
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNatural, modified and conjugated carbohydrates in nucleic acids
Debashis
Dhara
ab,
Laurence A.
Mulard
 b and
Marcel
Hollenstein
*a
b and
Marcel
Hollenstein
*a
aDepartment of Structural Biology and Chemistry, Laboratory for Bioorganic Chemistry of Nucleic Acids, Institut Pasteur, Université Paris Cité, CNRS UMR 352328, rue du Docteur Roux, 75724 Paris Cedex 15, France. E-mail: marcel.hollenstein@pasteur.fr
bDepartment of Structural Biology and Chemistry, Laboratory for Chemistry of Biomolecules, Institut Pasteur, Université Paris Cité, CNRS UMR 3523, 28, rue du Docteur Roux, 75724 Paris Cedex 15, France
First published on 12th February 2025
Abstract
Storage of genetic information in DNA occurs through a unique ordering of canonical base pairs. However, this would not be possible in the absence of the sugar–phosphate backbone which is essential for duplex formation. While over a hundred nucleobase modifications have been identified (mainly in RNA), Nature is rather conservative when it comes to alterations at the level of the (deoxy)ribose sugar moiety. This trend is not reflected in synthetic analogues of nucleic acids where modifications of the sugar entity is commonplace to improve the properties of DNA and RNA. In this review article, we describe the main incentives behind sugar modifications in nucleic acids and we highlight recent progress in this field with a particular emphasis on therapeutic applications, the development of xeno-nucleic acids (XNAs), and on interrogating nucleic acid etiology. We also describe recent strategies to conjugate carbohydrates and oligosaccharides to oligonucleotides since this represents a particularly powerful strategy to improve the therapeutic index of oligonucleotide drugs. The advent of glycoRNAs combined with progress in nucleic acid and carbohydrate chemistry, protein engineering, and delivery methods will undoubtedly yield more potent sugar-modified nucleic acids for therapeutic, biotechnological, and synthetic biology applications.
 Debashis Dhara | Debashis Dhara is a postdoctoral research associate in the Department of Chemistry at the University of Oxford, UK. He obtained his BSc in Chemistry (Honours) from the University of Calcutta in 2009 and completed his MSc in Chemistry there in 2011. He pursued his PhD at Bose Institute, Kolkata, from 2012 to 2017, followed by postdoctoral research at the University of Galway, Ireland (2017–2018), and the Institut Pasteur, Paris (2018–2023). His research focuses on carbohydrates, nucleic acids, glycoconjugate vaccines, therapeutic oligonucleotides, and sustainable chemistry. |
 Laurence A. Mulard | Laurence A. Mulard graduated as an engineer from the Ecole Supérieure de Physique et Chimie de la ville de Paris (ESPCI, Paris, France). She obtained her PhD in Chemistry from the Université Pierre et Marie Curie (UPMC, Paris, France). After a post-doctoral stay at the National Institutes of Health (NIH, Bethesda, MD, USA), she joined Institut Pasteur (Paris, France) where she was appointed Head of the Chemistry of Biomolecules Laboratory in 2008. Whether involving peptide science or glycoscience, her research interests focus on the development of chemical tools and bioactive compounds aimed at studying and interfering with molecular phenomena governing infectious diseases. The focus is on peptide/carbohydrate-based diversity oriented synthesis and bioconjugates, on investigating synthetic glycans as surrogates of bacterial polysaccharides, and on advancing synthetic carbohydrate-based conjugate vaccines up to clinics. |
 Marcel Hollenstein | Marcel Hollenstein received his PhD in Chemistry (2004) from the University of Bern (Switzerland) under the supervision of Prof. Christian Leumann. He then moved to the group of Prof. David Perrin (University of British Columbia, Vancouver) as a postdoctoral fellow. In 2009, he moved back to the University of Bern as a Swiss National Science Foundation Ambizione Fellow. He then moved to Institut Pasteur in Paris, France where he was first an independent junior research group leader (2016–2022) and then a senior research group leader (from 2022 on). Research activities in his lab focus on the selection of modified aptamers, the enzymatic synthesis of modified nucleic acids, and the development of biocatalytic approaches for the synthesis of modified oligonucleotides, particularly with therapeutically relevant modifications. |
1. Introduction
Oligonucleotides are one of the most important classes of biomolecules that are present in all domains of life. DNA serves as the storage of genetic information while RNA relays this information to the protein synthesis machinery. Both DNA and RNA are made of nucleotide building blocks consisting of a phosphate backbone, a pentose, and a heterocyclic nucleobase (Fig. 1). Besides these structural key features, DNA and RNA can be transiently modified mainly at the level of the nucleobase. These epigenetic modifications allow regulation and alteration of gene expression without requiring a change in the sequence composition.1–5 Interestingly, so far, over 160 such chemical alterations have been identified in RNA and 17 in DNA.1 These modifications range from simple methylation patterns to more intricate substitution patterns.6–9 On the other hand, naturally occurring chemical modifications at the level of the sugar unit are rather scarce and are mainly restricted to the 2′-position of the ribose moiety.10–12 This dearth of chemical diversity of the sugar unit is in stark contrast with the chemical and structural diversity present in natural mono- and oligosaccharides.13,14 Another intriguing question is Nature's choice of five- rather than six-membered sugar building blocks.15–18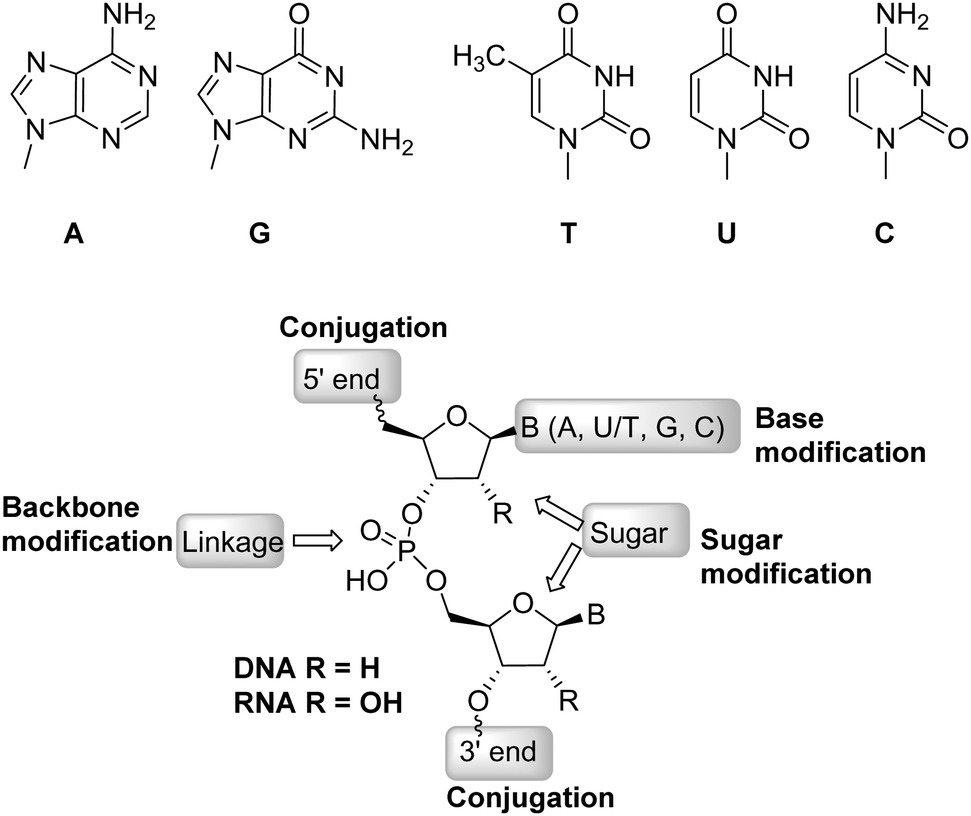 | ||
| Fig. 1 Top: Structures of nucleobases. Bottom: Representation of nucleic acids and their scope of various modifications. | ||
This question can be addressed by synthesizing nucleic acid analogues with building blocks containing modified sugar moieties and comparing their chemical, biochemical, and structural properties to those of canonical DNA and RNA. Experiments towards this aim, pioneered by Albert Eschenmoser, have shown that the furanose plays an important role in maintaining an adequate structure for helix formation by imposing conformational restraints on nucleotides and represents the best balance between duplex stability, structural diversity and flexibility.19,20 On the other hand, six-membered hexopyranosyl-(4′ → 6′) oligonucleotides (homo-DNA; vide infra) display much stronger Watson–Crick base pairing capacities than their natural, five-membered ring containing counterparts and are unable to interact with canonical DNA and RNA.16 The origin of the enhanced thermodynamic stability of homo-DNA is believed to be entropic in nature and to originate from the higher rigidity imposed by the pyranose ring.16 These studies aiming at understanding the origin of the structure of nucleic acids have spearheaded an immense research activity striving to improve the stability of oligonucleotides in biological media.
Chemical modification of nucleic acids originally stems from the advent of the antisense oligonucleotide therapeutic strategy. Antisense oligonucleotides (ASOs) consist of short sequences of DNA or RNA that are designed to recognize specific mRNA target regions and either sterically block the protein machinery or recruit endonucleases to hydrolyze mRNA.21–27 In the absence of chemical modifications, short oligonucleotides are rapidly degraded by nucleases, suffer from high renal excretion and poor cellular penetration, and often induce important off-target binding toxicities.28–34 Alleviation of these shortcomings can be achieved for instance by altering the chemical nature of the sugar moiety of nucleotides or coupling to ligands that act as vectors carrying oligonucleotides to their targets.11,35–39 Inspired by the chemistry of nucleic acids both to improve the therapeutic efficiency of oligonucleotides and understanding of the chemical etiology of the nucleic acid structure, a new research field was born which strives to identify artificial genetic polymers called xenonucleic acids (XNAs).20,40–44 XNAs differ from their natural counterparts DNA and RNA by the nature of the sugar moiety solely and retain the structure of the original nucleobases (i.e. A, C, G, and T) and phosphodiester linkages found in canonical nucleic acids (Fig. 1).40 Such a chemical differentiation allows XNAs to store genetic information while eluding interaction with DNA or RNA.
Besides the choice for furanoses rather than pyranoses as salient structural elements of nucleotides, glycosylation of nucleic acids is another, fundamental question that still needs to be further addressed. Until the recent discovery that certain small, non-coding RNAs were found to be equipped with sialylated glycans,45 it was believed that glycosylation did not affect nucleic acids. This is in stark contrast with all other natural biopolymers since glycosylation is one of the most common post-transcriptional modifications of proteins14,46 and glycolipids play important roles in numerous cellular processes.14,47
Overall, these considerations highlight the presence of an intricate and deep interconnection between nucleic acids and carbohydrates. In this review article, we describe the nature and properties of carbohydrate-modified nucleic acids. In the first section, we highlight recent progress made towards the synthesis of nucleosides and nucleotides with modified sugar residues for the improvement of the in vivo properties of therapeutic oligonucleotides and the development of orthogonal XNAs. In the second section, we discuss nucleic acid modification with glycans and in the last section, we summarize progress on the conjugation of carbohydrates and (poly)saccharides to oligonucleotides. This review article, however, addresses modifications of phosphate and/or nucleobases other than carbohydrates, and the interested reader is redirected to recent reviews covering these aspects.37,42,48–50
2. Chemical modification of the (deoxy)ribose of nucleosides and nucleotides
Each unit of the backbone of natural nucleic acids consists of six-covalent bonds (except for 2′-5′-branched RNA which contains seven bonds51). Changes in the backbone induce important alterations in the sugar pucker conformation which in turn can affect the base pairing strength and thus the (bio) physical/chemical properties of duplexes. In addition, the Watson–Crick base pairing rule (i.e. pairs with T (U) and G with C) governs most of the fundamental properties of DNA and RNA and modifying the structure of the backbone of DNA/RNA building blocks can alter the hydrogen bonding capacity and hence duplex stability.19,52,53 On the other hand, the ribose/deoxyribose is the main constituting part of nucleic acids involved in creating structural and functional diversity, and thus their chemical modification can lead to analogues with improved functional and/or biological properties. Indeed, changing the nature of the sugar moiety can improve the resistance against nuclease-mediated degradation, modulate or lock the conformation of the sugar puckering to reduce entropic penalty upon duplex formation, target binding efficiency and specificity, favor cellular penetration, and improve pharmacokinetics of nucleic acid drugs.Realization of the tremendous possibilities offered by sugar alterations, especially in the context of developing more potent therapeutic agents, stimulated intensive research activities. At first, chemical alterations consisted in minute changes of the (deoxy)ribose. For instance, replacing the 2′-OH of ribose with NH2, 2′-methoxy (2′-OMe, 2), fluorine (2′-F, 3), methoxyethyl (2′-MOE, 4), and aminopropyl (2′-O-AP) led to the development of potent first-generation chemistries that have been included in a number of therapeutic oligonucleotides such as antisense oligonucleotides (ASOs) or small interfering RNAs (siRNAs) (Fig. 2).37,39,54,55 In addition to these rather minor alterations, more drastic modifications can be made to the backbone by changing the nature of the sugar moiety to trioses, tetroses, other pentoses, hexoses including pyranose and furanose forms or even seven-membered sugar rings (Fig. 3).41,56
 | ||
| Fig. 2 Chemical structures of representative first generation modified carbohydrate backbones used in therapeutic oligonucleotides. | ||
 | ||
| Fig. 3 Chemical structures of representative, modified carbohydrate backbones used as DNA and RNA analogues (blue highlighting indicates six bond backbone units and green highlighting indicates altered backbone units). | ||
Such alterations of the natural ribose/deoxyribose can improve the properties of therapeutic oligonucleotides but also confer a certain degree of orthogonality to canonical nucleic acids which is one of the main prerequisites for the crafting of XNAs.20,40,42,43 In this section, we emphasize the main artificial sugar scaffolds that have been integrated into oligonucleotides and their effect on duplex/triplex stability and potential usefulness in practical applications. Interested readers on the topic of other therapeutic relevant modifications (e.g. at the level of phosphate) are redirected to recent review articles of this field.25,26,37,39,55,57–60
2.1 Glycol nucleic acid (GNA)
The sugar unit of nucleic acids can be replaced by short spacers such as glycerol. Indeed, in glycol nucleic acid (GNA) the (deoxy)ribose is replaced by a more flexible, acyclic glycol-phosphate backbone which lacks one bond compared to canonical DNA/RNA (6 in Fig. 3).61–65 In addition, the 2,3-dihydroxypropyl-based scaffold of GNA is reminiscent of the triose glyceraldehyde and can thus be considered as the smallest possible XNA scaffold.66 Even though the connecting glycerol moiety in GNA is achiral, it is also prochiral with respect to substitution at the primary hydroxyl group. Hence, there are two diastereoisomers of GNA (6), namely (R)-GNA and (S)-GNA (Fig. 4). Oligomers formed by both (S)- and (R)-GNA form antiparallel self-pairs that display highly improved thermal stabilities compared to the corresponding, unmodified RNA and DNA duplexes (ΔTm of 22 °C and 20 °C, respectively).64 This result is remarkable considering the fact that the GNA backbone is acyclic and hence markedly deviates from the structure of ribose/deoxyribose. However, (S)-GNA and (R)-GNA oligomers do not undergo antiparallel cross-pairing with each other and fail to form duplexes with complementary DNA sequences. Interestingly, only (S)-GNA is capable of cross-pairing with complementary RNA and follows canonical Watson–Crick base pairing rules. Overall, the combination of structural simplicity and enhanced thermal stability of GNA (6) makes this modification an attractive scaffold for the crafting of therapeutic agents.62 | ||
| Fig. 4 Chemical structures of (R)-GNA and (S)-GNA (6), D- and L-aTNA (13), and SNA (14). The color code highlights structural differences between members of acyclic nucleic acid analogues. | ||
Storage of genetic information and retrieving thereof rely on the possibility of enzymatically synthesizing and replicating nucleic acid polymers.67 This important prerequisite is only met when stable duplexes are formed and when polymerases can efficiently read through and faithfully copy the information encoded in template sequences by incorporating the correct (deoxy)nucleoside monophosphates ((d)NMPs) into primers. Interestingly, even though GNA and DNA do not form stable duplexes, DNA polymerases such as Bst are capable of catalyzing DNA synthesis on short dodecamer GNA templates.68 GNA triphosphates on the other hand are only moderately tolerated by the terminator polymerase since DNA primers are extended by only a few glycol nucleotides.69
The favorable assets of GNA spawned the design of other acylic DNA analogues such as ZNA (which consists of a phosphonate rather than a phosphate connector and has an additional methylene unit compared to GNA),70 acyclic D- and L-threoninol nucleic acid (D- and L-aTNA, 13 in Fig. 4)71,72 and serinol nucleic acid (SNA, 14 in Fig. 4)73 which also form highly stable homo-duplexes but display a higher propensity to form stable duplexes with DNA and RNA than GNA.74 Interestingly, the methyl group dictates the stereochemistry in aTNA (13) monomers, oligomers, and duplexes. In the absence of this kind of methyl moiety, the stereochemistry is only controlled by the sequence in SNA (14) oligomers.73 Nonetheless, GNA and related analogues are interesting scaffolds for the development of bio-orthogonal genetic systems (XNAs) since they fulfill most of the requirements.70
 | ||
| Fig. 5 Chemical structures of GNA equipped with glycose-nucleobase surrogates and schematic representation of the putative H-bonding interaction between 6-deoxyglucose in 16 and deoxyguanine in duplex DNA (16:dG). The modified oligonucleotides comprise in particular (S)-2,3-dihydroxypropyl glucose (15), (S)-2,3-dihydroxypropyl 6-deoxyglucose (16), (S)-2,3-dihydroxypropyl permethylated glucose (17), (R)-2,3-dihydroxypropyl permethylated glucose (18), and (S)-1,4-dihydroxybutyl-2-(6-deoxyglucose) (19). | ||
2.2 Three-membered ring analogues
The smallest possible cyclic surrogate of the deoxyribose/ribose moiety is a three-membered cyclopropane ring. Such a three-membered ring system is heavily strained and displays an intermediate sp3- and sp2-hybridization on the constituting carbon atoms.77 Consequently, substitution of the (deoxy)ribose moiety by cyclopropane imparts a near-linear ring structure as well as a strong rigidity to the scaffold.78,79 These alluring structural and chemical features have attracted attention for the development of potent antiviral nucleoside agents. Nonetheless, the properties of the cyclopropyl ring system did not hold to promises and the antiviral activity of such analogues (see e.g. nucleoside 20 in Fig. 6) was usually low to moderate.78–80 A noticeable exception is synadenol (21 in Fig. 6) and the corresponding guanosine analogue, synguanol, which display important antiviral activity against human cytomegalovirus and the Epstein–Barr virus.81,82 So far, cyclopropyl nucleosides have not been introduced into nucleic acids by solid-phase oligonucleotide synthesis (SPOS) and all nucleotides with this altered sugar moiety were designed to inhibit polymerases and transcriptases.83–85 | ||
| Fig. 6 Chemical structures of cyclopropyl nucleoside analogues. A (±)-2,2-bis(hydroxymethyl)cyclopropyl nucleoside 20 and B synadenol 21. | ||
2.3 Four-membered ring analogues
Increasing the ring size by a single carbon atom results in cyclobutyl-containing nucleoside analogues which, unlike their three-membered counterparts, often display interesting antiviral properties. For instance, such an altered scaffold has been identified in the naturally occurring antiviral agent oxetanocin A (7 in Fig. 3 and 22 in Fig. 7)86 which acts as a DNA chain terminator.87–89 The unusual three-dimensional structure conveyed by the presence of the oxetane motif90 combined with its highly potent pharmacological properties have led to the synthesis of numerous oxetanocin variants including carbacylic 25 (Fig. 7) and 3′-substituted analogues (23 in Fig. 7).86 In the 1990s, Katagiri and colleagues have reported the synthesis of oligonucleotides containing oxetanocin nucleotides. Homopolymeric dodecamers containing oxetanocin analogues (24 in Fig. 7) formed duplexes with complementary DNA sequences albeit with a marked thermal destabilization (ΔTm ∼ −7 °C per modification).91 Interestingly, when the ring oxygen was replaced by a carbon atom (25 in Fig. 7), the oligomer properties changed drastically.92 Indeed, homopolymers containing enantiomeric carbocyclic oxetanocin nucleotides display a strong tendency to form stable triple helices with complementary DNA and RNA sequences even under low salt conditions. These modified oligonucleotides feature an extended seven-bond backbone and display a more marked preference for RNA than DNA.92,93 Finally, monophosphorylated oxetanocin species have been reported in the literature (22 in Fig. 7).89 Nucleoside triphosphates might thus be accessible and when combined with engineered polymerases, enzymatic synthesis of oxetanocin oligonucleotides could be envisioned in the future. Such nucleotide analogues represent alluring alternatives for the construction of XNA systems. | ||
| Fig. 7 Chemical structures of four membered ring containing nucleosides, nucleotides, and oligonucleotides. | ||
2.3 Five-membered ring analogues
The most common and intuitive approach for the generation of nucleic acid analogues consists in either decorating the pentose with additional and/or different substituents or by changing the nature of the sugar without altering the size of the ring. Such modification patterns allow for fine tuning of the sugar pucker as well as improving the in vivo properties of the ensuing analogues without deviating substantially from the structure of canonical nucleic acids.52 In this section, we will discuss the most relevant examples of both approaches.C2′-modifications. The chemical/biological differences between DNA and RNA arise from the presence of a 2′-OH or substituent on the ribose moiety. Chemical alteration at the C2′ position is a potent strategy to increase the biological stability of therapeutic oligonucleotides. Currently, all FDA-approved or in late stage clinical trials oligonucleotide drugs are fully modified at the C2′-position.37,39,55 In addition to improved biological stability, appendage of electron withdrawing substituents at the 2′-position will favor a C3′-endo (North type) conformation while a C2′-endo (South type) sugar pucker is favored in DNA or constrained systems such as locked nucleic acids (vide infra). Lastly, depending on the nature of the C2′-substituent, the binding affinity to target RNA can also be improved by this simple modification pattern.
Amongst the C2′-substituents, 2′-fluorine is one of the most popular and effective modifications (3 in Fig. 2). The 2′-fluorine substituent induces a strong gauche effect which is reflected by a marked preference for a C3′-endo sugar pucker. RNA oligonucleotides equipped with 2′-fluoro riboses display enhanced thermal duplex stabilities when paired with complementary RNA sequences (ΔTm of +2 to 3 °C per modification).94 This enhancement in thermal stability is presumably caused by an enthalpic gain generated by favorable stacking interactions and base-pairing due the presence of the electronegative substituent.95,96 Expectedly, the presence of 2′-fluoro modifications also offers a strong resistance to nucleases both in vitro and in vivo.96,97 Nonetheless, the multiple advantageous assets of 2′-fluoro-modified RNAs are impinged by unfavorable pharmacokinetic properties.98,99
Replacement of the 2′-hydroxy moieties with 2′-OMe is another potent strategy to increase nuclease resistance and binding affinity, and also to reduce the immune response to therapeutic nucleic acids (2 in Fig. 2).100,101 As for the 2′-F substituent, the presence of a methoxy moiety at the C2′ raises duplex stability (ΔTm of +1 °C per modification) and locks the sugar pucker in a C3′-endo conformation. The increase of stabilization is caused by favorable hydrophobic interactions of the methoxy moiety in the duplex minor groove and by reduced entropic penalty upon duplex formation due to partial pre-organization of the sugar pucker.58,101–104 Given the favorable properties of the 2′-OMe modification strategy, most FDA-approved oligonucleotide therapeutics contain such nucleotide analogues.39
The 2′-OMe represents the smallest of all 2′-O-alkyl modifications reported to date. With the exception of 2′-MOE (4 in Fig. 2), it is also the most efficient in terms of improvement of RNA biological and therapeutic properties. Indeed, when the size of the alkyl moiety was varied from 2′-OMe to larger units such as 2′-O-ethyl, 2′-O-propyl, 2′-O-butyl, 2′-O-pentyl, and 2′-O-nonyl (28 in Fig. 8), a clear correlation was observed for the stability of DNA–RNA duplexes with the smaller substituents stabilizing and larger substituents destabilizing the duplex, respectively.105,106 Nuclease resistance increases with the size of the 2′-O-alkyl chain, suggesting that fine-tuning of the bulkiness and length of the 2′-O-modification can produce an adequate mix of thermal stability and nuclease resistance.101 However, these observations are also sequence-dependent since the presence of a 3′-terminal 2′-O-[2-(benzyloxy)ethyl] substituent (31 in Fig. 8) is rapidly degraded by exonucleases despite its rather important size.106
 | ||
| Fig. 8 Chemical structures of modification patterns at the level of the 2′-O-position of ribose. | ||
Over the past decades, the effect of a vast number of other substituents at the C2′-position has been explored (structures 26–31 in Fig. 8)107 including 2′-O-alkyl, 2′-O-alkyl ether,108,109 2′-O-amino alkyls,110,111 2′-O-ethylene glycol or 2′-O-glycerol,112 2′-O-carbamate,113 2′-O-(N-methylcarbamate),114 2′-O-allyl/alkenyl,106 2′-O-aminohexyl,115 and 2′-O-(2-(methylthio)ethyl).116
Amongst all the modifications that have been explored, the 2′-MOE substitution pattern displays the best balance between nuclease resistance and induced thermal stability (ΔTm of +1 to 2 °C per modification).112,117 While less pronounced than in the case of 2′-fluoro, these properties arise in part due to an additional conformational pre-organization imparted by the gauche effect due to the impaired rotation around the ethylene carbon–carbon bond.118 Hence, 2′-MOE substitution has advanced as a popular, second generation modification found in many therapeutic oligonucleotides.39
In terms of synthesis, C2′-modified nucleotides can be introduced by SPOS using activated phosphoramidite building blocks but also by enzymatic, polymerase-mediated synthesis. Indeed, some wild-type polymerases such as the T7 RNA polymerase or RNA polymerases with terminal transferase activity do have a propensity for accepting ribonucleotides equipped with C2′-susbtituents.119–123 Nonetheless, this capability remains limited and engineered polymerases are required to tolerate bulkier modifications such as 2′-MOE and achieve efficient synthesis.124–127 In order to alleviate these synthetic limitations, alternative chemoenzymatic methods involving for instance ligases are currently in development.108,128–130
Modifications at positions C2′ and C4′ of the ribose. The simultaneous introduction of substituents at the C2′ and C4′ positions was also explored even though not as extensively as single 2′-O-modifications.
Nuclease resistance of oligonucleotides can also be improved by introducing chemical modifications at C4′. This is because modifications at this position can insert between the 3′,5′-phosphodiesters and thus hinder cleavage activity mediated by nucleases.131 C4′-substitution also changes the sugar puckering and shifts towards a North-East conformation are often observed.132
Recently, several C4′-modified ribonucleotides were incorporated into RNA sequences and their effect on biostability and thermal stability was assessed. Prominent examples include 4′-C-methyl-2′-deoxy-2′-fluoro (32 in Fig. 9),133 4′-C-methoxy-2′-deoxy-2′-fluoro (33),132 4′-C-aminomethyl-2′-O-methyl (34 with n = 1),134 4′-C-aminoalkyl-2′-O-methyl (34),135 4′-C-aminoalkyl-2′-O-fluoro (34 with n = 1–3),136 4′-C-guanidinomethyl-2′-O-methyl (35),137 4′-C-guanidinocarbohydrazidomethyl-5-methyl (36),138 and (S)-5′-C-aminopropyl-2′-O-methyl139 (37). Collectively, these modifications induced a strong protection against nuclease-mediated degradation and were well-tolerated within RNA duplexes. Importantly, this modification pattern does not interfere with siRNA-mediated gene silencing activity and interacts favorably with human Argonaute 2 (Ago-2).133 These studies suggest that C2′/C4′-dual modification patterns represent alternative scaffolds that can be implemented in oligonucleotide therapeutics. Such nucleotide analogues also display potent antiviral activity as showcased by a nucleoside triphosphate equipped with fluorine substituents at the C2′- and C4′-positions which efficiently inhibited RNA synthesis by acting as a chain terminator.140
 | ||
| Fig. 9 Modifications at the 2′- and 4′-C-positions of the ribose. | ||
Instead of modifying the ribose moiety by appending additional functional groups at position C4′, the O4′ oxygen atom can be substituted by other heteroelements (Fig. 10). In this context, Matsuda et al. first reported the synthesis of 4′-thio-2′-deoxyribonucleosides (38) and 4′-thioribonucleosides (39).141,142 After conversion to phosphoramidite building blocks and incorporation into oligonucleotides by SPOS, 4′-thio-modified DNA and RNA were shown to increase nuclease resistance and thermal stability of duplexes with complementary sequences.143,144
 | ||
| Fig. 10 Modifications at the 2′-C and 4′-O-positions. | ||
Later, the same group synthesized and characterized the properties of 2′-modified-4′-thio-RNAs.145 In a comprehensive study, the authors demonstrated that 2′-fluoro-4′-thio-RNA (40) showed an impressive increase in thermal stability (ΔTm of +16 °C) with complementary RNA compared to an unmodified RNA duplex. In addition, 2′-fluoro-4′-thio-RNA (40) and 2′-OMe-4′-thio-RNA (41) sequences preferred binding to complementary RNA rather than DNA. Lastly, 2′-OMe-4′-thio-RNA (41) displayed improved resistance against endo- and exonuclease-mediated degradation when compared to oligonucleotides decorated with other modifications (i.e. 2′-fluoro, 2′-fluoro-4′-thio, 2′-OMe-4′-thio, and 4′-thio) or natural DNA and RNA.145
Besides SPOS, 4′-thio-modifed ribo- and deoxyribonucleotides are well-tolerated by polymerases and can thus be incorporated enzymatically into oligonucleotides.146–149 Interestingly, the corresponding nucleoside triphosphate, 4′-seleno-TTP, was prepared and could be incorporated into DNA using various polymerases, albeit with reduced efficiency compared to the corresponding 4′-thio nucleotide or unmodified dTTP.150
Carbocyclic nucleosides have increased chemical and metabolic stability compared to natural nucleosides. They are unaffected by phosphorylases and hydrolases due to the absence of glycosidic bonds. This important feature has been exploited for the construction of numerous nucleoside antiviral agents,78 some of which (e.g. 2′-deoxyaristeromycin,15142 in Fig. 10) have been incorporated into oligonucleotides by SPOS. In addition, carbocyclic analogues can also be used to improve the properties of therapeutic oligonucleotides. Indeed, the inclusion of such analogues into DNA sequences mostly results in duplexes with improved thermal stability.152,153 The additional degree of stabilization provided by a C2′-carbocyclic nucleotide containing oligonucleotides mainly results from a preferred North-type conformation of the cyclopentane moieties which imposes an A-like conformation on the duplexes.154
The nuclease resistance of carbocyclic-modified oligonucleotides can be further fine-tuned by the addition of substituents at position C6′ of the carbacyclic unit and/or at position C2′.155 To prove this hypothesis, Altmann et al. prepared a series of DNA oligonucleotides equipped with 2′- or 6′-alkoxy substituted carbocyclic nucleotide units (43) and evaluated their binding affinity to complementary RNA as well as their resistance to nucleases.155 The presence of both types of substitution patterns led to decreased thermal stability of the resulting duplexes, but as expected, the presence of an additional 6′-substituent massively increased biostability.
The last realistic heteroelement exchange possible on a five-membered ring system is swapping oxygen with a nitrogen atom. Many iminoribitol containing nucleoside and nucleotide analogues such as immucilin A (44 in Fig. 10) are good inhibitors of various enzymes because they are good transition state mimics. Yet, they have barely been incorporated into nucleic acids by SPOS.156–159 Related to iminoribitol are the pyrrolidine C-nucleoside analogues developed by the Leumann group (45 in Fig. 10). Such analogues were conceived to enhance the properties of triplex forming oligonucleotides (in the context of the antigene strategy160), particularly their capacity at recognizing duplexes via salt bridge formation.161,162
Combination of 2′-O- and 5′-C/O-ribose modifications. As mentioned previously, an important prerequisite for the development of efficient RNA/DNA therapeutics is biological stability. In particular, this implies that the nucleotidic scaffold needs to be chemically altered to resist against the hydrolytic degradation mediated predominantly by 3′-exonucleases.163,164 In addition, the presence of a high density of negative charges carried by the phosphate linkages denies oligonucleotides from penetrating cellular membranes. In order to alleviate these shortcomings, a number of modifications were introduced at the level of the phosphodiester moiety. An important example is 5′-methyl-DNA (5′-Me-DNA) where an additional methyl moiety was introduced at position 5′ (46 and 47 in Fig. 11).165 The presence of such a substituent reduces recognition by nucleases and does not alter the propensity of base pairing with complementary sequences (ΔTm of −1 to −3 °C per modification). Interestingly, unlike phosphorothioates or methylphosphates, the chiral center in 5′-Me-DNA is not located on the phosphorous atom but on the vicinal carbon atom of the sugar moiety, whose stereochemistry can be controlled.166 A similar strategy was applied to locked nucleic acids (LNA) where the effect of a 5′-(S)-methyl group (48) was shown to be beneficial for the recognition of complementary sequences, while the exact opposite was observed in sequences containing (R)-substituents (49).167
 | ||
| Fig. 11 Modification at the 2′-O, 5′-C position of ribose and internucleotidic modification patterns. | ||
Obviously, the nature of the 5′-substituent is not restricted to a short methyl moiety. In a recent work by Prakash and colleagues various 5′-C-modification patterns were introduced in a stereoselective manner and the resulting ribonucleosides were introduced into siRNA sequences by SPOS (50 in Fig. 11). This bulk of work demonstrated that a single stranded siRNA (ss-siRNA) sequence equipped with a single (R)-methyl group located on the first nucleotide (i.e. at the 5′-end of the sequence) displayed a more potent gene silencing activity than the corresponding sequence modified with an (S)-methyl group. This differential activity is presumably caused by the conformational preference around the torsion angle γ which is in a favorable ap range in the (R)-isomer. Interestingly, various substituents of larger steric bulk than a short methyl group were well-tolerated and did not negatively affect the gene silencing activity of both modified ss-siRNAs and siRNAs. On the other hand, introducing side-chains with anionic or cationic characteristics was detrimental for the activity of ss-siRNAs but not for that of siRNAs. This negative impact was deemed to originate either from negative charge repulsion or salt bridge formation with critical amino acid residues in the binding pocket of Ago-2.168
Numerous synthetic campaigns have been directed at modifying the internucleosidic linkage.169 For instance, a simple 5′-O to 5′-CH2 transposition results in the installation of a phosphonate backbone (51). The introduction of such a phosphodiester isostere improved the biostability of oligonucleotides but led to a destabilization of duplexes (ΔTm ∼−3 °C per modification).170 Increasing the length of the internucleosidic backbone either at the 3′- (52) or the 5′-termini (51) results in similar duplex destabilization.171 On the other hand, when the negatively charged phosphate is substituted with neutral mimics such as triazole (54),172,173 sulfamate (55),174–176 or amide (56),177 or with positively charged alternatives such as amines (57),178 both biostability and affinity for complementary sequences can be enhanced.
ANA-modified oligonucleotides are mainly produced by SPOS since the corresponding arabinonucleoside triphosphates are only poor substrates for naturally occurring archaeal DNA polymerases.185 On the hand, various polymerases have been engineered to better tolerate arabinonucleotides and permit molecular evolution experiments to generate ANA-containing aptamers and enzymes.67,186–188
 | ||
| Fig. 12 Chemical structures of RNA, 2′-fluoro-RNA (3) and FANA (58). | ||
FANA-containing oligonucleotides can be obtained by the sequential assembly of phosphoramidite building blocks using SPOS but also enzymatically by the polymerase-mediated incorporation of the corresponding nucleoside triphosphates. Indeed, 2′-deoxy-2′fluoro-arabinonucleoside triphosphates are tolerated as substrates by a number of Family B DNA polymerases such as 9°N and Vent (exo−).203–205 However, while these polymerases can be used for the construction of oligonucleotides containing FANA modifications, engineered polymerases need to be used to synthesize fully-modified FANA sequences.67,185 The possibility of enzymatic synthesis combined with the properties of this type of chemical modification have led to the identification of aptamers and XNAzymes by application of SELEX.67,185,186,206,207
Due to their capacity at recognizing complementary DNA and RNA sequences, α-L-threofuranosyl nucleoside triphosphates are well incorporated into TNA or DNA primers by modified and specifically engineered polymerases.185,215–218 The possibility of reverse transcribing DNA into TNA and then converting TNA into DNA using polymerases permits molecular evolution experiments to raise potent affinity reagents (aptamers) as well as TNA-based enzymes.219–224 The combination of chemical dissimilarity from the backbone of canonical nucleic acids with the propensity at adopting A-DNA conformations makes TNA an attractive analogue for the development of therapeutic oligonucleotides. What is more, TNAs are highly stable in human serum and acidic media, even more than RNA structural mimics such as 2′-F-RNA and FANA (see Section 2.3.3), which is an important prerequisite for therapeutic applications.225,226 Given these favorable assets, stability and high efficiency, siRNAs227 as well as antisense oligonucleotides223,228,229 have been constructed using TNA building blocks.
 | ||
| Fig. 13 Chemical structures of five-membered xylose and pentulose phosphate derived nucleic acids. | ||
2.4 Six membered ring containing nucleic acids
As for xylulo- and ribulo-nucleic acids, expanding the ring size from five-carbon sugars (pentoses and pentuloses) to larger and conformationally more rigid hexoses (mainly pyranoses) stems from efforts in understanding the chemical etiology of nucleic acids.15,16,239 In addition to these prebiotic considerations, sugar units with expanded size have attracted attention for the construction of alternative genetic polymers capable of storage of information due to their orthogonal base pairing capacities.67 Also, the additional atom in the ring offers the possibility of introducing more substituents and hence gives rise to a larger number of isomers and analogues thereof. These monomers display different structural conformations which cannot be accessed by regular pentoses.19 Lastly, an important and interesting serendipitous finding stemming from the introduction of six-membered ring systems in nucleic acids is the concept of conformational restriction which allowed the potency of therapeutic oligonucleotides to be significantly improved.17(4′ → 6′)-2,3-Dideoxy-D-glucopyranosyl nucleic acid (homo-DNA). The first system that was explored consisted of a single addition of a methylene unit on the pentose which resulted in oligonucleotides containing 4′ → 6′ connected 2′,3′-dideoxy-β-D-glucopyranosyl nucleotides, the so-called β-homo-DNA (64 in Fig. 14).244,245
 | ||
| Fig. 14 Chemical structures of DNA and β-homo-DNA 64. | ||
β-Homo-DNA is not capable of recognizing single-stranded, complementary DNA or RNA sequences but forms antiparallel self-duplexes that are yet more stable than the corresponding canonical duplexes.246 This increase in thermal stability originates from a favorable entropic contribution itself stemming from the higher rigidity of six-membered hexoses. Interestingly, the preferences observed in canonical duplexes where G–C pairs are more stable than A–T pairs are not strictly obeyed in β-homo-DNA where base pairing follows the order: G–C > A–A ≈ G–G > A–T.246 Resolution of the crystal structure of a β-homo-DNA oligomer has revealed a ladder-like pairing conformation and a large backbone-base inclination along with a chair conformation of the sugar moiety.247
Subsequently, several hexopyranosyl-containing oligonucleotides, featuring (6′ → 4′)-allopyranosyl-, (6′ → 4′)-altropyranosyl-, (6′ → 4′)-mannopyranosyl- and (6′ → 4′)-glucopyranosyl-containing units were explored.239 Most analogues performed poorly at recognizing complementary sequences and the steric hindrance was especially severe in the (6′ → 4′)-glucopyranosyl system. In this system, the 2′- and 3′-OH moieties clash with the backbone as well as the edges of the bases which might explain why Nature's most abundant carbohydrate, D-glucose, is not preferred over D-ribose in genetic systems.239
Collectively, these results tend to suggest that Nature opted for a more flexible pentose rather than the rigid nature of six-membered sugars which imposes structural constraints. These in turn explain why oligonucleotides with six-membered sugar units cannot cross-talk with canonical DNA and RNA and display altered base pairing properties. Interestingly, by a simple change of the nucleobase from a β- to an α-orientation (64 in Fig. 15), partial communication to complementary RNA (but not DNA) could be restored.248
 | ||
| Fig. 15 Chemical structures of the most common hexitol-based nucleic acid analogues. | ||
(6′ → 4′)-1′,5′-Anhydro-D-arabino-hexitol nucleic acid (HNA). In the course of developing novel nucleoside antiviral agents, the group of Piet Herdewijn observed a good structural correlation between 2′-deoxynucleosides and 1,5-anhydrohexitol nucleosides which stem from a formal insertion of a methylene moiety between the carbon connecting the nucleobase to the sugar and the ring oxygen (66 in Fig. 15).249 Based on these considerations, the same laboratory expanded on these observations and incorporated 1′,5′-anhydro-D-arabino-hexitol nucleosides into oligomers via 6′ → 4′ connections. The resulting HNA sequences form stable complexes with complementary DNA and RNA.250 Importantly, HNA displayed an important affinity for RNA (ΔTm of ∼ +3 °C per modification) due to the preferred conformation which is highly reminiscent of the C3′-endo sugar ring pucker found in canonical RNA.250–252 Substitution of single natural 2′-deoxynucleotides with the corresponding anhydrohexitol nucleotides led to duplexes with similar Tm values (ΔTm of ∼ +0.2 to −0.8 °C per modification) except for the dT to hT substitution which leads to a substantial decrease in melting temperature (ΔTm of ∼−2 °C). Interestingly, introducing a hydroxyl group at the 3′-position of the sugar (D-mannitol nucleic acids) decreases the stability of duplexes with complementary RNA and abrogates DNA recognition. This is due to the formation of strong hydrogen bonding interactions between the 3′-OH and 6′-oxygen of the phosphate backbone which causes a large deviation from the A-RNA like structure adopted by unmodified HNA.253,254 HNA has also been shown to display a propensity at forming duplexes with β-homo-DNA (64), suggesting that sequences containing different six-membered sugar units could cross-talk.20,255
HNAs represent interesting scaffolds to improve the efficiency and properties of oligonucleotide therapeutics including antisense oligonucleotides,256 aptamers,67,257 and nucleic acid catalysts186 but also to consolidate the architecture of nanotechnology objects.258 It is noteworthy mentioning that functional nucleic acids (i.e. binders and catalysts) obtained with HNA chemistry in the SELEX process require engineered polymerases that tolerate such an altered backbone.67
D-Altritol nucleic acid (D-AlNA). The strong preference of HNA for complementary RNA over DNA is caused by a differential minor groove solvation.259 In order to improve the hydration of HNA/DNA duplexes, Herdewijn and colleagues prepared D-altritol nucleic acids (here we have used the abbreviation D-AlNA to avoid confusion with ANA 9 in Fig. 3 even though both structures often bear the same abbreviation) which is similar to HNA but bears an additional hydroxy moiety at position 3′ of the sugar unit (11 in Fig. 3 and 15).260 While the introduction of such an additional hydroxyl moiety resulted in yet more stable complexes with RNA and DNA compared to those formed by HNA (66), the preference for DNA could not be increased. Interestingly, fully modified D-AlNA sequences form extremely stable homo-duplexes (ΔTm of +10 °C per modification when compared to dsDNA). Unlike β-homo-DNA (64) which adopts near linear structures, both HNA (66) and D-AlNA (11) adopt A-form helical structures which explains not only their capacity at interacting with canonical nucleic acids but also their strong preference for RNA.260,261 As stated for HNA, D-AlNA is also a promising scaffold to improve the properties of oligonucleotide therapeutics262 as well as functional nucleic acids.67
D-Mannitol nucleic acid (D-MNA). A simple change in the stereochemistry of the carbon atom at position 3′ from the (S)- in D-AlNA (11) to the (R)-configuration leads to D-mannitol nucleic acids (D-MNA, 67 in Fig. 15).253 This seemingly minute alteration has a profound impact on the molecular recognition capacity of the resulting oligonucleotides. Indeed, D-MNA oligonucleotides lose the capacity to interact with complementary RNA which is the exact opposite of its epimer D-AlNA (11). This differential behaviour was ascribed to unfavorable hydrogen bonding interactions and/or steric hinderance.
Cyclohexene nucleic acid (CeNA). Removal of the ring oxygen of the hexopyranosyl sugar of β-homo-DNA (64) not only results in a carbocyclic analogue but also in a chiral scaffold. The resulting cyclohexanyl nucleic acids (CNA) are capable of strongly pairing to complementary RNA (ΔTm of +1 °C per modification) and to a certain extent to DNA but only when in the D-isomer and not in the L-isomer configuration.263 However, CNA forms more stable self-complementary (i.e. CNA–CNA) than hetero-duplexes and thus displays a behavior that is intermediate between β-homo-DNA (64) and canonical nucleic acids, presumably due to the possibility of a chair flip of the cyclohexane moiety. The introduction of a double bond yields a cyclohexene sugar mimic which is yet more flexible than the cyclohexane in CNA and approaches the conformational flexibility of (deoxy)ribose. Cyclohexene nucleic acids (CeNA, 68 in Fig. 15) are better tolerated segments than CNAs within DNA than RNA sequences and lead to stabilization of duplexes formed with complementary RNA (ΔTm of around +2 °C per modification).264 Importantly, unlike most hexopyranosyl nucleic acid analogues, DNA sequences containing CeNA (68) building blocks are capable of efficiently recruiting RNase H, which is an important feature for the development of antisense oligonucleotide therapeutics (besides the enhanced resistance against nucleases induced by the modified sugar backbone).265 Given the success of CeNAs, several cyclohexene nucleotide analogues containing fluorine or hydroxyl substituents have been prepared.266–268
In terms of synthetic access, CNA and CeNA (68) containing oligonucleotides are mainly produced by SPOS even though an engineered polymerase supports enzymatic CeNA synthesis.67
(6′ → 4′)- and (6′ → 3′)-Glucosamine nucleic acids (GANA). D-Glucosamine is one of the most abundant monosaccharides. In contrast, examples of D-glucosamine-containing nucleic acids are scarce. Recently, Kitabe et al. reported the synthesis of (6′ → 4′)- and (6′ → 3′)-linked D-glucosamine nucleic acids that contain thymine in the anomeric position of the pyranose residue (69 and 70 in Fig. 16, respectively).269 Despite promising assets, the incorporation of zwitterionic glucosamine nucleotides at the 5′/3′-terminal positions or in the middle of a 15-mer DNA sequence led to the destabilization of DNA–DNA and DNA/RNA duplexes.269
 | ||
| Fig. 16 Structures of glucosaminonucleic acids (GANAs) and comparison with β-homo-DNA (64). | ||
 | ||
| Fig. 17 Chemical structures of p-RNA (10) and other representative members of the pentopyranosyl nucleic acid family (colour coding indicates differences in stereochemistry). | ||
The Eschenmoser lab has also investigated other members of the (2′ → 4′)-connected pyranosyl nucleic acids (71) and all maintain strong base pairing capacities but none of these analogues can interact with canonical RNA. The strongest base pairing capacity, not only in this family but in general for all types of nucleic acids, has been observed in the α-arabinopyranosyl system.276
(3′ → 4′)-α-L-Lyxopyranosyl nucleic acid. The aforementioned (2′ → 4′)-connected pyranosyl nucleic acid (71) displays the same number of bonds per backbone unit as canonical DNA and RNA. Changing the connectivity from 2′ → 4′ to 3′ → 4′ will reduce the number of bonds from six to five and hence affect recognition of complementary sequences. In this context, the Eschenmoser laboratory reported α-L-lyxopyranosyl-(3′ → 4′) nucleic acid (72).277 Interestingly, this pyranosyl nucleic acid analogue with a shorter backbone unit displayed a propensity to form weak interactions with canonical DNA. In addition, α-L-lyxopyranosyl-(3′ → 4′) nucleic acids (72) formed weakly stable homo-duplexes and exhibited a capacity to form triplex structures. This is in stark contrast with other 3′ → 4′ connected ribopyranosyl oligonucleotide analogues such as β-D-ribopyranosyl nucleic acid (10), which do not form any stable complexes.
2.5 Conformationally restricted nucleic acids
An important observation made with β-homo-DNA (64) is that the increased thermal stabilization of duplexes is of entropic rather than enthalpic origin. This decrease in entropy in turn stems from the decrease in flexibility of six-membered ring systems compared to the canonical five-membered (deoxy)ribose.244,290 In other words, the ring system in single stranded β-homo-DNA is already pre-organized in the conformation it adopts in duplexes which alleviates the entropic penalty associated with a change in structure upon binding. Conceptually, this feature can be hijacked to improve the efficiency of therapeutics, particularly of antisense oligonucleotides. Indeed, pre-organizing nucleic acid analogues in a conformation similar to that observed in duplex DNA, DNA–RNA, or RNA will yield thermodynamically more stable complexes. Efficient analogues obeying this strategy include bicyclo- (73) and tricyclo-DNA (74), LNA (75), CeNA (68) and HNA (66).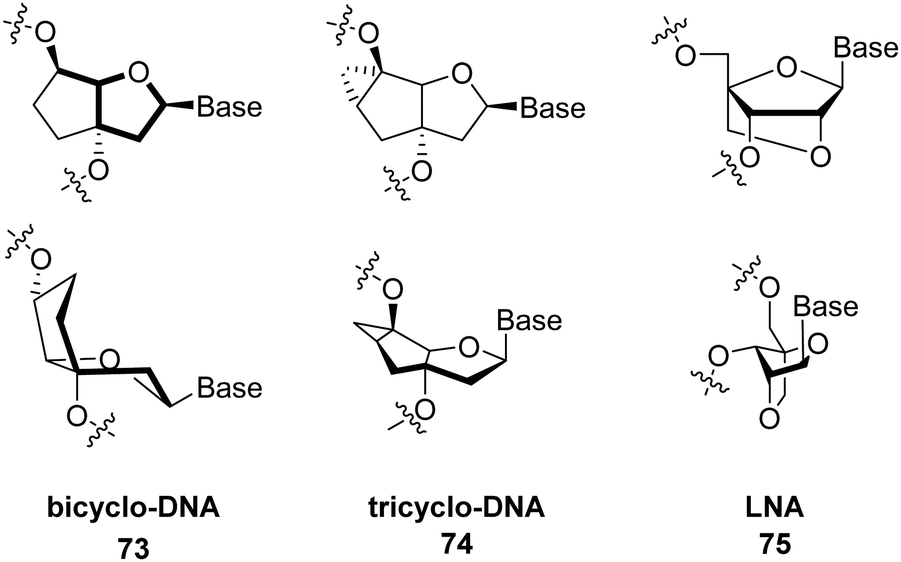 | ||
| Fig. 18 Chemical structures of bicyclo-DNA (73), tricyclo-DNA (74), and LNA (75) as well as their preferred conformations (adapted from ref. 17, 292 and 295). | ||
The introduction of bicyclo-DNA nucleotides (73) into oligonucleotide sequences generates a loss in enthalpy which is compensated by a decrease in entropy. In addition to favourable base pairing properties and enhanced nuclease resistance, the additional ethylene bridge allows for the introduction of substituents such as an amine296 or a fluorine atom.297 An interesting substitution pattern on bicyclo-DNA (73) is a cyclopropyl ring between C5′ and C6′. The resulting system, coined tricyclo-DNA (74 in Fig. 18), was conceived to further restrict the overall conformation of the nucleoside and correct the unfavourable geometry of torsion angle γ observed in bicyclo-DNA (C2′-exo sugar pucker).298–300 Fully modified tricyclo-DNA (74) sequences display remarkable binding properties since they form very stable duplexes with complementary DNA (ΔTm of +1.2 °C per modification) and even more so with complementary RNA (ΔTm of +2.4 °C per modification).301 As a result, tricyclo-DNA (74) has been used extensively to improve the properties of therapeutics, particularly of exon-skipping antisense oligonucleotides which resulted successful in mouse testing.302,303 Various substituted versions of the tricyclo-DNA have then been reported to improve properties such as cellular delivery304 and recruiting of RNase H.305
Given the chemical nature of bicyclo- and tricyclo-DNA, polymerases do not readily incorporate the corresponding nucleotides into DNA and hence synthesis of sequences containing such modifications proceeds only via SPOS.306,307
Although the increased binding provided by the inclusion of locked nucleotides is usually highly advantageous, if duplex hybridisation becomes too high, it can cause non-specific binding effects. As a result, LNAs (75) must be designed specifically to balance thermodynamic properties allowing for selective targeting while preventing toxic off-target effects.324,325
2.6 Seven membered ring containing nucleic acids
Increasing the size of the sugar from five to six (Section 2.4) abrogated the capacity of nucleic acids to interact with canonical nucleic acids due to the decreased flexibility of pyranose compared to furanose. Expanding the sugar moiety to seven membered rings was expected to restore flexibility and hence cross-talk with canonical nucleic acids.326 Indeed, molecular modeling of oxepane nucleic acids (ONAs, Fig. 19) revealed that these analogues would adopt a more compact and slightly twisted chair conformation that would be compatible with complementary DNA and even more with RNA.326 | ||
| Fig. 19 Chemical structures of seven membered ring derived nucleic acids. | ||
However, once incorporated into homopurine and homopyrimidine sequences, (5′ → 7′)-connected ONAs (12) did not show any capacity at interacting with complementary DNA and only weakly with RNA.326 In order to improve the molecular recognition capacity of seven membered ring analogues for complementary DNA/RNA, additional hydroxyl groups were included in the scaffold and the connectivity was altered.56,327 The resulting oxepane derivatives were incorporated into DNA and RNA strands by SPOS.56 Biophysical studies revealed that oxepane-modified RNA and DNA sequences (76) could cross-pair with complementary DNA and RNA. Nonetheless, single incorporation of OxNAs into RNA sequences massively reduced their affinity for complementary DNA and RNA (ΔTm > −10 °C per modification). On the other hand, when introduced into DNA sequences, only moderate (ΔTm of ∼−4 °C per modification) or no destabilization was observed for (4′ → 7′)- (77) and (3′ → 7′)- (78) OxNA with complementary DNA and RNA, respectively. (5′ → 7′)-OxNA (76) was strongly destabilizing also in a DNA sequence context. The conformation analysis of these seven membered nucleoside analogues showed that the heptose unit adopts a chair conformation with the pyrimidine base in the equatorial position. While there are some structural differences between oxepane nucleic acids and (deoxy)ribose oligonucleotides, molecular modelling and dynamics studies revealed that base–base stacking and sugar–phosphate H-bond interactions are responsible for the tolerance of OxNA in DNA–RNA duplexes.
3. Nucleic acids containing glycosylated nucleobases
In addition to the canonical nucleobases that make up genomic DNA and RNA, naturally occurring variations of these canonical nucleobases affect the biophysical, biological, and chemical properties of nucleic acids. Until now, it has not been demonstrated that natural (epigenetic) modifications can change the specificity of Watson–Crick base pairing or reverse to another binding mode. In contrast, they were shown to affect gene expression, regulate a broad range of different biological pathways, and permit the interaction with cellular and viral encoded proteins.1–5,328 Advances in analytical tools and methodologies have enabled the genome-wide mapping of these modified bases and led to the discovery that naturally occurring carbohydrates were covalently attached to the pyrimidine or purine bases, particularly in the genome of phages.329,330 Glycosylation of proteins and lipids is ubiquitous. It plays fundamental roles in most cellular functions and is involved in various diseases.331 Until recently, only DNA of bacteriophages was believed to be part of the glycome, mainly to elude protective mechanisms of hosts.332 Nonetheless, the constant identification of additional and more complex post-transcriptional chemical modifications in RNA combined with the recent discovery of glycosylated RNA45 suggest that this initial assumption might be inaccurate. Indeed, more than 140 RNA modifications have been discovered and these structural motifs are present at a variety of positions on all four canonical nucleobases.333 It is assumed that all RNA species in cells of all living organisms contain modified RNA nucleosides and the largest fraction of them are present in tRNAs. The chemical diversity displayed by epigenetic modifications ranges from small alterations such as methylation patterns to complete remodeling of the nucleobases (Fig. 20). One of the most common and ubiquitous modifications is pseudouridine (79, Ψ) which is present in all forms of RNAs and is often considered the fifth RNA base.334 More complex modifications are found on the so-called hyper-modified nucleobases. Two interesting representatives of the above are queuosine (81, Q)6,335 and wybutosine (80, yW).336–338 | ||
| Fig. 20 Chemical structures of hypermodified nucleotides, including examples of glycosylation on the nucleobase. | ||
The basic structure of the Q base features the central N7-deazaguanosine heterocycle bearing a carbon atom instead of a nitrogen in position 7 and a cyclopentenediol moiety attached via a methylamine moiety to the C7-position. Queuosine (81) is mainly found in tRNAs of bacteria and eukaryotes where it helps regulate translation and decode fidelity.339,340 In addition to these roles, the cyclopentenediol moiety of the Q base can be glycosylated in higher vertebrates with sugars such as D-galactose (Gal) and D-mannose (Man) to form the corresponding D-galactosyl-Q (82, galQ) and D-mannosyl-Q (83, manQ) hypermodified bases. The exact structures of manQ and galQ bases have recently been elucidated by the Carell group and revealed that the sugar is attached via β-glycosidic and α-glycosidic linkages in galQ (82) and manQ (83), respectively.6,8,339 Recently, the reaction mechanism of Q glycosylation was identified. It appears to involve RNA glycosylases and activated nucleotide diphosphate sugar precursors.341 Despite extensive research over the last few years, the exact biological function of the Q base, and even more so of the corresponding glycosylated nucleobases galQ (82) and manQ (83), is still not fully known. It is believed though that Q may be important to preventing stop codon readthrough from occurring.341,342
Besides galQ and manQ, no other hypermodified glycosylated nucleobases have yet been identified in RNA.343
However, the group of Bertozzi has reported in a groundbreaking study that RNA can be glycosylated with a variety of glycans.45 This study found that small (<200 nt) noncoding RNAs contain covalently linked sialylated glycans which were dubbed as ‘glycoRNAs’. Besides, sialic acid, fucose was identified to a certain extent. The exact chemical nature of these glycans as well as the structure of the linker and modified base have not yet been elucidated but the biosynthesis of glycoRNAs is unlikely to involve a direct glycosylation of RNA. Nonetheless, the sensitivity of glycoRNAs to treatment with amidase PNGase F which strips N-linked oligosaccharides off from glycoproteins suggests the presence of an amide bond between glycans and RNA. In this context, a recent study by Flynn and colleagues suggested that the modified RNA nucleobase 3-(3-amino-3-carboxypropyl)uridine (acp3U) acts as a connector between RNA and N-glycans via a carboxamide bond that would be generated on acp3U prior to conjugation to glycans (Fig. 21).344
 | ||
| Fig. 21 Representation of glycoRNAs and connection to acp3U. | ||
Interestingly, most glycoRNAs are located at the cell surface, which raises important questions on their function, the mechanisms that permit trafficking of these glycoconjugates to this cellular location, and on how glycoRNAs are inserted into membranes. While these questions remain unanswered, a recent study revealed that in a mice model, cell surface glycoRNAs are involved in neutrophil recruitment to accelerate the response to a tissue injury.345 More generally, glycans appear to assume multiple roles including protection of RNA against degradation and mediating binding to the lectin Selp which is important for neutrophil–endothelial interactions.
Ultimately, the expression of glycoRNAs on the cell surface indicates their significant biological importance, and further research will continue to uncover the many unknowns surrounding their function and structure.
3.1 O-Glycosylated nucleobases in DNA: hypermodified deoxycytidine
The chemical diversity of epigenetic modifications is much larger for RNA (>160) than in DNA (17, vide supra), which is possibly due to their highly complex and diverse functions. Despite this discrepancy, several glycosylated nucleobases have been identified in DNA. The most common epigenetic DNA modifications (Fig. 22) are the deoxycytidine analogues 5-methyl-dC (5mC, 84), 5-hydroxymethyl-dC (5hmC, 85), 5-formylcytosine (5fC, 86), and 5-carboxylcytosine (5caC, 87) along with the deoxyuridine derivative 5-hydroxymethyluracil (5hmU). Unsurprisingly, glycosylation of DNA often involves a sugar-attachment to one of the side chains of these common modification patterns. In this context, glycerol is the simplest carbohydrate that was found attached to a nucleobase. Indeed, 5-glyceryl-methylcytosine (5gmC, 88) was detected in the genome of the green algae of Chlamydomonas reinhardii.346 This modification is believed to act as a transcription repressor. It emanates from the addition of a glycerol unit to the exocyclic methyl group of 5mC (84) catalyzed by an analogue of the TET enzyme with vitamin C as a co-factor. Similarly, several hypermodified analogues of 5hmC (85) have been identified mainly in the genome of phages (89–91 in Fig. 22).347 The attached sugars consist mainly of D-glucose,332,348–350 but arabinosylation of 5hmC has also been reported.351 Going beyond monosaccharide modifications, a small fraction of the 5hmC sites of the DNA of coliphages T2 and T6 has been found to be further modified with disaccharides, mainly β-linked diglucose units (gentiobiosyl, 91).352,353 | ||
| Fig. 22 Chemical structures of epigenetic DNA modifications (84–87) and carbohydrate modified nucleotides in DNA (89–93) and of the natural product septacidin (94). 5hmC (85): 5-hydroxymethyl-deoxycytidine; 5fC (86): 5-formyl-deoxycytidine: 5caC (87): 5-carboxy-deoxycytidine; 5gmC (88): 5-glyceryl-methyldeoxycytidine; α/β-D-glc-5hmC (89): α/β-D-glucosyl-5-(hydroxymethyl)deoxycytidine; 5hmU: 5-(hydroxymethyl)uracil; β-D-glc-5hmU (92): β-D-glucosyl-5-(hydroxymethyl)uracil. | ||
3.2 O-Glycosylated nucleobases in DNA: hypermodified deoxyuracil
β-D-Glucopyranosyloxymethyluracil (base J, 92 in Fig. 22) was the first discovered hypermethylated DNA nucleobase. It was detected in telomeres of trypanosomatids.354–356 Since its first discovery, base J has been identified in many other parasites where it acts as a terminator of transcription by regulating RNA polymerase II.357–359More complex hypermodification patterns have been identified in rare occasions. For instance, the DNA of bacteriophage SP-15 has been shown to contain deoxyuridine nucleotides modified with a 4,5-dihydroxypentyl moiety attached at position C5 of the nucleobase (5dhpU, 93). What is more, each of the hydroxyl groups of this linker were further modified with a glucuronolactone connected through a phosphodiester bond and a glucose residue (93 in Fig. 22). The exact connectivity of both hydroxyl units to each of these modifications has not yet been elucidated.360,361 Hocek et al. recently reported the enzymatic synthesis of DNA containing glucosylated uridine and cytidine nucleotides. The presence of the modified residues protects DNA against the activity of type II restriction endonucleases (Res). Their capacity at serving as templates in transcription by the RNA polymerase of E. coli depends on the nature of the nucleobase to which they are connected.362 Indeed, glucosylated 5hmU units completely inhibited transcription while similarly modified 5hmC did not block RNA synthesis. Recently, this approach was extended to nucleotides equipped with other carbohydrates.363
3.3 Glycosylated bacterial nucleosides
An interesting family of compounds are the septacidins (94 in Fig. 22). These natural products consist of L-heptosamine moieties which are decorated with a nucleobase, an amino acid residue, and a lipid, thus combining elements of all naturally existing biopolymers.364–366 While such nucleoside analogues have not yet been incorporated into nucleic acids, they display potent antibiotic and anticancer activities.4. Carbohydrate–nucleic acid conjugates for efficient cellular delivery
Since the discovery of RNA interference (RNAi) by Fire and Mello,367 followed by a seminal clinical trial that demonstrated that siRNAs could specifically target human genes,368 there has been an exponential increase in interest in the synthesis of efficient and biostable siRNAs.37,39 These short RNA duplexes display several advantages compared to traditional drugs such as monoclonal antibodies or small molecules. Indeed, siRNAs recognize their mRNA target via Watson–Crick base pairing and do not need to specifically identify target proteins by binding to intricate three-dimensional folds. Also, siRNAs can be administered only a few times a year at most and are easier to prepare and store than antibodies for instance.369–371Even though the clinical success of siRNA-based therapeutics has been less than satisfactory, 22 different nucleic acid therapeutics have been approved by the FDA. Included are 6 siRNAs and 11 ASOs, while many more are in different stages of clinical trials.372 Nucleic acid drugs and RNAs particularly have several limitations that are in part connected to their rather high molecular weight and the high density of negative charge on the phosphate backbone which massively restrict efficient cellular uptake of naked oligonucleotides. In addition, as mentioned previously, all nucleic acids are rather unstable in the presence of nucleases and can display undesired off target effects and in vivo toxicity. The low cellular uptake is caused by a repulsion of the phosphodiester backbone of oligonucleotides and the negatively charged lipids and (glyco)proteins on cell membranes.373 To overcome the challenges of delivering oligonucleotides to their intended cellular targets, various chemical strategies have been designed. A very popular strategy consists in attaching small peptides (<40 amino acids), coined cell-penetrating peptides (CPPs), to the termini of oligonucleotides. These CPPs are capable of penetrating cellular membranes, mainly by endocytosis, and hence will help attached oligonucleotides crossing these biological barriers.373 Similarly, lipophilic modifications such as fatty acids or cholesterol, are capable of interacting with the phospholipid bilayers of cellular membranes and when attached to oligonucleotides promote their internalization.374,375 Attachment of such modifications can be covalent and permanent or bioreversible.376 In this context, carbohydrate–oligonucleotide conjugates have also been shown to improve cell-specific delivery through receptor-mediated endocytosis. Indeed, the benefit brought by the appendage of carbohydrates, often designed in the form of clusters, is reflected by the FDA-approved siRNA drug inclisiran used for the treatment of hypercholesterolemia.39,377 Equipped with an essential trivalent N-acetyl-D-galactosamine ligand (D-GalNAc, vide infra) that is preferentially recognized by the asialoglycoprotein receptor (ASGPR), inclisiran is specifically and efficiently delivered to hepatic cells without the need for any other vector or agent. Altogether, important research activity has been dedicated to producing and identifying (non-)natural carbohydrates conjugated with therapeutic oligonucleotides to help cellular delivery via carbohydrate-cell surface protein interactions. While factors such as the nature and valence of the selected carbohydrate clusters, and that of the connecting linker attached to the oligonucleotide are crucial for improved cellular delivery, we only discuss the general properties of carbohydrate–oligonucleotide bioconjugates. We defer the interested reader to recent review articles dealing with more specific therapeutic or design-oriented aspects of such hybrid constructs.378–380
4.1 Monosaccharide nucleic acid conjugates
Conjugation of nucleic acids with carbohydrates can be quite challenging. To overcome synthetic hurdles, several approaches have been developed.380,381 Most existing methods to facilitate the crafting of nucleic acid–carbohydrate conjugates rely on the use of modified phosphoramidite building blocks in SPOS. For this, monosaccharides (such as glucose, galactose, fucose) or disaccharides (such as lactose, cellobiose) can either be directly converted to an phosphoramidite or first connected to a linker moiety. Alternatively, carbohydrates can also be conjugated to oligonucleotides in solution using suitably modified precursors and well-established attachment reactions such as SN2, oxime bond formation, and CuAAC (copper catalyzed azide alkyne cycloaddition).36,381–385 On the other hand, while biocatalytic approaches are currently explored for the production of (modified) oligonucleotides,119,386–389 biocatalysts have been underexplored for the synthesis of nucleic acid–carbohydrate conjugates (vide infra). Interesting enzymatic bioconjugation methods that could be explored in this context could encompass using glycogen phosphorylases and phosphorylated glycans,390 glycosyltransferases,391,392 or polymerase-based approaches with carbohydrate triphosphates.393 In this section, we describe illustrative examples of oligonucleotides equipped with mono- or multi-valent saccharides consisting of a single type of carbohydrate.Berzal-Herranz et al. reported the conjugation of a single glucose moiety at the 5′-end of the fully phosphorothioate-modified antisense oligonucleotide GEM 91 (registered as Trecovirsen) which targets the translation initiation site of the HIV gag mRNA.394 The presence of a single glucose moiety further protected the antisense oligonucleotide against nuclease-mediated degradation. Importantly, this modification improved the therapeutic index of the antisense drug since, in addition to avoiding degradation, a significant reduction of the innate immune response and potent anti-HIV-1 activity were both observed.395 Yet, the attachment of a glucose unit did not increase the cellular delivery of GEM 91, but this is presumably due to the presence of a short connecting linker arm. Indeed, similar siRNA–carbohydrate conjugates albeit featuring longer connectors (15–18 atoms) displayed enhanced cell penetration capacities.382 Interestingly, a similar trend was observed when the glucose appendage was per-methylated, even though the cellular penetration capacity was reduced compared to unmodified glucose.396 The same authors then investigated the influence of the nature of the sugar moiety and the number of attached carbohydrates on the cellular uptake and capacity of siRNAs at regulating the expression of a target gene (Fig. 23).
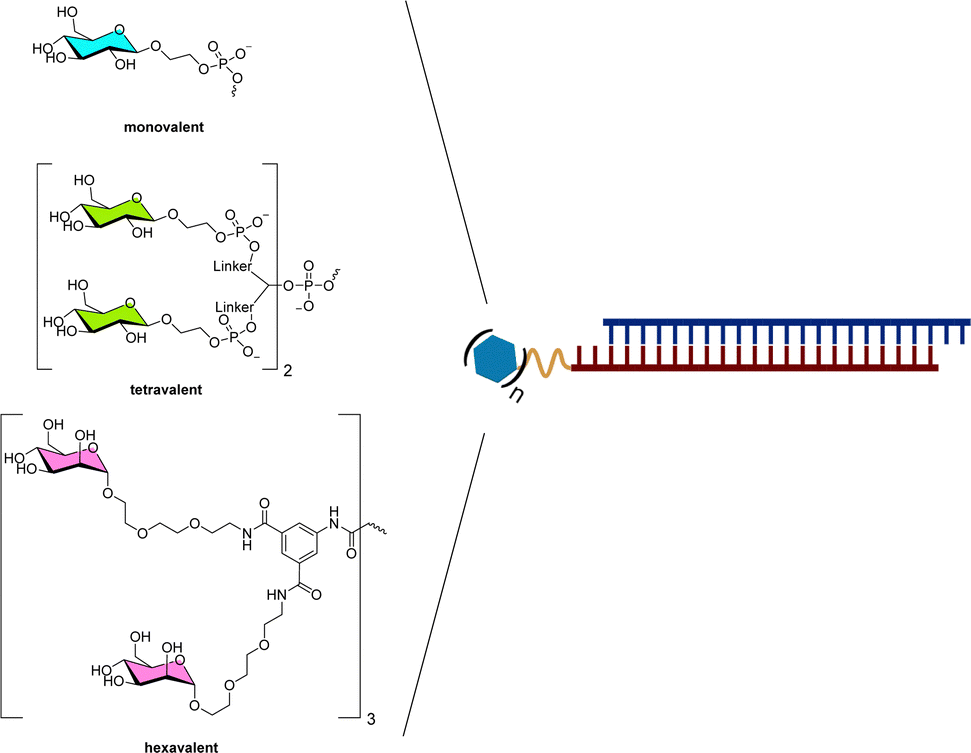 | ||
| Fig. 23 Representative examples of monovalent, tetravalent, and hexavalent carbohydrate–siRNA conjugates. | ||
Indeed, Eritja et al. synthesized siRNAs designed to inhibit the expression of the tumor necrosis factor (TNF-α) and modified with one to four glucose and galactose residues covalently linked to the 5′-end of the passenger strand.383 siRNAs equipped with galactose instead of glucose produced better gene silencing activities. D-Glucose-modified oligonucleotides did not display any significant capacity at penetrating the membrane of HeLa cells. siRNAs decorated with galactose units could enter HuH-7 cells, albeit with moderate efficiency, most likely via interaction with ASGPRs. The number of ligands appended to the siRNAs did not appear to influence the outcome of all these experiments.
Macrophages represent one of the major producers of the cytokine TNF-α and hence perform numerous and essential immune reactions under various pathophysiological conditions. Due to their biological relevance and involvement in several pathologies, macrophages have advanced as alluring therapeutic targets. Particularly, macrophages can be coerced to produce TNF-α by stimulating some Toll-like receptors (TLR) such as TLR-9. In this context, oligonucleotides that contain CpG motifs (CpG ODN) are good TLR-9 ligands and upon binding can elicit the production of significant amounts of cytokines like TNF-α, IL-6, and IFN-γ. In order to improve cellular delivery, CpG ODNs can be directly conjugated with D-mannosyl residues which can interact with the D-mannose receptor CD 206 mainly found on macrophages.397 Such a simple construct improved the cellular delivery two-fold compared to the unmodified counterparts. Moreover, the presence of D-mannose did not interfere with TNF-α release. A similar strategy was employed for the targeted delivery of siRNAs and silencing of the hypoxanthine phosphoribosyl transferase 1 and beta-2 microglobulin genes.398 The cellular uptake of the siRNA–carbohydrate conjugates was strongly improved by using tetra- instead of monovalent D-mannose ligands due to strong (KD value of 2.9 nM) and specific binding to the CD 206 receptor. The presence of four rather than one D-mannose moieties also improved gene silencing activity due to preferential internalization mediated by the former. The number of D-mannoses that are strictly required for efficient cellular delivery is still a matter of debate. Notably, a trivalent cluster was found to be sufficient for efficient cellular delivery and gene silencing activity both in vitro and in vivo.399 On the other hand, while increasing the valency to six D-mannose units further improved receptor binding and internalization efficiency, the presence of octavalent D-mannose complexes appeared to be slightly detrimental.400
Collectively, these observations clearly showcase the complex interplay between each constituting element of oligonucleotide–carbohydrate conjugates. The length of the linker arms as well as the number of and the nature of the sugar moieties can all have a profound impact on the biological properties and functions of such conjugates. These studies also demonstrate the possibility of site-specific delivery of therapeutic oligonucleotides by using sugar–receptor interactions. This concept was further developed and resulted in one of the major discoveries in the field of therapeutic oligonucleotide delivery in the last decades: triantennary N-acetyl-D-galactosamine (D-GalNAc) conjugation at the 3′-terminus of a preferred nucleic acid segment (Fig. 24).36
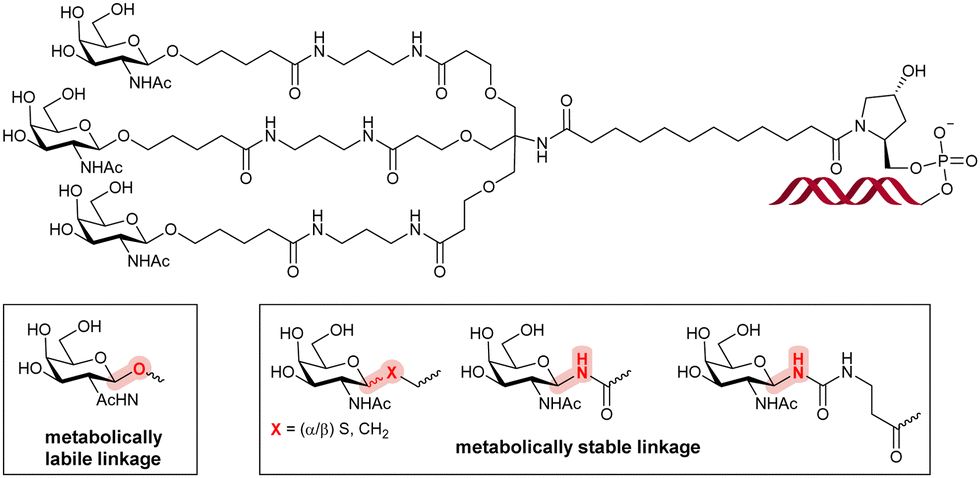 | ||
| Fig. 24 Chemical structures of a triantennary D-GalNAc ligand and examples of metabolically stable glycosidic linkages incorporated in siRNAs. | ||
Liver cells (hepatocytes) assume multiple essential functions including converting metabolites such as carbohydrates into energy, metabolizing toxic agents, or removing aging proteins and cells from the blood stream.378 They use multiple glycan-specific surface receptors to distinguish glycoproteins that need to be degraded from those which should be left intact. One of the most expressed receptors on the surface of hepatocytes (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 500000 per cell401) is ASGPR.402 The C-type lectin ASGPR is specific for the terminal galactose of glycoproteins and recognizes preferentially D-GalNAc (KD value of 2.5 nM) over D-Gal or D-Gal-like moieties such as D-fucose or D-lactose.401,403–405 Early studies have also demonstrated that monovalent sugars display low binding affinity for ASGPR and multivalent ligands are necessary for potent recognition.403,404 Based on these considerations, Manoharan and colleagues equipped the 3′-termini of the sense strand of siRNAs with bi- and tri-antennary D-GalNAc ligands.36 Trivalent D-GalNAc systems were much more efficient at cellular delivery of siRNAs36 and antisense406 oligonucleotides than the corresponding biantennary ligands. In addition, since binding of a D-GalNAc to ASGPR is pH-dependent (due to release of the required Ca2+ cofactor which ensures binding to the glycan),21,406 the D-GalNAc–siRNA conjugate dissociates from ASGPR in the endosome.407 Upon internalization into hepatic cells, the D-GalNAc–oligonucleotide conjugates are further metabolized, mainly by glycosidases, at various sites including the glycosidic and amide bonds of the D-GalNAc cluster as well as the connecting phosphodiester linkage.408 This enzymatic degradation eventually liberates the therapeutic oligonucleotide. Interestingly, when the glycosidic bonds of D-GalNAc ligands were stabilized by replacing the anomeric C–O linkages with C–S, C–N, or C–C bonds (Fig. 24), the metabolic stability of the resulting carbohydrates clusters was improved without affecting ASGPR-binding.409 Besides increasing the biostability of the triantennary D-GalNAc ligand, this study also revealed that the stereochemistry and the nature of the glycosidic bonds are not crucial for specific and strong interaction with ASGPR. On the other hand, increasing the metabolic stability of the D-GalNAc delivery system did not correlate with improved duration of gene silencing activity suggesting that this is governed by the intrinsic stability of siRNAs rather than by that of the ligand. Building on decades of research, Alnylam pharmaceuticals conjugated a trimer of D-GalNAc to several siRNAs that were driven through clinical trials. Subsequently, several of these D-GalNAc-conjugated siRNAs, including givosiran, lumasiran, inclisiran, and vutrisiran, have been approved for clinical use by the FDA.39
000 to 500000 per cell401) is ASGPR.402 The C-type lectin ASGPR is specific for the terminal galactose of glycoproteins and recognizes preferentially D-GalNAc (KD value of 2.5 nM) over D-Gal or D-Gal-like moieties such as D-fucose or D-lactose.401,403–405 Early studies have also demonstrated that monovalent sugars display low binding affinity for ASGPR and multivalent ligands are necessary for potent recognition.403,404 Based on these considerations, Manoharan and colleagues equipped the 3′-termini of the sense strand of siRNAs with bi- and tri-antennary D-GalNAc ligands.36 Trivalent D-GalNAc systems were much more efficient at cellular delivery of siRNAs36 and antisense406 oligonucleotides than the corresponding biantennary ligands. In addition, since binding of a D-GalNAc to ASGPR is pH-dependent (due to release of the required Ca2+ cofactor which ensures binding to the glycan),21,406 the D-GalNAc–siRNA conjugate dissociates from ASGPR in the endosome.407 Upon internalization into hepatic cells, the D-GalNAc–oligonucleotide conjugates are further metabolized, mainly by glycosidases, at various sites including the glycosidic and amide bonds of the D-GalNAc cluster as well as the connecting phosphodiester linkage.408 This enzymatic degradation eventually liberates the therapeutic oligonucleotide. Interestingly, when the glycosidic bonds of D-GalNAc ligands were stabilized by replacing the anomeric C–O linkages with C–S, C–N, or C–C bonds (Fig. 24), the metabolic stability of the resulting carbohydrates clusters was improved without affecting ASGPR-binding.409 Besides increasing the biostability of the triantennary D-GalNAc ligand, this study also revealed that the stereochemistry and the nature of the glycosidic bonds are not crucial for specific and strong interaction with ASGPR. On the other hand, increasing the metabolic stability of the D-GalNAc delivery system did not correlate with improved duration of gene silencing activity suggesting that this is governed by the intrinsic stability of siRNAs rather than by that of the ligand. Building on decades of research, Alnylam pharmaceuticals conjugated a trimer of D-GalNAc to several siRNAs that were driven through clinical trials. Subsequently, several of these D-GalNAc-conjugated siRNAs, including givosiran, lumasiran, inclisiran, and vutrisiran, have been approved for clinical use by the FDA.39
Besides siRNAs, D-GalNAc conjugation has been employed for the successful delivery to hepatocytes of antisense agents,408,410 miRNAs,411 anti-miRNAs,412 and D-GalNAc-lipid nanoparticles for CRISPR base editing therapy.413
4.2 Disaccharide/oligosaccharide–nucleic acid conjugates
Besides monosaccharides or polysaccharides consisting of repeats of the same carbohydrate unit, more complex oligosaccharide systems begin to be investigated as potential delivery systems for therapeutic oligonucleotides.An interesting example stems from the glycopeptide-derived antibiotic bleomycin (BLM) which is used for cancer treatment. The therapeutic activity of BLM stems from its capacity at inducing damage to both ssDNA and dsDNA when in the presence of defined cofactors (i.e. Fe2+ and O2). BLM consists of several key elements, notably a six amino acid peptide chelator of Fe2+ (for the DNA cleavage activity), a bithiazole tail which is involved in DNA intercalation, and a disaccharide moiety which is responsible for tumor cell selectivity.414–416 The 2-O-(3-carbamoyl-α-D-mannopyranosyl)-L-gulose disaccharide motif of BLM can thus represent a potential tool for the specific delivery of therapeutic oligonucleotides to cancer cells. In this context, trimers of the BLM disaccharide were conjugated with splice-switching antisense oligonucleotides (Fig. 25). These glycoconjugates were then assembled together on spherical nucleic acids (SNAs) by Watson–Crick base-pairing with a complementary sequence present on the SNA surface. The resulting carbohydrate-decorated spherical nucleic acids were taken up by the prostate cancer cell line PC3 more efficiently than the corresponding carbohydrate–oligonucleotide conjugates.417
 | ||
| Fig. 25 Chemical structures of representative examples of disaccharide/oligosaccharide–nucleic acid conjugates. | ||
Sialic acid-carrying ligands represent another interesting example of oligosaccharide-based systems used to enhance the pharmacokinetics of therapeutic oligonucleotides. Indeed, the CD22 receptor (Siglec 2) which is highly expressed on B-cells and B cell lymphomas, modulates B-cell signalling. The inhibitory activity of CD22 is regulated by an extracellular ligand-binding domain which exclusively recognizes sialylated carbohydrates, mainly the sequence Neu5Acα2–6Galβ1–4GlcNAc.418 The appendage of trimers of this trisaccharide to the 3′-end of the sense strand of an siRNA targeting hypoxanthine phosphoribosyl transferase 1, resulted in strong binding of the conjugates to CD22 receptors (KD values in the low nM range). After binding to CD22, the glycosylated siRNAs were efficiently internalized into cells via endocytosis and the modifications did not preclude gene knockdown of Hprt-1.419 Even though further optimization of the ligand structure and valency will be necessary to improve the knockdown activity, this study clearly highlights the potential of using sialylated-nucleic acids to promote cellular delivery via binding to CD22. It is noteworthy mentioning that the chemical integrity of the oligosaccharide is of high importance since small changes in composition can be deleterious to cellular internalization.420
Besides delivery systems, the binding affinity of oligosaccharides such as oligomannose (Man9) glycans can be raised from low (KD of μM to mM range) to high (KD in the low nM range) by combining grafting on oligonucleotides and in vitro evolution methods.421,422
5. Future perspectives and applications
Chemical modifications of nucleic acids have been used to elucidate, manipulate, and stabilize non-canonical structures and ultimately have permitted the intrusion of oligonucleotides into a plethora of applications. Importantly, the introduction of modified nucleoside analogues has also been crucial for the development of potent oligonucleotide therapeutics, including antisense, RNAi, and aptamer-based candidates, without which the field would not have gained such important momentum. Carbohydrate-based modifications slowly infiltrate the field of nucleic acids. At first, modifications were targeted at the constituting sugar moiety of the (deoxy)ribose scaffold. With the advent of glycoRNAs and D-GalNAc-based delivery agents, more complex carbohydrate modifications introduced at multiple locations of the scaffold are being considered such as oligomannose or bleomycin-based systems. This trend will undoubtedly continue and in this section, we summarize other existing applications of carbohydrates in the field of nucleic acids and highlight possible future directions in production and development of such constructs.5.1 Stabilization of structural motifs
Modified nucleosides, particularly guanosine analogues, can be strategically placed to stabilize and modulate the folding topology of G-quadruplexes. Small sugar-modified guanosine analogues such as FANA (2′-F-araG, 58) and LNA (LNA-G, 75) have been introduced in G-quadruplex structures.425,426 Generally speaking, when introduced in anti positions, such modified nucleotides stabilize G-quadruplexes while substitution in syn positions is often detrimental.427 The use of 2′-F-araG has also proven beneficial in studying telomerase activity by inducing an exclusive and stable parallel topology in the otherwise polymorphic telomeric G-quadruplex, as demonstrated by single-molecule FRET studies.428 Interestingly, TNA (8) can also adopt G-quadruplex structures of similar stability and structure to those formed by canonical DNA and RNA, except that both Na+ or K+ are equally well tolerated.429 While more complex sugar modified residues such as those involved in β-homo-DNA (64) or 5′-7′-ONA (12) have not been introduced in G-quadruplexes yet, the recent identification and characterization of an HNA (66) aptamer suggests that this functional nucleic acid adopts stable G-quadruplex-like structures.430 In the near future, other more complex, mainly XNA analogues, will undoubtedly be explored for their capacity to form and/or stabilize such structures.
Finally, carbohydrates have been found to interact with and stabilize G-quadruplexes mainly via stacking, hydrogen bonding, and hydrophobic contacts.431 These observations open up the possibility of using carbohydrate-G-quadruplex interactions to develop novel drugs,432,433 identify specific ligands,434 or modulate their structures. Ultimately, oligosaccharides such as D-GalNAc ligands could also be employed to facilitate the cellular delivery of functional nucleic acids adopting G-quadruplex structures.435,436
Alternatively, this structural motif can also be stabilized by introducing sugar-modified cytidine analogues. However, this is not an easy undertaking since the nucleotides should be preorganized in a C3′-endo conformation which is favored for i-motif formation without including electronegative substituents at position 2′ of the ribose since these are detrimental for this structural motif.37,444 Nonetheless, some stabilization of i-motifs could be achieved when ara-C (9), 2′-F-araC (58), and LNA-C (75) were introduced at specific locations.37 Indeed, 2′-fluoro-ribonucleotides moderately stabilize i-motifs by reducing the solvation effect.445,446 LNA-C substitutions can have varying effects on i-motif stability depending on the number and their localization within the sequences.444,447 In particular, LNA-C nucleotides stabilize i-motifs at specific locations due to a favorable C3′-endo conformation and the formation of additional C–H⋯O interactions that compensate for unfavorable van der Waals contacts.444,447 In contrast, araC and 2′-F-araC adopt both C2′-endo (South) conformations but still induce a degree of stabilization because their 2′-substituent points towards the major groove which thus avoids steric clashes.445,448 Of all nucleosides featuring a substituted sugar analogue, 2′-F-araC revealed to be the most effective at stabilizing i-motifs at neutral pH. Notably, an impressive ΔTm of over 17 °C was observed for an i-motif containing four 2′-F-araC analogues instead of dC units.445 Interestingly, even though HNAs (66) display a favorable C3′-endo conformation they do not seem to be capable of forming stable i-motifs.449
As mentioned in the section on G-quadruplexes, more stable i-motifs will undoubtedly be identified in the future by screening other (more complex) sugar modifications especially those stabilizing C3′-endo sugar puckers such as variants of HNA (66), CeNA (68), and bicyclo- (73) or tricyclo- (74) DNA. Furthermore, the addition of carbohydrates on nucleobases or at terminal locations of sequences might also contribute to the overall stability of i-motifs.
5.2 Carbohydrate modifications in therapeutic oligonucleotides
Therapeutic nucleic acids have tremendously benefited from the introduction of modified sugar moieties and the appendage of oligosaccharide-based ligands to increase their therapeutic index. Indeed, many (deoxy)ribose modifications such as 2′-OMe (2), 2′-F (3), 2′-MOE (4), LNA (75), and FANA (58) are routine chemical alterations of oligonucleotides and drastically improve oligonucleotide nuclease stability and binding affinity to complementary RNA. Conjugation with carbohydrate ligands such as triantennary D-GalNAc, D-Man, or sialic acid-containing moieties permits efficient and specific cellular delivery of therapeutic nucleic acids without negatively impacting their biological function.So far, mainly siRNAs and ASOs have benefited from such modification patterns, but lessons learned from these treatment modalities will certainly be applied to other purposes in the future. In this context, the first examples of modified CRISPR RNA (crRNA) sequences for CRISPR/Cas9 and Cas12 applications450,451 and RNA segments for endogenous adenosine deaminases acting on RNA (ADAR)-mediated RNA editing have already been reported.452 Appendage of carbohydrates at the 5′-termini of triplex forming oligonucleotides could enhance the stability of triplexes under physiological pH via stacking and van der Waals interactions which would be of importance for more efficient antigene therapy.160,453 Finally, mRNA vaccines are relatively new players in the field of FDA-approved therapeutic nucleic acids and display a huge potential for vaccination and therapy.454 These agents will certainly benefit from sugar modifications and/or conjugation to carbohydrates. Specifically, mRNA vaccines could be encapsulated in polysaccharides rather than lipid nanoparticles or equipped with D-GalNAc ligands to facilitate their delivery and more importantly convey target specificity.
Lastly, all (therapeutic) oligonucleotides will benefit from the identification of additional and optimized receptor–ligand pairs to favor specific cellular delivery. These additional ligand systems would remediate one of the long-standing challenges in the field of therapeutic oligonucleotides: specific and efficient cellular delivery.
5.3 Alternative XNA-based functional nucleic acids
XNA building blocks have been included in SELEX experiments and related combinatorial methods of in vitro selection for the identification of aptamers and catalysts to improve their biological stability but also their functions.42,126,207,222,455 Initially, nucleoside triphosphates equipped with small sugar alterations such as 2′-OMe (2), 2′-F (3), and 2′-NH2 were used to construct naïve libraries for SELEX. Progress in protein engineering and directed evolution has led to the discovery of polymerases and reverse transcriptases capable of synthesizing XNA sequences from DNA templates and complementary DNA from XNAs.127,456,457 In turn, these more tolerant mutant enzymes have permitted the introduction of more complex sugar modifications such as TNA (8), HNA (66), or charge-neutral sugar backbones into SELEX protocols.206,224,458 This trend is expected to continue or even increase to include other alternative sugar modification patterns in functional nucleic acids. An interesting example consists of L-stereoisomer oligonucleotides or mirror-image DNA/RNA also coined Spiegelmers.459 These mirror image nucleic acids do not cross-talk with canonical D-oligonucleotides (but form Watson–Crick base pairs with complementary L-sequences) and are not recognized by natural enzymes including nucleases. L-aptamers and L-oligonucleotides thus display a remarkable biostability.460 However, since these analogues are not recognized by natural, L-amino acid containing enzymes, they cannot be copied or transcribed by existing polymerases or reverse transcriptases. Recently, a mirror-image version of the T7 RNA polymerase was produced and shown to be capable of in vitro transcription of short L-RNA sequences, thus opening up the possibility of direct in vitro selection of Spiegelmers.4615.4 Biocatalytic methods to produce carbohydrate–nucleic acid conjugates and sugar-modified sequences
Most sugar-modified oligonucleotides as well as all carbohydrate conjugates are mainly produced by SPOS. While this is a robust and versatile method that is clearly suitable for an immense diversity of modified phosphoramidites, the identification of more potent sugar modifications, ligands, or longer sequences would benefit from alternative synthetic protocols. For instance, an efficient version of SPOS but with suitable carbohydrate building blocks would certainly facilitate the production of oligosaccharides.462,463 In addition, alternative biocatalytic approaches would aid in the production of longer oligonucleotides containing modified sugar and/or nucleobases decorated with carbohydrates. Such approaches could involve polymerase-based methods,386,389,457,464 ligases,129,465 or nucleic acid enzymes.466 Lastly, glycosyltransferases and related enzymes could be used for the production of glycoRNAs to help understand their structures and biological functions.6. Conclusions
Besides being an integral and central element of the structure of DNA and RNA, carbohydrates can be modified or added to the scaffold of nucleic acids to alter their physicochemical properties and biological functions. In the first part of this review article, we have described the most common (deoxy) ribose modifications of nucleotides that were introduced into oligonucleotides. Such synthetic campaigns were mainly undertaken to understand the etiology of nucleic acids, improve the properties of therapeutics, and develop synthetic and orthogonal genetic polymers. In the second part of this review article, we have described the most common carbohydrate–nucleic acid conjugates either naturally occurring or chemically designed on purpose. These hybrids are mainly employed in therapeutic applications to improve cellular delivery both in terms of specificity and efficiency. Lastly, we have provided insights into future potential developments of such modified nucleic acids and alternative methods for their production. This field has profited from interesting and vast chemistry programs aimed at developing nucleoside and nucleotide antiviral agents in the 1970s and 1980s and has now emerged as an increasingly important area of research. It is expected that the development of alternative sugar modifications, ligand–receptor systems based on carbohydrates, and synthetic protocols will yield potent FDA-approved drugs, robust binders and catalysts, and might shed more light into the origin of life.7. Summary
7.1 Sugar modifications
Numerous synthetic campaigns were launched to identify sugar modifications that could help oligonucleotides resist against nuclease-mediated hydrolysis and thus increase their therapeutic usefulness. In addition, these synthetic efforts were guided by early research dedicated to identifying potent nucleoside antiviral agents. This combination led to the development of three generations of chemical modifications commonly employed in oligonucleotides undergoing clinical trials and in the >20 FDA-approved nucleic acid therapeutics.39 The most common sugar modifications are summarized in Table 1, along with their most striking properties.| Chemical structure | Sugar pucker | Main properties | Selected references |
|---|---|---|---|
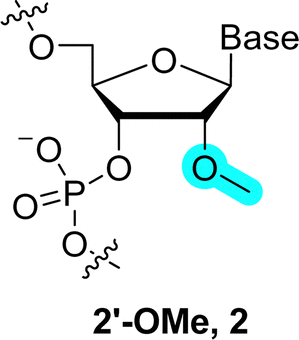
|
C3′-endo | Enhanced duplex stability (ΔTm of +1 °C per modification) opposite RNA (i.e. increased target recognition), enhanced nuclease resistance and chemical resistance to base, increased in vivo half-lives and stability in plasma, no recruitment of RNase H and no enhanced delivery | 39, 95, 101 and 104 |

|
C3′-endo | Enhanced duplex stability (ΔTm of +2–3 °C per modification) opposite RNA (i.e. increased target recognition), enhanced nuclease resistance and chemical resistance to base, increased in vivo half-lives and stability in plasma, no recruitment of RNase H and no enhanced delivery | 39 and 94–96 |

|
C3′-endo | Enhanced duplex stability (ΔTm of +1–2 °C per modification) opposite RNA (i.e. increased target recognition), enhanced nuclease resistance and chemical resistance to base, increased in vivo half-lives and stability in plasma, no recruitment of RNase H and no enhanced delivery | 39, 112, 117 and 118 |

|
n.a. | Charge neutral, enhanced cellular delivery and nuclease resistance, stabilization of duplexes and triplexes, do not elicit RNase H, common backbone modification in ASOs and siRNAs, four ASOs based on PMOs have been approved by the FDA, no structural information | 287 and 467 |

|
C4′-exo | Stems from questions on the chemical etiology of nucleic acids. Capable of cross-pairing with DNA and RNA, but destabilize duplexes. Enhanced nuclease resistance, used in siRNAs, aptamers, and the design of XNAs | 209, 210, 224, 227 and 468 |
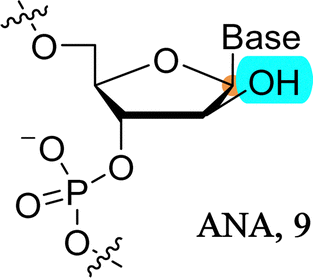
|
C2′-endo | Reduced duplex stability (ΔTm of −1 to −1.5 °C per modification) opposite RNA, form triplexes, enhanced nuclease resistance and chemical resistance to base, increased in vivo half-lives and stability in plasma, capacity of recruiting RNase H | 181, 183 and 184 |
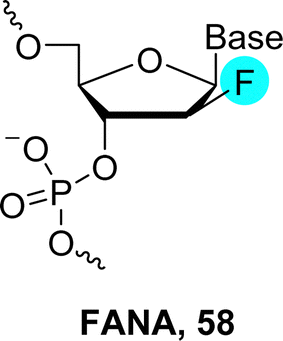
|
O4′-endo | Enhanced duplex stability (ΔTm of ∼1 °C per modification) opposite RNA (i.e. increased target recognition), enhanced nuclease resistance and chemical resistance to base, increased in vivo half-lives and stability in plasma, capacity of recruiting RNase H; part of XNAs and used in aptamers and DNAzyme selections | 67, 186, 193, 196, 198 and 199 |
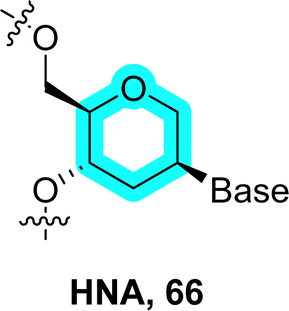
|
C3′-endo | Enhanced duplex stability (ΔTm of ∼3 °C per modification) opposite RNA (i.e. increased target recognition), enhanced nuclease resistance and chemical resistance to base, increased in vivo half-lives and stability in plasma, no capacity to recruit RNase H; part of XNAs and used in aptamers and DNAzyme selections | 67, 186, 250, 252 and 430 |

|
C1′-exo/C2′-endo | Conformationally restricted nucleotide, slight stabilization of duplexes with complementary DNA and RNA, triplex formation, enhanced nuclease resistance and chemical resistance to base, increased in vivo half-lives and stability in plasma, has been introduced in exon skipping ASOs, some chemical variants of 73 can recruit RNase H469 | 292–294 |

|
C2′-exo | Conformationally restricted nucleotide, enhanced duplex stability (ΔTm of ∼2 °C per modification) opposite RNA (i.e. increased target recognition), enhanced nuclease resistance and chemical resistance to base, increased in vivo half-lives and stability in plasma, has been introduced in exon skipping ASOs, some chemical variants of 74 can recruit RNase H | 298, 299, 301 and 302 |

|
C3′-endo | Conformationally restricted nucleotide, enhanced duplex stability (ΔTm of ∼4–10 °C per modification) opposite RNA (i.e. increased target recognition) and DNA (ΔTm of ∼2–5 °C per modification), enhanced nuclease resistance and chemical resistance to base, increased in vivo half-lives and stability in plasma, capacity to recruit RNase H, used in gapmers and siRNAs, and in XNA aptamers and catalysts | 67, 186, 309, 310, 312, 313 and 315 |
Other sugar modification patterns stem from questions related to chemical etiology of nucleic acids and the origin of life. For instance, structures such as β-homo-DNA (64) and p-RNA (10) have shed light onto Nature's choice for five rather than six-membered rings. Though not initially planned, studies towards the understanding of chemical etiology have also spawned interesting scaffolds for therapeutic applications via conformational restriction of nucleotides (highlighted in Table 1). In addition, given the inherent incapacity of certain surrogates to cross-talk with natural nucleic acids, these questions have also led to the development of XNAs. XNAs are sugar modified nucleic acids orthogonal to DNA/RNA that are capable of heredity and evolution. The most common XNA scaffolds are also highlighted in Table 1.
7.2 Glycosylated nucleobases
Glycosylated DNA nucleobases are found in the genome of bacteriophages and parasites and help elude immune defense responses during infection.470 The recent advent of glycosylated-RNA further underscores that nucleic acids are an integral part of the glycome on par with proteins and lipids. The identification of novel glycosylation patterns and the understanding of the structure and biological function of glycan-bearing DNA and RNA are of high relevance and gaining increased attention. The most relevant glycosylated nucleobase patterns are summarized in Table 2.| Chemical structure | Main properties | Selected references |
|---|---|---|

|
Hypermodified, natural RNA modification mainly found in tRNAs, plays a role in preventing stop codon readthrough. | 8, 339 and 341 |
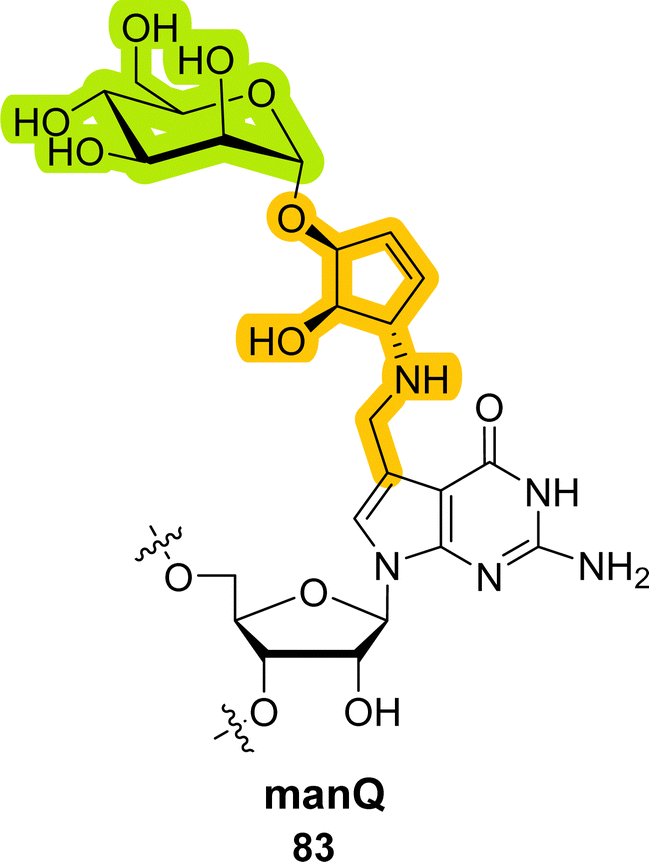
|
Hypermodified, natural RNA modification mainly found in tRNAs, exact biological role is not yet known. | 339 and 342 |

|
First hypermodified DNA nucleobase to be identified, terminates transcription, eludes defence mechanisms. | 362 and 470 |
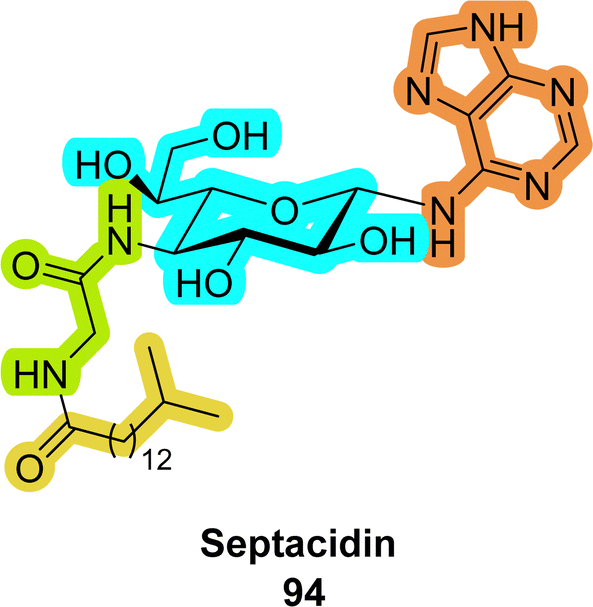
|
L-Heptosamine containing a nucleobase, an amino acid residue, and a lipid; potent antiviral and anticancer activities. | 364 and 365 |
7.3 Carbohydrate conjugates
In addition to modifying the nature of the deoxyribose/ribose of natural nucleic acids and adding glycans on the nucleobases, carbohydrates can be conjugated to the 5′/3′-termini of oligonucleotides. These appended carbohydrates guide therapeutic oligonucleotides to their target and favor their cellular delivery after receptor binding. Moreover, the addition of multiple rather than single carbohydrate units often enhances receptor recognition and specificity of oligonucleotides by the principle of multivalency. Carbohydrate–oligonucleotide conjugates are essential to improve the therapeutic index of nucleic acids. The most prominent member of this type of modification undoubtedly is the triantennary GalNAc ligand.Data availability
No primary research results and no new data were generated or analysed in the context of this review article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge generous funding from DARRI and Institut Carnot ‘Pasteur Microbes and Health’ Call 2021 (grant # INNOV-100-21, including a postdoctoral fellowship to D. D.).Notes and references
- X. Chen, H. Xu, X. Shu and C.-X. Song, Cell Death Differ., 2025, 32, 56–65 CrossRef CAS PubMed
.
- Y. Chen, T. Hong, S. Wang, J. Mo, T. Tian and X. Zhou, Chem. Soc. Rev., 2017, 46, 2844–2872 RSC
- J. Song and C. Yi, ACS Chem. Biol., 2017, 12, 316–325 CrossRef CAS PubMed
- P. J. McCown, A. Ruszkowska, C. N. Kunkler, K. Breger, J. P. Hulewicz, M. C. Wang, N. A. Springer and J. A. Brown, Wiley Interdiscip. Rev.: Mech. Dis., 2020, 11, e1595 CrossRef CAS PubMed
- F. Gracias, O. Ruiz-Larrabeiti, V. Vaňková Hausnerová, R. Pohl, B. Klepetářová, V. Sýkorová, L. Krásný and M. Hocek, RSC Chem. Biol., 2022, 3, 1069–1075 RSC
- A. Pichler, M. Hillmeier, M. Heiss, E. Peev, S. Xefteris, B. Steigenberger, I. Thoma, M. Müller, M. Borsò, A. Imhof and T. Carell, J. Am. Chem. Soc., 2023, 145, 25528–25532 CrossRef CAS PubMed
- M. Witzenberger, S. Burczyk, D. Settele, W. Mayer, L. M. Welp, M. Heiss, M. Wagner, T. Monecke, R. Janowski, T. Carell, H. Urlaub, S. M. Hauck, A. Voigt and D. Niessing, Nucleic Acids Res., 2023, 51, 8691–8710 CrossRef CAS PubMed
- P. Thumbs, T. T. Ensfelder, M. Hillmeier, M. Wagner, M. Heiss, C. Scheel, A. Schön, M. Müller, S. Michalakis, S. Kellner and T. Carell, Angew. Chem., Int. Ed., 2020, 59, 12352–12356 CrossRef CAS PubMed
- Š. Pospíšil, A. Panattoni, F. Gracias, V. Sýkorová, V. V. Hausnerová, D. Vítovská, H. Šanderová, L. Krásný and M. Hocek, ACS Chem. Biol., 2022, 17, 2781–2788 CrossRef PubMed
- J. R. Androsavich, Nat. Rev. Drug Discovery, 2024, 23, 421–444 CrossRef CAS PubMed
- S. Delaunay, M. Helm and M. Frye, Nat. Rev. Genet., 2024, 25, 104–122 CrossRef CAS PubMed
- R. J. Ontiveros, J. Stoute and K. F. Liu, Biochem. J., 2019, 476, 1227–1245 CrossRef CAS PubMed
- E. Fadda, Curr. Opin. Chem. Biol., 2022, 69, 102175 CrossRef CAS PubMed
- S. S. Shivatare, V. S. Shivatare and C.-H. Wong, Chem. Rev., 2022, 122, 15603–15671 CrossRef CAS PubMed
- A. Eschenmoser and M. Dobler, Helv. Chim. Acta, 1992, 75, 218–259 CrossRef CAS
- A. Eschenmoser, Science, 1999, 284, 2118–2124 CrossRef CAS PubMed
- C. J. Leumann, Bioorg. Med. Chem., 2002, 10, 841–854 CrossRef CAS PubMed
- D. M. Fialho, T. P. Roche and N. V. Hud, Chem. Rev., 2020, 120, 4806–4830 CrossRef CAS PubMed
- E. Lescrinier, M. Froeyen and P. Herdewijn, Nucleic Acids Res., 2003, 31, 2975–2989 CrossRef CAS PubMed
- J. C. Chaput, P. Herdewijn and M. Hollenstein, ChemBioChem, 2020, 21, 1408–1411 CrossRef CAS PubMed
- T. C. Roberts, R. Langer and M. J. A. Wood, Nat. Rev. Drug Discovery, 2020, 19, 673–694 CrossRef CAS PubMed
- S. T. Crooke, X.-H. Liang, B. F. Baker and R. M. Crooke, J. Biol. Chem., 2021, 296, 100416 CrossRef CAS PubMed
- M. C. Lauffer, W. van Roon-Mom and A. Aartsma-Rus, Commun. Med., 2024, 4, 6 CrossRef CAS PubMed
- B. R. Leavitt and S. J. Tabrizi, Science, 2020, 367, 1428–1429 CrossRef CAS PubMed
- S. T. Crooke, B. F. Baker, R. M. Crooke and X.-H. Liang, Nat. Rev. Drug Discovery, 2021, 20, 427–453 CrossRef CAS PubMed
- E. Çakan, O. D. Lara, A. Szymanowska, E. Bayraktar, A. Chavez-Reyes, G. Lopez-Berestein, P. Amero and C. Rodriguez-Aguayo, Cancers, 2024, 16, 2940 CrossRef PubMed
- A. Vaillant, Viruses, 2022, 14, 2052 CrossRef CAS PubMed
- M. Herkt and T. Thum, Mol. Ther., 2021, 29, 521–539 CrossRef CAS PubMed
- R. S. Geary, D. Norris, R. Yu and C. F. Bennett, Adv. Drug Delivery Rev., 2015, 87, 46–51 CrossRef CAS PubMed
- C. F. Bennett, Annu. Rev. Med., 2019, 70, 307–321 CrossRef CAS PubMed
- D. C. Guenther, S. Mori, S. Matsuda, J. A. Gilbert, J. L. S. Willoughby, S. Hyde, A. Bisbe, Y. Jiang, S. Agarwal, M. Madaoui, M. M. Janas, K. Charisse, M. A. Maier, M. Egli and M. Manoharan, J. Am. Chem. Soc., 2022, 144, 14517–14534 CrossRef CAS PubMed
- A. Akinc, M. A. Maier, M. Manoharan, K. Fitzgerald, M. Jayaraman, S. Barros, S. Ansell, X. Du, M. J. Hope, T. D. Madden, B. L. Mui, S. C. Semple, Y. K. Tam, M. Ciufolini, D. Witzigmann, J. A. Kulkarni, R. van der Meel and P. R. Cullis, Nat. Nanotechnol., 2019, 14, 1084–1087 CrossRef CAS PubMed
- P. Mangla, Q. Vicentini and A. Biscans, Cells, 2023, 12, 2253 Search PubMed
- M. Bege and A. Borbás, Pharmaceuticals, 2022, 15, 909 CrossRef CAS PubMed
- A. J. Debacker, J. Voutila, M. Catley, D. Blakey and N. Habib, Mol. Ther., 2020, 28, 1759–1771 CrossRef CAS PubMed
- J. K. Nair, J. L. S. Willoughby, A. Chan, K. Charisse, M. R. Alam, Q. Wang, M. Hoekstra, P. Kandasamy, A. V. Kel’in, S. Milstein, N. Taneja, J. O’Shea, S. Shaikh, L. Zhang, R. J. van der Sluis, M. E. Jung, A. Akinc, R. Hutabarat, S. Kuchimanchi, K. Fitzgerald, T. Zimmermann, T. J. C. van Berkel, M. A. Maier, K. G. Rajeev and M. Manoharan, J. Am. Chem. Soc., 2014, 136, 16958–16961 CrossRef CAS PubMed
- L. K. McKenzie, R. El-Khoury, J. D. Thorpe, M. J. Damha and M. Hollenstein, Chem. Soc. Rev., 2021, 50, 5126–5164 RSC
- C. Fàbrega, A. Aviñó and R. Eritja, Chem. Rec., 2022, 22, e202100270 CrossRef PubMed
- M. Egli and M. Manoharan, Nucleic Acids Res., 2023, 51, 2529–2573 CrossRef CAS PubMed
- J. C. Chaput and P. Herdewijn, Angew. Chem., Int. Ed., 2019, 58, 11570–11572 CrossRef CAS PubMed
- P. Herdewijn and P. Marlière, Chem. Biodiversity, 2009, 6, 791–808 CrossRef CAS PubMed
- N. Freund, M. J. L. J. Fürst and P. Holliger, Curr. Opin. Biotechnol, 2022, 74, 129–136 Search PubMed
- A. Kowalczyk, M. Piotrowicz, M. Gapińska, D. Trzybiński, K. Woźniak, T. M. Golding, T. Stringer, G. S. Smith, R. Czerwieniec and K. Kowalski, Bioorg. Chem., 2022, 125, 105847 CrossRef CAS PubMed
- G. S. Ivanov, V. G. Tribulovich, N. B. Pestov, T. I. David, A.-S. Amoah, T. V. Korneenko and N. A. Barlev, Biol. Direct, 2022, 17, 39 CrossRef CAS PubMed
- R. A. Flynn, K. Pedram, S. A. Malaker, P. J. Batista, B. A. H. Smith, A. G. Johnson, B. M. George, K. Majzoub, P. W. Villalta, J. E. Carette and C. R. Bertozzi, Cell, 2021, 184, 3109–3124 CrossRef CAS PubMed
- K. T. Schjoldager, Y. Narimatsu, H. J. Joshi and H. Clausen, Nat. Rev. Mol. Cell Biol., 2020, 21, 729–749 CrossRef CAS PubMed
- C. D. Hunter, T. Guo, G. Daskhan, M. R. Richards and C. W. Cairo, Chem. Rev., 2018, 118, 8188–8241 CrossRef CAS PubMed
- M. Hocek, Acc. Chem. Res., 2019, 52, 1730–1737 CrossRef CAS PubMed
- V. Roy and L. A. Agrofoglio, Drug Discovery Today, 2022, 27, 1945–1953 Search PubMed
- M. Chowdhury and R. H. E. Hudson, Chem. Rec., 2023, 23, e202200218 CrossRef CAS PubMed
- Y. Wang and S. K. Silverman, Angew. Chem., Int. Ed., 2005, 44, 5863–5866 CrossRef CAS PubMed
- G. Das, S. Harikrishna and K. R. Gore, Chem. Rec., 2022, 22, e202200174 Search PubMed
- C. J. Leumann, Chimia, 2005, 59, 776–779 Search PubMed
- J. A. Kulkarni, D. Witzigmann, S. B. Thomson, S. Chen, B. R. Leavitt, P. R. Cullis and R. van der Meel, Nat. Nanotechnol., 2021, 16, 630–643 Search PubMed
- R. Obexer, M. Nassir, E. R. Moody, P. S. Baran and S. L. Lovelock, Science, 2024, 384, eadl4015 Search PubMed
- S. K. Jana, S. Harikrishna, S. Sudhakar, R. El-Khoury, P. I. Pradeepkumar and M. J. Damha, J. Org. Chem., 2022, 87, 2367–2379 Search PubMed
- A. Khvorova and J. K. Watts, Nat. Biotechnol., 2017, 35, 238–248 CrossRef CAS PubMed
- H. Abou Assi, A. K. Rangadurai, H. Shi, B. Liu, M. C. Clay, K. Erharter, C. Kreutz, C. L. Holley and H. M. Al-Hashimi, Nucleic Acids Res., 2020, 48, 12365–12379 CrossRef PubMed
- W. Yuan, X. Shi and L. T. O. Lee, Mol. Ther.--Nucleic Acids, 2024, 35, 102195 CrossRef CAS PubMed
- K. Yadav, K. K. Sahu, S. P. E. Gnanakani, P. Sure, R. Vijayalakshmi, V. D. Sundar, V. Sharma, R. Antil, M. Jha, S. Minz, A. Bagchi and M. Pradhan, Int. J. Biol. Macromol., 2023, 241, 124582 CrossRef CAS PubMed
- N. Ueda, T. Kawabata and K. Takemoto, J. Heterocycl. Chem., 1971, 8, 827–829 Search PubMed
- M. Egli, M. Schlegel and M. Manoharan, RNA, 2023, 29, 402–414 CrossRef CAS PubMed
- P. Nielsen, L. H. Dreiøe and J. Wengel, Bioorg. Med. Chem., 1995, 3, 19–28 Search PubMed
- L. Zhang and E. Meggers, J. Am. Chem. Soc., 2005, 127, 74–75 Search PubMed
- L. Zhang, A. Peritz and E. Meggers, J. Am. Chem. Soc., 2005, 127, 4174–4175 CrossRef CAS PubMed
- K. Kowalski, Bioorg. Chem., 2023, 141, 106921 Search PubMed
- V. B. Pinheiro, A. I. Taylor, C. Cozens, M. Abramov, M. Renders, S. Zhang, J. C. Chaput, J. Wengel, S.-Y. Peak-Chew, S. H. McLaughlin, P. Herdewijn and P. Holliger, Science, 2012, 336, 341–344 Search PubMed
- C.-H. Tsai, J. Chen and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14598–14603 CrossRef CAS PubMed
- J. J. Chen, C.-H. Tsai, X. Cai, A. T. Horhota, L. W. McLaughlin and J. W. Szostak, PLoS One, 2009, 4, e4949 Search PubMed
- M. Luo, E. Groaz, M. Froeyen, V. Pezo, F. Jaziri, P. Leonczak, G. Schepers, J. Rozenski, P. Marlière and P. Herdewijn, J. Am. Chem. Soc., 2019, 141, 10844–10851 Search PubMed
- H. Asanuma, T. Toda, K. Murayama, X. Liang and H. Kashida, J. Am. Chem. Soc., 2010, 132, 14702–14703 CrossRef CAS PubMed
- K. Murayama, H. Kashida and H. Asanuma, Chem. Commun., 2015, 51, 6500–6503 RSC
- H. Kashida, K. Murayama, T. Toda and H. Asanuma, Angew. Chem., Int. Ed., 2011, 50, 1285–1288 Search PubMed
- F. Sato, Y. Kamiya and H. Asanuma, J. Org. Chem., 2023, 88, 796–804 Search PubMed
- E. Vengut-Climent, I. Gómez-Pinto, R. Lucas, P. Peñalver, A. Aviñó, C. Fonseca Guerra, F. M. Bickelhaupt, R. Eritja, C. González and J. C. Morales, Angew. Chem., Int. Ed., 2016, 55, 8643–8647 Search PubMed
- E. Vengut-Climent, P. Peñalver, R. Lucas, I. Gómez-Pinto, A. Aviñó, A. M. Muro-Pastor, E. Galbis, M. V. de Paz, C. Fonseca Guerra, F. M. Bickelhaupt, R. Eritja, C. González and J. C. Morales, Chem. Sci., 2018, 9, 3544–3554 Search PubMed
- A. de Meijere, Angew. Chem., Int. Ed. Engl., 1979, 18, 809–826 Search PubMed
- K. L. Seley-Radtke and M. K. Yates, Antiviral Res., 2018, 154, 66–86 Search PubMed
- C. Hyun Oh and J. H. Hong, Int. J. Pharm. Chem., 2006, 339, 507–512 Search PubMed
- T. Izawa, S. Nishiyama, S. Yamamura, K. Kato and T. Takita, J. Chem. Soc., Perkin Trans. 1, 1992, 2519–2525 Search PubMed
- Y.-L. Qiu, A. Hempel, N. Camerman, A. Camerman, F. Geiser, R. G. Ptak, J. M. Breitenbach, T. Kira, L. Li, E. Gullen, Y.-C. Cheng, J. C. Drach and J. Zemlicka, J. Med. Chem., 1998, 41, 5257–5264 Search PubMed
- R. J. Rybak, C. B. Hartline, Y.-L. Qiu, J. Zemlicka, E. Harden, G. Marshall, J.-P. Sommadossi and E. R. Kern, Antimicrob. Agents Chemother., 2000, 44, 1506–1511 Search PubMed
- R. Wang, S. Harada, H. Mitsuya and J. Zemlicka, J. Med. Chem., 2003, 46, 4799–4802 CrossRef CAS PubMed
- N. Ono, S. Iwayama, K. Suzuki, T. Sekiyama, H. Nakazawa, T. Tsuji, M. Okunishi, T. Daikoku and Y. Nishiyama, Antimicrob. Agents Chemother., 1998, 42, 2095–2102 Search PubMed
- R. Csuk and Y. von Scholz, Tetrahedron, 1994, 50, 10431–10442 Search PubMed
- Y.-C. Yan, H. Zhang, K. Hu, S.-M. Zhou, Q. Chen, R.-Y. Qu and G.-F. Yang, Bioorg. Med. Chem., 2022, 72, 116968 CrossRef CAS PubMed
- H. Zhang, P.-F. Liu, Q. Chen, Q.-Y. Wu, A. Seville, Y.-C. Gu, J. Clough, S.-L. Zhou and G.-F. Yang, Org. Biomol. Chem., 2016, 14, 3482–3485 Search PubMed
- A. Zhong, Y.-H. Lee, Y.-N. Liu and H.-W. Liu, Biochemistry, 2021, 60, 537–546 Search PubMed
- J. Bridwell-Rabb, A. Zhong, H. G. Sun, C. L. Drennan and H.-W. Liu, Nature, 2017, 544, 322–326 CrossRef CAS PubMed
- J. An, L. P. Riel and A. R. Howell, Acc. Chem. Res., 2021, 54, 3850–3862 Search PubMed
- A. Kakefuda, A. Masuda, Y. Ueno, A. Ono and A. Matsuda, Tetrahedron, 1996, 52, 2863–2876 Search PubMed
- N. Katagiri, Y. Morishita, I. Oosawa and M. Yamaguchi, Tetrahedron Lett., 1999, 40, 6835–6840 Search PubMed
- S. Honzawa, S. Ohwada, Y. Morishita, K. Sato, N. Katagiri and M. Yamaguchi, Tetrahedron, 2000, 56, 2615–2627 Search PubMed
- A. M. Kawasaki, M. D. Casper, S. M. Freier, E. A. Lesnik, M. C. Zounes, L. L. Cummins, C. Gonzalez and P. D. Cook, J. Med. Chem., 1993, 36, 831–841 Search PubMed
- G. F. Deleavey and M. J. Damha, Chem. Biol., 2012, 19, 937–954 Search PubMed
- M. Manoharan, A. Akinc, R. K. Pandey, J. Qin, P. Hadwiger, M. John, K. Mills, K. Charisse, M. A. Maier, L. Nechev, E. M. Greene, P. S. Pallan, E. Rozners, K. G. Rajeev and M. Egli, Angew. Chem., Int. Ed., 2011, 50, 2284–2288 Search PubMed
- R. V. Fucini, H. J. Haringsma, P. Deng, W. M. Flanagan and A. T. Willingham, Nucleic Acid Ther., 2012, 22, 205–210 Search PubMed
- J. M. Layzer, A. P. McCaffrey, A. K. Tanner, Z. Huang, M. A. Kay and B. A. Sullenger, RNA, 2004, 10, 766–771 CrossRef CAS PubMed
- M. Manoharan, Curr. Opin. Chem. Biol., 2004, 8, 570–579 Search PubMed
- J. L. Hyde and M. S. Diamond, Virology, 2015, 479–480, 66–74 CrossRef CAS PubMed
- L. L. Cummins, S. R. Owens, L. M. Risen, E. A. Lesnik, S. M. Freier, D. McGee, C. J. Guinosso and P. D. Cook, Nucleic Acids Res., 1995, 23, 2019–2024 CrossRef CAS PubMed
- I. Yildirim, E. Kierzek, R. Kierzek and G. C. Schatz, J. Phys. Chem. B, 2014, 118, 14177–14187 Search PubMed
- E. T. Kool, Chem. Rev., 1997, 97, 1473–1488 Search PubMed
- P. Lubini, W. Zürcher and M. Egli, Chem. Biol., 1994, 1, 39–45 CrossRef CAS PubMed
- E. A. Lesnik, C. J. Guinosso, A. M. Kawasaki, H. Sasmor, M. Zounes, L. L. Cummins, D. J. Ecker, P. D. Cook and S. M. Freier, Biochemistry, 1993, 32, 7832–7838 Search PubMed
- M. Egli, G. Minasov, V. Tereshko, P. S. Pallan, M. Teplova, G. B. Inamati, E. A. Lesnik, S. R. Owens, B. S. Ross, T. P. Prakash and M. Manoharan, Biochemistry, 2005, 44, 9045–9057 CrossRef CAS PubMed
- E. Pfund, C. Dupouy, S. Rouanet, R. Legay, C. Lebargy, J.-J. Vasseur and T. Lequeux, Org. Lett., 2019, 21, 4803–4807 CrossRef CAS PubMed
- K.-H. Altmann, D. Fabbro, N. M. Dean, T. Geiger, B. P. Monia, M. Müllert and P. Nicklin, Biochem. Soc. Trans., 1996, 24, 630–637 CrossRef CAS PubMed
- T. P. Prakash, A. M. Kawasaki, E. V. Wancewicz, L. Shen, B. P. Monia, B. S. Ross, B. Bhat and M. Manoharan, J. Med. Chem., 2008, 51, 2766–2776 CrossRef CAS PubMed
- R. H. Griffey, B. P. Monia, L. L. Cummins, S. Freier, M. J. Greig, C. J. Guinosso, E. Lesnik, S. M. Manalili, V. Mohan, S. Owens, B. R. Ross, H. Sasmor, E. Wancewicz, K. Weiler, P. D. Wheeler and P. D. Cook, J. Med. Chem., 1996, 39, 5100–5109 CrossRef CAS PubMed
- M. Manoharan, T. P. Prakash, I. Barber-Peoc'h, B. Bhat, G. Vasquez, B. S. Ross and P. D. Cook, J. Org. Chem., 1999, 64, 6468–6472 Search PubMed
- P. Martin, Helv. Chim. Acta, 1995, 78, 486–504 Search PubMed
- M. Prhavc, E. A. Lesnik, V. Mohan and M. Manoharan, Tetrahedron Lett., 2001, 42, 8777–8780 Search PubMed
- R. Pattanayek, L. Sethaphong, C. Pan, M. Prhavc, T. P. Prakash, M. Manoharan and M. Egli, J. Am. Chem. Soc., 2004, 126, 15006–15007 Search PubMed
- J. Winkler, M. Gilbert, A. Kocourková, M. Stessl and C. R. Noe, ChemMedChem, 2008, 3, 102–110 Search PubMed
- T. P. Prakash, M. Manoharan, A. M. Kawasaki, A. S. Fraser, E. A. Lesnik, N. Sioufi, J. M. Leeds, M. Teplova and M. Egli, Biochemistry, 2002, 41, 11642–11648 CrossRef CAS PubMed
- V. Tereshko, S. Portmann, E. C. Tay, P. Martin, F. Natt, K.-H. Altmann and M. Egli, Biochemistry, 1998, 37, 10626–10634 Search PubMed
- M. Teplova, G. Minasov, V. Tereshko, G. B. Inamati, P. D. Cook, M. Manoharan and M. Egli, Nat. Struct. Mol. Biol., 1999, 6, 535–539 Search PubMed
- M. Pichon, F. Levi-Acobas, C. Kitoun and M. Hollenstein, Chem. – Eur. J., 2024, 30, e202400137 CrossRef CAS PubMed
- R. Tomecki, K. Drazkowska, R. Madaj, A. Mamot, S. Dunin-Horkawicz and P. J. Sikorski, ChemBioChem, 2024, 25, e202400202 CrossRef CAS PubMed
- H. Aurup, D. M. Williams and F. Eckstein, Biochemistry, 1992, 31, 9636–9641 CrossRef CAS PubMed
- J. B. J. Pavey, A. J. Lawrence, I. A. O'Neil, S. Vortler and R. Cosstick, Org. Biomol. Chem., 2004, 2, 869–875 RSC
- Y. Masaki, H. Ito, Y. Oda, K. Yamazaki, N. Tago, K. Ohno, N. Ishii, H. Tsunoda, T. Kanamori, A. Ohkubo, M. Sekine and K. Seio, Chem. Commun., 2016, 52, 12889–12892 Search PubMed
- V. Siegmund, T. Santner, R. Micura and A. Marx, Chem. Commun., 2012, 48, 9870–9872 RSC
- J. Chelliserrykattil and A. D. Ellington, Nat. Biotechnol., 2004, 22, 1155–1160 Search PubMed
- N. Freund, A. I. Taylor, S. Arangundy-Franklin, N. Subramanian, S.-Y. Peak-Chew, A. M. Whitaker, B. D. Freudenthal, M. Abramov, P. Herdewijn and P. Holliger, Nat. Chem., 2023, 15, 91–100 CrossRef CAS PubMed
- T. Chen, N. Hongdilokkul, Z. Liu, R. Adhikary, S. S. Tsuen and F. E. Romesberg, Nat. Chem., 2016, 8, 556–562 Search PubMed
- D. Kestemont, M. Renders, P. Leonczak, M. Abramov, G. Schepers, V. B. Pinheiro, J. Rozenski and P. Herdewijn, Chem. Commun., 2018, 54, 6408–6411 RSC
- N. Sabat, A. Stämpfli, S. Hanlon, S. Bisagni, F. Sladojevich, K. Püntener and M. Hollenstein, Nat. Commun., 2024, 15, 8009 Search PubMed
- A. Hoose, R. Vellacott, M. Storch, P. S. Freemont and M. G. Ryadnov, Nat. Rev. Chem., 2023, 7, 144–161 Search PubMed
- R. Liboska, J. Snášel, I. Barvík, M. Buděšínský, R. Pohl, Z. Točík, O. Páv, D. Rejman, P. Novák and I. Rosenberg, Org. Biomol. Chem., 2011, 9, 8261–8267 Search PubMed
- E. Malek-Adamian, D. C. Guenther, S. Matsuda, S. Martínez-Montero, I. Zlatev, J. Harp, M. Burai Patrascu, D. J. Foster, J. Fakhoury, L. Perkins, N. Moitessier, R. M. Manoharan, N. Taneja, A. Bisbe, K. Charisse, M. Maier, K. G. Rajeev, M. Egli, M. Manoharan and M. J. Damha, J. Am. Chem. Soc., 2017, 139, 14542–14555 CrossRef CAS PubMed
- J. M. Harp, D. C. Guenther, A. Bisbe, L. Perkins, S. Matsuda, G. R. Bommineni, I. Zlatev, D. J. Foster, N. Taneja, K. Charisse, M. A. Maier, K. G. Rajeev, M. Manoharan and M. Egli, Nucleic Acids Res., 2018, 46, 8090–8104 CrossRef CAS PubMed
- K. R. Gore, G. N. Nawale, S. Harikrishna, V. G. Chittoor, S. K. Pandey, C. Höbartner, S. Patankar and P. I. Pradeepkumar, J. Org. Chem., 2012, 77, 3233–3245 Search PubMed
- K. Koizumi, Y. Maeda, T. Kano, H. Yoshida, T. Sakamoto, K. Yamagishi and Y. Ueno, Bioorg. Med. Chem., 2018, 26, 3521–3534 CrossRef CAS PubMed
- T. Kano, Y. Katsuragi, Y. Maeda and Y. Ueno, Bioorg. Med. Chem., 2018, 26, 4574–4582 Search PubMed
- A. Uematsu, R. Kajino, Y. Maeda and Y. Ueno, Nucleosides, Nucleotides Nucleic Acids, 2020, 39, 280–291 Search PubMed
- G. N. Nawale, S. Bahadorikhalili, P. Sengupta, S. Kadekar, S. Chatterjee and O. P. Varghese, Chem. Commun., 2019, 55, 9112–9115 Search PubMed
- R. Kajino, Y. Maeda, H. Yoshida, K. Yamagishi and Y. Ueno, J. Org. Chem., 2019, 84, 3388–3404 Search PubMed
- S. Martínez-Montero, G. F. Deleavey, A. Kulkarni, N. Martín-Pintado, P. Lindovska, M. Thomson, C. González, M. Götte and M. J. Damha, J. Org. Chem., 2014, 79, 5627–5635 CrossRef PubMed
- N. Inoue, D. Kaga, N. Minakawa and A. Matsuda, J. Org. Chem., 2005, 70, 8597–8600 Search PubMed
- T. Naka, N. Minakawa, H. Abe, D. Kaga and A. Matsuda, J. Am. Chem. Soc., 2000, 122, 7233–7243 Search PubMed
- N. Inoue, N. Minakawa and A. Matsuda, Nucleic Acids Res., 2006, 34, 3476–3483 CrossRef CAS PubMed
- S. Hoshika, N. Minakawa and A. Matsuda, Nucleic Acids Res., 2004, 32, 3815–3825 CrossRef CAS PubMed
- M. Takahashi, N. Minakawa and A. Matsuda, Nucleic Acids Res., 2009, 37, 1353–1362 Search PubMed
- N. S. Tarashima, A. Matsuo and N. Minakawa, J. Am. Chem. Soc., 2020, 142, 17255–17259 Search PubMed
- T. Kojima, K. Furukawa, H. Maruyama, N. Inoue, N. Tarashima, A. Matsuda and N. Minakawa, ACS Synth. Biol., 2013, 2, 529–536 Search PubMed
- N. Minakawa, M. Sanji, Y. Kato and A. Matsuda, Bioorg. Med. Chem., 2008, 16, 9450–9456 Search PubMed
- Y. Kato, N. Minakawa, Y. Komatsu, H. Kamiya, N. Ogawa, H. Harashima and A. Matsuda, Nucleic Acids Res., 2005, 33, 2942–2951 CrossRef CAS PubMed
- N. Tarashima, T. Sumitomo, H. Ando, K. Furukawa, T. Ishida and N. Minakawa, Org. Biomol. Chem., 2015, 13, 6949–6952 Search PubMed
- S. Smirnov, F. Johnson, R. Marumoto and C. de los Santos, J. Biomol. Struct. Dyn., 2000, 17, 981–991 Search PubMed
- M. Perbost, M. Lucas, C. Chavis, A. Pompon, H. Baumgartner, B. Rayner, H. Griengl and J. L. Imbach, Biochem. Biophys. Res. Commun., 1989, 165, 742–747 CrossRef CAS PubMed
- J. Sági, A. Szemzõ, J. Szécsi and L. Ötvös, Nucleic Acids Res., 1990, 18, 2133–2140 Search PubMed
- Y. Xu, K. Kino and H. Sugiyama, J. Biomol. Struct. Dyn., 2002, 20, 437–446 Search PubMed
- K.-H. Altmann, M.-O. Bévierre, A. De Mesmaeker and H. E. Moser, Bioorg. Med. Chem. Lett., 1995, 5, 431–436 CrossRef CAS
- R. W. Miles, P. C. Tyler, G. B. Evans, R. H. Furneaux, D. W. Parkin and V. L. Schramm, Biochemistry, 1999, 38, 13147–13154 Search PubMed
- G. B. Evans, P. C. Tyler and V. L. Schramm, ACS Infect. Dis., 2018, 4, 107–117 Search PubMed
- G. B. Evans, R. H. Furneaux, G. J. Gainsford, J. C. Hanson, G. A. Kicska, A. A. Sauve, V. L. Schramm and P. C. Tyler, J. Med. Chem., 2003, 46, 155–160 CrossRef CAS PubMed
- J. M. Mason, H. Yuan, G. B. Evans, P. C. Tyler, Q. Du and V. L. Schramm, Eur. J. Med. Chem., 2017, 127, 793–809 CrossRef CAS PubMed
- S. Buchini and C. J. Leumann, Curr. Opin. Chem. Biol., 2003, 7, 717–726 Search PubMed
- A. Häberli and C. J. Leumann, Org. Lett., 2002, 4, 3275–3278 Search PubMed
- A. Häberli and C. J. Leumann, Org. Lett., 2001, 3, 489–492 Search PubMed
- D. M. Tidd and H. M. Warenius, Br. J. Cancer, 1989, 60, 343–350 Search PubMed
- R. J. Boado and W. M. Pardridge, Bioconjugate Chem., 1992, 3, 519–523 CrossRef CAS PubMed
- A. K. Saha, C. Waychunas, T. J. Caulfield, D. A. Upson, C. Hobbs and A. M. Yawman, J. Org. Chem., 1995, 60, 788–789 CrossRef CAS
- G. Wang and P. J. Middleton, Tetrahedron Lett., 1996, 37, 2739–2742 Search PubMed
- P. P. Seth, C. R. Allerson, A. Siwkowski, G. Vasquez, A. Berdeja, M. T. Migawa, H. Gaus, T. P. Prakash, B. Bhat and E. E. Swayze, J. Med. Chem., 2010, 53, 8309–8318 Search PubMed
- T. P. Prakash, W. F. Lima, H. M. Murray, W. Li, G. A. Kinberger, A. E. Chappell, H. Gaus, P. P. Seth, B. Bhat, S. T. Crooke and E. E. Swayze, Nucleic Acids Res., 2015, 43, 2993–3011 Search PubMed
- G. Clavé, M. Reverte, J.-J. Vasseur and M. Smietana, RSC Chem. Biol., 2021, 2, 94–150 RSC
- D. Hutter, M. O. Blaettler and S. A. Benner, Helv. Chim. Acta, 2002, 85, 2777–2806 Search PubMed
- T. Kofoed, P. B. Rasmussen, P. ValentinHansen and E. B. Pedersen, Acta Chem. Scand., 1997, 51, 318–324 Search PubMed
- S. Epple, A. Modi, Y. R. Baker, E. Węgrzyn, D. Traoré, P. Wanat, A. E. S. Tyburn, A. Shivalingam, L. Taemaitree, A. H. El-Sagheer and T. Brown, J. Am. Chem. Soc., 2021, 143, 16293–16301 CrossRef CAS PubMed
- N. Z. Fantoni, A. H. El-Sagheer and T. Brown, Chem. Rev., 2021, 121, 7122–7154 CrossRef CAS PubMed
- B. Zengin Kurt, D. Dhara, A. H. El-Sagheer and T. Brown, Org. Lett., 2024, 26, 4137–4141 CrossRef CAS PubMed
- E. M. Huie, M. R. Kirshenbaum and G. L. Trainor, J. Org. Chem., 1992, 57, 4569–4570 CrossRef CAS
- K. J. Fettes, N. Howard, D. T. Hickman, S. Adah, M. R. Player, P. F. Torrence and J. Micklefield, J. Chem. Soc., Perkin Trans. 1, 2002, 485–495, 10.1039/B110603C
- E. Rozners, D. Katkevica, E. Bizdena and R. Strömberg, J. Am. Chem. Soc., 2003, 125, 12125–12136 CrossRef CAS PubMed
- C. Pal, M. Richter and E. Rozners, ACS Chem. Biol., 2024, 19, 249–253 CrossRef CAS PubMed
- R. L. Momparler, Biochem. Biophys. Res. Commun., 1969, 34, 465–471 CrossRef CAS PubMed
- O. Rechkoblit, R. E. Johnson, A. Buku, L. Prakash, S. Prakash and A. K. Aggarwal, Sci. Rep., 2019, 9, 16400 CrossRef PubMed
- A. M. Noronha, C. J. Wilds, C.-N. Lok, K. Viazovkina, D. Arion, M. A. Parniak and M. J. Damha, Biochemistry, 2000, 39, 7050–7062 Search PubMed
- P. A. Giannaris and M. J. Damha, Can. J. Chem., 1994, 72, 909–918 CrossRef CAS
- M. J. Damha, C. J. Wilds, A. Noronha, I. Brukner, G. Borkow, D. Arion and M. A. Parniak, J. Am. Chem. Soc., 1998, 120, 12976–12977 CrossRef CAS
- A. Noronha and M. J. Damha, Nucleic Acids Res., 1998, 26, 2665–2671 CrossRef CAS PubMed
- E. Medina, E. J. Yik, P. Herdewijn and J. C. Chaput, ACS Synth. Biol., 2021, 10, 1429–1437 CrossRef CAS PubMed
- A. I. Taylor, V. B. Pinheiro, M. J. Smola, A. S. Morgunov, S. Peak-Chew, C. Cozens, K. M. Weeks, P. Herdewijn and P. Holliger, Nature, 2015, 518, 427–430 CrossRef CAS PubMed
- K. B. Wu, C. J. A. Skrodzki, Q. Su, J. Lin and J. Niu, Chem. Sci., 2022, 13, 6873–6881 RSC
- L. Sun, X. Ma, B. Zhang, Y. Qin, J. Ma, Y. Du and T. Chen, RSC Chem. Biol., 2022, 3, 1173–1197 RSC
- P. Kois, Z. Tocik, M. Spassova, W.-Y. Ren, I. Rosenberg, J. F. Soler and K. A. Watanabe, Nucleosides, Nucleotides Nucleic Acids, 1993, 12, 1093–1109 CrossRef CAS
- I. Rosenberg, J. F. Soler, Z. Tocik, W.-Y. Ren, L. A. Ciszewski, P. Kois, K. W. Pankiewicz, M. Spassova and K. A. Watanabe, Nucleosides, Nucleotides Nucleic Acids, 1993, 12, 381–401 CrossRef CAS
- R. El-Khoury and M. J. Damha, Acc. Chem. Res., 2021, 54, 2287–2297 Search PubMed
- C. J. Wilds and M. J. Damha, Nucleic Acids Res., 2000, 28, 3625–3635 CrossRef CAS PubMed
- F. Li, S. Sarkhel, C. J. Wilds, Z. Wawrzak, T. P. Prakash, M. Manoharan and M. Egli, Biochemistry, 2006, 45, 4141–4152 Search PubMed
- J.-F. Trempe, C. J. Wilds, A. Y. Denisov, R. T. Pon, M. J. Damha and K. Gehring, J. Am. Chem. Soc., 2001, 123, 4896–4903 CrossRef CAS PubMed
- S. A. Ingale, P. Leonard, Q. N. Tran and F. Seela, J. Org. Chem., 2015, 80, 3124–3138 CrossRef CAS PubMed
- C.-N. Lok, E. Viazovkina, K.-L. Min, E. Nagy, C. J. Wilds, M. J. Damha and M. A. Parniak, Biochemistry, 2002, 41, 3457–3467 CrossRef CAS PubMed
- M. E. Østergaard, J. Nichols, T. A. Dwight, W. Lima, M. E. Jung, E. E. Swayze and P. P. Seth, Mol. Ther.--Nucleic Acids, 2017, 7, 20–30 CrossRef PubMed
- T. Dowler, D. Bergeron, A.-L. Tedeschi, L. Paquet, N. Ferrari and M. J. Damha, Nucleic Acids Res., 2006, 34, 1669–1675 CrossRef CAS PubMed
- G. F. Deleavey, J. K. Watts, T. Alain, F. Robert, A. Kalota, V. Aishwarya, J. Pelletier, A. M. Gewirtz, N. Sonenberg and M. J. Damha, Nucleic Acids Res., 2010, 38, 4547–4557 CrossRef CAS PubMed
- D. O’Reilly, Z. J. Kartje, E. A. Ageely, E. Malek-Adamian, M. Habibian, A. Schofield, C. L. Barkau, K. J. Rohilla, L. B. DeRossett, A. T. Weigle, M. J. Damha and K. T. Gagnon, Nucleic Acids Res., 2018, 47, 546–558 Search PubMed
- M. J. Donde, A. M. Rochussen, S. Kapoor and A. I. Taylor, Commun. Biol., 2022, 5, 1010 CrossRef CAS PubMed
- A. I. Taylor, C. J. K. Wan, M. J. Donde, S.-Y. Peak-Chew and P. Holliger, Nat. Chem., 2022, 14, 1295–1305 CrossRef CAS PubMed
- C. G. Peng and M. J. Damha, J. Am. Chem. Soc., 2007, 129, 5310–5311 CrossRef CAS PubMed
- C. G. Peng and M. J. Damha, Can. J. Chem., 2008, 86, 881–891 CrossRef CAS
- F. Levi-Acobas, A. Katolik, P. Röthlisberger, T. Cokelaer, I. Sarac, M. J. Damha, C. J. Leumann and M. Hollenstein, Org. Biomol. Chem., 2019, 17, 8083–8087 RSC
- I. Alves Ferreira-Bravo, C. Cozens, P. Holliger and J. J. DeStefano, Nucleic Acids Res., 2015, 43, 9587–9599 Search PubMed
- Y. Wang, A. K. Ngor, A. Nikoomanzar and J. C. Chaput, Nat. Commun., 2018, 9, 5067 CrossRef PubMed
- J. C. Chaput, Acc. Chem. Res., 2021, 54, 1056–1065 CrossRef CAS PubMed
- K.-U. Schöning, P. Scholz, S. Guntha, X. Wu, R. Krishnamurthy and A. Eschenmoser, Science, 2000, 290, 1347–1351 CrossRef PubMed
- K.-U. Schöning, P. Scholz, X. Wu, S. Guntha, G. Delgado, R. Krishnamurthy and A. Eschenmoser, Helv. Chim. Acta, 2002, 85, 4111–4153 CrossRef
- P. S. Pallan, C. J. Wilds, Z. Wawrzak, R. Krishnamurthy, A. Eschenmoser and M. Egli, Angew. Chem., Int. Ed., 2003, 42, 5893–5895 CrossRef CAS PubMed
- H. H. Lackey, E. M. Peterson, Z. Chen, J. M. Harris and J. M. Heemstra, ACS Synth. Biol., 2019, 8, 1144–1152 CrossRef CAS PubMed
- C. J. Wilds, Z. Wawrzak, R. Krishnamurthy, A. Eschenmoser and M. Egli, J. Am. Chem. Soc., 2002, 124, 13716–13721 CrossRef CAS PubMed
- M.-O. Ebert, C. Mang, R. Krishnamurthy, A. Eschenmoser and B. Jaun, J. Am. Chem. Soc., 2008, 130, 15105–15115 CrossRef PubMed
- J. C. Chaput and J. W. Szostak, J. Am. Chem. Soc., 2003, 125, 9274–9275 CrossRef CAS PubMed
- L. N. Jackson, N. Chim, C. Shi and J. C. Chaput, Nucleic Acids Res., 2019, 47, 6973–6983 CrossRef CAS PubMed
- M. R. Dunn, C. Otto, K. E. Fenton and J. C. Chaput, ACS Chem. Biol., 2016, 11, 1210–1219 CrossRef CAS PubMed
- N. Chim, C. Shi, S. P. Sau, A. Nikoomanzar and J. C. Chaput, Nat. Commun., 2017, 8, 1810 CrossRef PubMed
- H. Yu, S. Zhang and J. C. Chaput, Nat. Chem., 2012, 4, 183–187 CrossRef CAS PubMed
- D. Wei, Y. Wang, D. Song, Z. Zhang, J. Wang, J.-Y. Chen, Z. Li and H. Yu, ACS Synth. Biol., 2022, 11, 3874–3885 CrossRef CAS PubMed
- Y. Wang, Y. Wang, D. Song, X. Sun, Z. Zhang, X. Li, Z. Li and H. Yu, J. Am. Chem. Soc., 2021, 143, 8154–8163 CrossRef CAS PubMed
- Y. Wang, K. Nguyen, R. C. Spitale and J. C. Chaput, Nat. Chem., 2021, 13, 319–326 CrossRef CAS PubMed
- M. Gao, D. Wei, S. Chen, B. Qin, Y. Wang, Z. Li and H. Yu, ChemBioChem, 2023, 24, e202200651 CrossRef CAS PubMed
- A. Lozoya-Colinas, Y. Yu and J. C. Chaput, J. Am. Chem. Soc., 2023, 145, 25789–25796 CrossRef CAS PubMed
- M. C. Culbertson, K. W. Temburnikar, S. P. Sau, J.-Y. Liao, S. Bala and J. C. Chaput, Bioorg. Med. Chem. Lett., 2016, 26, 2418–2421 CrossRef CAS PubMed
- E. M. Lee, N. A. Setterholm, M. Hajjar, B. Barpuzary and J. C. Chaput, Nucleic Acids Res., 2023, 51, 9542–9551 CrossRef CAS PubMed
- S. Matsuda, S. Bala, J.-Y. Liao, D. Datta, A. Mikami, L. Woods, J. M. Harp, J. A. Gilbert, A. Bisbe, R. M. Manoharan, M. Kim, C. S. Theile, D. C. Guenther, Y. Jiang, S. Agarwal, R. Maganti, M. K. Schlegel, I. Zlatev, K. Charisse, K. G. Rajeev, A. Castoreno, M. Maier, M. M. Janas, M. Egli, J. C. Chaput and M. Manoharan, J. Am. Chem. Soc., 2023, 145, 19691–19706 CrossRef CAS PubMed
- F. Wang, L. S. Liu, C. H. Lau, T. J. Han Chang, D. Y. Tam, H. M. Leung, C. Tin and P. K. Lo, ACS Appl. Mater. Interfaces, 2019, 11, 38510–38518 CrossRef CAS PubMed
- F. Wang, L. S. Liu, P. Li, C. H. Lau, H. M. Leung, Y. R. Chin, C. Tin and P. K. Lo, Mater. Today Bio, 2022, 15, 100299 CrossRef CAS PubMed
- M. Maiti, M. Maiti, C. Knies, S. Dumbre, E. Lescrinier, H. Rosemeyer, A. Ceulemans and P. Herdewijn, Nucleic Acids Res., 2015, 43, 7189–7200 CrossRef CAS PubMed
- H. Rosemeyer and F. Seela, Helv. Chim. Acta, 1991, 74, 748–760 CrossRef CAS
- F. Seela, M. Heckel and H. Rosemeyer, Helv. Chim. Acta, 1996, 79, 1451–1461 CrossRef CAS
- D. J. Sharpe, K. Röder and D. J. Wales, J. Phys. Chem. B, 2020, 124, 4062–4068 CrossRef CAS PubMed
- S. Ivanov, Y. Alekseev, J. R. Bertrand, C. Malvy and M. B. Gottikh, Nucleic Acids Res., 2003, 31, 4256–4263 CrossRef CAS PubMed
- B. Ravindra Babu, Raunak, N. E. Poopeiko, M. Juhl, A. D. Bond, V. S. Parmar and J. Wengel, Eur. J. Org. Chem., 2005, 2297–2321 CrossRef
- N. E. Poopeiko, M. Juhl, B. Vester, M. D. Sørensen and J. Wengel, Bioorg. Med. Chem. Lett., 2003, 13, 2285–2290 CrossRef CAS PubMed
- B. Bauwens, J. Rozenski, P. Herdewijn and J. Robben, ChemBioChem, 2018, 19, 2410–2420 CrossRef CAS PubMed
- M. Maiti, V. Siegmund, M. Abramov, E. Lescrinier, H. Rosemeyer, M. Froeyen, A. Ramaswamy, A. Ceulemans, A. Marx and P. Herdewijn, Chem. – Eur. J., 2012, 18, 869–879 CrossRef CAS PubMed
- A. Eschenmoser, Angew. Chem., Int. Ed., 2011, 50, 12412–12472 CrossRef CAS PubMed
- M. Stoop, G. Meher, P. Karri and R. Krishnamurthy, Chem. – Eur. J., 2013, 19, 15336–15345 CrossRef CAS PubMed
- T. Efthymiou, J. Gavette, M. Stoop, F. De Riccardis, M. Froeyen, P. Herdewijn and R. Krishnamurthy, Chem. – Eur. J., 2018, 24, 12811–12819 CrossRef CAS PubMed
- A. Eschenmoser and E. Loewenthal, Chem. Soc. Rev., 1992, 21, 1–16 RSC
- N. Nogal, M. Sanz-Sánchez, S. Vela-Gallego, K. Ruiz-Mirazo and A. de la Escosura, Chem. Soc. Rev., 2023, 52, 7359–7388 RSC
- M. Böhringer, H.-J. Roth, J. Hunziker, M. Göbel, R. Krishnan, A. Giger, B. Schweizer, J. Schreiber, C. Leumann and A. Eschenmoser, Helv. Chim. Acta, 1992, 75, 1416–1477 CrossRef
- K. Augustyns, A. van Aerschot, C. Urbanke and P. Herdewijn, Bull. Soc. Chim. Belg., 1992, 101, 119–130 CrossRef CAS
- J. Hunziker, H.-J. Roth, M. Böhringer, A. Giger, U. Diederichsen, M. Göbel, R. Krishnan, B. Jaun, C. Leumann and A. Eschenmoser, Helv. Chim. Acta, 1993, 76, 259–352 CrossRef CAS
- M. Egli, P. S. Pallan, R. Pattanayek, C. J. Wilds, P. Lubini, G. Minasov, M. Dobler, C. J. Leumann and A. Eschenmoser, J. Am. Chem. Soc., 2006, 128, 10847–10856 CrossRef CAS PubMed
- M. Froeyen, E. Lescrinier, L. Kerremans, H. Rosemeyer, F. Seela, B. Verbeure, I. Lagoja, J. Rozenski, A. Van Aerschot, R. Busson and P. Herdewijn, Chem. – Eur. J., 2001, 7, 5183–5194 CrossRef CAS PubMed
- I. Verheggen, A. Van Aerschot, S. Toppet, R. Snoeck, G. Janssen, J. Balzarini, E. De Clercq and P. Herdewijn, J. Med. Chem., 1993, 36, 2033–2040 CrossRef CAS PubMed
- A. Aerschot Van, I. Verheggen, C. Hendrix and P. Herdewijn, Angew. Chem., Int. Ed. Engl., 1995, 34, 1338–1339 CrossRef
- C. Paolella, D. D'Alonzo, G. Schepers, A. Van Aerschot, G. Di Fabio, G. Palumbo, P. Herdewijn and A. Guaragna, Org. Biomol. Chem., 2015, 13, 10041–10049 RSC
- C. Hendrix, H. Rosemeyer, B. De Bouvere, A. Van Aerschot, F. Seela and P. Herdewijn, Chem. – Eur. J., 1997, 3, 1513–1520 CrossRef CAS
- N. Hossain, B. Wroblowski, A. Van Aerschot, J. Rozenski, A. De Bruyn and P. Herdewijn, J. Org. Chem., 1998, 63, 1574–1582 CrossRef CAS
- E. Lescrinier, R. Esnouf, J. Schraml, R. Busson, H. A. Heus, C. W. Hilbers and P. Herdewijn, Chem. Biol., 2000, 7, 719–731 CrossRef CAS PubMed
- L. Kerremans, G. Schepers, J. Rozenski, R. Busson, A. Van Aerschot and P. Herdewijn, Org. Lett., 2001, 3, 4129–4132 CrossRef CAS PubMed
- H. Kang, M. H. Fisher, D. Xu, Y. J. Miyamoto, A. Marchand, A. Van Aerschot, P. Herdewijn and R. L. Juliano, Nucleic Acids Res., 2004, 32, 4411–4419 CrossRef CAS PubMed
- E. Eremeeva, A. Fikatas, L. Margamuljana, M. Abramov, D. Schols, E. Groaz and P. Herdewijn, Nucleic Acids Res., 2019, 47, 4927–4939 CrossRef CAS PubMed
- A. I. Taylor, F. Beuron, S.-Y. Peak-Chew, E. P. Morris, P. Herdewijn and P. Holliger, ChemBioChem, 2016, 17, 1107–1110 CrossRef CAS PubMed
- H. D. Winter, E. Lescrinier, A. V. Aerschot and P. Herdewijn, J. Am. Chem. Soc., 1998, 120, 5381–5394 CrossRef
- B. Allart, K. Khan, H. Rosemeyer, G. Schepers, C. Hendrix, K. Rothenbacher, F. Seela, A. Van Aerschot and P. Herdewijn, Chem. – Eur. J., 1999, 5, 2424–2431 CrossRef CAS
- M. Ovaere, J. Sponer, J. E. Sponer, P. Herdewijn and L. Van Meervelt, Nucleic Acids Res., 2012, 40, 7573–7583 CrossRef CAS PubMed
- B. T. Le, S. X. Chen, M. Abramov, P. Herdewijn and R. N. Veedu, Chem. Commun., 2016, 52, 13467–13470 RSC
- Y. Maurinsh, H. Rosemeyer, R. Esnouf, A. Medvedovici, J. Wang, G. Ceulemans, E. Lescrinier, C. Hendrix, R. Busson, P. Sandra, F. Seela, A. Van Aerschot and P. Herdewijn, Chem. – Eur. J., 1999, 5, 2139–2150 CrossRef CAS
- J. Wang, B. Verbeure, I. Luyten, E. Lescrinier, M. Froeyen, C. Hendrix, H. Rosemeyer, F. Seela, A. Van Aerschot and P. Herdewijn, J. Am. Chem. Soc., 2000, 122, 8595–8602 CrossRef CAS
- B. Verbeure, E. Lescrinier, J. Wang and P. Herdewijn, Nucleic Acids Res., 2001, 29, 4941–4947 CrossRef CAS PubMed
- J. Wang, D. Viña, R. Busson and P. Herdewijn, J. Org. Chem., 2003, 68, 4499–4505 CrossRef CAS PubMed
- Y. Moai and T. Kodama, Tetrahedron Lett., 2020, 61, 151708 CrossRef CAS
- P. P. Seth, J. Yu, A. Jazayeri, P. S. Pallan, C. R. Allerson, M. E. Østergaard, F. Liu, P. Herdewijn, M. Egli and E. E. Swayze, J. Org. Chem., 2012, 77, 5074–5085 CrossRef CAS PubMed
- Y. Kitamura, S. Moribe and Y. Kitade, Bioorg. Med. Chem. Lett., 2018, 28, 3174–3176 CrossRef CAS PubMed
- S. Pitsch, S. Wendeborn, B. Jaun and A. Eschenmoser, Helv. Chim. Acta, 1993, 76, 2161–2183 CrossRef CAS
- S. Pitsch, R. Krishnamurthy, M. Bolli, S. Wendeborn, A. Holzner, M. Minton, C. Lesueur, I. Schlönvogt, B. Jaun and A. Eschenmoser, Helv. Chim. Acta, 1995, 78, 1621–1635 CrossRef CAS
- I. Schlönvogt, S. Pitsch, C. Lesueur, A. Eschenmoser, B. Jaun and R. M. Wolf, Helv. Chim. Acta, 1996, 79, 2316–2345 CrossRef
- M. O. Ebert and B. Jaun, Chem. Biodiversity, 2010, 7, 2103–2128 CrossRef CAS PubMed
- P. S. Pallan, P. Lubini, M. Bolli and M. Egli, Nucleic Acids Res., 2007, 35, 6611–6624 CrossRef CAS PubMed
- M. Bolli, R. Micura, S. Pitsch and A. Eschenmoser, Helv. Chim. Acta, 1997, 80, 1901–1951 CrossRef CAS
- M. Beier, F. Reck, T. Wagner, R. Krishnamurthy and A. Eschenmoser, Science, 1999, 283, 699–703 CrossRef CAS PubMed
- F. Reck, H. Wippo, R. Kudick, M. Bolli, G. Ceulemans, R. Krishnamurthy and A. Eschenmoser, Org. Lett., 1999, 1, 1531–1534 CrossRef CAS PubMed
- E. P. Stirchak, J. E. Summerton and D. D. Weller, Nucleic Acids Res., 1989, 17, 6129–6141 CrossRef CAS PubMed
- J. Summerton, Biochim. Biophys. Acta, Gene Struct. Expression, 1999, 1489, 141–158 CrossRef CAS PubMed
- L. Lacroix, P. B. Arimondo, M. Takasugi, C. Hélène and J.-L. Mergny, Biochem. Biophys. Res. Commun., 2000, 270, 363–369 CrossRef CAS PubMed
- H. Torigoe, K. Sasaki and T. Katayama, J. Biochem., 2009, 146, 173–183 CrossRef CAS PubMed
- R. M. Hudziak, E. Barofsky, D. F. Barofsky, D. L. Weller, S.-B. Huang and D. D. Weller, Antisense Nucleic Acid Drug Dev., 1996, 6, 267–272 CrossRef CAS PubMed
- D. Stein, E. Foster, S.-B. Huang, D. D. Weller and J. Summerton, Antisense Nucleic Acid Drug Dev., 1997, 7, 151–157 CrossRef CAS PubMed
- Y. Miao, C. Fu, Z. Yu, L. Yu, Y. Tang and M. Wei, Acta Pharm. Sin. B, 2024, 14, 3802–3817 CrossRef CAS PubMed
- A. Ghosh, A. Banerjee, S. Gupta and S. Sinha, J. Am. Chem. Soc., 2024, 146, 32989–33001 CrossRef CAS PubMed
- K. W. Knouse, J. N. deGruyter, M. A. Schmidt, B. Zheng, J. C. Vantourout, C. Kingston, S. E. Mercer, I. M. Mcdonald, R. E. Olson, Y. Zhu, C. Hang, J. Zhu, C. Yuan, Q. Wang, P. Park, M. D. Eastgate and P. S. Baran, Science, 2018, 361, 1234–1238 CrossRef CAS PubMed
- J. Rihon, C. A. Mattelaer, R. W. Montalvão, M. Froeyen, V. B. Pinheiro and E. Lescrinier, Nucleic Acids Res., 2024, 52, 2836–2847 CrossRef CAS PubMed
- H. K. Langner, K. Jastrzebska and M. H. Caruthers, J. Am. Chem. Soc., 2020, 142, 16240–16253 CrossRef CAS PubMed
- B. T. Le, S. Paul, K. Jastrzebska, H. Langer, M. H. Caruthers and R. N. Veedu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2207956119 CrossRef CAS PubMed
- M. Tarköy, M. Bolli, B. Schweizer and C. Leumann, Helv. Chim. Acta, 1993, 76, 481–510 CrossRef
- M. Tarköy and C. Leumann, Angew. Chem., Int. Ed. Engl., 1993, 32, 1432–1434 CrossRef
- M. Bolli, H. U. Trafelet and C. Leumann, Nucleic Acids Res., 1996, 24, 4660–4667 CrossRef CAS PubMed
- M. Bolli, J. Christopher Litten, R. Schütz and C. J. Leumann, Chem. Biol., 1996, 3, 197–206 CrossRef CAS PubMed
- M. Tarköy, M. Bolli and C. Leumann, Helv. Chim. Acta, 1994, 77, 716–744 CrossRef
- P. S. Pallan, D. Ittig, A. Héroux, Z. Wawrzak, C. J. Leumann and M. Egli, Chem. Commun., 2008, 883–885, 10.1039/B716390H
- R. Meier, S. Grüschow and C. Leumann, Helv. Chim. Acta, 1999, 82, 1813–1828 CrossRef CAS
- B. Dugovic and C. J. Leumann, J. Org. Chem., 2014, 79, 1271–1279 CrossRef CAS PubMed
- R. Steffens and C. Leumann, Helv. Chim. Acta, 1997, 80, 2426–2439 CrossRef CAS
- R. Steffens and C. J. Leumann, J. Am. Chem. Soc., 1999, 121, 3249–3255 CrossRef CAS
- A. Istrate, S. Johannsen, A. Istrate, R. K. O. Sigel and C. J. Leumann, Nucleic Acids Res., 2019, 47, 4872–4882 CrossRef CAS PubMed
- D. Renneberg and C. J. Leumann, J. Am. Chem. Soc., 2002, 124, 5993–6002 CrossRef CAS PubMed
- A. Goyenvalle, G. Griffith, A. Babbs, S. E. Andaloussi, K. Ezzat, A. Avril, B. Dugovic, R. Chaussenot, A. Ferry, T. Voit, H. Amthor, C. Bühr, S. Schürch, M. J. A. Wood, K. E. Davies, C. Vaillend, C. Leumann and L. Garcia, Nat. Med., 2015, 21, 270–275 CrossRef CAS PubMed
- F. Bizot, T. Tensorer, L. Garcia and A. Goyenvalle, Nucleic Acid Ther., 2023, 33, 374–380 CrossRef CAS PubMed
- J. Lietard and C. J. Leumann, J. Org. Chem., 2012, 77, 4566–4577 CrossRef CAS PubMed
- A. Istrate, A. Katolik, A. Istrate and C. J. Leumann, Chem. – Eur. J., 2017, 23, 10310–10318 CrossRef CAS PubMed
- M. Hollenstein and C. J. Leumann, ChemBioChem, 2014, 15, 1901–1904 CrossRef CAS PubMed
- S. Diafa, D. Evéquoz, C. J. Leumann and M. Hollenstein, Chem. – Asian J., 2017, 12, 1347–1352 CrossRef CAS PubMed
- M. A. Campbell and J. Wengel, Chem. Soc. Rev., 2011, 40, 5680–5689 RSC
- S. Obika, D. Nanbu, Y. Hari, J.-I. Andoh, K.-I. Morio, T. Doi and T. Imanishi, Tetrahedron Lett., 1998, 39, 5401–5404 CrossRef CAS
- S. K. Singh, A. A. Koshkin, J. Wengel and P. Nielsen, Chem. Commun., 1998, 455–456, 10.1039/A708608C
- A. A. Koshkin, S. K. Singh, P. Nielsen, V. K. Rajwanshi, R. Kumar, M. Meldgaard, C. E. Olsen and J. Wengel, Tetrahedron, 1998, 54, 3607–3630 CrossRef CAS
- A. A. Koshkin, P. Nielsen, M. Meldgaard, V. K. Rajwanshi, S. K. Singh and J. Wengel, J. Am. Chem. Soc., 1998, 120, 13252–13253 CrossRef CAS
- J. Duschmalé, H. F. Hansen, M. Duschmalé, E. Koller, N. Albaek, M. R. Møller, K. Jensen, T. Koch, J. Wengel and K. Bleicher, Nucleic Acids Res., 2019, 48, 63–74 CrossRef PubMed
- M. Shin, I. L. Chan, Y. Cao, A. M. Gruntman, J. Lee, J. Sousa, T. C. Rodríguez, D. Echeverria, G. Devi, A. J. Debacker, M. P. Moazami, P. M. Krishnamurthy, J. M. Rembetsy-Brown, K. Kelly, O. Yukselen, E. Donnard, T. J. Parsons, A. Khvorova, E. J. Sontheimer, R. Maehr, M. Garber and J. K. Watts, Nucleic Acids Res., 2022, 50, 8418–8430 CrossRef CAS PubMed
- J. Elmén, H. Thonberg, K. Ljungberg, M. Frieden, M. Westergaard, Y. Xu, B. Wahren, Z. Liang, H. Ørum, T. Koch and C. Wahlestedt, Nucleic Acids Res., 2005, 33, 439–447 CrossRef PubMed
- H. Iribe, K. Miyamoto, T. Takahashi, Y. Kobayashi, J. Leo, M. Aida and K. Ui-Tei, ACS Omega, 2017, 2, 2055–2064 CrossRef CAS PubMed
- C. R. Cromwell, K. Sung, J. Park, A. R. Krysler, J. Jovel, S. K. Kim and B. P. Hubbard, Nat. Commun., 2018, 9, 1448 CrossRef PubMed
- M. Kuwahara, S. Obika, J.-I. Nagashima, Y. Ohta, Y. Suto, H. Ozaki, H. Sawai and T. Imanishi, Nucleic Acids Res., 2008, 36, 4257–4265 CrossRef CAS PubMed
- R. N. Veedu, B. Vester and J. Wengel, New J. Chem., 2010, 34, 877–879 RSC
- R. N. Veedu, B. Vester and J. Wengel, J. Am. Chem. Soc., 2008, 130, 8124–8125 CrossRef CAS PubMed
- N. Sabat, D. Katkevica, K. Pajuste, M. Flamme, A. Stämpfli, M. Katkevics, S. Hanlon, S. Bisagni, K. Püntener, F. Sladojevich and M. Hollenstein, Front. Chem., 2023, 11, 1161462 CrossRef CAS PubMed
- M. Flamme, S. Hanlon, H. Iding, K. Puentener, F. Sladojevich and M. Hollenstein, Bioorg. Med. Chem. Lett., 2021, 48, 128242 CrossRef CAS PubMed
- H. Hoshino, Y. Kasahara, M. Kuwahara and S. Obika, J. Am. Chem. Soc., 2020, 142, 21530–21537 CrossRef CAS PubMed
- S. A. Burel, C. E. Hart, P. Cauntay, J. Hsiao, T. Machemer, M. Katz, A. Watt, H.-H. Bui, H. Younis, M. Sabripour, S. M. Freier, G. Hung, A. Dan, T. P. Prakash, P. P. Seth, E. E. Swayze, C. F. Bennett, S. T. Crooke and S. P. Henry, Nucleic Acids Res., 2015, 44, 2093–2109 CrossRef PubMed
- E. E. Swayze, A. M. Siwkowski, E. V. Wancewicz, M. T. Migawa, T. K. Wyrzykiewicz, G. Hung, B. P. Monia and A. C. F. Bennett, Nucleic Acids Res., 2006, 35, 687–700 CrossRef PubMed
- D. Sabatino and M. J. Damha, J. Am. Chem. Soc., 2007, 129, 8259–8270 CrossRef CAS PubMed
- M. Habibian, S. Martínez-Montero, G. Portella, Z. Chua, D. S. Bohle, M. Orozco and M. J. Damha, Org. Lett., 2015, 17, 5416–5419 CrossRef CAS PubMed
- Y. Y. Zheng, Y. Wu, T. J. Begley and J. Sheng, RSC Chem. Biol., 2021, 2, 990–1003 RSC
- C.-X. Song, K. E. Szulwach, Y. Fu, Q. Dai, C. Yi, X. Li, Y. Li, C.-H. Chen, W. Zhang, X. Jian, J. Wang, L. Zhang, T. J. Looney, B. Zhang, L. A. Godley, L. M. Hicks, B. T. Lahn, P. Jin and C. He, Nat. Biotechnol., 2011, 29, 68–72 CrossRef CAS PubMed
- J. Josse and A. Kornberg, J. Biol. Chem., 1962, 237, 1968–1976 CrossRef CAS PubMed
- C. Reily, T. J. Stewart, M. B. Renfrow and J. Novak, Nat. Rev. Nephrol., 2019, 15, 346–366 CrossRef PubMed
- Y.-J. Lee, N. Dai, S. E. Walsh, S. Müller, M. E. Fraser, K. M. Kauffman, C. Guan, I. R. Corrêa and P. R. Weigele, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E3116–E3125 CAS
- T. Carell, C. Brandmayr, A. Hienzsch, M. Müller, D. Pearson, V. Reiter, I. Thoma, P. Thumbs and M. Wagner, Angew. Chem., Int. Ed., 2012, 51, 7110–7131 CrossRef CAS PubMed
- E. K. Borchardt, N. M. Martinez and W. V. Gilbert, Annu. Rev. Genet., 2020, 54, 309–336 CrossRef CAS PubMed
- H. Kasai, K. Nakanishi, R. D. Macfarlane, D. F. Torgerson, Z. Ohashi, J. A. McCloskey, H. J. Gross and S. Nishimura, J. Am. Chem. Soc., 1976, 98, 5044–5046 CrossRef CAS PubMed
- J. C. Salazar, A. Ambrogelly, P. F. Crain, J. A. McCloskey and D. Söll, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7536–7541 CrossRef CAS PubMed
- M. Blaise, H. D. Becker, J. Lapointe, C. Cambillau, R. Giegé and D. Kern, Biochimie, 2005, 87, 847–861 CrossRef CAS PubMed
- A. Hienzsch, C. Deiml, V. Reiter and T. Carell, Chem. – Eur. J., 2013, 19, 4244–4248 CrossRef CAS PubMed
- M. Hillmeier, M. Wagner, T. Ensfelder, E. Korytiakova, P. Thumbs, M. Müller and T. Carell, Nat. Commun., 2021, 12, 7123 CrossRef CAS PubMed
- Y. Sun, M. Piechotta, I. Naarmann-de Vries, C. Dieterich and A. E. Ehrenhofer-Murray, Nucleic Acids Res., 2023, 51, 11197–11212 CrossRef CAS PubMed
- X. Zhao, D. Ma, K. Ishiguro, H. Saito, S. Akichika, I. Matsuzawa, M. Mito, T. Irie, K. Ishibashi, K. Wakabayashi, Y. Sakaguchi, T. Yokoyama, Y. Mishima, M. Shirouzu, S. Iwasaki, T. Suzuki and T. Suzuki, Cell, 2023, 186, 5517–5535.e5524 CrossRef CAS PubMed
- T. Suzuki, A. Ogizawa, K. Ishiguro and A. Nagao, Cell Chem. Biol., 2024 DOI:10.1016/j.chembiol.2024.11.004
- A. Biela, A. Hammermeister, I. Kaczmarczyk, M. Walczak, L. Koziej, T.-Y. Lin and S. Glatt, J. Biol. Chem., 2023, 299, 104966 CrossRef CAS PubMed
- Y. Xie, P. Chai, N. A. Till, H. Hemberger, C. G. Lebedenko, J. Porat, C. P. Watkins, R. M. Caldwell, B. M. George, J. Perr, C. R. Bertozzi, B. A. Garcia and R. A. Flynn, Cell, 2024, 187, 5228–5237 CrossRef CAS PubMed
- N. Zhang, W. Tang, L. Torres, X. Wang, Y. Ajaj, L. Zhu, Y. Luan, H. Zhou, Y. Wang, D. Zhang, V. Kurbatov, S. A. Khan, P. Kumar, A. Hidalgo, D. Wu and J. Lu, Cell, 2024, 187, 846–860 CrossRef CAS PubMed
- J.-H. Xue, G.-D. Chen, F. Hao, H. Chen, Z. Fang, F.-F. Chen, B. Pang, Q.-L. Yang, X. Wei, Q.-Q. Fan, C. Xin, J. Zhao, X. Deng, B.-A. Wang, X.-J. Zhang, Y. Chu, H. Tang, H. Yin, W. Ma, L. Chen, J. Ding, E. Weinhold, R. M. Kohli, W. Liu, Z.-J. Zhu, K. Huang, H. Tang and G.-L. Xu, Nature, 2019, 569, 581–585 CrossRef CAS PubMed
- A. A. Hossain, Y. Z. Pigli, C. F. Baca, S. Heissel, A. Thomas, V. K. Libis, J. Burian, J. S. Chappie, S. F. Brady, P. A. Rice and L. A. Marraffini, Nature, 2024, 629, 410–416 CrossRef CAS PubMed
- P. Weigele and E. A. Raleigh, Chem. Rev., 2016, 116, 12655–12687 CrossRef CAS PubMed
- G. Hutinet, Y.-J. Lee, V. D. Crécy-Lagard and P. R. Weigele, EcoSal Plus, 2021, 9, eESP–0028 CrossRef CAS PubMed
- D. Swinton, S. Hattman, R. Benzinger, V. Buchanan-Wollaston and J. Beringer, FEBS Lett., 1985, 184, 294–298 CrossRef CAS PubMed
- J. A. Thomas, J. Orwenyo, L.-X. Wang and L. W. Black, Viruses, 2018, 10, 313 CrossRef PubMed
- I. R. Lehman and E. A. Pratt, J. Biol. Chem., 1960, 235, 3254–3259 CrossRef CAS PubMed
- C. L. Bair, D. Rifat and L. W. Black, J. Mol. Biol., 2007, 366, 779–789 CrossRef CAS PubMed
- J. Gommers-Ampt, J. Lutgerink and P. Borst, Nucleic Acids Res., 1991, 19, 1745–1751 CrossRef CAS PubMed
- P. Borst and R. Sabatini, Annu. Rev. Microbiol., 2008, 62, 235–251 CrossRef CAS PubMed
- J. H. Gommers-Ampt, F. Van Leeuwen, A. L. de Beer, J. F. Vliegenthart, M. Dizdaroglu, J. A. Kowalak, P. F. Crain and P. Borst, Cell, 1993, 75, 1129–1136 CrossRef CAS PubMed
- Henri G. A. M. van Luenen, C. Farris, S. Jan, P.-A. Genest, P. Tripathi, A. Velds, Ron M. Kerkhoven, M. Nieuwland, A. Haydock, G. Ramasamy, S. Vainio, T. Heidebrecht, A. Perrakis, L. Pagie, B. van Steensel, Peter J. Myler and P. Borst, Cell, 2012, 150, 909–921 CrossRef CAS PubMed
- F. van Leeuwen, E. R. Wijsman, E. Kuyl-Yeheskiely, G. A. van der Marel, J. H. van Boom and P. Borst, Nucleic Acids Res., 1996, 24, 2476–2482 CrossRef CAS PubMed
- R. Kieft, Y. Zhang, A. P. Marand, J. D. Moran, R. Bridger, L. Wells, R. J. Schmitz and R. Sabatini, PLoS Genet., 2020, 16, e1008390 CrossRef CAS PubMed
- M. Ehrlich and K. C. Ehrlich, J. Biol. Chem., 1981, 256, 9966–9972 CrossRef CAS PubMed
- A. M. Kropinski, D. Turner, J. H. E. Nash, H.-W. Ackermann, E. J. Lingohr, R. A. Warren, K. C. Ehrlich and M. Ehrlich, Viruses, 2018, 10, 217 CrossRef PubMed
- A. Chakrapani, O. Ruiz-Larrabeiti, R. Pohl, M. Svoboda, L. Krásný and M. Hocek, Chem. – Eur. J., 2022, 28, e202200911 CrossRef CAS PubMed
- M. Krömer, L. P. Slavětínská and M. Hocek, Chem. – Eur. J., 2024, 30, e202402318 CrossRef PubMed
- D. Dhara, L. A. Mulard and M. Hollenstein, Carbohydr. Res., 2023, 534, 108985 CrossRef CAS PubMed
- M. Chen, Z. Guo, J. Sun, W. Tang, M. Wang, Y. Tang, P. Li, B. Wu and Y. Chen, Acta Pharm. Sin. B, 2023, 13, 765–774 CrossRef CAS PubMed
- M. Chen, Z. Cao, W. Tang, M. Wang, Y. Chen and Z. Guo, J. Antibiot., 2022, 75, 172–175 CrossRef CAS PubMed
- A. Fire, S. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Nature, 1998, 391, 806–811 CrossRef CAS PubMed
- M. E. Davis, J. E. Zuckerman, C. H. J. Choi, D. Seligson, A. Tolcher, C. A. Alabi, Y. Yen, J. D. Heidel and A. Ribas, Nature, 2010, 464, 1067–1070 CrossRef CAS PubMed
- J. M. Sasso, B. J. B. Ambrose, R. Tenchov, R. S. Datta, M. T. Basel, R. K. DeLong and Q. A. Zhou, J. Med. Chem., 2022, 65, 6975–7015 CrossRef CAS PubMed
- V. Genna, L. Reyes-Fraile, J. Iglesias-Fernandez and M. Orozco, Curr. Opin. Struct. Biol., 2024, 87, 102838 CrossRef CAS PubMed
- B. Hu, L. Zhong, Y. Weng, L. Peng, Y. Huang, Y. Zhao and X.-J. Liang, Signal Transduction Targeted Ther., 2020, 5, 101 CrossRef CAS PubMed
- J. Belgrad, H. H. Fakih and A. Khvorova, Nucleic Acid Ther., 2024, 34, 52–72 CrossRef CAS PubMed
- D. M. Copolovici, K. Langel, E. Eriste and Ü. Langel, ACS Nano, 2014, 8, 1972–1994 CrossRef CAS PubMed
- D. Kodr, E. Kužmová, R. Pohl, T. Kraus and M. Hocek, Chem. Sci., 2023, 14, 4059–4069 RSC
- E.-A. Kusznir, J.-C. Hau, M. Portmann, A.-G. Reinhart, F. Falivene, J. Bastien, J. Worm, A. Ross, M. Lauer, P. Ringler, F. Sladojevich, S. Huber, K. Bleicher and M. Keller, Bioconjugate Chem., 2023, 34, 866–879 CrossRef CAS PubMed
- B. R. Meade, K. Gogoi, A. S. Hamil, C. Palm-Apergi, A. V. D. Berg, J. C. Hagopian, A. D. Springer, A. Eguchi, A. D. Kacsinta, C. F. Dowdy, A. Presente, P. Lönn, M. Kaulich, N. Yoshioka, E. Gros, X.-S. Cui and S. F. Dowdy, Nat. Biotechnol., 2014, 32, 1256–1261 CrossRef CAS PubMed
- K. K. Ray, R. S. Wright, D. Kallend, W. Koenig, L. A. Leiter, F. J. Raal, J. A. Bisch, T. Richardson, M. Jaros, P. L. J. Wijngaard and J. J. P. Kastelein, N. Engl. J. Med., 2020, 382, 1507–1519 CrossRef CAS PubMed
- V. Kumar and W. B. Turnbull, Chem. Soc. Rev., 2023, 52, 1273–1287 RSC
- V. Jadhav, A. Vaishnaw, K. Fitzgerald and M. A. Maier, Nat. Biotechnol., 2024, 42, 394–405 CrossRef CAS PubMed
- A. Novoa and N. Winssinger, Beilstein J. Org. Chem., 2015, 11, 707–719 CrossRef CAS PubMed
- N. Spinelli, E. Defrancq and F. Morvan, Chem. Soc. Rev., 2013, 42, 4557–4573 RSC
- B. Ugarte-Uribe, S. Pérez-Rentero, R. Lucas, A. Aviñó, J. J. Reina, I. Alkorta, R. Eritja and J. C. Morales, Bioconjugate Chem., 2010, 21, 1280–1287 CrossRef CAS PubMed
- A. Aviñó, S. M. Ocampo, R. Lucas, J. J. Reina, J. C. Morales, J. C. Perales and R. Eritja, Mol. Diversity, 2011, 15, 751–757 CrossRef PubMed
- G. Pourceau, A. Meyer, J.-J. Vasseur and F. Morvan, J. Org. Chem., 2009, 74, 1218–1222 CrossRef CAS PubMed
- A. Kiviniemi, P. Virta, M. S. Drenichev, S. N. Mikhailov and H. Lönnberg, Bioconjugate Chem., 2011, 22, 1249–1255 CrossRef CAS PubMed
- M. Pichon and M. Hollenstein, Commun. Chem., 2024, 7, 138 CrossRef CAS PubMed
- M. Hollenstein, Nat. Biotechnol., 2024 DOI:10.1038/s41587-024-02322-z
- D. J. Wiegand, J. Rittichier, E. Meyer, H. Lee, N. J. Conway, D. Ahlstedt, Z. Yurtsever, D. Rainone, E. Kuru and G. M. Church, Nat. Biotechnol., 2024 DOI:10.1038/s41587-024-02244-w
- E. R. Moody, R. Obexer, F. Nickl, R. Spiess and S. L. Lovelock, Science, 2023, 380, 1150–1154 CrossRef CAS PubMed
- C. Nybro Dansholm, S. Meier and S. R. Beeren, ChemBioChem, 2024, 25, e202300832 CrossRef CAS PubMed
- D. Larsen and S. R. Beeren, Chem. – Eur. J., 2020, 26, 11032–11038 CrossRef CAS PubMed
- A. Debon, E. Siirola and R. Snajdrova, JACS Au, 2023, 3, 1267–1283 CrossRef CAS PubMed
- P. Röthlisberger, F. Levi-Acobas, I. Sarac, R. Ricoux, J. P. Mahy, P. Herdewijn, P. Marlière and M. Hollenstein, Tetrahedron Lett., 2018, 59, 4241–4244 CrossRef
- S. Agrawal, Nucleic Acid Ther., 2024, 34, 37–51 CrossRef CAS PubMed
- J. A. Reyes-Darias, F. J. Sánchez-Luque, J. C. Morales, S. Pérez-Rentero, R. Eritja and A. Berzal-Herranz, ChemBioChem, 2015, 16, 584–591 CrossRef CAS PubMed
- E. Vengut-Climent, M. Terrazas, R. Lucas, M. Arévalo-Ruiz, R. Eritja and J. C. Morales, Bioorg. Med. Chem. Lett., 2013, 23, 4048–4051 CrossRef CAS PubMed
- H. Jung, G. Yu and H. Mok, Appl. Biol. Chem., 2016, 59, 759–763 CrossRef CAS
- K. Uehara, T. Harumoto, A. Makino, Y. Koda, J. Iwano, Y. Suzuki, M. Tanigawa, H. Iwai, K. Asano, K. Kurihara, A. Hamaguchi, H. Kodaira, T. Atsumi, Y. Yamada and K. Tomizuka, Nucleic Acids Res., 2022, 50, 4840–4859 CrossRef CAS PubMed
- K. Yamazaki, K. Kubara, Y. Suzuki, T. Hihara, D. Kurotaki, T. Tamura, M. Ito and K. Tsukahara, Eur. J. Pharm. Biopharm., 2023, 183, 61–73 CrossRef CAS PubMed
- X. Ye, R. Holland, M. Wood, C. Pasetka, L. Palmer, E. Samaridou, K. McClintock, V. Borisevich, T. W. Geisbert, R. W. Cross and J. Heyes, Mol. Ther., 2023, 31, 269–281 CrossRef CAS PubMed
- T. Onizuka, H. Shimizu, Y. Moriwaki, T. Nakano, S. Kanai, I. Shimada and H. Takahashi, FEBS J., 2012, 279, 2645–2656 CrossRef CAS PubMed
- A. G. Morell, R. A. Irvine, I. Sternlieb, I. H. Scheinberg and G. Ashwell, J. Biol. Chem., 1968, 243, 155–159 CrossRef CAS PubMed
- K. Ozaki, R. T. Lee, Y. C. Lee and T. Kawasaki, Glycoconjugate J., 1995, 12, 268–274 CrossRef CAS PubMed
- M. Spiess, Biochemistry, 1990, 29, 10009–10018 CrossRef CAS PubMed
- J. U. Baenziger and Y. Maynard, J. Biol. Chem., 1980, 255, 4607–4613 CrossRef CAS PubMed
- K. Schmidt, T. P. Prakash, A. J. Donner, G. A. Kinberger, H. J. Gaus, A. Low, M. E. Østergaard, M. Bell, E. E. Swayze and P. P. Seth, Nucleic Acids Res., 2017, 45, 2294–2306 CrossRef CAS PubMed
- A. A. D'Souza and P. V. Devarajan, J. Controlled Release, 2015, 203, 126–139 CrossRef PubMed
- T. P. Prakash, M. J. Graham, J. Yu, R. Carty, A. Low, A. Chappell, K. Schmidt, C. Zhao, M. Aghajan, H. F. Murray, S. Riney, S. L. Booten, S. F. Murray, H. Gaus, J. Crosby, W. F. Lima, S. Guo, B. P. Monia, E. E. Swayze and P. P. Seth, Nucleic Acids Res., 2014, 42, 8796–8807 CrossRef CAS PubMed
- P. Kandasamy, S. Mori, S. Matsuda, N. Erande, D. Datta, J. L. S. Willoughby, N. Taneja, J. O’Shea, A. Bisbe, R. M. Manoharan, K. Yucius, T. Nguyen, R. Indrakanti, S. Gupta, J. A. Gilbert, T. Racie, A. Chan, J. Liu, R. Hutabarat, J. K. Nair, K. Charisse, M. A. Maier, K. G. Rajeev, M. Egli and M. Manoharan, J. Med. Chem., 2023, 66, 2506–2523 CrossRef CAS PubMed
- T. P. Prakash, J. Yu, M. T. Migawa, G. A. Kinberger, W. B. Wan, M. E. Østergaard, R. L. Carty, G. Vasquez, A. Low, A. Chappell, K. Schmidt, M. Aghajan, J. Crosby, H. M. Murray, S. L. Booten, J. Hsiao, A. Soriano, T. Machemer, P. Cauntay, S. A. Burel, S. F. Murray, H. Gaus, M. J. Graham, E. E. Swayze and P. P. Seth, J. Med. Chem., 2016, 59, 2718–2733 CrossRef CAS PubMed
- C. Diener, A. Keller and E. Meese, Trends Genet., 2022, 38, 613–626 CrossRef CAS PubMed
- Y. Huang, Mol. Ther.--Nucleic Acids, 2017, 6, 116–132 CrossRef CAS PubMed
- L. N. Kasiewicz, S. Biswas, A. Beach, H. Ren, C. Dutta, A. M. Mazzola, E. Rohde, A. Chadwick, C. Cheng, S. P. Garcia, S. Iyer, Y. Matsumoto, A. V. Khera, K. Musunuru, S. Kathiresan, P. Malyala, K. G. Rajeev and A. M. Bellinger, Nat. Commun., 2023, 14, 2776 CrossRef CAS PubMed
- J.-C. Chapuis, R. M. Schmaltz, K. S. Tsosie, M. Belohlavek and S. M. Hecht, J. Am. Chem. Soc., 2009, 131, 2438–2439 CrossRef CAS PubMed
- J. Chen, M. K. Ghorai, G. Kenney and J. Stubbe, Nucleic Acids Res., 2008, 36, 3781–3790 CrossRef CAS PubMed
- B. R. Schroeder, M. I. Ghare, C. Bhattacharya, R. Paul, Z. Yu, P. A. Zaleski, T. C. Bozeman, M. J. Rishel and S. M. Hecht, J. Am. Chem. Soc., 2014, 136, 13641–13656 CrossRef CAS PubMed
- V. Tähtinen, V. Gulumkar, S. K. Maity, A.-M. Yliperttula, S. Siekkinen, T. Laine, E. Lisitsyna, I. Haapalehto, T. Viitala, E. Vuorimaa-Laukkanen, M. Yliperttula and P. Virta, Bioconjugate Chem., 2022, 33, 206–218 CrossRef PubMed
- W. Peng and J. C. Paulson, J. Am. Chem. Soc., 2017, 139, 12450–12458 Search PubMed
- T. Harumoto, H. Iwai, M. Tanigawa, T. Kubo, T. Atsumi, K. Tsutsumi, M. Takashima, G. Destito, R. Soloff, K. Tomizuka, C. Nycholat, J. Paulson and K. Uehara, ACS Chem. Biol., 2022, 17, 292–298 Search PubMed
- G. St-Pierre, S. Pal, M. E. Østergaard, T. Zhou, J. Yu, M. Tanowitz, P. P. Seth and S. Hanessian, Bioorg. Med. Chem., 2016, 24, 2397–2409 CrossRef CAS PubMed
- R. L. Redman and I. J. Krauss, J. Am. Chem. Soc., 2021, 143, 8565–8571 CrossRef CAS PubMed
- I. S. MacPherson, J. S. Temme, S. Habeshian, K. Felczak, K. Pankiewicz, L. Hedstrom and I. J. Krauss, Angew. Chem., Int. Ed., 2011, 50, 11238–11242 CrossRef CAS PubMed
- O. Doluca, J. M. Withers and V. V. Filichev, Chem. Rev., 2013, 113, 3044–3083 CrossRef CAS PubMed
- D. Varshney, J. Spiegel, K. Zyner, D. Tannahill and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2020, 21, 459–474 CrossRef CAS PubMed
- H. Abou Assi, R. El-Khoury, C. González and M. J. Damha, Nucleic Acids Res., 2017, 45, 11535–11546 CrossRef CAS PubMed
- M. Marušič, R. N. Veedu, J. Wengel and J. Plavec, Nucleic Acids Res., 2013, 41, 9524–9536 CrossRef PubMed
- Z. Li, C. J. Lech and A. T. Phan, Nucleic Acids Res., 2013, 42, 4068–4079 CrossRef PubMed
- B. P. Paudel, A. L. Moye, H. Abou Assi, R. El-Khoury, S. B. Cohen, J. K. Holien, M. L. Birrento, S. Samosorn, K. Intharapichai, C. G. Tomlinson, M.-P. Teulade-Fichou, C. González, J. L. Beck, M. J. Damha, A. M. van Oijen and T. M. Bryan, eLife, 2020, 9, e56428 Search PubMed
- J.-y Liao, I. Anosova, S. Bala, W. D. Van Horn and J. C. Chaput, Biopolymers, 2017, 107, e22999 CrossRef PubMed
- P. Schofield, A. I. Taylor, J. Rihon, C. D. Peña Martinez, S. Zinn, C.-A. Mattelaer, J. Jackson, G. Dhaliwal, G. Schepers, P. Herdewijn, E. Lescrinier, D. Christ and P. Holliger, Nucleic Acids Res., 2023, 51, 7736–7748 CrossRef CAS PubMed
- I. Gómez-Pinto, E. Vengut-Climent, R. Lucas, A. Aviñó, R. Eritja, C. González and J. C. Morales, Chem. – Eur. J., 2013, 19, 1920–1927 CrossRef PubMed
- S. Yin, W. Lan, X. Hou, Z. Liu, H. Xue, C. Wang, G.-L. Tang and C. Cao, J. Med. Chem., 2023, 66, 6798–6810 Search PubMed
- J. L. Childs-Disney, I. Yildirim, H. Park, J. R. Lohman, L. Guan, T. Tran, P. Sarkar, G. C. Schatz and M. D. Disney, ACS Chem. Biol., 2014, 9, 538–550 Search PubMed
- E. Belmonte-Reche, M. Martínez-García, A. Guédin, M. Zuffo, M. Arévalo-Ruiz, F. Doria, J. Campos-Salinas, M. Maynadier, J. J. López-Rubio, M. Freccero, J.-L. Mergny, J. M. Pérez-Victoria and J. C. Morales, J. Med. Chem., 2018, 61, 1231–1240 CrossRef CAS PubMed
- L. Yao, L. Wang, S. Liu, H. Qu, Y. Mao, Y. Li and L. Zheng, Chem. Sci., 2024, 15, 13011–13020 RSC
- P. Travascio, Y. Li and D. Sen, Chem. Biol., 1998, 5, 505–517 CrossRef CAS PubMed
- K. Gehring, J.-L. Leroy and M. Guéron, Nature, 1993, 363, 561–565 CrossRef CAS PubMed
- I. Zanin, E. Ruggiero, G. Nicoletto, S. Lago, I. Maurizio, I. Gallina and S. N. Richter, Nucleic Acids Res., 2023, 51, 8309–8321 CrossRef CAS PubMed
- Y. Feng, X. Ma, Y. Yang, S. Tao, A. Ahmed, Z. Gong, X. Cheng and W. Zhang, Nucleic Acids Res., 2024, 52, 1243–1257 CrossRef CAS PubMed
- H. Abou Assi, M. Garavís, C. González and M. J. Damha, Nucleic Acids Res., 2018, 46, 8038–8056 CrossRef PubMed
- H. A. Day, P. Pavlou and Z. A. E. Waller, Bioorg. Med. Chem., 2014, 22, 4407–4418 CrossRef CAS PubMed
- B. Mir, I. Serrano-Chacón, P. Medina, V. Macaluso, M. Terrazas, A. Gandioso, M. Garavís, M. Orozco, N. Escaja and C. González, Nucleic Acids Res., 2024, 52, 3375–3389 CrossRef CAS PubMed
- V. B. Tsvetkov, T. S. Zatsepin, E. S. Belyaev, Y. I. Kostyukevich, G. V. Shpakovski, V. V. Podgorsky, G. E. Pozmogova, A. M. Varizhuk and A. V. Aralov, Nucleic Acids Res., 2018, 46, 2751–2764 CrossRef CAS PubMed
- N. Kumar, J. T. Nielsen, S. Maiti and M. Petersen, Angew. Chem., Int. Ed., 2007, 46, 9220–9222 CrossRef CAS PubMed
- H. A. Assi, R. W. Harkness, N. Martin-Pintado, C. J. Wilds, R. Campos-Olivas, A. K. Mittermaier, C. González and M. J. Damha, Nucleic Acids Res., 2016, 44, 4998–5009 CrossRef PubMed
- C. P. Fenna, V. J. Wilkinson, J. R. P. Arnold, R. Cosstick and J. Fisher, Chem. Commun., 2008, 3567–3569, 10.1039/B804833A
- N. Kumar, M. Petersen and S. Maiti, Chem. Commun., 2009, 1532–1534, 10.1039/B819305C
- H. Abou Assi, Y. C. Lin, I. Serrano, C. González and M. J. Damha, Chem. – Eur. J., 2018, 24, 471–477 CrossRef CAS PubMed
- M. Ghezzo, L. Grigoletto, R. Rigo, P. Herdewijn, E. Groaz and C. Sissi, Biochimie, 2023, 214, 112–122 CrossRef CAS PubMed
- E. A. Ageely, R. Chilamkurthy, S. Jana, L. Abdullahu, D. O’Reilly, P. J. Jensik, M. J. Damha and K. T. Gagnon, Nat. Commun., 2021, 12, 6591 CrossRef CAS PubMed
- B. Li, W. Zhao, X. Luo, X. Zhang, C. Li, C. Zeng and Y. Dong, Nat. Biomed. Eng., 2017, 1, 0066 CrossRef CAS PubMed
- P. Monian, C. Shivalila, G. Lu, M. Shimizu, D. Boulay, K. Bussow, M. Byrne, A. Bezigian, A. Chatterjee, D. Chew, J. Desai, F. Favaloro, J. Godfrey, A. Hoss, N. Iwamoto, T. Kawamoto, J. Kumarasamy, A. Lamattina, A. Lindsey, F. Liu, R. Looby, S. Marappan, J. Metterville, R. Murphy, J. Rossi, T. Pu, B. Bhattarai, S. Standley, S. Tripathi, H. Yang, Y. Yin, H. Yu, C. Zhou, L. H. Apponi, P. Kandasamy and C. Vargeese, Nat. Biotechnol., 2022, 40, 1093–1102 CrossRef CAS PubMed
- J. Hennessy, B. McGorman, Z. Molphy, N. P. Farrell, D. Singleton, T. Brown and A. Kellett, Angew. Chem., Int. Ed., 2022, 61, e202110455 CrossRef CAS PubMed
- A. Saraf, R. Gurjar, S. Kaviraj, A. Kulkarni, D. Kumar, R. Kulkarni, R. Virkar, J. Krishnan, A. Yadav, E. Baranwal, A. Singh, A. Raghuwanshi, P. Agarwal, L. Savergave, S. Singh, H. Pophale, P. Shende, R. B. Shinde, V. Vikhe, A. Karmalkar, B. Deshmukh, K. Giri, S. Deshpande, A. Bulle, M. S. Siddiqui, S. Borthakur, V. R. Tummuru, A. V. Rao, D. Shukla, M. K. Jain, P. Bhardwaj, P. D. Supe, M. K. Das, M. Lahoti and V. Barge, Nat. Med., 2024, 30, 1363–1372 CrossRef CAS PubMed
- M. Hollenstein, Curr. Opin. Chem. Biol., 2019, 52, 93–101 CrossRef CAS PubMed
- L. B. Huber, K. Betz and A. Marx, ChemBioChem, 2023, 24, e202200521 CrossRef CAS PubMed
- M. Brunderová, V. Havlíček, J. Matyašovský, R. Pohl, L. Poštová Slavětínská, M. Krömer and M. Hocek, Nat. Commun., 2024, 15, 3054 CrossRef PubMed
- S. Arangundy-Franklin, A. I. Taylor, B. T. Porebski, V. Genna, S. Peak-Chew, A. Vaisman, R. Woodgate, M. Orozco and P. Holliger, Nat. Chem., 2019, 11, 533–542 CrossRef CAS PubMed
- D. Oberthür, J. Achenbach, A. Gabdulkhakov, K. Buchner, C. Maasch, S. Falke, D. Rehders, S. Klussmann and C. Betzel, Nat. Commun., 2015, 6, 6923 CrossRef PubMed
- A. Vater and S. Klussmann, Drug Discovery Today, 2015, 20, 147–155 CrossRef CAS PubMed
- Y. Xu and T. F. Zhu, Science, 2022, 378, 405–412 CrossRef CAS PubMed
- O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523–1527 CrossRef CAS PubMed
- A. Pardo-Vargas, M. Delbianco and P. H. Seeberger, Curr. Opin. Chem. Biol., 2018, 46, 48–55 CrossRef CAS PubMed
- N. Sabat, A. Stämpfli, M. Flamme, S. Hanlon, S. Bisagni, F. Sladojevich, K. Püntener and M. Hollenstein, Chem. Commun., 2023, 59, 14547–14550 RSC
- M. Vanmeert, J. Razzokov, M. U. Mirza, S. D. Weeks, G. Schepers, A. Bogaerts, J. Rozenski, M. Froeyen, P. Herdewijn, V. B. Pinheiro and E. Lescrinier, Nucleic Acids Res., 2019, 47, 7130–7142 CrossRef CAS PubMed
- A. R. Hesser, B. M. Brandsen, S. M. Walsh, P. Wang and S. K. Silverman, Chem. Commun., 2016, 52, 9259–9262 RSC
- F. Maksudov, E. Kliuchnikov, D. Pierson, M. L. Ujwal, K. A. Marx, A. Chanda and V. Barsegov, Mol. Ther.–Nucleic Acids, 2023, 31, 631–647 CrossRef CAS PubMed
- I. Anosova, E. A. Kowal, M. R. Dunn, J. C. Chaput, W. D. Van Horn and M. Egli, Nucleic Acids Res., 2015, 44, 1007–1021 CrossRef PubMed
- D. Evéquoz, I. E. C. Verhaart, D. van de Vijver, W. Renner, A. Aartsma-Rus and C. J. Leumann, Nucleic Acids Res., 2021, 49, 12089–12105 CrossRef PubMed
- J. D. Pyle, S. R. Lund, K. H. O’Toole and L. Saleh, Cell Rep., 2024, 43, 114631 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |