
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmpere-level electroreduction of CO2 and CO†
Qian
Sun
 a,
Chen
Jia
a,
Haochen
Lu
a,
Mengmeng
Yang
a,
Ruirui
Liu
a,
Dan M.
Villamanca
b,
Yong
Zhao
*b and
Chuan
Zhao
*a
a,
Chen
Jia
a,
Haochen
Lu
a,
Mengmeng
Yang
a,
Ruirui
Liu
a,
Dan M.
Villamanca
b,
Yong
Zhao
*b and
Chuan
Zhao
*a
aSchool of Chemistry, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: chuan.zhao@unsw.edu.au
bGlobal Innovative Centre for Advanced Nanomaterials, School of Engineering, The University of Newcastle, Callaghan, NSW 2308, Australia. E-mail: yong.zhao@newcastle.edu.au
First published on 16th June 2025
Abstract
Electroreduction of carbon dioxide (CO2RR) and carbon monoxide (CORR) is promising to reduce the global carbon footprint and obtain high-value products. However, both reactions are limited by the intrinsically low activity of catalysts and mass transport of reactants at the catalyst/electrolyte interface. Recent progress has highlighted the need of rational catalyst design and mass transport engineering for improving the reaction kinetics and operating the CO2RR/CORR at current densities at ampere levels (>500 mA cm−2). This review introduces recent advances in the CO2RR/CORR at ampere-level current densities, especially the catalytic mechanisms and the principles for catalyst design and mass transport manipulation. The strategies for catalyst design including alloying and doping, single atom effects, regulating the morphology and structure, oxidation state control, and organic molecule functionalization are reviewed together with the mass transfer manipulation through electrode engineering and electrolyzer optimization. The challenges and perspectives are discussed for further industrial development in this field.
 Qian Sun | Qian Sun obtained her PhD degree in Chemistry from the University of New South Wales (UNSW) in 2023, under the supervision of Prof. Chuan Zhao. She then did postdoctoral research in Prof. Chuan Zhao's group, focusing on single atom catalysts for electroreduction of CO2 and CO (2023–2024). After that, she joined Prof. David Sinton's group as a postdoctoral fellow at the University of Toronto in Canada. Her current research is production of multi-carbon species from CO2 and CO electroreduction. |
 Chen Jia | Chen Jia received his PhD degree in Chemistry from the University of New South Wales (UNSW), Sydney, in 2021. He is currently a postdoctoral research fellow at UNSW, working under the supervision of Prof. Chuan Zhao. His research focuses on the design and synthesis of nanomaterials and single-atom catalysts for energy conversion. |
 Haochen Lu | Haochen Lu received his BSc and MSc degrees from Nanjing University of Science and Technology in 2017 and 2020. He is currently a PhD candidate in Prof. Chuan Zhao's group at the University of New South Wales in Sydney. His research interests focus on the electrocatalytic reduction of CO2. |
 Mengmeng Yang | Mengmeng Yang received her BSc degree from Qiqihar University in 2018 and her MSc degree from Beijing University of Chemical Technology in 2021. She is currently a PhD candidate in Prof. Chuan Zhao's group at the University of New South Wales. Her research interests focus on electrocatalytic CO2 reduction. |
 Yong Zhao | Yong Zhao is a Lecturer and DECRA Fellow in the School of Engineering at the University of Newcastle, Australia. His research focuses on electrochemical conversion of small molecules (e.g., CO2, O2, N2, and H2), with an emphasis on energy-efficient catalysts and systems. He recieved his PhD in Chemistry from the University of Wollongong in 2019 and conducted postdoctoral research at the University of New South Wales, the University of Sydney, and the University of Toronto until 2022. Prior to joining the University, he was a Research Scientist at the CSIRO Energy Centre from 2023 to 2025. |
 Chuan Zhao | Chuan Zhao is a professor at the School of Chemistry at the University of New South Wales (UNSW), Sydney. He is currently the Deputy Director of the Australian Research Council (ARC) Training Centre for the Global Hydrogen Economy, and the Deputy Research Chair and Flagship Program Director of the ARC Centre of Excellence on Green Electrochemical Transformation of Carbon Dioxide. He is interested in discovering novel electrochemical methodologies and nanomaterials for energy applications, including water splitting, hydrogen fuel cells, CO2 and N2 reduction, batteries, and sensors. |
1. Introduction
The carbon dioxide electroreduction reaction (CO2RR), powered by renewable electricity, is an attractive strategy to convert CO2 into valuable chemicals and fuels, thus mitigating global warming and relieving the strong dependence on traditional fossil fuels.1–9 Multicarbon C2+ products (e.g. ethylene, ethanol, acetic acid, and n-propanol), compared to C1 products (e.g. CO, formate, methane, and methanol), exhibit higher energy densities and economic value and are important feedstock for developing long-chain hydrocarbon fuels.10 Over the past decade, numerous efforts have been devoted to improving the selectivity of multi-carbon products by using highly alkaline electrolytes to suppress the hydrogen evolution and boost the yield of the target products.11,12 However, the alkaline CO2RR suffers from the carbonate formation issue, which results in depletion of CO2 and OH−, catalytic performance degradation,13,14 and high energy penalty for CO2 recovery from the generated carbonates.14 An alternative route for CO2-to-C2+ conversion is to reduce CO2 initially to CO and subsequently to C2+. As a carbonate-formation-free approach, the CO electroreduction reaction (CORR) operates stably in high-alkalinity electrolytes that favors C–C coupling kinetics.14 Moreover, the CO2RR and CORR share common reduction pathways.15 Exploring the CORR is also beneficial for further understanding of the mechanism of the CO2RR.Despite recent advances in CO2/CO upgrading to valuable fuels and feedstock, the reaction productivity is typically restricted by the poor activity of catalysts and mass transport issues. Most related studies are conducted in the lab-scale H-cells, which deliver low current densities (<50 mA cm−2) due to the low gas solubilities in aqueous electrolytes (CO2: ∼34 mM in H2O; CO: ∼1 mM in H2O),2,16–18 whereas industrial electrolyzers typically require current densities >200 mA cm−2.19 For practical applications, CO2/CO electrolysis must perform at ampere-level current densities (>0.5 A cm−2) with high energy efficiency to minimize operating and capital costs.20–26 This requires active catalysts for efficient CO2/CO reduction and electrode/electrolyzer engineering for improved mass transfer of the reactants, intermediates (e.g. *CO, *COH, *CHOH, and *CHO), and electrons.
Many effective methods have been devoted to optimizing catalysts, such as developing single atom catalysts,27 element doping,25 alloying,28 molecule functionalization,12 hydrophobicity manipulation,29etc. Mass transport is another critical factor. Normally, the application of gas diffusion electrodes (GDEs), consisting of gas diffusion layers (GDLs), microporous layers (MPLs) and catalyst layers, affords maximization of CO2/CO solubility and sufficient mass transport directly to the catalyst surface and further reduction at the gas–solid–liquid triple-phase boundaries.30 Optimization of GDEs should consider the key factors of high porosity, low electrical resistance, catalyst accessibility, chemical and mechanical stability, and scalability.31,32 Approaches proposed to facilitate mass transfer for the CO2RR/CORR include porous micro-structured GDE construction, wettability control, polymer engineering, quasi-two-phase interface modification, optimization of flow cells and MEA (gas channels, membranes), cell-stack deployment, etc.33–35
A significant challenge and an important topic of research in the field of CO2RR/CORR is how to enhance the current densities for the CO2RR/CORR to an ampere level for industrial applications.36–41 There have been many reviews focusing on catalyst design, electrolyzer design, and mechanisms in traditional H-cells and flow-cell electrolyzers, respectively.42–44 Nevertheless, few review articles to date have focused on a holistic consideration for boht catalysts and mass transport for realizing the CO2RR/CORR with ampere-level productivity. A review paper is therefore timely and will offer straightforward understanding and insights into the ampere-level CO2RR/CORR.
This review focuses on CO2/CO reduction with high selectivity in the ampere-level current density regime. First, we provide a brief overview of the CO2RR/CORR, followed by introduction of the reaction mechanism and the status of ampere-level CO2/CO electrolysis in terms of productivity and selectivity. Next, the key principles of designing catalysts and controlling mass transport to promote CO2/CO reduction at ultrahigh reaction rates are summarized. Finally, we highlight the challenges for the ampere-level CO2RR/CORR and offer our views on some important and promising research directions to address these challenges (Scheme 1).
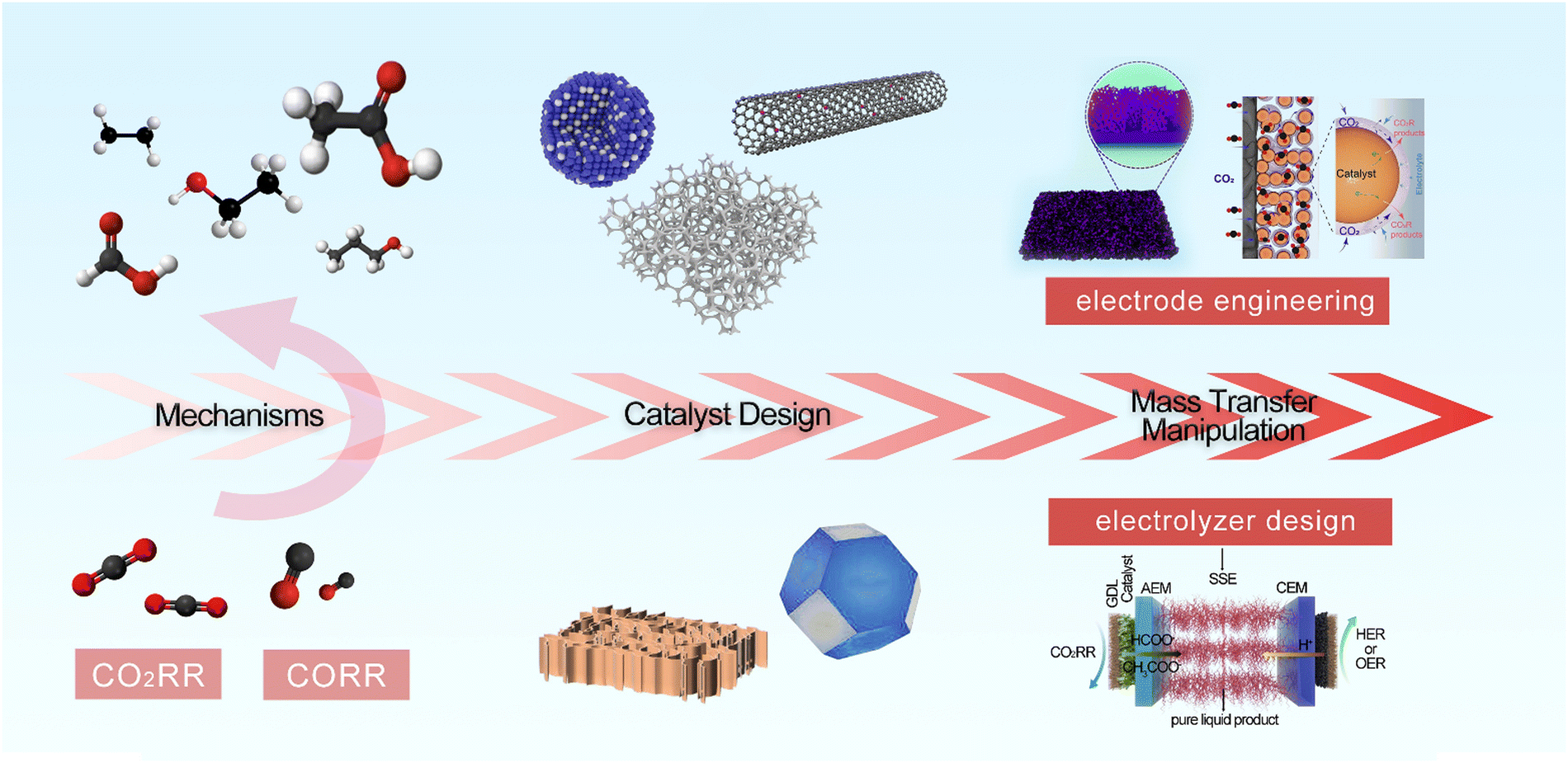 | ||
| Scheme 1 Schematic diagram of the main content for CO2RR/CORR at ampere-level current densities. Bi-ene-NW. Reproduced with permission.45 Copyright 2021, Royal Society of Chemistry. The mode for managing the CO2/H2O balance at the CO2RR reaction interface. Reproduced with permission.46 Copyright 2024, Springer Nature. | ||
2. CO2RR/CORR mechanism
2.1. CO2RR mechanism
CO2 can be converted into various products through different multi-electron/proton transfer pathways. Fig. 1a summarizes the faradaic efficiency (FE) of the reported CO2RR products at different production rates (current density), representing the selectivity of the reaction at ampere-level current. Notably, the state-of-the-art catalysts can facilitate C1 products (e.g., CO and formate) formation with >95% FE at high current density over 1 A cm−2,20,47,48 while the specific C2+ products (e.g., C2H4 and C2H5OH) have not achieved such a high FE (>80%) so far,49,50 although the FE of total C2+ products has reached over 80%.51 This originates from the adsorption of several CO2 molecules, stepwise conversion, and spatial positioning for C–C coupling.52 Propelling deep research on the CO2RR mechanisms favors designing more efficient catalysts and achieving more significant breakthroughs. | ||
| Fig. 1 CO2RR/CORR mechanism. (a) Summary of the CO2RR at ampere-level current (data from Table S1, ESI†). (b) Most possible C2+ pathways during the CO2RR. (c) Summary of the CORR at ampere-level current (data from Table S2, ESI†). | ||
Thermodynamically, CO2 molecules are difficult to activate due to the high dissociation energy of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (750 kJ mol−1).53 Kinetically, the CO2RR proceeds on practical electrodes for various products, powered by an overpotential relative to the standard potentials. According to the number of electrons transferred, CO2 can be converted to CO/formate, methanol, methane, and ethylene/ethanol via 2, 6, 8, and 12 electron transfer, respectively. As for the generation of the most common product CO, the reduction pathway involves either three or four kinetic steps. Normally, the first step of CO2 adsorption to form COOH* occurs via a proton-coupled electron transfer (PCET),54 or two uncoupled steps with CO2 conversion to COO* and further to COOH*.55 Then, the COOH* intermediate transforms into CO* and H2O over a H+/e− transfer, followed by CO desorption. Different from the traditional PCET pathway, H adsorption strength can be modulated to enable tuning of hydrogen radical (H˙) formation whilst simultaneously inhibiting the HER,56 thereby enhancing the CO2-to-CO reaction kinetics.
O bond (750 kJ mol−1).53 Kinetically, the CO2RR proceeds on practical electrodes for various products, powered by an overpotential relative to the standard potentials. According to the number of electrons transferred, CO2 can be converted to CO/formate, methanol, methane, and ethylene/ethanol via 2, 6, 8, and 12 electron transfer, respectively. As for the generation of the most common product CO, the reduction pathway involves either three or four kinetic steps. Normally, the first step of CO2 adsorption to form COOH* occurs via a proton-coupled electron transfer (PCET),54 or two uncoupled steps with CO2 conversion to COO* and further to COOH*.55 Then, the COOH* intermediate transforms into CO* and H2O over a H+/e− transfer, followed by CO desorption. Different from the traditional PCET pathway, H adsorption strength can be modulated to enable tuning of hydrogen radical (H˙) formation whilst simultaneously inhibiting the HER,56 thereby enhancing the CO2-to-CO reaction kinetics.
The formate formation is dependent on pH, which can take place at low pH (<3) or higher values (weak alkaline).57 The intermediate (HCOO*) is formed as CO2 molecules gain an electron, while formate is generated by combining a proton and another electron following three pathways. Pathway 1: a radical anion is formed by transferring a proton to CO2, where the oxygens are connected to the electrode surface. Thus, the protonation takes place on the carbon atom to produce the intermediate (HCOO), and further conversion into formate via a second electron transfer. Pathway 2: the OCHO intermediate is formed after transferring an electron to HCOO, which further protonates to formic acid. Pathway 3: the carbon atom in the radical anion is bound to the electrode surface, while the oxygen atom undergoes protonation to produce a COOH intermediate, which further combines protons and electrons to formic acid. Among these three pathways, pathway 1 is more favorable in product selection, due to its lower energy and overpotential requirements. Furthermore, the OCHO intermediate generation is revealed as the rate-determining step (RDS) for CO2 reduction,58 which further protonates to formate.
Carbene species (*CH2) derived from *CO is considered as the common intermediate for CH4 and C2H4 formation.59 *CH2 transforms into CH4 through double proton–electron transfer, while C2H4 is produced via the *CH2 dimerization or CO insertion in a Fischer–Tropsch-like step, which is also the pathway towards alcohols. Differently, the second C–O bond is broken at a late stage in the thermodynamically most favorable pathway to CH4.60 The methoxy intermediate (*HCO, *H2CO, and *H3CO) derived from *CO hydrogenation is converted to CH4 and *O and finally to H2O. C2H4 is produced by the HxCO species dimerization and subsequent deoxygenation. The binding ability to OH species is considered the determining factor for methanol production over methane.61 Specially, methanol is preferred on the catalysts with weakly bound OH which facilitates removing –OCH3 from the surface (*OCH3 + H+ + e− → * + CH3OH), while the ones with strong OH binding promote methane formation after breaking the *O–CH3 bond (*OCH3 + H+ + e− → *O + CH4).
As for the C2+ production, the *CH2 intermediate derived from *CO via PCET is for generating C2H6 and CH3COO− (Fig. 1b), which is further protonated to *CH3 and finally dimmerized to C2H6, whereas CH3COO− is produced by CO insertion into *CH2. Noteworthily, the RDS for C2H4, CH3CH2OH, and n-propanol production is the *CO dimerization step. The *CH2CHO intermediate derived from the *CO–CO dimer determines the selectivity of C2H4 and C2H5OH. Besides, n-propanol is formed by CO insertion into the stabilized C2 intermediates.62
Apart from the above intermediates, proton transfer also plays a crucial role in the CO2 electrolysis process.3 Typically, hydrogen transfer proceeds via the Eley–Rideal mechanism with proton-coupled electron transfer directly from solvent water,63 while hydrogenation can also happen via the Langmuir–Hinshelwood mechanism using surface-adsorbed *H. These two mechanisms are responsible for C–H bond and O–H bond formation, respectively, with the former one being critical to C2+ production and H2 suppression and the latter one being the dominant hydrogenation route. H+ reduction is negligible in neutral and alkaline electrolytes, while H2O reduction is pH independent. H+ serves as an additional proton source for the hydrogen evolution reaction (HER) in acid, whose reduction rate can be decided by the local pH near the cathode. Suppression of H+ mass transfer benefits constructing a local high-pH environment around the catalyst and restricting the H+ reduction rate. To facilitate the CO2RR in acid, mass transfer and electrode reactions can be modulated by various strategies, such as introducing alkali cations in electrolytes to inhibit H+ migration, suppressing H+ diffusion by catalyst surface decoration, regulating the electronic structure of catalysts, and controlling the interfacial H+ microenvironment via adding sulfonate-based electrolyte additives.64
2.2. CORR mechanism
Recently, the CORR has attracted much interest for the green synthesis of C2+ species, due to its stable operation in alkaline electrolytes, circumvention of carbonate formation, and higher reaction rates than those of the CO2RR.14,40,65,66Fig. 1c displays the FE of the reported CORR products at ampere-level current density. Obviously, most FEC2+ values approach 80–90%,41,65 but the specific C2+ product (acetate, C2H4) exhibits a FE less than 70%,28,40 while n-propanol exhibits only <37%.67 The CORR shares the common reduction pathways to the CO2RR after CO2 is converted to the absorbed *CO. The CO-to-methanol conversion is promoted by Rh1Cu4 alloy with homogeneous distribution of isolated Rh sites inside the Cu framework.68 The isolated Rh active sites enriched *H coverage on the catalyst and favored *CH2OH hydrogenation, resulting in efficient methanol production.69 The lowest energy pathway for CO-to-C2H5OH reveals that CO is firstly reduced at 0 and −0.40 V vs. RHE, followed by the intermediate protonation via proton and electron transfer and finally the product desorption. During which, the dehydration of OH groups makes them more active than the oxygen atoms in the carbonyl group. Sharing the same pathway as C2H5OH, the CO reduction to C2H4 is formed at the sixth H+/e− transfer step, which is inclined from C2H5OH by approximately 0.2 eV.The pH effects on the CORR reduction pathways were studied theoretically.70 CHO and COH are produced from the protonation of absorbed CO at acidic pH, which transforms into C1 species by removing OH or the CHOH path, whereas the oxygen atoms in carbonyl are hydrogenated to C1 products via CHO. In comparison, C2+ species are favored at neutral and high pH. The C–C coupling is facilitated at neutral pH through a novel CO–COH pathway, while the dimerization of adsorbed CO is dominantly responsible for generating C2/C3 products at high pH. Thus, modulating pH and applied potential play crucial roles in boosting C2+ selectivity. Protons in electrolytes also present critical effects on the surface adsorbed *H species and the production of C2+ species such as ethanol. However, the selectivity of C2+ oxygenates is challenged by the hindered coupling of *H and carbon-containing intermediates due to their inappropriate distributions in catalytic interfaces. Therefore, efficient water dissociation and proper *H distribution benefit the coupling with the carbon-containing species to generate C2+ oxygenates.71
The adsorbed CO initially dimerizes at neutral pH and then transforms into *(OH)CCOH, followed by protonation to *CCOH,72,73 which is further reduced to ethylene and alcohol via parallel pathways. Besides, a surface ketene *CCO derived from *(OH)CCOH at high pH is attacked by OH− to produce acetate.74 After the dimerization of adsorbed CO, the *CO–COH intermediate is formed, which further transforms into acetate.
Currently, most works are devoted to the CO2RR/CORR for liquid alcohols, due to their high energy density, transportation readiness and well-established utilization infrastructure, especially the monohydric alcohols (methanol, ethanol). Apart from this, the higher-valued diols (ethylene glycol (EG), (CH2OH)2) attract increasing attention. The densely arrayed Cu nanopyramids could retain two oxygen atoms for hydroxyl formation fed by CO2 or CO.75 The unique spatial-confinement structure facilitates C–C coupling by lowering the reaction barrier, leading to EG generation. This is achieved by maintaining oxygen to the *COH–CHO pathway to EG, and inhibiting C–O breaking in *CH2OH–CH2O to further hydrogenation to EG.
2.3. In situ/operando characterization
To clearly understand the CO2RR/CORR mechanism at ampere-level current density, in situ/operando investigations are powerful to provide valuable insights.76–78 X-ray absorption spectroscopy (XAS) can determine the electronic structure and coordination environment of the absorbed element, which is classified into the X-ray absorption near edge structure (XANES) and the extended X-ray absorption fine structure (EXAFS) regions. XANES mainly probes the oxidation states and electronic configurations of active species, while EXAFS sheds light on the interatomic distances and the coordination with neighbouring atoms. XAS demonstrates the activity–structure relationship, probes the active sites, and reveals the structural evolution of active sites during the CO2RR/CORR process. Very recently, operando XAS demonstrated that the active Cu+ sites are stable during the industrial-current–density CO2RR for C2+ species production in both acidic (77.0% at 700 mA cm−2) and alkaline (82.6% at 900 mA cm−2) electrolytes.77 The dendritic morphology of copper oxide catalysts is effective in simultaneously enriching K+, increasing local pH, and promoting *CO adsorption. This updated mechanism indicated that the sharp surface microarchitecture other than the oxidation state contributes to advancing the CO2RR industrialization. Attractively, in situ XAS favors detecting electron donation onto the support and reactants and proposes a transient poisoning mechanism on Ni SAC for CO production in consideration of impurities (NOx, and CN−).79 Besides, in situ XAS discovered that the F dopant with high electronegativity,80 among the Cu NPs supported on a series of carbon doped with a heteroatom with varying electronegativity (B < P < S < N < F), contributes to reduced electron density on Cu and facilitates C2+ formation, showing 82.5% FEC2+ at 400 mA cm−2 with 44 h stability, due to the decreased C–C coupling energy barrier by F doping.In situ and quasi in situ X-ray photoelectron spectroscopy (XPS) can characterize the changes in composition and chemical and electronic states of the catalysts during the CO2RR/CORR process. Because of the different measured depths of XPS and XAS, these techniques are normally complementary to one another for investigating the bulk material properties. For instance, quasi in situ XPS demonstrated the surface chemical state changes from initial Cu2+/Cu+ to Cu+/Cu0 at applied negative potentials and the asymmetrical sites of Cu+/Cu0 remaining stable during the CO2RR,81 while operando XAS indicated the gradual evolution of the initial Cu3P particles into Cu clusters upon increased negative potential under CO2RR conditions. This reconstructed structure exhibited abundant asymmetrical sites facilitating C–C coupling, and the Cu cluster–support interaction promoted *CHCOH hydrogenation towards ethanol at industrial current density.
In situ X-ray diffraction (XRD) sheds light on the crystal phase of catalysts in real time, benefiting the analysis of the catalyst stability and phase transition. However, in situ XRD can only characterize the crystalline samples, and the local sites of components are impossible to detect due to the low spatial resolution. Time-dependent, synchrotron-based XRD probed the structural evolution of SnO2 nanospheres during long-term CO2 electrolysis at −1.2 V vs. RHE,82 with transformation of SnO2 nanocrystals into metallic Sn (ca. 23 nm) and maintaining the crystallite size over 30 h. Besides, a minor residual oxide phase was detected, due to the reoxidation in air exposure between electrochemical and XRD tests. The metallic Sn worked as active sites during the long-term CO2-to-formate conversion. Moreover, in situ XRD of the CuAu1%Ag0.2%N-based GDE demonstrated the reduction of Cu3N to Cu0 during the CORR,83 evidenced by no detection of the Cu3N signal in in situ XRD. This transformation was also confirmed by the in situ Cu K-edge XANES with shift to low energies at applied CORR potentials.
Vibrational spectroscopy (UV/visible/IR photons) helps to elucidate the electrochemical performance by testing the molecular vibrations, such as the solvent, catalyst and reactants/intermediates/products, interpreting the catalytic mechanism. In addition, in situ vibrational spectroscopy also detects the transient changes of catalysts (redox transformation, oxide metallization). In situ UV-vis absorption spectroscopy is powerful to identify the reaction intermediates, such as in the reduction of Co(II) to Co(I) during the CO2RR,84 and the recovery of the molecular phthalocyanine species after the electrode washing with trifluoroethanol.85 Similar intermediate detection ability was also demonstrated by in situ XPS. Compared with them, in situ Raman and in situ IR are more widely applied for probing the real-time intermediates, because of the easier conversion of traditional Raman and FTIR analyses into in situ characterization techniques by rational design of the in situ cells, and more sensitive and accurate intermediate detection based on chemical bond vibration in the molecules.
In situ Raman helps to elucidate the catalytic mechanism by monitoring the catalyst structural evolution and the intermediate adsorption. When combined with in situ XRD, the catalyst structure evolution (In2O3 → In → In(OH)3) and variations in intermediate adsorption were discovered to be related to overpotential and selectivity changes during the CO2RR.86 The innovative small current pre-evolution helps to construct a stable and porous catalytic interface, resulting in 88.7% FEformate at 500 mA cm−2. In situ Raman not only tracked the structural evolution of the COC-NPs,77 including the reduction of COC-NPs with anion leaching and the incomplete reduction of Cu2+ in Cu dendrites, but also verified that the CuOx-dendrites promoted COatop formation for C–C coupling towards C2+ species. Similarly, high *CO coverage around the partially reduced Cu+/Cu0 active sites at the Ag/Cu2O interfaces was tracked by in situ Raman, explaining the high production of C2+ (FEC2+ 73.6%, jC2+ 478.4 mA cm−2) and C2H4 (FEC2H4 66%, jC2H4 429.1 mA cm−2).87 Additionally, in situ Raman indicated COatop on Cu changed from static high-frequency band CO to dynamic low-frequency band CO by incorporating Pd,88 achieving increased C2+ production (FEC2+ 66.2%, jC2+ 463.2 mA cm−2).
In situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATRSEIRAS) detects the adsorbed molecule–catalyst surface interactions, evaluates the existence of individual metal atoms and quantifies their percentage, in situ collects interfacial information, reveals the persistent alkalinity near electrode surfaces during the CO2RR, and probes the reduction pathways.89–91 For example, in situ ATR-IR demonstrated the HCOO− presence and absence of other intermediates on the InNCN catalyst during the CO2RR, elucidating the high formate production (FEformate 96%, jformate 400 mA cm−2).92 Besides, in situ FTIR also detected the enhanced *OCHO coverage on Sn–C/SiO2 for CO2-to-foramte conversion,93 pronounced COOH formation and CO2 consumption on CoPc-POP-c upon increasing overpotential for CO production,94 and showed a strong *CHO signal on Cu-BIF/NO3 for CO2-to-C1 and intensified *OCCHO and *CH3CH2O signals on Cu-BIF/Cl for CO2-to-C2+,10 respectively. Additionally, operando IR confirmed that increased K+ concentration intensified the adsorption of CO2 and *CO on Ni-NC,95 which changed the interfacial water structure (H2O to H3O+), accelerating CO production in strong acid.
Combination of in situ Raman and in situ IR helps to probe the key intermediates, revealing the catalytic mechanism.96,97 For example, in situ Raman and ATR-SEIRAS verified that the Ag–Cu–nitrogen-doped carbon tandem catalysis system effectively enhances the linear adsorption of *CO and H2O dissociation, promotes C–C coupling, and stabilizes *OCCOH for C2+ species production.98In situ Raman and in situ IR indicated that Cu NPs supported on electron-donating amine group modified single-walled carbon nanotubes (SWCNTs) presented enhanced *CHO coverage towards C–C coupling,99 while Cu NPs on electron-withdrawing cyano modified SWCNTs preferred producing C1 species. Besides, in situ electrochemical attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) and in situ Raman confirmed the *CO stabilization and promoted C–C coupling by C–Cu2O NPs,100 which contributed to CO2-to-C2+ conversion with a high FEC2+ (76.2% at 800 mA cm−2). To understand the relationship between the surface microenvironment and the CO2RR performance, in situ surface-enhanced Raman spectroscopy (SERS) and in situ ATRSEIRAS were deployed to detect the key intermediates and molecular structures of functional groups and reveal the underlying mechanism.101 The results demonstrated that the *CO coverage is considerably higher on the Cu nanorod developed by isopropanol (Cu-NRIPA) than that by dimethyl sulfoxide (Cu-NRDMSO) and EAC (Cu-NREAC), and the enhanced C–C coupling by Cu-NRIPA was confirmed by the intense IR signal of *COOH and *OCCOH intermediates. Moreover, the Nafion ionomer exhibits various aggregation behaviours in solvents with different dielectric constants (ε), and increasingly aggregates under smaller ε, posing the *CO-incompatible –SO3H groups away from the Cu surface, alongside the aggregated hydrophobic –(CF2)n– chains. This arrangement enhances *CO adsorption and promotes the HER, suggesting that the CO2RR product distribution can be controlled by adjusting the dispersion solvent. For example, changing low-ε DMSO into moderate-ε IPA contributed to increasing FEC2+ from 67.5% to 90.5% at 800 mA cm−2 on Cu nanosheets.
In situ electron spin resonance (ESR) derived from magnetic resonance mainly detects the unpaired electron transitions under an applied magnetic field, and is appropriate to identify the existence of unpaired electrons or single electrons in molecular orbitals.102 Radicals generated on a catalyst surface can be monitored by in situ ESR, providing insights into the reaction pathway and catalytic mechanism.
Apart from the structural information obtained from the above-mentioned techniques, operando electrochemical liquid-cell scanning transmission electron microscopy (EC-STEM) demonstrated the structural evolution of Cu nanocatalysts into active metallic Cu nanograins for the CO2RR,103 while operando 4D STEM in liquids,104 with co-existing H2 bubbles formed during the CO2RR to create a thin liquid layer to improve spatial resolution for nanoscale dynamic evolution of catalysts, revealed the complex structure of active polycrystalline metallic Cu nanograins at solid/liquid interfaces. In situ environmental TEM (ETEM) was powerful to track the formation of a Bi–N–C SAC with Bi–N4 sites on porous carbon networks,105 with evolution from Bi nanoparticles to single atoms during the CO2RR.
In situ mass spectrometry (MS) can monitor the catalytic products in real time, which helps to determine the onset potential and the product species at various potentials. This strategy has been devoted to detecting gaseous hydrocarbon products of the CO2RR,106 with utilization of pervaporation to separate and collect products continuously. In more details, according to the potential sweep from 0 to −2 V vs. SHE and the intensities of the different fragments, the products of H2, CH4, and C2H4 are recorded at a mass to charge ratio of 2, 15, and 26, respectively. Their corresponding onset potential is ∼−1.12 V, −1.64 V, and −1.53 V vs. SHE, respectively, indicating a much lower overpotential of hydrogen evolution products than the other CO2RR products. In this work, a combination of an electrochemical scanning flow cell with an online mass spectrometer together with a gas purging system was designed to enable analyzing the volatile reaction products to screen the electrochemical reactions, which accelerates the catalyst material research and optimal condition determination for fabricating practical devices. However, analyzing the liquid products in real time is complex due to the ion suppression by the non-volatile salts in electrolytes. To successfully detect the liquid products, the online electrochemical mass spectrometry method (EC/MS) was deployed to investigate the CO2 reduction in sulfuric and perchloric acid on polycrystalline Cu.107 Apart from identifying gas (CH4, C2H4) and highly volatile product (CH3OH, HCHO) formation, EC/MS also demonstrates that the formation of methane and formaldehyde is kinetically favored than that of ethene and alcohol in both electrolytes. The absorbed anion from the electrolyte determines the potential at which HCHO generates. The CO2RR/CORR proceeds via an electrocatalytic hydrogenation process. Various products (H2, CH4, C2H4, CH3OH, and C2H5OH) can be detected within one minute, but there are challenges because of the difficulty of delineating all the CO2RR products due to the ionized and broken-down samples, and the product quantification issue by the low and/or ill-defined product collection. Despite these problems, a novel flow cell was designed to extract gas and liquid through a membrane,108 achieving quantitation of H2, C2H4, and propanol. This novel differential electrochemical mass spectrometer (DEMS) cell demonstrated CO2RR on polycrystalline copper. The real-time quantification of products is determined by the function of the applied potential during linear sweep voltammetry within 1 h. Moreover, some other applications involving MS such as online inductively coupled mass spectrometry (ICP–MS) have demonstrated significant potential in investigating the catalyst degradation, validating the atomic elemental compositions of catalysts, and elucidating the effects of impurities, while synchrotron-radiation-based vacuum ultraviolet photoionization mass spectrometry (SVUV-PIMS) plays a crucial role in resolving the dynamic interfacial species evolution during the CO2RR on Cu. In situ MS not only directly tracks the generation of gas/liquid intermediates and final products in real time, but can also be used to observe and quantify the local reaction environment.
Therefore, in situ characterization benefits identifying the intermediates/active sites/reaction pathways, studying the reaction environment effects, and probing the catalyst evolution, which shed light on the CO2RR/CORR mechanism. In situ XAS/XRD/XPS monitors the element structural information, in situ UV/Raman/IR identifies the intermediates/functional groups/reduction routines, in situ MS focuses on determining the intermediates/final products, while in situ TEM tracks the morphological evolution of catalysts and the distribution of single atoms. Each technique exhibits advantages and disadvantages (Table 1), based on which, combining multiple in situ techniques promotes understanding the CO2RR/CORR mechanism and enriching the structure database for machine learning, guiding to design more efficient catalysts for practical applications.
| Techniques | Functions | Sample requirements | Measurement conditions | Advantages | Limitations | Ref. |
|---|---|---|---|---|---|---|
| XAS | Oxidation state | Gas, liquid and solid samples | X-ray beam; | Element specificity | Average information on local structures | 77 and 103 |
| Electronic structure | A custom flow cell | Sensitivity | Difficult to analyze complex systems and light elements | |||
| Spatial distribution | Electrochemical workstation | Not limited by the sample state | ||||
| Coordination environment | Controlled atmosphere, temperature and pressure | |||||
| XPS | Catalyst surface composition | Solid samples | Ultrahigh vacuum | Surface sensitivity | Hard to uncover the inner structural changes | 109 |
| Chemical states | Controlled temperature | Excellent quantitative accuracy | Limited spatial resolution | |||
| Almost all elements (except H, He). | Limited time resolution | |||||
| Pressure gap | ||||||
| XRD | Crystal structure (phase composition, lattice parameters, preferred orientation) | Solid samples | Controlled atmosphere, temperature and pressure | Direct determination of size, shape, and orientation of the unit cell | Low spatial resolution | 82 |
| Compatible with high temperature and high pressure | Hard to detecting amorphous/weakly crystalline samples | |||||
| UV | Light absorption capabilities | Solid or liquid samples | Controlled atmosphere, temperature and pressure | Non-destructive sampling | Low accuracy | 84 |
| Electronic transitions (bandgap, excitons) | Simplify experimental process | Light source impact | ||||
| Organic species or reactive radical | Controlled-temperature detection | |||||
| Raman | Identification of surface structural changes and adsorbed intermediates | Solid or liquid samples | Electrochemical workstation | Non-destructive sampling | Spectroscopic limits | 109 and 110 |
| An EC-RS cell | Not interfered by water | Hard to direct quantitative analysis | ||||
| Controlled atmosphere, temperature and pressure | The high-speed acquisition | Limited intensity and spatial resolution | ||||
| Wide spectra region | ||||||
| IR | Functional groups; | Solid samples | Electrochemical workstation | Low cost, facile optical design and operation | Limited lR transmission; Electrolyte interference | 109 |
| Monitor adsorption and reaction processes | Three electrodes cell | High sensitivity | Limited absorption range. | |||
| Infrared spectrophotometer with a built-in mercury cadmium telluride (MCT) detector | Fast characterization. | |||||
| Controlled atmosphere, temperature and pressure | ||||||
| ESR | Detect unpaired electrons (or single electrons) in molecular orbitals | Solid samples | Controlled atmosphere, temperature and pressure | Non-destructive | Low resolution | |
| Specific detection of unpaired electrons | Difficulty of quantitative analysis | |||||
| TEM | Morphology changes of catalyst | Solid samples (<100 nm) | Ultrahigh vacuum | Real-time high-resolution imaging | The electron beam may damage the sample | 103 and 104 |
| Distribution of single atoms | Controlled temperature | Atomic resolution | Strict operating environment and sample preparation | |||
| MS | Qualitatively and quantitatively determine the product composition | Gas or liquid samples | Ultrahigh vacuum | High sensitivity | Hard to detect liquid products in real-time | 106–108 |
| Controlled temperature | Real-time monitoring of reaction products |
3. Principles of catalyst design for the CO2RR/CORR at ampere-level current
Some works reported remarkable CO2RR/CORR at real ampere-level current density (>1 A cm−2, Fig. 1a and c). Such high reaction rates are technically significant and promising for practical applications. Herein, in-depth investigation into CO2RR/CORR beyond 1 A cm−2 is discussed. Two aspects for achieving high current density should be considered. (1) Catalyst design – the scientific principles; (2) mass transfer management – technically.Catalyst design for CO2-to-CO conversion beyond 1 A cm−2 can be achieved by modulating the d-band of a metal center for efficient *COOH adsorption and *CO desorption, especially using square-pyramidal Ni–N5,47 NiN4–O sites with an axial oxygen,111 and Hg-CoTPP combined with N-doped graphene.112 The Ni SAC derived from Ni-doped ZIF-8 exhibits abundant defective sites and a mesoporous structure for CO2 adsorption and mass transfer, affording 96% FECO and 1.06 A cm−2.113 While Ni dual-atom sites facilitate hydroxyl adsorption to generate electron-rich active centers,48 resulting in a moderate reaction barrier for *COOH formation and *CO desorption. This further results in over 99% FECO, 77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 h−1 turnover frequency and jCO approaching ∼1 A cm−2. Other strategies to achieve CO2-to-CO conversion beyond 1 A cm−2 include applying external magnetic field to promote CO2 adsorption at Ni sites and suppress the HER,114 depositing CoPc crystals on carbon paper to enhance surface charge transfer,115 and introducing Ru atoms on the Ce0.8Sm0.2O2−δ surface to alternate the catalyst electronic structure for achieving favorable oxygen vacancy formation and improved CO2 adsorption and activation.116 While modifying the Cu(200) nanocube surface by Cs atoms provides an effective strategy for ampere-level CO2-to-ethanol conversion,117 attributed to the stabilized C–O bond in oxygenate intermediates towards ethanol.
500 h−1 turnover frequency and jCO approaching ∼1 A cm−2. Other strategies to achieve CO2-to-CO conversion beyond 1 A cm−2 include applying external magnetic field to promote CO2 adsorption at Ni sites and suppress the HER,114 depositing CoPc crystals on carbon paper to enhance surface charge transfer,115 and introducing Ru atoms on the Ce0.8Sm0.2O2−δ surface to alternate the catalyst electronic structure for achieving favorable oxygen vacancy formation and improved CO2 adsorption and activation.116 While modifying the Cu(200) nanocube surface by Cs atoms provides an effective strategy for ampere-level CO2-to-ethanol conversion,117 attributed to the stabilized C–O bond in oxygenate intermediates towards ethanol.
To promote *CO intermediate and *H coupling while minimizing side reactions (C–C coupling, H–H coupling) for CO2-to-CH4 at high current density,118 Cu-based catalysts comprising a Cu–N coordination polymer and a CuO component were developed which can manage the key intermediates (*CO, *H). The Cu–N coordination polymer exhibited increased Cu–Cu distance favoring *CO hydrogenation over the dimerization, while the CuO component afforded sufficient *H supply, resulting in a fast CH4 formation rate (3.14 mmol cm−2 h−1) and 51.7% FECH4 at 1300 mA cm−2.
Doping (Rh doped Cu,119 S–Li co-doped Sn,120 Eu-doped CuOx,121 S-doped BiS,122 F-doped Cu,123 Pb1Cu single atom ally (SAA),124 Mo1Cu SAA,125 and SAA-Zn1Bi126) and alloying (Cu6Sn5,127 Cu9Ga4128) indicated high activity and selectivity for the CO2RR beyond 1 A cm−2. Among which, the Rh dopant exhibits a stronger oxygen affinity than Cu and facilitates *CH2CHO adsorption and weakens the C–O bond, favoring ethylene production, Au dopants with weaker oxygen affinity than Cu promoted ethanol production by the C–O bond stabilization, while Ru with too strong oxygen affinity suffers from slow *O removal from the dopant site. Li dopants incur electron localization and lattice strains on Sn, which boosted CO2 conversion to formate with enhanced activity and selectivity. The introduced Eu to Cu, compared to other lanthanide metals, undergoes Eu2+ reduction from Eu3+, which prevents the nanoparticle agglomeration, achieving ∼80% FEC2+ at 1.25 A cm−2. Apart from the metal dopants, non-metal dopants such as S and F effectively modulated the electronic structure of catalysts (Bi, Cu), promoted water activation and formation of the key intermediates (HCOO*, CHO) for formate and further C–C coupling towards C2+ species, respectively. Different from the promoted formation of HCOO* by Pb1Cu and the moderate *OCHO adsorption by SAA-Zn1Bi for formate, Mo1Cu achieved atom-scale cascade catalysis for CO2-to-C2+ conversion with remarkable FEC2+ (86.4%) and maximum jC2+ (1.33 A cm−2). Mo sites facilitated water dissociation to *H, the Cu sites (Cu0) far from the Mo atom promoted CO2 conversion to CO, while the adjacent Cu sites (Cu&+) near the Mo atom captured CO and *H to enhance CO conversion and C–C coupling. As for the Cu6Sn5 and Cu9Ga4 alloys, the former one exhibits strong *OCHO affinity and weak *H binding, delivering 91% FEformicacid at 1.2 A cm−2 in acid, while the latter one presents an elongated Cu–Cu distance for simultaneous reduced *CO repulsion and increased *CO coverage, which promoted C–C coupling towards C2+ species with maximum jC2+ (1.27 A cm−2) and 71% FEC2+.
Alternation of the catalyst particle size or structure resulted in CO2RR beyond 1 A cm−2. By solely controlling the Cu nanocluster size from 200 to 0.5 nm, the selectivity shifted from ethylene to methane,129 approaching 85% FECH4 and maximum jCH4 (1.2 A cm−2). This originated from the dominant Cu(111) facets at such extremely small size. While other works on lattice-distorted Bi,130 highly exposed Bi(110),24 and tensile strain engineering on non-defective Bi sites131 or Cu(100),132 and 5 nm Cu quantum dots with dominant Cu(100),133 achieved outstanding CO2RR with partial current density over 1 A cm−2, due to the structure modulation effects on the intermediates of *OCHO for formate, and the enhanced *CO coverage for C–C/C–CHO coupling towards C2+/C2H4, respectively. Interestingly, the in situ reconstructed Bi-based catalysts20,23,25 optimized *OCHO adsorption and further conversion to *HCOOH towards formate, resulting in remarkable jformate over 1 A cm−2, while the reconstructed CuO regulated by carbonate shell presents Cu(0) generation with abundant grain boundaries and small particles,134 which stabilized *CO and facilitated C–C coupling, leading to 82.8 ± 2.2% FEC2+ at 2.0 A cm−2.
Tandem catalysts (Cu/Fe–N–C s-GDE, Cu/Ag s-GDE,135 CuO/Ni SAC,51 Cu2O/Al2O3,87 and CuO/AgIO3136) exhibited good control over the *CO intermediate and promoted C–C coupling at ampere-level current density, where the CO-selective catalysts enriched local CO coverage and enabled rapid consumption of the in situ generated CO, subsequently, C2+ species formation was promoted on the Cu-based catalysts.
Surface modification of catalysts/electrodes by an indigo-based polymer (Id),137 polymeric ionic liquids (PILs),138 alkaline ionic liquids (AILs),139 long alkyl chain (toluene),49 sodium citrate,140 and tetrabutylammonium cations (TBA+)141 is highly active and selective for CO2/CO electrolysis beyond 1 A cm−2, attributed to the optimized interfacial CO2 binding affinity and accelerated formation of *CO2− and *COOH by Id, the alternated H+ transfer, enriched K+, and promoted C–C coupling by the PIL adlayer, the multi-roles of AILs (CO2 accumulator/activator, intermediate stabilizer, and CO dimerization promoter), the remained and enriched CO2 but blocked water transfer by toluene, Cu+ stabilization and promoted *CO hydrogenation to *CHO by citrate anion, and the enhanced CORR activity and improved ethylene production by TBA+, respectively.
Efficient mass transfer is a must for the CO2RR/CORR at ultra-high current density (>1 A cm−2). This can be achieved by a catalyst:ionomer bulk heterojunction to decouple gas, ion, and electron transport,12 a GDE with a Cu-based ultrathin superhydrophobic macroporous layer142 and a porous organic cage additive for highly enhanced CO2 diffusion,143 as well as the MgAl LDH nanosheet ‘house-of-cards’ scaffold to disperse CuO-derived Cu,39 and a GDE with a mesopore-rich hydrophobic copper catalyst layer for fast CO transfer.144 Moreover, Cu/Ag/Ni/Bi/Zn-based hollow fiber penetration electrodes (HFPEs), characteristic of a unique micron-scale hollow finer structure, oriented mass transfer from the inside to outside of the HFPEs, and construction of stable tri-phase interfaces, facilitating CO2/CO activation and favoring key intermediate formation, are effective in promoting desorption of adsorbed species at ultra-high current densities over 1 A cm−2, heightening the CO2RR/CORR and suppressing the HER simultaneously. As a result, ultra-high partial current density (>1 A cm−2) was achieved, together with outstanding product selectivity145–147 and stability (>100 h).148
Furthermore, K+ accumulation can be achieved by Cu nanoneedles,149,150 which enables highly efficient CO2/CO electrolysis.
At the device/system level, the PiperION membrane with high carbonate conductance demonstrates record high activity and selectivity for industrial CO2-to-CO conversion in a tailed MEA,151 while the utilization of a robust and efficient catalyst, stable three-phase interface and durable membrane afforded producing formic acid over 5200 h in the CO2RR–hydrogen oxidation coupling system.152 Optimization of the components (oxide-derived Cu, carbon-based GDE, and their assembly) demonstrates ultra-high-rate CO2 electrolysis.153 Another significant factor of CO2/CO coverage,154,155 controlled by CO pressure, can also be tuned for ultra-high rate CO2RR/CORR.
Therefore, the key to enable the CO2RR/CORR at ampere-level current is to develop efficient catalysts with improved activity and selectivity,156 which can be realized by increasing the number of active sites and the intrinsic activity of each active site.157 Another key to ampere-level CO2/CO electrolysis is the mass transfer manipulation. Representative principles for catalyst design are summarized in this section, including alloying and doping, single atom effects, regulation of the morphology and structure, oxidation state control, and organic molecule modification, while mass transfer will be discussed in the next section.
3.1. Alloying and doping
Bimetallic catalysts are effective in breaking the scaling relationship during the CO2RR/CORR process (Fig. 2a), thereby stabilizing the intermediate and decreasing the overpotential and further improving the selectivity (Fig. 2c). For comparison, the pure Cu-based catalysts for CO2/CO electrolysis at ampere-level are summarized (Fig. 2b). With CO2 feed, pure Cu catalysts presented a FEC2+ ranging from 40% to 90.9% with more accumulation at 70% to 80%, lower than those of the alloys or doped Cu (80% to 94%, Fig. 2b). Similar trends were detected for C2H4 (32% to 67% vs. 60% to 70%) and ethanol (37% to 44% vs. 45% to 79%). Both pure Cu catalysts and alloys/doped Cu contributed to high C1 species production (CH4, formate) with FECH4 at 57% to 85% and FEformate over 90% to unity, respectively. CO-to-C2+ conversion by pure Cu catalysts exhibited FEC2+ at 75% to 93%, inferior to those of the alloys/doped Cu (80% to 96%). Besides, pure Cu catalysts facilitated acetate production (43% to 68%), while the alloys/doped Cu exhibits better capability of producing alcohols (ethanol, propanol, and C2+ alcohols) beyond acetate. Therefore, pure Cu-based catalysts are relatively difficult to achieve high activity and selectivity to C2+ species, due to their low adsorption ability to the key intermediates and the non-specific selective CO2RR/CORR pathways. This can be addressed by doping/alloying to alternate the electronic circumstance of Cu-based catalysts. Normally, the dopants include non-metal elements (C, N, O, F,123,158 P, S) and metal elements (Na, Mg, K,159 Sc,160 Mn, Co,161 Ni, Cu,162 Zn, Ga, Nb, Ru, Pd, Ag,110,144,163 In, Sb, Te, Cs,117 La,164 Sm,121 Eu, Gd, Au, Pb). The non-metal doping plays crucial roles in manipulating the electronic structure of active metal species and thus enhancing the electrochemical performance.165,166 For example, among a series of heteroatom-engineered Cu (X–Cu, X = N, P, S, O),38 N–Cu demonstrated the optimal CO2-to-C2+ performance with 73.7% FEC2+ and 909 mA cm−2jC2+, due to its enhanced *CO coverage, favorable *CO adsorption, and much slower local H proton consumption than Cu. S doping into Bi modulates the HCOO* formation towards formate,122 achieving ∼95% FEformate at 2000 mA cm−2 in both alkaline and acidic flow cells, while S doping into Cu167 and Sn168 enhances the *OCHO coverage and promotes CO2-to-HCOOH at industrial reaction rates, contributing to 92% FEformate with 321 mA cm−2jformate and 92.15% FEformate with 730 mA cm−2jformate, respectively. Non-metal (S) and metal (Na, Ag) co-doped catalysts (Bi(110)–S–Na,24 Ag–In–S169) illustrated outstanding formate production, due to the co-doping induced improved Bi–Bi metallic bonds, stabilized *OCHO and promoted water dissociation to H*, as well as the decorated interface strain for stabilizing the key intermediate and accelerating charge transfer. | ||
| Fig. 2 Alloying and doping effects. (a) Summary of reported alloying and doping for ampere-level CO2RR/CORR. (b) Summary of pure Cu-based materials for CO2RR/CORR at ampere current density (data from Tables S1 and S2, ESI†). (c) Summary of reported FE by alloying and doping (data from Tables S1 and S2, ESI†). (d) The proposed reaction mechanism for CO2RR over TeBi NTs. The purple and brown balls represent Bi and Te, respectively. Reproduced with permission.170 Copyright 2024, John Wiley and Sons. | ||
Incorporating metal dopants into a second metal such as Cu–Bi,25,171 Sn–Bi,172 Pb–Cu,124 Zn–Bi,126 Sb–Bi,172 Cu–Sb/Pd,173 CuSb,174 Co1Cu,175 Cu–Au,176 and Cu–Sn,127 are highly selective to CO2-to-C1 conversions. The interaction effects in the alloys Cu–Bi,25 Sn–Bi,172 Zn–Bi,126 and Sb–Bi172 selectively stabilize *OCHO towards formate, while Pb–Cu124 modulates the first protonation step of the CO2RR and facilitates formate formation via HCOO* rather than COOH*. Apart from which, the unique high-curvature Te–Bi nanotips incur enhanced electric field to steer the *HCOOH formation (Fig. 2d),170 thus decreasing the energy barrier for *OCHO and *HCOOH and affording high formate selectivity at low overpotential. As for CO2-to-CH4 conversion, the incorporated Cu single atom into Cu enhances water activation and dissociation,175 reducing the energy barrier for *CO hydrogenation. The modulated *CO adsorption configuration exhibited stronger bridge-binding, benefiting CH4 production over the C–C coupling or CO desorption pathways.
The alloy effects on C2+ production are more complicated, either by Ag–Cu,177,178 Cu–Pd,88 Cu–Ag/Au,179 Cu–Sn,180,181 Cu–Ga,22,128 Cu–Mg,182,183 Gd–Cu,184 Cu–Zn,185 and Mo–Cu125 from the CO2RR, or by Cu–Sn,186 Cu–Cd,187 Cu–Au,1 Cu–Ag,67,188 Cu–Zn,189 and Cu–Pd28 from the CORR. The introduced metals into the alloys contribute to enriching the *CO coverage and promoting C–C coupling. For instance, the Ag–Cu single atom alloy (SAA) enhances the *CO adsorption energy on Cu sites,177 because of the compressive strain between the Cu atom and the adjacent Ag atom, and C–C coupling is promoted to produce C2+ species with high FEC2+ (94 ± 4%) under ∼720 mA cm−2 at −0.65 V in an alkaline flow cell. While the introduced Mo sites promote water dissociation to *H,125 the active Cu sites (Cu0) far from the Mo atom activate CO2 into CO, and the adjacent Cu sites (Cu&+) near the Mo atom capture CO and *H, thus promoting C–C coupling. The Mo–Cu single atom alloy demonstrated remarkable FEC2+ (86.4% at 800 mA cm−2) and maximum jC2+ (1330 mA cm−2).
The p–d orbital hybridization works as well in promoting C2+ production. The characteristic Ga–Cu interaction enhances the active site concentration and provides strong binding to the *CO intermediate and facilitates C–C coupling where a high FEC2+ (81.5%) at 900 mA cm−2 and −1.07 V vs. RHE were achieved.22 This strategy can be extended to other p-block metal-doped Cu catalysts (CuAl, CuGe). Similarly, a Cu–Ga atomic alloy with both active (Cu(100), Cu(111)) and inert (Ga) *CO binding sites for locally enriched *CO coverage and reduced *CO repulsion was developed,128 which delivered a peak jC2+ (1207 mA cm−2), high FEC2+ (71%), and high-power capability (∼200 W).
The product distribution of the CO2RR can be modulated by varying the doping amount of the alien element.88 For example, a small amount of Pd (0.25% and 0.5% Pd) doping to Cu facilitates C2+ production, while a higher Pd content (1–5%) leads to a 3–5-fold increase in FECO. Doping Sn atoms to Cu has been confirmed effective in compressing Cu atoms and enriching *CO on Cu sites. Low-entropy state CuSn alloy outperforms high-entropy state CuSn alloys (Cu6Sn5) with enhanced *CO adsorption capability and facilitates CO2 reduction to ethanol.180 Cu3Sn exhibits substantially enhanced adsorption of the key intermediates (*CO and *CHCHOH) for ethanol formation, thus enabling high FEethanol (>40%) at industrial current (900 mA cm−2).
Stabilizing the Cu+ sites by Mg modification promotes CO2 electroreduction to C2+.182 Assisted by a fast-screening platform, Cu–Mg stands out among the 109 Cu-based bimetallic catalysts. The Mg modified Cu achieves 80% FEC2+ with 1000 mA cm−2 at –0.77 V vs. RHE, attributed to the stabilization of Cu+ sites by Mg species for enhanced *CO coverage to facilitate C–C coupling. Different from the bimetallic alloys, copper co-alloyed with isolated Sb and Pd was designed for effective CO2-to-CO conversion,173 where Sb and Pd single atoms work synergistically to shift the Cu electronic structure to promote CO generation, suppress HER and enhance the catalyst stability by inhibiting atom aggregation. Almost unity CO selectivity at 402 mA cm−2 and high activity (1 A cm−2) in neutral electrolyte, as well as ultrastability of 528 h at 100 mA cm−2 with over 95% FECO were achieved.
The alloy effect also applies for the CORR.1,28 Modulating the active sites by developing atomic interfaces or grain boundary sites is able to boost n-propanol synthesis from the CORR. An asymmetric C–C coupling active site was constructed wherein the adjacent Cu atoms exhibit different electronic structures for interaction with two adsorbates to activate the asymmetric reaction.67 Ag doping into Cu results in arising strain and ligand effects, which determine the asymmetry among the neighbors’ energetics. The Ag-doped Cu provides the optimal activity to C1–C1 and C1–C2 coupling for C2 and C3 production, respectively. FEn-proponal of 33 ± 1% and a cathodic energy conversion efficiency of 21% for CO-to-n-propanol conversion are achieved.
3.2. Single atom effects
Single atom catalysts (SACs) featured by isolated metal atoms distribution on various supports are effective in catalyzing the CO2RR/CORR,43,83,190–198 due to their advantages of maximum metal utilization, well-defined active sites, strong atom–support interaction, suppressed HER, and outstanding catalytic activity. In this section, we mainly focus on the representative M–N–C structured SACs,199,200 which are mostly studied both experimentally and theoretically.Different metal elements (Al,201 Mn, Co,112,202–205 Ni,111,198,206–210 Zn, Cu,211–216 Ga,217 Se, Rh, Ag,218,219 Cd, In,220 Sn, Cs,221 Bi, Ru,116 La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu196) have been selected as metal centers (Fig. 3a), among which, the typical Ni, Co, Ag SACs are highly selective to CO production at ampere-level current density (Fig. 3c); Bi, In, Zn, Ag and Sn SACs favor generating formate; while Cu SACs are unique for hydrocarbon and multi-carbon formation. The typical Ni–N4 sites can enhance the adsorption energy of the key intermediates (CO2* and COOH*),27 and afford abundant defective sites,222 assisted by the mesoporous architecture for fast electron and mass transfer,223 boosting the CO2-to-CO conversion. Interestingly, the NiNx sites proceed structural evolution upon varying the thermal temperature (Fig. 3b).224 The shortened Ni–N bonds in compressively strained NiN4 sites produced at 900 °C are intrinsically active and selective to CO, while the NiN3 sites activated at higher temperature (1200 °C) exhibit not only the optimal local structure but also superior CO activity and selectivity to NiN4. Thus, a high FECO (>90%) and an industrial jCO (726 mA cm−2) are achieved in a flow cell. In comparison, the square-pyramidal Ni–N5 site outperforms the planar Ni–N4 site for industrial-scale CO2-to-CO conversion,47 because of the increased and decreased energy levels in dz2 and dxz/yz orbitals, respectively, which facilitate CO2 activation, lower the energy barrier and benefit CO desorption.
 | ||
| Fig. 3 Single atom effects. (a) Summary of reported active elements in SACs for CO2RR/CORR at ampere current density. (b) Synthesis scheme of Ni–N–C. Reproduced with permission.224 Copyright 2022, Royal Society of Chemistry. (c) Summary of reported FE by SACs (data from Tables S1 and S2, ESI†). | ||
Apart from Ni SACs, Zn SAC225 and Co SAC226 also contribute to industrial-scale CO2-to-CO conversion, due to the COOH* intermediate stabilization and HER suppression by the unsaturated Zn–N3 site with the electron-rich environment of Zn, and the isolated CoN4 sites, respectively. Introducing heteroatoms such as S, P, F into the active sites is one of the main methods to modulate the electronic structure.227 For instance, a series of Co–SxN4−x (x = 0, 1, 2, 3) SACs were designed by a thermal replacement of coordinated N with S,228 with Co–S1N3 exhibiting the balanced bindings to COOH*and CO*, thus delivering the optimal FECO (>90%). This coordination tailoring provides a rational approach to control the catalytic performance. Increasing the metal loading in SACs is considered promising to enrich active sites, thus promoting the CO2RR. The high loading Ni SAC (4.3 wt%) outperformed the low loading one (0.8 wt%) towards CO production at industrial-level current.229
A few studies are devoted to SACs for the industrial-scale CORR.1,187 For instance, Cu atoms can be anchored on Ti3C2Tx nanosheets for CO-to-C2+ conversion.191 The atomically dispersed Cu–O3 sites facilitate C–C coupling to form the key *CO–CHO species and further lower the energy barrier of the potential-determining step, thereby contributing to high activity and selectivity for C2+. Cu can also support other single atoms (Ni,230 Pd,28 Ag,231 Au1) for efficient CO-to-C2+ conversion as well.
Apart from SAC, dual atom catalysts (DACs) are also effective for CO2-to-CO, where the atom pairs cooperate to facilitate *COOH formation and *CO desorption. For example, the hydroxyl adsorbate-induced Ni dual-atom sites (Ni2N6OH) afford fast CO2RR kinetics,48 suppress the HER, and promote hydroxyl adsorption to generate the electron-rich active centres, which favor CO2 activation, lower the energy barrier for *COOH formation and facilitate *CO desorption.
Introducing another metal atom into SACs can effectively modify the electronic structure of the simple active sites and further improve the catalytic activity. The Ni–Fe DAC with neighboring Ni–N4 and oversaturated Fe–N5 moieties232 and Co–Cu hetero-diatomic pairs233 facilitate CO2 activation and *COOH formation, due to the effective synergistic electronic modification of the dual atom sites, thus promoting CO production with almost unity selectivity at industrial current density.
3.3. Morphology and structure regulation
Regulating the morphology and structure is an alternative route to influence the CO2RR/CORR performance (Fig. 4a). Different morphologies have been engineered to enhance their catalytic behaviors, such as nanoparticles,27,100,234–236 nanotubes97,222 or nanowires,230 nanosheets,237–239 needles,149 cavities,240,241 3D structure,242,243 core–shell,244,245 hierarchical,223 nanospheres,47 hollow spheres,246 accordion-like layered,247 flower-like,248 nanotips,170 nanocubes,179 multi-shell,249 nanocages,213 nanoribbons,250 arrays,251 polygon facets,252 snowflake-like,253 dendritic,141,180 nanorod/nanosheet hierarchical,254 and mesostructures.255 The well-established morphology results in efficient mass and electron transfer and active site exposure.94 For example, electron and mass transfer can be facilitated by developing a nanoporous Cu with 3D interconnected pores of 100–200 nm in diameter,256 hydrophobic porous Cu2O spheres with varying pore sizes,257 and an ordered macroporous carbon skeleton with mesoporous “wall” supported P modified Bi atomic site,243 which promoted gas transfer across the electrode–electrolyte interface, afforded fast CO2 transfer and trapped compressed CO2 to construct abundant and stable triple-phase interfaces, and facilitated CO diffusion, thus contributing to exceptional production of C2+ (≈62% FEC2+, 653 mA cm−2jC2+), 75.3% FEC2+, and CO (>90% FECO, 414 mA cm−2jCO). Especially, to enrich the concentration of the limited K+/H2O and facilitate water uptake via electro-osmosis,149 needle-array catalysts were designed with intensified electric fields at the tips. This unique interface overcomes the restricted K+/H2O migration in the catholyte-free MEA for the CORR, where no aqueous electrolyte is maintained to afford the three-phase interface formation. A large current density of 2500 mA cm−2 at a very low potential of 2.7 V was achieved. | ||
| Fig. 4 Morphology and structure effects. (a) Summary of morphology and structure control over catalysts. (Nanowire, reproduced with permission.230 Copyright 2024, John Wiley and Sons. Needles, reproduced with permission.149 Copyright 2024, Elsevier. Wrinkles, reproduced with permission.258 Copyright 2023, John Wiley and Sons. Cavities, reproduced with permission.240 Copyright 2023, American Chemical Society. 3D structure, reproduced with permission.242 Copyright 2023, John Wiley and Sons. Core–shell, reproduced with permission.244 Copyright 2024, Springer Nature. Nanosphere, reproduced with permission.47 Copyright 2022, John Wiley and Sons. Accordion-like layered, reproduced with permission.247 Copyright 2025, John Wiley and Sons. Polygon facets, reproduced with permission.252 Copyright 2024, John Wiley and Sons. Nanotip, reproduced with permission.170 Copyright 2024, John Wiley and Sons. Nanocube, reproduced with permission.179 Copyright 2020, John Wiley and Sons. Multi-shell, reproduced with permission.249 Copyright 2023, John Wiley and Sons. Nanoribbon, reproduced with permission.250 Copyright 2023, Elsevier.) (b) Summary of the reported FE by regulated structure (data from Tables S1 and S2, ESI†). | ||
By regulating the shape (confinement degree) and dimension of the confined space of the wrinkle structured Cu,258 the CO2RR product selectivity can be alternated without varying the intrinsic properties of Cu. The selectivity of CH4 and EtOH was affected by the shape, with favorable production on non-folded and folded structures, respectively, while the C2H4 selectivity was dominantly influenced by dimension of geometry which increased upon higher depth of wrinkle. This product selectivity difference came from the changes in structure induced local pH and local intermediate confinement, where the non-folded structure favors protonation while the folded structure favors C–C coupling. For example, the Cu2O catalyst featuring a nanosheet-stacked sphere architecture with abundant open and deep conical cavities facilitates timely refreshing of active sites in its depths259 and the fast efflux of the product C2H4 with high local concentration from the elongated channels within the cavity. This design outperforms the hollow Cu2O sphere with single cavities and solid Cu2O spheres, exhibiting maximum FEC2+ (81.7%) with jC2+ (286 mA cm−2) and over 40.0% cathode power conversion efficiency.
Apart from morphology control, regulating the catalyst structure (vacancy, grainy boundary, phase, facets, defects, lattice) also boosts the CO2RR/CORR performances (Fig. 4a).251,260–266 Among which, the catalysts with regulated structure are highly selective to C1 species (CO, formate, Fig. 4b), followed by C2+ ones (C2H4, ethanol, propanol). To target the post-C–C coupling reaction intermediates other than C–C coupling,267 a series of core–shell vacancy engineering catalysts were designed for efficient CO2RR with sulfur atoms in the nanoparticle core and copper vacancies in the shell for propanol and ethanol production. The engineered catalysts, compared to Cu NPs, exhibited a 16-fold larger alcohol-to-ethylene ratio, directing to alcohols other than alkenes. Creating lithium vacancies (VLi) in [CuO4] sites contributed to 90.6 ± 7.6% FEC2+ with 706 ± 32 mA cm−2jC2+,50 as VLi shortens the distance between adjacent [CuO4] layers and decreases the coordination number of Li+ around each Cu, thus promoting CO–CO coupling. Introducing interfacial oxygen vacancies in neighboring Cu (Ov–Cu) pair sites promoted CO2-to-ethanol conversion,268 realizing 48.5% FEethanol and 344 mA cm−2jethanol in acid.
Exposing the selective crystal facets of electrocatalysts is a potential approach to generate specific CO2RR products.269In situ electrodeposition of copper during CO2 reduction preferentially exposed Cu(100) facets,270 due to the strong interactions between the Cu(100) facets and CO2RR intermediates such as CO2*, COOH*, CO*, H*. Using the Cu(100) facet rich catalyst, high FEC2+ (90%), jC2+ (520 mA cm−2), full-cell C2+ power conversion efficiency (37%), and stable C2H4 selectivity over 65 h at 350 mA cm−2 are obtained in MEA. Ni doping into the Cu(100) surface reduced the kinetic barrier of the ethanol path via facilitating *H formation,230 achieving 86.1% FEC2+ and 60.8% FEC2H4 at 700 mA cm−2, and 54% FEethanol and a 300-h stability, respectively. The Cu(100)/Cu(111) interfaces afford a favorable local electronic configuration to improve *CO adsorption and reduce C–C coupling energy barriers,271 surpassing respective Cu(100) and Cu(111) surfaces. Therefore, efficient CO2-to-C2+ conversion is facilitated with 74.9 ± 1.7% FEC2+ at 300 mA cm−2.
Lattice engineering contributes significantly to producing hydrocarbon/multi-carbon. Introducing lattice tension enhances CO chemisorption on the Cu surface by mitigating the dipole–dipole repulsion,272 which favors C–C coupling at high CO coverages. A spindle-shaped copper with 4% lattice tension is highly active and selective to multi-carbon olefins and oxygenates, exhibiting 1.0 A cm−2 current density with 84% FEC2+ at 2.4 V in MEA. Similar tensile strain engineering is obtained by electro-reducing CuO precursors with different crystallinity,273 guiding Bi-MOF-TS catalyst reconstruction to generate continuous vacancies and activate more inert sites,131 and introducing tension strain heteatom dopants (Gd,184 Pd, Au, Ag,274 Li120). The former strategy enables the co-adsorption of *CO and *OH in high concentrations to promote *CO dimerization and suppresses the HER, resulting in 90.9% FEC2+ and 486.1 mA cm−2jC2+. The second strategy introduced weak tensile strain on the whole scale non-defective Bi sites, which enhanced *OCHO adsorption and decreased the reaction barrier, resulting in impressive jformate (995 ± 93 mA cm−2) and FEformate (96 ± 0.64). Gd doping into Cu2O promoted CO2 activation, stabilized O*CCO and favored C–C coupling to give 81.4% FEC2+ and 444.3 mA cm−2jC2+. The Ag doping into Cu outperformed Pd and Au with better ability to affect the d-band center of Cu to stabilize *CO and facilitate C–C coupling, thus delivering 77.9% FEC2+ at 300 mA cm−2. In contrast, the regulated Cu(111) lattice strain with 11.4% compression exhibited reduced surface energies for lower C–C coupling reaction free energy,275 compared to the pristine lattice and 10% compressed lattice, which promotes spontaneous *O splitting after OC–CHO coupling and the *CCH formation for C2H4.
Grain boundary (GB) engineering provides a powerful approach to enhance the CO2RR/CORR performance. For example, owing to the abundant GBs on Bi-based catalysts, the CO2 adsorption is promoted and CO2˙− intermediate is stabilized, leading to facilitated CO2 activation and enhanced HCOOH production (91.9% FEHCOOH at 600 mA cm−2) in a flow cell,276 as well as ∼97% FEHCOOH with ∼450 mA cm−2jHCOOH in an all-solid-state CO2RR system.277 Similarly, the abundant GBs on Cu nanoribbons,250 developed by stacking tiny nanoparticles with exposing Cu(111), Cu(200) and Cu(220) facets, enhanced CO2 activation and promoted *CO formation and adsorption, thus promoting C–C coupling into *OCCO and *OCCOH and enhancing C2H4 and other C2+ species production. Introducing numerous atomic Pb-concentrated GBs to Cu helps to stabilize abundant low-coordinated Cu sites,278 which improved *CO coverage, and created a structurally flexible Pb–Cu surface to adaptively stabilize the key intermediates (*COCOH, *COCOHCO, *COCCH2), thus boosting CO-to-n-propanol conversion with 47 ± 3% FEpropanol with 25% EEhalf in a flow cell. Especially, CO2/CO-assisted generation of GBs has been demonstrated effective in promoting CO adsorption and C–C coupling for C2+ species production. The GBs in perovskite oxide-derived Cu induced by CO2-assisted La leaching helped trapping in situ generated defective sites,279 illustrating maximum FEC2+ (80.3%) with over 400 mA cm−2jC2+. Very recently, the low-coordinated amorphous GB Cu defect sites exhibit strong adsorption to *OH and CH2CHO* which facilitates C–O breaking and producing C2H4,280 while the medium-coordinated GB rich Cu presents weak *OH and CH2CHO* adsorption, favouring C2H5OH production through the protonation on β-C of CH2CHO*. Therefore, high full-cell EEethanol (29%) and EEC2H4 (25.6%) were achieved, as well as 63.4 ± 1.5% FEC2H4 at 12.5 A in a 25 cm2 MEA.
3.4. Oxidation state alternation and oxide derived Cu modification
Manipulation of the oxidation states,281 especially copper species (Cu0,282 Cu+, Cuδ+ state, Cu2+), as one of the major principles to tune the catalyst performance (Fig. 5a and b), has effects on the local electronic structure of materials. For instance, sufficient polyvinyl pyrrolidone (PVP) capped Cu NPs contributes to presence of complete Cu0 species,234 while the deficient PVP capped Cu NPs (DPVP-Cu) demonstrate an oxide structure with face-centered cubic Cu and partial Cu2O species in the inner and outer layer, respectively. The former favors CH4 production, displaying a 70% FECH4 at >200 mA cm−2, while the latter mainly converts CO2 into C2+ species (C2H4, C2H5OH, CH3COOH, and C3H7OH) with >80% FEC2+ at 300 mA cm−2. | ||
| Fig. 5 Regulated oxidation state and modified OD-Cu/CuO. (a) Summary of the regulated oxidation states and modified OD-Cu/CuO. (Reconstructed Cu/Au, reproduced with permission.283 Copyright 2024, American Chemical Society.) (b) Summary of the reported FE of the CO2RR/CORR products in this section (data from Tables S1 and S2, ESI†). | ||
The mixed Cu+/Cu0 states facilitate CO adsorption and benefit C–C coupling towards C2+ species. The Cu+/Cu0 interface controlled by physically mixing Cu nanoparticles and CuI powders,284 or I2 addition involved strategy285 boosted CO2-to-C2+ conversion with over 70% FEC2+ at industrial-scale current density. The Cu(I)/Cu(0) interfaces with an optimal electronic structure, engineered by a fluoride-assisted pulse-sequence method, play crucial roles in the CO2-to-ethnaol conversion.286 The close Cu(0)–Cu(I) coupling promotes COH* formation and directs the key intermediate OCCOH* toward ethanol, while the Cu(I) sites afford CO to generate ethylene on the Cu(0) sites. This coexistence of Cu0 and Cu+ during the CO2RR can also be achieved by grain refining,287 fluoride-assisted pulse-sequence method,286in situ electrochemical reduction of CuO nanosheet/graphene oxide dot hybrids,288 co-feeding H2O and CO2 on the Cu2O–Cu0 interfaces,289 applying nitrogen-doped carbon (CN) coating,246 constructing a cyanamide-coordinated isolated copper framework (Cuδ+NCN) with both Cu0 and Cu+,290 modification by pyroglutamic acid,291 introducing Ag single atoms to stabilize Cu+ in Cu/Cu2O,292 and constructing Ag/Cu+/Cu0 interfaces.110
The Cuδ+ state (0 < δ < 1) can be stabilized by surface coordination via Cu(II) carboxylate,21 doping with interstitial carbon atoms,100 modification by electron-withdrawing molecule,293 creating dynamically stable Cu0Cu0.18+ OCa motifs by insoluble carbonate,294 and developing the unique Pr–O–Cu linkage in the stable oxide heterointerfaces.295 Attributed to their roles of generating rich active sites, preserving stable tri-phase interface, optimizing the *CO adsorption for its dimerization, creating abundant unsaturated Cu–O bonds, alternating the Cu oxidation state to retain Cuδ+, reducing the C–C energy barrier, and alternating the binding strength and binding type of *CO, the CO2/CO-to-C2+ conversion at industrial-scale current densities is significantly boosted. These five strategies result in 90.6% FEC2+ with 453 mA cm−2jC2+, 76.9% FEC2+ with 615.2 mA cm−2jC2+, 89% FEC2+ with 397 mA cm−2jC2+, 83.7% FEC2+ with 393 mA cm−2jC2+, and 71.3% FEC2+alcohols, respectively. In addition, higher oxidated Cu+/Cu2+ interfaces demonstrated 81% FEC2+ at 900 mA cm−2.296
Oxide-derived copper (OD-Cu) has been reported highly selective to C2+ production. However, various CuOx precursors can lead to their reconstruction and product selectivity. Introducing new dopants is a favorable strategy to promote the catalytic activity of OD-Cu. Incorporation of Al dopants to OD-Cu induces CuO reconstruction into OD-Cu,297 forming nanoflakes, which affords high hydrophobicity and high electrochemically active surface area. The doped catalyst exhibits high FEC2+ (68.4%) and large jC2+ (478.7 mA cm−2), far surpassing those of the undoped one, while La doping contributes to alternating the phase composition of OD-La–CuOx and higher *H concentrations,235 controlling the electron selectivity and steering the CO2RR products. Differently, the intercalated I into OD Cu boosts CO2 and CO adsorption,298 during the CO2RR process, the excess I release enhances the surface roughness, while the remaining I regulates the chemical state of the surface Cu. Therefore, the I modified OD Cu exhibited 79.5% FEC2+ and 43.5% EEC2+ at 300 mA cm−2. Interestingly, when introducing Au NPs during the CORR, CuO nanosheets in situ transformed into undercoordinated Cu sites around Au NPs,283 which facilitate CO binding and stabilize C2 species to n-propanol. The Au directed Cu reconstruction contributes to high FEn-propanol (∼48.6%) and jn-propanol (800 mA cm−2).
3.5. Modification with organic molecules
Modifying the catalyst surface using small molecules contributes to significant improvement in the CO2RR/CORR performance (Fig. 6a and b).299–301 Apart from alternating the oxidation state of surface copper,140,234,302 molecules such as sodium citrate, AEI,303 TBA+, toluene, hexanethiol,304n-butylamine, PVDF, fluoric polymer, and hexaethynylbenzene modulate the reaction microenvironment for C2+ species production. Among which, toluene with long-chain alkyl helps increase the catalyst stability, block the water transport, and build a hydrophobic interface to inhibit cathode corrosion.49 The toluene-modified Cu nanosheets presented maximum FEC2+ (78%), jC2+ (1.81 A cm−2), and cathodic EEC2+ (42%), as well as a 400-h stability, substantially superior to those of pristine or stearic-acid-modified Cu. Adding a PVDF binder to catalyst ink can enhance the interfacial *CO/*H coverage to alternate the alcohol/alkene ratio from 0.69 to 1.35 with notable 37.5% FEalcohol at 800 mA cm−2.305 Fluoric polymers limit the water wettability,306 increase the *CO coverage, and decrease the *COCOH formation energy induced by the electron withdrawing –CF3, thereby contributing to remarkable jC2+ (355.4 mA cm−2) and high FEC2+ (71.08%). Hexaethynylbenzene organic layers reduce the coordinated K·H2O coverage and enhance the CO intermediate interaction with surface, thus facilitating efficient C–C coupling and enhancing the C2+ yield.307 | ||
| Fig. 6 Organic molecule modified catalysts. (a) Summary of the organic molecule modifications for catalysts. (Cu-poly-1, reproduced with permission.306 Copyright 2024, Elsevier. Cu-NBA, reproduced with permission.308 2024, John Wiley and Sons. C18S–CuNPs, reproduced with permission.309 Copyright 2024, Springer Nature. Metal–ligand coordination, reproduced with permission.310 Copyright 2023, American Chemical Society. TA-Cu, reproduced with permission.311 Copyright 2023, John Wiley and Sons. BS/VC, reproduced with permission.312 Copyright 2023, Springer Nature. Cu2O@Cu–TCPP(Co), reproduced with permission.313 Copyright 2024, John Wiley and Sons. Cu/PPy, reproduced with permission.314 Copyright 2025, Royal Society of Chemistry. Cu-nanorod/CC3, reproduced with permission.143 Copyright 2022, John Wiley and Sons. Cu@AIL, reproduced with permission.139 Copyright 2023, American Chemical Society.) (b) Summary of the reported FE by organic molecule modification (data from Tables S1 and S2, ESI†). | ||
The rate-determining step (RDS) of the CO-to-acetate conversion can be alternated by thiols with optimal alkyl chains (C18 and C12).309 The nucleophilic S-intermediate interaction facilitated the RDS (CO* to CHO*) by increasing the CO(ad) sp2 hybridization. The ligands stabilized the HOOC–CH2* intermediate, thus directing the CORR to acetate. Another interesting example focuses on Ag crystal-triazole with dynamically reversible transformation between adsorbed triazole and adsorbed triazolyl on Ag(111) during the CO2RR,310 included by the strong metal–ligand conjugation, presented a 98% FECO with 802.5 mA cm−2jCO. The dynamic metal–ligand coordination promotes CO2 protonation and branches this rate-determining step to the C–OH breakage in adsorbed COOH.
The organic molecule coating layer, such as polyaniline,315N-aryl-substituted tetrahydro-bipyridine and a related oligomeric film,316 and poly(2-aminoazulene)317 can stabilize the key intermediate *CO and enrich the *CO intermediates on active Cu sites, thus facilitating C–C coupling to C2+ species. Meanwhile *CHO stabilized by hydrogen bonding can be achieved by N–H-rich molecules (3,5-diamino-1,2,4-triazole (DAT)), pyroglutamic acid (Pyr), tannic acid, Vitamin C, and C8F18. Among which, DAT steers the CO2RR selectivity from C2+ in acid to C1.318 The Pyr modified Cu+ sites are reconstruction-resistant,291 while the unmodified regions transformed into Cu, creating stable Cu+/Cu0 interfaces to lower the C–C coupling energy barrier. Similar effects are obtained by tannic acid but with the hydroxyl species in molecules to stabilize Cuδ+ towards ethylene.311 The modified Cu presented excellent FEC2H4 (63.6%) and jC2H4 (497.2 mA cm−2) in a flow cell. Vitamin C affords strong hydrogen bonding to afford favorable electron and proton transfer for *CO formation and dimerization for ethylene production,319 achieving 60.7% FEC2H4 and 539 mA cm−2jC2H4. The 1-ethyl-3-methylimidazolium modification299 improved the adsorption of multi-site *CO2/*CO-related intermediates on Mo sites, leading to C3H8 production at jC3H8 395 mA cm−2 with an FEC3H8 of 91% at −0.8 V vs. RHE.
Engineering the catalyst with organic molecules leads to structure modulation, such as defects, edge sites, and facets. By introducing an antioxidant passivation layer (oxyphilic ascorbic acid vitamin C, VC) to prevent the defective Bi sites from OH− poisoning,312 the modified Bi2S3 (BS) nanowires exhibited ultrastability of 120 h for CO2-to-formate conversion and high formate production at ampere current density. This excellent performance originates from the VC molecules which isolate OH− and defective Bi sites, while the VC layer traps OH− to stabilize the defective sites. The poly(methacrylic acid) ligand in situ transformed into a carbon shell under CO2RR conditions to confine and limit the Bi–BiOx nanodots growth,320 preserving abundant active edge sites for formate production. Besides, the carboxyl group in a ligand maintained on nanodots works as oxygen donors to Bi–BiOx, responsible for formate selectivity at high reaction rates up to 1 A cm−2. Similar confinement effects and seeding-growth manipulation can be achieved by the MOF overlayer to encapsulate Cu2O preserving the valence state and crystalline (200) for *CO–*COH coupling,313 and leveraging the synchronous leaching of ligand tannic acid during CO2RR reconstruction and exposing favorable (100) facets to enrich *CO and facilitate *CO dimerization.251
Mass transfer can be alternated by porous organic cages,143 and 2D sulfonated COF nanosheet based ionomers.321 They promoted CO2 transfer, accumulated CO2 and K+ near the catalyst surface, enhanced *CO coverage, and facilitated H2O dissociation, and incorporating an oppositely charged anionic ionomer (perfluorinated sulfonic acid, PFSA) into a cationic COF on a Cu surface to release the hidden positive charge within the COF322 facilitated gas transfer and enhanced K+ immobilization which impede both outward and inward migration of generated OH− and cations. Meanwhile a local proton-transport promoter,314 developed by hybridizing Cu catalytic sites with proton hopping sites from dual-conductive polypyrrole, modifies the Cu surface for extraordinary CO2-to-C2+ performance (FEC2+ 80.0% at 700 mA cm−2). Where the protons transfer via the Grotthuss mechanism, and proton conductivity is up to the formation and breakage of the hydrogen bond (“–HN1⋯H N2H–” to “–HN1 H⋯N2H–”) at the hopping site. The proton availability can also be controlled by tuning molecule electrophilicity on Cu to steer the CO2RR selectivity.323 1,2-Bis(4-pyridyl)ethane with low electrophilicity promotes proton transfer towards *CO hydrogenation and further to CH4 (58.2% FECH4), while trans-1,2-bis(4-pyridyl)ethylene with high electrophilicity exhibits stronger hydrogen binding to stabilize *CO and further dimerization into C2H4 with 65.9% FE.
Alkaline ionic liquids on Cu play multiple roles of a CO2 concentrator,139 an adsorbed CO2 activator, an intermediate stabilizer, and a CO dimerization promoter for C2+ production. High FEC2+ (81.4%) at 900 mA cm−2 and 47.4% half-cell EE at −0.76 V vs. RHE are achieved, as well as 71.6% FEC2+ at 1.8 A cm−2. Poly(ionic liquids) (PILs) can be confined into Cu for acidic CO2RR performance (83.1% FEC2+ at –700 mA cm−2, 37.6% EEC2+),324 due to the abundant semi-rigid porous structure and dense cationic–anionic network to afford a moderate local alkaline microenvironment and enrich K+ at the active sites. Regulating the functionality and morphology of PILs directs the CO2RR pathway via different C–C coupling at high reaction rates, while the PIL adlayer on the Cu surface boosts the CO2-to-C2+ conversion at ampere-level current density and low K+ concentration (1 M),138 delivering 82.2% and 75.8% FEC2+ at 1 and 1.5 A cm−2, respectively. This excellent performance originated from the PIL layer for inhibited proton diffusion to the catalyst surface, enriched K+, and facilitated C–C coupling. Hydrophobic ionic liquid (HIL) modification on oxide derived Cu also demonstrated prominent C2+ production with 85.1% FEC2+ at 800 mA cm−2, maximum jC2+ of 779 mA cm−2, and an impressive formation rate (2512 μmol h−1 cm−2),96 attributed to the formation of the most stable adsorption site and COatop dimerization, increased *CO coverage, and enlarged electrochemical surface charge by the HIL, thus promoting the CO2RR.
3.6. Tandem catalyst design
Tandem CO2 reduction, with integration of cascade CO2-to-CO and CO-to-C2+ in series on two distinct but complementary catalysts, is effective in circumventing the linear scaling relationship limitations, improves the *CO coverage for C–C coupling, thus enhancing C2+ species production.325,326 Tandem CO2 reduction enables high current density operation. Besides, tandem catalysts, compared to traditional catalysts, are more thermodynamically and kinetically favorable, which enhanced the catalytic efficiency. Cu-based tandem catalysts contributed to improved FE and current density for the CO2RR by optimizing the reduction pathway. Additionally, changes in local environments (intensified active sites, enriched local CO, retained pH, and led to synergistic effects of different elements in a tandem system) also improve the CO2-to-C2+ conversion.The keys of catalysts/electrodes design for tandem CO2 reduction should confirm efficient CO transport, balance the CO production and utilization, and match the overpotential of CO generation and C2+ production. Ag, Au and Zn were introduced to Cu and formed a tandem catalyst, which facilitates CO2-to-CO conversion, enriches CO on the Cu surface and promotes C–C coupling. Due to the high activity in CO generation, Ag is mostly used as a component in tandem catalysts from economic perspective, although its CO selectivity is lower than Au selectivity. Three kinds of Ag–Cu Janus nanostructures with (100) facets (JNS-100) are highly selective to CO2-to-C2+ conversion (Fig. 7a),327 especially, Ag65–Cu35 JNS-100 produced 54% FEC2H4 and 72% FEC2+, attributed to its suitable electronic structure by the optimal Ag/Cu ratio and the tandem effect by CO spillover (Fig. 7b). The tandem electrode of sputtered Ag nanoparticle layers on hydrophobic polytetrafluoroethylene (PTFE) and N-doped carbon(NC)-modified Cu nanowire arrays exhibits a hierarchical configuration,98 where CO is generated on the Ag layer and spills over and transports to the NC-modified Cu layer, and the Cu/NC interfaces are responsible for *CO trapping and adsorption (Fig. 7c). The optimal electrode exhibited 53.5% FEC2H4 and 87.5% FEC2+ at 519.0 mA cm−2, respectively, as well as a high C2+/C1 ratio of 10.42 and 50-h stability. The segmented Cu/Ag s-GDE tandem catalyst also achieved well management of *CO by the Ag layer, increased CO utilization and jC2+.135 Similar to Ag, Au attracts much interest for its high CO selectivity, which weakens CO2 bonds and boosts the CO intermediate formation. Bimetallic CuAu tandem catalysts have exhibited excellent CO2RR performance with Au and Cu facilitating CO production and subsequent C–C coupling, respectively. For example, the deposited Au nanoparticles on Cu foil enhanced the CO concentration on Cu and facilitated C–C coupling,328 while the Au@Cu core–shell with different Cu shell thicknesses only produced hydrocarbons or formate,329 indicating the effects of arrangements of the catalyst components on CO2RR products. It is quite challenging to retain highly active tandem sites due to the potentiodynamic structural evolution, to mediate this issue, self-reducing ions (iodate) that leverage the dissolved iodate self-reduction were deployed to achieve directional reconstruction of the tandem catalyst.136 The CuO/AgIO3 tandem catalyst presents rapid iodate ion dissolution during the CO2RR to generate defective Ag. The dissolved iodate self-reduction drags the CuO reconstruction rate and directs transformation into Cu(100) (Fig. 7d). The defective Ag with asymmetric charge distribution promotes *COOH formation and enriches local CO for C–C coupling on Cu(100). Therefore, the reconstructed tandem catalyst demonstrates 82% FEC2+ at 1.2 A cm−2 and maximum jC2+ (1024 mA cm−2) in strong acid.
 | ||
| Fig. 7 Tandem catalyst design. (a) Summary of the organic molecule modifications for catalysts. Schematic illustration of the synthesis of three kinds of Ag–Cu JNS-100 via confined growth of Cu on one of the six equal faces of Ag NCs. (b) Schematic illustration of a plausible CO2RR mechanism on Ag65–Cu35 JNS-100. Reproduced with permission.327 Copyright 2022, John Wiley and Sons. (c) Schematic illustration of the PTFE-based tandem electrocatalysis for CO2 reduction in a flow-cell system. Reproduced with permission.98 Copyright 2025, American Chemical Society. (d) Schematic illustration of directing reconstruction of the CuO/AgIO3 tandem catalyst in the acidic CO2RR reaction. Reproduced with permission.136 Copyright 2024, John Wiley and Sons. | ||
Although bimetallic tandem systems of Cu and other metals (Ag, Au) have been extensively studied, these catalysts suffer from the changes in the composition and morphology during the CO2RR/CORR process, altering their physical and electronic properties and further complicating the understanding of the catalytic mechanism and dragging the catalyst development. In this regard, significant efforts have been devoted to designing tandem catalysts that use CO-selective molecular catalysts330 and SACs.331 For example, coupling CoPc on Cu GDE enabled high *CO coverage and enriched *COtop, reducing the energy barrier for OCCO formation.332 This tandem catalyst gave a 1.8-time higher FEC2+ (82% FEC2+ at 480 mA cm−2) than the Cu GDE. Similarly, the tandem catalyst consisting of CuO and Ni SACs exhibited superior FEC2+ (81.4%), FEC2H4 (54.1%) and significantly high jC2+ (1220.8 mA cm−2),51 attributed to the unique co-loaded nanostructures which realizes the vicinity of the two catalyst units. The developed tandem catalyst not only achieves efficient cascade CO2–CO–C2+ conversion, but also integrates the merits of Ni SAC which in situ produced CO and its fast consumption, guiding the way of tandem catalysts for large-scale synthesis of C2+. Moreover, the CO-producing Fe–N–C SAC layer segment locates at the inlet to prolong the CO residence time in the C2+-selective Cu catalyst layer,135 contributing to 90% FEC2+ and jC2+ over 1 A cm−2. Instead of working as the CO-formation catalyst, the Ni–N–C catalyst in the tandem system mainly modulates the local pH near the Cu catalyst, contributing to the maximum FEC2+ (68% ± 5%) and jC2+ (356 ± 18 mA cm−2) in acid.333
4. Principles of mass transport manipulation for the CO2RR/CORR at ampere-level current
Much effort has been devoted to constructing highly selective and active catalysts for decades. However, satisfactory performance was only observed at low current density due to the increased mass transfer of reactants and products at higher current. Although high CO2RR/CORR selectivity based on micro-design of catalysts modulates the active sites' amount for the target product, the industrial-level CO2/CO electrolysis focuses more on mass transfer promotion. Engineering the electrode and electrolyzer presents an important principle to facilitate efficient mass transport for improving CO2RR/CORR at ampere current.334 Significant progress has been achieved in developing novel electrodes and optimizing electrolysis systems. In this section, principles of mass transport manipulation by electrode/electrolyzer engineering will be discussed.4.1. Electrode engineering
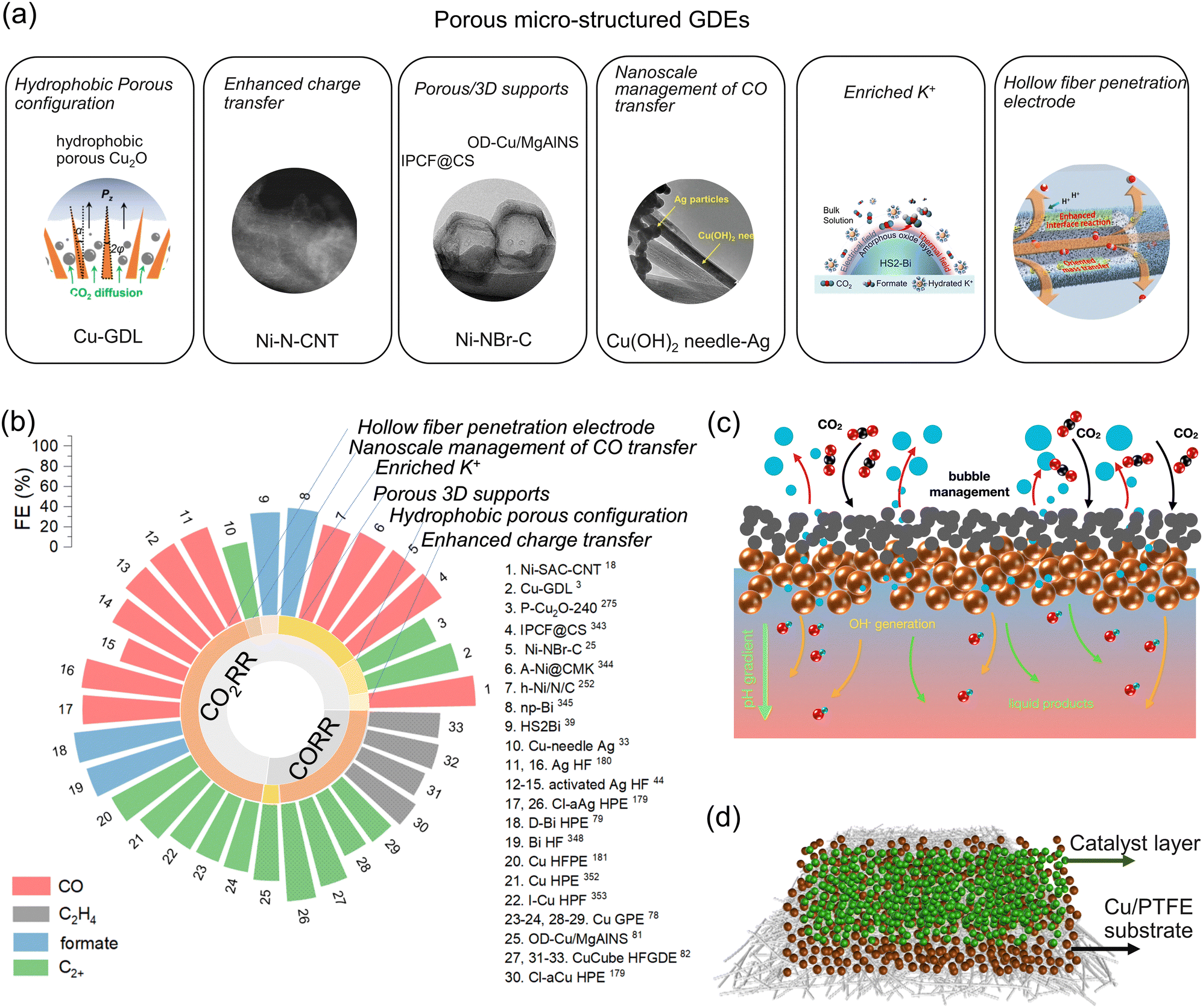 | ||
| Fig. 8 Porous micro-structured GDEs. (a) Summary of porous micro-structured GDEs. (Cu-GDL, reproduced with permission.142 Copyright 2024, Springer Nature. Ni-N-CNT, reproduced with permission.222 Copyright 2021, Elsevier. Ni-NBr-C, reproduced with permission.335 Copyright 2024, John Wiley and Sons. Cu(OH)2 needle-Ag, reproduced with permission.336 Copyright 2023, John Wiley and Sons. K+ enrichment, reproduced with permission.337 Copyright 2024, Journal of Energy Chemistry. HFPE, reproduced with permission.145 Copyright 2022, Springer Nature.) (b) Summary of the reported FE for CO2RR/CORR products by micro-structured GDEs. (data from Tables S1 and S2, ESI†) (c) illustration of the electrode–electrolyte interface on GDL. Reproduced with permission.256 Copyright 2018, John Wiley and Sons. | ||
Enhanced charge transfer and intrinsic catalytic activity can be facilitated by using conductive substrates such as carbon nanotubes.222,338 Derived from the hierarchically structured Ni-imidazolate coordination polymer,222 the hierarchical nanostructured Ni-SACs exhibited flower-like micro-structures assembled from nanoribbons, which converted into hierarchical nanocomposites after introducing the multi-functional substrate CNTs. The substrate helps to establish nanostructured N-doped carbon with large surface pores for fast etchant diffusion to remove Ni NPs, while the high-density Ni single atom sites within the hierarchical structures have facile access to CO2, thereby maximizing the active site utilization at ultra-high current densities.
To reveal the mass transfer-performance relationship, our group242 designed a Fe SAC with a highly ordered porous structure involving hierarchical micropores, mesopores, and macropores, which effectively facilitates mass transfer to Fe atom sites, thus contributing to excellent CO2RR performance. The large mesopores afford strong CO2 diffusion ability inside the porous structured catalyst, constructing a favorable CO2 concentrated environment for CO production. Applying porous or 3D supports facilitates smooth CO2 transfer, such as an interconnected mesoporous carbon nanofiber and carbon nanosheet network (IPCF@CS),339 the hollow Br/N co-doped carbon nanocages,335 and the unique house-of-cards structured scaffolds derived from MgAl LDH,39 hierarchical supports,223 mesoporous carbon,340 poriferous 3D frameworks,113 and 3D interconnected ligament-channel networks.341 Among these, the IPCF@CS structure exhibited highly mesoporous IPCF to hinder CS stacking, affording additional completely exposed sites and abundant bicontinuous mesochannels of IPCF to ensure efficient CO2 transfer. The unique configuration maximized the utilization of active sites and enriched the local CO2 concentration.
Nanoscale management of CO transport for CO2-to-C2+ conversion can be achieved via a 3D tandem catalyst electrode design such as anchoring CO-formation Ag NPs on Cu nanoneedles.336 This configuration was effective in prolonging the CO diffusion path length to enhance CO utilization, thus leading to 70% FEC2+ and 350 mA cm−2jC2+ in a flow cell.
An efficient formate-selective Bi catalyst-GDE, hierarchical Bi (HS2-Bi), was constructed,337 characteristic of a hierarchical nanostructure with an amorphous shell and a high-curvature. The amorphous layer (8–15 nm) not only boosted the kinetics for CO2 adsorption/activation and reduced the formation energy barrier for *OCHO, but also improved the electric-thermal field and K+ adsorption, hence accelerating CO2 reduction to formate. As a result, maximum jformate (677.7 mA cm−2) and over 94% formate selectivity were achieved by HS2-Bi, significantly outperforming the hierarchical structured sample without the amorphous layer. Similar K+ enrichment has been demonstrated by the carbon coated tip-like In2O3 for efficient CO2-to-HCOOH conversion.342
Hollow fiber penetration electrodes (HFPEs) featuring oriented mass transfer, favorable triple phase formation, and facilitated desorption of adsorbed species contributed to CO2/CO electrolysis at ultra-high reaction rates over 1 A cm−2, such as the Ag-HFPE145–147 and Zn HPE343 for CO2-to-CO conversion, Bi HFPEs,37,344 SnO2@Ni HF,345 chlorine-doped SnO2 nanoflowers on 3D Ni HFPEs346 for CO2-to-formate conversion, La(OH)3@Cu HPE for CO2-to-ethanol,347 Cu HFPEs for CO2-to-C2+ conversion36,148,348,349 and CO-to-C2+ conversion,40,146 respectively. Especially, the hierarchical micro/nanostructured Ag hollow fiber exhibited 90.3% FECO with 3.2 A cm−2jCO.146 By optimizing the K+/H+ concentration and CO2 flow rate, the tensile-strained Cu nanosheet layer-coated Cu HPE exhibited 84.5% FEC2+, 3.1 A cm−2jC2+, and 240-h ultrastability in acid.148
Different from the above mentioned GDL-based GDE (Fig. 8c) and HFPE, hydrophobic PTFE with sputtered Cu (Cu/PTFE) has also been used to support catalysts for CO2RR/CORR (Fig. 8d), due to the construction of a favorable hydrophobic microenvironment and robust interface against flooding at high current densities. Assisted by Cu/PTFE,350 the Pd-modified GDE exhibited a high FEC2+ (89 ± 4%) and single-pass carbon efficiency (SPCE, 60 ± 2%) at 500 mA cm−2 in acid. Some other Cu/PTFE based GDEs also demonstrated their potential for industrial-scale CO2/CO electrolysis.12,187,351–356
Alternating the porosity and thickness of the microporous layer (MPL) in GDEs is one of the major principles to control the mass transport for CO-to-acetate conversion. Our group optimized the GDE configuration to maximize the CO transfer through GDLs to the catalyst surface and further reduction at the solid–liquid–gas triple phase boundary.1 By depositing different carbon black (CB) loadings of 1, 2, and 3 mg cm−2 into MPL, the 2 mg cm−2 CB exhibits the best activity and selectivity to acetate production, attributed to its suitable porosity and thickness, with optimal porosity and thickness favoring efficient mass transfer to boost the CORR. In addition, the 2 mg cm−2 CB affords a strong support for the catalyst layer, confirmed by the low penetration of the catalyst layer within the GDL but good penetration within the MPL.
Regulating the pore size distribution of the MPL and the hydrophobicity of carbon fiber substrates (CFSs) has also been confirmed as effective in promoting the mass transfer of electron transfer, CO2, and water,357 thus favoring the C2+ production. The hydrophobicity modification is effective in regulating the local CO2 concentration and *H coverage by controlling the thickness of the localized water layer between the MPL and CL, which contributes to enhanced current density and ethylene selectivity. The optimal Cu-based GDEs presented respective 10% and 30% PTFE in CFS and MPL, and numerous 30–150 nm pores, contributing to jethylene (697 mA cm−2), jC2+ (885 mA cm−2), and jtotal (1.36 A cm−2) at −1.44 V vs. RHE.
 | ||
| Fig. 9 GDEs with molecule/polymer engineering. (a) Summary of molecule/polymer engineered GDEs. (GDE-Nafion, reproduced with permission.358 Copyright 2023, John Wiley & Sons. PT/Cu, reproduced with permission.46 Copyright 2024, Springer Nature. Tailored water and hydroxide, reproduced with permission.33 Copyright 2023, Elsevier. Gemini surfactant, reproduced with permission.359 Copyright 2024, John Wiley and Sons. C/Cu/MOF/PTFE, reproduced with permission.351 Copyright 2022, John Wiley and Sons.) (b) Summary of the reported FE by molecule/polymer engineered GDEs (data from Tables S1 and S2, ESI†). (c) Schematic illustration of the CO2 mass transport inside the catalyst layer with added PTFE, including gas-phase diffusion (solid red arrows) and aqueous-phase diffusion (dashed blue arrows). Reproduced with permission.248 Copyright 2021, American Chemical Society. | ||
Wettability control for a favorable CO2RR/CORR microenvironment can be achieved by applying PT coating,46 alkanethiols,360 polymer binders,361 and PTFE NPs.248 The thin PT coatings on Cu GDEs alternate the local H2O/CO2 concentrations,46 attributed to its hydrophobicity, low water-uptake ability, high porosity and gas permeability for efficient mass transfer. As a result, the modified GDEs presented a high FEC2+ (>87%) at 2 A cm−2, EEcathode over 50% because of the substantially reduced cathodic potential, and long-term stability (>150 h at 200 mA cm−2, 10 h at 1 A cm−2) due to the robust reaction interface. Meanwhile the alkanethiol coatings with different alkyl chain lengths can block water absorption and facilitate CO2 transfer,360 thus alternating the H2O/CO2 transfer to modulate the local H2O/CO2 concentrations. The optimal equilibrium of kinetic-controlled *CO/*H affects the CO2RR to ethylene and ethanol pathways. By changing the hydrophilic interface to superhydrophobic one, there exists limitations of insufficient supply of *CO to that of *H. The ethanol to ethylene ratio is alternated from 0.9 to 1.92 with remarkable FEethannol (53.7%) and FEC2+ (86.1%), respectively. Additionally, a multifunctional conductive polymer, polyaniline modified by p-aminobenzenesulfonic acid (ABSA-PANI),362 was deployed to construct an ideal microenvironment for CO2-to-C2+ conversion and meanwhile improving the charge transfer and ion transport of K+/H+/OH−. Therefore, the CO2RR kinetics was boosted in acids, giving 81% FEC2+ at 600 mA cm−2. The conductive ABSA-PANI decreased the electrode ohmic resistance, resulting in low overpotential and enhanced cathode energy efficiency.
Introducing polymer binders plays crucial roles in facilitating CO2 mass transfer and meanwhile mitigating the competitive HER. Different hydrophilic polymers (polyacrylic acid (PAA), Nafion, and fluorinated ethylene propylene (FEP)) as binders are deployed for Cu to modulate the CO2 accessibility relative to H2O at the catalyst vicinity.361 FEP with hydrophobic (aerophilic) properties prefers to decrease the local H2O concentration and enrich the reactant (CO2) and intermediate (CO) concentration, while the PAA binder renders a highly hydrophilic electrode surface, which hinders CO2 access to the catalyst surface. High FEC2+ (≈77%) and jC2+ (>600 mA cm−2) at −0.76 V vs. RHE are obtained. Directly introducing hydrophobic PTFE NPs into the Bi-based catalyst layer (CL) can also tune the microenvironment for enhanced CO2 electrolysis.248 A moderate hydrophobicity facilitates the balance between the CO2 gas and the liquid electrolyte inside the CL (Fig. 9c), accelerating CO2 mass transfer by reducing the diffusion layer thickness. Therefore, the PTFE modified GDE approached high jacetate (677 mA cm−2) and 35% single-pass CO2 conversion at −0.7 V vs. RHE.
Tailoring water and hydroxide transfer at a quasi-two-phase interface of MEA helps to boost CO-to-C2+ conversion. The CuO–ionomer structure assembled into a MEA helps to uncover the water/hydroxide transfer-performance relationship.33 Attractively, a remarkable FEC2+ (>90%) and low cell voltage (2.4 V) at 1000 mA cm−2 are achieved by using the CuO-Nafion configuration, due to the optimized H2O and OH− transfers. Compared with flow cells where the CORR happens at the catalyst-CO-catholyte three-phase interface and catholyte flow excludes the possible transfer of H2O and OH−, MEA presents limited water supply by humidified CO and membrane crossover. When the supply and consumption of H2O approach an equilibrium with increasing current, the cathodic reductions prefer to occur at the catalysts-CO quasi-two-phase interface with water vapor existence and the polymer membrane as a solid electrolyte. Therefore, applying sufficient H2O transport from the anolyte through the membrane to the dry cathode is vital to proton donation for CO reduction and oppositely moves the in situ-generated OH−. The OH− migration is accompanied by solvated H2O, which leads to lower water content near the cathode, especially at high reaction rates. The interfacial water activity can be enhanced by using anion ionomers,363 which decrease the near-electrode local concentration of K+ (via Donnan exclusion), favoring ethanol production over acetate. Remarkable FEethanol (42.5%) and FEalcohol (55.1%) at 700 mA cm−2 were obtained, with jethanol and jalcohol approaching 698 and 942 mA cm−2 at 2000 mA cm−2, respectively.
By stabilizing the key intermediates via the surface-intermediate interaction, cationic Gemini surfactant modification on the catalyst surface effectively boosts the CO2RR.359 Combination of the double quaternary ammonium cations and alkyl chains of the Gemini surfactants facilitates *CO enrichment on the surface and HCOO* stabilization and affords facile availability to CO2, promoting CO2 reduction. The modified Cu exhibits 96% FEformate and 71% energy efficiency (EE), and the modified commercial Bi2O3 nanosheets present a high FEformate up to 91% at 510 mA cm−2. Similarly, 4-dimethylaminopyridine (DMAP) works as a molecular-additive to functionalize Cu GDEs for enhancing *CO on the Cu surface,364 due to its hydrogen bonding interaction with *COOH. The *CO coverage improvement on GDEs can also be realized by applying dodecanethiol365 and aromatic heterocycles,293,366 due to the thiol-stabilized Cu(100) and active Cu site stabilization by the electron-withdrawing molecule, respectively, thus boosting the production of C2+ species, while polyethylene glycol on the Cu GDE results in Nafion relaxation,367 which affords facile availability of active sites, enhances the *CO and *OH adsorption, and reduces the active hydrogen species, thus promoting C–C coupling and inhibiting the HER.
Sandwiched polymers contribute to high-rate CO2/CO-to-C2+ conversion.351,368 The inserted MOF-induced organic layers in GDEs with a catalyst:MOFs:hydrophobic substrate configuration enriched the local CO2 concentration near the active Cu sites,351 favoring ethylene production. While the sandwiched chitosan (CS) layer works as a “transition layer” between the Cu catalyst and the GDL,368 the developed 3D Cu–CS-GDL presented a highly interconnected network to induce the 3D Cu film growth, which facilitates fast electron transfer and overcomes mass diffusion limitations during CO2 electrolysis. As a result, 88.2% FEC2+ with 900 mA cm−2 current density at −0.87 V was achieved, including 51.4% FEC2+alcohols with 462.6 mA cm−2jC2+alcohols. Apart from the sandwiched polymer, a nanoconfined ionomer was deployed to develop GDEs with uniform ionomer distribution for improving the local mass transfer at the active centers,369 boosting CO2-to-CO conversion. This nanoconfined ionomer not only ensures the CO2RR happening at the active sites and facilitates ion transfer within the CL, but also renders more average distributions of pores on GDEs and prevents ionomer accumulations to avoid high local mass transfer resistance. Therefore, the optimal GDE presented over 90% FECO at a wide current density range and ultrastability over 220 h.
Ionically conductive bifunctional ionomers benefit the CO2 activation at the catalyst–electrolyte interface and promote ethylene production while operating the pure-water fed MEA.370 Specifically, the quaternary ammonia poly(ether ether ketone) (QAPEEK) contributed to industrial-scale jethylene (420 mA cm−2 at 3.54 V), and jtotal up to 1000 mA cm−2 at 3.73 V.
Constructing heterojunctions benefits efficient mass transfer for the CO2RR at ampere-level current density.371 For example, the superfine ionomer layer in the catalyst:ionomer bulk heterojunction (CIBH) architecture exhibits both hydrophobic and hydrophilic properties to extend gas and ion transport from tens of nanometers to the micrometer scale.12 The ionomer intersperses sulfonate-lined paths for the H2O and fluorocarbon channels for CO2, decoupling gas, ion, and electron transport. The constructed CIBH is composed of Cu NPs blended with perfluorinated sulfonic acid, which was spray-coated on a porous PTFE/Cu/ionomer (CIPH) GDE to generate a 3D architecture with metal and ionomer percolation paths. Assisted by this CIBH, an impressive jethylene (1.3 A cm−2) with 45% EEcathodic was achieved in 7 M KOH. Another polymer/catalyst/ionomer heterojunction,372 (perfluorosulfonic acid) PFSA@Cu/PTFE electrode, was designed by combining hydrophobic and highly charged hydrophilic domains to diminish the impurity (SO2) mass transfer to the Cu surface and facilitate unimpeded CO2 transport, respectively. The SO2-tolerant electrode achieved high FEC2+ (84%), jC2+ (790 mA cm−2), and EEC2+ (∼25%). The COF:PFSA heterojunction is effective in suppressing the HER and promoting CO2-to-C2+ conversion in acid.353 The imine and carbonyl-modified COF modulates the ionomer structure to generate homogeneously distributed cation-carrying and hydrophilic–hydrophobic nanochannels to confine proton transport and enrich K+. This regulated proton flux and cation distribution constructs a favorable local environment for the CO2RR.
4.2. Electrolyzer design and optimization
Normally, H-cells are deployed to rapidly screen the desired catalyst for CO2RR/CORR, because of their low cost, facile assembly and operation.30 Typically, H-cell consists of two independent cathodic and anodic chambers separated by ion-exchange membranes, using a reference electrode and an aqueous electrolyte. The disadvantages of H-cells include low CO2/CO solubility, limited mass transfer, a restricted jtotal (<100 mA cm−2) and electrode area, which make it challenging to meet the requirements of industrial CO2RR/CORR applications, especially at ampere-level current. These issues can be addressed by applying the GDEs with GDLs to afford gas mass transfer directly to the cathode to improve the mass transfer and reaction rates.Flow cells with applying GDEs are widely investigated for CO2RR/CORR at ampere-scale current (Fig. 10a). CO2 is supplied directly to the cathode where the catholyte is circulated, leading to much faster mass diffusion and production rates than H-cells. In the anode side, the anolyte is also circulated for the oxygen evolution reaction. Circulation of electrolytes helps to balance the pH variation and attain a stable concentration. Therefore, flow cells contribute to better electrochemical performance than H-cells, due to the enhanced CO2 transfer, suppressed HER, and the constructed local gas–electrolyte–catalyst triple-interface.267,373,374 Different from the traditional flow cell, a novel configuration was proposed,375 which uses a filter press reactor in a continuous mode, with a Sustainion membrane for formate transfer from the cathodic to anodic component, and operating at 600 mA cm−2 on Bi-based catalysts. This reactor afforded high FEformate (73.7%) and production rate (22.9 mmol m−2 s−1), demonstrating its potential for practical applications.
 | ||
| Fig. 10 Electrolyzer engineering. (a) Schematic illustration of a flow cell. Reproduced with permission.1 Copyright 2023, American Chemical Society. (b) Schematic illustration of MEA. (c) A comparison of different electrolysis systems for CO2RR. Reproduced with permission.376 Copyright 2024, Springer Nature. (d) Illustration of a solid electrolyte cell. | ||
MEA as the zero-gap cell is another type of electrolyzer to enhance mass transfer for efficient CO2/CO electrolysis (Fig. 10b), which eliminates the catholyte flow channel in flow cells, with two electrodes pressed together and separated by ion-exchange membranes. Compared to flow cells, the MEA configuration exhibits significantly decreased resistance for mass transfer and electron transfer, thereby boosting the energy efficiency.377
Many works have been devoted to the MEA-based CO2RR in alkaline electrolytes for efficient product formation. However, the serious carbonate formation by the CO2 reaction with OH− is detrimental to the catalytic performance, which precipitates within the catalyst and GDLs and blocks the pores for CO2 transfer, degrading the catalytic system and precluding the stable CO2RR. To overcome these issues, a pure-water-fed (alkali-cation-free) MEA was designed for CO2-to-ethylene conversion.376 An anion-exchange membrane (AEM) + proton-exchange membrane (PEM) MEA (APMA) system involves an AEM at the cathode and a PEM at the anode, respectively. The APMA does not involve a water dissociation catalyst, different from the bipolar membrane (BPM) system (Fig. 10c), wherein water dissociation occurs at the cathode and anode to produce OH− and H+ spontaneously, which are driven through AEM/PEM to take part in the oxygen evolution reaction/CO2RR. The AEM helps to create an alkaline cathode environment, while PEM circumvents the crossover of all anions and facilitates CO2 reduction at low cell voltages, meanwhile suppressing the HER and maintaining high conversion efficiencies. The BPM system exhibits the junction/bonding layer at the anion-exchange layer/cation-exchange layer (AML/CEL) interface, where H2O forms, leading to delamination and further instability.
Other strategies to address carbonate formation include applying acid electrolytes,214,352,378,379 using bipolar membranes,380 proposing a self-cleaning CO2 reduction strategy with short, periodic reductions in applied voltage,381 dividing CO2-to-C2+ conversion in alkaline electrolytes into two steps of CO2-to-CO followed by CO-to-C2+,1 physical washing, pulsed operation,152 replacing K+ to Cs+ in the electrolyte due to the higher solubility of CsHCO3 and Cs2CO3 than potassium salts, using solid electrolytes,382 increasing the operating temperature (60–80 °C) to improve carbonate salt solubility, lowering the K+ concentration in electrolytes,383 performing direct CO2 conversion from carbonate,384 and applying the proton-exchange membrane system.152
Optimizing the flow fields in MEA contributes to developing high-current-density CO2 electrolyzers.385 The typical three flow fields of parallel, serpentine, and interdigitated exhibited weak, moderate, and excessive flow-through transport, respectively. Based on these, the CO2 distribution uniformity is enhanced, no CO2 starvation is ensured, CO2 flow-through transport is provided, and suppressed drainage behavior is ensured. As a result, a multi-serpentine flow field, compared to the conventional parallel flow field, achieved high CO selectivity (95% at 0–350 mA cm−2) and maximum jCO (409 mA cm−2). Meanwhile, the CO2 mass transfer properties in MEA can be improved by controlling the cell compression via varying the gasket thickness,386 which alternates the porosity and thickness of GDEs and affects the electrolyzer performance. Demonstrated by Ag-deposited GDEs, high and low compressed MEA present similar FECO and jCO at low voltages (<2.9 V), while the low compressed device illustrated outstanding selectivity and activity to CO and inhibited the HER.
Solid electrolyte cells have been proposed to promote ampere-level CO2RR/CORR (Fig. 10d). For instance, assembled into a MEA with a porous solid electrolyte, the strongly coupled nanosheets with Ag NPs and Sn–SnO2 grains (Ag/Sn–SnO2 NSs) demonstrated continuous production of ∼0.12 M pure HCOOH solution at 100 mA cm−2 over 200 h.26 The porous solid electrolytes (PSEs) afford direct generation of pure liquid acid solution, which efficiently delivers ions between the cathode and anode without introducing additional solutes. Humidified CO2 is supplied to the cathode to generate HCOO− which is driven by electrical field to the PSE layer through AEM, while water oxidation occurs in the anode with 0.1 M H2SO4 circulation to produce H+ which transfers through CEM to PSE. The generated HCOOH via ionic recombination is flushed out of the PSE layer by deionized (DI) water or N2 vapor. The solid electrolyte cell not only avoids the carbonate formation in the alkaline CO2RR in a flow cell, but also directly generates formic acid solutions (3.5 M) with 93% FEformicacid and 1.1 A partial current at 4.2 V.387 Moreover, using solid electrolyte cells, liquid C2H5OH with 90% relative purity was generated on the Cu catalyst over 50 h continuous CO2RR at 600 mA cm−2,21 as well as >13 wt% ethanol on Cu over 80 h CO2RR at 200 mA cm−2.388 Recently, a pure water fed MEA was deployed with a positively charged polyelectrolyte as an alternative to alkali cations and to modify Ag to avoid bicarbonate formation and achieve high-performance CO2-to-CO conversion.389 This enabled a 78% FECO at 100 mA cm−2 and 55% EE at 200 mA cm−2 at room temperature (RT) but with much larger voltages. Increasing the temperature to mitigate the ohmic impedance from the diffusion limitation and voltage increase by using pure water resulted in larger current densities at the cell voltage, approaching 200 mA cm−2 at 3.5 V at 60 °C which dropped from 5 V at RT, together with 30% EE at 100 mA cm−2, slightly smaller than the 35% in the acid-fed reactor. This strategy can also be applied universally for other CO-selective catalysts (Ni–N–C, CoPc).
Apart from the commercial CO2RR applications, solid electrolyte cells also demonstrate continuous production of high-purity (96%) acetic acid solutions from the CORR on Cu nanocubes,390 with a current density of 1 A cm−2. Moreover, 90 mM pure acetic acid was generated over 120 h CORR around −4.45 V on Cu(25 nm)-CN-3 in a PSE device, with 55.6% FEacetate at 100 mA cm−2.391 Another exciting MEA-based CORR study with a solid-state electrolyte (SSE) illustrates that the highly lattice-disordered Cu3N with abundant twin structures generates 17.4 vol% ethylene stream (1.45 M) and liquid C2+ products (0.23 M) at the outlet of the cathode and SSE layer.392 Additionally, the MEA using a SSE with a tandem catalyst,393 a covalent organic framework and a metal–organic framework for respective CO2-to-CO and CO-to-acetate, achieved 44% acetic acid selectivity at 160 mA cm−2 and 3.6 V, producing high-purity (95 wt%) 20 mM acetic acid solution over 200 h operation with stable FEaceticacid (43%) and current density (>150 mA cm−2). All these works highlight the crucial roles of solid electrolyte cells in commercial CO2RR/CORR applications.
Scaling-up of CO2RR/CORR is conducted in cell-stacks based on the single MEA cells, with enlarged electrode area and more electrolyzer units. To improve the CO2, electron, proton and product (CEPP) transfer at high current densities, an electrolyzer using the forced convection of the CO2 saturated catholyte throughout the porous cathode (in situ electrodeposited Ag NPs on carbon fibers) was designed.394 Induced by the CO2 exsolution from dissolved CO2 and bicarbonate, the CO2RR device with flow-through induced dynamic triple-phase boundaries (FTDT) presented a high FECO (92.0 ± 3.0% at 1.78 A cm−2 and 3.5 V) and maximum current density (3.37 A cm−2 at 4.5 V) on Ag-TEL. Differently, the GDE cells,12 with created triple-phase boundaries involving inlets of the gas-phase CO2 and liquid-phase alkaline catholyte, display maximum current density (jmax, 1.4 A cm−2) but lose input CO2 to bicarbonate/carbonate,395 wherease the GDE cells using the humidified CO2 inlet and MEA are effective in improving the stability,396,397 but challenging to achieve jmax over 1 A cm−2, due to the reduced ionic conductivity and low proton availability. When integrating a Cu-based catalyst, the FTDT cell exhibited 0.57 A cm−2jC2+ at 3.1 V with stable FEC2+ (>55% at 2.7–3.1 V). Further amplification was conducted in the 4 × 100 cm2 electrolyzer stack based on FTDT which produces CO with a yield of 90.6 L h−1 at 59.0 ± 2.6 A and 14 V. Impressively, the large-scale 140 cm2 4-MEA cell stack using NiFe DAC232 presents a high FECO (>97%) over 6 h continuous CO2RR, a stable cell potential (12.2 V at 200 mA cm−2) and exclusive CO yield (∼45 L h−1). As for C2+ production, the practicality is confirmed in a scaled-up CO2 electrolyser stack consisting of six MEA cells,376 wherein ultra-stability over 1000 h and 50% FEC2H4 at 10 A are realized. Besides, the electrolyser stack of four 100 cm2 MEA cells demonstrates the practical application of CO2/CO electrolysis on CuO nanosheets,154 delivering the largest formation rates of 457.5 mL min−1 at 150 A and 2.97 g min−1 at 250 A for ethylene and acetate, respectively.
Very recently, a stable and scalable electrode substrate was designed to resist flooding and operate stably over 400 h for CO2-to-C2H4 conversion.398 The enhanced stability originated from the hydrophobic PTFE percolating network in the microporous layer. Further scale tests were conducted in 800 cm2 cell and 8000 cm2 stack (ten 800 cm2 MEA cells) with stability of 240 h at 100 A (125 mA cm−2) and 240 h at 800 A (100 mA cm−2), respectively. Impressively, a total charge transfer of 6.9 × 108 C was obtained, the largest reported CO2RR demonstration to date. More impressively, a 1000 cm2 CO electrolyzer at 0.71 kW and a 500 cm2 CO2 electrolyzer at 0.40 kW were designed successfully,399 with the former one producing high-yield acetate (1.2 M with 96% purity at 300 A over 125 h) and ethylene.
4.3. Tandem/hybrid device systems
Most fundamental research focuses on catalyst design, overlooking the carbonate formation problem caused by the CO2 reaction with OH− which results in huge voltage loss, significant CO2 consumption, and deteriorated catalytic performance. This should be tackled for CO2RR applications in real devices. To maximize the CO2 utilization, tandem reaction systems are designed to enable two-step reduction of CO2-to-CO and CO-to-C2+ in two separate devices. This tandem system is more attractive and effective in addressing the CO2 loss issue and enhancing the stability. Tandem CO2 electrolysis promotes FEC2+ by 25% compared to direct CO2 electrolysis.400 Other merits of tandem CO2 electrolysis include affording engineered separate reaction environments (neutral conditions for CO2-to-CO and alkaline for CO-to-C2+) against carbonate formation,401 kilowatt scale demonstration towards acetate and ethylene,265,399 and production of highly pure liquid products (acetate) with significantly reduced separation costs.Beyond the above traditional CO2/CO electrolyzers, CO2/CO electrolyzers have been combined with other electrolyzers (cascade MEA,402,403 second CO-to-C2H4 MEA,404 tandem CO-to-C2+ MEA405) for producing C3–C6 acetate esters, ethylene and C2+ at high rates, respectively. Moreover, the hybrid systems have been developed successfully, which consist of CO2/CO electrolyzers and subsequently a thermochemical hydroformylation device,406 a C2H4 dimerizer,407,408 a cascade C2H4 oxidation reactor,409 a solid oxide water electrolyzer,410 a syngas fermentation device,411 a biological upgrading device,412 a thermal catalytic device,413 a thermochemical CO reduction device,414 following a two-step cascade system (C2H4-bromoethanol-ethylene carbonate),415 and a formaldehyde and alcohol dehydrogenase device416 for generating respective propionaldehyde, butane, ethylene glycol, synthetic fuel, mid-chain fatty acid, butanol/hexanol, methanol, 3D-printed carbon nanocomposites, ethylene carbonate, and methanol, respectively. The tandem and hybrid strategy affords new opportunities to target products with higher value than that produced in a single-step process.
The typical two-step CO2RR was operated in two flow cells with Ag and Cu GDEs for CO and C2+ production in non-alkaline and alkaline electrolytes, respectively.417 The gas products of the first flow cell (CO, H2) were purified to remove the unreacted CO2 and then fed into the second flow cell for C2+ production. This configuration overcomes the issues of CO2 loss and KOH consumption, delivering a cumulative FEC2+ of 62% at 300 mA cm−2, 30% higher than that of a single-step electrolysis at the same current density. A similar two-flow cell structured tandem system was demonstrated by 3D Ni SAC and multi-hollow Cu2O nanoparticles for CO and propanol production, respectively,418 suggesting the feasibility of the tandem system, because of their unique capability of combining various catalysts and facile control over each step. Additionally, a tandem system consisting of two MEA cells exhibits significant potential for CO2-to-C2+ conversion,402,403 as well as a hybrid cascade system of an electrochemical reactor connected by thermochemical407,408/bological upgrading412,419,420/sygnas fermentation411/hydroformylation406 devices in series. These systems afford renewable electricity-activated routines for slective produciton of valuable long-chain chemicals/fuels that can not be generated by a single reactor meanwhile circumventing the separaiton of unreacted CO2 between the two reactors. However, these systems are challenged by high-concentration CO stream to the downstream CO electrolyser. The existence of unreacted CO2 in the CO electrolyser results in carbonate generation, reducing the C2+ species production. The CO2 electrolyzer must achieve highly efficient CO2 consumption, and high gas separations costs are demanded to afford pure CO feed into the downstream. Therefore, developing robust and efficient Cu-based catalysts with high activity and selectivity, and their integration into tandem/hybrid membrane electrolyzers with avoidance of electrolyte and liquid product separation are promising to achieve green conversion of CO2 and practical applications.
5. Conclusions and perspectives
CO2RR/CORR, powered by renewable electricity, affords a promising approach for closing the carbon cycle and producing high-value fuels. Although multi-scale research on CO2RR/CORR at ampere current has achieved significant progress, its large-scale deployment still faces challenges. To commercialize CO2RR/CORR, specific criteria should be considered. For instance, continuous electrolysis for 5000 h with a high energy efficiency (>60%),421 operation at ampere current densities,139,154 high pressure,422 large electrode area (100 cm2),154etc. Achieving these goals demands seriously addressing the crucial issues of low selectivity of specific C2+ products, unsatisfactory full cell energy efficiency, insufficient stability, high subsequent separation costs, etc. These challenges are in close relationship with the catalyst design, characterization of reaction mechanisms, and mass transport control. The main challenges and perspectives for ampere-level CO2RR/CORR are listed as follows (Scheme 2). | ||
| Scheme 2 Perspectives and strategies to accelerate CO2RR/CORR at ampere-level current and the large-scale commercialization. | ||
5.1. Challenges in achieving ampere-level CO2RR/CORR
5.2. Perspectives
Author contributions
Qian Sun: investigation, analysis, validation, and writing – review and editing. Chen Jia: writing – review and editing. Haochen Lu: writing – review and editing, Schemes 1 and 2 and TOC drawing. Mengmeng Yang: literature search, writing – review and editing, and summarizing Table 1. Ruirui Liu: literature search, writing – review and editing. Dan M. Villamanca: writing – review and editing. Yong Zhao: conception, resources, funding, writing – review and editing, and supervision. Chuan Zhao: conception, resources, funding, writing– review and editing, and supervision.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was supported by the Australian Research Council (FT170100224, CE230100017, IC200100023, DE250101462).References
- Q. Sun, Y. Zhao, X. Tan, C. Jia, Z. Su, Q. Meyer, M. I. Ahmed and C. Zhao, ACS Catal., 2023, 13, 5689–5696 CrossRef CAS.
- Y. Ji, A. Guan and G. Zheng, Cell Rep. Phys. Sci., 2022, 3, 101072 CrossRef CAS.
- X. Zou and J. Gu, Chin. J. Catal., 2023, 52, 14–31 CrossRef CAS.
- Q. Sun, K. Dastafkan and C. Zhao, Conversion of Water and CO2 to Fuels using Solar Energy, 2024, pp. 233–284 Search PubMed.
- J. Jiao, X. Kang, J. Yang, S. Jia, Y. Peng, S. Liu, C. Chen, X. Xing, M. He and H. Wu, J. Am. Chem. Soc., 2024, 146, 15917–15925 CrossRef CAS PubMed.
- A. Xu, S. F. Hung, A. Cao, Z. Wang, N. Karmodak, J. E. Huang, Y. Yan, A. Sedighian Rasouli, A. Ozden, F. Y. Wu, Z. Y. Lin, H. J. Tsai, T. J. Lee, F. Li, M. Luo, Y. Wang, X. Wang, J. Abed, Z. Wang, D. H. Nam, Y. C. Li, A. H. Ip, D. Sinton, C. Dong and E. H. Sargent, Nat. Catal., 2022, 5, 1081–1088 CrossRef CAS.
- Y. Ma, J. Wang, J. Yu, J. Zhou, X. Zhou, H. Li, Z. He, H. Long, Y. Wang and P. Lu, Matter, 2021, 4, 888–926 CrossRef CAS.
- J. Yu, J. Wang, Y. Ma, J. Zhou, Y. Wang, P. Lu, J. Yin, R. Ye, Z. Zhu and Z. Fan, Adv. Funct. Mater., 2021, 31, 2102151 CrossRef CAS.
- J. Yu, J. Yin, R. Li, Y. Ma and Z. Fan, Chem. Catal., 2022, 2, 2229–2252 CrossRef CAS.
- H. Guan, Y. Zhang, W. Fan, K. Yang, G. Li, S. Chen, L. Li and J. Duan, Small, 2025, 21, 2406605 CrossRef CAS PubMed.
- C. T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. G. De Arquer, A. Kiani, J. P. Edwards, P. De Luna and O. S. Bushuyev, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- F. P. G. De Arquer, C. T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D. H. Nam, C. Gabardo, A. Seifitokaldani and X. Wang, Science, 2020, 367, 661–666 CrossRef PubMed.
- S. Verma, Y. Hamasaki, C. Kim, W. Huang, S. Lu, H. R. M. Jhong, A. A. Gewirth, T. Fujigaya, N. Nakashima and P. J. Kenis, ACS Energy Lett., 2017, 3, 193–198 CrossRef.
- M. Jouny, G. S. Hutchings and F. Jiao, Nat. Catal., 2019, 2, 1062–1070 CrossRef CAS.
- K. Schouten, Y. Kwon, C. Van der Ham, Z. Qin and M. Koper, Chem. Sci., 2011, 2, 1902–1909 RSC.
- Q. Sun, Y. Zhao, W. Ren and C. Zhao, Appl. Catal., B, 2022, 304, 120963 CrossRef CAS.
- W. Ren, X. Tan, X. Chen, G. Zhang, K. Zhao, W. Yang, C. Jia, Y. Zhao, S. C. Smith and C. Zhao, ACS Catal., 2020, 10, 13171–13178 CrossRef CAS.
- S. Park, D. T. Wijaya, J. Na and C. W. Lee, Catalysts, 2021, 11, 253 CrossRef CAS.
- J. Chen, C. Hu, Y. Liu, Y. Wei, K. Shen, L. Chen and Y. Li, Angew. Chem., Int. Ed., 2024, e202422775 Search PubMed.
- Z. Chen, D. Zhang, Q. Li, H. Zhang, Y. Zhao, Q. Ke, Y. Yan, L. Liu, M. Liu and X. He, Appl. Catal., B, 2024, 341, 123342 CrossRef CAS.
- M. Fang, M. Wang, Z. Wang, Z. Zhang, H. Zhou, L. Dai, Y. Zhu and L. Jiang, J. Am. Chem. Soc., 2023, 145, 11323–11332 CrossRef CAS PubMed.
- P. Li, J. Bi, J. Liu, Y. Wang, X. Kang, X. Sun, J. Zhang, Z. Liu, Q. Zhu and B. Han, J. Am. Chem. Soc., 2023, 145, 4675–4682 CrossRef CAS PubMed.
- X. D. Liang, Q. Z. Zheng, N. Wei, Y. Y. Lou, S. N. Hu, K. M. Zhao, H. G. Liao, N. Tian, Z. Y. Zhou and S. G. Sun, Nano Energy, 2023, 114, 108638 CrossRef CAS.
- C. Peng, S. Yang, G. Luo, S. Yan, N. Chen, J. Zhang, Y. Chen, X. Wang, Z. Wang and W. Wei, Chem, 2023, 9, 2830–2840 CAS.
- H. Shen, Y. Zhao, L. Zhang, Y. He, S. Yang, T. Wang, Y. Cao, Y. Guo, Q. Zhang and H. Zhang, Adv. Energy Mater., 2023, 13, 2202818 CrossRef CAS.
- M. Zhang, A. Cao, Y. Xiang, C. Ban, G. Han, J. Ding, L. Y. Gan and X. Zhou, Nano-Micro Lett., 2024, 16, 1–15 CrossRef PubMed.
- Z. Chen, X. Zhang, W. Liu, M. Jiao, K. Mou, X. Zhang and L. Liu, Energy Environ. Sci., 2021, 14, 2349–2356 RSC.
- Y. Ji, Z. Chen, R. Wei, C. Yang, Y. Wang, J. Xu, H. Zhang, A. Guan, J. Chen and T. K. Sham, Nat. Catal., 2022, 5, 251–258 CrossRef CAS.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12, 136 CrossRef CAS PubMed.
- Q. Sun, C. Jia and C. Zhao, Encycl. Ionic Liq., 2022, 676–696 Search PubMed.
- Y. Wu, L. Charlesworth, I. Maglaya, M. N. Idros, M. Li, T. Burdyny, G. Wang and T. E. Rufford, ACS Energy Lett., 2022, 7, 2884–2892 CrossRef CAS.
- D. Ma, T. Jin, K. Xie and H. Huang, J. Mater. Chem. A, 2021, 9, 20897–20918 RSC.
- W. Ren, W. Ma and X. Hu, Joule, 2023, 7, 2349–2360 CrossRef CAS.
- L. Lin, X. He, S. Xie and Y. Wang, Chin. J. Catal., 2023, 53, 1–7 CrossRef CAS.
- R. Shi, J. Guo, X. Zhang, G. I. Waterhouse, Z. Han, Y. Zhao, L. Shang, C. Zhou, L. Jiang and T. Zhang, Nat. Commun., 2020, 11, 3028 CrossRef CAS PubMed.
- C. Zhu, Y. Song, X. Dong, G. Li, A. Chen, W. Chen, G. Wu, S. Li, W. Wei and Y. Sun, Energy Environ. Sci., 2022, 15, 5391–5404 RSC.
- A. Chen, C. Zhu, J. Mao, S. Li, G. Wu, Y. Wei, X. Liu, X. Dong, Y. Song and G. Li, Appl. Catal., B, 2024, 343, 123493 CrossRef CAS.
- M. Zheng, P. Wang, X. Zhi, K. Yang, Y. Jiao, J. Duan, Y. Zheng and S. Z. Qiao, J. Am. Chem. Soc., 2022, 144, 14936–14944 CrossRef CAS PubMed.
- S. Kwon, J. Zhang, R. Ganganahalli, S. Verma and B. S. Yeo, Angew. Chem., Int. Ed., 2023, 62, e202217252 CrossRef CAS PubMed.
- H. Rabiee, J. K. Heffernan, L. Ge, X. Q. Zhang, P. H. Yan, E. Marcellin, S. H. Hu, Z. H. Zhu, H. Wang and Z. G. Yuan, Appl. Catal., B, 2023, 330, 12258 CrossRef.
- X. Wang, P. F. Ou, A. Ozden, S. F. Hung, J. Tam, C. M. Gabardo, J. Y. Howe, J. Sisler, K. Bertens, F. P. G. de Arquer, R. K. Miao, C. P. O'Brien, Z. Y. Wang, J. Abed, A. S. Rasouli, M. J. Sun, A. H. Ip, D. Sinton and E. H. Sargent, Nat. Energy, 2022, 7, 170–176 CrossRef CAS.
- C. Jia, K. Dastafkan, W. Ren, W. Yang and C. Zhao, Sustainable Energy Fuels, 2019, 3, 2890–2906 RSC.
- Q. Sun, C. Jia, Y. Zhao and C. Zhao, Chin. J. Catal., 2022, 43, 1547–1597 CrossRef CAS.
- W. Yang, K. Dastafkan, C. Jia and C. Zhao, Adv. Mater., 2018, 3, 1700377 Search PubMed.
- M. Zhang, W. Wei, S. Zhou, D. D. Ma, A. Cao, X. T. Wu and Q. L. Zhu, Energy Environ. Sci., 2021, 14, 4998–5008 RSC.
- J. Chen, H. Qiu, Y. Zhao, H. Yang, L. Fan, Z. Liu, S. Xi, G. Zheng, J. Chen and L. Chen, Nat. Commun., 2024, 15, 5893 CrossRef CAS PubMed.
- J. R. Huang, X. F. Qiu, Z. H. Zhao, H. L. Zhu, Y. C. Liu, W. Shi, P. Q. Liao and X. M. Chen, Angew. Chem., Int. Ed., 2022, 61, e202210985 CrossRef CAS PubMed.
- Q. Hao, H. X. Zhong, J. Z. Wang, K. H. Liu, J. M. Yan, Z. H. Ren, N. Zhou, X. Zhao, H. Zhang and D. X. Liu, Nat. Synth., 2022, 1, 719–728 CrossRef CAS.
- Z. Liu, X. Lv, S. Kong, M. Liu, K. Liu, J. Zhang, B. Wu, Q. Zhang, Y. Tang and L. Qian, Angew. Chem., Int. Ed., 2023, 62, e202309319 CrossRef CAS PubMed.
- C. Peng, X. Zhu, Z. Xu, S. Yan, L. Y. Chang, Z. Wang, J. Zhang, M. Chen, T. K. Sham and Y. Li, Small, 2022, 18, 2106433 CrossRef CAS PubMed.
- Y. Zhang, P. Li, C. Zhao, G. Zhou, F. Zhou, Q. Zhang, C. Su and Y. Wu, Sci. Bull., 2022, 67, 1679–1687 CrossRef CAS PubMed.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- J. Tang, E. Weiss and Z. Shao, Carbon Neutralization, 2022, 1, 140–158 CrossRef.
- A. Klinkova, P. De Luna, C.-T. Dinh, O. Voznyy, E. M. Larin, E. Kumacheva and E. H. Sargent, ACS Catal., 2016, 6, 8115–8120 CrossRef CAS.
- N. J. Firet and W. A. Smith, ACS Catal., 2017, 7, 606–612 CrossRef CAS.
- Z. Yao, H. Cheng, Y. Xu, X. Zhan, S. Hong, X. Tan, T. S. Wu, P. Xiong, Y. L. Soo and M. M. J. Li, Nat. Commun., 2024, 15, 9881 CrossRef CAS PubMed.
- S. Zhao, S. Li, T. Guo, S. Zhang, J. Wang, Y. Wu and Y. Chen, Nano-Micro Lett., 2019, 11, 1–19 CrossRef PubMed.
- B. Zhang, Y. Chang, Y. Wu, Z. Fan, P. Zhai, C. Wang, J. Gao, L. Sun and J. Hou, Adv. Energy Mater., 2022, 12, 2200321 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- S. Back, H. Kim and Y. Jung, ACS Catal., 2015, 5, 965–971 CrossRef CAS.
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2020, 6, eaay3111 CrossRef CAS PubMed.
- J. Zhang, C. Zhang, M. Wang, Y. Mao, B. Wu, Q. Yang, B. Wang, Z. Mi, M. Zhang and N. Ling, Nat. Chem., 2025, 1–10 Search PubMed.
- W. Ge, L. Dong, C. Wang, Y. Zhu, Z. Liu, H. Jiang and C. Li, ACS Catal., 2024, 14, 10529–10537 CrossRef CAS.
- M. Jouny, W. Luc and F. Jiao, Nat. Catal., 2018, 1, 748–755 CrossRef CAS.
- Q. Xu, B. Ó. Joensen, N. C. Kani, A. Sartori, T. Wilson, J. R. Varcoe, L. Riillo, A. Ramunni, J. Drnec and I. Chorkendorff, Angew. Chem., Int. Ed., 2025, e202501505 CAS.
- L. J. Richter, A. R. Kirmani, X. Wang, W. Ziyuan, D. Cao-Thang, L. Jun, N. Dae-Hyun, L. Fengwang, H. Chun-Wei and T. Chih-Shan, Nat. Commun., 2019, 10, 5186 CrossRef PubMed.
- J. Zhang, P. Yu, C. Peng, X. Lv, Z. Liu, T. Cheng and G. Zheng, ACS Catal., 2023, 13, 7170–7177 CrossRef CAS.
- F. Calle-Vallejo and M. T. Koper, Angew. Chem., Int. Ed., 2013, 125, 7423–7426 CrossRef.
- H. Xiao, T. Cheng, W. A. Goddard III and R. Sundararaman, J. Am. Chem. Soc., 2016, 138, 483–486 CrossRef CAS PubMed.
- C. Yang, Y. Yan, Y. Hu, Y. Chen, A. Guan, C. Hu, L. Zhang and G. Zheng, Small Methods, 2024, 9, 2400393 CrossRef PubMed.
- T. Cheng, H. Xiao and W. A. Goddard III, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS.
- Y. Lum, T. Cheng, W. A. Goddard III and J. W. Ager, J. Am. Chem. Soc., 2018, 140, 9337–9340 CrossRef CAS PubMed.
- T. Cheng, A. Fortunelli and W. A. Goddard III, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7718–7722 CrossRef CAS PubMed.
- L. Chen, C. Tang, K. Davey, Y. Zheng, Y. Jiao and S. Z. Qiao, Chem. Sci., 2021, 12, 8079–8087 RSC.
- A. Prajapati, C. Hahn, I. M. Weidinger, Y. Shi, Y. Lee, A. N. Alexandrova, D. Thompson, S. R. Bare, S. Chen and S. Yan, Nat. Commun., 2025, 16, 2593 CrossRef CAS PubMed.
- R. Yang, M. Wu, D. Huang, Y. Yang, Y. Liu, L. Zhang, F. Lai, B. You, J. Fang and T. Liu, Energy Environ. Sci., 2024, 17, 2897 RSC.
- J. Wang, H. Y. Tan, C. S. Hsu, Y. C. Chu, C. W. Chan, K. H. Chen, X. R. Lin, Y. C. Lee, H. C. Chen and H. M. Chen, J. Am. Chem. Soc., 2025, 147, 13027–13038 CrossRef CAS PubMed.
- J. Leverett, G. Baghestani, T. Tran-Phu, J. A. Yuwono, P. Kumar, B. Johannessen, D. Simondson, H. Wen, S. L. Chang and A. Tricoli, Angew. Chem., Int. Ed., 2025, e202424087 CAS.
- M. Wang, Y. Li, J. Jia, T. Ghosh, P. Luo, Y.-J. Shen, S. Wang, J. Zhang, S. Xi and Z. Mi, Sci. Adv., 2025, 11, eado5000 CrossRef CAS PubMed.
- Y. Zang, S. Wang, J. Sang, P. Wei, X. Zhang, Q. Wang and G. Wang, Nano Lett., 2024, 24, 7261–7268 CrossRef CAS PubMed.
- T. D. Nguyen-Phan, L. Hu, B. H. Howard, W. Xu, E. Stavitski, D. Leshchev, A. Rothenberger, K. C. Neyerlin and D. R. Kauffman, Sci. Rep., 2022, 12, 8420 CrossRef CAS PubMed.
- H. Phong Duong, J. G. Rivera de la Cruz, D. Portehault, A. Zitolo, J. Louis, S. Zanna, Q. Arnoux, M. W. Schreiber, N. Menguy and N.-H. Tran, Nat. Mater., 2025, 1–7 Search PubMed.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang and O. M. Yaghi, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- N. Morlanes, K. Takanabe and V. Rodionov, ACS Catal., 2016, 6, 3092–3095 CrossRef CAS.
- L. Ma, D. Geng, M. Tan, J. Li, R. Guo, Z. Zhai, X. Liu, J. Chen and Q. Li, Appl. Catal., B, 2025, 369, 125153 CrossRef CAS.
- Z. Wei, W. Wang, T. Shao, S. Yang, C. Liu, D. Si, R. Cao and M. Cao, Angew. Chem., Int. Ed., 2024, 6, e202417066 Search PubMed.
- X. Li, M. Qin, X. Wu, X. Lv, J. Wang, Y. Wang and H. B. Wu, Small, 2023, 19, 2302530 CrossRef CAS PubMed.
- Y. Chang, Z. Tan, J. Han, C. Ji, Q. Gao, J. Zhang and C. Pan, available at SSRN 5168331, http://dx.doi.org/10.2139/ssrn.5168331.
- L. Li, S. Yao, Y. Feng, Z. Gao, J. Wang, P. Yin, H. Liu and Z. Zhang, Appl. Catal., B, 2025, 372, 125302 CrossRef CAS.
- K. Yang, R. Kas and W. A. Smith, J. Am. Chem. Soc., 2019, 141, 15891–15900 CrossRef CAS PubMed.
- B. Jia, Z. Chen, C. Li, Z. Li, X. Zhou, T. Wang, W. Yang, L. Sun and B. Zhang, J. Am. Chem. Soc., 2023, 145, 14101–14111 CrossRef CAS PubMed.
- Z. Wang, H. Li, T. Dong, Y. Geng, X. Tian, R. Chang, J. Lai, S. Feng and L. Wang, Chem. Eng. J., 2024, 489, 151238 CrossRef CAS.
- W. Ji, B. Liu, J. Zhang, J. Zhu, W. Zhou, T. Guo, L. Guo, X. Jiang, M. Ya and Z. Zhang, Green Chem., 2025, 27, 4679–4687 RSC.
- H. Yun, S. Yoo, J. Son, J. H. Kim, J. Wu, K. Jiang, H. Shin and Y. J. Hwang, Chem, 2025, 102461 Search PubMed.
- R. Chen, Q. Wu, J. Zhu, S. Wang, Z. Hu, J. Hu, J. Zhu, H. Zhang, B. Ye and Y. Sun, J. Am. Chem. Soc., 2025, 147, 7921–7931 CrossRef CAS PubMed.
- Q. Huang, Z. Qian, N. Ye, Y. Tan, M. Li, M. Luo and S. Guo, Adv. Mater., 2024, 37, 2415639 CrossRef PubMed.
- L. Bian, Y. Bai, J. Y. Chen, H. K. Guo, S. Liu, H. Tian, N. Tian and Z. L. Wang, ACS Nano, 2025, 19, 9304–9316 CrossRef CAS PubMed.
- K. Wang, K. Huang, Z. Wang, G. An, M. Zhang, W. Liu, S. Fu, H. Guo, B. Zhang and C. Lian, Small, 2025, 2502733 CrossRef CAS PubMed.
- H. Wang, R. Sun, P. Liu, H. Hu, C. Ling, X. Han, Y. Shi, X. Zheng, G. Wu and X. Hong, Nano Res., 2024, 17, 7013–7019 CrossRef CAS.
- Y. Yin, Z. Ling, S. Liu, J. Jiao, M. Zhou, P. Zhang, X. Tong, Y. Fan, J. Yang and H. Liu, The Innovation, 2025, 100882 CrossRef CAS.
- X. Li, S. Wang, L. Li, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2020, 142, 9567–9581 CAS.
- Y. Yang, S. Louisia, S. Yu, J. Jin, I. Roh, C. Chen, M. V. Fonseca Guzman, J. Feijóo, P.-C. Chen and H. Wang, Nature, 2023, 614, 262–269 CrossRef CAS PubMed.
- Y. Yang, Y.-T. Shao, J. Jin, J. Feijóo, I. Roh, S. Louisia, S. Yu, M. V. Fonseca Guzman, C. Chen and D. A. Muller, ACS Sustainable Chem. Eng., 2023, 11, 4119–4124 CrossRef CAS.
- E. Zhang, T. Wang, K. Yu, J. Liu, W. Chen, A. Li, H. Rong, R. Lin, S. Ji and X. Zheng, J. Am. Chem. Soc., 2019, 141, 16569–16573 CrossRef CAS PubMed.
- J. P. Grote, A. R. Zeradjanin, S. Cherevko and K. J. J. Mayrhofer, Rev. Sci. Instrum., 2014, 85, 104101 CrossRef PubMed.
- P. Dubé and G. M. Brisard, J. Electroanal. Chem., 2005, 582, 230–240 CrossRef.
- E. L. Clark, M. R. Singh, Y. Kwon and A. T. Bell, Anal. Chem., 2015, 87, 8013–8020 CrossRef CAS PubMed.
- Z. Li, L. Wang, L. Sun and W. Yang, J. Am. Chem. Soc., 2024, 146, 23901–23908 CrossRef CAS PubMed.
- Y. Jiang, C. Lv, B. Lu, Y. Song, T. Liu, X. Zhang, D. Gao, K. Ye and G. Wang, ACS Nano, 2025, 19, 11263–11272 CrossRef CAS PubMed.
- Y. Sun, X. Liu, J. Tian, Z. Zhang, Y. Li, Y. Xie, M. Hao, Z. Chen, H. Yang and G. I. Waterhouse, ACS Nano, 2025, 19, 4528–4540 CrossRef CAS PubMed.
- M. Fang, L. Xu, H. Zhang, Y. Zhu and W.-Y. Wong, J. Am. Chem. Soc., 2022, 144, 15143–15154 CrossRef CAS PubMed.
- M. Wen, N. N. Sun, L. Jiao, S. Q. Zang and H. L. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202318338 CrossRef CAS PubMed.
- J. Song, D. He, X. Ma, P. Liu, W. Guo, R. Sun, F. Li, Z. Zhong, H. Zhou and J. Tang, J. Am. Chem. Soc., 2025, 147, 16198–16206 CrossRef CAS PubMed.
- T. Liu, D. Zhang, Y. Hirai, K. Ito, K. Ishibashi, N. Todoroki, Y. Matsuo, J. Yoshida, S. Ono and H. Li, Adv. Sci., 2025, 2501459 CrossRef PubMed.
- Y. Song, J. Min, Y. Guo, R. Li, G. Zou, M. Li, Y. Zang, W. Feng, X. Yao and T. Liu, Angew. Chem., Int. Ed., 2024, 63, e202313361 CrossRef CAS PubMed.
- C. Peng, S. Yang, G. Luo, S. Yan, M. Shakouri, J. Zhang, Y. Chen, Z. Wang, W. Wei and T. K. Sham, Small, 2023, 19, 2207374 CrossRef CAS PubMed.
- S. Hu, Y. Chen, Z. Zhang, H. Liu, X. Kang, J. Liu, S. Li, Y. Luo and B. Liu, Angew. Chem., Int. Ed., 2025, 64, e202423915 CrossRef CAS PubMed.
- Z. Li, P. Wang, X. Lyu, V. K. R. Kondapalli, S. Xiang, J. D. Jimenez, L. Ma, T. Ito, T. Zhang and J. Raj, Nat. Chem. Eng., 2024, 1, 159–169 CrossRef.
- S. Yan, C. Peng, C. Yang, Y. S. Chen, J. B. Zhang, A. X. Guan, X. M. Lv, H. Z. Wang, Z. Q. Wang, T. K. Sham, Q. Han and G. F. Zheng, Angew. Chem., Int. Ed., 2021, 133, 25945–25949 CrossRef.
- Y. Shen, Z. Wang, Y. Wang and C. Wang, Artif. Intell. Chem., 2024, 2, 100056 CrossRef.
- Z. Jiang, S. Ren, X. Cao, Q. Fan, R. Yu, J. Yang and J. Mao, Angew. Chem., Int. Ed., 2024, 136, e202408412 CrossRef.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- T. Zheng, C. Liu, C. Guo, M. Zhang, X. Li, Q. Jiang, W. Xue, H. Li, A. Li and C.-W. Pao, Nat. Nanotechnol., 2021, 16, 1386–1393 CrossRef CAS PubMed.
- C. Jin, Y. Lin, Y. Wang, J. Shi, R. Li, Y. Liu, Z. Yue, K. Leng, Y. Zhao and Y. Wang, Adv. Mater., 2025, 2412658 CrossRef CAS PubMed.
- H. Shen, T. Wang, H. Jiang, P. Zhao, Z. Chen, Y. Feng, Y. Cao, Y. Guo, Q. Zhang and H. Zhang, Appl. Catal., B, 2023, 339, 123140 CrossRef CAS.
- X. Yu, Y. Xu, L. Li, M. Zhang, W. Qin, F. Che and M. Zhong, Nat. Commun., 2024, 15, 1711 CrossRef CAS PubMed.
- S. Yan, Z. Chen, Y. Chen, C. Peng, X. Ma, X. Lv, Z. Qiu, Y. Yang, Y. Yang and M. Kuang, J. Am. Chem. Soc., 2023, 145, 26374–26382 CrossRef CAS PubMed.
- M. Salehi, H. Al-Mahayni, A. Farzi, M. McKee, S. Kaviani, E. Pajootan, R. Lin, N. Kornienko and A. Seifitokaldani, Appl. Catal., B, 2024, 353, 124061 CrossRef CAS.
- C. Zhang, X. Hao, J. Wang, X. Ding, Y. Zhong, Y. Jiang, M. C. Wu, R. Long, W. Gong and C. Liang, Angew. Chem., Int. Ed., 2024, 136, e202317628 CrossRef.
- X. Chen, R. Lu, C. Li, W. Luo, R. Yu, J. Zhu, L. Lv, Y. Dai, S. Gong, Y. Zhou, W. Xiong, J. Wu, H. Cai, X. Wu, Z. Deng, B. Xing, L. Su, F. Wang, F. Chao, W. Chen, C. Xia, Z. Wang and L. Mai, Nat. Commun., 2025, 16, 1927 CrossRef CAS PubMed.
- Q. Liu, Y. Tan, Q. Chen, X. Zi, Z. Mei, Q. Wang, K. Liu, J. Fu, C. Ma and L. Chai, Nano Lett., 2024, 24, 13741–13746 CrossRef CAS PubMed.
- Y. Wang, J. Wang, R. Cai, J. Zhang, S. Xia, Z. Li, C. Yu, J. Wu, P. Wang and Y. Wu, Adv. Funct. Mater., 2024, 35, 2417764 CrossRef.
- X. Ma, T. Yang, D. He, X. Gao, W. Jiang, D. Li, Y. Sun, X. Lin, J. Xu and H. Wang, Nat. Synth., 2025, 4, 53–66 CrossRef CAS.
- T. Zhang, J. C. Bui, Z. Li, A. T. Bell, A. Z. Weber and J. Wu, Nat. Catal., 2022, 5, 202–211 CrossRef CAS.
- R. Yang, Q. Wen, Y. Yang, Y. Liu, Y. Yang, M. Wu, Y. Wei, B. Mei, Y. Liu and H. Li, Adv. Mater., 2025, 37, 2414642 CrossRef CAS PubMed.
- Z. Li, X. Li, R. Wang, A. Campos Mata, C. S. Gerke, S. Xiang, A. Mathur, L. Zhang, D. Z. Lin and T. Li, Nat. Commun., 2025, 16, 3206 CrossRef CAS PubMed.
- Z. Tan, J. Zhang, Y. Yang, J. Zhong, Y. Zhao, Y. Teng, B. Han and Z. Chen, Nat. Commun., 2025, 16, 1843 CrossRef CAS PubMed.
- Z. H. Tan, J. L. Zhang, Y. S. Yang, J. J. Zhong, Y. Z. Zhao, J. Y. Hu, B. X. Han and Z. J. Chen, J. Am. Chem. Soc., 2023, 145, 21983–21990 CrossRef CAS PubMed.
- J. Q. Feng, Y. L. Wang, G. L. Li, Q. Z. Xue, M. Wang, X. F. Sun, S. J. Zeng and X. P. Zhang, Appl. Catal., B, 2025, 366, 125014 CrossRef CAS.
- M. P. L. Kang, H. Ma, R. Ganganahalli and B. S. Yeo, ACS Catal., 2023, 14, 116–123 CrossRef.
- M. Sun, J. Cheng and M. Yamauchi, Nat. Commun., 2024, 15, 491 CrossRef CAS PubMed.
- C. J. Chen, X. P. Yan, Y. H. Wu, S. J. Liu, X. D. Zhang, X. F. Sun, Q. G. Zhu, H. H. Wu and B. X. Han, Angew. Chem., Int. Ed., 2022, 61, e202202607 CrossRef CAS PubMed.
- S. Chen, B. Rowley, R. Ganganahalli and B. S. Yeo, Adv. Sci., 2024, 11, 2405938 CrossRef CAS PubMed.
- S. Li, W. Chen, X. Dong, C. Zhu, A. Chen, Y. Song, G. Li, W. Wei and Y. Sun, Nat. Commun., 2022, 13, 3080 CrossRef CAS PubMed.
- X. Dong, S. J. Li, C. Zhu, J. N. Mao, G. F. Wu, G. H. Li, G. H. Feng, A. H. Chen, Y. H. Wei, X. H. Liu, J. J. Wang, Y. F. Song, W. Chen and W. Wei, Appl. Catal., B, 2023, 336, 122929 CrossRef CAS.
- S. J. Li, X. Dong, Y. H. Zhao, J. N. Mao, W. Chen, A. H. Chen, Y. F. Song, G. H. Li, Z. Jiang, W. Wei and Y. H. Sun, Angew. Chem., Int. Ed., 2022, 61, e202210432 CrossRef CAS PubMed.
- S. Li, G. Wu, J. Mao, A. Chen, X. Liu, J. Zeng, Y. Wei, J. Wang, H. Zhu, J. Xia, X. Wang, G. Li, Y. Song, X. Dong, W. Wei and W. Chen, Angew. Chem., Int. Ed., 2024, 63, e202407612 CrossRef CAS PubMed.
- W. Ren, H. Zhang, M. Chang, N. Chen, W. Ma, J. Gu, M. Lin and X. Hu, Chem, 2024, 11, 102352 Search PubMed.
- X. Zi, Y. Zhou, L. Zhu, Q. Chen, Y. Tan, X. Wang, M. Sayed, E. Pensa, R. A. Geioushy and K. Liu, Angew. Chem., Int. Ed., 2023, 62, e202309351 CrossRef CAS PubMed.
- B. Endrődi, E. Kecsenovity, A. Samu, T. Halmágyi, S. Rojas-Carbonell, L. Wang, Y. Yan and C. Janáky, Energy Environ. Sci., 2020, 13, 4098–4105 RSC.
- W. S. Fang, W. Guo, R. H. Lu, Y. Yan, X. K. Liu, D. Wu, F. M. Li, Y. S. Zhou, C. H. He, C. F. Xia, H. T. Niu, S. C. Wang, Y. W. Liu, Y. Mao, C. Y. Zhang, B. You, Y. J. Pang, L. L. Duan, X. Yang, F. Song, T. Y. Zhai, G. X. Wang, X. P. Guo, B. Tan, T. Yao, Z. Y. Wang and B. Y. Xia, Nature, 2024, 626, 86–91 CrossRef CAS PubMed.
- A. Inoue, T. Harada, S. Nakanishi and K. Kamiya, EES. Catal., 2023, 1, 9–16 RSC.
- P. F. Wei, D. F. Gao, T. F. Liu, H. F. Li, J. Q. Sang, C. Wang, R. Cai, G. X. Wang and X. H. Bao, Nat. Nanotechnol., 2023, 18, 299–306 CrossRef CAS PubMed.
- X. Lu, T. Shinagawa and K. Takanabe, ACS Catal., 2023, 13, 1791–1803 CrossRef CAS.
- Y. Chen, R. K. Miao, C. Yu, D. Sinton, K. Xie and E. H. Sargent, Matter, 2024, 7, 25–37 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- J. Zhou, B. He, P. Huang, D. Wang, Z. Zhuang, J. Xu, C. Pan, Y. Dong, D. Wang and Y. Wang, Angew. Chem., Int. Ed., 2024, 137, e202418459 CrossRef.
- C. Peng, S. Yang, G. Luo, S. Yan, M. Shakouri, J. Zhang, Y. Chen, W. Li, Z. Wang and T. K. Sham, Adv. Mater., 2022, 34, 2204476 CrossRef CAS PubMed.
- R. Chen, Y. Jiang, Y. Zhu, L. Zhang, Y. Li and C. Li, Adv. Funct. Mater., 2024, 2415940 Search PubMed.
- B. Kim, Y. C. Tan, Y. Ryu, K. Jang, H. G. Abbas, T. Kang, H. Choi, K. S. Lee, S. Park and W. Kim, ACS Energy Lett., 2023, 8, 3356–3364 CrossRef CAS.
- Y. Jiang, J. Shan, P. Wang, L. Huang, Y. Zheng and S. Z. Qiao, ACS Catal., 2023, 13, 3101–3108 CrossRef CAS.
- P. Wang, H. Yang, C. Tang, Y. Wu, Y. Zheng, T. Cheng, K. Davey, X. Huang and S. Z. Qiao, Nat. Commun., 2022, 13, 3754 CrossRef CAS PubMed.
- J. Feng, L. Wu, X. Song, L. Zhang, S. Jia, X. Ma, X. Tan, X. Kang, Q. Zhu and X. Sun, Nat. Commun., 2024, 15, 4821 CrossRef CAS PubMed.
- C. Jia, X. Tan, Q. Sun, R. Liu, R. K. Hocking, S. Wang, L. Zhong, Z. Shi, S. Smith and C. Zhao, Adv. Mater., 2025, 2417443 CrossRef CAS PubMed.
- J. Lu, Y. Ren, J. Liang, L. Zou, Y. Gao, F. Li, J. Gao and J. Liu, Small, 2024, 20, 2402879 CrossRef CAS PubMed.
- Y. Wang, H. Xu, Y. Liu, J. Jang, X. Qiu, E. P. Delmo, Q. Zhao, P. Gao and M. Shao, Angew. Chem., Int. Ed., 2024, 63, e202313858 CrossRef CAS PubMed.
- H. Shen, H. Jin, H. Li, H. Wang, J. Duan, Y. Jiao and S. Z. Qiao, Nat. Commun., 2023, 14, 2843 CrossRef CAS PubMed.
- J. Zhang, T. Fan, P. Huang, X. Lian, Y. Guo, Z. Chen and X. Yi, Adv. Funct. Mater., 2022, 32, 2113075 CrossRef CAS.
- Y. Li, J. Li, W. Ai, J. Chen, T. Lu, X. Liao, W. Wang, R. Huang, Z. Chen and J. Wu, Angew. Chem., Int. Ed., 2024, 63, e202407772 CrossRef CAS PubMed.
- B. Liu, Y. Xie, X. Wang, C. Gao, Z. Chen, J. Wu, H. Meng, Z. Song, S. Du and Z. Ren, Appl. Catal., B, 2022, 301, 120781 CrossRef CAS.
- W. Yang, C. Si, Y. Zhao, Q. Wei, G. Jia, G. Cheng, J. Qin and Z. Zhang, Appl. Catal., B, 2022, 316, 121619 CrossRef CAS.
- J. Xue, X. Dong, C. Liu, J. Li, Y. Dai, W. Xue, L. Luo, Y. Ji, X. Zhang and X. Li, Nat. Commun., 2024, 15, 5998 CrossRef CAS PubMed.
- P. Li, J. Liu, Y. Wang, X. D. Zhang, Y. Hou, Y. Zhang, X. Sun, X. Kang, Q. Zhu and B. Han, J. Am. Chem. Soc., 2024, 146, 26525–26533 CrossRef CAS PubMed.
- J. Li, M. Wei, B. Ji, S. Hu, J. Xue, D. Zhao, H. Wang, C. Liu, Y. Ye and J. Xu, Angew. Chem., Int. Ed., 2025, e202417008 CAS.
- X. Zhou, A. Zhang, B. Chen, S. Zhu, Y. Cui, L. Bai, J. Yu, Y. Ge, Q. Yun and L. Li, Adv. Mater., 2023, 35, 2304414 CrossRef CAS PubMed.
- C. Du, J. P. Mills, A. G. Yohannes, W. Wei, L. Wang, S. Lu, J.-X. Lian, M. Wang, T. Guo and X. Wang, Nat. Commun., 2023, 14, 6142 CrossRef CAS PubMed.
- Y. Yang, J. Zhang, Z. Tan, J. Yang, S. Wang, M. Li and Z. Su, Angew. Chem., Int. Ed., 2024, 136, e202408873 CrossRef.
- L. K. Xiong, X. Zhang, H. Yuan, J. Wang, X. Z. Yuan, Y. B. Lian, H. D. Jin, H. Sun, Z. Deng, D. Wang, J. P. Hu, H. M. Hu, J. Choi, J. Li, Y. F. Chen, J. Zhong, J. Guo, M. H. Rummerli, L. Xu and Y. Peng, Angew. Chem., Int. Ed., 2021, 60, 2508–2518 CrossRef CAS PubMed.
- L. Shang, X. Lv, L. Zhong, S. Li and G. Zheng, Small Methods, 2022, 6, 2101334 CrossRef CAS PubMed.
- Y. Liu, Z. Yue, C. Jin, L. Zheng, J. Shi, D. Li, Y. Wang, J. Bai, K. Leng and W. Wang, Small, 2025, 2409259 CrossRef CAS PubMed.
- M. Xie, Y. Shen, W. Ma, D. Wei, B. Zhang, Z. Wang, Y. Wang, Q. Zhang, S. Xie and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202213423 CrossRef CAS PubMed.
- C. Peng, J. Ma, G. Luo, S. Yan, J. Zhang, Y. Chen, N. Chen, Z. Wang, W. Wei and T. K. Sham, Angew. Chem., Int. Ed., 2024, 63, e202316907 CrossRef CAS PubMed.
- J. Feng, L. Wu, S. Liu, L. Xu, X. Song, L. Zhang, Q. Zhu, X. Kang, X. Sun and B. Han, J. Am. Chem. Soc., 2023, 145, 9857–9866 CrossRef CAS PubMed.
- J. Zhang, C. Guo, S. Fang, X. Zhao, L. Li, H. Jiang, Z. Liu, Z. Fan, W. Xu, J. Xiao and M. Zhong, Nat. Commun., 2023, 14, 1298 CrossRef CAS PubMed.
- Y. J. Chen, X. Y. Wang, X. Y. Li, R. K. Miao, J. C. Dong, Z. L. Zhao, C. H. Liu, H. Z. Liu, J. E. Huang, J. H. Wu, S. Chu, C. Yu, J. Yu, W. Y. Ni, P. Y. Wang, R. Xia, P. F. Ou, K. Xie, B. J. Xu, Y. Hou, D. Sinton and E. H. Sargent, Nat. Catal., 2025, 1–9 Search PubMed.
- X. Wang, Y. Chen, F. Li, R. K. Miao, J. E. Huang, Z. Zhao, X.-Y. Li, R. Dorakhan, S. Chu and J. Wu, Nat. Commun., 2024, 15, 616 CrossRef CAS PubMed.
- D. Wang, H. D. Jung, S. Liu, J. Chen, H. Yang, Q. He, S. Xi, S. Back and L. Wang, Nat. Commun., 2024, 15, 4692 CrossRef CAS PubMed.
- Z. Wu, N. Meng, R. Yang, M. Chen, J. Pan, S. Chi, C. Wu, S. Xi, Y. Liu and Y. Ou, Angew. Chem., Int. Ed., 2025, 64, e202420283 CrossRef CAS PubMed.
- W. Ren, X. Tan, C. Jia, A. Krammer, Q. Sun, J. Qu, S. C. Smith, A. Schueler, X. Hu and C. Zhao, Angew. Chem., Int. Ed., 2022, 134, e202203335 CrossRef.
- H. Bao, Y. Qiu, X. Peng, J.-A. Wang, Y. Mi, S. Zhao, X. Liu, Y. Liu, R. Cao and L. Zhuo, Nat. Commun., 2021, 12, 238 CrossRef CAS PubMed.
- Y. Zhao, Z. Shi, F. Li, C. Jia, Q. Sun, Z. Su and C. Zhao, ACS Catal., 2024, 14, 3926–3932 CrossRef CAS.
- Y. Li, X. Cao, Q. Chen, R. Pan, J. Zhang, G. Meng, Y. Yang, Y. Li, J. Mao and W. Chen, Small, 2024, 20, 2405367 CrossRef CAS PubMed.
- T. Zheng, K. Jiang, N. Ta, Y. Hu, J. Zeng, J. Liu and H. Wang, Joule, 2019, 3, 265–278 CrossRef CAS.
- Y. Zhang, F. Chen, X. Y. Yang, Y. R. Guo, X. H. Zhang, H. Dong, W. H. Wang, F. Lu, Z. M. Lu, H. Liu, Y. Xiao and Y. H. Cheng, Nat. Commun., 2025, 16, 1956 CrossRef CAS PubMed.
- Q. Wang, T. Luo, X. Cao, Y. Gong, Y. Liu, Y. Xiao, H. Li, F. Gröbmeyer, Y. R. Lu and T. S. Chan, Nat. Commun., 2025, 16, 2985 CrossRef CAS PubMed.
- X. P. Yang, Z. Z. Wu, Y. C. Li, S. P. Sun, Y. C. Zhang, J. W. Duanmu, P. G. Lu, X. L. Zhang, F. Y. Gao and Y. Yang, Nat. Commun., 2025, 16, 2811 CrossRef CAS PubMed.
- W. Sun, S. Liu, H. Sun, H. Hu, J. Li, L. Wei, Z. Tian, Q. Chen, J. Su and L. Chen, Adv. Energy Mater., 2025, 2500283 CrossRef.
- X. Wang, W. Ju, L. Liang, M. Riyaz, A. Bagger, M. Filippi, J. Rossmeisl and P. Strasser, Angew. Chem., Int. Ed., 2024, 63, e202401821 CrossRef CAS PubMed.
- Y. Zang, Y. Liu, R. Lu, Q. Yang, B. Wang, M. Zhang, Y. Mao, Z. Wang and Y. Lum, Adv. Mater., 2025, 2417034 CrossRef CAS PubMed.
- Z. Ma, B. Wang, X. Yang, C. Ma, W. Wang, C. Chen, F. Liang, N. Zhang, H. Zhang and Y. Chu, J. Am. Chem. Soc., 2024, 146, 29140–29149 CrossRef CAS PubMed.
- X. Lv, Q. Liu, H. Yang, J. Wang, X. Wu, X. Li, Z. Qi, J. Yan, A. Wu and T. Cheng, Adv. Funct. Mater., 2023, 33, 2301334 CrossRef CAS.
- Q. Zhang, C. B. Musgrave III, Y. Song, J. Su, L. Huang, L. Cheng, G. Li, Y. Liu, Y. Xin and Q. Hu, Nat. Synth., 2024, 3, 1231–1242 CrossRef CAS.
- H. Yang, N. Guo, S. Xi, Y. Wu, B. Yao, Q. He, C. Zhang and L. Wang, Nat. Commun., 2024, 15, 7703 CrossRef CAS PubMed.
- N. Han, X. Xiong, M. Noufal, B. Weng and A. R. Puente-Santiago, Chem, 2025, 11, 102458 CAS.
- H. Li, H. Li, P. Wei, Y. Wang, Y. Zang, D. Gao, G. Wang and X. Bao, Energy Environ. Sci., 2023, 16, 1502–1510 RSC.
- Y. Ma, T. Xiao, K. Zhu, W. Zhang, Z. Yin, A. Dong, Z. Sun, D. Zhao and W. Li, Angew. Chem., Int. Ed., 2025, 64, e202416629 CrossRef CAS PubMed.
- H. Zheng, H. Wu, L. Qiu, M. Yu, J. Zhou, H. Xu, C. Lv, P. Tian, J. Wang and L. Ling, ACS Appl. Nano Mater., 2024, 7, 27275–27286 CrossRef CAS.
- F. Wu, S. Q. Wang, S. Kunze, X. P. Gao, P. Q. Yin and Y.-E. Wu, Rare Met., 2024, 1–8 Search PubMed.
- C. Wang, B. Chen, H. Ren, X. Wang, W. Li, H. Hu, X. Chen, Y. Liu, Q. Guan and W. Li, Appl. Catal., B, 2025, 125151 CrossRef CAS.
- X. Chen, S. Jia, J. Zhai, J. Jiao, M. Dong, C. Xue, T. Deng, H. Cheng, Z. Xia and C. Chen, Nat. Commun., 2024, 15, 7691 CrossRef CAS PubMed.
- Y. Rong, T. Liu, J. Sang, R. Li, P. Wei, H. Li, A. Dong, L. Che, Q. Fu and D. Gao, Angew. Chem., Int. Ed., 2023, 135, e202309893 CrossRef.
- J. Yu, J. Xiao, L. Guo, Z. Xie, K. Wang, Y. Wang, F. Hao, Y. Ma, J. Zhou and P. Lu, ACS Nano, 2024, 18, 33602–33613 CrossRef CAS PubMed.
- D. Kim, S. Park, J. Lee, Y. Chen, F. Li, J. Kim, Y. Bai, J. E. Huang, S. Liu and E. D. Jung, J. Am. Chem. Soc., 2024, 146, 27701–27712 CrossRef CAS PubMed.
- R. Wang, J. Liu, Q. Huang, L. Z. Dong, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2021, 60, 19829–19835 CrossRef CAS PubMed.
- Y. Liang, J. Zhao, Y. Yang, S.-F. Hung, J. Li, S. Zhang, Y. Zhao, A. Zhang, C. Wang and D. Appadoo, Nat. Commun., 2023, 14, 474 CrossRef CAS PubMed.
- J. Wang, Q. Ji, H. Zang, Y. Zhang, C. Liu, N. Yu and B. Geng, Adv. Funct. Mater., 2024, 34, 2404274 CrossRef CAS.
- K. Pellumbi, D. Krisch, C. Rettenmaier, H. Awada, H. Sun, L. Song, S. A. Sanden, L. Hoof, L. Messing and K. Junge Puring, Cell Rep. Phys. Sci., 2023, 4, 101746 CrossRef CAS.
- H. Chen, Q. Huang, K. Yang, S. Chen, H. Feng, C. Jia, Q. Li, C. Zhao and J. Duan, Nano Energy, 2024, 131, 110265 CrossRef CAS.
- M. Li, K. Yang, Y. Sun, T. Gao, Z. Nie, S. Chen, Q. Li and J. Duan, Adv. Energy Mater., 2024, 14, 2303073 CrossRef CAS.
- Y. Sun, J. Chen, X. Du, J. Cui, X. Chen, C. Wu, X. Yang, L. Liu and J. Ye, Angew. Chem., Int. Ed., 2024, 136, e202410802 CrossRef.
- G. H. Jeong, Y. C. Tan, J. T. Song, G. Y. Lee, H. J. Lee, J. Lim, H. Y. Jeong, S. Won, J. Oh and S. O. Kim, Chem. Eng. J., 2021, 426, 131063 CrossRef.
- Y. Chen, J. Zhang, J. Tian, Y. Guo, F. Xu, Y. Zhang, X. Wang, L. Yang, Q. Wu and Z. Hu, Adv. Funct. Mater., 2023, 33, 2214658 CrossRef CAS.
- Y. Li, N. M. Adli, W. Shan, M. Wang, M. J. Zachman, S. Hwang, H. Tabassum, S. Karakalos, Z. Feng and G. Wang, Energy Environ. Sci., 2022, 15, 2108–2119 RSC.
- S. Li, S. Zhao, X. Lu, M. Ceccato, X. M. Hu, A. Roldan, J. Catalano, M. Liu, T. Skrydstrup and K. Daasbjerg, Angew. Chem., Int. Ed., 2021, 60, 22826–22832 CrossRef CAS PubMed.
- C. Wang, Y. Liu, H. Ren, Q. Guan, S. Chou and W. Li, ACS Catal., 2022, 12, 2513–2521 CrossRef CAS.
- C. Jia, X. Tan, Y. Zhao, W. Ren, Y. Li, Z. Su, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2021, 60, 23342–23348 CrossRef CAS PubMed.
- J. Pei, H. Shang, J. Mao, Z. Chen, R. Sui, X. Zhang, D. Zhou, Y. Wang, F. Zhang and W. Zhu, Nat. Commun., 2024, 15, 416 CrossRef CAS PubMed.
- S. Wang, Z. Qian, Q. Huang, Y. Tan, F. Lv, L. Zeng, C. Shang, K. Wang, G. Wang and Y. Mao, Adv. Energy Mater., 2022, 12, 2201278 CrossRef CAS.
- X. Zhang, C. Ling, S. Ren, H. Xi, L. Ji, J. Wang and J. Zhu, Adv. Mater., 2025, 37, 2413111 CrossRef CAS PubMed.
- T.-U. Wi, Y. Xie, Z. H. Levell, D. Feng, J. Y. T. Kim, P. Zhu, A. Elgazzar, T. H. Jeon, M. Shakouri and S. Hao, Nat. Synth., 2024, 3, 1392–1403 CrossRef CAS.
- H. Han, S. Lee, J. Im, M. Lee, T. Lee, S. T. Hyun, J. Hong, T. Seok and D. Choo, Chem. Eng. J., 2024, 479, 147603 CrossRef CAS.
- J. d Yi, X. Gao, H. Zhou, W. Chen and Y. Wu, Angew. Chem., Int. Ed., 2022, 61, e202212329 CrossRef CAS PubMed.
- Q. Fan, X. Zhang, X. Ge, L. Bai, D. He, Y. Qu, C. Kong, J. Bi, D. Ding and Y. Cao, Adv. Energy Mater., 2021, 11, 2101424 CrossRef CAS.
- Z. Guo, H. Zhu, Z. Yan, L. Lei, D. Wang, Z. Xi, Y. Lian, J. Yu, K. L. Fow and H. Do, Appl. Catal., B, 2025, 364, 124839 CrossRef CAS.
- W. Yang, Y. Zhao, Y. Chen, H. Ren, J. Sun, Z. Shi, X. Jin, Z. Zhang and X. Wang, Angew. Chem., Int. Ed., 2025, e202422082 CAS.
- X. Wang, Z. Jiang, P. Wang, Z. Chen, T. Sheng, Z. Wu and Y. Xiong, Angew. Chem., Int. Ed., 2023, 62, e202313646 CrossRef CAS PubMed.
- H. Zhao, Y. Xie, B. Lv, G. Jing and Y. Li, Appl. Catal., B, 2025, 125234 CrossRef CAS.
- D. Wang, Y. Li, S. Geng, R. Li and K. Chen, Adv. Funct. Mater., 2025, 2503497 CrossRef.
- L. X. Liu, Y. Cai, H. Du, X. Lu, X. Li, F. Liu, J. Fu and J. J. Zhu, ACS Appl. Mater., 2023, 15, 16673–16679 CrossRef CAS PubMed.
- H. Zang, Y. Zhao, C. Liu, H. Lu, N. Yu and B. Geng, Adv. Funct. Mater., 2025, 2504400 CrossRef.
- C. Jia, Y. Zhao, S. Song, Q. Sun, Q. Meyer, S. Liu, Y. Shen and C. Zhao, Adv. Energy Mater., 2023, 13, 2302007 CrossRef CAS.
- D. Chen, F. Wang, Y. Liu, W. Lyu, X. Zhao, R. Fang, L. Chen and Y. Li, Angew. Chem., Int. Ed., 2025, e202421149 CAS.
- Y. Shi, J. Li, Z. Min, X. Wang, M. Hou, H. Ma, Z. Zhuang, Y. Qin, Y. Sun and D. Wang, Sci. China Mater., 2025, 68, 173–179 CrossRef CAS.
- J. Pei, L. Yang, J. Lin, Z. Zhang, Z. Sun, D. Wang and W. Chen, Angew. Chem., Int. Ed., 2024, 136, e202316123 CrossRef.
- N. Zhang and Y. Zhang, J. Mater. Chem. A, 2025, 13, 2902–2910 RSC.
- J. Chen, C. Hu, Y. Liu, Y. Wei, K. Shen, L. Chen and Y. Li, Angew. Chem., Int. Ed., 2025, 64, e202422775 CrossRef CAS PubMed.
- Z. Xing, X. Hu and X. Feng, ACS Energy Lett., 2021, 6, 1694–1702 CrossRef CAS.
- Y. K. Xiao, M. Wang, H. Z. Yang, H. R. Qiu, H. T. Lu, Y. Da, G. W. Chen, T. Y. Jiang, W. W. Fu, B. H. Hu, J. M. Chen, L. Chen, Y. S. Ding, B. H. Cui, C. L. Jiang, Z. J. Sun, Y. Long, H. T. Yang, Z. L. Tian, L. Wang and W. Chen, Adv. Energy Mater., 2024, 14, 2302556 CrossRef CAS.
- L. Bian, Z. Y. Zhang, H. Tian, N. N. Tian, Z. Ma and Z. L. Wang, Chin. J. Catal., 2023, 54, 199–211 CrossRef CAS.
- M. Wu, D. Huang, F. Lai, R. Yang, Y. Liu, J. Fang, T. Zhai and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2302851120 CrossRef CAS PubMed.
- W. S. Fang, R. H. Lu, F. M. Li, C. H. He, D. Wu, K. H. Yue, Y. Mao, W. Guo, B. You, F. Song, T. Yao, Z. Y. Wang and B. Y. Xia, Angew. Chem., Int. Ed., 2024, 63, e202319936 CrossRef CAS PubMed.
- Z. Wei, W. Wang, T. Shao, S. Yang, C. Liu, D. Si, R. Cao and M. Cao, Angew. Chem., Int. Ed., 2024, 6, e202417066 Search PubMed.
- X. Zheng, Y. Hu, X. Wang, J. Zhu, X. Zhang, T. Sheng and Z. Wu, Angew. Chem., Int. Ed., 2025, 64, e202415273 CrossRef CAS PubMed.
- Z. Li, B. Sun, D. Xiao, H. Liu, Z. Wang, Y. Liu, Z. Zheng, P. Wang, Y. Dai and B. Huang, Angew. Chem., Int. Ed., 2025, 137, e202413832 CrossRef.
- J. J. Lv, M. Jouny, W. Luc, W. Zhu, J. J. Zhu and F. Jiao, Adv. Mater., 2018, 30, 1803111 CrossRef PubMed.
- Q. Geng, L. Fan, H. Chen, C. Zhang, Z. Xu, Y. Tian, C. Yu, L. Kang, Y. Yamauchi and C. Li, J. Am. Chem. Soc., 2024, 146, 10599–10607 CrossRef CAS PubMed.
- Y. Kim, G. T. Yun, M. Kim, A. Jamal, I. Gereige, J. W. Ager, W. B. Jung and H. T. Jung, Angew. Chem., Int. Ed., 2024, 63, e202316264 CrossRef CAS PubMed.
- C. Li, T. Zhang, H. Liu, Z. Guo, Z. Liu, H. Shi, J. Cui, H. Li, H. Li and C. Li, Adv. Mater., 2024, 36, 2312204 CrossRef CAS PubMed.
- G. Zhang, Z. J. Zhao, D. F. Cheng, H. M. Li, J. Yu, Q. Z. Wang, H. Gao, J. Y. Guo, H. Y. Wang, G. A. Ozin, T. Wang and J. L. Gong, Nat. Commun., 2021, 12, 5745 CrossRef CAS PubMed.
- J. Q. Xu, S. H. Yang, L. Ji, J. W. Mao, W. Zhang, X. L. Zheng, H. Y. Fu, M. L. Yuan, C. K. Yang, H. Chen and R. X. Li, Nano Res., 2023, 16, 53–61 CrossRef CAS.
- Y. P. Zang, T. F. Liu, H. F. Li, P. F. Wei, Y. P. Song, C. F. Cheng, D. F. Gao, Y. F. Song, G. X. Wang and X. H. Bao, Chem. Eng. J., 2022, 446, 137444 CrossRef CAS.
- Z. Q. Wang, X. L. Zu, X. D. Li, L. Li, Y. Wu, S. M. Wang, P. Q. Ling, Y. Zhao, Y. F. Sun and Y. Xie, Nano Res., 2022, 15, 6999–7007 CrossRef CAS.
- Q. Q. Wu, R. A. Du, P. Wang, G. I. N. Waterhouse, J. Li, Y. C. Qiu, K. Y. Yan, Y. Zhao, W. W. Zhao, H. J. Tsai, M. C. Chen, S. F. Hung, X. Wang and G. X. Chen, ACS Nano, 2023, 17, 12884–12894 CrossRef CAS PubMed.
- H. F. Li, P. F. Wei, T. F. Liu, M. R. Li, C. Wang, R. T. Li, J. Y. Ye, Z. Y. Zhou, S. G. Sun, Q. Fu, D. F. Gao, G. X. Wang and X. H. Bao, Nat. Commun., 2024, 15, 4603 CrossRef CAS PubMed.
- S. H. Ruan, B. Zhang, J. H. Zou, W. F. Zhong, X. Y. He, J. H. Lu, Q. H. Zhang, Y. Wang and S. J. Xie, Chin. J. Catal., 2022, 43, 3161–3169 CrossRef CAS.
- T. T. Zhuang, Z. Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. L. Che, F. Meng, Y. M. Min, R. Quintero-Bermudez, C. T. Dinh, Y. J. Pang, M. Zhong, B. Zhang, J. Li, P. N. Chen, H. Y. Liang, W. N. Ge, B. J. Ye, D. Sinton, S. H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS.
- Y. Qiao, S. Shen, C. Mao, Y. Xiao, W. Lai, Y. Wang, X. Zhong, Y. Lu, J. Li and J. Ge, Angew. Chem., Int. Ed., 2024, e202424248 Search PubMed.
- Y.-C. Zhang, X.-L. Zhang, Z.-Z. Wu, Z.-Z. Niu, L.-P. Chi, F.-Y. Gao, P.-P. Yang, Y.-H. Wang, P.-C. Yu and J.-W. Duanmu, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2400546121 CrossRef CAS PubMed.
- Y. H. Wang, Z. Y. Wang, C. T. Dinh, J. Li, A. Ozden, M. G. Kibria, A. Seifitokaldani, C. S. Tan, C. M. Gabardo, M. C. Luo, H. Zhou, F. W. Li, Y. Lum, C. McCallum, Y. Xu, M. X. Liu, A. Proppe, A. Johnston, P. Todorovic, T. T. Zhuang, D. Sinton, S. O. Kelley and E. H. Sargent, Nat. Catal., 2020, 3, 98–106 CrossRef CAS.
- Z. Z. Wu, X. L. Zhang, Z. Z. Niu, F. Y. Gao, P. P. Yang, L. P. Chi, L. Shi, W. S. Wei, R. Liu, Z. Chen, S. J. Hu, X. Zheng and M. R. Gao, J. Am. Chem. Soc., 2022, 144, 259–269 CrossRef CAS PubMed.
- W. Ma, S. Xie, B. Zhang, X. He, X. Liu, B. Mei, F. Sun, Z. Jiang, L. Lin and Q. Zhang, Chem, 2023, 9, 2161–2177 CAS.
- J. P. Jiao, X. C. Kang, J. H. Yang, S. Q. Jia, X. Chen, Y. G. Peng, C. J. Chen, X. Q. Xing, Z. J. Chen, M. Y. He, H. H. Wu and B. X. Han, Angew. Chem., Int. Ed., 2024, 63, e202409563 CrossRef CAS PubMed.
- Z. Y. Zhai, D. L. Li, X. Lu, H. Z. Cai, Q. Hu, H. P. Yang and C. X. He, Carbon Energy, 2024, 6, e648 CrossRef CAS.
- Z. Li, W. Wei, X. Hu, Z. Zhang, Y. Hu, Y. Wu, Y. Wang, J. Xu and M. Ding, Adv. Funct. Mater., 2024, 2422898 Search PubMed.
- Y. L. Xing, H. H. Chen, Y. Liu, Y. L. Sheng, J. Zeng, Z. G. Geng and J. Bao, Chem. Commun., 2021, 57, 1502–1505 RSC.
- L. Fan, C. Xia, P. Zhu, Y. Y. Lu and H. T. Wang, Nat. Commun., 2020, 11, 3633 CrossRef CAS PubMed.
- W. Z. Niu, Z. Chen, W. Guo, W. Mao, Y. Liu, Y. N. Guo, J. Z. Chen, R. Huang, L. Kang, Y. W. Ma, Q. S. Yan, J. Y. Ye, C. Y. Cui, L. Q. Zhang, P. Wang, X. Xu and B. Zhang, Nat. Commun., 2023, 14, 4882 CrossRef CAS PubMed.
- Z.-Z. Niu, L.-P. Chi, Z.-Z. Wu, P.-P. Yang, M.-H. Fan and M.-R. Gao, Natl. Sci. Open, 2023, 2, 20220044 CrossRef.
- D. Zhong, Q. Fang, R. Du, Y. Jin, C. Peng, D. Cheng, T. Li, T. Zhao, S. Zhang and Y. Zheng, Angew. Chem., Int. Ed., 2025, e202501773 Search PubMed.
- Z. Wang, L. Xu, Y. Zhou, Y. Liang, J. Yang, D. Wu, S. Zhang, X. Han, X. Shi and J. Li, Chem. Soc. Rev., 2024, 53, 6295–6321 RSC.
- M. G. Kibria, C. T. Dinh, A. Seifitokaldani, P. De Luna, T. Burdyny, R. Quintero-Bermudez, M. B. Ross, O. S. Bushuyev, F. P. G. de Arguer, P. D. Yang, D. Sinton and E. H. Sargen, Adv. Mater., 2018, 30, 1804867 CrossRef PubMed.
- C. Long, K. Wan, Y. Chen, L. Li, Y. Jiang, C. Yang, Q. Wu, G. Wu, P. Xu and J. Li, J. Am. Chem. Soc., 2024, 146, 4632–4641 CrossRef CAS PubMed.
- H. Li, T. Liu, P. Wei, L. Lin, D. Gao, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2021, 60, 14329–14333 CrossRef CAS PubMed.
- Y. L. Jiang, H. B. Li, C. J. Chen, Y. Zheng and S. Z. Qiao, ACS Catal., 2024, 14, 8310–8316 CrossRef CAS.
- R. M. Cai, M. Z. Sun, F. Yang, D. Y. Gu, M. Ju, Y. P. Chen, M. D. Gu, B. L. Huang and S. H. Yang, Chem, 2024, 10, 211–233 CAS.
- X. Z. Lv, Q. Liu, J. H. Wang, X. J. Wu, X. T. Li, Y. Yang, J. H. Yan, A. J. Wu and H. B. Wu, Appl. Catal., B, 2023, 324, 122272 CrossRef CAS.
- R. A. Du, Q. Q. Wu, S. Y. Zhang, P. Wang, Z. J. Li, Y. C. Qiu, K. Y. Yan, G. I. N. Waterhouse, P. Wang, J. Li, Y. Zhao, W. W. Zhao, X. Wang and G. X. Chen, Small, 2023, 19, 2301289 CrossRef CAS PubMed.
- X. Y. He, L. Lin, X. Y. Li, M. Z. Zhu, Q. H. Zhang, S. J. Xie, B. B. Mei, F. F. Sun, Z. Jiang, J. Cheng and Y. Wang, Nat. Commun., 2024, 15, 9923 CrossRef CAS PubMed.
- K. H. Yue, Y. Y. Qin, H. H. Huang, Z. R. Lv, M. Z. Cai, Y. Q. Su, F. Q. Huang and Y. Yan, Nat. Commun., 2024, 15, 7820 CrossRef CAS PubMed.
- H. Y. Jing, K. R. Lu, J. X. Li, Z. D. Wu, X. F. Xia, M. Z. Xia, B. Y. Liu, C. Su, C. Liu, J. Lei, W. Lei and Q. L. Hao, Appl. Catal., B, 2025, 365, 124977 CrossRef CAS.
- Z. N. Zhang, Q. Fang, X. Yang, S. W. Zuo, T. Cheng, Y. Yamauchi and J. Tang, Adv. Mater., 2025, 2411498 CrossRef CAS PubMed.
- Q. Sun, X. Tan, C. Jia, C. L. Rong, S. H. Wang, C. Han, Y. Xiao, H. Q. Qi, S. C. Smith and C. Zhao, Adv. Funct. Mater., 2024, 34, 2406281 CrossRef CAS.
- R. R. Zhang, H. B. Ma, S. H. Han, Z. T. Wu, X. Zhou, Z. X. Chen, J. Liu, Y. K. Xiao, W. Chen and K. P. Loh, Angew. Chem., Int. Ed., 2025, e202421860 CAS.
- J. Liu, P. Li, S. Jia, Y. Wang, L. Jing, Z. Liu, J. Zhang, Q. Qian, X. Kang and X. Sun, Nat. Synth., 2025, 1–14 Search PubMed.
- W. H. Guo, S. W. Zhang, J. J. Zhang, H. R. Wu, Y. B. Ma, Y. Song, L. Cheng, L. Chang, G. Li, Y. Liu, G. D. Wei, L. Gan, M. H. Zhu, S. B. Xi, X. Wang, B. I. Yakobson, B. Z. Tang and R. Q. Ye, Nat. Commun., 2023, 14, 7383 CrossRef PubMed.
- J. Jang, K. Lee, H. Shin, H. S. Lee, B. H. Lee, J. Jeong, J. Kim, W. Hwang, S. Park, M. S. Bootharaju, S. Back, J. Shim, J. H. Kim, T. Hyeon and Y. E. Sung, J. Mater. Chem. A, 2023, 11, 19066–19073 RSC.
- Z. J. Yan, M. Liu, Z. Y. Guo, Q. H. Chen, Z. Y. Xi, X. Z. Sun, J. H. Yu and T. Wu, Adv. Funct. Mater., 2025, 2420493 CrossRef CAS.
- M. Esmaeilirad, Z. Jiang, A. M. Harzandi, A. Kondori, M. T. Saray, C. U. Segre, R. Shahbazian-Yassar, A. M. Rappe and M. Asadi, Nat. Energy, 2023, 8, 891–900 CrossRef CAS.
- B. S. Crandall, M. Naughton, S. Park, J. Yu, C. Y. Zhang, S. Mahtabian, K. Y. Wang, X. H. Liang, K. Fu and F. Jiao, Nat. Commun., 2024, 15, 1–9 Search PubMed.
- J. E. Huang, F. W. Li, A. Ozden, A. S. Rasouli, F. P. G. de Arquer, S. J. Liu, S. Z. Zhang, M. C. Luo, X. Wang, Y. W. Lum, Y. Xu, K. Bertens, R. K. Miao, C. T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- H. Wu, L. Huang, J. Timoshenko, K. Qi, W. Wang, J. Liu, Y. Zhang, S. Yang, E. Petit and V. Flaud, Nat. Energy, 2024, 9, 422–433 CrossRef CAS.
- Y. Zhao, X. L. Zu, R. H. Chen, X. D. Li, Y. W. Jiang, Z. Q. Wang, S. M. Wang, Y. Wu, Y. F. Sun and Y. Xie, J. Am. Chem. Soc., 2022, 144, 10446–10454 CrossRef CAS PubMed.
- H. Cai, H. Yang, D. Li, S. He, X. Zhang, Q. Hu and C. He, Angew. Chem., Int. Ed., 2025, e202425325 CAS.
- C. J. Chen, L. S. Huang, Y. L. Jiang, Y. Zheng and S. Z. Qiao, Nano Energy, 2024, 126, 109656 CrossRef CAS.
- T. Zhao, X. Zong, J. Liu, J. Chen, K. Xu, X. Wang, X. Chen, W. Yang, F. Liu and M. Yu, Appl. Catal., B, 2024, 340, 123281 CrossRef CAS.
- S. Ren, X. Cao, Q. K. Fan, Z. M. Yang, F. Wang, X. Wang, L. C. Bai and J. Yang, Nano-Micro Lett., 2024, 16, 262 CrossRef CAS PubMed.
- Y. Wu, C. Chen, S. Liu, Q. Qian, Q. Zhu, R. Feng, L. Jing, X. Kang, X. Sun and B. Han, Angew. Chem., Int. Ed., 2024, 63, e202410659 CrossRef CAS PubMed.
- E. Shirzadi, Q. Jin, A. S. Zeraati, R. Dorakhan, T. J. Goncalves, J. Abed, B. H. Lee, A. S. Rasouli, J. Wicks and J. Zhang, Nat. Commun., 2024, 15, 2995 CrossRef CAS PubMed.
- X. Kong, J. Zhao, Z. Xu, Z. Wang, Y. Wu, Y. Shi, H. Li, C. Ma, J. Zeng and Z. Geng, J. Am. Chem. Soc., 2023, 145, 14903–14911 CrossRef CAS PubMed.
- S. Chen, C. Ye, Z. Wang, P. Li, W. Jiang, Z. Zhuang, J. Zhu, X. Zheng, S. Zaman and H. Ou, Angew. Chem., Int. Ed., 2023, 135, e202315621 CrossRef.
- J. Zhu, J. Li, R. Lu, R. Yu, S. Zhao, C. Li, L. Lv, L. Xia, X. Chen and W. Cai, Nat. Commun., 2023, 14, 4670 CrossRef CAS PubMed.
- M. Ma, L. Xiong, Y. Dong, Q. Bai, W. Hua, Z. Zheng, F. Lyu, Y. Lian, Z. Wei and H. Yuan, Adv. Funct. Mater., 2024, 34, 2315667 CrossRef CAS.
- H. Guo, Q. Huang, D. Li, S. Dai, K. Yang, S. Chen, W. Ma, Q. Li and J. Duan, J. Mater. Chem. A, 2025, 13, 348–355 RSC.
- L. Ma, Q. H. Geng, L. L. Fan, J. X. Li, D. W. Du, J. L. Bai and C. L. Li, Nano Res., 2023, 16, 9065–9072 CrossRef CAS.
- F. W. Li, A. Thevenon, A. Rosas-Hernandez, Z. Y. Wang, Y. L. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. H. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Z. Tao, Z. Q. Liang, M. C. Luo, X. Wang, H. H. Li, C. P. O'Brien, C. S. Tan, D. H. Nam, R. Quintero-Bermudez, T. T. Zhuang, Y. G. C. Li, Z. J. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- C. Lu, Q. He, S. Huang, P. Shi, C. Yang, J. Zhang, J. Zhu, J. Zhang, T. Wang and X. Zhuang, Adv. Mater., 2024, 9, 2415092 Search PubMed.
- Z. Y. Du, S. B. Li, G. H. Liang, Y. M. Xie, Y. L. A, Y. Zhang, H. Zhang, J. H. Tian, S. S. Zheng, Q. N. Zheng, Z. Chen, W. F. Ip, J. X. Liu and J. F. Li, J. Am. Chem. Soc., 2024, 146, 32870–32879 CrossRef CAS PubMed.
- J. Kim, T. Lee, H. D. Jung, M. Kim, J. Eo, B. Kang, H. Jung, J. Park, D. Bae, Y. Lee, S. Park, W. Kim, S. Back, Y. Lee and D. H. Nam, Nat. Commun., 2024, 15, 192 CrossRef PubMed.
- Y. H. Li, R. Z. Chen, J. Z. Liu, L. Cheng, J. W. Zhao, Y. Lu, J. Q. Feng, Z. M. Gong and C. Z. Li, Chem. Eng. Sci., 2023, 267, 118354 CrossRef CAS.
- Z. J. J. Zhu, Y. H. Zhu, Z. X. Ren, D. Liu, F. Y. Yue, D. F. Sheng, P. P. Shao, X. Y. Huang, X. Feng, A. X. Yin, J. Xie and B. Wang, J. Am. Chem. Soc., 2024, 146, 1572–1579 CrossRef CAS PubMed.
- M. W. Fang, X. Miao, Z. H. Huang, M. L. Wang, X. C. Feng, Z. W. Wang, Y. Zhu, L. M. Dai and L. Jiang, J. Am. Chem. Soc., 2024, 146, 27060–27069 CrossRef CAS PubMed.
- X. L. Zhou, J. Q. Shan, M. Zheng, H. Li, B. Y. Xia and Y. Zheng, Sci. China Mater., 2024, 67, 1858–1865 CrossRef CAS.
- Y. Pan, X. Q. Li, G. Y. Duan, J. Fang and B. H. Xu, Appl. Catal., B, 2025, 361, 124681 CrossRef CAS.
- T. Zhang, Z. Li, A. K. Ummireddi and J. Wu, Trends Chem., 2023, 5, 252–266 CrossRef CAS.
- Q. Qin, H. Suo, L. Chen, Y. X. Wang, J. Z. Wang, H. K. Liu, S. X. Dou, M. Lao and W. H. Lai, Adv. Mater. Interfaces, 2024, 11, 2301049 CrossRef CAS.
- Y. Ma, J. Yu, M. Sun, B. Chen, X. Zhou, C. Ye, Z. Guan, W. Guo, G. Wang and S. Lu, Adv. Mater., 2022, 34, 2110607 CrossRef CAS PubMed.
- C. Morales-Guio, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- J. Monzó, Y. Malewski, R. Kortlever, F. J. Vidal-Iglesias, J. Solla-Gullón, M. Koper and P. Rodriguez, J. Mater. Chem. A, 2015, 3, 23690–23698 RSC.
- Y. Chen, X.-Y. Li, Z. Chen, A. Ozden, J. E. Huang, P. Ou, J. Dong, J. Zhang, C. Tian and B.-H. Lee, Nat. Nanotechnol., 2024, 19, 311–318 CrossRef CAS PubMed.
- M. Liu, Q. Wang, T. Luo, M. Herran, X. Cao, W. Liao, L. Zhu, H. Li, A. Stefancu and Y.-R. Lu, J. Am. Chem. Soc., 2023, 146, 468–475 CrossRef PubMed.
- X. Kong, J. Zhao, J. Ke, C. Wang, S. Li, R. Si, B. Liu, J. Zeng and Z. Geng, Nano Lett., 2022, 22, 3801–3808 CrossRef CAS PubMed.
- F.-Z. Li, H.-G. Qin, H.-L. Zhang, X. Yue, L.-K. Fu, B. Xu, M. Lin and J. Gu, Joule, 2024, 8, 1772–1789 CrossRef CAS.
- M. Alfath and C. W. Lee, Catalysts, 2020, 10, 859 CrossRef CAS.
- Y. Lin, C. Xia, Z. Zhu, J. Wang, H. Niu, S. Gong, Z. Li, N. Yang, J. Song Chen and R. Wu, Angew. Chem., Int. Ed., 2024, 137, e202414569 CrossRef.
- C. Wei, Y. Yang, H. Ma, G. Sun, X. Wang, Y. Cheng, C. Zhang, B. S. Yeo, C. He and A. B. Wong, Adv. Funct. Mater., 2023, 33, 2214992 CrossRef CAS.
- B. Yang, J. Zeng, Z. Zhang, L. Meng, D. Shi, L. Chen and Y. Huang, J. Energy Chem., 2024, 90, 233–243 CrossRef CAS.
- Q. Sun, W. H. Ren, Y. Zhao and C. Zhao, Chem. Commun., 2021, 57, 1514–1517 RSC.
- H. Y. Li, L. Z. Fang, T. Wang, R. Bai, J. Zhang, T. Li, Z. Y. Duan, K. J. Chen and F. P. Pan, Adv. Mater., 2024, 2416337 Search PubMed.
- B. T. Chen, B. R. Li, Z. Q. Tian, W. B. Liu, W. P. Liu, W. W. Sun, K. Wang, L. Chen and J. Z. Jiang, Adv. Energy Mater., 2021, 11, 2102152 CrossRef CAS.
- Q. R. Wei, J. Y. Qin, G. X. Jia, Y. Zhao, Z. Y. Guo, G. H. Cheng, W. S. Ma, W. F. Yang and Z. H. Zhang, J. Phys. Chem. Lett., 2022, 13, 9058–9065 CrossRef CAS PubMed.
- Z. Wang, D. Liu, C. Xia, X. Shi, Y. Zhou, Q. Liu, J. Huang, H. Wu, D. Zhu and S. Zhang, Nat. Commun., 2025, 16, 1754 CrossRef CAS PubMed.
- X. Liu, S. Li, A. Chen, X. Dong, J. Mao, C. Zhu, G. Wu, Y. Wei, J. Xia and H. Zhu, ACS Catal., 2025, 15, 4259–4269 CrossRef.
- A. H. Chen, X. Dong, J. N. Mao, W. Chen, C. Zhu, S. J. Li, G. F. Wu, Y. H. Wei, X. H. Liu, G. H. Li, Y. F. Song, Z. Jiang, W. Wei and Y. H. Sun, Appl. Catal., B, 2023, 333, 122768 CrossRef CAS.
- Y. Wei, Y. Song, C. Zhu, J. Mao, A. Chen, G. Feng, G. Wu, X. Liu, S. Li and G. Li, The Innovation, 2025, 6, 100844 CrossRef CAS.
- Y. Wei, X. Wang, J. Mao, Y. Song, H. Zhu, X. Liu, C. Luo, S. Li, A. Chen and G. Li, Angew. Chem., Int. Ed., 2025, 137, e202423370 CrossRef.
- J. Xia, S. Li, X. Liu, X. Dong, J. Mao, A. Chen, H. Zhu, X. Wang, Z. Xu and Y. Wei, Appl. Catal., B, 2025, 371, 125202 CrossRef CAS.
- C. Zhu, G. F. Wu, A. H. Chen, G. H. Feng, X. Dong, G. H. Li, S. J. Li, Y. F. Song, W. Wei and W. Chen, Energy Environ. Sci., 2024, 17, 510–517 RSC.
- C. Zhu, G. F. Wu, J. N. Mao, A. H. Chen, Y. H. Zhao, G. H. Feng, Y. H. Wei, X. H. Liu, S. J. Li, G. H. Li, X. Dong, Y. F. Song, W. Wei and W. Chen, Chem. Eng. J., 2024, 485, 150040 CrossRef CAS.
- Y. Xie, P. F. Ou, X. Wang, Z. Y. Xu, Y. C. Li, Z. Y. Wang, J. E. Huang, J. Wicks, C. McCallum, N. Wang, Y. H. Wang, T. X. Chen, B. T. W. Lo, D. Sinton, J. C. Yu, Y. Wang and E. H. Sargent, Nat. Catal., 2022, 5, 564–570 CrossRef CAS.
- D. H. Nam, O. Shekhah, A. Ozden, C. McCallum, F. Li, X. Wang, Y. Lum, T. Lee, J. Li and J. Wicks, Adv. Mater., 2022, 34, 2207088 CrossRef CAS PubMed.
- M. Y. Fan, J. E. Huang, R. K. Miao, Y. Mao, P. F. Ou, F. Li, X. Y. Li, Y. F. Cao, Z. S. Zhang, J. Q. Zhang, Y. Yan, A. Ozden, W. Y. Ni, Y. Wang, Y. Zhao, Z. Chen, B. Khatir, C. P. O'Brien, Y. Xu, Y. C. Xiao, G. I. N. Waterhouse, K. Golovin, Z. Y. Wang, E. H. Sargent and D. Sinton, Nat. Catal., 2023, 6, 763–772 CrossRef CAS.
- Y. Zhao, L. Hao, A. Ozden, S. J. Liu, R. K. Miao, P. F. Ou, T. Alkayyali, S. Z. Zhang, J. Ning, Y. X. Liang, Y. Xu, M. Y. Fan, Y. J. Chen, J. E. Huang, K. Xie, J. Q. Zhang, C. P. O'Brien, F. W. Li, E. H. Sargent and D. Sinton, Nat. Synth., 2023, 2, 403–412 CAS.
- Y. Cao, Z. Chen, P. Li, A. Ozden, P. Ou, W. Ni, J. Abed, E. Shirzadi, J. Zhang and D. Sinton, Nat. Commun., 2023, 14, 2387 CrossRef CAS PubMed.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden and S.-F. Hung, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- M. Luo, Z. Wang, Y. C. Li, J. Li, F. Li, Y. Lum, D.-H. Nam, B. Chen, J. Wicks and A. Xu, Nat. Commun., 2019, 10, 5814 CrossRef CAS PubMed.
- L. Yuan, Q. Q. Wan, W. X. Jiang, J. B. Hou, X. D. Zhuang, J. L. Zhang and C. C. Ke, Electrochim. Acta, 2024, 475, 143662 CrossRef CAS.
- T. Shao, K. Yang, S. Chen, M. Zheng, Y. Zhang, Q. Li and J. Duan, Carbon Energy, 2024, 6, e416 CrossRef CAS.
- X. Zhang, X. Yan, P. Chen, P. Zhang, X. Kang, J. Ma, C. Chen and B. Han, Angew. Chem., Int. Ed., 2024, 136, e202315822 CrossRef.
- Y. Lin, T. Wang, L. L. Zhang, G. Zhang, L. L. Li, Q. F. Chang, Z. F. Pang, H. Gao, K. Huang, P. Zhang, Z. J. Zhao, C. L. Pei and J. L. Gong, Nat. Commun., 2023, 14, 3575 CrossRef CAS PubMed.
- T. H. Pham, J. Zhang, M. Li, T. H. Shen, Y. Ko, V. Tileli, W. Luo and A. Züttel, Adv. Energy Mater., 2022, 12, 2103663 CrossRef CAS.
- L. Su, Q. Hua, G. Feng, Y. Yang, H. Mei, Y. Yu, X. Chang and Z. Huang, Adv. Funct. Mater., 2025, 2425636 CrossRef.
- J. Liu, B. Zhang, D. R. Chen, O. W. Peng, H. B. Ma, S. B. Xi, C. Wu, Q. K. Hu, K. Zhang, J. Y. Feng and K. P. Loh, Angew. Chem., Int. Ed., 2024, 63, e202412266 CrossRef CAS PubMed.
- W. W. Fu, Y. K. Li, J. Y. Chen, J. Y. Chen, S. B. Xi, J. Zhang and L. Wang, Angew. Chem., Int. Ed., 2024, 63, e202407992 CrossRef CAS PubMed.
- Y. C. Yao, T. Shi, W. X. Chen, J. H. Wu, Y. Y. Fan, Y. C. Liu, L. Cao and Z. Chen, Nat. Commun., 2024, 15, 1257 CrossRef CAS PubMed.
- H. L. Wu, J. Li, K. Qi, Y. Zhang, E. Petit, W. S. Wang, V. Flaud, N. Onofrio, B. Rebiere, L. Q. Huang, C. Salameh, L. Lajaunie, P. Miele and D. Voiry, Nat. Commun., 2021, 12, 7210 CrossRef CAS PubMed.
- Y. Wang, Y. Cheng, S. Liu, Y. Yin, J. Yang, H. Wang, K. Li, M. Zhou, J. Jiao and P. Zhang, Angew. Chem., Int. Ed., 2025, e202420661 CAS.
- J. H. Bi, P. S. Li, J. Y. Liu, S. Q. Jia, Y. Wang, Q. G. Zhu, Z. M. Liu and B. X. Han, Nat. Commun., 2023, 14, 2823 CrossRef CAS PubMed.
- X. W. Du, P. Zhang, G. Zhang, H. Gao, L. L. Zhang, M. M. Zhang, T. Wang and J. L. Gong, Natl. Sci. Rev., 2024, 11, nwad149 CrossRef CAS PubMed.
- W. Z. Li, Z. L. Yin, Z. Y. Gao, G. W. Wang, Z. Li, F. Y. Wei, X. Wei, H. Q. Peng, X. T. Hu, L. Xiao, J. T. Lu and L. Zhuang, Nat. Energy, 2022, 7, 835–843 CrossRef CAS.
- A. Ozden, F. Li, F. P. García de Arquer, A. Rosas-Hernández, A. Thevenon, Y. Wang, S.-F. Hung, X. Wang, B. Chen and J. Li, ACS Energy Lett., 2020, 5, 2811–2818 CrossRef CAS.
- P. Papangelakis, R. K. Miao, R. H. Lu, H. Q. Liu, X. Wang, A. Ozden, S. J. Liu, N. Sun, C. P. O'Brien, Y. F. Hu, M. Shakouri, Q. F. Xiao, M. S. Li, B. Khatir, J. E. Huang, Y. K. Wang, Y. C. Xiao, F. Li, A. S. Zeraati, Q. Zhang, P. Y. Liu, K. Golovin, J. Y. Howe, H. Y. Liang, Z. Y. Wang, J. Li, E. H. Sargent and D. Sinton, Nat. Energy, 2024, 9, 1011–1020 CrossRef CAS.
- T. Möller, W. Ju, A. Bagger, X. L. Wang, F. Luo, T. N. Thanh, A. S. Varela, J. Rossmeisl and P. Strasser, Energy Environ. Sci., 2019, 12, 640–647 RSC.
- J. Chen, X. Peng, Z. Li, B. Yang, Q. Zhang, J. Lu, L. Lei and Y. Hou, Adv. Mater., 2024, 2409106 CrossRef PubMed.
- G. Díaz-Sainz, M. Alvarez-Guerra and A. Irabien, J. CO2 Util., 2022, 56, 101822 CrossRef.
- X. She, L. Zhai, Y. Wang, P. Xiong, M. M. J. Li, T. S. Wu, M. C. Wong, X. Guo, Z. Xu and H. Li, Nat. Energy, 2024, 9, 81–91 CrossRef CAS.
- C. P. O’Brien, R. K. Miao, A. Shayesteh Zeraati, G. Lee, E. H. Sargent and D. Sinton, Chem. Rev., 2024, 124, 3648–3693 CrossRef PubMed.
- K. Yang, M. Li, T. Gao, G. Xu, D. Li, Y. Zheng, Q. Li and J. Duan, Nat. Commun., 2024, 15, 7060 CrossRef CAS PubMed.
- J. Yu, J. Xiao, Y. Ma, J. Zhou, P. Lu, K. Wang, Y. Yan, J. Zeng, Y. Wang and S. Song, Chem. Catal., 2023, 3, 100670 CrossRef CAS.
- Z. F. Yan, J. L. Hitt, Z. C. Zeng, M. A. Hickner and T. E. Mallouk, Nat. Chem., 2021, 13, 33–40 CrossRef CAS PubMed.
- Y. Xu, J. P. Edwards, S. J. Liu, R. K. Miao, J. E. Huang, C. M. Gabardo, C. P. O'Brien, J. Li, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 809–815 CrossRef CAS.
- Y. Xu, R. K. Miao, J. P. Edwards, S. J. Liu, C. P. O'Brien, C. M. Gabardo, M. Y. Fan, J. E. Huang, A. Robb, E. H. Sargent and D. Sinton, Joule, 2022, 6, 1333–1343 CrossRef CAS.
- W. H. Ren, A. N. Xu, K. R. Chan and X. L. Hu, Angew. Chem., Int. Ed., 2022, 61, e202214173 CrossRef CAS PubMed.
- G. H. Lee, A. S. Rasouli, B. H. Lee, J. Q. Zhang, D. H. Won, Y. C. Xiao, J. P. Edwards, M. G. Lee, E. D. Jung, F. Arabyarmohammadi, H. Z. Liu, I. Grigioni, J. Abed, T. Alkayyali, S. J. Liu, K. Xie, R. K. Miao, S. Park, R. Dorakhan, Y. Zhao, C. P. O'Brien, Z. Chen, D. Sinton and E. Sargent, Joule, 2023, 7, 1277–1288 CrossRef CAS.
- S. Yuan, R. Y. Wang, R. Xue, L. Z. Wu, G. R. Zhang, H. Y. Li, Q. Wang, J. W. Yin, L. X. Luo, S. Y. Shen, L. An, X. H. Yan and J. L. Zhang, ACS Energy Lett., 2024, 9, 5945–5954 CrossRef CAS.
- D. U. Lee, B. Joensen, J. Jenny, V. M. Ehlinger, S. W. Lee, K. Abiose, Y. Xu, A. Sarkar, T. Y. Lin, C. Hahn and T. F. Jaramillo, ACS Sustainable Chem. Eng., 2023, 11, 16661–16668 CrossRef CAS.
- L. Lin, X. Y. He, X. G. Zhang, W. C. Ma, B. Zhang, D. Y. Wei, S. J. Xie, Q. H. Zhang, X. D. Yi and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202214959 CrossRef CAS PubMed.
- R. K. Miao, Y. Xu, A. Ozden, A. Robb, C. P. O'Brien, C. M. Gabardo, G. Lee, J. P. Edwards, J. E. Huang, M. Y. Fan, X. Wang, S. J. Liu, Y. Yan, E. H. Sargent and D. Sinton, Joule, 2021, 5, 2742–2753 CrossRef CAS.
- J. Fan, B. B. Pan, J. L. Wu, C. C. Shao, Z. Y. Wen, Y. C. Yan, Y. H. Wang and Y. G. Li, Angew. Chem., Int. Ed., 2024, 63, e202317828 CrossRef CAS PubMed.
- P. Zhu, C. Xia, C. Y. Liu, K. Jiang, G. H. Gao, X. Zhang, Y. Xia, Y. J. Lei, H. N. Alshareef, T. P. Senftle and H. T. Wang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2010868118 CrossRef CAS PubMed.
- X. P. Yan, M. L. Zhang, Y. Z. Chen, Y. H. Wu, R. Z. Wu, Q. Wan, C. X. Liu, T. T. Zheng, R. J. Feng, J. Zhang, C. J. Chen, C. Xia, Q. G. Zhu, X. F. Sun, Q. L. Qian and B. X. Han, Angew. Chem., Int. Ed., 2023, 62, e202301507 CrossRef CAS PubMed.
- C. M. Zhao, G. Luo, X. K. Liu, W. Zhang, Z. J. Li, Q. Xu, Q. H. Zhang, H. J. Wang, D. M. Li, F. Y. Zhou, Y. T. Qu, X. Han, Z. Z. Zhu, G. Wu, J. Wang, J. F. Zhu, T. Yao, Y. F. Li, H. J. M. Bouwmeester and Y. E. Wu, Adv. Mater., 2020, 32, 2002382 CrossRef CAS PubMed.
- H. L. Zhu, J. R. Huang, M. D. Zhang, C. Yu, P. Q. Liao and X. M. Chen, J. Am. Chem. Soc., 2024, 146, 1144–1152 CrossRef CAS PubMed.
- G. Wen, B. Ren, X. Wang, D. Luo, H. Dou, Y. Zheng, R. Gao, J. Gostick, A. Yu and Z. Chen, Nat. Energy, 2022, 7, 978–988 CrossRef CAS.
- C. P. O'Brien, R. K. Miao, S. J. Liu, Y. Xu, G. Lee, A. Robb, J. E. Huang, K. Xie, K. Bertens, C. M. Gabardo, J. P. Edwards, C. T. Dinh, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 2952–2959 CrossRef.
- B. Endrödi, A. Samu, E. Kecsenovity, T. Halmágyi, D. Sebök and C. Janáky, Nat. Energy, 2021, 6, 439–448 CrossRef PubMed.
- S. X. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed.
- C. P. O'Brien, D. McLaughlin, T. Boehm, Y. C. Xiao, J. P. Edwards, C. M. Gabardo, M. Bierling, J. Wicks, A. S. Rasouli, J. Abed, D. Young, C. T. Dinh, E. H. Sargent, S. Thiele and D. Sinton, Joule, 2024, 8, 2903–2919 CrossRef.
- H. Zhang, Matter, 2024, 7, 421–429 Search PubMed.
- S. Overa, B. S. Crandall, B. Shrimant, D. Tian, B. H. Ko, H. Shin, C. Bae and F. Jiao, Nat. Catal., 2022, 5, 738–745 CrossRef CAS.
- W. Deng, A. Lee, W. Dai, L. Cherniack, B. S. Crandall, H. Li and F. Jiao, Nat. Rev. Clean Technol., 2025, 1–14 Search PubMed.
- Y. S. Zhou, R. Ganganahalli, S. Verma, H. R. Tan and B. S. Yeo, Angew. Chem., Int. Ed., 2022, 61, e202202859 CrossRef CAS PubMed.
- T. Moeller, M. Filippi, S. Brueckner, W. Ju and P. Strasser, Nat. Commun., 2023, 14, 5680 CrossRef CAS PubMed.
- A. Ozden, Y. H. Wang, F. W. Li, M. C. Luo, J. Sisler, A. Thevenon, A. Rosas-Hernández, T. Burdyny, Y. W. Lum, H. Yadegari, T. Agapie, J. C. Peters, E. H. Sargent and D. Sinton, Joule, 2021, 5, 706–719 CrossRef CAS.
- S. Overa, T. G. Feric, A. H. A. Park and F. Jiao, Joule, 2021, 5, 8–13 CrossRef.
- J. Zhang, X. S. Kang, Y. C. Yan, X. Ding, L. He and Y. G. Li, Angew. Chem., Int. Ed., 2024, 63, e202315777 CrossRef CAS PubMed.
- M. G. Lee, S. Kandambeth, X. Y. Li, O. Shekhah, A. Ozden, J. Wicks, P. F. Ou, S. S. Wang, R. Dorakhan, S. Park, P. M. Bhatt, V. S. Kale, D. Sinton, M. Eddaoudi and E. H. Sargent, J. Am. Chem. Soc., 2024, 146, 14267–14277 CrossRef CAS PubMed.
- M. G. Lee, X. Y. Li, A. Ozden, J. Wicks, P. F. Ou, Y. H. Li, R. Dorakhan, J. K. Y. Lee, H. K. Park, J. W. Yang, B. Chen, J. Abed, R. dos Reis, G. H. Lee, J. E. Huang, T. Peng, Y. H. Chin, D. Sinton and E. H. Sargent, Nat. Catal., 2023, 6, 310–318 CrossRef CAS.
- L. Fan, Y. L. Zhao, L. Chen, J. Y. Chen, J. M. Chen, H. Z. Yang, Y. K. Xiao, T. Y. Zhang, J. Y. Chen and L. Wang, Nat. Catal., 2023, 6, 585–595 CrossRef CAS.
- S. van Bavel, S. Verma, E. Negro and M. Bracht, ACS Energy Lett., 2020, 5, 2597–2601 CrossRef CAS.
- Y. Pu, Y. Wang, G. Y. Wu, X. B. Wu, Y. L. Lu, Y. Y. Yu, N. Chu, X. H. He, D. P. Li, R. J. Zeng and Y. Jiang, Environ. Sci. Technol., 2024, 58, 7445–7456 CrossRef CAS PubMed.
- T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Nat. Catal., 2018, 1, 32–39 CrossRef CAS.
- S. Fernández, E. A. Assaf, S. Ahmad, B. D. Travis, J. B. Curley, N. Hazari, M. Z. Ertem and A. J. Miller, Angew. Chem., Int. Ed., 2025, 137, e202416061 CrossRef.
- B. S. Crandall, M. Naughton, S. Park, J. Yu, C. Y. Zhang, S. Mahtabian, K. Y. Wang, X. H. Liang, K. Fu and F. Jiao, Nat. Commun., 2024, 15, 1–9 Search PubMed.
- Z. Y. Wen, M. J. Wang, C. L. Liang, B. J. Fan, Y. C. Yan, J. Fan, N. Han, Y. H. Wang and Y. G. Li, J. Am. Chem. Soc., 2024, 146, 32575–32581 CrossRef CAS PubMed.
- P. P. Wang, X. Wang, S. Chandra, A. Lielpetere, T. Quast, F. Conzuelo and W. Schuhmann, Angew. Chem., Int. Ed., 2025, e202422882 CAS.
- N. R. Cuellar, C. Scherer, B. Kaçkar, W. Eisenreich, C. Huber, K. Wiesner-Fleischer, M. Fleischer and O. Hinrichsen, J. CO2 Util., 2020, 36, 263–275 CrossRef.
- G. Wu, Y. Song, Q. Zheng, C. Long, T. Fan, Z. Yang, X. Huang, Q. Li, Y. Sun and L. Zuo, Adv. Energy Mater., 2022, 12, 2202054 CrossRef CAS.
- T. Zheng, M. Zhang, L. Wu, S. Guo, X. Liu, J. Zhao, W. Xue, J. Li, C. Liu, X. Li, Q. Jiang, J. Bao, J. Zeng, T. Yu and C. Xia, Nat. Catal., 2022, 5, 388–396 CrossRef CAS.
- E. C. Hann, S. Overa, M. Harland-Dunaway, A. F. Narvaez, D. N. Le, M. L. Orozco-Cárdenas, F. Jiao and R. E. Jinkerson, Nat. Food, 2022, 3, 461–471 CrossRef CAS PubMed.
- A. J. Martín, G. O. Larrazábal and J. Pérez-Ramírez, Green Chem., 2015, 17, 5114–5130 RSC.
- S. S. He, F. L. Ni, Y. J. Ji, L. E. Wang, Y. Z. Wen, H. P. Bai, G. J. Liu, Y. Zhang, Y. Y. Li, B. Zhang and H. S. Peng, Angew. Chem., Int. Ed., 2018, 57, 16114–16119 CrossRef CAS PubMed.
- R. Xia, J.-J. Lv, X. Ma and F. Jiao, J. Catal., 2021, 398, 185–191 CrossRef CAS.
- J. Li, Z. Y. Wang, C. McCallum, Y. Xu, F. W. Li, Y. H. Wang, C. M. Gabardo, C. T. Dinh, T. T. Zhuang, L. Wang, J. Y. Howe, Y. Ren, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 1124–1131 CrossRef CAS.
- W. Lai, Y. Qiao, J. Zhang, Z. Lin and H. Huang, Energy Environ. Sci., 2022, 15, 3603–3629 RSC.
- S. Verma, S. Lu and P. J. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- M. Zeng, W. Fang, Y. Cen, X. Zhang, Y. Hu and B. Y. Xia, Angew. Chem., Int. Ed., 2024, 63, e202404574 CrossRef CAS PubMed.
- W. Lai, Y. Qiao, Y. Wang and H. Huang, Adv. Mater., 2023, 35, 2306288 CrossRef CAS PubMed.
- Y. Wen, C. Zhan, J. Liu, X. Zhuang, S. Liu, T. Yang, W. Liu, X. Liu, C. W. Kao, Y. C. Huang, T. S. Chan, Z. Hu, D. Su, J. Han, N. Chen and X. Huang, Nat. Nanotechnol., 2025, 1–8 Search PubMed.
- L. Li, A. Ozden, S. Guo, F. P. García de Arquer, C. Wang, M. Zhang, J. Zhang, H. Jiang, W. Wang and H. Dong, Nat. Commun., 2021, 12, 5223 CrossRef CAS PubMed.
- S. You, J. Xiao, S. Liang, W. Xie, T. Zhang, M. Li, Z. Zhong, Q. Wang and H. He, Energy Environ. Sci., 2024, 17, 5795–5818 RSC.
- L. B. Zhang, J. Q. Feng, S. J. Liu, X. X. Tan, L. M. Wu, S. H. Jia, L. Xu, X. D. Ma, X. N. Song, J. Ma, X. F. Sun and B. X. Han, Adv. Mater., 2023, 35, 2209590 CrossRef CAS PubMed.
- H. L. Zhu, H. Y. Chen, Y. X. Han, Z. H. Zhao, P. Q. Liao and X. M. Chen, J. Am. Chem. Soc., 2022, 144, 13319–13326 CrossRef CAS PubMed.
- J. M. Heng, H. L. Zhu, Z. H. Zhao, D. S. Huang, J. Y. Li, P. Q. Liao and X. M. Chen, Research, 2022, 2022, 0008 CrossRef CAS PubMed.
- S. H. Chen, X. B. Zheng, P. Zhu, Y. P. Li, Z. C. Zhuang, H. J. Wu, J. X. Zhu, C. H. Xiao, M. Z. Chen, P. S. Wang, D. S. Wang and Y. L. He, Angew. Chem., Int. Ed., 2024, 63, e202411591 CrossRef CAS PubMed.
- L. Fan, F. Li, T. Liu, J. E. Huang, R. K. Miao, Y. Yan, S. Feng, C.-W. Tai, S.-F. Hung and H.-J. Tsai, Nat. Synth., 2024, 4, 262–270 CrossRef.
- Z. J. Min, C. F. Shao, B. Chang, N. Wang, Y. Zhao, S. Q. Gao, X. G. Zhang, M. H. Fan, S. J. Zhang and J. J. Wang, Chem. Eng. J., 2024, 502, 157813 CrossRef CAS.
- D. S. Huang, H. L. Zhu, Z. H. Zhao, J. R. Huang, P. Q. Liao and X. M. Chen, ACS Catal., 2022, 12, 8444–8450 CrossRef CAS.
- M. Zhang, S. H. Zhou, W. B. Wei, D. D. Ma, S. G. Han, X. F. Li, X. T. Wu, Q. Xu and Q. L. Zhu, Chem. Catal., 2022, 2, 3528–3545 CrossRef CAS.
- X. Jiang, L. Lin, Y. Rong, R. Li, Q. Jiang, Y. Yang and D. Gao, Nano Res., 2023, 16, 12050–12057 CrossRef CAS.
- M. Zhang, M. Lu, M. Y. Yang, J. P. Liao, Y. F. Liu, H. J. Yan, J. N. Chang, S. L. Li and Y. Q. Lan, eScience, 2023, 3, 100116 CrossRef.
- X. Cao, S. Ren, X. Zhang, Q. K. Fan, Q. Q. Chen, J. Yang and J. J. Mao, Chem, 2024, 10, 2089–2102 CAS.
- J. Liu, Y. Wen, W. Yan, Z. Huang, X. Liu, X. Huang, C. Zhan, Y. Zhang, W.-H. Huang, C.-W. Pao, Z. Hu, D. Su, S. Xie, Y. Wang, J. Han, H. Xiong, X. Huang and N. Chen, Energy Environ. Sci., 2025, 18, 4396–4404 RSC.
- N. Ye, K. Wang, Y. Tan, Z. Qian, H. Guo, C. Shang, Z. Lin, Q. Huang, Y. Liu, L. Li, Y. Gu, Y. Han, C. Zhou, M. Luo and S. Guo, Nat. Synth., 2025, 1–9 Search PubMed.
- N. Han, X. Y. Guo, J. L. Cheng, P. Y. Liu, S. G. Zhang, S. P. Huang, M. R. Rowles, J. Fransaer and S. M. Liu, Matter, 2021, 4, 1720–1734 CrossRef CAS.
- Y. Ma, M. Sun, H. Xu, Q. Zhang, J. Lv, W. Guo, F. Hao, W. Cui, Y. Wang, J. Yin, H. Wen, P. Lu, G. Wang, J. Zhou, J. Yu, C. Ye, L. Gan, D. Zhang, S. Chu, L. Gu, M. Shao, B. Huang and Z. Fan, Adv. Mater., 2024, 36, 2402979 CrossRef CAS PubMed.
- K. Liu, P. Lin, J. Li, Y. Liu, M. Wang, H. Cui and S. Yi, Adv. Funct. Mater., 2025, 2424357 CrossRef.
- J. Yu, J. Wang, Y. Ma, J. Zhou, Y. Wang, P. Lu, J. Yin, R. Ye, Z. Zhu and Z. Fan, Adv. Funct. Mater., 2021, 31, 2102151 CrossRef CAS.
- J. Ding, Q. Song, L. Xia, L. Ruan, M. Zhang, C. Ban, J. Meng, J. Ma, Y. Feng, Y. Wang, X. Tao, D. Yu, J. Y. Dai, L. Gan and X. Zhou, Nano Energy, 2024, 128, 109945 CrossRef CAS.
- Z. Guo, T. Wang, H. Liu, X. Jia, D. Zhang, L. Wei, J. Xu and H. Li, ACS Catal., 2025, 15, 3173–3183 CrossRef CAS.
- Z. Guo, Y. Yu, C. Li, E. Campos dos Santos, T. Wang, H. Li, J. Xu, C. Liu and H. Li, Angew. Chem., Int. Ed., 2024, 63, e202319913 CrossRef CAS PubMed.
- Y. Wang, D. Zhang, B. Sun, X. Jia, L. Zhang, H. Cheng, J. Fan and H. Li, Angew. Chem., Int. Ed., 2024, e202418228 Search PubMed.
- Z. L. Song, L. F. Fan, S. H. Lu, C. Y. Ling, Q. H. Zhou and J. L. Wang, Nat. Commun., 2025, 16, 1053 CrossRef CAS PubMed.
- N. Han and B. L. Su, Nat. Sci. Rev., 2025, nwaf110 CrossRef PubMed.
- Y. Shen, N. Fang, X. Liu, Y. Ling, Y. Su, T. Tan, F. Chen, H. Lin, B. Zhao, J. Wang, D. Si, S. Xie, Y. Wang, D. Zhou, T. Zhang, R. Cao and C. Wang, Nat. Commun., 2025, 16, 2073 CrossRef CAS PubMed.
- X. Yang, H. Ding, S. Li, S. Zheng, J. F. Li and F. Pan, J. Am. Chem. Soc., 2024, 146, 5532–5542 CrossRef CAS PubMed.
- H. Ding, S. Zheng, X. Yang, J. Pan, Z. Chen, M. Zhang, S. Li and F. Pan, ACS Catal., 2024, 14, 14330–14338 CrossRef CAS.
- C. X. Cui, Y. Shen, J. R. He, Y. Fu, X. Hong, S. Wang, J. Jiang and Y. Luo, J. Am. Chem. Soc., 2024, 146, 34551–34559 CrossRef CAS PubMed.
- X. Q. Han, X. D. Wang, M. Y. Xu, Z. Feng, B. W. Yao, P. J. Guo, Z. F. Gao and Z. Y. Lu, Chin. Phys. Lett., 2025, 42, 027403 CrossRef.
- Y. N. Xu, J. Li, J. C. Wu, W. Li, Y. Yang, H. Wu, H. Q. Fu, M. Zhu, X. L. Wang, S. Dai, C. Lian, P. F. Liu and H. G. Yang, Adv. Mater., 2025, 2500343 CrossRef CAS PubMed.
- T. Alkayyali, A. S. Zeraati, H. Mar, F. Arabyarmohammadi, S. Saber, R. K. Miao, C. P. O'Brien, H. S. Liu, Z. Xie, G. Y. Wang, E. H. Sargent, N. N. Zhao and D. Sinton, ACS Energy Lett., 2023, 8, 4674–4683 CrossRef CAS.
- K. Xie, R. K. Miao, A. Ozden, S. J. Liu, Z. Chen, C. T. Dinh, J. E. Huang, Q. C. Xu, C. M. Gabardo, G. Lee, J. P. Edwards, C. P. O'Brien, S. W. Boettcher, D. Sinton and E. H. Sargent, Nat. Commun., 2022, 13, 3609 CrossRef CAS PubMed.
- J. P. Edwards, Y. Xu, C. M. Gabardo, C. T. Dinh, J. Li, Z. B. Qi, A. Ozden, E. H. Sargent and D. Sinton, Appl. Energy, 2020, 261, 114305 CrossRef CAS.
- Y. Yang, F. He, X. Z. Lv, Q. Liu, A. J. Wu, Z. F. Qi and H. B. Wu, Small Methods, 2025, 9, 2400786 CrossRef CAS PubMed.
- X. Zhang, H. X. Lu, Y. Miao, Y. S. Zhang and J. Y. Wang, J. Environ. Chem. Eng., 2024, 12, 112369 CrossRef CAS.
- J. Shi, F. Shi, N. Song, J. X. Liu, X. K. Yang, Y. J. Jia, Z. W. Xiao and P. Du, J. Power Sources, 2014, 259, 50–53 CrossRef CAS.
- J. Jin, J. Wicks, Q. H. Min, J. Li, Y. F. Hu, J. Y. Ma, Y. Wang, Z. Jiang, Y. Xu, R. H. Lu, G. Z. Si, P. Papangelakis, M. Shakouri, Q. F. Xiao, P. F. Ou, X. Wang, Z. Chen, W. Zhang, K. S. Yu, J. Y. Song, X. H. Jiang, P. Qiu, Y. H. Lou, D. Wu, Y. Mao, A. Ozden, C. D. Wang, B. Y. Xia, X. B. Hu, V. P. Dravid, Y. M. Yiu, T. K. Sham, Z. Y. Wang, D. Sinton, L. Q. Mai, E. H. Sargent and Y. J. Pang, Nature, 2023, 617, 724–729 CrossRef CAS PubMed.
- A. S. Zeraati, F. Li, T. Alkayyali, R. Dorakhan, E. Shirzadi, F. Arabyarmohammadi, C. P. O'Brien, C. M. Gabardo, J. A. T. Kong, A. Ozden, M. Zargartalebi, Y. Zhao, L. Z. Fan, P. Papangelakis, D. Kim, S. Park, R. K. Miao, J. P. Edwards, D. Young, A. H. Ip, E. H. Sargent and D. Sinton, Nat. Synth., 2025, 4, 75–83 CrossRef.
- A. Ozden, J. Li, S. Kandambeth, X. Y. Li, S. J. Liu, O. Shekhah, P. F. Ou, Y. Z. Finfrock, Y. K. Wang, T. Alkayyali, F. P. G. de Arquer, V. S. Kale, P. M. Bhatt, A. H. Ip, M. Eddaoudi, E. H. Sargent and D. Sinton, Nat. Energy, 2023, 8, 179–190 CrossRef CAS.
- C. Kim, J. C. Bui, X. Y. Luo, J. K. Cooper, A. Kusoglu, A. Z. Weber and A. T. Bell, Nat. Energy, 2021, 6, 1026–1034 CrossRef CAS.
- M. Y. Fan, R. K. Miao, P. F. Ou, Y. Xu, Z. Y. Lin, T. J. Lee, S. F. Hung, K. Xie, J. E. Huang, W. Y. Ni, J. Li, Y. Zhao, A. Ozden, C. P. O'Brien, Y. J. Chen, Y. C. Xiao, S. J. Liu, J. Wicks, X. Wang, J. Abed, E. Shirzadi, E. H. Sargent and D. Sinton, Nat. Commun., 2023, 14, 3314 CrossRef CAS PubMed.
- C. L. Rong, S. H. Wang, X. Shen, C. Jia, Q. Sun, Q. Zhang and C. Zhao, Energy Environ. Sci., 2024, 17, 4196–4204 RSC.
- N. Han, X. Zhang, C. Zhang, S. H. Feng, W. Zhang, W. Guo, R. T. Zheng, R. J. Zheng, P. Y. Liu, Y. Li, J. Fransaer and B. L. Su, Matter, 2024, 7, 1330–1343 CrossRef CAS.
- C. L. Rong, X. J. Shen, Y. Wang, L. Thomsen, T. W. Zhao, Y. B. Li, X. Y. Lu, R. Amal and C. Zhao, Adv. Mater., 2022, 34, 2110103 CrossRef CAS PubMed.
- C. L. Rong, K. Dastafkan, Y. Wang and C. Zhao, Adv. Mater., 2023, 35, 2211884 CrossRef CAS PubMed.
- N. Han, S. H. Feng, Y. Liang, J. Wang, W. Zhang, X. L. Guo, Q. R. Ma, Q. Liu, W. Guo, Z. Y. Zhou, S. J. Xie, K. Wan, Y. Z. Jiang, A. Vlad, Y. Z. Guo, E. M. Gaigneaux, C. Zhang, J. Fransaer and X. Zhang, Adv. Funct. Mater., 2023, 33, 2208399 CrossRef CAS.
- N. Han, W. Zhang, W. Guo, H. Pan, B. Jiang, L. B. Xing, H. Tian, G. X. Wang, X. Zhang and J. Fransaer, Nano-Micro Lett., 2023, 15, 185 CrossRef CAS PubMed.
- X. Li, Q. S. Chen, W. Sun, C. C. He and Z. H. Wen, Angew. Chem., Int. Ed., 2024, 63, e202412410 CrossRef CAS PubMed.
- H. Yadegari, A. Ozden, T. Alkayyali, V. Soni, A. Thevenon, A. Rosas-Hernández, T. Agapie, J. C. Peters, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 3538–3544 CrossRef CAS.
- B. van den Bosch, B. Rawls, M. B. Brands, C. Koopman, M. F. Phillips, M. C. Figueiredo and G. J. M. Gruter, ChemPlusChem, 2023, 88, e202300112 CrossRef CAS PubMed.
- X. Y. Tan, C. Yu, Y. W. Ren, S. Cui, W. B. Li and J. S. Qiu, Energy Environ. Sci., 2021, 14, 765–780 RSC.
- H. Xiong, D. Wu, R. Jiang, H. Li, Q. Hu, X. Lu, B. Xu and Q. Lu, Angew. Chem., Int. Ed., 2025, e202504782 CAS.
- A. N. Biswas, Z. H. Xie, R. Xia, S. Overa, F. Jiao and J. G. Chen, ACS Energy Lett., 2022, 7, 2904–2910 CrossRef CAS.
- H. Wu, H. Yu, Y. L. Chow, P. A. Webley and J. Zhang, Adv. Mater., 2024, 36, 2403217 CrossRef CAS PubMed.
- W. Wu, L. Xu, Q. Lu, J. Sun, Z. Xu, C. Song, J. C. Yu and Y. Wang, Adv. Mater., 2025, 37, 2312894 CrossRef CAS PubMed.
- X. Lu, C. S. Zhou, R. S. Delima, E. W. Lees, A. Soni, D. J. Dvorak, S. X. Ren, T. X. Ji, A. Bahi, F. Ko and C. P. Berlinguette, Nat. Chem., 2024, 16, 979–987 CrossRef CAS PubMed.
- Y. H. Qu, K. C. Yang, W. Z. Li, G. Z. Wang, L. Xiao, G. W. Wang and L. Zhuang, ACS Energy Lett., 2024, 9, 3042–3048 CrossRef CAS.
- Y. D. Sun, F. H. Bai, J. P. Liu, S. Q. Sun, Y. L. Mao, X. J. Liu, Y. Huang and Y. H. Chen, J. Phys. Chem. Lett., 2024, 15, 9122–9128 CrossRef CAS PubMed.
- H. Warkentin, C. P. O'Brien, S. Holowka, B. Maxwell, M. Awara, M. Bouman, A. S. Zeraati, R. Nicholas, A. H. Ip, E. S. Elsahwi, C. M. Gabardo and D. Sinton, ChemSusChem, 2023, 16, e202300657 CrossRef CAS PubMed.
- J. Disch, L. Bohn, S. Koch, M. Schulz, Y. Y. Han, A. Tengattini, L. Helfen, M. Breitwieser and S. Vierrath, Nat. Commun., 2022, 13, 6099 CrossRef CAS PubMed.
- D. Shin, H. Karasu, K. Jang, C. Kim, K. Kim, D. Kim, Y. J. Sa, K. B. Lee, K. H. Chae, I. Moon, D. H. Won, J. Na and U. Lee, ACS Catal., 2025, 15, 2158–2170 CrossRef CAS.
- S. Hao, A. Elgazzar, N. Ravi, T.-U. Wi, P. Zhu, Y. Feng, Y. Xia, F.-Y. Chen, X. Shan and H. Wang, Nat. Energy, 2025, 1–12 CAS.
- S. Hao and H. Wang, Nat. Energy, 2025, 10, 164–165 Search PubMed.
- J. Biemolt, J. Singh, G. Prats Vergel, H. M. Pelzer and T. Burdyny, ACS Energy Lett., 2025, 10, 807–814 CrossRef CAS.
- Y. N. Guan, Y. Z. Li, Z. J. Li, Y. Hou, L. C. Lei and B. Yang, Adv. Mater., 2025, 2417567 CrossRef CAS PubMed.
- L. Yuan, X. Li, G. Li, K. Peng, H. Zhang, S. Zeng, X. Sun and X. Zhang, Adv. Sci., 2025, 2500368 CrossRef CAS PubMed.
- C. M. Gabardo, C. P. O'Brien, J. P. Edwards, C. McCallum, Y. Xu, C. T. Dinh, J. Li, E. H. Sargent and D. Sinton, Joule, 2019, 3, 2777–2791 CrossRef CAS.
- T. Kose, C. P. O'Brien, J. Wicks, J. Abed, Y. C. Xiao, B. Sutherland, A. Sarkar, S. A. Jaffer, E. H. Sargent and D. Sinton, Catal. Sci. Technol., 2022, 12, 6239–6245 RSC.
- J. P. Edwards, T. Alerte, C. P. O'Brien, C. M. Gabardo, S. J. Liu, J. Wicks, A. Gaona, J. Abed, Y. C. Xiao, D. Young, A. S. Rasouli, A. Sarkar, S. A. Jaffer, H. L. MacLean, E. H. Sargent and D. Sinton, ACS Energy Lett., 2023, 8, 2576–2584 CrossRef CAS.
- V. E. Nelson, C. P. O'Brien, J. P. Edwards, S. J. Liu, C. M. Gabardo, E. H. Sargent and D. Sinton, ACS Appl. Mater. Interfaces, 2024, 16, 50818–50825 CrossRef CAS PubMed.
- M. Wang, B. Q. Wang, J. G. Zhang, S. B. Xi, N. Ling, Z. Y. Mi, Q. Yang, M. S. Zhang, W. R. Leow, J. Zhang and Y. Lum, Nat. Commun., 2024, 15, 1218 CrossRef CAS PubMed.
- T. Alerte, J. P. Edwards, C. M. Gabardo, C. P. O'Brien, A. Gaona, J. Wicks, A. Obradovi, A. Sarkar, S. A. Jaffer, H. L. MacLean, D. Sinton and E. H. Sargent, ACS Energy Lett., 2021, 6, 4405–4412 CrossRef CAS.
- T. Alerte, A. Gaona, J. P. Edwards, C. M. Gabardo, C. P. O'Brien, J. Wicks, L. Bonnenfant, A. S. Rasouli, D. Young, J. Abed, L. Kershaw, Y. C. Xiao, A. Sarkar, S. A. Jaffer, M. W. Schreiber, D. Sinton, H. L. MacLean and E. H. Sargent, ACS Sustainable Chem. Eng., 2023, 11, 15651–15662 CrossRef CAS.
- K. Xie, A. Ozden, R. K. Miao, Y. H. Li, D. Sinton and E. H. Sargent, Nat. Commun., 2022, 13, 3070 CrossRef CAS PubMed.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cs00863d |
| This journal is © The Royal Society of Chemistry 2025 |