
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetrically tailored catalysts towards electrochemical energy conversion with non-precious materials
Lei
Zhang
 a,
Qiaoling
Xu
a,
Lu
Xia
c,
Wulyu
Jiang
c,
Kaiwen
Wang
c,
Pengfei
Cao
d,
Qiang
Chen
e,
Ming
Huang
f,
F. Pelayo
García de Arquer
*c and
Yingtang
Zhou
*b
a,
Qiaoling
Xu
a,
Lu
Xia
c,
Wulyu
Jiang
c,
Kaiwen
Wang
c,
Pengfei
Cao
d,
Qiang
Chen
e,
Ming
Huang
f,
F. Pelayo
García de Arquer
*c and
Yingtang
Zhou
*b
aSchool of Materials Science and Engineering, Anhui Province Key Laboratory of Specialty Polymers, Anhui University of Science and Technology, Huainan, Anhui 232001, P. R. China
bZhejiang Key Laboratory of Petrochemical Environmental Pollution Control, Marine Science and Technology College, Zhejiang Ocean University, Zhoushan, Zhejiang Province 316004, P. R. China. E-mail: zhouyingtang@zjou.edu.cn
cICFO-Institut de Ciències Fotòniques, The Barcelona Institute of Science and Technology, Castelldefels (Barcelona), 08860, Spain. E-mail: pelayo.garciadearquer@icfo.eu
dForschungszentrum Jülich GmbH, ER-C, 52425 Jülich, Germany
eInstitute of Functional Nano & Soft Materials (FUNSOM), Soochow University, Suzhou 215123, P. R. China
fInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 611731, P. R. China
First published on 25th April 2025
Abstract
Electrocatalytic technologies, such as water electrolysis and metal–air batteries, enable a path to sustainable energy storage and conversion into high-value chemicals. These systems rely on electrocatalysts to drive redox reactions that define key performance metrics such as activity and selectivity. However, conventional electrocatalysts face inherent trade-offs between activity, stability, and scalability particularly due to the reliance on noble metals. Asymmetrically tailored electrocatalysts (ATEs) – systems that are being exploited for non-symmetric designs in composition, size, shape, and coordination environments – offer a path to overcome these barriers. Here, we summarize recent developments in ATEs, focusing on asymmetric coupling strategies employed in designing these systems with non-precious transition metal catalysts (TMCs). We explore tailored asymmetries in composition, size, and coordination environments, highlighting their impact on catalytic performance. We analyze the electrocatalytic mechanisms underlying ATEs with an emphasis on their roles in water-splitting and metal–air batteries. Finally, we discuss the challenges and opportunities in advancing the performance of these technologies through rational ATE designs.
 Lei Zhang | Lei Zhang received his PhD from Nanjing University in 2011. From 2016 to 2017, he conducted postdoctoral research at Tsinghua University. He is currently a full professor at the School of Materials Science and Engineering, Anhui University of Science and Technology, P. R. China. His research interests focus on the design and preparation of functional micro- and nanomaterials for novel energy conversion and storage applications. |
 Qiaoling Xu | Qiaoling Xu received her BS degree from Anhui University of Science and Technology, China, in 2019. Since 2022, she has been pursuing her doctoral studies under the supervision of Prof. Zhang at the School of Materials Science and Engineering, Anhui University of Science and Technology. Her current research focuses on the design, synthesis, and performance optimization of highly efficient electrocatalytic materials. |
 F. Pelayo García de Arquer | F. Pelayo García de Arquer has been a Professor and group leader at ICFO since 2021. His research spans the design and implementation of materials for energy and optoelectronic applications, including the development of decarbonizing technologies such as CO2 capture and solar fuels. |
 Yingtang Zhou | Yingtang Zhou is a senior professor in Zhejiang Ocean University, China. He obtained his PhD Degree in Biology Engineering from KU. Leuven in 2008. Currently he has interests in theoretical study, computational design and experimental synthesis of novel light-thermal materials as catalysts for light irradiation thermal treatment, renewable clean energy and environmental protection applications. |
1. Introduction
The demand for renewable energy accelerates the need to develop cost-effective and environmentally friendly energy conversion and storage technologies.1–3 Some strategies to this end involve batteries, fuel cells, and water electrolyzers.4–6 This work focuses on water electrolysis and rechargeable metal–air batteries, highlighting how advances in catalyst design could overcome existing technology bottlenecks.Both water electrolysis and rechargeable metal–air batteries face limitations related to their three critical supporting reactions: the hydrogen evolution reaction (HER), the oxygen evolution reaction (OER), and the oxygen reduction reaction (ORR) (Fig. 1).7–9 The HER and the OER are crucial in water electrolysis and extended electrosynthesis systems,10 which may combine alternative anodic oxidation reactions (e.g., alcohol and biomass valorization), or direct cathodic conversion reactions such as CO2 and N2 reduction.11–13
 | ||
| Fig. 1 Overview of HER, OER, and ORR applications in electrochemical energy systems. The illustration highlights critical reaction pairings, like anodic oxidation paired with the HER and cathodic CO2 and N2 reduction with the OER, showcasing their importance in diverse applications. These processes usually rely on noble metals to achieve high performance, posing a major challenge for their large-scale deployment due to high cost and resource scarcity. | ||
The OER and the ORR are the primary polarization processes in rechargeable metal–air batteries, fuel cells, and electrochemical carbon capture technologies based on electrocatalysis.14–18 These half reactions involve multi-electron transfer processes and exhibit sluggish kinetics.19–21 Therefore, designing electrocatalysts to speed up and improve energy conversion efficiency in these processes is important.22–25
Noble metal catalysts are widely used to support these reactions due to their activity and stability. However, their high cost and scarcity pose barriers to large-scale deployment.26–30 In contrast, non-precious TMCs offer a promising alternative, as they can be structurally modified through techniques such as size control, heteroatom introduction, defect engineering, and substrate effects.31–36 These modifications enhance the conductivity and electronic states of TMCs, improving their catalytic performance and positioning them as potential alternatives for noble metal materials.37–39
Asymmetricity is a common feature in natural systems, offering functional advantages.40,41 For instance, asymmetrical leaf arrangements in plants optimize sunlight absorption, while the asymmetric shells of mollusks enhance self-protection and buoyancy regulation. Inspired by these principles, introducing asymmetry into material structures presents an attractive approach to improving catalytic activity.42 Achieving this requires precise control over component selection, size, coordination environments, and the design of catalyst assembly units.43
Several works have provided evidence regarding the potential of asymmetric coupling designs in electrocatalysts.44,45 Examples include the use of single-atom (SA) electrocatalysts featuring asymmetric coordination,46 the use of metal–nitrogen–carbon (M–N–C) supported SAs with asymmetric coordination,47 and the use of axially coordinated SAs.48 These works have focused on particular aspects of individual asymmetric coupling strategies. The present work bridges the gaps between these individual views, providing an in-depth overview of the entire field of asymmetric coupling engineering.49–51
First, we present an overview of the broader development of asymmetric catalyst design strategies, categorizing them into compositional, size, coordination, and multiple asymmetric coupling approaches (Fig. 2). Next, we discuss their impact on physicochemical properties and electronic structure, focusing on how these factors regulate the adsorption–desorption processes of intermediates and enhance catalytic performance. Finally, we identify key challenges and outline perspectives for advancing ATEs toward improved functionality and scalability.
 | ||
| Fig. 2 Asymmetric coupling design strategies for non-precious TMCs. Illustration of asymmetric coupling design strategies for non-precious TMCs: size-asymmetric coupling among SAs, clusters, and particles, coordination-asymmetric coupling highlighting low-, lateral-, and axial-coordination, compositional-asymmetric coupling achieved through heterojunction design, and multiple asymmetric couplings combining these. | ||
2. Development of asymmetric coupling design strategies
Asymmetric coupling strategies provide versatile approaches to enhancing electrocatalyst performance.52–55 Compositional-asymmetric engineering involves integrating heterostructures with distinct components to modulate the resulting electronic structure.56 This, in turn, facilitates introducing novel functionalities and optimizing catalytic intermediates, thereby boosting their catalytic performance. One important advance in recent years is the introduction of asymmetry in SA catalysts, which are characterized by a planar symmetry metal (M)–nitrogen (N4) sites where the metal center is tetra-coordinated with nitrogen atoms.57 Breaking the coordination symmetry of M-N4 sites disrupts their charge distribution, offering an efficient method to boost catalytic activity.58,59In catalytic systems featuring size asymmetry, integrating clusters (particles) to regulate the electronic structure of SAs/clusters presents a promising strategy to optimize electrochemical performance.60 This approach utilizes inherent disparities in electronic states among SAs, clusters, and nanoparticles, resulting in multiscale nano-hybrids with size-asymmetric coupling.61 Another example involves material systems with coexisting forms of asymmetric couplings,62 such as size asymmetry combined with coordination asymmetry, size asymmetry coupled with composition asymmetry, or a tripartite integration of size asymmetry, coordination asymmetry, and composition asymmetry. These synergistic interactions between multiple asymmetry mechanisms enhance catalytic performance.63,64Fig. 3 overviews the evolution and recent advancements in ATEs, highlighting key milestones and offering a roadmap for future research and innovation.
 | ||
| Fig. 3 Timeline of evolution and breakthroughs in asymmetric coupling strategies for electrocatalysts. An overview of key milestones from 2013 to 2024 in the development of asymmetric coupling strategies, including coordination, composition, size, and multiple asymmetric designs. Examples showcase advancements such as FePc-Py-CNT (2013), MoS2/CoSe2 (2014), Co-Co3O4@NC (2015), Co SAs/N-C (2016), and Cu-S3N3/Cux (2023). Each entry highlights pivotal innovations driving the progression of asymmetric tailored electrocatalysts (ATEs), offering insights into the field's evolution and future directions. Copyright 2013,65 2014.66 Reproduced with permission. Copyright 2013 and 2014, Springer Nature. 2015,67 2017.68 Reproduced with permission. Copyright 2015 and 2017, American Chemical Society. 2016,69 2018,70 2020,71 2021,72 2023,73 2024.74 Reproduced with permission. Copyright 2016, 2018, 2020, 2021, 2023 and 2024, John Wiley and Sons. 2019.75 Reproduced with permission. Copyright 2019, Royal Society of Chemistry. 2022.76 Reproduced with permission. Copyright 2022, Elsevier. | ||
3. Regulation strategies of asymmetric coupling
3.1 Compositional-asymmetric coupling
Materials are typically classified into conductors, semiconductors, and insulators based on electrical conductivity.77,78 When different materials are coupled, they form diverse heterogeneous interfaces with unique physicochemical properties arising from variations in electronic structures, band gaps, and other inherent characteristics among constituent units.79–83 These interfaces enable precise control over atomic configurations, allowing catalytic reaction pathways to be fine-tuned, thereby enhancing efficiency and selectivity.84–86 Here, we categorize heterointerfaces into three main types: metal–metal heterojunctions, metal–semiconductor heterojunctions, and semiconductor–semiconductor heterojunctions. Building on the assembly strategies discussed earlier, we have organized and summarized recent advances and their key performance metrics in these heterojunctions (Fig. 4).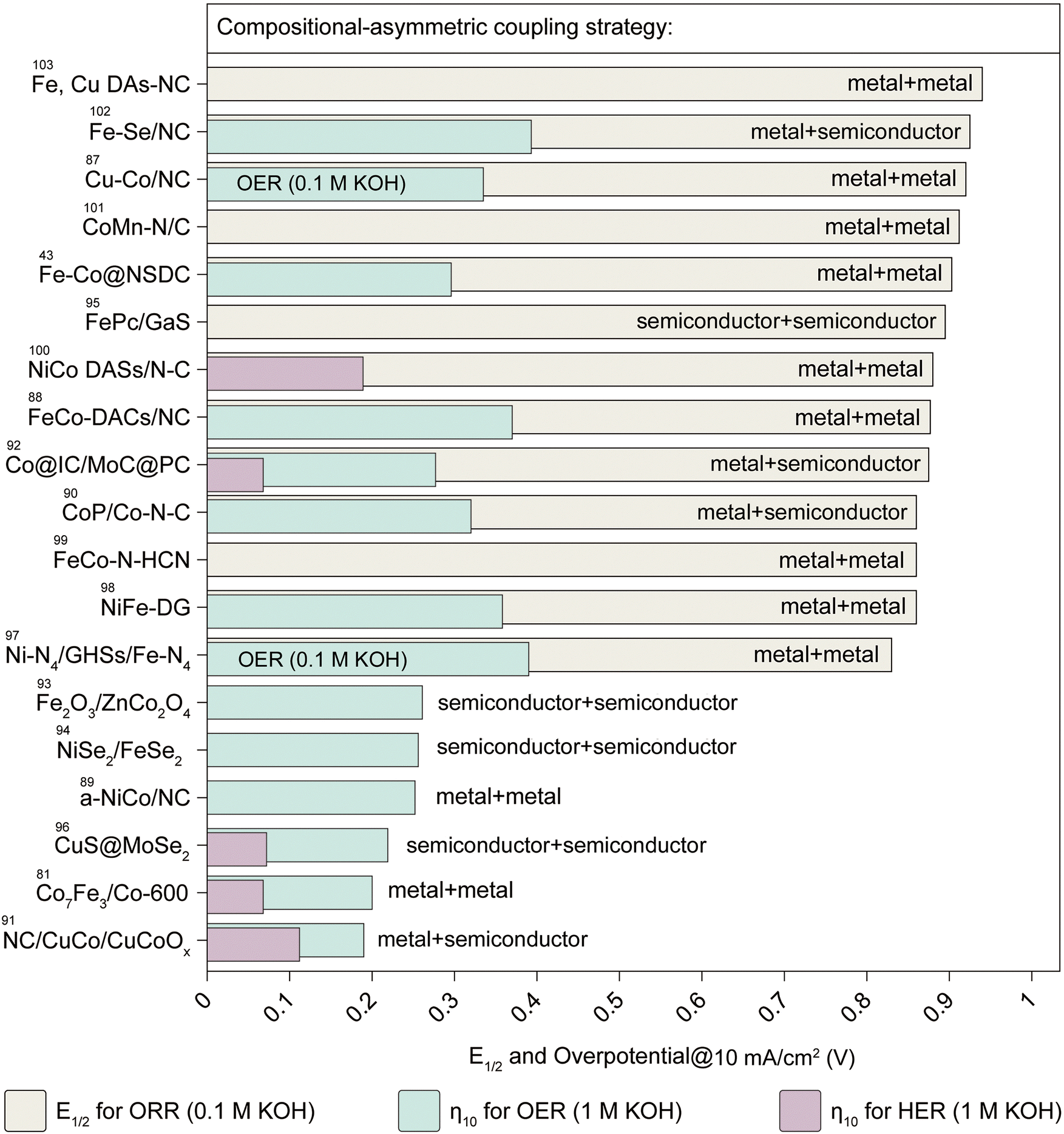 | ||
| Fig. 4 Summary of the performances for the designed catalysts based on the compositional-asymmetric coupling strategy. The plot categorizes catalysts into metal–metal, metal–semiconductor, and semiconductor–semiconductor heterojunctions, highlighting their respective catalytic performance indicators for the ORR, OER, and HER under alkaline conditions (0.1 M KOH or 1 M KOH). The figure emphasizes the half-wave potential (E1/2) for the ORR and overpotential at 10 mA cm−2 (η10) for the OER and HER as key benchmarks. This diagram provides a summary of the performance metrics reported in ref. 43, 81 and 87–103. | ||
The structural advantages of compositional-asymmetric coupling electrocatalysts can be summarized as follows. (i) Tailored material properties: the integration of diverse materials enables precise modulation of properties such as conductivity, hydrophilicity, chemical stability, and active site density, collectively enhancing catalytic performance; (ii) lattice strain engineering: distinct chemical compositions and crystal structures induce lattice strain, which modifies the intermediate adsorption energies at active sites, leading to improved catalytic selectivity; (iii) enhanced charge transfer: variations in band alignments across different phases facilitate efficient charge transfer at interfaces, enabling precise modulation of surface electronic states within heterogeneous structures; and (iv) material versatility: the broad range of material types provides extensive opportunities for optimizing and assembling catalytic building units, fostering the design of advanced ATEs, and enriching the “gene library” of electrocatalysts.
A compelling realization of this concept is heteronuclear dual-metal atom catalysts (DMACs), where two different metal atoms form a dimer structure on a support material.62,104 Homonuclear DMACs do not merely amplify intrinsic activity through ensemble effects but also disrupt the local environment, leading to distinct electronic structures. Asymmetric DMACs may induce localized atomic orbital distortions, which impact π-orbital interactions and back-donation effects.105 This can disrupt traditional linear scaling relationships, which often impose fundamental activity–stability trade-offs in homogeneous catalytic systems.106 DMACs may impact selectivity and kinetics in multielectron transfer reactions, beyond SACs,107 making these sites effective in activating small molecules like N2, CO2, and H2O.108
For instance, atomically dispersed Cu and Co bimetallic sites anchored on a porous nitrogen-doped carbon (NC) framework exhibit exceptional bifunctional oxygen catalytic activity due to their optimal geometry and electronic structure (Fig. 5a and b).87 Both experimental and theoretical findings indicated that the synergistic effects between the metal–N4 coordination structures of Cu–Co bimetallic centers induced a charge asymmetry distribution, contributing to moderate the adsorption–desorption ability of the catalytic centers with oxygen intermediates. Under alkaline conditions, the electrocatalyst yielded remarkable ORR/OER catalytic activities, with an ORR half-wave potential of 0.920 V and an overpotential of just 0.335 V at 10 mA cm−2 (Fig. 5c and d). It also delivered outstanding ORR activities under acidic (0.850 V) and neutral (0.740 V) conditions. When applied in Zn–air batteries, this catalyst yielded excellent performance and durability, with a longevity of 510 hours and 1000 cycles. Following this, a “pre-constrained metal twins” approach was introduced, effectively incorporating Fe and Co into the NC skeleton.88 This approach led to the construction of a bifunctional catalyst with high-quality Fe–Co heteronuclear pairs, facilitating efficient oxygen electrocatalysis (Fig. 5e and f). Specifically, the Fe- and Co-containing binuclear phthalocyanine was initially introduced into the ZIF-8 matrix. Subsequently, through high-temperature carbonization, Zn species in ZIF-8 were volatilized, and ligands were transformed into NC materials. The adjacent Fe and Co atoms form proximal active sites known as Fe–N4 and Co–N4 complexes within this material. Electrochemical investigations demonstrated the exceptional catalytic performance of these heteronuclear pairs in both the OER/ORR. Theoretical simulations indicated that the collaborative effects of neighboring Fe and Co metals optimized the d-band center of metallic sites, equilibrating the free energy of *O species and consequently boosting the electrocatalytic performance towards oxygen.
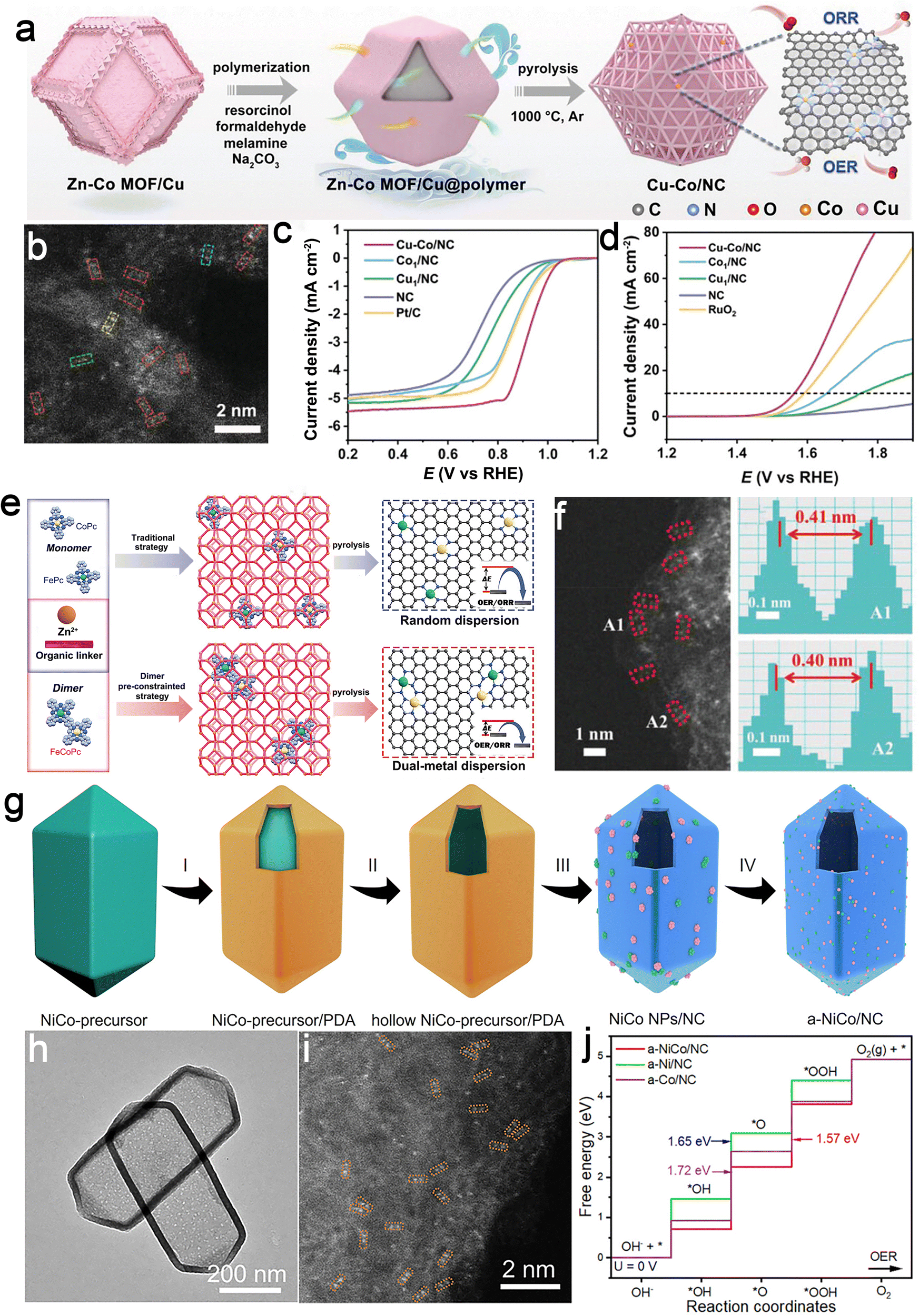 | ||
| Fig. 5 Examples of metal–metal coupled catalysts. Cu-Co@NC catalyst: (a) synthetic scheme; (b) high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) image showcasing atomic dispersion; and (c) ORR and (d) OER polarization curves. Reproduced with permission from ref. 87. Copyright 2023, John Wiley and Sons. Fe–Co heteronuclear pairs catalyst: (e) schematic illustration comparing the traditional strategy and “pre-constrained metal twins” strategy; (f) HAADF-STEM image and intensity profiles for metal pairs. Reproduced with permission from ref. 88. Copyright 2022, John Wiley and Sons. Ni–Co dual sites catalyst: (g) Schematic illustration of the synthesis route; (h) transmission electron microscopy (TEM) image illustrating structural features and (i) HAADF-STEM image indicating dual-site dispersion; (j) free energy diagram of OER. Reproduced with permission from ref. 89. Copyright 2022, John Wiley and Sons. | ||
As for water-splitting, compositional asymmetry through the coupling of different metals offers substantial advantages. For instance, a pioneering multistep templating method involving atomic migration and capture processes was developed, leveraging atomic migration and capture processes to synthesize atomically dispersed Ni and Co sites confined within a nitrogen-doped carbon (NC) hollow prism (Fig. 5g–i).89 Theoretical calculations substantiated the enhanced electrocatalytic activity resulting from the synergistic catalytic effects between dual-nuclear sites (Fig. 5j). Through a similar synthesis approach, nanoscale asymmetric ATEs can also be constructed. For example, a spatial segregation strategy was employed to produce an N/S doped carbon nanobox hosting Fe–Co pair-sites with compositional asymmetric coupling.43 This unique nanostructure displayed outstanding electrocatalytic characteristics, delivering a half-wave potential of 0.903 V for the ORR and an overpotential of 0.296 V at 10 mA cm−2 for the OER. In summary, metal–metal asymmetric coupling leverages the electronic and geometric disparity between distinct metal centers to modulate adsorption energies and intermediate stabilization beyond conventional SACs.
For example, a Mott–Schottky heterojunction composed of a porous cobalt–nitrogen–carbon (Co–N–C) polyhedron and abundant CoP nanoparticles was successfully designed (Fig. 6a and b).90 For the ORR, the half-wave potential of CoP/Co–N–C was 0.860 V, along with an overpotential of 0.320 V at 10 mA cm−2 for the OER. Density functional theory (DFT) calculations indicated that the Mott–Schottky heterojunction formed an intrinsic electric field to facilitate interface charge transfer. This led to the movement of the d-band, thereby regulating the adsorption/desorption behavior of oxygen intermediates and achieving enhanced dual functionality for reversible oxygen electrocatalysis. Another metal–semiconductor Mott–Schottky hybrid, featuring CuCo nanoalloys on arrays of defective CuCoOx semiconductor nanowires, was also recently fabricated.91 To establish an uninterrupted electron transport route between a metal and a semiconductor, an NC matrix has been incorporated into the nanowire array of the metal–semiconductor, forming an integrated core–shell architecture (Fig. 6c–f). This innovative NC/CuCo/CuCoOx core–shell array demonstrated exceptional electrocatalytic OER–HER performances in a 1 M KOH medium (Fig. 6g). It achieved a noteworthy oxygen evolution current density of 10 mA cm−2 with a mere 0.190 V overpotential and a hydrogen evolution current density of 10 mA cm−2 with a 0.112 V overpotential. When employed as both an anode and cathode for the overall water splitting, the core–shell composite array electrodes exhibited a remarkable current density of 10 mA cm−2 at an impressively low voltage of only 1.53 V, demonstrating their exceptional electrochemical performance. These electrodes demonstrated excellent stability during continuous operation for 100 hours under high current density conditions. Experimental findings indicated that the three-dimensional hierarchical structure provided an extensive electrochemical active surface area, abundant active sites, and rapid gas transport. The in situ formation of metal–semiconductor Mott–Schottky catalysts generated rich oxygen vacancies, enhancing conductivity and promoting electron transfer. The NC skeleton promoted the adsorption of intermediates and constructed optimized pathways for electron transfer between the semiconductor, metal, and carbon layers by redistributing electron density. Recently, an ORR/OER/HER tri-functional electrocatalyst, comprising distinct Co nanoparticles and MoC components on hollow carbon structure inner and outer layers, has been reported (Fig. 6h).92 Due to the electronic polarization interface between these two components, charge redistribution at the interface was facilitated. Subsequent investigations demonstrated that Zn–air batteries and water splitting assembled with this catalyst exhibited outstanding catalytic performance and stability (Fig. 6i). Therefore, the coupling strategy between a metal and a semiconductor affords a feasible avenue for designing and developing multifunctional catalysts for the next generation of reversible energy conversion systems.
 | ||
| Fig. 6 Examples of metal–semiconductor coupled catalysts. CoP/Co–N–C catalyst: (a) schematic illustration of the synthesis process and (b) high-resolution TEM (HRTEM) images of the sample highlighting lattice fringes of CoP and Co. Reproduced with permission from ref. 90. Copyright 2023, John Wiley and Sons. NC/CuCo/CuCoOx catalyst: (c) schematic illustration of the fabrication procedure; (d) scanning electron microscopy (SEM) and (e and f) TEM images showing the structural details; (g) OER polarization curves. Reproduced with permission from ref. 91. Copyright 2018, John Wiley and Sons. Co@IC/MoC@PC catalyst: and (h) design idea and preparation path, as well as (i) linear sweep voltammetry (LSV) plots. Reproduced with permission from ref. 92. Copyright 2021, American Chemical Society. | ||
An n–n heterojunction was successfully fabricated at the biphasic interface by coupling oxygen-vacancy-rich Fe2O3 and ZnCo2O4 using a cation-exchange method as an example (Fig. 7a and b).93 This heterojunction exhibited exceptional performance with a minimal overpotential of 0.261 V to achieve a current density of 10 mA cm−2. It displayed remarkable stability at high current densities, exceeding that of the majority of Co/Fe-based OER catalysts reported to date (50 hours). DFT calculations validated that the formation of the nanojunction fine-tuned the d-band center, improving the adsorption of oxygen intermediates at the active sites and consequently optimizing the Gibbs free energy of the OER reaction (Fig. 7c). Another illustrative example involves the construction of a NiSe2/FeSe2 p–p junction prepared through a solid-state selenization method (Fig. 7d–g).94 The intrinsic electric field formed at the NiSe2/FeSe2 heterointerface enhanced charge transfer, leading to localized charge redistribution. This increased the conductivity and facilitated the adsorption of relevant reactants driven by electrostatic interactions. Integrating hetero-type semiconductors with p–n heterojunctions can provide highly efficient catalytic active sites, thereby accelerating the kinetics of electrochemical reaction processes. Taking the FePc/GaS p–n junction as a model, atomic geometric characterization revealed a distortion of the Fe–N4 moiety, thus disrupting the Fe–N coordination configuration and modulating the local charge density and spin configuration of the Fe center (Fig. 7h–k).95 As a result, the p–n junction catalyst exhibited a comparative ORR activity at 0.850 V, more than twofold that of the original FePc and over twelvefold that of commercial Pt/C catalysts. This strategy could be extended to other n-type sulfide carriers with different WFs, revealing a linear relationship between the ORR activity of loaded Fe–N4 and the rectification degree. This suggested the continuous tunability of SACs by altering n-type carriers.
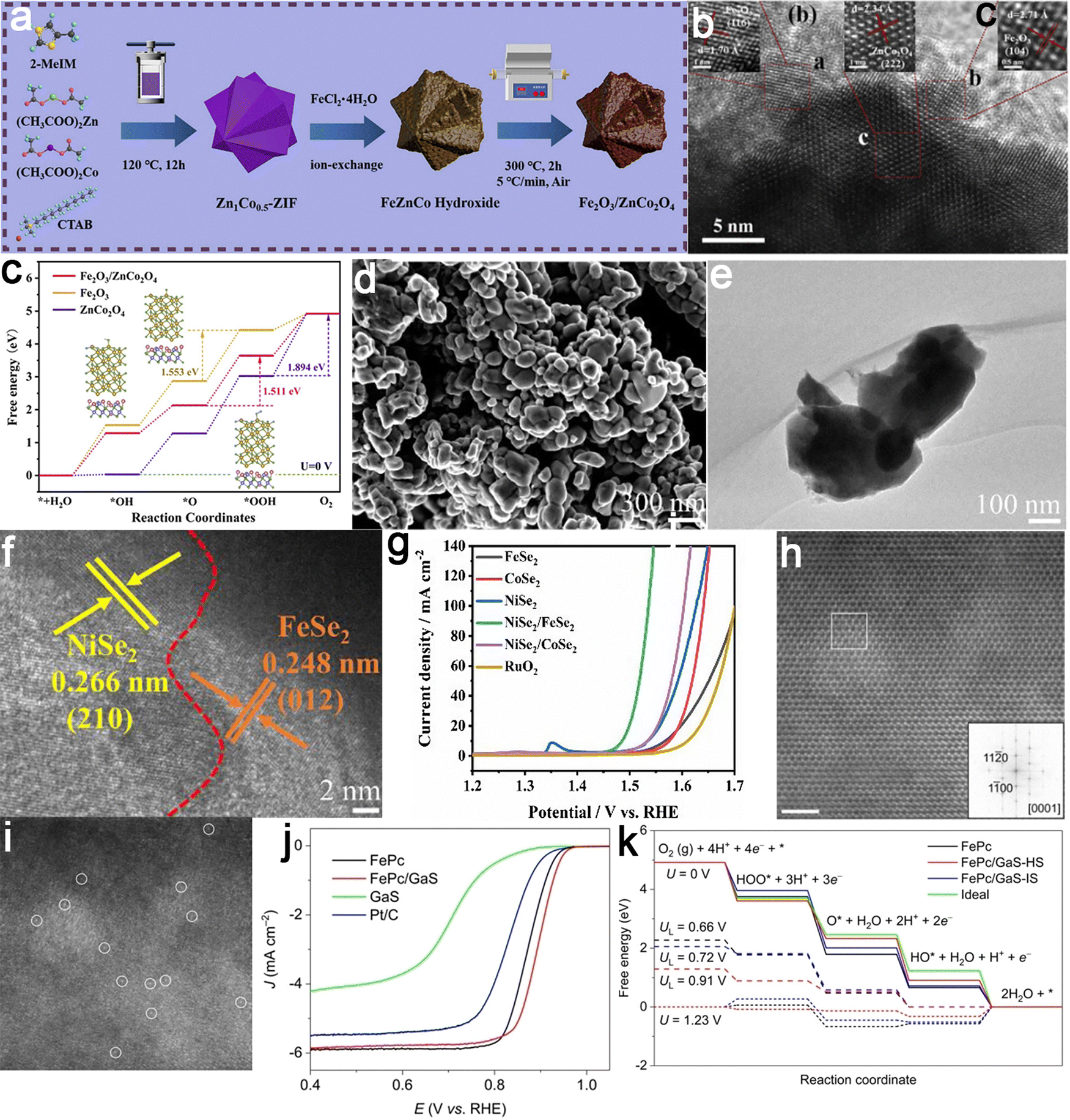 | ||
| Fig. 7 Examples of semiconductor–semiconductor coupled electrocatalysts. Fe2O3/ZnCo2O4 catalyst: (a) schematic illustration for the synthetic process; (b) HRTEM images showing the well-defined heterojunction interface; (c) the energetic pathway of the OER, highlighting the reduced energy barriers enabled by the heterojunction design. Reproduced with permission from ref. 93. Copyright 2023, Elsevier. NiSe2/FeSe2 heterojunction catalyst: (d) SEM, (e) TEM, and (f) HRTEM images showing distinct lattice planes of NiSe2 (210) and FeSe2 (012); (g) LSV curves comparing the electrocatalytic performance of NiSe2/FeSe2 against benchmark catalysts for the OER. Reproduced with permission from ref. 94. Copyright 2021, Elsevier. FePc/GaS catalyst: (hand i) HAADF-STEM image; and (j) LSV curves illustrating the enhanced ORR performance of FePc/GaS over individual components and (k) free energy diagrams for ORR showing optimized intermediate adsorption energies, confirming the role of electronic coupling in boosting catalytic efficiency. Reproduced with permission from ref. 95. Copyright 2023, John Wiley and Sons. | ||
3.2 Coordination-asymmetric coupling
Asymmetric coordination offers a promising approach to enhance catalytic activity, distinct from the traditional metal–N4 active site model.119 Incorporating asymmetric ligands or varied coordination environments provides a novel means to precisely regulate the electronic structure and charge distribution of active sites.120 Based on the coordination attributes, asymmetrical atomic sites can be categorized into three types: low coordination, lateral coordination, and axial coordination.121 These classifications clearly demonstrate the diverse structural arrangements inherent in coordination-asymmetric coupling. Fig. 8 presents recent research findings using these strategies, offering a concise visual summary of advancements in the field. The advantages of asymmetric coordination structures over conventional M–N4 sites in electrocatalysis primarily include lower overpotential, higher selectivity, and more excellent catalytic performance. SA centers with low coordination display enhanced intrinsic electrocatalytic activity attributed to the optimized electron structure, which lowers the adsorption of oxygen intermediates, consequently improving the activity of metal sites.47 Structural distortions can induce charge transfer and asymmetric charge distribution in lateral coordination SA systems, optimizing adsorption and desorption behaviors towards oxygen intermediates and reducing energy barriers for the ORR and OER.48 Axial coordination can also disrupt the symmetry of catalytic centers, optimizing the local coordination environment of active metals, boosting the charge polarization effects, and facilitating charge transfer between metal centers and adsorbed intermediates, thus decreasing the Gibbs free energy of the rate-limiting steps in catalytic reactions. | ||
| Fig. 8 Summary of catalytic performances using coordination-asymmetric coupling strategies. The chart categorizes catalysts based on coordination types, including axial coordination, lateral coordination, and low coordination. The performance metrics include the half-wave potential (E1/2) for the ORR (0.1 M KOH), the overpotential at 10 mA cm−2 (η10) for the OER (1 M KOH), and η10 for the HER (1 M KOH). Catalysts with unique coordination environments, such as FePcP/C-I/C with axial coordination or Co–N, B-CSs with lateral coordination, exhibit notable catalytic efficiencies. These findings emphasize the influence of coordination asymmetry in optimizing catalytic sites for specific reactions. Data points also highlight catalysts such as Ni–B/N–C and Co-SAs@N-CNFs for their remarkable activity across multiple coordination designs. This diagram provides a summary of the performance metrics reported in ref. 10, 51, 70 and 122–139. | ||
 | ||
| Fig. 9 Examples of metal–Nx sites with low coordination. Ni-SA/NC catalyst: (a) schematic illustration of the fabrication process; (b and c) HAADF-STEM images showcasing atomic dispersion; (d) HER polarization curves illustrating catalytic performance and (e) the Gibbs free energy diagram highlighting the thermodynamic favorability of HER. Reproduced with permission from ref. 122. Copyright 2021, John Wiley and Sons. Fe SAs/NC catalyst: (f) schematic drawing of the operando synchrotron radiation characterizations; (g) HAADF-STEM image capturing isolated Fe sites and (h) LSV curves demonstrating excellent catalytic activity. Reproduced with permission from ref. 123. Copyright 2021, American Chemical Society. Cu–N–C SAC catalyst: (i) Schematic illustration of the two-step synthesis strategy; (j) HAADF-STEM image; (k) ORR polarization curves showing enhanced catalytic behavior and (l) free energy diagram for the ORR indicating reduced reaction barriers. Reproduced with permission from ref. 124. Copyright 2021, American Chemical Society. | ||
In recent years, attention has been directed towards the rational design of Fe SACs with asymmetrical coordination. Taking the Fe–N2 active site as an example, it was observed that the interaction of OH species occurred with Fe atoms characterized by unsaturated coordination structures (Fig. 9f and g).123 This led to the creation of the OH–Fe–N2 site, which facilitated O2 adsorption, promoted the rapid formation of *OOH species at low operating potentials, and contributed to the cleavage of the O–O bond with a lower barrier, thus enhancing the kinetics of the multi-electron transfer reaction. As a result, the resulting catalyst demonstrated excellent ORR activity, exhibiting a high kinetic current density (Jk) of 81.3 mA cm−2 and an elevated high turnover frequency (TOF) value of 5804 h−1 (Fig. 9h). In addition to Fe and Ni, Cu-based SAC has also shown remarkable ORR activity (Fig. 9i–k).124 Extended X-ray absorption fine structure (EXAFS) and electron energy loss spectra (EELS) verified that Cu SA closely resembled Cu–N4. Electrochemical in situ XANES identified the unsaturated Cu+–N3 as the active site for the ORR, with Cu2+–N4 functioning primarily as its precursor. Under reducing potential, water dissociated into OH− and protons, facilitating the hydrogenation of pyridinic nitrogen, which in turn drove the transition from Cu2+–N4 to Cu+–N3. These Cu+–N3 SA sites activated O2 by donating electrons to its antibonding orbitals, thereby weakening the O–O bond and promoting its cleavage, ultimately leading to the formation of OH−. During the ORR, Cu–N3 further evolved into Cu–N2, likely due to ligand reorganization under reducing conditions. However, upon removal of the applied potential, the system reverted to its stable Cu–N4 configuration, suggesting a reversible structural evolution (Fig. 9l). Therefore, the SACs with low coordination configuration have demonstrated their potential for practical applications in electrochemical systems.
 | ||
| Fig. 10 Examples of metal–NxAy sites with lateral coordination. Co-SA/P-in situ catalyst: (a) scheme of the formation and (b) HAADF-STEM images of the sample showing atomic dispersion of Co sites. Reproduced with permission from ref. 125. Copyright 2020, American Chemical Society. Ni–B/N–C catalyst: (c) schematic diagram of the preparation process, (d and e) HAADF-STEM images confirming the presence of Ni–B sites, and (f) ORR polarization curves highlighting catalytic performance improvements due to lateral coordination. Reproduced with permission from ref. 127. Copyright 2023, John Wiley and Sons. Mn/C–NO catalyst: (g) schematic illustration of the synthetic process for Mn Sas and (h) HAADF-STEM images of Mn SAs visualizing Mn atomic sites; and (i) free-energy diagram of the ORR on Mn–N1O3, Mn–N2O2, and Mn–N3O1 surfaces demonstrating their catalytic advantages. Reproduced with permission from ref. 65. Copyright 2018, John Wiley and Sons. | ||
Apart from S and P, incorporating B into the carbon skeleton decreases the d-band center of the metal atom and the adjacent position of the initial peak concerning the Fermi level.142 This alteration improves the adsorption and desorption of intermediates, thereby promoting the catalytic reaction. Inspired by this, a B-doped Ni–N–C catalyst was developed for efficient ORR (Fig. 10c–e).127 This catalyst featured atomically dispersed Ni atoms anchored on a carbon support, coordinated with one B atom and three N atoms, forming a Ni–B1N3 configuration. The prepared Ni–B/N–C catalyst demonstrated enhanced ORR performance (Fig. 10f), with a half-wave potential (E1/2) of 0.870 V, surpassing that of the original Ni–N–C (0.760 V). Even after 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, E1/2 only degraded by 21 mV, outperforming most reported Fe–N–C and Co–N–C catalysts, indicating remarkable stability. DFT calculations verified that, compared to Ni–N4, the Ni–B1N3 configuration increased the electron density at the Ni site, improving the adsorption of ORR intermediates and lowering the thermodynamic barrier, thus expediting ORR kinetics. As an illustrative example, O and N atoms were coordinated with Mn sites to fabricate a metal–N3O1 configuration, employing conductive 3D graphene frameworks as the substrate.76 Inspired by enzyme coordination effects, Mn d-electron energy levels were adjusted optimally (Fig. 10g and h). Computational results revealed that Mn–N1O3, Mn–N2O2, and Mn–N3O1 emerged as active sites for the ORR (Fig. 10i). The deliberate downward shift of the d-band center and the proximity of the initial peak to the Fermi level within the Mn–N3O1 site promoted the desorption of reaction intermediates. This, in turn, optimized the kinetics for the ORR.
000 cycles, E1/2 only degraded by 21 mV, outperforming most reported Fe–N–C and Co–N–C catalysts, indicating remarkable stability. DFT calculations verified that, compared to Ni–N4, the Ni–B1N3 configuration increased the electron density at the Ni site, improving the adsorption of ORR intermediates and lowering the thermodynamic barrier, thus expediting ORR kinetics. As an illustrative example, O and N atoms were coordinated with Mn sites to fabricate a metal–N3O1 configuration, employing conductive 3D graphene frameworks as the substrate.76 Inspired by enzyme coordination effects, Mn d-electron energy levels were adjusted optimally (Fig. 10g and h). Computational results revealed that Mn–N1O3, Mn–N2O2, and Mn–N3O1 emerged as active sites for the ORR (Fig. 10i). The deliberate downward shift of the d-band center and the proximity of the initial peak to the Fermi level within the Mn–N3O1 site promoted the desorption of reaction intermediates. This, in turn, optimized the kinetics for the ORR.
The above discussion focused on the lateral coordination of single metal centers. However, the efficiency of these strategies is often constrained by limited electron transfer pathways, particularly in multi-electron transfer processes, where a single-site configuration may struggle to efficiently accommodate reaction intermediates with varying electronic structures and binding affinities. While homonuclear dual-atom catalysts have been proposed to enhance multi-electron reactions, such as the ORR and OER, they still face similar limitations—two identical metal atoms may not fundamentally overcome the constraints of single-site configurations. Instead, alternative approaches involving out-of-plane architectures or DMACs may provide a more effective means to modulate electronic structures, facilitate charge delocalization, and optimize intermediate stabilization, ultimately improving catalytic activity and stability.
For example, a high-density asymmetric dual-iron atom catalyst, A–Fe2S1N5@N/S co-doped carbon, was synthesized using a multilayer stabilization strategy involving defect capture and coordination riveting.144 The asymmetric nitrogen–sulfur coordination around the dual-iron sites modulated the electronic structure, leading to enhanced adsorption and activation of oxygen intermediates. Specifically, this asymmetric coordination strengthened Fe–O interactions, promoting more efficient reactant conversion, lowering energy barriers, and accelerating reaction kinetics. These findings highlight the potential of asymmetric coupling strategies to further advance single-atom and dual-atom catalysts, offering new pathways for designing efficient electrocatalysts.
Coordination asymmetry strategies also play a crucial role in the design of molecular ORR catalysts. For example, metal porphyrins consist of a central metal ion coordinated to a ring of four nitrogen atoms.145,146 This coordination enables strong catalytic activity for oxygen reduction. Specifically, iron and cobalt porphyrins promote oxygen molecule adsorption and reduction, demonstrating catalytic activity comparable to that of platinum-based catalysts.147 However, the catalytic performance and stability of metal porphyrins are highly sensitive to the electronic structure of the metal center, the ligand environment, and interactions with reactants. Optimizing their coordination structure is thus important to enhance ORR activity. For instance, introducing substituents at the side coordination sites of the porphyrin structure can tune the electronic properties of the catalyst, improving both its activity and selectivity.148 Notably, the asymmetric Pacman-type dinuclear cobalt porphyrin catalyst exhibited higher activity compared to its symmetric counterpart,149,150 highlighting the advantages of asymmetric design in promoting the ORR and further underscored the potential of asymmetric structures in molecular catalysis. This research provides valuable insights for the future development of small-molecule activation catalysts, particularly in improving catalytic performance, selectivity, and stability.
Typically, a porous FeN4–O–NCR electrocatalyst, rich in catalytically accessible FeN4–O sites (wherein the Fe SAs are coordinated to four in-plane N atoms and one subsurface axial O atom), supported on N-doped carbon nanorods (NCR), has been reported (Fig. 11a and b).128 The resulting FeN4–O–NCR electrocatalyst exhibited a hierarchically porous architecture, featuring a substantial specific surface area (1159 m2 g−1) and an abundance of FeN4–O active sites conducive to optimal oxygen adsorption and activation. In Fig. 11c, the FeN4–O–NCR catalyst displayed remarkable ORR activity and stability in alkaline media (E1/2 = 0.942 V). DFT calculations unveiled a precise tailoring of the d-orbital electronic structure of Fe cations, effectively reducing the thermodynamic barrier for the ORR (Fig. 11d). Considering the impact of OH doping on axial coordination and electron transfer, a self-sacrificial strategy was proposed for synthesizing asymmetrically N and S-coordinated FeSA sites.129 This strategy embeds an axial fifth hydroxy (OH) coordination (Fe–N3S1OH) within N, S co-doped porous carbon nanospheres (Fe–N/S–C) (Fig. 11e). The distinctive structure of Fe-N3S1OH, characterized by inherent high activity and a large specific surface area (2539 m2 g−1), ensured sufficient exposure to active sites. Coupled with an interconnected layered porous structure facilitating rapid ion/mass transfer, the fabricated Fe–N/S–C exhibited outstanding ORR catalytic activity.
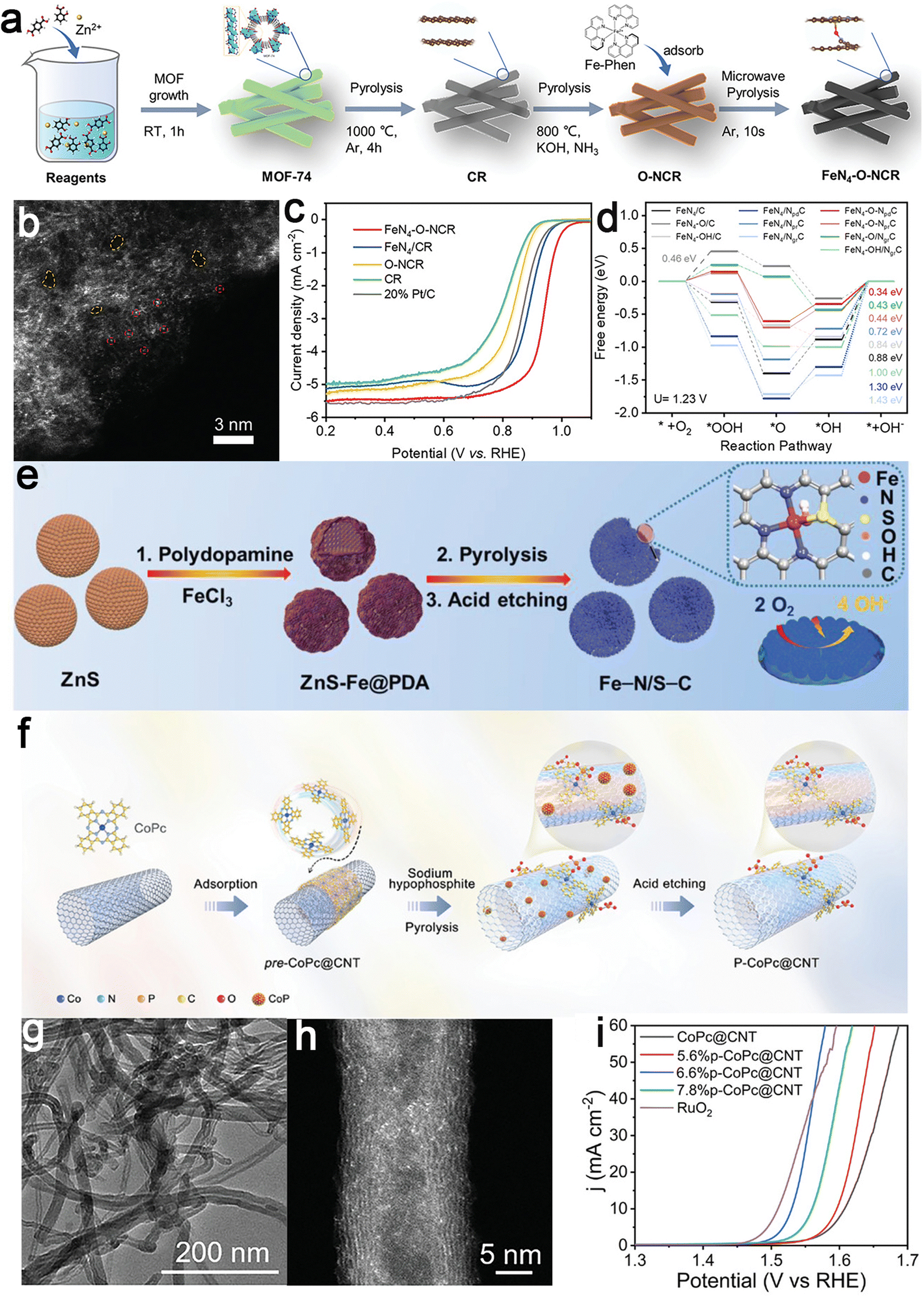 | ||
| Fig. 11 Examples of axial heteroatom coordination catalysts. FeN4–O–NCR catalyst: (a) schematic synthesis illustration and (b) HAADF-STEM image showing atomic dispersion; (c) LSV curves in 0.1 M KOH highlighting catalytic activity and (d) free energy diagram comparing various FeN4-based models. Reproduced with permission from ref. 128. Copyright 2022, John Wiley and Sons. Fe–N/S–C catalyst: (e) Schematic illustration of the synthetic procedure of the Fe–N/S–C catalyst. Reproduced with permission from ref. 129. Copyright 2022, John Wiley and Sons. 6.6%P–CoPc@CNT catalyst: (f) the schematic illustration of the preparation process and (g) the TEM image depicting the overall structure; (h) the HAADF-STEM image showing atomic resolution of active sites; and (i) polarization curves of various catalysts in 0.1 M KOH, demonstrating performance enhancement through axial coordination. Reproduced with permission from ref. 130. Copyright 2023, John Wiley and Sons. | ||
In a recent study, metal phthalocyanine (M-Pc) was used as a model for M–N4 electrocatalysts to explore the positive impact of axial ligands on catalytic activity. M–Pc complexes consist of metal ions coordinated with phthalocyanine ligands, forming distinct M–N4 active centers that exhibit unique electronic structures and stability, making them valuable molecular catalysts for the ORR.155 However, their planar, symmetric structure weakens O2 adsorption and activation, resulting in low ORR catalytic efficiency. Breaking this symmetry is an effective strategy to enhance performance. For example, modifying the axial direction of the metal center by coordinating it with a single metal atom on graphene via a bridging oxygen atom induces symmetry-breaking, promoting out-of-plane polarization, and facilitating O2 adsorption, thereby improving ORR catalytic efficiency.156 In parallel, axial PO4 coordination was introduced at the Co1N4 site in CoPc@CNT (CNT stands for carbon nanotube, Fig. 11f–h).130 This regulation of reaction pathways demonstrated the capability to enhance intrinsic OER activity (Fig. 11i). A similar phenomenon has been observed in metal porphyrin systems. By introducing imidazole groups axially to the metal center, the original square-planar coordination was disrupted. This axial ligand induced a “push-electron effect”, which increased the electron density at the metal site, thereby facilitating the binding and activation of O2 at the metal center.157 Comparable to heteroatoms coordinated laterally, axially coordinated heteroatoms impact the catalytic efficacy of central metal sites via the regulation of the electronic structure. The axial coordination of heteroatoms may also initiate the displacement of metal atoms along the axial direction, consequently altering the length of coordination bonds and influencing the behavior of adsorption/desorption for intermediates.
3.3 Size-asymmetric coupling
Electrocatalysts are classified into three groups based on size: SA-type (0.1–0.2 nm), cluster-type (0.2–1 nm), and particle-type (>1 nm).158 Earlier research on hydrogen and oxygen electrocatalysis focused on particle-type TMCs as alternatives to precious metal benchmarks.159 However, clusters and SAs have recently gained attention due to their unique physicochemical properties. Size reduction can enhance the electrocatalyst surface area and atomic utilization efficiency. Smaller particles will alter the electronic structure, surface morphology, and metal–support interactions, impacting catalytic selectivity. Reduced cluster size can increase coordinatively unsaturated atoms, enhancing reactivity, while sub-nanometer catalysts generate diverse crystal facets and defects.54 Despite recent progress, SAs and clusters exhibit limitations in providing multiple active sites for complex multi-step catalytic reactions, impeding their practical application. Recent studies highlight the synergistic interaction among SAs, clusters, and particles in interface regulation to enhance electrocatalytic performance.55 Incorporating metal clusters or particles near SA centers can modify the rate-determining step of catalytic reactions, lowering the energy barrier associated with these steps. Simultaneously, the non-coherent vibrations induced by these clusters or particles can reduce the bond length of the SA, thereby boosting its stability (Fig. 12).56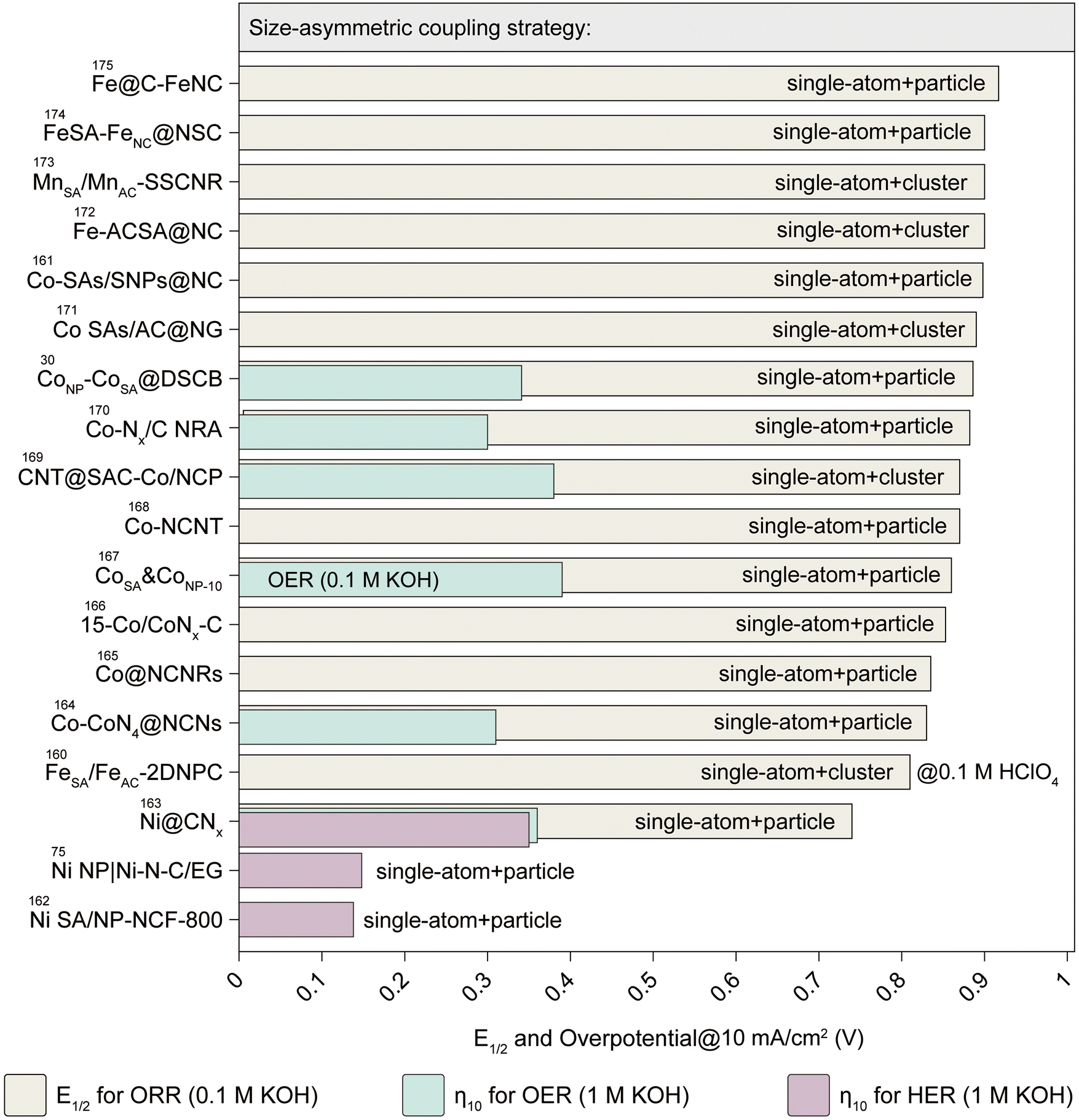 | ||
| Fig. 12 Performance summary of catalysts designed with size-asymmetric coupling strategies. The figure categorizes catalysts based on coupling configurations such as single-atom + particle and single-atom + cluster. Performance metrics include half-wave potential (E1/2) for the ORR in 0.1 M KOH, overpotential (η10) at 10 mA cm−2 for the OER in 1 M KOH, and the HER in 1 M KOH. It highlights the impact of size-asymmetric coupling on catalytic activity and efficiency across various reaction types. This diagram provides a summary of the performance metrics reported in ref. 30, 75 and 160–175. | ||
For example, a Fe–N–C electrocatalyst exhibiting size asymmetry was developed. N-coordinated Fe clusters were positioned adjacent to closely surrounding Fe–N4 sites, utilizing two-dimensional N-doped porous carbon as a substrate (2DNPC) (Fig. 13a–f).160 The rapid electron transfer pathway and short interaction distance facilitated robust electronic interactions between the Fe clusters and adjacent Fe–N4 moieties (Fig. 13g and h). The incorporation of Fe clusters serves a dual purpose: restructuring rate-determining steps and fortifying stability in SAs through reduced bond lengths. Consequently, this composite catalyst exhibited strong adsorption of OH, leading to the permanent grafting of the OH ligand onto the Fe–N4 site. This OH ligand optimized the binding strength of the Fe–N4 site with the ORR intermediates on the opposite side, lowering the limiting energy barrier. In contrast, nanoparticles also play a pivotal role in adjusting the local structures of SA sites, refining intermediate adsorption/activation, and enhancing electrocatalytic performance. For example, a binuclear site consisting of Co SAs in the configuration of Co–N4 moieties and Co nanoparticles (SNPs) was co-anchored on a porous NC nanocage (denoted as Co-SAs/SNPs@NC, Fig. 13i–k).161 Taking advantage of the combined effects of Co SAs and Co particles, along with the improved anti-corrosion ability of the carbon matrix, this product exhibited remarkable ORR activity (E1/2 = 0.898 V) and exceptional stability in alkaline environments, as illustrated in Fig. 13l. DFT calculations revealed that the strong interaction between Co particles and Co–N4 sites increased the valence state of active Co atoms and mitigated the adsorption-free energy of intermediates in the ORR, thereby promoting the reduction of O2 (Fig. 13m and n). The assembled Zn–air battery showed a maximum power density of 223.5 mW cm−2, a substantial specific capacity of 742 W h kg−1 at 50 mA cm−2, and great cycling stability.
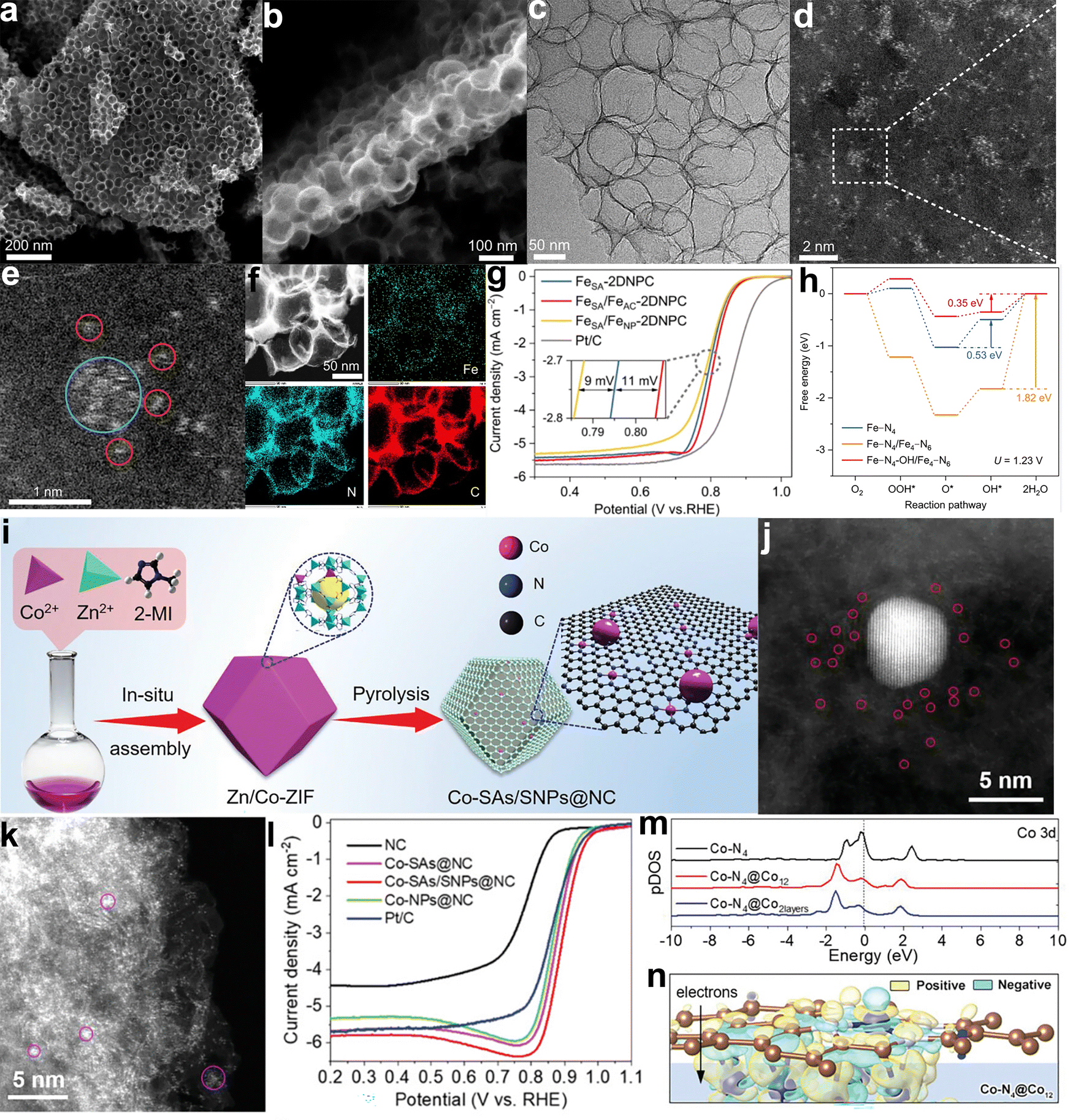 | ||
| Fig. 13 Examples of size-asymmetric coupling catalysts. FeSA/FeAC–2DNPC catalyst: (a) SEM and (b and c) TEM images; (d and e) HAADF-STEM images with zoom-in image showing an iron cluster (cyan circle) and its satellite iron atoms (red circles); (f) energy-dispersive spectroscopy mapping image illustrating elemental distribution; (g) ORR polarization curves comparing catalytic performance and (h) free energy diagrams over three types of active sites. Reproduced with permission from ref. 160. Copyright 2022, Springer Nature. Co-SAs/SNPs@NC catalyst: (i) schematic illustration of the synthetic process, (j and k) HAADF-STEM images displaying cobalt single atoms and nanoparticles and (l) ORR polarization curves highlighting enhanced activity; (m) calculated projected density of states of Co d band in Co–N4, Co–N4@Co12, and Co–N4@Co2 layers; and (n) the charge density difference for Co–N4@Co12, where yellow and blue regions represent electron accumulation and depletion, respectively. Reproduced with permission from ref. 161. Copyright 2021, John Wiley and Sons. | ||
In another illustration, a facile synthetic strategy was presented to co-anchor FeN4, and Fe-nanoclusters/nanoparticles on a multichannel carbon nanofiber (FeN4–FeNCP@MCF).74 This configuration led to the development of a highly efficient and robust ORR catalyst. The exceptional ORR activity and stability demonstrated by the synthesized FeN4–FeNCP@MCF catalyst could be ascribed to the synergistic effects between the FeN4 sites and Fe nanoclusters/nanoparticles, along with the high-porosity structure of the carbon substrate. Indeed, employing the size-asymmetric coupling strategy to integrate individual atoms and clusters/nanoparticles within a unified system affords a promising avenue for achieving synergistic coupling. This approach optimizes the electronic structure and enriches oxygen catalytic sites, thus enhancing catalytic performance.
3.4 Multiple asymmetric coupling
Multiple asymmetric coupling refers to two or more asymmetric configurations within material structures, offering a versatile approach for fabricating highly efficient electrocatalytic materials.176 The recent development described in Fig. 14 shows the performance summary of this strategy in catalyst design, as evidenced by its enhanced performance. The utilization of this multiple asymmetric coupling can enhance both the activity and stability of catalysts. By applying this method to the design of bifunctional catalysts, active sites with different compositions and catalytic functionalities can be initially coupled. Moreover, introducing concepts of size or coordination asymmetry can further optimize the local electronic structure of active metals, thereby promoting charge delocalization. This optimization provides operational space for accelerating the kinetics of electrocatalytic reactions. | ||
| Fig. 14 Performance summary of catalysts designed using multiple asymmetric coupling strategies. The chart categorizes catalysts based on combinations of composition, size, and coordination asymmetries, illustrating their electrocatalytic performance. The metrics include half-wave potential (E1/2) for the ORR in 0.1 M KOH and overpotentials (η10) for the OER and the HER at 10 mA cm−2 in 1 M KOH, highlighting the enhanced activity achieved through strategic coupling approaches. This diagram provides a summary of the performance metrics reported in ref. 49, 73, 76 and 176–190. | ||
For instance, a dual-metal NiFe–N–C SA catalyst coupling with Fe nanoclusters was prepared by simply pyrolyzing metal phthalocyanine and NC precursors (Fig. 15a and b).177 This catalyst demonstrated size asymmetry, featuring Fe nanoclusters and Fe SAs and heteroatomic coupling involving Ni SAs. As a result, NiFe–N–C delivered notable ORR/OER catalytic activity and durability (Fig. 15c and d). After 50000 and 90000 potential cycles, minimal ΔE was merely 0.680 V, with negligible degradation observed for E1/2 and Ej10. In-depth characterization and DFT calculations suggest the presence of Ni–N4 and Fe–N4 coordination structures, along with electron synergistic effects from adjacent Fe nanoclusters (Fig. 15e). The key active sites were Fe–N4, Ni–N4, and Fe nanoclusters, with Fe4 at the Ni–N4 site exhibiting the lowest overpotential. The synergy between Fe–N4 and Ni–N4 optimized the electronic structure, improving oxygen intermediate adsorption/desorption and enhancing OER/ORR activity. Fe nanoclusters further accelerated reaction kinetics, while spin-state modulation refined the reaction pathway. This synergy efficiently optimized oxygen intermediate formation and dissociation, boosting catalytic efficiency. Employing this dual asymmetry in the component and size, analogous electrocatalytic materials, such as Fe2O3/FeSA@NC, NiSAFeSA–Ni50Fe@carbon nanotube, and WSA–W3N4@NC, have been successively synthesized and applied in electrolysis for water electrolysis and Zn–air batteries.178,191,192 Similarly, the simultaneous exploitation of asymmetry in both component and coordination emerges as a viable strategy for developing robust electrocatalysts. For example, a two-dimensional metal–organic framework (MOF) nanosheet containing Fe and Cu components was initially prepared, followed by calcination to produce a distinctive asymmetric FeN3O–CuN4 dual-atomic coordination configuration (Fig. 15f and g).179 Introducing non-metallic oxygen atoms into the carbon substrate disrupted the electronic/geometric symmetry of M–N4, inducing charge distribution polarization. This enabled the adjustment of the d-band centers of the catalyst, thereby lowering the binding energy of the ORR intermediates. Cu–N4 disrupted the localization of Fe-3d orbital electrons, speeding up electron transfer and optimizing the adsorption of OH* intermediates, thus promoting the ORR process. Similar operative mechanisms have also been validated at asymmetric Cu–N–S/Co–N–S and Fe–B–Co/NC sites.180,181
 | ||
| Fig. 15 Examples of multiple asymmetric coupling catalysts. NiFe–N–C catalyst: (a) preparation strategy and (b) HAADF-STEM images (the inset in (b) shows the interplanar spacing of the metal nanoclusters); (c) ORR polarization curves and (d) OER polarization curves of various catalysts; and (e) Gibbs free energy diagrams of the ORR on Fe4@Fe–N4, Ni4@Ni–N4 and Fe4@Ni/Fe–N4 (Fe4 on the Ni–N4 site). Reproduced with permission from ref. 177. Copyright 2023, Royal Society of Chemistry. FeCu–NC catalyst: (f) synthetic procedure and (g) HAADF-STEM image highlighting SAs (green dashed circles) and dual atoms (red dashed circles). Reproduced with permission from ref. 179. Copyright 2023, John Wiley and Sons. FeN3P1–Fe2P@NC catalyst: (h) schematic of the preparation and (i) HAADF-STEM image; (j) ORR polarization curves of various catalysts and (k) free energy diagram for Feac–FeN4, Fe2Pac–FeN4, FeN3P1, and Fe2Pac–FeN3P1 at 1.23 V. Reproduced with permission from ref. 182. Copyright 2023, John Wiley and Sons. | ||
Using the synergistic coordination of asymmetry- and size-asymmetric coupling, the local microenvironment of catalysts can also be precisely modulated in two dimensions. For example, an atomic catalyst with adjustable coordination environments, featuring single copper sites (CuN4 or CuS1N3) and adaptable copper clusters (Cux), was developed for efficient electrocatalytic ORR.73 Theoretical analyses indicated that the asymmetric coordination environment and charge redistribution induced by Cux effectively enhanced the *OOH intermediate adsorption. The regulation of intermediate adsorption rendered CuS1N3/Cux great in the ORR process compared to catalysts lacking S atoms or Cux. The adsorption energy of *OOH on the single Cu site, regulated by the coordination environment and interface interactions, exhibited a linear relationship with the d-band center. This suggests that intermediate adsorption strength and d-band center are robust indicators of catalytic performance. This study provides a comprehensive regulatory strategy for electron configuration and intermediate adsorption behavior, potentially applicable to other proton-coupled electron transfer reactions.
The multiple asymmetric coupling strategy was recently employed to fabricate a FeN3P1–Fe2P@NC nanohybrid.182 This material comprised Fe2P nanoclusters, nanoparticles, and an NC substrate for accommodating the asymmetric N, P coordination of FeSA centers, thus exhibiting a tripartite asymmetric coupling structure in component, size, and coordination environment (Fig. 15h and i). Owing to its unique asymmetric electronic/geometric configuration, the synthesized catalyst demonstrated outstanding ORR activity, with decent E1/2 values of 0.76 V (0.5 M H2SO4) and 0.90 V (0.1 M KOH) and great performance in Zn–air batteries. Theoretical analysis showed that the synergistic interaction between P-coordination and Fe2P nanoclusters lowered the adsorption energy and overpotential of *O hydrogenation, optimized OH adsorption, and facilitated the reaction. Furthermore, the introduction of Fe2P induced electron redistribution, breaking the localized electronic structure of the Fe sites, thereby accelerating electron transfer and improving reaction kinetics (Fig. 15j and k). Therefore, employing a multiple asymmetric coupling strategy offers convenience and various dimensions for effectively modulating the phase, component, structure, morphology, and defects of catalysts. This approach provides advantages in developing high-performance and stable electrode materials.
4. Enhancement mechanism of performance in the asymmetric coupling
4.1 Charge transfer
Charge transfer involves the redistribution and directed charge transport between a “donor” and an “acceptor”. A prevalent strategy in electrocatalysis is optimizing components and structure, facilitating electron transfer to promote catalytic activity and stability. Electrocatalysts characterized by compound structures incorporating heterojunctions may exhibit behavior associated with charge transfer, thus boosting catalytic activity.193 Constructing heterojunctions is an efficient method for modifying the electron structure to modulate catalytic activity, employing the inherent diversity in electron structures among individual components within the composite catalyst.194 A compositionally asymmetric W2N/WC heterogeneous catalyst was prepared through simultaneous nitridation and carbonization processes (Fig. 16a).195 The resulting W2N/WC catalyst demonstrated highly efficient and durable catalytic ability for the ORR, OER, and HER. Notably, the ORR half-wave potential reached 0.810 V, with OER overpotential at a mere 0.320 V, and HER overpotential at only 0.149 V. When employed as the air electrode in Zn–air batteries, the W2N/WC catalyst enabled the battery to achieve a high-power density of 172 mW cm−2. DFT calculations elucidated that the well-regulated W2N/WC heterointerface facilitated the charge transfer and separation (Fig. 16b and c), enhancing ORR, OER, and HER activities. | ||
| Fig. 16 Four typical examples of asymmetric coupling catalysts regulated by charge transfer. W2N/WC catalyst: (a) HRTEM image; (b) density of states and (c) electronic local function of the (010) surface of the W2N (111)/WC (100) heterostructure. Reproduced with permission from ref. 195. Copyright 2019, John Wiley and Sons. Fe-ACSA@NC catalyst: (d) synthesis procedure, (e) X-ray photoelectron spectroscopy (XPS) of C 1s, (f) normalized Fe K-edge XANES, and (g) free energy diagrams of the ORR. Reproduced with permission from ref. 172. Copyright 2022, John Wiley and Sons. Co4/Fe1@NC catalyst: (h) HAADF-STEM image with active sites marked, (i) ORR polarization curves, and (j) Gibbs free energy diagrams. Reproduced with permission from ref. 196. Copyright 2023, John Wiley and Sons. S–Cu–ISA/SNC catalyst: (k) SEM image showing the hierarchical morphology, (l) polarization curves for the ORR, and (m) free-energy diagrams for different Cu-centered moieties. Reproduced with permission from ref. 197. Copyright 2020, Springer Nature. | ||
In recent times, extensive research has focused on the asymmetric coupling of heterogeneous components. These instances exploited variations in physicochemical properties, including electronegativity and WF, to prompt interface charge redistribution, effectively influencing electrochemical performance. Relevant examples include Er2O3-Co, Mo5N6-MoS2, Co/CoxMy (M = P, N), Ni/CeO2, and other similar cases.113,117,198,199 Utilizing the coupling of homogeneous metal SAs and clusters provides an avenue for achieving interface charge transfer. For example, a multi-step synthesis strategy involving coating, thermal decomposition, and etching was designed to prepare Fe cluster-modified Fe–N–C atomic centers characterized by a distinctive size-asymmetric feature (Fig. 16d).172 The interaction between Fe–N–C and the Fe clusters induced charge redistribution, with electrons from the Fe center transferring to the adjacent carbon substrate. This increased the positive charge density on the Fe center, weakening the adsorption strength of *OH (Fig. 16e and f). As a result, the desorption energy barrier of the *OH intermediate on the Fe active sites was lowered, accelerating ORR kinetics. Additionally, the presence of both atomic Fe–N–C and metallic Fe clusters caused elongation of the Fe–N bond, altering the coordination environment of Fe–N–C. This structural adjustment facilitated electron transfer (Fig. 16g), promoting the 4-electron transfer pathway and enhancing overall catalytic efficiency.
Recent investigations have broadened the scope of interaction dynamics between SAs and clusters, including homonuclear and heteronuclear systems. By employing a multi-asymmetric strategy, Co atomic clusters were strategically incorporated to finely tune the electronic structure of Fe–N4, enhancing ORR activity (Fig. 16h and i).196 These findings revealed that, in the Co4/Fe1@NC and Fe4/Fe1@NC systems, the O–O bond stretching was more pronounced than in the FeN4 system, facilitating O2 dissociation at the FeN4 active sites. This improvement was largely driven by electron transfer induced by the coupling of Co4 clusters with FeN4 sites, which redistributed charge and altered the electronic structure of the FeN4 sites, ultimately enhancing their catalytic activity. Specifically, the downward shift of the d-band center of the Fe 3d orbitals in Co4/Fe1@NC compared to Fe1@NC reduced the binding strength of *O, promoting easier desorption and accelerating the ORR process. Additionally, the conversion from O* to OH* was identified as the rate-determining step, where the Co4/Fe1@NC system exhibited a smaller free energy change than Fe4/Fe1@NC and Fe1@NC, indicating that Co4/Fe1@NC effectively weakened O adsorption, optimizing the ORR pathway (Fig. 16j). Interestingly, a CuSA ORR catalyst comprised of asymmetric Cu–S1N3 coordination moieties anchored within a hierarchically porous carbon framework was fabricated through atomic interface engineering.197 In this configuration, copper is directly coordinated with sulfur and nitrogen atoms, designated as S–Cu-ISA/SNC (Fig. 16k). Due to the rational construction of active sites, the S–Cu-ISA/SNC sample exhibited notable ORR performance in alkaline media (Fig. 16l). Both experimental and theoretical analyses revealed that the introduction of sulfur altered the electronic structure of Cu atoms in the Cu–S1N3 site, particularly increasing electron occupancy in the dx2−y2 orbitals. After O* adsorption, the Cu–S1N3 site exhibited stronger π-bonding contributions, which enhanced the weak binding of ORR intermediates, thus improving the catalytic performance of the Cu center (Fig. 16m). This localized structural regulation strategy shows potential for catalyzing advanced O-containing reactions and other electrochemical processes.
4.2 Strain engineering
Strain effects are induced by altering atom-atom bond lengths or inducing lattice mismatches, resulting in diverse electronic structures and modified adsorption capabilities towards reactants, intermediates, and products.200–202 This tunability facilitates the ability of the strain-rich electrocatalysts to catalyze the HER, ORR, and OER processes.203–208 For instance, a heterostructure involving NiFe layered double hydroxides (LDH) and Co1−xS in a sandwich-like configuration was fabricated (Fig. 17a–d). This design showed synergistic catalytic effects from compositionally asymmetric coupling, leading to efficient OER performance.209 The surface lattice mismatch within NiFe LDH induced substantial strain interfaces in the electrodeposited Co1−xS, rearranging local electron structures. This restructuring enhanced charge transfer efficiency and the chemisorption of intermediates containing oxygen (Fig. 17e). In another investigation, a facile hydrothermal method was introduced for synthesizing asymmetric heterostructure nanomaterials involving Co(OH)2 and Ni(OH)2, effectively activating the basal plane of Co(OH)2.210 The heterogeneous interfaces introduced lattice strains, leading to a downward shift in the d-band center of the Co site within the Co(OH)2 structure. This facilitated the efficient desorption of oxygen during the OER process, accelerating reaction kinetics and improving catalytic activity. | ||
| Fig. 17 Examples of asymmetric coupling catalysts regulated by strain engineering. NiFe LDH/Co1−xS catalyst: (a) schematic representation of the synthesis procedure, (b and c) SEM images depicting the morphology, (d) TEM image showing the interface between NiFe LDH and Co1−xS, and (e) free energy diagrams comparing the catalytic pathways for different active sites. Reproduced with permission from ref. 209. Copyright 2020, Elsevier. FePc-{PW12}@nanotube catalyst: (f) schematic illustration of the preparation steps, (g) HAADF-STEM image revealing structural details, and (h) the structure of the nanohybrids; (i) ORR polarization curves of various catalysts and (j) free energy diagrams of ORR pathways. Reproduced with permission from ref. 211. Copyright 2023, John Wiley and Sons. | ||
Recently, a host–guest chemical approach was also employed to anchor FePc onto carbon nanotubes incorporating polyoxometalate, successfully fabricating an asymmetrical heterostructure designed for the ORR electrocatalyst (FePc-{PW12}@nanotube).211 The result unveiled that polyoxometalate induced localized tensile strain in single-walled nanotubes, strengthening their interaction with FePc (Fig. 17f–h). Thus, the strain and curvature effects stemming from the {PW12}@nanotube substrate adeptly regulated the geometric configuration of the Fe–N4 center and the dynamics of local electrons, ultimately enhancing the ORR catalytic performance (Fig. 17i and j). As expected, this heterostructure exhibited exceptional durability, methanol tolerance, and ORR activity in an alkaline environment. Thus, these findings contribute to a comprehensive understanding of the strain-adsorption-activity relationships and offer novel insights into the rational design of multifunctional catalysts tailored for various energy conversion applications.
4.3 Spin-state regulation
The importance of spin states or magnetic moments in catalysis is increasingly evident, especially given the prevalence of active sites on surfaces, edges, or defects where unsaturated bonding can induce local spins and magnetic moments.212 Transition metals with local magnetic moments can enhance electrocatalytic performances. These phenomena have been observed across various reactions, including the HER, CO2 reduction, N2 reduction, and other catalytic processes. However, the most extensively studied reactions involve O2, specifically the OER and ORR.213 Transition metal oxides and hydroxides like FexNi1−xOOH, FeOOH, and NiOOH are prominent examples that improve their efficacy in the OER process.214–216 Additionally, intermediates in catalytic reactions may be associated with spin states.217 For instance, the O2 molecule exhibits a ground paramagnetic triplet state, while oxygenated compounds and intermediates often assume a singlet state during the OER process.218 Given the spin selection rule, spin-involved electron transfer plays a crucial role in reaction kinetics, particularly in the presence of the O2 molecule.219 Manipulating spin arrangement and magnetic moments is beneficial for reducing energy barriers or overcoming spin suppression, known as spin catalysis, which has been widely recognized in various reactions. Notably, spin-related catalysis has garnered increased attention for influencing adsorption, reactant and intermediate formation, rate-determining steps, electron transfer, and catalytic activity. Hence, catalyst design should prioritize guiding spin-selective regulation to enhance catalytic performance.For example, a spin state regulation strategy was proposed by introducing axial Fe–O–Ti ligands to enhance the ORR activity of coordinatively asymmetric Fe centers (Fig. 18a and b).220 Experimental and theoretical calculations indicated that Fe–O–Ti ligands induced a transition of the Fe center from a low-spin to a medium-spin state. This transition facilitated the appropriate filling of the eg orbitals of the Fe center, optimizing the O2 adsorption energy, which promoted the activation of oxygen molecules and enhanced the ORR activity (Fig. 18c–e). An asymmetric structure of Fe–Mn/NC bimetal SACs was also recently investigated.221 The proximity of atomically dispersed Mn–N sites effectively activated FeIII centers within the FeN4 moiety, leading to single-electron filling (t2g4eg1) and promoting electron transfer into the antibonding orbitals of oxygen (Fig. 18f–i). Both magnetization measurements and theoretical calculations revealed that these adjacent Mn–N species induced spin-state transitions and modulated the electronic structure of FeIII, resulting in ORR activity and stability under both alkaline and acidic conditions. Furthermore, when O2 was adsorbed by Fe/Mn atom pairs, the optimized bond lengths and binding energies reduced the dissociation energy barrier. This configuration facilitated the capture of oxygen-containing intermediates and rapidly cleaved M–OH bonds, ensuring the regeneration of *O and *OH. As a result, it accelerated ORR kinetics by suppressing the formation of peroxides.
 | ||
| Fig. 18 Examples of asymmetric coupling catalysts regulated by spin-state. O-MQFe catalyst: (a) schematic of the synthesis process and (b) HRTEM image showing active sites; (c) 57Fe Mössbauer spectroscopy highlighting spin states, (d) 1/χm plots revealing spin configurations, and (e) O2-temperature-programmed desorption curves. Reproduced with permission from ref. 220. Copyright 2022, John Wiley and Sons. Fe–Mn/NC catalyst: (f) synthesis procedure, (g) room-temperature 57Fe Mössbauer spectrum showing distinct components; (h) magnetic susceptibility of Fe–Mn/NC indicating high-spin Fe3+ states with μeff = 3.75μB; and (i) low-spin states in Fe/N–C with μeff = 2.16μB. Reproduced with permission from ref. 221. Copyright 2021, Springer Nature. | ||
In another work, the incorporation of single Cu atoms onto Fe–N–C aerogels in asymmetrically assembled binuclear sites resulted in reduced magnetic moments and enhanced ORR activity of the Fe center within a broad pH range of 0 to 14.222 Specifically, Fe atoms located at the nanopore edges contributed more electron density compared to those on the basal plane, thereby increasing their catalytic activity. Additionally, in the Cu1/Fe1-2 system, the electronic states of the Cu atoms were close to the Fermi level, providing more active electrons and promoting the reduction reaction. Through redistribution of electron density, the dz2 orbital electron density of Fe increased, reducing its magnetic moment and further optimizing the ORR reaction pathway, particularly by weakening the adsorption strength of *O and lowering the reaction energy barrier. It was also observed that the spin state of single atomic tungsten induced spin delocalization in the heterogeneous W-NiS0.5Se0.5 structure, resulting in the spin-state delocalization of Ni.223 This effect increased the electron density in Ni d-orbitals, effectively facilitating the Heyrovsky step and promoting the formation of *OOH intermediates during the OER process, thereby enhancing the overall catalytic performance of NiS0.5Se0.5. Specifically, at the Ni active sites, *OH was initially generated, followed by its conversion into *OOH, which was subsequently released as O2 through a proton-coupled electron transfer process. The incorporation of single-atomic W accelerated this reaction sequence, markedly improving OER kinetics.
4.4 Tandem catalytic effect
Electrocatalytic reactions often include a series of reaction steps and intermediates. However, the linear scaling relationships that govern adsorption energy between intermediates present a difficulty in independently regulating adsorption energy for specific steps, particularly for single-type sites, thus hindering the achievement of optimal catalytic effects.224 Conversely, introducing asymmetrically coupled multi-active sites is expected to facilitate the independent optimization of the conversion of specific reaction intermediates. This structural arrangement reduces the overall reaction process through a synergistic catalytic effect. Tandem catalysis involves a continuous process in which reaction intermediates undergo conversion at one active site, followed by diffusion to another nearby active site, initiating subsequent reactions and collaboratively completing the entire catalytic process.60 Therefore, the tandem catalytic effect has the potential to expedite the whole reaction by sequentially cascading multiple reaction steps across various active sites. The elastic structure of asymmetrically coupled catalysts facilitates the development of tandem catalysis tailored for complex electrocatalytic reactions.225For example, an efficient electrocatalyst with asymmetrically coupled dual active sites containing Fe–NiOOH and NiC4 single atoms (Ni-SAs/Fe–NiOOH) was prepared (Fig. 19a–c).226 Experimental results and DFT calculations indicated that the collaborative action of NiC4-SAs and Fe–NiOOH dual active sites endowed the Ni-SAs/Fe–NiOOH catalyst with outstanding OER activity (Fig. 19d–f). Further investigations revealed that the initial formation of *OH and *O occurs on Fe–NiOOH, followed by the migration of *O to Ni–C4 SAs, leading to the subsequent formation of *OOH and O2. In the tandem catalytic mechanism, the potential-determining step from *OH to *O on Fe–NiOOH (Fe) required a potential of 0.110 V. In contrast, the potential-determining step from *O to *OOH on Ni–C4 SAs necessitated a potential of 0.150 V. Consequently, the tandem catalysis between the two active sites efficiently optimized the potential-determining steps, substantially lowering the electrocatalytic overpotential for the OER process. This study provides fundamental insights into the rational design of OER catalysts and underscores the advantages of asymmetrically coupled active sites in enhancing catalytic performance. Following these findings, an innovative catalyst named Fe, Cu/N–C, featuring the asymmetric coupling of atomically dispersed Fe and Cu, was reported recently (Fig. 19g–i).227 Using the advantages of electron spin-state regulation and tandem catalysis mechanisms, the ORR activity of the resulting catalyst was improved. Theoretical calculations and detailed analysis revealed that the transformation of O2 molecules into *OOH and *O intermediates primarily occurred at the Fe center within the Fe, Cu/N–C catalyst due to the more favorable reaction barriers at the Fe sites in the initial two steps compared to those at the Cu sites. Next, owing to the reduced barrier of the Cu site, *OH species generated in the third step systematically migrated from the Fe domain to the Cu region, following an expected tandem catalysis pathway. The stepwise procedure rapidly hastened the breakdown of the metal–OH linkage. As a result, the simultaneous presence of dual active sites within Fe, Cu/N–C overcame the limitations of the negative scaling relationship in O2 reduction. This contribution paves an intriguing path for overcoming the scale-related constraints of multi-stage reactions and unveils an avenue for developing advanced catalysts.
 | ||
| Fig. 19 Examples of asymmetric coupling catalysts regulated by the tandem catalytic effect. Ni–SAs/Fe–NiOOH catalyst: (a and b) HRTEM and (c) HAADF-STEM images showing the structural features and active sites; (d) reaction-free energies illustrating the catalytic pathway for Ni–C4 SAs and Fe–NiOOH; (e) tandem catalysis mechanism depicting the interaction of Ni SAs and Fe–NiOOH during the OER; and (f) schematic diagram of the OER highlighting oxygen migration as a key step. Reproduced with permission from ref. 226. Copyright 2021, Elsevier. Fe, Cu/N–C catalyst: (g) HAADF-STEM image displaying the catalyst morphology; (h) ORR free energy diagrams for Fe and Cu active sites; and (i) schematic illustrating the tandem reaction mechanism for ORR on Fe, Cu/N–C. Reproduced with permission from ref. 227. Copyright 2023, Elsevier. | ||
4.5 Vacancy defect regulation
Incorporating diverse defects spanning multiple dimensions has been employed to create highly active sites driven by the unordered atomic arrangement, distinct electron distribution, and energy fluctuations.228 These voids can be classified into three types based on the charge polarity of initially existing ions: anionic, cationic, and mixed voids.229 Due to the greater atomic mass and stronger chemical bonding of cations, forming cationic voids requires highly aggressive physical bombardment or chemical etching treatment, posing challenges in obtaining high-purity mono-typic voids.230 Conversely, constructing a series of anionic voids is more feasible and prevalent due to the generally lighter atomic mass and looser coordination structure of anions. Among these, voids involving O, C, and S are the most adopted group of anionic voids.231As a typical practice, a CoP@CoOOH/CP electrocatalyst was synthesized in situ on carbon paper (CP) through a hydrothermal process followed by phosphorization and oxidation (Fig. 20a).232 The CoOOH shell, rich in oxygen vacancies, could actively modulate surface–oxygen interactions, thereby exposing more active sites and reducing the energy barrier for the rate-determining step. This structural optimization enhanced OER activity and accelerated O2 evolution (Fig. 20b–d). The introduction of oxygen vacancies facilitated an increase in the concentration of Co3+ species with lower coordination numbers, further boosting the catalytic activity for the OER. As an illustrative example, a defect engineering strategy was developed to introduce carbon vacancies near Fe–N4 (Fe–N4VC), disrupting the localized electronic structural symmetry of Fe–N4 (Fig. 20e).233 This enhanced the intrinsic activity of Fe–N4 and reduced the reaction energy barrier (Fig. 20f–g). Similarly, a universal sub-nanoreactor strategy was designed to synthesize a yolk–shell nanostructure characterized by a distinctive asymmetric coupling of MoS2 support and metal SA sites (Fig. 20h).234 Co SA was coordinated with C and Mo atoms in this catalytic system, establishing a dual-anchored coordination microenvironment in the C–Co–MoS2 composite (Fig. 20i–k). These findings (Fig. 20l) indicated that the optimized C–Co–MoS2 exhibited the lowest overpotential (η10 = 0.017 V) for the HER. DFT calculations demonstrated that metal SAs could stabilize the dual carrier system with the effective electron lock at sulfur vacancies and induce tunable electron transfer at the C–M–Mo interface. The presence of sulfur vacancies and intercalated carbon, serving as dual carriers, facilitated the formation of electron lock and directed electron transfer channel, promoting the stabilization and activation of metal SAs. Therefore, this research strategy provides a new perspective for designing efficient electrocatalysts.
 | ||
| Fig. 20 Examples of asymmetric coupling catalysts regulated by vacancy defects. CoP@CoOOH/CP catalyst: (a) schematic of the synthesis strategy involving core–shell design with oxygen vacancies, (b) O 1s XPS spectra showing oxygen-related surface states, and (c) electron paramagnetic resonance (EPR) spectra highlighting electronic structure changes; (d) mechanism diagram of OER catalysis facilitated by oxygen vacancies. Reproduced with permission from ref. 232. Copyright 2022, John Wiley and Sons. Fe–N4VC catalyst: (e) schematic of the synthesis route for generating vacancy-enriched Fe–N–C structures and (f) the N peak analysis of Fe–N–C-2 in EELS and its corresponding FeN bond; (g) ORR free energy diagrams. Reproduced with permission from ref. 233. Copyright 2023, John Wiley and Sons. C–Co–MoS2 catalyst: (h) illustration for the sub-nanoreactor synthesis strategy; (i) TEM image, (j) Raman profiles, (k) EPR data, and (l) HER polarization curves of various catalysts. Reproduced with permission from ref. 234. Copyright 2023, John Wiley and Sons. | ||
4.6 Oxophilicity regulation
Previous investigations have underscored the pivotal role played by the adsorption strength of reactants and intermediates in the catalyst surface in determining catalytic activity. The recent focus on regulating oxygen affinity has proven advantageous in optimizing the adsorption configuration of intermediates and accelerating the kinetics of chemical reactions.235 As an illustration, an efficient three-dimensional nanostructured comprising Cr-doped Ni nanohelixes was fabricated. Notably, incorporating oxygen-affinitive chromium disrupted the structural symmetry, optimizing the H and OH binding energies on nickel nanohelixes.236 This alteration accelerated the processes of water dissociation and adsorption, consequently enhancing the overall kinetics of the HER (Fig. 21a and b). By introducing the element Sn, which is characterized by lower oxygen affinity and weaker hydrogen adsorption capability compared to Ni, an asymmetric Ni3Sn2–NiSnOx nanocomposite was prepared (Fig. 21c).237 The optimized Ni3Sn2–NiSnOx composite exhibited overpotentials of 0.014 and 0.165 V at 10 and 1000 mA cm−2 in 1 M KOH, respectively, along with long-term stability. Computational analyses indicated that the intermetallic compound Ni3Sn2 component exhibited appropriate hydrogen binding energy and low OH adsorption capacity, while the NiSnOx component facilitated the water dissociation and OH transfer processes (Fig. 21d and e). The interaction between these components resulted in effective dissociation adsorption of water, facile OH detachment, and hydrogen bond formation, enhancing the HER kinetics. In another work, asymmetrically coupled Co SAs and Co nanoparticles into N-doped carbon nanotubes were developed (Co@NCNT/Co-SA@NCMT, Fig. 21f–h).238 Experimental and theoretical calculations indicated that Co SAs could catalyze oxygen reduction to H2O2, while the oxygen-affine Co nanoparticles exhibited enhanced efficiency in further reducing it to H2O (Fig. 21i–k). Thus, regulating oxygen affinity emerges as a feasible approach to boost activity and stability, paving the way for developing advanced hydrogen/oxygen electrocatalysts. | ||
| Fig. 21 Examples of asymmetric coupling catalysts regulated by oxophilicity. Cr-doped Ni nanohelix catalyst: (a) Schematic illustration showing reaction paths in alkaline HER; (b) theoretical calculation of the HER energy profile on Ni (111) and Cr–Ni (111). Reproduced with permission from ref. 236. Copyright 2021, American Chemical Society. Ni3Sn2–NiSnOx nanocomposite catalyst: (c) HRTEM images highlighting structural features, (d) charge density difference visualizing electron interactions, and (e) energy profiles for alkaline HER showing the role of oxophilic sites. Reproduced with permission from ref. 237. Copyright 2023, John Wiley and Sons. Co@NCNT/Co-SA@NCMT catalyst: (f) TEM image illustrating the hierarchical structure, (g) HAADF-STEM image showing Co single atoms; (h) energy dispersive X-ray spectroscopy (EDS) mapping demonstrating elemental distributions; (i) EXAFS spectra revealing Co coordination environments; (j) density of state showing electronic interactions; and (k) free energy diagrams of the OER for different catalysts. Reproduced with permission from ref. 238. Copyright 2022, Elsevier. | ||
5. Conclusions and perspectives
5.1 Conclusions
The urgent demand for green, low-carbon, and highly efficient energy conversion technologies highlights the necessity for low-cost, high-efficiency energy conversion materials. This review systematically categorizes and summarizes the component and structural characteristics of ATEs. Additionally, it provides a detailed analysis of their electrocatalytic activity regulation mechanisms in the ORR, OER, and HER, including charge transfer, strain engineering, spin-state regulation, tandem catalytic effects, vacancy defect modulation, and oxophilicity regulation. Design principles, unique advantages, and drawbacks are depicted to deepen the understanding of related modulation methods (Fig. 22). Although current asymmetric coupling designs have exhibited considerable advantages and reached considerable milestones, this review acknowledges the remaining challenges. Addressing these challenges requires the integration of advanced synthesis methods, operando probes, and machine learning-driven approaches to accelerate the development of next-generation electrocatalysts.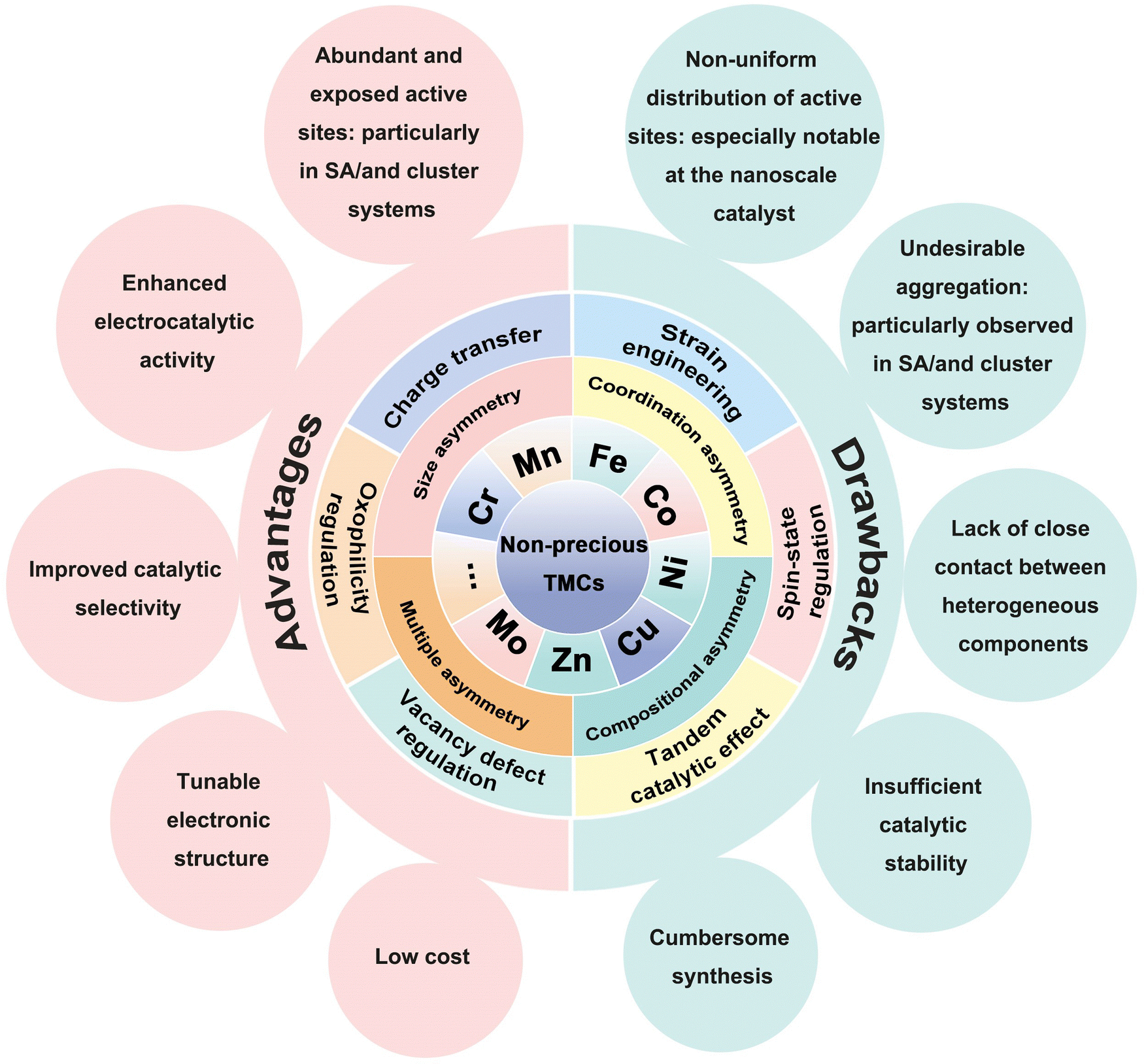 | ||
| Fig. 22 The advantages and drawbacks of asymmetrically tailored non-precious TMCs for hydrogen and oxygen electrocatalysis. This schematic summarizes the benefits and limitations of using asymmetrically engineered TMCs. Key advantages include enhanced electrocatalytic activity, improved selectivity, tunable electronic structure, abundant active sites, and low cost. However, challenges persist, such as non-uniform active site distribution, undesirable aggregation, lack of close contact between heterogeneous components, insufficient stability, and cumbersome synthesis processes. These insights highlight both the opportunities and hurdles in advancing TMC-based electrocatalysis. | ||
5.2 Challenges and perspectives
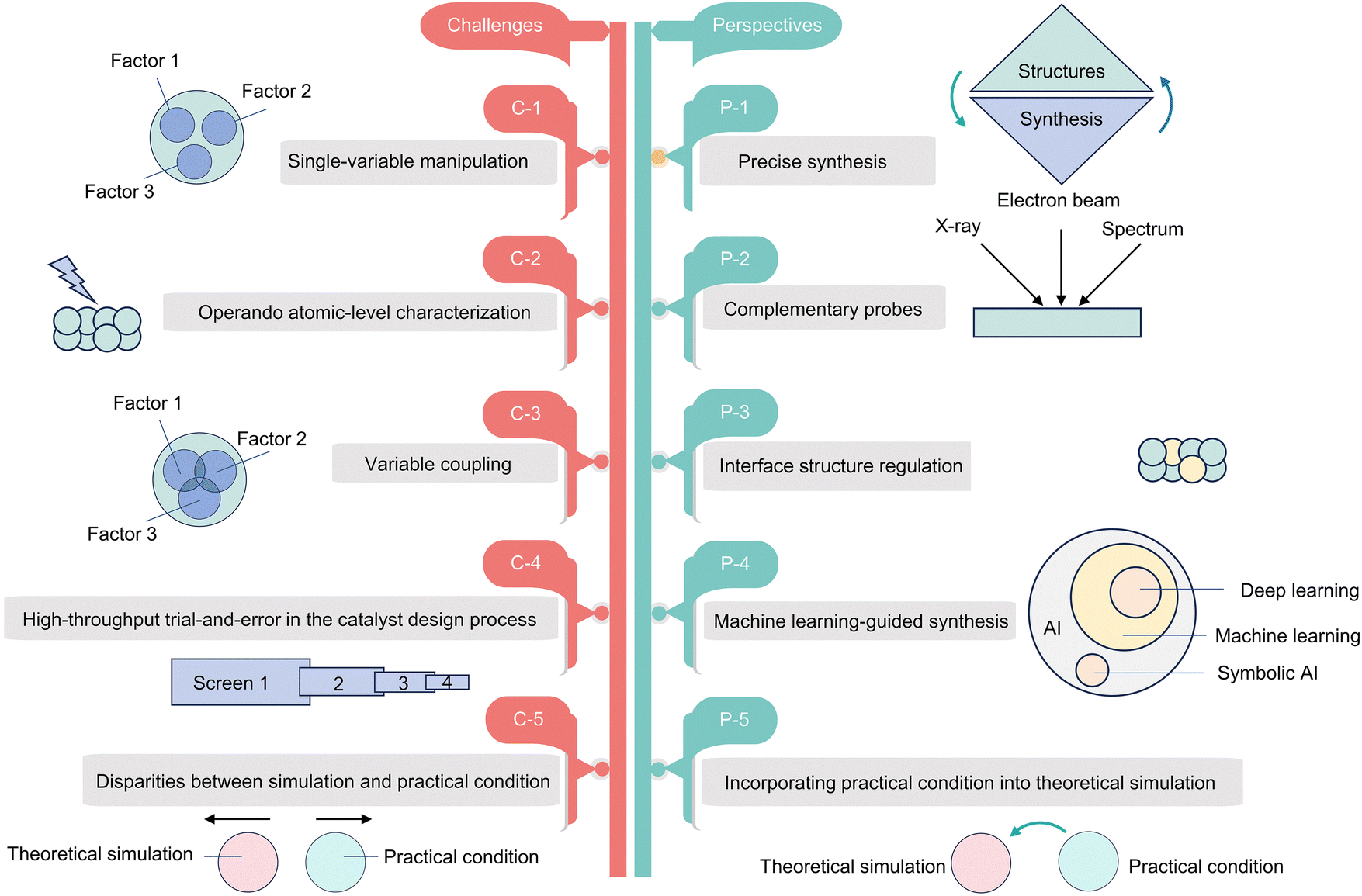 | ||
| Fig. 23 Challenges and future strategies in the development of ATEs. This illustration outlines the key challenges (C-1 to C-5) and corresponding perspectives (P-1 to P-5) for advancing ATEs. Challenges include the complexity of single-variable manipulation (C-1), the need for operando atomic-level characterization (C-2), variability in coupling effects (C-3), reliance on high-throughput trial-and-error methods (C-4), and the gap between theoretical simulations and practical conditions (C-5). Suggested strategies involve precise synthesis (P-1), integrating complementary probes (P-2), regulating interface structures (P-3), leveraging machine learning-guided synthesis (P-4), and incorporating practical conditions into theoretical simulations (P-5). These strategies aim to drive innovation in catalyst design, efficiency, and sustainability. | ||
(i) When investigating coordination asymmetry using M–N4 and M–N3P1 models, achieving consistency in the number of metal center atoms between the two material models poses a challenge in experimental synthesis. Beyond that, ensuring that each metal atom is precisely coordinated with only one heteroatom presents a considerable obstacle.239,240
(ii) Exploring the mechanism of size asymmetry based on MAC–MSA, precise control of the same metal center density across different samples is challenging.171 Moreover, ensuring that each atom is exclusively coupled with adjacent clusters, thereby achieving a 100% formation of size-asymmetric active sites within the material structure, remains a hurdle.
(iii) In exploring multiple asymmetric strategies, the complex catalyst structures pose challenges in elucidating specific catalytic mechanisms. The accurate assessment of the distinct contributions of various effects to the activity and stability of the catalysts becomes difficult. The improved catalytic activity is often attributed to the synergistic catalysis effects, leaving the actual catalytic mechanism unclear. These challenges reflect technological limitations in achieving univariate control over components, size, and coordination, thus requiring the development of more sophisticated experimental techniques and novel design strategies to better understand and regulate the inherent catalytic mechanism in ATEs. Overcoming these obstacles will broaden the research landscape for the future development of asymmetric catalysis, propelling the field toward more excellent controllability and efficiency.
(i) Incorporating heteroatoms into the axial positions of metal centers within the M–N4 structure illustrates the potential to achieve axially asymmetric catalytic configurations.241 However, accurately verifying whether ligands bridge each axial position of the metal center presents a hurdle in characterizing electrocatalyst structures, necessitating the advancement of sophisticated and high-resolution instruments.
(ii) Exploring the correlations between strain, defects, oxygen affinity, and performance encounters challenges due to the absence of quantitative assessments. To overcome this, it is crucial to explore more precise research methods to enhance our comprehension of the practical impact of these factors on catalytic activity.
(iii) The investigation of charge-directed transfer phenomena underscores the necessity for advanced imaging techniques to visualize charge distribution. The conventional characterization methods include X-ray photoelectron spectroscopy, ultraviolet photoelectron spectroscopy, and theoretical modeling.242 Similarly, Mössbauer spectroscopy, magnetic susceptibility analysis, and theoretical calculations are predominant in elucidating spin regulation mechanisms.243 However, the absence of solid experimental evidence makes the proposed catalytic mechanisms less convincing.
(iv) Observing the dynamic changes in asymmetric structures during the catalytic process necessitates advanced in situ characterization techniques. This involves overcoming both the inherent challenges of conventional asymmetric structure characterization and the additional complexities posed by in situ experimental systems, including the introduction of gases, voltage variations, and catalyst structure evolution. Therefore, we suggest intensified cutting-edge research in this domain, especially on advancing instrument technology.244
Author contributions
The manuscript was written with contributions from all authors, who have all approved the final version.Data availability
No primary research results, software, or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the Outstanding Youth Scientific Research Project for Colleges and Universities of Anhui Province of China (2022AH020054), Anhui Provincial Natural Science Foundation (2208085Y06), National Natural Science Foundation of China (No. 21975001) and Open Research Fund Program of Anhui Province Key Laboratory of Specialty Polymers (Anhui University of Science & Technology, AHKLSP23-06). The Institut de Ciències Fotòniques (ICFO) acknowledges CEX2019-000910-S, Fundació Cellex, Fundació Mir-Puig, La Caixa Foundation and the European Union's Horizon 2023 research and innovation program under the Marie Sklodowska-Curie grant agreement No. 101150688. The authors would like to thank Dr Chunhui Zhou from the Analysis and Testing Center of Anhui University of Science and Technology for his valuable suggestions on revising the manuscript.References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- Z. G. Yang, J. L. Zhang, M. C. W. Kintner-Meyer, X. C. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- W. J. Li, S. Ghosh, N. Liu and B. Poudel, Joule, 2024, 8, 1274–1311 CrossRef CAS.
- S. Li, C. H. Zhou, J. S. Hu, A. J. Duan, C. M. Xu and X. L. Wang, J. Catal., 2023, 426, 153–161 CrossRef CAS.
- K. Liu, H. M. Li, M. H. Xie, P. F. Wang, Z. Y. Jin, Y. T. Liu, M. Zhou, P. P. Li and G. H. Yu, J. Am. Chem. Soc., 2024, 146, 7779–7790 CrossRef CAS PubMed.
- H. Y. Shi, L. Zhang, X. H. Huang, Q. Q. Kong, A. Abdukayum, Y. T. Zhou, G. Y. Cheng, S. S. Gao and G. Z. Hu, Small, 2025, 2409129 CrossRef CAS PubMed.
- Z. F. Ke, L. Zhang, Q. Liu, Q. L. Zhu, C. L. Liu and G. Z. Hu, Int. J. Hydrogen Energy, 2024, 49, 862–874 CrossRef CAS.
- X. Y. Zhang, S. Y. Liang, F. Li and Y. P. Bai, Chem. Eng. J., 2025, 503, 158603 CrossRef CAS.
- J. T. Ren, L. Chen, H. Y. Wang, W. W. Tian and Z. Y. Yuan, Energy Environ. Sci., 2024, 17, 49–113 RSC.
- K. M. Zhao, S. Q. Liu, Y. Y. Li, X. L. Wei, G. Y. Ye, W. W. Zhu, Y. K. Su, J. Wang, H. T. Liu, Z. He, Z. Y. Zhou and S. G. Sun, Adv. Energy Mater., 2022, 12, 2103588 CrossRef CAS.
- D. Bao, Q. Zhang, F. L. Meng, H. X. Zhong, M. M. Shi, Y. Zhang, J. M. Yan, Q. Jiang and X. B. Zhang, Adv. Mater., 2016, 29, 1604799 CrossRef PubMed.
- P. L. Deng, F. Yang, Z. T. Wang, S. H. Chen, Y. Z. Zhou, S. Zaman and B. Y. Xia, Angew. Chem., Int. Ed., 2020, 59, 10807–10813 CrossRef CAS PubMed.
- H. R. Zhang, H. J. Wang, X. Q. Cao, M. S. Chen, Y. L. Liu, Y. T. Zhou, M. Huang, L. Xia, Y. Wang, T. S. Li, D. D. Zheng, Y. S. Luo, S. J. Sun, X. Zhao and X. P. Sun, Adv. Mater., 2024, 36, 2312746 CrossRef CAS PubMed.
- Z. Y. Li, Z. Li, H. Y. Yao, Y. Wei and J. S. Hu, J. Colloid Interface Sci., 2024, 653, 857–866 CrossRef CAS PubMed.
- Y. Liu, L. Zhang, Q. L. Xu, S. J. Zhang, Y. T. Zhou and G. Z. Hu, Adv. Funct. Mater., 2025, 35, 2413134 CrossRef CAS.
- Y. Z. D. Tong, X. H. Huang, N. Liu, R. K. Zhao and G. Z. Hu, Desalination, 2025, 600, 118535 CrossRef CAS.
- Z. Y. Li, M. S. Chen, L. Zhang, R. Xing, J. S. Hu, X. H. Huang, C. H. Zhou, Y. T. Zhou, T. Wågberg and G. Z. Hu, J. Mater. Chem. A, 2023, 11, 2155–2167 RSC.
- Z. R. Wang, X. H. Huang, Y. Z. D. Tong, T. Wang, M. Xu, R. Li and G. Z. Hu, Chem. Eng. J., 2025, 504, 158899 CrossRef CAS.
- Y. T. Liu, W. X. Qiu, P. F. Wang, R. Li, K. Liu, K. M. Omer, Z. Y. Jin and P. P. Li, Appl. Catal., B, 2024, 340, 123228 CrossRef CAS.
- J. S. Hu, Q. L. Xu, X. Y. Wang, X. H. Huang, C. H. Zhou, Y. Ye, L. Zhang and H. Pang, Carbon Energy, 2023, 5, e315 CrossRef CAS.
- Q. L. Xu, L. Zhang, L. Li, S. J. Zhang, Y. T. Zhou and G. Z. Hu, Adv. Funct. Mater., 2025, 35, 2414379 CrossRef CAS.
- Y. Ye, L. Zhang, Q. L. Zhu, Z. A. Du, T. Wågberg and G. Z. Hu, J. Colloid Interface Sci., 2023, 650, 1350–1360 CrossRef CAS PubMed.
- J. W. Wu, Y. Z. D. Tong, X. H. Huang, D. Y. Qin, R. K. Zhao, R. Li and G. Z. Hu, Desalination, 2025, 600, 118479 CrossRef CAS.
- Y. Z. Li, T. T. Xu, Q. Huang, L. T. Zhu, Y. Y. Yan, P. Peng and F. F. Li, ACS Catal., 2023, 13, 7597–7605 CrossRef CAS.
- S. Y. Lin, X. Li, D. Y. Yuan, Y. T. Liu, Z. Y. Jin and P. P. Li, Small, 2024, 20, 2406235 CrossRef CAS PubMed.
- K. S. Sun, L. Zhang, S. J. Zhang and X. W. Li, J. Alloys Compd., 2025, 1011, 178419 CrossRef CAS.
- Q. R. Liang, W. Q. Li, L. Xie, Y. J. He, B. L. Qiu, H. Zeng, S. Zhou, J. Zeng, T. Y. Liu, M. Yan, K. Liang, O. Terasaki, L. Jiang and B. Kong, Nano Lett., 2022, 22, 2889–2897 CrossRef CAS PubMed.
- D. B. Liu, X. Y. Li, S. M. Chen, H. Yan, C. D. Wang, C. Q. Wu, Y. A. Haleem, S. Duan, J. L. Lu, B. H. Ge, P. M. Ajayan, Y. Luo, J. Jiang and L. Song, Nat. Energy, 2019, 4, 512–518 CrossRef CAS.
- R. L. Zhang, Y. Z. Li, X. Zhou, A. Yu, Q. Huang, T. T. Xu, L. T. Zhu, P. Peng, S. Y. Song, L. Echegoyen and F. F. Li, Nat. Commun., 2023, 14, 2460 CrossRef CAS PubMed.
- J. Hong, M. S. Chen, L. Zhang, L. Qin, J. S. Hu, X. H. Huang, C. H. Zhou, Y. T. Zhou, T. Wågberg and G. Z. Hu, Chem. Eng. J., 2023, 455, 140401 CrossRef CAS.
- Q. L. Xu, J. S. Hu, C. H. Zhou, L. Zhang, Q. Q. Kong, S. S. Gao and G. Z. Hu, Chem. Eng. J., 2024, 499, 156425 CrossRef CAS.
- P. P. Li, L. Liao, Z. W. Fang, G. H. Su, Z. Y. Jin and G. H. Yu, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2305489120 CrossRef PubMed.
- Q. Q. Cheng, S. B. Han, K. Mao, C. Chen, L. J. Yang, Z. Q. Zou, M. Gu, Z. Hu and H. Yang, Nano Energy, 2018, 52, 485–493 CrossRef CAS.
- M. B. Stevens, L. J. Enman, A. S. Batchellor, M. R. Cosby, A. E. Vise, C. D. M. Trang and S. W. Boettcher, Chem. Mater., 2016, 29, 120–140 CrossRef.
- Q. L. Xu, J. S. Hu, H. Y. Yao, J. Lei, C. H. Zhou, L. Zhang and H. Pang, ACS Appl. Mater. Interfaces, 2024, 16, 42352–42362 CrossRef CAS PubMed.
- Z. C. Nie, M. S. Chen, L. Zhang, Q. Feng, J. S. Hu, X. H. Huang, C. H. Zhou, Y. T. Zhou, T. Wågberg and G. Z. Hu, Chem. Eng. J., 2023, 463, 142411 CrossRef CAS.
- X. H. Lv, Z. X. Xiao, H. Y. Wang, X. L. Wang, L. L. Shan, F. L. Wang, C. Y. Wei, X. J. Tang and Y. L. Chen, J. Energy Chem., 2021, 54, 626–638 CrossRef CAS.
- H. J. Niu, Y. P. Chen, R. M. Sun, A. J. Wang, L. P. Mei, L. Zhang and J. J. Feng, J. Power Sources, 2020, 480, 229107 CrossRef CAS.
- J. Zhang, J. W. Chen, Y. Luo, Y. H. Chen, C. Y. Zhang, Y. J. Luo, Y. L. Xue, H. G. Liu, G. Wang and R. L. Wang, J. Energy Chem., 2021, 60, 503–511 CrossRef CAS.
- H. H. Xie, B. Du, X. X. Huang, D. H. Zeng, H. Meng, H. J. Lin, W. Li, T. Asefa and Y. Y. Meng, Small, 2023, 19, 2303214 CrossRef CAS PubMed.
- L. B. Zong, K. C. Fan, P. Li, F. H. Lu, B. Li and L. Wang, Adv. Energy Mater., 2023, 13, 2203611 CrossRef CAS.
- Y. Ye, L. Zhang, Z. C. Nie, N. P. Li, S. X. Zhou, H. S. Wang, T. Wågberg and G. Z. Hu, Chem. Eng. J., 2022, 446, 137210 CrossRef CAS.
- Z. Y. Li, L. Zhang, Q. L. Zhu, Z. F. Ke and G. Z. Hu, J. Colloid Interface Sci., 2024, 653, 1577–1587 CrossRef CAS PubMed.
- F. H. Lu, K. C. Fan, L. X. Cui, B. Li, Y. Yang, L. B. Zong and L. Wang, Appl. Catal., B, 2022, 313, 121464 CrossRef CAS.
- K. Srinivas, Z. Chen, F. Ma, A. R. Chen, Z. H. Zhang, Y. Wu, M. Q. Zhu and Y. F. Chen, Appl. Catal., B, 2023, 335, 122887 CrossRef CAS.
- Y. C. Wang, Q. C. Wang, J. Wu, X. Zhao, Y. Xiong, F. H. Luo and Y. P. Lei, Nano Energy, 2022, 103, 107815 CrossRef CAS.
- W. J. Xu, H. Tang, H. F. Gu, H. Y. Xi, P. F. Wu, B. L. Liang, Q. Q. Liu and W. X. Chen, J. Mater. Chem. A, 2022, 10, 14732–14746 RSC.
- L. J. Zhang, N. Jin, Y. B. Yang, X. Y. Miao, H. Wang, J. Luo and L. L. Han, Nano-Micro Lett., 2023, 15, 228 CrossRef CAS PubMed.
- Y. Y. Wu, C. C. Ye, L. Yu, Y. F. Liu, J. F. Huang, J. B. Bi, L. Xue, J. W. Sun, J. Yang, W. Q. Zhang, X. Wang, P. Xiong and J. W. Zhu, Energy Storage Mater., 2022, 45, 805–813 CrossRef.
- W. Ye, S. M. Chen, Y. Lin, L. Yang, S. J. Chen, X. S. Zheng, Z. M. Qi, C. M. Wang, R. Long, M. Chen, J. F. Zhu, P. Gao, L. Song, J. Jiang and Y. J. Xiong, Chem, 2019, 5, 2865–2878 CAS.
- Y. Y. Guo, P. F. Yuan, J. N. Zhang, Y. F. Hu, I. S. Amiinu, X. Wang, J. G. Zhou, H. C. Xia, Z. B. Song, Q. Xu and S. C. Mu, ACS Nano, 2018, 12, 1894–1901 CrossRef CAS PubMed.
- L. L. Zhang, J. Rong, Y. X. Lin, Y. Q. Yang, H. Z. Zhu, X. H. Yu, X. D. Kang, C. L. Chen, H. M. Cheng and G. Liu, Adv. Funct. Mater., 2023, 33, 2307947 CrossRef CAS.
- F. Mo, Q. X. Zhou, W. D. Xue, W. T. Liu, S. Z. Xu, Z. L. Hou, J. L. Wang and Q. Wang, Adv. Energy Mater., 2023, 13, 2301711 CrossRef CAS.
- X. P. Han, X. F. Ling, Y. Wang, T. Y. Ma, C. Zhong, W. B. Hu and Y. D. Deng, Angew. Chem., Int. Ed., 2019, 58, 5359–5364 CrossRef CAS PubMed.
- T. P. Zhou, H. Shan, H. Yu, C. A. Zhong, J. K. Ge, N. Zhang, W. S. Chu, W. S. Yan, Q. Xu, H. A. Wu, C. Z. Wu and Y. Xie, Adv. Mater., 2020, 32, 2003251 CrossRef CAS PubMed.
- X. Y. Yang, W. Q. Song, T. Zhang, Z. C. Huang, J. F. Zhang, J. Ding and W. B. Hu, Adv. Energy Mater., 2023, 13, 2301737 CrossRef CAS.
- J. W. Hu, D. Y. Wu, C. Zhu, C. Hao, C. C. Xin, J. W. Zhang, J. Y. Guo, N. N. Li, G. F. Zhang and Y. T. Shi, Nano Energy, 2020, 72, 104670 CrossRef CAS.
- J. W. Wang, C. G. Hu, L. G. Wang, Y. Yuan, K. Zhu, Q. Zhang, L. Yang, J. Lu and Z. Y. Bai, Adv. Funct. Mater., 2023, 33, 2304277 CrossRef CAS.
- X. G. Fu, G. P. Jiang, G. B. Wen, R. Gao, S. Li, M. Li, J. B. Zhu, Y. Zheng, Z. Q. Li, Y. F. Hu, L. Yang, Z. Y. Bai, A. P. Yu and Z. W. Chen, Appl. Catal., B, 2021, 293, 120176 CrossRef CAS.
- Y. G. Wu, X. N. Tang, K. Yuan and Y. W. Chen, Energy Environ. Sci., 2023, 16, 5663–5687 RSC.
- J. B. Liu, J. W. Liao, K. Huang, J. C. Dong, G. C. He, Z. C. Gong and H. L. Fei, Adv. Mater., 2023, 35, 2211398 CrossRef CAS PubMed.
- C. L. Xu, C. Z. Guo, J. P. Liu, B. H. Hu, J. Y. Dai, M. Wang, R. Jin, Z. L. Luo, H. L. Li and C. G. Chen, Energy Storage Mater., 2022, 51, 149–158 CrossRef.
- Y. Pan, X. L. Ma, M. M. Wang, X. Yang, S. J. Liu, H. C. Chen, Z. W. Zhuang, Y. H. Zhang, W. C. Cheong, C. Zhang, X. Cao, R. G. Shen, Q. Xu, W. Zhu, Y. Q. Liu, X. D. Wang, X. J. Zhang, W. S. Yan, J. Li, H. M. Chen, C. Chen and Y. D. Li, Adv. Mater., 2022, 34, 2203621 CrossRef CAS PubMed.
- Y. Y. Qiao, P. F. Yuan, Y. F. Hu, J. N. Zhang, S. C. Mu, J. H. Zhou, H. Li, H. C. Xia, J. He and Q. Xu, Adv. Mater., 2018, 30, 1804504 CrossRef PubMed.
- R. G. Cao, R. Thapa, H. Kim, X. D. Xu, M. Gyu Kim, Q. Li, N. Park, M. L. Liu and J. Cho, Nat. Commun., 2013, 4, 2076 CrossRef PubMed.
- M. R. Gao, J. X. Liang, Y. R. Zheng, Y. F. Xu, J. Jiang, Q. Gao, J. Li and S. H. Yu, Nat. Commun., 2015, 6, 5982 CrossRef CAS PubMed.
- H. Y. Jin, J. Wang, D. F. Su, Z. Z. Wei, Z. F. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688–2694 CrossRef CAS PubMed.
- J. Wang, Z. Q. Huang, W. Liu, C. R. Chang, H. L. Tang, Z. J. Li, W. X. Chen, C. J. Jia, T. Yao, S. Q. Wei, Y. Wu and Y. D. Li, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed.
- P. Q. Yin, T. Yao, Y. Wu, L. R. Zheng, Y. Lin, W. Liu, H. X. Ju, J. F. Zhu, X. Hong, Z. X. Deng, G. Zhou, S. Q. Wei and Y. D. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- P. Q. Yin, T. Yao, Y. Wu, L. R. Zheng, Y. Lin, W. Liu, H. X. Ju, J. F. Zhu, X. Hong, Z. X. Deng, G. Zhou, S. Q. Wei and Y. D. Li, Adv. Mater., 2018, 30, 1801732 CrossRef PubMed.
- D. F. Qi, Y. F. Liu, M. Hu, X. Y. Peng, Y. Qiu, S. S. Zhang, W. Liu, H. Y. Li, G. Z. Hu, L. C. Zhuo, Y. J. Qin, J. He, G. C. Qi, J. Q. Sun, J. Luo and X. J. Liu, Small, 2020, 16, 2004855 CrossRef CAS PubMed.
- M. J. Liu, J. Lee, T. C. Yang, F. Y. Zheng, J. Zhao, C. M. Yang and L. Y. S. Lee, Small Methods, 2021, 5, 2001165 CrossRef CAS PubMed.
- F. S. Yu, J. Y. Zhan, D. T. Chen, J. Y. Guo, S. B. Zhang and L. H. Zhang, Adv. Funct. Mater., 2023, 33, 2214425 CrossRef CAS.
- Z. Wang, Z. Lu, Q. T. Ye, Z. B. Yang, R. J. Xu, K. X. Kong, Y. F. Zhang, T. Yan, Y. P. Liu, Z. J. Pan, Y. Z. Huang and X. H. Lu, Adv. Funct. Mater., 2024, 34, 2315150 CrossRef CAS.
- C. J. Lei, Y. Wang, Y. Hou, P. Liu, J. Yang, T. Zhang, X. D. Zhuang, M. W. Chen, B. Yang, L. C. Lei, C. Yuan, M. Qiu and X. L. Feng, Energy Environ. Sci., 2019, 12, 149–156 RSC.
- M. Jiang, F. Wang, F. Yang, H. He, J. Yang, W. Zhang, J. Y. Luo, J. Zhang and C. P. Fu, Nano Energy, 2022, 93, 106793 CrossRef CAS.
- P. Kumari, N. Bahadur, L. X. Kong, L. A. O’Dell, A. Merenda and L. F. Dumée, Adv. Mater., 2022, 3, 2309–2323 RSC.
- J. Brownless, J. W. Zhang and A. M. Song, Carbon, 2020, 168, 201–208 CrossRef CAS.
- Z. Zhang and J. T. Yates, Chem. Rev., 2012, 112, 5520–5551 CrossRef CAS PubMed.
- R. T. Tung, Appl. Phys. Rev., 2014, 1, 011304 Search PubMed.
- Z. C. Zhang, G. G. Liu, X. Y. Cui, B. Chen, Y. H. Zhu, Y. Gong, F. Saleem, S. B. Xi, Y. H. Du, A. Borgna, Z. C. Lai, Q. H. Zhang, B. Li, Y. Zong, Y. Han, L. Gu and H. Zhang, Adv. Mater., 2018, 30, 1801741 CrossRef PubMed.
- H. M. Li, S. P. Li, R. J. Guan, Z. Y. Jin, D. Xiao, Y. Guo and P. P. Li, ACS Catal., 2024, 14, 12042–12050 CrossRef CAS.
- X. Y. Zhang, F. Li, Z. Z. Li and Y. P. Bai, J. Mater. Chem. A, 2025, 13, 5304–5314 RSC.
- L. Zhang, B. Wang, J. S. Hu, X. H. Huang, W. Y. Ma, N. P. Li, T. Wågberg and G. Z. Hu, J. Colloid Interface Sci., 2022, 625, 521–531 CrossRef CAS PubMed.
- X. Y. Wang, X. Q. Xu, Y. Nie, R. H. Wang and J. L. Zou, Adv. Sci., 2023, 10, 2301961 CrossRef CAS PubMed.
- B. W. Wang, J. X. Zou, X. C. Shen, Y. C. Yang, G. Z. Hu, W. Li, Z. M. Peng, D. Banham, A. G. Dong and D. Y. Zhao, Nano Energy, 2019, 63, 103851 CrossRef CAS.
- Z. J. Li, S. Q. Ji, C. Wang, H. X. Liu, L. P. Leng, L. Du, J. C. Gao, M. Qiao, J. H. Horton and Y. Wang, Adv. Mater., 2023, 35, 2300905 CrossRef CAS PubMed.
- M. Liu, N. Li, S. F. Cao, X. M. Wang, X. Q. Lu, L. J. Kong, Y. H. Xu and X. H. Bu, Adv. Mater., 2022, 34, 2107421 CrossRef CAS PubMed.
- Z. H. Pei, X. F. Lu, H. B. Zhang, Y. X. Li, D. Y. Luan and X. W. Lou, Angew. Chem., Int. Ed., 2022, 61, e202207537 CrossRef CAS PubMed.
- C. H. He, Q. Q. Liu, H. M. Wang, C. F. Xia, F. M. Li, W. Guo and B. Y. Xia, Small, 2023, 19, 2207474 CrossRef CAS PubMed.
- J. G. Hou, Y. Q. Sun, Y. Z. Wu, S. Y. Cao and L. C. Sun, Adv. Funct. Mater., 2018, 28, 1704447 CrossRef.
- L. Zhang, Y. X. Zhu, Z. C. Nie, Z. Y. Li, Y. Ye, L. H. Li, J. Hong, Z. H. Bi, Y. T. Zhou and G. Z. Hu, ACS Nano, 2021, 15, 13399–13414 CrossRef CAS PubMed.
- S. Q. Fu, Y. R. Ma, X. C. Yang, X. Yao, Z. Jiao, L. L. Cheng and P. D. Zhao, Appl. Catal., B, 2023, 333, 122813 CrossRef CAS.
- S. Ni, H. N. Qu, Z. H. Xu, X. Y. Zhu, H. F. Xing, L. Wang, J. M. Yu, H. Z. Liu, C. M. Chen and L. R. Yang, Appl. Catal., B, 2021, 299, 120638 CrossRef CAS.
- Z. C. Zhuang, L. X. Xia, J. Z. Huang, P. Zhu, Y. Li, C. L. Ye, M. G. Xia, R. H. Yu, Z. Q. Lang, J. X. Zhu, L. R. Zheng, Y. Wang, T. Y. Zhai, Y. Zhao, S. Q. Wei, J. Li, D. S. Wang and Y. D. Li, Angew. Chem., Int. Ed., 2022, 62, e202212335 CrossRef PubMed.
- M. Z. Gu, L. Jiang, S. R. Zhao, H. Wang, M. Lin, X. Y. Deng, X. M. Huang, A. Gao, X. D. Liu, P. Sun and X. J. Zhang, ACS Nano, 2022, 16, 15425–15439 CrossRef CAS PubMed.
- J. Y. Chen, H. Li, C. Fan, Q. W. Meng, Y. W. Tang, X. Y. Qiu, G. T. Fu and T. Y. Ma, Adv. Mater., 2020, 32, 2003134 CrossRef CAS PubMed.
- K. Khan, X. X. Yan, Q. M. Yu, S. H. Bae, J. J. White, J. X. Liu, T. C. Liu, C. J. Sun, J. Kim, H. M. Cheng, Y. Wang, B. L. Liu, K. Amine, X. Q. Pan and Z. T. Luo, Nano Energy, 2021, 90, 106488 CrossRef CAS.
- H. X. Li, Y. L. Wen, M. Jiang, Y. Yao, H. H. Zhou, Z. Y. Huang, J. W. Li, S. Q. Jiao, Y. F. Kuang and S. L. Luo, Adv. Funct. Mater., 2021, 31, 2102420 CrossRef.
- M. Li, H. Y. Zhu, Q. Yuan, T. Y. Li, M. M. Wang, P. Zhang, Y. L. Zhao, D. L. Qin, W. Y. Guo, B. Liu, X. Yang, Y. Q. Liu and Y. Pan, Adv. Funct. Mater., 2023, 33, 2210867 CrossRef CAS.
- X. X. Yan, D. Liu, P. F. Guo, Y. F. He, X. Q. Wang, Z. L. Li, H. G. Pan, D. L. Sun, F. Fang and R. B. Wu, Adv. Mater., 2023, 35, 2210975 CrossRef CAS PubMed.
- Y. Wang, J. Wu, S. H. Tang, J. R. Yang, C. L. Ye, J. Chen, Y. G. Lei and D. S. Wang, Angew. Chem., Int. Ed., 2023, 62, e202219191 CrossRef CAS PubMed.
- F. T. Kong, M. Wang, Y. F. Huang, G. Meng, M. X. Chen, H. Tian, Y. F. Chen, C. Chen, Z. W. Chang, X. Z. Cui and J. L. Shi, Energy Storage Mater., 2023, 54, 533–542 CrossRef.
- S. P. Zhang, G. Wang, B. B. Wang, J. M. Wang, J. T. Bai and H. Wang, Adv. Funct. Mater., 2020, 30, 2001592 CrossRef CAS.
- D. Y. Chung, S. B. Park, H. Lee, H. Kim, Y. H. Chung, J. M. Yoo, D. Ahn, S. H. Yu, K. S. Lee, M. Ahmadi, H. X. Ju, H. D. Abruña, S. J. Yoo, B. S. Mun and Y. E. Sung, ACS Energy Lett., 2020, 5, 2827–2834 CrossRef CAS.
- D. C. Duan, J. L. Huo, J. X. Chen, B. Chi, Z. S. Chen, S. H. Sun, Y. Zhao, H. Zhao, Z. M. Cui and S. J. Liao, Small, 2024, 20, 2310491 CrossRef CAS PubMed.
- M. M. Tong, F. F. Sun, Y. Xie, Y. Wang, Y. Q. Yang, C. G. Tian, L. Wang and H. G. Fu, Angew. Chem., Int. Ed., 2021, 60, 14005–14012 CrossRef CAS PubMed.
- L. Cheng, X. Y. Yue, L. X. Wang, D. N. Zhang, P. Zhang, J. J. Fan and Q. J. Xiang, Adv. Mater., 2021, 33, 2105135 CrossRef CAS PubMed.
- Y. H. Xie, X. Chen, K. A. Sun, J. Q. Zhang, W. H. Lai, H. Liu and G. X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202301833 CrossRef CAS PubMed.
- D. Liang, C. Lian, Q. C. Xu, M. M. Liu, H. L. Liu, H. Jiang and C. Z. Li, Appl. Catal., B, 2020, 268, 118417 CrossRef CAS.
- P. F. Zhang, Y. D. Liu, T. T. Liang, E. H. Ang, X. Zhang, F. Ma and Z. F. Dai, Appl. Catal., B, 2021, 284, 119738 CrossRef CAS.
- Z. X. Xu, S. Jin, M. H. Seo and X. L. Wang, Appl. Catal., B, 2021, 292, 120168 CrossRef CAS.
- T. F. Li, J. W. Yin, D. M. Sun, M. Y. Zhang, H. Pang, L. Xu, Y. W. Zhang, J. Yang, Y. W. Tang and J. M. Xue, Small, 2022, 18, 2106592 CrossRef CAS PubMed.
- L. Q. Deng, K. Zhang, D. Shi, S. F. Liu, D. Q. Xu, Y. L. Shao, J. X. Shen, Y. Z. Wu and X. P. Hao, Appl. Catal., B, 2021, 299, 120660 CrossRef CAS.
- S. L. Jiao, X. W. Fu and H. W. Huang, Adv. Funct. Mater., 2022, 32, 2107651 CrossRef CAS.
- M. M. Wang, M. K. Zhang, W. W. Song, W. T. Zhong, X. Y. Wang, J. Wang, T. M. Sun and Y. F. Tang, Chem. Commun., 2021, 57, 785–788 RSC.
- C. R. Pi, X. X. Li, X. M. Zhang, H. Song, Y. Zheng, B. Gao, A. Kızılaslan, P. K. Chu and K. F. Huo, Small, 2022, 18, 2201137 CrossRef CAS PubMed.
- C. J. Gu, G. Y. Zhou, J. Yang, H. Pang, M. Y. Zhang, Q. Zhao, X. F. Gu, S. Tian, J. B. Zhang, L. Xu and Y. W. Tang, Chem. Eng. J., 2022, 443, 136321 CrossRef CAS.
- G. B. Chen, Y. An, S. W. Liu, F. F. Sun, H. Y. Qi, H. F. Wu, Y. H. He, P. Liu, R. Shi, J. Zhang, A. Kuc, U. Kaiser, T. Zhang, T. Heine, G. Wu and X. L. Feng, Energy Environ. Sci., 2022, 15, 2619–2628 RSC.
- Y. Zhang, C. X. Li, J. Li, X. N. Liu, G. J. Li, B. Li and L. Wang, J. Mater. Chem. A, 2023, 11, 11326–11333 RSC.
- T. Zhang, X. Han, H. Liu, M. Biset-Peiró, J. Li, X. Zhang, P. Y. Tang, B. Yang, L. R. Zheng, J. R. Morante and J. Arbiol, Adv. Funct. Mater., 2022, 32, 2111446 CrossRef CAS.
- W. J. Zang, T. Sun, T. Yang, S. B. Xi, M. Waqar, Z. K. Kou, Z. Y. Lyu, Y. P. Feng, J. Wang and S. J. Pennycook, Adv. Mater., 2021, 33, 2003846 CrossRef CAS PubMed.
- W. L. Zhou, H. Su, Y. L. Li, M. H. Liu, H. Zhang, X. X. Zhang, X. Sun, Y. Z. Xu, Q. H. Liu and S. Q. Wei, ACS Energy Lett., 2021, 6, 3359–3366 CrossRef CAS.
- J. Yang, W. G. Liu, M. Q. Xu, X. Y. Liu, H. F. Qi, L. L. Zhang, X. F. Yang, S. S. Niu, D. Zhou, Y. F. Liu, Y. Su, J. F. Li, Z. Q. Tian, W. Zhou, A. Q. Wang and T. Zhang, J. Am. Chem. Soc., 2021, 143, 14530–14539 CrossRef CAS PubMed.
- J. W. Wan, Z. H. Zhao, H. S. Shang, B. Peng, W. X. Chen, J. J. Pei, L. R. Zheng, J. C. Dong, R. Cao, R. Sarangi, Z. L. Jiang, D. N. Zhou, Z. B. Zhuang, J. T. Zhang, D. S. Wang and Y. D. Li, J. Am. Chem. Soc., 2022, 142, 8431–8439 CrossRef PubMed.
- Y. L. Zhao, H. C. Chen, X. L. Ma, J. Y. Li, Q. Yuan, P. Zhang, M. M. Wang, J. X. Li, M. Li, S. F. Wang, H. Guo, R. B. Hu, K. H. Tu, W. Zhu, X. N. Li, X. Yang and Y. Pan, Adv. Mater., 2024, 36, 2308243 CrossRef CAS PubMed.
- F. Q. Wang, R. Zhang, Y. Y. Zhang, Y. Li, J. Zhang, W. H. Yuan, H. Liu, F. Wang and H. L. Xin, Adv. Funct. Mater., 2023, 33, 2213863 CrossRef CAS.
- L. S. Peng, J. Yang, Y. Q. Yang, F. R. Qian, Q. Wang, D. X. Sun-Waterhouse, L. Shang, T. R. Zhang and G. I. N. Waterhouse, Adv. Mater., 2022, 34, 2202544 CrossRef CAS PubMed.
- L. B. Li, S. H. Huang, R. Cao, K. Yuan, C. B. Lu, B. Y. Huang, X. N. Tang, T. Hu, X. D. Zhuang and Y. W. Chen, Small, 2022, 18, 2105387 CrossRef CAS PubMed.
- Y. Liu, S. S. Zhang, C. Jiao, H. M. Chen, G. Wang, W. J. Wu, Z. W. Zhuo and J. J. Mao, Adv. Sci., 2022, 10, 2206107 CrossRef PubMed.
- S. C. Ding, J. A. Barr, Q. R. Shi, Y. C. Zeng, P. Tieu, Z. Y. Lyu, L. Z. Fang, T. Li, X. Q. Pan, S. P. Beckman, D. Du, H. F. Lin, J. C. Li, G. Wu and Y. H. Lin, ACS Nano, 2022, 16, 15165–15174 CrossRef CAS PubMed.
- S. K. Zhang, Q. X. Zhou, L. Y. Fang, R. Wang, T. Y. Lu, Q. Zhao, X. F. Gu, S. Tian, L. Xu, H. Pang, J. Yang, Y. W. Tang and S. H. Sun, Appl. Catal., B, 2023, 328, 122489 CrossRef CAS.
- M. R. Wang, W. J. Yang, X. Z. Li, Y. S. Xu, L. R. Zheng, C. L. Su and B. Liu, ACS Energy Lett., 2021, 6, 379–386 CrossRef CAS.
- H. J. Shen, E. Gracia-Espino, J. Y. Ma, H. D. Tang, X. Mamat, T. Wagberg, G. Z. Hu and S. J. Guo, Nano Energy, 2017, 35, 9–16 CrossRef CAS.
- K. Yuan, D. Lützenkirchen-Hecht, L. B. Li, L. Shuai, Y. Z. Li, R. Cao, M. Qiu, X. D. Zhuang, M. K. H. Leung, Y. W. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS PubMed.
- Y. M. Zhao, P. C. Zhang, C. Xu, X. Y. Zhou, L. M. Liao, P. J. Wei, E. Liu, H. Q. Chen, Q. G. He and J. G. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 17334–17342 CrossRef CAS PubMed.
- X. K. Wang, X. K. Zhou, C. Li, H. X. Yao, C. H. Zhang, J. Zhou, R. Xu, L. Chu, H. L. Wang, M. Gu, H. Q. Jiang and M. H. Huang, Adv. Mater., 2022, 34, 2204021 CrossRef CAS PubMed.
- B. L. Yang, X. L. Li, Q. Cheng, X. D. Jia, Y. J. Liu and Z. H. Xiang, Nano Energy, 2022, 101, 107565 CrossRef CAS.
- Y. J. Chen, R. Gao, S. F. Ji, H. J. Li, K. Tang, P. Jiang, H. B. Hu, Z. D. Zhang, H. G. Hao, Q. Y. Qu, X. Liang, W. X. Chen, J. C. Dong, D. S. Wang and Y. D. Li, Angew. Chem., Int. Ed., 2020, 60, 3212–3221 CrossRef PubMed.
- J. B. Liu, D. S. Wang, K. Huang, J. C. Dong, J. W. Liao, S. Dai, X. Tang, M. M. Yan, H. S. Gong, J. J. Liu, Z. C. Gong, R. Liu, C. Y. Cui, G. L. Ye, X. L. Zou and H. L. Fei, ACS Nano, 2021, 15, 18125–18134 CrossRef CAS PubMed.
- Y. J. Deng, J. M. Luo, B. Chi, H. B. Tang, J. Li, X. C. Qiao, Y. J. Shen, Y. J. Yang, C. M. Jia, P. Rao, S. J. Liao and X. L. Tian, Adv. Energy Mater., 2021, 11, 2101222 CrossRef CAS.
- R. X. Qin, K. L. Liu, Q. Y. Wu and N. F. Zheng, Chem. Rev., 2020, 120, 11810–11899 CrossRef CAS PubMed.
- B. T. Hu, A. J. Huang, X. J. Zhang, Z. Chen, R. Y. Tu, W. Zhu, Z. B. Zhuang, C. Chen, Q. Peng and Y. D. Li, Nano Res., 2021, 14, 3482–3488 CrossRef CAS.
- L. L. Zhang, N. Zhang, H. S. Shang, Z. Y. Sun, Z. H. Wei, J. T. Wang, Y. T. Lei, X. C. Wang, D. Wang, Y. F. Zhao, Z. T. Sun, F. Zhang, X. Xiang, B. Zhang and W. X. Chen, Nat. Commun., 2024, 15, 9440 CrossRef CAS PubMed.
- J. L. Zhang, Y. H. Mou, W. R. Suo, S. J. Yang, J. Shen, H. Xu, Z. Q. Zeng, R. Zhang, Z. Z. Liang, Y. Wang, H. Q. Zheng, J. H. Cao and R. Cao, Adv. Funct. Mater., 2025, 35, 2417621 CrossRef CAS.
- Z. Y. Jin, R. J. Guan, X. Li, D. Y. Yuan and P. P. Li, Chin. Chem. Lett., 2025 DOI:10.1016/i.cclet.2024.110506.
- J. X. Han, H. Tan, K. Guo, H. Y. Lv, X. Y. Peng, W. Zhang, H. P. Lin, U. P. Apfel and R. Cao, Angew. Chem., Int. Ed., 2024, 63, e202409793 CrossRef CAS PubMed.
- Y. J. Liu, G. J. Zhou, Z. Y. Zhang, H. T. Lei, Z. Yao, J. F. Li, J. Lin and R. Cao, Chem. Sci., 2020, 11, 87–96 RSC.
- X. L. Li, H. T. Lei, L. S. Xie, N. Wang, W. Zhang and R. Cao, Acc. Chem. Res., 2022, 55, 878–892 CrossRef CAS PubMed.
- M. Yi, H. M. Li, M. H. Xie, P. P. Li, Z. Y. Jin and G. H. Yu, Sci. China: Chem., 2024, 67 DOI:10.1007/s11426-024-2286-7.
- Y. J. Wu, X. Y. Wang, B. B. Tian, W. Shuang, Z. Y. Bai and L. Yang, Inorg. Chem. Front., 2023, 10, 4209–4220 RSC.
- Y. H. Han, Y. G. Wang, R. R. Xu, W. X. Chen, L. R. Zheng, A. J. Han, Y. Q. Zhu, J. Zhang, H. B. Zhang, J. Luo, C. Chen, Q. Peng, D. S. Wang and Y. D. Li, Energy Environ. Sci., 2018, 11, 2348–2352 RSC.
- B. X. Zhang, J. L. Zhang, J. B. Shi, D. X. Tan, L. F. Liu, F. Y. Zhang, C. Lu, Z. Z. Su, X. N. Tan, X. Y. Cheng, B. X. Han, L. R. Zheng and J. Zhang, Nat. Commun., 2019, 10, 2980 CrossRef PubMed.
- M. Huang, B. W. Deng, X. L. Zhao, Z. Y. Zhang, F. Li, K. L. Li, Z. H. Cui, L. X. Kong, J. M. Lu, F. Dong, L. L. Zhang and P. Chen, ACS Nano, 2022, 16, 2110–2119 CrossRef CAS PubMed.
- S. X. Yang, Y. H. Yu, X. J. Gao, Z. P. Zhang and F. Wang, Chem. Soc. Rev., 2021, 50, 12985–13011 RSC.
- X. Y. Li, X. S. Wu, Y. Zhao, Y. X. Lin, J. H. Zhao, C. Q. Wu, H. J. Liu, L. Shan, L. Yang, L. Song and J. Jiang, Adv. Mater., 2023, 35, 2302467 CrossRef CAS PubMed.
- X. L. Li, P. Li, J. D. Yang, L. S. Xie, N. Wang, H. T. Lei, C. C. Zhang, W. Zhang, Y. M. Lee, W. Q. Zhang, S. Fukuzumi, W. Nam and R. Cao, J. Energy Chem., 2023, 76, 617–621 CrossRef CAS.
- L. C. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- Y. Feng, K. X. Song, W. Zhang, X. Y. Zhou, S. J. Yoo, J. G. Kim, S. F. Qiao, Y. G. Qi, X. Zou, Z. J. Chen, T. T. Qin, N. L. Yue, Z. Z. Wang, D. B. Li and W. T. Zheng, J. Energy Chem., 2022, 70, 211–218 CrossRef CAS.
- X. Wan, Q. T. Liu, J. Y. Liu, S. Y. Liu, X. Y. Liu, L. R. Zheng, J. X. Shang, R. H. Yu and J. L. Shui, Nat. Commun., 2020, 13, 2963 CrossRef PubMed.
- Z. Wang, C. Zhu, H. Tan, J. Liu, L. L. Xu, Y. Q. Zhang, Y. P. Liu, X. X. Zou, Z. Liu and X. H. Lu, Adv. Funct. Mater., 2021, 31, 2104735 CrossRef CAS.
- J. Yu, J. Li, C. Y. Xu, Q. Liu, J. Y. Liu, R. R. Chen, J. H. Zhu, R. M. Li and J. Wang, Carbon, 2021, 185, 96–104 CrossRef CAS.
- J. Quílez-Bermejo, S. García-Dalí, A. Daouli, A. Zitolo, R. L. S. Canevesi, M. Emo, M. T. Izquierdo, M. Badawi, A. Celzard and V. Fierro, Adv. Funct. Mater., 2023, 33, 2300405 CrossRef.
- K. X. Ding, J. G. Hu, J. Luo, L. M. Zhao, W. Jin, Y. P. Liu, Z. H. Wu, G. Q. Zou, H. S. Hou and X. B. Ji, Adv. Funct. Mater., 2022, 32, 2207331 CrossRef CAS.
- Q. Yu, J. S. Lv, J. T. Li, R. H. Yu, J. S. Wu, S. B. Xi, X. Y. Li, N. Xu, L. Zhou and L. Q. Mai, Energy Environ. Mater., 2022, 6, e12389 CrossRef.
- J. Zhang, Y. Xie, Q. K. Jiang, S. Guo, J. H. Huang, L. L. Xu, Y. Wang and G. Li, J. Mater. Chem. A, 2022, 10, 16920–16927 RSC.
- J. M. Guo, W. Q. Li, Y. C. Xu, Y. Q. Mao, Z. W. Mei, H. H. Li, Y. He, X. Y. San, K. Xu and X. G. Liang, Small Methods, 2023, 7, 2201371 CrossRef CAS PubMed.
- J. J. Chen, S. Gu, R. Hao, Z. Y. Wang, M. Q. Li, Z. Q. Li, K. Liu, K. M. Liao, Z. Q. Wang, H. Huang, Y. Z. Li, K. L. Zhang and Z. G. Lu, Rare Met., 2022, 41, 2055–2062 CrossRef CAS.
- J. C. Li, Y. Meng, L. L. Zhang, G. Z. Li, Z. C. Shi, P. X. Hou, C. Liu, H. M. Cheng and M. H. Shao, Adv. Funct. Mater., 2021, 31, 2103360 CrossRef CAS.
- I. S. Amiinu, X. B. Liu, Z. H. Pu, W. Q. Li, Q. D. Li, J. Zhang, H. L. Tang, H. N. Zhang and S. C. Mu, Adv. Funct. Mater., 2018, 28, 1704638 CrossRef.
- M. T. Zhang, H. Li, J. X. Chen, F. X. Ma, L. Zhen, Z. H. Wen and C. Y. Xu, Adv. Funct. Mater., 2023, 33, 2209726 CrossRef CAS.
- H. J. Huang, D. S. Yu, F. Hu, S. C. Huang, J. N. Song, H. Y. Chen, L. L. Li and S. J. Peng, Angew. Chem., Int. Ed., 2022, 61, e202116068 CrossRef CAS PubMed.
- Y. Y. Li, Z. H. Li, K. K. Shi, L. K. Luo, H. M. Jiang, Y. He, Y. L. Zhao, J. Y. He, L. Lin, Z. M. Sun and G. B. Sun, Small, 2024, 20, 2309727 CrossRef CAS PubMed.
- W. J. Zhai, S. H. Huang, C. B. Lu, X. N. Tang, L. B. Li, B. Y. Huang, T. Hu, K. Yuan, X. D. Zhuang and Y. W. Chen, Small, 2022, 18, 2107225 CrossRef CAS PubMed.
- S. Chandrasekaran, R. Hu, L. Yao, L. J. Sui, Y. P. Liu, A. Abdelkader, Y. L. Li, X. Z. Ren and L. B. Deng, Nano-Micro Lett., 2023, 15, 48 CrossRef CAS PubMed.
- Z. Y. Chen, X. Su, J. Ding, N. Yang, W. B. Zuo, Q. Y. He, Z. M. Wei, Q. Zhang, J. Huang and Y. M. Zhai, Appl. Catal., B, 2022, 308, 121206 CrossRef CAS.
- H. B. Meng, B. Wu, D. T. Zhang, X. H. Zhu, S. Z. Luo, Y. You, K. Chen, J. C. Long, J. X. Zhu, L. P. Liu, S. B. Xi, T. Petit, D. S. Wang, X. M. Zhang, Z. J. Xu and L. Q. Mai, Energy Environ. Sci., 2024, 17, 704–716 RSC.
- W. H. Luo, Y. Wang, L. X. Luo, S. Gong, M. N. Wei, Y. X. Li, X. P. Gan, Y. Y. Zhao, Z. H. Zhu and Z. Li, ACS Catal., 2022, 12, 1167–1179 CrossRef CAS.
- Y. R. Liu, S. Yuan, C. T. Sun, C. L. Wang, X. J. Liu, Z. H. Lv, R. Liu, Y. Z. Meng, W. X. Yang, X. Feng and B. Wang, Adv. Energy Mater., 2023, 13, 2302719 CrossRef CAS.
- Y. Q. Chen, J. N. Mao, H. Zhou, L. L. Xing, S. S. Qiao, J. L. Yuan, B. B. Mei, Z. X. Wei, S. L. Zhao, Y. H. Tang and C. B. Liu, Adv. Funct. Mater., 2024, 34, 2311664 CrossRef CAS.
- Q. Z. An, X. Zhang, C. Y. Yang, H. Su, W. L. Zhou, M. H. Liu, X. X. Zhang, X. Sun, S. W. Bo, F. F. Yu, J. J. Jiang, K. Zheng and Q. H. Liu, Small, 2023, 19, 2304303 CrossRef CAS PubMed.
- H. Liu, L. Z. Jiang, Y. Y. Sun, J. Khan, B. Feng, J. M. Xiao, H. D. Zhang, H. J. Xie, L. N. Li, S. Y. Wang and L. Han, Adv. Energy Mater., 2023, 13, 2301223 CrossRef CAS.
- T. H. Nguyen, P. K. L. Tran, D. T. Tran, T. N. Pham, N. H. Kim and J. H. Lee, Chem. Eng. J., 2022, 440, 135781 CrossRef CAS.
- M. J. Wu, X. H. Yang, X. Cui, N. Chen, L. Du, M. Cherif, F. K. Chiang, Y. Wen, A. Hassanpour, F. Vidal, S. Omanovic, Y. K. Yang, S. H. Sun and G. X. Zhang, Nano-Micro Lett., 2023, 15, 232 CrossRef CAS PubMed.
- Q. R. Zhang, P. Kumar, X. F. Zhu, R. Daiyan, N. M. Bedford, K. H. Wu, Z. J. Han, T. R. Zhang, R. Amal and X. Y. Lu, Adv. Energy Mater., 2021, 11, 2100303 CrossRef CAS.
- Y. Zhang, X. Yu Gao, Z. Wen, C. C. Yang and Q. Jiang, Chem. Eng. J., 2022, 446, 137441 CrossRef CAS.
- Z. Wang, X. Y. Jin, R. J. Xu, Z. B. Yang, S. D. Ma, T. Yan, C. Zhu, J. Fang, Y. P. Liu, S. J. Hwang, Z. J. Pan and H. J. Fan, ACS Nano, 2023, 17, 8622–8633 CrossRef CAS PubMed.
- H. Lv, P. Tong, H. Li, B. Sun, Y. Li, P. Qiao, H. Tian and H. Xia, Chem. Eng. J., 2023, 478, 147375 CrossRef CAS.
- H. Liu, L. Z. Jiang, J. Khan, X. X. Wang, J. M. Xiao, H. D. Zhang, H. J. Xie, L. N. Li, S. Y. Wang and L. Han, Angew. Chem., Int. Ed., 2022, 62, e202214988 CrossRef PubMed.
- J. R. Bai, Y. B. Lian, Y. Y. Deng, M. Xiang, P. Xu, Q. F. Zhou, Y. W. Tang and Y. Q. Su, Nano Res., 2024, 17, 2291–2297 CrossRef CAS.
- F. F. Zhang, Y. L. Zhu, Y. J. Zhong, J. Zou, Y. Chen, L. H. Zu, Z. Y. Wang, J. J. Hinsch, Y. Wang, L. Zhang, Z. P. Shao and H. T. Wang, J. Energy Chem., 2023, 85, 154–163 CrossRef CAS.
- Y. Y. Ma, Y. Yu, J. H. Wang, J. Lipton, H. N. Tan, L. R. Zheng, T. Yang, Z. L. Liu, X. J. Loh, S. J. Pennycook, L. Shen, Z. K. Kou, A. D. Taylor and J. Wang, Adv. Sci., 2022, 9, 2105192 CrossRef CAS PubMed.
- L. Tao, Y. Q. Wang, Y. Q. Zou, N. N. Zhang, Y. Q. Zhang, Y. J. Wu, Y. Y. Wang, R. Chen and S. Y. Wang, Adv. Energy Mater., 2019, 10, 1901227 CrossRef.
- P. Zhu, X. Xiong, D. S. Wang and Y. D. Li, Adv. Energy Mater., 2023, 13, 2300884 CrossRef CAS.
- J. X. Diao, Y. Qiu, S. Q. Liu, W. T. Wang, K. Chen, H. L. Li, W. Y. Yuan, Y. T. Qu and X. H. Guo, Adv. Mater., 2019, 32, 1905679 CrossRef PubMed.
- A. L. Han, W. M. Sun, X. Wan, D. D. Cai, X. J. Wang, F. Li, J. L. Shui and D. S. Wang, Angew. Chem., Int. Ed., 2023, 62, e202303185 CrossRef CAS PubMed.
- H. S. Shang, X. Y. Zhou, J. C. Dong, A. Li, X. Zhao, Q. H. Liu, Y. Lin, J. J. Pei, Z. Li, Z. L. Jiang, D. N. Zhou, L. R. Zheng, Y. Wang, J. Zhou, Z. K. Yang, R. Cao, R. Sarangi, T. T. Sun, X. Yang, X. S. Zheng, W. S. Yan, Z. B. Zhuang, J. Li, W. X. Chen, D. S. Wang, J. T. Zhang and Y. D. Li, Nat. Commun., 2020, 11, 3049 CrossRef CAS PubMed.
- X. Wang, M. Li, P. Wang, D. M. Sun, L. F. Ding, H. Li, Y. W. Tang and G. T. Fu, Small Methods, 2023, 7, 2300100 CrossRef CAS PubMed.
- J. Y. Chen, C. Fan, X. Y. Hu, C. Wang, Z. H. Huang, G. T. Fu, J. M. Lee and Y. W. Tang, Small, 2019, 15, 1901518 CrossRef PubMed.
- Z. C. Nie, L. Zhang, Q. L. Zhu, Z. F. Ke, Y. T. Zhou, T. Wågberg and G. Z. Hu, J. Energy Chem., 2024, 88, 202–212 CrossRef CAS.
- H. M. Liu, M. M. Jin, D. Zhan, J. M. Wang, X. Y. Cai, Y. T. Qiu and L. F. Lai, Appl. Catal., B, 2020, 272, 118951 CrossRef CAS.
- Y. Lin, X. M. Chen, Y. X. Tuo, Y. Pan and J. Zhang, J. Energy Chem., 2022, 70, 27–35 CrossRef CAS.
- X. B. Yang, Y. Y. Wang, X. L. Tong and N. J. Yang, Adv. Energy Mater., 2022, 12, 2102261 CrossRef CAS.
- B. You, M. T. Tang, C. Tsai, F. Abild-Pedersen, X. L. Zheng and H. Li, Adv. Mater., 2019, 31, 1807001 CrossRef PubMed.
- Z. H. Xia and S. J. Guo, Chem. Soc. Rev., 2019, 48, 3265–3278 RSC.
- J. Yang, Z. Y. Wang, C. X. Huang, Y. Zhang, Q. H. Zhang, C. Chen, J. Y. Du, X. Zhou, Y. Zhang, H. Zhou, L. X. Wang, X. S. Zheng, L. Gu, L. M. Yang and Y. Wu, Angew. Chem., Int. Ed., 2021, 60, 22722–22728 CrossRef CAS PubMed.
- R. Chattot, T. Asset, P. Bordet, J. Drnec, L. Dubau and F. Maillard, ACS Catal., 2016, 7, 398–408 CrossRef.
- Z. J. Li, H. Jang, D. N. Qin, X. L. Jiang, X. Q. Ji, M. G. Kim, L. J. Zhang, X. E. Liu and J. Cho, J. Mater. Chem. A, 2021, 9, 4036–4043 RSC.
- L. F. Gu, C. F. Li, J. W. Zhao, L. J. Xie, J. Q. Wu, Q. Ren and G. R. Li, J. Mater. Chem. A, 2021, 9, 13279–13287 RSC.
- F. Du, X. T. Ling, Z. Y. Wang, S. G. Guo, Y. T. Zhang, H. C. He, G. L. Li, C. H. Jiang, Y. Zhou and Z. G. Zou, J. Catal., 2022, 389, 132–139 CrossRef.
- S. Zhu, L. T. Ding, X. H. Zhang, K. Wang, X. Wang, F. Yang and G. Y. Han, Angew. Chem., Int. Ed., 2023, 62, e202309545 CrossRef CAS PubMed.
- D. P. Xue, P. F. Yuan, S. Jiang, Y. F. Wei, Y. Zhou, C. L. Dong, W. F. Yan, S. C. Mu and J. N. Zhang, Nano Energy, 2023, 105, 108020 CrossRef CAS.
- Y. Y. Jiang, K. Yang, M. G. Li, D. H. Xu and Z. H. Ma, Mater. Chem. Front., 2024, 8, 528–552 RSC.
- H. S. Ahn and A. J. Bard, J. Am. Chem. Soc., 2015, 138, 313–318 CrossRef PubMed.
- B. J. Trześniewski, O. Diaz-Morales, D. A. Vermaas, A. Longo, W. Bras, M. T. M. Koper and W. A. Smith, J. Am. Chem. Soc., 2015, 137, 15112–15121 CrossRef PubMed.
- J. Y. C. Chen, L. Dang, H. Liang, W. Bi, J. B. Gerken, S. Jin, E. E. Alp and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 15090–15093 CrossRef CAS PubMed.
- Z. Fang, W. T. Zhao, T. Shen, D. P. Qiu, Y. C. Lv, X. M. Hou and Y. L. Hou, Precis. Chem., 2023, 1, 395–417 CrossRef CAS.
- J. O. Bockris and T. Otagawa, J. Electrochem. Soc., 2019, 131, 290–302 CrossRef.
- R. Larsson and L. Y. Johansson, J. Power Sources, 1990, 32, 253–260 CrossRef CAS.
- Y. R. Liu, X. J. Liu, Z. H. Lv, R. Liu, L. H. Li, J. M. Wang, W. X. Yang, X. Jiang, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2022, 61, e202117617 CrossRef CAS PubMed.
- G. G. Yang, J. W. Zhu, P. F. Yuan, Y. F. Hu, G. Qu, B. A. Lu, X. Y. Xue, H. B. Yin, W. Z. Cheng, J. Q. Cheng, W. J. Xu, J. Li, J. S. Hu, S. C. Mu and J. N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed.
- T. He, Y. Chen, Q. M. Liu, B. Z. Lu, X. W. Song, H. T. Liu, M. Liu, Y. N. Liu, Y. Zhang, X. P. Ouyang and S. W. Chen, Angew. Chem., Int. Ed., 2022, 61, e202201007 CrossRef CAS PubMed.
- Y. Wang, X. P. Li, M. M. Zhang, J. F. Zhang, Z. L. Chen, X. R. Zheng, Z. L. Tian, N. Q. Zhao, X. P. Han, K. Zaghib, Y. S. Wang, Y. D. Deng and W. B. Hu, Adv. Mater., 2022, 34, 2107053 CrossRef CAS PubMed.
- C. X. Zhao, B. Q. Li, J. N. Liu and Q. Zhang, Angew. Chem., Int. Ed., 2020, 60, 4448–4463 CrossRef PubMed.
- X. Zhao, F. L. Wang, X. P. Kong, R. Q. Fang and Y. W. Li, J. Am. Chem. Soc., 2021, 143, 16068–16077 CrossRef CAS PubMed.
- H. B. Liu, X. C. Xu, H. X. Xu, S. T. Wang, Z. Q. Niu, Q. H. Jia, L. Yang, R. Cao, L. R. Zheng and D. P. Cao, Appl. Catal., B, 2021, 297, 120451 CrossRef CAS.
- L. L. Wang, Q. An, X. L. Sheng, Z. Y. Mei, Q. Jing, X. Y. Zhao, Q. J. Xu, L. Y. Duan, X. X. Zou and H. Guo, Appl. Catal., B, 2024, 343, 123509 CrossRef CAS.
- Z. L. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS PubMed.
- J. H. Hong, C. H. Jin, J. Yuan and Z. Zhang, Adv. Mater., 2017, 29, 1606434 CrossRef PubMed.
- D. F. Yan, Y. X. Li, J. Huo, R. Chen, L. M. Dai and S. Y. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef PubMed.
- M. Zheng and J. Wang, Chem. Res. Chin. Univ., 2022, 38, 1275–1281 CrossRef CAS.
- B. Zhang, J. W. Shan, W. L. Wang, P. Tsiakaras and Y. Y. Li, Small, 2022, 18, 2106012 CrossRef CAS PubMed.
- H. L. Tu, H. X. Zhang, Y. H. Song, P. Z. Liu, Y. Hou, B. S. Xu, T. Liao, J. J. Guo and Z. Q. Sun, Adv. Sci., 2023, 10, 2305194 CrossRef CAS PubMed.
- F. L. Gong, Y. H. Liu, Y. Zhao, W. Liu, G. Zeng, G. Q. Wang, Y. H. Zhang, L. H. Gong and J. Liu, Angew. Chem., Int. Ed., 2023, 62, e202308091 CrossRef CAS PubMed.
- M. C. Luo and S. J. Guo, Nat. Rev. Mater., 2017, 2, 17059 CrossRef CAS.
- J. Kim, H. Jung, S. M. Jung, J. Hwang, D. Y. Kim, N. Lee, K. S. Kim, H. Kwon, Y. T. Kim, J. W. Han and J. K. Kim, J. Am. Chem. Soc., 2020, 143, 1399–1408 CrossRef PubMed.
- X. M. Wang, G. F. Long, B. Liu, Z. L. Li, W. S. Gao, P. F. Zhang, H. Zhang, X. Zhou, R. Z. Duan, W. Hu and C. Li, Angew. Chem., Int. Ed., 2023, 62, e202301562 CrossRef CAS PubMed.
- J. X. Sun, P. Leng, Y. H. Xie, X. X. Yu, K. G. Qu, L. G. Feng, H. F. Bao, F. Luo and Z. H. Yang, Appl. Catal., B, 2022, 319, 121905 CrossRef CAS.
- Y. J. Chen, H. Cui, Q. Jiang, X. Bai, P. Y. Shan, Z. P. Jia, S. Lu, P. Song, R. Feng, Q. Kang, Z. Y. Liang and H. K. Yuan, ACS Appl. Nano Mater., 2023, 6, 7694–7703 CrossRef CAS.
- N. Wang, R. G. Mei, L. Q. Chen, T. Yang, Z. W. Chen, X. D. Lin and Q. X. Liu, Small, 2024, 20, 2400327 CrossRef CAS PubMed.
- X. Y. Li, X. S. Wu, Y. Zhao, Y. X. Lin, J. H. Zhao, C. Q. Wu, H. J. Liu, L. Shan, L. Yang, L. Song and J. Jiang, Adv. Mater., 2023, 35, 2302467 CrossRef CAS PubMed.
- Y. T. He, J. B. Yang, Y. Wang, Y. F. Jia, H. T. Li, Y. N. Liu, L. T. Liu and Q. Tan, ACS Appl. Mater. Interfaces, 2024, 16, 12398–12406 CrossRef CAS PubMed.
- S. Yadav, V. Yadav, M. A. Siegler, P. Moënne-Loccoz, G. N. L. Jameson and D. P. Goldberg, J. Am. Chem. Soc., 2024, 146, 7915–7921 CrossRef CAS PubMed.
- E. Pastor, Z. Lian, L. Xia, D. Ecija, J. R. Galán-Mascarós, S. Barja, S. Giménez, J. Arbiol, N. López and F. P. García de Arquer, Nat. Rev. Chem., 2024, 8, 159–178 CrossRef PubMed.
- D. H. Li, Y. Jia, G. J. Chang, J. Chen, H. W. Liu, J. C. Wang, Y. F. Hu, Y. Z. Xia, D. J. Yang and X. D. Yao, Chem, 2018, 4, 2345–2356 CAS.
- Z. Lian, F. Dattila and N. López, Nat. Catal., 2024, 7, 401–411 CrossRef CAS.
- X. R. Zheng, Y. H. Cao, H. Z. Wang, J. F. Zhang, M. H. Zhao, Z. Huang, Y. Wang, L. Zhang, Y. D. Deng, W. B. Hu and X. P. Han, Angew. Chem., Int. Ed., 2023, 62, e202302689 CrossRef CAS PubMed.
- L. Xia, B. F. Gomes, W. Jiang, D. Escalera-López, Y. Wang, Y. Hu, A. Y. Faid, K. Wang, T. Chen, K. Zhao, X. Zhang, Y. Zhou, R. Ram, B. Polesso, A. Guha, J. Su, C. M. S. Lobo, M. Haumann, R. Spatschek, S. Sunde, L. Gan, M. Huang, X. Zhou, C. Roth, W. Lehnert, S. Cherevko, L. Gan, F. P. García de Arquer and M. Shviro, Nat. Mater., 2025 DOI:10.1038/s41563-025-02128-7.
| This journal is © The Royal Society of Chemistry 2025 |