
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvancements and prospects of near-infrared-light driven CO2 reduction reaction†
Siheng
Yang‡
a,
Wei
Che‡
 b,
Yanhua
Shao
b,
Woo Jin
Byun
b,
Xiaodong
Li
c,
Xingchen
Jiao
d,
Ruixiang
Li
*a,
Jae Sung
Lee
b,
Jiaqi
Xu
*ae and
Jong-Beom
Baek
*b
b,
Yanhua
Shao
b,
Woo Jin
Byun
b,
Xiaodong
Li
c,
Xingchen
Jiao
d,
Ruixiang
Li
*a,
Jae Sung
Lee
b,
Jiaqi
Xu
*ae and
Jong-Beom
Baek
*b
aKey Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu, Sichuan 610064, P. R. China. E-mail: liruixiang@scu.edu.cn; jqxu@scu.edu.cn
bUlsan National Institute of Science and Technology (UNIST), 50 UNIST, Ulsan 44919, South Korea. E-mail: jbbaek@unist.ac.kr
cMax Planck Institute of Microstructure Physics, Weinberg 2, Halle 06120, Germany
dKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, P. R. China
eLaboratory of Photonics and Interfaces, École Polytechnique Fédérale de Lausanne, 1015 Lausanne, Switzerland. E-mail: jiaqi.xu@epfl.ch
First published on 30th June 2025
Abstract
In the realm of photoconversion of CO2 into high-value chemicals, the importance of near-infrared (NIR) light is gradually gaining recognition. Relative to ultraviolet (UV) and visible light, NIR light (700–2500 nm), accounting for ca. 50% of solar energy, offers unique advantages such as deeper penetration depth and stronger photothermal effects. Thus, utilizing NIR light can not only compensate for the inherent limitations of UV/visible light-based CO2 reduction systems, but also maximize the use of solar energy. However, efficiently harnessing NIR light remains challenging because of its low photon energy, making it difficult to drive CO2 reduction. Additionally, the limited knowledge of the reduction mechanism driven by low-energy photons further hinders progress in this field. In this review, we systematically introduce the motivation and fundamental principles of NIR-light-driven CO2 reduction, the design strategies for NIR-light-activated photocatalysts (including the energy band structure regulation strategy, the energy transfer strategy, and the photothermal utilization strategy), NIR-light absorption mechanisms of these catalysts, and representative applications of these strategies. Finally, we present our perspectives on the challenges facing NIR-light-driven CO2 reduction and provide suggestions for improving current photocatalysts, characterization techniques, evaluation procedures, and potential large-scale applications in future research. With further advancements in NIR-light-driven CO2 reduction, it holds great promise to maximize the exploitation of solar energy, ultimately achieving efficient CO2 photoconversion for industrial applications.
 Siheng Yang | Siheng Yang received his bachelor's and master's degrees in Chemistry from Sichuan University under the supervision of Dr Jiaqi Xu and Prof. Ruixiang Li. He is currently a research assistant at Sichuan University. His research interests include the design and fabrication of NIR-light-responsive photocatalysts and their applications in CO2 reduction. |
 Wei Che | Wei Che received her PhD in 2019 from the University of Science and Technology of China (USTC). After receiving her PhD, she joined the Guangdong University of Technology as a postdoctoral. She is currently a post-doctoral fellow in the School of Energy and Chemical Engineering, Center for Dimension-Controllable Organic Frameworks, at Ulsan National Institute of Science and Technology (UNIST), South Korea. Her current research focuses on photocatalysis for solar energy conversion and advanced organic catalysis. |
 Left to Right: Yanhua Shao; Woo Jin Byun; Xiaodong Li; Xingchen Jiao | Yanhua Shao obtained his PhD degree from Nanjing Tech University under the supervision of Prof. Rizhi Chen. His research focuses on the development of advanced carbon materials based on heterogeneous catalysis. Woo Jin Byun received his PhD from the School of Energy and Chemical Engineering from the UNIST. His research focuses on the development of advanced materials for solar fuel production, including photocatalytic hydrogen production and CO2 reduction. Xiaodong Li received his BS degree from Jilin University (2014) and PhD degree from the USTC (2019). Now he is a postdoc at Max Planck Institute for Microstructure Physics in Germany. His current interests include the theoretical computation, synthesis and characterization of nanostructures and their application in energy storage and conversion. Xingchen Jiao received his BS degree from the Hefei University of Technology (2010) and PhD degree from USTC (2019). In 2022, he joined Jiangnan University as a professor. His current interests include the synthesis and characterization of nanostructures, as well as their applications in CO2 photo-/electro-conversion into hydrocarbon fuels. |
 Ruixiang Li | Ruixiang Li obtained his master's degree and PhD degree from Lanzhou University and City University of Hong Kong, respectively. He is now a professor in the Department of Chemistry, Sichuan University. Prof. Li's research interests include the design and synthesis of homogeneous and heterogeneous catalysts, and green and industrial catalysis. |
 Jae Sung Lee | Jae Sung Lee received his BS from Seoul National University in 1975, MS from Korea Advanced Institute of Science and Technology (KAIST) in 1977, and PhD under Prof. Michel Boudart from Stanford University in 1984. Following a brief tenure at Catalytica Inc., he became a professor at POSTECH and later moved to Ulsan National Institute of Science and Technology (UNIST) in 2013. He leads the eco-friendly catalysis and energy laboratory, focusing on photocatalysis for solar energy conversion, electrocatalysis for fuel cells and electrolyzers, and heterogeneous catalysis for CO2 utilization. |
 Jiaqi Xu | Jiaqi Xu received his BS degree in Applied Chemistry from Tianjin University in 2014 and PhD degree in Inorganic Chemistry from the University of Science and Technology of China in 2019. He then worked as a postdoctoral fellow at Sichuan University. Currently, he is working as an associate research professor at Sichuan University. In 2024, he joined Prof. Michael Grätzel's lab at École Polytechnique Fédérale de Lausanne as a visiting postdoctoral researcher. His interests include the design and fabrication of low-dimensional materials and their applications in CO2 conversion. |
 Jong-Beom Baek | Jong-Beom Baek is a distinguished professor of the Department of Energy and Chemical Engineering, and a director of Center for Dimension Controllable Organic Frameworks (CDCOF), at Ulsan National Institute of Science and Technology (UNIST), South Korea. After receiving his PhD from the University of Akron, USA (Polymer Science, 1998), he joined the Wright-Patterson Air Force Research Laboratory (AFRL). He returned to South Korea to take a position as an assistant professor at Chungbuk National University in 2003, before moving to UNIST in 2008. His current research interests include the synthesis of 2D high-performance polymers, chemical modification of carbon-based materials for multifunctional applications, and mechanochemistry for sustainable applications. |
1. Introduction
Global fossil fuel consumption releases around 35 billion tons of CO2 into the atmosphere annually. This amount is nearly double that can be absorbed by the natural carbon sinks, such as forests and oceans (Fig. 1A).1 As a result, the average global CO2 level has reached 417.06 parts per million (ppm), showing an increase of 2.13 ppm compared to the last year, a rate consistent with the trend observed over the past decade. Currently, atmospheric CO2 levels are 50% higher than they were before the industrial era.2 The rapid increase of atmospheric CO2 levels has resulted in obvious issues: global warming, abnormal changes in the climate, and the alarming threat of species extinction, all posing serious risks to human existence. For instance, since 1890, the average temperature anomaly has shown a continuous upward trend (Fig. 1B). The issues of global temperature anomalies over land and oceans are primarily linked to the overemission of greenhouse gases from human activities.3 Therefore, it is crucial to reduce our dependence on fossil fuels and decrease CO2 emissions. While utilizing CO2 as a one-carbon (C1) feedstock for the production of value-added products is recognized as a promising strategy to mitigate these issues, CO2 is a chemically stable molecule with a strong C
with a strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond energy of 750 kJ mol−1, making it thermodynamically inert and challenging to be activated.4 Consequently, the chemical conversion of CO2 often requires harsh reaction conditions to drive the energetically uphill reactions. To accomplish efficient CO2 activation, many approaches have been explored, including thermal catalysis, electrocatalysis, photocatalysis, photoelectrocatalysis, mechanochemical catalysis, and enzymatic catalysis.5–13 Among these strategies, harnessing renewable solar energy for photochemically converting CO2 into fine products and fuels is a potential alternative to both mitigate CO2 emissions and meet the global energy demand.
O bond energy of 750 kJ mol−1, making it thermodynamically inert and challenging to be activated.4 Consequently, the chemical conversion of CO2 often requires harsh reaction conditions to drive the energetically uphill reactions. To accomplish efficient CO2 activation, many approaches have been explored, including thermal catalysis, electrocatalysis, photocatalysis, photoelectrocatalysis, mechanochemical catalysis, and enzymatic catalysis.5–13 Among these strategies, harnessing renewable solar energy for photochemically converting CO2 into fine products and fuels is a potential alternative to both mitigate CO2 emissions and meet the global energy demand.
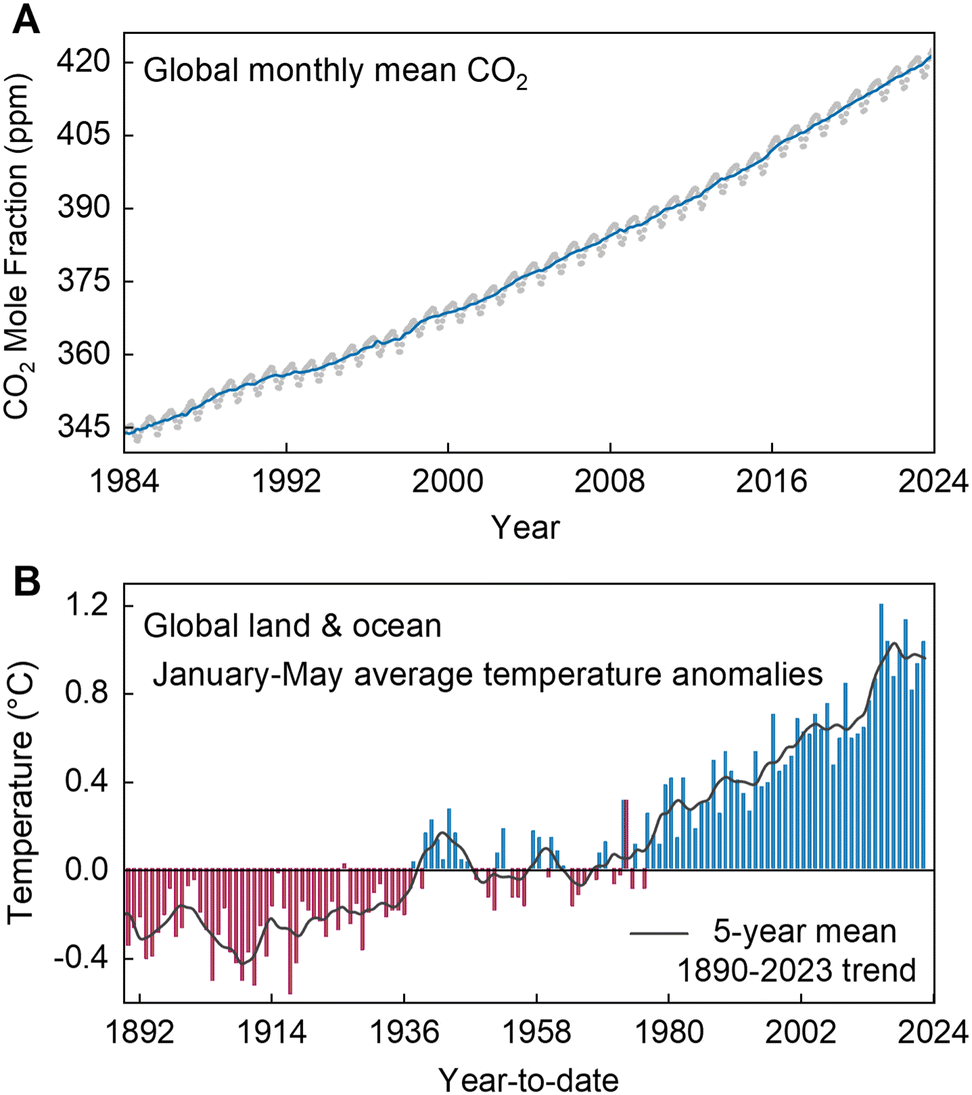 | ||
| Fig. 1 (A) Global trends in the increase in CO2 concentration are shown via globally averaged CO2 mole fraction since 1980. Reproduced with permission from ref. 2. Copyright 2024, NOAA. (B) Global land and ocean average temperature anomalies since 1850. Coordinate anomalies are concerning the 1890–2020 average. Data adapted from the National Ocean and Atmospheric Administration (NOAA) website. Reproduced with permission from ref. 3. Copyright 2024, NOAA. | ||
To fully harness solar energy for photocatalytic CO2 conversion, mastery is required not only in the design and synthesis of single component catalysts (composition, nanostructure, etc.) but also in the nanoscale assembly of these photocatalytic active units with effective auxiliary nanomaterials. It is reasonable to anticipate that, given the considerable contemporary emphasis on CO2 capture and utilization in the context of carbon neutrality, the coming years will witness significant advancements in CO2 photochemical fixation, including the development of effective light absorption models, synthetic approaches for tailored photocatalysts, and the design of high-efficiency photocatalytic reactors. All of these are developed for the maximal solar energy exploitation and efficient CO2 conversion. To reach the goal, one of the tasks is to fully utilize the low-energy photons from solar light. Although the concept of CO2 photoreduction has existed for a long time, unlike the well-established ultraviolet (UV) or visible light photocatalysis, the utilization of near-infrared (NIR) light for CO2 upgrading has gained prominence only recently. That is because, even though NIR light (700–2500 nm) accounts for ca. 50% of the solar spectrum, its low photon energy makes it difficult to drive the uphill CO2 reduction reaction (CO2RR). Consequently, studies on the utilization of NIR light to drive CO2RR are still rare. Meanwhile, the lack of basic understanding of the reaction mechanisms driven by low-energy photons also has resulted in slow progress in this field.
Photocatalysts serve as crucial “tools” in the process of NIR-light-driven CO2RR, where the inherent properties of incident photons dictate how these “tools” are designed and engineered. In this case, precisely matching the characteristics of incident photons and the electronic structures of the photocatalysts is particularly crucial. In this review, we outline the state-of-the-art photocatalysts for NIR-light-driven CO2RR, focusing on the light absorption mechanisms and design strategies of these NIR-light-responsive photocatalysts, and their applications in CO2RR. First of all, we begin with brief discussions on the basic principles of CO2 photoconversion, as well as the motivation and fundamentals of NIR-light-driven CO2RR, to offer a contextual backdrop. This is followed by a discussion of design strategies for NIR-light-responsive photocatalysts. For instance, NIR light can be directly harnessed to achieve CO2RR through energy band structure regulation strategies, including the construction of narrow-bandgap, intermediate-band, metallic, and heterojunction photocatalysts. Moreover, to enable the indirect utilization of NIR light for CO2RR, energy transfer strategies are proposed, including pure surface plasmon resonance (SPR) systems, SPR-semiconductor synergetic systems, and up-conversion systems. Especially, significant progress focused on photothermal utilization, which can improve conversion and/or yield in the NIR-light-driven CO2 reduction systems, has also been discussed. Finally, we highlight the future directions for constructing advanced NIR-photocatalytic CO2RR systems, including developing an advanced multifunctional photocatalytic system (referred to as a “more-in-one” system), constructing electron-rich sites and enhancing local CO2 concentration for deep CO2RR, and quantifying light and thermal contributions in photothermal CO2RR systems driven by NIR light. To push the advancement of NIR-light-driven CO2RR, we also provide insights into developing advanced techniques, conducting reliable evaluations of catalytic performance, and increasing the feasibility of CO2 photoreduction for practical applications. Throughout the review, we aim to identify the challenges in NIR-light-driven CO2RR, provide feasible design strategies for NIR-responsive photocatalysts, and propose strategic directions for future development in this area.
2. Principles and development of photocatalytic CO2 recycling
2.1. Principles of photocatalytic CO2 recycling
Utilization of solar energy to construct an efficient artificial carbon cycle holds immense promise for reducing fossil fuel consumption and mitigating climate change (Fig. 2A). In nature, photosynthetic organisms adeptly harness solar energy through intricate supramolecular assemblies to synthesize energy-rich compounds from water and atmospheric CO2.14 The robust stability and exceptional selectivity of photosynthesis hinge upon the precise spatial arrangement of chromophore molecules and catalytic centers achieved through self-assembly. However, the efficiency of natural photosynthesis is too low to effectively reduce current atmospheric CO2 level on its own. Inspired by photosynthetic organisms, the synthesized photocatalysts have been successfully integrated into the carbon cycle to achieve an artificial photosynthesis process. The widespread adoption of such artificial photosynthesis can facilitate CO2 utilization, lower atmospheric CO2 concentration, and transform CO2 into energy-dense fuels, thus diminishing the reliance on traditional fossil fuels. The underlying mechanism of photosynthesis in nature offers invaluable design principles for developing artificial photosynthesis systems aiming at capturing and storing CO2 on a global scale.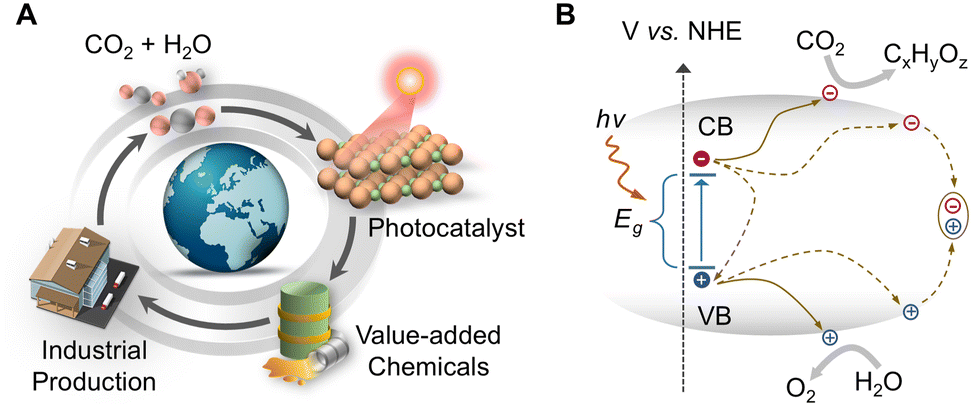 | ||
| Fig. 2 (A) Scheme of the artificial carbon cycle aiming at upgrading CO2 into value-added chemicals. (B) The diagram of a typical photocatalytic CO2 reduction process on semiconductors. | ||
Photocatalytic CO2 reduction (PCO2R) is a complicated process, which involves a variety of intricate elementary reactions that work together to facilitate CO2 conversion. In a typical PCO2R process, activated by the incident photons whose energy (hν) exceeds the energy bandgap (Eg) of the catalyst, the electrons in the valence band (VB) will transition to the conduction band (CB), producing active holes and electrons. Most of the electrons and holes suffer from severe recombination either within the bulk material or at the surface. However, a fraction of long-lived photogenerated carriers can successfully transfer to the surface, where they are engaged in the PCO2R process (Fig. 2B). To enable CO2 reduction, the conduction band minimum (CBM) of the catalyst must be more negative than the CO2RR potential (Ered), while its valence band maximum (VBM) is required to be more positive than the water (H2O) oxidation potential (Eox) or the oxidation potentials of other substituted agents (e.g., H217 and amines18) (Table 1). This enables photogenerated electrons to drive the CO2 reduction reaction (CO2RR), while the photogenerated holes drive the oxidation reaction.
| Product | Half reaction | E 0/V vs. NHE | ΔGhalf/kJ mol−1 |
|---|---|---|---|
| a Reproduced with permission from ref. 15. Copyright 2021, Wiley-VCH. Reproduced with permission from ref. 16. Copyright 2022, Wiley-VCH. | |||
| CO | CO2 + 2H+ + 2e− → CO + H2O | −0.53 | 102.29 |
| HCOOH | CO2 + 2H+ + 2e− → HCOOH | −0.61 | 117.73 |
| HCHO | CO2 + 4H+ + 4e− → HCHO + H2O | −0.48 | 185.28 |
| CH3OH | CO2 + 6H+ + 6e− → CH3OH + 2H2O | −0.38 | 220.02 |
| CH4 | CO2 + 8H+ + 8e− → CH4 + 2H2O | −0.24 | 185.28 |
| C2H4 | CO2 + 12H+ + 12e− → C2H4 + 4H2O | −0.34 | 393.72 |
| C2H5OH | CO2 + 12H+ + 12e− → C2H5OH + 3H2O | −0.33 | 382.14 |
| C2H6 | CO2 + 14H+ + 14e− → C2H6 + 4H2O | −0.27 | 364.77 |
| C3H7OH | CO2 + 18H+ + 18e− → C3H7OH + 5H2O | −0.32 | 555.84 |
| O2 | 1/2O2 + 2H+ + 2e− → H2O | +0.82 | −158.26 |
Ideally, H2O oxidation is the recommended oxidation half-reaction for PCO2R. Most cases discussed in this review are based on the water-assisted PCO2R. However, it is sometimes replaced by the oxidation reactions of the reagents that possess lower oxidation potential to enhance the PCO2R efficiency. One should be aware that the overall reaction of PCO2R varies according to the oxidation half-reaction, which suggests that the thermodynamics and kinetics may be different as well.19,20 Due to the diversity of oxidation half-reactions, we will refrain from delving into detailed discussions of specific oxidation half-reactions, in order not to distract the review's focus from the CO2RR half-reaction.
2.2. Development of photocatalytic CO2 recycling
Demonstrating artificial photocatalytic CO2 conversion systems with high efficiency, comparable to or even better than natural photosynthesis systems, has been a long-standing dream for scientists. This aspiration has also propelled photocatalysis to become one of the most dynamic fields in chemical research. However, replicating the natural light-harvesting superstructure through synthetic pathways is difficult to achieve and far from cost-effective. In this case, precise modulation of the photocatalyst structure is a highly attractive solution to this challenge. Particularly, regulating the light-harvesting ability by adjusting the electronic structures of catalysts is pivotal for artificial photosynthesis. To efficiently activate the photocatalysts for CO2RR, UV and visible light-driven CO2RR systems have been extensively studied in the past few decades. By contrast, although the concept of NIR photocatalysis has existed for a long time, there are few reports on using NIR irradiation, which comprises about 50% of the solar energy, to enable CO2RR as a result of the low energy of NIR photons (Fig. 3A). The first discovery of heterogeneous photoelectrochemical CO2RR over a p-type GaP semiconductor was reported by Halmann21 in 1978, and the first demonstration of pure photocatalytic CO2RR was presented by Inoue et al.22 in 1979. Since then, light-driven CO2 reduction has attracted broad scientific attention. As shown in Fig. 3B, a cursory search of the term “light driven CO2 reduction” in the Web of Science database indicates significant growth in related research over the last decade, driven by rapid advancements in nanotechnology and characterization techniques. | ||
| Fig. 3 (A) The solar spectrum primarily comprises visible and NIR light, constituting ∼45% and ∼50% of the total irradiance, respectively. (B) Publication and citation statistics of photocatalytic CO2 reduction reports for the topics “light driven CO2 reduction” and “infrared light driven CO2 reduction”. (Data adapted from the Web of Science, timespan from 2012 to 2023, collected on August 14, 2024.) | ||
Reactors and reaction modes are critical factors for PCO2R. So far, four fundamental reaction modes have been developed: solid–gas mode, solid–vapor mode, solid–gas–liquid mode, and liquid–gas mode (Fig. 4).23 For both solid–gas and solid–vapor modes, the solid photocatalyst is deposited on a substrate, such as quartz tray24–26 or foam-type metals,27,28 to form a catalyst film. Fig. 4A and B display that the solid–gas mode consists of CO2, a catalyst film, and reducing gas (e.g., H217,29), while the solid–vapor mode consists of CO2, a catalyst film, and water vapor,30,31 with the water vapor being generated from a trace of H2O at the bottom of the reactor. These modes are relatively simple catalytic systems and their CO2RR activity is strongly relevant to the intrinsic properties of the catalysts. Different from the solid–gas and solid–vapor modes, the solid–liquid–gas mode involves dispersing the photocatalyst powder in a solvent, where CO2RR occurs at the solid–liquid–gas interface (Fig. 4C).32 The solvent can be water or organic solvents (e.g., acetonitrile).18 In this mode, hole sacrificial agents or co-catalysts are sometimes added to the solvent to boost the CO2RR.33 The liquid–gas mode, on the other hand, is designed for homogeneous PCO2R, where molecular catalysts, organic photosensitizers, reducing agents, and dissolved CO2 are involved (Fig. 4D).34,35 Based on these four reaction modes, various reactors have been developed as well, including offline batch reactors, online batch reactors, and flow reactors, most of which are kettle type or tubular reactors.24,27,36–38
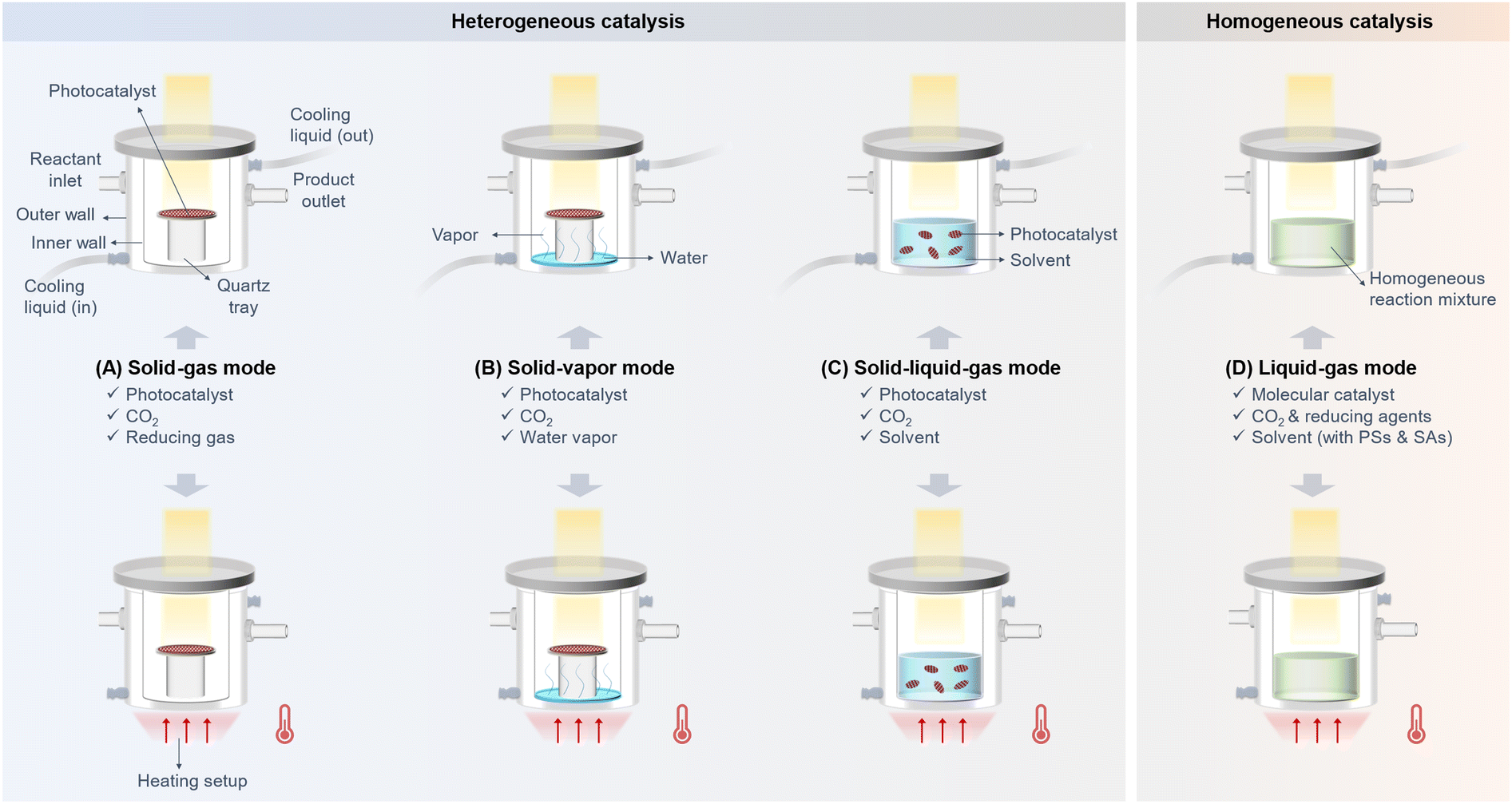 | ||
| Fig. 4 Different reaction modes for CO2 photoreduction: (A) solid–gas mode, (B) solid–vapor mode, (C) solid–liquid–gas mode, and (D) liquid–gas mode, where PS represents the photosensitizer and SA represents the sacrificial agents. | ||
Moreover, a circulating liquid is usually used to dissipate photoinduced heat and keep the system at a steady and low temperature (e.g., 10 °C), which is conducive to investigating the roles of heat and light separately. For instance, Xu et al.25 utilized this method to identify the effect of photoinduced heat on UiO-66/Co9S8 photocatalysts. As shown in Fig. 5A, after 5 h NIR irradiation, the central temperature of the photocatalyst reaches ca. 106 °C, exhibiting a strong photothermal effect. To investigate the role of light and heat, separately, two control experiments are performed: (i) eliminating the photoinduced heat by cycling cool water (Fig. 5B), and (ii) conducting the PCO2R at 120 °C without light irradiation. When the reaction temperature was held at 10 °C by cycling cool water, the UiO-66/Co9S8 photocatalyst still exhibited the ability of converting CO2 into CH4, although the CH4 production rate significantly decreased (Fig. 5C). On the other hand, when the PCO2R was performed at 120 °C without light irradiation, no gas product was observed (Fig. 5D), which indicated that the light illumination was the key factor in triggering the photoreduction of CO2 to CH4, while the photoinduced heat played a vital role in accelerating the reaction.
 | ||
| Fig. 5 Identifying the effect of light and heat on UiO-66/Co9S8. (A) The central temperature of the catalyst after NIR irradiation for 5 h. (B) Typical photoreactor configuration for excluding the photoinduced heat. (C) The influence of photoinduced heat on PCO2R performance. (D) The PCO2R performance under standard photothermal catalytic conditions and 120 °C in the dark. Reproduced with permission from ref. 25. Copyright 2024, Wiley-VCH. | ||
Also, to enhance photocatalytic efficiency, heating setups can be equipped to provide additional heat, thereby facilitating CO2 photoreduction.17,29 In addition, novel supports, such as monolith supports39 and optical fibers,40,41 have been recently introduced for PCO2R to ensure sufficient contact between CO2 and photocatalysts as well as uniform light irradiation. Besides, solar concentrators, like Fresnel lens,27 are usually employed to enhance light absorption and utilization, thereby improving the feasibility of utilizing natural sunlight for CO2RR.
The majority of the materials are only responsive to UV or visible light that possesses high photon energy, and these include metal chalcogenides,42–45 metal–organic frameworks (MOFs),18,36,46 and non-metallic materials.47,48 With the rapid development of reaction modes and reactors, diverse photocatalysts have been explored for PCO2R, such as metal complexes,34,49,50 metal oxides,51–53 and metal nitrides,54–56 for the utilization of NIR light. Given that NIR light constitutes ca. 50% of the solar spectrum, enhancing its utilization is crucial for making full use of solar energy during the PCO2R process. To this end, many researchers have turned their focus on harvesting NIR photons for PCO2R (Fig. 3B). However, the low energy of NIR light poses significant challenges to its effective use in photocatalysis for an extended period. In 2016, Ye et al. introduced a defect level into the band gap of BiOI by constructing oxygen defects, successfully achieving NIR-light-driven transformation of CO2 to CO for the first time.37 Since then, more and more materials, such as metallic materials,24,57 SPR materials,58,59 up-conversion nanoparticles (UCNPs)60,61 and intermediate-band semiconductors,62,63 have been developed for driving CO2 conversion by NIR light activation (Fig. 6). These photocatalysts can efficiently harness NIR photons for the transformation of CO2 to high-value chemicals. Besides light absorption ability, CO2 affinity is also a critical property of photocatalysts. With future practical applications in mind, PCO2R is supposed to be operated under low CO2 concentration atmospheres, which is unfavored by many traditional photocatalysts due to their weak CO2 affinity. Recent studies have demonstrated that photocatalysts with strong NIR light absorption ability, for instance, metallic materials, have the potential to achieve efficient CO2 activation and conversion in the air.29,64 Moreover, owing to the biocompatibility of NIR light, the NIR-light-driven CO2 reduction can be realized in vivo,65 gradually expanding the applications to biomedical research. Therefore, NIR-light responsive photocatalysts exhibit huge potential for future CO2 photoreduction.
 | ||
| Fig. 6 The timeline highlights key developments in NIR light harvesting and CO2 photoreduction over the past few decades. Reproduced with permission from ref. 21. Copyright 1978, Springer Nature. Reproduced with permission from ref. 22. Copyright 1979, Springer Nature. Reproduced with permission from ref. 66. Copyright 2012, Springer Nature. Reproduced with permission from ref. 67. Copyright 2013, Wiley-VCH. Reproduced with permission from ref. 37. Copyright 2015, Elsevier. Reproduced with permission from ref. 68. Copyright 2017, Springer Nature. Reproduced with permission from ref. 62. Copyright 2018, Elsevier. Reproduced with permission from ref. 57. Copyright 2018, American Chemical Society. Reproduced with permission from ref. 69. Copyright 2017, Wiley-VCH. Reproduced with permission from ref. 70. Copyright 2018, Wiley-VCH. Reproduced with permission from ref. 71. Copyright 2019, American Chemical Society. Reproduced with permission from ref. 72. Copyright 2021, American Chemical Society. Reproduced with permission from ref. 24. Copyright 2021, Wiley-VCH. Reproduced with permission from ref. 73. Copyright 2022, American Chemical Society. Reproduced with permission from ref. 18. Copyright 2022, American Chemical Society. Reproduced with permission from ref. 64. Copyright 2022, Wiley-VCH. Reproduced with permission from ref. 50 and 59. Copyright 2023, Springer Nature. Reproduced with permission from ref. 65. Copyright 2024, Wiley-VCH. | ||
3. Motivation and fundamentals of NIR photocatalytic CO2 recycling
3.1. NIR light versus UV/visible light
As a clean and sustainable energy source, solar light can be utilized to mitigate both energy and environmental challenges. The solar light is composed of about 5% UV light (300–400 nm), about 45% visible light (400–700 nm), and about 50% NIR light (700–2500 nm) (Fig. 3A).74 In the catalytic system of artificial photosynthesis, light absorption is the first step, directly affecting the solar-to-chemical conversion efficiency (SCC). To date, numerous catalysts have been widely explored for PCO2R, but the majority of them are triggered only by UV and visible light. Owing to the high energy of UV or visible photons, most photocatalysts can be efficiently activated to generate carriers. Furthermore, photocatalysts with a wide band gap can thermodynamically drive a broader range of reactions, making UV/visible-light photocatalytic systems the most extensively studied photocatalytic systems for CO2RR. Conversely, research on efficient NIR-light-driven CO2 reduction remains in its early stage. Currently, the energy conversion efficiency and economic viability of existing PCO2R systems fall short of industrialization requirements, primarily due to the insufficient utilization of solar energy. This has led researchers to increasingly focus on the exploration of novel NIR-photocatalysts to fully use solar energy. Additionally, there are inherent challenges associated with using UV/visible radiation as a light source. Therefore, it is crucial to thoroughly compare NIR light with UV and visible light, given their significant differences in energy distribution, physicochemical properties, and effects on the earth and life. This comparison is especially important for understanding their roles and significance in PCO2R, forming the foundation for maximizing the utilization of solar energy. | ||
| Fig. 7 Comparison of UV/visible light and NIR light. (A) Penetration depth: (i) catalyst penetration, (ii) medium penetration, and (iii) tissue penetration. (B) Light absorption competition. (C) Carbon contamination. (D) Photothermal effect. (E) Biocompatibility: (i) CO2RR in vivo and (ii) photobiohybrid CO2RR system. | ||
According to previous reports, wide-bandgap photocatalysts, which require high-energy photons from UV/visible light to generate photocarriers, are more prone to form the products from carbon impurities or even from the carbon-containing photocatalyst itself through oxidation processes rather than through CO2 photoreduction, resulting in false-positive CO2RR results.92,93 Therefore, complex and expensive isotope labeling experiments and control experiments are often indispensable to confirm whether the detected products originate from CO2 feedstock or not. By contrast, NIR light exhibits excellent photochemical compatibility and is less likely to generate those pollutants during PCO2R.
Although photocatalysts with narrow bandgaps and NIR light absorption properties have received considerable attention for CO2 photoreduction, there have been no reports of carbon contamination that severely affects the PCO2R results. The advantages of using NIR light include the following: (1) carbon impurities adsorbed on the surface are less likely to oxidize under low-energy NIR photons, ensuring that the PCO2R products originate from CO2RR; (2) VB holes excited by NIR light have weaker oxidation capabilities, reducing the likelihood of self-decomposition of carbon-containing photocatalysts and avoiding false-positive CO2RR signals; and (3) the photothermal effect of NIR light heats the photocatalyst, acting similarly to high-temperature annealing and removing surface-adsorbed carbon impurities.93 These impurities are rapidly consumed during stability tests at higher temperatures, thus reducing their potential interference with the PCO2R system.
Therefore, NIR light sources offer a cost-effective and time-efficient means to minimize and eliminate carbon contamination in CO2 photoconversion systems, making NIR-light-driven CO2 reduction a promising approach to avoid or mitigate carbon contamination in photocatalytic processes.
In short, using NIR light to promote photocatalytic CO2 recovery is mainly based on the following considerations.
Solar energy utilization. NIR light makes up a substantial part of solar light. Harnessing NIR light is conducive to maximizing the solar energy utilization for CO2 conversion.
Technological advancements. Advancements in synthetic chemistry and characterization techniques ensure the possibility of CO2 photoreduction by low-energy photons, further expanding the methods of CO2 reduction.
Photocatalytic efficiency. Broadening the spectrum responsiveness of photocatalysts from the UV to the NIR region will enhance their overall efficiency in converting CO2 into value-added products.
Environmental and energy concerns. Given the pressing environmental and energy challenges associated with CO2 emissions, there is a growing imperative to explore novel strategies for CO2 conversion. Utilizing NIR light for catalytic CO2 recovery presents a promising avenue for addressing these concerns while leveraging renewable energy sources.
Intrinsic advantages of NIR light. NIR light, with its ability to deeply penetrate reaction media and material surfaces, can uniformly activate the bulk of catalysts. The lower energy of NIR photons helps minimize competitive light absorption and unwanted side reactions. Moreover, NIR light exhibits a unique photothermal effect, which aids in activating CO2 molecules and modulating the selectivity-determining steps in the PCO2R process. This photothermal conversion enhances the reaction kinetics of CO2 transformation. Additionally, due to their excellent tissue penetration and biocompatibility, NIR photons have found successful applications in biological CO2 conversion systems.
In other words, developing NIR-light-driven photocatalytic systems for CO2 recovery displays advantages in energy utilization, conversion efficiency and technical feasibility. It has great potential to solve the environmental and energy challenges by fully using solar energy from UV to NIR regions.
3.2. Fundamentals and challenges of NIR-light-driven CO2 reduction
To accomplish NIR-light-driven CO2RR, photocatalysts must have extremely narrow bandgaps in addition to well-matched band edge positions. Nevertheless, it is hard to simultaneously satisfy both the bandgap and band edge position requirements for NIR-light-driven CO2RR when designing a photocatalyst (Fig. 8). Because the energy of NIR photons is too low, it cannot excite most of the photocatalysts, which seriously impedes the progress of NIR-light-driven CO2RR. As a result, only a limited number of narrow-bandgap photocatalysts are capable of directly achieving this objective.62 However, although the narrow-bandgap photocatalysts exhibit excellent NIR light absorption, they often possess inappropriate band edge positions, making them unable to drive the overall CO2RR.18 Yet, only a few photocatalysts have been reported to realize NIR-light-driven CO2RR, including oxygen-deficient WO3 and metallic CuS nanosheets, but the efficiencies of these systems are still quite low.56,57,62 Therefore, precisely tuning the band structures is key to achieving an efficient NIR-light-driven CO2RR. | ||
| Fig. 8 Paradoxes in NIR-light-driven CO2 reduction systems. (A) Unsuitable band edge position. (B) Unsuitable band gap for light absorption. | ||
It is noted that H2O oxidation is the most recommended half-reaction for CO2 photoreduction systems in overall photocatalysis due to its green and abundant features. For photocatalysts with narrow bandgaps, charge recombination occurs easily, which significantly impairs their overall efficiency. Moreover, such photocatalysts with small bandgaps usually fail to trigger sufficient potential for H2O oxidation due to their unsuitable VBM position, which further lowers the efficiency of NIR-light-driven CO2RR or even inhibits the reaction. To resolve these limitations, sacrificial reagents, like triethanolamine (TEOA), Na2S, Na2S2O3, etc., are often introduced into the NIR-light-driven CO2 reduction system.56,60 The oxidation of these sacrificial agents is able to be triggered by a potential lower than that of H2O oxidation, which can not only overcome the limitation of VBM position but also narrow the required band gap for CO2RR. These sacrificial agents facilitate charge separation by scavenging the holes, greatly promoting the reaction. However, the use of sacrificial agents is costly and environmentally unfriendly. Moreover, the use of sacrificial agents sometimes makes the system more complex, hindering the investigation of the mechanism of NIR-light-driven CO2 reduction. Therefore, careful selection of sacrificial agents is essential for PCO2R investigation.
Furthermore, the typical CO2 photoreduction assisted by H2O is an uphill process, and the efficiency of NIR-light-driven CO2 reduction is still undesirable. Two critical hindrances in NIR-light-driven CO2RR are poor utilization of low-energy photons and low energy conversion efficiency. To advance the progress of NIR-light-driven CO2 reduction, reasonable guidance should be developed, especially for the design of photocatalysts. Moreover, there are diverse possible products in CO2 photoreduction, such as CO,31,56 HCOOH,18 CH4,24,30 C2H4,49 CH3OH,72etc. To trigger NIR-light-driven CO2 reduction, the catalysts are required to efficiently absorb NIR light and generate free electrons for CO2RR. However, the main product in reported NIR-light-driven CO2 reduction systems is CO, a kinetically favored product, because of the low photon energy of NIR light (Tables 2 and 3). It is challenging to convert CO2 to hydrocarbon compounds or C2+ products. To obtain multi-electron transfer products, the photocatalysts should generate a high concentration of electrons. Additionally, to selectively produce C1 or C2+ products, the catalytic sites ought to be precisely designed to ensure appropriate adsorption intensity towards intermediates (Fig. 9). Thus, developing catalysts with excellent NIR light absorption is the key to achieving highly efficient NIR-light-driven CO2RR. Therefore, it is pivotal to explore innovative strategies and catalysts for NIR-light-driven CO2 reduction. Several strategies have been explored to accomplish NIR-light-driven CO2 reduction, including the energy band structure regulation strategy, the energy transfer strategy, and the photothermal strategy (Fig. 10). The energy band structure regulation strategy mainly focuses on band structure engineering to meet the requirements of efficient NIR light absorption and band edge position matching simultaneously. Typical examples include narrow-bandgap photocatalysts, intermediate-band photocatalysts, metallic photocatalysts, and heterojunction photocatalysts (Fig. 10A–D), which are able to directly utilize NIR light for CO2 conversion due to their narrow or relay band structure features. The energy transfer strategy is an indirect method for NIR light utilization. For traditional semiconductors that can only harvest UV or visible light, it is a very attractive way to combine them with up-conversion nanoparticles (UCNPs) or plasmonic components (Fig. 10E and F). UCNPs are capable of transforming NIR light into UV or visible light, which can be harvested by most semiconductors. Plasmonic components can absorb NIR light through the SPR effect and subsequently transfer hot electrons and resonance energy to traditional semiconductors via direct electron transfer (DET) and plasmon-induced resonant energy transfer (PIRET). As a result, the UCNP and SPR material-based photocatalytic systems can utilize NIR light effectively. In addition, NIR light possesses a unique photothermal effect, which is conducive to accelerating the photocatalytic reaction. Therefore, creating a photothermal synergetic catalysis system is also a good strategy to utilize NIR light (Fig. 10G). In the following sections, we will discuss the fundamental principles and applications of these strategies for NIR-light-driven CO2 reduction in detail.
| Photocatalyst | Catalyst type | Light source | Photocatalytic system | CO2RR performance (μmol g−1 h−1) | Ref. |
|---|---|---|---|---|---|
| V o-rich WO3 | Intermediate-band photocatalyst | 40 W SiN lamp, λ: 0.8–17 μm | CO2(g) + H2O(g), solid–vapor mode | CO: 2.75 | 62 |
| Cu2−xS/g-C3N4 | Intermediate-band photocatalyst | 300 W Xe lamp, λ > 800 nm | CO2(g) + H2O(g), solid–vapor mode | CO: 101.1, CH4: 21.0 | 103 |
| HCNT-NA | Intermediate-band photocatalyst | 5 W white LED lamp, λ: 780–850 nm | ACN + TEOA + CO2(g) + CoCl2 + 2,2′-bipyridine, solid–liquid–gas mode | CO: 6.31 | 48 |
| Cu2−xS/Ni–Al-LDH | Intermediate-band photocatalyst | 300 W Xe lamp, λ > 800 nm | CO2(g) + H2O(l), solid–liquid–gas mode | CO: 3.00, CH4: 2.83 | 42 |
| CuInS2 | Intermediate-band photocatalyst | 300 W Xe lamp, λ > 800 nm | CO2(g) + H2O(g), solid–vapor mode | CO: <10.00 | 63 |
| c-CSON | Intermediate-band photocatalyst | 71 mW cm−2 Xe lamp, λ > 800 nm | CO2(g) + H2O(g), solid–vapor mode | CO: 21.95, CH4: 4.11 | 50 |
| CuS | Metallic photocatalyst | 300 W Xe lamp, λ > 800 nm | CO2(g) + H2O(g), solid–vapor mode | CO: 14.5 | 57 |
| CoN | Metallic photocatalyst | 300 W Xe lamp, λ > 800 nm | CO2(g) + Na2S (aq.), solid–liquid–gas mode | CO: 14.5 | 56 |
| B13P2 | Metallic photocatalyst | 300 W Xe lamp, λ > 780 nm | DMF + TEOA + CO2(g) + CoCl2 + 2,2′-bipyridine, solid–liquid–gas mode | CO: 0.13 | 104 |
| Ni-doped CoS2 | Metallic photocatalyst | 300 W Xe lamp, λ > 800 nm | CO2(g) + H2O(g), solid–vapor mode | CO: 37.5, CH4: 101.8 | 24 |
| V o-rich MoO2−x | Metallic photocatalyst | 300 W Xe lamp, λ > 700 nm | CO2(g) + H2O(g), solid–vapor mode | CO: 1.9, CH4: 6.8 | 64 |
| Air + H2O(g), solid–vapor mode | CO: 6.5, CH4: 0.5 | ||||
| Pt/Ni-MOF | Metallic photocatalyst + photothermal utilization | 940 nm LED light | CO2(g) + H2(g), solid–gas mode | CO: 606, CH4: 135 | 29 |
| UiO-66/Co9S8 | Metallic photocatalyst + photothermal utilization | 300 W Xe lamp, λ > 800 nm | CO2(g) + H2O(g), solid–vapor mode | CH4: 25.7 | 25 |
| HO-Ru/TiN | Metallic photocatalyst + photothermal utilization | 300 W Xe lamp, λ > 800 nm | CO2(g) + H2O(g), solid–vapor mode | CO: 2.76 | 55 |
| Bismuthene | Metallic photocatalyst + photothermal utilization | 300 W Xe lamp, λ ≥ 700 nm | CO2(g) + H2O(g), solid–vapor mode | CO: 0.66, CH4: 0.11 | 105 |
| C@Fe2C/TiO | Metallic + heterojunction photocatalyst | 300 W Xe lamp, λ > 700 nm | CO2(g) + H2O(l) + TEOA, solid–liquid–gas mode | CH4: 18.315 | 106 |
| WS2/Bi2S3 | Heterojunction photocatalyst | 300 W Xe lamp, 0.45 mW cm−2, λ: 800–1100 nm | CO2(g) + H2O(l), solid–liquid–gas mode | CH3OH + C2H5OH: 10.5 | 107 |
| 10% Ag2S/Sb2S3 | Heterojunction photocatalyst | 300 W Xe lamp, NIR light | CO2(g) + H2O(g), solid–vapor mode | CO: 0.81, CH4: 4.11 | 108 |
| Bi2O3−x | SPR system | LED lamp, λ = 940 nm | CO2(g) + H2(g), solid–gas mode | CO: 4.5 | 17 |
| Ru1@H-MoO3−x | SPR system | 300 W Xe lamp, λ > 700 nm | CO2(g) + H2O(g), solid–vapor mode | CH4: 9.75 | 109 |
| 10-BP/WO | SPR system | 300 W Xe lamp, λ > 800 nm | CO2(g) + H2O(l), solid–liquid–gas mode | CO: 4.17 | 110 |
| Au rod@CuPd | SPR system | Xe lamp, 400 mW cm−2, λ > 600 nm | CO2(g) + H2O(g), solid–vapor mode | CH4: 13.0 | 59 |
| Aun/Au1-CMS | SPR system | 300 W Xe lamp, 200 mW cm−2, NIR light | CO2(g) + H2O(l), solid–liquid–gas mode | CH3COOH: 8.2 | 58 |
| NiAl–Ru-LDH | Narrow-bandgap photocatalyst | 300 W Xe lamp (79 mW cm−2), λ = 1200 nm | CO2(g) + H2O(l) + TEOA + ACN, solid–liquid–gas mode | CO: 110.8 | 111 |
| PSCN | Narrow-bandgap photocatalyst | 300 W Xe lamp, λ > 800 nm | CO2(g) + H2O(g), solid–vapor mode | CO: 58.5 | 112 |
| TNP-MOF | Narrow-bandgap photocatalyst | 300 W Xe lamp, λ > 730 nm | ACN + TEOA + CO2(g), solid–liquid–gas mode | HCOOH: 6630 | 18 |
| Photocatalyst | Catalyst type | Light source | Photocatalytic system | CO2RR performance (μmol g−1 h−1) | Ref. |
|---|---|---|---|---|---|
| UCNPs/ZIS | Up-conversion photocatalyst | 300 W Xe lamp, λ > 800 nm | ACN + TEOA + CO2(g), solid–liquid–gas mode | CO: 1.50, CH4: 0.22 | 60 |
| CQDs/Bi2WO6 | Up-conversion photocatalyst | 500 W Xe lamp, λ > 700 nm | CO2(g) + H2O(g), solid–vapor mode | CH4: 0.051 | 68 |
| ZnO1−x/C | Up-conversion photocatalyst | 400 W Xe lamp, λ: 715–900 nm | CO2(g) + H2O(g), solid–vapor mode | CO: 15.98 | 61 |
| OD-ZnO/C | Up-conversion photocatalyst | 400 W Xe lamp, λ: 715–900 nm | CO2(g) + H2O(g), solid–vapor mode | CO: 31.2 | 113 |
| Few-layered BiOI | Intermediate-band photocatalyst | 300 W Xe lamp, λ > 700 nm | CO2(g) + H2O(g), solid–vapor mode | CO: 0.119, CH4: 0.021 | 37 |
| Bi2WO6-OV | Intermediate-band photocatalyst | 500 W Xe lamp, λ > 700 nm | CO2(g) + H2O(g), solid–vapor mode | CH4: 0.049 | 114 |
| V-Bi19Br3S27 | Metallic photocatalyst | 300 W Xe lamp, λ > 720 nm | CO2(g) + H2O(l), solid–liquid–gas mode | CH3OH: 0.4 | 72 |
| CuNi/C | SPR system | 300 W Xe lamp, λ > 700 nm | H2O(l) + TEOA + CO2(g), solid–liquid–gas mode | CO: 11.205, CH4: 0.9 | 115 |
| 8Ni/TiO2 | Photothermal utilization | 375 W IR lamp, 1230 mW cm−2 | CO2(g) + H2(g), solid–gas mode | CH4: 271.9 | 116 |
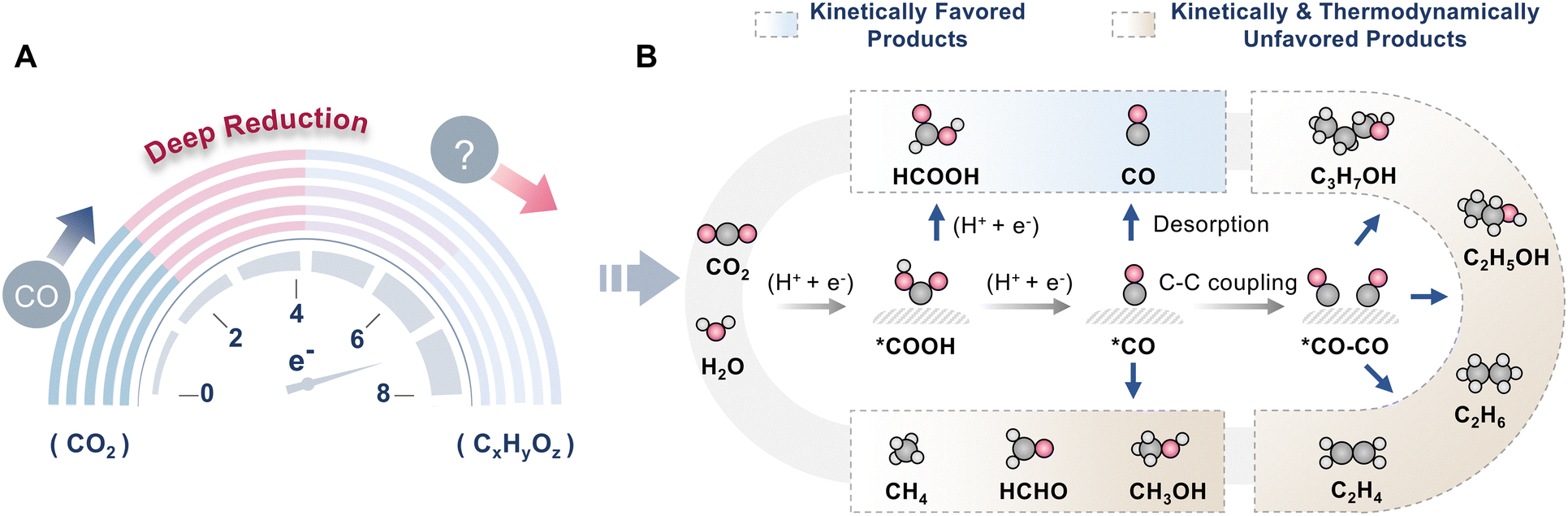 | ||
| Fig. 9 (A) Deep photocatalytic CO2 reduction. (B) Various pathways of photocatalytic CO2 reduction. | ||
 | ||
| Fig. 10 Schematic illustration of different NIR photocatalysis mechanisms. Energy band structures of (A) narrow bandgap photocatalysts, (B) intermediate-band photocatalysts, (C) metallic photocatalysts, and (D) heterojunction photocatalysts. Illustrations of the (E) SPR system, (F) up-conversion system, and (G) photothermal synergetic system. CB = conduction band, VB = valence band, IB = intermediate band, B−1 = the highest occupied band, B1 = the lowest unoccupied band, DET = direct electron transfer, PIRET = plasmon induced resonant energy transfer, ET = energy transfer, and ETU = energy transfer up-conversion. | ||
4. Strategies for harvesting NIR photons for photocatalytic CO2 reduction
4.1. Energy band structure regulation strategy
 | ||
| Fig. 11 Narrow-bandgap photocatalysts for harvesting NIR photons. (A) Molecular structure of the TNP linker. (B) and (C) Crystal structure of TNP-MOF. (D) Mott–Schottky curves and measured energy band structure of TNP-MOF. (E) NIR-light-driven CO2 reduction mechanism for TNP-MOF. (F) AQE results for TNP-MOF measured under monochromatic light of different wavelengths. Reproduced with permission from ref. 18. Copyright 2022, American Chemical Society. (G) Schematic illustration of the NiAl–Ru-LDH preparation route. (H) TDOS and PDOS of NiAl–Ru-LDH. (I) The CO2 photoreduction performance of NiAl–Ru-LDH at different irradiation wavelengths. Reproduced with permission from ref. 111. Copyright 2024, Wiley-VCH. | ||
Recently, Zhao et al.111 synthesized an organic–inorganic hybrid catalyst (NiAl–Ru-LDH) by introducing an anionic Ru coordination compound into the NiAl–NO3−-LDH interlayer, which successfully improved the spectral response of the LDH towards NIR light (Fig. 11G). The calculated total density of states (TDOS) and projected density of states (PDOS) indicate that introducing the Ru complex into the LDH interlayer significantly reduces its theoretical band gap from 2.41 to 0.74 eV (Fig. 11H). Consequently, this desirable band gap endows the NiAl–Ru-LDH catalyst with wide spectral response and impressive PCO2R performance. As shown in Fig. 11I, even at an irradiation wavelength of 1200 nm, the NiAl–Ru-LDH catalyst can still exhibit an activity of 1.108 μmol h−1 (ca. 110.8 μmol g−1 h−1, given a specific photocatalyst amount of 10 mg).
Hence, constructing narrow-bandgap photocatalysts represents a promising strategy for NIR-light-driven CO2RR.
Defect engineering is a powerful method for introducing an IB into the forbidden band of semiconductors, as the structural defects will lead to defect states within the forbidden band, enabling the semiconductor to absorb lower-energy photons. A common example is the creation of oxygen vacancies (Vo), which can be introduced during material synthesis in a reducing atmosphere. For instance, Liang et al.62 prepared defective WO3 atomic layers by calcining the WO3 precursor under a 20% H2/Ar atmosphere (Fig. 12A). The density of states (DOS) and electronic structures reveal that a new energy level forms in the band gap of WO3 when sufficient oxygen vacancies are introduced (Fig. 12B). The photoluminescence (PL) spectra of defective WO3 atomic layers indicate that the IB is ca. 1.4 eV above the VB. As the defect concentration in the WO3 atomic layers increases, the emission wavelength shifts to longer values, suggesting a narrower band gap between the IB and the VB. Consequently, Vo-rich WO3 atomic layers exhibit the best NIR-light-driven catalytic activity in the conversion of CO2 to CO, outperforming both the Vo-poor WO3 and the original WO3 atomic layers (Fig. 12C). Besides introducing oxygen vacancies, constructing metal-site defects is another feasible way to establish an IB.42,103 For example, Zhang and co-workers fabricated a Cu2−xS/g-C3N4 catalyst (CSCN) via the ethylene glycol-assisted solvothermal method.103 The Cu vacancies generated by reducing ethylene glycol lead to the formation of an IB in Cu2−xS, enhancing its absorption of NIR light (Fig. 12D). In the Cu2−xS/g-C3N4 composite, the band gap of g-C3N4 is too large to utilize NIR light (Fig. 12E), which is why Cu2−xS acts as the NIR light absorber. The NIR-light-driven CO2 reduction activity of the composite (CSCN) surpasses that of Cu2−xS alone (Fig. 12F). This increased activity stems from the enhanced CO2 adsorption ability of g-C3N4.
 | ||
Fig. 12 Intermediate-band (IB) photocatalysts for CO2RR under NIR illumination. (A) TEM image of the Vo-rich WO3 atomic layers. (B) Measured electronic structures of WO3 with oxygen defects. (C) NIR-light-driven CO2 reduction activity under silicon nitride lamp irradiation (800 nm to 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nm) for WO3 atomic layers with different amounts of oxygen vacancies. Reproduced with permission from ref. 62. Copyright 2018, Elsevier. (D) Mechanism of PCO2R over Cu2−xS/g-C3N4 by NIR light irradiation. (E) Electronic structures of Cu2−xS and g-C3N4. (F) Catalytic performance of Cu2−xS/g-C3N4 under NIR light irradiation. Reproduced with permission from ref. 103. Copyright 2020, Wiley-VCH. (G) Illustration of the band structures of bulk polymerized carbon nitride (BCN) and N-acetylethanolamine activated polymerized carbon nitride microtube (HCNT-NA). (H) Photocurrent response measurements over HCNT-NA. (I) AQE of HCNT-NA measured at different monochromatic wavelengths. The inset is the mass spectrum of a 13C-labelling experiment for CO2 photoreduction over HCNT-NA. Reproduced with permission from ref. 48. Copyright 2020, Royal Society of Chemistry. 000 nm) for WO3 atomic layers with different amounts of oxygen vacancies. Reproduced with permission from ref. 62. Copyright 2018, Elsevier. (D) Mechanism of PCO2R over Cu2−xS/g-C3N4 by NIR light irradiation. (E) Electronic structures of Cu2−xS and g-C3N4. (F) Catalytic performance of Cu2−xS/g-C3N4 under NIR light irradiation. Reproduced with permission from ref. 103. Copyright 2020, Wiley-VCH. (G) Illustration of the band structures of bulk polymerized carbon nitride (BCN) and N-acetylethanolamine activated polymerized carbon nitride microtube (HCNT-NA). (H) Photocurrent response measurements over HCNT-NA. (I) AQE of HCNT-NA measured at different monochromatic wavelengths. The inset is the mass spectrum of a 13C-labelling experiment for CO2 photoreduction over HCNT-NA. Reproduced with permission from ref. 48. Copyright 2020, Royal Society of Chemistry. | ||
Polymerized carbon nitrides (PCN) have drawn widespread interest in the area of photocatalysis because of their lamellar structures, chemical inertness, and affordable cost.118,119 However, in terms of PCO2R efficiency, the original PCN is far from satisfactory. An attractive strategy is to modify their structures with specific organic groups to tune their band structures for photocatalysis. For example, the amino groups on the edge of pristine PCN have a propensity to adsorb the polar water molecules over nonpolar CO2 molecules. This might potentially trigger the photocatalytic water-splitting process, thereby lowering the CO2 photoreduction efficiency. Thus, Liu et al. introduced hydroxyethyl groups into porous PCN using N-acetylethanolamine (NA) as a modifier to activate its edge sites.48 Compared to bulk PCN (BCN), the NA-modified PCN microtube (HCNT-NA) exhibits a slightly narrowed band gap while simultaneously forming an intermediate band (IB) (Fig. 12G). As a result, the spectral response range of HCNT-NA is expanded to the NIR region (Fig. 12H and I), thereby reinforcing its NIR photocatalytic activity.
Similarly, the empty d orbitals in certain transition-metal complexes, along with the intense interaction between the transition-metal ions and their associated ligands, have been reported to act as a natural intermediate band (Fig. 13A). These complexes absorb NIR light via d–d orbital transitions mediated by the vacant d orbitals. For instance, Li et al.50 synthesized ultrathin Cu4(SO4)(OH)6 nanosheets (CSON), which exhibited excellent NIR-photocatalytic performance, achieving the CO2RR with evolution rates of 4.11 and 21.95 μmol g−1 h−1 for CH4 and CO, respectively. The electronic structure and DOS of CSON reveal the presence of vacant d orbitals within the forbidden band (Fig. 13B and C). The Tauc curves and UV-vis-NIR diffuse reflectance spectra (DRS) demonstrate that both pristine CSON (p-CSON) and calcined CSON (c-CSON) samples display two narrow absorption bandgaps of 1.09 and 1.17 eV, which are attributed to electron transitions from the VB to vacant d orbitals and subsequent transfer to the CB (Fig. 13D–F). Moreover, the d–d electron transition mechanism has also been observed in other complexes similar to CSON, such as Cu2(NO3)(OH)3, Cu3(PO4)(OH)3, and Cu2(CO3)(OH)2 nanosheets. Therefore, utilizing d-d orbital transitions in transition-metal complexes is a feasible strategy for achieving NIR-light-driven CO2RR.
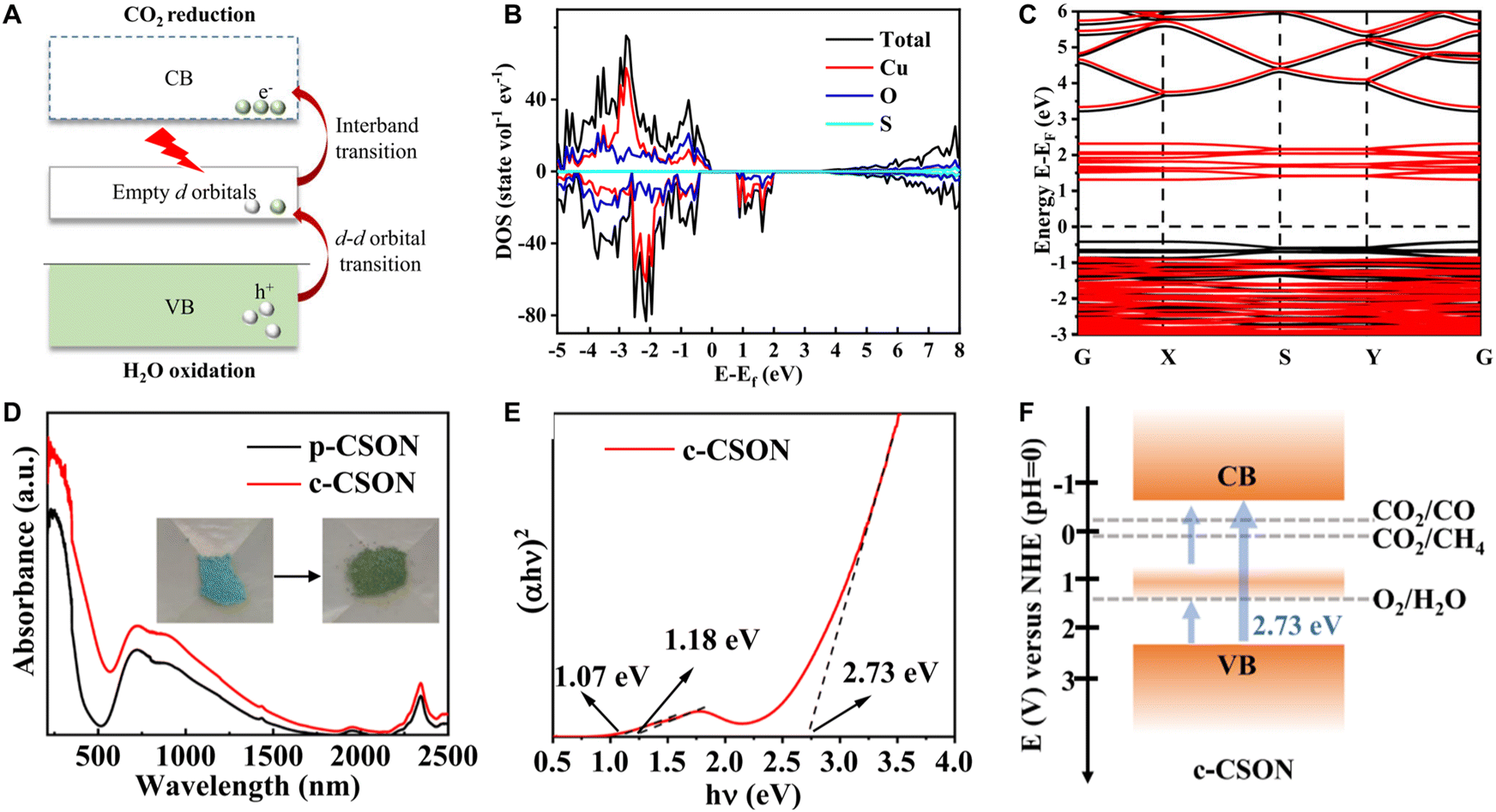 | ||
| Fig. 13 Harvesting NIR photons via the d–d orbital transition mechanism. (A) Illustration of the d–d orbital transition mechanism. (B) Calculated DOS and (C) electronic structure of the CSON slab model. (D) UV-vis-NIR diffuse reflectance spectra of p-CSON and c-CSON. (E) Tauc plot of c-CSON. (F) NIR-light-driven CO2 reduction mechanism over c-CSON. Reproduced with permission from ref. 50. Copyright 2023, Springer Nature. | ||
Hence, constructing an IB in photocatalysts—whether through defect engineering, structural modification, or d–d orbital transitions—presents a viable approach for fine-tuning their electronic structures to enable efficient NIR-light-driven CO2 reduction.
 | ||
| Fig. 14 Harvesting NIR photons via metallic photocatalysts. (A) Electron transition mechanisms in metallic CuS atomic layers under NIR light irradiation. (B) TEM image of CuS atomic layers. (C) Temperature dependence of resistivity and (D) VB-XPS spectrum of the ultrathin CuS conductor. (E) Calculated electronic band structure and (F) DOS of CuS nanosheets. Reproduced with permission from ref. 57. Copyright 2019, American Chemical Society. (G) HRTEM image of porous CoN atomic layers. (H) The electron transition mechanism within the CoN catalyst. (I) NIR-light-driven CO2 reduction performance over porous CoN atomic layers. Reproduced with permission from ref. 56. Copyright 2019, Elsevier. | ||
Among the metallic photocatalysts, cobalt-based catalysts demonstrate remarkable NIR-light-driven CO2 reduction performance. As depicted in Fig. 14G, Xie's group prepared metallic CoN atomic layers via nitriding CoO nanosheets, achieving conversion of CO2 into CO with around 100% product selectivity under NIR light irradiation.56 The femtosecond time-resolved transient absorption (fs-TA) spectra disclose interband recombination and intraband relaxation processes of charge carriers during NIR photocatalysis (Fig. 14H). Adding Na2S solution into the system significantly prolongs the lifetime of photo-generated electrons, increasing their intraband relaxation time by 9 times and interband recombination time by 1.6 times. Consequently, the CO release rate was dramatically enhanced to 14.5 μmol g−1 h−1 in the presence of Na2S, a 16-fold increase compared to that without Na2S (Fig. 14I).
In light of their low carrier concentration, traditional catalysts find it challenging to deeply transform CO2 into hydrocarbons via multiple proton–electron transfer processes under UV and visible light conditions, let alone under NIR light radiation. To achieve deep CO2 reduction, the photocatalysts are required to generate a sufficient concentration of electrons and possess adequate adsorption capacity to form reaction intermediates. Here, metallic catalysts, with their near-zero bandgap, can generate a high density of charge carriers even under NIR light, making them advantageous for facilitating deep CO2 reduction. Therefore, metallic photocatalysts show great potential for NIR-driven CO2 conversion into hydrocarbons. In this context, Xu et al. designed metallic Ni-doped CoS2 (Ni–CoS2) nanosheets via a NaCl-mediated strategy and realized a remarkable CH4 productivity of 101.8 μmol g−1 h−1 by NIR light excitation (Fig. 15A).24 Both Arrhenius plots and PL spectra indicate that doping Ni atoms in CoS2 nanosheets promotes carrier separation as well as reduces the activation energy required for CO2 photocatalysis. Furthermore, DFT calculations reveal that Ni doping effectively reduces the generation energy of intermediates (e.g., *CHO and *COOH), as well as the desorption energy of CH4, thus leading to the high selectivity of CH4 (90.6%) (Fig. 15B). To reinforce the CO2 affinity of catalysts, Xu et al.25 developed a UiO-66/Co9S8 composite photocatalyst, leveraging the CO2 adsorption ability of UiO-66 and the metallic feature of Co9S8. The extended X-ray absorption fine structure (EXAFS) spectra reveal that two inequivalent Co sites (Co1 and Co2 site) present in Co9S8 and the adjacent Co1 sites have strong metal–metal interaction, which also induces the metallic property of Co9S8 (Fig. 15C and D). The electron-rich Co1 sites help stabilize the intermediates and reduce the reaction barriers for CH4 production, leading to a high CH4 selectivity of ca. 100%. Besides, finite element (FEM) simulations reveal that the local CO2 concentration on Co9S8 is significantly increased (Fig. 15E), promoting the reaction kinetics of CO2RR. The control experiments verified that the generation of CH4 is attributed to the reduction of CO2, rather than the carbon contamination in the reaction system. To achieve alcohol production through CO2 photoreduction, Li et al.72 reported the metallic Bi19Br3S27 nanowires (V-Bi19Br3S27) with abundant Br and S defects, as well as Bi-O bonds at the surface (Fig. 15F and G). The cooperative effect of both O doping and vacancies diminishes the energy barriers of the entire reaction and helps transform the conversion process of *CO to *CHO from being endothermic to exothermic. As a result, the V-Bi19Br3S27 nanowires exhibit extraordinary activity for converting CO2 into methanol (CH3OH) without any sacrificial agent under NIR light excitation (Fig. 15H).
 | ||
| Fig. 15 Deep reduction of CO2 into hydrocarbon compounds over metallic photocatalysts. (A) Formation rates of CO and CH4 on Ni–CoS2 nanosheets under NIR light excitation. (B) Gibbs energy changes during the NIR-light-driven CO2RR over Ni–CoS2 nanosheets. Reproduced with permission from ref. 24. Copyright 2021, Wiley-VCH. (C) Structural information derived from Co K-edge EXAFS spectra. (D) Crystal structure of Co9S8. (E) FEM simulation for CO2 distribution around UiO-66/Co9S8. Reproduced with permission from ref. 25. Copyright 2024, Wiley-VCH. (F) Structural information derived from Bi K-edge EXAFS spectra and (G) corresponding wavelet transforms for k2-weighted EXAFS curves for V-Bi19Br3S27. (H) Photocatalytic CH3OH evolution over V-Bi19Br3S27 nanowires. Reproduced with permission from ref. 72. Copyright 2021, American Chemical Society. (I) The charge density difference of Vo-rich MoO2−x with adsorption of CO2. (J) CO2 adsorption isotherms of Vo-poor, Vo-moderate, and Vo-rich MoO2−x. (K) Yields of gas products from CO2RR under NIR light. Reproduced with permission from ref. 64. Copyright 2022, Wiley-VCH. | ||
Moreover, metallic photocatalysts are more likely to achieve a reduction process under low CO2 concentration atmospheres due to their ability to generate high carrier concentrations. Recently, Wu et al.64 reported a metallic MoO2 enriched with oxygen defects (Vo-rich MoO2−x), which is not only suitable for PCO2R in the UV to NIR wavelength range but also achieves NIR-light-driven CO2RR in ambient air. The existence of oxygen defects promotes the adsorption and activation of CO2, and induces the generation of the Mo-C-O-Mo intermediate. This intermediate further enables the deep reduction of CO2 to CH4 by NIR irradiation in a pure CO2 atmosphere (Fig. 15I). Additionally, the enhanced CO2 affinity contributes to highly selective photoconversion of CO2 into CO under NIR light in air (Fig. 15J and K). In light of the foregoing analysis, metallic materials are regarded as promising candidates for NIR-light-driven CO2 reduction.
 | ||
| Fig. 16 Heterojunction structure promotes NIR-light-driven CO2 reduction. (A) CuOx/Co3O4 composites. (B) Surface potential profiles of the Cu2O/Co3O4 heterojunction. (C) Electronic structure of Cu2O/Co3O4. Reproduced with permission from ref. 27. Copyright 2023, American Chemical Society. (D) HRTEM image of C@Fe2C/TiO. (E) The formation of CH4 on C@Fe2C/TiO by visible and NIR light irradiation. (F) Charge transfer pathway in C@Fe2C/TiO. Reproduced with permission from ref. 106. Copyright 2021, Elsevier. (G) TEM characterization of WS2@Bi2S3 nanotubes. (H) Synthetic procedure of the WS2@Bi2S3 heterojunction. (I) Cycle measurement of WS2@Bi2S3 under NIR irradiation. Reproduced with permission from ref. 107. Copyright 2019, Elsevier. | ||
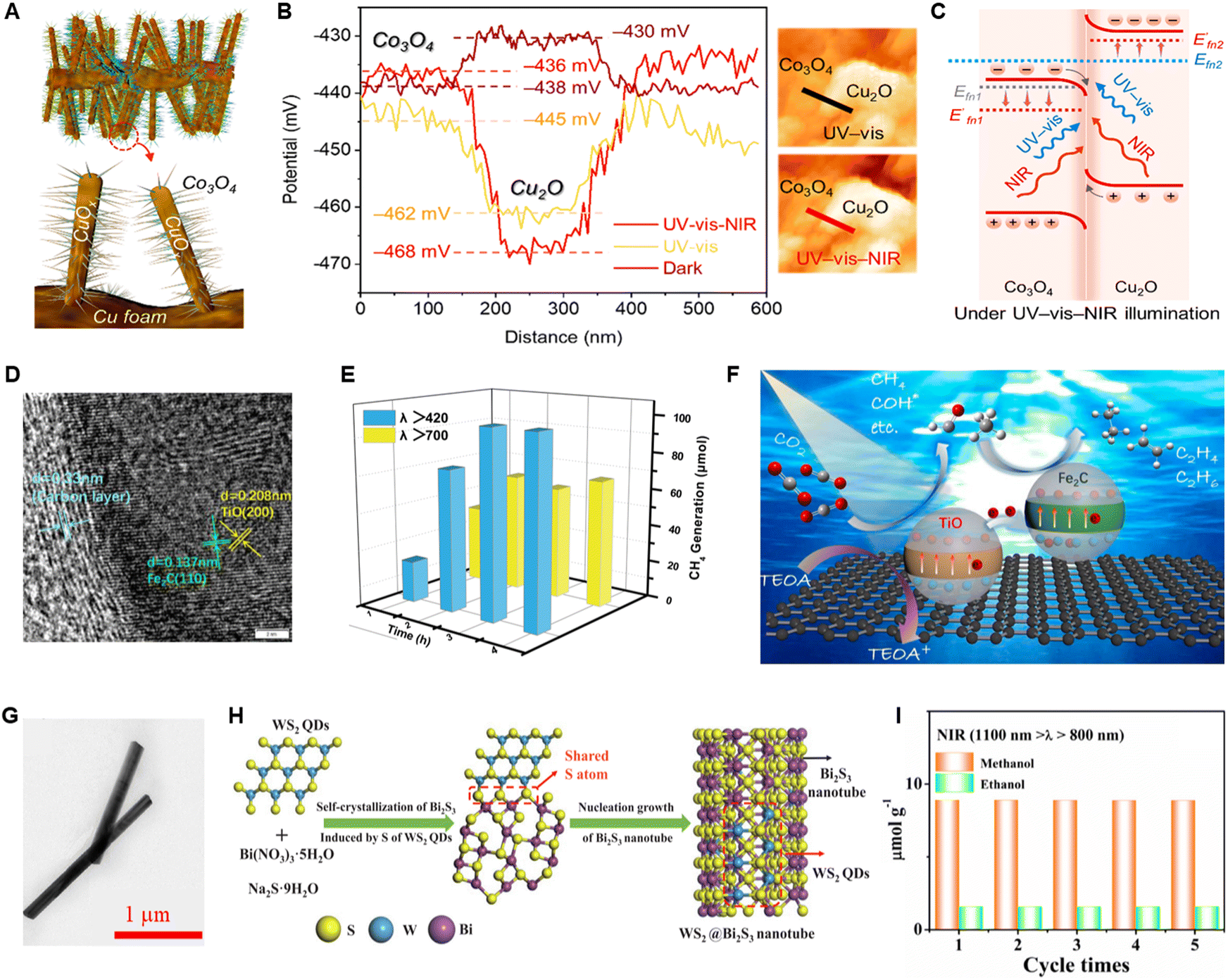
To deal with the problem of carrier recombination in metallic photocatalysts, Hao et al.106 prepared a carbon-coated Z-scheme heterojunction catalyst (C@Fe2C/TiO) using two metallic components, TiO and Fe2C (Fig. 16D). The Z-scheme heterojunction system significantly enhances the PCO2R activity of C@Fe2C/TiO under NIR light illumination (Fig. 16E). As shown in Fig. 16F, the holes in the VB of Fe2C will recombine with the excited electrons in the CB of TiO, further enhancing carrier separation. Dai et al.107 constructed a WS2@Bi2S3 heterojunction catalyst by doping WS2 quantum dots (QDs) into Bi2S3 nanotubes (Fig. 16G and H). The interface between WS2 and Bi2S3 matches very perfectly, minimizing resistance and facilitating rapid electron transfer while ensuring efficient charge separation. Although WS2 QDs (Eg = 3.85 eV) cannot be activated upon NIR illumination, the NIR-light induced electrons in the CB of Bi2S3 will efficiently migrate to the CB of WS2 QDs through the heterostructure. As a result, the WS2@Bi2S3 heterojunction catalyst effectively converts CO2 into CH3OH and C2H5OH under NIR illumination (Fig. 16I). In a word, constructing heterostructures to accelerate electron–hole separation in NIR-light-driven CO2 reduction is an effective method for improving catalytic performance.
4.2. Energy transfer strategy
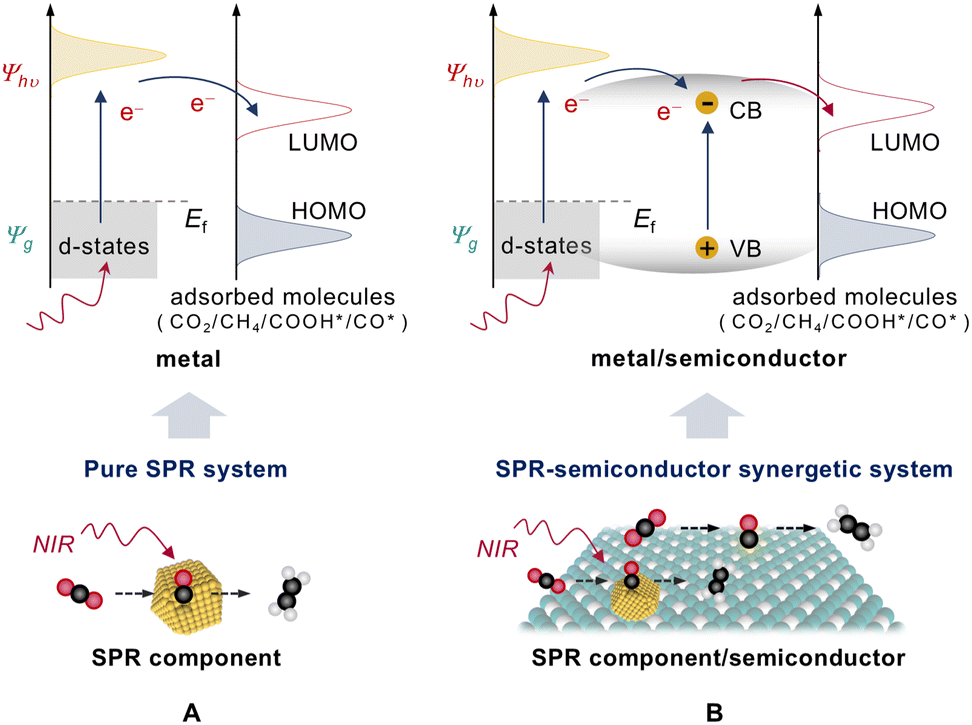 | ||
| Fig. 17 Different SPR systems promote NIR-light-driven CO2 reduction. (A) Pure SPR system. (B) SPR-semiconductor synergetic system. | ||
4.2.1.1. Pure SPR system. Unlike semiconductors, plasmonic materials, including precious metals (e.g., Au, Pt, and Ag), non-precious metals (e.g., Al and Cu), and specific carbon allotropes (e.g., carbon nanotube), can induce SPR effects when irradiated by light.59,125–128 These pure SPR systems can effectively utilize light energy to drive chemical reactions. By precisely modifying the size, morphology, and structure of nanomaterials, the SPR frequency of the plasmonic materials can be adjusted to match the frequency of low-energy photons, making these materials highly applicable for NIR-light-driven CO2RR. For instance, Xiong's group59 fabricated Au rod@CuPd composites through an alloying strategy, attaining the CH4 release rate of 0.55 mmol g−1 h−1 and the hydrocarbon selectivity of 100% by full-spectrum light irradiation. Moreover, this SPR system can achieve CO2RR with an AQE of 0.38% at 800 nm, exhibiting impressive NIR photocatalytic activity (Fig. 18A–C). The in situ near ambient pressure X-ray photoelectron spectra (NAP-XPS) reveal that illumination increases the electron densities of Cu and Pd atoms in the Au rod@CuPd composite (Fig. 18D), indicating that the hot electrons induced by the SPR effect transfer from Au to the CuPd alloy and subsequently to the adsorbed CO2. Meanwhile, the quasi-isolated trap states caused by the SPR effect prolong the lifespan of hot electrons, allowing their reactivation by additional low-energy photons (Fig. 18E). Moreover, the interaction between metal sites and CO2 molecules is enhanced by the strong local electric fields created by the localized SPR effect, thereby boosting the migration of the electrons from the metal sites to CO2 (Fig. 18F). To avoid the use of precious metals, Min et al. developed a CuNi/C bimetallic composite photocatalyst via pyrolysis of CuNi bimetallic MOF precursors (Fig. 18G and H).115 As a consequence of the SPR effect generated by Cu and Ni, the CuNi/C catalyst responds to a broad spectral range from 200 to 2000 nm. Compared to monometallic Ni/C and Cu/C, the CuNi/C catalysts obtained by an alloying strategy also exhibit a higher CO production rate during the NIR-light-driven CO2 reduction process (Fig. 18I).
 | ||
| Fig. 18 Pure SPR system promotes NIR-light-driven CO2 reduction. (A) The diagram of the CO2 capture on the CuPd co-catalyst. (B) AQY of the Au rod@CuPd catalyst, measured under the illumination of monochromatic light of different wavelength. (C) CH4 production over Au rod@CuPd with various CuPd/Au atomic ratios. (D) In situ NAP-XPS contour plot of Cu 2p3/2 over Au rod@CuPd. (E) Illustration of the SPR-mediated energy transfer mechanism during the PCO2R process. (F) Natural orbitals for chemical valence (NOCV) relate to the key deformation densities of CO2 and H2O on the CuPd(100) plane. Reproduced with permission from ref. 59. Copyright 2023, Springer Nature. (G) Synthetic procedures of CuNi/C catalysts. (H) Elemental mapping images of CuNi/C catalysts. (I) PCO2R performance of CuNi-T/C catalysts (T represents the calcination temperature, T = 450, 550, and 650 °C) under NIR irradiation (λ > 700 nm). Reproduced with permission from ref. 115. Copyright 2020, Royal Society of Chemistry. | ||
4.2.1.2. SPR-semiconductor synergetic system. Integrating SPR components with traditional semiconductor photocatalysts is an innovative tactic for constructing efficient NIR-light-driven CO2 reduction systems. In these SPR-semiconductor synergetic systems, the plasmonic component can efficiently harvest NIR photons and transfer hot electrons and resonance energy to the semiconductor via the SPR effect. Simultaneously, the semiconductor offers various active sites for CO2RR that can potentially be used to tune its photocatalytic activity and product selectivity. In addition, the metal/semiconductor contact could form a Schottky barrier, which improves photocatalytic efficiency by trapping and prolonging the lifespan of electrons and holes. Meanwhile, by adjusting the size and shape of SPR components, the SPR effect can be reinforced and the resonance frequency can be changed, leading to a wider light response suitable for the NIR photocatalytic system.129–132 Therefore, integrating SPR materials into a semiconductor is a promising way to synthesize NIR-light responsive catalysts.
Constructing a SPR-semiconductor composite system enables the spectral response range to be expanded to the long-wavelength region; nevertheless, the supply of active hydrogen (*H) species and the efficiency of hot electron transfer in this system are still insufficient for effective CO2 photoreduction. To address this, Xiao et al.109 fabricated hydrogenated MoO3−x nanosheets with Ru single atom substitution (Ru1@H-MoO3−x), achieving an efficient transformation of CO2 into CH4 with a production rate of 9.75 μmol g−1 h−1 and a high selectivity of ca. 96% by NIR light irradiation (Fig. 19A). The Ru substitution and oxygen vacancy engineering induce the generation of the doped/defect energy band, which narrows the bandgap of the catalyst for enhancing NIR photon harvest. The fitting results of fs-TA kinetics curves indicate that the states of excited electrons within Ru1@H-MoO3−x may undergo three relaxation processes (Fig. 19B): (i) from the excited state to the exciton state (τ1), (ii) from the exciton state to the defect trap state (τ2), and (iii) from the defect trap state to the ground state (τ3), where τ represents the consumed time for each transition process. As shown in Fig. 19B, Ru1@H-MoO3−x possesses a shorter τ1 (0.50 ps) than that of H-MoO3−x (2.95 ps), suggesting a faster transition from excited states to exciton states. Besides, the τ2 (7.74 ps) and τ3 (167.46 ps) of Ru1@H-MoO3−x are significantly higher than those of H-MoO3−x (τ2: 3.06 ps, τ3: 135.69 ps), verifying the longer carrier lifetime and higher charge separation efficiency. The surface photovoltage (SPV) analysis reveals that Ru1@H-MoO3−x exhibits a larger SPV signal change (8 mV) than that of H-MoO3−x (6.5 mV), demonstrating a higher photogenerated electron concentration on the Ru1@H-MoO3−x surface (Fig. 19C). Furthermore, theoretical calculations indicate that the doped Ru single atom can lower the energy barrier for *H generation and facilitate *H migration. With the synergy of Ru single atoms and oxygen vacancies, Ru1@H-MoO3−x achieves the outstanding activation of CO2 and H2O, which ultimately contributes to its high selectivity towards CH4.
 | ||
| Fig. 19 SPR-semiconductor synergetic system promotes NIR-light-driven CO2 reduction. (A) PCO2R performance over MoO3 and Rux@H-MoO3−x (x = 0, 0.5, 1, 2, 4) catalysts under NIR irradiation. (B) Transient absorption kinetics curves of Ru1@H-MoO3−x. (C) Surface photovoltage measurement of Ru1@H-MoO3−x. Reproduced with permission from ref. 109. Copyright 2024, Wiley-VCH. (D) The color-coded channel map of filtered atomic-resolution HAADF-STEM image for Aun/Au1-CMS catalysts. (E) NIR-light-driven CO2 reduction performance over Aun/Au1-CMS and the control catalysts. (F) Electronic structure of Aun/Au1-CMS and the possible PCO2R mechanism. Reproduced with permission from ref. 58. Copyright 2024, Springer Nature. | ||
The NIR-light-driven transformation of CO2 into C2+ products remains a challenge, primarily due to the low activation efficiency of CO2, the low C–C coupling probability of CO2RR intermediates, and insufficient carrier utilization.49,52,106,133 To accomplish this goal, Wu et al. developed a cross-scale heterojunction catalyst (denoted as Aun/Au1-CMS) by implanting Au nanoparticles (Aun) and single atoms (Au1) into unsaturated Mo atoms of edge-rich MoS2 (CMS) (Fig. 19D).58 Under NIR light irradiation, Aun/Au1-CMS presents high selectivity (95.1%) and activity (8.2 μmol g−1 h−1) for the photoreduction of CO2 into acetate (Fig. 19E). The impressive NIR photocatalytic performance results from the SPR effect of Au nanoparticles. Additionally, the measured energy band structure of the Aun/Au1-CMS catalyst displays that the plasmonic Au metal, CMS semiconductor, and Au single atom form a SPR-semiconductor system, where hot electrons can transfer from Au nanoparticles to Au1 through the CB of CMS, which remarkably inhibits the recombination of carriers (Fig. 19F). Hence, the SPR-semiconductor synergetic system exhibits huge potential in harvesting NIR photons for CO2RR.
In brief, plasmonic materials are capable of efficiently harvesting NIR photons via the SPR effect, enabling the constructed photocatalysts to realize NIR-light-driven CO2 reduction. Unfortunately, current plasmonic components are mainly based on noble metal nanoparticles, which are costly and obstructive to the development of SPR-photocatalytic systems. Besides, the carrier recombination is severe in the SPR-based CO2 photoreduction system, which has an adverse impact on improving the efficiency of NIR-light-driven CO2RR. Thus, novel and affordable plasmonic components and advanced SPR-semiconductor composite systems should be explored to reinforce SPR-promoted NIR-light-driven CO2RR.
Among Ln-doped materials, Ln3+-doped materials exhibit efficient up-conversion performance due to their extremely long-lived intermediate energy states. For instance, UCNPs are typically formed by doping Ln3+ ions into an appropriate host matrix, such as fluorides or oxides. Fluorides, particularly NaYF4, are the common matrices due to their low phonon energy, high structural stability, and low non-irradiative energy losses. Moreover, by varying the type and concentration of the doped Ln3+ ions, the wavelengths and intensities of the luminescence peaks of UCNPs can be fine-tuned to align with photocatalysts possessing different bandgaps. For instance, Yu et al. integrated UCNPs (NaYF4: Yb, Tm) with ZnIn2S4 nanorods (ZIS), achieving CO2 photoreduction into CO and CH4 through NIR light activation (Fig. 20A and B).60 As shown in Fig. 20C, ZIS displayed a spectral response in the region from 325 to 600 nm, while UCNPs emitted several peaks in the same range. Therefore, the up-conversion emission spectrum of UCNPs aligns well with the DRS spectrum of ZnIn2S4, indicating that the photons emitted by UCNPs are well-suited for exciting ZIS. However, since the VBM of ZIS was not sufficiently positive to drive water oxidation, a sacrificial agent (TEOA) was introduced to consume the holes and promote charge carrier separation, enhancing the overall photocatalytic performance.
 | ||
| Fig. 20 Up-conversion systems promote NIR-light-driven CO2 reduction. (A) Schematic diagram of the up-conversion mechanism and the CO2 reduction process on UCNPs/ZIS. (B) The activity of UCNPs/ZIS. (C) PL spectrum of UCNPs overlaps with the DRS spectra of ZIS. Reproduced with permission from ref. 60. Copyright 2022, Elsevier. (D) XRD spectra. (E) Up-conversion PL spectra for CQDs. (F) PCO2R mechanism on CQDs/UBW catalysts. (G) The yield of CH4 during CO2 reduction by NIR light irradiation over the sample catalysts. Reproduced with permission from ref. 68. Copyright 2016, Springer Nature. (H) The diagram of the synthetic procedure of the ZnO1−x/C composite catalyst. (I) UV-vis-NIR DRS spectra. (J) The CO evolution rate (5 h) on ZnO1−x/C samples. Reproduced with permission from ref. 61. Copyright 2018, Elsevier. | ||
While Ln-doped up-conversion materials show promise in utilizing NIR light, their low quantum efficiency (ca. 1–10%) and strong dependence on specific excitation wavelengths limit their practical applications.138,139 Recently, carbon quantum dots (CQDs) have garnered wide attention in photocatalysis because of their impressive up-conversion capabilities and conductivity.140 The conjugated π-electron transitions in CQDs enable increased spectral responsiveness to NIR light, effectively overcoming the limitations associated with Ln-doped materials that require specific excitation wavelengths. For instance, Kong et al. decorated ultrathin Bi2WO6 nanosheets with CQDs, which enhanced the CO2 reduction activity by broadening the spectral response range and enhancing the charge separation (Fig. 20D and E).68 Under NIR light irradiation, the Bi2WO6 nanosheets, with the assistance of CQDs, achieved a CH4 yield from 1 wt%-CQDs/Bi2WO6 that was 10.5-fold higher than that of original Bi2WO6 (Fig. 20F and G). Besides, CQDs/Bi2WO6 composites exhibited a larger transient photocurrent density and lower charge transfer resistance compared to pure Bi2WO6, possibly attributed to the exceptional electron conductivity of CQDs. In another case, Lin and co-workers synthesized a composite hollow sphere of ZnO1−x/carbon dots (ZnO1−x/C) for CO2 photoreduction in the entire UV-vis-NIR spectral range (Fig. 20H–J).61 Under NIR illumination alone, ZnO1−x/C could also demonstrate a high CO2RR activity, achieving a CO formation rate of 15.98 μmol g−1 h−1. This performance can be attributed to the strong up-conversion photoluminescence emission capability of CQDs. Notably, no sacrificial agents were used in the CQDs/Bi2WO6 and ZnO1−x/C systems mentioned above. The only reactants were CO2 and water vapor (or humid CO2 gas), rendering the photocatalytic system straightforward and environmentally friendly.
Consequently, up-conversion materials can be considered as a good medium for transforming long-wavelength light into short-wavelength light. By modifying the structures of these up-conversion materials, the energy of emission photons can be tailored to activate the catalysts with various bandgaps, thereby increasing the NIR-light responsiveness of the catalysts.
4.3. Photothermal utilization strategy
Compared with UV and visible light, NIR light exhibits a unique photothermal effect, and utilizing this effect is beneficial for promoting CO2 reduction. Both the photochemical process and the thermochemical process caused by the NIR light are conducive to accelerating CO2RR. The photochemical process can lower the activation barriers of CO2 and the intermediates by increasing the acidity or alkalinity of the catalyst surface (carrier accumulation or depletion). In contrast, the thermochemical process promotes the overall reaction by elevating the temperature at active sites, leading to an increase of kBT (kB represents the Boltzmann constant, T represents the reaction temperature), which is related to the kinetic energy of reaction molecules (Fig. 21).141–144 | ||
| Fig. 21 Photothermal strategy for promoting NIR-light driven CO2 conversion. Photochemistry contributes to reducing the reaction energy barrier, while thermochemistry helps the reactants overcome the barrier by elevating the temperature. | ||
Hence, maximizing the photothermal effect of NIR light is crucial for improving CO2 photoreduction. Below are the key advantages of photothermal utilization for CO2 reduction.
Accelerating carrier migration. In the solar-induced photothermal process, the extra heat induced by the photothermal effect could elevate the energy levels of charge carriers and accelerate their transport behavior.145 For example, Yu et al.146 synthesized TiO2 with abundant oxygen vacancies via a solvothermal method, introducing defects that generated deep energy levels within the bandgap. Subsequently, post-solution plasma processing was used to dope hydrogen into the oxygen-vacancy-rich TiO2, creating new shallow energy levels. These shallow energy levels were significantly closer to the CB of TiO2 than those associated with oxygen vacancies (Fig. 22A and B). As a result, the modified TiO2, enriched with mid-gap states, exhibited broad NIR absorption. During photothermal catalysis, photogenerated electrons can be extracted from deep energy levels through shallow-level bridges and then thermally released to the catalyst surface (Fig. 22B). This photothermal effect not only promotes the electron extraction but also favors charge utilization and surface reactions. Compared to conventional photocatalysis, the catalyst demonstrated significantly higher formation rates for CH4 (11.93 μmol g−1 h−1) and CO (38.99 μmol g−1 h−1) during photothermal reactions, representing 108-fold and 300-fold enhancements, respectively (Fig. 22C).
 | ||
| Fig. 22 The main potential advantages of the photothermal effect in CO2 reduction. (A) The diagram of the solution plasma processing (SPP) method. (B) Carrier transfer process promoted by the photothermal effect in OV-TiO2 and SPP-treated TiO2. (C) The photothermal catalytic performance (PTC) of OV-TiO2 (T-0 h) and SPP-treated TiO2 (T-2 h). Reproduced with permission from ref. 146. Copyright 2020, Wiley-VCH. (D) Temperature-dependent CO and H2 evolution rates over the Bi–Hx catalyst. (E) Photothermal effect facilitating proton/electron transfer over the Bi–Hx catalyst. Reproduced with permission from ref. 148. Copyright 2021, Springer Nature. (F) Influence of H2O on thermo-photo catalytic H2-assisted CO2 reduction over a Cu2O/graphene catalyst. Reproduced with permission from ref. 149. Copyright 2017, Royal Society of Chemistry. Temperature-dependent quasi in situ XPS spectra of the (G) Cu 2p core level and (H) Co 2p core level for Co3O4/CuOx. (I) Photothermal catalytic performance of Co3O4/CuOx under different temperatures. Reproduced with permission from ref. 27. Copyright 2023, American Chemical Society. (J) Temperature dependence of the redox potentials for CO2 reduction. Reproduced with permission from ref. 152. Copyright 2020, Elsevier. | ||
Promoting the electron/proton mitigation. The introduction of heat enhances proton/electron transfer, thereby boosting the CO2 conversion rate.147 For instance, Li et al. developed a hydrogen-stored bismuth (Bi–Hx) catalyst that significantly boosts CO2-to-CO conversion during the thermal-assisted photocatalytic process (Fig. 22D and E).148 Within the optimal temperature range of 150–180 °C, the additional thermal energy generated by the increasing temperature benefits the transfer of protons and electrons from Bi–Hx species to the adsorbed CO2, thereby contributing to the production of CO. However, above 180 °C, stored hydrogen in Bi–Hx tends to form H2, limiting proton/electron migration and reducing CO evolution.
Regulating the desorption/adsorption of CO2 and products. The local temperature of catalysts can be effectively increased under light illumination via lattice vibrations or relaxation of carriers.52,94 Within a specific temperature range, the elevated temperature promotes the desorption of products and by-products during CO2 conversion, ensuring the timely renewal of catalytic sites and maintaining activity and stability.149,150 For instance, photo-promoted H2-assisted CO2 reduction to produce CH4 can be realized at 250 °C using Cu2O/graphene as the photocatalyst. Studies have shown that in the gas-phase methanation process, the product H2O requires relatively high temperatures to desorb from the material surface.149 As shown in Fig. 22F, control experiments in which the temperature and the amount of added H2O were varied have revealed the impact of H2O adsorption/desorption behavior on CH4 production efficiency. These experiments indicate that the adsorbed H2O molecule on the catalyst surface inhibited CH4 formation. Conversely, the increased temperature favors H2O desorption, therefore, enhancing CH4 evolution. Generally, a moderate increase in the reaction temperature facilitates CO2 diffusion in the photocatalytic process, thereby enhancing mass transfer efficiency and improving reaction kinetics.4,151 However, excessively high temperatures can hinder CO2 adsorption on the catalyst surface, reducing overall catalytic efficiency. This occurs because elevated temperatures weaken the interplay between CO2 and active sites, resulting in desorption before the reaction can proceed effectively.153 Therefore, precisely controlling the local temperature of the photocatalysts to regulate the desorption and adsorption of CO2 and products is essential for enhancing photothermal catalytic efficiency.
Modulating the catalyst structure. The complex CO2RR pathways and the imperfect structure of catalysts often result in low selectivity for specific products in light-driven CO2 conversion. Recent studies indicate that during high-temperature photocatalysis, catalysts can undergo in situ restructuring under light and/or heat, refining their structure or surface properties to enhance activity and control product distribution.4,141,144,154 For example, Bai et al.27 used quasi in situ XPS to reveal that during thermal-assisted CO2 photoreduction, the Co3O4/CuOx precursor underwent an in situ transformation (Fig. 22G and H). With the temperature increasing from 25 to 240 °C, the Cu(I) content in CuOx increased while the Co(II) content in Co3O4 decreased. This suggests that strong photothermal effects facilitated the conversion of the Co3O4/CuOx composite into the Co3O4/Cu2O heterojunction. As shown in Fig. 22I, at 240 °C, the CO2-to-CH4 conversion rate reached 6.5 μmol h−1, with CH4 selectivity exceeding 99%, which was promoted by the in situ formation of the Co3O4/Cu2O catalyst. Thus, understanding the impact of photo-thermal effects on catalyst reconstruction is crucial for improving NIR-light-driven CO2 conversion efficiency.
Tuning the redox potentials of half-reactions. According to the Nernst equation, the standard redox potential can be significantly tuned by temperature.152,155 The photothermal effect induces local temperature variations on the catalyst surface, resulting in either a positive or a negative shift in the redox potential. These potential shifts enable a broader range of catalysts to satisfy the thermodynamic requirements of reactions. For instance, as the temperature rises, the potential of CO2RR shifts positively, indicating that this process is more thermodynamically favorable under elevated temperature conditions. Fig. 22J displays that when the reaction temperature is increased from 25 to 600 °C, the redox potential for CO2 reduction to CO shifts positively from −0.526 V (vs. SHE, pH = 7) to −0.162 V (vs. SHE, pH = 7), significantly improving the thermodynamic feasibility of CO2-to-CO conversion.155 Therefore, the photothermal effect may allow more catalysts to reach the thermodynamic requirements for CO2RR by tuning the standard redox potentials.
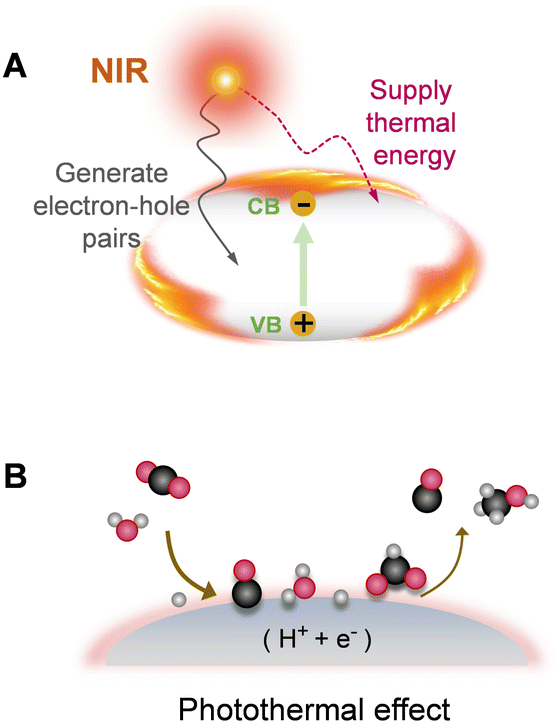 | ||
| Fig. 23 (A) Photothermal effect for supplying thermal energy and generating electron–hole pairs. (B) Photothermal strategy for promoting NIR-light driven PCO2R. | ||
For instance, Guo et al. reported a CO2RR process operating in a solid–vapor mode, in which a CuInS2 catalyst was fabricated as a thin film, allowing a CO2–H2O gas mixture to react on its surface.63 The material exhibited an intrinsic absorption edge at 860 nm, corresponding to a bandgap of 1.44 eV (Fig. 24A). In their study, while maintaining the same light intensity but varying the wavelength range, temperatures of this system were monitored via an infrared camera (Fig. 24B). CuInS2 rapidly heated within seconds, and its surface temperatures stabilized above 80 °C after five minutes of illumination, showing excellent photothermal performance. Notably, the photothermal response of CuInS2 under UV-visible light was slower than under NIR and full-spectrum light, suggesting the inferior photothermal effect of UV-visible light. Under UV-visible light, CO production was only 2.8 μmol g−1 h−1, whereas it rose to 19.9 μmol g−1 h−1 when NIR light was also incorporated (UV-visible-NIR), highlighting the significant role of NIR in boosting the CO2 conversion performance. Meanwhile, CO2RR was tested under IR wavelengths above 800 and 980 nm (Fig. 24C), which showed that NIR photons alone (above 800 nm) could also efficiently drive CO2RR. To differentiate between the thermal effect and the role of NIR photons, control experiments involving external heating were conducted (Fig. 24D). As shown, the activity increased from 80 to 200 °C, confirming the promoted effect of photothermal contribution. However, the activity under UV-vis light with external heating at 200 °C remained lower than that under full-spectrum illumination. Considering the photothermal temperature of ca. 90 °C, this emphasizes the crucial role of photon synergy over simple heating. It was found that UV-vis-NIR light led to a more negative surface potential than UV-vis alone, indicating that NIR light enhances electron accumulation. Moreover, in situ XPS and DRIFTS analyses further revealed that NIR improves CO2 adsorption and activation. Thus, the superior photothermal effect of IR light, combined with enhanced physical adsorption and activation of CO2 and H2O, collectively promotes CO2RR. Also, it has become increasingly important to explore innovative and highly effective strategies for achieving efficient NIR photothermal energy conversion to overcome the limitations of current catalyst designs.143,158 Recent studies have highlighted that incorporating components with strong NIR photothermal properties into catalytic systems significantly enhances the photothermal conversion efficiency of composite catalysts. For instance, Chen et al. successfully developed a sequence of POMs@GO-PEI samples through a combination of covalent grafting and electrostatic adsorption methods (Fig. 25A).73 In this system, the sheetlike graphene oxide (GO) matrix functions as a highly localized photothermal heater. Under mild and convenient reaction conditions, the synthesized POMs@GO-PEI catalysts efficiently promote the cycloaddition of CO2 using NIR irradiation, utilizing CO2 as a feedstock in organic synthesis. Notably, the heterogeneous catalysts display outstanding photothermal stability along with excellent recyclability. Among a sequence of POMs@GO-PEI samples, the designed SiWCo@GO-PEI materials leverage a low-energy photoredox-driven photothermal catalytic process to significantly boost the cycloaddition reaction (Fig. 25B and C). This approach achieves an outstanding 98.9% epoxide conversion and 99% selectivity, with an impressive TOF of up to 2718 h−1 under ambient pressure.
 | ||
| Fig. 24 Photothermal effect promotes CO2 conversion performance. (A) Optical properties of CuInS2 manifested by UV-vis-NIR DRS spectra. (B) Photothermal images of CuInS2 under UV-vis, NIR, and full spectrum light irradiation. (C) Photocatalytic CO2 reduction performance under different long-pass cut-off filters (800 nm and 980 nm). (D) CO2 reduction performance under light irradiation and additional heating. Reproduced with permission from ref. 63. Copyright 2024, American Chemical Society. | ||
 | ||
| Fig. 25 Photothermal effect facilitates NIR-light-driven CO2 reduction. (A) The preparation of POMs@GO-PEI catalysts and the diagram of photothermal catalytic cycloaddition of CO2 with epoxides. (B) Kinetic curves of CO2 cycloaddition with epichlorohydrin on SiWCo@GO-PEI under the three conditions of room temperature (R.T.), external heating (ex. heating), and photothermal condition, with NIR radiation. (C) Reusability of the SiWCo@GO-PEI catalyst. Reproduced with permission from ref. 73. Copyright 2022, American Chemical Society. | ||
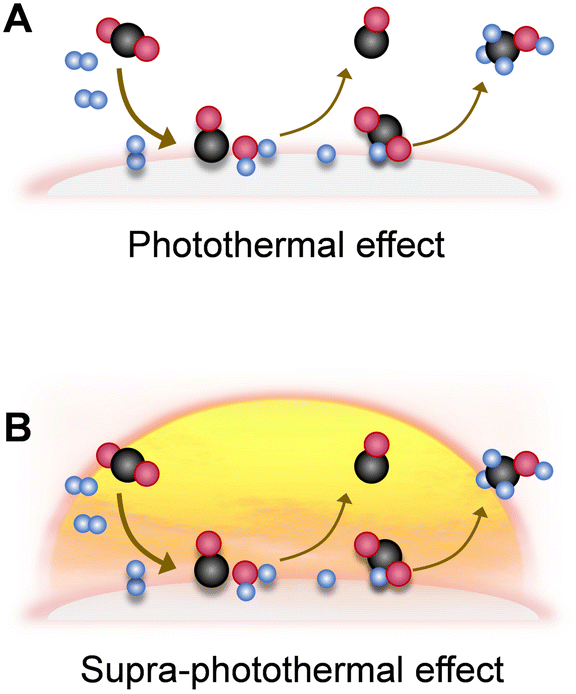 | ||
| Fig. 26 (A) Photothermal effect and (B) supra-photothermal effect for promoting NIR-light driven H2-assisted CO2 conversion. | ||
4.3.2.1. Photothermal effect. For instance, Jia et al. designed a series of TiO2 catalysts loaded with Ni NPs, named xNi/TiO2, which exhibited excellent activity for the conversion of CO2 and hydrogen under IR irradiation (Fig. 27A).116 Notably, the surface temperature of xNi/TiO2 is closely related to the nickel content, confirming that the Ni NPs are primarily responsible for photothermal conversion. In xNi/TiO2, the highly dispersed, small-sized Ni NPs are rich in oxygen vacancies, which enhance their exothermic plasmonic resonance behaviour. In this study, while pure TiO2 exhibited no photocatalytic or photothermal catalytic activity for H2-assisted CO2 conversion, the xNi/TiO2 catalyst was effectively activated under infrared light. As shown in Fig. 27B, the temperature of the 8Ni/TiO2 catalyst increases almost linearly with the intensity of the IR light, and the corresponding CH4 production rates also show a linear increase with the rise in the surface temperatures of 8Ni/TiO2. Under optimized conditions, the catalyst achieves nearly 100% selectivity toward methane and a high production rate of 463.9 mmol gNi−1 h−1. In situ DRIFTS studies suggest that methane formation on the 8Ni/TiO2 catalyst may proceed through a formate-mediated pathway (Fig. 27C). Hydrogen atoms generated from H2 dissociation on the highly dispersed, small-sized Ni nanoparticles rapidly diffuse through overflow processes. These hydrogen species facilitate the conversion of CO2 into formate intermediates, which subsequently lead to methane production. Further analysis revealed that the excellent photothermal catalytic performance of the 8Ni/TiO2 catalyst can be attributed to the uniform distribution and small size of the nickel NPs. These features enable strong infrared light absorption, high photothermal conversion efficiency, and excellent capabilities for CO2 and H2 adsorption and activation.
 | ||
| Fig. 27 H2-assisted photothermal CO2 conversion. (A) IR-light-driven photothermal conversion of CO2 to methane over Ni/TiO2. (B) Temperature and corresponding catalytic activity of Ni/TiO2 under different IR irradiation intensities. (C) Schematic description of the CO2 methanation pathways. Reproduced with permission from ref. 116. Copyright 2022, Elsevier. (D) SEM image of 10 nm Ru sputtered Si nanowires (SiNW). (E) The light absorption spectra of Ru/glass, Ru/Si, and Ru/SiNW catalysts. (F) The photon flux of different incident photons. (G) The relationship between the photon flux and CO2 methanation rate. (H) The relative absorption spectrum of the In2O3 nanoparticle catalyst is shown by the green curve. The shaded areas in different colors represent distinct wavelength ranges within the solar spectrum. (I) Temperature profile over the SiNW materials carried out in the dark and under the Xe lamp. Reproduced with permission from ref. 162. Copyright 2023, Wiley-VCH. | ||
Ozin et al.162 developed a Ru/silicon nanowire (Ru/SiNW, Fig. 27D) catalyst through the sputter deposition of ca. 10 nm Ru nanoparticles onto black silicon nanowires (SiNWs). In this system, the Ru nanoparticles function as the catalytically active sites for the Sabatier reaction (CO2 + 4H2 → CH4 + 2H2O), while the SiNWs act as a support and an efficient light-harvesting medium. Owing to its unique three-dimensional morphology, narrow electronic bandgap (1.1 eV), and inherently low optical reflectance, the Ru/SiNW catalyst achieves outstanding photon absorption across the ultraviolet, visible, and near-infrared spectral regions (Fig. 27E). Consequently, under an illumination intensity of 14.5 suns, the Ru/SiNW catalyst achieved a self-induced increase in temperature to approximately 125 °C at 15 psi solely through photothermal effects, without the need for external heating. To further elucidate the activation mechanism, the authors employed a series of high-pass optical filters during the Sabatier reaction (Fig. 27F). These controlled experiments demonstrated that the Ru/SiNW catalyst can facilitate the Sabatier reaction through a synergistic combination of photochemical and thermochemical pathways. More specifically, photons with energies below the SiNW bandgap (λ > 1100 nm) are incapable of directly driving the Sabatier reaction through photochemical pathways; instead, their absorption generates thermal energy that promotes the reaction via thermochemical activation. In contrast, as illustrated in Fig. 27G, a linear correlation was observed between the flux of photons with energies exceeding the SiNW bandgap (1.1 eV) and the rate of CO2 methanation, thereby providing clear evidence of a photochemical contribution. Notably, the slope of this linear relationship (∼4 × 10−9 CH4 molecules per photon) indicates that only a minor fraction of high-energy photons induces photochemical activity, while the majority of photogenerated charge carriers undergo thermalization and recombination, indirectly enhancing the reaction through heat generation. Despite the low quantum efficiency for photochemical activation, the contribution of these photons to the overall catalytic performance is nonetheless significant. In the synergistic process involving both photochemical and photothermal contributions, the Sabatier reaction rate over the Ru/SiNW catalyst was enhanced by a factor of five under photon irradiation within the spectral range of 615 nm < λ < 1100 nm, compared to the rate observed under dark conditions at 95 °C. Remarkably, the reaction rate also exhibited an approximate twofold increase when solely irradiated with NIR photons (850 nm < λ < 1100 nm). Furthermore, isotope labeling experiments utilizing 13CO2 unequivocally verified that the methane product originated from the reduction of CO2 rather than from adventitious carbon sources.
In addition, SiNWs hold significant potential as support materials capable of harvesting solar energy to provide heat to enhance the catalytic performance of various other catalysts immobilized on their surface. To prove this concept, Ozin et al. reported that indium oxide (In2O3) nanoparticles deposited on SiNWs (In2O3/SiNW) effectively catalyzed the reverse water-gas shift reaction (CO2 + H2 → CO + H2O). As shown in Fig. 27H, the yellow-shaded region of the solar spectrum delineates the fraction of solar irradiance capable of photochemically activating the In2O3 nanoparticle catalysts, whereas the red-shaded region corresponds to the portion of solar energy that contributes to heating the catalyst via photothermal effects. Notably, the catalytic reaction can be driven entirely by radiant energy without reliance on an external heating source. Specifically, by increasing the irradiation intensity of the xenon lamp to levels exceeding 15 suns, photons within the ultraviolet and visible spectral ranges initiated photochemical activation of the reaction, while sub-bandgap photons—predominantly in the near-infrared (NIR) region—supplied sufficient energy to elevate the temperature of the SiNW support to approximately 145 °C (Fig. 27I). These studies present an effective strategy for the utilization of low-energy NIR light through the photothermal effect to boost H2-assisted CO2 conversion without the need for external heating.
4.3.2.2. Supra-photothermal effect. Through modulating the composition, structures, and morphology of catalysts, the photothermal effect can be further reinforced, leading to a supra-photothermal effect. For instance, inspired by the greenhouse effect, Ozin et al.156 fabricated porous-SiO2-encapsulated Ni nanocrystals (named Ni@p-SiO2), which mimic the greenhouse gases, to trap IR photons and heat (Fig. 28A and B). Theoretical calculations demonstrate that the presence of the SiO2 shell reduces thermal radiation by 43%, thereby achieving a supra-photothermal effect and resulting in a temperature increase of 60 K for the Ni particles. The local temperature (Tlocal) of Ni@p-SiO2 is estimated to reach nearly 852 K under 2.8 W cm−2 illumination due to the supra-photothermal effect (Fig. 28C), which also contributes to its outstanding CO2RR performance and product selectivity in the photothermal catalytic transformation of CO2 to CO. In nature, animals such as polar bears usually use their fur to resist the cold environment. The fur can prevent heat exchange with the outside world, contributing to reducing the loss of heat. Inspired by this, Zou et al.163 fabricated biomimetic villous carbon frameworks (VCF) which consist of carbon nanotube (CNT) arrays embedded with FeNi3 alloys and carbon frameworks (CF) (Fig. 28D). The hair-like CNT arrays promote the efficient conversion of the energy of the broadband spectrum into heat by restricting heat convection and radiation, thereby achieving a supra-photothermal effect. Recently, leveraging the radiation trapping effect of array structures and the nano-greenhouse effect, He et al.157 prepared SiO2-coated ultrasmall Co nanoparticles (Co-ss@SiO2) on SiO2 nanorod arrays (SiO2 NRAs) (Fig. 28E and F), which exhibit supra-photothermal CO2 methanation activity with an impressive CH4 productivity of 2.3 mol gCo−1 h−1 and a selectivity of nearly 100%. The ultrasmall size of the Co nanoparticles and the silica shell endow Co-ss@SiO2 with greenhouse-like plasmonic superstructures, which remarkably improve the light-trapping capacity and photothermal conversion ability of this composite photocatalyst (Fig. 28G). In addition, the finite-difference time-domain (FDTD) approach demonstrates that the light-induced electric field around Co-ss@SiO2 is firmly confined within the gaps between adjacent Co atoms of Co-ss@SiO2 catalysts, and the peak intensity of the electric field (ΔE/ΔE0) over Co-ss@SiO2 is much higher than that of Co-L@SiO2 (SiO2-coated large Co particles) (Fig. 28H), which is owing to the strong SPR effect of the ultrasmall Co particles. Additionally, the simulated electric field indicates that SiO2 NRAs will promote the light absorption and heat retention abilities of this plasmonic superstructure. Consequently, the supra-photothermal effect contributes to the excellent catalytic performance and selectivity of the Co-ss@SiO2 catalyst. In brief, making full use of the (supra-)photothermal effect induced by low-energy photons holds significant potential in facilitating CO2 photoreduction.
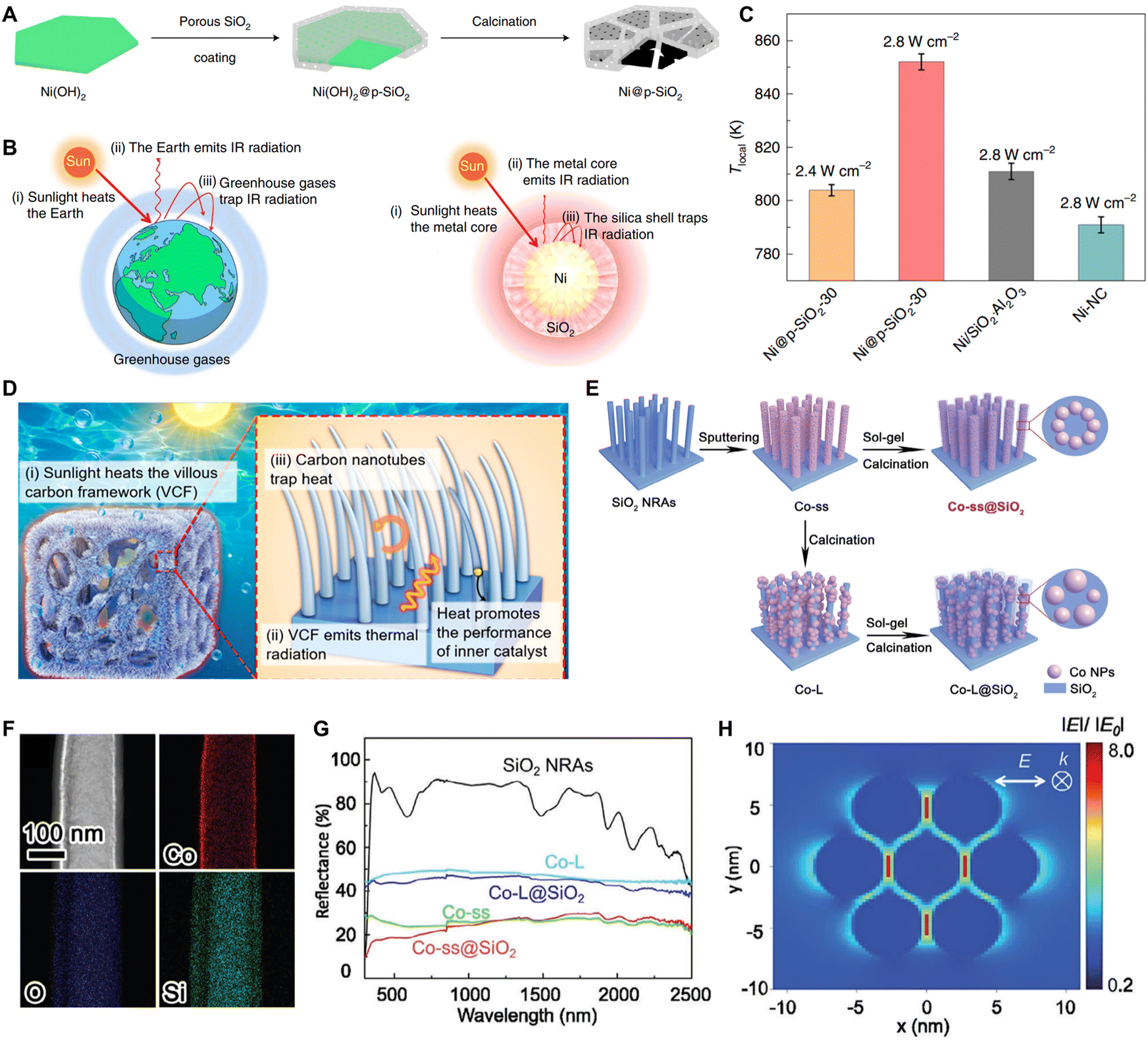 | ||
| Fig. 28 Supra-photothermal effect facilitates NIR-light-driven CO2 reduction. (A) The synthetic route for Ni@p-SiO2. (B) Diagrams of the natural greenhouse effect and supra-photothermal effect. (C) The estimated local temperatures of different catalysts under various irradiation intensities. Reproduced with permission from ref. 156. Copyright 2021, Springer Nature. (D) Schematic illustration of the polar bear hair-like effect for trapping thermal radiation. Reproduced with permission from ref. 163. Copyright 2022, American Chemical Society. (E) Illustration of the synthetic procedure of Co-ss, Co-ss@SiO2, Co-L, and Co-L@SiO2 samples. (F) The morphological analysis and the elemental distribution images of Co-ss@SiO2. (G) DRS spectra of SiO2 NRAs, Co-ss, Co-ss@SiO2, Co-L, and Co-L@SiO2 samples. (H) Simulated electric field distribution around tightly packed small Co particles. Reproduced with permission from ref. 157. Copyright 2023, Wiley-VCH. | ||
5. Summary and outlook
NIR-light-driven CO2 reduction has garnered substantial attention for mitigating climate warming and the energy crisis due to the superiority of NIR light, including deep penetration depth, weak competitive absorption, and photothermal effects. However, developing a photocatalyst with robust NIR light response and desirable conversion efficiency for NIR-light-driven CO2RR is still a formidable task. To tackle this challenge, the key is to design high-efficiency catalysts.In this review, several design strategies for NIR-responsive photocatalysts have been discussed, including energy band structure regulation strategy, energy transfer strategy, and photothermal utilization strategy. Although these strategies exhibit significant advantages in promoting NIR-light-driven CO2RR, they still have certain limitations (Table 4). To overcome these limitations, the photocatalysts for NIR-light-driven CO2RR should be improved according to the following aspects (Fig. 29).
| Design strategy | Photocatalyst type | Characteristics | Limitations |
|---|---|---|---|
| Energy band structure regulation | Narrow-bandgap catalysts | Direct NIR light absorption | (1) Difficult to possess suitable band edge positions |
| (2) Often requiring sacrificial agents | |||
| Intermediate-band catalysts | (1) Direct utilization of broad bandgap semiconductors | (1) Difficult to introduce the intermediate band | |
| (2) Cascaded electron transitions | (2) Often low efficiency | ||
| Metallic catalysts | (1) Zero band gap | Severe carrier recombination | |
| (2) Superb IR light absorption | |||
| (3) High carrier concentration | |||
| Heterojunction catalysts | (1) Multiple narrow bandgap components | Complex to construct | |
| (2) Effective charge separation | |||
| Energy transfer strategy | Pure SPR catalysts | (1) Local electric field manipulation | (1) Reliance on noble metals |
| (2) Hot-electron transfer | (2) Difficult to possess suitable band edge positions | ||
| SPR-semiconductor composites | (1) Local electric field manipulation | (1) Reliance on noble metals | |
| (2) Hot-electron transfer | (2) Matching between SPR components and semiconductors | ||
| (3) Energy transfer between SPR components with semiconductors | |||
| (4) Effective charge separation | |||
| Up-conversion catalysts | (1) Transforming low-energy photons into high-energy photons | (1) Low up-conversion quantum yield | |
| (2) Tunable emission wavelength | (2) Reliance on specific wavelength lasers | ||
| Photothermal utilization strategy | (Supra-)photothermal catalysts | (1) (Supra-)photothermal effect | Commonly limited by photothermal conversion efficiency |
| (2) Photothermal synergy |
 | ||
| Fig. 29 Future directions to advance the NIR-light-driven CO2RR system. | ||
5.1. Prospects for the next NIR-photocatalytic CO2 reduction systems
At present, converting CO2 into multi-carbon (C2+) products using NIR light is hindered by poor efficiency and productivity. Therefore, it is crucial to address strategic factors in designing the next generation of NIR-responsive catalysts to enhance the production of drop-in fuels through the CO2RR process: (1) developing a “more-in-one” NIR system: the efficiencies of single-component catalysts are often limited by factors such as unsuitable electronic structures, low carrier concentrations, weak CO2 adsorption, poor NIR-light absorption, etc. To overcome these challenges, it is urgent to design novel multi-component NIR photocatalysts. Integrating cocatalysts, NIR light absorbers, and other functional components into a single system may effectively enhance the NIR light absorption and boost the PCO2R efficiency. For example, incorporating UCNPs or plasmonic nanoparticles into photocatalysts can increase their ability to harvest low-energy NIR photons. Additionally, well-ordered redox-active architectures can improve charge transfer kinetics and the efficiency of surface redox reactions. Therefore, developing “more-in-one” NIR photocatalysts with spatially arranged and composition-tunable bandgaps is a compelling strategy to enhance SCC. (2) Constructing electron-rich sites: designing structures that provide electron-rich reactive sites is beneficial for the deep reduction of CO2 into high-value chemicals. In this regard, materials with metallic properties are particularly effective, as they not only enhance NIR light absorption but also demonstrate excellent activity in converting CO2 into C2+ compounds. These metallic materials, usually with zero energy gaps, are highly effective in NIR light utilization and facilitating multiple electron/proton transfer for selective C–C coupling, even under low CO2 concentration conditions. (3) Increasing local CO2 concentration: increasing the local concentration of CO2 near active sites has been shown to improve multi-carbon product selectivity.164 Enhancing CO2 adsorption, in turn, accelerates CO2 reduction kinetics. Various catalyst modification techniques, such as defect engineering, integration with porous framework materials, and the construction of Lewis acid/base sites, can be employed to elevate local CO2 concentrations around the active sites. (4) Quantify light and thermal contributions: to further optimize NIR-light-driven CO2RR, it is essential to disclose the photothermal mechanism by quantifying the contributions of both light and heat. Cooperatively, the photoinduced carriers and the local thermal effect play pivotal roles in driving the CO2RR process. The photothermal synergy may help steer the reactions by lowering the energy barriers for CO2RR and lead to a greater apparent catalytic efficiency in the photothermal system than the sum of photochemistry and thermochemistry. NIR light, rather than UV or visible light, offers strong photothermal conversion efficiency, especially when using photocatalysts with inherent photothermal conversion capabilities.However, accurately measuring the actual surface/local temperature of photocatalysts remains challenging, often leading to unreasonable evaluation of the contributions from photocatalytic and thermal catalytic processes.165 To make reasonable evaluations of the photothermal effect, advanced techniques for accurate reaction temperature measurements should be developed. Additionally, control experiments related to temperatures and light are required simultaneously.
5.2. Developing advanced techniques
Nowadays, the progress of NIR-light-driven CO2 reduction is very slow, due to a lack of understanding of its mechanism. To push the development of this field, advanced techniques are supposed to be explored to gain a deep insight into the structure–activity relationships of photocatalysts. Advanced characterization techniques can monitor the variations of CO2 and the photocatalysts, revealing the real CO2 reduction process on the catalyst surface. To make a better comprehension of NIR-light-driven CO2 reduction, it is recommended to develop advanced techniques in the following aspects (Fig. 30). | ||
| Fig. 30 Prospects of advanced characterization techniques for NIR-light-driven CO2 reduction. | ||
Overall, the progress of NIR-light-driven CO2 reduction requires the advancement of various characterization techniques, and these advanced techniques can offer a guidance for designing reasonable photocatalysts and photocatalytic systems.
5.3. Making a reasonable evaluation of NIR-light-driven CO2RR performance
Recent studies have revealed that some published research claims on NIR-light-driven CO2RR and its catalytic mechanisms may be thermodynamically unfeasible.91,92,174 These errors might arise from a limited understanding of NIR-light-driven CO2RR, improper experimental methods, overlooking key factors in the analysis, or carbon contamination interference. Any mistakes can lead to a range of issues, such as prolonged experimental durations, excessive resource wastes, incorrect interpretations of structure–activity relationships, and erroneous conclusions. Thus, to obtain reliable experimental results, researchers must critically assess the reported literature and perform experiments with meticulous care. In this section, two main components for promoting best practices toward reliable NIR-driven CO2RR are presented, namely general steps and key considerations (Fig. 31). | ||
| Fig. 31 General steps for reliable NIR-light-driven CO2 reduction. | ||
As shown in Fig. 31, step-by-step guides regarding catalyst preparation, experimental system building, performance evaluation, and mechanism investigation are summarized. When preparing catalysts, the key is to design catalysts that can effectively respond to NIR light, promote carrier separation, and facilitate CO2RR. The process typically involves several steps, including synthesis, basic structural and morphological characterization, and optical property analysis. Depending on their efficiency in capturing NIR light, the synthesized catalysts can be narrow-bandgap materials, or they can be functionally assembled components. Then, it is feasible to conduct some basic characterization, such as morphology, structure, and optical properties, which lays the foundation for understanding the characteristics of the catalyst and further elucidating its chemical state and electronic structure.
To build an experimental system, a suitable reactor, NIR light sources with different intensities, solvents with or without additives, gaseous CO2, and the designed photocatalyst must be properly assembled for the next step of performance testing. In this system setup, key operational details such as irradiation intensities and wavelengths, reaction temperature, CO2 feed pressure and concentration, cocatalyst presence, catalyst dosage, reaction time, and solvent pH are crucial for optimizing CO2 conversion processes. Precise control over these factors ensures experimental consistency, which is critical for reliable and robust fundamental research. Once these parameters are established, the photocatalytic CO2 reduction performance can then be evaluated.
For performance evaluation, important metrics include SCC, CO2 conversion efficiency, product selectivity, production rate, quantum efficiency, and stability of the photosynthetic system under NIR light. These metrics offer deep insights into efficiency benchmarking, enabling the comparison of state-of-the-art results across different photocatalysts.
The mechanistic investigation primarily focuses on interfacial charge behaviors and reaction pathways. In this step, detailed studies using excited-state spectroscopic techniques, such as TAS and time-resolved photoluminescence (TRPL), are essential for understanding the charge-carrier dynamics in semiconductors and identifying key efficiency-limiting factors in the possible mechanism under NIR light excitation. Moreover, advanced experimental methods like in situ/operando XPS, XAS, and RAIRS are essential for detecting and differentiating the information at the reaction interfaces. Additionally, modeling studies using DFT calculations are instrumental in exploring and understanding the structure–property–performance relationship to achieve high photocatalytic performance.
To enhance the reliability of NIR-light-driven CO2RR results, three key aspects should be considered. Firstly, before conducting experiments, it is crucial to fully understand the complexities associated with NIR-light-driven CO2RR processes and recognize the factors that can interfere with the results. Proper planning is essential to ensure that the experiment is carried out with care and consideration. The complexity arises from four main factors: (1) thermodynamic and kinetic complexity: long-wavelength light-activatable CO2RR has inherent challenges. The CO bond in CO2 is chemically inert, necessitating a huge input of energy to break it.4,23 The CO2RR process involves multiple steps with numerous electron/proton transfers, leading to various intermediates that may recombine at different stages. (2) Interaction of photo-reduction and -oxidation reactions: the interaction between the paired reactions adds additional complexity to the research. (3) Influence of various parameters: the reactivity is highly sensitive to parameters such as the type of photocatalyst, the choice of reaction device, pH, and the addition of cocatalysts and sacrificial agents (if used).23 (4) Challenges of excluding carbon contamination: because of the low yield of products in NIR-light-driven CO2RR, carbon contamination can lead to indistinguishable false and true positive results.92
It is widely acknowledged that understanding the thermodynamic and kinetic factors, along with their mechanisms, has always been a key challenge in PCO2R research. Thus, the first key consideration in performing NIR-light-driven CO2RR is to evaluate the thermodynamic and kinetic feasibility of the system. This can be estimated by identifying catalysts’ components, determining the energy band structure, evaluating charge transfer efficiency, and measuring carrier lifetimes. We have learned much from conventional H2-assisted CO2 reduction in terms of catalyst design. However, different catalysts used in photocatalytic systems may follow distinct mechanisms, which require specific thermodynamic forces to drive the reactions accordingly. Thus, to successfully achieve NIR-light-driven CO2RR, the catalysts’ components—including substrates, semiconductors, cocatalysts, and other light-absorbing components—must first be carefully checked. For example, photocatalysis in conventional semiconductor-based systems typically involves photon absorption, which requires the energy of incident light to be higher than the band gaps.18,62 The band edge positions of conventional semiconductors should be adjusted to provide sufficient thermodynamic driving force, allowing the paired reactions to occur concurrently. For another example, SPR-mediated catalysis (see Section 4.2.1) differs from conventional semiconductor-based systems, which cannot drive reactions without sufficient photon energy.98,175 Quite differently, the SPR-induced highly energetic hot electrons in plasmonic materials can directly drive redox reactions. The generated hot carriers are engaged in these processes including photo/thermo-conversion, and then may boost the reaction with CO2 under the lower thermodynamic driving force. For example, Hu et al. demonstrated that Au rod@CuPd core–shell composites have achieved CO2 reduction under near-infrared light illumination (800 nm, ca. 1.55 eV). A key issue in this study is that, without a sacrificial agent, the low-energy photons are insufficient to overcome the reaction barrier of CO2RR. To address this issue and better understand how low-energy photons interact with catalysts and CO2, the research team employed in situ XPS to study SPR-mediated light–matter interactions within the reaction system.59
Another important consideration is the exclusion of carbon contamination.91,92,176,177 The process of eliminating carbon contamination throughout the experiment involves lots of details. Carbon contamination from organic reagents present in reactors, lab coats, low-purity gases, etc., can be eliminated through rigorous cleaning procedures. Carbon contamination on the surface of photocatalysts, originating from solvents, reactants, and surfactants used during synthesis, can be mitigated by employing suitable treatment methods: washing the catalysts with ethanol/water, annealing the catalysts under high-temperature conditions, and using Ar or O2 plasma to clean the surfaces of catalysts. Carbon contamination originating from the light-induced self-degradation of carbon-containing catalysts can be eliminated or recognized by selecting stable carbon-based photocatalysts, using low-energy photon-driven systems, and conducting structural stability characterization before and after reactions. Further, carbon contamination can also be eliminated via blank/control experiments, long-term performance tests, and isotope labeling experiments. These solutions help enhance data reliability and avoid the false-positive results.
The third consideration is the identification of the catalytic sites as well as reaction mechanisms. The distribution, numbers, and nature of these active sites directly influence the overall performance of the catalyst. Identifying these active sites is crucial for understanding the parameters that determine activity and for uncovering potential catalytic mechanisms. This review does not delve further into this aspect.
Inspired by the above general steps and key considerations for conducting NIR-light-driven CO2RR studies, we hope this section also serves as a valuable reference for conducting PCO2R experiments that utilize the full spectrum of sunlight. A dependable breakthrough in the field of PCO2R can establish a robust basis for both fundamental studies and industrial applications.
5.4. Increasing the feasibilities in practical application for CO2 capture
In this section, the considerations for scaling up laboratory PCO2R to the industrial level are discussed, along with the relevant application examples. Current systems of artificial photosynthesis are far from meeting the requirements for practical application, because of their low efficiency (<1%) and productivity.23 To break through the low-efficiency bottleneck of the PCO2R system and improve its potential for practical application, it is crucial to consider the future strategic design of the PCO2R system. This includes many specific considerations for scaling up PCO2R-based applications or integrating them as part of a chain partnership with existing applications in the industry (Fig. 32): (i) regulating the paired half-reactions in NIR-light-driven CO2 reduction systems,20 (ii) using CO2 as a building block in direct organic synthesis,73 (iii) making full use of the photothermal effect to enhance CO2 conversion via the integration of CO2 conversion with existing high-temperature catalytic reactions, including Sabatier reaction,162 reverse water gas shift (RWGS) reaction,178 reverse Boudouard (RB) reaction,179 and (iv) engineering cascaded CO2 conversion systems.180 | ||
| Fig. 32 Future design directions of CO2 chemical utilization systems driven by NIR light. | ||
O bonds in CO2 molecules (750 kJ mol−1) limits the efficient conversion of CO2. Fortunately, many other chemical bonds possess much lower bond energies than that of CO in CO2 (e.g., C–H: 430 kJ mol−1, C–C: 336 kJ mol−1, C–O: 327 kJ mol−1).190,191 Introducing CO2 molecules into the activation processes of these bonds helps lower the activation barriers of CO2 and promote its conversion into fine chemicals. In other words, CO2 can be a building block for photocatalytic synthesis of organic compounds. In this approach, CO2 can be utilized from two main perspectives: (i) conducting an organic chemical reaction that involves CO2 as a reactant and (ii) conducting the catalyst synthesis that consumes CO2. As shown in Fig. 32, conducting an organic chemical reaction that involves CO2 as a reactant is a feasible approach for CO2 utilization.192 For instance, using NIR as the driving energy, the synthesis of cyclic carbonates from the cycloaddition of CO2 with epoxides has been successfully conducted on multicomponent catalysts, demonstrating the effectiveness of using NIR photons.73 From the perspective of sustainable chemistry, the onsite NIR-driven organic photoredox catalysis involving CO2 conversion becomes an extendable routine that consumes this greenhouse gas. The second direction is to synthesize the catalysts that consume CO2. Recently, it has been reported that CO2 can be inserted into the terminal alkynes or N–H bonds to form carboxylate ligands, which can then be used to prepare CO2-based MOFs.193,194 It should be noted that these CO2-based MOFs require a synthetic temperature of 25–70 °C. Therefore, because of the photothermal effect, NIR light can be utilized as an easily available heat source to elevate the reaction temperature to facilitate the synthesis of CO2-based MOFs, further promoting CO2 capture. In short, with the unique photothermal effect of NIR photons, CO2 can be involved in various organic reactions, consumed in the synthesis of functional catalysts, and applied in other applications. These developments in CO2 fixation are regarded as potential pathways to realize solar energy utilization and environmental sustainability.
:1 mole ratio by photocatalysis.198 Bi2S3/CdXZn1−XS catalysts have demonstrated their extremely high activity for the production of H2 and CO (33.10 and 32.11 μmol g−1 h−1).158 Being a mature route, syngas (a mixture of H2 and CO in different ratios) has been widely used as a reactant in the FTS procedures.4 Thus, more efforts should be devoted to the direct combination of the PCO2R and FTS technologies to scale up the solar-driven CO2 reforming. In another cascaded CO2 conversion system, CO can be utilized as a reactant in carbonylation reactions. For instance, during the carbonylation process to synthesize diethyltoluamide, the CO conversion rate can reach more than 85%. At the same time, these cascaded conversion approaches also provide easy and affordable strategies for labeling organic molecules with 13C isotopes. The successful achievement of cascaded reactions offers an important and feasible way for upgrading PCO2R products into fine products.180 We believe that, by ingeniously designing the cascaded catalytic systems, more exciting discoveries leading to practical and scalable CO2 chemical utilization systems driven by NIR light are on the horizon.
In summary, various significant advances and breakthroughs in NIR-light-driven CO2 reduction have been realized over the past decade, making a step towards the full utilization of sunlight for CO2RR. Several strategies, including energy band structure regulation strategy, energy transfer strategy, and photothermal utilization strategy, have been explored for synthesizing NIR-photocatalysts. Many NIR-photocatalysts fabricated using these strategies have expanded the spectral absorption range of the photocatalytic systems from the UV to the NIR wavebands, while simultaneously possessing suitable energy band structures for CO2RR and its paired half-reaction. However, the efficiency of PCO2R under NIR irradiation is still not satisfactory and falls short of requirements for practical applications. One of the main problems is the severe carrier recombination during the PCO2R process. To enhance the charge separation efficiency, strategies such as heterojunction engineering and energy transfer strategies are recommended for catalyst design. To enhance the PCO2R activity and product selectivity, we have also outlined potential directions for the next generation of NIR-light-responsive CO2RR systems. Additionally, the lack of deep understanding of the mechanisms underlying NIR-light-driven CO2RR limits the development of this field. Meanwhile, carbon contamination resulting from the photodegradation of organic additives can further complicate the mechanism. Thus, it is essential to explore advanced characterization methods and establish a reasonable workflow to avoid false-positive results and deepen the understanding of NIR-light-driven CO2RR. To increase the feasibility of PCO2R in practical applications, a promising approach is to combine the downstream products of PCO2R with other valuable reactions to achieve cascaded conversion of CO2. Besides, the economic value of the photocatalytic system can be effectively increased by coupling the CO2 reduction process with value-added paired half-reactions. Overall, the study of NIR-light-driven CO2 reduction is still in its early stage despite progress in recent years. As NIR-photocatalytic CO2 reduction is continuously developing, there is no doubt that it will find applications in diverse fields, such as biological therapy, photoelectrocatalysis, and organic synthesis. Hence, selecting suitable application scenarios and continuously developing NIR-photocatalysts will be crucial in advancing CO2 photoconversion toward industrial applications.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22105133), Natural Science Foundation of Sichuan Province of China (2023NSFSC1085), China Scholarship Council, Science and Technology Project of the State Administration for Market Regulation (2022MK111), Fundamental Research Funds for the Central Universities, and the Creative Research Initiative (CRI, RS-2023-00221668) program through the National Research Foundation (NRF) of Korea.References
- C. F. Shih, T. Zhang, J. Li and C. Bai, Joule, 2018, 2, 1925–1949 CrossRef CAS.
- Thoning: Trends in globally-averaged CO2 determined from NOAA Global Monitoring Laboratory measurements DOI:10.15138/9N0H-ZH07, (accessed May 2024).
- Climate at a Glance: Global Time Series, https://www.ncei.noaa.gov/access/monitoring/climate-ata-glance/global/time-series, (accessed May 2024).
- S. Fang and Y. H. Hu, Chem. Soc. Rev., 2022, 51, 3609–3647 RSC.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef CAS.
- S. Mori, W.-C. Xu, T. Ishidzuki, N. Ogasawara, J. Imai and K. Kobayashi, Appl. Catal., A, 1996, 137, 255–268 CrossRef CAS.
- I.-Y. Jeon, Y.-R. Shin, G.-J. Sohn, H.-J. Choi, S.-Y. Bae, J. Mahmood, S.-M. Jung, J.-M. Seo, M.-J. Kim, D. Wook Chang, L. Dai and J.-B. Baek, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5588–5593 CrossRef CAS.
- S. Bhattacharjee, M. Rahaman, V. Andrei, M. Miller, S. Rodríguez-Jiménez, E. Lam, C. Pornrungroj and E. Reisner, Nat. Synth., 2023, 2, 182–192 CrossRef CAS.
- J. Zhu, G. Zhou, Y. Tong, L. Chen and P. Chen, Adv. Funct. Mater., 2024, 2420177 Search PubMed.
- H. Wang, Y. Tong and P. Chen, Nano Energy, 2023, 118, 108967 CrossRef CAS.
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato, R. Haasch, P. Zapol, B. Kumar, R. F. Klie, J. Abiade, L. A. Curtiss and A. Salehi-Khojin, Science, 2016, 353, 467–470 CrossRef CAS PubMed.
- J. Tian, J. Yu, Q. Tang, J. Zhang, D. Ma, Y. Lei and Z.-T. Li, Mater. Futures, 2022, 1, 042104 CrossRef CAS.
- S. Wang, X. Han, Y. Zhang, N. Tian, T. Ma and H. Huang, Small Struct., 2021, 2, 2000061 CrossRef CAS.
- L. K. Putri, B.-J. Ng, W.-J. Ong, S.-P. Chai and A. R. Mohamed, Adv. Energy Mater., 2022, 12, 2201093 CrossRef CAS.
- Y. Li, M. Wen, Y. Wang, G. Tian, C. Wang and J. Zhao, Angew. Chem., Int. Ed., 2021, 60, 910–916 CrossRef CAS PubMed.
- J.-Y. Zeng, X.-S. Wang, B.-R. Xie, Q.-R. Li and X.-Z. Zhang, J. Am. Chem. Soc., 2022, 144, 1218–1231 CrossRef CAS PubMed.
- Y. Wang and T. He, J. Mater. Chem. A, 2021, 9, 87–110 RSC.
- Q. Hu, Z. Zhang, D. He, J. Wu, J. Ding, Q. Chen, X. Jiao and Y. Xie, J. Am. Chem. Soc., 2024, 146, 16950–16962 CrossRef CAS PubMed.
- M. Halmann, Nature, 1978, 275, 115–116 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- S. Fang, M. Rahaman, J. Bharti, E. Reisner, M. Robert, G. A. Ozin and Y. H. Hu, Nat. Rev. Methods Primers, 2023, 3, 61 CrossRef CAS.
- J. Xu, Z. Ju, W. Zhang, Y. Pan, J. Zhu, J. Mao, X. Zheng, H. Fu, M. Yuan, H. Chen and R. Li, Angew. Chem., Int. Ed., 2021, 60, 8705–8709 CrossRef CAS PubMed.
- S. Yang, W. J. Byun, F. Zhao, D. Chen, J. Mao, W. Zhang, J. Peng, C. Liu, Y. Pan, J. Hu, J. Zhu, X. Zheng, H. Fu, M. Yuan, H. Chen, R. Li, M. Zhou, W. Che, J.-B. Baek, J. S. Lee and J. Xu, Adv. Mater., 2024, 36, 2312616 CrossRef CAS PubMed.
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS.
- S. Bai, W. Jing, G. He, C. Liao, F. Wang, Y. Liu and L. Guo, ACS Nano, 2023, 17, 10976–10986 CrossRef CAS PubMed.
- L. Hurtado, A. Mohan, U. Ulmer, R. Natividad, A. A. Tountas, W. Sun, L. Wang, B. Kim, M. M. Sain and G. A. Ozin, Chem. Eng. J., 2022, 435, 134864 CrossRef CAS.
- W. Ma, J. Sun, S. Yao, Y. Wang, G. Chen, G. Fan and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202313784 CrossRef CAS PubMed.
- X. Shi, Y. Huang, Y. Bo, D. Duan, Z. Wang, J. Cao, G. Zhu, W. Ho, L. Wang, T. Huang and Y. Xiong, Angew. Chem., Int. Ed., 2022, 61, e202203063 CrossRef CAS PubMed.
- X. Zu, Y. Zhao, X. Li, R. Chen, W. Shao, L. Li, P. Qiao, W. Yan, Y. Pan, Q. Xu, J. Zhu, Y. Sun and Y. Xie, Angew. Chem., Int. Ed., 2022, 62, e202215247 CrossRef PubMed.
- Y. Shen, C. Ren, L. Zheng, X. Xu, R. Long, W. Zhang, Y. Yang, Y. Zhang, Y. Yao, H. Chi, J. Wang, Q. Shen, Y. Xiong, Z. Zou and Y. Zhou, Nat. Commun., 2023, 14, 1117 CrossRef CAS PubMed.
- C. Gao, S. Chen, Y. Wang, J. Wang, X. Zheng, J. Zhu, L. Song, W. Zhang and Y. Xiong, Adv. Mater., 2018, 30, 1704624 CrossRef PubMed.
- A. Mavridi-Printezi, A. Menichetti, M. Guernelli and M. Montalti, Nanoscale, 2021, 13, 9147–9159 RSC.
- H. Kumagai, Y. Tamaki and O. Ishitani, Acc. Chem. Res., 2022, 55, 978–990 CrossRef CAS PubMed.
- F. Yu, X. Jing, Y. Wang, M. Sun and C. Duan, Angew. Chem., Int. Ed., 2021, 60, 24849–24853 CrossRef CAS PubMed.
- L. Ye, H. Wang, X. Jin, Y. Su, D. Wang, H. Xie, X. Liu and X. Liu, Sol. Energy Mater. Sol. Cells, 2016, 144, 732–739 CrossRef CAS.
- T. Arai, S. Sato and T. Morikawa, Energy Environ. Sci., 2015, 8, 1998–2002 RSC.
- B. Tahir, M. Tahir and N. S. Amin, Appl. Surf. Sci., 2015, 338, 1–14 CrossRef CAS.
- J. C. S. Wu, H.-M. Lin and C.-L. Lai, Appl. Catal., A, 2005, 296, 194–200 CrossRef CAS.
- T.-V. Nguyen and J. C. S. Wu, Appl. Catal., A, 2008, 335, 112–120 CrossRef CAS.
- X. Ji, R. Guo, J. Tang, Y. Miao, Z. Lin, L. Hong, Y. Yuan, Z. Li and W. Pan, ACS Appl. Energy Mater., 2022, 5, 2862–2872 CrossRef CAS.
- J. Wang, T. Bo, B. Shao, Y. Zhang, L. Jia, X. Tan, W. Zhou and T. Yu, Appl. Catal., B, 2021, 297, 120498 CrossRef CAS.
- S. Si, H. Shou, Y. Mao, X. Bao, G. Zhai, K. Song, Z. Wang, P. Wang, Y. Liu, Z. Zheng, Y. Dai, L. Song, B. Huang and H. Cheng, Angew. Chem., Int. Ed., 2022, 61, e202209446 CrossRef CAS PubMed.
- S. Chakraborty, R. Das, M. Riyaz, K. Das, A. K. Singh, D. Bagchi, C. P. Vinod and S. C. Peter, Angew. Chem., Int. Ed., 2023, 62, e202216613 CrossRef CAS PubMed.
- L. Zhao, J. Bian, X. Zhang, L. Bai, L. Xu, Y. Qu, Z. Li, Y. Li and L. Jing, Adv. Mater., 2022, 34, 2205303 CrossRef CAS PubMed.
- H. Ou, S. Ning, P. Zhu, S. Chen, A. Han, Q. Kang, Z. Hu, J. Ye, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202206579 CrossRef CAS PubMed.
- Q. Liu, Z. Chen, W. Tao, H. Zhu, L. Zhong, F. Wang, R. Zou, Y. Lei, C. Liu and X. Peng, J. Mater. Chem. A, 2020, 8, 11761–11772 RSC.
- R. Xu, D.-H. Si, S.-S. Zhao, Q.-J. Wu, X.-S. Wang, T.-F. Liu, H. Zhao, R. Cao and Y.-B. Huang, J. Am. Chem. Soc., 2023, 145, 8261–8270 CrossRef CAS PubMed.
- X. Li, L. Li, G. Chen, X. Chu, X. Liu, C. Naisa, D. Pohl, M. Löffler and X. Feng, Nat. Commun., 2023, 14, 4034 CrossRef CAS PubMed.
- S. Wang, Z. Ding and X. Wang, Chem. Commun., 2015, 51, 1517–1519 RSC.
- J. Zhu, W. Shao, X. Li, X. Jiao, J. Zhu, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2021, 143, 18233–18241 CrossRef CAS PubMed.
- X. Chen, Y. Zhou, Q. Liu, Z. Li, J. Liu and Z. Zou, ACS Appl. Mater. Interfaces, 2012, 4, 3372–3377 CrossRef CAS PubMed.
- J. Di, C. Chen, C. Zhu, P. Song, M. Duan, J. Xiong, R. Long, M. Xu, L. Kang, S. Guo, S. Chen, H. Chen, Z. Chi, Y.-X. Weng, H. Li, L. Song, M. Wu, Q. Yan, S. Li and Z. Liu, Nano Energy, 2021, 79, 105429 CrossRef CAS.
- B. Su, Y. Kong, S. Wang, S. Zuo, W. Lin, Y. Fang, Y. Hou, G. Zhang, H. Zhang and X. Wang, J. Am. Chem. Soc., 2023, 145, 27415–27423 CrossRef CAS PubMed.
- L. Liang, X. Li, J. Zhang, P. Ling, Y. Sun, C. Wang, Q. Zhang, Y. Pan, Q. Xu, J. Zhu, Y. Luo and Y. Xie, Nano Energy, 2020, 69, 104421 CrossRef CAS.
- X. Li, L. Liang, Y. Sun, J. Xu, X. Jiao, X. Xu, H. Ju, Y. Pan, J. Zhu and Y. Xie, J. Am. Chem. Soc., 2019, 141, 423–430 CrossRef CAS PubMed.
- C. Chen, C. Ye, X. Zhao, Y. Zhang, R. Li, Q. Zhang, H. Zhang and Y. Wu, Nat. Commun., 2024, 15, 7825 CrossRef CAS PubMed.
- C. Hu, X. Chen, J. Low, Y.-W. Yang, H. Li, D. Wu, S. Chen, J. Jin, H. Li, H. Ju, C.-H. Wang, Z. Lu, R. Long, L. Song and Y. Xiong, Nat. Commun., 2023, 14, 221 CrossRef CAS PubMed.
- M. Yu, X. Lv, A. Mahmoud Idris, S. Li, J. Lin, H. Lin, J. Wang and Z. Li, J. Colloid Interface Sci., 2022, 612, 782–791 CrossRef CAS PubMed.
- L.-Y. Lin, S. Kavadiya, B. B. Karakocak, Y. Nie, R. Raliya, S. T. Wang, M. Y. Berezin and P. Biswas, Appl. Catal., B, 2018, 230, 36–48 CrossRef CAS.
- L. Liang, X. Li, Y. Sun, Y. Tan, X. Jiao, H. Ju, Z. Qi, J. Zhu and Y. Xie, Joule, 2018, 2, 1004–1016 CrossRef CAS.
- C. Liao, Z. He, F. Wang, Y. Liu and L. Guo, ACS Nano, 2024, 18, 35480–35489 CrossRef CAS PubMed.
- X. Wu, W. Zhang, J. Li, Q. Xiang, Z. Liu and B. Liu, Angew. Chem., Int. Ed., 2023, 62, e202213124 CrossRef CAS PubMed.
- F. Zhuang, L. Jing, H. Xiang, C. Li, B. Lu, L. Yan, J. Wang, Y. Chen and B. Huang, Adv. Sci., 2024, 11, 2402256 CrossRef CAS PubMed.
- X. Xu, C. Randorn, P. Efstathiou and J. T. S. Irvine, Nat. Mater., 2012, 11, 595–598 CrossRef CAS PubMed.
- G. Wang, B. Huang, X. Ma, Z. Wang, X. Qin, X. Zhang, Y. Dai and M.-H. Whangbo, Angew. Chem., Int. Ed., 2013, 52, 4810–4813 CrossRef CAS PubMed.
- X. Y. Kong, W. L. Tan, B.-J. Ng, S.-P. Chai and A. R. Mohamed, Nano Res., 2017, 10, 1720–1731 CrossRef CAS.
- Y. L. Wang, T. Nie, Y. H. Li, X. L. Wang, L. R. Zheng, A. P. Chen, X. Q. Gong and H. G. Yang, Angew. Chem., Int. Ed., 2017, 56, 7430–7434 CrossRef CAS PubMed.
- J. Li, X. Wu, W. Pan, G. Zhang and H. Chen, Angew. Chem., Int. Ed., 2018, 57, 491–495 CrossRef CAS PubMed.
- H. Jia, A. Du, H. Zhang, J. Yang, R. Jiang, J. Wang and C. Zhang, J. Am. Chem. Soc., 2019, 141, 5083–5086 CrossRef CAS PubMed.
- J. Li, W. Pan, Q. Liu, Z. Chen, Z. Chen, X. Feng and H. Chen, J. Am. Chem. Soc., 2021, 143, 6551–6559 CrossRef CAS PubMed.
- X. Chen, M. Wei, A. Yang, F. Jiang, B. Li, O. A. Kholdeeva and L. Wu, ACS Appl. Mater. Interfaces, 2022, 14, 5194–5202 CrossRef CAS PubMed.
- Y. Cai, F. Luo, Y. Guo, F. Guo, W. Shi and S. Yang, Molecules, 2023, 28, 2142 CrossRef CAS PubMed.
- V. R. Nair and V. S. Kodialbail, Environ. Sci. Pollut. Res., 2020, 27, 14441–14453 CrossRef CAS PubMed.
- Y. Pan, X. Yuan, L. Jiang, H. Wang, H. Yu and J. Zhang, Chem. Eng. J., 2020, 384, 123310 CrossRef CAS.
- C. Han, B. K. Kundu, Y. Liang and Y. Sun, Adv. Mater., 2024, 36, 2307759 CrossRef CAS PubMed.
- R. Chen, K. Qiu, D. C. Y. Leong, B. K. Kundu, C. Zhang, P. Srivastava, K. E. White, G. Li, G. Han, Z. Guo, C. G. Elles, J. Diao and Y. Sun, Biosens. Bioelectron., 2023, 239, 115604 CrossRef CAS PubMed.
- B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343–346 CrossRef CAS PubMed.
- B. K. Kundu, C. Han, P. Srivastava, S. Nagar, K. E. White, J. A. Krause, C. G. Elles and Y. Sun, ACS Catal., 2023, 13, 8119–8127 CrossRef CAS.
- A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef CAS PubMed.
- Pragti, B. K. Kundu, R. Chen, J. Diao and Y. Sun, Adv. Healthcare Mater., 2025, 14, 2403272 CrossRef CAS PubMed.
- X. Zhao, S. He, W. Chi, X. Liu, P. Chen, W. Sun, J. Du, J. Fan and X. Peng, Adv. Sci., 2022, 9, 2202885 CrossRef CAS PubMed.
- X. Wang, F. Wang, Y. Sang and H. Liu, Adv. Energy Mater., 2017, 7, 1700473 CrossRef.
- D. C. Cabanero and T. Rovis, Nat. Rev. Chem., 2025, 9, 28–45 CrossRef CAS PubMed.
- B. Kumar Kundu, N. Bashar, P. Srivastava, C. G. Elles and Y. Sun, Chem. – Eur. J., 2024, 30, e202402856 CrossRef CAS PubMed.
- B. K. Kundu, G. Han and Y. Sun, J. Am. Chem. Soc., 2023, 145, 3535–3542 CrossRef CAS PubMed.
- J. Hou, Z. Wang, W. Kan, S. Jiao, H. Zhu and R. V. Kumar, J. Mater. Chem., 2012, 22, 7291–7299 RSC.
- J. Dankar, V. Rouchon, C. Pagis, M. Rivallan and M. El-Roz, Inorg. Chem. Front., 2023, 10, 7155–7166 RSC.
- Y. Liu, D. Shen, Q. Zhang, Y. Lin and F. Peng, Appl. Catal., B, 2021, 283, 119630 CrossRef CAS.
- J. You, M. Xiao, S. Liu, H. Lu, P. Chen, Z. Jiang, W. Shangguan, Z. Wang and L. Wang, J. Mater. Chem. A, 2023, 11, 10149–10154 RSC.
- Y. Zhang, D. Yao, B. Xia, M. Jaroniec, J. Ran and S.-Z. Qiao, ACS Energy Lett., 2022, 7, 1611–1617 CrossRef CAS.
- A. M. Alotaibi, S. Sathasivam, B. A. D. Williamson, A. Kafizas, C. Sotelo-Vazquez, A. Taylor, D. O. Scanlon and I. P. Parkin, Chem. Mater., 2018, 30, 1353–1361 CrossRef CAS.
- J. Wang, Y. Li, L. Deng, N. Wei, Y. Weng, S. Dong, D. Qi, J. Qiu, X. Chen and T. Wu, Adv. Mater., 2017, 29, 1603730 CrossRef PubMed.
- M. Sun, B. Zhao, F. Chen, C. Liu, S. Lu, Y. Yu and B. Zhang, Chem. Eng. J., 2021, 408, 127280 CrossRef CAS.
- F. Fresno, A. Iglesias-Juez and J. M. Coronado, Top. Curr. Chem., 2023, 381, 21 CrossRef CAS PubMed.
- M. Evstigneev, in Introduction to Semiconductor Physics and Devices, ed. M. Evstigneev, Springer International Publishing, Cham, 2022, pp. 171–196 Search PubMed.
- X. Meng, L. Liu, S. Ouyang, H. Xu, D. Wang, N. Zhao and J. Ye, Adv. Mater., 2016, 28, 6781–6803 CrossRef CAS PubMed.
- S. Yang, W. Dai, W. Zheng and J. Wang, Coord. Chem. Rev., 2023, 475, 214913 CrossRef CAS.
- J. Kim, J.-A. Lin, J. Kim, I. Roh, S. Lee and P. Yang, Nat. Catal., 2024, 7, 977–986 CrossRef CAS.
- W. Ghosh and B. Dam, FEMS Microbiol. Rev., 2009, 33, 999–1043 CrossRef CAS PubMed.
- J. Li, Y. Tian, Y. Zhou, Y. Zong, N. Yang, M. Zhang, Z. Guo and H. Song, Trans. Tianjin Univ., 2020, 26, 237–247 CrossRef CAS.
- L. Jiang, K. Wang, X. Wu and G. Zhang, Sol. RRL, 2021, 5, 2000326 CrossRef CAS.
- L. Shi, X. Ren, Q. Wang, W. Zhou and J. Ye, J. Mater. Chem. A, 2021, 9, 2421–2428 RSC.
- B. Wang, H. Chen, W. Zhang, H. Liu, Z. Zheng, F. Huang, J. Liu, G. Liu, X. Yan, Y.-X. Weng, H. Li, Y. She, P. K. Chu and J. Xia, Adv. Mater., 2024, 36, 2312676 CrossRef CAS PubMed.
- J. Hao, D. Yang, J. Wu, B. Ni, L. Wei, Q. Xu, Y. Min and H. Li, Chem. Eng. J., 2021, 423, 130190 CrossRef CAS.
- W. Dai, J. Yu, S. Luo, X. Hu, L. Yang, S. Zhang, B. Li, X. Luo and J. Zou, Chem. Eng. J., 2020, 389, 123430 CrossRef CAS.
- Z. Zhang, X. Liu, Y. Li, H. Yu, W. Li and H. Yu, Appl. Catal., B, 2022, 319, 121960 CrossRef CAS.
- J. Li, X. Liu, X. Wu, Z. Liu, Z. Zhao, Y. Liu, S. Dou and Y. Xiao, Adv. Sci., 2024, 11, 2405668 CrossRef CAS PubMed.
- C. Lu, X. Li, J. Li, L. Mao, M. Zhu, Q. Chen, L. Wen, B. Li, T. Guo and Z. Lou, Chem. Eng. J., 2022, 445, 136739 CrossRef CAS.
- S. Li, Z. Li, J. Yue, H. Wang, Y. Wang, W. Su, G. I. N. Waterhouse, L. Liu, W. Zhang and Y. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202407638 CrossRef CAS PubMed.
- Z. Guo, B. Shen, Z. Jiang, N. Wu and Y. You, Ind. Eng. Chem. Res., 2024, 63, 18931–18939 CrossRef CAS.
- L.-Y. Lin, C. Liu and T.-T. Hsieh, J. Catal., 2020, 391, 298–311 CrossRef CAS.
- X. Y. Kong, Y. Y. Choo, S.-P. Chai, A. K. Soh and A. R. Mohamed, Chem. Commun., 2016, 52, 14242–14245 RSC.
- H. Jiang, S. Gong, S. Xu, P. Shi, J. Fan, V. Cecen, Q. Xu and Y. Min, Dalton Trans., 2020, 49, 5074–5086 RSC.
- Q. Li, Y. Gao, M. Zhang, H. Gao, J. Chen and H. Jia, Appl. Catal., B, 2022, 303, 120905 CrossRef CAS.
- X. Jiao, K. Zheng, Z. Hu, Y. Sun and Y. Xie, ACS Cent. Sci., 2020, 6, 653–660 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- B. V. Lotsch and W. Schnick, Chem. – Eur. J., 2007, 13, 4956–4968 CrossRef CAS PubMed.
- Z. Hu, G. Liu, X. Chen, Z. Shen and J. C. Yu, Adv. Mater., 2016, 26, 4445–4455 CAS.
- X. Jiao, Z. Hu, Y. Wu, K. Zheng, L. Li, S. Zhu, W. Shao, J. Zhu, Y. Pan and Y. Sun, Sci. China Mater., 2022, 65, 985–991 CrossRef CAS.
- W. L. Barnes, A. Dereux and T. W. Ebbesen, Nature, 2003, 424, 824–830 CrossRef CAS PubMed.
- S. A. Maier, Plasmonics: Fundamentals and Applications, Springer US, New York, NY, 2007 Search PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- M. Rycenga, C. M. Cobley, J. Zeng, W. Li, C. H. Moran, Q. Zhang, D. Qin and Y. Xia, Chem. Rev., 2011, 111, 3669–3712 CrossRef CAS PubMed.
- F. J. García de Abajo, ACS Photonics, 2014, 1, 135–152 CrossRef.
- K.-L. Lee, M.-L. You and P.-K. Wei, ACS Appl. Nano Mater., 2019, 2, 1930–1939 CrossRef CAS.
- S. Song, C. Xin, W. Liu, W. Shang, T. Liu, W. Peng, J. Hou and Y. Shi, Angew. Chem., Int. Ed., 2025, 64, e202415173 CrossRef CAS PubMed.
- T. Atay, J.-H. Song and A. Nurmikko, in Conference on Lasers and Electro-Optics/International Quantum Electronics Conference and Photonic Applications Systems Technologies, Optica Publishing Group, San Francisco, California, 2004, p. IPDB7.
- K. Kolwas, A. Derkachova and M. Shopa, J. Quant. Spectrosc. Radiat. Transfer, 2009, 110, 1490–1501 CrossRef CAS.
- J. J. Mock, M. Barbic, D. R. Smith, D. A. Schultz and S. Schultz, J. Chem. Phys., 2002, 116, 6755–6759 CrossRef CAS.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- S. Gong, Y. Niu, X. Liu, C. Xu, C. Chen, T. J. Meyer and Z. Chen, ACS Nano, 2023, 17, 4922–4932 CrossRef CAS PubMed.
- A. A. Ansari and M. Sillanpää, Renewable Sustainable Energy Rev., 2021, 151, 111631 CrossRef CAS.
- W. Yang, X. Li, D. Chi, H. Zhang and X. Liu, Nanotechnology, 2014, 25, 482001 CrossRef PubMed.
- B. S. Richards, D. Hudry, D. Busko, A. Turshatov and I. A. Howard, Chem. Rev., 2021, 121, 9165–9195 CrossRef CAS PubMed.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS PubMed.
- X. Zhao, Q. Liu, X. Li, H. Li, Z. Shen, H. Ji and T. Ma, Angew. Chem., Int. Ed., 2023, 62, e202219214 CrossRef CAS PubMed.
- Q. Zhang, F. Yang, Z. Xu, M. Chaker and D. Ma, Nanoscale Horiz., 2019, 4, 579–591 RSC.
- J. Xiao, X. Hou, L. Zhao and Y. Li, J. Catal., 2017, 346, 70–77 CrossRef CAS.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- A. Corma and H. Garcia, J. Catal., 2013, 308, 168–175 CrossRef CAS.
- F. Wang, Y. Huang, Z. Chai, M. Zeng, Q. Li, Y. Wang and D. Xu, Chem. Sci., 2016, 7, 6887–6893 RSC.
- M. Ghoussoub, M. Xia, P. N. Duchesne, D. Segal and G. Ozin, Energy Environ. Sci., 2019, 12, 1122–1142 RSC.
- L. Wang, Y. Wang, Y. Cheng, Z. Liu, Q. Guo, M. N. Ha and Z. Zhao, J. Mater. Chem. A, 2016, 4, 5314–5322 RSC.
- F. Yu, C. Wang, Y. Li, H. Ma, R. Wang, Y. Liu, N. Suzuki, C. Terashima, B. Ohtani, T. Ochiai, A. Fujishima and X. Zhang, Adv. Sci., 2020, 7, 2000204 CrossRef CAS PubMed.
- Y. Li, C. Wang, M. Song, D. Li, X. Zhang and Y. Liu, Appl. Catal., B, 2019, 243, 760–770 CrossRef CAS.
- Y. Li, D. Hui, Y. Sun, Y. Wang, Z. Wu, C. Wang and J. Zhao, Nat. Commun., 2021, 12, 123 CrossRef CAS PubMed.
- D. Mateo, J. Albero and H. García, Energy Environ. Sci., 2017, 10, 2392–2400 RSC.
- Y.-F. Xu, P. N. Duchesne, L. Wang, A. Tavasoli, F. M. Ali, M. Xia, J.-F. Liao, D.-B. Kuang and G. A. Ozin, Nat. Commun., 2020, 11, 5149 CrossRef CAS PubMed.
- S. Ning, H. Xu, Y. Qi, L. Song, Q. Zhang, S. Ouyang and J. Ye, ACS Catal., 2020, 10, 4726–4736 CrossRef CAS.
- Z. Ma, W. Liu, W. Yang, W. Li and B. Han, Fuel, 2021, 286, 119490 CrossRef CAS.
- L. Zhang, C. Li, Y. Liu, C. Xu and Y. Zhang, npj Comput. Mater., 2024, 10, 132 CrossRef CAS.
- C. Foo, Y. Li, K. Lebedev, T. Chen, S. Day, C. Tang and S. C. E. Tsang, Nat. Commun., 2021, 12, 661 CrossRef CAS PubMed.
- B. Han, W. Wei, L. Chang, P. Cheng and Y. H. Hu, ACS Catal., 2016, 6, 494–497 CrossRef CAS.
- M. Cai, Z. Wu, Z. Li, L. Wang, W. Sun, A. A. Tountas, C. Li, S. Wang, K. Feng, A.-B. Xu, S. Tang, A. Tavasoli, M. Peng, W. Liu, A. S. Helmy, L. He, G. A. Ozin and X. Zhang, Nat. Energy, 2021, 6, 807–814 CrossRef CAS.
- M. Cai, C. Li, X. An, B. Zhong, Y. Zhou, K. Feng, S. Wang, C. Zhang, M. Xiao, Z. Wu, J. He, C. Wu, J. Shen, Z. Zhu, K. Feng, J. Zhong and L. He, Adv. Mater., 2024, 36, 2308859 CrossRef CAS PubMed.
- H. He, X. Gao, K. Xu, H. Li, Y. Hu, C. Yang and F. Fu, Chem. Eng. J., 2022, 450, 138266 CrossRef CAS.
- B. An, Z. Li, Y. Song, J. Zhang, L. Zeng, C. Wang and W. Lin, Nat. Catal., 2019, 2, 709–717 CrossRef CAS.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS PubMed.
- Z. Feng, C. Tang, P. Zhang, K. Li, G. Li, J. Wang, Z. Feng and C. Li, J. Am. Chem. Soc., 2023, 145, 12663–12672 CrossRef CAS PubMed.
- P. G. O’Brien, A. Sandhel, T. E. Wood, F. M. Ali, L. B. Hoch, D. D. Perovic, C. A. Mims and G. A. Ozin, Adv. Sci., 2014, 1, 1400001 CrossRef PubMed.
- Y. Cao, L. Gao, B. Wang, Y. Yao, C. Wu, Q. Shen, J. Feng, Y. Zhou, Z. Li and Z. Zou, ACS Mater. Lett., 2022, 4, 1912–1920 CrossRef CAS.
- Y. C. Tan, K. B. Lee, H. Song and J. Oh, Joule, 2020, 4, 1104–1120 CrossRef CAS.
- X. Bian, Y. Zhao, G. I. N. Waterhouse, Y. Miao, C. Zhou, L.-Z. Wu and T. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304452 CrossRef CAS PubMed.
- C. Zhang, Z.-C. Shao, X.-L. Zhang, G.-Q. Liu, Y.-Z. Zhang, L. Wu, C.-Y. Liu, Y. Pan, F.-H. Su, M.-R. Gao, Y. Li and S.-H. Yu, Angew. Chem., Int. Ed., 2023, 62, e202305571 CrossRef CAS PubMed.
- B. Ren, Z. Zhang, G. Wen, X. Zhang, M. Xu, Y. Weng, Y. Nie, H. Dou, Y. Jiang, Y.-P. Deng, G. Sun, D. Luo, L. Shui, X. Wang, M. Feng, A. Yu and Z. Chen, Adv. Mater., 2022, 34, 2204637 CrossRef CAS PubMed.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- N. Kuhar, S. Sil and S. Umapathy, Spectrochim. Acta, Part A, 2021, 258, 119712 CrossRef CAS PubMed.
- R. Beranek, Adv. Phys. Chem., 2011, 2011, 786759 CrossRef.
- J. Kubota, Z. Ma and F. Zaera, Langmuir, 2003, 19, 3371–3376 CrossRef CAS.
- L. Li, H. Guo, G. Yao, C. Hu, C. Liu, Z. Tian, B. Li, Q. Zhang and L. Chen, J. Mater. Chem. A, 2020, 8, 22327–22334 RSC.
- H. Mai, T. C. Le, D. Chen, D. A. Winkler and R. A. Caruso, Chem. Rev., 2022, 122, 13478–13515 CrossRef CAS PubMed.
- J. Hong, W. Zhang, J. Ren and R. Xu, Anal. Methods, 2013, 5, 1086–1097 RSC.
- Y. Xin, K. Yu, L. Zhang, Y. Yang, H. Yuan, H. Li, L. Wang and J. Zeng, Adv. Mater., 2021, 33, 2008145 CrossRef CAS PubMed.
- J. Yu, K. Wang, W. Xiao and B. Cheng, Phys. Chem. Chem. Phys., 2014, 16, 11492–11501 RSC.
- R. Li, J. Hu, M. Deng, H. Wang, X. Wang, Y. Hu, H.-L. Jiang, J. Jiang, Q. Zhang, Y. Xie and Y. Xiong, Adv. Mater., 2014, 26, 4783–4788 CrossRef CAS PubMed.
- X.-B. Li, Z.-K. Xin, S.-G. Xia, X.-Y. Gao, C.-H. Tung and L.-Z. Wu, Chem. Soc. Rev., 2020, 49, 9028–9056 RSC.
- A. Velty and A. Corma, Chem. Soc. Rev., 2023, 52, 1773–1946 RSC.
- Y.-S. Xia, M. Tang, L. Zhang, J. Liu, C. Jiang, G.-K. Gao, L.-Z. Dong, L.-G. Xie and Y.-Q. Lan, Nat. Commun., 2022, 13, 2964 CrossRef CAS PubMed.
- S. Verma, S. Lu and P. J. A. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- S. Kar, M. Rahaman, V. Andrei, S. Bhattacharjee, S. Roy and E. Reisner, Joule, 2023, 7, 1496–1514 CrossRef CAS.
- E. Lam and E. Reisner, Angew. Chem., Int. Ed., 2021, 60, 23306–23312 CrossRef CAS PubMed.
- M. Li and S. Zhang, ACS Catal., 2024, 14, 6717–6727 CrossRef CAS.
- J. Fan, X. Yue, Y. Liu, D. Li and J. Feng, Chem. Catal., 2022, 2, 531–549 CrossRef CAS.
- M.-Y. Qi, Q. Lin, Z.-R. Tang and Y.-J. Xu, Appl. Catal., B, 2022, 307, 121158 CrossRef CAS.
- L. Hong, H. Zhang, L. Hu, R. Xiao and S. Chu, Sci. Adv., 2024, 10, eadn9441 CrossRef CAS PubMed.
- T. Wang, L. Tao, X. Zhu, C. Chen, W. Chen, S. Du, Y. Zhou, B. Zhou, D. Wang, C. Xie, P. Long, W. Li, Y. Wang, R. Chen, Y. Zou, X.-Z. Fu, Y. Li, X. Duan and S. Wang, Nat. Catal., 2022, 5, 66–73 CrossRef CAS.
- H.-L. Wu, X.-B. Li, C.-H. Tung and L.-Z. Wu, Adv. Mater., 2019, 31, 1900709 CrossRef PubMed.
- R. Zhai, L. Zhang, M. Gu, X. Zhao, B. Zhang, Y. Cheng and J. Zhang, Small, 2023, 19, 2207840 CrossRef CAS PubMed.
- Bhawna, S. Kumar, R. Sharma, S. J. Borah, A. Gupta, M. K. Gupta, R. Kumar, K. K. Dubey, Y. K. Mishra and V. Kumar, Sustainable Energy Fuels, 2023, 7, 4354–4395 RSC.
- L. Yuan, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Angew. Chem., Int. Ed., 2021, 60, 21150–21172 CrossRef CAS PubMed.
- B. Song, Y. Liang, Y. Zhou, L. Zhang, H. Li, N.-X. Zhu, B. Z. Tang, D. Zhao and B. Liu, J. Am. Chem. Soc., 2024, 146, 14835–14843 CrossRef CAS PubMed.
- K. Kadota, Y. Hong, Y. Nishiyama, E. Sivaniah, D. Packwood and S. Horike, J. Am. Chem. Soc., 2021, 143, 16750–16757 CrossRef CAS PubMed.
- F. Sastre, A. V. Puga, L. Liu, A. Corma and H. García, J. Am. Chem. Soc., 2014, 136, 6798–6801 CrossRef CAS PubMed.
- X. Meng, T. Wang, L. Liu, S. Ouyang, P. Li, H. Hu, T. Kako, H. Iwai, A. Tanaka and J. Ye, Angew. Chem., Int. Ed., 2014, 53, 11478–11482 CrossRef CAS PubMed.
- S. A. Chernyak, M. Corda, J.-P. Dath, V. V. Ordomsky and A. Y. Khodakov, Chem. Soc. Rev., 2022, 51, 7994–8044 RSC.
- M. Mao, Q. Zhang, Y. Yang, Y. Li, H. Huang, Z. Jiang, Q. Hu and X. Zhao, Green Chem., 2018, 20, 2857–2869 RSC.
Footnotes |
| † Dedicated to Prof. Michael Grätzel on the occasion of his 80th birthday. |
| ‡ These authors have contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2025 |