
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Antifreezing hydrogels: from mechanisms and strategies to applications
Dong
Zhang†
 a,
Hong
Chen†
b,
Yanxian
Zhang
c,
Jintao
Yang
d,
Qiang
Chen
e,
Jiang
Wu
f,
Yonglan
Liu
g,
Chao
Zhao
h,
Yijing
Tang
i and
Jie
Zheng
*i
a,
Hong
Chen†
b,
Yanxian
Zhang
c,
Jintao
Yang
d,
Qiang
Chen
e,
Jiang
Wu
f,
Yonglan
Liu
g,
Chao
Zhao
h,
Yijing
Tang
i and
Jie
Zheng
*i
aThe Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, Georgia 30332, USA
bCollege of Polymer Science and Engineering, Sichuan University, Chengdu 610065, China
cDivision of Endocrinology and Diabetes, Department of Pediatrics, School of Medicine, Stanford University, Palo Alto, CA 94304, USA
dCollege of Materials Science and Engineering, Zhejiang University of Technology, Hangzhou 310014, China
eWenzhou Institute, University of Chinese Academy of Sciences, Wenzhou, Zhejiang 352001, China
fSchool of Pharmaceutical Sciences, Key Laboratory of Biotechnology and Pharmaceutical Engineering, Wenzhou Medical University, Wenzhou, Zhejiang 325035, China
gCancer Innovation Laboratory, National Cancer Institute, Frederick, MD 21702, USA
hDeptartment of Chemical and Biological Engineering, The University of Alabama, Tuscaloosa, AL 35487, USA
iDepartment of Chemical, Biomolecular, and Corrosion Engineering, The University of Akron, Ohio 44325, USA. E-mail: zhengj@uakron.edu
First published on 21st May 2025
Abstract
Antifreezing hydrogels have emerged as an innovative solution for maintaining functional performance and mechanical integrity in subzero environments, offering a robust alternative to traditional water-free antifreezing materials that often fail under wet and cold conditions. These water-rich hydrogels leverage their porous, crosslinked, polymeric networks, which serve as the structural basis for implementing two parallel strategies: the incorporation of antifreezing additives (peptides/proteins, salts, ionic liquids, and organics) and the meticulous engineering of polymer systems and network structures for manipulating the water–ice phase equilibrium to significantly enhance antifreezing properties. This review synthesizes recent findings to provide a fundamental overview of the important advancements in antifreezing hydrogels, focusing on their designs, mechanisms, performances, and functional applications. Various types of antifreezing hydrogels have been developed, utilizing strategies like the incorporation of antifreeze agents, use of strongly water-bound polymers, and design of highly crosslinked networks to illustrate different antifreezing mechanisms: freezing point depression, ice recrystallization inhibition, and network freezing inhibition. This review also explores the diverse functions of antifreezing hydrogels in biomedical devices, soft robotics, flexible electronics, food industry, and environmental engineering. Finally, this review concludes with future directions, emphasizing the potential of integrating machine learning and advanced molecular simulations into materials design. This strategic vision is aimed at promoting continuous innovation and progress in the rapidly evolving field of antifreezing hydrogels.
 Dong Zhang | Dr Dong Zhang earned his PhD in Chemical, Biomolecular, and Corrosion Engineering from The University of Akron in 2022, under the supervision of Professor Jie Zheng. He is currently a postdoctoral fellow in Professor Younan Xia's group at the Georgia Institute of Technology. His research focuses on the rational design and synthesis of polymers and nanomaterials for applications in environmental, biological, and biomedical fields. |
 Hong Chen | Dr Hong Chen is an Associate Professor in the College of Polymer Science and Engineering at Sichuan University. She received a BS degree in Polymer Engineering from Sichuan University in 2012, an MS degree in Materials Science from the University of California, Los Angeles in 2013, and a PhD in Chemical Engineering from the University of Akron in 2017. Her research focuses on polymeric biomaterials, tough hydrogels, and biosensors. |
 Yanxian Zhang | Dr Yanxian Zhang earned her PhD in Chemical, Biomolecular, and Corrosion Engineering from the University of Akron, focusing on amyloid peptide cross-seeding and inhibitor development. Her work revealed critical interactions between antimicrobial human α-defensins and amyloid peptides, notably inhibiting amyloid aggregation. Since 2021, she has been a postdoctoral researcher at Stanford University, developing biopharmaceuticals by merging molecular biology, chemical engineering, and protein engineering to enhance stability and optimize pharmacological profiles. |
 Chao Zhao | Prof. Chao Zhao, an Assistant Professor in the Department of Chemical and Biological Engineering at the University of Alabama, earned his PhD from the University of Akron in 2013. His research interests include drug delivery, biomaterials, nanomedicine, and drug discovery for various biomedical applications, with an emphasis on pain management. He has authored over 70 journal papers, achieving an h-index of 34 and accumulating over 6000 citations. For further details of his achievements and publications, please visit his profile at https://sites.ua.edu/zhaolab/. |
 Yijing Tang | Dr Yijing Tang received her BE in Polymer Science and Materials from the Beijing University of Chemical Technology and her MS in Polymer Science from the University of Akron. After completing her PhD study in 2024 under the supervision of Prof. Jie Zheng at the University of Akron, she joined the groups of Prof. Brian Bacskai and Prof. Steven Hou at the Massachusetts General Hospital and Harvard Medical School as a Postdoc research fellow. Since then, her research has primarily focused on developing in vivo fluorescence labeling techniques to monitor real-time progression and dynamics of neurodegeneration. |
 Jie Zheng | Prof. Jie Zheng, a Full Professor of Dept. of Chemical, Biomolecular, and Corrosion Engineering at the University of Akron, earned his PhD from the University of Washington in 2005. His research spans disease-related proteins, biomaterials, and structural biology, incorporating experimental methods, biophysical analysis, molecular simulations, and machine-learning models. He has authored over 290 journal papers, achieving an h-index of 86 and accumulating over 25 |
1. Introduction
Icing, as a ubiquitous and unavoidable phenomenon in nature, is vital for all life on Earth. However, unwanted icing has long plagued our daily lives with severe economic, environmental, and safety issues, such as icing-induced mechanical failures in aircrafts, power lines, and wind turbine blades, optical malfunctions in telescope lens and glasses, and traffic accidents in cold weather.1–3 To combat these challenges, significant efforts have been devoted to developing various antifreezing polymers and strategies for applications in flexible robotics, electronic skin, wearable devices, and human–machine interfaces. Traditional water-free antifreezing polymers, such as superhydrophobic materials,4 elastomers,5 and organogels,6–8 have been the focus of extensive research. Superhydrophobic materials, however, can easily lose their superhydrophobic properties due to surface contamination with oils, dust, or other particulates, as well as environmental changes in humidity, light, and temperature, which reduce their antifreezing efficacy. While many elastomers can prevent freezing to some extent, they tend to become brittle at low temperatures, thereby losing their mechanical properties and structural integrity under freezing conditions.9 Organogels, which always contain organic solvents, can become volatile, leading to shrinkage, a loss of mechanical properties, and reduced antifreezing effectiveness over time. Additionally, organic solvents used in organogels can be toxic or flammable, posing environmental and health risks.Unlike traditional water-free antifreezing polymers, which often suffer from the loss of antifreezing properties upon exposure to wet environments and mechanical failures under cold conditions, antifreezing hydrogels have emerged as innovative solutions (Fig. 1).10–13 The advantages of antifreezing hydrogels are primarily attributed to their inherent properties,14–18 such as high water content, the ability to mimic soft tissue, integrated liquid–solid properties, porous structures conducive to transport and inclusion, and structural adaptability and flexibility.19–21 These features offer significant advantages over water-free antifreezing polymers, particularly in applications demanding flexibility, biocompatibility, and environmental responsiveness. However, from a materials design perspective, designing water-rich antifreezing hydrogels presents significant challenges due to their high-water content, which typically ranges from 50 to 95% within the hydrogel network. On the other hand, the confined water in polymer networks adapts more naturally to external wet and cold environments. This water adaptability allows them to function effectively without fracturing, a common issue with more rigid, water-free polymers when exposed to freezing temperatures. Consequently, this inherent capability of antifreezing hydrogels positions them as superior in maintaining their antifreezing properties, mechanical integrity, and other functionalities under adverse conditions.
 | ||
| Fig. 1 Historical timeline of antifreezing materials development. | ||
Given that conventional design wisdom prevents a large amount of water (50–95%) and antifreezing function to be both present in the same material, current antifreezing strategies predominantly rely on incorporating various antifreezing additives into polymer networks to modify the water–ice phase equilibrium during the different stages of ice condensation, nucleation, and growth.17 These additives include: (1) ionic compounds such as NaCl, ZnCl2, CaCl2, and ionic liquids (ILs);22–25 (2) natural biomolecules like ice-binding proteins (IBPs) or more specifically antifreeze proteins (AFPs), glycoproteins, and silk proteins,17,26–30 and (3) organic/water solvents such as dimethyl sulfoxide (DMSO), benzyltrimethylammonium hydroxide, betaine, proline, ethylene glycol (EG), and glycerol (Gly).31–36 Despite these advancements, there has been a notable lack of developments in pure polymeric antifreezing hydrogels that do not require any antifreezing additive. Recently, a study has reported the development of a highly hydrophilic crosslinker, which enables the crosslinking of a wide range of hydrogels with single-network (SN) and double-network (DN) structures. This development has led to hydrogels that exhibit strong antifreezing and mechanical properties at −20 °C for extended periods, both in aqueous solutions and gel states, without requiring any antifreezing additives.37
Building on these antifreezing strategies, various mechanisms have been identified or developed to inhibit ice formation and growth in water-rich hydrogels. The most common mechanism involves water freezing point depression through the incorporation of additives such as ionic compounds, organic solvents, or ILs. These additives (e.g., DMSO, Gly, EG, or CaCl2) disrupt the formation of ice crystals by altering the water–ice phase equilibrium, which results in a depression of the freezing point and prevents ice formation within the polymer matrix. Ice recrystallization inhibition is another key mechanism, achieved through the adsorption of certain polymers, incorporated additives, or AFPs onto ice crystal surfaces. This adsorption prevents ice crystals from coalescing and growing larger during freeze–thaw cycles. The modification of polymer networks by adjusting hydrophilic-to-hydrophobic domains or pore sizes facilitates antifreezing effects through combinational network freezing inhibition. Firstly, an increase in (super)hydrophobic regions creates microenvironments that displace water, thereby preventing contact and inhibiting the initial nucleation of ice. Secondly, a highly crosslinked network provides nanoscale confinement that can significantly restrict water mobility and limit the size of any forming ice crystals. Furthermore, the formation of a dense network of hydrogen bonds within the polymer matrix—referred to as hydrogen-bonding network inhibition—enables these reversible bonds to break and reform dynamically in response to temperature changes. This dynamic bonding behavior helps prevent the formation of stable ice crystals, enhancing the antifreezing capabilities of the hydrogels. From a mechanistic perspective, hydrogels containing antifreezing additives or cryoprotectants derive their freeze-resistance primarily from the strong interactions between the additives and water molecules. These interactions significantly reduce the amount of free water available to form ice nuclei. Unlike this cryoprotectant-induced mechanism, the crosslinked polymer network plays a multifaceted role in influencing their antifreezing properties. The chemical, physical, and structural attributes of polymer networks—including hydrophilicity, the chemistry of pendant groups, mesh size, polymer conformation, and network crosslinking density—profoundly impact the intricate balance between water–polymer interactions and the confinement effects on the dynamics and structures of water. These antifreezing mechanisms are not necessarily exclusive; instead, they can work synergistically to enhance antifreezing performance.
From a challenge perspective, maintaining mechanical properties at subzero temperatures remains a significant obstacle for antifreezing hydrogels. High mechanical strength and antifreezing properties are two highly desirable yet inherently distinct characteristics of hydrogels at the macroscale. Although antifreezing hydrogels can retain their overall structural integrity at subzero temperatures, even minimal ice formation can significantly compromise the polymer networks, leading to brittleness and mechanical failure. This deterioration is evidenced by a considerable loss of mechanical properties,38,39 and in some cases, these hydrogels may fail to withstand standard tensile or compression tests at subzero temperatures.33,35,40,41 Furthermore, there is a lack of in-depth molecular simulations exploring the behaviors of water (ice) confined within polymer networks at subzero temperatures. Such simulations are critical for a fundamental understanding of the structure-based design and functional mechanisms of new antifreezing hydrogels. More critically, the field faces the challenge of a scarcity of comprehensive datasets on antifreezing hydrogels. These datasets are essential for developing machine-learning models that can assess, predict, explain, and design antifreezing hydrogels based on a quantitative understanding of the components, structure, and performance of these hydrogels.
Antifreezing hydrogels have emerged as innovative and unprecedented materials and as a rapidly developing field. Given this rapid progression, it is timely to summarize the recent advancements in antifreezing hydrogels, focusing on design principles, fabrication strategies, and antifreezing mechanisms, all of which are still in their early stages and require further exploration. Consequently, as detailed in Tables 1 and 2, we systematically summarize and categorize notable antifreezing hydrogels, highlighting their distinctive design principles and synthesis strategies from both material and structural perspectives. While there are a few existing reviews on antifreezing hydrogels,10 this review specifically concentrates on the fundamental aspects, from antifreezing mechanisms and design and fabrication strategies to advanced and complementary functions for each section. Ultimately, the scope and limitations of these design strategies and antifreezing mechanisms are discussed in the Conclusions and Perspectives section, with the aim of accelerating transformative applications of antifreezing hydrogels in this promising field.
| Antifreezing mechanisms | Antifreezing reagents | Type and composition | Synthesis strategy | Antifreezing performance | Applications (or additional functions) | Ref. |
|---|---|---|---|---|---|---|
| IRI & WFD | AFPs | P(AAm-co-AMPS) | Radical copolymerization | Stable toughness and adhesion at −10 °C | Wearable strain and pressure sensors | 42 |
| IRI & WFD | AFPs | P(AAm-co-MAANa)/PVA | Radical copolymerization | Remain stretchable, conductive at −15 °C | Wearable strain sensors | 43 |
| IRI & WFD | AFP and NaCl | Agar/PAAm | Sol–gel transition and radical polymerization | Stable adhesion, stretchable and conductive at −20 °C | Wearable motion sensors, human–machine interaction systems | 44 |
| IRI & WFD | ISP and CaCl2 | PAAc | Radical polymerization | Remain stretchable and conductive at −20 °C | Strain/temperature sensors | 45 |
| IRI & WFD | ISPs and CNFs | P(AAm-co-AMPS)/PVA | Radical copolymerization | Stretchable conductive at −10 °C | Wearable human motion sensors | 46 |
| IRI | INPs | P(AAm-co-HEMA) | Radical copolymerization | Ice coverage fraction controlled by <30% at −20 °C | Long-term anti-icing coatings | 47 |
| IRI | PF patterns | PAAc | Radical polymerization and surface modification | Improved resistance to ice formation at −20 °C | Antifreezing coatings | 48 |
| NFI | EGINA network | PVA/(gelatin, agar, or BSA)/EGINA | Sol–gel transition/freeze–thaw and polymerization | Robust antifreezing and mechanical properties at −20 °C | Supercapacitors and smart windows | 37 |
| NFI | DMA/FOSA nanodomain | P(DMA-co-FOSA) | Radical copolymerization | Stable viscoelasticity at 205 K | High mechanical properties, σb 0.4 MPa; deicing surfaces | 49 and 50 |
| NFI | Hydrophobic FOSM | Poly(HEA-FOSM) | Radical copolymerization | Prevent nearly all the water from freezing at 128 K | Antifreezers | 40 |
| NFI | Dense H-bonds | P(AAm-co-MAAc) | Radical copolymerization | Transparent and flexible at −45 °C and −196 °C | High mechanical properties, σb 8 MPa; phosphorescence | 51 |
| WFD | LiCl and solketal | PAAm-TA-CMC | Radical copolymerization | Reliable freezing-tolerance at −30 °C | Wearable strain sensors | 52 |
| WFD | LiCl and EG | P(SBMA-HEMA) | Radical copolymerization | Remain transparent at −60 °C | Supercapacitors with 59% capacitance retention at −20 °C | 53 |
| WFD | LiCl and Gly | Silk fibroin/PVA | Cyclic freezing–thawing | Freezing point below −70 °C | Supercapacitors with 66.5% capacitance retention at −40 °C | 54 |
| WFD | LiClO4 and Gly | Hydroxypropyl cellulose/PVA | Cyclic freezing–thawing and solvent exchange | Stable tensile strength and superior conductivity at −40 °C | Supercapacitors | 55 |
| WFD | NaCl and Fe3+ | PAAm/PAAc-Fe3+ | Solvent exchange | Freezing point of −24.7 °C | Ionic skin strain/stress sensors (−14 °C) | 56 |
| WFD | NaCl | PAAm-DVB | Radical copolymerization | Flexible, conductive at −20 °C | Conductive (10.6 S m−1) | 57 |
| WFD | NaCl | PAAm-CGO | Radical copolymerization | Reliable freezing-tolerance at −56.8 °C | Flexible strain sensors (−20 °C) | 58 |
| WFD | NaCl/KCl/CaCl2 | PVA | Covalent crosslinking | Reduce ice adhesion strength at −48.4 °C | Self-deicing and antifrost coatings | 59 |
| WFD | NaCl/CaCl2/FeCl3 | P(VBIPS/HEAA)-SA | Radical copolymerization | Remain flexible at −60 °C | High ionic conductivity and switchable friction/lubrication | 60 |
| WFD | NaAc | MoS2-PDMAA/PVA | Radical polymerization and solvent exchange | Remain conductive at −56 °C | Supercapacitors with 96.9% retention at −40 °C | 61 |
| WFD | KCl | CMCS-OCS-Cu-CDs | Radical polymerization | Antifreezing point (−30 °C) | Weed germination and growth inhibition | 62 |
| WFD | KCl/PIL | PIL | Radical polymerization | Freezing point (−41 °C) and remain stretchable at −20 °C | Conductive (6.2 S m−1 at −30 °C) | 23 |
| WFD | KAc/ZnAc2 and DMSO | PVA OHG | Solvent exchange | Unfrozen at −77 °C, tensile stress/strain (23 MPa/400%) at −30 °C | Zinc-ion batteries | 63 |
| WFD | CaCl2 | Carboxymethyl chitosan | Salt solution equilibration | Remain flexible and transparent at −50 °C | Strain sensors | 64 |
| WFD | CaCl2 | PAAm/Alginate | Radical polymerization and ionic crosslinking | Excellent tensile stress/strain of 0.25 MPa/5 at −50 °C | Strain sensors, cryopreservation, and sustained release of drug | 22 and 65 |
| WFD | CaCl2 | Sodium alginate | Ionic crosslinking | Preferentially vitrified during cryopreservation | Cell cryopreservation and coatings | 66 and 67 |
| WFD | CaCl2 and betaine | Arabic gum/PAAc-CNCs | Radical copolymerization | Remain tough and stretchable at −30 °C | Self-healing at −20 °C | 68 |
| WFD | CaSO4 | Alginate-PAAm | Radical polymerization and ionic crosslinking | Antifrosting and deicing at subzero temperature | Lubricating and anti-adhesion | 69 |
| WFD | ILs and AlCl3 | Starch/PVA | Cyclic freezing–thawing | Remain flexible, conductive at −20 °C | Strain sensors and antibacterial coatings | 70 |
| WFD | ILs and Gly | Polyglutamic acid-lysine | EDC/NHS coupling | Conductive at −60 °C | Wound dressing and wearable strain sensors | 71 |
| WFD | DES | P(SBMA-AAc) | Radical copolymerization | Reliable freezing-tolerance at −60 °C | Wearable strain sensors (anti-swelling) | 72 |
| WFD | Proline | GG/PAAc/CNCs/PDA | Radical copolymerization and sol–gel transition | Remain stretchable at −30 °C | Self-healing at −30 °C | 73 |
| WFD | EG | PVA-PEDOT:PSS | Cyclic freezing–thawing | Stable flexibility and strain-sensitivity at −40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C °C |
Strain sensors | 8 |
| WFD | EG | PVA/PAAm | UV and thermal polymerization | Stable electrochemical performances at −40°C |
Electrolytes and supercapacitors | 74 |
| WFD | EG | Carrageenan/PAAm/CNTs | Radical copolymerization | Flexible, conductive, and stretchable at −85 °C | Self-healing at room temperature | 75 |
| WFD | EG | BP/PVA/borax/sodium alginate | 3D printing | Remain soft, elastic, and conductive at −80 °C | Self-healing at −80 °C | 76 |
| WFD | EG | APTES-SiO2/PVA | Sol–gel transition | Remain flexible at −25 °C | Oil–water separation | 77 |
| WFD | EG | P(HEA-AMPS) | Radical copolymerization | Reliable freezing-tolerance at −78 °C | Strain sensors/self-adhesive (10.2–27.8 kPa) | 78 |
| WFD | EG | P(NIPAM-co-DMAA) | Radical copolymerization | Tunable and excellent freezing resistance at −100 °C | Solar and thermal radiation regulation smart window | 79 |
| WFD | EG | CMCS/NVP | Radical copolymerization | Remain flexible, stable sensing performance at −70 °C | Self-healing at −35 °C | 80 |
| WFD | EG | GO/LAP PAAm | Radical polymerization and solvent exchange | Remain flexible and conductive at −20 °C | Strain sensors and wound dressing | 81 |
| WFD | EG and CaCl2 | PVA/SF/PPy | Radical copolymerization | Remain stretchable and strain sensible at −14 °C | Self-healing at room temperature | 82 |
| WFD | EG and MgCl2/CaCl2 | C-CS/SNFs/TA/PVA | Cyclic freezing–thawing | Flexible, conductive, and self-healable at −18 °C | Flexible strain sensors for wound monitoring | 83 |
| WFD | EG/Gly/sorbitol and CaCl2 | Alginate/PAAm | Radical polymerization and solvent exchange | Unfrozen, stable, and mechanically flexible at −70°C |
Non-drying and high weight retention at −70 °C | 84 |
| WFD | EG and LiCl | PAMPS/PAAm | Radical polymerizations and solvent exchange | Strong and flexible at −80 °C and conductive at −20 °C | Strain sensors | 85 |
| WFD | Gly | TA-CNTs- PVA | Cyclic freezing–thawing | Stable integrity and stretchability at −30 °C | Strain sensors and moist-electric generators | 86 |
| WFD | Gly | PDA-coated CNTs PAAm-co-PAAc | Radical polymerization | Strong tissue adhesion and excellent thermal tolerance at −20 °C | Strain sensors and self-adhesive bioelectronics | 87 |
| WFD | Gly | Guar gum | Sol–gel transition | Remain soft and elastic at −16 °C | Ultrafast self-healing ability at room temperature | 88 |
| WFD | Gly | PAAc | Radical copolymerization | Remain tough, stretchable, and twistable at −70 °C | Self-healing at −20 °C | 89 |
| WFD | Gly | PVA/PAAc | Radical copolymerization | Stretchable and twistable at −15 °C | Self-healing at room temperature | 90 |
| WFD | Gly | Gelatin | Solvent exchange | Remain flexible at −45 °C | High mechanical properties, σb ∼ 2 MPa and self-healing | 91 |
| WFD | Gly | PVA | Self-assembly | Remain flexible at −78.5 °C | Mechanically strong σb ∼ 2.5 MPa; self-healable | 92 |
| WFD | Gly | Sodium alginate | Ionic crosslinking | Preserve freshness of fresh-cut cantaloupes at 4 °C | Antifreezing coatings | 93 |
| WFD | Gly | B–O crosslinked sodium alginate | One-pot synthesis | Consistent lubrication at −40 to 60 °C | Heat resistance and switchable tribological performances | 94 |
| WFD | Gly | Silica | Condensation polymerization | Maintain lubricity at −80 °C | Anti-volatilization and anti-corrosion | 95 |
| WFD | Gly | PAAm-BSA | Radical copolymerization and sol–gel transition | Flexible, conductive at −20 °C | Strain sensors | 96 |
| WFD | Gly | Gelatin/PVA | Cyclic freezing–thawing | Reliable freezing-tolerance at −25 °C | Wearable strain sensors | 97 |
| WFD | Gly | Cellulose | Solvent exchange | Tough and flexible at −30 °C | Antimicrobial wound dressings | 98 |
| WFD | Gly | PSBMA-GelMA/QCS | Radical polymerization and solvent exchange | Remain transparent and flexible at −20 °C | Pressure sore prevention and wound dressings | 99 |
| WFD | Gly | Keratin/SA/CCS OHG | Sol–gel transition | Maintain good flexibility at −20 °C | Drug carriers | 100 |
| WFD | Gly | HA/QCS/P(AAm-THMA-SBMA) | Radical copolymerization | Stretchable at −18 °C | Wound dressings | 101 |
| WFD | Gly | BC/P(THMA-AASP) | Radical copolymerization | Flexible at −20 °C | Wound dressings | 101 |
| WFD | Gly and citrate | Gelatin | Solvent exchange | Remain flexible at −80 °C | Mechanically strong (σb ∼ 2 MPa) and conductive | 102 |
| WFD | Gly and ZnCl2 | Cellulose | Sol–gel transition | Reducing ice formation temperature to −100 °C | Stretchable, compressive, and conductive at −60 °C | 25 |
| WFD | DMSO | PAAc | Radical polymerization | Remain flexible, conductive, and adhesive at −40 °C | Flexible electronic devices | 103 |
| WFD | DMSO | GelMA | Sol–gel transition | Vitrification temperature of −111 °C | Cell cryopreservation | 104 |
| WFD | DMSO | L-Pro-GelMA | Sol–gel transition | Lower the freezing point of solution to −4.2 °C | Cell cryopreservation | 105 |
| WFD | Heptane | PDMA/PEGDMA | Radical polymerization and solvent exchange | Reversible elasticity without fracture at −78°C |
Anti-icing coatings | 6 |
| NFI | Sodium alginate | SA-g-DA | EDC/NHS coupling | Water in the coatings remain liquid-like at subzero temperatures | Antifreezing coatings | 106 |
| WFD | DMF and 1,4-dioxane | P[PEGMA-co-(PEGMA-UPy)] | RAFT polymerization | Maintain structural integrity down to −60 °C | Self-healable, injectable and cell cryopreservation | 107 |
| WFD | PROH/EG/trehalose/dextran | Alginate (core–shell structures) | Tube-in-tube capillary microfluidic | Preferentially vitrified during cryopreservation | Cell cryopreservation | 108 |
| IRI & WFD | PCBMA and nanostructures | PCL-b-PCBMA-b-P(PEGMA-UPy) | Sol–gel transition | Freezing point reaching −35.6 °C | Cryo-preservation and injectable drug storage and delivery | 109 |
| IRI & NFI | PDMS grafted polyelectrolyte | P(AAm-co-AAc-co-AAene) | Radical copolymerization, spin-coating and in situ polymerization | Slow ice formation, low ice adhesion at −25 °C | Icephobic surface coatings | 2 |
| WFD & NFI | EG-based polyurethane acrylates | EG-waPUA/PAAm | Radical polymerization | Lower the freezing point to −25 °C and stable storage modulus at −30 °C | Electrolytes for zinc manganese-dioxide battery | 110 |
| WFD & NFI | KOH and ChCl (or betaine) | ChCl/TA or betaine/TA | Supramolecular interaction | Durable and repeatable adhesion at −196 °C | Biomedical adhesives | 111 |
| WFD & NFI | LiBr/PILs | P(VBIMBr-co-AAm) | Radical copolymerization and solvent exchange | Conductive at −20 °C | Supercapacitors with 80% capacitance retention at −20 °C | 112 |
| WFD & NFI | PILs | P(AAm-co-VBIMBr-PEGDA) | Radical copolymerization | Adhesive, strong, and flexible at −20 °C | Antibacterial and antifungal wound dressings | 113 |
| WFD & NFI | PILs | PILs-chitosan | Radical copolymerization and supramolecular interaction | Conductive and compressive at −40 °C | Diabetic wound dressings for real-time monitoring | 114 |
| WFD & NFI | ILs | PVP/P(AAm-co-VBIMBr) | Radical copolymerization | Reliable freezing-tolerance at −50 °C | Strain sensors (ultra-stretchability (ca. 1750%)) | 115 |
| WFD & NFI | LiCl and ILs | PVA/PAS-ILs | Radical polymerization | Flexible and stretchable at −60 °C | Supercapacitors with 75.6% capacitance retention at −60 °C | 116 |
| Antifreezing hydrogels | Tensile strength | Stretchability | Biocompatibility | Environmental impact | Ref. | |
|---|---|---|---|---|---|---|
| Antifreezing reagent | Gel matrix | |||||
| AFPs | P(AAm-co-AMPS) | 25–80 kPa | 1000–2400% | (i) Non-toxic and biodegradable: AFP-based hydrogels support cell adhesion, proliferation, and viability | (i) Biodegradation: AFPs as proteins and naturally can degrade into non-toxic amino acids, reducing long-term environmental persistence | 42 |
| AFPs | P(AAm-co-MAANa)/PVA | 40–80 kPa | 900–2500% | (ii) Minimal immune response: AFPs generally do not trigger adverse immune reactions | (ii) Eco-friendly and wide environmental implications: AFP-based hydrogels can help protect crops from frost damage without harmful chemical residues | 43 |
| INPs | P(AAm-co-HEMA) | — | — | (iii) Network effect: in synthetic polymer networks, toxicity may be a concern, and thorough monomer removal is recommended before further application. In contrast, natural polymer networks (e.g., agar and gelatin) are typically highly biocompatible and biodegradable | (iii) Limited large-scale production challenges: AFPs are typically produced through recombinant biotechnology, which has a lower carbon footprint than chemical synthesis but may require optimization for scalability | 47 |
| AFP and NaCl | Agar/PAAm | 0.15–0.50 MPa | 500–2200% | (iv) Additional antifreezing agent effect: the introduction of salts into the networks can significantly alter their biocompatibility | (iv) Network/additional antifreezing agents concerns: chemically cross-linked networks (serving as the main matrix) and loaded salts (synergistical antifreezing agents), are typically non-degradable, sometimes raising significant environmental concerns | 44 |
| ISP and CaCl2 | PAAc | 0.05–0.9 MPa | 700–2100% | 45 | ||
| ISPs and CNFs | P(AAm-co-AMPS)/PVA | 0.02–0.1 MPa | 200–600% | 46 | ||
| PF patterns | PAAc | — | — | (i) Synthetic hydrophobic polymer coatings are non-biodegradable and exhibit stable antifreezing behavior | (i) Non-biodegradability: hydrophobic polymers are highly stable and resistant to natural degradation, leading to long-term environmental accumulation. However, they may degrade into micro- and nano-scale particles, contributing to microplastic pollution | 48 |
| EGINA network | PVA/(gelatin, agar, or BSA)/EGINA | 0.1–0.65 MPa | 200–3000% | (ii) Incorporating hydrophobic (nano)domains into a hydrogel network typically makes them vulnerable to external conditions, potentially leading to the loss of antifreezing capability due to structural mismatches | (ii) Bioaccumulation & toxicity: some hydrophobic nanodomains could interact with biological membranes, potentially resulting in bio-immune response | 37 |
| DMA/FOSA nanodomain | P(DMA-co-FOSA) | — | — | (iii) Chemically/ionically crosslinked networks formed using unique crosslinkers (e.g., EGINA) are non-biodegradable; however, they can exhibit good biocompatibility as they do not require additional additives | (iii) Water and soil contamination persistent pollutants: hydrophobic polymers tend to repel water, reducing their breakdown in natural environments, leading to long-term contamination of soil and water sources. Moreover, unlike natural polymers, these synthetic materials are difficult to degrade through microbial activity, necessitating energy-intensive treatments like incineration or advanced chemical degradation | 49 and 50 |
| Hydrophobic FOSM | Poly(HEA-FOSM) | — | — | (iv) Sustainable alternative modification for degradability: introducing biodegradable components (e.g., zwitterionic polymers, hydrolysable linkages) can reduce the long-term environmental impact | 40 | |
| Dense H-bonds | P(AAm-co-MAAc) | 30 MPa (−45 °C) | 35% (−45 °C) | 51 | ||
| LiCl and solketal | PAAm-TA-CMC | 48–60 kPa | 700–800% | (i) Salt effect: varies depending on salt type and concentration. Some salts, like NaCl, KCl, CaCl2, are naturally present in biological fluids and are more biocompatible at physiological concentrations. However, high salt concentrations can lead to (1) osmotic stress, which can dehydrate cells and tissues, (2) high cytotoxicity due to high ionic strength disrupting cellular homeostasis, and (3) pH imbalances that can interfere with enzymatic activities and cell viability | (i) Salt-based systems pose risks of long-term environmental accumulation and salinity stress in ecosystems. (1) Excessive salt leaching into soil and water bodies can lead to soil degradation, reduced agricultural productivity, and freshwater contamination. (2) High concentrations of salts, especially LiCl and CaCl2, can disrupt aquatic ecosystems by altering osmotic balance in marine organisms. (3) Salts persist in the environment and accumulate over time, making them difficult to remove. To reduce the environmental impact, we can use biodegradable or zwitterionic polymers to minimize salt leaching, optimize salt concentrations to balance performance and environmental safety, and explore recyclable or controlled-release hydrogel matrices to prevent excessive ion release | 52 |
| LiCl and EG | P(SBMA-HEMA) | ∼8.2MPa (−60 °C) |
∼500% (−60 °C) | (ii) Organic solvent effect: for biocompatibility, generally better than salts, especially with glycerol, which is widely used in cryopreservation. In particular, (1) Glycerol (Gly): commonly used as a cryoprotectant for cells, tissues, and organs. (2) Ethylene glycol (EG): moderately biocompatible but can be toxic at high concentrations. (3) DMSO and betaine compounds: more toxic, especially at higher doses. However, high concentrations of EG or Gly can disrupt cell membranes, with EG exhibiting cytotoxic effects at concentrations exceeding 10–20 wt% | (ii) Organic solvent-based systems vary in impact, with glycerol being the more eco-friendly choice. (1) EG is toxic to aquatic life and can cause water pollution if not properly managed. It is also classified as hazardous waste at high concentrations. (2) Gly (bio-based antifreeze) is considered environmentally friendly, biodegradable, and non-toxic. To minimize environmental impact, we can develop hybrid hydrogel systems that reduce solvent concentrations and incorporate controlled degradation or recycling strategies for long-term sustainability | 53 |
| LiCl and Gly | Silk fibroin/PVA | 0.8–1.8 MPa | 200–400% | (iii) A hybrid approach using biodegradable polymers, controlled-release mechanisms, or alternative green antifreezing agents could enhance sustainability | 54 | |
| LiClO4 and Gly | Hydroxypropyl cellulose/PVA | 130–180 kPa | 220–280% | 55 | ||
| NaCl and Fe3+ | PAAm/PAAc-Fe3+ | 0.69–1.36 MPa | 460–630% | 56 | ||
| NaCl | PAAm-DVB | ∼1.5 MPa | Up to 10200% |
57 | ||
| NaCl | PAAm-CGO | 0.12–1.5 MPa | 800–1400% | 58 | ||
| NaCl/KCl/CaCl2 | PVA | — | — | 59 | ||
| NaCl/CaCl2/FeCl3 | P(VBIPS/HEAA)-SA | 70–115 kPa | 200–255% | 60 | ||
| NaAc | MoS2-PDMAA/PVA | 2–6 MPa | 700–930% | 61 | ||
| KCl | CMCS-OCS-Cu-CDs | — | — | 62 | ||
| KCl/PIL | PIL | 60 kPa | 950% (−20 °C) | 23 | ||
| KAc/ZnAc2 and DMSO | PVA OHG | 4–16 MPa | 810–1040% | 63 | ||
| CaCl2 | Carboxymethyl chitosan | 10–50 kPa | 30–80% | 64 | ||
| CaCl2 | PAAm/alginate | 40 kPa (−20 °C) | 500% (−20 °C) | 22 and 65 | ||
| CaCl2 | Sodium alginate | — | — | 66 and 67 | ||
| CaCl2 and Betaine | Arabic gum/PAAc-CNCs | 0.5–1.1 MPa | 1100–1400% | 68 | ||
| CaSO4 | Alginate-PAAm | — | — | 69 | ||
| ILs and AlCl3 | Starch/PVA | 0.1–0.52 MPa | 110–580% | 70 | ||
| ILs and Gly | Polyglutamic acid-lysine | 0.15–1.1 MPa | 280–2700% | 71 | ||
| DES | P(SBMA-AAc) | 0.08–0.28 MPa | 600–1600% | 72 | ||
| Proline | GG/PAAc/CNCs/PDA | 2–3 MPa | 560–710% | 73 | ||
| EG | PVA-PEDOT:PSS | 0.05–2.1 MPa | 200–1000% | 8 | ||
| EG | PVA/PAAm | 3–15 MPa (Compression strength; −40–25 °C) | 80% (Compression strain) | 74 | ||
| EG | Carrageenan/PAAm/CNTs | 100–160% | 280–320% | 75 | ||
| EG | BP/PVA/borax/sodium alginate | 30–50 kPa | 1700–2200% | 76 | ||
| EG | APTES-SiO2/PVA | 200 kPa (−25 °C) | 250% (−25 °C) | 77 | ||
| EG | P(HEA-AMPS) | ∼320 kPa | 6185% | 78 | ||
| EG | P(NIPAM-co-DMAA) | 0.03–0.12 MPa (Compression strength) | 80% (Compression strain) | 79 | ||
| EG | CMCS/NVP | ∼18 kPa | ∼130% | 80 | ||
| EG | GO/LAP PAAm | — | — | 81 | ||
| EG and CaCl2 | PVA/SF/PPy | 0.02–0.32 MPa | 400∼850% | 82 | ||
| EG and MgCl2/CaCl2 | C-CS/SNFs/TA/PVA | 80–200 kPa | 260–340% | 83 | ||
| EG/Gly/sorbitol & CaCl2 | Alginate/PAAm | 10–40 kPa | 1200–1700% | 84 | ||
| EG and LiCl | PAMPS/PAAm | 0.05–1.32 MPa (−70 °C) | 100–210% (−70 °C) | 85 | ||
| Gly | TA-CNTs-PVA | 0.6–0.95 MPa | 280–360% | 86 | ||
| Gly | PDA-coated CNTs PAAm-co-PAAc | 40–80 kPa | 400–800% | 87 | ||
| Gly | Guar gum | 100–360 kPa | — | 88 | ||
| Gly | PAAc | 0.13–0.26 MPa | 600–2200% (<0 °C) | 89 | ||
| Gly | PVA/PAAc | ∼1.25 MPa | ∼400% | 90 | ||
| Gly | Gelatin | 0.1–2 MPa | 80–460% | 91 | ||
| Gly | PVA | 0.04–0.8 MPa | 280–520% | 92 | ||
| Gly | Sodium alginate | — | — | 93 | ||
| Gly | B–O crosslinked sodium alginate | ∼5.0 MPa | ∼220% | 94 | ||
| Gly | Silica | — | — | 95 | ||
| Gly | PAAm-BSA | 80–410 kPa | 1800–2300% | 96 | ||
| Gly | Gelatin/PVA | 0.47–2 MPa | 350–520% | 97 | ||
| Gly | Cellulose | — | — | 98 | ||
| Gly | PSBMA-GelMA/QCS | 2–6 MPa | 50–150% | 99 | ||
| Gly | Keratin/SA/CCS OHG | 200–600 kPa | 60–75% | 100 | ||
| Gly | HA/QCS/P(AAm-THMA-SBMA) | 150–350 kPa | 400–1500% | 101 | ||
| Gly | BC/P(THMA-AASP) | 200–350 kPa | 1100–1650% | 101 | ||
| Gly and citrate | Gelatin | ∼6 MPa (−30 °C) | ∼400% (−30 °C) | 102 | ||
| Gly and ZnCl2 | Cellulose | 0.12–0.33 MPa | 50–120% | 25 | ||
| DMSO | PAAc | 10–28 kPa | 300–1000% | 103 | ||
| DMSO | GelMA | — | — | 104 | ||
| DMSO | L-Pro-GelMA | — | — | 105 | ||
| Heptane | PDMA/PEGDMA | 15–200 kPa | 60–100% | 6 | ||
| Sodium alginate | SA-g-DA | — | — | 106 | ||
| DMF and 1,4-dioxane | P[PEGMA-co-(PEGMA-UPy)] | — | — | 107 | ||
| PROH/EG/trehalose/dextran | Alginate (core–shell structured) | — | — | 108 | ||
| PCBMA and nanostructures | PCL-b-PCBMA-b-P(PEGMA-UPy) | — | — | 109 | ||
| PDMS grafted polyelectrolyte | P(AAm-co-AAc-co-AAene) | — | — | 2 | ||
| EG-based polyurethane acrylates | EG-waPUA/PAAm | 21–120 kPa | 230–1040% | 110 | ||
| KOH and ChCl (or betaine) | ChCl/TA or betaine/TA | 1–10 kPa | — | 111 | ||
| LiBr/PILs | P(VBIMBr-co-AAm) | 20–40 kPa | 2–83% | (i) Biocompatible ILs (e.g., choline-based ILs and amino acid-derived ILs) generally exhibit low toxicity and good compatibility with biological systems. (1) Some ILs (e.g., choline chloride-glycerol deep eutectic solvents) have been explored for biomedical applications, including cryopreservation. (2) Chemical modification of these biocompatible ILs into PILs can enhance hydrophilicity, allowing them to integrate into the hydrogel network, further reducing cytotoxic effects | Some ILs persist in the environment and pose ecotoxic risks, but biodegradable ILs offer more sustainable options | 112 |
| PILs | P(AAm-co-VBIMBr-PEGDA) | 30–40 kPa | 48–100% | (ii) Toxic ILs (e.g., imidazolium-, pyridinium-based ILs) can disrupt cell membranes and exhibit cytotoxicity at high concentrations (1) Long-term exposure may affect cellular metabolism and enzymatic activity. (2) Higher toxicity in aquatic environments compared to traditional solvents | (1) Many conventional ILs (e.g., imidazolium- and pyridinium-based) are non-biodegradable and persist in the environment | 113 |
| PILs | PILs-chitosan | 0.32–0.86 MPa | 150–260% | (2) Anions such as PF4− (hexafluorophosphate) and BF4− (tetrafluoroborate) can hydrolyze into toxic byproducts, contributing to environmental hazards | 114 | |
| ILs | PVP/P(AAm-co-VBIMBr) | 28–150 kPa | 1800–2700% | (3) ILs can disrupt microbial ecosystems, affecting soil and water remediation processes | 115 | |
| LiCl and ILs | PVA/PAS-ILs | 0.2–1.9 MPa | 170–320% | Considering sustainability and green chemistry, we should prioritize: (1) using biodegradable ILs (e.g., choline- or amino acid-based), which can reduce environmental risks, (2) designing task-specific ILs with reduced toxicity and enhanced degradation potential, and (3) developing IL–polymer hybrid systems to limit free IL release into the environment | 116 | |
2. Antifreezing mechanisms of hydrogels
Since ice nucleation, which involves the formation of a stable nucleus of critical size, is the controlling step in ice formation, inhibiting ice nucleation at the early stage is crucial for designing effective antifreezing strategies. Although our understanding of antifreezing mechanisms at the molecular level remains incomplete, nature offers numerous examples of organisms, including certain fish, insects, and amphibians, which produce AFPs or IBPs within their bodies to protect themselves from freezing.30,117 These natural adaptations provide valuable insights into developing effective antifreezing strategies.AFPs, also known as a subclass of IBPs, function by modulating the growth of existing ice crystals in various ways. AFPs absorb onto the surface of ice crystals and prevent their growth, leading to the depression of the freezing point of water. Additionally, AFPs possess a strong capability to inhibit ice recrystallization (IRI). This IRI activity prevents many small, often harmless, crystals from ripening into large crystals. Another natural strategy for preventing freezing in organisms involves the accumulation of solutes such as Gly and sugars, which lower the freezing point of bodily fluids through colligative properties. Mechanistically, Gly and sugars act as effective cryoprotectants by forming hydrogen bonds with water molecules. This bonding reduces the mobility of water, thereby hindering ice formation. Additionally, these solutes increase the viscosity of the bodily fluids, which helps maintain fluidity of their cellular and extracellular fluids. This increased viscosity adds another layer of protection for vital biological functions even at subzero temperatures.
Both key mechanisms observed in nature—lowering the freezing point and inhibiting ice recrystallization—have inspired the development of synthetic antifreezing hydrogels. By mimicking these natural strategies, some design principles behind antifreezing hydrogels aim to effectively prevent ice formation and recrystallization by incorporating antifreezing additives, such as natural AFPs and cryoprotective solutes, into the polymer matrix. Parallel to biomimetic approaches, another design strategy for antifreezing hydrogels involves the design and engineering of polymer compositions and network structures, aiming to control the specific interactions between water molecules and polymer networks. These strategies include (1) the design of tightly water-bound polymers via hydrogen bonds for hydrophilic polymers, such as poly(acrylates), poly(acrylamides), and poly(ethylene glycol) (PEG), or through ionic solvation for zwitterionic polymers, such as poly(carboxybetaine) (PCB), poly(sulfobetaine) (PSB), and poly(phosphatidylcholine) (PPC), (2) the use of various network topologies, such as interpenetrating networks (IPNs), semi-IPNs, double networks, nanocomposite networks, branched networks, and gradient networks, and (3) the introduction of different crosslinking chemistries (chemical crosslinking and physical crosslinking). These synthetic design strategies primarily focus on intrinsic properties of polymer chemistry and physics, not on antifreezing additives, aiming to explore the antifreeze performance of hydrogels by optimizing their structural and dynamic interactions with water. Taken together, these antifreezing design strategies can be categorized into two main groups: the addition of antifreezing additives (such as AFPs, salts, ions, and organic solvents) and the engineering of polymer networks. These strategies specifically target the water-to-ice transformation by employing two primary antifreezing mechanisms: lowering the freezing point of water and inhibiting the crystallization of ice.
2.1. Water freezing point depression (WFD)
This mechanism directly impacts the thermodynamic properties of water to reduce its freezing point through both colligative and non-colligative methods. AFPs, inspired by natural processes, achieve this by lowering the freezing point without significantly altering the melting point, thus creating a temperature gap known as thermal hysteresis (TH). Temperatures within this TH gap represent a non-freezing range. The freezing point depression effect induced by AFPs is facilitated through a non-colligative, adsorption–inhibition mechanism.118,119 From the perspective of water transitioning to ice, the adsorption–inhibition mechanism reveals how water molecules adsorbed on the ice-binding face (IBF) facilitate the formation of an ice-like lattice structure, controlled by the geometric arrangement of hydrogen bonds, which promotes ice nucleation. In contrast, when water molecules adsorb on a non-IBF surface, this adsorption disrupts the hydrogen-bonding pattern between the adsorbed water molecules and adjacent non-adsorbed water molecules, inhibiting the formation of ice-like water structures on the non-IBF and thus depressing ice nucleation.To implement this mechanism in systems that include antifreezing additives, the optimal strategy should consider two key factors. The first involves using antifreeze-mimicking additives that selectively adsorb onto the IBF of ice-like surfaces. This selective adsorption induces local curvature on the ice-like surface, disrupting the regular lattice structures of adjacent ice-like water molecules. This disruption makes it harder for water molecules to integrate into the ice lattice, thereby effectively retarding ice nucleation. Meanwhile, the second factor ensures that water molecules on non-IBF surfaces remain unaffected by antifreezing additives, thus preserving their non-ice-like structure. Collectively, the antifreezing additives on the IBF and the water molecules on the non-IBF work together to prevent freezing, as the adsorption–inhibition mechanism and the formation of a curved ice surface reduce the freezing temperature through the Kelvin effect.119 For the colligative mechanism, this suggests that adding synthetic substances, such as salts, Gly, EG, propylene glycol, salts/ions, polymers, and two-dimensional materials,3,120 increases the solute concentration in water, which lowers the freezing point through colligative effects. This mechanism does not necessitate any specific interaction between the substance and ice; rather, it depends solely on the number of particles dissolved in the solution.
2.2. Ice recrystallization inhibition (IRI)
Fundamentally different from mechanisms that lower the freezing point, IRI does not alter the temperature at which ice forms. Instead, it significantly influences how ice crystals grow. Ice recrystallization is a process where large ice crystals grow at the expense of smaller ones, typically during the thawing process of a frozen substance. This results in an increase in the average crystal size and a reduction in the overall number of crystals, facilitating the overall ice growth. IRI molecules, including AFPs and synthetic mimics,6 adhere to small ice crystals and alter their normal growth patterns. This binding specifically inhibits the crystals’ ability to grow larger by preventing water molecules from attaching to their lattice structures. Moreover, different IRI molecules have distinct preferences and affinities for various crystallographic planes. This selective binding not only prevents recrystallization but also inhibits further growth from certain planes, while allowing other planes that do not absorb these molecules to continue growing freely, resulting in diverse ice shapes. By controlling ice recrystallization and shaping microstructural changes, IRI agents effectively prevent the formation of large, sharp ice crystals, thus modifying the structural characteristics of the ice. Furthermore, the adsorption–inhibition mechanism, initially derived from the antifreezing effects of AFPs in solutions or on substrates, may not comprehensively or precisely explain the transition from water to ice in confined networks when induced by additives. The structure of hydration water on solid surfaces or in solutions differs markedly from that within polymer networks. Therefore, the adsorption of IRI molecules on non-IBF surfaces could also impact the formation of a hexagonal ice-like structured hydration layer, thereby inducing IRI activity.2.3. Network freezing inhibition (NFI)
In parallel to traditional antifreezing mechanisms such as water freezing point depression and IRI that rely on antifreezing additives, there exists an alternative non-additive strategy. This involves engineering polymer chemistry, network structures, and crosslinking types to inhibit ice nucleation, recrystallization, and growth. This approach requires sophisticated consideration of how network chemistry, structure, and confinement influence water structures, dynamics, and interactions. Despite its potential, relatively less research has been conducted on this strategy. Inspired by the amphiphilic nature of AFPs in inhibiting ice, the design of amphiphilic polymers with an optimal hydrophilic–hydrophobic ratio is utilized to suppress ice crystal growth. In this strategy, hydrophilic polymer networks serve as scaffolds and integrate with the embedded hydrophobic domains. In these amphiphilic gels, highly hydrophilic polymers such as polysaccharides, PEG, and polyvinyl alcohol (PVA) strongly bind to water molecules, forming extensive hydrated networks to provide structural and mechanical support. Concurrently, hydrophobic domains form nanoscale cavities that isolate weakly bound water molecules from those tightly bound to hydrophilic compounds, enhancing the robustness of the confinement effect and further inhibiting ice propagation and growth. This freeze-tolerance mechanism functions similarly to the IRI activity of AFPs, which also feature spatially segregated hydrophobic and hydrophilic domains within cellular structures. In addition to amphiphilic polymers, polyampholyte gels made of positively and negatively charged monomer subunits, and polyzwitterionic gels with equal ratios of positively and negatively charged groups in one side chain, can promote strong ionic solvation around their charged sites.121 This ionic solvation effectively traps water molecules within gel networks, preventing them from forming ice crystals. Consequently, the growth of ice crystals is significantly hindered, enhancing the antifreezing properties of these gels.In addition to engineering polymer chemistry, such as adjusting the balance between hydrophobic and hydrophilic groups or varying the ratio between positive and negative groups, another strategy for engineering polymer physical properties involves regulating the size of porous structures to micro- or nano-scale dimensions within hydrogels. This approach focuses on adjusting porous sizes and crosslinking density to optimize the antifreezing performance. First, the micro-/nano-scale porous meshes confine water molecules, disrupting the efficient alignment of hydrogen bonds necessary for forming the crystalline structure of ice. Previous studies on water in nanopores have demonstrated that when confinement is less than 10 nm, water does not form ordered ice crystals and remains in an amorphous state instead.122,123 In water-swollen, cross-linked polymer networks, which contain microscopic pores comparable in scale to nanoconfinement, it has been observed that when the polymer mesh nanodomain is less than 3 nm, ice formation is almost completely inhibited in supramolecular hydrogels.40,49 Second, the crosslinking density of polymer networks can be adjusted through variations in crosslinker concentrations and network topologies. It has been observed that DN hydrogels exhibit better antifreezing efficiency compared to SN hydrogels when crosslinked by the same molecules.37,124 This is likely because DN hydrogels possess tighter and more narrowly configured network structures than SN hydrogels, which enhances the confinement effect on the water-to-ice transition. Due to the strong nanoscale confinement between polymer chains and the intense interactions between water molecules and these chains, confining water effectively minimizes contact with hydrophobic surroundings and maximizes interaction with hydrophilic areas of the materials, thereby preventing freezing.
Taken together, the three fundamental antifreezing mechanisms—water freezing point depression, IRI, and network freezing inhibition—differ in significant ways (Fig. 2). WFD directly lowers the water freezing point through colligative and non-colligative effects, changing the temperature at which water transitions into ice and thus effectively altering its natural freezing threshold. This adjustment of physical properties via solute addition impacts the water's colligative properties. WFD is particularly useful in applications requiring rapid thermal responses without significant alteration of the water's physical state, proving effective in various industrial processes where control over freezing temperature is essential. Unlike WFD, IRI does not modify the freezing point itself but influences the formation and growth patterns of ice crystals after freezing has commenced. IRI involves subtle interactions with ice crystals, affecting their size, shape, and structure, and thus controls the microstructural changes within the freezing environment. This mechanism is vital in cryopreservation and food storage, where it is crucial to maintain the integrity of cell structures and textures during thawing. IRI works by preventing the aggregation of small ice crystals into larger ones, thereby controlling the microstructure of ice. NFI employs a different strategy by utilizing the strategic engineering of polymer chemistries and network structures to directly inhibit ice nucleation and growth. This approach is particularly beneficial in biomedical applications where precise manipulation of ice formation is necessary to prevent damage to cellular structures. NFI's effectiveness stems from its ability to adapt hydrogel structures to prevent ice formation, leveraging the unique properties of amphiphilic polymers or polyampholyte gels to influence water interactions at the molecular level. These differences highlight the distinct approaches each mechanism employs in adapting hydrogel structures to prevent ice formation and underscore their unique contributions to antifreezing strategies. It should be noted that these three mechanisms are not mutually exclusive; instead, they can work together to achieve a synergistic antifreezing effect.
 | ||
| Fig. 2 Schematic illustrating the three fundamental antifreezing mechanisms. (a) Lowering the water freezing point, which changes the temperature at which water freezes, (b) ice recrystallization inhibition, which affects the growth and structure of ice crystals after freezing has occurred, and (c) network freezing inhibition, which utilizes polymer chemistry (hydrophobic–hydrophilic ratio and charged groups) and network structures (porous mesh size and crosslinking density) to enhance the confinement effect that prevents water from freezing. | ||
3. Design strategies for different types of antifreezing hydrogels
3.1. Incorporation of antifreezing biomolecules
Living organisms have evolved remarkable freeze tolerance to survive at extremely low temperatures, utilizing both colligative125 and non-colligative126 strategies to prevent osmotic stress and ice formation. They produce colligative metabolites such as glucose, Gly, trehalose, betaine, proline, and polyols to lower fluid freezing points and stabilize cellular structures, thereby protecting against ice formation and osmotic stress.127 Additionally, IBPs as non-colligative cryoprotectants, particularly AFPs, impede ice growth and enhance freeze tolerance by shielding cells, tissues, and organs from ice-induced damage like membrane disruption and osmotic shock during freezing and thawing.128 Given their unique antifreezing properties, it is a logical strategy to incorporate them into the polymeric matrix of hydrogels, thereby endowing the hydrogels with enhanced antifreezing capabilities.AFPs expressed in microbes, plants, fish, algae, and insects, exhibit structural and functional diversity, categorized into α-helical, β-strand-rich (β-clips and β-solenoids), lectin-like, and polyproline II helices based on structural characteristics (Fig. 3a).129 AFPs in different organisms play different roles, including freeze tolerance, ice structuring, and ice adhesion, extending beyond their original function of preventing organismal freezing.128 For example, fish AFPs typically appear as isoforms with varying antifreeze activities, distinguished by high alanine (Ala) content that contributes to a distinct α-helical structure.130,131 The Ala-rich face of these AFPs preferentially binds to ice through hydrophobic interactions, inhibiting ice crystal growth.132 In addition to inhibiting ice crystals directly, the hydrophilic residues or domains of AFPs can form hydrogen bonds with polymer chains, increasing crosslinking density. This enhanced crosslinking restricts water molecule mobility, limits water–water interactions, and inhibits the formation of ice-like lattice structures.37 Concurrently, the aggregation of hydrophobic residues or domains creates physical barriers that isolate small ice crystals, preventing them from coalescing into larger crystals. These naturally evolved antifreezing mechanisms provide a variety of strategies for developing biomolecule-incorporated hydrogels with enhanced antifreeze properties.12
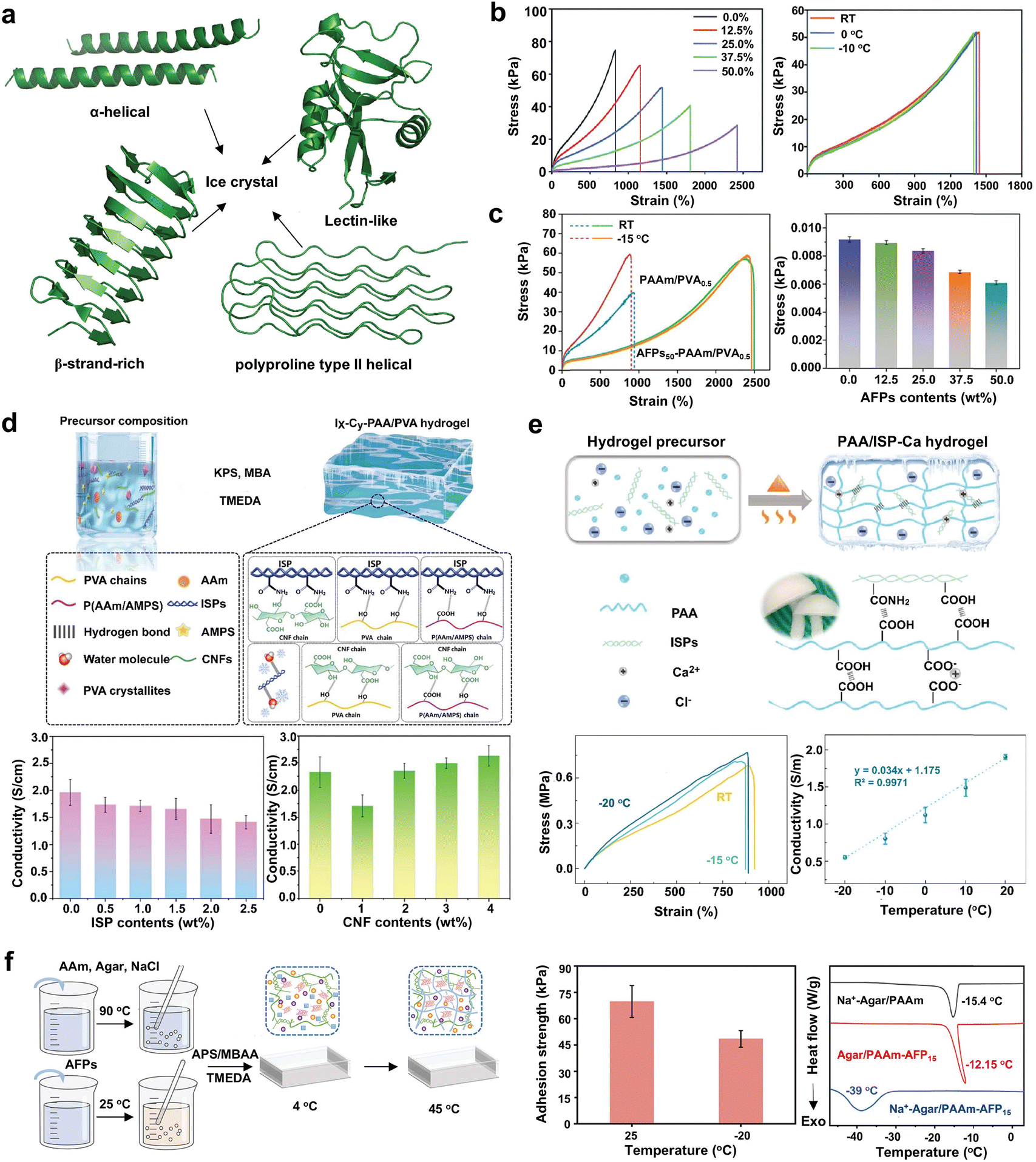 | ||
| Fig. 3 Bioinspired design of antifreezing hydrogels through natural antifreezing biomolecule integration. (a) Representative structures of AFPs showcasing their structural diversity: α-helical (PDB: 1WFA), β-strand-rich (PDB: 1M8N), lectin-like (PDB: 2PY2), and polyproline type II helices (PDB: 7JJV). Each type exhibits preferential ice-binding properties. (b) AFPs as plasticizers in the P(AAm/AMPS)-AFP network, with reduction in tensile strength and enhanced stretchability correlating with increased AFP content. Optimal mechanical performance at 25.0 wt% AFP, consistent across room temperature, 0, and −10 °C. Reproduced with permission from ref. 42, Copyright 2020 Royal Society of Chemistry. (c) AFP-PAAm/PVA hydrogels consisting of a chemically crosslinked P(AAm/MAANa) network and a physically crosslinked PVA network with natural AFPs. Stable mechanical properties between room temperature and −15 °C, and reduced conductivity at higher AFP content, due to the limited ion mobility within the compact hydrogel network. Reproduced with permission from ref. 43, Copyright 2022 Elsevier. (d) Natural ISPs and CNF-incorporated PAAc/PVA hydrogels, where CNFs mitigate conductivity reduction from compact gel structure due to ISPs. Reproduced with permission from ref. 46, Copyright 2024 Elsevier. (e) ISPs and CaCl2-incorporated PAAc hydrogels. Low-temperature adaptability with stable tensile strength across room temperature, −15, and −20 °C and a high conductivity of 0.50 S m−1 at −20 °C. Reproduced with permission from ref. 45, Copyright 2021 Elsevier. (f) AFP and NaCl-incorporated Agar/PAAm DN hydrogel, with enhanced adhesion at −20 °C and reduced ice crystal formation temperature at −39 °C. Reproduced with permission from ref. 44, Copyright 2024 Elsevier. | ||
The incorporation of AFPs into hydrogel systems has shown promise, particularly in an anti-icing hydrogel sensor characterized by its low-temperature adhesion and toughness. This was achieved by copolymerizing fish AFP with acrylamide (AAm) and 2-acrylamide-2-methylpropanesulfonic acid (AMPS), producing a P(AAm/AMPS)-AFP hydrogel that exhibited stable mechanical performance, including toughness and adhesiveness, at −10 °C.42 AFP acts as a plasticizer by forming multiple weak bonds within the network, which reduces tensile strength and enhances stretchability as its concentration increased (Fig. 3b). As the AFP content increases from 0 to 50 wt%, the crystallization temperature of the hydrogel decreases from −10.48 to −20.08 °C. The hydrogel exhibits optimal mechanical properties, including 510 N m−1 adhesive strength, ∼52 kPa fracture stress, and ∼1400% fracture strain at 25.0 wt% AFP content. These properties consistently maintain across various temperatures—room temperature, 0, and −10 °C—demonstrating their exceptional stability and freezing tolerance (Fig. 3b). In another study, fish AFPs are integrated into a hydrogel system composed of a chemically crosslinked poly(AAm/sodium methacrylate) (P(AAm/MAANa)) network and a physically crosslinked PVA network, conferring freeze tolerance at −15 °C.43 The inclusion of AFPs also enhances mechanical properties, achieving a peak fracture strength of 79 kPa and a fracture strain of 2358% with a formulation containing 37.5 wt% AFPs and 0.5 w/v% PVA (AFPs-PAAm/PVA). As the AFP content increases from 0 to 50 wt%, the crystallization temperature drops from −16.1 to −28.7 °C, while the mechanical performance remains stable between room temperature and −15 °C for the AFPs-PAAm/PVA hydrogel (Fig. 3c). The protein structure of AFPs, rich in amino acid groups, enhances the gel's adhesiveness and structural integrity, promoting swift recovery. However, higher concentrations of AFPs can decrease conductivity, due to the reduced ion mobility within the denser network. To improve conductivity and antifreezing properties in the conductive hydrogel, cellulose nanofibers (CNFs) with adjustable surface charge characteristics are utilized alongside ice structuring proteins (ISPs, another term for AFPs).46 CNFs, with their functional surface groups, provide numerous ion transfer sites, and their one-dimensional structures facilitate fast ion movement, enhancing ionic conductivity. When integrating into a chemically crosslinked network of PAAm and PAMPS, and physically crosslinked PVA, the resulting ISP-CNF-P(AAm/AMPS)/PVA hydrogels demonstrate high conductivity (reaching 2.63 S m−1) and maintain performances at the temperature as low as −10 °C (Fig. 3d).46
To further enhance the antifreezing performance of hydrogels, a dual approach utilizing colligative CaCl2 and non-colligative ISPs was developed to simultaneously inhibit ice nucleation and growth within an ionic conductive hydrogel.45 Through one-pot, in situ polymerization of acrylic acid (AAc) with CaCl2 and ISPs, the resulting hydrogel demonstrates an excellent stretchability of 890% and conductivity of 0.50 S m−1 at −20 °C, maintaining robust performance at both room temperature and −20 °C (Fig. 3e).45 Similarly, in another study, a combination of type I AFP from cod, NaCl, and an advanced Agar/PAAm double network design was utilized, achieving a robust balance of mechanical strength (50 kPa on aluminum substrates) and excellent electrical conductivity (ca. 1.8 S m−1) even at temperatures down to −20 °C.44 When AFP and NaCl are integrated into the Agar/PAAm double network to create a double cross-linked composite hydrogel (Na+-Agar/PAAm-AFP), they display a significantly lower phase transition point of −39 °C, compared to −15.4 °C for Na+-Agar/PAAm and −12.15 °C for Agar/PAAm-AFP hydrogels (Fig. 3f).44
Despite the enhanced antifreezing properties conferred by biomolecules like AFPs or ISPs, their integration into hydrogel networks presents challenges, including the high cost and complexity associated with protein extraction or total synthesis.133,134 Consequently, researchers are developing more cost-effective synthetic analogues of AFPs, unitizing water-soluble polymers to broaden potential applications.18,135,136 Understanding the mechanism of AFPs has guided the design of polymeric mimics and antifreezing hydrogels. AFPs inhibit ice crystal growth through preferential adsorption onto the ice surface, mediated by their hydrophilic domains, while their hydrophobic domains aggregate in water, creating spatial confinement to inhibit ice nucleation.137 The amphiphilic nature of AFPs simultaneously inhibits ice nucleation and reduces ice adhesion by managing interfacial water dynamics.3 This dual functionality has inspired the development of amphiphilic polymers that mimic AFPs, enabling the creation of antifreezing hydrogels.
To achieve multifaceted antifreezing activities, a novel approach integrates hydrophobic and hydrogen-bonding functionalities within a hydrophilic polyelectrolyte network—poly(AAm-co-AAc-co-N-allylacrylamide) (poly(AAm-co-AAc-co-AAene)). This network acts as a scaffold for grafting hydrophobic polydimethylsiloxane (PDMS) and cationic charged chains (poly[2-(methacryloyloxy)ethyl]trimethylammonium chloride, PMETAC),2 creating a multifunctional hybrid hydrogel designed to inhibit ice nucleation, prevent ice propagation, and reduce ice adhesion, thereby providing comprehensive anti-icing capabilities. By adjusting the hydrophobicity through variations in PDMS chain lengths and employing ion-specific effects via ion exchange reactions, this approach controls the properties of interfacial water, enhancing the hydrogel's anti-icing performance (Fig. 4a). The tailored hydrogel surface adjusts ice nucleation, propagation, and adhesion effectively, achieving functionalities akin to AFPs. This anti-icing hydrogel demonstrates impressive characteristics, including an ice nucleation temperature below −30 °C, an ice propagation rate less than 0.002 cm2 s−1, and an ice adhesion strength below 20 kPa, positioning it as a highly effective multifunctional anti-icing material.
 | ||
| Fig. 4 Bioinspired design of antifreezing hydrogels utilizing polymeric antifreezing biomolecule mimics. (a) AFP-inspired poly(AAm-co-AAc-co-AAene) hydrogels crosslinked by NG (PMETA), SG (PMETA-co-PDMS), and PG1, PG2, and PG3 [(PMETAC)-g-(PDMS-0.9 K, 5.7 K, 13.2 K)]. Reproduced with permission from ref. 2, Copyright 2020 Elsevier. Hydrophobic and hydrogen-bonding groups regulate interfacial water, controlling ice nucleation, propagation, and adhesion by tuning structure, mobility, and water interface amount. (b) AFP-mimetic acidic (PAAc-D-S) hydrogel electrolyte with chemically crosslinked PAAc, DMSO, and H2SO4, exhibiting stable storage modulus (E′), high ionic conductivity (12.0 mS cm−1), and strong interfacial adhesiveness on various surfaces at temperatures down to −40 °C. Reproduced with permission from ref. 103, Copyright 2020 Elsevier. | ||
Utilizing the ice nucleation inhibition mechanism of AFPs, a high-performance antifreezing hydrogel electrolyte was synthesized via a one-pot, photo-polymerization of AAc in the DMSO and water solvent.103 Mimicking AFPs, the carboxyl groups of PAAc and the thioacyl groups of DMSO act as hydrogen bond donors and acceptors, respectively, enhancing the hydrophobicity through the PAAc backbone and methyl groups in DMSO. The PAAc-DMSO hydrogel, featuring dense hydrogen bonds and hydrophobic domains, effectively impedes ice crystallization and binds water molecules to maintain ion conduction channels. This polymer network configuration lowers the operating temperature from −15 °C in the pure PAAc hydrogel to −40 °C, while preserving excellent flexibility, high ionic conductivity (up to 12.0 mS cm−1), and strong adhesion to various substrates (Fig. 4b). These properties render the hydrogel electrolyte ideal for constructing supercapacitors with significant capacitance at extremely low temperatures.
Despite advancements in AFP-incorporated antifreezing hydrogels, several challenges persist, particularly concerning the incorporation of antifreezing proteins. AFP activity is susceptible to environmental variables like pH and temperature,138,139 which can compromise the stability and efficacy of these hydrogels. Moreover, the limited availability of natural biomolecules constrains the development of robust and economically viable hydrogel materials.134 Consequently, there is a critical need to develop synthetic, stable AFP mimics that provide effective antifreezing functionality for hydrogel applications. Beyond polymeric AFP mimics, research has extended to small molecular AFP analogues, such as C-linked antifreeze glycoprotein analogues,140 which offer enhanced stability and easier synthesis. Additionally, potent non-ice-binding molecules such as carbohydrate derivatives and para-methoxyphenyl-β-D-glycosides141,142 have demonstrated their ability to decrease the mean ice grain size at millimolar concentrations, expanding the scope of antifreezing agents. Furthermore, novel AFP analogues, like de novo polypeptides,143,144 peptoids,145 and other AFP analogues, have shown thermal hysteresis and ice recrystallization inhibition activities, though their effectiveness remains lower compared to natural AFPs. The integration of these analogues into antifreezing hydrogels is still emerging, highlighting a gap in current research. This area calls for further exploration, especially to enhance the mechanical strength and robustness of hydrogels. Future research could focus on optimizing AFP mimics, refining hydrogel network design, and modifying composite scaffolds to overcome these limitations, aiming to develop advanced antifreezing hydrogels.
3.2. Incorporation of ionic components
A popular and straightforward strategy to endow hydrogels with antifreezing properties involves incorporating ionic compounds, such as antifreezing inorganic salts (e.g., NaCl, LiCl, CaCl2, ZnCl2, and KAc) and ILs.22,25,146,147 Fundamentally, the ionic compounds in hydrogels, such as LiCl, NaCl, CaCl2, and ZnCl2, lower the freezing point of water through freezing point depression, a process stemming from the colligative properties of highly concentrated ion solutions. Upon dissolution, these inorganic salts bind tightly to water molecules, disrupting hydrogen bonding among them and inhibiting ice formation. This mechanism, similar to how seawater remains liquid at subzero temperatures due to its salt content, prevents ice crystal formation within the polymer matrix, thereby enabling the hydrogels to remain flexible and stretchable even under subzero conditions. Dissociated Li+ and Cl−, for instance, can strongly coordinate with four and six water molecules,148 respectively, leading to a depression in freezing points compared to pure water, with a cryoscopic constant of 9.25 °C kg mol−1.149 Each salt exhibits a unique eutectic point, reflecting its antifreezing potential. For instance, NaCl can lower the freezing point of water to −21.6 °C,150 while ZnCl2 reaches as low as −73 °C.25 LiCl demonstrates a minimum freezing temperature of −75.5 °C at 25.3 wt% concentration, underscoring its excellent antifreezing capability. Given the high-water content of hydrogels, equilibrating them in these salt solutions is a straightforward and environmentally friendly strategy to impart antifreezing properties.Unlike other antifreezing reagents, high salt concentrations within hydrogels facilitate the formation of conductive pathways, enabling ionic conductivity. This unique property has led to the widespread use of salts in the development of antifreezing conductive hydrogels.58,151–155 For example, incorporating NaCl (2 M) into the PAAm/PAAc-Fe3+ hydrogel largely reduced the freezing point of water within the hydrogel from −12.4 to −24.7 °C, while also endowing a conductivity of ∼0.72 S m−1 (Fig. 5a).56 Similarly, adding LiCl directly into the hydrogel precursor solution of a PAAm-hydroxyl cellulose (HEC) hydrogel before polymerization lowered the phase transition temperature from −11 to −62 °C as the LiCl concentration increased from 0 to 3 M (Fig. 5b). This modification allows the hydrogel to function as a triboelectric nanogenerator, even at subzero temperatures as low as −20 °C.156 Additionally, CaCl2 can effectively reduce the freezing point of the carboxymethyl chitosan (CMCS) hydrogel from −27 to −51 °C with CaCl2 concentrations ranging from 1 to 3 M (Fig. 5c). The resulting CMCS–CaCl2 hydrogel remained flexible and transparent at an ultralow temperature of −50 °C, while CMCS hydrogels without CaCl2 became rigid and lost their deformability.64 However, the mechanical strength of the single CMCS hydrogels is limited, with a tensile fracture strength of 52 kPa at the strain of 50%.
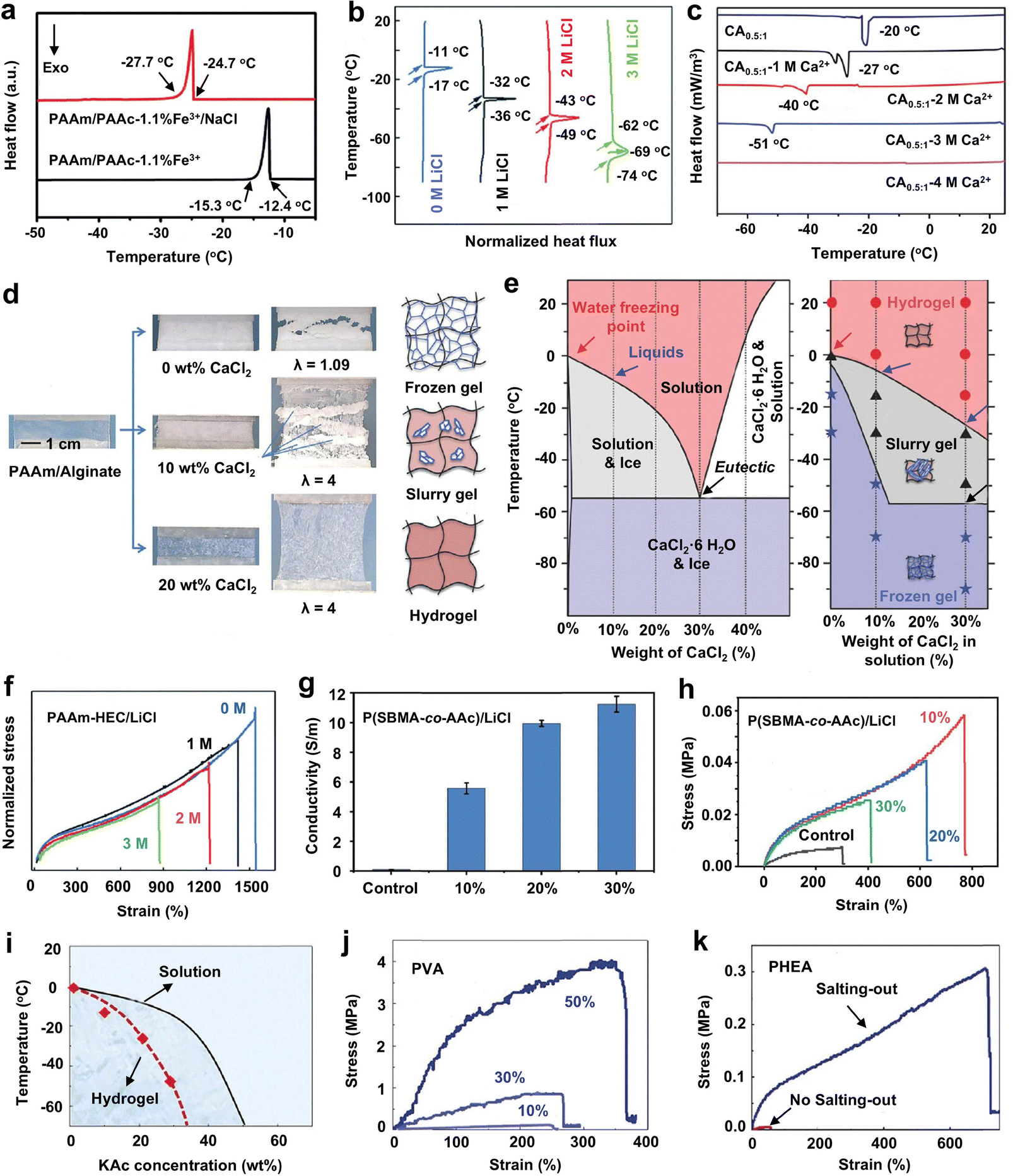 | ||
| Fig. 5 Inorganic salt-incorporated antifreezing hydrogels. (a) PAAm/PAAc-Fe3+ hydrogel: NaCl addition lowers the freezing point from −12.4 to −24.7 °C. Reproduced with permission from ref. 56, Copyright 2020 Royal Society of Chemistry. (b) PAAm-hydroxyl cellulose hydrogel with LiCl: freezing temperature drops from −11 to −62 °C as LiCl concentration increases from 0 to 3 M. Reproduced with permission from ref. 156, Copyright 2020 Royal Society of Chemistry. (c) Carboxymethyl chitosan hydrogel with CaCl2: freezing point decreases from −20 to −51 °C with increasing CaCl2 up to 4 M. Reproduced with permission from ref. 64, Copyright 2022 American Chemical Society. (d) Hydrogel states at −15 °C: 0 wt% CaCl2 fully frozen and fractures; 10 wt% CaCl2 in a semi-frozen slurry state remains stretchable; 30 wt% CaCl2 maintains a flexible, unfrozen state. (e) Phase diagram comparison: CaCl2 aqueous solution vs. CaCl2 incorporated PAAm/alginate hydrogel. Reproduced with permission from ref. 22, Copyright 2018 Wiley-VCH. (f) PAAm-HEC hydrogel with LiCl: “Salting-in” effect reduces mechanical strength as salt concentration increases. Reproduced with permission from ref. 156, Copyright 2020 Royal Society of Chemistry. (g) P(SBMA-co-AAc) hydrogel with LiCl: improved conductivity with increasing salt concentration. (h) PAAc with LiCl: optimal mechanical strength at 10 wt% LiCl “polyelectrolyte effect” on PAAc and “anti-polyelectrolyte effect” on PSBMA. Reproduced with permission from ref. 157, Copyright 2021 Elsevier. (i) Phase diagram: “Salting-out” KAc solution vs. KAc-incorporated PVA hydrogel shows effective freezing point depression. Both KAc-incorporated (j) PVA hydrogels and (k) PHEA hydrogels with improved mechanical strength. Reproduced with permission from ref. 53, Copyright 2022 Springer Nature. | ||
It is crucial to understand how the salt solution influences the balance between antifreezing performance and the mechanical properties of the corresponding hydrogels. Evidently, increasing the CaCl2 concentration from 0 to 30 wt% in PAAm-alginate hydrogels transitions the gel from a frozen state to a flexible state at −15 °C (Fig. 5d). This phase behavior analysis demonstrates that freezing temperatures decrease as salt concentration increases, thus enhancing antifreezing properties. The mechanical property of the hydrogel at a fixed concentration is dependent on the phase of CaCl2 aqueous solution: it remained flexible and tough when in the liquid phase (red regions in Fig. 5e). As the temperature decreases and CaCl2 transitions to a slurry phase, the hydrogel becomes semi-frozen yet still stretchable (gray regions). At even lower temperatures, the hydrogel becomes brittle as the aqueous phase is fully frozen (purple regions). This intermediate slurry state allows the hydrogel to maintain an excellent tensile stress of 0.25 MPa and a tensile strain of 500% at −50 °C with a 30 wt% CaCl2 solution.22
Importantly, various studies have reported conflicting effects of adding inorganic salts on the mechanical properties of hydrogels. In some instances, the addition of salts can compromise the mechanical properties of hydrogels.25,154,158,159 Salts not only disrupt the bonding among water molecules but also alter interactions between polymer chains, potentially affecting the overall network structure of the hydrogel. For example, in CaCl2-based PAAm-alginate hydrogels at 20 °C, the fracture stress decreases from 0.38 to 0.1 MPa as the CaCl2 concentration increases to 30 wt%, likely due to the “salting-in” effect of Cl− ions.22 Similarly, in PAAm-HEC hydrogels, increasing LiCl concentrations lead to reduced strength and toughness (Fig. 5f). Therefore, a moderate LiCl concentration (1 M) is recommended to achieve a balanced combination of antifreezing performance and mechanical durability in practical applications.156 But, in other instances, ionic compounds enhance both antifreezing performance and mechanical strength. For example, NaCl improves the mechanical strength of PAAm/PAAc-Fe3+ hydrogels by shielding the repulsive forces within the polyelectrolyte PAAc chains, leading to a denser network structure with fewer pores. This results in an “ionic skin” that functions effectively as a resistive sensor at −14 °C, capable of promptly monitoring strain up to 200% and pressure up to 40 kPa,56 due to the combined effects of freezing point depression, ionic conductivity, and the “polyelectrolyte effect”. In more complex systems, such as LiCl-based poly(sulfobetaine methacrylate (SBMA)-co-AAc) hydrogels, which contain both zwitterionic PSBMA chains and negatively charged PAAc chains, there is an enhanced antifreezing capability and conductivity as LiCl concentration increases (Fig. 5g). At 30 wt% LiCl, the hydrogel remains flexible even at −80 °C for up to 30 days. However, the LiCl induced “anti-polyelectrolyte effect” causes swelling in PSBMA chains, while the “polyelectrolyte effect” leads to aggregation in PAAc chains, with peak mechanical strength observed at 10 wt% LiCl and a decline at higher concentrations (Fig. 5h).157
The Hofmeister series illustrates specific ion effects on hydrophilic polymers, where kosmotropic “salting-out” ions such as acetate (Ac−) promote polymer aggregation, enhancing mechanical strength, while chaotropic “salting-in” ions like Cl− increase solubility, supporting antifreezing performance but potentially weakening mechanical properties.160 For example, salts such as LiCl, NaCl, and CaCl2 are known to induce “salting-in,” which benefits antifreezing characteristics but may cause swelling in the network, as observed in LiCl-based PAAm-HEC hydrogels156 and CaCl2-based PAAm-alginate hydrogels.22 Conversely, employing the “salting-out” effect can result in a denser hydrogel network that improves mechanical strength.63,158,161–164 For instance, the incorporation of the “salting-out” Ac− into a PVA hydrogel not only makes it tough and antifreezing but also allows it to withstand extremely low temperatures.53 Immersing a frozen PVA gel in a KAc solution lowers the freezing temperature from 0 to below −70 °C as the KAc concentration increases from 0 to 50 wt% (Fig. 5i). Moreover, the hydrogel's conductivity improves with KAc, remaining conductive at −60 °C, although the conductivity decreases at lower temperatures due to reduced ion transport. Notably, higher salt concentrations toughen the PVA hydrogel, with fracture stress increasing from 0.1 to 4 MPa and strain from 250 to 380% (Fig. 5j). This mechanical enhancement through the “salting-out” effect is also evident in poly(2-hydroxyethyl acrylate) (PHEA) hydrogels (Fig. 5k).
Inorganic salts can also be combined with other antifreezing strategies to enhance hydrogel performance at low temperatures. For example, the “salting-out” effect of binary salts like KAc/ZnAc2, when paired with the “nonsolvency effect” of PVA in a water/DMSO mixture, results in a robust, antifreezing PVA hydrogel. This hydrogel is produced by mixing aqueous and DMSO-based PVA solutions in a specific ratio, followed by freezing at −20 °C and a solvent exchange with a KAc/ZnAc2 aqueous solution. The final product displays exceptional mechanical strength and flexibility, with tensile properties of approximately 23 MPa and 400% strain at −30 °C, significantly outperforming water-based PVA hydrogels (about 1 MPa/10%). It also exhibits remarkable freeze tolerance, remaining functional at temperatures as low as −77 °C (Fig. 6a). When used as an electrolyte in coin cell batteries, the hydrogel maintains capacity over 30000 cycles at 2 A g−1 at −20 °C.63 Similarly, combining antifreezing salts with Gly can further lower the freezing point.55,165 For instance, a ZnCl2-incorporated cellulose hydrogel remains stable down to −70 °C, thanks to the low freezing point of ZnCl2 (−73 °C) and its interaction with cellulose chains. The addition of Gly intensifies this effect by forming hydrogen bonds with water molecules, disrupting water–water hydrogen bonding and decreasing the ice formation temperature from −80 to −100 °C (Fig. 6b).25
 | ||
| Fig. 6 Integration of ionic compounds with other antifreezing strategies in hydrogels. (a) Antifreezing PVA hydrogel designed using dual salts of KAc/ZnAc2 and the “nonsolvency effect” of water/DMSO solvent incorporation and its flexibility at −77 °C. Reproduced with permission from ref. 63, Copyright 2023 Wiley-VCH. (b) ZnCl2-cellulose hydrogels with Gly: the freezing point reduced from −80 °C (Gel-0 with only ZnCl2 and Gel-water with additional water) to −100 °C (Gel-Gly with added Gly). Reproduced with permission from ref. 25, Copyright 2019 Wiley-VCH. (c)–(e) Combined effects of PIL and inorganic salts on antifreezing hydrogels, where (c) antifreezing performance comparison between the PIL gel, frozen PAAm, and KCl-incorporated PAAm (PAAm/KCl) hydrogels at −20 °C, (d) freezing point comparison between the PIL gel and PIL gel with added KCl, and (e) conductivity comparison between the PIL gel and the PAAm-KCl gel across temperatures from −30 to 60 °C. Reproduced with permission from ref. 23, Copyright 2020 Royal Society of Chemistry. | ||
Another effective antifreezing strategy utilizes zwitterionic polyionic liquids (PILs), which balance positive and negative charges to form strong hydration layers. A PIL hydrogel, created by copolymerizing a zwitterionic ionic liquid monomer with AAm, remains unfrozen at −20 °C—unlike both pure PAAm and KCl-incorporated PAAm hydrogels, which freeze at this temperature (Fig. 6c). Further DSC analysis revealed a freezing point of −26 °C for the PIL hydrogel, which can be further lowered to −41 °C with the addition of KCl (Fig. 6d). This hydrogel is exceptionally flexible, demonstrating about 900% tensile strain and 60 kPa strength at −20 °C, while maintaining improved ionic conductivity (6.2 S m−1 at −30 °C) compared to the KCl-incorporated PAAm hydrogel (Fig. 6e), allowing this PIL gel for use in resistive and triboelectric sensing at −20 °C. Additionally, the PIL hydrogel also features self-healing capabilities due to its reversible ionic and hydrogen bonds, enhancing its durability and adaptability for use in extremely cold environments.23
Overall, utilizing ionic compounds as antifreezing agents in hydrogels provides several benefits, including effective freezing point depression, improved ionic conductivity, and compatibility with other antifreezing strategies. This approach allows hydrogels to retain flexibility and functionality at subzero temperatures, making them ideal for applications such as sensors and energy devices. However, high salt concentrations can compromise mechanical properties due to swelling effects. Achieving an optimal balance between antifreezing capabilities and mechanical performance necessitates meticulous control over the types and concentrations of ions used. By strategically utilizing the “polyelectrolyte” effect, the “salting-out” effect, and synergistic interactions with other antifreezing methods, it is possible to refine hydrogels for both antifreezing properties and mechanical robustness, thus expanding their practical applications under extreme conditions.
3.3. Organohydrogels
To address the drying-out issue in most of the antifreezing hydrogels, another common antifreezing strategy involves using nonvolatile organic solvents, such as EG,8,31,74,75,77,85,166,167 Gly,55,86,87,102,168,169 and sorbitol,84 to prepare organohydrogels either by directly submerging preformed hydrogels in organic solvents through a solvent-exchange method or by in situ polymerization in a binary water/organic solvent system. This alternative strategy enables organohydrogels to enhance water retention capacities and promote the formation of non-freezing water molecules at subzero temperatures, although organogels are not typically considered pure hydrogels, as they contain solvents other than water within their polymer networks.The solvent-exchange method is a widely used technique to prepare antifreezing hydrogels by replacing the water molecules in the hydrogel with organic molecules from an external source. This exchange is driven by the diffusion of molecules due to concentration gradients across the hydrogel boundary. In hydrogels, water exists in three states: “free water (bulk-like water)”, “intermediate water (weakly bound water)”, and “non-freezing water (strongly bound water)”. Free water molecules, not bound to the polymer networks, can freely and rapidly exchange with organic molecules. Intermediate water molecules, loosely bound to the polymer networks via hydrogen bonds, exchange more slowly. This strategy has been effectively employed to fabricate several organohydrogels, including (i) Ca-containing alginate/PAAm organohydrogels using a Gly, EG, or sorbitol exchange, which confers antifreezing property effective down to −70 °C,84 (ii) PAMPS/PAAm organohydrogels, doped with EG/LiCl, which maintain their mechanical flexibility at −80 °C and exhibit no detectable ice crystals even at temperature as low as −130 °C,85 (iii) gelatin organohydrogels prepared in a sodium citrate (Na3Cit) (water)-Gly solution, which maintain their mechanical flexibility at −80 °C or can be stored effectively up to 3 days,102 (iv) hydroxypropyl cellulose (HPC)/(PVA) organohydrogels in a LiClO4 water/Gly mixed solution, which maintains a tensile strength of 192 kPa and a tensile strain of 205% at −40 °C,55 (v) tannic acid (TA)-CNT-incorporated, water–Gly-treated PVA organohydrogels, which maintain their integrity under mechanical twisting and stretching without any observable damage at −30 °C (Fig. 7a).86 However, it is a notable challenge for the solvent-exchange method to replace the strongly bound non-rotational water molecules with organic solvents within the hydrogel matrix. This limitation often restricts the method's efficiency in fully enhancing the antifreezing capabilities of the hydrogels.
 | ||
| Fig. 7 Two typical synthesis routes of antifreezing organogels: (a) the solvent-exchange method for preparing Ca-alginate/PAAm-Gly,84 PAMPS/PAAm-EG,85 gelatin-EG,102 HPC/PVA-Gly,55 and TA-CNTs/PVA-Gly organogels.86 Reproduced with permission from ref. 84, Copyright 2018 Wiley-VCH. (b) In situ polymerization for preparing PVA/PEDOT:PSS-EG,8 PVA/PAAm-EG,74 and PDMA/PEGDMA-heptane organogels.6 Reproduced with permission from ref. 74, Copyright 2020 Royal Society of Chemistry. | ||
Alternatively, the in situ polymerization strategy addresses this limitation by directly integrating organic solvents into the hydrogel matrix during the polymerization process. This approach ensures a more uniform distribution and integration of both solvents throughout the hydrogel network, enhancing its antifreezing properties by effectively modifying the interaction between the polymer chains and the incorporated solvent molecules. In situ polymerized organohydrogels include: (i) PVA and poly(3,4-ethylenedioxythiophene)polystyrene sulfonate (PEDOT:PSS)-conjugated polymers in a binary solvent of water/EG, which exhibit notable flexibility and strain-sensitive performance down to −55 °C,8 (ii) EG-incorporated PVA/PAAm DN organohydrogels, which demonstrate a high compressive stress (15.5 MPa), excellent shape recovery property, and a high ionic conductivity (0.48 S m−1) at −40 °C,74 and (iii) poly(dimethylacrylamide) (PDMA)/poly(ethylene glycol dimethacrylate) (PEGDMA) organogels prepared in a heptane/water solvent mixture, which show stable elasticity across a wide temperature range of −78 to 80 °C (Fig. 7b).6
Upon incorporating both organic and water solvents into gel networks through both solvent-exchange and in situ polymerization methods, the primary antifreezing mechanism of these organic solvents derives from their ability to lower the freezing point of water through colligative properties. These solvents can form strong hydrogen bonds with water, which disrupt the native hydrogen bonding network among water molecules and interfere with the lattice structure necessary for ice crystal growth. This disruption of water interactions and structures hinders the ability of water molecules to organize into a solid ice structure, inhibiting the formation and growth of ice crystals. Moreover, organic solvents increase the viscosity of the solution, which slows down the mobility of water molecules. This reduced mobility also makes it more difficult for water molecules to align into ice crystalline structure, further complicating the process of ice formation. For instance, EG is highly effective as an antifreeze additive, commonly used in automobile antifreeze. While pure EG freezes at temperatures below −13 °C due to strong competitive inter- and intra-molecular hydrogen bonding interactions, its freezing point dramatically decreases to below −45 °C when mixed with water in a 6:4 volume ratio.170
3.4. Design of crosslinked network structures
As an alternative to antifreezing additive strategies, a more sophisticated and innovative approach relies on engineering polymer networks. This strategy encompasses adjusting the hydrophobic–hydrophilic ratio (amphiphilic nature) of polymer chains, the positive–negative charge ratios (electrolyte nature) of zwitterionic or polyelectrolyte chains, network topologies, and network crosslinkers. These elements of polymer network design enable strong compositional and spatial influences on antifreezing and mechanical enhancement by (i) enhancing polymer–water interactions to competitively suppress ice nucleation and growth and (ii) activating multiple energy dissipation pathways to improve mechanical resilience.Specifically, similar to the role of amphiphilicity in antifreeze proteins, the design of antifreezing polymers also requires a carefully balanced combination of hydrophobic and hydrophilic groups. This balance is essential for synergistically enhancing antifreezing properties while maintaining mechanical flexibility and functionality at low temperatures. Adjusting the hydrophobic–hydrophilic balance within the polymer network allows polymer chains to interact with water molecules in diverse ways. Hydrophilic chains form strong hydrogen bonds with water, disrupting the interactions and lattice structures while simultaneously reducing water mobility, both key factors that typically facilitate ice formation. In contrast, hydrophobic chains repel water molecules, effectively preventing the growth of ice crystals by isolating small ice fragments and stopping them from merging into large, continuous crystal domains within the polymer network. Inspired by AFPs, it is recognized that confining water to dimensions less than 10 nm is crucial in preventing water crystallization.122,123 This can be achieved by introducing AFP-mimetic polymer networks designed to confine water within hydrophobic nanodomains. Driven by developing AFP-mimetic polymer networks, the supramolecular poly(dimethylacrylamide-2-(N-ethylperfluorooctane sulfonamido)ethyl acrylate) (DMA-FOSA) hydrogel, where hydrophobic nanodomains are physically crosslinked by the association of copolymers of DMA and FOSA, effectively creates confinement regions smaller than 2 nm within the network, with the size of these regions tunable by altering the FOSA content (Fig. 8a).49 This hydrogel design retains 45% of its water unfrozen even at 205 K, enabling it to rearrange and relax under the stress of ice formation. This low freezing fraction helps prevent catastrophic fractures. Visually, the hydrogel transitions from transparent to translucent and maintains its structural integrity without cracking through multiple freeze–thaw cycles. Employing a similar strategy that combines water confinement and AFP mimicry, a highly effective antifreeze supramolecular hydrogel was developed using a copolymer of 2-hydroxyethyl acrylate (HEA) and 2-(N-ethylperfluorooctane sulfonamido)ethyl methacrylate (FOSM). This design successfully prevents up to 99 wt% of absorbed water from freezing, even under conditions of slow cooling to 128 K followed by reheating.40 The hydrophilic nature of HEA, akin to PVA—a known AFP mimic—contributes to ice growth inhibition,135 while the glassy, hydrophobic FOSM nanodomains confine water on a sub-3 nm scale,171 the critical size for effective ice inhibition. This nanostructured design of hydrophobically modified hydrogels provides a blueprint for controlling and manipulating ice formation in concentrated soft materials. It leverages the delicate balance between the length scale of hydrophobic domains and the hydrophilicity of water-soluble network components. This mechanism closely resembles the freezing tolerance strategies observed in AFPs and other biomolecules that feature a mix of hydrophobic and hydrophilic residues/domains.118
 | ||
| Fig. 8 Fully polymeric antifreezing hydrogels without antifreezing additives. (a) Water confinement-inspired supramolecular hydrogels of DMA and FOSA copolymers, with core–shell morphology that inhibits crystallization and ensures high water mobility in supercooled water. Average center-to-center distance between the supramolecular cross-links tuned by FOSA content creates favorable water confinement regions. Reproduced with permission from ref. 49, Copyright 2016 American Chemical Society. (b) A universal crosslinking strategy employed to develop a family of DN hydrogels, featuring agar, gelatin, PVA, and BSA as the first network, paired with zwitterions (SBAA, VBIPS, SBMA, and SVBA), electrolytes (DMAE, AAc, AMPS, and META), hydrophilies (AAm, HEAA, HEMA, and HEA), and macromolecules (PEGDA and GelMA) as the second network, with enhanced antifreezing and mechanical properties at −20 °C. Reproduced with permission from ref. 37, Copyright 2021 Wiley-VCH. | ||
Secondly, antifreezing hydrogels can be effectively designed using polymers that inherently possess a strong water-binding capacity, which enables them to engage in antifreezing activities through ice-recrystallization inhibition, thermal-hysteresis, and ice-crystal shaping by altering water structures and dynamics. These polymers are typically categorized into two groups: highly hydrophilic polymers and highly electrolytic polymers, with each type interacting with water molecules in distinct ways. Hydrophilic polymers, such as PEG,172 PAAm,173 and poly(acrylates),174 form a strong hydration layer with water molecules through hydrogen bonding,175 which is relatively easy to break and reform compared to ionic solvation.176 On the other hand, zwitterionic polymers, commonly derived from five standard zwitterionic moieties—carboxybetaine (CB), sulfobetaine (SB), phosphatidylcholine (PC), cysteine (Cys), and 3-(1-(4-vinylbenzyl)-1H-imidazol-3-ium-3-yl)propane-1-sulfonate (VBIPS)—create a tightly and stably bound water layer near the polymers through strong electrostatically induced hydration.177 This distinction in water interaction underpins the unique antifreezing properties of each polymer type.
Recently, only a few studies have reported antifreezing hydrogels that derive their intrinsic antifreezing properties directly from the polymer itself, rather than relying on antifreezing additives. Recently, a notable advancement has been the development of a novel crosslinking strategy using 4,9-dioxo-5,8-dioxa-3,10-diazadodecane-1,12-diyl diacrylate (EGINA), a universal antifreezing crosslinker.37,124 This crosslinker has enabled the creation of a family of DN hydrogels with intrinsic antifreezing and mechanical properties, thereby eliminating the need for additives. These DN hydrogels are synthesized using a diverse array of first networks made of agar, gelatin, PVA, or bovine serum albumin (BSA), paired with second networks composed of zwitterions (SBAA, VBIPS, SBMA, and 3-(dimethyl-(4-vinylbenzyl)ammonio)propyl sulfonate (SVBA)), electrolytes (DMAE, AAc, AMPS, and META), hydrophiles (AAm, HEAA, HEMA, and HEA), or macromolecules (PEGDA and methacryloyl gelatin (GelMA)). This wide variety of network compositions allows the EGINA-crosslinked DN hydrogels to maintain robust antifreezing and mechanical properties at −20 °C for prolonged periods (Fig. 8b).37 Furthermore, the application of EGINA extends beyond hydrogels. Materials containing EGINA exhibit antifreezing properties in various forms—including solutions, gels, and hydrogel/solid interfaces—demonstrating the versatility and effectiveness of this universal crosslinking strategy in imparting antifreezing properties to hydrogels. Molecular dynamics simulations have revealed that the superior antifreezing properties of EGINA-crosslinked hydrogels, regardless of polymer composition or network structure, likely stem from their highly hydrophilic and tightly crosslinked DN structures. These structures facilitate strong water-network binding, which is effective in preventing ice crystal formation within the hydrogel networks.178
Another example of polymeric antifreezing hydrogels that do not require antifreezing additives is the EG-based waterborne anionic polyurethane acrylate (EG-waPUA)/PAAm hydrogel.110 This hydrogel is synthesized through free radical polymerization, combining EG-waPUA precursors with AAm. The EG-waPUA/PAAm hydrogel can lower its freezing point to −25 °C while maintaining a nearly unchanged storage modulus across a temperature range from −30 to 30 °C. This robust antifreezing property is attributed to its dual crosslinking mechanism, which involves covalent interactions among laterally associated EG-waPUA polymer chains and physical entanglements of PAAm polymer helices within extended junction zones. However, the fabrication of this hydrogel involves a complex, multi-step process to achieve its intricate network structure, which complicates the derivation of a general, structural-dependent antifreezing mechanism. A comparison between alkalified PAAc hydrogels and PVA hydrogels, both containing 10% KOH, revealed different freezing points of −25 and −13 °C, respectively.179 This difference is attributed to the polarized terminal groups in A-PAAc, which have strong interactions with water molecules, thereby lowering the freezing temperature. With few hydrogel systems reported, it is evident that antifreezing hydrogels that derive their properties directly from the polymers and network structures themselves, without relying on antifreezing additives, are still far from reaching innovative design principles.180,181
3.5. Surface/interface engineering of antifreezing hydrogels
Unlike bulk antifreezing hydrogels aimed at inhibiting ice nucleation and restricting ice propagation, when using antifreezing hydrogels for surface coatings, the most effective strategy focuses on reducing ice adhesion on the gels, even prior to ice nucleation. Mechanistically, ice adhesion can be categorized into intrinsic and macroscopic types. Intrinsic adhesion stems from the atomistic attraction of water/ice molecules to the surface via coulombic and van der Waals interactions. A common strategy to reduce ice adhesion involves lowering surface energy and enhancing surface hydrophobicity, which weakens the atomistic interactions between the surface and water molecules. Given that real surfaces typically operate on macroscopic scales, macroscopic adhesion is largely influenced by surface roughness. Theoretically, strategies such as spatially controlled surface patterning or reducing surface roughness can effectively diminish ice adhesion strength.The design of micro/nano-patterned anti-icing gels with Janus properties effectively reduces water–substrate interactions by minimizing both intrinsic adhesion and macroscopic contact areas or points. The hydrophobicity of the patterned domains collaborates with the substrate to enhance water repellency, preventing ice formation by inhibiting the effective wetting of the surface by water droplets. In a representative example, an innovative chemically patterned binary coating was developed by combining hierarchical hydrophobic perfluoroalkyl methacrylate (PF) brushes with a hydrophilic substrate of PAAc hydrogels (Fig. 9a).48 This unique combination was achieved using a photomask technique, allowing for precise spatial control over the surface patterning. Importantly, such rational design enables the hydrophobic domains to repel water and minimize ice formation, while the hydrophilic polyelectrolyte hydrogel still maintains the integrity and functionality of the anti-icing surface over time. As a result, the coatings significantly extended the time to freezing and reduced the supercooling temperature on the modified copper surface. These effects were maintained over prolonged periods and through multiple frosting/defrosting cycles, even at temperatures as low as −20 °C. Similarly, when hydrophobic PDMS chains or other hydrophobic patterns were chemically grafted onto polyelectrolyte hydrogels, the modified substrates demonstrated remarkable control over the behavior of interfacial water, thus effectively modulating ice nucleation, propagation, and adhesion.2
 | ||
| Fig. 9 Design of antifreezing or anti-icing surfaces. (a) Design of PF patterned PAAc coatings coupling hierarchical nano/microstructures and dual hydrophobic/hydrophilic features for anti-icing applications. The PF-coated PAAc-Cu surfaces (the diameter of the micropatterns is ca. 800 μm) demonstrate enhanced anti-icing properties by significantly delaying the onset of freezing compared to unmodified Cu surfaces. This delay reduces the rapid temperature rise associated with recalescence, indicating improved resistance to ice formation at −20 °C. Reproduced with permission from ref. 48, Copyright 2018 Royal Society of Chemistry. (b) Schematic of PB-I polyelectrolyte surfaces with different grafting densities. Time-resolved images show ice growth on patterned PB surfaces at grafting densities of 0.50 and 0.02 chains per nm2 at a supersaturated humidity of 109%. Scale bar: 200 μm. “W”, “I”, and “RIF” are denoted as water, ice, and region of ice free, respectively. Reproduced with permission from ref. 182, Copyright 2020 American Chemical Society. | ||
In addition to utilizing hydrophobic patterns, designs inspired by ice-nucleating proteins (INPs) have emerged as an innovative method for localized control over ice formation in antifreezing hydrogels.11 These biologically inspired designs utilize a strategic combination of hydrophobic and hydrophilic properties to effectively influence ice nucleation and propagation. Two representative examples are patterned polyelectrolyte hydrogel (PH) surfaces182 and patterned hydrogel-encapsulated INPs (PHINPs),47 both of which have demonstrated capabilities to inhibit ice propagation. Specifically, the patterned PHs use ionic groups within their polymer network to influence water and ice behavior at the surface (Fig. 9b). By tuning the counterions within these hydrogels, it is possible to raise the ice nucleation temperature above normal levels. Ion specificity is crucial, as certain counterions interact with water in ways that either promote or hinder the early stages of ice crystallization. This adjustment leads to localized ice formation in specific patterns across the coated areas of the surface. Instead of allowing ice to form indiscriminately across the surface, the design directs ice formation to specific regions, such as designated ice stripes. This precise control over ice nucleation curbs widespread ice propagation, resulting in more manageable and easily removable ice patterns. On the other hand, encapsulating INPs within a hydrogel matrix, such as P(AAm-co-HEMA), enhances the system's ability to modulate ice nucleation temperatures. This allows the hydrogel to selectively initiate ice formation in predefined regions. This strategic design prevents ice from spreading to undesired areas, ensuring localized control. Additionally, the INPs within the hydrogel matrix actively inhibit the uncontrolled spread of ice across the surface, helping to maintain extensive ice-free zones.
While various antifreezing strategies have been employed to develop different types of antifreezing hydrogels, integrating mechanisms such as the incorporation of specific ions, cryoprotectant molecules, and AFPs into hydrogels or organohydrogels can often work synergistically to enhance antifreezing performance. It is important to note that when these strategies involve antifreezing additives, their physicochemical properties may differ from or oppose those of hydrophilic polymer chains, often leading to structural incompatibility and network heterogeneity at the nanoscale. This random distribution of additives can result in nonuniform energy dissipation and unstable mechanical performance under deformation, issues that can manifest even at room temperature. Moreover, the high-water content in hydrogels still presents significant challenges and opportunities for discovering new materials and phenomena, as well as for understanding novel antifreezing and toughening mechanisms under extreme cold and deformation conditions.
4. Molecular simulations and understanding of antifreezing hydrogels
Despite significant advances in molecular simulations of polymers in various solvents, the application of molecular dynamic (MD) simulations to study antifreezing hydrogels remains very limited, largely due to challenges in accurately modeling their complex structures. These computationally constructed hydrogel models often contain certain undesirable artifacts related to initial configuration, topology structures, and limited simulation time. The typical approach in these models involves duplicating monomers to achieve a desired polymer length while introducing conformational diversity through randomized torsional angles along the polymer backbone. However, this method often results in polymer chains that contain a limited number of monomeric units, leading to configurations with improbable entanglements or voids. Such issues may deviate from the accurate representation of polymer conformations and interactions.Simplified MD simulations have been conducted on hydrogels composed of LiCl-incorporated PAAm with CNFs.183 The simulation setup consists of just a single PAAm chain and a single CNF fiber. Despite this simplicity, both PAAm polymer chains and cellulose nanofibrils engaged in hydrogen bonding with water molecules, while the ions (Li+ and Cl−) contributed through electrostatic interactions. Analysis of potential mean force (PMF) and radial distribution function (g(r)) profiles indicated that the addition of LiCl significantly increased the binding affinity of water molecules to the polymer network and enhanced the structuring of water around the network (Fig. 10a), leading to the formation of a non-freezing bound water state, without any observable solid–liquid phase transition. MD simulations of NaCl-induced deicing properties of PVA hydrogels have shown that NaCl ions within the gel network migrate to the interface (Fig. 10b),59 where they prevent ice nucleation and contribute to the disintegration of ice crystals into liquid water at the boundary between the gel and ice/water. The antifreezing efficiency of these NaCl-PVA hydrogels is primarily attributed to the strong ionic solvation between ions and surrounding water molecules, which disrupts hydrogen bonding networks and structural arrangement among water molecules. This disruption not only facilitates the destruction of the ice crystal structure but also promotes the formation of a liquid-like lubricating layer on the hydrogels, enhancing their antifreezing capabilities.
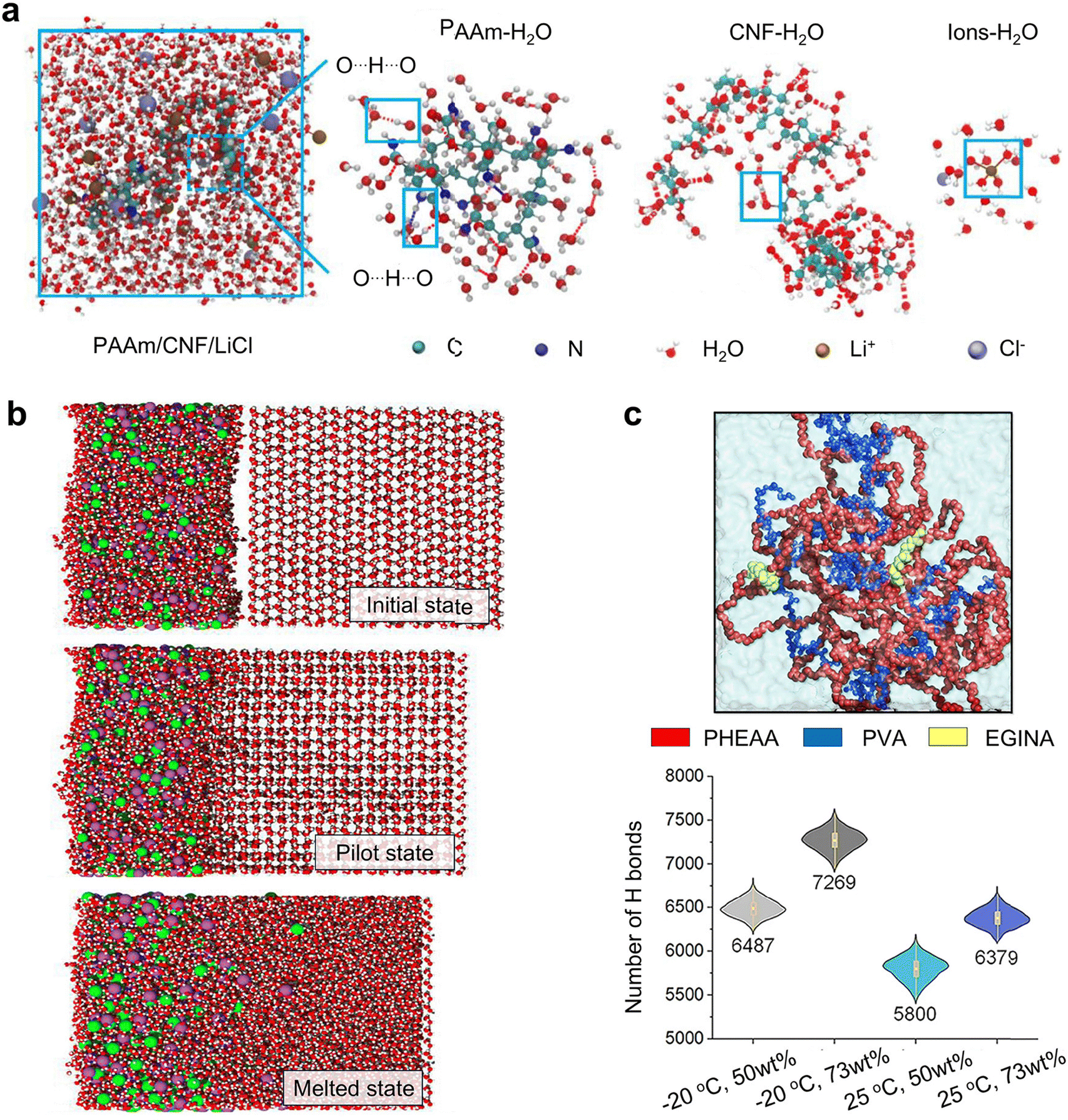 | ||
| Fig. 10 Molecular dynamics simulations of antifreezing hydrogels. (a) LiCl-incorporated PAAm/CNF hydrogels, demonstrating the ion-induced enhancement of water binding affinity and structuring around PAAm and CNF. Reproduced with permission from ref. 183, Copyright 2021 Elsevier. (b) NaCl-incorporated PVA hydrogels, illustrating a gradual deicing process on the PVA surface. Reproduced with permission from ref. 59, Copyright 2020 American Chemical Society. (c) PVA/PHEAA DN hydrogels, showing an increase in the number of hydrogen bonds between polymer and water molecules at subzero temperatures compared to room temperature. Reproduced with permission from ref. 178, Copyright 2023 Springer Nature. | ||
To address the abovementioned challenge of constructing realistic gel network structures without introducing artifacts, a multiscale simulation platform, integrating “random walk reactive polymerization” (RWRP) with MD simulations, was recently designed to computationally construct physically and chemically linked PVA/PHEAA DN hydrogels from monomers, closely mimicking the radical polymerization process.178 Different from previous simulations focusing on the ion-induced antifreezing mechanism, this simulation study concentrated on how the network structure of PVA SN, PHEAA SN, and PVA/PHEAA DN hydrogels influences water structures, dynamics, and interactions around polymers, in relation to antifreezing mechanisms. As compared to single-network hydrogels composed solely of PVA or PHEAA, both PVA and PHEAA chains in PVA/PHEAA DN hydrogels exhibited stronger binding to water molecules through hydrogen bonds, as evidenced by enhanced polymer–water interactions and an increased number of hydrogen bonds between polymer and water molecules (Fig. 10c). These strong interactions and hydrogen bonds contribute to the presence of strongly bound water molecules (non-freezing water molecules) around polymer chains, which obstruct the organization and alignment of water molecules necessary for ice crystal formation. Thus, the antifreezing properties of PVA/PHEAA hydrogels stem from two primary factors: (i) intrinsic, strong water-binding networks formed by crosslinkers and (ii) the tightly crosslinked and interpenetrating structures of the double-network. These characteristics significantly enhance the interactions between the polymer and water, effectively competing against ice nucleation and growth.
5. Advances in the functionality and performance of antifreezing hydrogels
In parallel to their antifreezing properties, we will explore the diverse functionalities of these antifreezing hydrogels, emphasizing their broad impact across various applications. These hydrogels not only prevent ice formation but also offer enhanced mechanical strength, self-healing, conductivity, surface adhesion, environmental and biomedical functions, making them suitable for use in biomedical devices, environmental sensors, and flexible electronics. Additionally, their unique properties facilitate applications in cryopreservation and low-temperature environments, demonstrating their versatility and potential to revolutionize materials science in cold climates. It should be noted that while some existing studies discuss additional functionalities of hydrogels at room temperature, separating these from their antifreezing properties, our discussion focuses exclusively on the additional functionalities exhibited at subzero temperatures. This focus aims to highlight the innovative adaptations and future potential of antifreezing hydrogels across various technological applications.5.1. Enhanced mechanical properties
Over the past few decades, addressing the intrinsic mechanical weakness of hydrogels has been a key focus in the development of various strategies for fabricating various types of tough hydrogels, including slide-ring hydrogels, nanocomposite hydrogels, DN hydrogels, and hydrogen bond-enhanced hydrogels.184,185 Among these advancements, the formation of crystalline or glassy domains not only creates high-strength hydrogels but also enhances their antifreeze properties. While these strategies may not always directly enhance the antifreezing properties of hydrogels, tough hydrogels can easily be converted into antifreezing hydrogels through simple post-treatment processes, such as immersion in solutions containing cryoprotectants or specific ions. Thus, this section will not delve into the synthesis strategies of high-strength antifreezing hydrogels due to their straightforward preparation. Instead, we will focus on the interplay between antifreezing properties and mechanical enhancement in hydrogels.Among various high-strength hydrogels, the fabrication of crystalline hydrogels based on the copolymerization of a hydrophilic monomer (such as DMA) and a hydrophobic monomer with a long alkyl chain (such as FOSM) has been reported. These hydrogels not only demonstrated a high strength of approximately 0.5 MPa,50 but also exhibited notable antifreezing properties, remaining functional down to −68 °C when formulated with a high FOSM content (22 mol%).186 The mechanical and antifreezing properties of the hydrogels were closely linked to their network microstructure. In the copolymer P(DMA-co-FOSM) gel, phase separation occurs in water, where FOSM nanodomains act as physical crosslinking points. Small-angle neutron scattering (SANS) revealed that a PDMA shell coats the FOSM nanodomains, forming a core–shell structure. It was also observed that hydrophilic regions, less than 10 nm in size (Fig. 11a), are present between the FOSM nanodomains, with their distribution varying depending on the FOSM content. The proximity of these nanodomains, with center-to-center spacing (dsp) of less than 3 nm, facilitates nearly complete ice inhibition due to the confinement effect, although some ice formation is still partially induced by the presence of the nanodomains. Another example is the statistical copolymer hydrogel, composed of hydroxyethyl acrylate and n-octadecyl acrylate (HEA-ODA) with 21.3 mol% ODA, which exhibited ODA crystalline nanodomains and antifreezing properties.40 This demonstrates that hydrogels lacking fluorinated hydrophobic moieties, yet possessing similar nanostructures, can effectively control water crystallization. Furthermore, it was observed that rubbery, hydrophobic, and physical crosslinks resulted in less effective ice inhibition compared to crystalline crosslinks.187 Although elastic domains are less conducive to antifreezing, it has been reported that hydrogels with glass domains exhibit intrinsic antifreeze capabilities. The poly(AAm-co-methacrylic acid (MAAc)) supramolecular hydrogel exhibits distinct mechanical behaviors at varying temperatures.51 At room temperature, the hydrogel remains in a glassy state with a moderate water content (∼50%) and demonstrates toughness and stretchability, featuring a Young's modulus (E) of 2.1 MPa, a tensile strength (σb) of 8 MPa, and a breaking strain (εb) of 627%. However, at −45 °C, the gel becomes significantly stronger and stiffer, yet maintaining ductility, with an E of 900 MPa, a σb of 30 MPa, and an εb of 35%. Despite these changes, the gel remains highly transparent even at −45 °C and continues to display transparency in liquid nitrogen (−196 °C), highlighting its antifreezing properties. Low-field nuclear magnetic resonance (NMR) analysis indicates that the proton spin–spin relaxation time (T2) of water in the gel is approximately 1 ms, significantly less than that of bulk water (∼3400 ms) and PAAm gel (∼200 ms) at 45 °C, suggesting severely restricted water mobility within the P(AAm-co-MAAc) gel (Fig. 11b).
 | ||
| Fig. 11 Mechanically enhanced antifreezing hydrogels through the formation of crystalline or glassy domains. (a) Microstructure of a supramolecular hydrogel formed from statistical copolymers of HEA and FOSM. Antifreezing mechanism relies on nanoconfinement and hydrogen bonding (i) to (iv) effectively retain >97% of water unfrozen within inter-nanodomain spacing below 3.0 nm at 203 K (iv). Reproduced with permission from ref. 40, Copyright 2019 American Chemical Society. (b) Antifreezing characterization of P(AAm-co-MAAc) gels by XRD curves at −45 °C, DSC and fractions of freezable water, low-field NMR curves of P(AAm-co-MAAc) gels, and the corresponding T2 and τc of the two gels. Reproduced with permission from ref. 51, Copyright 2021 Wiley-VCH. | ||
Suppressing chain dynamics in hydrogels with viscous cosolvents, such as EG and Gly, is a common antifreezing strategy. The viscosity (η) of EG and Gly is significantly higher compared to pure water. At 25 °C, the viscosity of pure water is 0.89 mPa s, but it increases twenty-fold to 17.12 mPa s for EG. Gly is even more viscous, with a value of approximately 1000 mPa s at the same temperature. The viscosity of a H2O/EG or H2O/Gly mixture varies with the concentration of EG or Gly (φ), increasing with the proportion of the cosolvent.188 Building on the concept of enhancing antifreezing properties, a series of antifreezing hydrogels have been developed, each demonstrating enhanced mechanical properties. Gelatin hydrogels enhanced through solvent exchange, with Gly demonstrating significant mechanical improvements, achieving a tensile strength of approximately 2 MPa and maintaining excellent antifreeze capabilities at temperatures as low as −45 °C.91 Similar enhancements were seen in PAAm gels, where chemical crosslinking combined with solvent swelling showed increased mechanical properties.189 Specifically, PAAm gels swollen in EG and those incorporating Gly both exhibited a substantial increase in modulus (E).190 The Gly-swollen PAAm hydrogels, in particular, not only showed higher Γ but also improved fracture energy, effectively transforming into organohydrogels with superior performance.
Because the Rouse time (τR) is proportional to η, the polymer dynamics in PAAm gels with EG or Gly are slowed down, leading to an enhancement of the E similar to that seen in physical gelatin gels containing EG, where polymer dynamics are suppressed by solvent viscosity. To further understand these dynamics, a novel dynamic light scattering micro-rheology technique was employed, revealing fast Rouse-like dynamics within PAAm hydrogels.191 These dynamics occur over an incredibly fast timescale, ranging from 10−3 to 10−5 seconds, highlighting the rapid response of these materials under dynamic conditions. Furthermore, efforts to extend the observed phenomena from SN hydrogels to DN hydrogels have been undertaken. It was discovered that the mechanical properties of a fully chemically crosslinked PAMPS/PAAm DN hydrogel decrease with an increase in solvent viscosity, although the tensile behavior of the gel remains unchanged.188 The dynamics of the PAAm second network, when combined with EG or Gly, plays a crucial role in the fracture behavior of the first, more brittle network. Consequently, the suppression of chain dynamics by viscous anti-freezers like EG or Gly can have varying impacts—either positive or negative—on the mechanical properties of hydrogels, dependent on the network structure.
Adding antifreezing additives, such as ions, can cause chain aggregation or assembly and increase chain entanglement, thereby improving the mechanical properties of hydrogels.158,161 The Hofmeister effect, which has been extensively studied, differentiates between well-hydrated ions called kosmotropes and poorly hydrated ions known as chaotropes. High concentrations of kosmotropes can cause a “salt-out” effect in certain synthetic polymers, significantly enhancing the mechanical properties of hydrogels. Although the exact mechanism of this enhancement is not fully understood, one possible explanation is that the ion-induced polymer chain aggregation enhances both mechanical and antifreezing properties. Gelatin, a natural protein sourced from collagen found in tissues, bones, and skins, forms hydrogels whose mechanical strength can reach approximately 4 MPa when soaked in a 30 wt% (NH4)2SO4 aqueous solution.192 However, the antifreezing properties of these gels are limited due to the salt-out effect induced by the Hofmeister series. The synergistic use of kosmotropes with other antifreezers such as Gly or betaine has proven effective. For instance, gelatin hydrogels treated with a citrate water/Gly solution demonstrated enhanced flexibility, strength, and toughness, even at temperatures as low as −70 °C (Fig. 12a). This combination not only achieves high mechanical strength and toughness but also exhibits superior surface adhesive strength to different substrates (e.g., copper sheets, glass slides, and PET) at −30 °C.102,193 Alternatively, using salt/EG aqueous solutions directly as solvents in hydrogel preparation provides another route to producing high-strength gels with excellent antifreezing properties. A specific concentration of salt promotes chain aggregation rather than precipitation, which enhances the mechanical properties through increased chain entanglement.194 Notably, the introduction of NaCl significantly improved the tensile strength of the poly(N-hydroxymethyl acrylamide) (PHA)/agar/EG DN gel. In addition to chain aggregation, macromolecules like PVA may assemble with antifreezers such as Gly92 and EG8 to enhance mechanical properties. These high-strength, antifreezing hydrogels benefit from the self-assembly of PVA with Gly to form nanostructures that significantly boost mechanical properties (Fig. 12b). The resulting gels exhibit high tensile strength up to 2 MPa and maintain low water crystallization temperatures as low as −96 °C. Similar mechanical enhancements are also observed in PVA/EG hydrogels. Consistently, self-assembly of PAAc with and without zwitterions in betaine/H2O and H2O systems resulted in self-assembled nanoclusters averaging 37.8 nm. In zwitterionic environments, these PAAc gels demonstrated remarkable mechanical strength, ranging from 10 to 16 MPa, and maintained flexibility at temperatures as low as −40 °C.195 Further research revealed that commercial zwitterionic monomers can form nanoparticles at high concentrations through liquid–liquid phase separation.196 It was discovered that zwitterionic monomers, including SBMA, 3-[[2-(methacryloyloyloxy)ethyl]dimethylammonio]propionate (CDME), and 2-(methacryloyloxy)ethyl 2-(trimethylammonio)ethyl phosphate (MPC), can form nanoparticles in 50 wt% aqueous solutions (Fig. 12c). This process is attributed to the strong dipole–dipole interactions among residual zwitterionic monomers under conditions with limited free water. Consequently, this leads to the creation of high-strength, high-toughness, and antifreezing pure polyzwitterionic hydrogels without the need for additional antifreezers, highlighting their potential for high-performance applications.
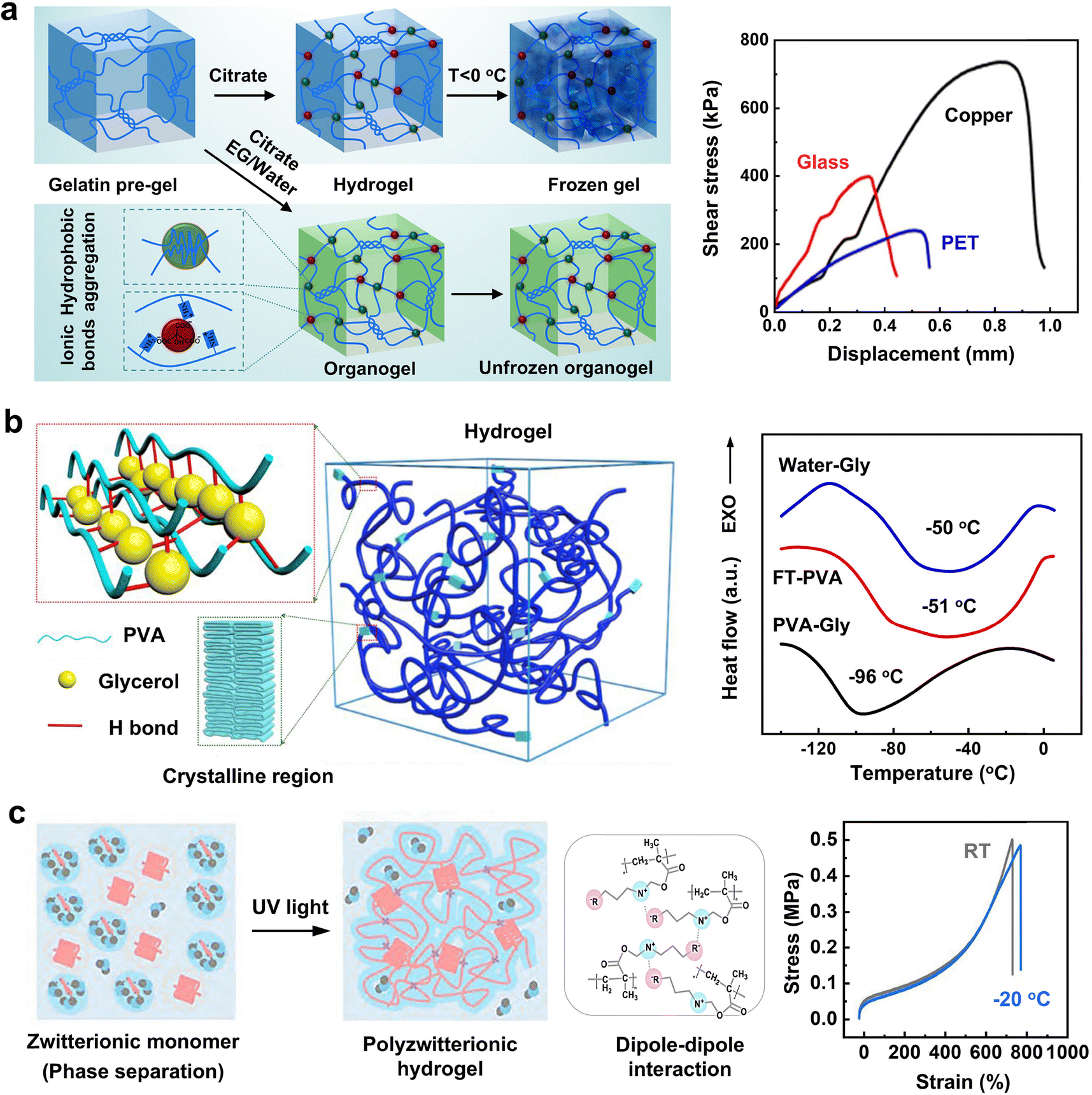 | ||
| Fig. 12 Mechanically enhanced antifreezing hydrogels through chain aggregation or assembly by antifreezing additives. (a) Network structure and antifreezing property comparison of gelatin hydrogels treated with a citrate vs. a citrate/Gly aqueous solution. Shear stress-displacement curves of the organohydrogel to different substrates (e.g., copper sheets, glass slides, and PET) at −30 °C. Reproduced with permission from ref. 102, Copyright 2019 American Chemical Society. (b) Network structure of the PVA-Gly organohydrogel. DSC curves of the water–Gly solution, freezing–thawing (FT)-PVA and PVA-Gly hydrogels (10 wt% PVA, water/glycerol). Reproduced with permission from ref. 92, Copyright 2017 Elsevier. (c) Preparation of zwitterionic hydrogels at high monomer (SBMA) concentrations. Stress–strain curves of PSBMA hydrogels at room temperature and −20 °C. Reproduced with permission from ref. 196, Copyright 2024 Royal Society of Chemistry. | ||
5.2. Conductive sensors
Conductive hydrogels have emerged as promising soft electrical materials due to their excellent flexibility, high conductivity, and adjustable mechanical properties, particularly showing great potential in wearable electronics as soft sensors. However, these hydrogels face challenges in harsh environments, especially in extreme cold, where their flexibility and conductivity are significantly compromised by freezing.197,198 Thus, preserving electrical conductivity and flexibility at subzero temperatures is essential for the reliable performance of conductive hydrogels in cold environments. The development of conductive hydrogels typically employs two primary strategies. The first strategy involves integrating intrinsically conductive materials, such as metal nanowires, CNTs, graphene, polypyrrole (PPy), polyaniline (PANI), and PEDOT:PSS, into conventional hydrogel matrices.87,115,167,199,200 The second strategy enhances conductivity by utilizing the conductive properties of ions, either through polyelectrolytes or by incorporating salts like LiCl into the hydrogel formulation.46,169,201 Although these strategies effectively improve conductivity and mechanical properties under cold conditions, they often do not introduce new material designs or explore innovative mechanisms.To achieve high sensing properties, conductive hydrogels must meet stringent requirements beyond those of traditional hydrogels. They need to exhibit not only exceptional mechanical properties, such as high stretchability and low hysteresis, but also high conductivity, strong adhesion to various substrates, and durability under harsh conditions. Achieving this comprehensive performance in antifreezing conductive hydrogels is challenging, as strategies to enhance certain characteristics often negatively impact others. For instance, while organic solvents can provide excellent freezing resistance, their increased presence typically reduces conductivity.202 Similarly, adding inorganic salts can improve conductivity and anti-freezing properties, but high saline concentrations often lead to polymer entanglement, such as with PVA and PAAm, which reduces flexibility due to the “salting-out” effect.203 Polyelectrolytes can enhance the ionic conductivity and lower the freezing point of water due to their high ionic content. However, their tendency to swell excessively and dissipate stress poorly often compromises mechanical strength.204 While employing a double network design can enhance stretchability and toughness, it typically involves using polyelectrolytes as a brittle or sacrificial first network. This results in the breakage of chemical bonds or network structures during stretching, leading to irreversible mechanical degradation, significant hysteresis, and reduced ionic conductivity.205 Furthermore, achieving a balance between mechanical properties, conductivity, and antifreezing capabilities often involves trade-offs. For instance, high stretchability typically requires relaxation of dangling chains or breakage of chemical bonds, which negatively impacts resilience, presenting a fundamental contradiction between high stretchability and low hysteresis. Due to the aforementioned tradeoffs, designing conductive hydrogels, particularly those with freezing tolerance for sensing applications, is more complex than designing hydrogels with singular antifreezing or conductive properties. Both the formula and structure require sophisticated design, with critical consideration of how properties and structures change with decreasing temperature. Advances in this field include strategies such as optimizing component synergy through formula adjustments, enhancing network construction via cross-linking or structural design and manipulating conductivity paths by developing topological structures.46
Generally, antifreezing conductive hydrogels consist of a hydrogel matrix, conductive materials, and additives, each contributing to mechanical strength, conductivity, and environmental tolerance, respectively. However, each component can also enhance other performances, allowing for synergistic effects through formula optimization.35 Polyelectrolytes, polyzwitterions, and their copolymers with AAm are the most commonly used matrices in these hydrogels. In these systems, polyelectrolytes and polyzwitterions provide abundant free ions or have an affinity for added ions, enhancing conductivity and antifreezing properties. Simultaneously, neutral PAAm limits excessive swelling and dissipates stress, thereby conferring high strength and toughness.206 Occasionally, other functional monomers featuring hydroxy, amine, and dopamine groups—such as methacrylate lysine, dopamine acrylamide, and N-[tris(hydroxymethyl)methyl]acrylamide—are copolymerized.207 The additional hydrogen bonds introduced by these monomers not only improve the mechanical and antifreezing properties of the hydrogels but also equip them with multifunctional capabilities, such as excellent adhesion and effective self-healing.
In copolymerized hydrogel matrices, chemical crosslinking is commonly employed to impart high strength, although this often results in reduced elasticity and compromised shape recovery. To address these drawbacks, materials such as PVA, gelatin, lignin, trehalose, cellulose, and other natural polysaccharides are frequently incorporated.43,208–210 These additions not only introduce physical crosslinking but also contribute abundant hydrogen bonds, enhancing the properties. This complementary network structure dramatically improves strength and durability while bolstering water binding, thereby naturally enhancing mechanical properties and freezing tolerance. When PVA is synergistically combined with PAAc, incorporating ZrOCl2 and sodium sulfate can further enhance their properties by exploiting metal ion coordination with PAAc and the salting-out effect of PVA.211 This approach leads to the fabrication of bionic PAAc/PVA hydrogel fibers, where ionic crosslinking from Zr4+-coordinated PAAc and crystalline domains formed via Na2SO4-treated PVA results in hydrogel fibers with exceptional mechanical properties, including a tensile stress of 24.43 ± 2.11 MPa, a strain of 844.44 ± 107.21%, an elastic modulus of 36.81 ± 1.58 MPa, and a toughness of 162.25 ± 21.99 MJ m−3. Together with EG, the inclusion of Na2SO4 in the hydrogel fiber enhances environmental tolerance, enabling it to withstand knotting at −40 °C. This bionic hydrogel fiber, featuring superior mechanical properties and ionic conductivity, is ideal as a flexible sensor, offering a high gauge factor of 0.88, a response time of 323 ms, and a recovery time of 374 ms (Fig. 13a).163
 | ||
| Fig. 13 Design of conductive, antifreezing hydrogels. (a) S-PAZr hydrogel-based fibers with enhanced sensing capabilities, rapid response, and fast recovery time. Reproduced with permission from ref. 163, Copyright 2024 Springer Nature. (b) The assembly scheme and sensing performance of a BSA/PAAm DN OHG flexible strain sensor. Reproduced with permission from ref. 96, Copyright 2024 Wiley-VCH. (c) PAMD-NaCl hydrogels with superior properties including soft compliance (low E), stretchability, fracture toughness, antifreezing capability, ionic conductivity, and reversible cyclic deformation. PAMD-NaCl hydrogels controlling LED brightness through deformation at room temperature and −20 °C. Reproduced with permission from ref. 212, Copyright 2023 Elsevier. | ||
To complement the primary components, additives specifically aimed at enhancing antifreezing and conductivity characteristics also play a crucial role in improving the comprehensive performance of conductive hydrogels. These additives introduce massive physical interactions through various mechanisms, including ionic associations from zwitterionic liquids, ionic liquids, salts, and PEDOT, as well as hydrogen bonding agents like PANI, PPy, and dopamine-modified PPy.10,213,214 Structural reinforcement is provided by high aspect ratio nanostructures such as MXenes, carbon nanotubes, and graphene.215 Often, these additives are synergistically combined to enhance mechanical, electrical, and antifreezing properties while enabling multifunctionality. For example, in PAAc/PVA hydrogels, the integration of Fe3+, EG, surfactant-functionalized multi-walled carbon nanotubes (F-MWCNT), and PEDOT provides low-temperature tolerance down to −25 °C, high healing efficiency, and robust piezoresistive performance.216
To enhance the comprehensive properties of antifreezing conductive hydrogels, synergistic component strategies are complemented by sophisticated network designs. Primarily, the DN structure is a prevalent method to bolster mechanical properties, using components like PVA, gelatin, and chitosan to improve stress dissipation, dynamic recoverability under strain, and overall toughness and resilience.217,218 Recently, BSA was doped into PAAm to construct a rigid DN structure upon heating, resulting in high mechanical strength and adjustable reversible adhesion. By further introducing Gly and a small quantity of PEDOT:PSS, the hydrogel was imparted with excellent antifreeze and anti-drying properties, as well as conductivity, making it suitable for all-weather flexible sensor devices (Fig. 13b).96 Additionally, innovative crosslinkers are engineered to optimize network structures, such as hybrid silica nanoparticles with multi-vinyl groups that act as covalent crosslinking and stress transfer centers. For example, when used in P(AA-co-AAm) hydrogels, these crosslinkers create a homogeneous network that excels in stress dissipation, leading to enhanced toughness and minimal mechanical hysteresis.219 Further advancements include using divinylbenzene (DVB), dispersed by sodium dodecyl sulfate (SDS) micelles, as a crosslinker in PAAm hydrogels prepared with saturated NaCl solution. This high-saline environment reduces the size of DVB droplets within micelles, diminishing hydrophobic associations and enhancing the gel's homogeneity and stretchability (up to 10200%). Additionally, the concentrated NaCl enhances ionic conductivity and antifreezing properties, maintaining high conductivity (106 mS cm−1 at 25 °C and 99 mS cm−1 at −30 °C). This enables the hydrogel sensors to exhibit exceptional sensing capabilities and robust performance across a range of temperatures, illustrating the potential of network design in developing advanced antifreezing conductive hydrogels (Fig. 13c).212
Taken together, the performance metrics crucial for antifreezing conductive hydrogels in sensor applications—such as conductivity, sensitivity, and response stability under harsh conditions—are intimately linked to the conductive network or ion transfer channels. Various strategies have been developed to manipulate electric conductive networks, including: (i) integrating dopants, employing post-processing treatments, or utilizing in situ polymerization to form crosslinked conductive networks within conductive polymers (Fig. 14a);220,221 (ii) modifying conductive fillers to enhance their dispersion within the hydrogel matrix (Fig. 14b);58,222,223 (iii) fabricating conductive nanofiller composite nanofibers via electrospinning to establish conductive nanochannels (Fig. 14c).224,225 These approaches not only boost the conductivity and efficiency of conductive paths within polymers and fillers but also enhance overall electrical conductivity. For improving ionic conductivity, strategies often involve using high concentrations of salts or engineering microphase-separated structures that offer stable and cohesive conductive pathways. Beyond microstructure manipulation, anisotropic architectures have been designed to optimize both mechanical and electrochemical properties. Sensors incorporating these sophisticated designs, such as those constructed with physically crosslinked, partially stretched PAAm-sodium alginate-MXene hydrogels or directionally frozen and thawed PVA with dopamine-modified PPy (DA-PPy), demonstrate exceptional sensitivity and stability.226 Natural polymers like soy protein, amylopectin, and gellan gum are used in hydrogel matrices for skin-contact sensors, ensuring excellent compatibility for physiological signal detection. Additionally, to address the reliance on external power sources, triboelectric nanogenerator technology is employed to create self-powered flexible sensors. Multi-functionalization is emerging as a key trend in the development of antifreezing conductive hydrogel sensors.
 | ||
| Fig. 14 Strategies for improving electrically conductive networks in antifreezing hydrogels. (a) In situ gelation process for six types of antifreezing hydrogels with improved conductivity. Reproduced with permission from ref. 220, Copyright 2022 Elsevier. (b) Design and fabrication of antifreezing hydrogels incorporating conductive fillers as flexible sensors, showcasing high GF values after 30 days at −20 °C. Reproduced with permission from ref. 58, Copyright 2022 Elsevier. (c) Integration of polyurethane/PANI conductive nanofibrous mats in hydrogels for superior sensing performance. Reproduced with permission from ref. 224, Copyright 2016 Elsevier. | ||
5.3. Flexible supercapacitors
Flexible supercapacitors (SCs), consisting of a polymer electrolyte sandwiched between two symmetric electrodes, have garnered significant attention in the field of wearable electronics, due to their high-power output, rapid charge/discharge cycles, and long lifespan.55,227–229 Hydrogel electrolytes are increasingly favored for SCs due to their unique blend of solid-like mechanical strength and liquid-like ionic transport capabilities, allowing them to effectively serve as both electrolytes and separators.206,230–232 However, one major challenge with hydrogel electrolytes is their tendency to freeze at low temperatures, which can impair their performance in cold environments. Therefore, enhancing the antifreezing properties of these electrolytes is crucial for the operation of flexible SCs under subzero conditions. Currently, most hydrogel-based supercapacitors utilize organohydrogels96,190 and ion-incorporated hydrogels,23,53 incorporating salts and organic solvents to realize intrinsic antifreezing properties.Incorporating ionic compounds into hydrogels generally enhances their antifreezing capabilities and ionic conductivity (see Section 3.2), making them well-suited for use in hydrogel-based supercapacitors.61,116,233,234 For example, LiCl-incorporated carrageenan/PAAm DN hydrogels exhibit an excellent conductivity of 1.9 S m−1 even at −40 °C. When employed as the electrolyte in a flexible supercapacitor with activated carbon (AC) electrodes on carbon cloth, this hydrogel demonstrates robust electrochemical performance, delivering a specific capacitance of 73.4 F g−1 and retaining 95.6% of its capacitance after 20000 cycles at −40 °C (Fig. 15a).235 Another example involves creating an NaAc-incorporated MoS2 hydrogel electrolyte-based mini supercapacitor using poly(N,N-dimethylacrylamide) (PDMAA) with PVA in the presence of MoS2 nanosheets to form a PDMAA/PVA hydrogel, which is then soaked in a saturated NaAc solution and encapsulated with the PET film.61 The “salting-out” effect of Ac− ions enabled this hydrogel-based supercapacitor to retain 96.2% of its capacity at −40 °C, even when bent to 180°, and to maintain 96.9% capacity retention after 10000 charge–discharge cycles (Fig. 15b). These properties position the NaAc-incorporated hydrogel as a promising candidate for energy storage devices in extremely cold environments.
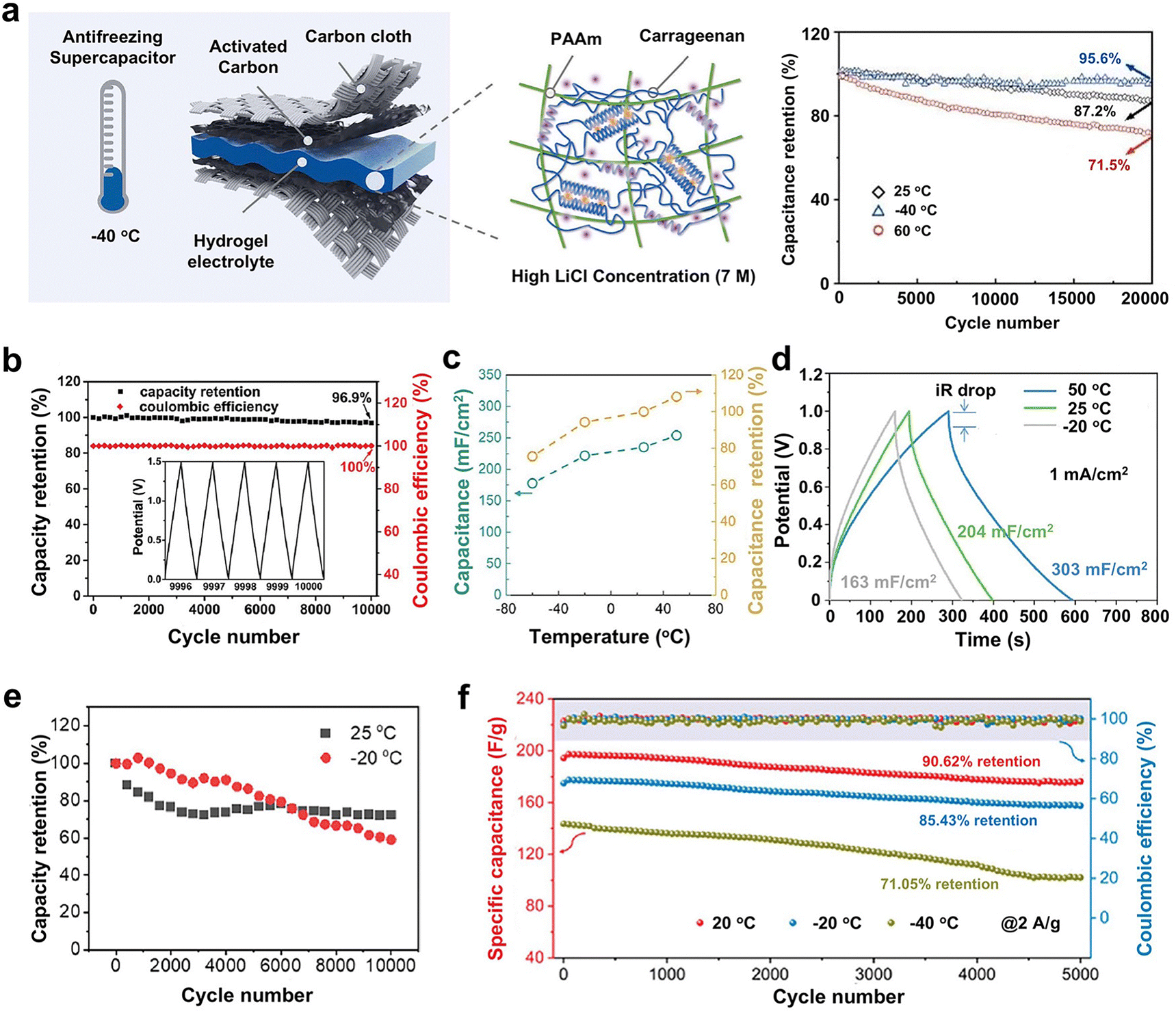 | ||
| Fig. 15 Electrochemical performance of antifreezing hydrogel-based supercapacitors. (a) Design and assembly scheme of the LiCl-incorporated carrageenan/PAAm DN hydrogel supercapacitor with high capacitance retention after 20000 cycles at −40 °C. Reproduced with permission from ref. 235, Copyright 2022 Elsevier. (b) NaAc-treated MoS2 hydrogel electrolyte supercapacitor, with high capacitance retention after 10000 cycles at −40 °C. Reproduced with permission from ref. 61, Copyright 2020 Wiley-VCH. (c) PVA/PAS-IL-based supercapacitors, with sustained capacitance and retention across temperatures from −60 to 60 °C. Reproduced with permission from ref. 116, Copyright 2022 Elsevier. (d) LiBr-PVBIMBr/PAAm-IL-based supercapacitors, with galvanostatic charge/discharge (GCD) curves at different temperatures (50, 25, and −20 °C) at a current density of 1 mA cm−2. Reproduced with permission from ref. 112, Copyright 2023 Elsevier. (e) LiCl and Gly-incorporated zwitterionic PSBMA-HEMA hydrogel-based supercapacitors, with sustained capacitance retention over 10000 cycles at 25 and −20 °C. Reproduced with permission from ref. 165, Copyright 2022 Elsevier. (f) LiClO4/Gly-treated HPC/PVA SC-OHE, with sustained capacitance over 5000 charging/discharging cycles at 2 A g−1 across temperatures of 20, −20, and −40 °C. Reproduced with permission from ref. 55, Copyright 2020 American Chemical Society. | ||
Alternatively, ILs and PILs offer a salt-free alternative for fabricating hydrogel electrolytes for supercapacitors, which combine the conductivity of ILs with the structural stability of polymer scaffolds, enhancing the overall functionality of the devices. A water-soluble ionic liquid (1-ethyl-3-methylimidazolium tetrafluoroborate, EMIM-BF4) incorporated into a PVA/(P(AAm-VBIPS)) (PVA/PAS-ILs) DN gel electrolyte has been used to assemble a supercapacitor with two layers of carbon cloth electrodes, demonstrating excellent antifreezing performance, flexibility, stretchability, and a high capacitance retention of 75.6% (177.6 mF cm−2) at −60 °C (Fig. 15c).116 Furthermore, an antifreezing PIL hydrogel electrolyte can be prepared by copolymerizing the ionic liquid monomer 1-vinyl-3-butyl imidazolium bromide (VBIMBr) with AAm, followed by the addition of a LiBr solution.112 This PIL hydrogel electrolyte achieves an excellent conductivity of 20.2 mS cm−1 at −20 °C. When assembled with two layers of AC electrodes, the resulting supercapacitor exhibits superior electrochemical properties, with a specific capacitance of 163 mF cm−2 at −20 °C, and retains about 80% of its room-temperature capacitance (Fig. 15d).
However, in some cases, incorporating ionic compounds alone may not provide adequate antifreezing properties, requiring the addition of organic solvents like EG and Gly to create salt-incorporated organohydrogel electrolytes.55,165,206,236,237 For instance, using a LiCl-incorporated silk fibroin/PVA-based Gly/water organohydrogel electrolyte in a supercapacitor retains 66.5% of its specific capacitance at −40 °C.54 Similarly, incorporating a zwitterionic P(SBMA-HEMA) hydrogel with EG and LiCl enhanced both conductivity and freezing resistance, enabling it to function as a supercapacitor that retains 59% of its initial specific capacitance at −20 °C after 10000 charging and discharging cycles, compared to 72.3% at 25 °C (Fig. 15e).165 Similarly, combining LiClO4 with Gly significantly improves antifreezing performance when integrated into a hydroxypropyl cellulose (HPC)/PVA organohydrogel electrolyte, prepared by immersing the HPC/PVA hydrogel in a LiClO4 water/Gly mixture. This organohydrogel electrolyte (SC-OHE), sandwiched between two AC electrodes, maintains nearly rectangular current–voltage (CV) curves at subzero temperatures down to −40 °C, closely mirroring its performance at 20 °C. This is in stark contrast to the significant performance decline observed in a supercapacitor using only a LiClO4-incorporated PVA hydrogel electrolyte without Gly (SC-HE). The SC-OHE's exceptional antifreezing capability is further underscored by its high capacitance retention through temperature cycling from 20 to −40 °C, achieving a specific capacitance of 143.6 F g−1 at 2 A g−1 at −40 °C—about 74% of its capacitance at 20 °C, markedly outperforming the SC-HE, which retains only 1.52% of its capacitance at −40 °C. Additionally, these flexible supercapacitors exhibit remarkable stability, retaining up to 71.05% of their specific capacitance after 5000 cycles at 2 A g−1 at −40 °C (Fig. 15f), showcasing the robustness of this design for applications in extreme cold environments.55
The development of antifreezing hydrogel electrolytes significantly enhances the functionality of SCs, allowing for their widespread use in cold climates and fulfilling the rigorous requirements of energy storage under extreme conditions. By incorporating ionic compounds such as organic salts and ILs, alongside antifreezing organic solvents, these hydrogels display robust antifreezing properties and improved ionic conductivity. Consequently, antifreezing hydrogel electrolytes sustain stable capacitance and remain flexible at subzero temperatures, providing reliable power output and enduring performance in harsh environments.
5.4. Self-healing and remoldability
Given the intrinsic mechanical weakness of antifreezing hydrogels at subzero temperatures, it is essential to design hydrogels with self-healing properties for applications that require sustained functionality and durability in cold environments. These properties allow the hydrogels to recover from mechanical damage and be reshaped as necessary, significantly enhancing their durability and longevity in practical applications. Currently, self-healing in hydrogels is primarily achieved through either dynamic covalent cross-linking or noncovalent cross-linking mechanisms.238 Dynamic covalent cross-linking methods include the formation of reversible bonds such as Schiff bases, oxime, acylhydrazone, disulfide bonds, and borate esters. These chemical bonds can autonomously reform under specific stimuli, enabling the hydrogel to self-repair after sustaining damage, even in low-temperature environments. Noncovalent cross-linking relies on reversible and adjustable interactions such as host–guest interactions, hydrophobic associations, electrostatic interactions, metal–ligand coordination, and hydrogen bonding.75,169 In these anti-freezing hydrogels, noncovalent cross-linking methods are more commonly used than dynamic covalent methods. On the other hand, when dynamic covalent methods are used, they are typically combined with noncovalent cross-linking methods to enhance the self-healing property.76 This preference stems from the fact that low temperatures may slow down or hinder the dynamic exchange of covalent bonds due to reduced kinetic energy. Conversely, noncovalent cross-linking methods remain more stable at low temperatures, as higher temperatures are more likely to disrupt these interactions by providing sufficient kinetic energy. Both types of cross-linking offer pathways to enhance the resilience and longevity of antifreezing hydrogels, making them viable for critical applications in outdoor sensors, biomedical devices, and other technologies operated under freezing conditions. Below, several representative examples of antifreezing hydrogels are presented, which incorporate noncovalent interactions such as hydrophobic associations, coordination bonds, hydrogen bonding, or a combination of these with covalent interactions to enhance the self-healing properties to varying degrees.Carboxymethyl chitosan (CMCS)/N-vinylpyrrolidone (NVP) hydrogels, formulated in a mixed solution of water, EG, and LiCl, showed exceptional freezing resistance down to −70 °C and highly efficient self-healing capabilities at both room and subzero temperatures.80 EG acts as an antifreezing agent, while LiCl serves as a conductive medium. Remarkably, the hydrogel fully recovers its structural integrity to support its own weight within just five minutes after cutting at temperatures from 25 to −35 °C. This rapid healing, with over 90% efficiency in tensile and compressive strength, is due to the robust hydrogen bonds between the carbonyl groups on CMCS and NVP, highlighting the hydrogel's versatility and resilience under harsh conditions. Another example of antifreezing hydrogels driven by hydrogen bonding is the multifunctional guar gum-Gly (GG-Gly) ionic hydrogels, synthesized from a mixture of Gly, water, and borax.88 This hydrogel retains antifreezing properties, primarily attributed to Gly. It also displays rapid self-healing facilitated by hydrogen bonding between Gly and water and chelation cross-linking involving borax. Mechanical tests using storage and loss moduli confirmed that the hydrogel regains mechanical integrity comparable to its pre-damaged state, indicating full recovery of its internal network. Notably, the hydrogel's self-healing is exceptionally rapid, with scratches disappearing in just 15 s. Similarly, carrageenan/PAAm DN hydrogels also demonstrate their self-healing capability in response to temperature via hydrogen bonding. Enhanced with CNTs and EG, carrageenan/PAAm hydrogels displayed unique self-healing properties.75 A simple heat–cool treatment enables the hydrogels to spontaneously reconnect, even after being segmented. This self-healing relies on the reversible structural transitions of carrageenan: at high temperatures, it shifts from a double-helical to a free-coil structure, which re-forms into double helices upon cooling. This dynamic mechanism not only maintains the hydrogel's structural integrity but also almost fully restores its conductivity after damage.
A hydrophobic associated polyacrylic acid (HAPAAc, copolymerized with lauryl methacrylate) hydrogel, formulated in a water/Gly mixture, demonstrates outstanding antifreezing properties down to −70 °C and impressive self-healing capabilities even at subzero temperatures. After three days at −20 °C, two separated sections of the hydrogel successfully self-repaired and could extend up to seven times their original length.89 The superior self-healing of HAPAAc hydrogels is driven by two key factors. Firstly, Gly modulates the hydrophobic associations within the hydrogel by reducing the interaction strength among micelles, allowing them to reorganize and repair the structure quickly after damage. Secondly, the abundant hydroxyl groups in Gly promote extensive hydrogen bonding with water, Gly, and HAPAAc. This hierarchical non-covalent crosslinking, which combines hydrophobic interactions and hydrogen bonding, significantly enhances the hydrogel's stretchability and self-healing capabilities.
Ionic interactions complement hydrogen bonding and hydrophobic interactions as another mechanism to endow hydrogels with self-healing properties. PVA/PAAc hydrogels, enhanced with Gly and Fe3+, exhibited good antifreezing and self-healing properties.90 Gly helps the hydrogel withstand temperatures down to −15 °C. Additionally, the self-healing capabilities are facilitated by dynamic ionic coordination bonds between Fe3+ ions and the carboxyl groups in PAAc. This effect is demonstrated by the rapid and near-complete erasure of cut marks within 12 h at room temperature after rejoining the separated sections.
In antifreezing hydrogels, employing multiple crosslinking mechanisms for self-healing properties is more prevalent than relying on single interaction. This approach is due to the complex nature of hydrogels, which often comprise various components interacting through different mechanisms. Meanwhile, this multiplicity enhances self-healing properties through synergistic effects, offering more robust and effective healing than strategies relying on single interactions. Arabic gum (GA)/PAAc-cellulose nanocrystal (CNC)/betaine/CaCl2 hydrogels exhibited anti-freezing and self-healing properties.68 Betaine and CaCl2 prevent ice formation, maintaining functionality down to −30 °C. The self-healing property relies on a network of noncovalent interactions—hydrogen bonds, ionic coordination, and electrostatic interactions. Upon contact, two segments immediately recombine at −20 °C, producing a robust structure that stretches without breaking, securely wraps around objects like a screw, and supports up to 500 g. Under an optical microscope, scratches visibly healed within 4 h. Substituting arabic gum with gellan gum and CNC with PANI-coated CNC preserves these properties.239
Ion- and Gly-incorporated PAAm organohydrogels demonstrated self-healing via electrostatic interactions and hydrogen bonding. Their healing efficiency, however, is influenced by temperature and Gly concentration, diminishing from 96% to 85% with temperature decreases or increases in Gly concentration.169 Similarly, gelatin organohydrogels prepared in a sodium citrate (Na3Cit) water/Gly solution merge upon heating and cooling, exhibiting enhanced stretchability due to the dynamic dissociation and re-association of physically cross-linked hydrophobic aggregates, ionic interactions, and hydrogen bonds.102 Similarly, nanocomposite hydrogels containing zwitterionic proline (ZP) and polydopamine-modified CNCs (CNCs@PDA) within a gellan gum/PAAc network exhibited outstanding stress tolerance and self-healing efficiency even at −30 °C.73 The antifreezing capability of these hydrogels is primarily due to hydrogen bonds formed by the imino groups in ZP with water. Their self-healing properties are enabled by a synergistic mix of coordination, electrostatic interactions, and hydrogen bonds (Fig. 16a and b).
 | ||
| Fig. 16 Self-healing mechanisms of antifreezing hydrogels using different types of noncovalent, dynamic covalent, or combined cross-linking approaches. (a) Synthesis process and the corresponding interactions of the hydrogels containing zwitterionic proline (ZP) and polydopamine modified cellulose nanocrystals (CNCs@PDA) integrated into a gellan gum/PAAc network via a combination of hydrogen bonding, coordination bonds, and electrostatic interactions. (b) Photo images demonstrate the cutting and healing of the hydrogels at ambient temperatures. (I) Different colored stretch strips; (II) two blocks resulting from cutting the stretch strips; (III) healing of two hydrogel strips with different combustion dyes without external stimulation; (IV) the self-healing hydrogel could lift a weight of 200 g. Reproduced with permission from ref. 73, Copyright 2023 Elsevier. (c) Schematic illustration shows the process of preparing the anti-freezing PAAm/PAAc-Fe3+/NaCl hydrogel via a combination of hydrogen bonding, coordination bonds, and hydrophobic interactions. Scale bar: 2 cm. (d) Photo images show the hydrogel after being cut into two pieces (i) and after complete healing (ii). Scale bar: 1 cm. (d) Stress–strain curves of the original hydrogel and the hydrogels healed for 3, 6, 12, 18, and 24 h after being cut into two pieces. Reproduced with permission from ref. 56, Copyright 2020 Royal Society of Chemistry. | ||
Furthermore, PAAm/Fe3+-chelated PAAc-NaCl hydrogels demonstrated the role of NaCl in lowering the freezing point to −24.7 °C, supporting self-healing through hydrogen bonding, ionic coordination, and hydrophobic interactions in high-humidity environments (Fig. 16c and d).56 Submerged in 2 M NaCl for 24 h, these hydrogels recover 90% of their mechanical strength, highlighting their durability. Additionally, PVA/silk fibroin (SF)/PPy hydrogels, prepared with water, EG, CaCl2, and formic acid, displayed antifreezing and self-healing properties from dynamic ionic and hydrogen bonding.82 When two pieces are rejoined at 30 °C and 25% humidity for an hour, their elongation at break fully restores. Similarly, black phosphorene (BP)/EG/PVA/sodium tetraborate/sodium alginate organogels demonstrated exceptional self-healing capabilities and can withstand temperatures below −120 °C.76 Their antifreezing property is primarily derived from EG, while rapid self-healing at −80 °C results from dynamic boron–diol bonds, hydrogen bonds, and supramolecular interactions within the gel network.
5.5. Surface adhesion
Reversible adhesion at subzero temperatures presents significant challenges primarily due to the adhesion of ice, which occurs as a result of water freezing at the interface. Common adhesion mechanisms that utilize stimuli-responsive polymers, dynamic covalent bonds, phase changes, topology transitions, and reversible non-covalent interactions, such as hydrogen bonds and host–guest interactions, often fail under these cold conditions. The low temperatures can disrupt the dynamic nature of these materials, limiting their ability to respond to stimuli and revert to their original state. Furthermore, the presence of ice can mechanically lock adhesive interfaces, preventing easy detachment and reattachment. Thus, developing adhesion systems that maintain reversible functionality at subzero temperatures requires innovative approaches that can overcome the limitations imposed by ice formation and the altered physical properties of materials at low temperatures.Reversible gel adhesives are typically engineered using: (i) stimuli-responsive polymers that react to external triggers like temperature or light,240 (ii) dynamic covalent bonds,241 such as those formed through Diels–Alder reactions or disulfide exchanges, which allow for the reversible breaking and reforming of bonds to tune adhesion, (iii) phase or topology transitions, where the adhesive alternates between gel and solid states in response to stimuli, facilitating easy application and removal,242 and (iv) non-covalent interactions including hydrogen bonding and host–guest interactions (e.g., cyclodextrin-based systems), which are pivotal for providing strong yet reversible binding capabilities.243,244 An example of an adhesive hydrogel features long-lasting moisture retention and exceptional temperature tolerance. This hydrogel is composed of a water/Gly mixture and reinforced with PDA-coated CNTs.87 The addition of CNTs enhances both mechanical strength and electrical conductivity of hydrogels. Thanks to the abundant catechol moieties in PDA and the viscosity of Gly, the resulting P(AAm-co-AAc) organohydrogel demonstrates strong tissue adhesiveness and excellent thermal tolerance, maintaining its properties across a broad temperature range from −20 to 60 °C. This stability enables long-term storage without degradation, maintaining a consistent adhesion strength of 57 ± 5.2 kPa to porcine skin at subzero temperatures (Fig. 17a). Additionally, natural polyphenols like TA, which are rich in quinones, catechol, and gallol groups, have become versatile components for crafting reversible adhesive hydrogels. Recently, TA has been combined with betaine or choline chloride to develop gel adhesives with low transition temperatures. This approach facilitates the self-assembly of guanosine into supramolecular viscoelastic materials, which exhibit remarkable adhesive properties (Fig. 17b).111 Molecular dynamics simulations have shown that strong hydrogen bonding between TA and betaine or choline chloride (ChCl) is critical in determining the structural properties of these materials. This bonding forms a robust network that boosts both adhesion and mechanical strength. The resulting hydrogel adhesive maintains durable, repeatable adhesion (i.e., 91.6 ± 9 kPa for 2% G-Bet/TA and 59.6 ± 6 kPa for 3% ChCl-Bet/TA hydrogels) even at extreme temperatures as low as −196 °C. This performance is attributed to the binding capabilities of the catechol and gallol groups in conjunction with the low glass transition temperatures of the TA–betaine and TA–choline chloride mixtures. Overall, these bio-inspired adhesive hydrogels demonstrate significant potential for diverse advanced applications, especially in fields such as biomedical devices, cryogenics, and materials suited for extreme environments.
 | ||
| Fig. 17 Design of surface-adhesive antifreezing hydrogels. (a) Mussel-inspired adhesive, tough, and conductive hydrogels utilizing a Gly–water binary solvent system for enhanced moisture retention and extreme temperature tolerance. The resultant P(AAm-co-AAc) hydrogels exhibited exceptional antifreezing properties, maintaining flexibility at −20 °C. The hydrogels remained pliable under various mechanical deformations, including twisting, compressing, and pulling, without losing structural integrity. Reproduced with permission from ref. 87, Copyright 2017 Wiley-VCH. (b) Schematic illustration of the preparation of reversible adhesive hydrogels (left) and the proposed interactions between TA and guanosine. The graph shows the statistical adhesion stress of the adhesive hydrogels (i.e., 2% G-Bet/TA and 3% ChCl-Bet/TA) on various substrates (left) and over different time periods at 25 °C (right), highlighting their strong and sustained adhesive properties. Reproduced with permission from ref. 111, Copyright 2024 American Chemical Society. | ||
ILs are exceptionally versatile materials ideal for achieving reversible bonding in various applications, thanks to their unique combinations of anions and cations, which enable tunable and responsive adhesion properties. A key advantage of ILs is their adjustable melting points, achieved by modifying the structure of the paired anions and cations, which allows for precise control over their thermal and functional properties. When integrated into reversible adhesive gels, ILs offer multiple benefits, particularly due to their adjustable nature and distinctive ionic interactions. By modifying the composition of ILs, these gels can display dynamic adhesion properties, enabling them to bond and de-bond in response to external stimuli such as temperature changes, electric fields, or mechanical stress. Recently, a temperature-responsive hydrogel made by using PDMAA and incorporating melted ILs was developed using in situ photo-crosslinking. This organohydrogel exhibits a remarkable surface adhesion strength of 5.82 MPa on glass, which can be attributed to the superior wetting properties of the melted gel and the formation of a thin, highly cohesive crystalline ionic layer on the glass surface.147 Notably, this adhesive layer is reversible, permitting easy detachment upon heating without leaving residue on the substrate. The adhesive can then be reactivated by cooling, facilitating re-adhesion over multiple cycles. Although IL-based hydrogels display ample mechanical strength, toughness, and strong adhesiveness even at subzero temperatures, their biocompatibility remains a significant challenge. ILs used in these hydrogels often exhibit cytotoxic properties, which can lead to adverse biological reactions in biomedical or clinical settings. Depending on their chemical structure, many ILs can impair cell viability, provoke inflammation, or initiate immune responses. Furthermore, the long-term impacts of ILs in the body, especially concerning their degradation products and potential for bioaccumulation, remain largely unexplored.
5.6. Wound dressing
Hydrogel itself has a strong ability to absorb exudate and excellent biocompatibility and promotes moist healing, making hydrogel dressings an indispensable part of wound dressings. Antifreezing hydrogels are emerging as a revolutionary solution in wound care, particularly in environments susceptible to extreme cold. These hydrogels maintain their flexibility and healing properties at subzero temperatures, preventing the gel from freezing and ensuring continuous, effective treatment of wounds. Their unique composition allows them to deliver therapeutic agents without losing structural integrity or therapeutic efficacy in cold climates. This makes them especially valuable in outdoor medical care, military applications, and emergency services in cold regions, providing a resilient and adaptable option for wound management.Early investigations into antifreezing hydrogels for wound dressings primarily explored their antibacterial properties. One of the initial strategies involved using VBIMBr ionic liquid to infuse solvent ions, providing antifreeze capabilities alongside antibacterial effects in hydrogel dressings.113 Simultaneously, Gly was incorporated as an organic solvent to enhance both antifreeze and antimicrobial features in hydrogels.98 As research evolved, a deep eutectic solvent (DES) approach combined [BMIM][BF4] ionic liquid with Gly, ensuring stable electromechanical properties in hydrogels capable of withstanding temperatures as low as −50 °C, even after self-repair.71 Subsequent advancements moved away from direct antifreeze agent additions towards creating hydrophobic/hydrophilic heterostructures (core–shell nanostructures) that effectively inhibited ice recrystallization down to −60 °C.107 Further developments utilized zwitterionic PCBMA in a similar hydrophobic/hydrophilic nanostructure, synergistically preventing ice crystallization and enhancing the cryoprotective capabilities.109
Despite significant advancements in hydrogel technology, research specifically focusing on antifreezing hydrogels as biological dressings remains limited. Key areas of investigation include developing hydrogels that not only exhibit antimicrobial properties and high mechanical performance but also enhanced adhesion to promote healing in infected wounds. Consequently, antifreezing hydrogels evolve their antimicrobial functions to heal full-thickness skin defect wounds in more specific and complex biological environments.113 Cellulose organohydrogels, derived from durian rind—a byproduct of the food industry—have shown promise in treating wounds with bacterial infections. Specifically, water-based cellulose hydrogels were prepared through a simple water–Gly exchange process, which not only incorporated Gly to enhance the hydrogels’ antifreezing properties and resistance to drying but also significantly improved their mechanical strength.98 Additionally, the inclusion of natural yeast phenolics endowed the hydrogels with notable antimicrobial activity, without exhibiting cytotoxic effects. When applied as wound dressings on porcine skin, these hydrogels demonstrated substantial antibacterial efficacy, establishing cellulose organohydrogels as promising candidates for antimicrobial wound dressings, especially effective under extreme temperature conditions (Fig. 18a). GQSG hydrogels, composed of GelMA, sulfobetaine, and quaternized chitosan (QCS), have proven highly effective for treating pressure ulcers due to their high strength, antibacterial, and antifreezing properties.99 After undergoing a solvent exchange with Gly—a recognized antifreeze agent—the hydrogels demonstrated excellent antifreeze properties and remained flexible and transparent even after being stored at −20 °C for three days. Furthermore, GQSG hydrogels resisted adsorption of E. coli at 37 °C, unlike a control hydrogel without Gly and commercially available dressings made of agar, PEG, and polyvinylpyrrolidone, which showed significant bacterial presence (Fig. 18b). This antibacterial effectiveness primarily stems from the zwitterionic PSBMA components, which inhibit bacterial adhesion. Chitosan hydrogels, which integrate a polymerized ionic liquid and a near-infrared (NIR) fluorescent probe (PIL-CS),114 display antifreezing properties that maintain excellent electrical and mechanical performance at −40 °C, attributable to the ILs. Critically, PIL-CS hydrogels are specific, sensitive, stable, and reversible in responding to pH changes at the wound site. These hydrogels enhance the visualization of wound pH through in vivo NIR fluorescence imaging and provide pH-responsive sustained drug release. In vivo studies have shown that PIL-CS hydrogels significantly accelerate diabetic wound healing, promote the production of vascular endothelial growth factor (VEGF), and reduce levels of reactive oxygen species (ROS) and tumor necrosis factor (TNF-α), underscoring their potential as effective treatments for diabetic wounds (Fig. 18c). Recently developed, the quaternized P(AAm-co-THMA-co-SBMA) (ATSHQ) hydrogel is stretchable, adhesive, antifreezing, and antibacterial, making it particularly well-suited for treating wounds on significantly deformed joints.101 The two formulations, ATSHQ-121 and ATSHQ-221, have shown remarkable self-healing capabilities, maintained mechanical stability at −18 °C, and demonstrated effective adhesive performance, along with good biocompatibility and notable antibacterial properties (Fig. 18d). Among them, the ATSHQ-121 hydrogel is notably softer, more stretchable, and has superior adhesive qualities, making it exceptionally suitable for wounds that undergo substantial deformation, particularly around joints.
 | ||
| Fig. 18 Antifreezing hydrogels for wound dressings. (a) Rind-derived cellulose organohydrogels as antifreezing and antimicrobial wound dressings on pig skin, showing reduced total viable bacteria as compared to moist gauze. Reproduced with permission from ref. 98, Copyright 2021 American Chemical Society. (b) GQSG hydrogels with antifreezing capacities at −20 °C for 3 days and strong surface resistance to E. coli at 37 °C. Reproduced with permission from ref. 99, Copyright 2021 Springer Nature. (c) PIL-CS hydrogels as a dressing for diabetic wounds, capable of both enhancing and monitoring skin repair and regeneration in real-time. Reproduced with permission from ref. 114, Copyright 2023 Wiley-VCH. (d) ATSHQ hydrogels with adhesive, antifreezing, and antibacterial properties for wound dressing. Reproduced with permission from ref. 101, Copyright 2024 Elsevier. | ||
However, studies in this area remain scarce due to several challenges: (i) the complexity of the physiological environment of wound surfaces necessitates hydrogels with superior healing effects; (ii) the specific disease characteristics and localized nature of wounds demand hydrogels with targeted treatment capabilities; and (iii) the shift from treating acute wounds to chronic wounds calls for hydrogels capable of sustained application. There is significant potential for further advancements in the application of antifreezing hydrogels within the biomedical field.
5.7. Cryopreservation
In tissue engineering and biomedical applications, antifreezing hydrogels are increasingly explored for cryopreservation due to their biocompatibility and ability to prevent damage from ice formation and recrystallization, common challenges during the freezing and thawing processes.181,245 These properties make antifreezing hydrogels ideal for developing implants and scaffolds that remain functional in cold storage or during transport, ensuring they can withstand low-temperature conditions—such as those in cryopreservation at −80 or −196 °C—without compromising their performance or the viability of the biological materials they contain.246GelMA hydrogels are extensively used in skin tissue engineering due to their biocompatibility, biodegradability, and tunable mechanical properties.247 To extend the shelf life and preserve the viability of engineered tissues for clinical use, it is crucial to optimize cryopreservation conditions. Several factors were carefully considered to modify the crystallization properties of the antifreezing GelMA hydrogel: (i) concentrations of the GelMA hydrogel up to 7% effectively inhibited ice crystal formation; (ii) adding a cryoprotectant (e.g., 10% DMSO) significantly enhanced the hydrogel's cryoprotective effects; (iii) sheets of the 10% DMSO–7% GelMA hydrogel crosslinked for up to 60 min showed no ice crystal formation or growth during cryopreservation. The protective efficacy of 10% DMSO–7% GelMA hydrogel sheets was substantiated in NIH 3T3 cell models, demonstrating a significant increase in cell viability by 67.6% and a proliferation efficiency of over 90% (Fig. 19a).104 In addition, the natural amino acid L-proline (L-Pro) has been explored as a non-toxic alternative to DMSO to mitigate DMSO-induced cytotoxicity. Optimal cryopreservation conditions were established using a 6% L-Pro-7% GelMA hydrogel, maintained at 37 °C for 3 h before undergoing rapid cooling for cryopreservation. The results showed that the L-Pro-GelMA complex hydrogel used for cryopreservation had no adverse effects on NIH 3T3 cells compared to a fresh group, as depicted in Fig. 19b. Unlike traditional DMSO-based cryoprotectants, cells revived from this hydrogel do not require extensive washing and can be prepared for culture with simple dilution.105
 | ||
| Fig. 19 Antifreezing hydrogels in cryopreservation. (a) GelMA hydrogel sheets with enhanced cell viability and proliferation in NIH 3T3 cryopreservation compared to unencapsulated cells. Reproduced with permission from ref. 104, Copyright 2024 Wiley-VCH. (b) L-Pro-GelMA complex hydrogels with comparable cell proliferation rates and morphologies to fresh (non-frozen) NIH 3T3 cells. Reproduced with permission from ref. 105, Copyright 2024 Elsevier. (c) Small alginate microcapsules (∼100 μm) with enhanced ice inhibition in cell cryopreservation using 0–0.15 M CPA of Ca2+. Reproduced with permission from ref. 66, Copyright 2010 Springer Nature. (d) Large alginate microcapsules (>500 μm) with increased cell viability and gene expression levels matching those of unencapsulated and fresh cells during cryopreservation. Reproduced with permission from ref. 108, Copyright 2017 Wiley-VCH. (e) UPy-based hydrogels with varied ice recrystallization inhibition based on UPy content. Ih, Ic, and Iam denote hexagonal, cubic, and amorphous/short-range ice structures, respectively. Reproduced with permission from ref. 107, Copyright 2021 American Chemical Society. | ||
Beyond traditional methods like programmable or controlled freezing for cryopreservation, vitrification—where a liquid transitions into a glass-like solid without forming ice crystals—is emerging as a promising alternative. This technique offers several key advantages, including the elimination of ice crystals, enhanced cell viability, and more rapid processing.248,249 However, conventional vitrification often necessitates the use of high cryoprotective agent (CPA) concentrations, up to 8 mol L−1, which can be highly cytotoxic. To mitigate this issue, researchers have developed anti-freezing alginate microcapsules, derived from natural seaweed polysaccharides, that enable effective cell vitrification with lower CPA levels. Specifically, reducing CPA concentrations to as low as 1.5 mol L−1 in vitreous cryopreservation allowed C3H10T1/2 cells encapsulated in small alginate microcapsules (∼100 μm) to achieve a significant increase in the survival rate, approximately 46.9%.66 This protective effect is primarily attributed to two factors: (i) the alginate hydrogel matrix vitrifies preferentially compared to the bulk solution66 and (ii) the alginate hydrogel's superior ice-control capabilities, which lower the ice-formation temperature and delay ice formation (Fig. 19c).66,250 Vitreous cryopreservation of cells can be further enhanced by encapsulating them in large-volume microcapsules (>500 μm) with a core–shell structure. This approach has been shown to increase cell viability by approximately 49% and does not significantly affect the expression of four typical stem cell genes (Nanog, Klf4, Sox2, and Oct4) compared to fresh cells (Fig. 19d).108
In addition to pure alginate, alginate chains can also be incorporated into a covalently crosslinked PAAm network, forming a DN hydrogel with enhanced mechanical properties. The addition of CaCl2 creates a hybrid PAAm/Alg-Ca DN hydrogel, which exhibits tunable antifreezing properties and exceptional recoverability at subzero temperatures. This hybrid hydrogel displays pH-responsive swelling within a pH range of 5–7, maintains mechanical flexibility at −20 °C, and demonstrates favorable biocompatibility. Notably, leveraging these properties, the multifunctional PAAm/Alg-Ca DN hydrogel has been used to encapsulate lomustine, an anti-cancer drug for glioblastoma, at −20 °C. This encapsulation not only achieves an excellent drug loading capacity of approximately 3.0%, but also ensures the preservation of drug potency and supports sustained drug release over a month.65
Recently, self-healing anti-freezing hydrogels have gained attention for cryopreservation applications, thanks to their capacity to self-repair damage from mechanical stress, dehydration, and ice crystal formation during freeze–thaw cycles. For instance, random copolymers containing a substantial portion (35–50 mol%) of 2-ureido-4[1H] pyrimidinone (UPy) moieties were developed to form stable hydrogels.107 These hydrogels maintain structural integrity down to −60 °C; at these low temperatures; the directional packing of UPy–UPy aggregates forms nano-scale gaps small enough to confine water molecules and inhibit ice nucleation. The extent of these gaps is closely related to the copolymer concentration and the proportion of UPy moieties, with higher UPy densities reducing the formation of crystalline ice peaks and causing water to crystallize into amorphous or short-range ice structures. This structure provides more effective inhibition of ice recrystallization within the hydrogels (Fig. 19e). Additionally, cell viability tests have shown negligible cytotoxicity, comparable to PVA (9.1 kDa)135—a well-known ice recrystallization inhibitor with minimal toxicity—thus underscoring its potential for use in drug storage and cell cryopreservation.
5.8. Environmental and industrial uses
Antifreezing hydrogels have potential applications in environmental and industrial settings, such as in antifreeze coatings, de-icing agents, and cold chain logistics, where their ability to prevent freezing and maintain functionality under harsh conditions is crucial. Additionally, these hydrogels can mitigate frost damage in agriculture, where ice formation within plant tissues at subfreezing temperatures causes cellular damage, potentially leading to cell death, stunted growth, or the loss of entire plants. To address this issue, sodium alginate-based hydrogels have been widely used as anti-freezing coatings, offering a protective barrier against frost and helping to preserve the quality of various fruits, such as apples,67,251 peaches,252 and cherries,253 during post-harvest storage. As an example, a protective coating created by cross-linking sodium alginate with CaCl2 effectively reduced chilling injury and preserved the postharvest quality of rose apples stored at low temperatures. Fruits coated with sodium alginate/CaCl2 exhibited lower respiration rates and reduced weight loss over 10 days of cold storage. Additionally, compared to uncoated fruits, the coated ones maintained a glossier appearance and showed fewer chilling injury-induced blemishes, such as brown flecks and shrinkage at the fruit's top (Fig. 20a).67 For fresh-cut cantaloupes, treatment with an alginate coating combined with repetitive pulsed light effectively preserves freshness by maintaining firmness and reducing fluid loss during cold storage at 4 °C. Microscopically, alginate interacts effectively with high pectin fractions, while repetitive pulsed light helps maintain hemicellulose, both of which contribute to preserving cell wall integrity in fresh-cut cantaloupes.93 Beyond directly protecting fruits, sodium alginate/CaCl2 coatings have also been shown to delay tree blooming, reducing the risk of frost damage during early spring and enhancing peach tree productivity. For instance, a combined treatment of spur pruning and sodium alginate gel effectively delayed primary shoot development in Chardonnay grapes, sparking widespread interest in the application of antifreezing alginate gels.254 In a subsequent study, a coating of varying concentrations of sodium alginate combined with CaCl2 was applied to peach flower buds to assess its effects on phenological development and bloom timing. The results showed that a 5% sodium alginate treatment was most effective for orchard application, delaying peach flowering by up to 9 days compared to untreated trees (Fig. 20b).255 Beyond fruits, seafood also requires careful management during cold chain storage and transportation to maintain freshness and taste. Alginate oligosaccharides have emerged as promising cryoprotectants in the seafood industry, aiding in the stabilization of muscle tissue structure, inhibition of protein denaturation, and reduction of myofibril damage caused by large ice crystals during freezing and frozen storage, as seen in products like peeled Pacific white shrimp.256 However, the use of full-length alginate hydrogels in seafood preservation remains an area of ongoing research and exploration. | ||
| Fig. 20 Environmental uses of different antifreezing hydrogels. (a) Sodium alginate/CaCl2 coatings to reduce chilling injury and preserve the postharvest quality of rose apples. Reproduced with permission from ref. 67, Copyright 2022 Elsevier. (b) Sodium alginate/CaCl2 coatings to delay flowering and fruit set in peach trees. Reproduced with permission from ref. 255, Copyright 2022 Springer Nature. (c) B–O/sodium alginate hydrogels as a lubricant for a drone propeller at −20 °C. Reproduced with permission from ref. 94, Copyright 2024 Wiley-VCH. (d) Mollusk-inspired organohydrogel dynamic interface as frost protection for a model car. (e) Organohydrogel dynamic interface as frost protection for a propeller. Reproduced with permission from ref. 69, Copyright 2020 American Chemical Society. (f) Electrolyte hydrogels with a self-generating lubricating layer. Reproduced with permission from ref. 59, Copyright 2020 American Chemical Society. (g) P(VBIPS/HEAA)-SA DN hydrogels with friction-to-lubrication transition between fresh and saltwater. Reproduced with permission from ref. 60, Copyright 2021 Elsevier. (h) PNDE hydrogel-based smart window for radiative cooling in hot climates and heat retention in cold climates. Reproduced with permission from ref. 79, Copyright 2023 Elsevier. (i) APTES-SiO2/PVA organohydrogel-coated mesh for oil/water separation. Reproduced with permission from ref. 77, Copyright 2020 Elsevier. (j) COCu–K hydrogels for weed germination and growth control. Reproduced with permission from ref. 62, Copyright 2023 Elsevier. | ||
Hydrogels with enhanced antifreeze, lubrication, and adaptive properties are being widely applied across challenging environments, including machinery, aerospace, and marine industries. In machinery systems exposed to cold and/or saline conditions, antifreeze hydrogels serve as effective lubricants to reduce wear and promote energy efficiency, while also acting as stable coatings that protect against environmental stress. Altogether, these properties enhance the functionality and durability of systems operating under harsh conditions. Notably, incorporating dynamic B–O bonds into the sodium alginate framework produced an adaptive lubricating hydrogel with tunable tribological performance, as well as excellent antifreeze and heat resistance properties.94 The resulting hydrogel's adjustable coefficient of friction (CoF) and strong environmental resilience make it suitable for applications such as connecting drone propellers to motor shafts, where it maintains stable lubrication across temperatures ranging from −40 to 60 °C (Fig. 20c). Focusing on applications in extremely cold environments, inorganic-based organohydrogels (IOOHG) were synthesized using a ternary mixture of commercially available colloidal silica nanoparticles, water, and Gly.95 The resulting IOOHG demonstrated an ultra-low CoF of approximately 0.02 at room temperature. Even at a low temperature of −80 °C, the addition of Gly provided the IOOHG with excellent antifreezing, anti-volatility, and anti-corrosion properties, allowing it to maintain a low CoF of around 0.08 without freezing. These characteristics support the IOOHG's potential for long-lasting lubrication in aerospace and other industries operating in polar regions. Inspired by (i) the mucus secretion of mollusks, a series of organohydrogel dynamic interfaces (ODIs) were prepared by incorporating CPAs, such as Gly and ethylene, into four types of tough hydrogels—gelatin, Ca2+-alginate/PAAm DN, PVA, and cellulose hydrogels—via a solvent displacement method.69 Additionally, (ii) inspired by the adhesive proteins in mussels, SA-g-DA was synthesized by introducing dopamine, the primary component of mussel adhesive proteins, into sodium alginate.106 Beyond antifreezing capabilities, as demonstrated in IOOHG, these bio-inspired hydrogels can be easily coated onto various surfaces, including glass, watches, smartphones, fabric, aluminum wire, model cars, leaves, and propellers, providing frost/ice-free surfaces and preserving their functional integrity (Fig. 20d and e).
For marine applications, an innovative electrolyte hydrogel (EH) was developed by infusing salted water into a hydrogel matrix, providing anti-icing and anti-frost capabilities for up to 5 days and achieving an ultralow (Pa-level) ice adhesion at temperatures as low as −48.4 °C.59 During the anti-icing process, ions from the EH diffuse to the interface between the EH and ice, preventing ice formation by maintaining a lubricating liquid layer. This layer facilitates rapid self-removal of ice by gravity, enabling efficient ice clearance within just 10 s at −10 °C (Fig. 20f). While ion diffusion may deplete ions in the EH and reduce its anti-icing performance over time, seawater simulation tests have shown that the EH can be sustainably recharged with salted water through coating or direct contact methods, thereby maintaining its anti-icing properties. These advantages make EH a promising solution for ice protection on offshore platforms and marine vessels. Additionally, a DN hydrogel of P(VBIPS/HEAA)-SA was synthesized as a promising material for marine applications.60 In addition to its robust mechanical properties and excellent recoverability, the P(VBIPS/HEAA)-SA hydrogel demonstrates unique environmental responsiveness: it shrinks and becomes opaque in fresh water but swells and turns highly transparent in saltwater, making it a potential material for optical camouflage. Furthermore, its transition from friction to lubrication between fresh and saltwater enables smoother movement for underwater equipment, reducing drag and improving energy efficiency (Fig. 20g). With exceptional antifreezing properties, high ionic conductivity, and flexibility at temperatures as low as −60 °C, the P(VBIPS/HEAA)-SA hydrogel is highly adaptable for applications under extreme conditions.
In energy-saving technologies, smart windows have become a promising solution for improving building efficiency and reducing energy consumption. NIPAM, a temperature-sensitive polymer, is widely used as a foundation for constructing antifreezing hydrogels, making it integral to smart window design. For instance, poly(NIPAM-co-N,N-dimethylacrylamide)/EG (PNDE) hydrogels were developed with impressive, tunable freezing resistance down to −100 °C.79 Building on PNDE hydrogels, combined with polyvinylidene fluoride and polymethyl methacrylate-silver nanowire panels, a solar and thermal radiation regulation (STR) smart window was developed. This STR smart window exhibited remarkable features: a high luminous transmittance of 68.2%, solar regulation efficiency of 62.6%, an optimal phase-change temperature of ∼30 °C, and excellent antifreezing capabilities down to −20 °C. Additionally, its dual-sided thermal emissivity allows the STR window to provide radiative cooling in warm climates and heat retention in cold climates (Fig. 20h), maintaining stable indoor temperatures, reducing energy consumption, and enhancing building sustainability. Alternatively, NIPAM was dissolved in a Gly–water binary solvent system to create a precursor solution, which was then directly sandwiched between glass panels to form smart windows.79 This smart window demonstrated a high luminous transmittance of 90%, effective solar regulation performance of 60.8%, and impressive antifreezing capabilities down to −18 °C. Additionally, it achieved an ultra-fast response time of only 0.16 min, more than an order of magnitude faster than other hydrogel devices.257,258 This energy-saving approach extends beyond environmental sources to include energy harvesting directly from the human body. A stretchable antifreezing hydrogel-based triboelectric nanogenerator (AH-TENG) was developed, consisting of an acrylamide-hydroxyethyl cellulose antifreezing hydrogel layer encased between two layers of silicone rubber.156 The AH-TENG effectively harnessed electrical energy from simple human motion, successfully charging wearable electronics even under harsh ice and snow conditions.
Environmentally, (3-aminopropyl)triethoxysilane-functionalized silica (APTES-SiO2) nanoparticles were incorporated into a PVA gel network to create APTES-SiO2/PVA organohydrogels.77 The resulting gel-coated stainless steel meshes demonstrated an oil–water separation efficiency exceeding 99% across various water environments, including deionized water, 1 M HCl solution, 1 M NaOH solution, saturated NaCl solution, artificial seawater, and even saturated NaCl solution at −20 °C. Notably, these gel-coated meshes are recyclable and can self-heal to restore their original oil–water separation capabilities through a simple heating-induced sol-to-gel process (Fig. 20i). To enhance crop production, a self-healing and biodegradable hydrogel mulching film (COCu–K) was developed to inhibit weed germination and growth. This film is composed of chitosan derivatives, copper-doped carbon dots (Cu–CDs), and KCl.62 Among various COCu–K2.5 formulations tested, the COCu0.08–K2.5 hydrogel emerged as the optimal formulation, achieving a low weed germination rate of 6.73% (Fig. 20j) while effectively promoting crop growth, such as peppers.
It is important to highlight that the hydrogels and antifreezing additives used for antifreezing hydrogels, including polymers, salts, and ionic liquids, are similar to those commonly found in various materials and everyday products. These components generally exhibit low toxicity and are highly biodegradable, with their environmental impact largely dependent on the polymeric backbone and specific additives incorporated. On the other hand, the environmental accumulation of antifreezing additives raises a valid concern. High concentrations of salt can lead to soil salinization and adversely affect microbial communities. Similarly, some ionic liquids may persist in aquatic systems, posing risks of bioaccumulation. Recent research has explored sustainable alternatives, such as bio-derived cryoprotectants and polymer-based antifreezing agents, to minimize environmental risks.259–261 To further this progress, the advancement of NFI is a promising greener alternative to traditional antifreezing additives. Unlike conventional methods that utilize salts, ionic liquids, or organic solvents, the NFI approach focuses on engineering polymer networks that intrinsically inhibit ice crystallization, offering a more environmentally friendly solution. There are three potential directions: (i) designing bio-based and degradable NFI hydrogels using renewable resources (e.g., polysaccharides and polyzwitterions) to enhance environmental sustainability; (ii) optimizing polymer–water interactions by tailoring the hydrophilic/hydrophobic balance and incorporating specific hydrogen-bonding motifs to improve ice inhibition efficiency; (iii) exploring synergistic strategies that combine NFI with mild, environmentally friendly cryoprotectants (e.g., glycerol and trehalose) to enhance performance while maintaining sustainability.
6. Conclusions and perspectives
Antifreezing hydrogels represent an innovative class of materials engineered to retain their flexibility, functionality, and mechanical integrity in harsh, subzero temperature environments, under which traditional hydrogels would typically freeze and become brittle. These hydrogels leverage different design and fabrication strategies by either incorporating antifreezing additives (ionic compounds, organic solvents, antifreezing proteins and peptides) or constructing highly crosslinked water-strong-binding polymer networks. The design of antifreezing hydrogels is driven by the challenge of preventing ice formation in high-water content materials, which paradoxically inspires new ideas for developing effective antifreeze materials and mechanisms. Such innovative designs represent a major advancement in materials science, offering promising solutions for applications in biomedical devices, tissue engineering, and flexible electronics, which require hydrogels to maintain their desired properties at subzero temperatures.Despite their significant advancements and promising potential, antifreezing hydrogels face several substantial challenges. A primary concern is their tendency to lose mechanical properties (i.e., strength, toughness, and durability) and become brittle at subzero temperatures. Even minimal ice formation within the polymer network can lead to mechanical failure and loss of functionality, severely limiting their usability in cold environments. From a materials design perspective, a long-standing issue lies in selecting and integrating appropriate network topologies (IPNs, semi-IPNs, double networks, nanocomposite networks, branched networks, and gradient networks), suitable crosslinkers (whether physical, chemical, reversible, or irreversible), and water-binding polymers with an optimal balance between hydrophilicity and hydrophobicity for achieving effective antifreeze properties. However, achieving this synergy and effectively optimizing these components remains a complex and challenging task. Thus, continued advancements in materials design and synthesis are crucial for driving innovative approaches in polymerization, crosslinking, and the incorporation of antifreeze agents. These developments will facilitate the creation of new hydrogels with enhanced antifreezing capabilities, long-term durability, and strong resistance to environmental factors, along with other tailored properties, ensuring their effectiveness over extended periods.
Another challenge is associated with the antifreezing additives incorporated in the hydrogels, which could introduce issues such as volatility, toxicity, and environmental hazards, raising concerns about the safe and sustainable use of these hydrogels. Consequently, the use of non-toxic and biodegradable materials in the development of biocompatible and environmentally friendly antifreezing hydrogels is essential to minimize adverse effects on the environment and human health. These hydrogels must also reliably perform under diverse environmental conditions, making this an important focus for ongoing research. It is interesting to note that designs incorporating two binary components with entirely opposite physicochemical properties at the nanoscale—such as hydrophobic versus hydrophilic, positively charged versus negatively charged, electron donor versus electron acceptor, organic versus water, brittle network versus elastic network—often yield high-performance materials. This approach, known as the cooperative complementary principle, leverages the contrasting characteristics of each component to enhance the complementary functionality and overall performance of the resulting materials.
Recent advancements in data-driven machine learning approaches have significantly influenced various research domains, from drug discovery, image recognition, and transportation analysis to materials informatics. However, the application of machine learning to antifreezing hydrogels, which require specific functional properties, is still in its early stages. A major hurdle is the lack of comprehensive, publicly available datasets for antifreezing hydrogels. Necessarily, researchers are often compelled to manually compile experimental data from disparate literature sources to create these databases. Additionally, the intrinsic structural complexity of hydrogel networks and the chemical diversity of polymers, coupled with their varied crosslinking properties, further complicate the compilation of reliable data. This issue is exacerbated by the fact that even identical antifreezing hydrogels can produce inconsistent data due to variations in experimental conditions, chemical/material purity, and synthesis methodologies across different laboratories. The absence of a reliable benchmarking dataset for antifreezing hydrogels poses a considerable challenge for developing robust machine-learning models, which can establish qualitative or quantitative frameworks for the rational assessment and design of antifreezing hydrogels. Deep learning, renowned for its efficacy in pattern recognition, could predict hydrogel properties by analyzing molecular structures through convolutional neural networks. Reinforcement learning could optimize hydrogel synthesis by iteratively adjusting synthesis parameters and learning from the results, aiming to perfect antifreeze capabilities and mechanical robustness. Transfer learning could mitigate data scarcity by applying knowledge from abundant datasets in related polymers, enhancing property predictions for antifreezing hydrogels. Additionally, graph neural networks could model complex polymer networks at the molecular level, providing insights into how different configurations influence water states and antifreeze properties. Coupling these machine learning strategies with molecular dynamics simulations could elucidate how different polymer networks (including linear, random, block, branched, and cross-linked) and how the functional groups affect water states (unfrozen water, weakly bound water, and free water), and consequently anti-icing ability. To date, there are no machine learning models that, along with experimental data, fully explore the behaviors of water confined in polymer networks at the atomic level or identify antifreezing structural groups. Consequently, this highlights the need for targeted efforts to generate standardized datasets and develop predictive models that can establish a composition–structure–antifreezing relationship for antifreezing hydrogels.
Finally, it must be acknowledged that much of the current literature on antifreezing hydrogels is filled with a simple method of adding functional components to the hydrogels to achieve desired properties. While this approach is straightforward and sometimes effective, it often lacks creativity in terms of materials design and mechanism exploration. To move beyond merely adding functional components to hydrogels, there is a need to develop hydrogels that inherently possess multifunctional properties without relying on the addition of numerous specific agents. This can be accomplished through the design of novel polymers or by modifying existing polymers to naturally integrate multiple functions. Furthermore, it is equally important to explore the design of hydrogels with hierarchical structures that can more closely mimic natural antifreeze mechanisms. These bio-inspired designs involve creating microscale and nanoscale architectures within the hydrogel, similar to those found in polar fish, Alaska wood frog, insects, and conifers, which have evolved to interact with ice formation on multiple levels. By incorporating such intricate structural features, the effectiveness of hydrogels in preventing ice formation and the underlying antifreezing mechanisms could be significantly enhanced. Overall, future research should focus on a deeper understanding of the fundamental interactions and bonding mechanisms within the hydrogel matrix. Investigating how different components interact at molecular and atomic levels will reveal how these interactions affect the overall properties of antifreezing hydrogels. Addressing these challenges requires ongoing research and innovative approaches, aiming not only to enhance the functionality of antifreezing hydrogels, but also to push the boundaries of materials science in this field.
Data availability
No primary research results, software or code have been included and no new data was generated or analyzed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank financial support from the NSF grant (DMR-2311985) and the ACS PRF grant (ND-65277).References
- J. Lv, Y. Song, L. Jiang and J. Wang, ACS Nano, 2014, 8, 3152–3169 CrossRef PubMed.
- Z. He, C. Wu, M. Hua, S. Wu, D. Wu, X. Zhu, J. Wang and X. He, Matter, 2020, 2, 723–734 CrossRef.
- Z. He, K. Liu and J. Wang, Acc. Chem. Res., 2018, 51, 1082–1091 CrossRef CAS PubMed.
- S. Wu, Y. Du, Y. Alsaid, D. Wu, M. Hua, Y. Yan, B. Yao, Y. Ma, X. Zhu and X. He, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 11240–11246 CrossRef CAS PubMed.
- K. Golovin and A. Tuteja, Sci. Adv., 2017, 3, e1701617 CrossRef PubMed.
- H. Gao, Z. Zhao, Y. Cai, J. Zhou, W. Hua, L. Chen, L. Wang, J. Zhang, D. Han, M. Liu and L. Jiang, Nat. Commun., 2017, 8, 15911 CrossRef CAS PubMed.
- Y. Ye, Y. Zhang, Y. Chen, X. Han and F. Jiang, Adv. Funct. Mater., 2020, 30, 2003430 CrossRef CAS.
- Q. Rong, W. Lei, L. Chen, Y. Yin, J. Zhou and M. Liu, Angew. Chem., Int. Ed., 2017, 56, 14159–14163 CrossRef CAS PubMed.
- S. Zhang, Y. Zhou, A. Libanori, Y. Deng, M. Liu, M. Zhou, H. Qu, X. Zhao, P. Zheng and Y.-L. Zhu, Nat. Electron., 2023, 6, 338–348 CrossRef.
- Y. Jian, S. Handschuh-Wang, J. Zhang, W. Lu, X. Zhou and T. Chen, Mater. Horiz., 2021, 8, 351–369 RSC.
- Y. Zhuo, J. Chen, S. Xiao, T. Li, F. Wang, J. He and Z. Zhang, Mater. Horiz., 2021, 8, 3266–3280 RSC.
- Y. Xu, Q. Rong, T. Zhao and M. Liu, Giant, 2020, 2, 100014 CrossRef.
- D. Zhou, F. Chen, S. Handschuh-Wang, T. Gan, X. Zhou and X. Zhou, ChemPhysChem, 2019, 20, 2139–2154 CrossRef CAS.
- C. Polge, A. U. Smith and A. S. Parkes, Nature, 1949, 164, 666 CrossRef CAS PubMed.
- J. E. Lovelock and M. W. H. Bishop, Nature, 1959, 183, 1394–1395 CrossRef CAS PubMed.
- L. DeVries Arthur and E. Wohlschlag Donald, Science, 1969, 163, 1073–1075 CrossRef PubMed.
- C. I. Biggs, T. L. Bailey, G. Ben, C. Stubbs, A. Fayter and M. I. Gibson, Nat. Commun., 2017, 8, 1546 CrossRef.
- B. Graham, T. L. Bailey, J. R. J. Healey, M. Marcellini, S. Deville and M. I. Gibson, Angew. Chem., Int. Ed., 2017, 56, 15941–15944 CrossRef CAS PubMed.
- S. H. M. Söntjens, D. L. Nettles, M. A. Carnahan, L. A. Setton and M. W. Grinstaff, Biomacromolecules, 2005, 7, 310–316 CrossRef.
- B. Balakrishnan and R. Banerjee, Chem. Rev., 2011, 111, 4453–4474 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS.
- X. P. Morelle, W. R. Illeperuma, K. Tian, R. Bai, Z. Suo and J. J. Vlassak, Adv. Mater., 2018, 30, 1801541 CrossRef.
- Z. Liu, Y. Wang, Y. Ren, G. Jin, C. Zhang, W. Chen and F. Yan, Mater. Horiz., 2020, 7, 919–927 RSC.
- P. Rao, T. Li, Z. L. Wu, W. Hong, X. Yang, H. Yu, T.-W. Wong, S. Qu and W. Yang, Extreme Mech. Lett., 2019, 28, 43–49 CrossRef.
- X.-F. Zhang, X. Ma, T. Hou, K. Guo, J. Yin, Z. Wang, L. Shu, M. He and J. Yao, Angew. Chem., Int. Ed., 2019, 58, 7366–7370 CrossRef CAS.
- X. Zhao, F. Chen, Y. Li, H. Lu, N. Zhang and M. Ma, Nat. Commun., 2018, 9, 3579 CrossRef.
- K. Meister, S. Strazdaite, A. L. DeVries, S. Lotze, L. L. C. Olijve, I. K. Voets and H. J. Bakker, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17732 CrossRef CAS.
- L. Wu, L. Li, M. Qu, H. Wang and Y. Bin, ACS Appl. Polym. Mater., 2020, 2, 3094–3106 CrossRef CAS.
- W. Wang, Y. Liu, S. Wang, X. Fu, T. Zhao, X. Chen and Z. Shao, ACS Appl. Mater. Interfaces, 2020, 12, 25353–25362 CrossRef CAS.
- I. K. Voets, Soft Matter, 2017, 13, 4808–4823 RSC.
- Q. Rong, W. Lei, J. Huang and M. Liu, Adv. Energy Mater., 2018, 8, 1801967 CrossRef.
- H. Chen, X. Ren and G. Gao, ACS Appl. Mater. Interfaces, 2019, 11, 28336–28344 CrossRef CAS PubMed.
- J. Wu, Z. Wu, X. Lu, S. Han, B.-R. Yang, X. Gui, K. Tao, J. Miao and C. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 9405–9414 CrossRef CAS PubMed.
- Y. Wang, L. Zhang and A. Lu, ACS Appl. Mater. Interfaces, 2019, 11, 41710–41716 CrossRef CAS PubMed.
- X. Sui, H. Guo, P. Chen, Y. Zhu, C. Wen, Y. Gao, J. Yang, X. Zhang and L. Zhang, Adv. Funct. Mater., 2020, 30, 1907986 CrossRef CAS.
- X. Su, H. Wang, Z. Tian, X. Duan, Z. Chai, Y. Feng, Y. Wang, Y. Fan and J. Huang, ACS Appl. Mater. Interfaces, 2020, 12, 29757–29766 CAS.
- D. Zhang, Y. Liu, Y. Liu, Y. Peng, Y. Tang, L. Xiong, X. Gong and J. Zheng, Adv. Mater., 2021, 33, 2104006 CrossRef CAS.
- J. Zhang, L. Zeng, Z. Qiao, J. Wang, X. Jiang, Y. S. Zhang and H. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 30247–30258 CrossRef CAS PubMed.
- H. Yu, N. Rouelle, A. Qiu, J.-A. Oh, D. M. Kempaiah, J. D. Whittle, M. Aakyiir, W. Xing and J. Ma, ACS Appl. Mater. Interfaces, 2020, 12, 37977–37985 CrossRef CAS PubMed.
- C. Wang, C. G. Wiener, P. I. Sepulveda-Medina, C. Ye, D. S. Simmons, R. Li, M. Fukuto, R. A. Weiss and B. D. Vogt, Chem. Mater., 2019, 31, 135–145 CrossRef CAS.
- Y. Tu, Q. Chen, S. Liang, Q. Zhao, X. Zhou, W. Huang, X. Huang and L. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 18746–18754 CrossRef CAS PubMed.
- J. Xu, R. Jing, X. Ren and G. Gao, J. Mater. Chem. A, 2020, 8, 9373–9381 RSC.
- Y. Wang, Y. Xia, P. Xiang, Y. Dai, Y. Gao, H. Xu, J. Yu, G. Gao and K. Chen, Chem. Eng. J., 2022, 428, 131171 CrossRef CAS.
- Z. Fu, D. Li, H. Liu, R. Liu, Q. Lyu, Y. Han, Y. Wang, K. Zhang, G. Chen and Y. Tian, Chem. Eng. J., 2024, 488, 150775 CrossRef CAS.
- C. Lu, J. Qiu, W. Zhao, E. Sakai, G. Zhang, R. Nobe, M. Kudo and T. Komiyama, Int. J. Biol. Macromol., 2021, 188, 534–541 CrossRef CAS.
- X. Gao, J. Wu, Y. Wang, Y. Wang, Y. Zhang, T. T. Nguyen and M. Guo, Int. J. Biol. Macromol., 2024, 265, 131118 CrossRef CAS PubMed.
- Z. Wang, B. Lin, S. Sheng, S. Tan, P. Wang, Y. Tao, Z. Liu, Z. He and J. Wang, CCS Chem., 2022, 4, 104–111 CrossRef CAS.
- S. Gam-Derouich, J. Pinson, A. Lamouri, P. Decorse, S. Bellynck, R. Herbaut, L. Royon and C. Mangeney, J. Mater. Chem. A, 2018, 6, 19353–19357 RSC.
- C. G. Wiener, M. Tyagi, Y. Liu, R. A. Weiss and B. D. Vogt, J. Phys. Chem. B, 2016, 120, 5543–5552 CrossRef CAS.
- J. Hao and R. A. Weiss, Macromolecules, 2011, 44, 9390–9398 CrossRef CAS.
- L. X. Hou, H. Ju, X. P. Hao, H. Zhang, L. Zhang, Z. He, J. Wang, Q. Zheng and Z. L. Wu, Adv. Mater., 2023, 35, 2300244 CrossRef CAS.
- L. Cui, W. Wang, J. Zheng, C. Hu, Z. Zhu and B. Liu, Carbohydr. Polym., 2024, 342, 122406 CrossRef CAS PubMed.
- S. Wu, T.-W. Wang, Y. Du, B. Yao, S. Duan, Y. Yan, M. Hua, Y. Alsaid, X. Zhu and X. He, NPG Asia Mater., 2022, 14, 65 CrossRef CAS.
- X. Tao, K. Zhu, H. Chen, S. Ye, P. Cui, L. Dou, J. Ma, C. Zhao, J. He and P. Feng, Mater. Today Chem., 2023, 32, 101624 CrossRef CAS.
- N. Lu, R. Na, L. Li, C. Zhang, Z. Chen, S. Zhang, J. Luan and G. Wang, ACS Appl. Energy Mater., 2020, 3, 1944–1951 CrossRef CAS.
- S. Li, H. Pan, Y. Wang and J. Sun, J. Mater. Chem. A, 2020, 8, 3667–3675 RSC.
- C. Wang, B. Yang, R. Xiang, J. Ji, Y. Wu and S. Tan, ACS Nano, 2023, 17, 23194–23206 CrossRef CAS PubMed.
- S.-N. Li, X.-F. He, Z.-F. Zeng, B. Jiang, Q. Wu, L.-X. Gong, Y. Li, J. Bae, S. Wang and L.-C. Tang, Nano Energy, 2022, 103, 107789 CrossRef CAS.
- T. Li, P. F. Ibáñez-Ibáñez, V. Håkonsen, J. Wu, K. Xu, Y. Zhuo, S. Luo, J. He and Z. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 35572–35578 CrossRef CAS.
- S. Xiao, X. He, Z. Zhao, G. Huang, Z. Yan, Z. He, Z. Zhao, F. Chen and J. Yang, Eur. Polym. J., 2021, 148, 110350 CrossRef CAS.
- J. Wei, G. Wei, Z. Wang, W. Li, D. Wu and Q. Wang, Small, 2020, 16, 2004091 CrossRef CAS.
- S.-S. Li, S.-B. Wang, Y. Chen, Q.-S. Zhu, L.-M. Lan, H. Bu, T. Hu and G.-B. Jiang, Chem. Eng. J., 2023, 462, 142211 CrossRef CAS.
- Y. Yan, S. Duan, B. Liu, S. Wu, Y. Alsaid, B. Yao, S. Nandi, Y. Du, T. W. Wang and Y. Li, Adv. Mater., 2023, 35, 2211673 CrossRef CAS.
- C. Zhao, X. Gong, L. Shen, Y. Wang and C. Zhang, ACS Appl. Polym. Mater., 2022, 4, 4025–4034 CrossRef CAS.
- L. Zeng, J. He, Y. Cao, J. Wang, Z. Qiao, X. Jiang, L. Hou and J. Zhang, Smart Mater. Med., 2021, 2, 229–236 CrossRef.
- W. Zhang, G. Yang, A. Zhang, L. X. Xu and X. He, Biomed. Microdevices, 2010, 12, 89–96 CrossRef PubMed.
- N. T. C. Duong, A. Uthairatanakij, N. Laohakunjit, P. Jitareerat and N. Kaisangsri, Sci. Hortic., 2022, 292, 110648 CrossRef CAS.
- W. Xiao, F. Jing, S. Zhang, H. Chen, L. Bai, W. Wang, H. Yang, L. Yang and D. Wei, Cellulose, 2023, 30, 7667–7680 CrossRef CAS.
- F. Chen, Z. Xu, H. Wang, S. Handschuh-Wang, B. Wang and X. Zhou, ACS Appl. Mater. Interfaces, 2020, 12, 55501–55509 CrossRef CAS.
- L. Lu, Z. Huang, X. Li, X. Li, B. Cui, C. Yuan, L. Guo, P. Liu and Q. Dai, Int. J. Biol. Macromol., 2022, 213, 791–803 CrossRef CAS.
- Y. Wang, Y. Jia, H. Ren, C. Lao, W. Peng, B. Feng and J. Wang, Mater. Today Bio, 2021, 12, 100138 CrossRef CAS PubMed.
- Z. Zhang, A. Yao and P. Raffa, Adv. Funct. Mater., 2024, 34, 2407529 CrossRef CAS.
- D. Yue, Y. Chen, Y. Wu, H. Chen, L. Bai, W. Wang, H. Yang, L. Yang and D. Wei, Sustainable Mater. Technol., 2023, 37, e00653 CrossRef CAS.
- Z. Liu, J. Zhang, J. Liu, Y. Long, L. Fang, Q. Wang and T. Liu, J. Mater. Chem. A, 2020, 8, 6219–6228 RSC.
- S. Han, C. Liu, X. Lin, J. Zheng, J. Wu and C. Liu, ACS Appl. Polym. Mater., 2020, 2, 996–1005 CrossRef CAS.
- J. Zhang, Y. Wang, Q. Wei, M. Li and X. Chen, J. Colloid Interface Sci., 2024, 653, 1514–1525 CrossRef CAS PubMed.
- Y. Liu, J. Yin, Y. Fu, P. Zhao, Y. Zhang, B. He and P. He, Chem. Eng. J., 2020, 382, 122925 CrossRef CAS.
- X. Huang, Z. Zheng, H. Wang, W. Xu, M. Wu, M. Wang, C. Chen, L. Wan, R. Du, T. Zhu, Z. Huang, X. Wang, X. Wang, Q. Zhang and X. Jia, Adv. Funct. Mater., 2024, 34, 2312149 CrossRef CAS.
- K. Wang, L. Zhang and X. Jiang, J. Colloid Interface Sci., 2023, 652, 663–672 CrossRef CAS PubMed.
- X. Gong, C. Zhao, Y. Wang, Y. Luo and C. Zhang, ACS Biomater. Sci. Eng., 2022, 8, 3633–3643 CrossRef CAS.
- X. Zhang, J. Wang, M. Wang, D. Liu and Z. Wang, Nano Res., 2024, 17, 4016–4022 CrossRef CAS.
- A. Abodurexiti and X. Maimaitiyiming, IEEE Sens. J., 2022, 22, 12588–12594 CAS.
- R. Si, Y. Wang, Y. Yang, Y. Wu, M. Wang and B. Han, Chem. Eng. J., 2024, 489, 151419 CrossRef CAS.
- F. Chen, D. Zhou, J. Wang, T. Li, X. Zhou, T. Gan, S. Handschuh-Wang and X. Zhou, Angew. Chem., Int. Ed., 2018, 57, 6568–6571 CrossRef CAS.
- D. Lou, C. Wang, Z. He, X. Sun, J. Luo and J. Li, Chem. Commun., 2019, 55, 8422–8425 RSC.
- P. He, J. Wu, X. Pan, L. Chen, K. Liu, H. Gao, H. Wu, S. Cao, L. Huang and Y. Ni, J. Mater. Chem. A, 2020, 8, 3109–3118 RSC.
- L. Han, K. Liu, M. Wang, K. Wang, L. Fang, H. Chen, J. Zhou and X. Lu, Adv. Funct. Mater., 2018, 28, 1704195 CrossRef.
- X. Pan, Q. Wang, D. Ning, L. Dai, K. Liu, Y. Ni, L. Chen and L. Huang, ACS Biomater. Sci. Eng., 2018, 4, 3397–3404 CrossRef CAS PubMed.
- S. Yin, G. Su, J. Chen, X. Peng and T. Zhou, Materials, 2021, 14, 6165 CrossRef CAS PubMed.
- L. Wu, M. Fan, M. Qu, S. Yang, J. Nie, P. Tang, L. Pan, H. Wang and Y. Bin, J. Mater. Chem. B, 2021, 9, 3088–3096 RSC.
- D. Zhou, F. Chen, J. Wang, T. Li, B. Li, J. Zhang, X. Zhou, T. Gan, S. Handschuh-Wang and X. Zhou, J. Mater. Chem. B, 2018, 6, 7366–7372 RSC.
- S. Shi, X. Peng, T. Liu, Y.-N. Chen, C. He and H. Wang, Polymer, 2017, 111, 168–176 CrossRef CAS.
- P. C. Koh, M. A. Noranizan, R. Karim, Z. A. Nur Hanani and N. L. Yusof, J. Food Sci. Technol., 2020, 57, 2206–2221 CrossRef CAS PubMed.
- H. Yang, L. Tan, J. Yan, J. Chen, X. Wu and S. Yang, Adv. Funct. Mater., 2024, 2411384 CrossRef CAS.
- W. Yu, J. Liu, Y. Yang, J. Hao and L. Xu, Tribol. Int., 2023, 180, 108302 CrossRef CAS.
- H. Chen, J. Yang, Z. Liu, Y. Li, Z. Tang, X. Shi and Q. Chen, Adv. Funct. Mater., 2024, 34, 2413171 Search PubMed.
- D. Lei, Y. Xiao, L. Shao, M. Xi, Y. Jiang and Y. Li, ACS Appl. Mater. Interfaces, 2023, 15, 24175–24185 CrossRef CAS.
- X. Cui, J. Lee, K. R. Ng and W. N. Chen, ACS Sustainable Chem. Eng., 2021, 9, 1304–1312 CrossRef CAS.
- L. Zhang, M. He, L. Chen, P. Xu, J. Bao and X. Hu, J. Mater. Sci., 2021, 56, 18697–18709 CrossRef CAS.
- L. Zhu, F. Ouyang, X. Fu, Y. Wang, T. Li, M. Wen, G. Zha and X. Yang, Sci. Rep., 2024, 14, 12864 CrossRef CAS.
- J. Liu, J. Deng, F. Zhao, T. Wu and J. Xing, Colloids Surf., A, 2024, 683, 133087 CrossRef CAS.
- Z. Qin, D. Dong, M. Yao, Q. Yu, X. Sun, Q. Guo, H. Zhang, F. Yao and J. Li, ACS Appl. Mater. Interfaces, 2019, 11, 21184–21193 CrossRef CAS.
- L. Yang, G. Zhou, Y. Jin, Y. Sun, Q. Liu and C. Chen, J. Power Sources, 2022, 548, 232015 CrossRef CAS.
- J. Tan, J. Li and X. Zhou, J. Biomed. Mater. Res., Part B, 2024, 112, e35408 CrossRef CAS PubMed.
- X. Li, Y. Cao, C. Liu, J. Tan and X. Zhou, Cryobiology, 2024, 116, 104942 CrossRef CAS.
- B. Huang, S. Jiang, Y. Diao, X. Liu, W. Liu, J. Chen and H. Yang, Molecules, 2020, 25, 3378 CrossRef CAS PubMed.
- J. Choi, S. Kim, J. Yoo, S.-H. Choi and K. Char, Macromolecules, 2021, 54, 6389–6399 CrossRef CAS.
- G. Zhao, X. Liu, K. Zhu and X. He, Adv. Healthcare Mater., 2017, 6, 1700988 CrossRef PubMed.
- J. Choi, S. Kim, H. J. Mun, J. Yoo, S. H. Choi and K. Char, Macromol. Rapid Commun., 2021, 43, 2100618 CrossRef PubMed.
- F. Mo, G. Liang, Q. Meng, Z. Liu, H. Li, J. Fan and C. Zhi, Energy Environ. Sci., 2019, 12, 706–715 RSC.
- P. A. Mercadal, M. d M. Montesinos, M. A. Macchione, S. D. Dalosto, K. L. Bierbrauer, M. Calderón, A. González and M. L. Picchio, ACS Mater. Lett., 2024, 6, 3726–3735 CrossRef CAS PubMed.
- X. He, T. Zhuang, S. Ruan, X. Xia, Y. Xia, J. Zhang, H. Huang, Y. Gan and W. Zhang, Chem. Eng. J., 2023, 466, 143209 CrossRef CAS.
- K. Wang, J. Wang, L. Li, L. Xu, N. Feng, Y. Wang, X. Fei, J. Tian and Y. Li, Chem. Eng. J., 2019, 372, 216–225 CrossRef CAS.
- Y. Zong, B. Zong, R. Zha, Y. Zhang, X. Li, Y. Wang, H. Fang, W.-L. Wong and C. Li, Adv. Healthcare Mater., 2023, 12, 2300431 CrossRef CAS.
- T. Lei, Y. Wang, Q. Zhang, H. Wang, X. Duan, J. Yan, Z. Xia, R. Wang, W. Shou, X. Li and J. Fan, Nano Energy, 2024, 126, 109633 CrossRef CAS.
- S. Wang, D. Zhang, X. He, J. Yuan, W. Que, Y. Yang, I. Protsak, X. Huang, C. Zhang and T. Lu, Chem. Eng. J., 2022, 438, 135607 CrossRef CAS.
- K. B. Storey and J. M. Storey, Physiol. Rev., 2017, 97, 623–665 CrossRef CAS PubMed.
- K. Liu, C. Wang, J. Ma, G. Shi, X. Yao, H. Fang, Y. Song and J. Wang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14739–14744 CrossRef CAS PubMed.
- J. A. Raymond and A. L. DeVries, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 2589–2593 CrossRef CAS PubMed.
- G. Bai, D. Gao, Z. Liu, X. Zhou and J. Wang, Nature, 2019, 576, 437–441 CrossRef CAS.
- X. Li, H. Charaya, G. M. Bernard, J. A. W. Elliott, V. K. Michaelis, B. Lee and H.-J. Chung, Macromolecules, 2018, 51, 2723–2731 CrossRef CAS.
- J. C. Rasaiah, S. Garde and G. Hummer, Annu. Rev. Phys. Chem., 2008, 59, 713–740 CrossRef CAS.
- K. Mochizuki and K. Koga, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8221–8226 CrossRef CAS PubMed.
- D. Zhang, Y. Liu, W. Gross, Y. Tang and J. Zheng, Giant, 2023, 16, 100203 CrossRef CAS.
- H. Oldenhof, M. Gojowsky, S. Wang, S. Henke, C. Yu, K. Rohn, W. F. Wolkers and H. Sieme, Biol. Reprod., 2013, 88, 68 Search PubMed.
- Y. Qiu, A. Hudait and V. Molinero, J. Am. Chem. Soc., 2019, 141, 7439–7452 CrossRef CAS PubMed.
- M. R. Michaud and D. L. Denlinger, J. Comp. Physiol., B, 2007, 177, 753–763 CrossRef CAS PubMed.
- P. L. Davies, Trends Biochem. Sci., 2014, 39, 548–555 CrossRef CAS.
- Y. Yeh and R. E. Feeney, Chem. Rev., 1996, 96, 601–618 CrossRef CAS PubMed.
- M. M. Harding, L. G. Ward and A. D. J. Haymet, Eur. J. Biochem., 1999, 264, 653–665 CrossRef CAS.
- F. Sicheri and D. S. C. Yang, Nature, 1995, 375, 427–431 CrossRef CAS.
- L. L. C. Olijve, K. Meister, A. L. DeVries, J. G. Duman, S. Guo, H. J. Bakker and I. K. Voets, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3740–3745 CrossRef CAS.
- R. Peltier, M. A. Brimble, J. M. Wojnar, D. E. Williams, C. W. Evans and A. L. DeVries, Chem. Sci., 2010, 1, 538–551 RSC.
- B. L. Wilkinson, R. S. Stone, C. J. Capicciotti, M. Thaysen-Andersen, J. M. Matthews, N. H. Packer, R. N. Ben and R. J. Payne, Angew. Chem., Int. Ed., 2012, 124, 3666–3670 CrossRef.
- R. C. Deller, M. Vatish, D. A. Mitchell and M. I. Gibson, Nat. Commun., 2014, 5, 3244 CrossRef.
- B. Graham, A. E. R. Fayter, J. E. Houston, R. C. Evans and M. I. Gibson, J. Am. Chem. Soc., 2018, 140, 5682–5685 CrossRef CAS.
- N. Du, X. Y. Liu and C. L. Hew, J. Biol. Chem., 2003, 278, 36000–36004 CrossRef CAS.
- O. García-Arribas, R. Mateo, M. M. Tomczak, P. L. Davies and M. G. Mateu, Protein Sci., 2007, 16, 227–238 CrossRef PubMed.
- E. Y. Chi, S. Krishnan, T. W. Randolph and J. F. Carpenter, Pharm. Res., 2003, 20, 1325–1336 CrossRef CAS PubMed.
- R. Y. Tam, C. N. Rowley, I. Petrov, T. Zhang, N. A. Afagh, T. K. Woo and R. N. Ben, J. Am. Chem. Soc., 2009, 131, 15745–15753 CrossRef CAS.
- S. Abraham, K. Keillor, C. J. Capicciotti, G. E. Perley-Robertson, J. W. Keillor and R. N. Ben, Cryst. Growth Des., 2015, 15, 5034–5039 CrossRef CAS.
- C. J. Capicciotti, M. Leclere, F. A. Perras, D. L. Bryce, H. Paulin, J. Harden, Y. Liu and R. N. Ben, Chem. Sci., 2012, 3, 1408–1416 RSC.
- W. Zhang and R. A. Laursen, FEBS Lett., 1999, 455, 372–376 CrossRef CAS.
- A. Wierzbicki, C. A. Knight, T. J. Rutland, D. D. Muccio, B. S. Pybus and C. S. Sikes, Biomacromolecules, 2000, 1, 268–274 CrossRef CAS.
- M. L. Huang, D. Ehre, Q. Jiang, C. Hu, K. Kirshenbaum and M. D. Ward, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19922–19927 CrossRef CAS.
- Y. Bai, B. Chen, F. Xiang, J. Zhou, H. Wang and Z. Suo, Appl. Phys. Lett., 2014, 105, 151903 CrossRef.
- L. Liu, Z. Liu, Y. Ren, X. Zou, W. Peng, W. Li, Y. Wu, S. Zheng, X. Wang and F. Yan, Angew. Chem., Int. Ed., 2021, 60, 8948–8959 CrossRef CAS PubMed.
- M. I. Barba, M. S. Larrechi and A. Coronas, Anal. Chim. Acta, 2016, 919, 20–27 CrossRef CAS PubMed.
- M. R. Conde, Int. J. Therm. Sci., 2004, 43, 367–382 CrossRef CAS.
- J. Lorimer and P. Gujral, in Alkaline Earth Hydroxides in Water and Aqueous Solutions, Pergamon Press, 1979 Search PubMed.
- H. Liu, X. Wang, Y. Cao, Y. Yang, Y. Yang, Y. Gao, Z. Ma, J. Wang, W. Wang and D. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 25334–25344 CrossRef CAS PubMed.
- L. Bai, Y. Jin, X. Shang, H. Jin, Y. Zhou and L. Shi, J. Mater. Chem. A, 2021, 9, 23916–23928 RSC.
- D. Chen, X. Zhao, X. Wei, J. Zhang, D. Wang, H. Lu and P. Jia, ACS Appl. Mater. Interfaces, 2020, 12, 53247–53256 CrossRef CAS.
- M. Zhu, X. Wang, H. Tang, J. Wang, Q. Hao, L. Liu, Y. Li, K. Zhang and O. G. Schmidt, Adv. Funct. Mater., 2020, 30, 1907218 CrossRef CAS.
- Q. Quan, T. Zhao, Z. Luo, B.-X. Li, H. Sun, H.-Y. Zhao, Z.-Z. Yu and D. Yang, ACS Appl. Mater. Interfaces, 2024, 16, 21133–21145 CrossRef CAS.
- D. Bao, Z. Wen, J. Shi, L. Xie, H. Jiang, J. Jiang, Y. Yang, W. Liao and X. Sun, J. Mater. Chem. A, 2020, 8, 13787–13794 RSC.
- X. Sui, H. Guo, C. Cai, Q. Li, C. Wen, X. Zhang, X. Wang, J. Yang and L. Zhang, Chem. Eng. J., 2021, 419, 129478 CrossRef CAS.
- S. Wu, M. Hua, Y. Alsaid, Y. Du, Y. Ma, Y. Zhao, C. Y. Lo, C. Wang, D. Wu and B. Yao, Adv. Mater., 2021, 33, 2007829 CrossRef CAS.
- K.-T. Huang, K. Ishihara and C.-J. Huang, Biomacromolecules, 2019, 20, 3524–3534 CrossRef CAS.
- S. Huang, L. Hou, T. Li, Y. Jiao and P. Wu, Adv. Mater., 2022, 34, 2110140 CrossRef CAS PubMed.
- M. Hua, S. Wu, Y. Ma, Y. Zhao, Z. Chen, I. Frenkel, J. Strzalka, H. Zhou, X. Zhu and X. He, Nature, 2021, 590, 594–599 CrossRef CAS PubMed.
- G. Cao, L. Zhao, X. Ji, Y. Peng, M. Yu, X. Wang, X. Li and F. Ran, Small, 2023, 19, 2207610 CrossRef CAS.
- S. Wu, Z. Liu, C. Gong, W. Li, S. Xu, R. Wen, W. Feng, Z. Qiu and Y. Yan, Nat. Commun., 2024, 15, 4441 CrossRef CAS.
- H. Fu, B. Wang, J. Li, D. Cao, W. Zhang, J. Xu, J. Li, J. Zeng, W. Gao and K. Chen, Mater. Horiz., 2024, 11, 1588–1596 RSC.
- W. Sun, J. Yang, X. Ji, H. Jiang, L. Gai, X. Li and L. Liu, Sustainable Mater. Technol., 2022, 32, e00437 CrossRef CAS.
- W. Xie, J. Duan, H. Wang, J. Li, R. Liu, B. Yu, S. Liu and J. Zhou, J. Mater. Chem. A, 2018, 6, 24114–24119 RSC.
- G. Ge, W. Yuan, W. Zhao, Y. Lu, Y. Zhang, W. Wang, P. Chen, W. Huang, W. Si and X. Dong, J. Mater. Chem. A, 2019, 7, 5949–5956 RSC.
- Q. Wang, X. Pan, C. Lin, D. Lin, Y. Ni, L. Chen, L. Huang, S. Cao and X. Ma, Chem. Eng. J., 2019, 370, 1039–1047 CrossRef CAS.
- Y. Yang, L. Guan, X. Li, Z. Gao, X. Ren and G. Gao, ACS Appl. Mater. Interfaces, 2019, 11, 3428–3437 CrossRef CAS.
- R. M. Kumar, P. Baskar, K. Balamurugan, S. Das and V. Subramanian, J. Phys. Chem. A, 2012, 116, 4239–4247 CrossRef CAS PubMed.
- T. Takamuku, M. Yamagami, H. Wakita, Y. Masuda and T. Yamaguchi, J. Phys. Chem. B, 1997, 101, 5730–5739 CrossRef CAS.
- J. M. Harris, Poly(ethylene glycol) chemistry: biotechnical and biomedical applications, Springer Science & Business Media, 2013 Search PubMed.
- H. Chen, C. Zhao, M. Zhang, Q. Chen, J. Ma and J. Zheng, Langmuir, 2016, 32, 3315–3330 CrossRef CAS PubMed.
- C. Zhao, J. Zhao, X. Li, J. Wu, S. Chen, Q. Chen, Q. Wang, X. Gong, L. Li and J. Zheng, Biomaterials, 2013, 34, 4714–4724 CrossRef CAS PubMed.
- S. Chen, L. Li, C. Zhao and J. Zheng, Polymer, 2010, 51, 5283–5293 CrossRef CAS.
- V. Gaberc-Porekar, I. Zore, B. Podobnik and V. Menart, Curr. Opin. Drug Discovery Dev., 2008, 11, 242 CAS.
- S. Chen, J. Zheng, L. Li and S. Jiang, J. Am. Chem. Soc., 2005, 127, 14473–14478 CrossRef CAS.
- Y. Liu, D. Zhang, Y. Tang, X. Gong and J. Zheng, npj Comput. Mater., 2023, 9, 209 CrossRef CAS.
- Z. Pei, Z. Yuan, C. Wang, S. Zhao, J. Fei, L. Wei, J. Chen, C. Wang, R. Qi, Z. Liu and Y. Chen, Angew. Chem., Int. Ed., 2020, 59, 4793–4799 CrossRef CAS PubMed.
- A. Tron, S. Jeong, Y. D. Park and J. Mun, ACS Sustainable Chem. Eng., 2019, 7, 14531–14538 CrossRef CAS.
- T. Chang and G. Zhao, Adv. Sci., 2021, 2002425 CrossRef CAS PubMed.
- Y. Jin, C. Wu, Y. Yang, J. Wu, Z. He and J. Wang, ACS Nano, 2020, 14, 5000–5007 CrossRef CAS PubMed.
- W. Ge, S. Cao, Y. Yang, O. J. Rojas and X. Wang, Chem. Eng. J., 2021, 408, 127306 CrossRef CAS.
- A. Bin Imran, K. Esaki, H. Gotoh, T. Seki, K. Ito, Y. Sakai and Y. Takeoka, Nat. Commun., 2014, 5, 5124 CrossRef CAS PubMed.
- J. A. Johnson, N. J. Turro, J. T. Koberstein and J. E. Mark, Prog. Polym. Sci., 2010, 35, 332–337 CrossRef CAS.
- M. Deserno and W. M. Gelbart, J. Phys. Chem. B, 2002, 106, 5543–5552 CrossRef CAS.
- P. I. Sepulveda-Medina, C. Wang, R. Li, M. Fukuto and B. D. Vogt, J. Phys. Chem. B, 2022, 126, 5544–5554 CrossRef CAS PubMed.
- Y. Zheng, T. Matsuda, T. Nakajima, W. Cui, Y. Zhang, C. Y. Hui, T. Kurokawa and J. P. Gong, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2111880118 CrossRef CAS.
- Y. Tanaka, R. Shimazaki, S. Yano, G. Yoshida and T. Yamaguchi, Soft Matter, 2016, 12, 8135–8142 RSC.
- J. Yang, Z. Liu, K. Li, J. Hao, Y. Guo, M. Guo, Z. Li, S. Liu, H. Yin, X. Shi, G. Qin, G. Sun, L. Zhu and Q. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 39299–39310 CrossRef CAS.
- B. A. Krajina, C. Tropini, A. Zhu, P. DiGiacomo, J. L. Sonnenburg, S. C. Heilshorn and A. J. Spakowitz, ACS Cent. Sci., 2017, 3, 1294–1303 CrossRef CAS.
- Q. He, Y. Huang and S. Wang, Adv. Funct. Mater., 2018, 28, 1705069 CrossRef.
- Z. Qin, X. Sun, H. Zhang, Q. Yu, X. Wang, S. He, F. Yao and J. Li, J. Mater. Chem. A, 2020, 8, 4447–4456 RSC.
- J. Yang, Q. Kang, B. Zhang, X. Fang, S. Liu, G. Qin and Q. Chen, Mater. Sci. Eng., C, 2021, 130, 112452 CrossRef CAS PubMed.
- W. Zhang, B. Wu, S. Sun and P. Wu, Nat. Commun., 2021, 12, 4082 CrossRef CAS.
- H. Yin, M. You, X. Shi, H. Yu and Q. Chen, Mater. Horiz., 2024, 11, 3946–3960 RSC.
- C. Liu, L. Jiang, O. Yue, Y. Feng, B. Zeng, Y. Wu, Y. Wang, J. Wang, L. Zhao, X. Wang, C. Shao, Q. Wu and X. Sun, Adv. Compos. Hybrid Mater., 2023, 6, 112 CrossRef CAS.
- Y. Zhou, S. Guo, Y. Zhou, L. Zhao, T. Wang, X. Yan, F. Liu, S. K. Ravi, P. Sun and S. C. Tan, Adv. Funct. Mater., 2025, 2425586 CrossRef.
- K. Liu, Z. Zhao, S. Zheng, A. Liu, Y. Wang, L. Chen and Q. Miao, Biomacromolecules, 2024, 25, 4384–4393 CrossRef CAS PubMed.
- S. Guo, S. De Wolf, M. Sitti, C. Serre and S. C. Tan, Adv. Mater., 2024, 36, 2311445 CrossRef CAS.
- Q. Yang, W. Yang, Z. Wang, R. Chen, M. Li, C. Qin, D. Gao and W. Chen, ACS Appl. Mater. Interfaces, 2023, 15, 51684–51693 CrossRef CAS.
- Z. Xu, F. Zhou, H. Yan, G. Gao, H. Li, R. Li and T. Chen, Nano Energy, 2021, 90, 106614 CrossRef CAS.
- Y. Feng, J. Yu, D. Sun, W. Ren, C. Shao and R. Sun, Chem. Eng. J., 2022, 433, 133202 CrossRef CAS.
- J. Yang, Z. Xu, J. Wang, L. Gai, X. Ji, H. Jiang and L. Liu, Adv. Funct. Mater., 2021, 31, 2009438 CrossRef CAS.
- Y. Liu, Q. Liu, L. Zhong, C. Chen and Z. Xu, Chem. Eng. J., 2023, 452, 139314 CrossRef CAS.
- W. Sun, Z. Xu, C. Qiao, B. Lv, L. Gai, X. Ji, H. Jiang and L. Liu, Adv. Sci., 2022, 9, 2201679 CrossRef CAS.
- Y. Yang, M. Zhou, J. Peng, X. Wang, Y. Liu, W. Wang and D. Wu, Carbohydr. Polym., 2022, 276, 118753 CrossRef CAS PubMed.
- M. Chen, W. Zhou, A. Wang, A. Huang, J. Chen, J. Xu and C.-P. Wong, J. Mater. Chem. A, 2020, 8, 6828–6841 RSC.
- J. Huang, X. Xu, F. Xu, J. Yang, M. Kharaziha, F. Sun and X. Zhang, Int. J. Biol. Macromol., 2023, 239, 124260 CrossRef CAS.
- Y. Zhou, R. a Li, L. Wan, F. Zhang, Z. Liu and Y. Cao, Int. J. Biol. Macromol., 2023, 240, 124171 CrossRef CAS PubMed.
- Y. Lu and W. Zhong, Chem. Eng. J., 2024, 490, 151691 CrossRef CAS.
- K. Xie, K. Ren, Q. Wang, Y. Lin, F. Ma, C. Sun, Y. Li, X. Zhao and C. Lai, eScience, 2023, 3, 100153 CrossRef.
- W. Zhao, J. Jiang, W. Chen, Y. He, T. Lin and L. Zhao, Chem. Eng. J., 2023, 468, 143660 CrossRef CAS.
- Y. Yang, Y. Ni, H. Wang, L. Chen, T. Zhu, Y. Zheng, Y. Cheng, Y. Lai, Y. Tang, W. Cai and J. Huang, Chem. Eng. J., 2024, 482, 148847 CrossRef.
- X. Li, L. Jin, A. Ni, L. Zhang, L. He, H. Gao, P. Lin, K. Zhang, X. Chu and S. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 30182–30191 CrossRef CAS.
- L. Wu, L. Li, M. Fan, P. Tang, S. Yang, L. Pan, H. Wang and Y. Bin, Composites, Part A, 2020, 138, 106050 CrossRef CAS.
- W. Zhu, Y. Zhang, S. Huang, L. Geng, J. Wu, G. Mao, X. Peng and Y. Cheng, Chem. Eng. J., 2024, 497, 154409 CrossRef CAS.
- Y. Wang, Y. Zhang, P. Ren, S. Yu, P. Cui, C. B. Nielsen, I. Abrahams, J. Briscoe and Y. Lu, Nano Energy, 2024, 125, 109599 CrossRef CAS.
- L. Zhou, B. Zhao, J. Liang, F. Lu, W. Yang, J. Xu, J. Zheng, Y. Liu, R. Wang and Z. Liu, Mater. Horiz., 2024, 11, 3856–3866 RSC.
- T. Yang, C. Xu, C. Liu, Y. Ye, Z. Sun, B. Wang and Z. Luo, Chem. Eng. J., 2022, 429, 132430 CrossRef CAS.
- H. Zheng, M. Chen, Y. Sun and B. Zuo, Chem. Eng. J., 2022, 446, 136931 CrossRef CAS.
- X. Dai, Y. Long, B. Jiang, W. Guo, W. Sha, J. Wang, Z. Cong, J. Chen, B. Wang and W. Hu, Nano Res., 2022, 15, 5461–5468 CrossRef CAS.
- Z.-F. Zeng, Y.-Q. Yang, X.-W. Pang, B. Jiang, L.-X. Gong, Z. Liu, L. Peng and S.-N. Li, Adv. Funct. Mater., 2024, 34, 2409855 CrossRef CAS.
- M. Tian, Y. Wang, L. Qu, S. Zhu, G. Han, X. Zhang, Q. Zhou, M. Du and S. Chi, Synth. Met., 2016, 219, 11–19 CrossRef CAS.
- C. Zhu, G. Chen, S. Li, H. Yang, J. Zheng, D. Wang, H. Yang, L. W. Y. Wong and J. Fu, Adv. Funct. Mater., 2024, 34, 2411725 CrossRef CAS.
- S. Chen, B. Guo, J. Yu, Z. Yan, R. Liu, C. Yu, Z. Zhao, H. Zhang, F. Yao and J. Li, Chem. Eng. J., 2024, 486, 150182 CrossRef CAS.
- Y. Huang, M. Zhong, Y. Huang, M. Zhu, Z. Pei, Z. Wang, Q. Xue, X. Xie and C. Zhi, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- P. Li, Z. Jin, L. Peng, F. Zhao, D. Xiao, Y. Jin and G. Yu, Adv. Mater., 2018, 30, 1800124 CrossRef.
- G. Zhang, X. Yang, H. Shu and W. Zhong, J. Mater. Chem. A, 2023, 11, 9097–9111 RSC.
- Q. Fu, S. Hao, L. Meng, F. Xu and J. Yang, ACS Nano, 2021, 15, 18469–18482 CrossRef CAS PubMed.
- X. Li, D. Lou, H. Wang, X. Sun, J. Li and Y. N. Liu, Adv. Funct. Mater., 2020, 30, 2007291 CrossRef CAS.
- Y. Huang, M. Zhong, F. Shi, X. Liu, Z. Tang, Y. Wang, Y. Huang, H. Hou, X. Xie and C. Zhi, Angew. Chem., Int. Ed., 2017, 56, 9141–9145 CrossRef CAS.
- H. Cai, D. Zhang, H. Zhang, M. Tang, Z. Xu, H. Xia, K. Li and J. Wang, Chem. Eng. J., 2023, 472, 144849 CrossRef CAS.
- A. K. Mondal, D. Xu, S. Wu, Q. Zou, W. Lin, F. Huang and Y. Ni, Int. J. Biol. Macromol., 2022, 214, 77–90 CrossRef CAS PubMed.
- S. Wu, D. Lou, H. Wang, D. Jiang, X. Fang, J. Meng, X. Sun and J. Li, Chem. Eng. J., 2022, 435, 135057 CrossRef CAS.
- D. Wang, F. Yang, C. Wang, F. Chu, J. Nan and R. Chen, Chem. Eng. J., 2023, 455, 140949 CrossRef CAS.
- J. Chen, Q. Yu, D. Shi, Z. Yang, K. Dong, D. Kaneko, W. Dong and M. Chen, ACS Appl. Energy Mater., 2021, 4, 9353–9361 CrossRef CAS.
- M. D. Hager, P. Greil, C. Leyens, S. van der Zwaag and U. S. Schubert, Adv. Mater., 2010, 22, 5424–5430 CrossRef CAS.
- P. Li, T. Zhou, L. Bai, H. Chen, W. Wang, H. Yang, L. Yang and D. Wei, ACS Appl. Polym. Mater., 2023, 5, 7621–7630 CrossRef CAS.
- A. Abdollahi, H. Roghani-Mamaqani, B. Razavi and M. Salami-Kalajahi, Polym. Chem., 2019, 10, 5686–5720 RSC.
- X. Li, J. Lai, Y. Deng, J. Song, G. Zhao and S. Dong, J. Am. Chem. Soc., 2020, 142, 21522–21529 CrossRef CAS.
- D. Zhao, Y. Zhu, W. Cheng, G. Xu, Q. Wang, S. Liu, J. Li, C. Chen, H. Yu and L. Hu, Matter, 2020, 2, 390–403 CrossRef CAS.
- G. Sinawang, M. Osaki, Y. Takashima, H. Yamaguchi and A. Harada, Chem. Commun., 2020, 56, 4381–4395 RSC.
- S. M. Mantooth, B. G. Munoz-Robles and M. J. Webber, Macromol. Biosci., 2019, 19, 1800281 CrossRef.
- C. Zhang, Y. Zhou, L. Zhang, L. Wu, Y. Chen, D. Xie and W. Chen, Int. J. Mol. Sci., 2018, 19, 3330 CrossRef.
- T. H. Jang, S. C. Park, J. H. Yang, J. Y. Kim, J. H. Seok, U. S. Park, C. W. Choi, S. R. Lee and J. Han, Integr. Med. Res., 2017, 6, 12–18 CrossRef PubMed.
- S. Bupphathong, C. Quiroz, W. Huang, P.-F. Chung, H.-Y. Tao and C.-H. Lin, Pharmaceuticals, 2022, 15, 171 CrossRef CAS.
- G. M. Fahy and B. Wowk, Cryopreservation and freeze-drying protocols, 2015, pp. 21–82 Search PubMed.
- U. Demirci and G. Montesano, Lab Chip, 2007, 7, 1428–1433 RSC.
- W. Lu, C. Ma, D. Zhang, X. Le, J. Zhang, Y. Huang, C.-F. Huang and T. Chen, J. Phys. Chem. C, 2018, 122, 9499–9506 CAS.
- G. Olivas, D. Mattinson and G. Barbosa-Cánovas, Postharvest Biol. Technol., 2007, 45, 89–96 CrossRef CAS.
- N. Maftoonazad, H. S. Ramaswamy and M. Marcotte, Int. J. Food Sci. Technol., 2008, 43, 951–957 CrossRef CAS.
- H. M. Díaz-Mula, M. Serrano and D. Valero, Food Bioprocess Technol., 2012, 5, 2990–2997 CrossRef.
- A. Friend, M. C. Trought, C. Stushnoff and G. Wells, Aust. J. Grape Wine Res., 2011, 17, 378–382 CrossRef.
- Y. Park and H. Shin, Hortic., Environ. Biotechnol., 2022, 63, 643–650 CrossRef CAS.
- B. Zhang, H.-X. Wu, H.-C. Yang, X.-W. Xiang, H.-B. Li and S.-G. Deng, Food Chem., 2017, 228, 257–264 CrossRef CAS PubMed.
- X.-H. Li, C. Liu, S.-P. Feng and N. X. Fang, Joule, 2019, 3, 290–302 CrossRef CAS.
- Y. Zhou, S. Wang, J. Peng, Y. Tan, C. Li, F. Y. C. Boey and Y. Long, Joule, 2020, 4, 2458–2474 CrossRef CAS.
- M. I. Gibson, Polym. Chem., 2010, 1, 1141–1152 RSC.
- J. Zhao, M. Johnson, R. Fisher, N. Burke and H. Stöver, Langmuir, 2018, 35, 1807–1817 CrossRef PubMed.
- Z. Wang, C. Valenzuela, J. Wu, Y. Chen, L. Wang and W. Feng, Small, 2022, 18, 2201597 CrossRef CAS.
Footnote |
| † These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2025 |