Chemical Society, respectively. | ||
The same authors have as well reported the use of their Janus-structures as potential drug carriers.98,100 Amphiphilic polymer-cyclic peptides were able to originate NTs with a hydrophobic internal channel (diameter: 16 nm) and a hydrophilic shell. The barrel-shaped alignment of single peptide NTs into a large tubisome supported the perforation of the lysosomal membrane in cells and the consequent release of calcein into the cytosol. Very recently, they have also described an efficient approach to create polymeric tubular structures from cyclic peptide-bridged amphiphilic diblock copolymers with photoresponsive abilities.101 The tubisomes obtained display good biocompatibility, high drug loading content and rapid drug release upon UV irradiation.
Perrier's group has also recently published a compelling work based on the stabilization of polymer–peptide hybrid NTs in water. In order to improve their self-assembly, a hydrophobic domain was formed in the periphery of the cyclic peptide through the connection of an amphiphilic diblock copolymer (compound 14 in Fig. 7b).99 Thus, a secondary driving force created a hydrophobic region surrounding the peptide core, which prevented the competition of water molecules with the H-bonds participating in the stacking of the cyclic peptides. The formation of NTs was confirmed by TEM and small-angle neutron scattering (SANS), and the presence of a hydrophobic core was supported by the emission merged from the encapsulation of the fluorescent dye 1,6-diphenylhexatriene, whose emission is normally quenched in water solutions. The investigation of the stability of the ensembles was carried out by comparing static light scattering (SLS) measurements in different experimental conditions. The dynamic behaviour of the ensembles was probed by Föster resonance energy transfer (FRET) studies. For that purpose, FRET donor Cy3 or acceptor Cy5 dyes, were linked to the periphery of the peptide structure. Only when the two dyes are assembled together in the same NT, the proximity between them favours the energy transfer event. If the cyclic peptides are highly dynamic, i.e., they disassemble and re-assemble readily, they would progressively form mixed NTs. However, when the pre-assembled Cy3- and Cy5-diblock conjugates were co-injected together, the change in FRET ratio was extremely slow, as opposed to the control experiment with free dyes where a constant moderate FRET ratio was observed. These results corroborated the kinetic stability of the NTs formed. Finally, in order to visualize this exchange, the group imaged the NTs using super-resolution fluorescence microscopy. By this technique, they could compare the images obtained from the mixture of Cy3 and Cy5 conjugates with the ones for the mixture of the separately pre-assembled Cy3 and Cy5 peptides. In the first experiment, the images showed aggregates composed of the combination of both conjugates, whilst the second experiment resulted in more aggregates exhibiting a single colour, showing that the Cy3 and Cy5 dyes were not as well mixed as in the non-preassembled compounds solutions. The kinetic stability of the hydrophobic core restricts the free interchange of cyclic peptide-conjugates between NTs, which accounts for the observation that Cy3 and Cy5 are not randomly distributed within the NTs.
This group has also described a method for creating efficient artificial light-harvesting systems using supramolecular peptide NTs in water.94 A two-step sequential energy transfer process is produced in the co-assembly of three hydrophobic chromophore (pyrene, naftalene monoimide and Cy3 in compounds 15a, 15b and 15c, respectively, in Fig. 7c) cyclic peptides. An efficient FRET process takes place in which light energy is transferred from 15a to 15b, and subsequently transmitted to 15c. This phenomenon leads to an energy transfer efficiency of up to 95%, a fluorescence quantum yield of 30% and highly stable system. Additionally, the emitted light can be adjusted from blue to green to orange and can even produce white light with a fluorescence quantum yield of 29.9%.
Xu and co-workers have as well used this convergent methodology, in which the connection of polymeric chains is performed before NT formation, in their investigations. The growth of NTs in a confined geometry can be modulated by the regulation of the interactions between NTs and medium through the selection of a polymer conjugate with a specific solubility in the copolymer microdomain. In one of their works, they were able to conjugate low molecular weight poly(ethylene oxide) (PEO) to cyclic peptides.102,103 After that, they assembled the cyclic peptides vertically by annealing the PEO–peptide conjugates with a hexagonally packing poly(styrene-b-methyl methacrylate) block copolymer. With this method, they created nanochannels in free standing polymeric films able to transport gases.102
2.2. Aromatic cycles
The first group to introduce cyclic oligoamides to create NTs was Gong's research group, which has subsequently explored this molecular design in manifold ways. In their attempt of extending their family of helical polymers based on the backbone rigidification of aromatic oligoamides, they discovered that one of their compounds originated a macrocycle and they reported the highly efficient one-pot synthesis of this new compound (16) and its subsequent aggregation into NTs (Fig. 8a).104,105 The formation of the macrocycle is favoured by the three-centered intramolecular H-bond interaction, which preorganizes the uncyclized oligomer precursors. Taking advantage of this finding, the group has published numerous examples showing the strong aggregation of related macrocycles into tubular structures.106,107 Due to the flatness of the structure and its large aromatic surface area, the directional assembly is most likely mediated by intermolecular aromatic stacking interactions, generating unidimensional objects with a long channel. They have also reported the use of these NTs for the incorporation of metal ions,108 as transmembrane systems,109–112 and as liquid crystals.113
 | ||
| Fig. 8 (a) Chemical structure of cyclic oligoamide 16 and proposed stacking of macrocycles into long tubes. (b) Structure of cyclic oligoamide 17 and non-cyclic oligoamide 18 (top), fluorescence spectra of 17 recorded at different concentrations in CHCl3 (bottom left) and schematic drawing of the columnar packing and the hexagonal lattice formed by 17 (bottom right). (c) Structure of oligoamide macrocycle 19, illustration of its assembly into tubes and selective permeation to different ions, and AFM image of a solution of 19 in CHCl3 deposited on freshly cleaved mica (right) (scale bar = 300 nm). Adapted with permission from ref. 105, 110 and 111. Copyright 2015 – Royal Society of Chemistry, and 2013 and 2016 – American Chemical Society, respectively. | ||
As previously mentioned, the control of the length in supramolecular polymers constitutes a challenge because monomers tend to associate into polydisperse aggregates. Gong and co-workers have described the assembly of oligoamide macrocycles into discrete stacks without the use of any template. Specifically, their compounds can stack into tetramers with a defined length and a fixed pore diameter.114 Concentration dependent 1H NMR and NOESY experiments in solution revealed the characteristic changes involved in the formation of supramolecular aggregates. Additional diffusion-ordered spectroscopY (DOSY) experiments suggested that the macrocycles associated into discrete oligomers. These findings were further supported by computational analysis and the X-ray structure of the single crystal of one of the compounds of the family, confirming the aggregation into “quadruple-decker hamburger” with two planar “inner cycles” and two bowl-shaped “capping cycles” that position protruding electronegative amide oxygen atoms at both ends of the tetrameric structure. The discrete stacks of these cycles closely pack into two dimensional (2D) layers that further stack on top of each other.
The same authors have also reported extremely strong tubular stacking processes with this kind of oligoamide macrocycles.110 In a new generation of cyclic systems, they attached amide side chains to the backbone of the cycle (compound 17 in Fig. 8b). 1H NMR studies carried out in CDCl3 and more polar solvents like DMSO-d6 and DMF-d7, proved the aggregation of oligoamide 17. Additional experiments performed in CDCl3 with increasing amounts of DMSO-d6 and the comparison of the same experiments with non-cyclic compound 18, allowed to extract information about the exposure of the different amides to the solvent media. The corresponding results suggested that amide side chains in 17 are oriented to solvent molecules as in the molecularly dissolved compound 18, whilst the aggregation in compound 17 prevents the contact of the amide groups of the skeleton of the cycle with solvent molecules. Emission spectra collected at reduced concentrations in CHCl3 (Fig. 8b bottom left) indicate that 17 remains aggregated at concentrations as low as 1 pM and it is at the monomeric state only at 0.1 pM, which disclose beyond doubt a huge aggregation propensity in this solvent. The columnar assembly was also demonstrated by XRD on solid samples. Reflections typical of columnar stacks of disc-like molecules that packed in a hexagonal (colh) lattice were observed. Bearing in mind these findings, it was possible to calculate the diameter of the ensemble as 29.8 Å. Moreover, a peak ascribed to π–π interactions at 3.66 Å was also found. Considering the Scherrer's equation,115 the length of the NTs could be estimated as 22 nm, corresponding to the stacking of 60 cycles. This data remarks the high-order association into long columns.
Recently, the authors have tuned the inner pore of the cycles by a structural modification of one of the macrocycles previously reported.108 This structural alteration comprised the replacement of part of the conventional aromatic oligoamide backbone used by the research group by a diethynylarene unit with an inward pointing functional group (X) that permitted the modulation of the pore properties (Fig. 8c).111 Furthermore, a secondary amide side chain was also incorporated to strengthen the directionality of the aggregation. The formation of NTs was supported by the results obtained by 1H NMR and emission and UV-vis spectroscopies. Moreover, AFM images exhibited nanofilaments that constituted and additional evidence of the organization into nanotubular structures. The assessment of the capacity of the family of macrocycles toward transmembrane proton transport led to different results as a function of the X group incorporated. A vesicle-based stopped-flow kinetic assay of the emission intensity was adopted to monitor proton transport across lipid bilayers. While significant enhancement of proton transport was observed in the presence of 19a and 19c, no transport above background was detected with 19b or 19d. This dissimilar behaviour could be attributed to the different arrangement of the corresponding NTs that gave rise to diverse pore properties. In order to check if the absence of transport could be due to the blocking of the channel by some X groups, chloride ion transport was also examined. Thus, the pores of 19b and 19d, even being impermeable to protons, allowed the transport of chloride ions, indicating that the pore was not physically blocked. Effective anion binding and transport has also been recently reported by Gong's research group for new C5-symmetric aromatic pentaamide macrocycles that were incorporated in biomembranes.112
Focused on the same research purpose, Zeng and co-workers have also synthesized aromatic oligoamide macrocycles with the ability to form 1D-nanostructures capable to encapsulate solvent molecules and gelate.116 In their work, they designed similar macrocycles to those described by Gong and co-workers in which fluorine atoms were incorporated at the aromatic rings facing towards the interior of the cycle (Fig. 9a). The introduction of F at these positions created very strong intramolecular C–F⋯H–N bonds and did not alter the planar conformation characteristic of the aromatic oligoamide macrocycles. X-Ray measurements of the single crystal obtained for compound 20a revealed the existence of intermolecular H-bonds and π–π stacking between cycles. Considering these results, compounds 20b and 20c, in which hydrocarbon chains were anchored to the cyclic structure, were designed as possible organogelators. Thus, the tendency of these cycles to stack that was previously probed for compound 20a, and the intercolumnar interactions through hydrocarbon chains, derived in the formation of 3D networks able to entrap organic solvents and gelate. The gelation behaviour of both compounds in a variety of organic solvents was investigated. UV-Vis spectroscopy informed about the arrangement of the molecules in the gel structure. The blue shift observed from CHCl3, where no gelation takes place, in comparison to the rest of the solvents in which both compounds form gels, was ascribed to the presence of H-aggregates, which was also in accordance with the crystal structure of 20a. TEM studies resulted in the observation of nanofibers and the corresponding SEM images of the gels showed their characteristic porous structure. Moreover, XRD analysis indicated the generation of 3D entangled nanofibers by the penetration of the alkyl side chains into each other.
 | ||
| Fig. 9 (a) Structures of fluoropentamers 20a–c (left), top and side views of crystal structure of 20a (middle) and TEM and SEM images of the gels formed by 20a in n-hexane (right). (b) Molecular structures of macrocycle 21 and schematic representation of the Cs+-directed self-assembly and the consequent UV-promoted polymerization. (c) Chemical structure of cyclic compound 22 and schematic illustration of the protonation-induced self-assembly process and consequent UV irradiation. Adapted with permission from ref. 116–118. Copyright 2011, 2019 and 2020 – American Chemical Society, respectively. | ||
Kim's research group have recently published an example of oligoamide macrocycle that combines amides, pyridines and polydiacetylenes (compound 21 in Fig. 9b).117 On one hand, the pyridine nitrogen and the amide groups provide donor sites to the cycle for binding metal ions. On the other hand, the polydiacetylenes facilitate the directional organization of the macrocycles into hollow 1D-dimensional ensembles and permit their topochemical polymerization to form rigid NTs. Macrocycle 21 crystallized into a chair-like conformation that stacked in a columnar arrangement generating the expected tubular structures. The system showed efficient Cs+ coordination and topochemical polymerization upon exposure to 254 nm UV light. As a result, Cs+-containing polydiacetylene NTs with thermochromic properties were generated.
A very similar compound to this one was recently published by the same authors (22 in Fig. 9c).118 In this case, the chemical structure was varied just in the length of the alkyl chains that act as flexible spacer between amides and diacetylenes. In this work, the protonation of the pyridine was used to promote the self-assembly of these flexible macrocycles. An aqueous H2SO4 solution was added to a solution of macrocycle 22 in THF to protonate the pyridine rings. The supramolecular salts generated were drop-casted on a glass substrate to afford the final one-dimensional structures, where protonation was confirmed by FTIR studies. Additional 1H NMR experiments in CDCl3 were performed to evaluate the appropriate ratio of H2SO4 for protonation of macrocycle 22. The ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (macrocycle 22:H2SO4) in which all the protons of the macrocycle disappeared due to the precipitation of the protonated salt, was found optimal. A mass peak attributed to the 1:2 adduct was also observed by MALDI-TOF spectrometry. Moreover, X-ray diffraction analysis provided information about the organization of the ensemble. Comparing the results obtained with those found for compound 21, the authors suggested the stacking of macrocycle 22 in a non-planar stair-like conformation. Taking advantage of the presence of polymerizable groups and as in the example previously described, the monomeric protonated macrocycle was polymerized under UV light to afford robust columnar nanoarchitectures with multi-stimuli colorimetric responses: reversible thermochromism, selective solvatochromism for dimethyl sulfoxide and dimethylformamide, and organic/inorganic base sensing. The differences in the morphology of the non-polymerized and the polymerized nanostructures were investigated by SEM. A very similar fibrous structure was observed for the nanostructures formed by protonated macrocycle 22 and by the polymerized structure.
:2 (macrocycle 22:H2SO4) in which all the protons of the macrocycle disappeared due to the precipitation of the protonated salt, was found optimal. A mass peak attributed to the 1:2 adduct was also observed by MALDI-TOF spectrometry. Moreover, X-ray diffraction analysis provided information about the organization of the ensemble. Comparing the results obtained with those found for compound 21, the authors suggested the stacking of macrocycle 22 in a non-planar stair-like conformation. Taking advantage of the presence of polymerizable groups and as in the example previously described, the monomeric protonated macrocycle was polymerized under UV light to afford robust columnar nanoarchitectures with multi-stimuli colorimetric responses: reversible thermochromism, selective solvatochromism for dimethyl sulfoxide and dimethylformamide, and organic/inorganic base sensing. The differences in the morphology of the non-polymerized and the polymerized nanostructures were investigated by SEM. A very similar fibrous structure was observed for the nanostructures formed by protonated macrocycle 22 and by the polymerized structure.
Shimizu and co-workers have synthesized different macrocycles constituted by oligoureas. In their first example, the smallest symmetrical member of the family containing two urea groups separated by two rigid aromatic units 23 was designed (Fig. 10a).119 After that, the aromatic spacers were varied from m-xylene, 4,4′dimethyl-diphenylether, to 4,4′-dimethylbenzophenone.119–121 The whole family crystallized into the expected columnar structures. The solid structure of compound 23 showed both intermolecular urea H-bonds and aryl interactions between the cycles.
 | ||
| Fig. 10 (a) Chemical structure of rigid macrocyclic bis-urea 23 and corresponding stacking into columnar structures (right). (b) Structure of flexible bis-ureas 24–28 and (c) normalized FT-IR spectra of macrocycles 24 (left), 25 (middle), and 26 (right) at different concentrations. Adapted with permission from ref. 122. Copyright 2009 – American Chemical Society. | ||
Inspired by the more flexible oligourea systems capable to crystallize into columnar ensembles described by Guichard's and Ranganathan's research groups,123,124 Shimizu's research group considered the creation of unsymmetrical oligoureas by the replacement of one of the aromatic spacers in compound 23 by an aliphatic spacer (compounds 24–28 in Fig. 10b).122 As a consequence, the solubility was improved and experiments in solution could be performed. Unexpectedly, some dissimilarities were found in the organization of the compounds in the solid state. Only oligoureas 24 and 28 were able to crystallize into columnar arrangements with strong urea–urea interactions, although the intermolecular π–π interactions unveiled for 23 and 28 were substituted by CH⋯π interactions in compound 24. However, compound 25 did not crystallize and compounds 26 and 27 led to solid structures in which the organization was not columnar. The influence of the heteroatoms in the strength of the macrocycle–macrocycle association constant was evaluated through solution studies. The calculation of the association constants was performed by FTIR (Fig. 10c) following the method used by Jadzyn and Bouteiller for the aggregation of acyclic bisureas.125 The corresponding association constants for the oligomerization of the macrocyclic oligoureas 24–26 in tetrachloroethane resulted much lower (K = 5–600 M−1) than those obtained for related acyclic bisureas (K = 1900 M−1) in CHCl3. Moreover, compounds equipped with ethylene glycol chains 24 and 25 showcased lower association constants compared to compound 26, probably due to the formation of intramolecular H-bonds with the ether oxygens, which compete with those promoting the cycle stacking.
Also, based on Ranganathan's model, Martin's research group designed a bisurea macrocyle with flexible alkyl spacers to link two urea groups to two electron-donor di(methylthio)tetrathiafulvalene (DMTTF) electroactive moieties (29 in Fig. 11a).126 Comparison of the room temperature 1H NMR experiments of 29 in solvents of different polarity, as well as variable temperature and depolymerisation experiments by the addition of a strong H-bond acceptor solvent, confirmed the formation of oligomers of 29 through H-bonding. Besides, DLS measurements of solutions of 29 in CH3OH permitted to do an estimation of the length of the oligomers, disclosing a distribution from 110 to 150 nm, centered at 135 nm. The organization of the molecules in the assembly was further investigated by cyclic voltammetry (CV; Fig. 11b, left) and differential pulse voltammetry (DPV; Fig. 11b, right) experiments, taking advantage of the presence of the electroactive DMTTF groups. Voltammograms recorded in DMF solutions disclosed two different quasi-reversible and one irreversible oxidation processes. The appearance of a third oxidation process at an intermediate potential, E2 = 0.39 V, between the first and third potentials, E1 = 0.30 V and E3 = 0.67 V, and the broadening of all waves in the DPV experiments are a trademark of TTF in close spatial proximity. This closeness can only be reached if the DMTTF units of the macrocycle stack forming the NT. In a control experiment, E2 was not detected when the supramolecular structure was disassembled by addition of DMSO. Considering this model, the group was able to control the self-assembly process electrochemically (Fig. 11c). For that purpose, CV experiments at different scan rates (100–1000 mV s−1) were performed. At potential E1 involving n electrons, (29)n+n would be formed (where n is the number of molecules of 29 that constitute the NT-mixed valence species). The stabilization of the aggregates would be explained by the charge-transfer interactions between vicinal oxidized and neutral DMTTF units. In a second more energetic oxidation process (E2) involving n electrons, (29)2n+n species would be generated. In a full oxidation at E3 involving 2n electrons, free n × 294+ macrocycles would be obtained due to Coulombic repulsion.
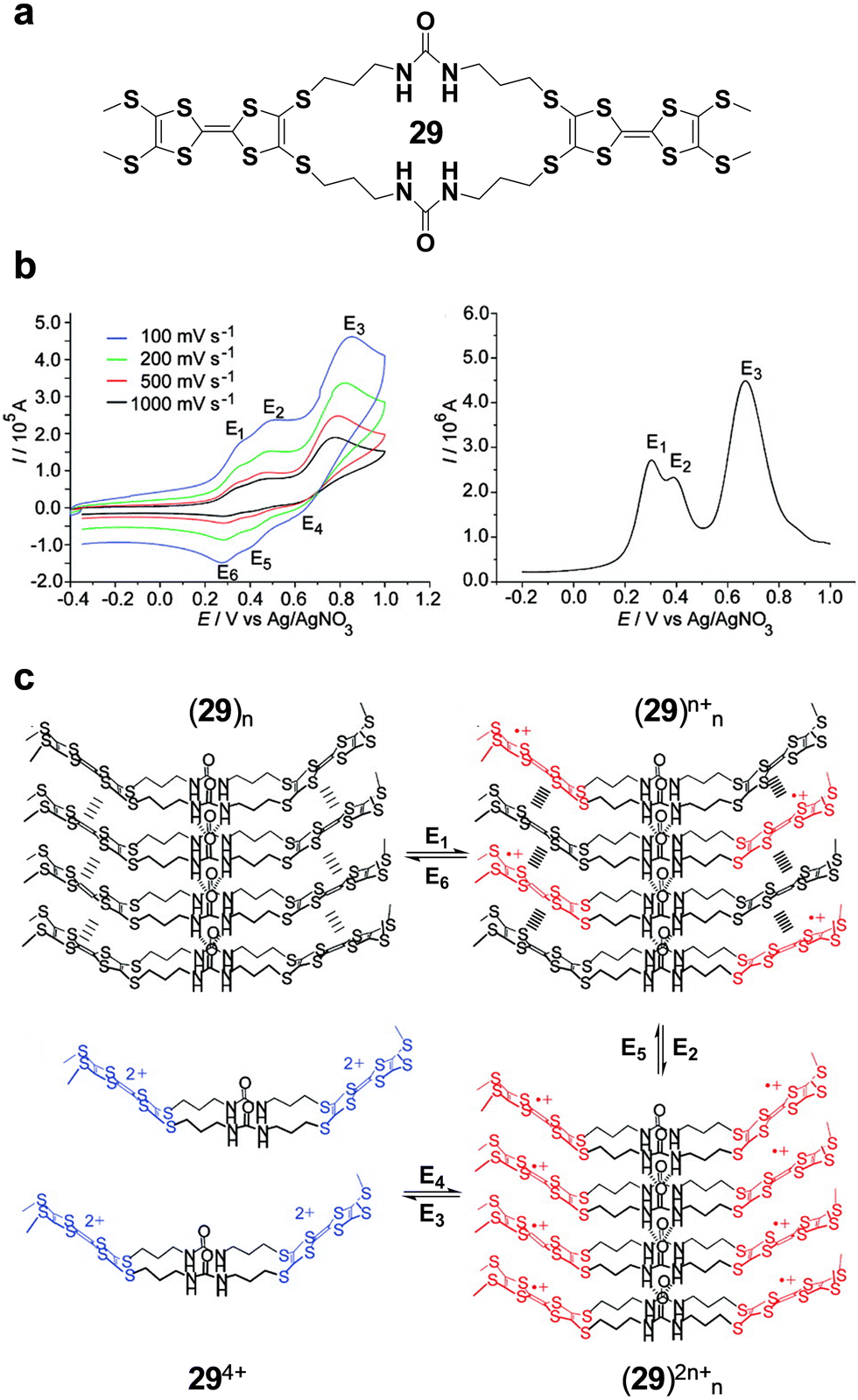 | ||
| Fig. 11 (a) Chemical structure of DMTTF-containing bis-urea 29. (b) Cyclic voltammograms of 29 (5 × 10−5 M, DMF, Ag/AgNO3 as reference electrode, glassy carbon as working electrode, Bu4NClO4 as supporting electrolyte, 298 K) at several scan rates (left) and differential pulse voltammetry of 29 under identical conditions (right). (c) Proposed redox-controlled assembly–disassembly mechanism. Adapted with permission from ref. 126. Copyright 2009-American Chemical Society. | ||
Rigid π-conjugated spacers have as well been incorporated to bisurea macrocycles. For instance, Greytak and Shimizu introduced two rigid phenyl spacers to link two urea groups (compound 30 in Fig. 12).127 XRD of the single crystals obtained from DMSO solution unveiled a columnar arrangement with a channel filled with DMSO molecules. In this work, the research group investigated the encapsulation and orientation of a guest within the pore of the NT formed. To permit the uptake of the fluorescent push–pull dye selected 31, the exchange of the DMSO molecules through activation of the NTs by heating was required. Then, the activated crystals were treated with an acetonitrile solution of 31. The first indication of the encapsulation of the dye came from the colour change from white to purple observed due to the solvatochromism of 31. A control experiment performed on non-activated crystals where no colour change was detected, allowed discarding surface absorption, and demonstrated the incorporation of 31 within the channels. With the objective of quantifying the filling of the dye, the complex was disassembled with DMSO-d6 and analysed by 1H NMR. The comparison of specific integrals of 30 and 31 provided a 11:1 ratio. Based on this result and the structural model, the loading ratio represented about 50% of the available space. Additional X-ray measurements indicated that the high crystallinity was not reduced by the incorporation of dye molecules into the pores. Moreover, the sensitivity of the dye to solvent polarity provided information about the nature of the pore. Within the channels, the dye exhibits an absorbance peak position comparable to that discerned in a highly polar solvent environment. Concerning the emission properties, even though 31 is almost nonfluorescent in polar solvents, prominent fluorescence was observed as an enhancement of emission in confined spaces. Regarding the orientation of the dye, the emission was polarized along the fiber axis, achieving polarization values as high as 0.729, which indicates a significant level of directional alignment within the one-dimensional channels.
 | ||
| Fig. 12 Chemical structure of rigid macrocycle bis-urea 30, schematic illustration of its crystallization into columnar stacks, and encapsulation of dye 31 with dipole moment orientation. Adapted with permission from ref. 127. Copyright 2017-American Chemical Society. | ||
More recently, Gale and co-workers have published an interesting example of a tetraurea macrocycle consisting of four urea groups connected by ortho-phenylene rings.128 The combination of urea H-bonds and π–π stacking interactions enabled the formation of nanotubular channels in which solvent-excluding “sandwich-like” binding sites between the macrocycles for selective binding of chloride from aqueous solutions were created. This approach offers the possibility of working in aqueous environments with anion-binding classical urea receptors that usually operate in organic solvents.
Aggregation of phenylene-ethynylene macrocycles was firstly investigated by Moore et al. Their studies revealed that the stacking of conjugated aromatic cycles is highly influenced by the polarity of the solvent used.131 Thus, polar solvents favour the aggregation whilst low-polar solvents attenuate the assembly. Likewise, the aggregation process is also affected by the properties of the side chains incorporated to the backbone, being enhanced by electron-withdrawing groups and weakened by electron-donating groups.132
In order to obtain well-defined, monodimensional assemblies built-up from arylene-ethynylene molecules, Moore and co-workers have also explored the substitution of phenylene groups by another aromatic derivative with large planar molecular surface. For instance, the incorporation of carbazole derivatives allowed the fabrication of a new type of nanomaterials with well-defined, non-collapsible internal channels and interesting potential applications in nanoscale optoelectronic devices.133,134
Following a similar strategy, Kobayashi's research group reported the combination of anthracene derivatives and phenylene ethynylene units for the synthesis of rigid and planar cycles suitable for π-stacking into supramolecular architectures (compound 32; Fig. 13a).135 The high dependence of the 1H NMR chemical-shifts of macrocycle 32 as a function of the concentration or temperature in CDCl3 (Fig. 13b) were indicative of the presence of π–π interactions. The corresponding fit of the data to the isodesmic mechanism resulted in an association constant of Ka = 6980 M−1 in CDCl3. This value was higher than the one obtained for simple cyclic meta-phenylene-ethynylene and meta-phenylene-diethynylene hexamer analogues and demonstrated the enhancement of π–π stacking interactions assisted by anthracene rings. SEM images of drop-casted films and XRD analysis of the powder of 32 from evaporated CHCl3 solutions confirmed the formation of nanofibers with hexagonal packing structure (Fig. 13c).
 | ||
| Fig. 13 (a) Structure of macrocyle 32. (b) Concentration dependence of the 1H NMR spectra of 32 in CDCl3 at 303 K (the signals marked ‘s’ are the satellite signals (13C–1H coupling) of the residual solvent). (c) Schematic illustration of the self-assembly of 32, where π-stacked columns form a hexagonal structure. Adapted with permission from ref. 135. Copyright 2016-Wiley-VCH GmbH. | ||
An alternative strategy to strengthen the formation of organic NTs from the self-assembly of phenylene-ethynylene macrocycles was developed by Gong's research group, who introduced cooperative interactions between π–π stacking and H-bonding. Their design consisted in planar, π-conjugated hexa(m-phenylene-ethynylene) cores decorated with amide side chains. Driven by the formation of the maximum number of H-bonds, the macrocycles were forced to stack cofacially, forming columnar assemblies that are confined in H-bonded networks. In their first article published on these chemical structures, they assessed the critical importance of H-bonds and the internal cavities in the formation of nanotubular structures.136 With this objective in mind, they synthesized macrocycles 33a–d with planar π-conjugated cores and six amide side chains (Fig. 14a). Compounds 33a and 33b possess chains of different length but similar internal cavity, whilst compound 33c shares the same side chain as 33a and differs from it in the substituents oriented to the internal cavity. Compound 33d bears tertiary amide side chains and cannot form H-bonds. The presence of long fibers was observed on solid surfaces for 33a and 33b by SEM and TEM microscopies. Additionally, AFM measurements revealed an interfilament distance for 33a of 3.2 nm, in agreement with the intercolumnar distance detected by XRD on the bulk sample of 33b. The AFM images of 33c (Fig. 14a), including two methyl groups in its cavity, also showed fibrillar structures and from this it can be inferred that there is no effect of the methylation of the inner cavity on the assembly of the cycles. On the contrary, compound 33d, devoid of the NH amide group, was unable to aggregate into tubular architectures and consequently, no fibers were observed by AFM. This fact was accompanied by the lack of CD response in conditions in which compound 33b exhibited strong CD signals indicative of the presence of chiral assemblies, and provides additional support for the significance of H-bonds in the formation of these NTs. The resultant hydrophobic pore created in the NTs formed displayed highly selective ion transport and efficient water transport across lipid membranes.
 | ||
| Fig. 14 (a) Chemical structures of macrocycles 33a–d, model of the helical stack of the macrocycles by QMD simulation (right) and AFM image (bottom) of 33c assembled in CCl4 and deposited on mica (scale bar for the main image and the inset is 500 nm and 40 nm, respectively). (b) Structure of macrocycles 34 (top) and measured (bottom, blue solid line) and simulated (bottom, red dash line) CD spectra of the 1:1 mixture of (left) 33b and 34a, (middle) 33b and 34b, and (right) 34a and 34b in CCl4 (5 μM per compound). (c) Structure of macrocycle 35 and rod-like hydrophobic guests with a defined length 36, and representation of the guest-dependent size of the tubular assemblies of 35. Adapted with permission from ref. 136–138. Copyright 2012-Springer Nature, 2017 and 2020 – American Chemical Society, respectively. | ||
Further modifications on the inner cavities of rigid macrocyclic building blocks permitted the authors to investigate the effect of simple structural modifications on the strength of the association.139 The comparison of absorption and dichroic spectra in solvents of different polarity and at different temperatures provided information about the stacking strength of the macrocycles. Thus, the introduction of different inward-pointing functional groups modified not only the cavities but also the electronic features that influenced the tendency of the cycles to stack. Electron-withdrawing groups diminish the electron density within the cycle, promoting π–π stacking interactions. Conversely, electron-donating groups increase the electron density, reducing the strength of the stacking interactions.
Taking advantage of the different electronic properties of the backbones, hetero-association can be promoted to form hybrid NTs.137 With this objective in mind, macrocycles derived from compound 33b in Fig. 14a were synthesized having electron-withdrawing fluorine substituents, as in compound 34a, or electron-donating amino substituents, as in compound 34b (Fig. 14b). The formation of nanotubular stacks of identical outer dimensions by the self-assembly of compounds 34a and 34b was confirmed by NMR, UV, CD and fluorescence spectroscopies and by AFM. The propensity of the different macrocycles to heteroassociate was first probed computationally by density functional theory (DFT) method considering stacks of 12 macrocycles. The binding energy obtained between two macrocyclic molecules pointed out that, with the exception of the most stable heterodimer, consisting of 33b and 34a, the remaining five dimeric stacks show little variation in their binding energies. This indicates that these macrocycles do not exhibit a strong tendency for self-association. The three macrocycles (33b, 34a and 34b) were then mixed in pairs in CCl4, leading to three 1:1 mixtures, 33b + 34a, 33b + 34b, and 34a + 34b. The CD spectrum of each pair was then measured and compared with the simulated spectra of a hypothetical 1:1 mixture in which the component macrocycles do not mix-assemble (Fig. 14b bottom). For the mixture of 33b and 34b, the calculated and observed spectra show some resemblance, although they have distinct features. The spectra obtained from the 1:1 mixtures of 33b and 34a, and 34a and 34b, were clearly dissimilar from the simulated ones and suggested the existence of favourable hetero-association processes.
In a recent article, the aggregation of this kind of structures in aqueous solution was studied.138 The self-association of compound 35 induced the formation of tubular structures with hydrophobic inner pores that served as supramolecular hosts for binding hydrophobic guests in water (Fig. 14c). Rigid oligo(p-phenylene ethynylene) segments 36a–d of different lengths were chosen as guests. The interaction of the four guests with 35 was probed by spectroscopic, NMR and computational studies. The mixtures of host and guests and host alone in D2O were analysed with DOSY. The average diffusion coefficients (D) obtained followed a trend that was inversely correlated to the length of the guests. These results indicated that the length of the guest used defined the number of macrocycles in each complex forming discrete tubular assemblies, i.e., pseudorotaxanes. Moreover, average diffusion coefficients of all four complexes resulted smaller than that of 35, which confirmed the control of the stack growth towards the formation of discrete templated assemblies.
The vast amount of examples highlighted in this first section proves the relevance of this strategy, where new structural motifs as well as emerging and novel functions derived from those are still under development.
3. Non-covalent cycles
The formation of NTs through the stacking of supramolecular macrocycles represents a significant advancement in the field of nanochemistry (Fig. 15).35,36,140 Macrocycles constructed through non-covalent interactions offer several advantages over the ones described in the previous section, built by covalent bonds. Non-covalent interactions, such as H-bonds, solvophobic forces or metal–ligand (M–L) interactions, among others, are reversible and highly dynamic, which brings about multiple novel properties to supramolecular materials, like stimuli responsiveness. Due to this reversibility, these interactions can be regulated smoothly through changes in concentration, pH or temperature, permitting quick responses to environmental changes. Moreover, supramolecular forces like H-bonds or M–L interactions, can provide high directionality and specificity in molecular recognition. For example, complementary supramolecular patterns can lead to selective binding without the need for permanent changes in covalent structure. In summary, while covalent bonds are crucial for creating stable and strong connections within molecules, non-covalent interactions provide flexibility, specificity, and dynamic properties to the final structure.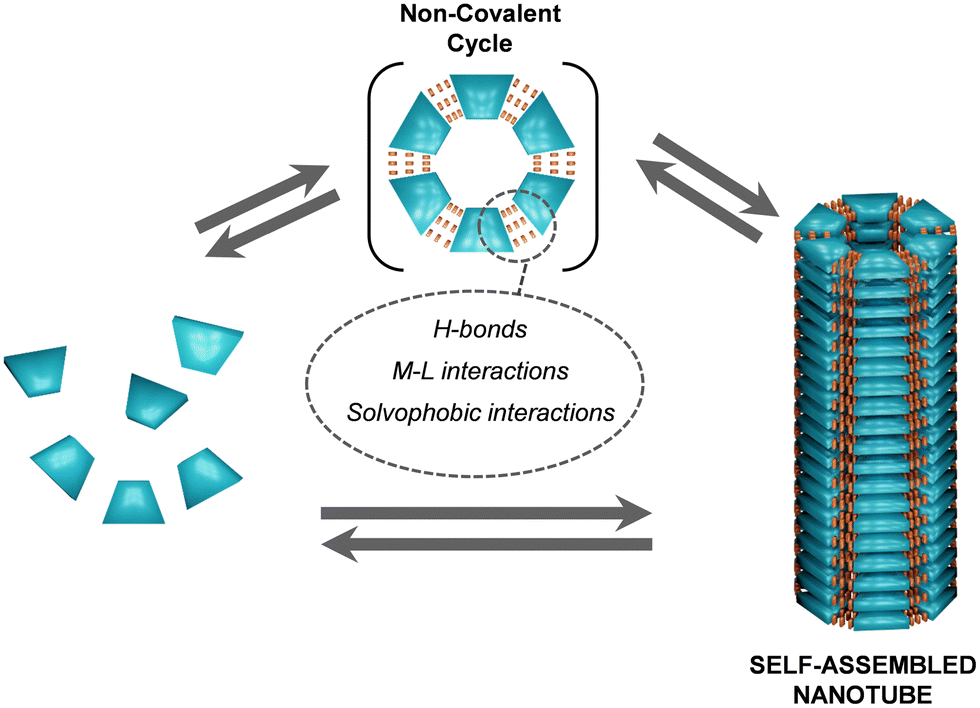 | ||
| Fig. 15 Schematic representation of the reversible association of non-covalent cycles from individual units and subsequent reversible stacking of these cyclic entities into self-assembled NTs. | ||
The formation of supramolecular macrocycles, in equilibrium with non-cyclic oligomeric species, is favoured as long as the associated chelate cooperativity is high enough.141–147 In this ring-chain equilibria, the cyclization process, or in general the formation of intramolecular bonds, competes with analogous intermolecular interactions. This competition is quantified by the effective molarity (EM), defined as the ratio between intramolecular and intermolecular equilibrium constants for thermodynamically controlled processes (EM = Kintra/Kinter). Therefore, in addition to a strong association constant, a careful structural design of the self-assembling units with the aim to maximize EM results crucial for the high-fidelity formation of cyclic structures.
Numerous examples in the literature demonstrate their success in forming stable supramolecular macrocycles under specific conditions of monomer concentration, temperature, and solvent polarity.143,144 In this section we focus on those that further allow the formation of nanotubular structures. The most common non-covalent interactions considered for the construction of non-covalent macrocycles for NT formation in the literature comprise highly directional H-bonds and M–L interactions, but also low directional interactions like solvophobic forces. Similarly to the covalent cycles, in the examples gathered in this section, H-bonds and/or π–π stacking are the primary interactions that promote the stacking of the cycles into the nanotubular structure.
3.1. Cycles formed by H-bonds
Many scientists have directed their efforts to the non-covalent synthesis of cyclic systems from molecules that interact through complementary recognition units. To achieve this goal, molecules must be meticulously designed with the necessary chemical information to form specific designed structures. This approach seeks to replicate the “smart” collective behavior found in nature, where information is embedded in the covalent bonds of biomolecules, guiding the structure and function of complex biological systems at the nano- and mesoscale.148–151 In this context, H-bonding offers a high degree of control over supramolecular assembly due to its selectivity and directionality. Despite typically exhibiting lower fidelity and cooperativity compared to M–L interactions, as well as faster dynamics, many examples in the bibliography prove the utility of H-bonds to create stable supramolecular rosettes and cycles142,145 as good candidates for the creation of self-assembled NTs.Examples of rosette macrocycles that provide internal pores that are too small to host any molecule are those formed by homo- or heteroassociation of cyanuric/barbituric acid and melamine/diaminotriazine derivatives. The group of Yagai has widely explored the hierarchical self-assembly of barbiturate derivatives decorated with π-conjugated units into six-membered H-bonded macrocycles that further stack and evolve into complex supramolecular structures with different topologies, including columnar structures.152,153 Despite their fascinating control over the topology of these superstructures, the authors have not proved the presence of a lumen within the columnar aggregates, and consequently, they typically define the nanostructures formed as fibres, and not as NTs.
A similar strategy was followed by Meijer and collaborators. The group reported the assembly of chiral π-conjugated oligo-(p-phenylenevinylene) into rosette-like hexameric supermacrocycles via double H-bonding.154 Scanning tunneling microscopy (STM) images revealed chiral hexameric rosette structures with a cavity of 0.7 nm of diameter. The rosettes further stack and form unidimensional objects with lengths up to 10 μm as shown by atomic force microscopy.
The guanine or guanosine (G)-quartet motif constitutes one of the most outstanding examples of the construction of discrete cyclic assemblies through H-bonds.155–158 These species are macrocycles that are assembled by double H-bonding (DD–AA) (A = H-bond acceptor, D = H-bond donor) association of guanosine derivatives. This cyclic arrangement typically exists in balance with linear formations. However, when certain cations, particularly Na+ or K+ salts, are present, the cyclic structures become more stable through cation complexation. This stability arises because the quartets feature four coordinating carbonyl groups oriented towards their central cavity. Furthermore, these quartets frequently organize into layered structures, with the complexed cation positioned between two layers of quartets and coordinated to eight carbonyl groups. These intricate multicyclic stacked arrangements are referred to as G-quadruplexes. These entities are formed through a hierarchical self-assembly process, influenced by a combination of π–π stacking, solvophobic interactions, and cation–dipole interactions.159–163 However, other than small cations, the inner cavity of G-quartet oligomers or polymers is too small to host common molecules.
Contrarily, some H-bonded rosette macrocycles can stack into unidimensional polymers in which the presence of a pore has been proven. That is the case of heterobicyclic fragments in which the Watson–Crick H-bonded interfaces of complementary nucleobases are formally fused. As opposed to the previous systems, the two H-bonding faces are here separated apart so that a central void space is created upon rosette cyclization. Fenniri's research group has made significant advancements in the use of heterocycles formed from fused guanine (G) and cytosine (C) nucleobases (G^C) for creating supramolecular macrocycles.146,164–170 The self-complementary nature of the ADD H-bonding pattern of guanine and the DAA pattern of cytosine drives the self-assembly into cyclic structures via H-bonds. While previous work by Mascal, Lehn and colleagues confirmed the formation of cyclic hexamers,171,172 it was Fenniri's group the one which first illustrated the stacking of these supramolecular macrocycles, so-called rosettes, into NTs in aqueous environments.164
In their seminal work in 2001, Fenniri's group demonstrated the ability of G^C motifs to self-assemble into stable six-membered rosettes maintained by 18 H-bonds (Fig. 16a).164 These macrocycles formed by compound 37a further stack into rosette NTs (RNTs), with a central channel that is approximately 1.1 nm in diameter, driven by hydrophobic interactions and π–π stacking. NOESY experiments conducted in a solvent mixture of 90% H2O and D2O revealed NOE cross-peaks between protons from G and C motifs, which implies the occurrence of intermolecular interactions. Additionally, in electrospray ionization mass spectrometry (ESI-MS) analyses of dilute aqueous solutions, diverse peaks were detected that corresponded to non-covalent intermediate species ranging from 1-mer to 6-mer of the final hexameric structure. Various spectroscopic and imaging techniques, such as TEM, confirmed the formation of nanotubular structures having a ∼4 nm outer diameter (Fig. 16e). Finally, CD measurements revealed dichroic activity of these systems in aqueous solution, ascribed to the formation of helical NTs.
 | ||
| Fig. 16 (a) Molecular structure of GC derivatives (37a–d) and representation of rosette structure (X = CF3CO2−). (b) TEM images and (c) DLS measurements of 37c. (d) Hexameric rosettes and NT models corresponding to bicyclic 37a and tricyclic variant 37b. (e) TEM of the rosette NTs obtained from the hierarchical self-assembly of 37a. (f) AFM image of an aqueous solution of 37b aged for 7 days. Adapted with permission from ref. 164, 165 and 169. Copyright 2001, 2002 and 2010 American Chemical Society. | ||
With this G^C molecule as reference model, the research group has introduced some alterations in its chemical structure with the objective of enhancing or modifying its intrinsic properties. Thus, in one of their works, they developed new water-soluble rosette NTs with giant molar ellipticity just incorporating a fused pyridine unit between both G and C nucleobases (compound 37b in Fig. 16a).169 The extended π system of the tricyclic self-assembling module 37a facilitated the formation of hexameric rosettes in water that stacked to create J-type NTs. The inner diameter increases by 0.4 nm and the outer diameter by 0.9 nm when transitioning from the bicyclic structure 37a to the tricyclic structure 37b (Fig. 16d). The CD signal of the assemblies of compound 37b increased ca. 40 times over 7 days reaching a superior molar ellipticity, with respect to what was reported until that moment. The formation of NTs was confirmed by TEM and AFM microscopies (Fig. 16f). This novel design permitted an enhanced intermodular electronic communication and resulted in a nanotubular architecture with unprecedented optical properties.
Other alteration that the group considered was the attachment of an achiral 4-aminobenzo-18-crown-6-ether to the G^C general structure (compound 37c in Fig. 16a).165 By this modification, NTs formed from 37c were able to encapsulate chiral molecules and exhibited chiral induction effects, leading to a shift from racemic to homochiral helical structures. In particular, whilst compound 37c was found to spontaneously form a racemic mixture of P and M helical NTs, the entrapment of chiral amino acid L-alanine into the crown ether resulted in a rearrangement to the formation of helical NTs with a dominant chirality, which was indicated by the appearance of CD activity.
In a subsequent work, the incorporation of an additional G^C motif to the general model led to twin G^C monomers able to form twin-rosette structures which relied on programmed H-bond information, stacking interactions and solvophobic forces (compound 37d in Fig. 16a). By this approach, rosette “cages” formed from compound 37d can be obtained, which can further self-organize into NTs.166 In another related work, twin G^C-bases were designed and synthesized to self-assemble into twin-rosette “cage” structures utilizing only programmed H-bonding information.173 In a similar way, the formation of NTs from the stacking of these rosette “cages” could be achieved.
 | ||
| Fig. 17 (a) Scheme of the two-step self-assembly of dinucleobase monomers into NTs through supramolecular macrocycles and chemical structure of G–C derivative 38a. Changes observed in the CD spectra as a function of the volume fraction of heptane (Vh) in mixtures with THF for (b) 38a, and (c) 38b. Adapted with permission from ref. 180 and 181. Copyright 2019 American Chemical Society, respectively. | ||
Due to the presence of rotatable σ-bonds in the central core, which allow the existence of multiple conformations where the Watson–Crick edges could point to the same or opposite sides of the monomer, these cyclic species can establish an equilibrium with linear oligomers.182 When monomers substituted with G and C bases at the edges were studied, the corresponding tetrameric macrocycles displayed high thermodynamic and kinetic stability due to the strong chelate cooperativity of the system (EM = 102–103 M).177 The group also demonstrated that the macrocyclization processes strongly relies on the symmetry of the H-bonding pattern by comparing the stability of cyclic assemblies made from monomers with G and C bases and from monomers with 2-aminoadenine (A):uracil (U) bases.182,183 The unsymmetric ADD–DAA pattern from the G:C interaction provided not only a higher association constant, but also an increased EM compared to the symmetric DAD–ADA pattern of the A:U interaction, which is due to entropic considerations. Furthermore, the impact of the length of the linear oligo(p-ethynylene-phenylene) spacer connecting the bases on cycle formation was also assessed.184 As the number of σ-bonds in this spacer increased, the number of rotational and torsional degrees of freedom that are lost during cyclotetramerization also increased, which led to a reduced chelate cooperativity. Consequently, shorter monomers exhibited stronger chelate cooperativities and thus a greater propensity to associate in cyclic structures versus linear oligomers.182,184
Taking advantage of the thermodynamic and kinetic stability of the G–C cyclic tetramers, the group then investigated the stacking of these cycles into tubular structures in organic solvents (Fig. 17).180,181,185 For this purpose, the heterocycles were designed with benzylic wedges that have long alkyl chains to improve solubility in non-polar solvents, along with a peripheral amide group to guide macrocycle stacking (compound 38a in Fig. 17b).180 This molecular design enabled independent control of two consecutive cooperative hierarchical processes that occurred as the volume fraction of apolar solvent increased in THF/heptane mixtures: an initial macrocyclization through Watson–Crick interactions between nucleobases, and subsequent polymerization directed by π–π stacking interactions among the extensive π-conjugated surfaces of the tetramers, as well as H-bonding interactions among the surrounding amide groups placed at the pyrimidine base (Fig. 17a). Along the first process, base pairing within the cyclotetramer was confirmed by the characteristic downfield shift of the G-amide and C-amine proton signals, which were found in slow exchange with the corresponding monomer protons in THF, a feature of highly cooperative H-bonded cyclic assemblies.142 On the other hand, UV-Vis, fluorescence, and CD techniques at low heptane volume fractions (Vh in Fig. 17b and c), showed a red-shifted band in absorbance, decreased emission, and a Cotton effect in CD (green spectrum in Fig. 17b), respectively. These spectroscopic features are attributed to the freezing of conformational motions in the chiral monomer upon cyclization. At higher heptane content (Vh > 0.7), the group observed a higher order aggregation process: a broadening and loss of all proton signals from the cyclic tetramer, along with additional red shifts in absorption, further reduction in emission intensity, a slight blue shift of the emission peak, and the emergence of a new Cotton effect (blue spectrum in Fig. 17b). These characteristics were linked to a supramolecular polymerization process that leads to the formation of chiral NTs. The dimensions of the self-assembled nanostructures generated were measured by TEM studies, unveiling heavily bundled longitudinal objects with a measured diameter of approximately 4 nm, consistent with the calculated aromatic section of the cyclic tetramers.
After that, the role of peripheral amide groups in the tubular self-assembly of dinucleobase monomers was analyzed.181 Interestingly, monomers with a peripheral amide group (38a) and without peripheral amide group (38b) differ enormously in their supramolecular behaviour. Specifically, 38b, devoid of these pendant H-bonding groups, only showed the corresponding CD signal attributed to the cyclotetramer unit, and remained associated as cyclic tetramers even in pure heptane (Fig. 17c). No CD signal corresponding to NT formation could be detected. These findings indicated that the presence of peripheral amides is essential for the formation of polymeric NTs.
In addition to studies with G and C nucleobases, variants employing A and U as complementary bases were also explored (compounds 38a and 39 in Fig. 18a and b),185 in order to elucidate the impact of the nature of the DNA bases, on the whole self-assembly process. Regarding the cyclotetramerization process, important differences in the NMR spectra were found as a consequence of the extremely different chelate cooperativities of the G:C- and A:U-bound cyclic tetramers, as introduced above.182 While the G:C-bound tetramers formed by 38a exhibited the characteristic downfield-shifted and slowly exchanging G-amide and C-amine protons (Fig. 18a), NMR experiments obtained in similar conditions from A–U compound 39, suggested the presence of a fast-exchanging mixture of non-cyclic oligomers that grew in length as the volume fraction of cyclohexane-d12 in THF-d8 (Vcy in Fig. 18a and b) was increased, as evidenced in the gradual downfield shift of the U-imine proton signal (Fig. 18a and b). Upon further increasing Vcy, both G–C and A–U NMR signals broaden and then disappear due to polymeric aggregation. However, whereas TEM results indicated that both compounds were able to form tubular aggregates with similar diameters, matching the cyclic tetramer section, the absorption, emission, and chirooptical features of the final NTs were considerably different. In particular, A–U supramolecular polymers unveiled CD and circularly polarized luminiscence (CPL) spectra that are shifted and of opposite sense to the ones displayed by G–C polymers. This suggests that the 38a or 39 molecules in each NT were internally arranged in a different manner. Besides, mixtures of 38a and 39 were found to aggregate narcissistically: each compound tend to self-associate in their own assemblies, and no mixing could be detected. Supported by molecular dynamics studies, which could simulate the experimental CD acquired, it was concluded that A–U compound 39 tends to form assemblies from open oligomers that fold into helical NTs with coiled internal arrangements, instead of involving the stacking of cyclotetramers, as it happens in the related G–C compound 38a. These results highlight the importance of the H-bonding pattern, and the derived propensity to form cycles as a function of chelate cooperativity, in the self-assembly pathway and the organization of the molecules within the tubular supramolecular polymers.
 | ||
| Fig. 18 Representation of the NTs formed by (a) the stacking of the macrocycles constituted by G–C derivative 38a or (b) the NTs formed through folding of linear oligomers formed by the A–U derivative 39, and corresponding NMR solvent-dependent experiment as a function of the volume fraction of cyclohexane-d12 (Vcy) in mixtures with THF-d8. (c) Chemical structure of G–C derivatives with different lengths (38a, 38c and 38d). (d) NT diameter distribution measured by TEM, and corresponding TEM images of (e) 38a, (f) 38c and (g) 38d. Adapted with permission from ref. 185 and 186. Copyright 2023 American Chemical Society and 2024 Wiley-VCH, respectively. | ||
A precise control of the diameter of self-assembled NTs was recently achieved by the same group through the stepwise extension of the oligomeric central blocks in these dinucleobase monomers.186 Thus, the self-assembly of G–C monomers with π-conjugated oligo(p-phenylene-ethynylene) (OPE) central blocks of different lengths, ranging from n = 1 to n = 5, was explored (Fig. 18c). The formation of NTs was confirmed by TEM, which showed longitudinal aggregates consisting of grouped individual NTs, with diameters matching well with those calculated for the rigid core of each cyclic tetrameric section. Monomers with a single, three or five central p-phenylene-ethynylene blocks (38a, 38c and 38d) formed NTs with average diameters of 3.9 nm, 4.6 nm and 5.7 nm, respectively. AFM and SAXS studies were in agreement with this dimensional control. These findings highlight the possibility to finely tune the pore diameter of self-assembled NTs by modifying the length of the corresponding monomer.
González-Rodríguez's research group has also extensively investigated the self-assembly of related dinucleobase monomers that are adapted to provide water-soluble NTs.187–189 Inspired by the previous strategy for organic solvents, different amphiphilic dinucleobase derivatives with small structural variations to guide their self-assembly in water were synthesized (Fig. 19).187 These variations included the attachment of hydrophilic chains with short hydrophobic segments connected to N-1/N-9 in the G/C bases, to impart solubility in water, as well as the incorporation of amphiphilic central blocks, with a chiral lipophilic tail in one side, and neutral or ionic hydrophilic groups at the other. As shown in Fig. 19a, in the final assembly, all the hydrophobic components would be oriented toward the centre of the structure, while the hydrophilic chains would be exposed to the surrounding water, resulting in a micellar NT. The spectral properties observed for 40a in water were very similar to those detected for the creation of NTs from macrocycles in organic solvents and were consistent with a chirally organized supramolecular structure. 1H NMR experiments at different H2O:THF-d8 compositions unravelled 1H chemical shifts ascribable to G:C base pairing, which were in slow exchange with the monomer signals. Additionally, the morphology and size of the aggregates were examined using AFM (Fig. 19c, top) revealing unidimensional objects with heights of ∼2.0–3.5 nm. Finally, TEM experiments confirmed the presence of tubular structures with widths of ∼5.8 ± 0.7 nm. The smaller values found by AFM may result from the compression of side chains due to the force exerted by the AFM probe,190 as well as the attraction of the assemblies to the surface.
 | ||
| Fig. 19 (a) Schematic illustration of the self-assembly of amphiphilic G–C derivatives into NTs in aqueous solution. (b) Molecular structures of compounds 40a–c. (c) AFM images of 40a in Milli-Q water (top) or acidic aqueous solution (bottom). (d) Chemical structure of compounds 40a and 41 and corresponding CPK Models of NT sections. (e) Chemical structure of NR, schematic representation of the encapsulation of NR within NTs and PAINT images of NT bundles formed by 41 (in red) in the presence of encapsulated NR molecules (bright dots) in Mili-Q water. Hydrophobic and hydrophilic parts are represented in red and blue colors, respectively. Adapted with permission from ref. 188 and 189. Copyright 2021 Royal Society of Chemistry and 2024, respectively. | ||
In a subsequent article, the group looked into the impact that different conditions exert in the self-assembly of dinucleobase amphiphilic monomers in aqueous solutions.188 These monomers feature hydrophilic groups of various types attached to the p-conjugated central block: anionic (carboxylate group in the already described compound 40a), neutral (glycol chain in compound 40b), or cationic (ammonium group in compound 40c). The study of aggregation processes was conducted by varying temperature, concentration, and environmental factors such as solvent composition and pH. Successful formation of NTs was discerned for all compounds under different experimental conditions. However, it was found that minor adjustments in specific parameters or alterations in the compound structure led to particularly noteworthy results. For example, at high temperatures, the CD signal of compound 40a reversed its sign, which suggests a structural reorganization. This was further supported by TEM and AFM images, which revealed the presence of NT bundles and poorly-defined structures, likely resulting from the dehydration of polar groups. Likewise, the alteration in the solvent composition or the pH also led to significant modifications in the self-assembly process. For instance, compound 40a exhibited very distinct spectroscopic characteristics when comparing acidic (pH = 3) and basic (pH = 9–5) levels. AFM and TEM images from these anomalous acidic solutions permitted to discern the presence of spherical objects (Fig. 19c bottom), in contrast to the long NTs observed at higher pH. These results highlight how subtle changes can have a significant impact on assembly morphology, underlining the importance of fine-tuning experimental setups to optimize self-assembly into the desired nanostructure.
Recently, the same group also explored the behaviour of dinucleobase monomers endowed with a slightly longer p-diethynylene-biphenylene core (41), instead of the p-diethynylene-phenylene core in compound 40a (Fig. 19d).189 Compound 41 was capable to form NTs in aqueous media with enhanced supramolecular stability compared to its related compound 40a, likely due to the larger aromatic surface of the biphenyl core. Furthermore, the incorporation of a longer spacer connecting the nucleobases resulted in NTs with an extended diameter to over 4 nm (Fig. 19d). To assess the capacity of the 40a and 41 NTs for extracting and retaining nonpolar substances, the authors examined their ability to encapsulate the well-known hydrophobic dye Nile red (NR). When NR, which is not soluble in water, was mixed with 40a or 41 NTs, an increase in absorption and emission of this dye was observed, indicating the solubilization within the NT pore, together with the characteristic blue shift in emission when surrounded by apolar environments. The encapsulation efficiency, that is, the maximum amount of dye extracted by the NTs, was calculated through titration experiments with increasing amounts of NR, resulting in values of 0.2.–0.4 equivalents of added dye per 40a/41 monomer. Subsequently, the aggregates could be observed using fluorescence microscopy by detecting the emitted light from the NR molecules encapsulated within the NT. By using point accumulation for imaging in nanoscale topography (PAINT), alignment of NR emission points in the form of lines was observed, corroborating the presence of dye molecules within the longitudinal hydrophobic lumen of 41 NTs (Fig. 19e).
Very recently, González-Rodríguez's research group has investigated the use of carboxylate:amidinium “salt bridges”, in which H-bonds are reinforced by the oppositely charged interface, as strong directional motifs that are compatible with polar environments for the construction of cyclic structures, with the aim to stack them in water to build NTs of controlled diameter.191
 | ||
| Fig. 20 (a) Molecular structure of compound 42 and schematic representation of its assembly in CDCl3. (b) 1H NMR spectra of monomer 42 in CDCl3, toluene-d8 and CDCl3 in the presence of C70. (c) Top view of H-bonding interface. (d) AFM image of 42 deposited on a silicon surface. (e) Chemical structure of monomer 43 and schematic representation of self-sorting, selective encapsulation of C60 guest and capsule-tube isomerization. Adapted with permission from ref. 195. Copyright 2017 Springer Nature. | ||
Successful NT formation was achieved by introducing an unsubstituted urea group into the ureidopyrimidinone motif of an enantiopure monomer (compound 42 in Fig. 20a).195 This urea group provided an additional H-bonding site placed orthogonally to the H-bonds responsible for the cyclotetramerization, promoting the aggregation of cycles into polymeric tubes. Moreover, a pyrrole spacer was fused between the bicyclic system and the ureidopyrimidinone motif, in order to avoid steric hindrance between the H-bonds responsible of the tetramerization and the solubilizing groups located at the bicyclic core. The formation of the cyclic tetramer in CDCl3 was confirmed by NMR techniques and gel permeation chromatography (GPC) experiments. 1H NMR experiments in this solvent showed well defined peaks that were assigned to the cyclic structure (Fig. 20b). By decreasing the polarity of the solvent, as in toluene, or by using a shape-complementary guest in CHCl3, such as C70, the formation of polymeric aggregates was triggered (Fig. 20b). The polymerization process was analysed by 1H NMR, AFM, DLS and GPC. NMR experiments disclosed a broadening of the proton signals and complete disappearance of the NH resonances, indicative of the existence of polymeric materials. AFM images corroborated the aggregation of the monomer into fibre-like superstructures (Fig. 20d). Moreover, FT-IR experiments demonstrated the involvement of both the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and NH2 groups of the terminal urea moiety in H-bonding (Fig. 20c). On the other hand, UV-Vis analysis verified that C70 molecules were integrated into the pore of the tube, as evidenced by the hypochromic effect and the 6 nm bathochromic shift observed in the absorption band of C70 at 470 nm. Due to the organized arrangement of electron-conducting C70 molecules inside the tube, these structures could be explored for use in organic photovoltaic devices. Conversely, when C60 was employed as guest in chloroform, a conformational change was induced, from an open-ended cyclic structure to a C60@42 closed shell capsule (Fig. 20e top). In addition to this, monomer 43, which is not able to form NTs, was explored as a host for C60 molecules (Fig. 20e). It was proved that, in chloroform, compound 43 cyclotetramerized and no interaction with C60 took place. When monomers 42 and 43 were mixed in CDCl3, a self-sorting phenomenon took place. Then, the addition of C60 molecules resulted in the selective formation of the C60@42 inclusion complex, leaving tetramer 43 intact as a cyclotetramer. When CDCl3 was replaced by toluene, disaggregation of the complex C60@42 occurred and NTs of 42 were formed, whereas cyclic tetramers of 43 and C60 guest formed a new complex.
O and NH2 groups of the terminal urea moiety in H-bonding (Fig. 20c). On the other hand, UV-Vis analysis verified that C70 molecules were integrated into the pore of the tube, as evidenced by the hypochromic effect and the 6 nm bathochromic shift observed in the absorption band of C70 at 470 nm. Due to the organized arrangement of electron-conducting C70 molecules inside the tube, these structures could be explored for use in organic photovoltaic devices. Conversely, when C60 was employed as guest in chloroform, a conformational change was induced, from an open-ended cyclic structure to a C60@42 closed shell capsule (Fig. 20e top). In addition to this, monomer 43, which is not able to form NTs, was explored as a host for C60 molecules (Fig. 20e). It was proved that, in chloroform, compound 43 cyclotetramerized and no interaction with C60 took place. When monomers 42 and 43 were mixed in CDCl3, a self-sorting phenomenon took place. Then, the addition of C60 molecules resulted in the selective formation of the C60@42 inclusion complex, leaving tetramer 43 intact as a cyclotetramer. When CDCl3 was replaced by toluene, disaggregation of the complex C60@42 occurred and NTs of 42 were formed, whereas cyclic tetramers of 43 and C60 guest formed a new complex.
In a more recent article, polymerization of the tautoleptic tetramers composed of isocytosine and bicyclo[3.3.1]nonane was achieved by changing the length, position or connectivity of the solubilizing groups to the bicyclic core.196 Both chloroform and toluene afforded the polymerization of the monomers into long NTs which led to gels able to encapsulate fullerene molecules. These new gels filled with fullerenes could be used to construct soft, gel-based electronic devices.
3.2. Cycles formed by metal–ligand interactions
Over the years, M–L coordination has been widely utilized to build supramolecular structures.197,198 These interactions are characterized by its binding strength, directionality, reversibility, and versatility, since they allow access to manifold geometries, including nanotubular structures. In this section, we have only considered examples of NTs formed from the supramolecular stacking of metal–organic cycles. Examples of the use of additional substances inserted between cycles to favour the vertical growth, the construction of discrete tubules or metal–organic cyclic structures that are intrinsically tubular and not planar have been reported in detail in specific reviews199,200 and are not included here.Lee's research group has exploited this interaction to build nanotubular structures. In a representative work, they described a stimuli-responsive supramolecular assembly able to be rearranged into metal–organic NTs, taking advantage of the specific coordination of pyridine to Ag(I) ions (Fig. 21).201 It was demonstrated that molecule 44 (Fig. 21a) can self-organize in aqueous solution to form flat sheets featuring a zigzag arrangement of the aromatic segments. Although molecule 44 has chiral centers, no dichroic activity was detected. However, when silver triflate was added, the pyridine protons experienced a downshift in the NMR spectrum as a result of their interaction with metal ions, leading to the emergence of a CD signal that indicated the formation of chiral superstructures. This CD signal could be eliminated by adding tetra-n-butylammonium fluoride (Bu4NF) to disrupt the complex, but it could be restored again by the addition of more silver triflate. The formation of helical NTs in the presence of Ag+ ions was confirmed by TEM (Fig. 21d) and AFM (Fig. 21e). Further DLS experiments conducted with diluted solutions of 44 with Ag+, with concentrations ranging from 0.03 to 0.01 wt%, disclosed a significant reduction in size from 200 nm to 8 nm. Additionally, TEM and AFM analyses at 0.01 wt% revealed toroidal structures, confirming that helical NTs were formed through the stacking of macrocycles. This indicates that the final assemblies resulted from the helical stacking of smaller objects, like dimeric macrocycles, rather than from the helical folding of polymer chains, producing tubular structures exhibiting left-handed supramolecular chirality (Fig. 21b and c).
 | ||
| Fig. 21 (a) Molecular structures of the bent-shaped amphiphile 44. (b) Schematic representation of the switch between Ag(I) ion-mediated dimeric macrocyclic structure and the tubular structure. (c) Schematic representation of reversible transformation through flat sheets, helical tubules and discrete toroids in response to external stimuli. (d) TEM and (e) AFM image of an aqueous solution of 44 with Ag(I) ions (0.03 wt%). Adapted with permission from ref. 201. Copyright 2013-American Chemical Society. | ||
The multidisciplinary Aida's research team also reported the formation of metal–organic NTs by M–L coordination between a tetratopic pyridyl ligand with a ferrocene core and AgBF4 (Fig. 22a).202 Large decagonal nanorings with a diameter of 7.5 nm were made to stack together through π–π and metallophilic (Ag:Ag) interactions from compound 45 and AgBF4 in MeCN/water solutions. TEM and AFM confirmed the presence of tubular structures with a consistent diameter of 7.3 nm and a height of 8.5 nm, respectively. In addition, the observed SAXS pattern was nicely fitted by the form factor of a hollow cylinder model with a 7.5 nm diameter, and a more detailed analysis of the 2D X-ray diffraction (2D XRD) image of magnetically aligned NTs suggests that there may be a slight helical twist in the arrangement of their stacking.
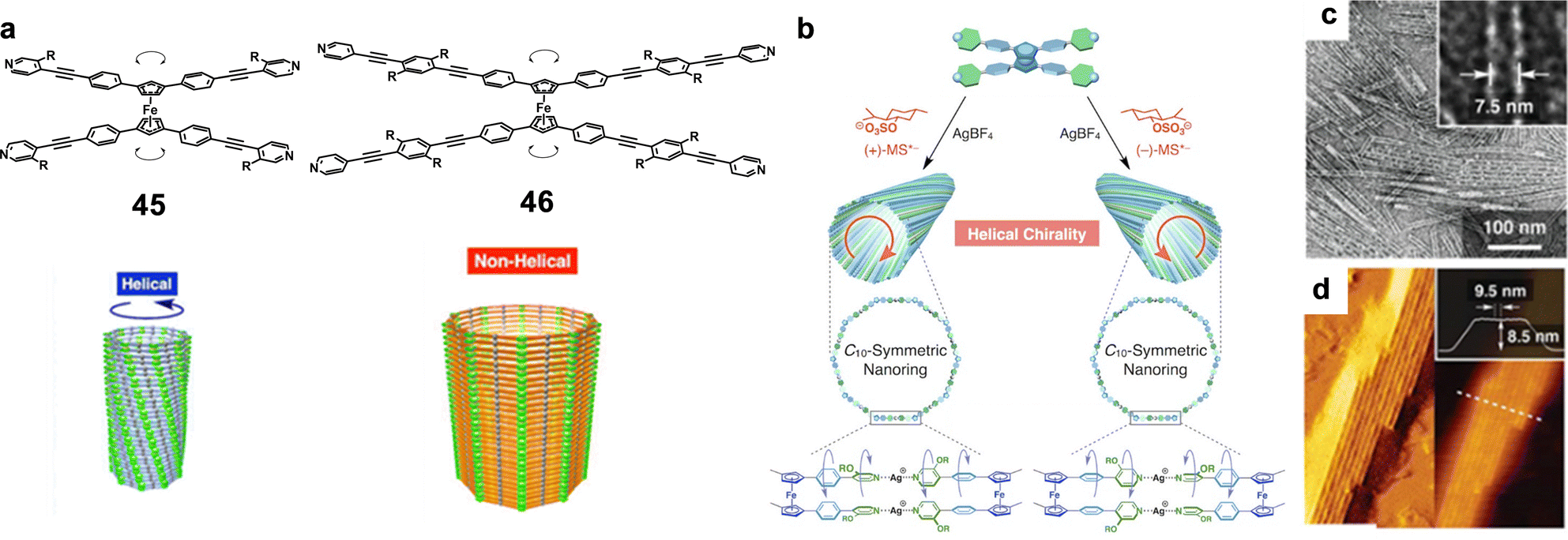 | ||
| Fig. 22 (a) Chemical structure of ferrocene-based tetratopic pyridyl ligands 45 and 46, and representations of metal–organic NTs formed by 45 (left) and 46 (right). (b) Schematic representations of the co-assembly of ferrocene-cored tetratopic pyridyl ligand (45) and AgBF4 in the presence of (+)- or (−)-menthylsulfate (MS*−), and possible helical motifs in the resulting helical metal–organic NT. (c) TEM image of 45 in the presence of (+)-Bu4N + MS*−, stained with uranyl acetate, and (d) corresponding AFM onto HOPG. Adapted with permission from ref. 203 and 204. Copyright 2015 and 2016-American Chemical Society, respectively. | ||
Making use of these highly symmetric structures, Aida et al. achieved to build optically active chiral NTs when they mixed 45 with AgBF4 in the presence of (+)- or (−)-menthylsulfate (Fig. 22b).203 When the co-assembly process was conducted using a mixture of dichloromethane and acetonitrile (7:3 v/v) with (+)- or (−)-menthylsulfate (MS*−), a red shift was observed in the absorption band. Moreover, distinct mirror-image CD signals were recorded in the presence of both (+)- and (−)-menthylsulfate, further validating the formation of helical NTs with a preferred chiral orientation (Fig. 22b), induced by the chiral ion. The formation of tubes in the presence of menthylsulfate was confirmed by TEM and AFM microscopies (Fig. 22c and d). The analogous monomer 46 with large π-aromatic ligands afforded hollow non-helical NTs with large and uniform diameter of 14 nm and height of 10.9 nm in similar conditions. Moreover, due to the nearly comparable binding affinities of 45 and 46 toward Ag(I), the group tried to create mixed-ligand NTs.204 They found that no heteromeric NTs were formed, and only homomeric ones were observed by TEM and SAXS analysis. This self-sorting phenomenon could be led by the preference of 45 to form helical NTs, contrary to 46, which tended to stack up with no discernible helical twist.
Other significant example of NTs formed by M–L interactions was described by the MacLachlan's group, involving this time the formation of Pt-pyridyl-type metallocycles. In 2010, they investigated the formation of head-to-tail tetrameric platinum-bound (Pt4) macrocycles 47, which self-assembled into columnar structures (Fig. 23a).205 Data from light scattering, XRD, and TEM supported the formation of 1D columnar aggregates from the Pt4 rings 47. The incorporation of bulky trityl-derived groups around the edges of the Pt4 macrocycles restricted the otherwise limitless one-dimensional columnar stacking, resulting in the formation of distinct short NTs constituted by the stacking of four or six metallocycles. In contrast, when the conjugation of the ligand was extended, as in 48, triangular Pt3 macrocycles were formed instead (Fig. 23b).206 This was achieved by the introduction of a ligand based on the pyridylsalicylaldehyde system, bearing more flexible acetylene groups, which contributed to the formation of macrocycles with unexpected geometry. The structure and aggregation of Pt3 macrocycles were investigated using computational techniques, UV-vis spectroscopy, variable-temperature NMR, as well as DOSY and NOESY NMR analyses. In the solid state, studies using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry and TEM measurements unravelled that the cyclic structures organized into tubular nanostructures (Fig. 23b).
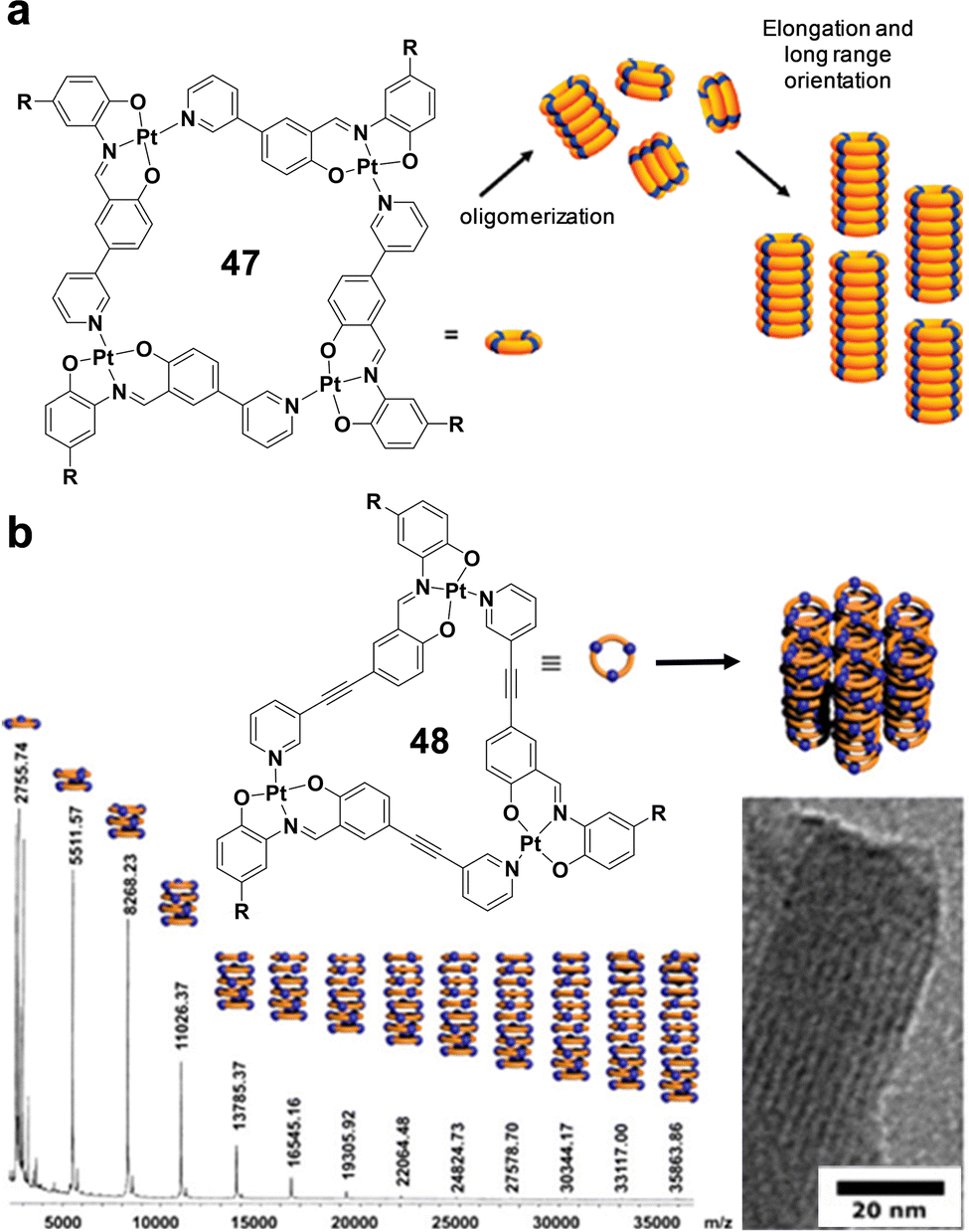 | ||
| Fig. 23 (a) Dynamic assembly of ring 47 into columns. (b) Chemical structure of macrocycle 48 and its stacking into NTs (top), MALDI-TOF MS of macrocycle 48 (bottom left) and TEM image of the NT formed by 48 (bottom right). Adapted with permission from ref. 205 and 206. Copyright 2010 and 2017-American Chemical Society. | ||
Recently, Wang and co-workers have reported the assembly of discrete metallo-macrocycles with double-layer and triple-layer concentric structures.207 In their work, a terpyridine (tpy) motif is used as the central backbone of the ligand and the complexation of metal ions results in the creation of heptagonal metallo-macrocycles. The planarity and rigidity of this scaffold favour its self-assembly into tubular structures. A variety of supramolecular forces may participate in the packing of the cycles towards NTs, such as π–π stacking, hydrophobic/hydrophilic interactions, electrostatic interactions, etc. However, investigations on the self-assembly as a function of the charge of the counter anions incorporated allowed the authors to hypothesize that electrostatic forces may play a crucial role in the stacking of these giant macrocycles.
3.3. Cycles formed by solvophobic interactions
Beyond the directional H-bonds and M–L interactions, non-directional solvophobic forces can be used as well for the formation of cyclic structures. Specifically, hydrophobic interactions can give rise to manifold organizations, including NTs, e.g., when hydrophobic molecules are dissolved in water. Actually, most self-assembled NTs are made from amphiphilic molecules in water.33,208 As aforementioned, this type of interaction shows low directional guidance along the self-assembly process and, consequently, the structural design of the monomers that constitute the supramolecular assemblies can be challenging.One of the most relevant research groups that incorporate solvophobic forces as key interactions in the formation of supramolecular nanostructures formed by the stacking of ring-like entities is Lee's group. In addition to other morphologies, tubular structures assembled from the π–π stacking of hexameric macrocycles, which are at the same time formed by weak nonspecific aromatic interactions between six aromatic rigid v-shaped segments (120° of internal angle), have been studied in the solid state.209 The monomers generally comprise a central arene meta-substituted with p-oligophenylene rods, with an oligoether chain pointing to the inner core of the ensemble. As a function of the length or flexibility of the oligoether side chains, different supramolecular organizations could be obtained. When this kind of structures were studied in aqueous solution, supramolecular NTs in which the oligoether chains pointed outwards, instead of inwards as in the previous examples, were formed.210 The careful design of the monomer structure and the addition of silver salts have led both to the dissociation of NTs into shorter aggregates,210 or to the promotion of a morphological change from sheets to helical NTs (see previous section: cycles formed by metal–ligand interactions).201 Moreover, it was proved that the NT self-assembly from hexameric cyclic sections was enhanced in monomers with nitrile edge groups, driven by complementary electrostatic interactions between nitrile and phenoxy groups (electron-withdrawing and electron-donating groups, respectively).211
In another relevant article, Lee and coworkers performed a comparative study of the self-assembly in water of three related monomers, 49a–c, in which the central aromatic unit in 49a is replaced by a pyridine unit in compounds 49b,c (Fig. 24a).212 Molecule 49a self-assembles into NTs with an external diameter of 7 nm and a hollow interior of 3 nm, as measured by TEM. Moreover, the blue-shifted absorption maximum and the reduced fluorescence intensity in water compared to chloroform solution, revealed the presence of H-type aggregates, while the Cotton effect observed by CD reflected that the monomers organized into one-handed helical ensembles. When the two pyridine-derivative isomers 49b,c were investigated, TEM images of toroids with outer and inner diameters of 11 nm and 4 nm, respectively, dimensions that corresponded to single stacks of hexameric macrocycles, were obtained. The increase in the concentration produced the formation of elongated tubules that were visualized by TEM. Contrary to 49a, J-type stacking of the aromatic segments was inferred from the red-shifted absorption and the enhanced fluorescence intensity in compounds 49b,c. The growth of the hexameric cycles can be attributed to the emergence of water clusters surrounding the nitrogen at the pyridine unit. This leads to a more spacious packing configuration, helping to diminish the steric crowding at the valley position within the internal pore. CD spectra showed increased intensity with increasing concentration, indicating the formation of chiral assemblies with a preferred handedness, which was inverted when employing the opposite enantiomer. The presence of ethylene oxide chains and pyridine units, which can get dehydrated when heated, imparted thermoresponsive properties to the system. When compounds 49b and 49c were heated, TEM images showed a reduction in the diameter of the NTs (Fig. 24b and c). Furthermore, spectral alterations were noted upon increasing the temperature, including an absorption shift towards shorter wavelengths, a decrease in fluorescence intensity, and an inversion in dichroic response. Finally, spectroscopic experiments were carried out to test the encapsulation of hydrophobic guests in the tubes of compound 49c. Indeed, fullerene C60 could be encapsulated and released as a function of temperature through the hydration or dehydration of the glycol chains and pyridine units (Fig. 24d).
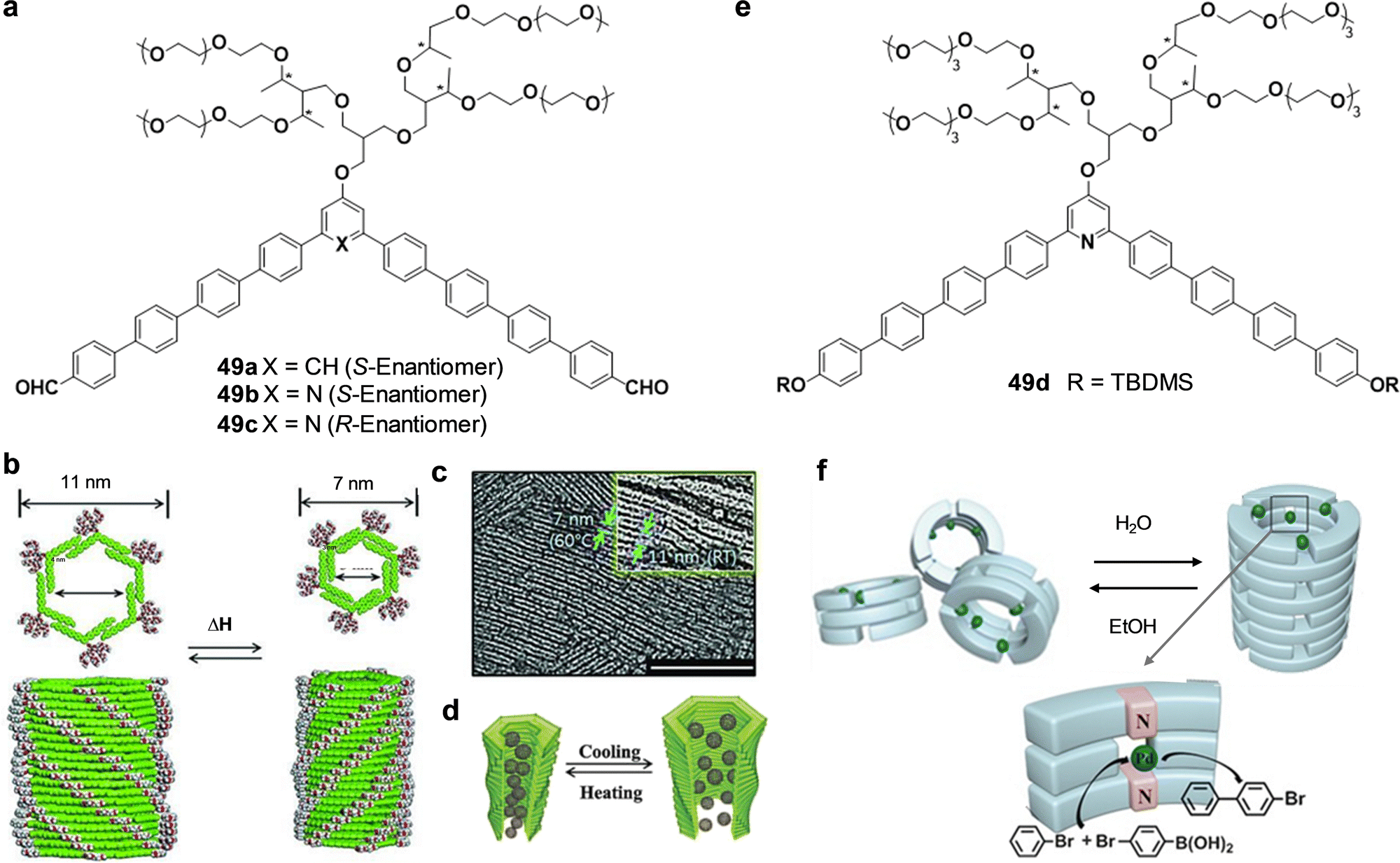 | ||
| Fig. 24 (a) Molecular structure of bent-shaped rod amphiphiles 49a–c. (b) Schematic representation of the reversible switching of the tubules between expanded and contracted states with chirality inversion. (c) TEM image of 49b from 0.01 wt% aqueous solutions prepared at 60 °C versus room temperature (inset). (d) Schematic representation of the regulation of C60–C60 interactions within the tubular cavities when the tubule contains 0.4 equiv. of C60. (e) Molecular structures of compound 49d. (f) Representation of the tubular regulator for the catalytic reaction based on reversible stacking of macrocycles. Adapted with permission from ref. 212 and 213. Copyright 2012-American Association for the Advancement of Science and 2017 Wiley-VCH, respectively. | ||
By utilizing similar pyridine monomers with shorter p-oligophenylene units, Lee and his team were able to create nanofibers composed of dimers.214 These dimers were then transformed into hollow NTs exhibiting a one-handed helical configuration when a guest molecule was present. This expansion process entailed the adaptation of compounds from dimers to hexameric macrocycles, facilitated by reversible H-bonding interactions between the pyridine units in the aromatic cores and the p-phenylphenol guest molecule. Remarkably, the weak Cotton effect seen in the aqueous solution of the nanofibers became much more pronounced with the introduction of the guest, implying a more compact helical arrangement of the aromatic segments.
This kind of pyridine monomers have been also used for selective catalysis in aqueous media.213 For this purpose, a monomer equipped with TBDMS groups, compound 49d, was synthetized (Fig. 24e). Vapor phase osmometry (VPO) experiments in ethanol suggested the formation of macrocycles constituted by three molecules, instead of the six-membered cycles commonly obtained with this type of structures, which would further self-assemble into NTs with the addition of water. TEM and STEM images permitted to discern 1D fibers with an external diameter of 4 nm and an interior of 2.3 nm in accordance with the dimensions of a trimeric cyclic section. Accommodation of Pd ions within the NTs formed by 49d was successfully accomplished and monitored by the observation of a red-shifted absorption band. Also, XPS experiments corroborated the interaction between N and Pd atoms. Finally, owing to the higher-ordered structure and stability of the Pd atoms at the porous walls, the Suzuki–Miyaura coupling reaction was carried out inside the corresponding tube in high yields and selectivities. It was proven that the heterogeneous catalyst could be reused and reinitiated through its supramolecular reversible assembly (Fig. 24f).
Recently, Lee and co-workers reported the simultaneous switching of DNA when exposed to a synthetic coat in response to a change in pH.215 This “synthetic coat” was obtained by the co-assembly of neutral pyridine and cationic pyridinium-based monomers. The encapsulation of double-stranded DNA in water was accomplished through electrostatic interactions between pyridinium cations and the phosphate anions present in the DNA. Furthermore, when the pH value reached physiological levels (pH = 7.4), optical and microscopy experiments revealed that the native right-handed helicity of the encapsulated DNA was transmitted to the surrounding coat assembly. However, at lower pH values, helicity reversal took place.
Apart from this biological application, this kind of monomers have been also used to remove and release micropollutants from water.216 For that purpose, a pH-responsive artificial pump from left-handed porous tubules into right-handed solid fibres was designed. These structures were formed by the stacking of hexameric macrocycles or dimeric disks, respectively. The expansion of right-handed solid fibers into left-handed tubules for pollutant removal was significantly stimulated by the (−)-malic acid enantiomer. In contrast, the contraction that leads to complete desorption of pollutants was sensitive to alkaline (+)-2-amino-1-butanol configuration.
Other remarkable example of the use of solvophobic forces for the formation of tubular assemblies composed of macrocyclic sections was reported by Rybtchinski's research group. In one of their works, they studied the formation of supramolecular NTs from perylene diimide (PDI) derivatives in water (Fig. 25a).217 The general monomer structure possesses two hydrophobic PDI cores connected by a bipyridine linker. Additionally, each PDI contains one PEG chain, in order to afford solubility in water. Self-assembly could be controlled by temperature, solvent or PEG chain length. When an aqueous solution of compound 50 was studied by cryo-TEM (Fig. 25c), NTs of approximately 5 nm wide were observed. Moreover, tubular cross sections showed the ordered hexagonal structure of the PDI core (Fig. 25d). Molecular modelling suggested that three molecules of 50 were stacked on top of another layer of trimers, which agreed with the cryo-TEM measurements (Fig. 25b). This article constitutes an elegant example of how symmetric amphiphilic molecules can be arranged in an organized manner only considering non-directional solvophobic forces.
 | ||
| Fig. 25 (a) Chemical structure of amphiphilic compound 50. (b) Proposed molecular model of self-assembled NTs formed by 50 in water. (c) Cryo-TEM images of a 10−3 M solution of 50 and (d) high-resolution hexagonal cross section. The tube outer dimensions are 5.0 × 4.6 nm. Adapted with permission from ref. 217. Copyright 2018-American Chemical Society. | ||
The wide variety of non-covalent interactions, combined with the degree of tunability of the constituting units of the NT, results in a very rich landscape from which we highlighted some of the most notorious examples in this second section. However, innovative examples keep appearing very frequently in the literature, proving the appeal of this particular type of supramolecular systems.
4. Dynamic covalent cycles
Although less explored than covalent bonds and non-covalent interactions, dynamic covalent chemistry (DCvC) has also been exploited to build macrocycles that subsequently stack into NTs of defined sizes. DCvC permits to construct complex architectures based on building blocks connected through reversible covalent bonds, producing structures lying in the thermodynamic minimum. This intrinsic dynamicity provides adaptable entities with abilities like self-healing and stimuli-responsiveness. Thus, DCvC encompass the advantages of covalent and non-covalent approaches previously described, yielding structures which are more robust than those based on supramolecular cycles due to their covalent character and yet conserving the hallmark adaptability (at the expense of slower kinetics) of non-covalent interactions.218–220DCvC comprises different chemistries which have been extensively discussed in several reviews.218–221 Regarding the construction of NTs through the stacking of macrocycles previously organized by DCvC, we can identify three main categories depending on the functional groups involved: imine-, disulfide-, and boron-based cycles (Fig. 26). In the examples presented in this section, the main contributors to the arrangement of the cycles into nanotubular structures are π–π stacking interactions or electrostatic interactions.
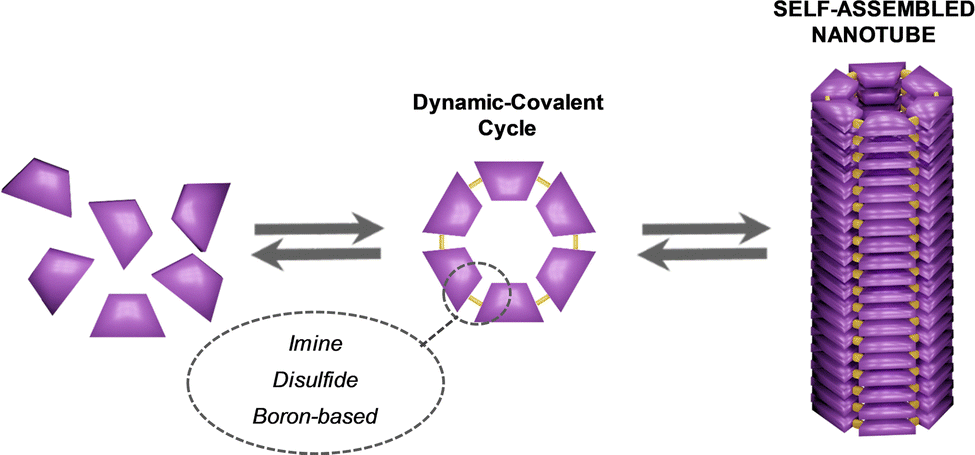 | ||
| Fig. 26 Schematic representation of the reversible formation of dynamic-covalent cycles from individual units and subsequent reversible self-assembly of cyclic entities into self-assembled NTs. | ||
4.1. Imine-based cycles
Traditionally used to build macrocycles and cages,222 polymer networks,223 and more recently, covalent organic frameworks (COFs),224 imine chemistry is probably the type of DCvC which has been the subject of more studies. The first report of an imine-based macrocycle with the ability to stack into NTs comprised the study of compound 51, as described by Moore et al. (Fig. 27).225 The aim was to compare the properties of these tubular assemblies with those exhibited by the arylene-ethynylene macrocycles 33–35. 1H NMR at different concentrations and temperatures proved that the self-assembly of the macrocycles occurred in an isodesmic manner though π–π stacking. The ability of these entities to stack was further supported by means of the presence of birefringence by POM, as well as by wide- and small-angle X-ray scattering (WAXS and SAXS, respectively), unveiling a peak at 3.6 Å, characteristic of π–π stacking, and longer distance peaks which are attributable to hexagonal lattices. | ||
| Fig. 27 Chemical structure of the imine containing m-phenylene ethynylene macrocycle 51 and its stacking into supramolecular NTs. | ||
The group of K. Kim reported the formation of NTs in the solid state due to the stacking of truncated cone-shaped porphyrin-based macrocycles.226 The formation of the macrocycles is driven through the condensation of three diaminoporphyrins and three dibenzaldehydes into imines. The authors determined the size of the pore through XRD and confirmed their stability by gas sorption experiments.
In the last years, research on imine based macrocycles able to yield NTs with high aspect ratios has been broadly capitalized by the group of Dichtel.227 After mastering the formation of the imine based cycle 52 (Fig. 28) with bifunctional diarylamines and terephtaldehydes, the authors found that the addition of a high number of equivalents of CF3CO2H (>2000 eq.) led to the unexpected formation of iminium ions and a counterintuitive stable material with gel-like properties.228 A closer inspection of the nanostructures by TEM and AFM revealed the presence of NTs with aspect ratios, comparable to those of single walled carbon NTs (SWCNTs). Notably, the addition of NEt3 provoked the breaking of these NTs, thus affording stimuli responsive systems. In contrast, the use of a low number of equivalents of CF3CO2H (6 eq.) gave rise to the expected hydrolysis of the imines and hence the decomposition of the macrocycle. Molecular dynamics calculations revealed that the main driving force for the formation of these NTs was the electrostatic attractions between the iminium groups and the CF3COO− ions in solution, whereas neutral macrocycles exhibited lower cohesive forces. Further modifications of these structures permitted to attain chiral NTs and unravel the presence of kinetic traps in their self-assembly process.229
 | ||
| Fig. 28 (a) Chemical structure of macrocycle 52. (b) Decomposition of macrocycle 52 by the hydrolysis of imine groups at a low number of equivalents of CF3CO2H (6 eq.). (c) Formation of NTs by electrostatic attractions between iminium atoms and CF3COO− counterions at a high number of equivalents of CF3CO2H. Adapted with permission from ref. 228. Copyright 2018 National Academy of Sciences. | ||
Taking into account that the main driving force for NT formation are electrostatic interactions, and considering the high number of equivalents of acid needed, which hampered potential applications of these systems, the authors incorporated a pyridine group in the triphenylbenzenediamine unit, with the objective of forming NTs with a lower amount of acid (Fig. 29).227,230 The authors discovered that substoichiometric amounts of acid led to partial formation of pyridinium ions and yet to the formation of stable NTs, unravelling a cooperative supramolecular polymerization mechanism. The robustness of this novel approach permitted the authors to accomplish edge or pore functionalization by the introduction of substituents in the 5- (53a) or 2-(53b) positions of the isophthalaldehyde moiety, respectively, with the aim to: (1) yield modular NTs with lipophilic functionalities through the incorporation of dialdehyde scaffolds with fluorine atoms (53c); (2) tune pore sizes and shapes of the NTs by the incorporation of isophthalaldehyde or terephthalaldehyde moieties (53d and 53e); and (3) generate modular NTs with complex functionalities by including phenanthroline ligands (53f), among others (Fig. 29a). The group usually proved the formation of these NTs as well as their pore sizes by usual microscopy techniques such as AFM (Fig. 29c) and SEM, in addition to XRD, since the self-assembly of the macrocycles led to the emergence of notable diffraction patterns. Thus, the macrocycles of bigger sizes as 53d showcased lower q values in the region between 0.1–0.2 A−1, when compared to those of 53e or 53f, demonstrating hence the versatility of the self-assembled macrocycles (Fig. 29b).231
 | ||
| Fig. 29 (a) Schematic representation of the modular nature of the imine-based macrocycle formed by pyridine containing diamine, enabling to tune the outer and inner part of the pore, as well as the size and shape of the resulting NT by tuning the dialdehyde unit, producing macrocycles 53a–f. (b) XRD pattern of the different NTs formed by 53d–f. (c) AFM images of the NTs formed by (i) 53a, (ii) 53b, (iii) 53c and (iv) 53f. Scale bar = 7 μM. Adapted with permission from ref. 227 and 231. Copyright 2022-American Chemical Society and 2021-American Chemical Society, respectively. | ||
In a following work in this research line, the group exploited the chemical information installed in the interior of the NTs to explore Li-ion conductivity within them.232 The inclusion of polyethylene glycol chains in the dialdehyde moiety afforded NTs with an inner part displaying a high propensity to coordinate Li ions. After characterizing the assemblies by XRD, AFM and SEM, the authors demonstrated through electrochemical impedance spectroscopy (EIS) their superior conductivity when compared to reference compounds, as well as their stability after the essays. Finally, their suitability in battery cells was demonstrated by lineal sweep voltammetry (LSV) and galvanostatic Li-platting-stripping experiments. This approach highlights how the controlled synthesis of NTs with defined properties can provide emerging materials with outstanding properties.
4.2. Disulfide-based cycles
Disulfide bonds and their thiol–thiol exchange properties have also been employed to build discrete macrocycles and cages.233 However, the deliberate formation of NTs utilizing this type of DCvC chemistry is less exploited. Haridas et al. reported the hierarchical organization of self-assembling disulfide-based macrocycles (Fig. 30a).234 Macrocycle 54 was constructed though the initial formation of disulfide bonds between cysteine units that were subsequently combined with an aromatic unit through a 1,3 dipolar cycloaddition, in order to generate the final structure. Compound 54 initially formed helical structures in MeOH that after time produced hollow NTs that further striated, as shown by TEM images (Fig. 30b). However, the width of these NTs was much bigger than the size of the macrocycle, demonstrating the difficulties encountered by this approach to control the stacking of macrocycles to yield tubes of defined sizes. | ||
| Fig. 30 (a) Chemical structure of disulfide-based macrocycle 54, and schematic representation of NT formation. (b) TEM images of (left) NTs and (right) striated NTs. (c) Schematic illustration of a small dynamic combinatorial library from dithiol building block 55. (d) and (e) Cryo-TEM images of (d) 556; (e) 557, (f) schematic representation of the proposed formation of fibers of 556. The benzenedithiol core of building block is shown in yellow and the peptide chain in blue. Adapted with permission from ref. 234 and 235. Copyright 2022-Royal Society of Chemistry and 2010-The American Association for the Advancement of Science. | ||
In this context, it is noteworthy to mention the work of Otto and coworkers, which have vastly explored dynamic covalent libraries of thiol-based peptide scaffolds able to form β-sheets (Fig. 30c–f).235,236 In these libraries, macrocycles of different sizes are formed by thiol–thiol exchange shift (Fig. 30c), and the most populated is the most stable one, which is the one with the ability to stack vertically and hence self-replicate (Fig. 30f). These facts have been proven by different techniques, such as cryo-TEM microscopy (Fig. 30d and e). This kind of supramolecular structures have been the subject of intensive research by the group in the last 15 years and, although different catalysts and processes have been carried out with these entities, the authors have not exploited the pore of the stack to our knowledge.
4.3. Boron-based cycles
Although not as thoroughly studied as the previous examples, boron-based cycles have also been exploited to build discrete macrocycles. In this context, boronate esters, which result from the condensation between boronic acids and alcohols, are a promising option. Dichtel and coworkers employed boronate esters to construct discrete macrocycles using different alcohols and boronic acids as scaffolds. This approach permitted to tune the size of the NT by featuring different spacer units in the boronic acid fragment such as phenyl, biphenyl and pyrenyl.237 The aggregation of the phenyl-containing macrocycle 56 (Fig. 31a) into NTs was confirmed by means of fluorescence experiments due to a red shift and a quenching of the signal of the aggregate with respect to the non-aggregated species. Additionally, AFM experiments evidenced that these NTs could disassemble back into the macrocycles by stimuli such as sonication. Notably, if left undisturbed afterwards, the macrocycles recover the nanotubular architecture (Fig. 31b). | ||
| Fig. 31 (a) Alcohol and boronic acids which yield NTs after the formation and stacking of the corresponding boronate ester-based macrocycle 56. (b) AFM images obtained via drop casting into mica of (A) the initial boronate ester-based NTs, (B) after sonication during 1 h and (C), and after leaving the previous sonicated solution undisturbed for 12 h. Adapted with permission from ref. 237. Copyright 2016-American Chemical Society. | ||
All the examples featured up to this point exhibit a bottom-up approach with respect to the formation of the NT. However, there are cases in which several dynamic covalent bonds may be combined to achieve a higher order structure that is broken apart into simpler ones in a top-down manner. In a beautiful example by Xin Zhao et al., a COF is first formed by (a) combination of boroxine and hydrazone bonds that arise from the pairing of an aromatic compound equipped with boronic acid and aldehyde groups (DETH in Fig. 32) and an aromatic dihydrazide derivative (DFPBA in Fig. 32), yielding the macrocycle 57. The generation of this COF was confirmed by XRD as well as by TEM measurements. Afterwards, selective hydrolysis of the boroxine units by means of concentrated hydrochloric acid in water breaks the COF apart into the corresponding NTs.238 The morphology as well as the dimensions of the tube's pore were elucidated by TEM. Successive stimulation of the NTs through sonication produced the decomposition of the NT structure, but after leaving the resulting suspension undisturbed, the tubular structure was not recovered, showing that the NTs were formed directly from the channels of the COF.
 | ||
| Fig. 32 Top-down approach for the obtention of NTs through the formation of hydrazone and boroxine bonds to yield a COF based on macrocycle 57, and successive treatment with acid for the breaking of the boroxines and the generation of NTs. Adapted with permission from ref. 238. Copyright 2020-American Chemical Society. | ||
The dynamic covalent systems presented in this final section very clearly show that, although not as prolific as their covalent and non-covalent counterparts, they serve as a solid and promising foundation to further develop tubular nanostructures based on these reversible bonds.
Conclusions
The development of supramolecular NTs from discrete macrocyclic entities constitutes a powerful tool to create porous unidimensional nanobjects of defined sizes and high aspect ratios. In this review we have covered most of the achievements described so far, emphasizing the tools available for chemists to create these fascinating tubular structures. An appropriate molecular design and the exploitation of different types of bonding to build them – covalent, non-covalent and dynamic covalent – has widen the scope of available supramolecular NTs, which have already been applied in molecular encapsulation or ion transport, among other applications. When compared to other type of NTs like 1D inorganic or carbon NTs, the modular nature of self-assembled NTs and the inherent reversibility of the forces driving their formation constitute an advantage that can lead to novel applications in the fields of catalysis and drug delivery, which have yet not been extensively explored.Regardless of the substantial number of strategies and methodologies explored to assemble these unidimensional containers, the chemistry within the NT pores has not been exploited to a large extent. Supramolecular NTs assembled from cyclic entities display an extraordinary structural versatility, and their internal lumens could in principle be custom-tailored with a given diameter and chemical coating so as to achieve the selective encapsulation of substances. This selective encapsulation and the dynamic nature of the self-assembled NT could be especially beneficial in the field of drug delivery. We also envisage that the field of catalysis could widely capitalize the modular unidimensional chemical inner environment of the NT, and profit from the well-defined confined 1D nanospace to bring together reactants and stabilize chemical intermediates.
Despite the significant achievements remarked in this review article, the field of self-assembled NTs still has to face some current structural limitations in order to broaden the range of applications. One significant hindrance involves the characterization of the NT itself, which is normally attained through a sum of spectroscopic (UV Vis, CD, DLS, FTIR, fluorescence), microscopic (AFM, SEM or TEM; among others) and scattering techniques (SAXS, WAXS, DLS). However, in many cases these measurements do not permit unequivocally to discern the hollow interior, making imperative the incorporation of novel techniques in the characterization of supramolecular NTs. Further on this vein, the structural integrity of the NT after using it for a given application has not been studied in detail. Although this structural integrity is expected to be inferior to that of NTs which grow through covalent or dynamic covalent interactions,239,240 we consider that the versatility and ease of design of supramolecular NTs offer substantial advantages when compared to those. Still more studies are necessary in order to clarify this point. Besides, the preparation method of the self-assembled NT is strongly dependent on factors like solvent mixture, temperature and preparation protocols, which make the description of the preparation method imperative in order to obtain the desired structure.
In general, the control over the length of the self-assembled NTs is scarce. Many studies have achieved it by combining supramolecular interactions with covalent bonds in the Z-direction of the assembly. Notable examples describe the use of π–π stacking between hexakis(m-phenylene ethynylene) covalent cycles,241 and the vertical assembly of alternately arranged positively and negatively charged covalent cycles formed from pillar[5]arene motifs through electrostatic interactions,242 strategies that give access to tubular structures with defined lengths. Additionally, the incorporation of dynamic covalent bonds, such as imine bond239,243 intercalated between macrocycles, has also proved itself successful to define the length of the resulting NT.
Although controlling the growth of the NT by these strategies or the use of pure covalent approaches117,118,244–248 or host–guest chemistry39–42,138 could constitute fruitful methods, we envisage that for the case of self-assembled NTs it might be convenient to take advantage of concepts like living supramolecular polymerization (LSP).249,250 This strategy could aid to control the length of the hollow polymers and thus to assess its impact in different fields, like transmembrane ion channels, or to control the size of nanorods in templated synthesis. Additionally, given the dynamic nature of non-covalent interactions, obtaining block or blocky type NTs remains challenging. The application of the concepts of LSP could also yield block supramolecular NTs, in which each part of the NT includes different scaffolds than in turn enable different functionality.251,252 Finally, although DCvC has been successfully applied to build NTs, their responsive properties have not been explored that much. In this context, we also envision that the application of chemical reaction networks to the formation of covalent cycles could help to improve their properties and give rise to novel behavior, as well as to control in time the formation of the supramolecular NT,253,254 which can be especially appealing in drug delivery.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by MCIN (PID2020-116921GB-I00, TED2021-132602B-I00, PID2023-148548NB-I00) and AEI (PID2020-116112RJ-I00). F. A. is grateful to MCIN and Next Generation EU funding for a “Ramon-y-Cajal” fellowship (RyC-2021-031538-I). J. V.-H. and P. B. C. are grateful to the Comunidad de Madrid for the PIPF-2023/TEC-30164 grant and the PEJ-2017-AI/IND-6246 contract, respectively. J. S. V. is grateful to EU funding from MSCA-IEF actions (101150870-Lightheal).Notes and references
- B. Eisenberg, Acc. Chem. Res., 1998, 31, 117–123 CrossRef CAS.
- P. B. Sigler, Z. Xu, H. S. Rye, S. G. Burston, W. A. Fenton and A. L. Horwich, Annu. Rev. Biochem., 1998, 67, 581–608 CrossRef CAS PubMed.
- A. L. Horwich, E. U. Weber-Ban and D. Finley, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11033–11040 CrossRef CAS PubMed.
- R. T. Hunt, K. A. Nasmyth, J. Diffley, P. Zwickl, D. Voges and W. Baumeister, Philos. Trans. R. Soc. London, Ser. B, 1999, 354, 1501–1511 CrossRef.
- D. Voges, P. Zwickl and W. Baumeister, Annu. Rev. Biochem., 1999, 68, 1015–1068 CrossRef CAS PubMed.
- D. A. Fletcher and R. D. Mullins, Nature, 2010, 463, 485–492 CrossRef CAS PubMed.
- J. Avila, FASEB J., 1990, 4, 3284–3290 CrossRef CAS PubMed.
- A. P. Kalra, B. B. Eakins, S. D. Patel, G. Ciniero, V. Rezania, K. Shankar and J. A. Tuszynski, ACS Nano, 2020, 14, 16301–16320 CrossRef CAS PubMed.
- S. Forth and T. M. Kapoor, J. Cell Biol., 2017, 216, 1525–1531 CrossRef CAS PubMed.
- M. Serra, R. Arenal and R. Tenne, Nanoscale, 2019, 11, 8073–8090 RSC.
- R. Tenne and G. Seifert, Annu. Rev. Mater. Res., 2009, 39, 387–413 CrossRef CAS.
- S. A. Miners, G. A. Rance and A. N. Khlobystov, Chem. Soc. Rev., 2016, 45, 4727–4746 RSC.
- S. Rathinavel, K. Priyadharshini and D. Panda, Mater. Sci. Eng., B, 2021, 268, 115095 CrossRef CAS.
- T. Shimizu, W. Ding and N. Kameta, Chem. Rev., 2020, 120, 2347–2407 CrossRef CAS PubMed.
- D. T. Bong, T. D. Clark, J. R. Granja and M. R. Ghadiri, Angew. Chem., Int. Ed., 2001, 40, 988–1011 CrossRef CAS PubMed.
- L. S. Shimizu, S. R. Salpage and A. A. Korous, Acc. Chem. Res., 2014, 47, 2116–2127 CrossRef CAS PubMed.
- W. Si, P. Xin, Z.-T. Li and J.-L. Hou, Acc. Chem. Res., 2015, 48, 1612–1619 CrossRef CAS PubMed.
- V. S. Saji, Mater. Today Adv., 2022, 14, 100239 CrossRef CAS.
- D. Yoshida, K. Kim, I. Takumi, F. Yamaguchi, K. Adachi and A. Teramoto, Med. Mol. Morphol., 2013, 46, 86–91 CrossRef CAS PubMed.
- J. Montenegro, M. R. Ghadiri and J. R. Granja, Acc. Chem. Res., 2013, 46, 2955–2965 CrossRef CAS PubMed.
- T. Fan, X. Yu, B. Shen and L. Sun, J. Nanomater., 2017, 2017, 4562474 Search PubMed.
- Q. Jin, L. Zhang, H. Cao, T. Wang, X. Zhu, J. Jiang and M. Liu, Langmuir, 2011, 27, 13847–13853 CrossRef CAS PubMed.
- Y. Zhou, M. Kogiso, M. Asakawa, S. Dong, R. Kiyama and T. Shimizu, Adv. Mater., 2009, 21, 1742–1745 CrossRef CAS.
- A. Saeki, Y. Koizumi, T. Aida and S. Seki, Acc. Chem. Res., 2012, 45, 1193–1202 CrossRef CAS PubMed.
- T. Lobovkina, P. G. Dommersnes, S. Tiourine, J. F. Joanny and O. Orwar, Eur. Phys. J. E:Soft Matter Biol. Phys., 2008, 26, 295–300 CrossRef CAS PubMed.
- A. Bernet, M. Behr and H.-W. Schmidt, Soft Matter, 2011, 7, 1058–1065 RSC.
- R. Chapman, M. Danial, M. L. Koh, K. A. Jolliffe and S. Perrier, Chem. Soc. Rev., 2012, 41, 6023–6041 RSC.
- Y. Liu, T. Wang, Y. Huan, Z. Li, G. He and M. Liu, Adv. Mater., 2013, 25, 5875–5879 CrossRef CAS PubMed.
- C.-Z. Liu, M. Yan, H. Wang, D.-W. Zhang and Z.-T. Li, ACS Omega, 2018, 3, 5165–5176 CrossRef CAS PubMed.
- H. Mamad-Hemouch, H. Ramoul, M. Abou Taha, L. Bacri, C. Huin, C. Przybylski, A. Oukhaled, B. Thiébot, G. Patriarche, N. Jarroux and J. Pelta, Nano Lett., 2015, 15, 7748–7754 CrossRef CAS PubMed.
- R.-G. Lin, L.-S. Long, R.-B. Huang and L.-S. Zheng, Cryst. Growth Des., 2008, 8, 791–794 CrossRef CAS.
- T. Sendai, S. Biswas and T. Aida, J. Am. Chem. Soc., 2013, 135, 11509–11512 CrossRef CAS PubMed.
- T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401–1444 CrossRef CAS PubMed.
- R. García-Fandiño, M. Amorín and J. R. Granja, in Supramol. Chem., ed. P. A. Gale and J. W. Steed, Wiley, Hoboken, 2012, pp. 1–34 Search PubMed.
- T. Shimizu, Bull. Chem. Soc. Jpn., 2018, 91, 623–668 CrossRef CAS.
- A. Nitti, A. Pacini and D. Pasini, Nanomaterials, 2017, 7, 167 CrossRef PubMed.
- T. Komatsu, Nanoscale, 2012, 4, 1910–1918 RSC.
- S. Rinaldi, Molecules, 2020, 25, 3276 CrossRef CAS PubMed.
- Y. Ohishi, J. Chiba and M. Inouye, J. Org. Chem., 2022, 87, 10825–10835 CrossRef CAS PubMed.
- T. A. Sobiech, Y. Zhong, D. P. Miller, J. K. McGrath, C. T. Scalzo, M. C. Redington, E. Zurek and B. Gong, Angew. Chem., Int. Ed., 2022, 61, e202213467 CrossRef CAS PubMed.
- D. Suzuki, H. Abe and M. Inouye, Org. Lett., 2016, 18, 320–323 CrossRef CAS PubMed.
- I. Popov, T.-H. Chen, S. Belyakov, O. Daugulis, S. E. Wheeler and O. Š. Miljanić, Chem. – Eur. J., 2015, 21, 2750–2754 CrossRef CAS PubMed.
- D. Pasini and M. Ricci, Curr. Org. Synth., 2007, 4, 59–80 CrossRef CAS.
- A. Ohira, M. Sakata, I. Taniguchi, C. Hirayama and M. Kunitake, J. Am. Chem. Soc., 2003, 125, 5057–5065 CrossRef CAS PubMed.
- G. Li and L. B. McGown, Science, 1994, 264, 249–251 CrossRef CAS PubMed.
- D. Whang, Y.-M. Jeon, J. Heo and K. Kim, J. Am. Chem. Soc., 1996, 118, 11333–11334 CrossRef CAS.
- S. Pipaón, M. Gragera, M. T. Bueno-Carrasco, J. García-Bernalt Diego, M. Cantero, J. Cuéllar, M. R. Fernández-Fernández and J. M. Valpuesta, Nanomaterials, 2021, 11, 503 CrossRef PubMed.
- N. Kameta, M. Masuda and T. Shimizu, ACS Nano, 2012, 6, 5249–5258 CrossRef CAS PubMed.
- Á. L. Fuentes de Arriba and J. R. Granja, Peptide Self-Assembly and Engineering, 2024, pp. 109–141 Search PubMed.
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS PubMed.
- M. R. Ghadiri, J. R. Granja and L. K. Buehler, Nature, 1994, 369, 301–304 CrossRef CAS PubMed.
- S. Fernandez-Lopez, H.-S. Kim, E. C. Choi, M. Delgado, J. R. Granja, A. Khasanov, K. Kraehenbuehl, G. Long, D. A. Weinberger, K. M. Wilcoxen and M. R. Ghadiri, Nature, 2001, 412, 452–455 CrossRef CAS PubMed.
- O. Silberbush, M. Engel, I. Sivron, S. Roy and N. Ashkenasy, J. Phys. Chem. B, 2019, 123, 9882–9888 CrossRef CAS PubMed.
- K. Rosenthal-Aizman, G. Svensson and A. Undén, J. Am. Chem. Soc., 2004, 126, 3372–3373 CrossRef CAS PubMed.
- H. Eagle, Science, 1959, 130, 432–437 CrossRef CAS PubMed.
- N. Ercal, X. Luo, R. H. Matthews and D. W. Armstrong, Chirality, 1996, 8, 24–29 CrossRef CAS PubMed.
- T. Kimura, S. Nada, N. Takegahara, T. Okuno, S. Nojima, S. Kang, D. Ito, K. Morimoto, T. Hosokawa, Y. Hayama, Y. Mitsui, N. Sakurai, H. Sarashina-Kida, M. Nishide, Y. Maeda, H. Takamatsu, D. Okuzaki, M. Yamada, M. Okada and A. Kumanogoh, Nat. Commun., 2016, 7, 13130 CrossRef CAS PubMed.
- A. Ghorai, B. Achari and P. Chattopadhyay, Tetrahedron, 2016, 72, 3379–3387 CrossRef CAS.
- D. Seebach, J. L. Matthews, A. Meden, T. Wessels, C. Baerlocher and L. B. McCusker, Helv. Chim. Acta, 1997, 80, 173–182 CrossRef CAS.
- L. Li, H. Zhan, P. Duan, J. Liao, J. Quan, Y. Hu, Z. Chen, J. Zhu, M. Liu, Y.-D. Wu and J. Deng, Adv. Funct. Mater., 2012, 22, 3051–3056 CrossRef CAS.
- M. Amorín, L. Castedo and J. R. Granja, J. Am. Chem. Soc., 2003, 125, 2844–2845 CrossRef PubMed.
- W.-H. Hsieh and J. Liaw, J. Food Drug Anal., 2019, 27, 32–47 CrossRef CAS PubMed.
- N. Rodríguez-Vázquez, M. Amorín and J. R. Granja, Org. Biomol. Chem., 2017, 15, 4490–4505 RSC.
- I. W. Hamley, Angew. Chem., Int. Ed., 2014, 53, 6866–6881 CrossRef CAS PubMed.
- W. S. Horne, C. D. Stout and M. R. Ghadiri, J. Am. Chem. Soc., 2003, 125, 9372–9376 CrossRef CAS PubMed.
- A. D. Pehere, C. J. Sumby and A. D. Abell, Org. Biomol. Chem., 2013, 11, 425–429 RSC.
- K. Sato, Y. Itoh and T. Aida, Chem. Sci., 2014, 5, 136–140 RSC.
- S. M. Darnall, C. Li, M. Dunbar, M. Alsina, S. Keten, B. A. Helms and T. Xu, J. Am. Chem. Soc., 2019, 141, 10953–10957 CrossRef CAS PubMed.
- F. Fujimura, T. Hirata, T. Morita, S. Kimura, Y. Horikawa and J. Sugiyama, Biomacromolecules, 2006, 7, 2394–2400 CrossRef CAS PubMed.
- A. Ghorai, E. Padmanaban, C. Mukhopadhyay, B. Achari and P. Chattopadhyay, Chem. Commun., 2012, 48, 11975–11977 RSC.
- F. Fujimura, Y. Horikawa, T. Morita, J. Sugiyama and S. Kimura, Biomacromolecules, 2007, 8, 611–616 CrossRef CAS PubMed.
- B. Jagannadh, M. S. Reddy, C. L. Rao, A. Prabhakar, B. Jagadeesh and S. Chandrasekhar, Chem. Commun., 2006, 4847–4849 RSC.
- H. Uji, H. Kim, T. Imai, S. Mitani, J. Sugiyama and S. Kimura, Pept. Sci., 2016, 106, 275–282 CrossRef CAS PubMed.
- M. Li, M. Ehlers, S. Schlesiger, E. Zellermann, S. K. Knauer and C. Schmuck, Angew. Chem., Int. Ed., 2016, 55, 598–601 CrossRef CAS PubMed.
- M. Cuerva, R. García-Fandiño, C. Vázquez-Vázquez, M. A. López-Quintela, J. Montenegro and J. R. Granja, ACS Nano, 2015, 9, 10834–10843 CrossRef CAS PubMed.
- A. Méndez-Ardoy, J. R. Granja and J. Montenegro, Nanoscale Horiz., 2018, 3, 391–396 RSC.
- A. Pizzi, H. L. Ozores, M. Calvelo, R. García-Fandiño, M. Amorín, N. Demitri, G. Terraneo, S. Bracco, A. Comotti, P. Sozzani, C. X. Bezuidenhout, P. Metrangolo and J. R. Granja, Angew. Chem., Int. Ed., 2019, 58, 14472–14476 CrossRef CAS PubMed.
- I. Insua and J. Montenegro, J. Am. Chem. Soc., 2020, 142, 300–307 CrossRef CAS PubMed.
- I. Insua, A. Cardellini, S. Díaz, J. Bergueiro, R. Capelli, G. M. Pavan and J. Montenegro, Chem. Sci., 2023, 14, 14074–14081 RSC.
- A. Bayón-Fernández, A. Méndez-Ardoy, C. Alvarez-Lorenzo, J. R. Granja and J. Montenegro, J. Mater. Chem. B, 2023, 11, 606–617 RSC.
- R.-V. Nuria, H. L. Ozores, G. Arcadio, G.-F. Eva, F. Alberto, P. Michele, M. P. Juan, O. Juan, M. Javier, G.-F. Rebeca, A. Manuel and R. G. Juan, Curr. Top. Med. Chem., 2014, 14, 2647–2661 Search PubMed.
- M. Amorín, R. J. Brea, L. Castedo and J. R. Granja, Org. Lett., 2005, 7, 4681–4684 CrossRef PubMed.
- R. J. Brea, L. Castedo and J. R. Granja, Chem. Commun., 2007, 3267–3269 RSC.
- A. Lamas, A. Guerra, M. Amorín and J. R. Granja, Chem. Sci., 2018, 9, 8228–8233 RSC.
- A. Fuertes, H. L. Ozores, M. Amorín and J. R. Granja, Nanoscale, 2017, 9, 748–753 RSC.
- A. Mendez-Ardoy, A. Bayon-Fernandez, Z. Yu, C. Abell, J. R. Granja and J. Montenegro, Angew. Chem., Int. Ed., 2020, 59, 6902–6908 CrossRef CAS PubMed.
- M. Vilela-Picos, F. Novelli, A. Pazó, A. Méndez-Ardoy, G. Marafon, M. Amorín, A. Moretto and J. R. Granja, Chem, 2023, 9, 3365–3378 CAS.
- M. Vilela-Picos, F. Novelli, A. Méndez-Ardoy, A. Moretto and J. R. Granja, STAR Protoc., 2024, 5, 103031 CrossRef CAS PubMed.
- F. Novelli, M. Vilela, A. Pazó, M. Amorín and J. R. Granja, Angew. Chem., Int. Ed., 2021, 60, 18838–18844 CrossRef CAS PubMed.
- J. Couet, J. D. J. S. Samuel, A. Kopyshev, S. Santer and M. Biesalski, Angew. Chem., Int. Ed., 2005, 44, 3297–3301 CrossRef CAS PubMed.
- M. G. J. ten Cate, N. Severin and H. G. Börner, Macromolecules, 2006, 39, 7831–7838 CrossRef CAS.
- S. Catrouillet, J. C. Brendel, S. Larnaudie, T. Barlow, K. A. Jolliffe and S. Perrier, ACS Macro Lett., 2016, 5, 1119–1123 CrossRef CAS PubMed.
- E. D. H. Mansfield, M. Hartlieb, S. Catrouillet, J. Y. Rho, S. C. Larnaudie, S. E. Rogers, J. Sanchis, J. C. Brendel and S. Perrier, Soft Matter, 2018, 14, 6320–6326 RSC.
- Q. Song, S. Goia, J. Yang, S. C. L. Hall, M. Staniforth, V. G. Stavros and S. Perrier, J. Am. Chem. Soc., 2021, 143, 382–389 CrossRef CAS PubMed.
- J. Y. Rho, J. C. Brendel, L. R. MacFarlane, E. D. H. Mansfield, R. Peltier, S. Rogers, M. Hartlieb and S. Perrier, Adv. Funct. Mater., 2018, 28, 1704569 CrossRef.
- S. C. Larnaudie, J. Sanchis, T.-H. Nguyen, R. Peltier, S. Catrouillet, J. C. Brendel, C. J. H. Porter, K. A. Jolliffe and S. Perrier, Biomaterials, 2018, 178, 570–582 CrossRef CAS PubMed.
- M. Hartlieb, S. Catrouillet, A. Kuroki, C. Sanchez-Cano, R. Peltier and S. Perrier, Chem. Sci., 2019, 10, 5476–5483 RSC.
- M. Danial, C. My-Nhi Tran, P. G. Young, S. Perrier and K. A. Jolliffe, Nat. Commun., 2013, 4, 2780 CrossRef PubMed.
- J. Y. Rho, H. Cox, E. D. H. Mansfield, S. H. Ellacott, R. Peltier, J. C. Brendel, M. Hartlieb, T. A. Waigh and S. Perrier, Nat. Commun., 2019, 10, 4708 CrossRef CAS PubMed.
- J. C. Brendel, J. Sanchis, S. Catrouillet, E. Czuba, M. Z. Chen, B. M. Long, C. Nowell, A. Johnston, K. A. Jolliffe and S. Perrier, Angew. Chem., Int. Ed., 2018, 57, 16678–16682 CrossRef CAS PubMed.
- J. Yang, J.-I. Song, Q. Song, J. Y. Rho, E. D. H. Mansfield, S. C. L. Hall, M. Sambrook, F. Huang and S. Perrier, Angew. Chem., Int. Ed., 2020, 59, 8860–8863 CrossRef CAS PubMed.
- T. Xu, N. Zhao, F. Ren, R. Hourani, M. T. Lee, J. Y. Shu, S. Mao and B. A. Helms, ACS Nano, 2011, 5, 1376–1384 CrossRef CAS PubMed.
- C. Zhang and T. Xu, Nanoscale, 2015, 7, 15117–15121 RSC.
- L. Yuan, W. Feng, K. Yamato, A. R. Sanford, D. Xu, H. Guo and B. Gong, J. Am. Chem. Soc., 2004, 126, 11120–11121 CrossRef CAS PubMed.
- B. Gong, A. R. Sanford and J. S. Ferguson, Adv. Polym. Sci., 2007, 206, 1–29 CAS.
- B. Gong and Z. Shao, Acc. Chem. Res., 2013, 46, 2856–2866 CrossRef CAS PubMed.
- K. Yamato, M. Kline and B. Gong, Chem. Commun., 2012, 48, 12142–12158 RSC.
- Y. Yang, W. Feng, J. Hu, S. Zou, R. Gao, K. Yamato, M. Kline, Z. Cai, Y. Gao, Y. Wang, Y. Li, Y. Yang, L. Yuan, X. C. Zeng and B. Gong, J. Am. Chem. Soc., 2011, 133, 18590–18593 CrossRef CAS PubMed.
- A. J. Helsel, A. L. Brown, K. Yamato, W. Feng, L. Yuan, A. J. Clements, S. V. Harding, G. Szabo, Z. Shao and B. Gong, J. Am. Chem. Soc., 2008, 130, 15784–15785 CrossRef CAS PubMed.
- M. A. Kline, X. Wei, I. J. Horner, R. Liu, S. Chen, S. Chen, K. Y. Yung, K. Yamato, Z. Cai, F. V. Bright, X. C. Zeng and B. Gong, Chem. Sci., 2015, 6, 152–157 RSC.
- X. Wei, G. Zhang, Y. Shen, Y. Zhong, R. Liu, N. Yang, F. Y. Al-mkhaizim, M. A. Kline, L. He, M. Li, Z.-L. Lu, Z. Shao and B. Gong, J. Am. Chem. Soc., 2016, 138, 2749–2754 CrossRef CAS PubMed.
- R. Cao, R. B. Rossdeutcher, Y. Zhong, Y. Shen, D. P. Miller, T. A. Sobiech, X. Wu, L. S. Buitrago, K. Ramcharan, M. I. Gutay, M. F. Figueira, P. Luthra, E. Zurek, T. Szyperski, B. Button, Z. Shao and B. Gong, Nat. Chem., 2023, 15, 1559–1568 CrossRef CAS PubMed.
- X. Li, B. Li, L. Chen, J. Hu, C. Wen, Q. Zheng, L. Wu, H. Zeng, B. Gong and L. Yuan, Angew. Chem., Int. Ed., 2015, 54, 11147–11152 CrossRef CAS PubMed.
- X. Wu, R. Liu, B. Sathyamoorthy, K. Yamato, G. Liang, L. Shen, S. Ma, D. K. Sukumaran, T. Szyperski, W. Fang, L. He, X. Chen and B. Gong, J. Am. Chem. Soc., 2015, 137, 5879–5882 CrossRef CAS PubMed.
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- C. Ren, S. Xu, J. Xu, H. Chen and H. Zeng, Org. Lett., 2011, 13, 3840–3843 CrossRef CAS PubMed.
- G. Shin, M. I. Khazi, U. Kundapur, B. Kim, Y. Kim, C. W. Lee and J.-M. Kim, ACS Macro Lett., 2019, 8, 610–615 CrossRef CAS PubMed.
- G. Shin, M. I. Khazi and J.-M. Kim, Macromolecules, 2020, 53, 149–157 CrossRef CAS.
- L. S. Shimizu, M. D. Smith, A. D. Hughes and K. D. Shimizu, Chem. Commun., 2001, 1592–1593 RSC.
- J. Yang, M. B. Dewal, S. Profeta, M. D. Smith, Y. Li and L. S. Shimizu, J. Am. Chem. Soc., 2008, 130, 612–621 CrossRef CAS PubMed.
- M. B. Dewal, Y. Xu, J. Yang, F. Mohammed, M. D. Smith and L. S. Shimizu, Chem. Commun., 2008, 3909–3911 RSC.
- J. Yang, M. B. Dewal, D. Sobransingh, M. D. Smith, Y. Xu and L. S. Shimizu, J. Org. Chem., 2009, 74, 102–110 CrossRef CAS PubMed.
- V. Semetey, C. Didierjean, J.-P. Briand, A. Aubry and G. Guichard, Angew. Chem., Int. Ed., 2002, 41, 1895–1898 CrossRef CAS PubMed.
- D. Ranganathan, C. Lakshmi and I. L. Karle, J. Am. Chem. Soc., 1999, 121, 6103–6107 CrossRef CAS.
- V. Simic, L. Bouteiller and M. Jalabert, J. Am. Chem. Soc., 2003, 125, 13148–13154 CrossRef CAS PubMed.
- J. L. López, E. M. Pérez, P. M. Viruela, R. Viruela, E. Ortí and N. Martín, Org. Lett., 2009, 11, 4524–4527 CrossRef PubMed.
- P. Kittikhunnatham, B. Som, V. Rassolov, M. Stolte, F. Würthner, L. S. Shimizu and A. B. Greytak, J. Phys. Chem. C, 2017, 121, 18102–18109 CrossRef CAS.
- X. Wu, P. Wang, P. Turner, W. Lewis, O. Catal, D. S. Thomas and P. A. Gale, Chem, 2019, 5, 1210–1222 CAS.
- D. Zhao and J. S. Moore, Chem. Commun., 2003, 807–818 RSC.
- C. Grave and A. D. Schlüter, J. Org. Chem., 2002, 2002, 3075–3098 Search PubMed.
- S. Lahiri, J. L. Thompson and J. S. Moore, J. Am. Chem. Soc., 2000, 122, 11315–11319 CrossRef CAS.
- A. S. Shetty, J. Zhang and J. S. Moore, J. Am. Chem. Soc., 1996, 118, 1019–1027 CrossRef CAS.
- K. Balakrishnan, A. Datar, W. Zhang, X. Yang, T. Naddo, J. Huang, J. Zuo, M. Yen, J. S. Moore and L. Zang, J. Am. Chem. Soc., 2006, 128, 6576–6577 CrossRef CAS PubMed.
- L. Zang, Y. Che and J. S. Moore, Acc. Chem. Res., 2008, 41, 1596–1608 CrossRef CAS PubMed.
- Y. Takaki, R. Ozawa, T. Kajitani, T. Fukushima, M. Mitsui and K. Kobayashi, Chem. – Eur. J., 2016, 22, 16760–16764 CrossRef CAS PubMed.
- X. Zhou, G. Liu, K. Yamato, Y. Shen, R. Cheng, X. Wei, W. Bai, Y. Gao, H. Li, Y. Liu, F. Liu, D. M. Czajkowsky, J. Wang, M. J. Dabney, Z. Cai, J. Hu, F. V. Bright, L. He, X. C. Zeng, Z. Shao and B. Gong, Nat. Commun., 2012, 3, 949 CrossRef PubMed.
- Y. Zhong, Y. Yang, Y. Shen, W. Xu, Q. Wang, A. L. Connor, X. Zhou, L. He, X. C. Zeng, Z. Shao, Z.-L. Lu and B. Gong, J. Am. Chem. Soc., 2017, 139, 15950–15957 CrossRef CAS PubMed.
- Q. Wang, Y. Zhong, D. P. Miller, X. Lu, Q. Tang, Z.-L. Lu, E. Zurek, R. Liu and B. Gong, J. Am. Chem. Soc., 2020, 142, 2915–2924 CrossRef CAS PubMed.
- Y. Zhong, Q. Wang, Y. Yang, Z. Lu, L. He and B. Gong, Org. Lett., 2016, 18, 2094–2097 CrossRef CAS PubMed.
- P. B. Chamorro and F. Aparicio, Chem. Commun., 2021, 57, 12712–12724 RSC.
- C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488–7499 CrossRef CAS PubMed.
- F. Aparicio, M. J. Mayoral, C. Montoro-García and D. González-Rodríguez, Chem. Commun., 2019, 55, 7277–7299 RSC.
- P. Ballester and J. d Mendoza, in Modern Supramolecular Chemistry, ed. F. Diederich, P. J. P. J. Stang and R. R. Tykwinski, Wiley-VCH, 2008, pp. 69–111 Search PubMed.
- E. A. Kataev, Non-covalent Interactions in the Synthesis and Design of New Compounds, 2016, pp. 63–82 Search PubMed.
- M. J. Mayoral, N. Bilbao and D. González-Rodríguez, ChemistryOpen, 2016, 5, 10–32 CrossRef CAS PubMed.
- R. L. Beingessner, Y. Fan and H. Fenniri, RSC Adv., 2016, 6, 75820–75838 RSC.
- D. Serrano-Molina, A. de Juan and D. González-Rodríguez, Chem. Rec., 2021, 21, 480–497 CrossRef CAS PubMed.
- C. C. Gregorio, A. Weber, M. Bondad, C. R. Pennise and V. M. Fowler, Nature, 1995, 377, 83–86 CrossRef CAS PubMed.
- B. Berger and P. W. Shor, J. Struct. Biol., 1998, 121, 285–294 CrossRef CAS PubMed.
- W. F. Marshall and J. L. Rosenbaum, J. Cell Biol., 2001, 155, 405–414 CrossRef CAS PubMed.
- N. C. Seeman, Nature, 2003, 421, 427–431 CrossRef PubMed.
- B. Adhikari, X. Lin, M. Yamauchi, H. Ouchi, K. Aratsu and S. Yagai, Chem. Commun., 2017, 53, 9663–9683 RSC.
- S. Yagai, Y. Kitamoto, S. Datta and B. Adhikari, Acc. Chem. Res., 2019, 52, 1325–1335 CrossRef CAS PubMed.
- P. Jonkheijm, A. Miura, M. Zdanowska, F. J. M. Hoeben, S. De Feyter, A. P. H. J. Schenning, F. C. De Schryver and E. W. Meijer, Angew. Chem., Int. Ed., 2004, 43, 74–78 CrossRef PubMed.
- J. T. Davis and G. P. Spada, Chem. Soc. Rev., 2007, 36, 296–313 RSC.
- J. T. Davis, Angew. Chem., Int. Ed., 2004, 43, 668–698 CrossRef CAS PubMed.
- S. Lena, S. Masiero, S. Pieraccini and G. P. Spada, Chem. – Eur. J., 2009, 15, 7792–7806 CrossRef CAS PubMed.
- G. M. Peters and J. T. Davis, Chem. Soc. Rev., 2016, 45, 3188–3206 RSC.
- J. E. Betancourt, M. Martín-Hidalgo, V. Gubala and J. M. Rivera, J. Am. Chem. Soc., 2009, 131, 3186–3188 CrossRef CAS PubMed.
- D. González-Rodríguez, J. L. J. van Dongen, M. Lutz, A. L. Spek, A. P. H. J. Schenning and E. W. Meijer, Nat. Chem., 2009, 1, 151–155 CrossRef PubMed.
- K. B. Sutyak, P. Y. Zavalij, M. L. Robinson and J. T. Davis, Chem. Commun., 2016, 52, 11112–11115 RSC.
- E. Fadaei, M. Martín-Arroyo, M. Tafazzoli and D. González-Rodríguez, Org. Lett., 2017, 19, 460–463 CrossRef CAS PubMed.
- M. Martín-Arroyo, A. del Prado, R. Chamorro, N. Bilbao and D. González-Rodríguez, Angew. Chem., Int. Ed., 2020, 59, 9041–9046 CrossRef PubMed.
- H. Fenniri, P. Mathivanan, K. L. Vidale, D. M. Sherman, K. Hallenga, K. V. Wood and J. G. Stowell, J. Am. Chem. Soc., 2001, 123, 3854–3855 CrossRef CAS PubMed.
- H. Fenniri, B.-L. Deng and A. E. Ribbe, J. Am. Chem. Soc., 2002, 124, 11064–11072 CrossRef CAS PubMed.
- J. G. Moralez, J. Raez, T. Yamazaki, R. K. Motkuri, A. Kovalenko and H. Fenniri, J. Am. Chem. Soc., 2005, 127, 8307–8309 CrossRef CAS PubMed.
- R. S. Johnson, T. Yamazaki, A. Kovalenko and H. Fenniri, J. Am. Chem. Soc., 2007, 129, 5735–5743 CrossRef CAS PubMed.
- G. Borzsonyi, A. Alsbaiee, R. L. Beingessner and H. Fenniri, J. Org. Chem., 2010, 75, 7233–7239 CrossRef CAS PubMed.
- G. Borzsonyi, R. L. Beingessner, T. Yamazaki, J.-Y. Cho, A. J. Myles, M. Malac, R. Egerton, M. Kawasaki, K. Ishizuka, A. Kovalenko and H. Fenniri, J. Am. Chem. Soc., 2010, 132, 15136–15139 CrossRef CAS PubMed.
- T. Yamazaki, H. Fenniri and A. Kovalenko, ChemPhysChem, 2010, 11, 361–367 CrossRef CAS PubMed.
- A. Marsh, M. Silvestri and J.-M. Lehn, Chem. Commun., 1996, 1527–1528 RSC.
- M. Mascal, N. M. Hext, R. Warmuth, M. H. Moore and J. P. Turkenburg, Angew. Chem., Int. Ed. Engl., 1996, 35, 2204–2206 CrossRef CAS.
- C. Igci, O. Karaman, Y. Fan, A. A. Gonzales, H. Fenniri and G. Gunbas, Sci. Rep., 2018, 8, 15949 CrossRef PubMed.
- A. del Prado, D. González-Rodríguez and Y.-L. Wu, ChemistryOpen, 2020, 9, 409–430 CrossRef CAS PubMed.
- M. J. Mayoral, C. Montoro-García and D. González-Rodríguez, in Comprehensive Supramolecular Chemistry II, ed. J. L. Atwood, Elsevier, Oxford, 2017, vol. 4, pp. 191–257 Search PubMed.
- J. Camacho-García, C. Montoro-García, A. M. López-Pérez, N. Bilbao, S. Romero-Pérez and D. González-Rodríguez, Org. Biomol. Chem., 2015, 13, 4506–4513 RSC.
- C. Montoro-García, J. Camacho-García, A. M. López-Pérez, N. Bilbao, S. Romero-Pérez, M. J. Mayoral and D. González-Rodríguez, Angew. Chem., Int. Ed., 2015, 54, 6780–6784 CrossRef PubMed.
- N. Bilbao, I. Destoop, S. De Feyter and D. González-Rodríguez, Angew. Chem., Int. Ed., 2016, 55, 659–663 CrossRef CAS PubMed.
- D. Serrano-Molina, C. Montoro-García, M. J. Mayoral, A. de Juan and D. González-Rodríguez, J. Am. Chem. Soc., 2022, 144, 5450–5460 CrossRef CAS PubMed.
- V. Vázquez-González, M. J. Mayoral, R. Chamorro, M. M. R. M. Hendrix, I. K. Voets and D. González-Rodríguez, J. Am. Chem. Soc., 2019, 141, 16432–16438 CrossRef PubMed.
- V. Vázquez-González, M. J. Mayoral, F. Aparicio, P. Martínez-Arjona and D. González Rodríguez, ChemPlusChem, 2021, 86, 1087–1096 CrossRef PubMed.
- C. Montoro-García, N. Bilbao, I. M. Tsagri, F. Zaccaria, M. J. Mayoral, C. Fonseca Guerra and D. González-Rodríguez, Chem. – Eur. J., 2018, 24, 11983–11991 CrossRef PubMed.
- C. Montoro-García, J. Camacho-García, A. M. López-Pérez, M. J. Mayoral, N. Bilbao and D. González-Rodríguez, Angew. Chem., Int. Ed., 2016, 55, 223–227 CrossRef PubMed.
- C. Montoro-García, M. J. Mayoral, R. Chamorro and D. González-Rodríguez, Angew. Chem., Int. Ed., 2017, 56, 15649–15653 CrossRef PubMed.
- M. González-Sánchez, M. J. Mayoral, V. Vázquez-González, M. Paloncýová, I. Sancho-Casado, F. Aparicio, A. de Juan, G. Longhi, P. Norman, M. Linares and D. González-Rodríguez, J. Am. Chem. Soc., 2023, 145, 17805–17818 CrossRef PubMed.
- M. González-Sánchez, M. J. Mayoral, F. Aparicio, V. Vázquez-González, I. Sancho-Casado, E. Anaya-Plaza and D. González-Rodríguez, Angew. Chem., Int. Ed., 2024, e202413321 Search PubMed.
- F. Aparicio, P. B. Chamorro, R. Chamorro, S. Casado and D. González-Rodríguez, Angew. Chem., Int. Ed., 2020, 59, 17091–17096 CrossRef CAS PubMed.
- P. B. Chamorro, F. Aparicio, R. Chamorro, N. Bilbao, S. Casado and D. González-Rodríguez, Org. Chem. Front., 2021, 8, 686–696 RSC.
- F. Aparicio, I. Sancho-Casado, P. B. Chamorro, M. Gonzalez-Sanchez, S. Pujals, V. Vega-Mayoral and D. Gonzalez-Rodriguez, Chem. – Eur. J., 2024, 30, e202402365 CrossRef CAS PubMed.
- M. A. Beuwer, M. F. Knopper, L. Albertazzi, D. van der Zwaag, W. G. Ellenbroek, E. W. Meijer, M. W. J. Prins and P. Zijlstra, Polym. Chem., 2016, 7, 7260–7268 RSC.
- I. Lopez-Martin, J. Veiga-Herrero, F. Aparicio and D. Gonzalez-Rodriguez, Chem. – Eur. J., 2023, 29, e202302279 CrossRef CAS PubMed.
- E. Orentas, C. J. Wallentin, K. E. Bergquist, M. Lund, E. Butkus and K. Warnmark, Angew. Chem., Int. Ed., 2011, 50, 2071–2074 CrossRef CAS PubMed.
- Q. Shi, K.-E. Bergquist, R. Huo, J. Li, M. Lund, R. Vácha, A. Sundin, E. Butkus, E. Orentas and K. Wärnmark, J. Am. Chem. Soc., 2013, 135, 15263–15268 CrossRef CAS PubMed.
- D. Rackauskaite, R. Gegevicius, Y. Matsuo, K. Warnmark and E. Orentas, Angew. Chem., Int. Ed., 2016, 55, 208–212 CrossRef CAS PubMed.
- Q. Shi, T. Javorskis, K.-E. Bergquist, A. Ulčinas, G. Niaura, I. Matulaitienė, E. Orentas and K. Wärnmark, Nat. Commun., 2017, 8, 14943 CrossRef PubMed.
- A. Neniškis, D. Račkauskaitė, Q. Shi, A. J. Robertson, A. Marsh, A. Ulčinas, R. Valiokas, S. P. Brown, K. Wärnmark and E. Orentas, Chem. – Eur. J., 2018, 24, 14028–14033 CrossRef PubMed.
- C. Bazzicalupi, A. Bianchi, E. García-España and E. Delgado-Pinar, Inorg. Chim. Acta, 2014, 417, 3–26 CrossRef CAS.
- B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022–2043 CrossRef CAS PubMed.
- J.-G. Jia and L.-M. Zheng, Coord. Chem. Rev., 2020, 403, 213083 CrossRef CAS.
- P. Thanasekaran, T.-T. Luo, C.-H. Lee and K.-L. Lu, J. Mater. Chem., 2011, 21, 13140–13149 RSC.
- S. Shin, S. Lim, Y. Kim, T. Kim, T.-L. Choi and M. Lee, J. Am. Chem. Soc., 2013, 135, 2156–2159 CrossRef CAS PubMed.
- T. Fukino, H. Joo, Y. Hisada, M. Obana, H. Yamagishi, T. Hikima, M. Takata, N. Fujita and T. Aida, Science, 2014, 344, 499 CrossRef CAS PubMed.
- H. Yamagishi, T. Fukino, D. Hashizume, T. Mori, Y. Inoue, T. Hikima, M. Takata and T. Aida, J. Am. Chem. Soc., 2015, 137, 7628–7631 CrossRef CAS PubMed.
- M. Obana, T. Fukino, T. Hikima and T. Aida, J. Am. Chem. Soc., 2016, 138, 9246–9250 CrossRef CAS PubMed.
- P. D. Frischmann, S. Guieu, R. Tabeshi and M. J. MacLachlan, J. Am. Chem. Soc., 2010, 132, 7668–7675 CrossRef CAS PubMed.
- Z. Chen, B. J. Sahli and M. J. MacLachlan, Inorg. Chem., 2017, 56, 5383–5391 CrossRef CAS PubMed.
- H. Zhao, A. M. S. D. Wijerathna, Q. Dong, Q. Bai, Z. Jiang, J. Yuan, J. Wang, M. Chen, M. Zirnheld, R. Li, D. Liu, P. Wang, Y. Zhang and Y. Li, Angew. Chem., Int. Ed., 2024, 63, e202318029 CrossRef CAS PubMed.
- T. G. Barclay, K. Constantopoulos and J. Matisons, Chem. Rev., 2014, 114, 10217–10291 CrossRef CAS PubMed.
- H.-J. Kim, Y.-H. Jeong, E. Lee and M. Lee, J. Am. Chem. Soc., 2009, 131, 17371–17375 CrossRef CAS PubMed.
- H.-J. Kim, S.-K. Kang, Y.-K. Lee, C. Seok, J.-K. Lee, W.-C. Zin and M. Lee, Angew. Chem., Int. Ed., 2010, 49, 8471–8475 CrossRef CAS PubMed.
- H.-J. Kim, F. Liu, J.-H. Ryu, S.-K. Kang, X. Zeng, G. Ungar, J.-K. Lee, W.-C. Zin and M. Lee, J. Am. Chem. Soc., 2012, 134, 13871–13880 CrossRef CAS PubMed.
- Z. Huang, S.-K. Kang, M. Banno, T. Yamaguchi, D. Lee, C. Seok, E. Yashima and M. Lee, Science, 2012, 337, 1521–1526 CrossRef CAS PubMed.
- S. Wu, Y. Li, S. Xie, C. Ma, J. Lim, J. Zhao, D. S. Kim, M. Yang, D. K. Yoon, M. Lee, S. O. Kim and Z. Huang, Angew. Chem., Int. Ed., 2017, 56, 11511–11514 CrossRef CAS PubMed.
- Y. Wang, Z. Huang, Y. Kim, Y. He and M. Lee, J. Am. Chem. Soc., 2014, 136, 16152–16155 CrossRef CAS PubMed.
- Y. Kim, H. Li, Y. He, X. Chen, X. Ma and M. Lee, Nat. Nanotechnol., 2017, 12, 551–556 CrossRef CAS PubMed.
- S. Bao, S. Wu, L. Huang, X. Xu, R. Xu, Y. Li, Y. Liang, M. Yang, D. K. Yoon, M. Lee and Z. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 31220–31226 CrossRef CAS PubMed.
- E. Cohen, H. Weissman, I. Pinkas, E. Shimoni, P. Rehak, P. Král and B. Rybtchinski, ACS Nano, 2018, 12, 317–326 CrossRef CAS PubMed.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- F. B. L. Cougnon, A. R. Stefankiewicz and S. Ulrich, Chem. Sci., 2024, 15, 879–895 RSC.
- S. Otto and J. N. H. Reek, Dynamic Combinatorial Chemistry, 2010, pp. 185–194 Search PubMed.
- A. Herrmann, Chem. Soc. Rev., 2014, 43, 1899–1933 RSC.
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Acc. Chem. Res., 2014, 47, 1575–1586 CrossRef CAS PubMed.
- T. Maeda, H. Otsuka and A. Takahara, Prog. Polym. Sci., 2009, 34, 581–604 CrossRef CAS.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- D. Zhao and J. S. Moore, J. Org. Chem., 2002, 67, 3548–3554 CrossRef CAS PubMed.
- I. Kim, A. Dhamija, I.-C. Hwang, H. Lee, Y. H. Ko and K. Kim, Chem. – Asian J., 2021, 16, 3209–3212 CrossRef CAS PubMed.
- M. J. Strauss, A. M. Evans, E. K. Roesner, R. J. Monsky, M. I. Bardot and W. R. Dichtel, Acc. Mater. Res., 2022, 3, 935–947 CrossRef CAS.
- C. Sun, M. Shen, A. D. Chavez, A. M. Evans, X. Liu, B. Harutyunyan, N. C. Flanders, M. C. Hersam, M. J. Bedzyk, M. Olvera de la Cruz and W. R. Dichtel, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8883 CrossRef CAS PubMed.
- S. Wang, A. D. Chavez, S. Thomas, H. Li, N. C. Flanders, C. Sun, M. J. Strauss, L. X. Chen, A. J. Markvoort, J.-L. Bredas and W. R. Dichtel, Chem. Mater., 2019, 31, 7104–7111 CrossRef CAS.
- M. J. Strauss, D. Asheghali, A. M. Evans, R. L. Li, A. D. Chavez, C. Sun, M. L. Becker and W. R. Dichtel, Angew. Chem., Int. Ed., 2019, 58, 14708–14714 CrossRef CAS PubMed.
- M. J. Strauss, M. Jia, A. M. Evans, I. Castano, R. L. Li, X. Aguilar-Enriquez, E. K. Roesner, J. L. Swartz, A. D. Chavez, A. E. Enciso, J. F. Stoddart, M. Rolandi and W. R. Dichtel, J. Am. Chem. Soc., 2021, 143, 8145–8153 CrossRef CAS PubMed.
- M. J. Strauss, I. Hwang, A. M. Evans, A. Natraj, X. Aguilar-Enriquez, I. Castano, E. K. Roesner, J. W. Choi and W. R. Dichtel, J. Am. Chem. Soc., 2021, 143, 17655–17665 CrossRef CAS PubMed.
- A. G. Orrillo and R. L. E. Furlan, Angew. Chem., Int. Ed., 2022, 61, e202201168 CrossRef CAS PubMed.
- V. Haridas, S. Sahu and A. R. Sapala, Chem. Commun., 2012, 48, 3821–3823 RSC.
- J. M. A. Carnall, C. A. Waudby, A. M. Belenguer, M. C. A. Stuart, J. J.-P. Peyralans and S. Otto, Science, 2010, 327, 1502–1506 CrossRef CAS PubMed.
- S. Otto, Acc. Chem. Res., 2022, 55, 145–155 CrossRef CAS PubMed.
- A. D. Chavez, B. J. Smith, M. K. Smith, P. A. Beaucage, B. H. Northrop and W. R. Dichtel, Chem. Mater., 2016, 28, 4884–4888 CrossRef CAS.
- R.-R. Liang, R.-H. A, S.-Q. Xu, Q.-Y. Qi and X. Zhao, J. Am. Chem. Soc., 2020, 142, 70–74 CrossRef CAS PubMed.
- K. Koner, S. Karak, S. Kandambeth, S. Karak, N. Thomas, L. Leanza, C. Perego, L. Pesce, R. Capelli, M. Moun, M. Bhakar, T. G. Ajithkumar, G. M. Pavan and R. Banerjee, Nat. Chem., 2022, 14, 507–514 CrossRef CAS PubMed.
- K. Dey, K. Koner, R. D. Mukhopadhyay, D. Shetty and R. Banerjee, Acc. Chem. Res., 2024, 57, 1839–1850 CrossRef CAS PubMed.
- C.-Y. Wu, S. Su, X. Zhang, R. Liu, B. Gong and Z.-L. Lu, Angew. Chem., Int. Ed., 2023, 62, e202303242 CrossRef CAS PubMed.
- Y.-H. Fu, Y.-F. Hu, T. Lin, G.-W. Zhuang, Y.-L. Wang, W.-X. Chen, Z.-T. Li and J.-L. Hou, Nat. Chem., 2024, 16, 1418–1426 CrossRef CAS PubMed.
- Y. Tian, Y. Guo, X. Dong, X. Wan, K.-H. Cheng, R. Chang, S. Li, X. Cao, Y.-T. Chan and A. C. H. Sue, Nat. Synth., 2023, 2, 395–402 CrossRef CAS.
- Y. Xu, M. D. Smith, M. F. Geer, P. J. Pellechia, J. C. Brown, A. C. Wibowo and L. S. Shimizu, J. Am. Chem. Soc., 2010, 132, 5334–5335 CrossRef CAS PubMed.
- T.-J. Hsu, F. W. Fowler and J. W. Lauher, J. Am. Chem. Soc., 2012, 134, 142–145 CrossRef CAS PubMed.
- S. Rondeau-Gagné, J. R. Néabo, M. Desroches, J. Larouche, J. Brisson and J.-F. Morin, J. Am. Chem. Soc., 2013, 135, 110–113 CrossRef PubMed.
- K. Bae, D. G. Lee, M. I. Khazi and J. M. Kim, Macromolecules, 2022, 55, 2882–2891 CrossRef CAS.
- J.-M. Heo, Y. Son, S. Han, H.-J. Ro, S. Jun, U. Kundapur, J. Noh and J.-M. Kim, Macromolecules, 2019, 52, 4405–4411 CrossRef CAS.
- M. Wehner and F. Würthner, Nat. Rev. Chem., 2020, 4, 38–53 CrossRef CAS.
- J. Matern, Y. Dorca, L. Sánchez and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 16730–16740 CrossRef CAS PubMed.
- W. Wagner, M. Wehner, V. Stepanenko and F. Würthner, J. Am. Chem. Soc., 2019, 141, 12044–12054 CrossRef CAS PubMed.
- A. Sarkar, R. Sasmal, C. Empereur-mot, D. Bochicchio, S. V. K. Kompella, K. Sharma, S. Dhiman, B. Sundaram, S. S. Agasti, G. M. Pavan and S. J. George, J. Am. Chem. Soc., 2020, 142, 7606–7617 CrossRef CAS PubMed.
- N. Singh, G. J. M. Formon, S. De Piccoli and T. M. Hermans, Adv. Mater., 2020, 32, 1906834 CrossRef CAS PubMed.
- N. Cissé and T. Kudernac, ChemSystemsChem, 2020, 2, e2000012 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |