
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantum life science: biological nano quantum sensors, quantum technology-based hyperpolarized MRI/NMR, quantum biology, and quantum biotechnology
Hiroshi
Yukawa
 *,
Hidetoshi
Kono
*,
Hitoshi
Ishiwata
,
Ryuji
Igarashi
,
Yoichi
Takakusagi
,
Shigeki
Arai
,
Yu
Hirano
,
Tetsuya
Suhara
and
Yoshinobu
Baba
*
*,
Hidetoshi
Kono
*,
Hitoshi
Ishiwata
,
Ryuji
Igarashi
,
Yoichi
Takakusagi
,
Shigeki
Arai
,
Yu
Hirano
,
Tetsuya
Suhara
and
Yoshinobu
Baba
*
Institute for Quantum Life Science, National Institutes for Quantum Science and Technology (QST), Anagawa 4-9-1, Inage-ku, Chiba 263-8555, Japan. E-mail: yukawa.hiroshi@qst.go.jp; kono.hidetoshi@qst.go.jp; baba.yoshinobu@qst.go.jp
First published on 28th January 2025
Abstract
The emerging field of quantum life science combines principles from quantum physics and biology to study fundamental life processes at the molecular level. Quantum mechanics, which describes the properties of small particles, can help explain how quantum phenomena such as tunnelling, superposition, and entanglement may play a role in biological systems. However, capturing these effects in living systems is a formidable challenge, as it involves dealing with dissipation and decoherence caused by the surrounding environment. We overview the current status of the quantum life sciences from technologies and topics in quantum biology. Technologies such as biological nano quantum sensors, quantum technology-based hyperpolarized MRI/NMR, high-speed 2D electronic spectrometers, and computer simulations are being developed to address these challenges. These interdisciplinary fields have the potential to revolutionize our understanding of living organisms and lead to advancements in genetics, molecular biology, medicine, and bioengineering.
 Hiroshi Yukawa | Dr Hiroshi Yukawa is a Project Director, Institute for Quantum Life Science in National Institutes for Quantum Science and Technology, Japan, and a Designated Professor, Institute of Nano-Life-Systems, Institutes of Innovation for Future Society, Nagoya University. He received his PhD in Medical Science from Nagoya University in 2010 under the supervision of Professor M. Hamaguchi, and received his PhD in Engineering from Tokyo University in 2011 under the supervision of Professor K. Araki. His current research focus is the application of quantum nano sensors to advanced medical sciences such as regenerative medicine, cancer medicine, and cranial nerves medicine. |
 Hidetoshi Kono | Dr Hidetoshi Kono earned his PhD from The University of Tokyo. After postdoctoral work at RIKEN's Tsukuba Life Science Center and the University of Pennsylvania, he joined the Japan Atomic Energy Agency. In 2007, he established a research group studying protein–DNA interactions through molecular modeling. Now Deputy Director General at the Institute for Quantum Life Science, National Institutes for Quantum Science and Technology, he investigates gene regulation mechanisms through molecular modeling, simulation, and single-molecule measurements using optical tweezers. |
 Hitoshi Ishiwata | Dr Hitoshi Ishiwata currently holds the position of Chief Researcher at the Institute for Quantum Life Science (iQLS), a part of the National Institutes for Quantum Science and Technology (QST). He completed his PhD at Stanford University in 2016, where he specialized in the use of diamondoids for photonics application and diamond growth. Following his doctoral studies, he focused on the fabrication of quantum sensors and the development of quantum measurement systems designed for the label-free detection of molecules within nanoscale detection volume. His expertise spans the fields of quantum sensor fabrication and quantum measurements of biological molecules. |
 Ryuji Igarashi | Dr Ryuji Igarashi is the Team Leader at the Institute for Quantum Life Science, part of the National Institutes for Quantum Science and Technology. He earned his PhD in Engineering from Kyoto University. Specializing in nanobioscience based on quantum sensing, Dr Igarashi has pioneered various measurement techniques using nanodiamond NV centers. He has developed selective fluorescence imaging, pH nanosensors, and three-dimensional rotational measurements. Furthermore, Dr Igarashi has been at the forefront of applying these techniques to biometric measurements, leading to notable advancements in the field. |
 Yoichi Takakusagi | Dr Yoichi Takakusagi is currently the Team Leader at the National Institutes for Quantum Science and Technology (QST), and a visiting Professor in Chiba University. He received his PhD in Pharmaceutical Science with honors from Tokyo University of Science in 2006. He joined the group of Dr Murali K. Cherukuri at NCI/NIH from 2011 to 2014. He belonged to Kyushu University (2014) and The University of Tokyo (2015) as a research fellow in the JST/CREST hyperpolarized NMR/MRI research project (Prof. Sando's team). His research interest lies in the development and application of theranostics. |
 Shigeki Arai | Dr Shigeki Arai received his PhD in 1999 from Gunma University, Japan. After postdoctoral research (1999–2004) in Neutron Science Research Center at Japan Atomic Energy Research Institute, he took up a position in Quantum Beam Science Directorate at Japan Atomic Energy Agency (2004–2016). In 2016, he moved to National Institutes for Quantum Science and Technology in Japan as a senior principal researcher. His research interests include quantum biology, biophysics, and quantum beam science. |
 Yu Hirano | Dr Yu Hirano received his PhD in 2007 from Graduate School of Science, Kyoto University, Japan. After postdoctoral research in Ibaraki University (2007–2009), in Kyoto University (2009–2013) and in Japan Atomic Energy Agency (2013–2016), he moved to National Institutes for Quantum Science and Technology as a senior researcher. His research interests include reaction mechanism catalyzed by redox proteins with high-resolution structural information. |
 Tetsuya Suhara | Dr Tetsuya Suhara is a psychiatrist and received PhD degree in 1991 from Jikei University School of medicine. He is now a Managing Director of Quantum Life and Medical Science Directorate, National Institutes for Quantum Science and Technology (QST). He has been using positron emission tomography (PET) to investigate the mechanism of neuropsychiatric disorders. Then he participated in the establishment of Quantum Life Science field in QST as a Deputy Director General of Institute for Quantum Life Science. He is author or co-author of 400 research publications. |
 Yoshinobu Baba | Dr Yoshinobu Baba received PhD degree from Kyushu University. He is now a Director General of Institute for Quantum Life Science, QST and a Professor of Nagoya University. He has been admitted as a Fellow of the Royal Society of Chemistry and received over 100 awards, prizes, and Medal. He is the author or co-author of 1160 publications and also an inventor of over 131 patents. He has delivered more than 1112 plenary and invited lectures. His work has been cited on 522 occasions by newspapers and television. |
1. Introduction
Quantum life science is an emerging field that combines principles from quantum physics and biology to study and understand various fundamental life processes such as photosynthesis, respiration, and magnetoreception at the molecular level. It explores how quantum phenomena, such as the tunnelling effect, quantum superposition, and quantum entanglement, may play a role in biological processes and systems. However, capturing such quantum effects is still challenging because the surrounding environment quickly causes dissipation and decoherence of the quantum system in warm and wet living systems. Quantum mechanics is the fundamental theory that can describe the properties of small particles such as electrons, atoms, and even molecules. All living systems are made up of molecules, and fundamentally, all molecules are described by quantum mechanics. However, there is a remarkable size and time scale gap between the system in which quantum mechanics operates and the living system. The technologies that will help solve these problems include biological nano quantum sensors, quantum technology-based hyperpolarized MRI/NMR, and molecular dynamics simulation.Quantum sensors are quantum devices that respond to external stimuli. Quantum sensors possess three characteristics: (1) discrete energy levels; (2) initialization, coherent manipulation, and readout; and (3) interaction with physical quantities. In particular, the nitrogen-vacancy-center (NV center), a fluorescent lattice defect that exists only slightly in diamond crystals, is attracting attention as a biological nano quantum sensor that functions at room temperature and pressure because of its high affinity to cells and living organisms. Quantum technology-based hyperpolarized MRI/NMR is the only method that employs quantum phenomena based on nuclear spin manipulation as the detection principle to detect the relaxation process among the various available medical diagnostic imaging techniques. This signal is expected to make it possible to monitor molecular and structural changes in many metabolites derived from cells and living bodies using the nuclear frequency of the NMR-positive nuclei as the chemical shift alteration at arbitrary magnetic field strength. In addition, practically and effectively, molecular dynamics simulation with the atomic model based on biomolecules’ 3D structures has contributed a lot to understanding how they function to achieve biological processes based on classical molecular mechanics. However, classical molecular mechanics cannot treat quantum effects because they employ empirical force fields that define fixed covalent bond topologies and typically fixed atomic charges. An open quantum systems (OQS) approach was recently applied to study proton tunnelling in the base pair of DNA.1 The OQS suggests that the proton transfer rate between the G–C base pair is several orders of magnitude larger than that calculated d by the classical mechanics, indicating DNA base mutation is more likely to occur in the living system.
These interdisciplinary fields in quantum life sciences have the strong potential to revolutionize our understanding of living organisms and will lead to advancements in genetics, molecular biology, medicine, and bioengineering (Fig. 1).
 | ||
| Fig. 1 Conceptual diagram of biological nano quantum sensors. Realization of nanodiamond quantum sensors to measure physicochemical parameters such as temperature, pH, magnetic field, and electric field in cells and living organisms. | ||
2. Biological nano quantum sensors
2.1. Quantum sensing and quantum systems
Quantum sensing harnesses quantum properties, including coherence, interference, and entanglement, to measure physical parameters with unparalleled precision and sensitivity.2–5 These quantum sensors often outperform their classical counterparts, enhancing accuracy across multiple applications. Traditionally, quantum sensors are built using superconductive devices,6 OPM,7 and trapped ions.8 However, these systems come with challenges. They demand extreme conditions, such as ultra-low temperatures and near-perfect vacuums, making them less feasible for biological measurements. Solid state spins such as NV centers in diamonds have emerged as brilliant solutions for applying quantum sensing to biological applications.9,10 Wide bandgap materials are perfect hosts for these spin states, producing isolated energy levels within solid-state crystals. Consequently, this results in electron spin states with coherence times spanning several microseconds, which are manipulable even at room temperature. The creation of new color centers has gained significant traction over the past decade, with researchers identifying over 500 such centers in materials like diamond,11 silicon carbide (SiC),12 hexagonal boron nitride (h-BN),13 aluminum nitride (AlN)14 and gallium nitride (GaN).15 Owing to its low spin–orbit coupling and the optimal photo cycle for spin state initialization and readout, the NV center in the diamond exhibits an extended coherence time and pronounced contrast. Moreover, a notable advantage of diamond NV center-based quantum sensors is their photostability16 and single-spin sensitivity.17 Conventional biological sensors, such as those using fluorescent dyes or proteins, require ensembles of molecules to achieve a reliable signal. This, in turn, demands larger measurement volumes, often in the micrometer range, due to optical instability. In contrast, NV centers in diamonds exhibit exceptional photostability and can function as highly sensitive sensors, even as individual lattice defects within molecular-sized particles.18 This enables fluorescent nanodiamonds to serve as nanoscale sensors, allowing precise measurements of physical and chemical parameters in biological microenvironments like mitochondria19 or membrane proteins.20 Consequently, it has been increasingly recognized as a color center for biological application, finding applications in areas such as in vivo thermometry21 and in-cell organelle measurements.19 The NV center's precise positioning is pivotal for advancing biological applications. Advancements like single-protein NMR (Fig. 2a–c)22 and single-molecule ESR (Fig. 2d–f)23 owe their success to accurately positioning an NV center nanometers away from the target spin. Upon integration into biological systems, quantum sensors can detect a range of physical attributes – magnetic24 and electric fields,25 temperature,26 pressure,27 and molecular diffusion28,29 with a groundbreaking sensitivity.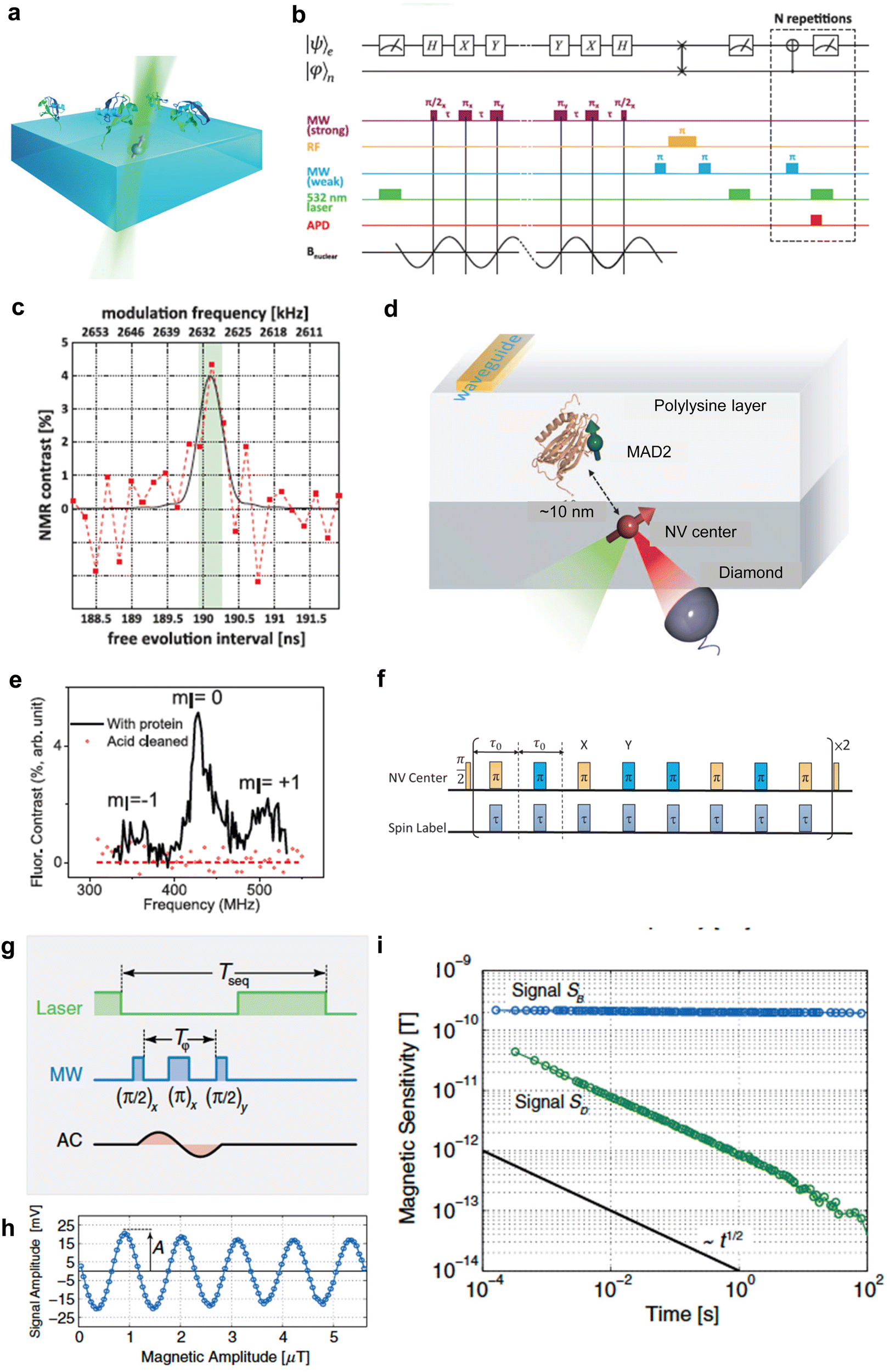 | ||
Fig. 2 Measurement of a single NV center. (a) Schematic depicting the experimental setup of ubiquitin proteins covalently tethered to the diamond's surface above a single NV center. (b) Pulse sequence for quantum sensing for detection of nuclear spin from single protein. (c) NMR spectra of 13C originating from a single ubiquitin protein. (d) Schematic depicting the experimental setup of MAD2 proteins labeled with nitroxide spin labels above a single NV center. (e) Pulse sequence for quantum sensing for detection of electron spin from a single protein. (f) Single spin ESR spectra under ambient conditions. (g) AC magnetic-field measurement scheme with pulsed sensor readout (h). Magnetic measurement of the test field with varying field amplitude retrieving sensor response A (i). Scaling of magnetic sensitivity (standard deviation over time) of signal SB (blue line) and SD (green line). The slope of the black line again indicates the aspired scaling behavior with  . Reproduced from ref. 22, 23 and 30 with permission from AAAS and APS. . Reproduced from ref. 22, 23 and 30 with permission from AAAS and APS. | ||
2.2. Sensitivity of quantum sensors
The sensitivity of a quantum sensor is fundamentally governed by spin projection noise. The sensitivity of the NV center in diamond is distinctively characterized by its optical readout.31 This is quantitatively represented by the shot-noise limited sensitivity, . Here T represents the coherent duration of a singular measurement. γ is the linear rate at which the spin state population changes concerning the unit target signal. N signifies the number of quantum sensors deployed in the measurement. The choice of measurement protocol directly influences the value of T. For instance, T is derived from T2* when employing the Ramsey measurement, whereas the Hahn-echo measurement necessitates deriving T from T2. Coherence enhancement achieved predominantly by mitigating spin noise sources like surrounding electrons and nuclear spins can augment the value of T. The parameter C is contingent upon the NV center's orientation within the diamond crystal lattice and its charge state. Conversely, N is influenced by the density of the diamond's NV centers and the optical excitation volume. The latter can be modulated by varying the optical excitation techniques, transitioning from, for example, confocal to bulk excitation methods. Subsequent sections will elucidate processes tailored to augment the intrinsic quality of the host crystal, methodologies for producing NV centers in diamond, and protocols for diamond surface treatments, all aimed at optimizing the parameters mentioned above.
. Here T represents the coherent duration of a singular measurement. γ is the linear rate at which the spin state population changes concerning the unit target signal. N signifies the number of quantum sensors deployed in the measurement. The choice of measurement protocol directly influences the value of T. For instance, T is derived from T2* when employing the Ramsey measurement, whereas the Hahn-echo measurement necessitates deriving T from T2. Coherence enhancement achieved predominantly by mitigating spin noise sources like surrounding electrons and nuclear spins can augment the value of T. The parameter C is contingent upon the NV center's orientation within the diamond crystal lattice and its charge state. Conversely, N is influenced by the density of the diamond's NV centers and the optical excitation volume. The latter can be modulated by varying the optical excitation techniques, transitioning from, for example, confocal to bulk excitation methods. Subsequent sections will elucidate processes tailored to augment the intrinsic quality of the host crystal, methodologies for producing NV centers in diamond, and protocols for diamond surface treatments, all aimed at optimizing the parameters mentioned above.
Quantitative analysis of the sensitivity of an NV center in diamond was achieved by applying an AC magnetic field synchronized with quantum manipulation pulses. The sensitivity was quantified by measuring the amplitude of the signal as a function of measurement time, and analyzing the resulting Allan deviation, as illustrated in Fig. 2g. An Allan deviation corresponding to a sensitivity of  at a frequency of 20 kHz was obtained.
at a frequency of 20 kHz was obtained.
2.3. Host crystal
The quality, inherent, and purity of the host material are pivotal in enhancing the coherence time of the NV center. Historically, the synthesis of diamond crystals has been challenging due to the greater stability of sp3-bonded carbon compared to sp2-bonded carbon at both atmospheric and lower temperatures. Consequently, it is difficult to produce diamond without the concurrent formation of sp2-bonded carbon, such as graphite. This distinction was notably illustrated in the Bachmann diagram during the early phases of diamond research.32 Industrial synthetic single-crystal diamonds are primarily synthesized using the high-pressure-high-temperature (HPHT) method. While diamonds produced via HPHT exhibit low strain, this technique grapples with issues related to precise impurity concentration control.33 Furthermore, managing the thickness and exact positioning of the quantum sensor remains a challenge with HPHT. An alternative approach, plasma-enhanced chemical vapor deposition (CVD), facilitates the homoepitaxial growth of diamond on HPHT substrates using CH4 as a carbon source. H2 serves as a carrier gas, producing a plasma that preferentially etches sp2 carbon formed during growth, ensuring the retention of sp3 carbon for premium diamond synthesis. Commercially, diamonds produced through CVD growth are available with nitrogen and boron concentrations of less than one part per billion.34 Further advancements include the isotopic purification of diamond crystals. Here, the naturally occurring 1% of 13C is refined to levels approaching 99.999% using highly isotopically purified gases.35 Employing high-quality diamond substrates, especially those with extreme isotopic purity, has resulted in record-setting coherence times of ∼1 ms for single NV centers in diamond (Fig. 3a–c).36 | ||
| Fig. 3 Spin coherence time of single spins in isotopically engineered diamond. (a) Fluorescence microscopy image of high-purity CVD diamond containing single nitrogen-vacancy defects. (b) FID signal measured on a single nitrogen-vacancy electron spin for diamond with a natural abundance of 13C isotope (black) and isotopically engineered crystal (red). And Fourier-transform spectra of FID signals. The satellites indicated by asterisks are related to the hyperfine interaction with the nitrogen and carbon nuclei. (c) Spin coherence time of single spins in isotopically engineered diamond measured using a two-pulse electron spin echo. (d) Detonation NDs produced by the detonation of carbon-containing explosives. The phase diagram shows that the most stable carbon phases for graphite and diamond are at low and high pressures, melting at temperatures above 4500 K. The phase diagrams for nanoscale carbon shows liquid phase at lower temperatures. In detonation, the pressure and temperature rise instantaneously, reaching the Jouguet point (point A), which falls within the region of liquid carbon clusters of 1–2 nm in size. As the temperature and pressure decrease along the red line, the growth of the diamond is replaced by the formation of graphite. (e) NDs produced by the HPHT methods. (f) Plots of temperature sensitivity versus diamond size Reproduced from ref. 36–39 with permission from Nature Portfolio, AIP Publishing and IOP Publishing Ltd. | ||
2.4. Fabrication of NV center
The fabrication of the NV center in diamond requires the precise introduction of nitrogen atoms and adjacent vacancies. This process is particularly challenging due to the extreme hardness of diamond, which complicates the insertion of impurities in close proximity40 to vacancies and necessitates specialized fabrication techniques. Conventional methods employ ion implantation to introduce nitrogen, electron irradiation to induce vacancies, and thermal annealing to promote the diffusion and consequent formation of the NV center within the diamond lattice.41 Through ion implantation, one can exert meticulous control over the spatial positioning of the introduced nitrogen, choose between the 14N or 15N isotopes, and regulate the concentration of NV centers formed in the host crystal.42 Leveraging this technique, a singular NV center has been successfully positioned as close as 10 nm from the diamond's surface,43 proving invaluable for single protein and single electron spin detection applications. The distinctiveness of the incorporated nitrogen isotope further aids in the unequivocal identification of the introduced species.23 To achieve precise spatial control over NV center location, integration with nanofabricated masks,44 focused ion beams,45 or nanometric apertures in AFM tips46 has been pursued. However, the exact depth control remains constrained to an approximation of 10 nm, a limitation arising from ion scattering and channeling phenomena.47 An alternative technique, laser writing, has demonstrated potential in generating vacancies and enabling three-dimensional localization of NV centers.48 Nevertheless, its in-plane accuracy remains tethered to the average inter-nitrogen spacing due to inherent challenges with vacancy diffusion.CVD growth is a prominent technique for precisely controlling the position, isotope, and alignment of the NV center within the diamond. Given that the nitrogen-to-carbon ratio can be adjusted by varying the gas ratio of nitrogen (N2) to methane (CH4), CVD offers unparalleled control over the NV center's density and nitrogen incorporation into the diamond matrix. The most substantial NV center densities within the diamond have been achieved by combining CVD growth, which introduces a high nitrogen concentration into the diamond, with electron irradiation at either the MeV range or 200 keV using an electron source from TEM.49,50 A lower acceleration voltage introduces vacancies more gently, resulting in enhanced coherence times and favorable NV center generation ratios. Recent innovations have facilitated the creation of high-density NV centers in diamonds, enabling the observation of spin interactions between adjacent NV centers. Furthermore, controlling the timing of gas introduction during CVD growth affords precision over the thickness and location of the resulting NV center. Through delta doping of nitrogen gas, CVD growth can confine the formation of an NV center within a tight 1–2 nm region.51
In the process where electron irradiation is paired with nitrogen implantation, the formation of the NV center in a diamond generally assumes a random orientation among the four possible [111] directions ([111], [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1], [1], [1]). To date, CVD growth remains the sole method allowing for the deliberate alignment of the NV center in the diamond crystal towards one of these four directions, enhancing the NV center's contrast to an optimal photo-cycle limit of approximately 30%. Alignment of the NV center within the diamond lattice via step-flow growth using a reduced methane concentration has been experimentally demonstrated by T. Fukui et al.,52 with corroborating theoretical calculations by T. Miyazaki et al.53 Subsequent research has pursued objectives like achieving a high-density ensemble of perfectly aligned NV centers,54 precisely aligned shallow NV centers suitable for nanoscale NMR measurements,55 and a 100 μm-thick diamond crystal tailored for high sensitivity applications.56 Incorporating tert-butylphosphine as a gaseous precursor during CVD growth has been explored to enhance the coherence time of these perfectly aligned NV centers.57 Furthermore, phosphorous doping via CVD growth has been employed to optimize the charge state of the NV center in diamonds, setting new benchmarks for the T2 and T2* values observed in individual NV centers.58
1], [1], [1]). To date, CVD growth remains the sole method allowing for the deliberate alignment of the NV center in the diamond crystal towards one of these four directions, enhancing the NV center's contrast to an optimal photo-cycle limit of approximately 30%. Alignment of the NV center within the diamond lattice via step-flow growth using a reduced methane concentration has been experimentally demonstrated by T. Fukui et al.,52 with corroborating theoretical calculations by T. Miyazaki et al.53 Subsequent research has pursued objectives like achieving a high-density ensemble of perfectly aligned NV centers,54 precisely aligned shallow NV centers suitable for nanoscale NMR measurements,55 and a 100 μm-thick diamond crystal tailored for high sensitivity applications.56 Incorporating tert-butylphosphine as a gaseous precursor during CVD growth has been explored to enhance the coherence time of these perfectly aligned NV centers.57 Furthermore, phosphorous doping via CVD growth has been employed to optimize the charge state of the NV center in diamonds, setting new benchmarks for the T2 and T2* values observed in individual NV centers.58
2.5. Nanodiamond
Nanodiamonds have been fabricated through many approaches, encompassing the detonation technique,59 high-pressure high-temperature (HPHT) synthesis,60 laser ultrasound cavitation,61 ablation,62 and microfabrication.63 The key metrics of interest for NV centers in nanodiamonds encompass the NV center's stability (attributed to charge state management), the quantity of NV centers, and the coherence time of the NV center within the diamond. This review delineates the distinctions between nanodiamonds synthesized via the detonation technique and HPHT synthesis, as these methods dominate commercial nanodiamond production.Detonation nanodiamonds (DNDs) derive from the detonation process. Specifically, the explosion of trinitrotoluene (TNT) and hexogen (RDX) blend within a confined metal chamber, leading to the high-pressure, high-temperature formation of 4–5 nm nanodiamonds (Fig. 3d and e).37 The hallmark of this method is its consistent yield of uniformly sized 4–5 nm nanodiamonds. However, DNDs often manifest as polycrystalline or ultra-nanocrystalline diamonds, typified by a patchwork of minute crystalline domains interspersed with sp2 carbon (such as graphite) grain boundaries. This results in elevated surface tension.64 The nitrogen concentration (P1 center) in DNDs, at approximately 1000 ppm, markedly exceeds that of HPHT-derived nanodiamonds, which hover around 100 ppm. This trait appears to favor the generation of NV centers in DNDs relative to HPHT nanodiamonds.65 Despite their high NV center content, DNDs are frequently sheathed in graphitic layers, necessitating rigorous cleaning protocols, such as acid treatments supplemented with sonication, to ensure stable emission of NV centers.18,64–66
Conversely, the predominant method for producing quantum sensing-applicable nanodiamonds entails milling HPHT micro-diamonds. Within the HPHT reactor, metal catalysts (like Fe, Ni, or Co) operate under extreme conditions to engender micro-diamonds infused with nitrogen (Fig. 3f).38,67 Post-fabrication, NV centers within these micro-diamonds are realized through electron irradiation, annealing, mechanical milling, and oxidation (a regimen designed to purge surface contaminants).68 Despite its prevalence in commercial nanodiamond production, this procedure has flaws. The mechanical milling phase tends to diminish the count of NV centers in the resultant nanodiamonds. Therefore, innovations that miniaturize micro-diamonds without compromising NV center quantities are actively sought. Recent research spotlighting HPHT diamond synthesis using molecular diamonds, termed “diamondoids” (e.g., adamantane), has renewed the field.69 The introduction of new catalysts has enabled reduced pressure and temperature conditions for HPHT synthesis stemming from adamantane.70 However, this method faces a significant challenge: adamantane, a diminutive diamond structure with a mere ten carbon atoms, is highly susceptible to disintegration. Any reaction undermining its structure will obliterate this diamond configuration, making reactions seeded by such a molecular diamond inherently unstable. Exploring larger molecular diamonds, like pentamantane, might pave the way for innovative nanodiamond production methodologies.71
The sensitivity of nanodiamonds, as discussed in Section 2.2, is influenced by several factors including longer coherence times, larger contrast, and a higher number of NV centers, which collectively enhance the measurement sensitivity of nanodiamonds. However, substantial variations exist in the actual values of these parameters, even when identical fabrication methods are used. Fig. 3f illustrates the dependence of sensitivity on the size of the nanodiamonds.39 Despite the inherent limitations in the characteristics of nanodiamonds, various measurement techniques have been developed to enhance sensitivity. The highest sensitivity has been achieved by integrating Magnetic Nano Particles (MNP) with nanodiamonds, a method that now rivals the sensitivity achieved by the MNP + ODMR methods due to recent advancements in nanodiamond fabrication.72
2.6. Diamond surface
Both single NV centers in diamond films and nanodiamonds offer material interfaces beneficial for biological applications. This is underscored by extensive research into the alteration of surface conductivity due to changes in surface termination.73 The charge state of the NV center that determines the stability of emission, contrast, and location of the NV center from the target of detection is controlled by the surface termination of diamonds. For single NV centers in diamond films, their precise localization relative to the diamond surface could be well defined by quantum measurement. Extensive studies have been performed to elucidate the effect of surface termination on location,43 contrast,74 and coherence75 of the NV center. Correlating quantum measurements with surface analysis via NEXAFS has deepened our understanding of diverse oxidation methodologies for diamond surfaces. Optimal conditions for a single NV center on a diamond film surface have been identified: air oxidation at 450 °C, which results in an optimized coherence time of 100 μs for centers located within 10 nm from the diamond surface.76 In contrast, nanodiamond surfaces often contend with contaminants like metal impurities, soots, and sp2 carbons during fabrication. These are typically removed through oxidizing acids or air oxidation, leading to carbonyl and carboxyl groups forming on the nanodiamonds.77–79 Much research has been dedicated to the surface functionalization of diamond films and nanodiamonds, specifically for the conjugation of molecules and proteins, offering a biological interface tailored for diverse applications.80–83 The prevailing challenge lies in precisely positioning the NV center within 5 nm from the surface, all while maintaining extended coherence times. Moreover, there's a need to accurately position biological targets on nanodiamonds or diamond films for prospective applications.2.7. Sensing by NV center
Nitrogen-vacancy centers (NV centers) are fluorescent lattice defects with impurity nitrogen and vacancies next to each other. Negatively charged NV centers have a stable triplet electron pair at room temperature. They are well known to emit strong near-infrared fluorescence with a quantum yield >0.7 upon photoexcitation at around 520–540 nm.84 Hereafter, NV centers with a negative charge will be called “NV centers.”An NV center functions as a quantum sensor by acquiring various physical and chemical information from the microenvironment as perturbations to the state of triplet electron spins. The energy levels of the triplet electron spins are approximated by the following spin Hamiltonian:
 | (1) |
![[S with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0053_20d1.gif) is the spin operator of the electron spin, D is the zero-field splitting width based on the spin–spin interaction, d‖ and d⊥ are the parallel and perpendicular components of the electric dipole moment,
is the spin operator of the electron spin, D is the zero-field splitting width based on the spin–spin interaction, d‖ and d⊥ are the parallel and perpendicular components of the electric dipole moment,  is the electric field, μB is the Bohr magneton, ge is the Landé g-factor for the NV center electron,
is the electric field, μB is the Bohr magneton, ge is the Landé g-factor for the NV center electron, ![[B with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0042_20d1.gif) is the magnetic field. Thus, it can be seen that the energy levels of the NV center change under various perturbations, such as electric and magnetic fields or thermal expansion of the crystal lattice, which can affect the spin–spin interaction.
is the magnetic field. Thus, it can be seen that the energy levels of the NV center change under various perturbations, such as electric and magnetic fields or thermal expansion of the crystal lattice, which can affect the spin–spin interaction.
The electron spin state of the NV center can be determined based on the fluorescence intensity because the spin magnetic quantum number of the triplet electron spin is strictly conjugated to its electronic transition pathway in the NV center. Fig. 4a shows the paths of electronic and spin transitions of the electron spins. Almost all the occupied electrons in the |0〉 emit photons through vibrational relaxation of the normal fluorescence pathway. On the other hand, about half of the electrons in the |±1〉 undergo vibrational relaxation through the intersystem crossing, in which they transit to and shelved in the singlet state and then return to the triplet state without emitting any fluorescence.85 The lifetime of vibrational relaxation in the fluorescence pathway is about 10 ns, whereas the lifetime of vibrational relaxation via intersystem crossing is more than 10 times longer.85,86 This means that the photoexcited electrons in the |±1〉 state cannot be ready for the next fluorescence cycle for a much longer time than those in the |0〉 state, emitting weaker fluorescence. Therefore, the spin population is determined based on the fluorescence intensity, and the magnetic resonance is measured by optical detection. This measurement, called optically detected magnetic resonance (ODMR), is the basis of quantum sensing using NV centers.
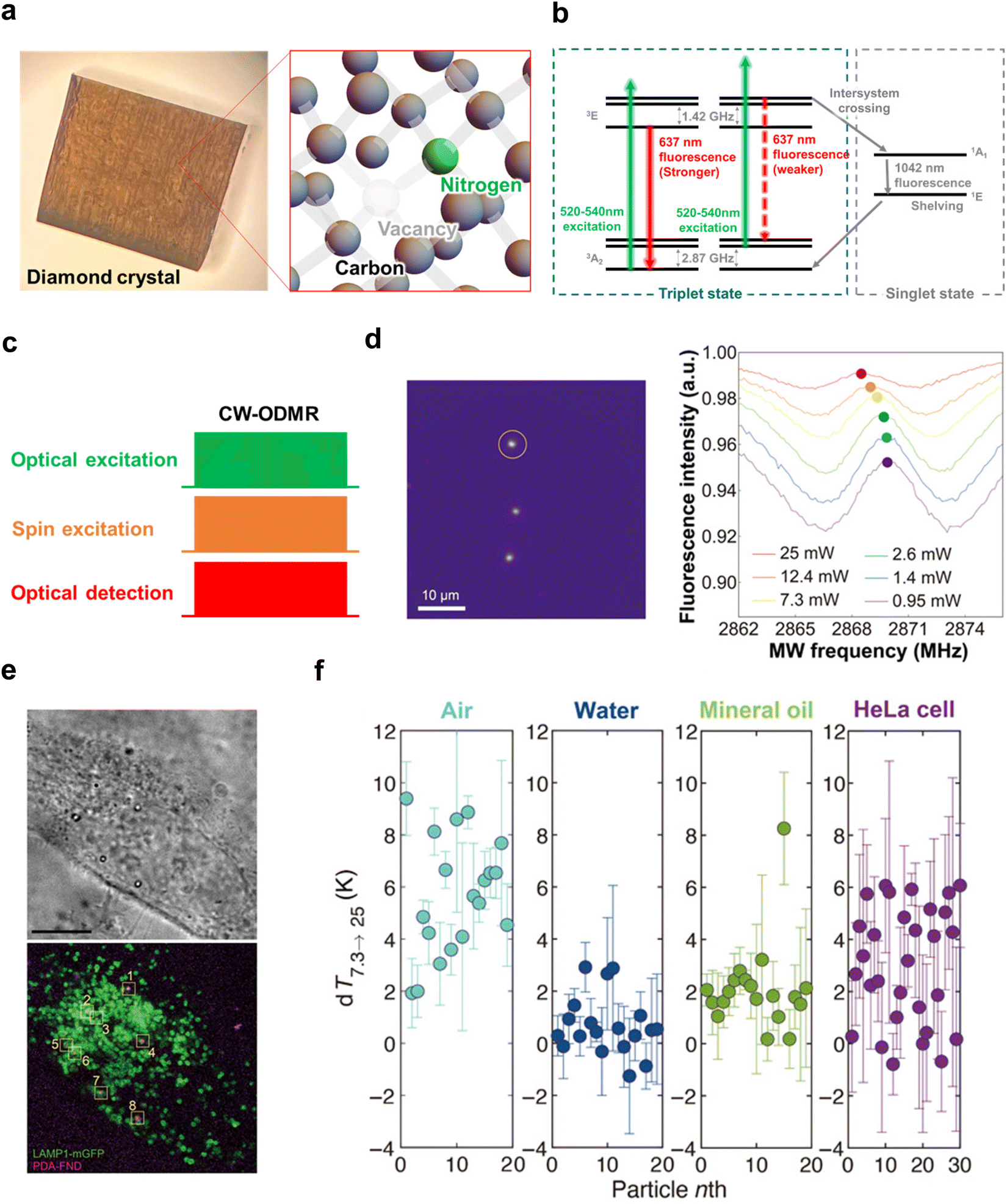 | ||
| Fig. 4 Characteristics of the diamond NV center and measurement example using CW-ODMR. (a) Left: Photograph of a Type Ib diamond substrate after enrichment of NV centers through electron irradiation. Right: Schematic representation of the lattice defect structure of the NV center in diamond. (b) Energy level diagram of the NV center, illustrating various electron transition processes corresponding to different values of the spin magnetic quantum number, ms. (c) A typical pulse sequence of CW-ODMR. (d) Fluorescent image of thermogenic fluorescent nanodiamonds based on polydopamine surface coating. The ODMR frequency spectra detected from the NV center of the nanodiamond marked by the yellow circle in the image at different excitation laser powers are also shown. A temperature increase with rising laser power is observed as a low-frequency shift in ODMR. (e) Bright-field image (top) and fluorescence image (bottom) of HeLa cells overexpressing LAMP1-mGFP incubated for 4 hours with polydopamine-coated fluorescent nanodiamonds. (f) Thermal conductivity measurements in air, water, mineral oil, and HeLa cells. Circles with error bars represent the mean and standard deviation (SD) of changes in excitation power from 7.3 mW to 25 mW (denoted as dT7.3 → 25) measured for individual particles under different conditions. The numbers on the x-axis refer to the identifiers given to particles used in measurements for each medium.87Fig. 4d–f were reproduced from ref. 87 with permission from AAAS, copyright 2021. | ||
However, this mechanism cannot fully explain why NV centers can function as highly sensitive sensors. In fact, the triplet electron spin in the NV center has an energy level difference of only 2.87 GHz between |0〉 and |±1〉 at zero magnetic fields, and the Zeeman energy follows the electron magnetic rotation ratio of 2.8 MHz Gauss−1. In other words, the electron spin polarization ratio based on the Boltzmann distribution is hopelessly tiny, and the measurement sensitivity in thermal equilibrium should be almost the same as that of ordinary electron spin resonance. However, as shown in Fig. 4b, most photo-excited electrons in the |±1〉 state relax to the ground state of |0〉 via intersystem crossing. The photoexcited electrons of |±1〉 also relax to the ground state of |0〉 via spin-allowed transitions, indicating that the photoexcitation cycle polarizes the triplet electron spin to the ground state of |0〉. The polarization rate exceeds 80% after photoexcitation cycles lasting from a few hundred nanoseconds to several microseconds.83 This optical polarization process, often called “initialization,” is generally used to reset the state to its initial state, |0〉. Consequently, magnetic resonance measurements of optically polarized NV centers can achieve sensitivity levels comparable to fluorescence detection. Indeed, the spin resonance of individual NV centers can be easily observed using a confocal fluorescence microscope stage.17
2.8. NV centers as a quantum sensor
To use NV centers as a quantum sensor, the perturbation to NV centers from the microenvironment is measured as spin population after certain spin manipulations and time evolutions.88 For examples of spin manipulation, the temperature is determined by using the spin resonance frequency spectrum obtained by continuous-wave ODMR (CW-ODMR).18,21,26,87 DC magnetic field is evaluated by measuring the spin magnetization precession by Ramsey fringe measurement (so-called “free induction decay (FID)”).36,89 Microenvironmental pH and radical are obtained from the longitudinal relaxation time T1 measured by the inversion/saturation recovery method (T1 relaxometry).90–93 AC magnetic field is detected based on transverse relaxation time T2 by Hahn echo or dynamic decoupling (T2 relaxometry).10,30The acquisition of spin magnetic frequency spectra (Fig. 4c–f) is currently the most frequently used method in life measurements. The measurement procedure is simple: continuous microwaves (or microwave pulses with a pulse width equivalent to a π pulse) are irradiated while sweeping the frequency. When the NV center is at a non-resonant microwave frequency (off-resonance), the spin population of |0〉 is more than 80%, whereas at a resonant microwave frequency (on-resonance), the spin population of |0〉 decreases due to spin excitation from |0〉 to |±1〉. The resonance frequency can be obtained by plotting the fluorescence intensity against the applied microwave frequencies (this plot is called “spin resonance frequency spectrum” or simply “ODMR spectrum”) because a decrease in fluorescence intensity is observed at on-resonance. Since the resonance frequency is equivalent to the energy level derived from Hamiltonian (1), this measurement allows us to know the external electric and magnetic fields, as well as the temperature that affects the spin–spin interaction through the expansion and contraction of the crystal lattice, from the ODMR spectrum.94
2.9. Application of NV centers to magnetic field detection
In addition to obtaining ODMR spectra, another method for obtaining the resonance frequency of NV centers is the Ramsey fringe measurement (Fig. 5a–c), in which a microwave pulse orthogonal to the external magnetic field (z-axis) is applied to obtain information on the precession of spin magnetization in the xy-plane. The information obtained is similar to FID's in pulsed Fourier NMR (nuclear magnetic resonance) when an π/2 pulse is applied. However, unlike NMR, in the Ramsey fringe measurement of NV centers, the spin precession is detected based on the fluorescence intensity, not the induced electromotive force generated in the coil by electromagnetic induction. For this purpose, information on the spin precession should be converted into information on the number of spin occupations, which can be detected as the fluorescence intensity with high sensitivity. This is achieved by applying a second π/2 pulse to the electron spins after the free precession time τ and transferring the x-axis (or y-axis) component of the spin magnetization vector to the z-axis component (= spin quantization axis). This makes it possible to detect the phase of the spin magnetization at time τ based on the population of each spin magnetic quantum number, and by sweeping time τ as well, an NMR-like FID can be obtained. The Fourier transform of this FID yields the resonance frequency of the NV centers. However, while pulse Fourier NMR is overwhelmingly more useful than CW-NMR, in the case of NV centers, the use of ODMR spectra or FID depends on the purpose and experimental conditions. This is because, in the case of pulsed Fourier NMR, it is possible to obtain a spectrum of the required frequency band in a single shot of radio frequency pulse, which is a forward-compatible alternative to CW-NMR. In contrast, it is difficult to excite the required frequency band by a single shot of microwave pulse in the case of NV centers. Moreover, a sweep of time τ is also required because it is necessary to break the precession and convert the phase information into spin magnetic quantum number information. In addition, when using a nanodiamond as a probe, it is often necessary to acquire spectra in the absence of an external magnetic field (= zero magnetic field) for temperature measurements because the resonance frequency changes due to the free rotation of the nanodiamond. Therefore, Ramsey fringe measurements cannot alternate the acquisition of ODMR spectra by CW-ODMR. On the other hand, Ramsey fringe measurement has become an indispensable technique in high-sensitivity quantum sensor technology, for example, the D-Ramsey pulse sequence that cancels the effect of magnetic field fluctuations95,96 and the use of two-quantum pulse sequences and spin-bath driving (decoupling from bus spins)89,97 for high sensitivity magnetic field measurement. Its application to life measurements is expected to accelerate in the future. However, Ramsey fringe measurements can be used to achieve higher sensitivity in temperature measurements, for example, by using D-Ramsey pulse sequences that cancel the effects of magnetic field fluctuations,89,90 and higher sensitivity in magnetic field measurements by using two-quantum pulse sequences and decoupling from bus spins (spin-bath driving),89,97 have become indispensable techniques in high-sensitivity quantum sensing, and their application to life measurements is expected to accelerate in the future. | ||
| Fig. 5 Examination of biometric measurements using Ramsey fringe. (a) A typical pulse sequence of Ramsey fringe. (b) Schematic representation of the magnetic field generated by a neuron. The illustration depicts an axon positioned on a diamond substrate. The “Potential” refers to the vertical change in membrane potential (Vm) of a crayfish lateral axon, as cited from Watanabe et al. (J. Gen. Physiol. 1961, 45, 267–308). The excitation propagates along the axon, resulting in time-dependent changes in the magnetic field at a distance of 100 nm. (c) Outline of neural measurements utilizing the NV center. The NV center's spin, controlled by a 2.88 GHz microwave, is perturbed by the magnetic field generated by the axon. These changes can be monitored based on the intensity of red fluorescence using a CCD or CMOS camera. Fig. 5b and c were reproduced from ref. 98 with permission from Springer Nature, copyright 2012. | ||
2.10. Application of NV center to pH detection
Inversion/saturation recovery T1 relaxometry (Fig. 6a–d) is a measurement method currently being applied to life science. The inversion/saturation recovery method is a measurement technique that monitors the relaxation from the thermal equilibrium state until the inverted or saturated spins return to the thermal equilibrium state again (spin–lattice relaxation, longitudinal relaxation). This measurement is often called T1 relaxometry because the time constant for this relaxation has a name: longitudinal relaxation time T1. T1 is particularly sensitive to external magnetic and electrical influences and is known to be useful for measuring magnetic and radical concentrations (and related protein and cellular function).19,99,100 It has also been recently reported that T1 of NV centers in a nanodiamond can be used to measure microenvironmental chemical parameters such as pH with appropriate chemical modifications on the surface of the nanodiamond.90,91 Because of its applicability to various measurements and simplicity of measurement, T1 relaxometry is one of the most widely used measurement methods for life measurements using NV centers. In particular, simplicity of measurement is important for life measurements because biological samples such as cultured cells and animal tissues are generally delicate, and their conditions change from moment to moment. Therefore, it is necessary to start measurement as quickly as possible, omitting trial-and-error in advance as much as possible, and to obtain highly immediate information in a short measurement time. In addition, prolonged application of excitation light and microwaves can cause changes in the state of the samples, although the light and microwaves are low toxic to biological samples. Therefore, in pulsed ODMR measurements, such as Ramsey fringe measurements, for example, the determination of the π/2 pulse before the measurement is one of the obstacles to the development of the application to biological measurements. To determine the π/2 pulse, it is necessary to determine the resonance frequency in an appropriate magnetic field and perform a Rabi measurement. In contrast, T1 relaxometry requires neither an external magnetic field nor microwave irradiation but only laser pulses to initialize the spins to |0〉 and read out the spin population to track the relaxation process. Furthermore, contrast imaging-like techniques provide high temporal resolution on the order of seconds,92,101 making it an ideal measurement technique for biological measurements, with low toxicity to the biological sample and short measurement time. | ||
| Fig. 6 Example of pH measurement using T1 relaxometry. (a) A typical pulse sequence for T1 relaxometry. (b) Mechanism of T1 shortening with the rise in pH in surface-carboxylated fluorescent nanodiamonds. The carboxyl groups become negatively charged due to deprotonation as the pH increases.91 (c) Decay of T1 in the NV centers of carboxylated fluorescent nanodiamonds under different pH conditions.91 The inset shows the fluorescent nanodiamonds with a scale bar of 1 μm. (d) T1-Enhanced fluorescence images of carboxylated nanodiamonds (particle diameter 100 nm) under various pH conditions (top row) and corresponding signal intensity plots (middle row). The red line represents the results of curve fitting using a Gaussian function. The bottom row shows the integral of the fitted Gaussian function for each pH condition. Error bars represent the standard error from Gaussian fitting.92Fig. 6b–c and d were reproduced with permission from ref. 91 (ACS, copyright 2019) and ref. 92 (MDPI, copyright 2020), respectively. | ||
2.11. Biological application of NV centers in nanodiamonds
Cellular temperature, pH, viscosity, and electric and magnetic fields are known as very important physicochemical parameters of the intracellular environment, as it has been pointed out that they may play a significant role in cell activity by affecting the dynamics and reactivity of biomolecules. Indeed, the increased activity of macrophages and immune cells during fever is one such example. In addition, cell division, gene expression, protein production, and metabolic activity have also been shown to be very closely related to cell temperature, pH, viscosity, and electric and magnetic fields,102 and cell physicochemical parameters have become very important for understanding cell biological processes.Therefore, NV centers in nanodiamonds that can measure physicochemical parameters (temperature, pH, viscosity, electric and magnetic fields, etc.), which largely reflect the cellular state, are useful. NV centers in nanodiamonds exhibit ODMR and can measure physicochemical parameters such as temperature from fluorescence intensity, pH from spin lifetime,103 viscosity from fluorescence bright spot mobility, and magnetic field from the optical detection of Zeeman splitting. It has already been reported that it is possible to measure physicochemical parameters using the optical detection of Zeeman splitting. This method can be accelerated with widefield imaging104 or integrated with chip-based microwave architecture for bioassay platforms.105 Specifically, by measuring the temperature of stem cells using NV centers in nanodiamonds, it has been shown that the ability of stem cells to produce regenerative factors such as HGF and VEGF and their differentiation function changes significantly with temperature.106 In addition, 3D tracking of the motor rotation of a motor protein (F1-ATPase), which is related to the efficacy of drugs, has been achieved by utilizing the magnetic field measurement capability of the NV center in nanodiamonds. Furthermore, nanodiamonds, including NV centers with carboxy groups on the surface, can measure pH in a tiny area inside a cell using the spin relaxation induced by pH change. By taking advantage of these quantum characteristics, the effects of physicochemical parameters of cells on the expression of their functions have not yet been fully elucidated, and the application to advanced biomedicine, such as cancer medicine, immunology, neuroscience, and regenerative medicine, is strongly anticipated. Recently, optoelectronical nanoparticles and 2D semiconductor nanomaterials are proactively utilized for the development of wearable health monitoring systems due to their unique properties, such as high sensitivity, flexibility, and biocompatibility.107–109 Considering this trend, diamond NV centers have potential as they can be incorporated into wearable devices to provide real-time, non-invasive monitoring of key physiological parameters. Their stable fluorescence, ability to measure physical and chemical parameters, and integration with flexible sensing systems suggest that diamond NV centers could be adapted for health tracking in applications like continuous temperature monitoring, biochemical sensing, and real-time physiological assessments.
3. Quantum technology-based hyperpolarized MRI/NMR
3.1. Nuclear magnetic resonance spectroscopy and imaging
Since the first report on the nuclear magnetic resonance (NMR) phenomenon in the 1940s,110,111 NMR spectroscopy has been established and used in various scientific fields such as chemical structure elucidation and drug/protein interaction analysis.112 Magnetic resonance imaging (MRI), initially developed in 1973,113 supports medical diagnostics, and its practical applications have been realized in (bio)chemical experiments and disease diagnosis by the manipulation of nuclear spins in the magnetic field using radio frequency (RF) pulses to obtain spectra or images. As a versatile quantum phenomenon-based technology, NMR/MRI has contributed to the progress of modern life sciences and medicine.MR essentially employs an RF pulse, which exhibits high penetration in living organisms, to mainly excite the spins of water or lipid protons (1H, nuclear spin I = 1/2) oriented in two possible directions within the magnetic field by Zeeman splitting.114 The resulting relaxation process is detected from the resonator coil as a weak current (free induction decay (FID)) and then amplified and converted to NMR spectra or 2D/3D images using the Fourier transform. It should be noted that “low sensitivity” in the NMR phenomenon is a fundamental technical drawback, which can be attributed to the small difference in the population of the two energy states of nuclear spins following the Boltzmann distribution under thermal equilibrium, involved in MR signal detection (Fig. 7a and b).114
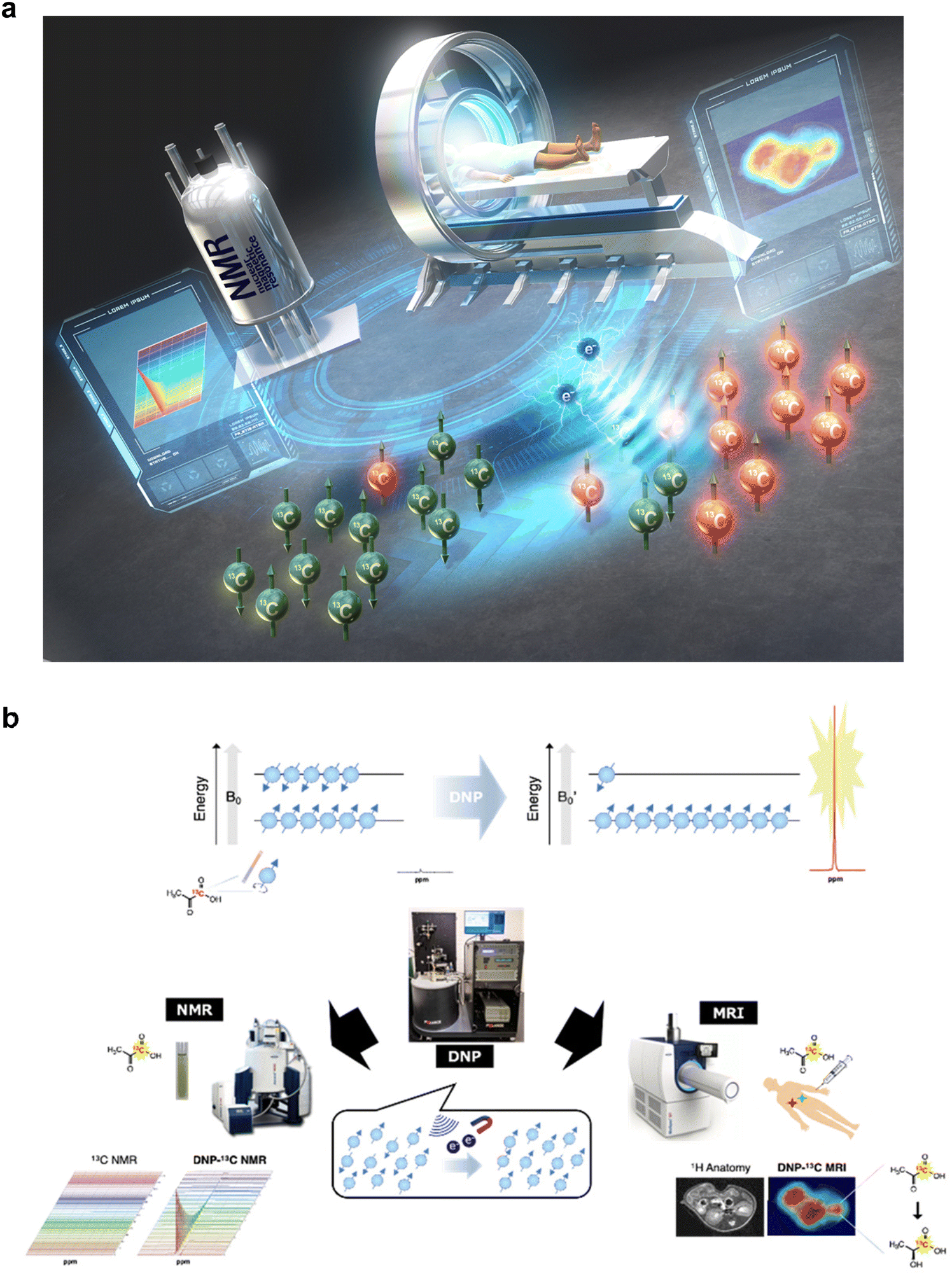 | ||
Fig. 7 Conceptual diagram of quantum technology-based hyperpolarized MRI/NMR and schematic of nuclear HP and subsequent measurement by NMR/MRI. (a) For the establishment of hyperpolarized NMR/MRI, benchtop-sized nuclear hyperpolarization (HP) machines without cryogen and much more sophisticated HP sites for clinical dissolution DNP (dDNP) technologies. (b) Nuclear spins (I = 1/2) align two possible directions within the magnetic field (B0) by Zeeman splitting. The slight difference in the two energy states following the Boltzmann distribution under thermal equilibrium contributes to the resulting intrinsically “low-sensitivity” MR signals. The proliferation of nuclear spins by DNP increases the population difference in the two energy states, and the dissolution of the following sample generates aqueous samples without polarization loss. Quick sample delivery to detectors allows in vitro spectroscopy (left) or in vivo spectroscopic imaging (right) based on >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold enhanced MR signals (hypersensitive NMR/MRI). Fig. 7b was reproduced from Fig. 2 and 3 in Takakusagi Y. Brain Neural Networks, 30(4), 179–188, with permission from the Japanese Neural Network Society, copyright 2023. 000-fold enhanced MR signals (hypersensitive NMR/MRI). Fig. 7b was reproduced from Fig. 2 and 3 in Takakusagi Y. Brain Neural Networks, 30(4), 179–188, with permission from the Japanese Neural Network Society, copyright 2023. | ||
3.2. Nuclear hyperpolarization
Various techniques have been developed to improve the intrinsically low-sensitivity signals in MR to maximize its applications in the biochemical and biomedical domains, which can be categorized into “nuclear hyperpolarization (HP)” by dynamic nuclear polarization (DNP).115 DNP is a powerful method for enhancing the sensitivity of NMR/MRI, and it has garnered significant attention in recent years. The original concept of spin diffusion can be backtracked to the theoretical background proposed by Albert Overhauser in 1953.116 This interaction occurs between unpaired electrons and NMR-positive nuclei upon irradiation at the electron paramagnetic resonance (EPR) frequency, following the transfer of the excitation of electron spin energy to the nuclei and significantly enhancing the NMR signals of the nuclei. Subsequently, this phenomenon was experimentally demonstrated117 and applied to 1D and 2D NMR techniques for inter- and intramolecular analyses. DNP in solid samples, mixed with unpaired radicals and NMR-positive nuclei, was accomplished in the 1980s and applied for molecular structural elucidation (nuclear Overhauser effect (NOE))118 or solid-state NMR.119 The DNP process can be described by three commonly accepted mechanisms, depending on experimental conditions, i.e., the solid effect (SE), cross effect (CE), and thermal mixing (TM).120 Applications of DNP have been extended to medical diagnostic imaging. Spin exchange optical pumping (SEOP) is a pioneering method in clinical HP measurement used to excite the nuclear spin of noble gas atoms (3He, 129Xe, etc.) by the direct collision of alkali metal atoms (Rb, etc.) whose unpaired electrons are excited by laser irradiation, thereby enhancing nuclear spin polarization by several tens of percent.121,122 This gas-based HP method has been successfully approved as medical diagnostics for chronic obstructive pulmonary disease (COPD) or other lung-related diseases where the 1H density is relatively low in vivo. Overhauser-enhanced MRI (OMRI), also known as proton electron double resonance imaging (PEDRI),123 is a DNP-based method for enhancing intrinsically low-sensitivity NMR signals. A paramagnetic contrast agent with a narrow EPR linewidth is used as a host radical during exogenous injection and irradiated to excite its spin polarization; then, the polarization is transferred to ambient nuclear spins to enhance the NMR sensitivity. This imaging provides highly resolved anatomical information with EPR spectroscopic functional information such as partial pressure of oxygen (pO2),124 redox status,125 microvascular permeability,126etc.Parahydrogen-induced polarization (PHIP), which is not categorized as an Overhauser-type DNP, is an alternative way to induce nuclear spin polarization by the chemical adduction of parahydrogen.127
Parahydrogen is a nuclear spin isomer of hydrogen with magnetization. Owing to the hydrogenation reaction with unsaturated bonds in molecular probes with a catalyst and via field cycling in zero magnetic field chambers, the magnetization is transferred to labeled nuclear spins (13C, 15N, etc.) in the molecular probe to enhance the following MR signals.128 The recent developments by SABRE or SABRE-SHEATH have accomplished PHIP in a reversible catalytic cycle at lower concentration levels in microtesla fields within a magnetic shield, thereby reducing the HP substrate required for NMR/MRI measurements.129–132 A commercial PHIP polarizer has recently been developed for preclinical and clinical studies (POLARIS, NVision Imaging Technologies GmbH, Germany). In addition, various molecular probes containing intramolecular unsaturated chemical bonds or introducing unsaturated bonds using a “side arm” for hydrogenation (SAH) in molecules have contributed to the expansion of PHIP utilization.133
3.3. Dissolution-DNP (dDNP)
In 2003, Ardenkjær-Larsen et al. developed an innovative apparatus for obtaining HP aqueous samples. This technique allows the rapid dissolution of highly polarized solid samples as a liquid with suitable temperature and minimal polarization loss, enabling the injection of living bodies to detect spectroscopic signals or metabolic reactions based on >10000-fold enhanced signals compared to the thermal equilibrium value.134 For a nucleus with spin = 1/2, the polarization level can be expressed by the Maxwell–Boltzmann statistics: | (2) |
Here, N+ and N− denote the number of spins in the up and down directions, respectively; moreover, B0 is the magnetic field strength, γ is the gyromagnetic ratio of the nucleus, T is the temperature, k is the Boltzmann constant, and ħ is the Dirac constant.114 To increase the population difference in two energy states, i.e., to create an HP state, a stronger magnetic field and cryogenic temperature are required.
Experimentally, low-molecular-mass compounds (MW ∼200) labeled with NMR-positive nuclei, such as 13C or 15N, are dissolved in a glassing agent doped with an electron paramagnetic agent (EPA). The mixture is conditioned in a cryogenic magnetic environment (3.35–7 T, 0.7–1.5 K); subsequently, owing to the irradiation of unpaired electrons with microwaves, the spin polarization is transferred from the electrons to the nuclei, which can be monitored as polarization buildup via solid-state NMR. These polarized samples must be dissolved in a superheated buffer to warm up to around the biological temperature, maintaining the polarization, physiological pH, and isotonicity. The resulting solution is subjected to NMR/MRI for in vitro spectroscopy or in vivo imaging using enhanced MR signals (Fig. 7a and b).
Unlike other MRI techniques, longitudinal relaxation by HP magnetization is nonrecoverable after dissolution from the magnetic field in the hyperpolarizer. The longitudinal magnetization, Mz(t), can be represented as:
 | (3) |
Thus far, several polarizers have been commercially developed to realize dDNP in typical laboratory environments. The apparatus of the prototype polarizer (3.35 T) contained a manual dissolution system with a dissolution wand.134 In 2006, HyperSenseTM, an automated hyperpolarizer (HyperSenseTM, Oxford Instruments, 3.35 T, 94 GHz, 1.4 K), was developed, and by 2009, over 40 machines had been sold. In 2011, a polarizer for viewing real-time cell activity (SPINlabTM, GE Research Circle Technology, 5 T, 140 GHz, 0.7 K) was introduced; this machine exhibited high throughput and was fully automated with fluid-path sample dissolution equipped with four channels (i.e., up to four samples can be simultaneously polarized in parallel).135 SPINlabTM was later transformed into a clinical hyperpolarizer with a quality control (QC) unit, and there are currently over 25 machines worldwide.136 More than 100 clinical studies have been conducted on the dissolution of DNP for future metabolic imaging.137,138 In 2018, a state-of-the-art cryogen-free hyperpolarizer (SpinAlignerTM, 6.7 T, 1.3 K) was commercially launched by Polarize IVS (Denmark).139 As of 2023, 15 SpinAligner machines have been installed globally. Furthermore, homemade polarizers140–143 or brute force polarization that enforces nuclear spin polarization in the sub-milli K temperature range has been developed and used for HP studies.144
3.4. Application for metabolic imaging
The advent of the dDNP technique has resulted in forming a new academic discipline, i.e., real-time metabolic imaging. Its application was first reported in rats and pigs. Golman et al. reported real-time monitoring of the metabolic fate of 13C1-enriched hyperpolarized pyruvate by detecting lactate, alanine, and bicarbonate as metabolites.145,146 Pyruvate is produced at the intersection of oxidative phosphorylation (OXPHOS) and anaerobic fermentation. In normal tissues, pyruvate enters mitochondrial metabolism, generating 36 ATPs in total via OXPHOS. In cancerous or inflamed tissues, pyruvate metabolism shifts from OXPHOS to lactate generation in the cytosol by increasing aerobic glycolysis due to elevated lactate dehydrogenase (LDH) activity, known as the Warburg effect.147 This metabolic shift is highly correlated with malignant transformation and therapeutic response in cancer, and a significant increase in the conversion of HP [1-13C]pyruvate to lactate can be a potential metabolic biomarker.148In vivo imaging studies have been conducted for healthy and diseased rodent models as preclinical research.149 Furthermore, this metabolic imaging has been successfully integrated into preclinical and clinical cancer research for the diagnosis of aggressiveness or prognosis of prostate cancer, renal cell carcinoma, hepatocellular carcinoma, glioma, pancreatic cancer, breast cancer, therapeutic response upon treatment with inhibitors, anti-angiogenesis, hypoxia-activated prodrugs, X-irradiation, androgen cessation therapy, and cancer immunotherapy.135,137,138,150 Its neurological applications in healthy human brains, age, sex, effects of anesthesia, cellular studies of immunometabolism, and primary brain diseases have also been investigated.151 Moreover, the metabolism in other lifestyle diseases, such as heart, kidney, diabetes, liver, and non-alcoholic fatty liver disease and liver fibrosis, has been examined to validate the disease state.1513.5. Molecular probes
Since the demonstration of [1-13C]pyruvate as a promising molecular probe for detecting metabolic reprogramming as a hallmark of cancer, studies on potential metabolic biomarkers have generated various functional molecular probes.152–154 These include [1,4-13C2]fumarate for detecting tumor necrosis, [1-13C]dehydroascorbic acid for redox monitoring, [1-13C]alpha-ketoglutarate for IDH1 activity in glioma, [1-13C]glycerate for assessing glycolysis, 13C-urea and 15N+-α-trideuteromethylglutamate for HP perfusion imaging, and [13C]bicarbonate as a pH sensor.152–154 Furthermore, the de novo design of molecular probes has generated [1-13C]pyruvate ethyl ester for metabolic imaging in rat brains, 13C-benzoyl formic acid and 13C-thiourea for H2O2 detection, ion chelators as in vitro metal sensors, and in vitro and in vivo pH sensors.152–154 Amino acids are components of the body and are attractive templates of the HP molecular probe with higher solubility and less toxicity.155 The amino acid-based molecular probes that maintain a longer HP lifetime in carbonyl carbon (position 1) and modified side chains were generated. These include [5-13C]glutamine for probing the glutaminase activity and treatment response, [5-13C]glutamate for imaging biomarker for IDH1 mutational status in glioma, aspartate, glycine, serine and their derivatives labeled with 13C at the C-1 position and [1-13C]lysine ethyl ester for pH sensor, [6-13C,15N3]arginine for in vivo arginase activity.152–154 Furthermore, [1-13C]alanine was applied for non-invasive assessment of telomere maintenance mechanisms in brain tumors,156 [1-13C]alanine amide for detecting aminopeptidase N (APN) activity157 and simultaneous evaluation of renal physiological state,158 and [1-13C]N-acetyl cysteine to monitor glutathione redox chemistry.159 The recent development of peptide-based molecular probes (γ-Glu-[1-13C]Gly) has enabled the direct monitoring of γ-glutamyl transpeptidase (GGT) activity.160,161 In particular, Ala-[1-13C]Gly-d2-NMe2, the second generation of APN probes that enabled in vivo APN activity, possess “self-glassing” characteristics, similar to pyruvate;162 in other words, glassing agents (glycerol, DMSO, or others) necessary for DNP, which are possibly toxic to living organisms in some cases, are not required. Recently, an optimal glutamine probe, [5-13C,4,4-d2,5-15N]-L-glutamine, which prolonged 13C relaxation at position 5, successfully monitored the in vivo glutamine metabolism in pancreatic cancer.163To generate functional HP molecular probes, it is important to design molecules with longer HP lifetimes and target specificity, including enzymes or various physiological states. The T1 value is generally correlated with the HP lifetime and is often used as its index. The relationship between the nuclear spin state and the molecular structure surrounding the nuclei can be understood according to the relaxation theory, as indicated by the following equation:
 | (4) |
Here, DD represents the dipole–dipole interaction between HP 13C and 1H, CSA is the chemical anisotropy that depends on the orientation of the molecule in the static magnetic field, SR is the spin rotation of a covalent bond connected to labeled and other atoms, and SC is the Scholar coupling with neighboring atoms. It should be noted that the precise investigation of the extent of each relaxation factor is a guideline for minimizing the relaxation factors and rationally designing functional molecular probes that can maintain longer HP lifetimes for labeled nuclear spins for in vivo imaging.152 Following this theory, 13C-labeled t-Bu benzene164 and 15N-labeled trimethylphenyl ammonium (TMPA)165,166 were rationally designed, indicating the longest HP lifetime (sulfonated 13C t-Bu benzene in D2O: 209 ± 7 s (9.4 T, 37 °C), [15N, D14]TMPA in D2O: 1177 ± 52 s (9.4 T, 30 °C)) among the HP probes of small molecules.
3.6. Other applications
In addition to developing instrumentation, various techniques and applications have been designed to maximize HP efficiency and reduce the complexity of HP measurements. These include automatic sample transfer from the polarizer to detector,167 construction of magnetic shield tunnels to maintain HP signals in molecular probes during transfer,168 and transferring hyperpolarized solid samples.169 Furthermore, the addition of trace amount of Gd ions to DNP sample formulation has been studied for enhancing the polarization,170,171 deuteration of EPA and glassy matrix to discard dipole–dipole interactions,172 DNP without glassing agent,173 UV irradiation for 13C probes to generate non-persistent radicals used for DNP,174,175 porous functional materials (HYPOP or HYPSO) to immobilize EPA for DNP and eliminate the process of EPA removal before injection into the living body,176,177 and transport metabolites from large-scale HP centers to end users in remote dedicated sites.178,179 Challenges to in vitro use were also analyzed, which could not otherwise be experimentally investigated, including the direct monitoring of cell metabolism, use of cell bioreactors, or tissue slice bioreactors, spheroids, brain organoids, and organs.151,180–182 Moreover, drug–protein interactions have been validated via DNP-NMR.183,1843.7. HP at room temperature
As mentioned earlier, the development of SEOP or PHIP enabled the nuclear HP of noble gases or functional molecular probes at room temperature without expensive cryogens.185 The development of dDNP with cryogen-free techniques has helped expand the use of HP metabolic imaging in preclinical and clinical sites with access to existing MRI scanners. Although the electron spin polarization is only 0.76% at 298 K in 3.35 T, photoexcited electron triplet states, which do not follow the Boltzmann distribution, resulted in HP at room temperature.186,187 Using lasers with an appropriate wavelength, pentacene in the excited singlet state could be converted to the triplet state through intersystem crossing, thereby exhibiting more polarization than the thermal equilibrium state.188 The polarization was then transferred to short T1 nuclei (e.g., 1H) following the labeled nuclei (e.g., 13C) in benzoic acid, regardless of the range of temperature or strength of magnetic field.189 While signal amplification using this technique was generally achieved with single or co-crystals of host–guest molecules, the recent developments allowed sample dissolution without polarization loss, thus broadening the scope of this technique.190,191 Nevertheless, triplet-DNP measurements are limited by the magnitude of signal amplification, mainly due to the low solubility of pentacene or the limited characteristics of guest molecules available for this polarization. To expand the utility of these techniques, several challenges have been reported via material development, such as the use of a metal–organic framework (MOF) composite containing pentacene, the generation of more dispersible pentacene derivatives, and the use of porphyrin as a biocompatible polarizing agent.192,193 Meanwhile, host medicinal agents or biological components can be used as the guest molecule for measuring triplet-DNP.194–196 Recently, the spin polarization of quintet multiexcitons generated by singlet fission (SF) has been reported to improve the sensitivity of MR of water molecules through DNP, indicating the utility of SF as a polarized spin generator in the bio-quantum technology.197 Moreover, drug–protein interaction analysis has also been achieved via triplet DNP-NMR.198 Further studies can improve the performance of the HP technique at room temperature.3.8. Perspectives
Among the various available medical diagnostic imaging techniques, MRI is the only method that employs a quantum phenomenon as the detection principle based on nuclear spin manipulation and detects the relaxation process. This signal allows the monitoring of molecular structural changes in metabolites using the nuclear frequency of the NMR-positive nuclei as the chemical shift alteration at arbitrary magnetic field strength. Combined use with nuclear HP is only possible in this quantum-based imaging technique to improve the sensitivity. Moreover, utilizing the difference in the observed spin frequency, single or multiple molecules can be simultaneously detected within several tens of seconds of a single scan.199–201 Further, combined use with other modalities (e.g., HyperPET combined with FDG-PET)199,202 will improve the clinical diagnostic accuracy by monitoring the tissue metabolism in the disease state or therapeutic response (metabolotheranostics).203 Even in the field of life sciences, dDNP will contribute to basic studies in various fields such as cancer, neurological studies, lifestyle diseases, regenerative medicine, and drug discovery studies targeting intrinsically disordered proteins in cellular milieus.204,205 The development of HP molecular probes in each type of nuclear HP modality will also expand the utility of the approach.206 Technical developments, e.g., benchtop-sized HP machines without cryogen or much more sophisticated HP sites, will facilitate the application of preclinical and clinical dDNP technologies to establish hypersensitive NMR/MRI.4. Quantum biology and quantum biotechnology
4.1. Quantum biology
Quantum biology is a new interdisciplinary scientific field that applies quantum principles such as the tunnelling effect, quantum superposition, quantum entanglement, and coherence to study processes in biological systems that are difficult or impossible to describe using classical physical laws alone. At present, magnetoreception, energy transfer in photosynthesis, olfaction, and enzymatic reactions are the main research subjects of quantum biology (Fig. 8). However, it is challenging to detect such effects because quantum effects may have little to no impact on biological processes in warm and wet living systems because the biological environment quickly causes dissipation and decoherence of the quantum system. Previously, olfaction was considered to depend on the shape and size of odorants fitting an olfactory receptor. However, the odorant of which hydrogen was substituted with deuterium resulted in odor change, suggesting vibrational frequency might play a role in the olfaction.207 Experiments with inelastic electron tunneling spectroscopy were carried out to validate and examine the proposed vibrational theory of olfaction. However, it is still controversial.208,209 In this chapter, we focus on the magnetoreception of migratory birds and photosynthesis in plants, which are currently well-studied as representative examples of research topics in quantum biology. | ||
| Fig. 8 Conceptual diagram of quantum biology and quantum biotechnology. Quantum effects which play a critical role in a variety of biological functions, including photosynthesis, magnetoreception, and enzymatic reactions. Their mechanisms are not well understood and are a hot topic in quantum biology. | ||
4.2. Radical pair mechanism in magnetoreception
Magnetoreception is the ability of organisms to receive geomagnetic information used for orientation, migratory, homing, feeding, breeding, etc. Many species, including birds, mammals, reptiles, amphibians, fish, crustaceans, insects, and plants, have been reported to have the magnetoreception.210 Since the 2000s, cryptochromes (CRYs), which are highly conserved blue light–absorbing flavoproteins containing a flavin adenine dinucleotide (FAD), have been attracting researcher's attention as possible quantum sensors of magnetoreception.211,212 CRYs in many species, such as CRY4s of Erithacus rubecula (European robin),213Columba livia (pigeon),214Gallus gallus (chicken),215–217 CRY1a of Sylvia borin (garden warbler),218 CRY of Drosophila melanogaster (fruit fly),219,220 CRY1 and CRY2 of Danaus plexippus (monarch butterfly),219 CRY2 of Periplaneta americana (cockroach),221 CRY1 of Arabidopsis thaliana,222–224etc. have been proposed to act as magnetosensors.The fundamental principle of the CRY magnetosensor is that the long-lived spin-correlated radical pair (RP) generated in CRY detects the angle of the magnetic field.212–214,216,217,220,223,225–227 The reaction schemes of avian CRY4s proposed from previous studies are summarized in Fig. 9a. When the blue light or UV irradiates the oxidized FAD (FADox) of the grand state in CRY, an excited singlet state of FAD (s[FAD*]) is produced. Then, the electron transfer from three or four tryptophans (TrpA395, TrpB372, TrpC318, and TrpD369 conserved in CRY4s of E. rubecula, C. livia, and G. gallus) to FAD is induced sequentially, and radical pairs (RPA, RPB, RPC and RPD) consisting of FAD and Trps are formed. In this reaction scheme, high-rate constants of RPA → RPB and RPB → RPC lead RPC and RPD that form the composite RP due to the dynamic equilibrium of RPC ↔ RPD. This composite RP is in a spin-correlated state consisting of an anion FAD radical (FAD˙−) and a cationic Trp radical (TrpH˙+),213 which can oscillate between electronic singlet (S) state (S[FAD˙− TrpH˙+]) and triplet (T) state (T[FAD˙− TrpH˙+]) under the influence of intramolecular electron–nuclear hyperfine interactions (“Intersystem crossing A” in Fig. 9a). The relative angle between the external magnetic field line and the composite RP can affect the extent and frequency of the S–T mixing process, which could alter the quantum yield of the subsequent, stabilized RP (S,T[FADH˙ Trp˙] in Fig. 9a) that consists of a neutral semiquinone radical (FADH˙) and a neutral Trp radical (Trp˙).218,228,229 Several spectroscopic analyses, such as transient absorption analysis near 50 μT, revealed that the reaction process from the composite RP to the stabilized RP proceeds over a time scale of 1 to 10 μs.213,225 This time scale is longer than required to receive geomagnetic information (approximately one μs in vivo).225 The quantum yield of the stabilized RP altered by the magnetic field effect could trigger the magnetic signaling from CRY4 to the nervous system, probably through the assistance from Tyr319, the conformational change in the C-terminal tail accompanying the change in the interaction to unknown CRY receptors.216,217,230 This RP mechanism also called the “CRY hypothesis,” is now widely believed to be the first step in geomagnetic sensing.213,216,231–233
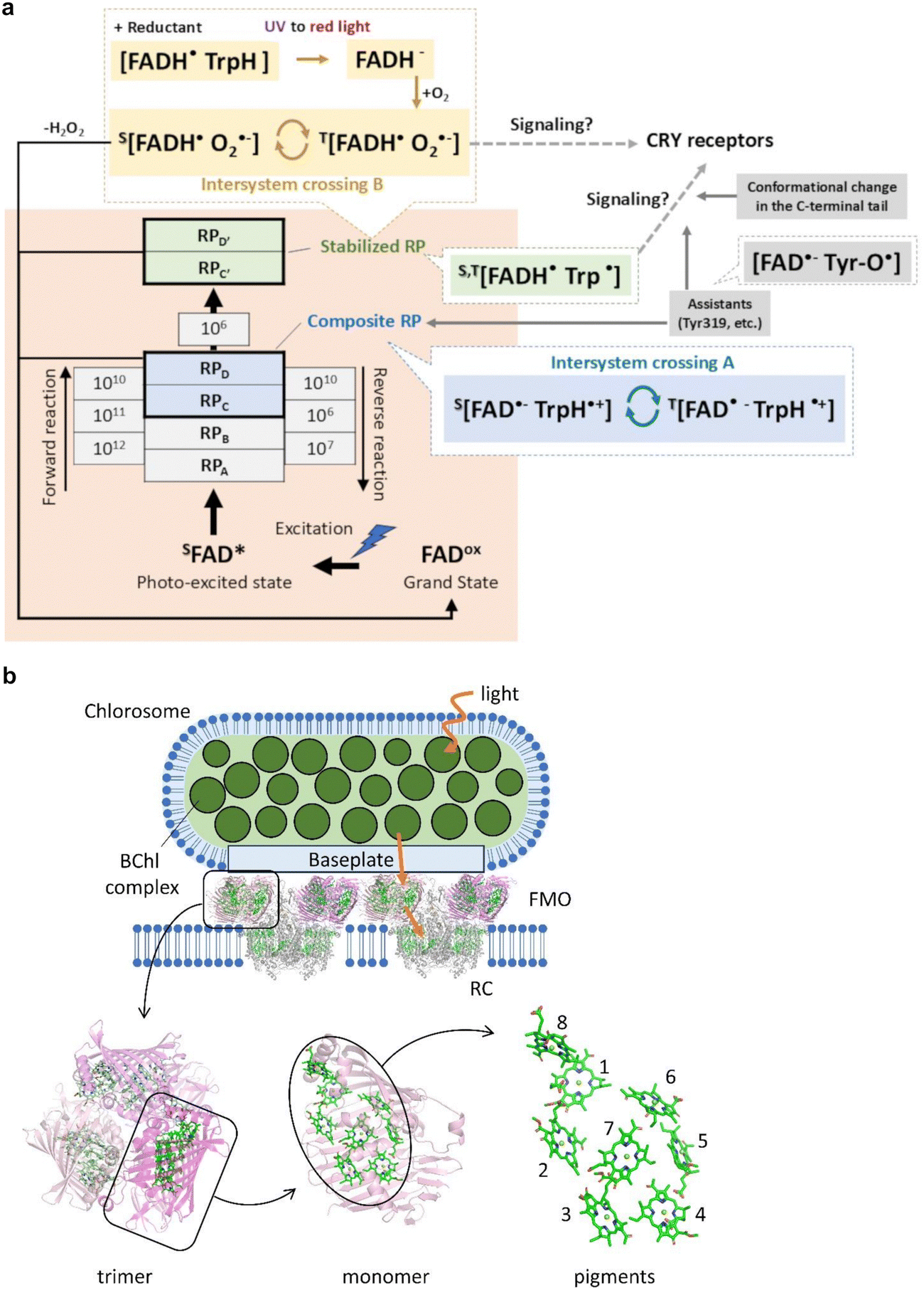 | ||
| Fig. 9 Reaction scheme of avian CRY4 and light-harvesting system in green sulfur bacteria. (a) Superscripts S and T indicate singlet and triplet states, respectively. The orange area shows the reaction scheme determined from E. rubecula CRY4 recently.213 The stabilized RP (green) converted from the composite RP (blue) would trigger the signaling. The curve arrows indicate the coherent, magnetic field-dependent intersystem crossing. The broken arrows indicate the unknown processes. The magnetic signaling could be proceeded by the assistance from Tyr319 and/or the conformational change in the C-terminal tail (grey).216,217,230 Another process (yellow) was also proposed.225,227,234 The CRY receptors have not yet been identified, but many candidates have been proposed.221,235–238 (b) Chlorosomes contain 100000–200000 BChl c molecules, and self-aggregated BChls are packed inside a lipid envelope. The chlorosome protein CmsA forms the oligomeric BChl a-CmsA complexes in the chlorosome envelope called baseplate. Homotrimers of the FMO proteins associate with the baseplate of chlorosome. The FMO proteins connect the chlorosome with the RCs embedded in the cytoplasmic membrane. The structural models for the FMO-RC complex (PDB ID: 7Z6Q)239 and the FMO protein (PDB ID: 3ENI)240 are shown as ribbons for protein moieties and sticks for pigments. Fig. 9a was reproduced from Fig. 4a from ref. 213 with permission form Springer Nature, copyright 2024. | ||
4.3. Universality of CRY hypothesis in magnetoreception
Even focusing on the avian CRY4s again, processes other than those described in Section 4.1 have been reported. For example, the highly conserved Tyr319 (colored in grey in Fig. 9a) may lead to the formation of RPs consisting of [FAD˙− Tyr-O˙] with highly long lifetimes (tens of microseconds or more).216,227 However, it remains unclear whether these RPs constitute entangled states and how they contribute to magnetoreception. The existence of RP formed by FADH˙ and superoxide O2˙−, which can oscillate between S–T states (“Intersystem crossing B” in Fig. 9a), was also reported.227,234,241 This process may relate to the signaling in the dark condition;227 however, there may be another additional process before the signaling because the extremely fast spin relaxation in O2˙− is unsuitable as a magnetosensor.234 Moreover, not only CRY4 but also CRY1a in birds have a magnetic sensitivity,242,243 whereas the identity of RP of CRY1a has not yet been elucidated.224,244–249 Also, vertebrate CRY1a, including birds, seems not to bind FAD strongly in vitro.248,250In invertebrates, the situation is more complicated. The magnetic-dependent binary-choice behavioral assay using D. melanogaster revealed that the CRY-defective mutant flies lost their magnetoreception, indicating that CRY is required for the magnetoreception of D. melanogaster.251 Surprisingly, a recent study showed that the 52 C-terminal amino acid residues (CT) of D. melanogaster CRY, lacking the canonical FAD-binding domain and Trp residues, were sufficient to facilitate magnetoreception in vivo.252 Moreover, several studies suggested that molecules other than CRY, probably high levels of FAD alone, could generate magnetically sensitive RPs and produce a biological effect under appropriate conditions.252–254 These observations suggested that D. melanogaster CRY-CT acts as an adaptor, bringing photoactivated FAD close to other effector molecules, including unknown CRY receptors.235–238,252
Despite many continuous discoveries, it still needs to entirely explain the magnetoreception based only on current knowledge. The perceptual mechanism of magnetic information is likely to be diverse across species and may be difficult to understand in line with human sensory modalities. Although we have gradually accumulated physical and biological knowledge of CRY-based magnetoreception, many mysteries remain, including how sensed magnetic information is propagated to the brain and contributes to decision-making for the direction.
4.4. Light-dependent reactions in photosynthesis
Photosynthesis is the fundamental biological process of converting light into chemical energy, essential for living organisms. Photosynthetic organisms are classified into the oxygenic and anoxygenic types. The former is observed in higher plants, algae, and cyanobacteria, and the latter in green sulfur and purple bacteria. Photosynthesis for both types consists of two separate processes: the light-dependent reactions and the carbon fixation reactions. In higher plants, the light-dependent reactions are conducted through pigment–protein complexes embedded in thylakoid membranes inside chloroplasts.255 The light-dependent reactions start with the capture of sunlight, which is mainly performed by light-harvesting complexes (LHC I & II), and excitation energy by sunlight is transferred to the reaction centers (RC) of photosystems (PS I & II).256 The energy transfer process from LHCs to the RCs achieves high quantum efficiency.257,258 The excitation energy, transferred from LHC, is absorbed by chlorophyll (Chl) molecules bound with the RC, and an electronically excited state of Chls results in a charge-separated state.259,260 Sequential electron transfer steps stabilize the charge-separated state via the cofactors in the RC. The light-induced electron transfer leads to the linear electron transfer and the proton transfer driven by the four membrane protein complexes, PS II, cytochrome b6f, PS I, and ATP synthase. Finally, the light-dependent reactions produce the energy-carrying molecule ATP and the reducing-power NADPH, and they are utilized in the subsequent carbon fixation reactions where carbon dioxides (CO2) are converted to carbohydrates.In oxygenic photosynthesis, PS II is the center of water oxidation, which produces dioxygen (O2) at the oxygen-evolving complex (OEC). The water oxidation cycle contains electron and proton removing steps during four light absorption events.261 A high-resolution X-ray crystal structure of PS II has made significant progress in understanding the water-splitting mechanism.262 The three-dimensional structure clarified the atomic arrangement of OEC as the Mn4CaO5 cluster. However, later theoretical works demonstrated that the Mn4CaO5 complex was a mixture of oxidation states because of photoreduction by exposure to intense X-rays of synchrotron.263,264 Recent advances in X-ray free-electron lasers (XFEL) enable solving the structure with less X-ray damage using femtosecond (fs) pulsed X-rays,265 and various states of the catalytic cycle in water oxidation have been determined by a pump–probe method using an optical laser in combination with XFEL.266,267 Based on the structural information, the electronic structure of the Mn4CaO5 complex has been studied by spectroscopic analyses and quantum chemical calculations to understand the catalytic process of water oxidation.268
4.5. Energy transfer in the light-harvesting system in photoreception
The absorption of sunlight by pigment molecules in light-harvesting systems causes electronic excitation states of the pigments, and then the excitation energy is efficiently funneled to the RC. Förster's theory describes the excitation energy transfer in which the coulombic interaction mediates the energy transfer between spatially separated pigments, and the excitation energy incoherently hops through adjacent pigment molecules.269 In photosynthetic light-harvesting systems, the pigment molecules are densely assembled in protein scaffolds, and electronic solid coupling between pigments results in a delocalized excitation state among pigments.270 The Redfield theory showed that extensive electronic coupling causes excitation relaxation.271 The delocalized states in light-harvesting systems were positioned in the intermediate regime between weak (Förster's case) and large (Redfield's case) electronic coupling, implying that quantum coherence might participate in efficient energy transfers.271,272 A direct observation of quantum coherence of electronic excitation states in the Fenna–Matthews–Olson (FMO) protein, the light-harvesting complex of green sulfur bacteria, was reported by Engel et al.273 Their 2D spectroscopic analysis at the cryogenic temperature (77 K) demonstrated long-lived “quantum beating” for >600 fs that was longer than the dephasing timescale of electronic coherence in the FMO protein predicted by theoretical studies.274 Since then, many spectroscopic and theoretical works have been reported to investigate whether quantum coherences contribute to efficient energy transfers by light-harvesting systems.271,275–277 In particular, the high-speed 2D electronic spectrometer has become an invaluable tool because it can capture ultrafast coherent and non-coherent processes with high temporal and spectral resolution, allowing us to observe quantum effects occurring in complex biological systems with multiple chromophores or energy transfer pathways. When combined with molecular beam techniques, 2D electronic spectroscopy can study biological molecules with minimal environmental perturbations, allowing for the observation of intrinsic quantum effects.278 The spectrometers disentangle vibrational coherence by resolving both excitation and detection frequencies. Cross-peaks of the 2D spectra can provide information about coupling between different energy levels, enabling us to identify vibrational coherences. In addition, measuring the 2D spectra over time also provides a dephasing time for each of the coherences.279 Thus, the high-speed 2D electronic spectrometer has unique advantages for studying quantum effects in biological systems, such as energy transfer pathways and coherent dynamics that may be responsible for the high efficiency of specific processes and observation of intrinsic quantum effects. However, many challenges remain in understanding 2D electron spectra. It is still difficult to interpret the origins of spectra because they can contain contributions from multiple chromophores and energy transfer processes. Collecting data on specimens that differ slightly, for instance, the environment or the number of chromophores, will help us develop analysis methods and build theoretical frameworks, finally understanding quantum effects in biomolecules.Green sulfur bacteria have large light-harvesting antennae called chlorosomes (Fig. 9b).280 The light energy absorbed by bacteriochlorophyll (BChl) molecules in chlorosomes is transferred to the RCs via the FMO proteins. The FMO proteins form a homotrimer, and each monomer contains eight BChl molecules with a specific geometrical arrangement.240 The eight BChls showed partially delocalized excitation states,240,281 and spectroscopic data suggested that the BChl 3 is the linker pigment between the FMO protein and the RC.240 The three-dimensional structures of the FMO-RC complex by cryo-electron microscopy (cryo-EM) revealed that the two BChls 3 in each FMO trimer are located close to the RC.239,282,283 Suppose quantum coherence plays a role in efficient excitation energy transfer in photosynthetic apparatus. In that case, coherence must also be present at physiological temperatures, even if thermal vibrations in the protein bath accelerate decoherence. The FMO protein showed that long-lived quantum coherence was preserved even at physiological temperature (277 K).284 Correlated motions within protein environments were thought to assist the preservation of long-lived quantum coherence in the pigment–protein complexes.285,286 Spectroscopic and theoretical studies have suggested how quantum coherence influences efficient energy transfers that vibronic coupling, the interaction between electronic and nuclear vibrational motion, drives excitation energy transfers in photosynthesis.287,288
The interpretation of quantum coherence in photosynthesis has raised considerable debate.275–277 Spectroscopic analyses of some groups demonstrated that the dephasing of electronic coherences in the FMO protein progressed faster than that observed in the previous study.275,289,290 Their works suggested that the observed long-lived beating derives from vibrational modes on the electronic ground state.275 Further experimental and theoretical studies will continue to prove the involvement of coherences in photosynthetic energy transfers. Spectroscopic and theoretical studies become necessary to deal with larger molecular assemblies, such as light-harvesting and RC complexes. Three-dimensional structural information of the complexes would greatly help these studies, and several structures have been determined for the complexes, including PS II-LHC II and PS I-LHC I, by X-ray crystallography and cryo-EM.239,283,291–293 However, it has been reported that high-dose electron beams in cryo-EM caused damage (structural changes) in protein samples.294 Therefore, intact structural information is needed to understand the role of coherences.
4.6. Proton tunnelling and enzyme reaction
In 1944, Schrödinger postulated a significant role for hydrogen (nuclear) tunnelling in biological reactions in What is Life?295 He introduced the concept of C–H bond activation, which entails the movement of the nucleus as a wave through the reaction barrier. A notable biological process that involves C–H bond cleavage, essential for life, is the metabolism of molecules like glucose. In the 1980s, researchers began to explore the contribution of hydrogen tunnelling to enzyme reactions by measuring kinetic hydrogen isotope effects (KIEs). They incorporated hydrogen isotopes, such as deuterium (D), into stable, non-exchanging positions within samples to study the isotopic effects in enzyme-mediated reactions.KIEs are observed in many enzymes, including soybean lipoxygenase (SLO), a paradigmatic example of quantum mechanical tunnelling in biological C–H bond cleavage reactions.296 SLO catalyzes the production of fatty acid hydroperoxides from (Z,Z)-1,4-pentadienoic moieties through a rate-limiting proton-coupled electron transfer (net hydrogen atom transfer) from the reactive carbon (C11) of the substrate to an FeIII(OH) cofactor. The oxygenated lipids and their by-products produced by this reaction are essential for signalling structural and metabolic changes within the cell, contributing to processes such as seed growth and plant development.
KIEs can be classified into two distinct types: semi-classical and full tunnelling or wave function overlap regimes. In the semi-classical regime, the activation energy varies for different isotopes due to changes in the ground state energy resulting from the isotope effect. Conversely, in the tunnelling or wave function overlap regime, the activation energy remains consistent across all isotopes, attributed to the minimal distance over the energy barrier required for tunnelling. This critical distance, the hydrogen donor–acceptor distance (DAD), is theoretically calculated to range between 2.7 to 2.8 Å. In SLO, DAD has been empirically measured using electron–nuclear double resonance (ENDOR) spectroscopy.297 Changes in DAD have been correlated with variations in activation energy caused by amino acid mutations.298
Temperature-dependent hydrogen–deuterium exchange coupled to mass spectrometry (TDHDX-MS) has recently been recognized as a widely accessible and practical experimental method for investigating heat-induced changes in local protein flexibility. TDHDX-MS provides a time-averaged spatial resolution of activated networks extending from protein–solvent interfaces to enzyme-active sites when combined with function-altering protein mutations. This approach has identified local networks within SLO that correlate with the activation energy for hydrogen–deuterium exchange and the enthalpic barrier for catalytic C–H bond cleavage.299–303 Recent studies have also established a connection between the catalytic reaction and a nanosecond stoke shift observed in SLO, enhancing our understanding of the molecular mechanisms at the solvent interface.304
4.7. Proton tunnelling and mutation in DNA
Spontaneous mutations can arise from tautomeric shifts in nucleotide bases, a phenomenon first proposed by Watson and Crick. These shifts can lead to mismatched base pairing during DNA replication and translation.305 Later hypotheses suggested that proton tunnelling could significantly increase the formation of rare tautomeric enol forms, leading to mismatches in nucleobase interstrand pairing and potential errors during DNA replication.306 These mismatches, which mimic the geometry of correct Watson–Crick base pairs, are challenging to detect due to subtle chemical shifts, such as the movement of a single proton and a π-bond. Current structure determination methods like X-ray crystallography and cryo-EM analysis struggle to visualize these mismatches since protons are generally invisible by these techniques.307 However, by integrating NMR relaxation dispersion with misincorporation rate measurements, researchers have mapped a kinetic network involving two tautomeric and one anionic Watson–Crick-like mismatches.308 This research underscores the complexity of replication fidelity and the challenges in pinpointing the sources of errors that could lead to mutations and, potentially, cause cancer.309Hydrogen transfer in DNA bases can produce transient, biologically relevant mutagens called tautomers. Keto–enol and amino–imino tautomerism are implicated in the spontaneous DNA mutation during replication and repair processes. The energy landscapes of both canonical (amino–keto) and tautomeric (imino–enol) forms of adenine–thymine (A–T) and guanine–cytosine (G–C) monomers and base pairs have been modeled using the ab initio quantum mechanical approach of density functional theory (DFT).310 Understanding the time scale of these processes is crucial for comprehending the role of proton tunnelling in DNA mutation. Previous research has indicated that the lifetime of tautomers is shorter than the timescale for helicase cleavage.311,312 Using the open quantum system (OQS) approach, where the quantum system (the H-bond proton in the double-well potential) undergoes dissipation and decoherence due to its coupling with a surrounding heat bath, it is suggested that tautomers could reach equilibrium concentration.313 These studies propose that proton transfer through quantum tunneling can occur within a few hundred femtoseconds, potentially leading to mismatches during DNA replication. Experimental tests using neutron scattering, which can directly observe hydrogen/deuterium atoms,314 are essential for advancing our understanding of this mechanism.
5. Conclusion
Quantum life science is still in its infancy, but it will provide new perspectives and contribute to resolving many biological phenomena that cannot be described and understood by conventional molecular mechanics or molecular biology. However, observing the quantum effect in living organisms is still challenging because it is very subtle, quickly dissipates, and gets decoherent under physiological conditions. To overcome the problems, it is essential to develop quantum sensors with more NV centers in nanodiamonds and hyperpolarized MRI/NMR molecules for higher accuracy. Moreover, new device development, such as two-dimensional electronic spectroscopy using a high-speed and stable 2D electronic spectrometer that can disentangle the vibrational coherence,315 helps deepen our understanding of the role of quantum effects in living systems. Such technologies and knowledge bring about the development of novel devices that utilize or mimic biological systems like photosynthesis, magnetoreception, and so on. It is strongly expected that the development of Quantum Life Sciences will elucidate a series of life phenomena that could not be elucidated by molecular biology and bring about innovative technological innovations that mimic these phenomena.Through further quantum life science research in the future, we will contribute to the development of a paradigm shift in life science, as well as the development of new cancer diagnosis and treatment methods, contributions to regenerative medicine, elucidation of brain function, elucidation of the aging process, and other innovations in the medical and healthcare fields. In addition, we will contribute to developing a peaceful and fulfilling human society by bringing about innovations in information, energy, agriculture, the environment, and space. However, quantum life science is a new academic discipline, and few researchers specialize in this field, so growing young researchers and experts is an extremely important issue. For this reason, establishing and improving specialized courses to teach quantum life science at universities and research institutions will be extremely important in the future.
Author contributions
Y. B. conceived the idea. Y. B., H. Y., and H. K. designed and decided the outline of this paper. H. Y., H. K., H. I., R. I., Y. T., S. A., and Y. H. conducted the literature study, interpreted the collected data, drew the figures, and wrote the manuscript. All authors checked and approved the manuscript and figures.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This paper was supported by the MEXT Quantum Leap Flagship Program (MEXT Q-LEAP, JPMXS0120330644).References
- L. Slocombe, M. Sacchi and J. Al-Khalili, Commun. Phys., 2022, 5, 109 CrossRef CAS.
- C. L. Degen, F. Reinhard and P. Cappellaro, Rev. Mod. Phys., 2017, 89, 035002 CrossRef.
- D. Budker and M. Romalis, Nat. Phys., 2007, 3, 227–234 Search PubMed.
- J. F. Barry, J. M. Schloss, E. Bauch, M. J. Turner, C. A. Hart, L. M. Pham and R. L. Walsworth, Rev. Mod. Phys., 2020, 92, 015004 CrossRef CAS.
- R. Schirhagl, K. Chang, M. Loretz and C. L. Degen, Annu. Rev. Phys. Chem., 2014, 65, 83–105 CrossRef CAS PubMed.
- M. Simmonds, W. Fertig and R. Giffard, IEEE Trans. Magn., 1979, 15, 478–481 CrossRef.
- H. B. Dang, A. C. Maloof and M. V. Romalis, Appl. Phys. Lett., 2010, 397, 151110 CrossRef.
- M. Brownnutt, M. Kumph, P. Rabl and R. Blatt, Rev. Mod. Phys., 2015, 87(4), 1419 CrossRef CAS.
- G. Balasubramanian, I. Y. Chan, R. Kolesov, M. Al-Hmoud, J. Tisler, C. Shin, C. Kim, A. Wojcik, P. R. Hemmer, A. Krueger, T. Hanke, A. Leitenstorfer, R. Bratschitsch, F. Jelezko and J. Wrachtrup, Nature, 2008, 455, 648–651 CrossRef CAS PubMed.
- J. R. Maze, P. L. Stanwix, J. S. Hodges, S. Hong, J. M. Taylor, P. Cappellaro, L. Jiang, M. V. Gurudev Dutt, E. Togan, A. S. Zibrov, A. Yacoby, R. L. Walsworth and M. D. Lukin, Nature, 2008, 455, 644–647 CrossRef CAS.
- A. M. Zaitsev, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 12909 CrossRef CAS.
- S. Castelletto and A. Boretti, J. Phys. Photonics, 2020, 2, 022001 CrossRef CAS.
- A. R.-P. Montblanch, M. Barbone, I. Aharonovich, M. Atatüre and A. C. Ferrari, Nat. Nanotechnol., 2023, 18, 555–571 CrossRef CAS PubMed.
- S. G. Bishop, J. P. Hadden, F. D. Alzahrani, R. Hekmati, D. L. Huffaker, W. W. Langbein and A. J. Bennett, ACS Photonics, 2020, 7, 1636 CrossRef CAS.
- A. M. Berhane, K.-Y. Jeong, Z. Bodrog, S. Fiedler, T. Schröder, N. V. Triviño, T. Palacios, A. Gali, M. Toth, D. Englund and I. Aharonovich, Adv. Mater., 2017, 29, 1605092 CrossRef.
- P. Reineck, A. Francis, A. Orth, D. W. M. Lau, R. D. V. Nixon-Luke, I. D. Rastogi, W. A. W. Razali, N. M. Cordina, L. M. Parker, V. K. A. Sreenivasan, L. J. Brown and B. C. Gibson, Adv. Opt. Mater., 2016, 4, 1549 CrossRef.
- A. Gruber, A. Dräbenstedt, C. Tietz, L. Fleury, J. Wrachtrup and C. von Borczyskowski, Science, 1997, 276, 2012–2014 CrossRef CAS.
- D. Terada, T. F. Segawa, A. I. Shames, S. Onoda, T. Ohshima, E. Ōsawa, R. Igarashi and M. Shirakawa, ACS Nano, 2019, 13, 6461 CrossRef CAS.
- L. Nie, A. C. Nusantara, V. G. Damle, R. Sharmin, E. P. P. Evans, S. R. Hemelaar, K. J. van der Laan, R. Li, F. P. P. Martinez, T. Vedelaar, M. Chipaux and R. Schirhagl, Sci. Adv., 2021, 7, 573 Search PubMed.
- R. Igarashi, T. Sugi, S. Sotoma, T. Genjo, Y. Kumiya, E. Walinda, H. Ueno, K. Ikeda, H. Sumiya, H. Tochio, Y. Yoshinari, Y. Harada and M. Shirakawa, J. Am. Chem. Soc., 2020, 142, 7542–7554 CrossRef CAS PubMed.
- M. Fujiwara, S. Sun, A. Dohms, Y. Nishimura, K. Suto, Y. Takezawa, K. Oshimi, L. Zhao, N. Sadzak, Y. Umehara, Y. Teki, N. Komatsu, O. Benson, Y. Shikano and E. Kage-Nakadai, Sci. Adv., 2020, 6, eaba9636 CrossRef CAS.
- I. Lovchinsky, A. O. Sushkov, E. Urbach, N. P. de Leon, S. Choi, K. De Greve, R. Evans, R. Gertner, E. Bersin, C. Müller, L. McGuinness, F. Jelezko, R. L. Walsworth, H. Park and M. D. Lukin, Science, 2016, 351, 836 CrossRef CAS PubMed.
- F. Shi, Q. Zhang, P. Wang, H. Sun, J. Wang, X. Rong, M. Chen, C. Ju, F. Reinhard, H. Chen, J. Wrachtrup, J. Wang and J. Du, Science, 2015, 347, 1135–1138 CrossRef CAS PubMed.
- C. Zhang, F. Shagieva, M. Widmann, M. Kübler, V. Vorobyov, P. Kapitanova, E. Nenasheva, R. Corkill, O. Rhrle, K. Nakamura, H. Sumiya, S. Onoda, J. Isoya and J. Wrachtrup, Phys. Rev. Appl., 2021, 15, 064075 CrossRef CAS.
- F. Dolde, H. Fedder, M. W. Doherty, T. Nöbauer, F. Rempp, G. Balasubramanian, T. Wolf, F. Reinhard, L. C. L. Hollenberg, F. Jelezko and J. Wrachtrup, Nat. Phys., 2011, 7, 459–463 Search PubMed.
- G. Kucsko, P. C. Maurer, N. Y. Yao, M. Kubo, H. J. Noh, P. K. Lo, H. Park and M. D. Lukin, Nature, 2013, 500, 54–58 CrossRef CAS.
- S. Hsieh, P. Bhattacharyya, C. Zu, T. Mittiga, T. J. Smart, F. Machado, B. Kobrin, T. O. Höhn, N. Z. Rui, M. Kamrani, S. Chatterjee, S. Choi, M. Zaletel, V. V. Struzhkin, J. E. Moore, V. I. Levitas, R. Jeanloz and N. Y. Yao, Science, 2019, 366, 1349–1354 CrossRef CAS.
- T. Staudacher, N. Raatz, S. Pezzagna, J. Meijer, F. Reinhard, C. A. Meriles and J. Wrachtrup, Nat. Commun., 2015, 6, 8527 CrossRef CAS PubMed.
- H. Ishiwata, H. C. Watanabe, S. Hanashima, T. Iwasaki and M. Hatano, Adv. Quantum Technol., 2021, 4(4), 2000106 CrossRef CAS.
- T. Wolf, P. Neumann, K. Nakamura, H. Sumiya, T. Ohshima, J. Isoya and J. Wrachtrup, Phys. Rev. X, 2015, 5, 041001 Search PubMed.
- J. M. Taylor, P. Cappellaro, L. Childress, L. Jiang, D. Budker, P. R. Hemmer, A. Yacoby, R. Walsworth and M. D. Lukin, Nat. Phys., 2008, 4(10), 810 Search PubMed.
- P. K. Bachmann, D. Leers and H. Lydtin, Diamond Relat. Mater., 1991, 1(1), 1 CrossRef CAS.
- Y. N. Palyanov, Y. M. Borzdov, A. F. Khokhryakov, I. N. Kupriyanov and A. G. Sokol, Cryst. Growth Des., 2010, 10(7), 3169 CrossRef CAS.
- E. Six, The Element Six CVD Diamond Handbook, https://e6cvd.com/media/wysiwyg/pdf/E6_CVD_Diamond_Handbook_A5_v10X.pdf.
- T. Teraji, T. Yamamoto, K. Watanabe, Y. Koide, J. Isoya, S. Onoda, T. Ohshima, L. J. Rogers, F. Jelezko, P. Neumann, J. Wrachtrup and S. Koizumi, Phys. Status Solidi A, 2015, 212(11), 2365 CrossRef CAS.
- G. Balasubramanian, P. Neumann, D. Twitchen, M. Markham, R. Kolesov, N. Mizuochi, J. Isoya, J. Achard, J. Beck, J. Tissler, V. Jacques, P. R. Hemmer, F. Jelezko and J. Wrachtrup, Nat. Mater., 2009, 8, 383–387 CrossRef CAS PubMed.
- V. N. Mochalin, O. Shenderova, D. Ho and Y. Gogotsi, Nat. Nanotechnol., 2012, 7, 11 CrossRef CAS.
- O. A. Shenderova, A. I. Shames, N. A. Nunn, M. D. Torelli, I. Vlasov and A. Zaitsev, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2019, 37, 030802 Search PubMed.
- M. Fujiwara and Y. Shikano, Nanotechnology, 2021, 32, 482002 CrossRef CAS PubMed.
- A. Haque and S. Sumaiya, J. Manuf. Mater. Process, 2017, 1(1), 6 CAS.
- V. M. Acosta, E. Bauch, M. P. Ledbetter, C. Santori, K.-M. C. Fu, P. E. Barclay, R. G. Beausoleil, H. Linget, J. F. Roch, F. Treussart, S. Chemerisov, W. Gawlik and D. Budker, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 115202 CrossRef.
- S. Sangtawesin, T. O. Brundage, Z. J. Atkins and J. R. Petta, Appl. Phys. Lett., 2014, 105, 063107 CrossRef.
- L. M. Pham, S. J. DeVience, F. Casola, I. Lovchinsky, A. O. Sushkov, E. Bersin, J. Lee, E. Urbach, P. Cappellaro, H. Park, A. Yacoby, M. Lukin and R. L. Walsworth, Phys. Rev. B, 2016, 93, 04542 CrossRef.
- D. M. Toyli, C. D. Weis, G. D. Fuchs, T. Schenkel and D. D. Awschalom, Nano Lett., 2010, 10(8), 3168 CrossRef CAS.
- A. Sipahigil, R. E. Evans, D. D. Sukachev, M. J. Burek, J. Borregaard, M. K. Bhaskar, C. T. Nguyen, J. L. Pacheco, H. A. Atikian, C. Meuwly, R. M. Camacho, F. Jelezko, E. Bielejec, H. Park, M. Lončar and M. D. Lukin, Science, 2016, 354(6314), 847 CrossRef CAS PubMed.
- S. Pezzagna, D. Wildanger, P. Mazarov, A. D. Wieck, Y. Sarov, I. Rangelow, B. Naydenov, F. Jelezko, S. W. Hell and J. Meijer, Small, 2010, 6(19), 2117 CrossRef CAS PubMed.
- T. Luhmann, N. Raatz, R. John, M. Lesik, J. Rodiger, M. Portail, D. Wildanger, F. Kleißler, K. Nordlund, A. Zaitsev, J. F. Roch, A. Tallaire, J. Meijer and S. Pezzagna, J. Phys. D: Appl. Phys., 2018, 51(48), 483002 CrossRef.
- Y. C. Chen, B. Griffiths, L. Weng, S. S. Nicley, S. N. Ishmael, Y. Lekhai, S. Johnson, C. J. Stephen, B. L. Green, G. W. Morley, M. E. Newton, M. J. Booth, P. S. Salter and J. M. Smith, Optica, 2019, 6(5), 662 CrossRef CAS.
- D. R. Glenn, D. B. Bucher, J. Lee, M. D. Lukin, H. Park and R. L. Walsworth, Nature, 2018, 555, 351 CrossRef CAS PubMed.
- L. B. Hughes, Z. Zhang, C. Jin, S. A. Meynell, B. Ye, W. Wu, Z. Wang, E. J. Davis, T. E. Mates, N. Y. Yao, K. Mukherjee and A. C. Bleszynski Jayich, APL Mater., 2023, 11, 021101 CrossRef CAS.
- K. Ohno, F. J. Heremans, L. C. Bassett, B. A. Myers, D. M. Toyli, A. C. Bleszynski Jayich, C. J. Palmstrøm and D. D. Awschalom, Appl. Phys. Lett., 2012, 101(8), 082413 CrossRef.
- T. Fukui, Y. Doi, T. Miyazaki, Y. Miyamoto, H. Kato, T. Matsumoto, T. Makino, S. Yamasaki, R. Morimoto, N. Tokuda, M. Hatano, Y. Sakagawa, H. Morishita, T. Tashima, S. Miwa, Y. Suzuki and N. Mizuochi, Appl. Phys. Express, 2014, 7, 055201 CrossRef CAS.
- T. Miyazaki, R. Miyamoto, T. Makino, H. Kato, S. Yamasaki, T. Fukui, Y. Doi, N. Tokuda, M. Hatano and N. Mizuochi, Appl. Phys. Lett., 2014, 105, 261601 CrossRef.
- H. Ozawa, K. Tahara, H. Ishiwata, M. Hatano and T. Iwasaki, Appl. Phys. Express, 2017, 10, 04550 Search PubMed.
- H. Ishiwata, M. Nakajima, K. Tahara, H. Ozawa, T. Iwasaki and M. Hatano, Appl. Phys. Lett., 2017, 111, 04310 CrossRef.
- T. Tsuji, T. Iwasaki and M. Hatano, Appl. Phys. Express, 2023, 16, 025503 CrossRef.
- R. Kawase, H. Kawashima, H. Kato, N. Tokuda, S. Yamasaki, M. Ogura, T. Makino and N. Mizuochi, J. Appl. Phys., 2022, 132, 174504 CrossRef CAS.
- E. D. Herbschleb, H. Kato, T. Makino, S. Yamasaki and N. Mizuochi, Nat. Commun., 2021, 12, 306 CrossRef CAS PubMed.
- N. R. Greiner, D. S. Phillips, J. D. Johnson and F. Volk, Nature, 1988, 333, 440 CrossRef CAS.
- J.-P. Boudou, P. A. Curmi, F. Jelezko, J. Wrachtrup, P. Aubert, M. Sennour, G. Balasubramanian, R. Reuter, A. Thorel and E. Gaffet, Nanotechnology, 2009, 20, 235602 CrossRef PubMed.
- A. K. Khachatryan, S. G. Aloyan, P. W. May, R. Sargsyan, V. A. Khachatryan and V. S. Baghdasaryan, Diamond Relat. Mater., 2008, 17, 931 CrossRef CAS.
- D. Amans, M. Diouf, J. Lam, G. Ledoux and C. Dujardin, J. Colloid Interface Sci., 2017, 489, 114 CrossRef CAS.
- M. E. Trusheim, L. Li, A. Laraoui, E. H. Chen, H. Bakhru, T. Schröder, O. Gaathon, C. A. Meriles and D. Englund, Nano Lett., 2013, 14, 32 CrossRef PubMed.
- J. Tisler, G. Balasubramanian, B. Naydenov, R. Kolesov, B. Grotz, R. Reuter, J. P. Boudou, P. A. Curmi, M. Sennour, A. Thorel, M. Börsch, K. Aulenbacher, R. Erdmann, P. R. Hemmer, F. Jelezko and J. Wrachtrup, ACS Nano, 2009, 3, 1959 CrossRef CAS PubMed.
- F. T.-K. So, A. I. Shames, D. Terada, T. Genjo, H. Morishita, I. Ohki, T. Ohshima, S. Onoda, H. Takashima, S. Takeuchi, N. Mizuochi, R. Igarashi, M. Shirakawa and T. F. Segawa, J. Phys. Chem. C, 2022, 126, 5206 CrossRef CAS.
- C. Bradac, T. Gaebel, N. Naidoo, M. J. Sellars, J. Twamley, L. J. Brown, A. S. Barnard, T. Plakhotnik, A. V. Zvyagin and J. R. Rabeau, Nat. Nanotechnol., 2010, 5, 345–349 CrossRef CAS.
- S. Kumar, M. Nehra, D. Kedia, N. Dilbaghi, K. Tankeshwar and K. H. Kim, Carbon, 2019, 143, 678 CrossRef CAS.
- O. A. Shenderova and G. E. McGuire, Biointerphases, 2015, 10, 030802 CrossRef CAS.
- R. H. Wentorf, J. Phys. Chem., 1965, 69, 3063 CrossRef CAS.
- E. A. Ekimov, O. S. Kudryavtsev, N. E. Mordvinova, O. I. Lebedev and I. I. Vlasov, ChemNanoMat, 2018, 4, 269 CrossRef CAS.
- M. A. Gebbie, H. Ishiwata, P. J. McQuade, V. Petrak, A. Taylor, C. Freiwald, J. E. Dahl, R. M. K. Carlson, A. A. Fokin, P. R. Schreiner, Z. X. Shen, M. Nesladek and N. A. Melosh, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(33), 8284–8289 CrossRef CAS.
- K. Oshimi, H. Nakashima, S. Mandić, H. Kobayashi, M. Teramoto, H. Tsuji, Y. Nishibayashi, Y. Shikano, T. An and M. Fujiwara, ACS Nano, 2024, 18(52), 35202–35313 CrossRef CAS PubMed.
- F. Maier, M. Riedel, B. Mantel, J. Ristein and L. Ley, Phys. Rev. Lett., 2000, 85, 3472 CrossRef CAS PubMed.
- S. Kawai, H. Yamano, T. Sonoda, K. Kato, J. J. Buendia, T. Kageura, R. Fukuda, T. Okada, T. Tanii, T. Higuchi, M. Haruyama, K. Yamada, S. Onoda, T. Ohshima, W. Kada, O. Hanaizumi, A. Stacey, T. Teraji, S. Kono, J. Isoya and H. Kawarada, J. Phys. Chem. C, 2019, 123(6), 3594–3604 CrossRef CAS.
- B. A. Myers, A. Das, M. C. Dartiailh, K. Ohno, D. D. Awschalom and A. C. B. Jayich, Phys. Rev. Lett., 2014, 113, 027602 CrossRef CAS PubMed.
- S. Sangtawesin, B. L. Dwyer, S. Srinivasan, J. J. Allred, L. V. H. Rodgers, K. De Greve, A. Stacey, N. Dontschuk, K. M. O’Donnell, D. Hu, D. A. Evans, C. Jaye, D. A. Fischer, M. L. Markham, D. J. Twitchen, H. Park, M. D. Lukin and N. P. de Leon, Phys. Rev. X, 2019, 9(3), 031052 CAS.
- Y. V. Butenko, V. L. Kuznetsov, E. A. Paukshtis, A. I. Stadnichenko, I. N. Mazov, S. I. Moseenkov, A. I. Boronin and S. V. Kosheev, Fullerenes, Nanotubes, Carbon Nanostruct., 2006, 14, 557 CrossRef CAS.
- V. Pichot, M. Comet, E. Fousson, C. Baras, A. Senger, F. Le Normand and D. Spitzer, Diamond Relat. Mater., 2008, 17, 13 CrossRef CAS.
- S. Osswald, G. Yushin, V. Mochalin, S. O. Kucheyev and Y. Gogotsi, J. Am. Chem. Soc., 2006, 128, 11635 CrossRef CAS.
- K. S. Liu, A. Henning, M. W. Heindl, R. D. Allert, J. D. Bart, I. D. Sharp, R. Rizzato and D. B. Bucher, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(5), e2111607119 CrossRef CAS.
- J. M. Abendroth, K. Herb, E. Janitz, T. Zhu, L. A. Völker and C. L. Degen, Nano Lett., 2022, 22, 7294–7303 CrossRef CAS.
- M. Xie, X. Yu, L. V. H. Rodgers, D. Xu, I. Chi-Duran, A. Toros, N. Quack, N. P. de Leon and P. C. Maurer, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(8), e2114186119 CrossRef CAS PubMed.
- Y. Wu and T. Weil, Adv. Sci., 2022, 9, 2200059 CrossRef CAS PubMed.
- F. Jelezko and J. Wrachtrup, Phys. Status Solidi A, 2006, 203, 3207–3225 CrossRef CAS.
- G. Thiering and A. Gali, Phys. Rev. B: Condens. Matter Mater. Phys., 2018, 98, 085207 CrossRef CAS.
- A. T. Collins, M. F. Thomaz and M. I. B. Jorge, J. Phys. C: Solid State Phys., 1983, 16, 2177 CrossRef CAS.
- S. Sotoma, C. Zhong, J. C. Y. Kah, H. Yamashita, T. Plakhotnik, Y. Harada and M. Suzuki, Sci. Adv., 2021, 7, eabd7888 CrossRef CAS.
- E. V. Levine, M. J. Turner, P. Kehayias, C. A. Hart, N. Langellier, R. Trubko, D. R. Glenn, R. R. Fu and R. L. Walsworth, Nanophotonics, 2019, 8(11), 1945–1973 CrossRef CAS.
- E. Bauch, C. A. Hart, J. M. Schloss, M. J. Turner, J. F. Barry, P. Kehayias, S. Singh and R. L. Walsworth, Phys. Rev. X, 2018, 8, 031025 CAS.
- T. Rendler, J. Neburkova, O. Zemek, J. Kotek, A. Zappe, Z. Chu, P. Cigler and J. Wrachtrup, Nat. Commun., 2017, 8, 14701 CrossRef CAS.
- T. Fujisaku, R. Tanabe, S. Onoda, R. Kubota, T. F. Segawa, F. T.-K. So, T. Ohshima, I. Hamachi, M. Shirakawa and R. Igarashi, ACS Nano, 2019, 13, 11726–11732 CrossRef CAS PubMed.
- T. Fujisaku, R. Igarashi and M. Shirakawa, Chemosensors, 2020, 8, 68 CrossRef CAS.
- T. F. Segawa and R. Igarashi, Prog. Nucl. Magn. Reson. Spectrosc., 2023, 134–135, 20–38 CrossRef CAS PubMed.
- V. M. Acosta, E. Bauch, M. P. Ledbetter, A. Waxman, L.-S. Bouchard and D. Budker, Phys. Rev. Lett., 2010, 104, 070801 CrossRef CAS.
- P. Neumann, I. Jakobi, F. Dolde, C. Burk, R. Reuter, G. Waldherr, J. Honert, T. Wolf, A. Brunner and J. H. Shim, Nano Lett., 2013, 13, 2738–2742 CrossRef CAS.
- P. Konzelmann, T. Rendler, V. Bergholm, A. Zappe, V. Pfannenstill, M. Garsi, F. Ziem, M. Niethammer, M. Widmann, S.-Y. Lee, P. Neumann and J. Wrachtrup, New J. Phys., 2018, 20, 123013 CrossRef CAS.
- H. J. Mamin, M. H. Sherwood, M. Kim, C. T. Rettner, K. Ohno, D. D. Awschalom and D. Rugar, Phys. Rev. Lett., 2014, 113, 030803 CrossRef CAS.
- L. T. Hall, G. C. G. Beart, E. A. Thomas, D. A. Simpson, L. P. McGuinness, J. H. Cole, J. H. Manton, R. E. Scholten, F. Jelezko, J. Wrachtrup, S. Petrou and L. C. L. Hollenberg, Sci. Rep., 2012, 2, 401 CrossRef CAS.
- A. Ermakova, G. Pramanik, J.-M. Cai, G. Algara-Siller, U. Kaiser, T. Weil, Y.-K. Tzeng, H. C. Chang, L. P. McGuinness, M. B. Plenio, B. Naydenov and F. Jelezko, Nano Lett., 2013, 13, 3305–3309 CrossRef CAS PubMed.
- J. Barton, M. Gulka, J. Tarabek, Y. Mindarava, Z. Wang, J. Schimer, H. Raabova, J. Bednar, M. B. Plenio, F. Jelezko, M. Nesladek and P. Cigler, ACS Nano, 2020, 14, 12938–12950 CrossRef CAS.
- T. Yanagi, K. Kaminaga, M. Suzuki, H. Abe, H. Yamamoto, T. Ohshima, A. Kuwahata, M. Sekino, T. Imaoka, S. Kakinuma, T. Sugi, W. Kada, O. Hanaizumi and R. Igarashi, ACS Nano, 2021, 15, 12869–12879 CrossRef CAS PubMed.
- M. Lobikin, B. Chernet, D. Lobo and M. Levin, Phys. Biol., 2012, 9, 065002 CrossRef CAS.
- M. Fujiwara, R. Tsukahara, Y. Sera, H. Yukawa, Y. Baba, S. Shikata and H. Hashimoto, RSC Adv., 2019, 9, 12606–12614 RSC.
- Y. Nishimura, K. Oshimi, Y. Umehara, Y. Kumon, K. Miyaji, H. Yukawa, Y. Shikano, T. Matsubara, M. Fujiwara, Y. Baba and Y. Teki, Sci. Rep., 2021, 11, 4248 CrossRef CAS PubMed.
- K. Oshimi, Y. Nishimura, T. Matsubara, M. Tanaka, E. Shikoh, L. Zhao, Y. Zou, N. Komatsu, Y. Ikado, Y. Takezawa, E. Kage-Nakadai, Y. Izutsu, K. Yoshizato, S. Morita, M. Tokunaga, H. Yukawa, Y. Baba, Y. Teki and M. Fujiwara, Lab Chip, 2022, 22, 2519–2530 RSC.
- H. Yukawa, M. Fujiwara, K. Kobayashi, Y. Kumon, K. Miyaji, Y. Nishimura, K. Oshimi, Y. Umehara, Y. Teki, T. Iwasaki, M. Hatano, H. Hashimoto and Y. Baba, Nanoscale Adv., 2020, 2, 1859–1868 RSC.
- L.-R. Zou, X.-D. Lyu, D.-D. Sang, Y. Yao, S.-H. Ge, X.-T. Wang, C.-D. Zhou, H.-L. Fu, H.-Z. Xi, J.-C. Fan, C. Wang and Q.-L. Wang, Rare Met., 2023, 42, 3201–3211 CrossRef CAS.
- L.-R. Zou, D.-D. Sang, Y. Yao, X.-T. Wang, Y.-Y. Zheng, N.-Z. Wang, C. Wang and Q.-L. Wang, Rare Met., 2022, 42, 17–38 CrossRef.
- R. Jha, P. Mishra and S. Kumar, Biosens. Bioelectron., 2024, 254, 116232 CrossRef CAS.
- E. M. Purcell, H. C. Torrey and R. V. Pound, Phys. Rev., 1946, 69, 1–2 CrossRef.
- F. Bloch, W. W. Hansen and M. Packard, Phys. Rev., 1946, 70, 7–8 Search PubMed.
- B. Meyer and T. Peters, Angew. Chem., Int. Ed., 2003, 42, 864–890 CrossRef CAS PubMed.
- P. Lauterbur, Nature, 1973, 242, 190–191 CrossRef CAS.
- S. Mansson, E. Johansson, P. Magnusson, C. M. Chai, G. Hansson, J. S. Petersson, F. Stahlberg and K. Golman, Eur. Radiol., 2006, 16, 57–67 CrossRef PubMed.
- J. Eills, D. Budker, S. Cavagnero, E. Y. Chekmenev, S. J. Elliott, S. Jannin, A. Lesage, J. Matysik, T. Meersmann, T. Prisner, J. A. Reimer, H. Yang and I. V. Koptyug, Chem. Rev., 2023, 123, 1417–1551 CrossRef CAS.
- A. W. Overhauser, Phys. Rev., 1953, 92, 411 CrossRef CAS.
- T. R. Carver and C. P. Slichter, Phys. Rev., 1953, 92, 212 CrossRef CAS.
- A. Kumar, R. R. Ernst and K. Wüthrich, Top. Catal., 1980, 95, 1–6 CAS.
- L. R. Becerra, G. J. Gerfen, R. J. Temkin, D. J. Singel and R. G. Griffin, Phys. Rev. Lett., 1993, 71, 3561–3564 CrossRef CAS.
- A. Abragam and M. Goldman, Rep. Prog. Phys., 1978, 41, 395–467 CrossRef CAS.
- H. E. Moller, X. J. Chen, B. Saam, K. D. Hagspiel, G. A. Johnson, T. A. Altes, E. E. de Lange and H. U. Kauczor, Magn. Reson. Med., 2002, 47, 1029–1051 CrossRef PubMed.
- M. S. Albert, G. D. Cates, B. Driehuys, W. Happer, B. Saam, C. S. Springer, Jr. and A. Wishnia, Nature, 1994, 370, 199–201 CrossRef CAS PubMed.
- D. J. Lurie, D. M. Bussell, L. H. Bell and J. R. Mallard, J. Magn. Reson., 1988, 76, 366–370 CAS.
- M. C. Krishna, S. English, K. Yamada, J. Yoo, R. Murugesan, N. Devasahayam, J. A. Cook, K. Golman, J. H. Ardenkjaer-Larsen, S. Subramanian and J. B. Mitchell, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2216–2221 CrossRef CAS.
- H. Utsumi, K. Yamada, K. Ichikawa, K. Sakai, Y. Kinoshita, S. Matsumoto and M. Nagai, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1463–1468 CrossRef CAS PubMed.
- S. Matsumoto, H. Yasui, S. Batra, Y. Kinoshita, M. Bernardo, J. P. Munasinghe, H. Utsumi, R. Choudhuri, N. Devasahayam, S. Subramanian, J. B. Mitchell and M. C. Krishna, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17898–17903 CrossRef CAS.
- J. B. Hovener, A. N. Pravdivtsev, B. Kidd, C. R. Bowers, S. Gloggler, K. V. Kovtunov, M. Plaumann, R. Katz-Brull, K. Buckenmaier, A. Jerschow, F. Reineri, T. Theis, R. V. Shchepin, S. Wagner, P. Bhattacharya, N. M. Zacharias and E. Y. Chekmenev, Angew. Chem., Int. Ed., 2018, 57, 11140–11162 CrossRef PubMed.
- J. Natterer and J. Bargon, Prog. Nucl. Magn. Reson. Spectrosc., 1997, 31, 293–315 CrossRef.
- K. V. Kovtunov, E. V. Pokochueva, O. G. Salnikov, S. F. Cousin, D. Kurzbach, B. Vuichoud, S. Jannin, E. Y. Chekmenev, B. M. Goodson, D. A. Barskiy and I. V. Koptyug, Chem. – Asian J., 2018, 13, 1857–1871 CrossRef CAS.
- P. J. Rayner and S. B. Duckett, Angew. Chem., Int. Ed., 2018, 57, 6742–6753 CrossRef CAS PubMed.
- J. F. P. Colell, M. Emondts, A. W. J. Logan, K. Shen, J. Bae, R. V. Shchepin, G. X. Ortiz, Jr., P. Spannring, Q. Wang, S. J. Malcolmson, E. Y. Chekmenev, M. C. Feiters, F. Rutjes, B. Blumich, T. Theis and W. S. Warren, J. Am. Chem. Soc., 2017, 139, 7761–7767 CrossRef CAS PubMed.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. Elliott, S. B. Duckett, G. G. Green, I. G. Khazal, J. Lopez-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS.
- F. Reineri, T. Boi and S. Aime, Nat. Commun., 2015, 6, 5858 CrossRef CAS PubMed.
- J. H. Ardenkjaer-Larsen, B. Fridlund, A. Gram, G. Hansson, L. Hansson, M. H. Lerche, R. Servin, M. Thaning and K. Golman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10158–10163 CrossRef CAS.
- M. Vaeggemose, R. F. Schulte and C. Laustsen, Metabolites, 2021, 11, 219 CrossRef CAS.
- J. H. Ardenkjaer-Larsen, J. Magn. Reson., 2016, 264, 3–12 CrossRef CAS.
- J. Kurhanewicz, D. B. Vigneron, J. H. Ardenkjaer-Larsen, J. A. Bankson, K. Brindle, C. H. Cunningham, F. A. Gallagher, K. R. Keshari, A. Kjaer, C. Laustsen, D. A. Mankoff, M. E. Merritt, S. J. Nelson, J. M. Pauly, P. Lee, S. Ronen, D. J. Tyler, S. S. Rajan, D. M. Spielman, L. Wald, X. Zhang, C. R. Malloy and R. Rizi, Neoplasia, 2019, 21, 1–16 CrossRef.
- Z. J. Wang, M. A. Ohliger, P. E. Z. Larson, J. W. Gordon, R. A. Bok, J. Slater, J. E. Villanueva-Meyer, C. P. Hess, J. Kurhanewicz and D. B. Vigneron, Radiology, 2019, 291, 273–284 CrossRef PubMed.
- J. H. Ardenkjaer-Larsen, S. Bowen, J. R. Petersen, O. Rybalko, M. S. Vinding, M. Ullisch and N. C. Nielsen, Magn. Reson. Med., 2019, 81, 2184–2194 CrossRef CAS.
- T. Cheng, A. P. Gaunt, I. Marco-Rius, M. Gehrung, A. P. Chen, J. J. van der Klink and A. Comment, NMR Biomed., 2020, 33, e4264 CrossRef.
- M. Baudin, B. Vuichoud, A. Bornet, G. Bodenhausen and S. Jannin, J. Magn. Reson., 2018, 294, 115–121 CrossRef CAS PubMed.
- L. L. Lumata, R. Martin, A. K. Jindal, Z. Kovacs, M. S. Conradi and M. E. Merritt, MAGMA, 2015, 28, 195–205 CrossRef CAS PubMed.
- S. Jannin, A. Comment, F. Kurdzesau, J. A. Konter, P. Hautle, B. van den Brandt and J. J. van der Klink, J. Chem. Phys., 2008, 128, 241102 CrossRef CAS.
- M. L. Hirsch, N. Kalechofsky, A. Belzer, M. Rosay and J. G. Kempf, J. Am. Chem. Soc., 2015, 137, 8428–8434 CrossRef CAS.
- K. Golman, R. in 't Zandt and M. Thaning, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11270–11275 CrossRef CAS PubMed.
- K. Golman, R. I. Zandt, M. Lerche, R. Pehrson and J. H. Ardenkjaer-Larsen, Cancer Res., 2006, 66, 10855–10860 CrossRef CAS PubMed.
- M. G. Vander Heiden, L. C. Cantley and C. B. Thompson, Science, 2009, 324, 1029–1033 CrossRef CAS.
- B. T. Scroggins, M. Matsuo, A. O. White, K. Saito, J. P. Munasinghe, C. Sourbier, K. Yamamoto, V. Diaz, Y. Takakusagi, K. Ichikawa, J. B. Mitchell, M. C. Krishna and D. E. Citrin, Clin. Cancer Res., 2018, 24, 3137–3148 CrossRef CAS PubMed.
- A. Comment, J. Magn. Reson., 2016, 264, 39–48 CrossRef CAS PubMed.
- Y. Takakusagi, R. Kobayashi, K. Saito, S. Kishimoto, M. C. Krishna, R. Murugesan and K. I. Matsumoto, Metabolites, 2023, 13, 69 CrossRef CAS PubMed.
- P. E. Z. Larson, Hyperpolarized Carbon-13 Magnetic Resonance Imaging and Spectroscopy, Academic Press, 2021 Search PubMed.
- Y. Kondo, H. Nonaka, Y. Takakusagi and S. Sando, Angew. Chem., Int. Ed., 2021, 60, 14779–14799 CrossRef CAS.
- C. Mu, D. E. Korenchan, S. Wang, D. M. Wilson and R. R. Flavell, Mol. Imaging Biol., 2021, 23, 323–334 CrossRef CAS.
- K. R. Keshari and D. M. Wilson, Chem. Soc. Rev., 2014, 43, 1627–1659 RSC.
- P. R. Jensen, M. Karlsson, S. Meier, J. O. Duus and M. H. Lerche, Chemistry, 2009, 15, 10010–10012 CrossRef CAS.
- P. Viswanath, G. Batsios, J. Mukherjee, A. M. Gillespie, P. E. Z. Larson, H. A. Luchman, J. J. Phillips, J. F. Costello, R. O. Pieper and S. M. Ronen, Nat. Commun., 2021, 12, 92 CrossRef CAS PubMed.
- R. Hata, H. Nonaka, Y. Takakusagi, K. Ichikawa and S. Sando, Angew. Chem., Int. Ed., 2016, 55, 1765–1768 CrossRef CAS PubMed.
- A. Radaelli, D. Ortiz, A. Michelotti, M. Roche, R. Hata, S. Sando, O. Bonny, R. Gruetter and H. A. I. Yoshihara, ACS Sens., 2022, 7, 2987–2994 CrossRef CAS PubMed.
- K. Yamamoto, A. Opina, D. Sail, B. Blackman, K. Saito, J. R. Brender, R. M. Malinowski, T. Seki, N. Oshima, D. R. Crooks, S. Kishimoto, Y. Saida, Y. Otowa, P. L. Choyke, J. H. Ardenkjaer-Larsen, J. B. Mitchell, W. M. Linehan, R. E. Swenson and M. C. Krishna, Sci. Rep., 2021, 11, 12155 CrossRef CAS.
- G. Batsios, C. Najac, P. Cao, P. Viswanath, E. Subramani, Y. Saito, A. M. Gillespie, H. A. I. Yoshihara, P. Larson, S. Sando and S. M. Ronen, Sci. Rep., 2020, 10, 6244 CrossRef CAS.
- T. Nishihara, H. A. Yoshihara, H. Nonaka, Y. Takakusagi, F. Hyodo, K. Ichikawa, E. Can, J. A. Bastiaansen, Y. Takado, A. Comment and S. Sando, Angew. Chem., Int. Ed., 2016, 55, 10626–10629 CrossRef CAS PubMed.
- Y. Saito, H. Yatabe, I. Tamura, Y. Kondo, R. Ishida, T. Seki, K. Hiraga, A. Eguchi, Y. Takakusagi, K. Saito, N. Oshima, H. Ishikita, K. Yamamoto, M. C. Krishna and S. Sando, Sci. Adv., 2022, 8, eabj2667 CrossRef CAS.
- R. Eskandari, N. Kim, A. Mamakhanyan, M. Saoi, G. Zhang, M. Berisaj, K. L. Granlund, A. J. Poot, J. Cross, C. B. Thompson and K. R. Keshari, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2120595119 CrossRef CAS.
- Y. Imakura, H. Nonaka, Y. Takakusagi, K. Ichikawa, N. R. Maptue, A. M. Funk, C. Khemtong and S. Sando, Chem. – Asian J., 2018, 13, 280–283 CrossRef CAS PubMed.
- H. Nonaka, M. Hirano, Y. Imakura, Y. Takakusagi, K. Ichikawa and S. Sando, Sci. Rep., 2017, 7, 40104 CrossRef CAS PubMed.
- H. Nonaka, R. Hata, T. Doura, T. Nishihara, K. Kumagai, M. Akakabe, M. Tsuda, K. Ichikawa and S. Sando, Nat. Commun., 2013, 4, 2411 CrossRef.
- A. Comment and M. E. Merritt, Biochemistry, 2014, 53, 7333–7357 CrossRef CAS PubMed.
- J. Milani, B. Vuichoud, A. Bornet, P. Mieville, R. Mottier, S. Jannin and G. Bodenhausen, Rev. Sci. Instrum., 2015, 86, 024101 CrossRef.
- K. Kouril, H. Kourilova, S. Bartram, M. H. Levitt and B. Meier, Nat. Commun., 2019, 10, 1733 CrossRef.
- A. Capozzi, S. Patel, W. T. Wenckebach, M. Karlsson, M. H. Lerche and J. H. Ardenkjaer-Larsen, J. Phys. Chem. Lett., 2019, 10, 3420–3425 CrossRef CAS PubMed.
- L. Lumata, M. E. Merritt, C. R. Malloy, A. D. Sherry and Z. Kovacs, J. Phys. Chem. A, 2012, 116, 5129–5138 CrossRef CAS PubMed.
- L. Lumata, M. E. Merritt and Z. Kovacs, Phys. Chem. Chem. Phys., 2013, 15, 7032–7035 RSC.
- B. Lama, J. H. Collins, D. Downes, A. N. Smith and J. R. Long, NMR Biomed., 2016, 29, 226–231 CrossRef CAS PubMed.
- A. P. Gaunt, J. S. Lewis, F. Hesse, T. Cheng, I. Marco-Rius, K. M. Brindle and A. Comment, Angew. Chem., Int. Ed., 2022, 61, e202112982 CrossRef CAS PubMed.
- A. Capozzi, S. Patel, C. P. Gunnarsson, I. Marco-Rius, A. Comment, M. Karlsson, M. H. Lerche, O. Ouari and J. H. Ardenkjaer-Larsen, Angew. Chem., Int. Ed., 2019, 58, 1334–1339 CrossRef CAS.
- T. El Darai, S. F. Cousin, Q. Stern, M. Ceillier, J. Kempf, D. Eshchenko, R. Melzi, M. Schnell, L. Gremillard, A. Bornet, J. Milani, B. Vuichoud, O. Cala, D. Montarnal and S. Jannin, Nat. Commun., 2021, 12, 4695 CrossRef CAS PubMed.
- D. Gajan, A. Bornet, B. Vuichoud, J. Milani, R. Melzi, H. A. van Kalkeren, L. Veyre, C. Thieuleux, M. P. Conley, W. R. Gruning, M. Schwarzwalder, A. Lesage, C. Coperet, G. Bodenhausen, L. Emsley and S. Jannin, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14693–14697 CrossRef CAS.
- A. S. Kiryutin, B. A. Rodin, A. V. Yurkovskaya, K. L. Ivanov, D. Kurzbach, S. Jannin, D. Guarin, D. Abergel and G. Bodenhausen, Phys. Chem. Chem. Phys., 2019, 21, 13696–13705 RSC.
- X. Ji, A. Bornet, B. Vuichoud, J. Milani, D. Gajan, A. J. Rossini, L. Emsley, G. Bodenhausen and S. Jannin, Nat. Commun., 2017, 8, 13975 CrossRef CAS.
- R. L. Hesketh and K. M. Brindle, Curr. Opin. Chem. Biol., 2018, 45, 187–194 CrossRef CAS PubMed.
- T. B. Rodrigues, E. M. Serrao, B. W. Kennedy, D. E. Hu, M. I. Kettunen and K. M. Brindle, Nat. Med., 2014, 20, 93–97 CrossRef CAS PubMed.
- S. E. Day, M. I. Kettunen, F. A. Gallagher, D. E. Hu, M. Lerche, J. Wolber, K. Golman, J. H. Ardenkjaer-Larsen and K. M. Brindle, Nat. Med., 2007, 13, 1382–1387 CrossRef CAS PubMed.
- K. R. Keshari, J. Kurhanewicz, J. M. Macdonald and D. M. Wilson, Analyst, 2012, 137, 3427–3429 RSC.
- M. H. Lerche, S. Meier, P. R. Jensen, H. Baumann, B. O. Petersen, M. Karlsson, J. O. Duus and J. H. Ardenkjaer-Larsen, J. Magn. Reson., 2010, 203, 52–56 CrossRef CAS PubMed.
- P. Nikolaou, B. M. Goodson and E. Y. Chekmenev, Chemistry, 2015, 21, 3156–3166 CrossRef CAS.
- M. Iinuma, Y. Takahashi, I. Shaké, M. Oda, A. Masaike, T. Yabuzaki and H. M. Shimizu, Phys. Rev. Lett., 2000, 84, 171–174 CrossRef CAS.
- P. Bachert, H. Brunner, K. H. Hausser and J. P. Colpa, Chem. Phys., 1984, 91, 435–447 CrossRef CAS.
- A. Kagawa, M. Negoro, R. Ohba, N. Ichijo, K. Takamine, Y. Nakamura, T. Murata, Y. Morita and M. Kitagawa, J. Phys. Chem. A, 2018, 122, 9670–9675 CrossRef CAS.
- K. Tateishi, M. Negoro, S. Nishida, A. Kagawa, Y. Morita and M. Kitagawa, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7527–7530 CrossRef CAS.
- M. Negoro, A. Kagawa, K. Tateishi, Y. Tanaka, T. Yuasa, K. Takahashi and M. Kitagawa, J. Phys. Chem. A, 2018, 122, 4294–4297 CrossRef CAS PubMed.
- A. Kagawa, K. Miyanishi, N. Ichijo, M. Negoro, Y. Nakamura, H. Enozawa, T. Murata, Y. Morita and M. Kitagawa, J. Magn. Reson., 2019, 309, 106623 CrossRef CAS PubMed.
- T. Hamachi, K. Nishimura, H. Kouno, Y. Kawashima, K. Tateishi, T. Uesaka, N. Kimizuka and N. Yanai, J. Phys. Chem. Lett., 2021, 12, 2645–2650 CrossRef CAS.
- K. Nishimura, H. Kouno, Y. Kawashima, K. Orihashi, S. Fujiwara, K. Tateishi, T. Uesaka, N. Kimizuka and N. Yanai, Chem. Commun., 2020, 56, 7217–7232 RSC.
- H. Sato, K. Miyanishi, M. Negoro, A. Kagawa, Y. Nishiyama, S. Horike, K. Nakamura and M. Inukai, J. Phys. Chem. Lett., 2023, 14, 4560–4564 CrossRef CAS PubMed.
- S. Fujiwara, N. Matsumoto, K. Nishimura, N. Kimizuka, K. Tateishi, T. Uesaka and N. Yanai, Angew. Chem., Int. Ed., 2022, 61, e202115792 CrossRef CAS.
- K. Tateishi, M. Negoro, A. Kagawa and M. Kitagawa, Angew. Chem., Int. Ed., 2013, 52, 13307–13310 CrossRef CAS PubMed.
- Y. Kawashima, T. Hamachi, A. Yamauchi, K. Nishimura, Y. Nakashima, S. Fujiwara, N. Kimizuka, T. Ryu, T. Tamura, M. Saigo, K. Onda, S. Sato, Y. Kobori, K. Tateishi, T. Uesaka, G. Watanabe, K. Miyata and N. Yanai, Nat. Commun., 2023, 14, 1056 CrossRef CAS.
- K. Miyanishi, T. Sugiki, T. Matsui, R. Ozawa, Y. Hatanaka, H. Enozawa, Y. Nakamura, T. Murata, A. Kagawa, Y. Morita, T. Fujiwara, M. Kitagawa and M. Negoro, J. Phys. Chem. Lett., 2023, 14, 6241–6247 CrossRef CAS PubMed.
- A. Eldirdiri, A. Clemmensen, S. Bowen, A. Kjaer and J. H. Ardenkjaer-Larsen, NMR Biomed., 2017, 30, e3803 CrossRef.
- D. M. Wilson, K. R. Keshari, P. E. Larson, A. P. Chen, S. Hu, M. Van Criekinge, R. Bok, S. J. Nelson, J. M. Macdonald, D. B. Vigneron and J. Kurhanewicz, J. Magn. Reson., 2010, 205, 141–147 CrossRef CAS.
- H. Qin, V. Zhang, R. A. Bok, R. D. Santos, J. A. Cunha, I. C. Hsu, J. D. Santos Bs, J. E. Lee, S. Sukumar, P. E. Z. Larson, D. B. Vigneron, D. M. Wilson, R. Sriram and J. Kurhanewicz, Int. J. Radiat. Oncol., Biol., Phys., 2020, 107, 887–896 CrossRef.
- H. Gutte, A. E. Hansen, M. M. Larsen, S. Rahbek, S. T. Henriksen, H. H. Johannesen, J. Ardenkjaer-Larsen, A. T. Kristensen, L. Hojgaard and A. Kjaer, J. Nucl. Med., 2015, 56, 1786–1792 CrossRef CAS.
- Z. M. Bhujwalla, S. Kakkad, Z. Chen, J. Jin, S. Hapuarachchige, D. Artemov and M. F. Penet, J. Magn. Reson., 2018, 291, 141–151 CrossRef CAS.
- C. Hilty, D. Kurzbach and L. Frydman, Nat. Protoc., 2022, 17, 1621–1657 CrossRef CAS PubMed.
- K. K. Frederick, V. K. Michaelis, B. Corzilius, T. C. Ong, A. C. Jacavone, R. G. Griffin and S. Lindquist, Cell, 2015, 163, 620–628 CrossRef CAS.
- G. Angelovski, B. J. Tickner and G. Wang, Nat. Chem., 2023, 15, 755–763 CrossRef CAS PubMed.
- M. I. Franco, L. Turin, A. Mershin and E. M. Skoulakis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3797–3802 CrossRef CAS PubMed.
- E. Block, S. Jang, H. Matsunami, S. Sekharan, B. Dethier, M. Z. Ertem, S. Gundala, Y. Pan, S. Li, Z. Li, S. N. Lodge, M. Ozbil, H. Jiang, S. F. Penalba, V. S. Batista and H. Zhuang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E2766–E2774 CAS.
- R. D. Hoehn, D. E. Nichols, H. Heven and S. Kais, Front. Phys., 2018, 6, 25 CrossRef.
- S. Johnsen and K. J. Lohmann, Nat. Rev. Neurosci., 2005, 6, 703–712 CrossRef CAS.
- H. Mouritsen and T. Ritz, Curr. Opin. Neurobiol., 2005, 15, 406–414 CrossRef CAS.
- T. Ritz, S. Adem and K. Schulten, Biophys. J., 2000, 78, 707–718 CrossRef CAS PubMed.
- J. Xu, L. E. Jarocha, T. Zollitsch, M. Konowalczyk, K. B. Henbest, S. Richert, M. J. Golesworthy, J. Schmidt, V. Déjean, D. J. C. Sowood, M. Bassetto, J. Luo, J. R. Walton, J. Fleming, Y. Wei, T. L. Pitcher, G. Moise, M. Herrmann, H. Yin, H. Wu, R. Bartölke, S. J. Käsehagen, S. Horst, G. Dautaj, P. D. F. Murton, A. S. Gehrckens, Y. Chelliah, J. S. Takahashi, K.-W. Koch, S. Weber, I. A. Solov'yov, C. Xie, S. R. Mackenzie, C. R. Timmel, H. Mouritsen and P. J. Hore, Nature, 2021, 594, 535–540 CrossRef CAS PubMed.
- T. Hochstoeger, T. Al Said, D. Maestre, F. Walter, A. Vilceanu, M. Pedron, T. D. Cushion, W. Snider, S. Nimpf, G. C. Nordmann, L. Landler, N. Edelman, L. Kruppa, G. Dürnberger, K. Mechtler, S. Schuechner, E. Ogris, E. P. Malkemper, S. Weber, E. Schleicher and D. A. Keays, Sci. Adv., 2020, 6, eabb9110 CrossRef CAS PubMed.
- H. Mitsui, T. Maeda, C. Yamaguchi, Y. Tsuji, R. Watari, Y. Kubo, K. Okano and T. Okano, Biochemistry, 2015, 54, 1908–1917 CrossRef CAS PubMed.
- H. Otsuka, H. Mitsui, K. Miura, K. Okano, Y. Imamoto and T. Okano, Biochemistry, 2020, 59, 3615–3625 CrossRef CAS PubMed.
- R. Watari, C. Yamaguchi, W. Zemba, Y. Kubo, K. Okano and T. Okano, J. Biol. Chem., 2012, 287, 42634–42641 CrossRef CAS PubMed.
- M. Liedvogel, K. Maeda, K. Henbest, E. Schleicher, T. Simon, C. R. Timmel, P. J. Hore and H. Mouritsen, PLoS One, 2007, 2, e1106 Search PubMed.
- R. J. Gegear, L. E. Foley, A. Casselman and S. M. Reppert, Nature, 2010, 463, 804–807 CrossRef CAS.
- D. M. W. Sheppard, J. Li, K. B. Henbest, S. R. T. Neil, K. Maeda, J. Storey, E. Schleicher, T. Biskup, R. Rodriguez, S. Weber, P. J. Hore, C. R. Timmel and S. R. Mackenzie, Sci. Rep., 2017, 7, 42228 CrossRef CAS PubMed.
- O. Bazalova, M. Kvicalova, T. Valkova, P. Slaby, P. Bartos, R. Netusil, K. Tomanova, P. Braeunig, H.-J. Lee, I. Sauman, M. Damulewicz, J. Provaznik, R. Pokorny, D. Dolezel and M. Vacha, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1660–1665 CrossRef CAS PubMed.
- H. G. Hiscock, S. Worster, D. R. Kattnig, C. Steers, Y. Jin, D. E. Manolopoulos, H. Mouritsen and P. J. Hore, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4634–4639 CrossRef CAS.
- K. Maeda, J. G. Storey, P. A. Liddell, D. Gust, P. J. Hore, C. J. Wedge and C. R. Timmel, Phys. Chem. Chem. Phys., 2015, 17, 3550–3559 RSC.
- M. Pooam, L.-D. Arthaut, D. Burdick, J. Link, C. F. Martino and M. Ahmad, Planta, 2019, 249, 319–332 CrossRef CAS.
- K. Maeda, A. J. Robinson, K. B. Henbest, H. J. Hogben, T. Biskup, M. Ahmad, E. Schleicher, S. Weber, C. R. Timmel and P. J. Hore, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4774–4779 CrossRef CAS PubMed.
- C. T. Rodgers and P. J. Hore, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 353–360 CrossRef CAS.
- B. D. Zoltowski, Y. Chelliah, A. Wickramaratne, L. Jarocha, N. Karki, W. Xu, H. Mouritsen, P. J. Hore, R. E. Hibbs, C. B. Green and J. S. Takahashi, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19449–19457 CrossRef CAS PubMed.
- B. Brocklehurst, Chem. Soc. Rev., 2002, 31, 301–311 RSC.
- U. E. Steiner and T. Ulrich, Chem. Rev., 1989, 89, 51–147 CrossRef CAS.
- C. Kerpal, S. Richert, J. G. Storey, S. Pillai, P. A. Liddell, D. Gust, S. R. Mackenzie, P. J. Hore and C. R. Timmel, Nat. Commun., 2019, 10, 3707 CrossRef PubMed.
- F. Cintolesi, T. Ritz, C. W. M. Kay, C. R. Timmel and P. J. Hore, Chem. Phys., 2003, 294, 385–399 CrossRef CAS.
- P. J. Hore and H. Mouritsen, Annu. Rev. Biophys., 2016, 45, 299–344 CrossRef CAS PubMed.
- K. Maeda, K. B. Henbest, F. Cintolesi, I. Kuprov, C. T. Rodgers, P. A. Liddell, D. Gust, C. R. Timmel and P. J. Hore, Nature, 2008, 453, 387–390 CrossRef CAS.
- H. J. Hogben, O. Efimova, N. Wagner-Rundell, C. R. Timmel and P. J. Hore, Chem. Phys. Lett., 2009, 480, 118–122 CrossRef CAS.
- S. Arai, R. Shimizu, M. Adachi and M. Hirai, Protein Sci., 2022, 31, e4313 CrossRef CAS.
- S. Qin, H. Yin, C. Yang, Y. Dou, Z. Liu, P. Zhang, H. Yu, Y. Huang, J. Feng, J. Hao, J. Hao, L. Deng, X. Yan, X. Dong, Z. Zhao, T. Jiang, H.-W. Wang, S.-J. Luo and C. Xie, Nat. Mater., 2016, 15, 217–226 CrossRef CAS.
- H. Wu, A. Scholten, A. Einwich, H. Mouritsen and K.-W. Koch, Sci. Rep., 2020, 10, 7364 CrossRef CAS PubMed.
- C. Xie, The Innovation, 2022, 3, 100229 CrossRef CAS.
- H. Xie, A. Lyratzakis, R. Khera, M. Koutantou, S. Welsch, H. Michel and G. Tsiotis, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2216734120 Search PubMed.
- D. E. Tronrud, J. Wen, L. Gay and R. E. Blankenship, Photosynth. Res., 2009, 100, 79–87 CrossRef CAS PubMed.
- I. A. Solov'yov and K. Schulten, Biophys. J., 2009, 96, 4804–4813 CrossRef.
- C. Nießner, S. Denzau, K. Stapput, M. Ahmad, L. Peichl, W. Wiltschko and R. Wiltschko, J. R. Soc., Interface, 2013, 10, 20130638 CrossRef PubMed.
- C. Nießner, S. Denzau, J. C. Gross, L. Peichl, H.-J. Bischof, G. Fleissner, W. Wiltschko and R. Wiltschko, PLoS One, 2011, 6, e20091 CrossRef.
- P. Bolte, A. Einwich, P. K. Seth, R. Chetverikova, D. Heyers, I. Wojahn, U. Janssen-Bienhold, R. Feederle, P. Hore, K. Dedek and H. Mouritsen, Ethology Ecology Evolution, 2021, 33, 248–272 CrossRef.
- P. Müller and M. Ahmad, J. Biol. Chem., 2011, 286, 21033–21040 CrossRef PubMed.
- C. Nießner, S. Denzau, L. Peichl, W. Wiltschko and R. Wiltschko, J. Exp. Biol., 2014, 217, 4221–4224 CrossRef.
- C. Nießner, S. Denzau, L. Peichl, W. Wiltschko and R. Wiltschko, J. Comp. Physiol., A, 2018, 204, 977–984 CrossRef PubMed.
- T. C. Player and P. J. Hore, J. Chem. Phys., 2019, 151, 225101 CrossRef PubMed.
- R. Wiltschko, M. Ahmad, C. Nießner, D. Gehring and W. Wiltschko, J. R. Soc., Interface, 2016, 13, 20151010 CrossRef.
- R. J. Kutta, N. Archipowa, L. O. Johannissen, A. R. Jones and N. S. Scrutton, Sci. Rep., 2017, 7, 44906 CrossRef CAS.
- R. J. Gegear, A. Casselman, S. Waddell and S. M. Reppert, Nature, 2008, 454, 1014–1018 CrossRef CAS.
- A. A. Bradlaugh, G. Fedele, A. L. Munro, C. N. Hansen, J. M. Hares, S. Patel, C. P. Kyriacou, A. R. Jones, E. Rosato and R. A. Baines, Nature, 2023, 615, 111–116 CrossRef CAS PubMed.
- E. W. Evans, C. A. Dodson, K. Maeda, T. Biskup, C. J. Wedge and C. R. Timmel, Interface Focus., 2013, 3, 20130037 CrossRef PubMed.
- N. Ikeya and J. R. Woodward, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2018043118 CrossRef CAS.
- D. Shevela, J. F. Kern, G. Govindjee and J. Messinger, Photosynth. Res., 2023, 156, 279–307 CrossRef CAS.
- T. Mirkovic, E. E. Ostroumov, J. M. Anna, R. Van Grondelle, G. Govindjee and G. D. Scholes, Chem. Rev., 2017, 117, 249–293 CrossRef CAS PubMed.
- M. L. Groot, J. P. Dekker, R. Van Grondelle, F. T. H. Den Hartog and S. Völker, J. Phys. Chem., 1996, 100, 11488–11495 CrossRef CAS.
- E. Wientjes, H. Van Amerongen and R. Croce, J. Phys. Chem. B, 2013, 117, 11200–11208 CrossRef CAS PubMed.
- M. Mamedov, G. Govindjee, V. Nadtochenko and A. Semenov, Photosynth. Res., 2015, 125, 51–63 CrossRef CAS PubMed.
- E. Romero, I. H. M. Van Stokkum, V. I. Novoderezhkin, J. P. Dekker and R. Van Grondelle, Biochemistry, 2010, 49, 4300–4307 CrossRef CAS.
- W. Lubitz, M. Chrysina and N. Cox, Photosynth. Res., 2019, 142, 105–125 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS.
- A. Galstyan, A. Robertazzi and E. W. Knapp, J. Am. Chem. Soc., 2012, 134, 7442–7449 CrossRef CAS PubMed.
- S. Luber, I. Rivalta, Y. Umena, K. Kawakami, J.-R. Shen, N. Kamiya, G. W. Brudvig and V. S. Batista, Biochemistry, 2011, 50, 6308–6311 CrossRef CAS PubMed.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J.-R. Shen, Nature, 2015, 517, 99–103 CrossRef CAS PubMed.
- A. Bhowmick, R. Hussein, I. Bogacz, P. S. Simon, M. Ibrahim, R. Chatterjee, M. D. Doyle, M. H. Cheah, T. Fransson, P. Chernev, I.-S. Kim, H. Makita, M. Dasgupta, C. J. Kaminsky, M. Zhang, J. Gätcke, S. Haupt, I. I. Nangca, S. M. Keable, A. O. Aydin, K. Tono, S. Owada, L. B. Gee, F. D. Fuller, A. Batyuk, R. Alonso-Mori, J. M. Holton, D. W. Paley, N. W. Moriarty, F. Mamedov, P. D. Adams, A. S. Brewster, H. Dobbek, N. K. Sauter, U. Bergmann, A. Zouni, J. Messinger, J. Kern, J. Yano and V. K. Yachandra, Nature, 2023, 617, 629–636 CrossRef CAS.
- M. Suga, F. Akita, K. Yamashita, Y. Nakajima, G. Ueno, H. Li, T. Yamane, K. Hirata, Y. Umena, S. Yonekura, L.-J. Yu, H. Murakami, T. Nomura, T. Kimura, M. Kubo, S. Baba, T. Kumasaka, K. Tono, M. Yabashi, H. Isobe, K. Yamaguchi, M. Yamamoto, H. Ago and J.-R. Shen, Science, 2019, 366, 334–338 CrossRef CAS.
- V. Krewald, M. Retegan, N. Cox, J. Messinger, W. Lubitz, S. DeBeer, F. Neese and D. A. Pantazis, Chem. Sci., 2015, 6, 1676–1695 RSC.
- M. Şener, J. Strümpfer, J. Hsin, D. Chandler, S. Scheuring, C. N. Hunter and K. Schulten, ChemPhysChem, 2011, 12, 518–531 CrossRef.
- A. M. Van Oijen, M. Ketelaars, J. Köhler, T. J. Aartsma and J. Schmidt, Science, 1999, 285, 400–402 CrossRef CAS.
- A. Chenu and G. D. Scholes, Annu. Rev. Phys. Chem., 2015, 66, 69–96 CrossRef CAS.
- A. Ishizaki and G. R. Fleming, Annu. Rev. Condens. Matter Phys., 2012, 3, 333–361 CrossRef CAS.
- G. S. Engel, T. R. Calhoun, E. L. Read, T.-K. Ahn, T. Mančal, Y.-C. Cheng, R. E. Blankenship and G. R. Fleming, Nature, 2007, 446, 782–786 CrossRef CAS PubMed.
- S. I. E. Vulto, M. A. De Baat, S. Neerken, F. R. Nowak, H. Van Amerongen, J. Amesz and T. J. Aartsma, J. Phys. Chem. B, 1999, 103, 8153–8161 CrossRef CAS.
- J. Cao, R. J. Cogdell, D. F. Coker, H.-G. Duan, J. Hauer, U. Kleinekathöfer, T. L. C. Jansen, T. Mančal, R. J. D. Miller, J. P. Ogilvie, V. I. Prokhorenko, T. Renger, H.-S. Tan, R. Tempelaar, M. Thorwart, E. Thyrhaug, S. Westenhoff and D. Zigmantas, Sci. Adv., 2020, 6, eaaz4888 CrossRef CAS PubMed.
- F. Fassioli, R. Dinshaw, P. C. Arpin and G. D. Scholes, J. R. Soc., Interface, 2014, 11, 20130901 CrossRef PubMed.
- N. Lambert, Y.-N. Chen, Y.-C. Cheng, C.-M. Li, G.-Y. Chen and F. Nori, Nat. Phys., 2013, 9, 10–18 Search PubMed.
- S. Roeding and T. Brixner, Coherent two-dimensional electronic mass spectrometry, Nat. Commun., 2018, 9, 2519 Search PubMed.
- E. Collini, J 2D Electronic Spectroscopic Techniques for Quantum Technology Applications, J. Phys. Chem. C, 2021, 125, 13096–13108 CrossRef CAS.
- N.-U. Frigaard and D. A. Bryant, in Complex Intracellular Structures in Prokaryotes, ed. J. M. Shively, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, vol. 2, pp. 79–114 Search PubMed.
- J. Adolphs and T. Renger, Biophys. J., 2006, 91, 2778–2797 CrossRef CAS PubMed.
- J. Moix, J. Wu, P. Huo, D. Coker and J. Cao, J. Phys. Chem. Lett., 2011, 2, 3045–3052 CrossRef CAS.
- J.-H. Chen, H. Wu, C. Xu, X.-C. Liu, Z. Huang, S. Chang, W. Wang, G. Han, T. Kuang, J.-R. Shen and X. Zhang, Science, 2020, 370, eabb6350 CrossRef CAS.
- G. Panitchayangkoon, D. V. Voronine, D. Abramavicius, J. R. Caram, N. H. C. Lewis, S. Mukamel and G. S. Engel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20908–20912 CrossRef CAS PubMed.
- H. Lee, Y.-C. Cheng and G. R. Fleming, Science, 2007, 316, 1462–1465 CrossRef CAS PubMed.
- B. S. Rolczynski, H. Zheng, V. P. Singh, P. Navotnaya, A. R. Ginzburg, J. R. Caram, K. Ashraf, A. T. Gardiner, S.-H. Yeh, S. Kais, R. J. Cogdell and G. S. Engel, Chem, 2018, 4, 138–149 CAS.
- J. S. Higgins, L. T. Lloyd, S. H. Sohail, M. A. Allodi, J. P. Otto, R. G. Saer, R. E. Wood, S. C. Massey, P.-C. Ting, R. E. Blankenship and G. S. Engel, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2018240118 CrossRef CAS.
- L. Wang, M. A. Allodi and G. S. Engel, Nat. Rev. Chem., 2019, 3, 477–490 CrossRef.
- H.-G. Duan, V. I. Prokhorenko, R. J. Cogdell, K. Ashraf, A. L. Stevens, M. Thorwart and R. J. D. Miller, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8493–8498 CrossRef CAS PubMed.
- E. Thyrhaug, R. Tempelaar, M. J. P. Alcocer, K. Žídek, D. Bína, J. Knoester, T. L. C. Jansen and D. Zigmantas, Nat. Chem., 2018, 10, 780–786 CrossRef CAS PubMed.
- S. Niwa, L.-J. Yu, K. Takeda, Y. Hirano, T. Kawakami, Z.-Y. Wang-Otomo and K. Miki, Nature, 2014, 508, 228–232 CrossRef CAS PubMed.
- X. Qin, M. Suga, T. Kuang and J.-R. Shen, Science, 2015, 348, 989–995 CrossRef CAS PubMed.
- X. Wei, X. Su, P. Cao, X. Liu, W. Chang, M. Li, X. Zhang and Z. Liu, Nature, 2016, 534, 69–74 CrossRef CAS PubMed.
- K. Kato, N. Miyazaki, T. Hamaguchi, Y. Nakajima, F. Akita, K. Yonekura and J.-R. Shen, Commun. Biol., 2021, 4, 382 CrossRef CAS PubMed.
- E. Schrödinger, What Is Life? The Physical Aspect of the Living Cell, Cambridge University Press, Cambridge, 1944 Search PubMed.
- M. P. Meyer, D. R. Tomchick and J. P. Klinman, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1146–1151 CrossRef CAS PubMed.
- M. Horitani, A. R. Offenbacher, C. A. M. Carr, T. Yu, V. Hoeke, G. E. Cutsail III, S. H. Schiffer, J. P. Klinman and B. M. Hoffman, J. Am. Chem. Soc., 2017, 139(5), 1984–1997 CrossRef CAS PubMed.
- A. R. Offenbacher, A. Sharma, P. E. Doan, J. P. Klinman and B. M. Hoffman, Biochemistry, 2020, 59(7), 901–910 CrossRef CAS.
- Z.-X. Liang, T. Lee, K. A. Resing, N. G. Ahn and J. P. Klinman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9556 CrossRef CAS PubMed.
- A. R. Offenbacher, S. Hu, E. M. Poss, C. A. M. Carr, A. D. Scouras, D. M. Prigozhin, A. T. Iavarone, A. Palla, T. Alber, J. S. Fraser and J. P. Klinman, ACS Cent. Sci., 2017, 3, 570–579 CrossRef CAS PubMed.
- J. Zhang, J. L. Balsbaugh, S. Gao, N. G. Ahn and J. P. Klinman, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 10797 CrossRef CAS PubMed.
- S. Gao, E. J. Thompson, S. L. Barrow, W. Zhang, A. T. Iavarone and J. P. Klinman, J. Am. Chem. Soc., 2020, 142, 19936–19949 CrossRef CAS PubMed.
- E. J. Thompson, A. Paul, A. T. Iavarone and J. P. Klinman, J. Am. Chem. Soc., 2021, 143, 785–797 CrossRef CAS PubMed.
- E. J. Thompson, A. Paul, A. T. Iavarone and J. P. Klinman, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2211630120 CrossRef PubMed.
- J. D. Watson and F. H. C. Crick, The structure of DNA, Cold Spring Harbor Symp. Quant. Biol., 1953, 18, 123–131 CrossRef CAS PubMed.
- P. O. Löwdin, Rev. Mod. Phys., 1963, 35, 724–732 CrossRef.
- A. B. Loveland, G. Demo, N. Grigorieff and A. A. Korostelev, Nature, 2017, 546, 113–117 CrossRef CAS.
- I. J. Kimsey, E. S. Szymanski, W. J. Zahurancik, A. Shakya, Y. Xue, C. C. Chu, B. Sathyamoorthy, Z. Suo and H. M. Al-Hashimi, Nature, 2018, 554, 195–201 CrossRef CAS.
- C. Tomasetti, L. Li and B. Vogelstein, Science, 2017, 355, 1330–1334 CrossRef CAS.
- L. Slocombe, J. S. Al-Khalilib and M. Sacchi, Phys. Chem. Chem. Phys., 2021, 23, 4141 RSC.
- O. O. Brovarets and D. M. Hovorun, J. Biomol. Struct. Dyn., 2019, 37, 1880–1907 CrossRef CAS PubMed.
- A. Gheorghiu, P. Coveney and A. Arabi, Interface Focus, 2020, 10, 20190120 CrossRef CAS PubMed.
- L. Slocombe, M. Sacchi and J. Al-Khalili, Commun. Phys., 2022, 5, 109 CrossRef CAS.
- M. Maestre-Reyna, P.-H. Wang, E. Nango, Y. Hosokawa, M. Saft, A. Furrer, C.-H. Yang, E. P. G. N. Putu, W.-J. Wu, H.-J. Emmerich, N. Caramello, S. Franz-Badur, C. Yang, S. Engilberge, M. Wranik, H. L. Glover, T. Weinert, H.-Y. Wu, C.-C. Lee, W.-C. Huang, K.-F. Huang, Y.-K. Chang, J.-H. Liao, J.-H. Weng, W. Gad, C.-W. Chang, A. H. Pang, K.-C. Yang, W.-T. Lin, Y.-C. Chang, D. Gashi, E. Beale, D. Ozerov, K. Nass, G. Knopp, P. J. M. Johnson, C. Cirelli, C. Milne, C. Bacellar, M. Sugahara, S. Owada, Y. Joti, A. Yamashita, R. Tanaka, T. Tanaka, F. Luo, K. Tono, W. Zarzycka, P. Müller, M. A. Alahmad, F. Bezold, V. Fuchs, P. Gnau, S. Kiontke, L. Korf, V. Reithofer, C. J. Rosner, E. M. Seiler, M. Watad, L. Werel, R. Spadaccini, J. Yamamoto, S. Iwata, D. Zhong, J. Standfuss, A. Royant, Y. Bessho, L.-O. Essen and M.-D. Tsai, Science, 2023, 382, 1014 CrossRef PubMed.
- M. Tsubouchi, N. Ishii, Y. Kagotani, R. Shimizu, T. Fujita, M. Adachi and R. Itakura, Opt. Express, 2023, 31, 6890–6906 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |