
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Peptide design to control protein–protein interactions
Suzanne P.
van Wier
 and
Andrew M.
Beekman
*
and
Andrew M.
Beekman
*
School of Chemistry, Pharmacy & Pharmacology, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK. E-mail: a.beekman@uea.ac.uk
First published on 16th January 2025
Abstract
Targeting of protein–protein interactions has become of huge interest in every aspect of medicinal and biological sciences. The control of protein interactions selectively offers the opportunity to control biological processes while limiting off target effects. This interest has massively increased with the development of cryo-EM and protein structure prediction with tools such as RosettaFold and AlphaFold. When designing molecules to control protein interactions, either inhibition or stabilisation, a starting point is commonly peptide design. This tutorial review describes that process, highlighting the selection of an initial sequence with and without structural information. Subsequently, methods for how the sequence can be analysed for key residues and how this information can be used to optimise the ligand efficiency are highlighted. Finally a discussion on how peptides can be further modified to increase their affinity and cell permeability, improving their drug-like properties, is presented.
 Suzanne P. van Wier | Suzanne van Wier is currently a postdoctoral researcher at the University of East Anglia. She received her MChem degree from the University of York, and her PhD from the University of East Anglia under the supervision of Dr Andrew Beekman and Prof. Mark Searcey. Her research explores new peptide and small molecule inhibitors for protein–protein interactions for anti-cancer applications. |
 Andrew M. Beekman | Andrew Beekman is a group leader and Associate Professor of Medicinal Chemistry at the School of Chemistry, Pharmacy & Pharmacology, University of East Anglia. After completing BSc (Hons.) and PhD degrees at the Australian National University, he undertook an Endeavour Research Fellowship at the University of British Columbia, and postdoctoral research at the University of East Anglia. Andrew started as a group leader at the University of East Anglia in 2019, and current research interests include the discovery of medically relevant protein–protein interactions and the design of peptides and small molecules to control them. |
Key learning points1. Proven methods for identifying key interfaces in protein interactions.2. How to identify key sequences for peptide–protein binding. 3. How to design probes for protein interactions where structure is available. 4. How to design probes for protein interactions where structure is unknown. 5. How to modify peptides to improve their value as chemical tools. |
1. Introduction
Protein–protein interactions (PPIs) modulate many biological processes, and dysregulation of these interactions leads to aberrant biology. The selective control of protein interactions offers the opportunity to control biological processes in all kingdoms of life. For example, control of targets in cancer, bacterial infection and immunity, and crop protection and food security.1,2 However, targeting PPIs is challenging in comparison to classical drug targets, such as enzymes, receptors and ion channels, as PPIs often take place across large, flat surfaces (1500–3000 Å) compared to the smaller, well-defined binding pockets of classical targets (300–1000 Å).3 As such, PPIs have been classed as ‘undruggable’, leaving many PPIs still largely unexplored in drug discovery and it is thus important to find new methods and improve existing techniques to target PPIs.Proteins and their interacting partners can be identified in vitro with methods such as affinity chromatography using immobilised protein (Fig. 1A), and co-immunoprecipitation, a technique in which PPIs are captured from the cell lysate.4,5In vivo yeast two-hybrid screens can confirm PPIs in which the protein complex is required for the transcription of reporter genes, visualised with cell growth or colour change (Fig. 1B).6,7 To understand the importance of the discovered PPIs, silencing RNA or gene knockout technology can inhibit the expression of one of the protein partners and the resulting cellular effect observed.8,9 However, protein knockout abolishes all interactions for the targeted protein, and as such, modulators able to control individual protein interactions are vital chemical tools for understanding biology. With the progress in cryo-EM for protein complexes and machine learning for protein multimer structure prediction, such as Alphafold, tools for understanding protein–protein interactions are increasingly democratized.10,11 These tools and the information they present allow for greater understanding of PPIs in all kingdoms of life, and therefore greater control.
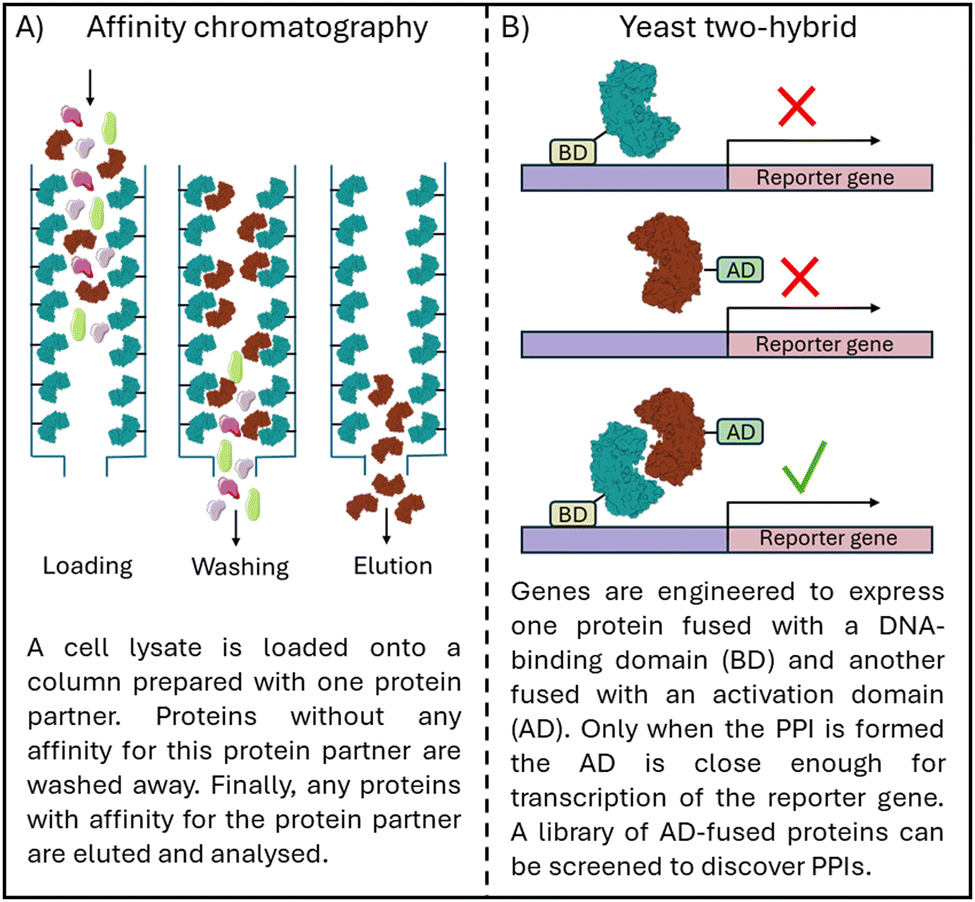 | ||
| Fig. 1 Schematic overview of (A) affinity chromatography to identify PPIs and (B) a yeast two-hybrid screen to confirm PPI formation. | ||
PPIs are commonly modulated using antibodies, peptides, and small molecules. Humanised antibodies are highly selective and effective, exemplified in targeting the PD-1/PD-L1 interaction in the human immune system, for which multiple antibody treatments have been approved.12 However, negatives to antibody approaches are their high production cost, low tissue penetration and adverse immune responses.13 Their use has been limited to extracellular targets, although development of intrabodies, intracellularly active antibodies, is ongoing.13,14
Peptides provide an alternative to antibodies with many benefits, including simpler production at lower cost, while providing specific targeting of interactions, unlike small molecules.
Identifying peptides from the secondary structures responsible for the targeted PPI promises efficacy and selectivity, and a conceptually simple starting point for modulator development. Compared to other peptide discovery techniques such a phage and mRNA display, it also requires less specialist knowledge and equipment. This review describes approaches for the selection of an initial peptide sequence, and the processes for optimising the peptide for affinity and cell permeability (Fig. 2).
 | ||
| Fig. 2 Schematic overview of the methods discussed in this review, from selecting the initial peptide sequence, to analysing this, increasing affinity for the target protein as well as cell permeability, leading to an optimised peptide. | ||
1.1 Biophysical/biochemical assays
To understand how peptide sequences are discovered, it is valuable to first appreciate techniques used to detect and measure peptide–protein interactions. Once lead molecules have been synthesised, they must be evaluated against the target protein. In vitro biophysical assays are routinely used in early drug discovery to investigate protein–ligand interactions. For the inhibition of PPIs, the ability of the ligand to disrupt the PPI formation is also of interest. In vitro assays to examine these properties are a useful tool in the development of PPI inhibitors (Fig. 3). | ||
| Fig. 3 Schematic overview techniques used to analyse peptide–protein interaction with (A) fluorescence polarisation (FP), (B) surface plasmon resonance (SPR), (C) isothermal titration calorimetry (ITC) and (D) enzyme linked immunosorbent assay (ELISA). | ||
Fluorescence polarisation (FP), microscale thermophoresis (MST), and surface plasmon resonance (SPR) can provide binding affinities, whereas differential scanning fluorimetry DSF is better used as a relative comparison tool between compounds. SPR provides information on the kinetics of the interaction. Isothermal titration calorimetry (ITC) can provide detailed information on kinetics and binding affinity but is poorly suited for high throughput application. However, it is the only technique that is label-free and in solution providing the most accurate information about the interaction. FP and MST rely on the use of a fluorescent label on the ligand or protein and DSF requires the addition of a fluorescent dye. Surface plasmon resonance (SPR) and enzyme linked immunosorbent assay (ELISA) requires an analyte to be immobilised, which could interfere with the binding.
Assays based on resonance energy transfer such as Förster resonance energy transfer (FRET) and homogeneous time resolved fluorescence (HTRF), can take place with all components in solution, allowing evaluation of PPI formation.
2. Selection of an initial peptide sequence
If structural information is available, the choice of peptide sequence starting point can be made by analysing the binding interface. However, without this structural information or knowledge of which part of the interaction is most important, methods including protein mutagenesis, sequence conservation analysis and peptide arrays can be used.2.1 Selection of peptides through structural information
When the structure of a protein is known a direct approach to designing a peptide inhibitor is through analysing the structure and identifying the interaction face of the protein partner. The interacting section can be taken from this as a starting point before further optimisations take place.If the interaction is mediated through an α-helix the whole helix can be used as a starting point for the interaction. For example, Kirsten rat sarcoma viral oncogene homolog (KRAS) is a small GTPase which is involved in cell survival and proliferation. KRAS mutations are found in cancers and lead to decreased GTPase activity which in turn leads to increased RAS pathway signalling.15 KRAS is negatively regulated by son of sevenless 1 (SOS1) and therefore the inhibition of the KRAS/SOS1 PPI can increase GTPase activity.16 The SOS1 derived section 929FFGIYLTNILKTEEGN944 (1) forming an α-helix (Fig. 4A) was identified from the crystal structure and used as the starting point for inhibitor development.17,18 Depending on the interaction site a region around the α-helix may also be included if the structure suggests it is part of the interacting sequence, exemplified in targeting the nuclear transcription factor Y (NF-Y) trimer formation. NF-Y activates the genes associated with cell cycle regulation and DNA repair with overexpression of NF-Y found in cancer.19 Residues 267VNAKQYHRILKRRQARAKLEAE-GKIPKER295 (2) were taken from NF-YA encompassing an α-helical section which continues into an interacting disordered section (Fig. 4B). This section was shown to bind to NF-YB/C dimer (KD = 0.7 μM) in a fluorescence polarisation (FP) assay. This sequence was used as a starting point allowing exploration of truncated sequences to determine if the disordered section is crucial for the interaction (see Section 3.2).20 This approach was also taken in targeting the RbAp48/MTA1 interaction which consists of both an α-helical and disordered section.21,22 The approach of deriving a peptide starting point from the sequence has now been taken for targeting many α-helix mediated PPIs, such as Axin/β-catenin,23 BCL9/β-catenin,24 Cav/Cavβ,25 Cullin3/KCTD1126 and SARS-CoV-2 spike/ACE2.27
 | ||
| Fig. 4 (A) Structure of SOS1 (grey)/KRAS (blue) (PDB: 1NVU) with the sequence 929FFGIYLTNILKTEEGN944 (1) in yellow, (B) structure of NFY trimer (PDB: 4AWL). NF-YA (grey) with peptide sequence 267VNAKQYHRILKRRQARAKLEAEGKIPKER295 (2) (yellow), NF-YB (blue) and NF-YC (burgundy) and (C) structure of the Nac1 dimer (PDB: 3GA1) with section 12FGNSILECLNEQR24 (3) in yellow and 44HRAVLAASSSYFRDLFN60 (4) in burgundy. | ||
The protein structure may also reveal more than one potential interaction site between the protein and its partner. Nucleus accumbens-associated protein 1 (Nac1) is a repressor protein that mediates the interactions between transcription factors with an essential role in carcinoma tumour growth.28 In targeting Nac1 homodimerization two separate sections were explored as initial sequences: 12FGNSILECLNEQR24 (3) and 44HRAVLAASSSYFRDLFN60 (4) with 3 showing weak binding in an FP assay (KD = 360 μM) and 4 showing no binding (Fig. 4C). Regardless, both sections were further explored to see whether their affinity could be improved.29
If the target protein has multiple protein partners, these could all be explored as potential starting points. The misregulations of Ras-related in brain (Rab) proteins are implicated in neurodegenerative diseases and cancer. In targeting these proteins crystal structures with multiple different protein partners were explored, identifying nine sequences.30,31 These were tested against a set of seven Rab proteins in an FP assay.
Four peptides (derived from R6IP, LidA, REP1 and Rabin8, Fig. 5) with low micromolar binding affinities were further explored.31 Using structural information is not limited to α-helical motifs with the same approach deployed for β-hairpin sections. This has been applied to targeting the epidermal growth factor receptor (EGFR) which is overexpressed in cancer.32 EGFR forms an asymmetric dimer and disruption of the dimerization could inactivate the kinase, which was explored by mimicking the EGFR dimerization arm residues 269YNPTTYQM278 (5) which form a β-hairpin (Fig. 6).33
 | ||
| Fig. 5 Structures of a Rab protein in complex with (A) R6IP (PDB: 3CWZ), (B) LidA (PDB: 3TNF), (C) Rep1 (PDB: 1VG0) and (D) Rabin8 (PDB: 4LHX) highlighting the peptide starting sequence in yellow. | ||
 | ||
| Fig. 6 Structure of EGFR dimer (PDB: 1IVO) with β-hairpin starting peptide sequence 269YNPTTYQM278 (5) highlighted in yellow. | ||
The continued democratization of cryo-EM will undoubtedly increase the experimental data for protein–protein interactions.
Similarly, the continued progress in protein structure prediction, including protein multimer, protein–DNA, and protein–RNA will provide new structural insight for identifying initial peptides for development. To date, this has been throttled by the poor accuracy of structure prediction tools34–36 for peptide–protein interactions, limiting confidence in peptide design. But recent reports into virtual array development37 and improvement in biomolecular structure prediction38 will continue to advance this area.
2.2 Using site-directed protein mutagenesis
Protein mutagenesis is a technique in which a mutation is introduced into the genes encoding for the desired protein. This can be used to analyse which residues within the protein are most important for the interaction. For example, this can be used for the expression of alanine mutants. The amino acid alanine has a methyl group as its side chain substituent. Compared to other amino acids, which have either larger hydrophobic groups in its place or polar or charged side chains, the methyl is not able to form strong interactions with the target protein. The expression of alanine mutants that result in a loss of binding affinity suggest a hot spot residue, the small subset of residues that contribute most of the binding energy in a PPI.39,40 Hot spots are defined as residues which upon mutation to alanine lead to a loss of 2.0 kcal mol−1 or more in binding free energy.41 Protein mutagenesis is a useful tool to determine the best starting point when the PPI is comprised of multiple binding interfaces. This approach can be combined with computational prediction to select which residues to mutate minimising the number of proteins generated.This approach was applied in targeting Leishmania infantum, a parasitic disease causing visceral leishmaniasis. Trypanothione reductase (TryR) maintains the intracellular redox state and is essential for parasitic activity.42 The homodimer interface of TryR was explored computationally to identify potential hot spots by examining the burial of solvent-accessible surface area, the van der Waals and electrostatic contributions of each residue to the interaction. Three mutant proteins (W81A, E436A and Q439A, Fig. 7A) were expressed and analysed using native polyacrylamide gel electrophoresis (PAGE), a technique used to separate proteins and protein complexes by size, showing that only the E436A mutation decreased dimer formation.43 This residue is part of an α-helix (435PEIIQSVGICMKM447 (6)) chosen as the starting point for peptide design. The cysteine was replaced with serine to avoid disulfide bond formation leading to peptide 7 (Ac-PEIIQSVGISMKM-NH2), which was able to disrupt the dimer formation in an Enzyme Linked Immunosorbent Assay (ELISA) with an IC50 of 40.8 μM.43
 | ||
| Fig. 7 (A) Structure of the Leishmania infantum TryR dimer (PDB: 2JK6) highlighting residues W81, E436 and Q439 in green with the starting peptide section 435PEIIQSVGICMKM447 (6) highlighted in yellow and (B) structure of CK2α (blue) in complex with CK2β1 (grey) and CK2β2 (purple) (PDB: 1JWH) highlighting residues M166, Y188 and F190 in green with the section 186RLYGFKIH193 (8) used as the starting point in yellow. | ||
Casein kinase 2 (CK2) is a protein kinase with functions in cell proliferation and apoptosis suppression. Overexpression of CK2 has been found in tumours, making it an attractive cancer target.44 CK2 consists of two catalytic (α) and two regulatory (β) subunits and as an inhibitory strategy the CK2α/CK2β PPI was targeted. Three residues of CK2β (M166, Y188 and F190, Fig. 7B) were identified as pointing directly towards CK2α with Y188 and F190 found together on one of the CK2β subunits and M166 found on the other CK2β subunit. A single F190A mutant showed significantly reduced binding and a double (F190A + Y188A) and triple (F190A + Y188A + M166A) mutants abolished binding as observed by surface plasmon resonance (SPR).45 Y188 and F190 are part of the same β-hairpin loop (186RLYGFKIH193 (8)) which was taken as the starting point for the design of cyclic peptides. The sequence was not tested as a linear peptide, but a disulfide cyclised 13-mer peptide GC*RLYGFKIHGC*G (9, *cyclised residues) demonstrated a of KD = 1.75 μM by Isothermal titration calorimetry (ITC).45,46
Site-directed mutagenesis can also confirm the importance of mutations found in disease, guiding the design of peptides. Phosphatidylinositol 3-kinase α (PI3Kα) is involved in cell proliferation forming interactions with adapter proteins such as insulin receptor substrate 1 (IRS1) in the presence of growth factor stimulation, stimulating the catalytic activity of PI3Kα.47 The two most common mutations found in cancer (E545K and H1047R) were explored experimentally identifying IRS1 as a binder of E545K mutant protein but not of H1074R mutant protein in pull-down experiments.48 It was also shown that the interaction of E545K mutant PI3Kα with IRS1 is required for the growth of colon cancer cells in vivo. A 30-residue section consisting of two α-helices around E545 (528EQLKAISTRDPLSEITEQEKDFLWSHRHYC558 (10)) was chosen for peptide design, which inhibited the PPI formation in cell lysate as investigated by immunoprecipitation. Following this, each helix was explored separately giving the N-terminal 21-mer peptide 528EQLKAISTRDPLSEITEQEKD549 (11) and the C-terminal 18-mer peptide 541SEITEQEKDFLWSHRHYC558 (12) with only the C-terminal peptide 12 able to disrupt the PPI.48
Deletion scanning mutagenesis can be used to investigate the binding regions of a PPI through the expression of mutant variants, where parts of the protein have been removed. Eukaryotic translation initiation factor 4E (eIF4E) plays a role in the initiation of translation with overexpression inducing tumorigenesis.49 eIF4E is negatively regulated by eIF4E-binding protein 1 (4E-BP1) through competition with eIF4G binding which is required for the formation of the active complex needed for translation.50 Peptides mimicking 4E-BP1 were explored to inhibit translation through stopping the eIF4E-eIF4G PPI formation. Mutagenesis was used to remove the genes encoding for sections across residues 1–118 of the 4E-BP1 protein (Fig. 8). Only the mutant without the 54–63 section of 4E-BP1 did not bind eIIF4E, showing this section to be crucial for binding. Therefore, it was chosen to start the peptide design from the longer 51RIIYDRKFLMECRNSPV67 (13) section encompassing residues 54–63. This peptide bound eIF4E with a KD of 50 nM as measured by ITC.51
 | ||
| Fig. 8 Sequences created in scanning deletion mutagenesis of 4E-BP1. | ||
2.3 Peptides spanning protein sequence
If the interacting site of the PPI is not known peptides probing the entire protein sequence can be generated. The synthesis and purification of many peptides can be time-consuming using solid phase peptide synthesis. Instead, techniques such as peptide arrays have been designed in which many peptides can be probed at the same time. | ||
| Fig. 9 Schematic overview of a peptide array. | ||
The same approach was also taken in targeting the BH3-interacting domain death agonist (BID) and mitochondrial carrier homologue 2 (MTCH2) PPI, an interaction involved in apoptosis. Two peptides (59WTDGNRSSHSRLGRIE73 (15) and 111WLQLRNTSRSEEDRNR125 (16)) were found to induce near complete cell death at 50 μM in osteosarcoma cells as penetratin, a cell-penetrating peptide, conjugates.56
It might not be necessary to probe the entire protein as was the case in targeting the β-barrel assembly machine (Bam), present in the outer membrane of Gram-negative bacteria. Bam contains the two essential proteins BamA and BamD. Here it was first determined through pull-down experiments (an experiment in which a protein is immobilised, and it is evaluated whether its protein partner can be captured from cell lysate) of BamD with urea-denatured fragments of BamA, that only the C-terminal residues 715–810 were able to bind BamD. Therefore five overlapping sequences spanning this region were designed with only the peptide spanning residues 765–779 effective at inhibiting the assembly of Bam.59
2.4 Conserved regions across proteins or species
Many proteins are conserved across species or across groups of proteins with similar functions, which can indicate the importance of these regions, and suggest an initial sequence for binding and inhibiting target PPIs.Targeting protein for Xklp2 (TPX2) is a spindle assembly factor which is required during cell mitosis. TPX2 localises Aurora-A, an essential mitotic kinase to spindle microtubules which has been highlighted as an oncogene.60 Residue conservation across human, Xenopus and pufferfish TPX2 is seen across residues 1–43 (19) (Fig. 10A) and immunoprecipitation and pull-down experiments showed the interaction of 19 with Aurora-A whilst also showing that the shorter 15–43 section (20) was not able to do so.61
 | ||
| Fig. 10 (A) Sequence alignment of human, Xenopus, and pufferfish TPX2 sequence highlighting in green the residues conserved across all three species and in orange the residues conserved across two species. Peptide 19 is highlighted in blue and peptide 20 in maroon. (B) Sequence alignment of human, mouse, chicken, and zebrafish Nrf2 sequence highlighting in green the residues conserved across all four species and underlined the sections 21 and 22. | ||
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription factor leading to the transcription of cytoprotective genes. Kelch-like ECH-associated protein 1 (Keap1) is a negative regulator of Nrf2 tagging Nrf2 for degradation by ubiquitination. To target the Nrf2/Keap1 PPI the human sequence of Nrf2 was compared to that in mouse, chicken, zebrafish, and drosophila with two highly conserved regions found: residues 17–32 (21) and residues 77–82 (22) containing an DEETGE motif (Fig. 10B). Through ITC measurements of mutant proteins missing either section, it was shown that the binding affinity of 22 was higher than 21.62 Therefore a 16-mer peptide (69AFFAQLQLDEETGEFL84 (23)) around the human 77DEETGE82 motif was used as an initial sequence for inhibitor development, binding Keap1 with a KD of 20 nM.62,63
Similar approaches have been deployed for DNA polymerases across eubacteria and archaea, leading to the identification of the QL[S/D]LF consensus sequence with further work based on this sequence discussed in multiple reviews.1,2,64,65 Similarly, the control of plant ethylene responses was achieved by investigating a conserved nuclear localization signal sequence found in Arabidopsis ethylene regulator ethylene insensitive-2 (EIN2).66–69
3. Analysing the sequence
Once an initial sequence has been identified, it is important to understand the importance of each of the amino acids within the peptide. This can be explored through truncation studies as well as by performing an alanine scan. In the same way as discussed for proteins in Section 2.2, an alanine scan provides information about the contribution of each residue in the sequence to the overall binding affinity of the peptide. Truncation studies can be used to explore a series of sequentially shortened peptides, to understand which section of the sequence is at the core of the binding interface. Together, these techniques provide further information about the sequence, which can then be used to increase the binding affinity (as discussed in Section 4).3.1 Alanine scan
An alanine scan can be used to highlight the hot spot residues within a peptide sequence. In an alanine scan, residues within the sequence are sequentially replaced with alanine and the binding affinity is analysed.Inducible nitric oxide (NO) synthase (iNOS) produces NO which is required for the intracellular killing of pathogens. iNOS is negatively regulated by SPRY domain-containing SOCS (suppressor of cytokine signalling) box protein 2 (SPSB2) and the linear peptide Ac-KEEKDINNNVKKT-NH2 (24) derived from iNOS was shown to bind SPSB2 with a KD of 13.3 nM (ITC). Through an alanine scan the most important residues in the sequence for binding were identified in the section 23DINNN27.70,71
Disruptor of telomeric silencing 1-like (DOT1L) catalyses the methylation of histone H3 at lysine 79 and has been implicated in leukaemia.72 In up to 10% of acute leukaemia cases the mixed lineage leukaemia (MLL) protein is fused to a partner protein such as AF9. The PPI of AF9 with DOT1L recruits DOT1L to AF9 targeted genes increasing their methylation therefore increasing their expression leading to leukaemia. An alanine scan was performed of peptide 865LPISIPLSTV874 (25) (IC50 = 0.49 μM by competitive SPR) derived from DOT1L showing that three C-terminal residues were not essential for binding. Therefore it was possible to generate the shortened 7-mer peptide 879LPVSIPL886 (26) with only a small loss in binding affinity (IC50 = 3.9 μM).73
An alanine scan may also lead to the discovery of a peptide with enhanced binding affinity. An alanine scan of Ac-DEETGEF-OH (27) (IC50 = 5.39 μM), a shortened version of the Nrf2 derived peptide 23, found that an E77A mutation increased binding (IC50 = 0.730 μM) nearly 10-fold.74 YAP residues 86MRLRKLPDSFFKPPE100 (28) form a twisted-coil with R87 and F96 forming a cation–π interaction. The section 81PQTVP85 (29) was previously reported to be essential to binding and therefore the linear peptide 81PQTVPMRLRKLPDSFFKPPE100 (30) was chosen as the starting point.7530 gave an IC50 of 49 μM by competitive SPR and an alanine scan of 30 revealed that a D93A mutation, giving 81PQTVPMRLRKLPASFFKPPE100 (31) increased binding (IC50 of 25 μM).76
Alternatively, an alanine scan can highlight unimportant residues which may then later be used to further improve the peptide such as through the introduction of mutations (see Section 4.1) or by peptide stapling (see Section 4.3).77
3.2 Sequence truncation
Shortening the peptide sequence to contain only the essential residues for the interaction improves ligand efficiency and simplifies synthesis.Truncation of the sequence derived from the structure of NF-YA (residues 267–295 (2)) was explored (Table 1). Six shortened peptides were generated, first removing residues from the C-terminus until no longer tolerated followed by shortening of the N-terminus until no longer tolerated (Table 1). In total, the sequence was shortened by 13 residues to 270–285 (32) with only a small loss in binding affinity in an FP assay (KD = 0.7 μM to KD = 2.9 μM).20
| Nr. | K D (μM) | Sequence |
|---|---|---|
| 2 | 0.7 | 267VNAKQYHRILKRRQARAKLEAEGKIPKER295 |
| 33 | 1.3 | 267VNAKQYHRILKRRQARAKLEAE288 |
| 34 | 2.0 | 267VNAKQYHRILKRRQARAKL285 |
| 35 | 9.5 | 267VNAKQYHRILKRRQAR282 |
| 36 | 2.1 | 268NAKQYHRILKRRQARAKL285 |
| 32 | 2.9 | 270KQYHRILKRRQARAKL285 |
| 37 | 45 | 271QYHRILKRRQARAKL285 |
Repressor/activator protein 1 (RAP1) is part of the shelterin complex which plays a role in the regulation of telomeres.78 The PPI of RAP1 with telomeric repeat-binding factor 2 (TRF2) represses the localisation of poly [ADP-ribose] polymerase 1 (PARP1) to telomeres resulting in catastrophic telomere loss.79 The original 41-residue TRF2 interacting sequence, consisting of two α-helices, bound RAP1 with a KD of 16.5 nM. This was shortened to a 16-mer peptide (281TTIGMMTLKAAFKTLS296 (38)) containing only one of the α-helices as a starting point for stapling (see Section 4.1).80
Apoptosis-inducing factor (AIF) regulates cellular survival through functions in the mitochondria and AIF mediates neuronal cell death under lethal cellular stress with inhibition of AIF having a neuroprotective effect.81 Cyclophilin A (CypA) binding to AIF initiates the translocation of both proteins into the nucleus.82 The linear AIF mimicking 25-mer peptide (370QSVGVSSGKLLIKLKDGRKVETDHI394 (39)) showed binding to CypA (KD = 12 μM by SPR) and blocked nuclear translocation.83 It was possible for 39 to be shortened by 16 amino acids to the 9-mer section 381LIKLKDGRKVE389 (40) as NMR studies had shown that was the most important section of the interaction.84 An increased binding affinity was seen for 40 (KD = 2.4 μM) compared to 39 (KD = 12 μM by SPR).85 Although removal of a large section may not be possible for most peptides, the removal of any residues is still beneficial to improve the drug-like properties of the peptide. The 16-mer peptide (69AFFAQLQLDEETGEFL84 (23)) derived from Nrf2 was truncated to a 14-mer, 12-mer, 10-mer, 9-mer, 8-mer and 7-mer. From the 16-mer to a 10-mer (75QLDEETGEFL84 (41)) the binding affinity was maintained (KD = 27.3 nM for 41 and KD = 23.9 nM for 23). The removal of another residue of the N-terminus to give the 9-mer peptide (76LDEETGEFL84 (42)) led to a big decrease in binding affinity (KD = 352 nM) with any further truncations abolishing binding.86,87
4. Increasing peptide affinity
Having identified the essential residues, the peptide can be optimised further for binding affinity, cell permeability and proteolytic stability. To increase the binding affinity, it may be possible to introduce mutations into the sequence or use peptide stapling or macrocyclization.88 Peptide stapling and/or macrocylisation can also provide an opportunity to increase their cell permeability and proteolytic stability.4.1 Introducing mutations into the sequence
Improving the binding affinity of the designed peptide can be achieved through the introduction of a mutation into the sequence with the possibility to explore unnatural amino acids. It may be possible to increase the size of a hydrophobic residue by introducing substituents or through introducing larger aromatic rings. As described in Section 3.1, an alanine scan may highlight the potential positions at which mutations may be introduced. The PPI between Suppressor of Mothers against Decapentaplegic (Smad) and Yes-associated protein (YAP) is involved in the activation of a signalling pathway leading to heterotopic ossification (HO) characterised by bone formation outside of the skeleton, making it a target for the treatment of HO.89 Using computational genetic evolution to sequentially vary the residues within the 205–214 section of Smad an optimised peptide was identified, 205DGWPPPYPRV214 (43) (KD = 2.5 μM). An alanine scan of 43 revealed three unimportant residues (G206, P212 and V214) which were computationally varied to all other natural amino acids. Six peptides were explored experimentally with the combination of two mutations (205DQWPPPYPRH214 (44)) leading to a nearly 10-fold improvement in binding (KD = 0.34 μM).90It is not only through an alanine scan that these positions may be highlighted, when the structural information is available, the PPI can be examined. A TRF2 derived peptide (38) has high affinity for RAP1 (Ki = 0.14 μM). The crystal structure of the RAP1/TRF2 complex showed a large hydrophobic space around I283 (Fig. 11A). Therefore different natural and unnatural hydrophobic mutations were attempted at I283 (2,2-dimethylpropylalanine, cyclohexylalanine, phenylalanine, tryptophan, naphthalene and 2, 3 and 4-chlorophenylananine) with the 2-chlorophenylalanine (45) found to increase binding the most (Ki = 7 nM).80 Similarly mutations were explored around F374 in a 35-mer linear peptide derived from BCL9 targeting β-catenin (residues 347–381 (46)). The binding pocket of F374 is shallow but wide which could accommodate a larger group (Fig. 11B). Smaller and larger hydrophobic groups were explored with the introduction of a 2-naphthylalanine (47) leading to a 3-fold increase in binding affinity.91
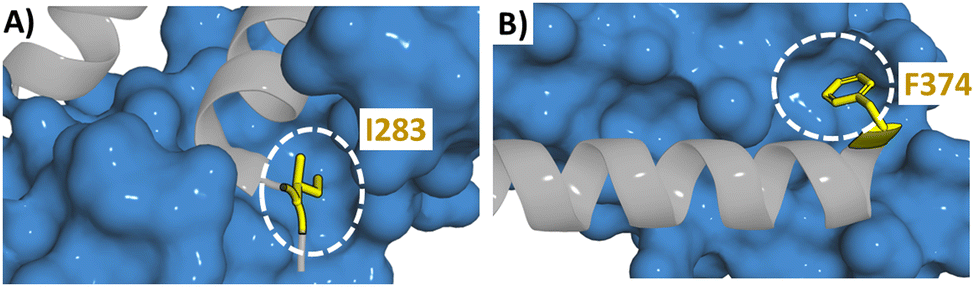 | ||
| Fig. 11 Structure of (A) RAP1 (blue) with TRF2 (grey) (PDB: 3K6G) highlighting the binding pocket of I283 with a white circle and (B) β-catenin (blue) with BCL9 (grey) (PDB: 2GL7) highlighting the binding pocket of F374 with a white circle. | ||
A CK2β derived cyclic 13-mer GCRLYGFKIHGCG (9) demonstrated moderate binding affinity for CK2α by ITC (KD = 1.75 μM). Docking experiments highlighted meta-substitution of F190 with hydrogen or halogen bond forming substituents reached a well-defined water molecule in the structure. Chloro and iodo-substituent analogues were synthesised with the meta-iodine (48) resulting in an improved binding affinity (KD = 0.239 μM by ITC).92
Multiple mutations can also be introduced into a sequence although the combination of individually beneficial mutations may not be additive. A FYCO1 derived 34-mer peptide spanning residues 1275GQGANTDYRPPDDAVFDIITDEELCQIQESGSSL1298 (49) was shown to bind LC3B (KD = 0.29 μM) in an FP assay.93 The binding pocket of F1280 was shown to be able to accommodate a bigger hydrophobic group and a 2-naphthylalanine substitution (50) increased the binding affinity (KD = 0.14 μM). A substitution of L1288 with a tert-butylalanine (50) led to a small improvement of binding affinity (KD = 0.25 μM). The N-terminal section of the peptide is located near various negatively charged residues of LC3B and an introduction of an Arg to the N-terminus (51) showed a slight improvement of the binding affinity (KD = 0.20 μM). A combination of these three mutations (52) led to a peptide with a KD of 0.12 μM which was similar to that of the single substituted 50.94
4.2 Stapling of α-helical peptides
Peptide stapling can increase the binding affinity of a peptide due to the preorganisation of a peptide secondary structure.95 In peptide stapling a bond is formed between two residues on the same side of the α-helix (Fig. 12A). This is achieved with canonical amino acids such as cysteines to form thioether stapled peptides, or lysine with aspartic or glutamic acid to create a lactam stapled peptide. Additionally, unnatural amino acids generate commonly used hydrocarbon (formation of an alkene bond via ring closing metathesis) and triazole staples. Fairlie and co-workers compared the α-helicity of pentapeptides using six stapling techniques demonstrating that lactam-stapling induced the highest level of α-helicity followed by hydrocarbon stapling.96 Hydrocarbon stapling is commonly used due to its ability to increase cell permeability.97 Hydrocarbon stapling has been extensively reviewed with many examples of its successful application in increasing binding affinity, proteolytic stability and cell permeability.23,88,98–100 Two cases will be highlighted here as examples of the use of a single hydrocarbon staple leading to the peptides with in vivo activity.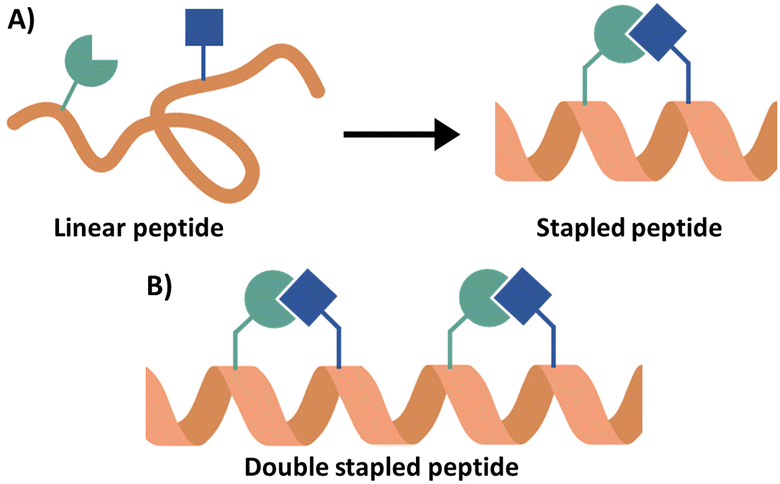 | ||
| Fig. 12 (A) Schematic overview of peptide stapling and (B) schematic overview of a double stapled peptide. | ||
The PPI between β-catenin and its cofactor B-cell lymphoma 9 (BCL9) is part of the Wnt signalling pathway which plays a key role in cell proliferation with activation of this pathway found in different cancers.101,102 From the crystal structure, a 35-mer linear peptide (residues 347–381 (53)) from BCL9 was shown to bind β-catenin with a KD of 616 nM in an FP assay. Truncation to a 24-mer (residues 351–375) (54) resulted in a small loss in binding inhibitory effect (Ki = 1.90 μM compared to 0.96 μM) as measured in a competitive FP assay.103 This shortened sequence was used in the design of hydrocarbon stapled peptides identifying 351LSQEQLEHRERSLS5TLRS5IQRMLF374 (55) which inhibited the BCL9/β-catenin complex at an IC50 of 135 nM. In vivo mouse xenograft models of Wnt-driven cancer showed suppression of tumour growth, angiogenesis and invasion with treatment of 55.104 Similarly, peptides were derived from Bcl-2-interacting mediator of cell death (BIM) to target antiapoptotic B-cell lymphoma 2 (BCL2) family proteins.105,106 The BIM BH3 helix (146IWIAQELRRIGDEFNAYYARR166 (56)) was modified with i, i + 4 hydrocarbon stapling at positions 154 and 158, giving peptide 57 (146IWIAQELRS5IGDS5FNAYYARR166). 57 showed nanomolar binding against a range of BCL-2 family proteins including BCL-XL, MCL-1 and BFL-1 and suppressed tumour growth in vivo.107
Hydrocarbon stapling may however not always prove the best option for structure restraint. In targeting Li-TryR dimer using peptides based on section 435PEIIQSVGISMKM447 (6) it was found that although hydrocarbon stapling gave the most α-helical peptides, only lactam stapled peptides were able to disrupt the dimer formation. Molecular dynamics studies suggested Q439 was not able to adopt the required conformation in the hydrocarbon stapled peptides.108–110 Other stapling methods may increase the solubility of the peptides, such when targeting the RAP1/TRF2 PPI in which triazole stapling was applied to peptide 38. The triazole stapled peptide, 281TTIGMMTLKZAFKXLS296 (58) was 10-fold more potent than the linear 16-mer peptide 38 (Ki = 0.14 compared to Ki = 2.0 μM in a competitive FP assay). The triazole staple increased the binding affinity (Ki = 0.14 compared to Ki = 2.0 μM) whilst improving its solubility.80
Multiple staples can also be introduced into the sequence, which can be beneficial for longer sequences (Fig. 12B). Double hydrocarbon i, i + 4 stapling was applied to a JAZ9 derived 21-mer peptide (residues 218–238) targeting MYC with both higher α-helicity (45% compared to 25%) and better binding affinity (KD = 0.10 μM compared to 0.88 μM and 2.4 μM) seen for the double over the single stapled analogues and inhibition of MYC-related gene expression in Arabidopsis thaliana was observed.111 The same approach taken in targeting the Rab protein, Rab6a, through deriving a peptide from R6IP,31 and for targeting DAP12 homodimerization,112 SNARE/synaptotagmin-1113 and the p53 PPI with MDM2/MDMX.114
4.3 Macrocyclisation of peptides
For α-helical structures stapling can be used to ensure the helical structure of the peptide. For other secondary structures, such as β-hairpin peptides, this can be achieved with macrocylisation. The EGFR dimerization arm residues 269YNPTTYQM278 (5) form a β-hairpin with a cyclic version of this peptide, CYNPTTYQMC (59), decreasing dimer formation by 20% at 1 μM (Fig. 13A).33 A triazole cyclised version (60) inhibited EGFR dimerization by 33% at 5 μM, similar to previous disulfide cyclised peptides. However, the proteolytic stability was improved for the triazolyl-cyclised peptide over the disulfide version.115 | ||
| Fig. 13 (A) The macrocyclisation of EGFR derived peptide 269YNPTTYQM278 (5) using disulfide (59) and triazole (60) linkers and (B) structure of bicyclic peptide 370QSCGVSSGZLLCKLKDGRKVXTDHI394 (74) derived from AIF. | ||
Vestigial-like protein (VGLL) is a coactivator of transcriptional enhanced associate domains (TEADs) and has been found to be a transcriptional repressor inhibiting tumorigenesis caused by YAP as part of the Hippo pathway with VGLL4 competing with YAP for binding to TEADs.116 VGLL4 is a small protein consisting of an α-helix linked to a double-stranded β-sheet through a loop linker. To mimic the double-stranded β-sheet cysteine residues were added to the peptide termini to allow for head-to-tail cyclisation across residues 19PKTEWNAGSVIFTY32 and this sequence was conjugated to the cell penetrating peptide TAT (see Section 5 for more information) to give peptide 61. 61 showed increased activity in a cell viability assay (IC50 = 18.1 μM) compared to its linear counterpart (IC50 = 87.4 μM) and the slightly shorter cyclised 20KTEWNAGSVIFT31 (62) showed a similar activity (IC50 = 25.0 μM) with its linear counterpart showing negligible activity (IC50 > 100 μM).117
The linear CK2β peptide 186RLYGFKIH193 (8) forms a β-hairpin loop with Y188 at the turn (see Section 2.2). The peptide was extended and Cys residues were added which facilitated cyclisation through a disulfide bridge giving the 13-mer peptide GCRLYGFKIHGCG (9). This peptide inhibited the CK2α/CK2β complex with an IC50 of 3 μM, a ten-fold increase on the linear version (IC50 = 30 μM).45 The replacement of the disulfide bridge with a triazole based bridge further increased binding (KD = 460 nM) compared to 9 (KD = 1000 nM) as measured by ITC.118
The linear AIF mimicking 25-mer peptide (370–394) (39) showed binding to CypA and blocked nuclear translocation.83 This section forms a β-hairpin and so disulfide and triazole cyclisation were applied to create mono and bicyclic peptides. A peptide with a combination of disulfide and a triazole bridge, 370QSCGVSSGZLLCKLKDGRKVXTDHI394 (63) (Fig. 13B) showed improved binding (KD = 0.85 μM) compared to the 39 (KD = 5.0 μM).119
Macrocyclisation strategies for β-hairpins as well as helix-turn-helix peptides have been recently reviewed.88
5. Increasing cell permeability
Key for the effectiveness of drugs with intracellular targets is their ability to cross the cell membrane. The main structure of a cell membrane is made up of a lipid bilayer formed by phospholipids, containing a hydrophilic head and hydrophobic tail. The bilayer is formed with the hydrophilic heads on the outside and the hydrophobic tails on the inside.120 Therefore for the passive absorption of drugs, they have to pass through this hydrophobic interior. The development of peptide therapeutics is often hindered by their lack of cell permeability due to the hydrophilic character of the backbone amide bonds as well as the side chains of some amino acids. Hydrocarbon stapling commonly increases the lipophilicity of peptides and can increase cell permeability. However, it may not be possible to insert a staple into a peptide sequence, especially in the development of non α-helical peptides. It has also been found that cyclic peptides may have enhanced cell permeability due to their conformation where the hydrogen bonds are formed within the peptide, leaving a hydrophobic exterior.121 Other methods include the methylation of the backbone nitrogen, removing its ability for hydrogen bonding.122,123 However, the increasing lipophilicity is not the only option in promoting cell permeability. Cell-penetrating peptides (CPPs) have been developed which can be conjugated to the main peptide, with this topic having been reviewed extensively.124–127 Many different CPPs have now been developed, but a similarity between them all is their highly positively charged nature. It is thought that their uptake may be not via passive absorption but through endocytosis or by direct translocation. Endocytosis is a process in which the peptide is surrounded by the hydrophilic outside of the cell membrane and then taken into the cell forming a vesicle with the peptide inside. In direct translocation the binding of the positively charged peptide to the negatively charged membrane causes instability in the membrane. This causes pore formation through which the peptide can enter the cell. This benefit may come at a cost, with toxicity sometimes observed at therapeutically relevant concentrations. Despite some conjugates progressing to phase III trials, no CPP containing peptides have progressed to clinical use.The conjugation of a CPP to a potent peptide to enhance cell permeability was successfully applied to the disulfide cyclised YAP derived peptide. Although the peptide showed high binding affinity (IC50 = 15 nM in competitive SPR), it was not able to penetrate cells. Therefore, the peptide was conjugated to TAT, a CPP derived from HIV, with cell permeability now seen.75,76 Alternatively, the addition of two or three arginine residues to the end of the peptide has also been shown to increase cell permeability by changing the overall charge without the need to add a large sequence of amino acids. For example, a hydrocarbon stapled peptide based on peptide 1 derived from SOS1 required the addition of two Arg residues, changing the overall charge from −1 to +1, for cellular uptake.18
Instead of improving the cell permeability of the peptides through the addition of a CPP or arginine residues, mutations can be introduced to change the overall charge. A double hydrocarbon-stapled R6IP derived peptide targeting Rab6a (900DDES5EWFS5YHLS5FFNS5V916 (64)) demonstrated a KD of 7.8 μM, but cellular uptake was limited. Introduction of D901N and E904Q (65) removed the negative charges and increased cellular uptake (cellular uptake in range of TAT) with only a small loss in binding affinity (KD = 12.7 μM).128 This method was also applied to a p53 peptide (17ETFSDLWKLLPE28 (66)). Through a double staple of hydrocarbon stapling at positions 17 and 21 and lactam stapling at positions 24 and 28 as well as mutations L22K and P27R, giving peptide 67 (Ac-S5TFSS5KWDLLRK-NH2), the overall charge was changed from −2 to +2. This peptide reduced cell viability whereas its counterpart without mutations (giving an overall neutral charge) did not affect cell viability.114
6. Conclusion and future outlook
Peptides for the inhibition of protein–protein interactions offer several advantages over antibodies and small molecules. Peptides are inexpensive, structurally designed, and readily modified, while offering exquisite selectivity for their target. Peptides cover large protein interaction surfaces and have demonstrated high levels of success for the inhibition of biomacromolecule interactions. We have highlighted the conceptually elegant and simple method of deriving a peptide to inhibit a PPI from one of the interacting protein partners. Using the information available about the interaction interface an initial sequence for investigation can be chosen. High amounts of structural information allow for the extraction of secondary motifs from the interface. With limited structural information available the interaction between the proteins can be analysed using protein mutagenesis to understand the importance of regions or residues for the interactions. When only sequence information is available peptide arrays allow for the synthesis of a large library of peptides which can span large sections of protein an identify binding motifs. Similarly, conserved regions across different proteins or species indicate the importance of that section of the sequence and offer an excellent initial sequence. After a binding sequence has been identified the sequence can be analysed using an alanine scan to provide information about the key residues for the interaction and the sequence may be shortened to improve the drug-like properties of the peptide. To further increase the binding affinity of the selected peptides mutations may be introduced or conformational constraints (peptide stapling or macrocyclization) may be applied. Further modifications can be made if necessary to improve the cellular uptake by increasing the positive charge of the peptide or by conjugation to a cell-penetrating peptide. These methods have resulted in many examples of peptide inhibitors for a wide array of interactions.Recent developments in the identification of protein–protein interactions, and with structural information increasingly available through advances in protein structure prediction and cryo-EM, this method of identifying peptide inhibitors for biomacromolecular interactions has become more accessible and can allow for the exploration of previously unexplored interactions including protein–protein, protein–DNA, and protein–RNA interactions.
Data availability
All data is presented in the manuscript or the appropriate citation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SPvW acknowledges the University of East Anglia Faculty of Science for a Postgraduate Studentship. AMB acknowledges funding from the Royal Society (RGS\R1\201008) and the Academy of Medical Science (SBF006/1096).Notes and references
- L. Carro, Beilstein J. Org. Chem., 2018, 14, 2881–2896 CrossRef CAS PubMed
.
- R. Kahan, D. J. Worm, G. V. De Castro, S. Ng and A. Barnard, RSC Chem. Biol., 2021, 2, 387–409 RSC
- M. C. Smith and J. E. Gestwicki, Expert Rev. Mol. Med., 2012, 14, 1–24 CrossRef PubMed
- S. Beeckmans, Methods, 1999, 19, 278–305 CrossRef CAS PubMed
-
J. S. Lin and E. M. Lai, in Bacterial Protein Secretion Systems: Methods and Protocols, ed. L. Journet and E. Cascales, Springer, New York, NY, 2017, pp. 211–219 Search PubMed
- S. Fields and O. Song, Nature, 1989, 340, 245–246 CrossRef CAS PubMed
- A. Brückner, C. Polge, N. Lentze, D. Auerbach and U. Schlattner, Int. J. Mol. Sci., 2009, 10, 2763–2788 CrossRef PubMed
-
C. D. Curtis and A. M. Nardulli, in The Nuclear Receptor Superfamily: Methods and Protocols, ed. I. J. McEwan, Humana Press, Totowa, NJ, 2009, pp. 187–204 Search PubMed
- M. Bouhaddou, M. Eckhardt, Z. Z. Chi Naing, M. Kim, T. Ideker and N. J. Krogan, Curr. Opin. Genet. Dev., 2019, 54, 110–117 CrossRef CAS PubMed
- X. Benjin and L. Ling, Protein Sci., 2020, 29, 872–882 CrossRef PubMed
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed
- J. Gong, A. Chehrazi-Raffle, S. Reddi and R. Salgia, J. Immunother. Cancer, 2018, 6, 1–18 Search PubMed
- P. Chames, M. Van Regenmortel, E. Weiss and D. Baty, Br. J. Pharmacol., 2009, 157, 220–233 CrossRef CAS PubMed
- T. Böldicke, Antibodies, 2022, 11, 49 CrossRef PubMed
- K. Scheffzek, M. R. Ahmadian, W. Kabsch, L. Wiesmuller, A. Lautwein, F. Schmitz and A. Wittinghofer, Science, 1997, 227, 333–338 CrossRef PubMed
- J. M. Rojas, J. L. Oliva and E. Santos, Genes Cancer, 2011, 2, 298–305 CrossRef CAS PubMed
- A. Patgiri, K. K. Yadav, P. S. Arora and D. Bar-Sagi, Nat. Chem. Biol., 2011, 7, 585–587 CrossRef CAS PubMed
- E. S. Leshchiner, A. Parkhitko, G. H. Bird, J. Luccarelli, J. A. Bellairs, S. Escudero, K. Opoku-Nsiah, M. Godes, N. Perrimon and L. D. Walensky, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1761–1766 CrossRef CAS PubMed
- D. Dolfini and R. Mantovani, Cell Death Differ., 2013, 20, 676–685 CrossRef CAS PubMed
- S. Jeganathan, M. Wendt, S. Kiehstaller, D. Brancaccio, A. Kuepper, N. Pospiech, A. Carotenuto, E. Novellino, S. Hennig and T. N. Grossmann, Angew. Chem., Int. Ed., 2019, 58, 17351–17358 CrossRef CAS PubMed
- E. E. Gallagher, J. M. Song, A. Menon, L. D. Mishra, A. F. Chmiel and A. L. Garner, J. Med. Chem., 2019, 62, 4967–4978 CrossRef CAS PubMed
- P. Hart, P. Hommen, A. Noisier, A. Krzyzanowski, D. Schüler, A. T. Porfetye, M. Akbarzadeh, I. R. Vetter, H. Adihou and H. Waldmann, Angew. Chem., Int. Ed., 2021, 60, 1813–1820 CrossRef PubMed
- T. N. Grossmann, J. T.-H. Yeh, B. R. Bowman, Q. Chu, R. E. Moellering and G. L. Verdine, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17942 CrossRef CAS PubMed
- K. Takada, D. Zhu, G. H. Bird, K. Sukhdeo, J.-J. Zhao, M. Mani, M. Lemieux, D. E. Carrasco, J. Ryan, D. Horst, M. Fulciniti, N. C. Munshi, W. Xu, A. L. Kung, R. A. Shivdasani, L. D. Walensky and D. R. Carrasco, Sci. Transl. Med., 2012, 4, 148ra117 Search PubMed
- F. Findeisen, M. Campiglio, H. Jo, F. Abderemane-Ali, C. H. Rumpf, L. Pope, N. D. Rossen, B. E. Flucher, W. F. DeGrado and D. L. Minor, ACS Chem. Neurosci., 2017, 8, 1313–1326 CrossRef CAS PubMed
- I. De Paola, L. Pirone, M. Palmieri, N. Balasco, L. Esposito, L. Russo, D. Mazzà, L. D. Marcotullio, S. Di Gaetano, G. Malgieri, L. Vitagliano, E. Pedone and L. Zaccaro, PLoS One, 2015, 10, 1–21 Search PubMed
- D. C. Morgan, C. Morris, A. Mahindra, C. M. Blair, G. Tejeda, I. Herbert, M. L. Turnbull, G. Lieber, B. J. Willett, N. Logan, B. Smith, A. B. Tobin, D. Bhella, G. Baillie and A. G. Jamieson, Pept. Sci., 2021, 113, e24217 CrossRef CAS PubMed
- K. Nakayama, N. Nakayama, B. Davidson, J. J.-C. Sheu, N. Jinawath, A. Santillan, R. Salani, R. E. Bristow, P. J. Morin, R. J. Kurman, T.-L. Wang and I.-M. Shih, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18739–18744 CrossRef CAS PubMed
- T. Wu, P. He, W. Wu, Y. Chen and F. Lv, Bioorg. Chem., 2018, 80, 1–10 CrossRef CAS PubMed
- S. Mitra, K. W. Cheng and G. B. Mills, Semin. Cell Dev. Biol., 2011, 22, 57–68 CrossRef CAS PubMed
- J. Spiegel, P. M. Cromm, A. Itzen, R. S. Goody, T. N. Grossmann and H. Waldmann, Angew. Chem., Int. Ed., 2014, 53, 2498–2503 CrossRef CAS PubMed
- N. Normanno, A. De Luca, C. Bianco, L. Strizzi, M. Mancino, M. R. Maiello, A. Carotenuto, G. De Feo, F. Caponigro and D. S. Salomon, Gene, 2006, 366, 2–16 CrossRef CAS PubMed
- T. Mizuguchi, H. Uchimura, T. Kakizawa, T. Kimura, S. Yokoyama, Y. Kiso and K. Saito, Bioorg. Med. Chem. Lett., 2009, 19, 3279–3282 CrossRef CAS PubMed
- S. Shanker and M. F. Sanner, J. Chem. Inf. Model., 2023, 63, 3158–3170 CrossRef CAS PubMed
- C. Y. Lee, D. Hubrich, J. K. Varga, C. Schafer, M. Welzel, E. Schumbera, M. Djokic, J. M. Strom, J. Schonfeld, J. L. Geist, F. Polat, T. J. Gibson, C. I. K. Valsecchi, M. Kumar, O. Schueler-Furman and K. Luck, Mol. Syst. Biol., 2024, 20, 75–97 CrossRef PubMed
- L. Chang and A. Perez, Angew. Chem., Int. Ed., 2023, 62, e202213362 CrossRef CAS PubMed
- A. Savinov, S. Swanson, A. E. Keating and G.-W. Li, 2023 DOI:10.1101/2023.12.19.572389.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C.-C. Hung, M. O’Neill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Žemgulytė, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve, A. I. Cowen-Rivers, A. Cowie, M. Figurnov, F. B. Fuchs, H. Gladman, R. Jain, Y. A. Khan, C. M. R. Low, K. Perlin, A. Potapenko, P. Savy, S. Singh, A. Stecula, A. Thillaisundaram, C. Tong, S. Yakneen, E. D. Zhong, M. Zielinski, A. Žídek, V. Bapst, P. Kohli, M. Jaderberg, D. Hassabis and J. M. Jumper, Nature, 2024, 1–3 Search PubMed
- T. Clackson and J. A. Wells, Science, 1995, 267, 383–386 CrossRef CAS PubMed
- A. A. Bogan and K. S. Thorn, J. Mol. Biol., 1998, 280, 1–9 CrossRef CAS PubMed
- K. S. Thorn and A. A. Bogan, Bioinformatics, 2001, 17, 284–285 CrossRef CAS PubMed
- C. Dumas, M. Ouellette, J. Tovar, M. L. Cunningham, A. H. Fairlamb, S. Tamar, M. Olivier and B. Papadopoulou, EMBO J., 1997, 16, 2590–2598 CrossRef CAS PubMed
- M. A. Toro, P. A. Sánchez-Murcia, D. Moreno, M. Ruiz-Santaquiteria, J. F. Alzate, A. Negri, M. J. Camarasa, F. Gago, S. Velázquez and A. Jiménez-Ruiz, ChemBioChem, 2013, 14, 1212–1217 CrossRef CAS PubMed
- J. H. Trembley, G. Wang, G. Unger, J. Slaton and K. Ahmed, Cell. Mol. Life Sci., 2009, 66, 1858–1867 CrossRef CAS PubMed
- B. Laudet, C. Barette, V. Dulery, O. Renaudet, P. Dumy, A. Metz, R. Prudent, A. Deshiere, O. Dideberg, O. Filhol and C. Cochet, Biochem. J., 2007, 408, 363–373 CrossRef CAS PubMed
- J. Raaf, B. Guerra, I. Neundorf, B. Bopp, O.-G. Issinger, J. Jose, M. Pietsch and K. Niefind, ACS Chem. Biol., 2013, 8, 901–907 CrossRef CAS PubMed
- M. F. White, Curr. Opin. Genet. Dev., 1994, 4, 47–54 CrossRef CAS PubMed
- Y. Hao, C. Wang, B. Cao, B. M. Hirsch, J. Song, S. D. Markowitz, R. M. Ewing, D. Sedwick, L. Liu, W. Zheng and Z. Wang, Cancer Cell, 2013, 23, 583–593 CrossRef CAS PubMed
- J. R. Graff, B. W. Konicek, J. H. Carter and E. G. Marcusson, Cancer Res., 2008, 68, 631–634 CrossRef CAS PubMed
- D. Rousseau, A. C. Gingras, A. Pause and N. Sonenberg, Oncogene, 1996, 13, 2415–2420 CAS
- J. Marcotrigiano, A.-C. Gingras, N. Sonenberg and S. K. Burley, Mol. Cell, 1999, 3, 707–716 CrossRef CAS PubMed
- L. C. Szymczak, H.-Y. Kuo and M. Mrksich, Anal. Chem., 2018, 90, 266–282 CrossRef CAS PubMed
- H. Amartely, A. Iosub-Amir and A. Friedler, J. Visualized Exp., 2014, e52097 Search PubMed
- A. Mehle, B. Strack, P. Ancuta, C. Zhang, M. McPike and D. Gabuzda, J. Biol. Chem., 2004, 279, 7792–7798 CrossRef CAS PubMed
- T. H. Reingewertz, E. Britan-Rosich, S. Rotem-Bamberger, M. Viard, A. Jacobs, A. Miller, J. Y. Lee, J. Hwang, R. Blumenthal, M. Kotler and A. Friedler, Bioorg. Med. Chem., 2013, 21, 3523–3532 CrossRef CAS PubMed
- C. Katz, Y. Zaltsman-Amir, Y. Mostizky, N. Kollet, A. Gross and A. Friedler, J. Biol. Chem., 2012, 287, 15016–15023 CrossRef CAS PubMed
- R. Craigie, Future Virol., 2012, 7, 679–686 CrossRef CAS PubMed
- H. Y. Li, Z. Zawahir, L. D. Song, Y. Q. Long and N. Neamati, J. Med. Chem., 2006, 49, 4477–4486 CrossRef CAS PubMed
- C. L. Hagan, J. S. Wzorek and D. Kahne, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2011–2016 CrossRef CAS PubMed
- G. Neumayer, C. Belzil, O. J. Gruss and M. D. Nguyen, Cell. Mol. Life Sci., 2014, 71, 3027–3047 CrossRef CAS PubMed
- R. Bayliss, T. Sardon, I. Vernos and E. Conti, Mol. Cell, 2003, 12, 851–862 CrossRef CAS PubMed
- K. I. Tong, Y. Katoh, H. Kusunoki, K. Itoh, T. Tanaka and M. Yamamoto, Mol. Cell. Biol., 2006, 26, 2887–2900 CrossRef CAS PubMed
- S.-C. Lo, X. Li, M. T. Henzl, L. J. Beamer and M. Hannink, EMBO J., 2006, 25, 3605–3617 CrossRef CAS PubMed
- B. P. Dalrymple, K. Kongsuwan, G. Wijffels, N. E. Dixon and P. A. Jennings, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11627–11632 CrossRef CAS PubMed
- P. Wolff, V. Oliéric, J. P. Briand, O. Chaloin, A. Dejaegere, P. Dumas, E. Ennifar, G. Guichard, J. Wagner and D. Y. Burnouf, J. Med. Chem., 2011, 54, 4627–4637 CrossRef CAS PubMed
- M. M. A. Bisson and G. Groth, Plant Signaling Behav., 2011, 6, 164–166 CrossRef CAS PubMed
- M. M. A. Bisson and G. Groth, Mol. Plant, 2015, 8, 1165–1174 CrossRef CAS PubMed
- M. M. A. Bisson, M. Kessenbrock, L. Müller, A. Hofmann, F. Schmitz, S. M. Cristescu and G. Groth, Sci. Rep., 2016, 6, 30634 CrossRef CAS PubMed
- M. Kessenbrock, S. M. Klein, L. Müller, M. Hunsche, G. Noga and G. Groth, Front. Plant Sci., 2017, 8, 1–12 Search PubMed
- B. K. Yap, E. W. W. Leung, H. Yagi, C. A. Galea, S. Chhabra, D. K. Chalmers, S. E. Nicholson, P. E. Thompson and R. S. Norton, J. Med. Chem., 2014, 57, 7006–7015 CrossRef CAS PubMed
- Z. Kuang, R. S. Lewis, J. M. Curtis, Y. Zhan, B. M. Saunders, J. J. Babon, T. B. Kolesnik, A. Low, S. L. Masters, T. A. Willson, L. Kedzierski, S. Yao, E. Handman, R. S. Norton and S. E. Nicholson, J. Cell Biol., 2010, 190, 129–141 CrossRef CAS PubMed
- A. T. Nguyen and Y. Zhang, Genes Dev., 2011, 25, 1345–1358 CrossRef CAS PubMed
- C. Shen, S. Y. Jo, C. Liao, J. L. Hess and Z. Nikolovska-Coleska, J. Biol. Chem., 2013, 288, 30585–30596 CrossRef CAS PubMed
- R. Hancock, H. C. Bertrand, T. Tsujita, S. Naz, A. El-Bakry, J. Laoruchupong, J. D. Hayes and G. Wells, Free Radical Biol. Med., 2012, 52, 444–451 CrossRef CAS PubMed
- L. Chen, S. W. Chan, X. Q. Zhang, M. Walsh, C. J. Lim, W. Hong and H. Song, Genes Dev., 2010, 24, 290–300 CrossRef CAS PubMed
- Z. Zhang, Z. Lin, Z. Zhou, H. C. Shen, S. F. Yan, A. V. Mayweg, Z. Xu, N. Qin, J. C. Wong, Z. Zhang, Y. Rong, D. C. Fry and T. Hu, ACS Med. Chem. Lett., 2014, 5, 993–998 CrossRef CAS PubMed
- H. Jo, N. Meinhardt, Y. Wu, S. Kulkarni, X. Hu, K. E. Low, P. L. Davies, W. F. Degrado and D. C. Greenbaum, J. Am. Chem. Soc., 2012, 134, 17704–17713 CrossRef CAS PubMed
- Y. Cai, V. Kandula, R. Kosuru, X. Ye, M. G. Irwin and Z. Xia, Cell Cycle, 2017, 16, 1765–1773 CrossRef CAS PubMed
- R. Rai, Y. Chen, M. Lei and S. Chang, Nat. Commun., 2016, 7, 10881 CrossRef CAS PubMed
- X. Ran, L. Liu, C. Y. Yang, J. Lu, Y. Chen, M. Lei and S. Wang, J. Med. Chem., 2016, 59, 328–334 CrossRef CAS PubMed
- E. C. C. Cheung, L. Melanson-Drapeau, S. P. Cregan, J. L. Vanderluit, K. L. Ferguson, W. C. McIntosh, D. S. Park, S. A. L. Bennett and R. S. Slack, J. Neurosci., 2005, 25, 1324–1334 CrossRef CAS PubMed
- C. Candé, N. Vahsen, I. Kouranti, E. Schmitt, E. Daugas, C. Spahr, J. Luban, R. T. Kroemer, F. Giordanetto, C. Garrido, J. M. Penninger and G. Kroemer, Oncogene, 2004, 23, 1514–1521 CrossRef PubMed
- N. Doti, C. Reuther, P. L. Scognamiglio, A. M. Dolga, N. Plesnila, M. Ruvo and C. Culmsee, Cell Death Dis., 2014, 5, 1–13 Search PubMed
- B. Farina, M. Sturlese, F. Mascanzoni, A. Caporale, A. Monti, G. Di Sorbo, R. Fattorusso, M. Ruvo and N. Doti, Biochem. J., 2018, 475, 2377–2393 CrossRef CAS PubMed
- L. Russo, F. Mascanzoni, B. Farina, A. M. Dolga, A. Monti, A. Caporale, C. Culmsee, R. Fattorusso, M. Ruvo and N. Doti, J. Med. Chem., 2021, 64, 11445–11459 CrossRef CAS PubMed
- Y. Chen, D. Inoyama, A. N. T. Kong, L. J. Beamer and L. Hu, Chem. Biol. Drug Des., 2011, 78, 1014–1021 CrossRef CAS PubMed
- K. I. Tong, B. Padmanabhan, A. Kobayashi, C. Shang, Y. Hirotsu, S. Yokoyama and M. Yamamoto, Mol. Cell. Biol., 2007, 27, 7511–7521 CrossRef CAS PubMed
- J. Cheng, J. Zhou, L. Kong, H. Wang, Y. Zhang, X. Wang, G. Liu and Q. Chu, RSC Med. Chem., 2023, 14, 2496–2508 RSC
- Y. Xu, M. Huang, W. He, C. He, K. Chen, J. Hou, M. Huang, Y. Jiao, R. Liu, N. Zou, L. Liu and C. Li, Front. Cell Dev. Biol., 2022, 10, 1–13 Search PubMed
- D. Chen, S. Liu, W. Zhang and L. Sun, J. Pept. Sci., 2015, 21, 826–832 CrossRef CAS PubMed
- S. A. Kawamoto, A. D. Thompson, A. Coleska, Z. Nikolovska-Coleska, H. Yi and S. Wang, Biochemistry, 2009, 48, 9534–9541 CrossRef CAS PubMed
- J. Hochscherf, D. Lindenblatt, M. Steinkrüger, E. Yoo, Ö. Ulucan, S. Herzig, O. G. Issinger, V. Helms, C. Götz, I. Neundorf, K. Niefind and M. Pietsch, Anal. Biochem., 2015, 468, 4–14 CrossRef CAS PubMed
- H. L. Olsvik, T. Lamark, K. Takagi, K. B. Larsen, G. Evjen, A. Øvervatn, T. Mizushima and X. T. Johansen, J. Biol. Chem., 2015, 290, 29361–29374 CrossRef CAS PubMed
- R. A. Cerulli, L. Shehaj, H. Brown, J. Pace, Y. Mei and J. A. Kritzer, ChemBioChem, 2020, 21, 2777–2785 CrossRef CAS PubMed
- J. A. Miles, D. J. Yeo, P. Rowell, S. Rodriguez-marin, C. M. Pask, S. L. Warriner, A. T. Edwards and A. J. Wilson, Chem. Sci., 2016, 7, 3694–3702 RSC
- A. D. De Araujo, H. N. Hoang, W. M. Kok, F. Diness, P. Gupta, T. A. Hill, R. W. Driver, D. A. Price, S. Liras and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 6965–6969 CrossRef CAS PubMed
- Q. Chu, R. E. Moellering, G. J. Hilinski, Y.-W. Kim, T. N. Grossmann, J. T.-H. Yeh and G. L. Verdine, MedChemComm, 2015, 6, 111–119 RSC
- C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef CAS PubMed
- L. D. Walensky, K. Pitter, J. Morash, K. J. Oh, S. Barbuto, J. Fisher, E. Smith, G. L. Verdine and S. J. Korsmeyer, Mol. Cell, 2006, 24, 199–210 CrossRef CAS PubMed
- P. Polakis, Genes Dev., 2000, 14, 1837–1851 CrossRef CAS PubMed
- M. Mani, D. E. Carrasco, Y. Zhang, K. Takada, M. E. Gatt, J. Dutta-Simmons, H. Ikeda, F. Diaz-Griffero, V. Pena-Cruz, M. Bertagnolli, L. L. Myeroff, S. D. Markowitz, K. C. Anderson and D. R. Carrasco, Cancer Res., 2009, 69, 7577–7586 CrossRef CAS PubMed
- S. A. Kawamoto, A. D. Thompson, A. Coleska, Z. Nikolovska-Coleska, H. Yi and S. Wang, Biochemistry, 2009, 48, 9534–9541 CrossRef CAS PubMed
- K. Takada, D. Zhu, G. H. Bird, K. Sukhdeo, J. Zhao, M. Lemieux, D. E. Carrasco, J. Ryan, D. Horst, N. C. Munshi, W. Xu, A. L. Kung, R. A. Shivdasani, L. D. Walensky and D. R. Carrasco, Sci. Transl. Med., 2012, 4, 117–136 Search PubMed
- L. Chen, S. N. Willis, A. Wei, B. J. Smith, J. I. Fletcher, M. G. Hinds, P. M. Colman, C. L. Day, J. M. Adams and D. C. S. Huang, Mol. Cell, 2005, 17, 393–403 CrossRef CAS PubMed
- M. Certo, V. D. G. Moore, M. Nishino, G. Wei, S. Korsmeyer, S. A. Armstrong and A. Letai, Cancer Cell, 2006, 9, 351–365 CrossRef CAS PubMed
- J. L. Labelle, A. L. Kung, L. D. Walensky, S. G. Katz, G. H. Bird, E. Gavathiotis, M. L. Stewart, C. Lawrence, J. K. Fisher, M. Godes and K. Pitter, J. Clin. Invest., 2012, 122, 2018–2031 CrossRef CAS PubMed
- P. A. Sánchez-Murcia, M. Ruiz-Santaquiteria, M. A. Toro, H. De Lucio, M. Á. Jiménez, F. Gago, A. Jiménez-Ruiz, M. J. Camarasa and S. Velázquez, RSC Adv., 2015, 5, 55784–55794 RSC
- M. Ruiz-Santaquiteria, P. A. Sánchez-Murcia, M. A. Toro, H. de Lucio, K. J. Gutiérrez, S. de Castro, F. A. C. Carneiro, F. Gago, A. Jiménez-Ruiz, M. J. Camarasa and S. Velázquez, Eur. J. Med. Chem., 2017, 135, 49–59 CrossRef CAS PubMed
- M. Ruiz-Santaquiteria, S. de Castro, M. A. Toro, H. de Lucio, K. J. Gutiérrez, P. A. Sánchez-Murcia, M. Á. Jiménez, F. Gago, A. Jiménez-Ruiz, M. J. Camarasa and S. Velázquez, Eur. J. Med. Chem., 2018, 149, 238–247 CrossRef CAS PubMed
- K. Suzuki, Y. Takaoka and M. Ueda, RSC Chem. Biol., 2021, 2, 499–502 RSC
- W. Tang, C. Wang, Y. Zhou, J. Luo, T. Ye and B. Yang, Chem. Pap., 2020, 74, 2153–2161 Search PubMed
- Y. Lai, G. Fois, J. R. Flores, M. J. Tuvim, Q. Zhou, K. Yang, J. Leitz, J. Peters, Y. Zhang, R. A. Pfuetzner, L. Esquivies, P. Jones, M. Frick, B. F. Dickey and A. T. Brunger, Nature, 2022, 603, 949–956 CrossRef CAS PubMed
- H. Li, X. Chen, M. Wu, P. Song and X. Zhao, Chin. Chem. Lett., 2022, 33, 1254–1258 CrossRef CAS
- L. E. Hanold, K. Oruganty, N. T. Ton, A. M. Beedle, N. Kannan and E. J. Kennedy, PLoS One, 2015, 10, 1–18 CrossRef PubMed
- S. Jiao, H. Wang, Z. Shi, A. Dong, W. Zhang, X. Song, F. He, Y. Wang, Z. Zhang, W. Wang, X. Wang, T. Guo, P. Li, Y. Zhao, H. Ji, L. Zhang and Z. Zhou, Cancer Cell, 2014, 25, 166–180 CrossRef CAS PubMed
- W. Zheng, J. Lan, L. Feng, Z. Chen, S. Feng, Y. Gao, F. Ren and Y. Chen, Int. J. Pept. Res. Ther., 2021, 27, 923–930 CrossRef CAS
- J. Iegre, P. Brear, D. J. Baker, Y. S. Tan, E. L. Atkinson, H. F. Sore, D. H. O’Donovan, C. S. Verma, M. Hyvönen and D. R. Spring, Chem. Sci., 2019, 10, 5056–5063 RSC
- A. Monti, M. Sturlese, A. Caporale, J. D. A. Roger, F. Mascanzoni, M. Ruvo and N. Doti, Biochim. Biophys. Acta, Gen. Subj., 2020, 1864, 129717 CrossRef CAS PubMed
- M. Bloom, E. Evans and O. G. Mouritsen, Q. Rev. Biophys., 1991, 24, 293–397 CrossRef CAS PubMed
- T. Rezai, B. Yu, G. L. Millhauser, M. P. Jacobson and R. S. Lokey, J. Am. Chem. Soc., 2006, 128, 2510–2511 CrossRef CAS PubMed
- W. M. Hewitt, S. S. F. Leung, C. R. Pye, A. R. Ponkey, M. Bednarek, M. P. Jacobson and R. S. Lokey, J. Am. Chem. Soc., 2015, 137, 715–721 CrossRef CAS PubMed
- Y. Hosono, S. Uchida, M. Shinkai, C. E. Townsend, C. N. Kelly, M. R. Naylor, H.-W. Lee, K. Kanamitsu, M. Ishii, R. Ueki, T. Ueda, K. Takeuchi, M. Sugita, Y. Akiyama, S. R. Lokey, J. Morimoto and S. Sando, Nat. Commun., 2023, 14, 1416 CrossRef CAS PubMed
- M. Lindgren, M. Hällbrink, A. Prochiantz, Ü. Langel, M. Lindgren, M. Hällbrink, A. Prochiantz and Ü. Langel, Trends Pharmacol. Sci., 2000, 21, 99–103 CrossRef CAS PubMed
- F. Heitz, M. C. Morris and G. Divita, Br. J. Pharmacol., 2009, 157, 195–206 CrossRef CAS PubMed
- D. M. Copolovici, K. Langel, E. Eriste and Ü. Langel, ACS Nano, 2014, 8, 1972–1994 CrossRef CAS PubMed
- G. Guidotti, L. Brambilla and D. Rossi, Trends Pharmacol. Sci., 2017, 38, 406–424 CrossRef CAS PubMed
- P. M. Cromm, J. Spiegel, P. Küchler, L. Dietrich, J. Kriegesmann, M. Wendt, R. S. Goody, H. Waldmann and T. N. Grossmann, ACS Chem. Biol., 2016, 11, 2375–2382 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |