
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC–H functionalization through benzylic deprotonation with π-coordination or cation–π-interactions
Hui
Zhu(s)
a,
Yu
Wu
 b,
Jianyou
Mao
c,
Jingkai
Xu
a,
Patrick J.
Walsh
*b and
Hang
Shi
*a
b,
Jianyou
Mao
c,
Jingkai
Xu
a,
Patrick J.
Walsh
*b and
Hang
Shi
*a
aDepartment of Chemistry, Zhejiang University, Hangzhou, Zhejiang Province 310058, China. E-mail: shihang@westlake.edu.cn
bRoy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6323, USA. E-mail: pwalsh@sas.upenn.edu
cSchool of Chemistry and Molecular Engineering, Nanjing Tech University, Nanjing 211816, China
First published on 6th February 2025
Abstract
Benzylic C–H functionalization is a valuable tool to make complex aromatic molecules from simple, readily available alkylbenzenes. While methods that involve benzylic radicals or cations generated by hydrogen atom transfer or oxidation have been well demonstrated, they often require oxidative conditions. In contrast, deprotonation methods offer a complementary approach to transform benzylic C–H bonds through a benzylic carbanion generated by deprotonation. Electrophilic transition metal complexes acidify benzylic protons upon π-coordination to the phenyl ring of substrates, facilitating deprotonation by stabilizing the corresponding benzylic carbanion. Cation-complexes with group(I) metals also acidify benzylic C–H bonds. These approaches enable a significant expansion of the scope and diversity of alkylarenes with various electrophilic reagents. In this review, we discuss the development of benzylic functionalization through deprotonation of η6-arene complexes of transition-metals and cation–π interactions with group(I) metals, as well as progress made in catalysis through reversible arene–metal interactions.
 Hui Zhu(s) | Hui Zhu is a PhD candidate in Professor Hang Shi's group in the Key Laboratory of Precise Synthesis of Functional Molecules of Zhejiang Province, Department of Chemistry, School of Science, Westlake University. She received her bachelor degrees from the School of Chemistry and Molecular Engineering, East China Normal University in 2020. Her research focuses on catalytic benzylic functionalization via η6-coordination. |
 Yu Wu | Yu Wu received her BA in Chemistry from Colorado College in 2020 and started to work with Prof. Patrick J. Walsh at the University of Pennsylvania in 2021. She completed her MS in Chemistry in 2023 and continued her doctoral studies in the same research group. Her work focuses on developing transition-metal-free alkyne synthesis using one-pot processes. Her broader research interests include advancing methodology development in small molecule synthesis and catalysis. |
 Jianyou Mao | Jianyou Mao is a professor at the Nanjing Tech University. He received his PhD from China Agricultural University with Prof. Min Wang and Prof. Qinghua Bian. During which time, he was a visiting student at the University of Pennsylvania (USA) under the guidance of Prof. Patrick J. Walsh (2013-2015). He finished his postdoctoral study with Prof. Patrick J. Walsh in the same group (2015-2016). His research focuses on alkali-metal-mediated functionalization of weakly acidic C–H bonds. |
 Jingkai Xu | Jingkai Xu completed his undergraduate studies in Chemistry at Nanjing University, China, in 2022, where he developed a solid foundation in scientific principles and research methodologies. After studying for half a year in Hang Shi's group at Westlake University, he further honed his expertise at King Abdullah University of Science and Technology in Jeddah, Saudi Arabia, earning an MS degree in Material Science and Engineering in 2024. His research interests now lie in spintronics, focusing on developing innovative materials with applications in sustainable technologies. |
 Patrick J. Walsh | Patrick J. Walsh is the Rhodes-Thompson Professor of Chemistry at the University of Pennsylvania (USA). He received his BA from UC San Diego with Prof. Charles Perrin and PhD from UC Berkeley with Prof. R. G. Bergman. He was an NSF postdoctoral fellow with Prof. K. B. Sharpless at Scripps Research Institute. He began his independent career in 1994 at San Diego State University and moved to Penn in 1999. His research is in the area of transition metal-catalyzed reactions, development of methods for organic synthesis and the study of reaction mechanism. |
 Hang Shi | Hang Shi earned his bachelor's degree from Hunan University in 2008, under the guidance of Prof. Shuangfeng Yin. He then pursued his PhD at Peking University under the mentorship of Prof. Zhen Yang, focusing on the synthesis of natural products. In 2013, Hang joined Prof. Tobias Ritter's lab at Harvard University as a postdoctoral fellow. In 2015, he moved to Scripps Research Institute to work with Prof. Jin-Quan Yu as a research associate. Hang began his independent research career at Westlake University in 2018. In 2023, he was promoted to Associate Professor. |
1. Introduction
Aryl rings are ubiquitous structural motifs in functional materials, pharmaceuticals, and natural products, underscoring the imperative to advance efficient methods for elevating the structural intricacy of readily accessible alkylarenes. Among these endeavors, the selective functionalization of benzylic C–H bonds emerges as a straightforward avenue toward accessing functionalized molecules bearing aromatic rings.1,2 Leveraging the relatively low bond dissociation energy of benzylic C–H bonds, a plethora of reactions have been unveiled involving the generation of benzyl radicals through hydrogen atom transfer under oxidative conditions.3–9 In contrast, deprotonation of benzylic C–H bonds offers a complementary approach to benzylic functionalization through reactions with electrophilic reagents.While this approach holds potential, the modest electronegativity difference between carbon and hydrogen atoms, and the mild stabilization of benzylic anions by electronically neutral aryl rings, imparts limited acidity to benzylic C(sp3)–H bonds (Scheme 1).10,11 The heterolytic cleavage of these bonds by deprotonation thus remains challanging.12–26 Therefore, conventional methods for benzylic deprotonation of toluene without directing groups require the use of strong bases, such as nBuLi and nBuLi/tBuOK.13–16,18,22,25 To circumvent this hurdle, the introduction of a functional group capable of stabilizing negative charge via strong delocalization and/or inductive effects (e.g., carbonyl, nitro, etc.) or the use of polycyclic compounds like indenes and fluorenes that achieve aromaticity upon deprotonation, have been employed to facilitate benzylic functionalization processes. The application of such activating strategies necessitates additional steps for installation of activating groups and post-functionalization conversion, which inevitably limit their applicability. This is particularly true when working with abundant raw materials like alkylbenzenes or complex arylated molecules. Moreover, deprotonation is generally less effective for secondary and tertiary benzylic C–H bonds due to greater steric hindrance (kinetic) and decrease stability of secondary and tertiary carbanions (thermodynamic).
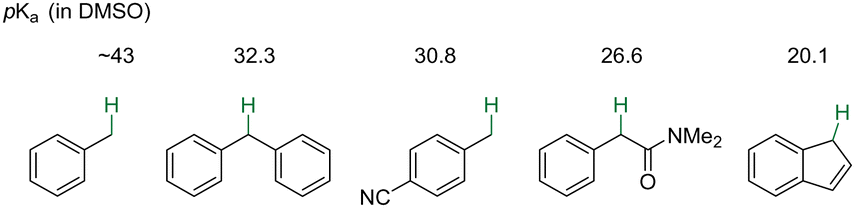 | ||
| Scheme 1 Benzylic acidity of aromatic compounds with pKa values in DMSO (compilation by Reich and Bordwell).11 | ||
In pursuit of strategies for benzylic deprotonation that avoid reliance on electron-withdrawing substituents, chemists have shifted their focus to activating the aromatic π-system. Unlike covalently bound substituents that facilitate benzylic deprotonation through resonance and/or inductive effects, electrophilic metal complexes activate aromatic methyl groups by removing electron density from the π-system through coordination with transition-metals or the formation of electrostatic cation–π complexes with main group metals. These interactions enhance the acidity of the benzylic C–H bonds by removing electron density from the π-cloud and stabilizing the resultant benzylic anion.
In this review, we survey the latest advances in benzylic C–H functionalization through deprotonation, bolstered by π-coordination with transition-metals or cation–π interactions. Coverage of this review is up to the end of 2023. Our focus is on the deprotonation of unbiased benzylic C–H bonds, highlighting illustrative examples featuring both alkyl and saturated cyclic ring systems.
2. Transition-metal-facilitated deprotonation through π-coordination
With the advancement of organometallic chemistry and its applications to organic synthesis, the interactions between transition-metals and unsaturated organic molecules have gained significant recognition.27–31 Since the 1950s, researchers have delved into the realm of sandwich and half-sandwich complexes, with a notable focus on η6-arene complexes.32–38 The discovery of bis(η6-benzene)chromium in 1955 marked a significant milestone.39 Subsequently, the preparation of half-sandwich (η6-arene)chromium tricarbonyl complexes demonstrated their electronically inverted reactivity of the bound aromatic ring, further advancing the field.40–43 This unique reactivity was rationalized by (arene) → M donation and electrostatic interactions.28,44,45 The activation model would not only increase the electrophilicity of bound aromatic rings, but also enhance the acidity of their benzylic C–H bonds if present.46–51 Upon the deprotonation of these C–H bonds, the resulting benzylic carbanions are stabilized through robust delocalization within the bound arene and onto the electrophilic metal center and its accompanying ligands (if π-acidic).The energy stabilization was quantitatively assessed through the reversible proton exchange reaction between a transition-metal η6-arene complex and phenylmethanide (Scheme 2).52–55 Consequently, the proton affinity of the benzyl anion is decreased by approximately 3.4–22.61 kcal mol−1 upon η6-coordination, depending on the specific metal and its ligands. Compared to toluene (pKa = 43 in DMSO), the energy barrier for benzylic deprotonation of a bound toluene molecule is significantly diminished.56–59 This section centers on the evolution of benzylic functionalization through deprotonation of η6-arene complexes with transition-metals, shedding light on the progress made in catalysis via reversible arene π-coordination.
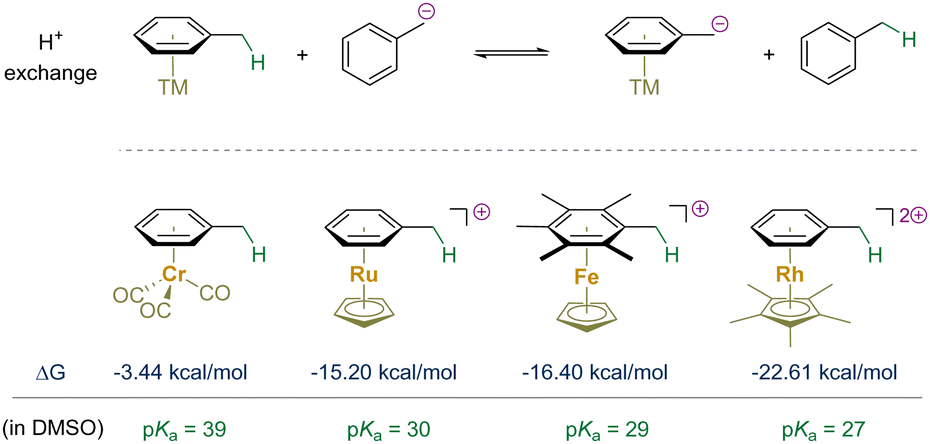 | ||
| Scheme 2 Computed stabilization energies of transition-metal complexes in the equilibrium above. | ||
2.1. Substitution and addition
Once deprotonated, the resulting benzylic anions readily react with electrophiles, providing a range of complexes diversified at the benzylic position (Scheme 3a).41,59,61–73 Besides various intermolecular alkylations, intramolecular cyclization reactions have been developed to generate the tetrahydrofuran derivative 1aa74 or tricyclic compounds 1ba and 1ca.75
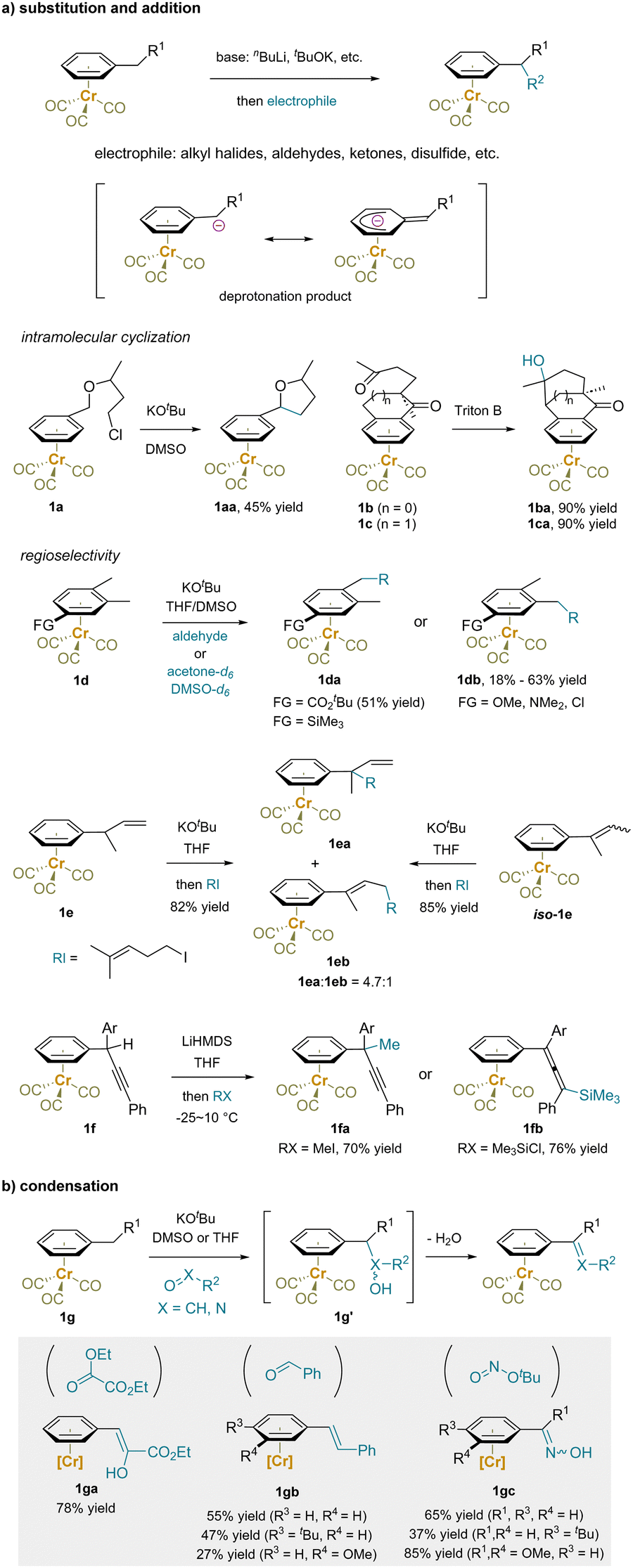 | ||
| Scheme 3 Functionalization of Cr(CO)3-complexed alkylarene: (a) substitution and addition reactions. (b) Condensations. | ||
The regioselectivity of benzylic deprotonation is influenced by substituents on the aromatic ring.76–80 For example, in the presence a carbonyl or silyl substituents, a para methyl group is preferentially functionalized over a meta methyl group, as shown in the reaction of 1d to produce 1da. Conversely, an electron donor group conjugated to the arene, such as methoxy or dimethylamino, steers reactions to occur with opposite selectivity, yielding 1db.65,76,79,80
(η6-arene)Cr(CO)3 complexes bearing an unsaturated bond at the benzylic site have also been examined. For example, alkylation of the but-3-en-2-ylbenzene (1e) preferentially takes place at the benzylic position (1ea![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1eb = 4.7:1), likely due to the anticipated stabilization of the benzylic anion.81 The isomer of 1e, but-2-en-2-ylbenzene complex (iso-1e) gave the same results under these conditions. Interestingly, regiodivergent substitution reactions of a complex bearing a benzylic triple bond (1f) were revealed, with the regioselectivity attributed to the size of the attacking electrophile. Specifically, methyl iodide gave the propargyl derivative 1fa, while trimethylsilyl chloride yielded the allenyl product 1fb.82
:1eb = 4.7:1), likely due to the anticipated stabilization of the benzylic anion.81 The isomer of 1e, but-2-en-2-ylbenzene complex (iso-1e) gave the same results under these conditions. Interestingly, regiodivergent substitution reactions of a complex bearing a benzylic triple bond (1f) were revealed, with the regioselectivity attributed to the size of the attacking electrophile. Specifically, methyl iodide gave the propargyl derivative 1fa, while trimethylsilyl chloride yielded the allenyl product 1fb.82
When compounds bearing an oxo motif were used as electrophiles with Cr(CO)3-complexed toluene and derivatives (1g) in the presence of KOtBu, a sequence of deprotonation, addition, and elimination occurred (Scheme 3b).63–65,68,83 Condensation products such as styrenes and benzaldehyde oximes were obtained, along with water as the byproduct.
In addition to reactions with carbon-based electrophiles, π-coordination-enabled deprotonation has also been utilized in the preparation of organometallic complexes (Scheme 4). Deprotonation of toluene complex 1h, followed by quenching with electrophilic metal reagents, such as CuI, ZnCl2, and Me3SnCl, provided the corresponding organocopper, organozinc, and organotin compounds, respectively.61,84–89 Notably, these metal reagents exhibited diverse reactivities. For instance, a benzylic copper complex underwent conjugate addition to methyl vinyl ketone, rather than 1,2-addition.85 Interestingly, the organozinc reagent (1ha) was further used to prepare a palladium complex (1hb), in which the distance between palladium and chromium is approximately 2.76 Å, indicating an interaction between the metal centers.84 The palladium motif further underwent oxidative addition with electrophiles, such as iodobenzene or acetyl chloride, followed by reductive elimination, installing a phenyl or an acetyl group at the benzylic position (1hc). Moreover, the benzylic carbanion reacted with a carbonyl ligand of iron pentacarbonyl, providing a bismetal complex 1ma. Directed ortho metalation of a proximal methyl substituent of 1ma by nBuLi was followed by nucleophilic attack by the benzylic anion. The second electrophile reacted first with the anionic iron center to form an Fe–C bond before undergoing reductive elimination yielding the difunctionalization products (1mb).90 The deprotonation of Cr(CO)3-complexed allylbenzene (1k) followed by transmetalation with a ligated palladium(II) complex resulted in the formation of a π-allyl palladium motif 1ka. This Pd(II) complex slowly decomposed to μ-allyl-bridged bis-Pd(I) complex 1kb (20–30% yield, characterized by X-ray crystallography) and a PdIIL2 (with the structure of L shown in the box). The reaction is accompanied by formation of a green precipitate, presumed to be Cr oxidation products generated on formation of the Pd(I) dimer.88 After prolonged stirring in toluene the tetranuclear bis-Pd(I) complex 1kb undergoes reductive coupling of the two allylbenzene moieties to furnish 1kc with precipitation of Pd black.
 | ||
| Scheme 4 Synthesis of bimetallic complexes and their use in C–C bond formation: (a) metallation-cross coupling reactions, (b) acylation reactions, (c) synthesis of Pd(I)-Pd(I) dimer, and (d) benzyl metallation and functionalization of η6-coordinated indene and fluorene complexes. | ||
Furthermore, the deprotonation of (η6-indene)Cr(CO)3 (1i) and transmetalation resulted in the formation of heterobimetallic indenyl complexes 1ia and 1ib, which were observed to form with Pd,91,92 Pt,91 Rh,93–99 and Ir100 complexes. The disposition of the two metal centers was dependent on the specific metal and ligands used. Interestingly, the syn-facial heterobimetallic indenyl complex 1ib was found to decompose upon exposure to air or excess PPh3, and exo-allylation products (1ic) as well as a homocoupling product of indenyl were obtained.100 Additionally, the transmetalation of bis(tricarbonylchromium)fluorene (1j) and a palladium or rhodium complex yielded the formation of organopalladium or organorhodium species (1ja) with metal–metal interactions.87,101
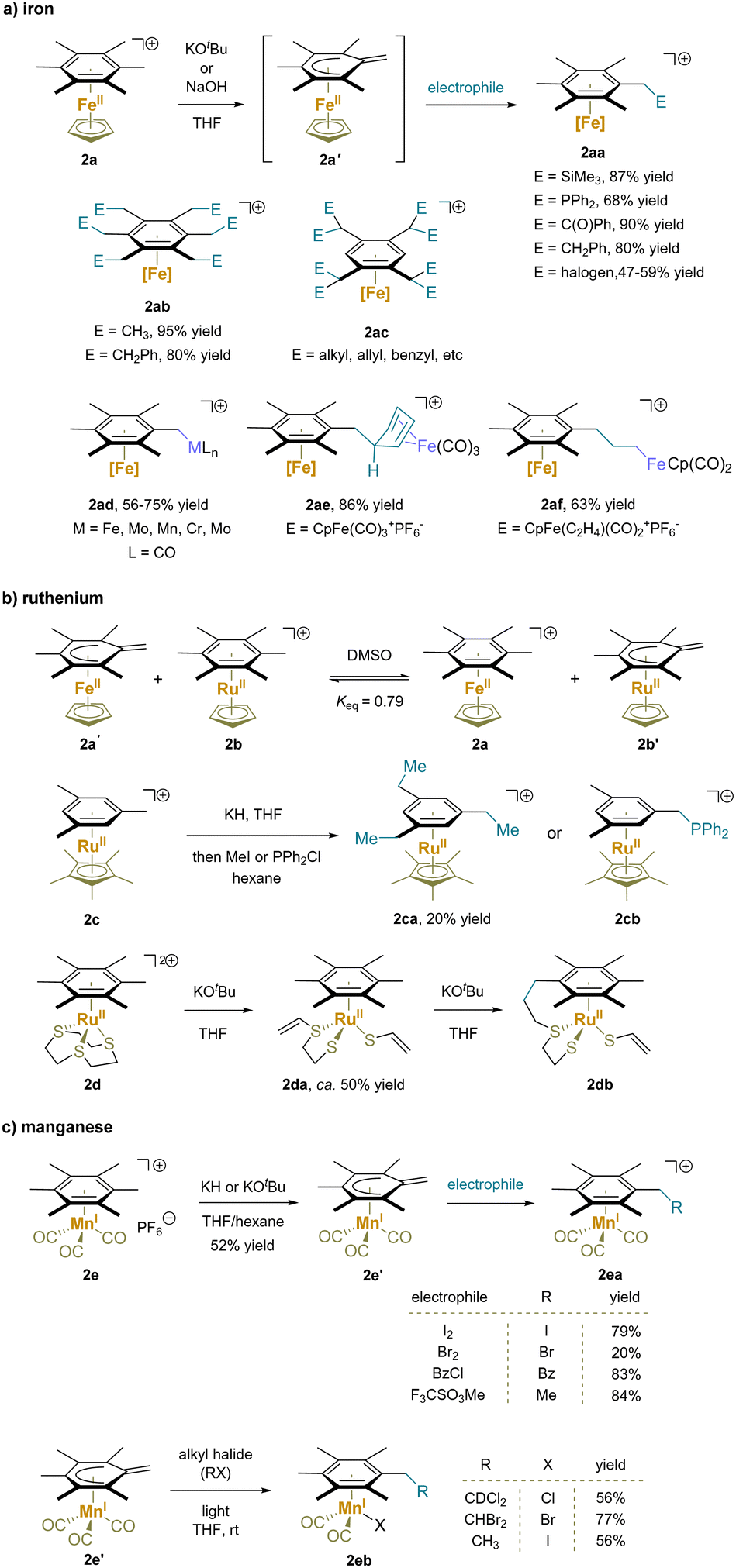 | ||
| Scheme 5 Functionalization of metal-complexed alkylarene: (a) iron-complex, (b) ruthenium-complex and (c) manganese-complex. | ||
Ruthenium(II) complexes have exhibited similar reactivity to the iron(II) analogues towards benzylic deprotonation due to the same valence d electronic structure and oxidation state (Scheme 5b).114–118 Specifically, the acidity of [(hexamethylbenzene)RuCp]+ (2b) was found to be very similar to that of the iron analogue 2a.59 In addition to intermolecular reactions (2ca, 2cb),116 an intramolecular addition reaction of the dicationic (hexamethylbenzene)Ru([9]aneS3) (2d) was also reported.119 Abstraction of a proton from [9]aneS3 caused cleavage of an adjacent C–S bond and formed the chelate vinylthioether-thiolate complex. Repetition of this process at a thioether CH2 group broke a second C–S bond giving complex 2da. Under basic conditions this intermediate then underwent benzylic deprotonation and intramolecular nucleophilic addition to form 2db.
Mono benzylic C–H functionalization of [(hexamethylbenzene)Mn(CO)3]+ (2e) has also been demonstrated (Scheme 5c).120–123 Compared to chromium(0) complexes, the benzylic protons of manganese(I) complexes are more acidic due to the stronger electron-withdrawing effect of the cationic Mn motif. Deprotonation with KH or KOtBu provided a stable neutral complex 2e′, which readily underwent substitution reactions with various electrophiles.120 Remarkably, under light irradiation, benzylic substitution and ligand exchange of complex 2e′ took place with alkyl halides. The author proposed a mechanism with light induced ejection of a CO ligand to open a coordination site at Mn. Binding of the electrophile X–R to Mn results in halide abstraction and generation of the radical R˙, which adds to the methylene, restoring aromaticity to the arene (2eb).
Cationic η6-multimethylbenzene complexes can undergo double deprotonation in the presence of excess strong base. For instance, treatment of η6-arene osmium(II) complexes (2f) with KOtBu yielded the cyclohexadiene-ligated complex of o-xylylene in the endo-coordinated form 2fa. Complexes 2fa isomerized quantitatively to the corresponding exo-isomer 2fa′ upon refluxing in toluene (Scheme 6a).124,125 Similar properties were observed for ruthenium(II) with phosphine ligands.115,117,124,126 On the other hand, double deprotonation of the dicationic bis(hexamethylbenzene)Ru complex (2g) exclusively resulted in an endocyclic complex 2ga that underwent double methylation to give 2gb. Interestingly, the combination of 2ga with a tetracarbonyl iron complex led to the addition of one methylene to an iron-bound carbonyl ligand followed by proton shuttling from the neighboring bound C6Me6 unit to afford cyclic 2gc.114,118 The cationic [(hexamethylbenzene)Mn(CO)3]+ (2h) also underwent double deprotonation by KOtBu or KH and alkylation with MeOTf to afford 2hb (Scheme 6c).121 It is noteworthy that π-coordination of arenes to the [IrCp*]2+ moiety results in highly electron deficient π-systems. An η6-phenol complex (2i) readily underwent deprotonation with Na2CO3 to generate an η5-phenoxo 2ia (Scheme 6d).127 Surprisingly, further deprotonation of the η5-phenoxo (2ia) occurred in the presence of KOtBu, yielding an η4-(o-quinone methide) complex (2ib). Michael addition of 2ib to an electrophilic alkyne generated the conjugate addition product 2ic; the addition of iodine to 2ib afforded iodination product 2idvia oxidation of the Ir through reaction with the methylene to install the C–I bond.128,129
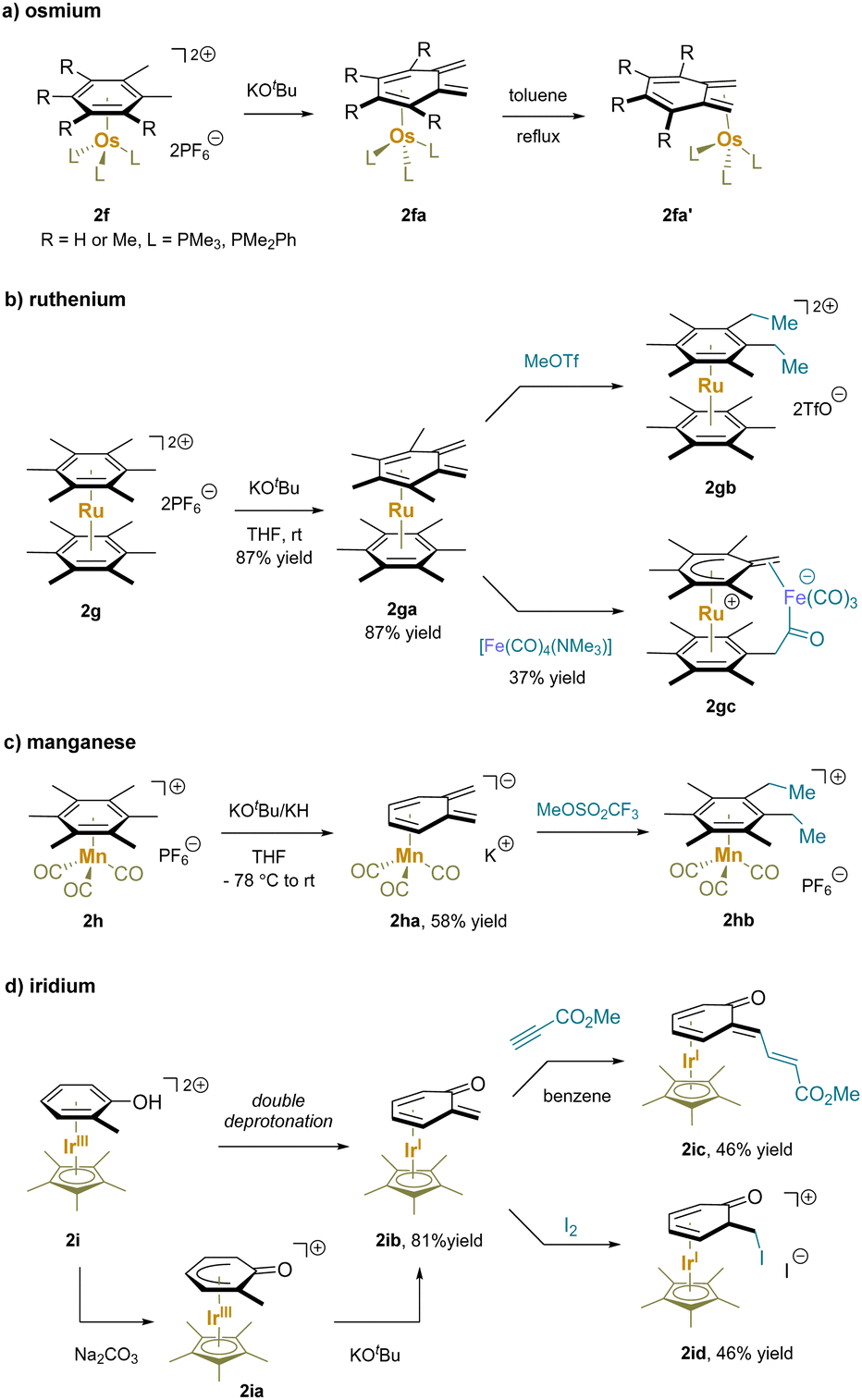 | ||
| Scheme 6 Functionalization via double deprotonation with: (a) osmium, (b) ruthenium, (c) manganese and (d) iridium complexes. | ||
2.2. Stereoselective reactions
Stereoselectivity is a crucial aspect of C–H functionalization at sp3-hybridized carbon centers. In reactions of an η6 complex, the ligated transition-metal motif differentiates the two faces of the bound aromatic ring, affecting both the benzylic deprotonation and the subsequent reaction of the resulting carbanions. This leads to endo/exo-selectivity of planar benzocyclic compounds and stereoselectivity of acyclic arenes.For example, the H/D exchange of (indane)Cr(CO)3 (3a) under basic conditions with DMSO-d6 yielded exclusively the exo-selective product 3aa without geminal-dideuterated product, suggesting a dominant anti-deprotonation/anti-reprotonation (Scheme 7a).130 In contrast, an electron-withdrawing group, such as an ester, led to benzylic deprotonation regardless of the stereochemistry of the α-C–H bonds. Reaction with electrophiles provided the exo-product 3ba.135 Moreover, the benzylic deprotonation of a (trimethylsilyl)dihydroisobenzofuran complex 3c with tBuLi as the base showed syn-deprotonation. This may be due to steric blocking of the benzylic anti-C–H by the bulky SiMe3 group and the α-silyl effect.136 Reaction of deprotonated 3c′ with an array of alkyl halides afforded exo-alkylated products 3ca. Subsequent desilylation of product 3ca with tetrabutyl ammonium fluoride (TBAF) most likely generated a benzylic anion that is delocalized into the arene-Cr system and is protonated from the exo-face to produced 3cb as pure endo-diastereomers. The regioselectivity of deprotonation of tetrahydroisoquinoline 3d was proposed to proceed through a chelated 6-membered cyclic transition state.62,137 Here again, electrophiles reacted on the exo-face of the ring system, including (η6-C6H5–F)Cr(CO)3 in an SNAr reaction.
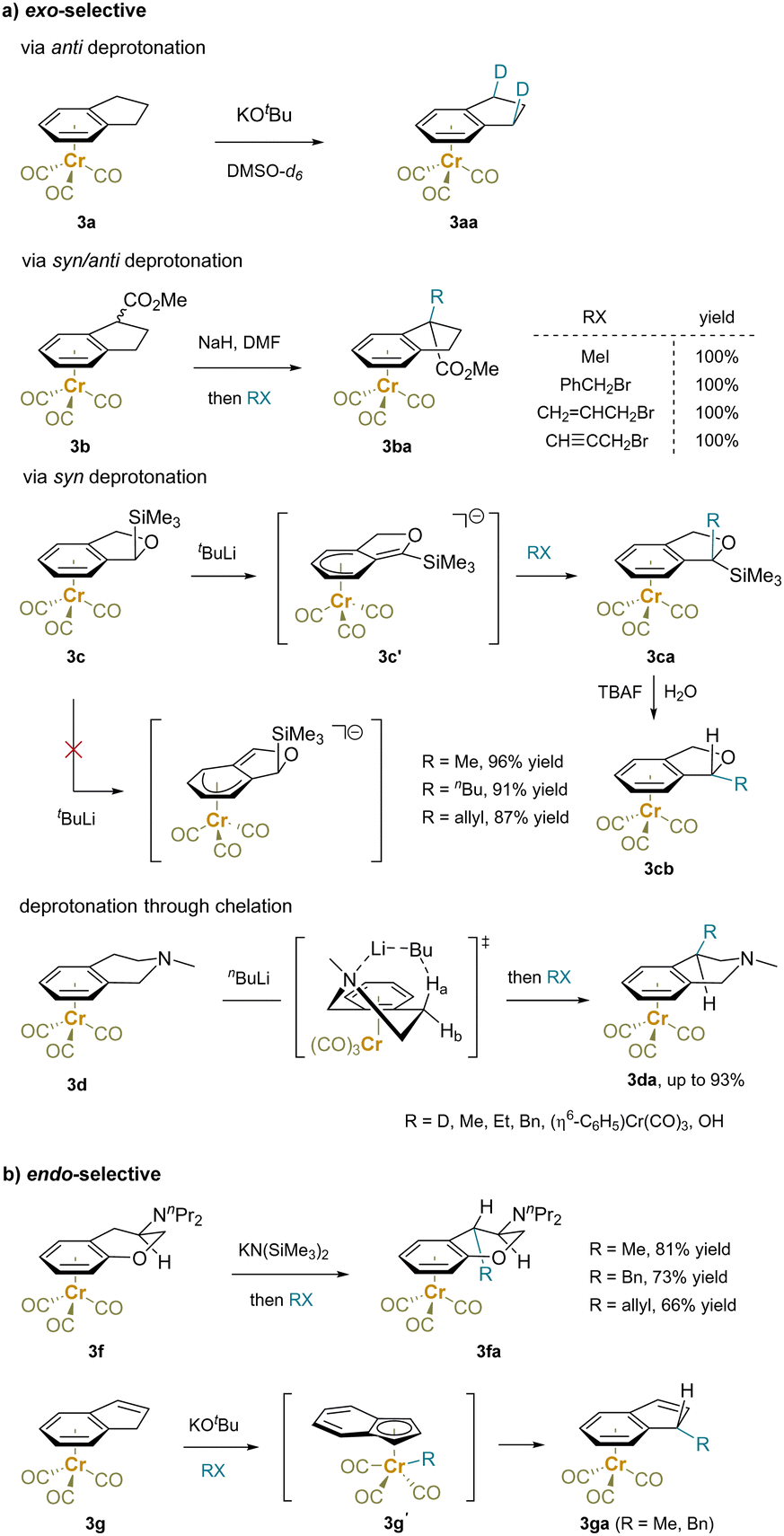 | ||
| Scheme 7 Stereoselective functionalization of exo-cyclic substrates: (a) exo-selective reactions and (b) endo-selective transformations. | ||
Some examples of endo-selectivity have also been demonstrated (Scheme 7b). For example, the endo-alkylation reactions of (η6-arene)chromium complexes 3f with the bulky NnPr2 group were reported. The selectivity was controlled by the amino substituent rather than the Cr(CO)3 moity.138 Moreover, an inner sphere mechanism was proposed to rationalize the endo-selectivity observed in the reactions of (η6-indene)Cr(CO)3 (3g) with electrophiles.139,140 Benzylic deprotonation is followed by η6- to η5-tautomerization to form the thermodynamically stable (η5-indenyl)Cr complex. The electrophile is then attacked by the Cr center, forming an intermediate with a Cr–R bond (3g′). Migration of the R group to the indenyl carbon and Cr back to the arene explains the endo-selective functionalization. Similar reactivity was observed with (η6-fluorene)Cr(CO)3, which also gives endo-alkylation products.139,140
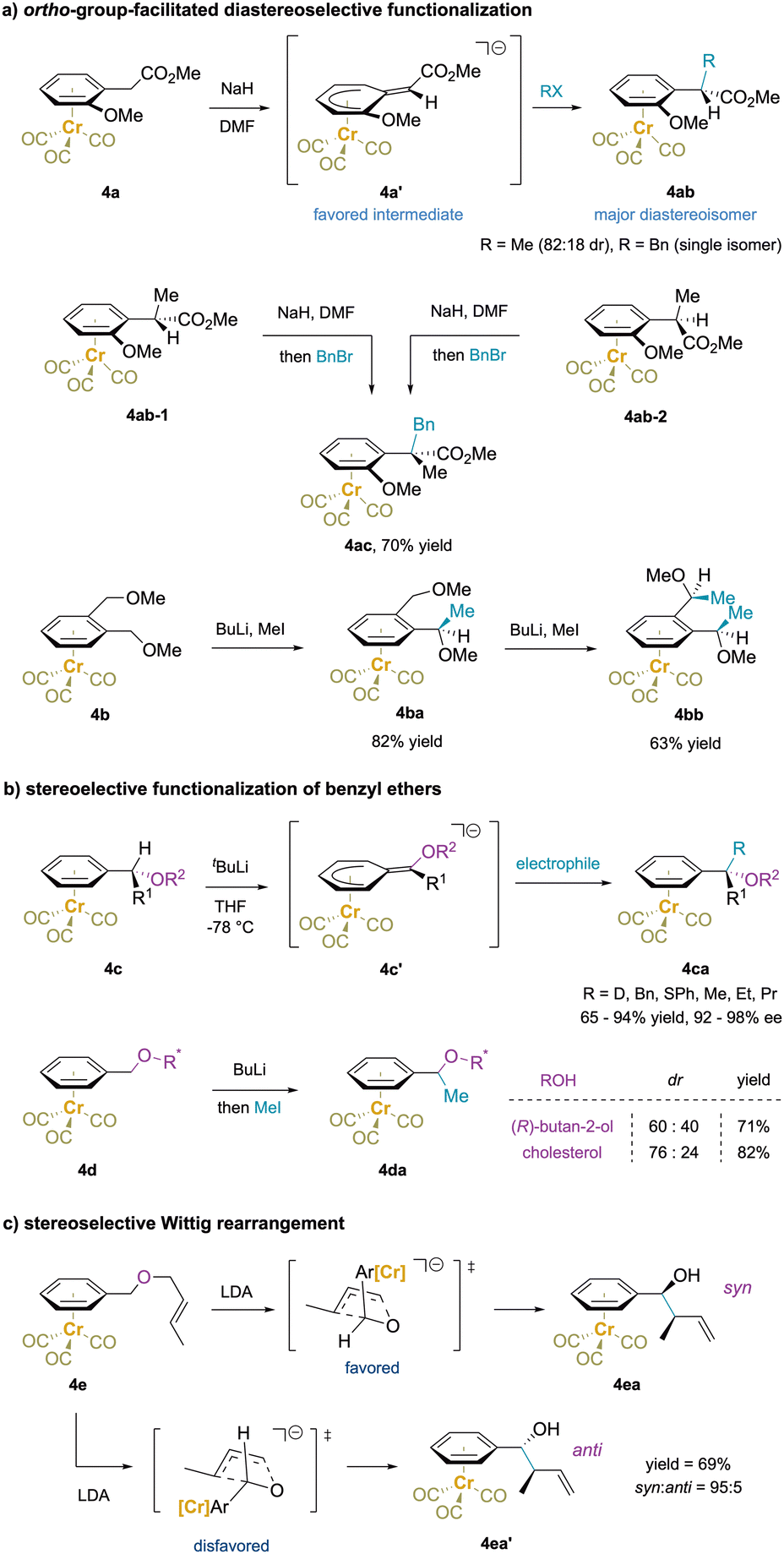 | ||
| Scheme 8 Stereoselective functionalization of acyclic substrates: (a) ortho-substituent-facilitated diastereoselective funtionalization, (b) stereoselective functionalization of benzyl ethers, and (c) stereoselective Wittig rearrangement. | ||
In addition to ortho substitution strategies outlined above, benzylic functionalization reactions of enantioenriched Cr(CO)3-complexed benzyl ethers have been shown to undergo stereospecific deprotonation. The resulting deprotonated intermediate (4c′) from complex 4c is planar chiral and reacts with electrophiles at the face opposite to Cr and provides enantioenriched product 4ca (Scheme 8b).74,141 A different strategy using enantioenriched ethers 4d (derived from Ar–CH2OR*) with the chirality located distal to the benzylic protons was employed with the goal of stereoselective benzylic methylations. Unfortunately, the methylations exhibited low levels of diastereoselectivity, likely because the chirality was located too far from the benzylic site.142–145
Additionally, reaction of (benzyl (E)-crotyl ether)Cr(CO)3 (4e) with LDA afforded a diastereomeric mixture of [2,3]-Wittig products in a ratio of 95:5.146 An envelope transition state was proposed for this anionic rearrangement in which the aryl motif lies in the pseudo-axial position to avoid the strong gauche interaction of the bulky (arene)Cr moiety. This leads to formation of the syn-product 4ea with high diastereoselectivity. In contrast, the corresponding Z isomer of 4e gave a 1:1 mixture of diastereomers.
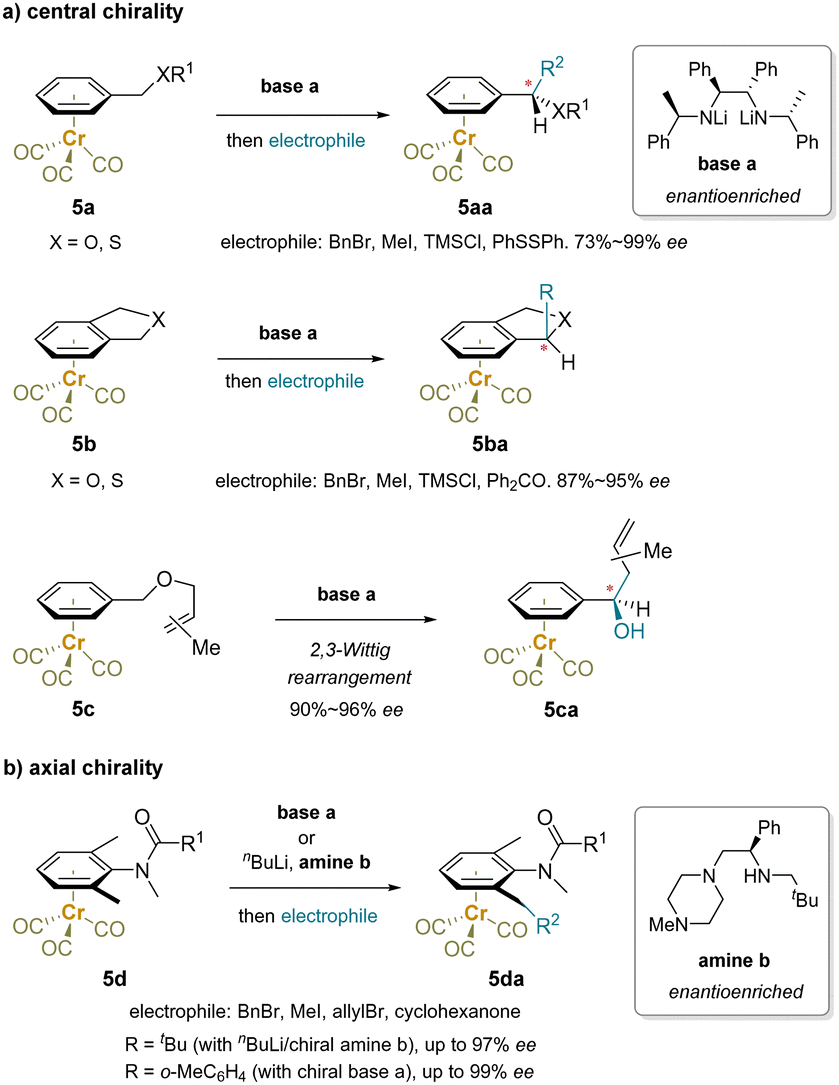 | ||
| Scheme 9 Enantioselective functionalization enabled by enantioenriched bases: (a) central chirality and (b) axial chirality. | ||
In Scheme 9b, a class of N-methylanilides containing axial and planar chirality were synthesized through desymmetrization of prochiral chromium tricarbonyl complexes using an enantioenriched base.150,151 The steric hindrance of the N-methyl amide and flanking methyl groups in 5d block rotation about the C–N bond and the benzylic methyl groups are prochiral. A chiral lithium amide discriminates the enantiotopic benzylmethyl groups of the Cr(CO)3-complexed anilide for the asymmetric deprotonation.
2.3. Palladium-catalyzed cross-coupling
Palladium-catalyzed cross-coupling is a versatile method for forming C–C bonds using various carbon-based nucleophiles, such as preformed organometallic compounds.152–159 In early studies, benzylic zinc derivatives (6aa) prepared from (alkylbenzene)Cr(CO)3 (6a) were found to be suitable coupling partners for acetylation with acetyl chloride (Scheme 10a).61 Arylation and vinylation were also achieved by varying the electrophiles to the corresponding halides using the same palladium catalyst, Pd(PPh3)4.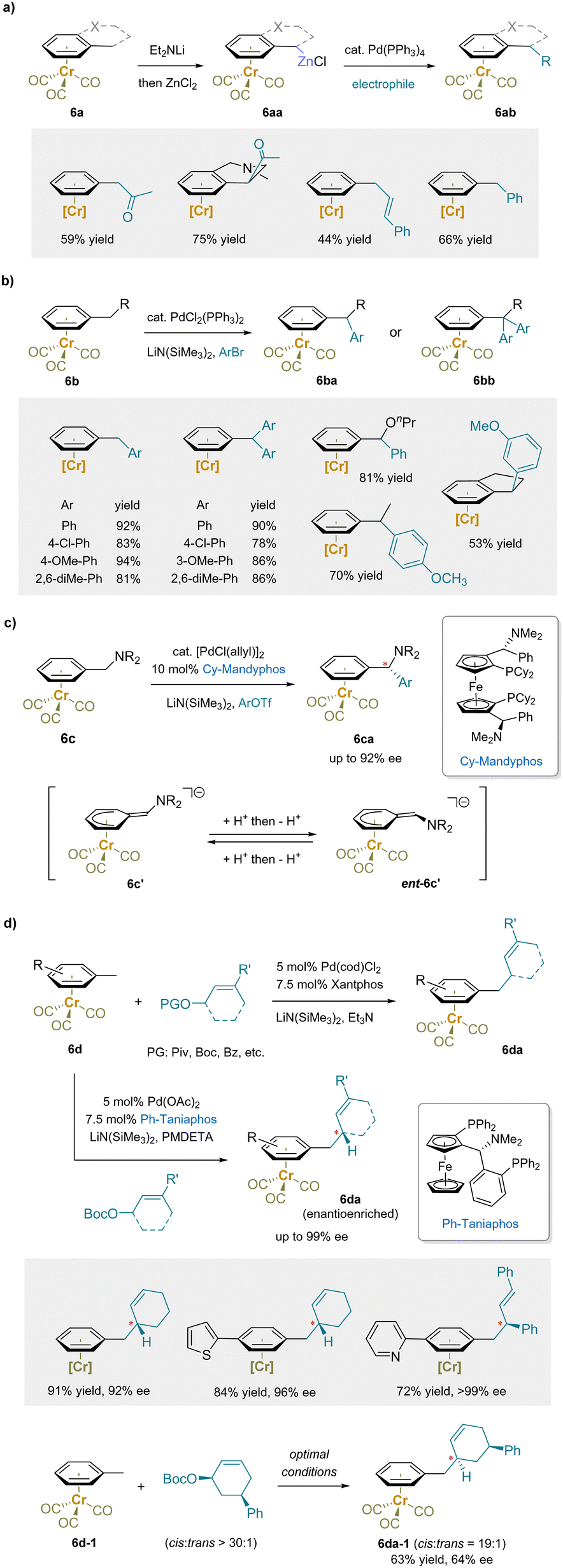 | ||
| Scheme 10 Pd-catalyzed cross-coupling and allylic substitution reactions: (a) Negishi reactions, (b) arylation reactions, (c) asymmetric arylation reactions, and (d) Tsuji–Trost reactions. | ||
Instead of preparing the main group organometallic nucleophiles beforehand, soft carbanions can be readily generated by deprotonation of pro-nucleophiles, including ketones and β-keto-esters, and used as nucleophiles in palladium-catalyzed cross-coupling reactions.160–176 In particular, an array of palladium-catalyzed C–C bond formation reactions of Cr(CO)3-complexed toluene derivatives with LiN(SiMe3)2 as the base has been reported. Arylation reactions with aryl bromides provided either diarylmethane or triarylmethane products in good yields depending on the ratio between (arene)chromium complexes (6b) and aryl bromides (Scheme 10b).177 Excellent exo-selectivity was observed in the reactions of the indane and tetrahydronaphthalene complexes. Notably, an asymmetric version of the above arylation was developed with an enantioenriched Cy-Mandyphos ligand, providing the diarylmethylamines (6ca) in high enantioselectivity.178 This reaction is particularly challenging, because the intermediate anionic [(Ar–CHNR2)Cr]− based intermediate is planar chiral and must be racemized under the reaction conditions by reversible protonation/deprotonation(Scheme 10c).
In addition to arylation, palladium-catalyzed Tsuji–Trost allylation reactions of (toluene)Cr(CO)3 complexes (6d) with allyl carbonates or pivalates were reported (Scheme 10d).179 Asymmetric allylation reactions with the enantioenriched ligand Ph-Taniaphos yielded allylic benzylation products (6da) in high enantioselectivities. Moreover, the stereoretentive substitution of a cis-disubstituted cyclohexene was obtained under the above reaction conditions (Scheme 10d), suggesting that the π-coordination with Li+ stabilized the benzylic anion, which served as a “soft” nucleophile and attacked the π-allyl palladium complex through an anti-addition mechanism (net double inversion) to give 6da-1.
2.4. Catalysis via π-coordination activation
Despite the successful π-coordination activation in benzylic functionalization of preformed η6-arene complexes, the utility of these reactions is limited by using stoichiometric amounts of transition-metals and ligands. To overcome this significant limitation, researchers are interested in developing methods using catalytic amount of arenophilic transition-metal complexes. This approach promises high atom economy by reducing the amount of transition-metals and ligands from stoichiometric to catalytic levels. Moreover, it would avoid the need for additional steps to prepare arene complexes and remove products from the metal centers. Scheme 11 outlines a general catalytic cycle, including an arene exchange step, following the benzylic functionalization step. | ||
| Scheme 11 Idealized catalytic cycle for benzylic C–H functionalization via π-coordination activation. | ||
The realization of a catalytic cycle, however, faces a major hurdle due to the strong (η6-arene)-transition-metal arene interactions that disfavors arene exchange. Additionally, the substrate may contain other weakly acidic protons that might be activated by stoichiometric amounts of strong base. Strong bases can react undesirably with electrophiles and may also decompose arene complexes or coordinatively unsaturated metal species, which are intermediates in arene exchange processes. To date, only a limited number of successful catalytic arene-activation reactions have been documented.
In 2016, a ruthenium-catalyzed dehydrative condensation between toluene and aldehydes was developed (Scheme 12).180 The reactions employed solvent quantities of toluene or xylene as the pro-nucleophile with [Cp*Ru]+ as catalyst, yielding either stilbenes or distyrylbenzenes. The narrow scope of this method, limited to primary C–H bonds, may be attributed to the inefficient activation by the Ru(II) catalyst. Nonetheless, this work represents a significant advance, demonstrating proof-of-concept of Scheme 11.
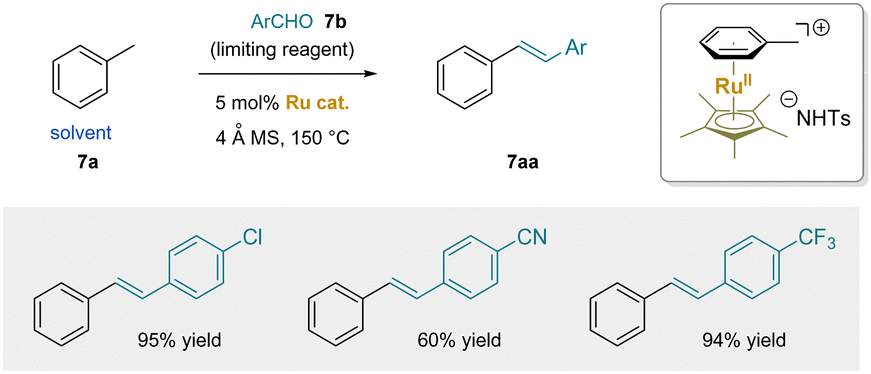 | ||
| Scheme 12 Ru-catalyzed condensation of toluene with aldehydes. | ||
In comparison to Ru(II) species, Rh(III) analogues exhibit stronger electron-withdrawing effects, increasing the acidity of the benzylic C–H's of the corresponding η6-arene complexes. This advantage of Rh(III) catalysts has been successfully applied to develop catalytic nucleophilic aromatic substitution of non-electrophilic aryl halides and phenols.51 In 2022, rhodium-catalyzed (5 mol% Rh) benzylic H/D exchange reactions with a wide range of alkylarenes (8a) were reported. Of note, these reactions featured primary, secondary, or tertiary C–H bonds (Scheme 13a).181 In the presence of the dicationic [CpRh]2+ catalyst with a catalytic amount of the weak base Li3PO4 in acetone-d6, or without any base in methanol-d4, deuterium exchange occurred exclusively at the benzylic positions. The mild reaction conditions allowed for tolerance of a diverse range of functional groups and enabled late-stage labeling of pharmaceuticals and their analogues. Shortly afterwards, this approach was extended to the addition of benzylic C–H bonds across electrophilic Michael acceptors (Scheme 13b).182 Notably, the rhodium-catalyzed addition reactions could overcome the steric hindrance of alkylarenes bearing a tertiary C–H bond, allowing for the construction of all-carbon quaternary centers. Beside Michael additions, 1,2-addition reactions of alkylbenzenes were also established under the conditions with electrophilic imines.183 The keys to these arene-activating transition-metal catalysts are facile exchange of arene pro-nucleophiles and acidification of the benzylic protons. The use of catalytic arene activation is an area of research that leaves much room for future development. In the next sections, main group metals are examined, which are known to rapidly swap arenes, potentially providing an alternative avenue for benzylic C–H functionalization.
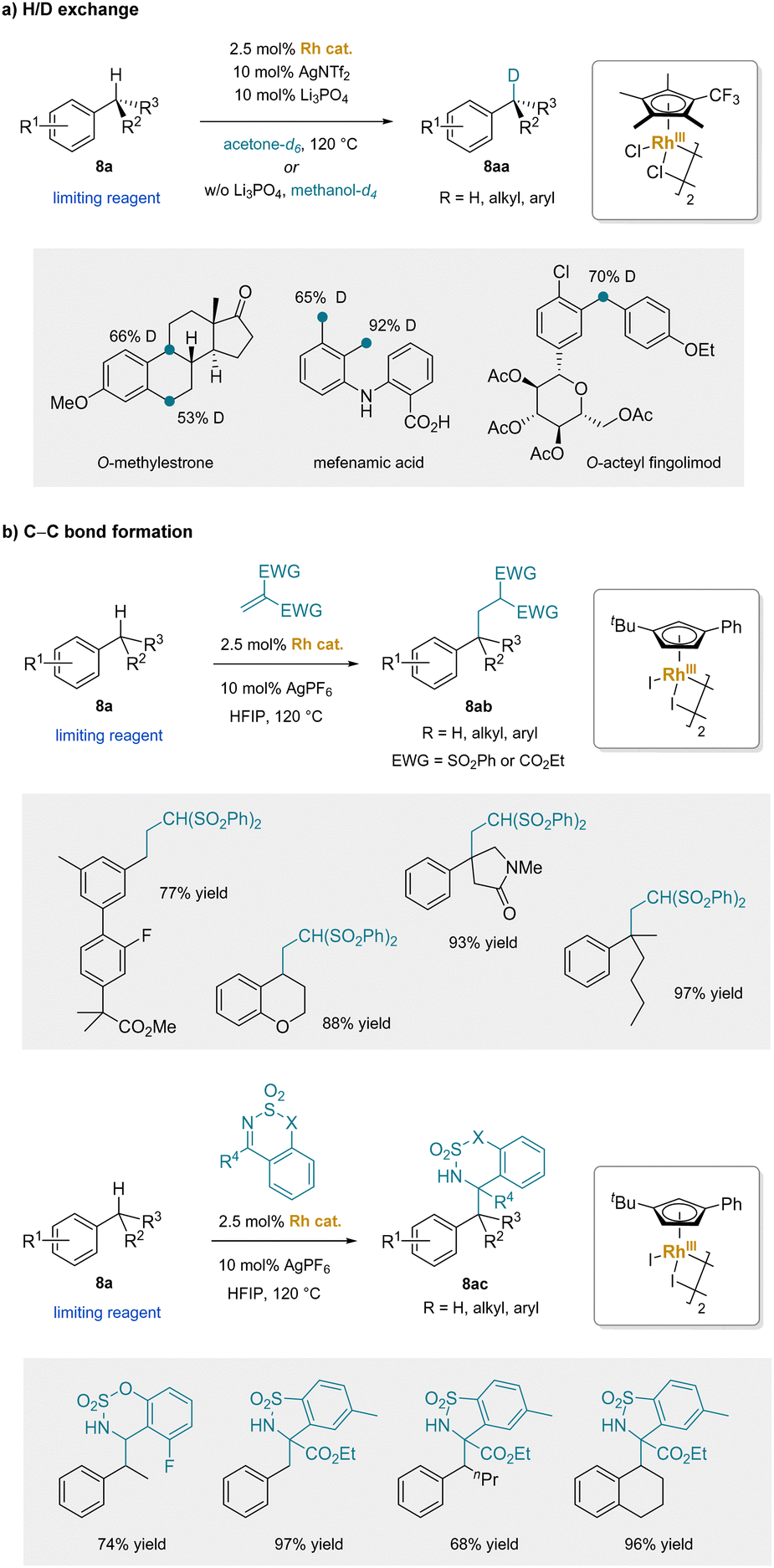 | ||
| Scheme 13 Rh-catalyzed benzylic C–H bond functionalization: (a) H/D exchange, and (b) C–C bond formation. | ||
3. Alkali-metal-facilitated deprotonation through cation–π-interactions
3.1. Cation–π interaction
Cation–π interactions are a type of noncovalent interaction between a cation and a π system, most commonly an aromatic ring. The electron-rich π system forms relatively strong electrostatic184 and induction185 (polarization) interactions (up to 20 kcal mol−1) with the positively charged cation. Cation–π interactions are different from the covalent interaction between a transition-metal and an arene π system, where the π system exhibits orbital overlap with the transition-metal orbitals and the interaction can be considerably stronger. Cation–π interactions can form between organic cations, like ammonium salts, with arene π-systems. Such interactions are an important component of organocatalysis and were recently reviewed.186 Here, the focus is on main group cations that can increase the acidity of benzylic C–H bonds by formation of cation–π interactions.In 2003, it was discovered that neither Zn(N(SiMe3)2)2 nor KN(SiMe3)2 could unilaterally metalate toluene. However, when KN(SiMe3)2 and Zn(N(SiMe3)2)2 (1:1) were added to toluene and stirred for 15 min, toluene was smoothly deprotonated at the benzylic site, generating benzylic metalation products {KZn(N(SiMe3)2)2(CH2Ph)}∞ (9aa) (Scheme 14).187 The structure of the product was determined by X-ray crystallography and contains cation–π interactions. The authors did not mention the possibility of cation–π interactions to acidify the benzylic hydrogens in this pioneering study with silylamide bases. The study confirms that the deprotonative metalation requires both bases and presumably cooperativity between them. Interestingly, the deprotonation failed when Zn(N(SiMe3)2)2 was replaced by Mg(N(SiMe3)2)2, which is presumably more basic due to the greater polarization of the Mg–N bonds over the Zn–N bonds in M(N(SiMe3)2)2. Subsequently, the authors demonstrated that toluene derivatives, such as m-xylene and mesitylene could be deprotonated.
 | ||
| Scheme 14 Synergic deprotonation using KN(SiMe3)2 and Zn(N(SiMe3)2)2. | ||
An early computational study188 elucidated details of the role of cation–π interactions in the deprotonation of benzylic C–H bonds by alkali metal amides (M–NHR, R = H, Cy). The calculations indicate that the metal-ring centroid distance decreases from a cation–π interaction moving toward the transition state for deprotonation and finally the deprotonated product (Scheme 15). This observation shows the polarization of electron density in the π system as the cation–π interaction assists in the C–H bond cleavage process. Hammett studies show that the bond cleavage is also facilitated by EWGs in both para- or meta-positions on the arene, and that resonance and inductive effects have similar influence on the barriers. It was found that Cs+ results in the largest decrease in the barrier to benzylic deprotonation relative to the smaller alkali metals. This is due to the greater basicity of nitrogen in the Cs–NH2 bond arising from the higher electropositivity of Cs+ and lower electron–electron repulsion. Overall, this important computational study documents the role that alkali metal cation–π interactions can play in benzylic deprotonations. It is also noteworthy that larger group(I) metal centers have a greater ability to interact with the π-system of the benzyl anion, as shown in both crystallographic and early computational studies.22,189 The implications of these studies for development of synthetic methods are highlighted below.
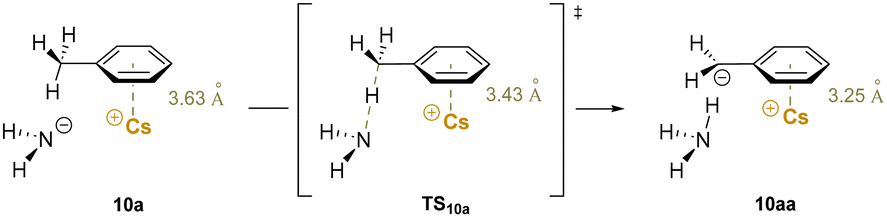 | ||
| Scheme 15 Calculated deprotonation of toluene. Cation π-interaction between Cs–NH2 and toluene (10a), transition state (TS10a) for deprotonation and deprotonated product (10aa). | ||
Another study22 arrived at similar conclusions in a more focused experimental system that employed Schlosser's base190 and related species in the deprotonation of toluene. The researchers reacted Schlosser's base (nBuLi/tBuOK) with toluene at −78 °C in THF, which is known to give the orange benzylic metalation product. Concentration and crystallization led to isolation of [(PhCH2K)3(THF)4] (11aa), which contains potassium coordinated to the benzylic anion in a variety of ways, including η6-coordination (Scheme 16). The authors also demonstrated that Schlosser's base would deprotonate benzene and isolated a Li/K bimetallic complex [(PhK)4(PhLi)(tBuOLi)(THF)6(C6H6)2] containing several phenyl anions and a tert-butoxide bonded to either Li or K. Using a truncated model for computational purposes [(PhK)(LiOtBu)(OMe2)4] (11b), they explored the deprotonation of toluene with a phenyl bridged bimetallic. The authors calculated that the exchange of a dimethyl ether on K+ for a free toluene was downhill by about 3 kcal mol−1. An interaction between K+ and toluene was shown to reduce the barrier to the deprotonation to about 18 kcal mol−1. Computational and experimental evidence suggest that the softer potassium cation is preferred over the hard lithium cation in the interaction with the delocalized π system. A four-membered ring Li–C–K–O is formed in the bimetallic cluster as the π system is added to the base system of nBuLi and tBuOK.
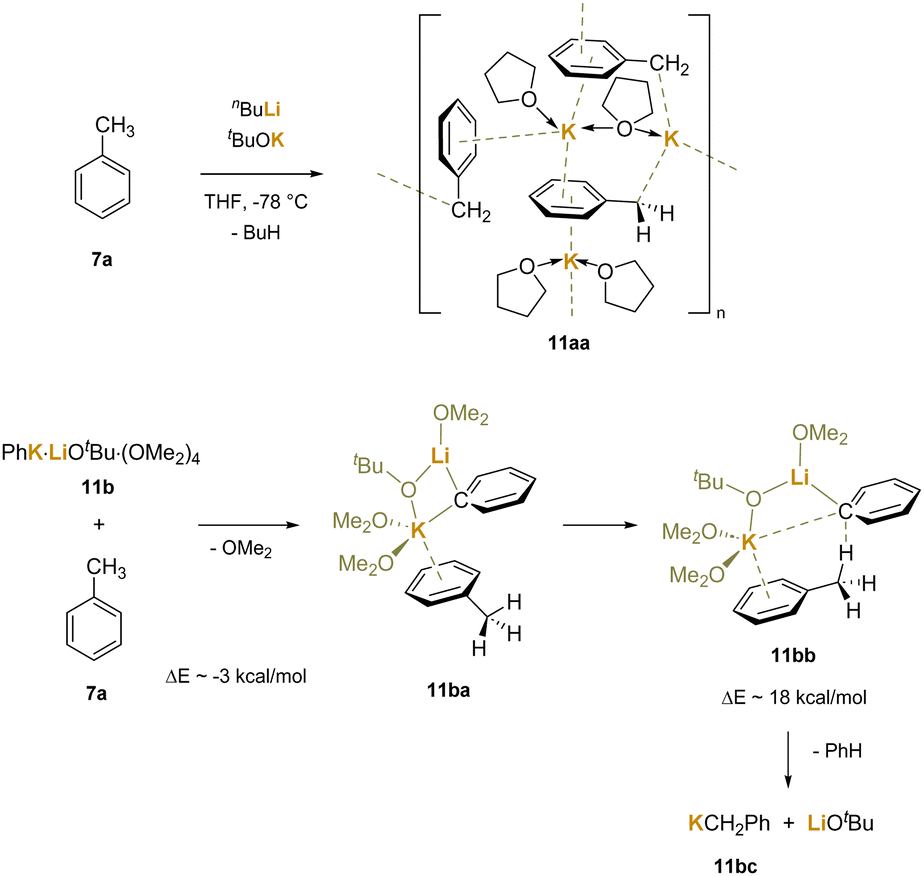 | ||
| Scheme 16 Calculated transition state for deprotonation of toluene with a model Schlosser base system via K+–π–toluene interactions. | ||
The importance of cation–π interactions in the cleavage of benzylic C–H bonds in the palladium catalyzed arylation of toluenes was shown both computationally and experimentally (Scheme 17).191 This study is significant for several reasons. The first is that the reaction was performed with KN(SiMe3)2, which is a considerably weaker base than Cs–NHR or Schlosser's base.190 Second, the reaction stops at the mono-arylation product (12aa), despite the enormous increase in acidity of diphenylmethane compared to toluene (32 vs. ∼43 in DMSO).11 The reaction employed van Leeuwen's NIXANTPHOS ligand192 which has a relatively acidic N–H (pKa ≈ 22)11 that is deprotonated under the reaction conditions.21 The reaction was found to have a significant dependency on the main group cation of the base (M = K, 80%; Na, 58% and Li, 0%). Experimental evidence for the involvement of the deprotonated ligand was gained using Xantphos and N-benzyl NIXANTPHOS. Both ligands lack sites that are easily deprotonated, and both gave no products.
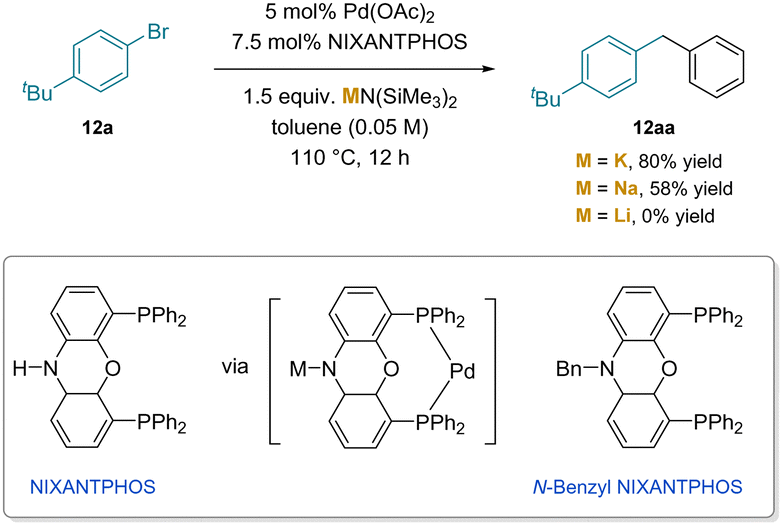 | ||
| Scheme 17 Arylation of toluene catalyzed by a (NIXANTPHOS)Pd catalyst. | ||
Calculations were performed using di-potassium systems, because KN(SiMe3)2 is predominantly oligomeric in low polarity solvents.193 The calculated transition states for deprotonation reveal that the coordination of the potassium cation to the π systems lowers the activation energy of the deprotonation (Scheme 18, TS13avs.TS13b). External activation had little impact on the energies (TS13c). Experimental support for the calculated TS in Scheme 18 was found in the reaction of toluene with KN(SiMe3)2 in the presence of trapping agent ArCH2Br at 110 °C. The reaction gave an unoptimized yield of 20% of the corresponding bibenzyl (PhCH2CH2Ar).
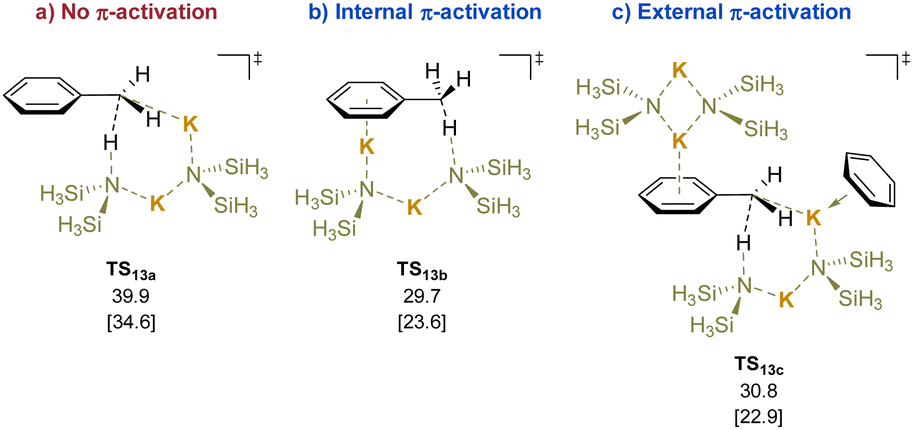 | ||
| Scheme 18 Calculated relative Gibbs free energies [and enthalpies] for the deprotonation of toluene (kcal mol−1): (a) no π-activation, (b) internal π-activation, and (c) external π-activation. | ||
Next, the deprotonation of toluene was calculated using the (NIXANTPHOS)Pd(II) system for comparison with the TS13c from Scheme 18c. A surprising K⋯Br–Pd interaction was found to assist the deprotonation transition state structure (Scheme 19b). Interestingly, and in support of the proposed K⋯Br interaction, the use of aryl chlorides in the (NIXANTPHOS)Pd-catalyzed arylation of toluenes was unsuccessful, despite the known ability of this catalyst system to activate aryl chlorides at room temperature.194 The results of the comparison between TS14 and TS13c are illustrated in Scheme 19a vs. b and suggest that, although both deprotonations are endothermic, Keq for TS14 is ∼109 times greater than for TS13c. Taken together, deprotonation viaTS14 is faster, providing a higher concentration of the benzyl organometallic, and the anion is held in the proximity of the Pd(II) and primed for transmetalation. Overall, this study provides experimental and computational support for the importance of cation–π interactions as a key activation mode to increase the acidity of benzylic C–H bonds. As outlined subsequently, this observation has been incorporated into a variety of synthetic methods with transition-metals and under transition-metal-free conditions.
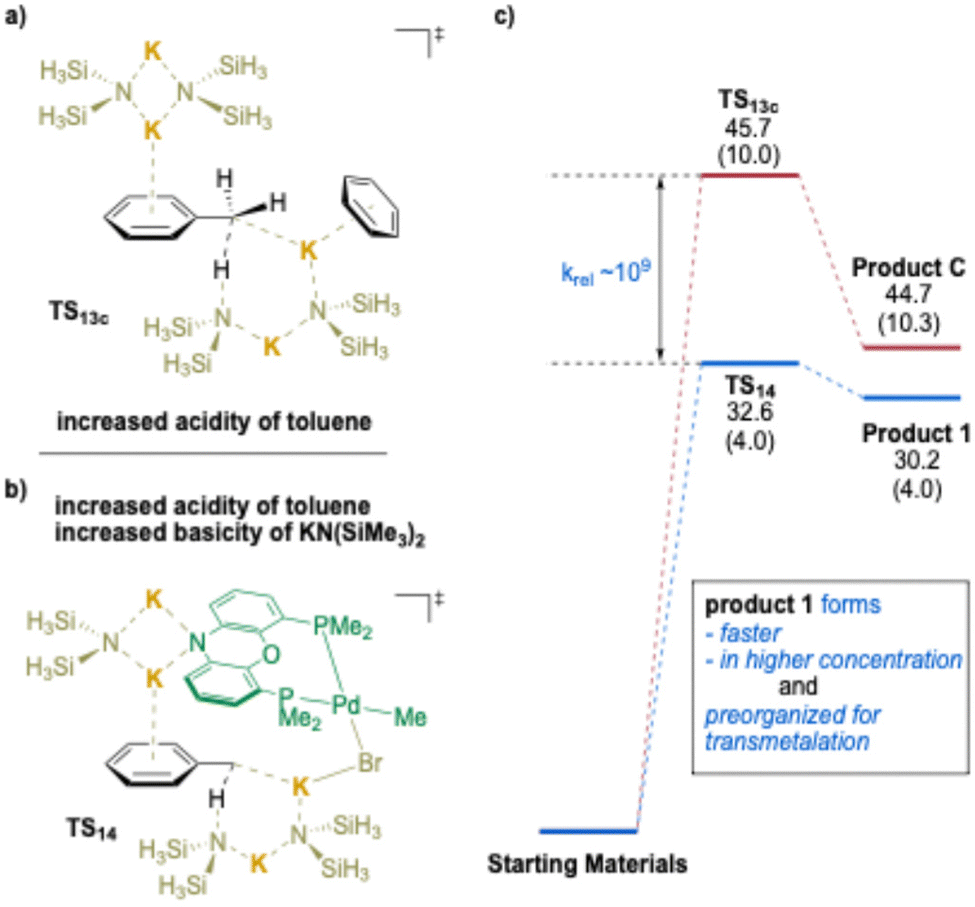 | ||
| Scheme 19 Calculated relative Gibbs free energies [and enthalpies] for the deprotonation of toluene (kcal mol−1) for (a). TS13c, (b). (NIXANTPHOS)Pd complex (TS14), and (c). relative rates. | ||
In a related investigation, a nickel catalyzed toluene arylation with aryl chlorides and bromides was studied.195 Like the palladium system above, this work used the NIXANTPHOS ligand and silyl amide bases (Scheme 20a). The more electropositive nickel center facilitates the oxidative addition step196 compared to palladium, so that both aryl bromides and chlorides were applicable in this system. Like the Pd catalyzed reaction above, this reaction also had a dependency on the main group cation of the silyl amide base (Scheme 20b). However, unlike the palladium system, which worked best with KN(SiMe3)2 (Scheme 17), the nickel catalyst gave the best results with NaN(SiMe3)2 (77% compared to KN(SiMe3)2 with 26% and LiN(SiMe3)2 with 8%).
 | ||
| Scheme 20 Arylation of toluene catalyzed by a (NIXANTPHOS)Ni-based catalyst: (a) arylation of toluene, and (b) dependency on the main group cation. | ||
The yields of coupling products with aryl chlorides and bromides with toluenes were similar (Scheme 20a). A comparison using Xantphos and N-benzyl NIXANTPHOS as in Scheme 17, resulted in only 15% yield with N-benzyl NIXANTPHOS and 0% with Xantphos, consistent with the need for the deprotonated ligand backbone (bearing the main group metal) to activate the toluene derivative via cation–π interactions. Further circumstantial evidence for this hypothesis was gained by conducting the reaction under the optimized conditions, but in the presence of 4 equiv. of 15-crown-5. The crown ether inhibited conversion to 7%, likely through coordination to Na+, blocking the cation–π interaction. While no computational work was done on the nickel system, a similar transition state for the deprotonation to the palladium system in Scheme 19 was assumed.
The selective C–H bond functionalization is challenging, because molecules often have a variety of C–H bonds with similar reactivity. To favor C–H functionalization of specific bonds, chemists often use directing groups to steer reactivity. Directing groups need to be added and removed after the functionalization step. It was found that site-selective arylation at the sp2vs. sp3 C–H bonds of 2-benzylfurans could be controlled by cation–π interactions.197 This research also used the NIXANTPHOS ligand and silyl amide bases to deprotonate the weakly acidic benzylic C–H bonds of 2-benzyl furan (pKa = 30.2 in DMSO11). The selectivity between formation of the benzylic (16aa) or C-3 arylation product (16ab) is controlled by the main group counterion of the base (Scheme 21a), as detailed below.
 | ||
| Scheme 21 Cation-controlled site-selective C3 and benzylic C-H arylation: (a) cation-controlled site-selective benzylic C–H arylation of furan, and (b) proposed reaction manifolds and intermediates for the benzylic and C3 arylations. | ||
The authors found that the main group cation of the silyl amide base MN(SiMe3)2 is crucial for the selectivity of the reaction between benzylic and C-3 arylation. With (NIXANTPHOS)Pd catalyst and KN(SiMe3)2, the C-3 arylation product (16ab) was generated exclusively. Under nearly identical conditions, but using LiN(SiMe3)2 with the addition of 12-crown-4, the benzylic arylation product (15a) was obtained.
Calculations were performed on the energy states of complex 16aa′ and 16ab′ from Scheme 21b, but without inclusion of the cation on the deprotonated NIXANTPHOS. The energy of Pd–CBn in complex 16aa′ is 5 kcal mol−1 lower than Pd–C-3 (16ab′), indicating the preference of forming the benzyl adduct and eventually the benzylic arylation product. Performing the calculations in the presence of the K+(dioxane), however, resulted in a K+–π interaction (Fig. 1) that shifts the equilibrium to the C-3 bound intermediate. It was proposed that this shift in the equilibrium toward C-3 guides the reductive elimination to provide the C-3 arylation product (16ab). More polarizable K+ is known to favor cation–π interactions compared to the smaller, harder Na+ and Li+ cations in solution.198,199 However, by adding 18-crown-6, the potassium center's coordination sites were blocked from forming cation–π interactions and the selectivity switched to favor the benzylic arylation (16aa).
 | ||
| Fig. 1 DFT geometry optimized structure with the bulky 3,5-di-tert-Bu phenyl ring. | ||
It was noted that steric and electronic properties of aryl bromide substrates did not influence the site-selectivity, but electronic properties of the furan substrates could impact the cation–π interactions. Indeed, use of (2-fural)–CH2(3,5-C6H3-(CF3)2) resulted in a shift back towards benzylic arylation, presumably partly due to the diminished strength of the cation–π interaction with the electron-poor aryl ring. This study provides experimental and computational support for the significance of cation–π interactions in controlling regioselectivity of 2-benzylfurans in bimetallic catalytic systems.
3.2. Cation–π complexes in the absence of transition-metals
The examples of benzylic functionalization through deprotonation above were conducted in the presence of transition-metals, which are well known to activate arenes. Although the transition-metals are not believed to be involved, it was viewed as prudent to explore benzylic functionalization without added transition metals, which will be discussed below. Of course, the use of strong bases to deprotonate benzylic C–H's and form benzyl organometallic species has been known for many decades.In an example, the superbasic mixed-metal “LIC-KOR” reagent (KOtBu + nBuLi) was used to metalate trialkylsilyl substituted toluenes (17). The authors compared the relative rates of metalation to the reaction of the parent toluene (Scheme 22a).200 It was found that the para-substituted trialkylsilyl groups enhanced the rate, while the ortho and meta groups exhibited rate retardation. To explain this observation, the authors proposed a cation–π interaction between the aromatic ring and the potassium in the transition state (TS16), as depicted in Scheme 22b. Given the distance between the benzylic methyl group undergoing deprotonation and the meta-SiR3 substituent, it was hypothesized that the decrease in rate was due to partial obstruction of the K+–cation–π interaction resulting from buttressing with the SiR3 substituent. In contrast, using nBuLi with the ligand N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDTA), which is coordinatively saturated and unable to form cation–π interactions, deprotonation reactions with the meta-trialkylsilyl toluenes were faster than with toluene itself. This system used superbasic mixed-metal “LIC-KOR”. Subsequent discussion will center on weaker bases where cation–π interactions can play a larger role.
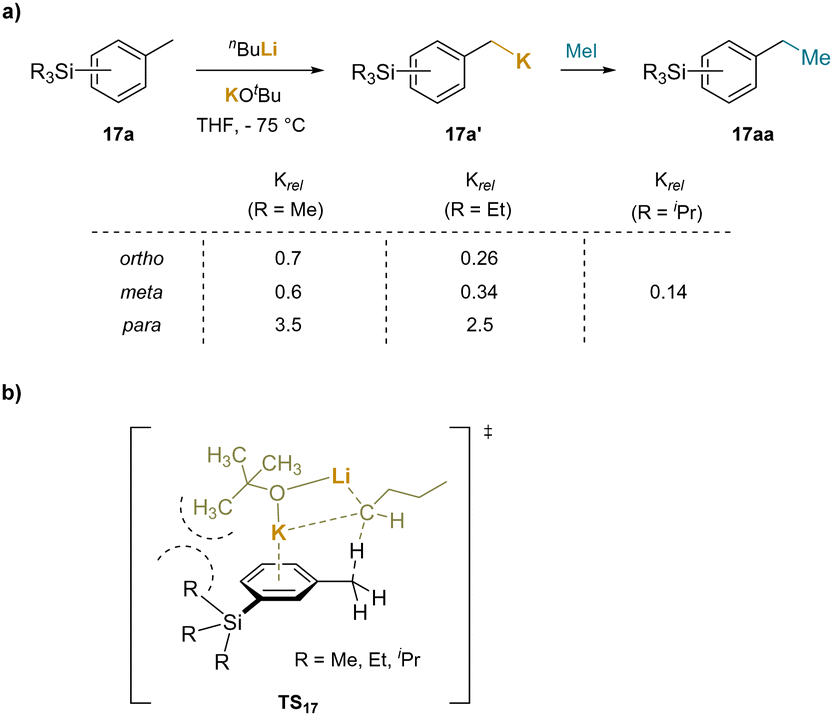 | ||
| Scheme 22 Metalation of trialkylsilyl substituted toluenes with “LIC-KOR” reagent: (a) relative metalation rates of trialkylsilyl substituted toluenes, and (b) proposed transtition state. | ||
The idea that weaker bases that benefit from cation–π interactions could be employed to reversibly deprotonate toluene derivatives was not put into practice until more recently (although computational support for the role of cation–π interactions on the deprotonation of heteroaromatic C(sp2)–H bonds had been reported201). In this regard, an early study that proposed the importance of cation–π interactions in the functionalization of toluenes involved the aminobenzylation of aldehydes (Scheme 23). In the first step, the non-enolizable aldehyde reacts with the silyl amide base via carbonyl addition to give 7b′. The resulting adduct undergoes an aza-Peterson olefination-type process to form the N-TMS imines (7ba).202 In the presence of toluene solvent, reversible deprotonation of the benzylic C–H bond by the silyl amide base gives the benzylic organometallic 18a′ which could be envisioned to have different hapticities (18a′′).189 The benzylic organometallic 18a′ could then undergo reaction with the N-TMS imine (7ba) to generate 18aa′, which upon workup affords the aminobenzylation product (18aa).
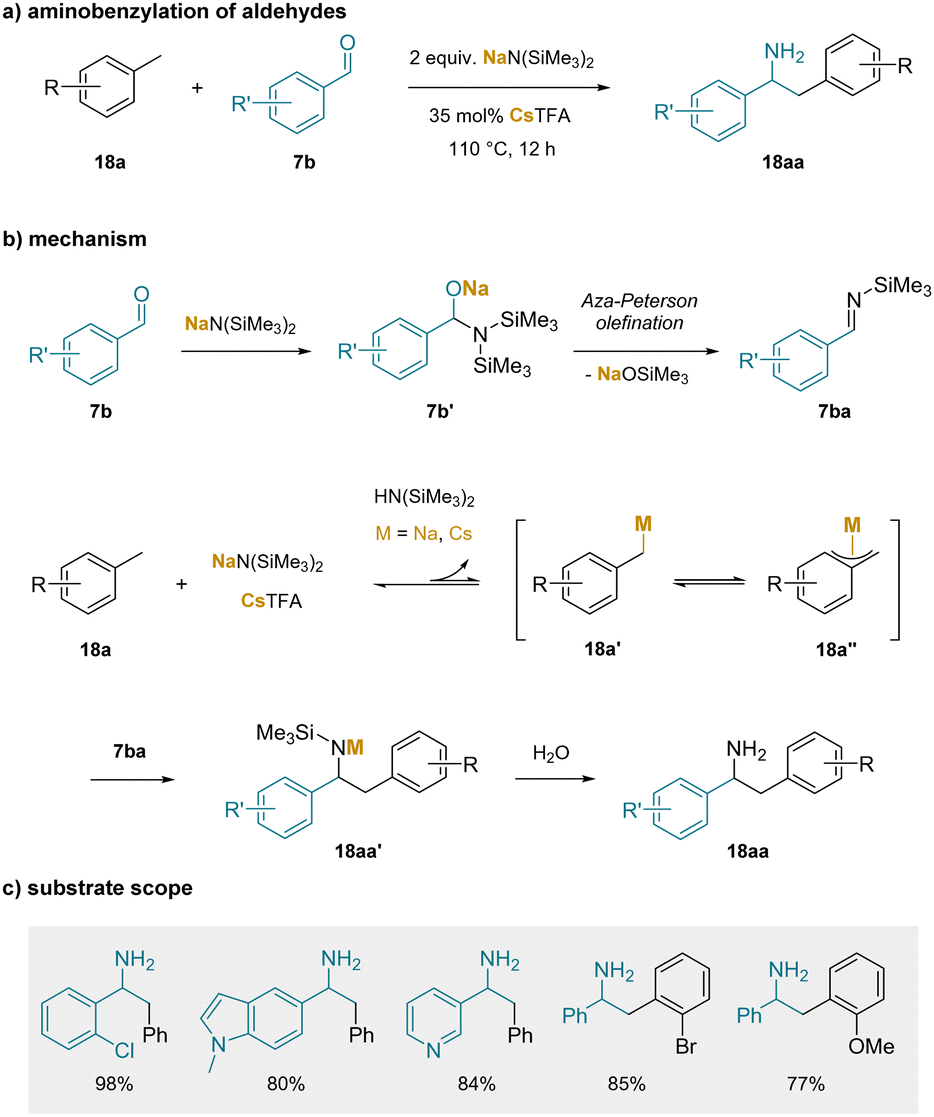 | ||
| Scheme 23 Aminobenzylation of aldehydes: (a) aminobenzylation of aldehydes, (b) proposed mechanism, and (c) selected substrate scope. | ||
It is noteworthy that of the MN(SiMe3)2 (M = Li, Na, K) bases, M = Li gave only the imine (no aminobenzylation) and M = Na gave the best results, albeit less than 50% yield of the amine (despite extensive optimization). The K+ salt, which would form the stronger cation–π interactions in solution, gave inferior results, possibly due to difficulty forming the imine. Of significance, it was found that addition of 35 mol% Cs(O2CCF3) resulted in the formation of the aminobenzylation products (18aa) in yields over 90%. In a related work, it was known that combining CsX (X = halide) with NaN(SiMe3)2 generated CsN(SiMe3)2,203 which could be crystallized from toluene to isolate the silyl amide with a cation–π complex between Cs+ and the toluene in the structure.
Overall, the scope of the aminobenzylation reaction was broad with respect to the starting aldehyde and the toluene derivatives, as shown in a few representative examples in Scheme 23c. It is noteworthy that the reaction could be conducted with 1.1 equiv. NaN(SiMe2)2 in the presence of 5 mol% Cs2CO3. Here, 1 equiv. NaN(SiMe2)2 was required to form the N-TMS imine (7ba) and the remaining 10 mol% catalyzed the aminobenzylation reaction.204
Two related strategies were developed based on cation–π interactions. In the first example, the synthesis of indoles (19ab) from 2-fluorotoluenes (19a) and benzonitrile derivatives (19b) was introduced (Scheme 24a). As with the aminobenzylation reaction, the indole synthesis provided much higher yields when a Cs+ source was present.205 Here, reversible deprotonation was followed by addition to the nitrile to give a metalated imine (19aa) that underwent SNAr.
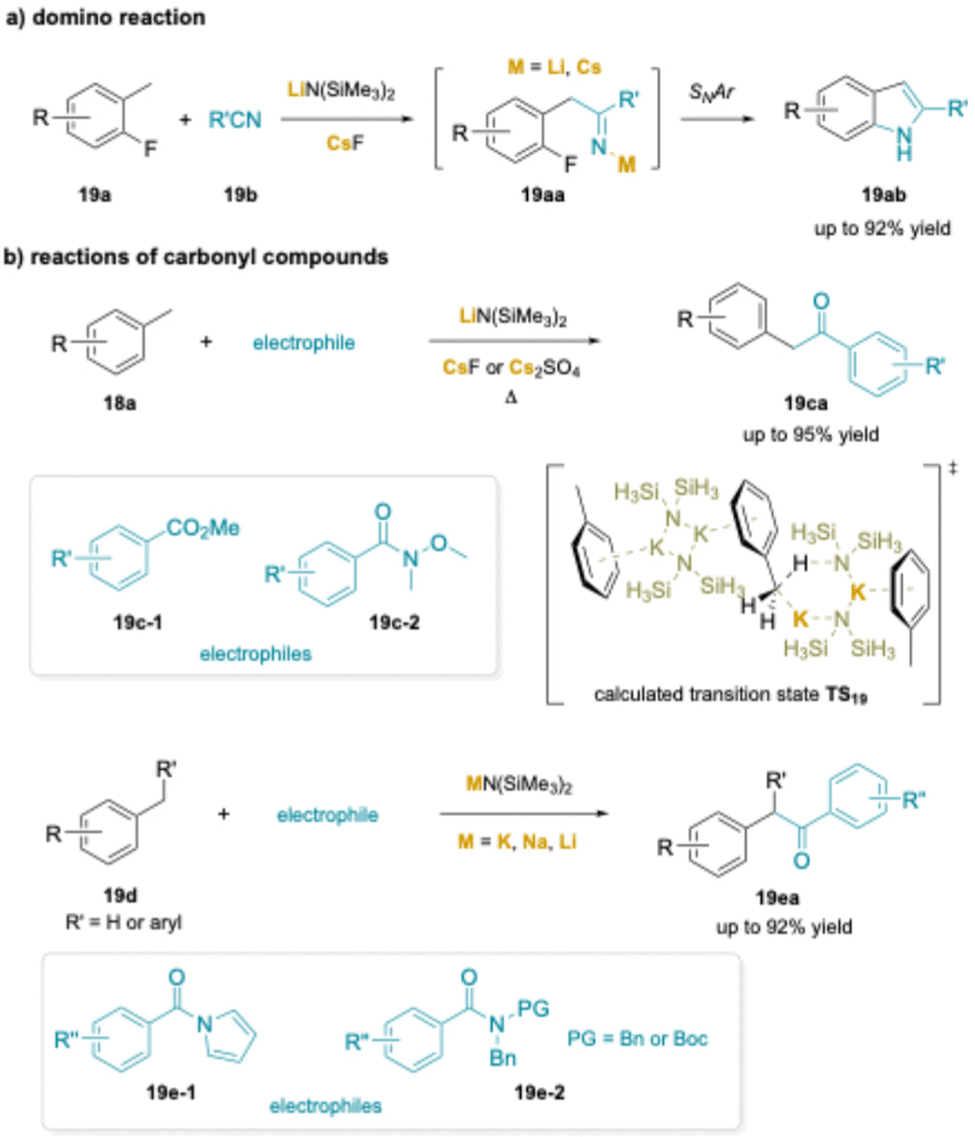 | ||
| Scheme 24 Applications of cation–π interactions: (a) an indole synthesis from 2-fluorotoluene, and (b) aroylation of toluenes. | ||
In the second line of work, toluene was reversibly deprotonated in the presence of methyl benzoate (19-c1) (Scheme 24b) or Weinreb amides (19-c2)206 in the presence of LiN(SiMe3)2 and CsF or 2,5-dimethyl N-acyl pyrroles207 (19-e1) in the presence of KN(SiMe3)2.26 In a related study, alkyl sodium NaCH2SiMe3 with chelating PMDTA, or mixing Zn(TMP)2 (TMP = 2,2,6,6-tetramethylpiperidinyl anion) with KOtBu, significantly enhanced metalating power compared to their organolithium, which can promote the benzylic aroylation of toluene derivatives and addition to Weinreb amides under mild conditions. In all cases, addition of the benzylic organometallic to the carbonyl group forms the tetrahedral intermediate that breaks down to generate an aryl benzyl ketone (19ca, 19ea). Rather than undergo a second addition of the benzyl organometallic, the ketone is rapidly deprotonated to afford the enolate. Upon workup, the enolate product is protonated to furnish the ketone.208,209
In a subsequent study, researchers found that toluene could be deprotonated in the presence of benzoate esters with KN(SiMe3)2.210 Most importantly, they performed a detailed study of the deprotonation using DFT calculations. Due to the oligomeric nature of MN(SiMe3)2 complexes in non-polar solvents, the authors modeled the deprotonation with a dimer of KN(SiMe3)2 and modeled the SiMe3 groups with SiH3 substituents. The barrier to deprotonation was calculated to be 32.5 kcal mol−1, which was found to be the rate determining step of the aroylation reaction. The transition state TS19 calculated by this team is shown in Scheme 24b.
Another application of cation–π interactions is the deuteration reaction. Since K+ could participate in cation–π interactions that enhance the acidity of the methyl protons to deprotonate toluene,211,212 phosphide anion [P(tBu)2]− generated from KH and phosphines was used to deprotonate toluene and afford HP(tBu)2 and KCH2Ph.213 The latter species can activate dihydrogen reversibly, promoting the hydrogen isotope exchange in the toluene methyl group via FLP (Frustrated Lewis pair) activation of H2 (Scheme 25a). A similar reaction mechanism can be applied to KH-catalyzed H/D exchange reaction benefiting from cation–π interactions between K+ and toluene (Scheme 25b). As with the phosphide, KCH2Ph was computed to exist mainly as the dimeric structure (KCH2Ph)2, with the monomeric form being 1.8 kcal mol−1 higher in free energy.
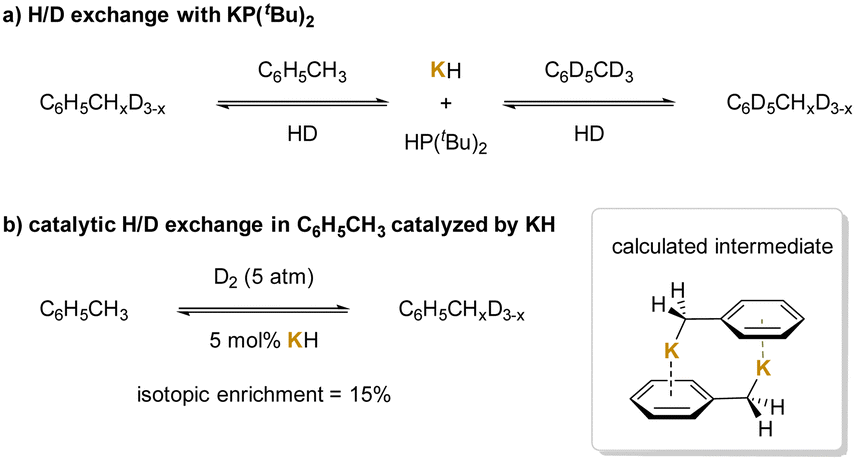 | ||
| Scheme 25 Deuteration of benzylic C–H bonds: (a) H/D exchange with KP(tBu)2, and (b) catalytic H/D exchange in C6H5CH3 catalyzed by KH. | ||
Cation–π interactions have also been used in the silylation of alkyl arenes under stoichiometric214 and catalytic conditions.211,212 Using 3 equiv. LiN(SiMe3)2 with 2 equiv. CsCl additive for most substrates, a series of methyl arenes (0.5 mL) were converted to benzylic silanes in 53–90% yields (Scheme 26a). As noted earlier, the Cs+ salt likely generates a CsN(SiMe3)2 species that forms cation–π interactions with the methyl arene during the deprotonation process. In the scope, a variety of silyl chlorides worked well, with the exception of Cl–SiMe3, which may react directly with the silyl amide base. Importantly, the chemistry was further extended to chlorogermanes to produced benzylgermanes in 60–90% yields. Even chlorotributyl tin reacted under the standard conditions to afford PhCH2–Sn(nBu)3 in 80%.
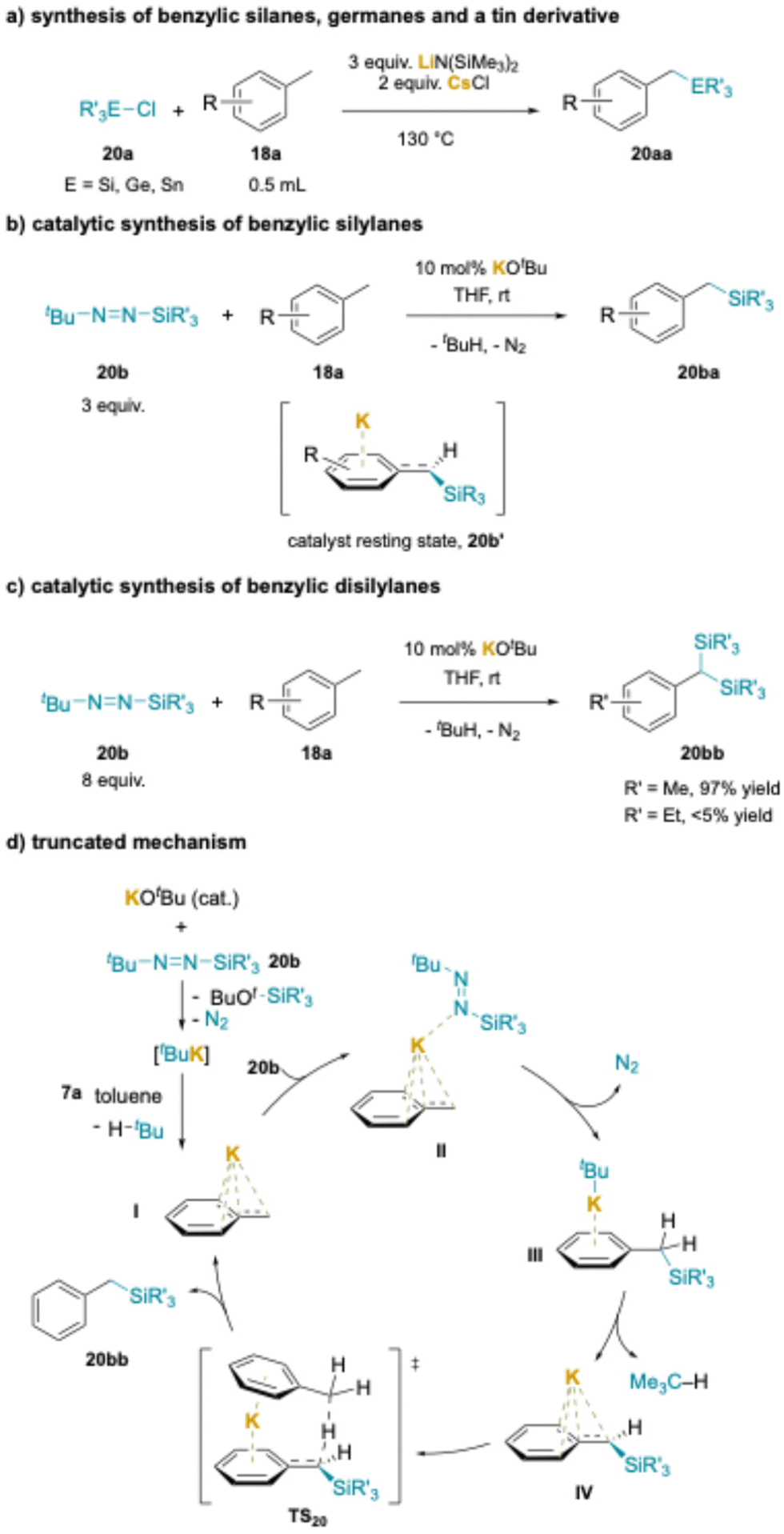 | ||
| Scheme 26 Synthesis of benzylic silanes, germanes and tin derivatives: (a) reaction of R3E-Cl with generated benzylic anions, (b) catalytic synthesis of benzylic silylanes, (c) catalytic synthesis of benzylic disilylanes and (d) truncated mechanism. | ||
Shortly after publication of the method in Scheme 26a, a related transformation was introduced.215 The benefits of this second method are the use of catalytic base, reactions conducted at RT and the employment of stoichiometric methyl arene, while the downside is the necessity for a specialized silylating reagent, tBu–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–SiR3 (20b) (Scheme 26b). The scope was demonstrated to be surprisingly good with respect to the alkyl arene and various functional groups were compatible with the reaction (F, Cl, CO2tBu, NHMe, alkene, alkyne, ether) despite the reactive nature of some of the proposed intermediates (see above). The authors also described the novel gem-disilylation of 4-phenyl toluene (Scheme 27b). Small silyl groups, like SiMe3 gave excellent results (97% yield), while use of larger diazine derived silanes resulted in lower yields (SiEt3, <5%). Unsymmetrical disilylanes could also be prepared.
N–SiR3 (20b) (Scheme 26b). The scope was demonstrated to be surprisingly good with respect to the alkyl arene and various functional groups were compatible with the reaction (F, Cl, CO2tBu, NHMe, alkene, alkyne, ether) despite the reactive nature of some of the proposed intermediates (see above). The authors also described the novel gem-disilylation of 4-phenyl toluene (Scheme 27b). Small silyl groups, like SiMe3 gave excellent results (97% yield), while use of larger diazine derived silanes resulted in lower yields (SiEt3, <5%). Unsymmetrical disilylanes could also be prepared.
 | ||
| Scheme 27 Carboxylation of methyl arenes under basic conditions. | ||
NMR studies demonstrated that the catalyst resting state is the deprotonated benzylic silane (Schemes 26b and 20b′), the only potassium-containing intermediate that was observed when monitoring the reaction progress. DFT computations were used to map this interesting mechanism, and a simplified version is drawn in Scheme 26d. Entry into the catalytic cycle is shown in the upper left portion. Attack of tBuOK on the silyl diazene forms a 5-coordinate silicon ate complex that expels N2 and generates tBuK. The tBuK readily deprotonates the toluene derivative to give KCH2Ph (I) and 2-methylpropane. N-Coordination of an equivalent of diazene to potassium and attack of the benzylic carbanion on silicon in II results in loss of N2, formation of the Si–C bond and generation of tBuK stabilized by a cation–π complex of the benzyl silane (III). The tBuK deprotonates the η6-benzyl silylane to afford IV. The authors then proposed activation of toluene through a cation–π complex, as shown in TS20, followed by intramolecular proton shuffling to regenerate KCH2Ph (I) and close the catalytic cycle. Comparison between the stoichiometric base in Scheme 26a and catalytic base in Scheme 26b is the diazene reagent reacts to generate base and sustain the catalytic reaction.
Carbon dioxide is a useful C1 source, but can be challenging to incorporate into organic scaffolds under mild conditions.216 Several methods for the carboxylation of organic compounds under basic conditions were developed (Scheme 27).217–220 The innovation of this work is that it circumvents the traditional two step approach that relies on use of very strong bases, such as nBuLi/KOtBu/TMP at −78 °C,221 followed by treatment of the resulting benzylic organometallics with CO2. A key feature of this work is that highly reactive bases will react directly with CO2. The authors found that use of LiOtBu and CsF in 1,3-dimethyl-2-imidazolidinone (DMI) under CO2 (1 atm) reacted reversibly, with generation of the adduct −OCO2tBu and leaving some unreacted tert-butoxide to deprotonate the methyl arene substrate. In some cases, LiOCEt3 proved more effective. A few of the most challenging examples are given in Scheme 27. In the second step, the 80 °C acidification of the reaction mixture is to decarbonylate the dicarboxylated products, ArCH(CO2−)2. The dicarboxylated product can also be methylated and isolated as the diester, ArCH(CO2Me)2.
Several observations were made on the carboxylation reactions that shed light on the reaction details. It was hypothesized that reaction of LiOtBu and CsF resulted in the formation of CsOtBu. Use of independently prepared CsOtBu in the absence of lithium salts, such as LiF, gave similar yields to the standard reactions. Furthermore, 133Cs NMR analysis of a solution of LiOtBu and CsF that had been heated for 1 h at 180 °C showed the formation of CsOtBu. The authors proposed that the Cs+ forms a cation–π interaction with the methylarene in the deprotonation of the benzylic methyl group. Addition of 18-crown-6 was found to decrease the yield of the carboxylation product (21) by sequestering K+ and inhibiting the formation of the cation–π complex.219
In another application of silylamide bases to toluene deprotonation and functionalization, the addition of in situ generated benzylic anions to non-enolazible ketones was examined.222 In this study, using DMI solvent, MN(SiMe3)2 (M = Li, Na, K) bases were examined with 4-bromotoluene (limiting reagent) as the pro-nucleophile and benzophenone (22a) as the trapping ketone. Acidic workup to eliminate the tertiary alkoxide provided the product olefin. Interestingly, the LiN(SiMe3)2 afforded the product in 94% assay yield compared to 39% for the Na+ and 6% for the K+ analogues. Using xanthone as the trapping ketone, a series of pro-nucleophiles were examined with different toluene derivatives in stoichiometric amounts, as outlined in Scheme 28a.
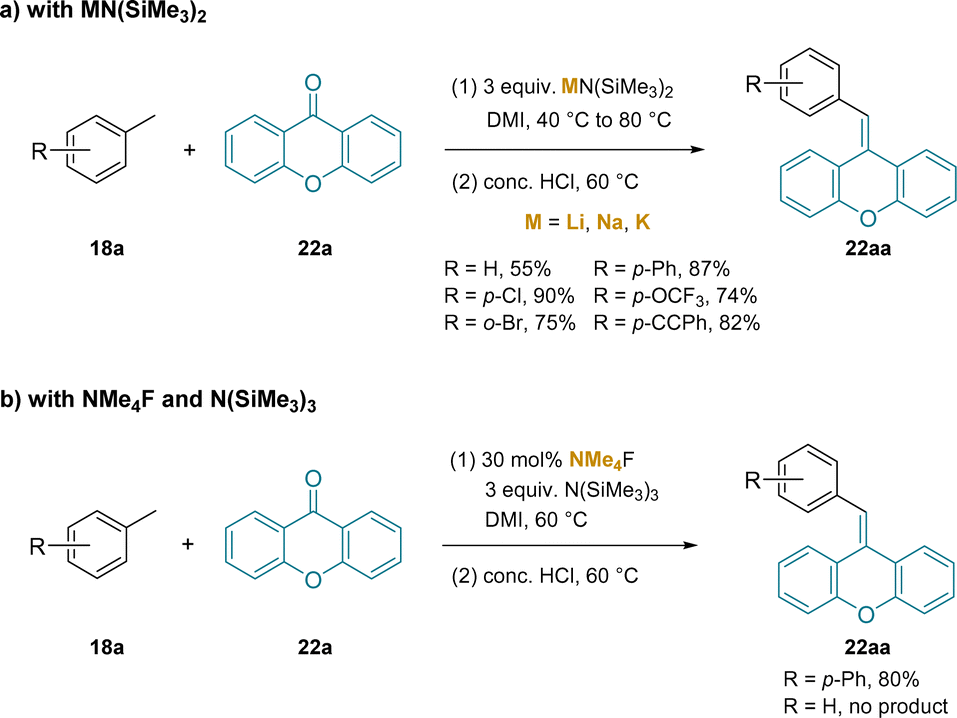 | ||
| Scheme 28 Alkene synthesis from toluene derivatives: (a) with MN(SiMe3)2, and (b) with NMe4F and N(SiMe3)3. | ||
In related work, the same group223 introduced an interesting method to generate the silyl amide base under conditions devoid of the main group counterion in the reactions in Scheme 28b. This approach enables a reactivity comparison between silyl amide bases with group(I) metal cations and the naked silyl amide. In the event, treatment of 3 equiv. N(SiMe3)3 with NMe4F resulted in fluoride induced removal of a SiMe3 group and generation of the ion pair +NMe4−N(SiMe3)2. Using 4-phenyl toluene, which has a pKa of 38.6 in THF224 (compared to HN(SiMe3)2 with a pKa 25.8 in THF225), the authors observed 80% yield of the olefin (22aa), compared to 87% in Scheme 28a. Of note, it was found that less acidic toluene was not a viable substrate with the ion pair base, +NMe4−N(SiMe3)2. In contrast, in the presence of LiN(SiMe3)2 (Scheme 28a)222 toluene was indeed a successful substrate. One interpretation of this data is that the cation–π interaction between the Li+ of LiN(SiMe3)2 and arene can account for the extra reactivity needed for this high pKa substrate. The impact of the +NMe4 cation on the reactivity of the toluene derivatives is not known, but it has been proposed that ammonium salts do form electrostatic interactions with arenes.199
There are likely several related deprotonations involving cation–π interactions that were not hypothesized to be important in the deprotonation process. Such interactions are difficult to characterize, often relying on computational results and with circumstantial experimental support. For instance, the study on NaN(SiMe3)2/CsTFA co-promoted aminobenzylation/cyclization of 2-isocyanobenzaldehydes (23a) with toluene derivatives (19d) offers one-pot access to dihydroquinazolines (23aa) and quinazolines226 (Scheme 29a). In this study, the authors proposed NaN(SiMe3)2 and CsTFA co-promoted deprotonation of the benzylic C–H bonds in toluene to form an η3-coordinated complex. In the report of benzylic aroylation of toluene derivatives with unactivated tertiary benzamides (23b),24 the researchers proposed an ortho-lithiation intermediate to be responsible for benzylic deprotonation through a σ-bond metathesis mechanism (Scheme 29b). In a more recent work, either NaN(SiMe3)2 or KN(SiMe3)2 was used in the addition reactions of toluenes to imines (23c), producing anilines (23ca) bearing branched alkyl motifs (Scheme 29c).227
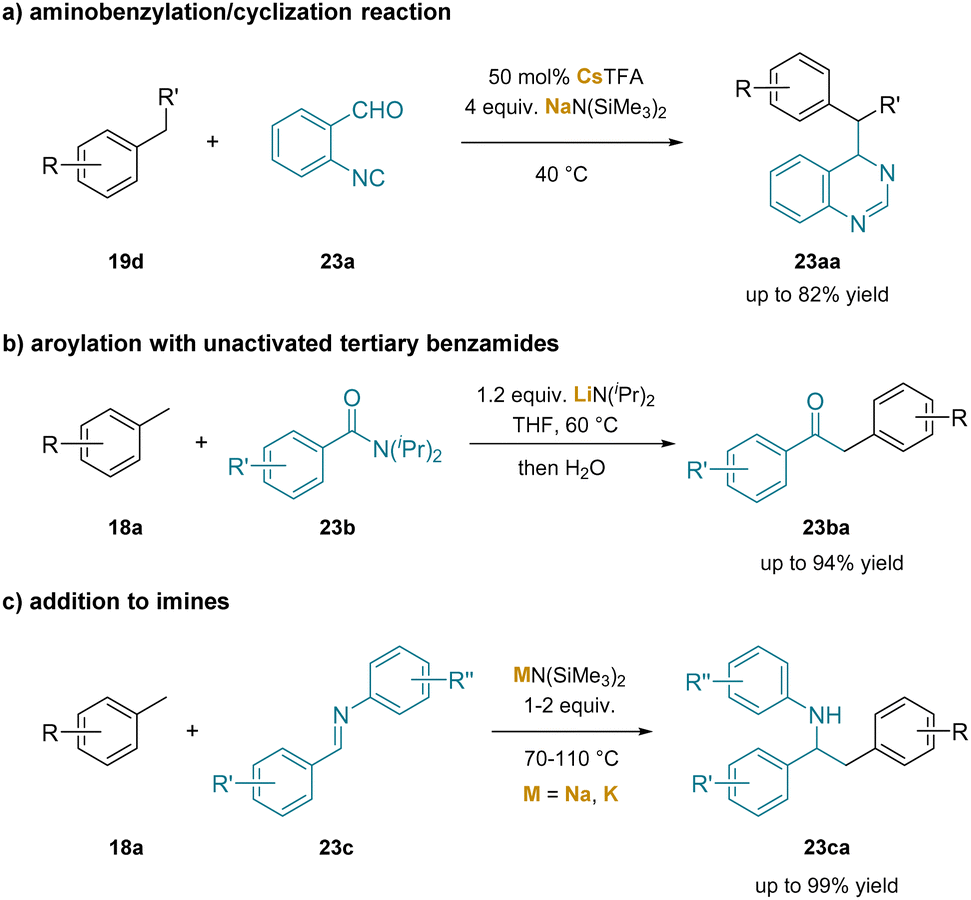 | ||
| Scheme 29 Other examples involving cation–π interactions: (a) aminobenzylation/cyclization reaction, (b) aroylation with unactivated tertiary benzamides and (c) addition to N-aryl imines. | ||
3.3. Base-catalyzed benzylic C–H bond functionalization
Several studies have implemented a catalytic amount of base to facilitate benzylic C–H bond functionalization through deprotonation. In the early stages, a catalytic amount of alkali metal was reported to generate benzylic carbanions, subsequently achieving side-chain functionalization of aromatics through the addition to styrene derivatives.228–230 In 2000, it was revealed that a mixture of nBuLi, LiK(OCH2CH2NMe2)2 and Mg(OCH2CH2OEt)2 could catalyze addition reactions of aromatic hydrocarbons bearing methyl groups (24aa) to ethylene, albeit with low functional group tolerance (Scheme 30a).231 In 2018, addition reactions of alkylarenes with imines (24b) and alkenes (24c, 24d), including four instances utilizing catalytic KOtBu and LiTMP were reported (Scheme 30b).232 The authors subsequently expanded the method's scope significantly by employing a combination of KCH2TMS and PMDTA as catalysts.233 Furthermore, they achieved asymmetric benzylic C–H bond addition with a chiral amine as the ligand, resulting in enantioenriched (62% to >99% ee) benzylic amine products (24ea) (Scheme 30c).234 In 2021, catalytic KOtBu was used to realize the hydrogen isotope exchange (HIE) of methylarenes (18a) with DMSO-d6 as a deuterium source (Scheme 30d).235 In 2023, a group of researchers developed cesium amide-catalyzed benzylic HIE reaction with D2 or T2 (Scheme 30e).236 They proposed that cesium amide could undergo a kinetic deprotonative process to generate the benzyl anion as a thermodynamically uphill intermediate at very low concentrations. Despite this, the benzylic metal species PhCH2Cs reacted with D2 to yield the HIE product (24ga) and the metal deuteride intermediate Cs–D. This intermediate could then undergo subsequent protonation with either HN(SiMe3)2 or toluene, resulting in CsN(SiMe3)2 or a benzylic anion intermediate, initiating another deuteration process.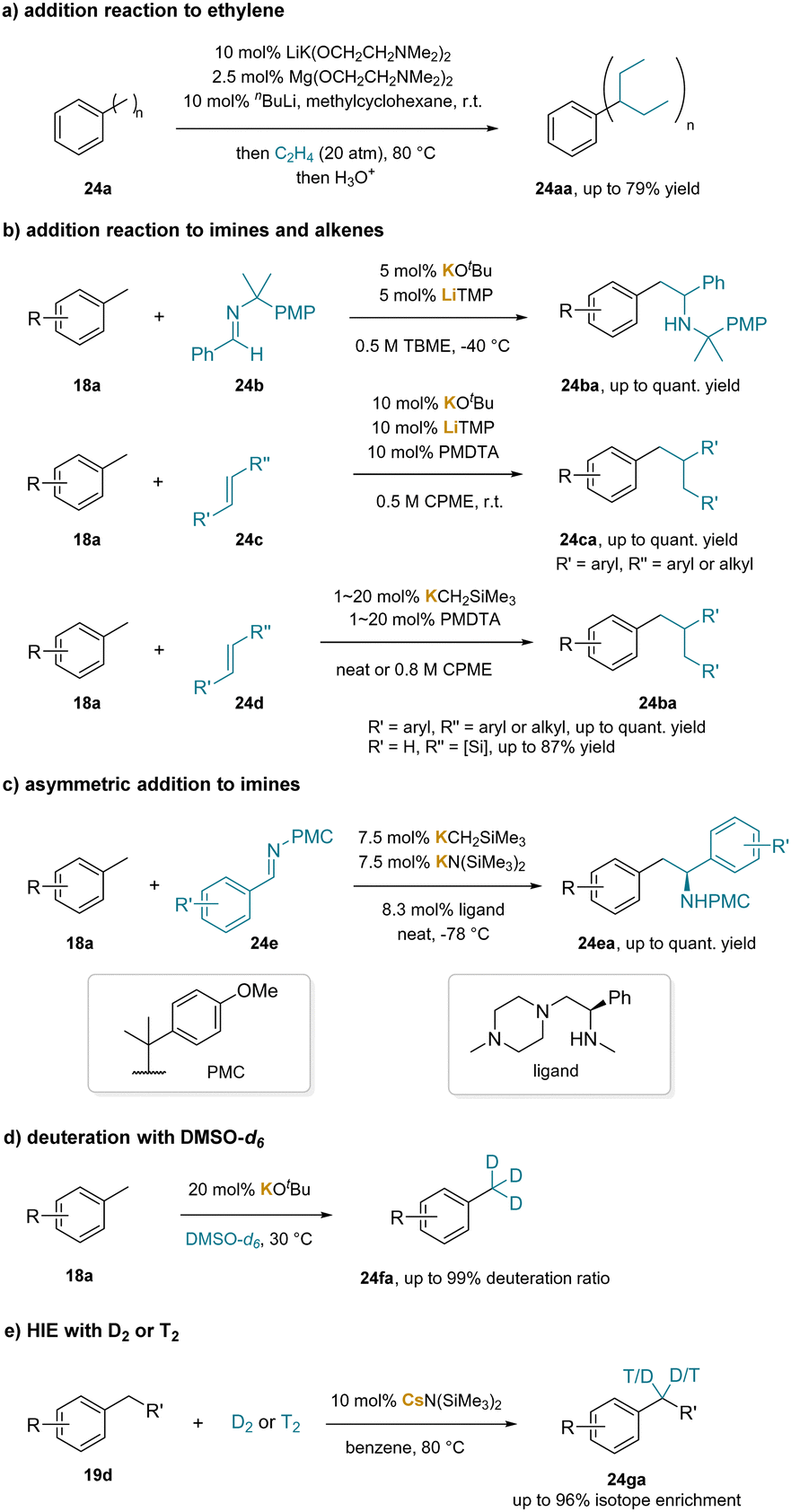 | ||
| Scheme 30 Base-catalyzed benzylic functionalization: (a) addition reaction to ethylene, (b) addition reaction to imines and alkenes, (c) asymmetric addition to imines, (d) deuteration with DMSO-d6, and (e) HIE with D2 or T2. | ||
Additionally, beyond alkylbenzenes, particularly toluene derivatives, base-catalyzed addition reactions of ethylbenzene derivatives, such as allylbenzene237,238 and diarylmethanes,227,239 have been established. These substrates feature benzylic C–H bonds with increased acidity owing to the benzylic substituent.
4. Conclusions
The π-coordination strategy utilizing transition-metals has emerged as an attractive approach for benzylic C–H bond functionalization via a combination of deprotonation and electrophilic attack. Since the 1970s, significant progress has been made in reactions of alkylarene complexes, particularly under conditions with stereocontrol. However, the use of strong bases in the deprotonation step of Cr(CO)3-complexed arenes has limited compatibility with functional groups, posing a challenge in the late-stage functionalization of complex molecules. While cationic metal units such as tricarbonyl manganese(I), cyclopentadienyl iron(II), and pentamethylcyclopentadienyl ruthenium(II) increase the acidity of the benzylic hydrogens of η6-alkyl arenes, their limited accessibility has hindered their use in studies. Future advancements in this field include the development of ligands that provide a general solution, establish asymmetric catalysis, and create new reactivities. Dual catalysis involving a Lewis acid, which is compatible with the acidic metal catalyst, would also expand the scope of benzylic C–H bond functionalizations. Ultimately, the progression from stoichiometric metal complexes to catalytic processes involving arene exchange steps is crucial for the advancement of this field. This review aims to promote a better understanding of the π-coordination-driven benzylic C–H bond activation and inspire future innovations in the field.Cation–π interactions can acidify the benzylic hydrogens, facilitating their deprotonation. Unlike transition-metals, which can form strong bonds to η6-arenes and result in dramatic decreases in the pKa of the benzylic C–H's, the impact on the cation–π interactions is more subtle. While cation–π interactions are often observed in crystal structures containing group(I) metals, the low strength of the interaction makes arene exchange rapid and difficult to characterize in solution. Further studies are necessary to both better understand these interactions and to develop guidelines for the design of systems that can maximize their beneficial impact to develop more synthetically useful complexes and catalysts.
Author contributions
The manuscript was written by all the authors, who approved the final version.Data availability
All data for the manuscript is contained in the original manuscripts cited in the text.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. S. thank the Zhejiang Provincial Natural Science Foundation of China (LR24B020001), the National Natural Science Foundation of China (22271235), and Westlake University high-performance computing center. P. J. W. thanks the US National Science Foundation (CHE-2154593) and the Petroleum Research Fund (67280-ND1) sponsored by the American Chemical Society for financial support.References
- Y. Zhang, T. Zhang and S. Das, Chem, 2022, 8, 3175–3201 CAS
.
- D. Z. Chenyi Cai, Chin. J. Org. Chem., 2022, 42, 1586–1608 CrossRef
- K. Qvortrup, D. A. Rankic and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 626–629 CrossRef CAS PubMed
- R. Vanjari and K. N. Singh, Chem. Soc. Rev., 2015, 44, 8062–8096 RSC
- W. Zhang, F. Wang, S. D. McCann, D. Wang, P. Chen, S. S. Stahl and G. Liu, Science, 2016, 353, 1014–1018 CrossRef CAS PubMed
- Q.-Y. Meng, T. E. Schirmer, A. L. Berger, K. Donabauer and B. König, J. Am. Chem. Soc., 2019, 141, 11393–11397 CrossRef CAS
- L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2020, 142, 17693–17702 CrossRef CAS PubMed
- M. Oliva, G. A. Coppola, E. V. Van der Eycken and U. K. Sharma, Adv. Synth. Catal., 2021, 363, 1810–1834 CrossRef CAS
- H. Yue, C. Zhu, L. Huang, A. Dewanji and M. Rueping, Chem. Commun., 2022, 58, 171–184 RSC
- F. G. Bordwell, D. Algrim and N. R. Vanier, J. Org. Chem., 1977, 42, 1817–1819 CrossRef CAS
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS
- C. D. Broaddus, J. Am. Chem. Soc., 1966, 88, 4174–4178 CrossRef CAS
- L. Lochmann, J. Pospíšil and D. Lím, Tetrahedron Lett., 1966, 7, 257–262 CrossRef
- C. D. Broaddus, J. Org. Chem., 1970, 35, 10–15 CrossRef CAS
- M. Schlosser and S. Strunk, Tetrahedron Lett., 1984, 25, 741–744 CrossRef CAS
- L. Assadourian, R. Faure and G. Gau, J. Organomet. Chem., 1985, 280, 153–158 CrossRef CAS
- M. Blangetti, P. Fleming and D. F. O'Shea, Beilstein J. Org. Chem., 2011, 7, 1249–1254 CrossRef CAS PubMed
- M. Iwasaki, T. Narita and Y. Umino, J. Organomet. Chem., 2011, 696, 2763–2766 CrossRef CAS
- M. Blangetti, P. Fleming and D. F. O’Shea, J. Org. Chem., 2012, 77, 2870–2877 CrossRef CAS PubMed
- C. Unkelbach, H. S. Rosenbaum and C. Strohmann, Chem. Commun., 2012, 48, 10612–10614 RSC
- J. Zhang, A. Bellomo, A. D. Creamer, S. D. Dreher and P. J. Walsh, J. Am. Chem. Soc., 2012, 134, 13765–13772 CrossRef CAS PubMed
- C. Unkelbach, D. F. O'Shea and C. Strohmann, Angew. Chem., Int. Ed., 2014, 53, 553–556 CrossRef CAS PubMed
- A. Manvar, P. Fleming and D. F. O’Shea, J. Org. Chem., 2015, 80, 8727–8738 CrossRef CAS PubMed
- C.-C. Bao, Y.-L. Luo, H.-Z. Du and B.-T. Guan, Sci. China: Chem., 2021, 64, 1349–1354 CrossRef CAS
- G. P. R. Freure, E. A. Skrotzki, J.-D. E. Lavertu and S. G. Newman, ACS Catal., 2021, 11, 12258–12263 CrossRef CAS
- H. Wang, J. Mao, S. Shuai, S. Chen, D. Zou, P. J. Walsh and J. Li, Org. Chem. Front., 2021, 8, 6000–6008 RSC
- Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals, ed. M. Beller and C. Bolm, 2004 Search PubMed.
-
J. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, 2010 Search PubMed
-
R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, John Wiley & Sons, Inc., 2014 Search PubMed
-
S. A. Patrick, L. Holland, D. P. Mills, S. T. Liddle, S. R. Daly, T. H. Warren, D. J. Liptrot, I. A. Tonks, D. P. Gates, L. J. Daumann and J. Daumann, Comprehensive Organometallic Chemistry IV, Elsevier, 2022 Search PubMed
-
E. W. Abel, F. G. A. Stone and G. Wilkinson, Comprehensive Organometallic Chemistry II, Elsevier, Oxford, 1995 Search PubMed
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS
- R. D. Pike and D. A. Sweigart, Coord. Chem. Rev., 1999, 187, 183–222 CrossRef CAS
- A. R. Pape, K. P. Kaliappan and E. P. Kündig, Chem. Rev., 2000, 100, 2917–2940 CrossRef CAS PubMed
-
F. Rose-Munch and E. Rose, Modern Arene Chemistry, 2002, pp. 368–399 Search PubMed
-
D. Astruc, S. Nlate and J. Ruiz, Modern Arene Chemistry, 2002, pp. 400–434 Search PubMed
-
E. P. Kündig, Transition Metal Arene π-Complexes in Organic Synthesis and Catalysis, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004 Search PubMed
- G. Pampaloni, Coord. Chem. Rev., 2010, 254, 402–419 CrossRef CAS
- E. O. Fischer and W. Hafner, Über Aromatenkomplexe von Metallen I, 1955, 10, 665–668.
- E. O. Fischer and K. Öfele, Chem. Ber., 1957, 90, 2532–2535 CrossRef CAS
- G. Jaouen, A. Meyer and G. Simonneaux, J. Chem. Soc., Chem. Commun., 1975, 813–814 RSC
-
M. F. Semmelhack and A. Chlenov, Transition Metal Arene π-Complexes in Organic Synthesis and Catalysis, ed. E. P. Kündig, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 43–69 Search PubMed
-
T. Song and Y. Mu, in Comprehensive Organometallic Chemistry IV, ed. G. Parkin, K. Meyer and D. O'hare, Elsevier, Oxford, 2022, pp. 81–173 Search PubMed
- V. M. Rayón and G. Frenking, Organometallics, 2003, 22, 3304–3308 CrossRef
-
Y. Jean, Molecular Orbitals of Transition Metal Complexes, Oxford University Press, 2005 Search PubMed
-
M. Uemura, in Transition Metal Arene π-Complexes in Organic Synthesis and Catalysis, ed. E. P. Kündig, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 129–156 Search PubMed
- D. Astruc, Tetrahedron, 1983, 39, 4027–4095 CrossRef CAS
- C. F. Pigge and J. J. Coniglio, Curr. Org. Chem., 2001, 5, 757–784 CrossRef
- H.-G. Schmalz and F. Dehmel, Transition Met. Org. Synth., 2004, 601–617 CAS
- L. J. Williams, Y. Bhonoah, L. A. Wilkinson and J. W. Walton, Chem. – Eur. J., 2021, 27, 3650–3660 CrossRef CAS PubMed
- K. Chen and H. Shi, Acc. Chem. Res., 2024, 57, 2194–2206 CrossRef CAS PubMed
- The ΔG values were calculated by the method published on the paper C. A. Merlic, J. C. Walsh, D. J. Tantillo and K. N. Houk, J. Am. Chem. Soc., 1999, 121, 3596–3606 CrossRef CAS
- C. A. Merlic, B. N. Hietbrink and K. N. Houk, J. Org. Chem., 2001, 66, 6738–6744 CrossRef CAS PubMed
- C. A. Merlic, M. M. Miller, B. N. Hietbrink and K. N. Houk, J. Am. Chem. Soc., 2001, 123, 4904–4918 CrossRef CAS PubMed
- C. A. Merlic, J. C. Walsh, D. J. Tantillo and K. N. Houk, J. Am. Chem. Soc., 1999, 121, 3596–3606 CrossRef CAS
- H. A. Trujillo, C. M. Casado and D. Astruc, J. Chem. Soc., Chem. Commun., 1995, 7–8 RSC
- G. Moutiers, A. Peigneux, D. Vichard and F. Terrier, Organometallics, 1998, 17, 4469–4476 CrossRef CAS
- A. Pfletschinger, T. K. Dargel, J. W. Bats, H.-G. Schmalz and W. Koch, Chem. – Eur. J., 1999, 5, 537–545 CrossRef CAS
- C. M. Casado, T. Wagner and D. Astruc, J. Organomet. Chem., 1995, 502, 143–145 CrossRef CAS
- M. Uemura, Organic Reactions, 2006, pp. 217–657 Search PubMed.
- V. N. Kalinin, I. y A. Cherepanov and S. K. Moiseev, J. Organomet. Chem., 1997, 536–537, 437–455 CrossRef
- J. Blagg, S. J. Coote, S. G. Davies and B. E. Mobbs, J. Chem. Soc., Perkin Trans. 1, 1986, 2257–2261 RSC
- D. Sénéchal, M.-C. Sénéchal-Tocquer, D. Gentric, J.-Y. Le Bihan, B. Caro, M. Gruselle and G. Jaouen, J. Chem. Soc., Chem. Commun., 1987, 632–633 RSC
- B. Caro, J.-Y. Le Bihan, J.-P. Guillot, S. Top and G. Jaouen, J. Chem. Soc., Chem. Commun., 1984, 602–603 RSC
- M. C. Senechal-Tocquer, D. Senechal, J. Y. Le Bihan, D. Gentric and B. Caro, J. Organomet. Chem., 1987, 321, 353–363 CrossRef CAS
- J. Brocard, J. Lebibi and D. Couturier, J. Chem. Soc., Chem. Commun., 1981, 1264–1265 RSC
- G. Simonneaux and G. Jaouen, Tetrahedron, 1979, 35, 2249–2254 CrossRef CAS
- M. C. Senechal-Tocquer, D. Senechal, J. Y. Le Bihan, D. Gentric and B. Caro, J. Organomet. Chem., 1985, 291, c5–c8 CrossRef CAS
- R. J. Card and W. S. Trahanovsky, J. Org. Chem., 1980, 45, 2555–2559 CrossRef CAS
- R. J. Card and W. S. Trahanovsky, J. Org. Chem., 1980, 45, 2560–2566 CrossRef CAS
- M. F. Semmelhack, G. R. Clark, J. L. Garcia, J. J. Harrison, Y. Thebtaranonth, W. Wulff and A. Yamashita, Tetrahedron, 1981, 37, 3957–3965 CrossRef CAS
- A. Ceccon, A. Gambaro and A. Venzo, J. Organomet. Chem., 1984, 275, 209–222 CrossRef CAS
- G. Jaouen, Ann. N. Y. Acad. Sci., 1977, 295, 59–78 CrossRef CAS
- J. Blagg, S. G. Davies, N. J. Holman, C. A. Laughton and B. E. Mobbs, J. Chem. Soc., Perkin Trans. 1, 1986, 1581–1589 RSC
- A. Meyer, Tetrahedron Lett., 1976, 17, 3547–3550 CrossRef
- J. Brocaed, A. Laconi, D. Couturier, S. Top and G. Jaouen, J. Chem. Soc., Chem. Commun., 1984, 475–476 RSC
- G. Jaouen, S. Top, A. Laconi, D. Couturier and J. Brocard, J. Am. Chem. Soc., 1984, 106, 2207–2208 CrossRef CAS
-
S. G. Davies, S. J. Coote and C. L. Goodfellow, in Advances in Metal-Organic Chemistry, ed. L. S. Liebeskind, Jai, 1991, vol. 2, pp. 1–57 Search PubMed
- J. Brocard, L. Pelinski and J. Lebibi, J. Organomet. Chem., 1986, 309, 299–305 CrossRef CAS
- J. Brocard and J. Lebibi, J. Organomet. Chem., 1987, 320, 295–306 CrossRef CAS
- N. Mathews and D. A. Widdowson, Synlett, 1990, 467–468 CrossRef CAS
- A. Netz and T. J. J. Müller, Organomet., 2000, 19, 1452–1454 CrossRef CAS
- M. C. Senechal-Tocquer, D. Senechal, J. Y. Le Bihan, D. Gentric, B. Caro, M. Gruselle and G. Jaouen, J. Organomet. Chem., 1992, 433, 261–278 CrossRef CAS
- V. N. Kalinin, I. Y. A. Cherepanov, S. K. Moiseev, A. S. Batsanov and Y. T. Struchkov, Mendeleev Commun., 1991, 1, 77–78 CrossRef
- V. N. Kalinin, I. Y. A. Cherepanov and S. K. Moiseev, Mendeleev Commun., 1993, 3, 43–45 CrossRef
- S. K. Moiseev, I. A. Cherepanov, P. V. Petrovskii, M. G. Ezernitskaya, H. Butenschön, M. Strotmann and V. N. Kalinin, Inorg. Chim. Acta, 1998, 280, 71–74 CrossRef CAS
- C. Werlé, L. Karmazin, C. Bailly and J.-P. Djukic, Eur. J. Inorg. Chem., 2019, 3301–3308 CrossRef
- C. Werlé, L. Karmazin, C. Bailly, L. Ricard and J.-P. Djukic, Organometallics, 2015, 34, 3055–3064 CrossRef
- V. N. Kalinin, I. Y. A. Cherepanov and S. K. Moiseev, Mendeleev Commun., 1994, 4, 53–55 CrossRef
- J.-J. Yaouanc, J.-C. Clement and H. des Abbayes, J. Chem. Soc., Chem. Commun., 1988, 1379–1380 RSC
- C. Werlé, C. Bailly, L. Karmazin-Brelot, X.-F. Le Goff, L. Ricard and J.-P. Djukic, J. Am. Chem. Soc., 2013, 135, 17839–17852 CrossRef PubMed
- C. Werlé, M. Hamdaoui, C. Bailly, X.-F. Le Goff, L. Brelot and J.-P. Djukic, J. Am. Chem. Soc., 2013, 135, 1715–1718 CrossRef PubMed
- A. Ceccon, A. Gambaro, S. Santi, G. Valle and A. Venzo, J. Chem. Soc., Chem. Commun., 1989, 51–53 RSC
- A. Ceccon, C. J. Elsevier, J. M. Ernsting, A. Gambaro, S. Santi and A. Venzo, Inorg. Chim. Acta, 1993, 204, 15–26 CrossRef CAS
- C. Bonifaci, A. Ceccon, A. Gambaro, P. Ganis, S. Santi, G. Valle and A. Venzo, Organometallics, 1993, 12, 4211–4214 CrossRef CAS
- C. Bonifaci, A. Ceccon, A. Gambaro, P. Ganis, S. Santi and A. Venzo, Organometallics, 1995, 14, 2430–2434 CrossRef CAS
- C. Bonifaci, A. Ceccon, S. Santi, C. Mealli and R. W. Zoellner, Inorg. Chim. Acta, 1995, 240, 541–549 CrossRef CAS
- C. Bonifaci, A. Ceccon, A. Gambaro, P. Ganis, S. Santi, G. Valle and A. Venzo, J.
Organomet. Chem., 1995, 492, 35–39 CrossRef CAS
- C. Werlé, C. Bailly, L. Karmazin-Brelot, X.-F. Le Goff, M. Pfeffer and J.-P. Djukic, Angew. Chem., 2014, 126, 9985–9989 CrossRef
- P. Cecchetto, A. Ceccon, A. Gambaro, S. Santi, P. Ganis, R. Gobetto, G. Valle and A. Venzo, Organometallics, 1998, 17, 752–762 CrossRef CAS
- C. Werlé, C. Bailly, L. Karmazin-Brelot, X.-F. Le Goff, M. Pfeffer and J.-P. Djukic, Angew. Chem., Int. Ed., 2014, 53, 9827–9831 CrossRef PubMed
- D. Astruc, E. Roman E, J. R. Hamon and P. Batail, J. Am. Chem. Soc., 1979, 101, 2240–2242 CrossRef CAS
- D. Astruc, J. R. Hamon, E. Roman and P. Michaud, J. Am. Chem. Soc., 1981, 103, 7502–7514 CrossRef CAS
- J. R. Hamon, D. Astruc, E. Roman, P. Batail and J. J. Mayerle, J. Am. Chem. Soc., 1981, 103, 2431–2433 CrossRef CAS
- J. R. Hamon, J. Y. Saillard, A. Le Beuze, M. J. McGlinchey and D. Astruc, J. Am. Chem. Soc., 1982, 104, 7549–7555 CrossRef CAS
- J. R. Hamon and D. Astruc, Organometallics, 1988, 7, 1036–1046 CrossRef CAS
- J.-R. Hamon, P. Hamon, S. Sinbandhit, P. Guenot and D. Astruc, J. Organomet. Chem., 1991, 413, 243–255 CrossRef CAS
-
D. Astruc, in Transition Metal Coordination Chemistry, ed. W. A. Herrmann, Springer Berlin Heidelberg, Berlin, Heidelgerg, 1992, pp. 47–95 Search PubMed
- F. Moulines, L. Djakovitch, J.-L. Fillaut and D. Astruc, Synlett, 1992, 57–58 CrossRef CAS
- V. Sartor, L. Djakovitch, J.-L. Fillaut, F. Moulines, F. Neveu, V. Marvaud, J. Guittard, J.-C. Blais and D. Astruc, J. Am. Chem. Soc., 1999, 121, 2929–2930 CrossRef CAS
- S. Rigaut, M.-H. Delville, J. Losada and D. Astruc, Inorg. Chim. Acta, 2002, 334, 225–242 CrossRef CAS
- A. C. Sander, J. Ruiz and D. Astruc, Organometallics, 2010, 29, 5722–5724 CrossRef CAS
- K. B. Wiberg, J. Am. Chem. Soc., 1979, 101, 2204–2205 CrossRef CAS
- J. W. Hull, Jr. and W. L. Gladfelter, Organometallics, 1982, 1, 1716–1718 CrossRef
- M. A. Bennett, L. Y. Goh, I. J. McMahon, T. R. B. Mitchell, G. B. Robertson, T. W. Turney and W. A. Wickramasinghe, Organometallics, 1992, 11, 3069–3085 CrossRef CAS
- U. Koelle and R. Pasch, Inorg. Chem. Commun., 1998, 1, 395–397 CrossRef CAS
- M. A. Bennett, T.-N. Huang, T. W. Matheson, A. K. Smith, S. Ittel and W. Nickerson, Inorg. Synth., 1982, 74–78 CrossRef CAS
- J. W. Hull, Jr., C. Mann and W. L. Gladfelter, Organometallics, 1992, 11, 3117–3121 CrossRef
- M. A. Bennett, L. Y. Goh and A. C. Willis, J. Chem. Soc., Chem. Commun., 1992, 1180–1182 RSC
- D. M. LaBrush, D. P. Eyman, N. C. Baenziger and L. M. Mallis, Organometallics, 1991, 10, 1026–1033 CrossRef CAS
- J. W. Hull, Jr., K. J. Roesselet and W. L. Gladfelter, Organometallics, 1992, 11, 3630–3635 CrossRef
- J. W. Johnson and P. M. Treichel, J. Chem. Soc., Chem. Commun., 1976, 688–689 RSC
- P. M. Treichel and J. W. Johnson, Inorg. Chem., 1977, 16, 749–753 CrossRef CAS
- M. A. Bennett, Coord. Chem. Rev., 1997, 166, 225–254 CrossRef CAS
- M. A. Bennett, M. Bown, L. Y. Goh, D. C. R. Hockless and T. R. B. Mitchell, Organometallics, 1995, 14, 1000–1007 CrossRef CAS
- M. A. Bennett, I. J. McMahon and T. W. Turney, Angew. Chem., 1982, 94, 373 CrossRef CAS
- H. Amouri, Y. Besace, J. L. Bras and J. Vaissermann, J. Am. Chem. Soc., 1998, 120, 6171–6172 CrossRef CAS
- H. Amouri, J. Vaissermann, M. N. Rager and D. B. Grotjahn, Organometallics, 2000, 19, 5143–5148 CrossRef CAS
- H. Amouri, J. Vaissermann, M. N. Rager and D. B. Grotjahn, Organometallics, 2000, 19, 1740–1748 CrossRef CAS
- W. S. Trahanovsky and R. J. Card, J. Am. Chem. Soc., 1972, 94, 2897–2898 CrossRef CAS
- H. Arzeno, D. H. R. Barton, R.-M. Bergé-Lurion, X. Lusinchi and B. M. Pinto, J. Chem. Soc., Perkin Trans. 1, 1984, 2069–2076 RSC
- D. Schinzer, U. Abel and P. Jones, Synlett, 1997, 632–634 CrossRef CAS
- A. Meyer and O. Hofer, J. Am. Chem. Soc., 1980, 102, 4410–4414 CrossRef CAS
- S. Top, A. Vessieres, J.-P. Abjean and G. Jaouen, J. Chem. Soc., Chem. Commun., 1984, 428–429 RSC
- H. Des Abbayes and M. A. Boudeville, J. Org. Chem., 1977, 42, 4104–4108 CrossRef CAS
- S. Zemolka, J. Lex and H.-G. Schmalz, Angew. Chem., Int. Ed., 2002, 41, 2525–2528 CrossRef CAS PubMed
- J. Blagg, S. G. Davies, C. L. Goodfellow and K. H. Sutton, J. Chem. Soc., Perkin Trans. 1, 1987, 1805–1811 RSC
- M. Brisander, P. Caldirola, A. M. Johansson and U. Hacksell, J. Org. Chem., 1998, 63, 5362–5367 CrossRef CAS
- A. N. Nesmeyanov, N. A. Ustynyuk, L. N. Novikova, T. N. Rybina, Y. A. Ustynyuk, Y. F. Oprunenko and O. I. Trifonova, J. Organomet. Chem., 1980, 184, 63–75 CrossRef CAS
- A. N. Nesmeyanov, N. A. Ustynyuk, L. N. Novikova, V. G. Andrianov, Y. T. Struchkov, Y. A. Ustynyuk, Y. F. Oprunenko and Y. N. Luzikov, J. Organomet. Chem., 1982, 226, 239–250 CrossRef CAS
- S. E. Gibson, P. C. V. Potter and M. H. Smith, Chem. Commun., 1996, 2757–2758 RSC
- E. L. M. Cowton, S. E. Gibson, M. J. Schneider and M. H. Smith, Chem. Commun., 1996, 839–840 RSC
- A. Ariffin, A. J. Blake, R. A. Ewin and N. S. Simpkins, Tetrahedron: Asymmetry, 1998, 9, 2563–2566 CrossRef CAS
- S. E. Gibson, P. O’Brien, E. Rahimian and M. H. Smith, J. Chem. Soc., Perkin Trans. 1, 1999, 909–912 RSC
- A. R. E. Brewer, A. F. Drake, S. E. Gibson and J. T. Rendell, Org. Lett., 2007, 9, 3487–3490 CrossRef CAS PubMed
- M. Uemura, H. Nishimura and Y. Hayashi, J. Organomet. Chem., 1989, 376, C3–C6 CrossRef CAS
- S. E. Gibson and E. G. Reddington, Chem. Commun., 2000, 989–996 RSC
- K. Abecassis and S. E. Gibson, Eur. J. Org. Chem., 2010, 2938–2944 CrossRef CAS
- S. E. Gibson, G. R. Jefferson, S. E. Gibson, P. Ham and G. R. Jefferson, Chem. Commun., 1998, 123–124 RSC
- T. Hata, H. Koide, N. Taniguchi and M. Uemura, Org. Lett., 2000, 2, 1907–1910 CrossRef CAS PubMed
- T. Hata, H. Koide and M. Uemura, Synlett, 2000, 1145–1147 CAS
- C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314–321 CrossRef CAS PubMed
- J. Terao and N. Kambe, Acc. Chem. Res., 2008, 41, 1545–1554 CrossRef CAS PubMed
- C. Fricke and F. Schoenebeck, Acc. Chem. Res., 2020, 53, 2715–2725 CrossRef CAS PubMed
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed
- G. C. Fu, Acc. Chem. Res., 2008, 41, 1555–1564 CrossRef CAS PubMed
- R. D. J. Froese, C. Lombardi, M. Pompeo, R. P. Rucker and M. G. Organ, Acc. Chem. Res., 2017, 50, 2244–2253 CrossRef CAS PubMed
- J. B. Johnson and T. Rovis, Acc. Chem. Res., 2008, 41, 327–338 CrossRef CAS PubMed
-
A. de Meijere and F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, 2004 Search PubMed
- D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234–245 CrossRef CAS PubMed
- J. M. Fox, X. Huang, A. Chieffi and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 1360–1370 CrossRef CAS
- S. Ge, S. I. Arlow, M. G. Mormino and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 14401–14404 CrossRef CAS PubMed
- T. Hamada, A. Chieffi, J. Åhman and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 1261–1268 CrossRef CAS PubMed
- B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1997, 119, 12382–12383 CrossRef CAS
- C. C. C. Johansson and T. J. Colacot, Angew. Chem., Int. Ed., 2010, 49, 676–707 CrossRef CAS PubMed
- M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 1473–1478 CrossRef CAS
- Z. Li, Y. Peng and T. Wu, Org. Lett., 2021, 23, 881–885 CrossRef CAS PubMed
- G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2002, 41, 953–956 CrossRef CAS
- N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS PubMed
- H. N. Nguyen, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 11818–11819 CrossRef CAS PubMed
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed
- M. Palucki and S. L. Buchwald, J. Am. Chem. Soc., 1997, 119, 11108–11109 CrossRef CAS
- T. Satoh, Y. Kawamura, M. Miura and M. Nomura, Angew. Chem., Int. Ed. Engl., 1997, 36, 1740–1742 CrossRef CAS
- T. Satoh, J.-i Inoh, Y. Kawamura, Y. Kawamura, M. Miura and M. Nomura, Bull. Chem. Soc. Jpn., 1998, 71, 2239–2246 CrossRef CAS
- M. S. Viciu, R. F. Germaneau and S. P. Nolan, Org. Lett., 2002, 4, 4053–4056 CrossRef CAS PubMed
- M. S. Viciu, R. A. Kelly, E. D. Stevens, F. Naud, M. Studer and S. P. Nolan, Org. Lett., 2003, 5, 1479–1482 CrossRef CAS PubMed
- G. I. McGrew, J. Temaismithi, P. J. Carroll and P. J. Walsh, Angew. Chem., Int. Ed., 2010, 49, 5541–5544 CrossRef CAS PubMed
- G. I. McGrew, C. Stanciu, J. Zhang, P. J. Carroll, S. D. Dreher and P. J. Walsh, Angew. Chem., Int. Ed., 2012, 51, 11510–11513 CrossRef CAS PubMed
- J. Zhang, C. Stanciu, B. Wang, M. M. Hussain, C.-S. Da, P. J. Carroll, S. D. Dreher and P. J. Walsh, J. Am. Chem. Soc., 2011, 133, 20552–20560 CrossRef CAS PubMed
- S. Takemoto, E. Shibata, M. Nakajima, Y. Yumoto, M. Shimamoto and H. Matsuzaka, J. Am. Chem. Soc., 2016, 138, 14836–14839 CrossRef CAS PubMed
- Q.-K. Kang, Y. Li, K. Chen, H. Zhu, W.-Q. Wu, Y. Lin and H. Shi, Angew. Chem., Int. Ed., 2022, 61, e202117381 CrossRef CAS PubMed
- Y. Li, W.-Q. Wu, H. Zhu, Q.-K. Kang, L. Xu and H. Shi, Angew. Chem., Int. Ed., 2022, 61, e202207917 CrossRef CAS PubMed
- Y. Li and H. Shi, Chin. Chem. Lett., 2024, 35, 108650 CrossRef CAS
- R. A. Kumpf and D. A. Dougherty, Science, 1993, 261, 1708–1710 CrossRef CAS PubMed
- S. Tsuzuki, M. Yoshida, T. Uchimaru and M. Mikami, J. Phys. Chem. A, 2001, 105, 769–773 CrossRef CAS
- C. R. Kennedy, S. Lin and E. N. Jacobsen, Angew. Chem., Int. Ed., 2016, 55, 12596–12624 CrossRef CAS PubMed
- W. Clegg, G. C. Forbes, A. R. Kennedy, R. E. Mulvey and S. T. Liddle, Chem. Commun., 2003, 406–407 RSC
- D. B. Pardue, S. J. Gustafson, R. A. Periana, D. H. Ess and T. R. Cundari, Comput. Theor. Chem., 2013, 1019, 85–93 CrossRef CAS
- D. Hoffmann, W. Bauer, F. Hampel, N. J. R. van Eikema Hommes, P. V. R. Schleyer, P. Otto, U. Pieper, D. Stalke, D. S. Wright and R. Snaith, J. Am. Chem. Soc., 1994,(116), 528–536 CrossRef CAS
- M. Schlosser, Pure Appl. Chem., 1988, 60, 1627–1634 CrossRef CAS
- S.-C. Sha, S. Tcyrulnikov, M. Li, B. Hu, Y. Fu, M. C. Kozlowski and P. J. Walsh, J. Am. Chem. Soc., 2018, 140, 12415–12423 CrossRef CAS PubMed
- P. W. N. M. van Leeuwen and P. C. J. Kamer, Catal. Sci. Technol., 2018, 8, 26–113 RSC
- B. L. Lucht and D. B. Collum, J. Am. Chem. Soc., 1994, 116, 6009–6010 CrossRef CAS
- J. Zhang, A. Bellomo, N. Trongsiriwat, T. Jia, P. J. Carroll, S. D. Dreher, M. T. Tudge, H. Yin, J. R. Robinson, E. J. Schelter and P. J. Walsh, J. Am. Chem. Soc., 2014, 136, 6276–6287 CrossRef CAS PubMed
- H. Jiang, S.-C. Sha, S. A. Jeong, B. C. Manor and P. J. Walsh, Org. Lett., 2019, 21, 1735–1739 CrossRef CAS PubMed
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed
- J. Zhang, S.-C. Sha, A. Bellomo, N. Trongsiriwat, F. Gao, N. C. Tomson and P. J. Walsh, J. Am. Chem. Soc., 2016, 138, 4260–4266 CrossRef CAS PubMed
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed
- D. A. Dougherty, Acc. Chem. Res., 2013, 46, 885–893 CrossRef CAS PubMed
- E. Masson and M. Schlosser, Org. Lett., 2005, 7, 1923–1925 CrossRef CAS PubMed
- S. Banerjee, Y.-F. Yang, I. D. Jenkins, Y. Liang, A. A. Toutov, W.-B. Liu, D. P. Schuman, R. H. Grubbs, B. M. Stoltz, E. H. Krenske, K. N. Houk and R. N. Zare, J. Am. Chem. Soc., 2017, 139, 6880–6887 CrossRef CAS PubMed
- M. Panunzio and P. Zarantonello, Org. Process Res. Dev., 1998, 2, 49–59 CrossRef CAS
- A. I. Ojeda-Amador, A. J. Martínez-Martínez, A. R. Kennedy and C. T. O’Hara, Inorg. Chem., 2016, 55, 5719–5728 CrossRef CAS PubMed
- G. Liu, P. J. Walsh and J. Mao, Org. Lett., 2019, 21, 8514–8518 CrossRef CAS PubMed
- J. Mao, Z. Wang, X. Xu, G. Liu, R. Jiang, H. Guan, Z. Zheng and P. J. Walsh, Angew. Chem., Int. Ed., 2019, 58, 11033–11038 CrossRef CAS PubMed
- Y. Gu, Z. Zhang, Y.-E. Wang, Z. Dai, Y. Yuan, D. Xiong, J. Li, P. J. Walsh and J. Mao, J. Org. Chem., 2022, 87, 406–418 CrossRef CAS PubMed
- F. Yang, D. Zou, S. Chen, H. Wang, Y. Zhao, L. Zhao, L. Li, J. Li and P. J. Walsh, Adv. Synth. Catal., 2020, 362, 3423–3430 CrossRef CAS
- D. E. Anderson, A. Tortajada and E. Hevia, Angew. Chem., Int. Ed., 2023, 62, e202218498 CrossRef CAS PubMed
- N. R. Judge and E. Hevia, Angew. Chem., Int. Ed., 2023, 62, e202303099 CrossRef CAS PubMed
- R. Sreedharan, P. K. Pal, P. K. R. Panyam, U. D. Priyakumar and T. Gandhi, Asian J. Org. Chem., 2022, 11, e202200372 CrossRef CAS
- P. C. Andrikopoulos, D. R. Armstrong, A. R. Kennedy, R. E. Mulvey, C. T. O’Hara and R. B. Rowlings, Eur. J. Inorg. Chem., 2003, 3354–3362 CrossRef CAS
- F. Feil and S. Harder, Organometallics, 2000, 19, 5010–5015 CrossRef CAS
- M. Xu, A. R. Jupp, Z.-W. Qu and D. W. Stephan, Angew. Chem., Int. Ed., 2018, 57, 11050–11054 CrossRef CAS PubMed
- Y. Yuan, Y. Gu, Y.-E. Wang, J. Zheng, J. Ji, D. Xiong, F. Xue and J. Mao, J. Org. Chem., 2022, 87, 13907–13918 CrossRef CAS PubMed
- B. Neil, L. Saadi, L. Fensterbank and C. Chauvier, Angew. Chem., Int. Ed., 2023, 62, e202306115 CrossRef PubMed
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed
- M. Shigeno, K. Hanasaka, K. Sasaki, K. Nozawa-Kumada and Y. Kondo, Chem. – Eur. J., 2019, 25, 3235–3239 CrossRef CAS PubMed
- M. Shigeno, I. Tohara, K. Nozawa-Kumada and Y. Kondo, Eur. J. Org. Chem., 2020, 1987–1991 CrossRef CAS
- M. Shigeno, K. Hanasaka, I. Tohara, K. Izumi, H. Yamakoshi, E. Kwon, K. Nozawa-Kumada and Y. Kondo, Org. Lett., 2022, 24, 809–814 CrossRef CAS PubMed
- M. Shigeno, I. Tohara, K. Sasaki, K. Nozawa-Kumada and Y. Kondo, Org. Lett., 2022, 24, 4825–4830 CrossRef CAS PubMed
- P. Fleming and D. F. O’Shea, J. Am. Chem. Soc., 2011, 133, 1698–1701 CrossRef CAS PubMed
- M. Shigeno, A. Kajima, E. Toyama, T. Korenaga, H. Yamakoshi, K. Nozawa-Kumada and Y. Kondo, Chem. – Eur. J., 2023, 29, e202203549 CrossRef CAS PubMed
- M. Shigeno, K. Nakaji, K. Nozawa-Kumada and Y. Kondo, Org. Lett., 2019, 21, 2588–2592 CrossRef CAS PubMed
- D. A. Bors, M. J. Kaufman and A. Streitwieser, Jr., J. Am. Chem. Soc., 1985, 107, 6975–6982 CrossRef CAS
- R. R. Fraser, T. S. Mansour and S. Savard, J. Org. Chem., 1985, 50, 3232–3234 CrossRef CAS
- X.-H. Meng, X.-C. Xu, Z. Wang, Y.-X. Liang and Y.-L. Zhao, J. Org. Chem., 2022, 87, 3156–3166 CrossRef CAS PubMed
- C. Weindl, S. L. Helmbrecht and L. Hintermann, J. Org. Chem., 2023, 88, 4155–4161 CrossRef CAS PubMed
- H. Pines, J. A. Vesely and V. N. Ipatieff, J. Am. Chem. Soc., 1955, 77, 554–559 CrossRef CAS
- J. Shabtai and H. Pines, J. Org. Chem., 1961, 26, 4225–4229 CrossRef CAS
- J. Shabtai, E. M. Lewicki and H. Pines, J. Org. Chem., 1962, 27, 2618–2621 CrossRef CAS
- B. R. Steele and C. G. Screttas, J. Am. Chem. Soc., 2000, 122, 2391–2392 CrossRef CAS
- Y. Yamashita, H. Suzuki, I. Sato, T. Hirata and S. Kobayashi, Angew. Chem., Int. Ed., 2018, 57, 6896–6900 CrossRef CAS PubMed
- I. Sato, Y. Yamashita and S. Kobayashi, Synthesis, 2019, 240–250 CAS
- T. Hirata, I. Sato, Y. Yamashita and S. Kobayashi, Commun. Chem., 2021, 4, 36 CrossRef CAS PubMed
- L. Tie, X.-H. Shan, J.-P. Qu and Y.-B. Kang, Org. Chem. Front., 2021, 8, 2981–2984 RSC
- H.-Z. Du, J.-Z. Fan, Z.-Z. Wang, N. A. Strotman, H. Yang and B.-T. Guan, Angew. Chem., Int. Ed., 2023, 62, e202214461 CrossRef CAS PubMed
- W. Bao, H. Kossen and U. Schneider, J. Am. Chem. Soc., 2017, 139, 4362–4365 CrossRef CAS PubMed
- X.-Y. Zhang, L. Zheng and B.-T. Guan, Org. Lett., 2018, 22, 7177–7181 CrossRef PubMed
- Y.-F. Liu, D.-D. Zhai, X.-Y. Zhang and B.-T. Guan, Angew. Chem., Int. Ed., 2018, 57, 8245–8249 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |