
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in the efficient synthesis of steroid natural products: emerging methods and strategies
Yu
Wang
and
Jinghan
Gui
 *
*
State Key Laboratory of Chemical Biology, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: guijh@sioc.ac.cn; Web: https://guigroup.sioc.ac.cn
First published on 11th June 2025
Abstract
Steroid natural products (SNPs) play an indispensable role in drug discovery owing to their remarkable structural features and biological activities. However, the inadequate amounts of steroid samples derived from natural sources has limited thorough assessment of SNP bioactivities. Accordingly, chemical synthesis of these compounds has become an important, practical way to obtain them in sufficient quantities. Chemists have been focusing on efficient synthesis of SNPs since the 1930s, and significant breakthroughs have been achieved in the past few decades. This review presents advances in this field over the past 20 years, highlighting key C–C bond formation and reorganization reactions in the construction of steroidal skeletons, as well as redox-relay events for the installation of complex oxidation states. We hope this review will serve as a timely reference to allow researchers to quickly learn about state-of-the-art achievements in SNP synthesis and will inspire the development of more powerful strategies for natural product synthesis.
 Yu Wang | Yu Wang received his BS degree from Yangzhou University in 2015, and his PhD degree from Shanghai Institute of Organic Chemistry (SIOC) in 2020 under the supervision of Prof. Jinghan Gui. From 2020 to 2021, he was a research associate in the same group. In 2021, he moved to Scripps Research and began his postdoctoral studies with Prof. Phil S. Baran. His research interests mainly focus on synthesis of bioactive complex natural products and invention of novel reagents for organic synthesis. |
 Jinghan Gui | Jinghan Gui received his BS degree from Anhui Normal University in 2007. After completing his PhD degree at Shanghai Institute of Organic Chemistry (SIOC) under the guidance of Prof. Weisheng Tian in 2012, he went on to pursue his postdoctoral research with Prof. Phil S. Baran at Scripps Research in February 2013. In March 2016, he moved back to SIOC to begin his independent research career. His research focuses on the efficient synthesis of biologically active steroid and terpenoid natural products, and the development of new synthetic methodologies. |
1. Introduction
Because of the scaffold diversity, structural complexity, and varied biological activities of natural products, they have long been targets pursued by synthetic and medicinal chemists.1 Natural products and their analogues are sources of therapeutic agents for various life-threatening diseases, including cancers, cardiovascular and cerebrovascular diseases, gastrointestinal diseases, central nervous system disorders, and immunological diseases.2 Natural product synthesis remains among the most exciting and challenging areas of organic chemistry.3 Newly developed synthetic methods and strategies have enabled significant progress in natural product synthesis, and the complex molecular architectures of natural products have motivated chemists to develop powerful new transformations, thus advancing organic chemistry.4Steroid natural products (SNPs) are found in a wide range of animals, plants, and microorganisms. SNPs and their analogues are valuable chemical resources with applications in medicine, agrochemistry, and the cosmetics industry, with examples including dexamethasone, brassinolide, and β-sitosterol. As reported in a chart by the Njardarson group at the University of Arizona, steroid-related compounds accounted for 13 of the top 200 small-molecule drugs by retail sales in 2024.5 Therefore, research on steroid chemistry has attracted significant attention in the field of organic chemistry, which can be traced back to the 18th century. In 1758, French physician François Poulletier de la Salle was the first to isolate crystalline cholesterol from gallstones.6 However, the correct structure of cholesterol was not elucidated until 1933. Notably, extensive research on the structural elucidation, biosynthesis, chemical synthesis, and molecular regulation of steroids has led to the awarding of several Nobel Prizes. For example, German chemist Heinrich Wieland received the Nobel Prize in Chemistry in 1927 for his investigations into the constitution of bile acids and related compounds. Later, American chemist Robert Burns Woodward was awarded the Nobel Prize in Chemistry in 1965 for his outstanding contributions to the field of organic synthesis, including the total syntheses of cholesterol and cortisone.7
Classical steroids are characterized by a 6/6/6/5 tetracyclic skeleton, and their isolation, structural elucidation, chemical synthesis, and biogenetic origins8 have been widely studied, starting in the twentieth century (Fig. 1(A)). Rearranged steroids, which are biosynthetically generated from classical steroids through one or more C–C bond migrations and scissions (Fig. 1(B)), have garnered increasing attention from synthetic chemists over the last two decades.9 Despite the structural complexity of SNPs, the past 20 years have witnessed tremendous achievements in their efficient synthesis, and thus a systematic review of these achievements is of significant importance. We hope this review will not only be a useful resource for researchers to quickly learn about the state-of-the-art achievements in SNP synthesis but also will stimulate the development of additional novel and efficient synthetic methods and strategies. Owing to limited space, some steroid and triterpenoid syntheses, such as List's synthesis of estrone,10 Lopchuk's synthesis of withanolides,11 our group's syntheses of aspersteroids12 and phomarol,13 Li's synthesis of phomarol,14 Dong's synthesis of phainanoid A,15 Micalizio's synthesis of octanorcucurbitacin B,16 as well as those that have been included in previous reviews,9,17 are not covered here.
 | ||
| Fig. 1 Selected steroid natural products (SNPs) with classical and rearranged skeletons. (A) Selected steroids that possess classical 6/6/6/5-tetracyclic skeletons; (B) Selected steroids that possess diverse rearranged skeletons. | ||
As summarized in Scheme 1, this review comprises two sections: one for SNPs constructed via convergent strategies, and the other for SNPs constructed via semisynthetic strategies. Convergent strategies typically require diastereoselective preparation of fragments, followed by convergent union of the fragments and intramolecular cyclization to build the polycyclic framework.18 Depending on where the key fragment-coupling and cyclization reactions take place (i.e., the A/B, B, C, or D ring), the first section of the review is divided into four subsections (Scheme 1, left). Semisynthetic strategies feature the utilization of inexpensive commercially available steroids, such as pregnenolone ($0.32 per gram) as starting materials (Scheme 1, right).19 Although these strategies benefit from the utilization of steroidal materials that have tetracyclic frameworks and some of the necessary stereogenic centers, successful implementation of these strategies necessitates precise installation of the requisite oxidation states within the aliphatic core skeleton and controllable skeletal reorganization to quickly forge the rearranged skeleton.17d,20
 | ||
| Scheme 1 Convergent and semisynthetic strategies for the synthesis of SNPs. | ||
2. Synthesis of SNPs via convergent strategies
Convergent strategies have been widely applied to the synthesis of SNPs and have served as crucial tools for increasing synthetic efficiency. Convergent strategies typically involve assembly of two fragments, followed by cyclization and further functionalization to access the target molecules. Because the coupling fragments can be highly functionalized, these strategies often lead to synthetic routes that are shorter than those that rely on linear strategies, wherein the rings are constructed in a stepwise manner. Therefore, reactions that can couple fragments chemo-, regio-, and diastereoselectively become highly appealing. In this section, examples of such reactions are categorized and summarized on the basis of the specific rings (A/B, B, C, or D) that are formed during the key steps (Scheme 2).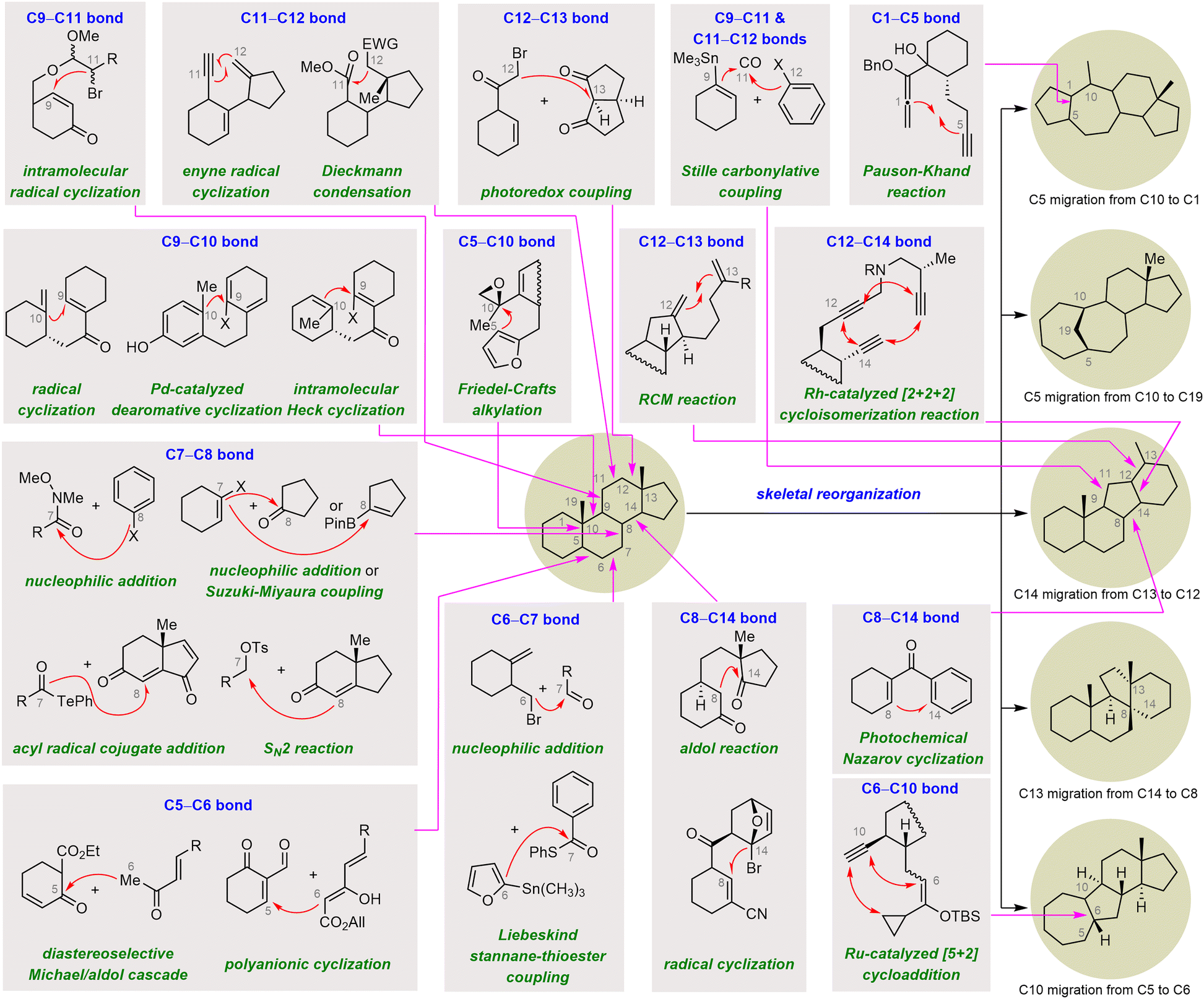 | ||
| Scheme 2 Strategic bond-forming reactions for efficient synthesis of SNPs. | ||
2.1. Convergent assembly of the A/B ring system
Strategic bond disconnection at the A/B ring system via a cycloaddition can markedly simplify the polycyclic skeleton of an SNP, leading to a simple intermediate that possesses the C/D ring system and can be derived from known chiral pool molecules, such as (+)-Hajos–Parrish ketone and sitolactone.21 Li's synthesis of bufospirostenin A via an alkoxyallene-yne Pauson–Khand reaction to construct the 5/7 bicyclic A/B ring system,22 and their synthesis of bufogargarizin A by means of a Ru-catalyzed [5+2] cycloaddition to install the 7/5 bicyclic system, have been selected as representative examples (Scheme 3).23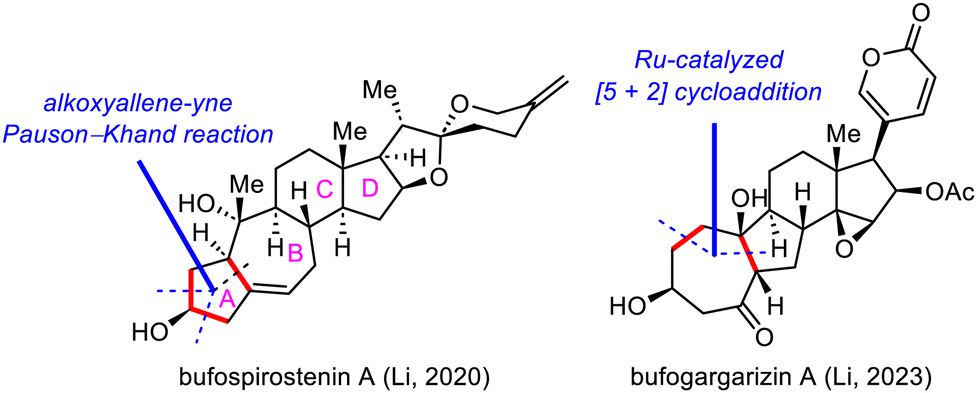 | ||
| Scheme 3 Retrosynthetic disconnections at the A/B ring system. | ||
Li's total synthesis commenced with the preparation of alkyne 17 in four steps from 16 (Scheme 4). A nucleophilic addition reaction between the ketone of 17 and an allene lithium reagent derived from 18 and n-butyllithium (nBuLi) delivered alkoxyallene-yne 19, setting the stage for the key intramolecular Pauson–Khand reaction.27 Extensive optimization experiments revealed that [RhCl(CO)2]2 was the optimal catalyst, and the reaction was run under a balloon of CO, giving rise to dienone 20, which has the desired A/B ring system. This compound was transformed into ketone 21 in seven steps, and the E ring of 22 was established in another five steps. Finally, the spiroketal moiety was installed via reaction of the lactone with a lithium reagent derived from iodide 23, followed by acid-promoted spiroketalization, completing the total synthesis of bufospirostenin A.
 | ||
| Scheme 4 Li's synthesis of bufospirostenin A (6). | ||
Central to Li's strategy was a ruthenium-catalyzed [5+2] cycloaddition reaction (Scheme 5).23 The synthesis started with the preparation of alkyne 25 in nine steps from sitolactone (24). Treatment of 25 with trimethylsilyl trifluoromethanesulfonate (TMSOTf) generated silyl enol ether 26. Crude 26 was subjected to the desired [5+2] cycloaddition reaction catalyzed by CpRu(CH3CN)3PF6 to yield 27, which possesses the tetracyclic skeleton of bufogargarizin A.29 Mukaiyama hydration of 27 produced alcohol 28, which was converted to enol triflate 29 in eight steps. Suzuki coupling of 29 with 30 provided 31, which was elaborated to bufogargarizin A (7) in seven steps.
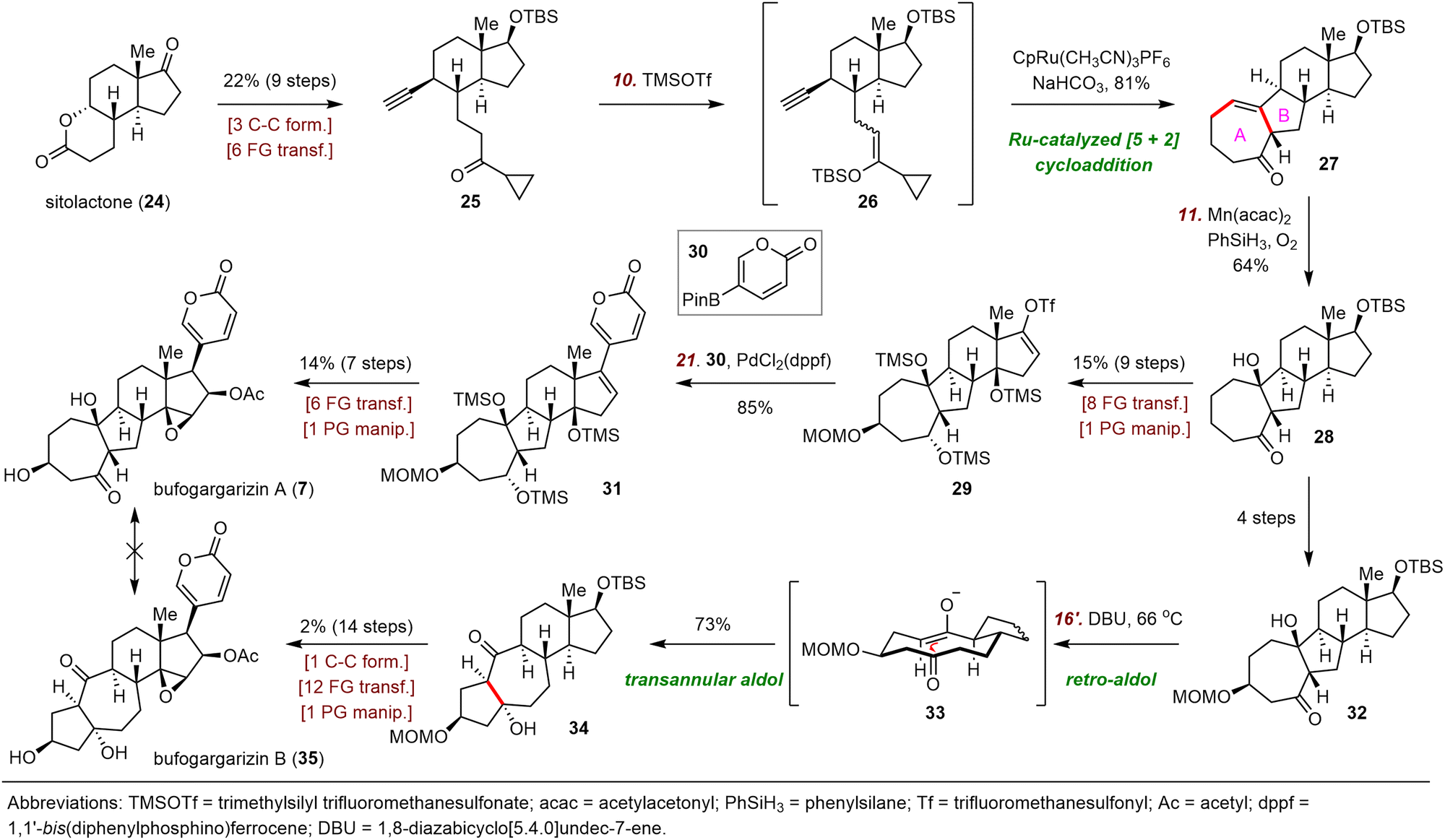 | ||
| Scheme 5 Li's syntheses of bufogargarizins A (7) and B (35). | ||
Li and co-workers then attempted a direct, bioinspired preparation of bufogargarizin B (35) from bufogargarizin A but were unable to accomplish this ideal transformation, despite many attempts; in most cases, undesired byproducts or decomposition of 7 were observed. Eventually, these investigators identified alcohol 32, which was prepared in four steps from intermediate 28, as a suitable intermediate for accessing bufogargarizin B via a retro-aldol/transannular aldol cascade. Specifically, heating a solution of 32 in tetrahydrofuran (THF) in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as a base triggered the desired retro-aldol reaction to afford anionic intermediate 33, which then underwent a transannular aldol reaction to produce ketone 34 in 73% yield. Notably, the entire sequence (32 → 34) was completely diastereoselective, an outcome that was supported by density functional theory calculations. The total synthesis of bufogargarizin B was then accomplished in fourteen steps from 34 by means of a route similar to that used for bufogargarizin A. Li et al. were unable to directly transform bufogargarizin B into bufogargarizin A.
2.2. Convergent assembly of the B ring
Strategic bond disconnection at the B ring leads to A-ring and C/D-ring fragments of similar structural complexity, thereby maximizing synthetic convergency and shortening the longest linear sequence of steps. Therefore, this disconnection is the one most often used by synthetic chemists. In this subsection of the review, representative work on building the B ring and associated stereogenic centers is described, and key bond-forming reactions are highlighted (Scheme 6).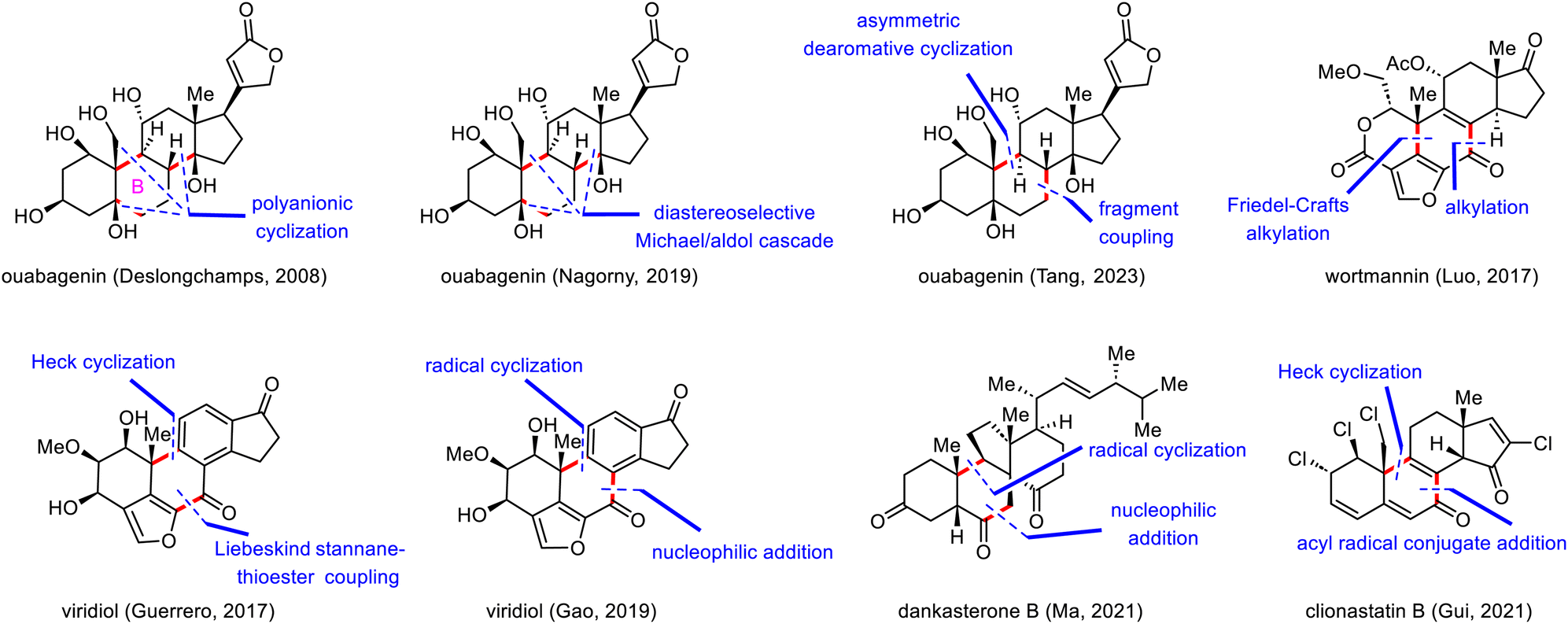 | ||
| Scheme 6 Retrosynthetic disconnections at the B ring. | ||
In 2008, Deslongchamps and co-workers completed the first total synthesis of ouabagenin by employing a polyanionic cyclization as the key skeletal construction step (Scheme 7).33 Their synthesis began with a diastereoselective double Michael addition reaction between Nazarov reagent 36 and chiral cyclohexenone 37 in the presence of Cs2CO3,37 which was followed by decarboxylation with Pd(PPh3)4 to provide aldehyde 38 with the desired cis-A/B ring junction. From intermediate 39, which was prepared from 38 in two steps, the C ring of ouabagenin was forged by means of a classical aldol condensation in the presence of potassium bis(trimethylsilyl)amide (KHMDS), a reaction that delivered key tetracyclic intermediate 40 in 83% yield. Finally, the butenolide side ring was installed via treatment of α-hydroxyketone 42 with Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCO, affording ouabagenin (1) after hydrolysis of the four ester groups.
CCO, affording ouabagenin (1) after hydrolysis of the four ester groups.
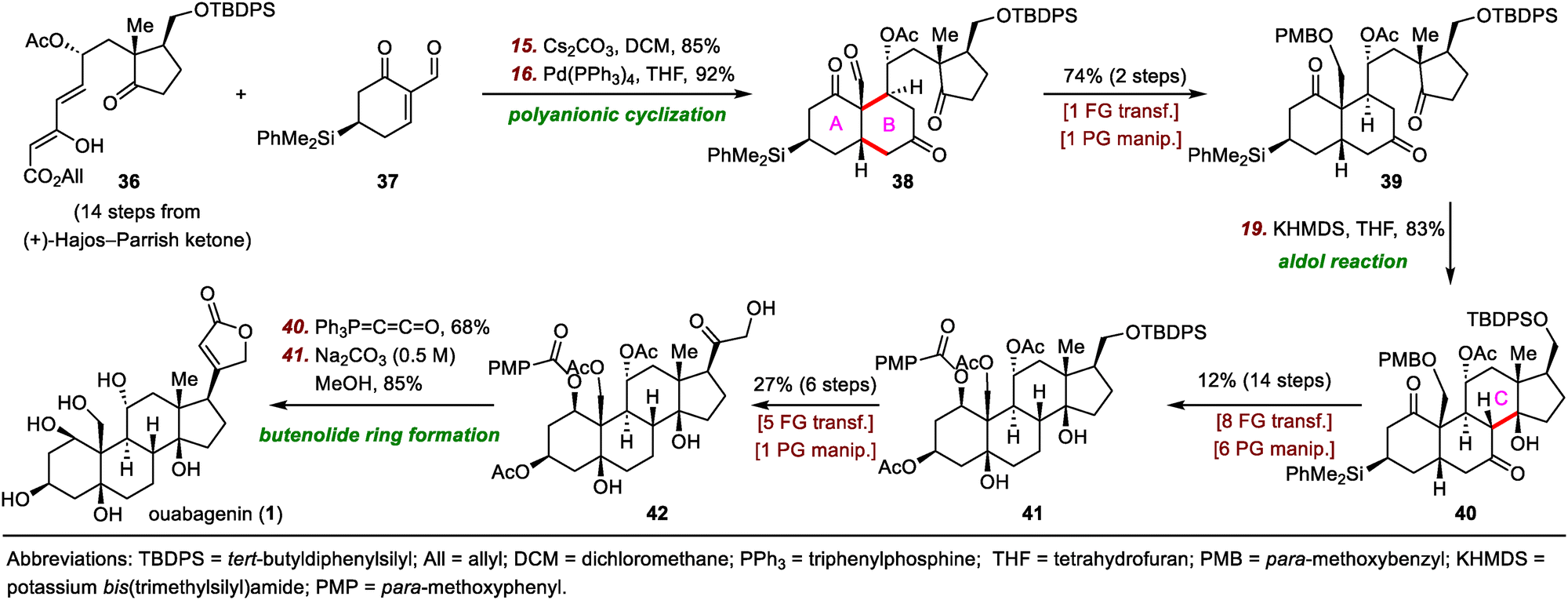 | ||
| Scheme 7 Deslongchamps's synthesis of ouabagenin (1). | ||
As shown in Scheme 8, the synthesis of ouabagenin commenced with a copper(II)-catalyzed diastereoselective Michael/aldol cascade reaction between enone 43 and β-ketoester 44, affording dienone 45 in 45% yield.35 After extensive optimization of the reaction parameters, β-hydroxy ketone 45 was eventually isomerized into 46 by treatment with sodium bis(trimethylsilyl)amide (NaHMDS) in toluene, possibly by means of a retro-aldol/intramolecular aldol cascade reaction. Reduction of 46 with diisobutylaluminium hydride (DIBAL-H) led to an allylic alcohol intermediate, which, upon treatment with aqueous HCO2H, spontaneously isomerized to produce dienone 47 in 72% yield. Finally, ouabagenin was synthesized in an additional sixteen steps via intermediate 48.
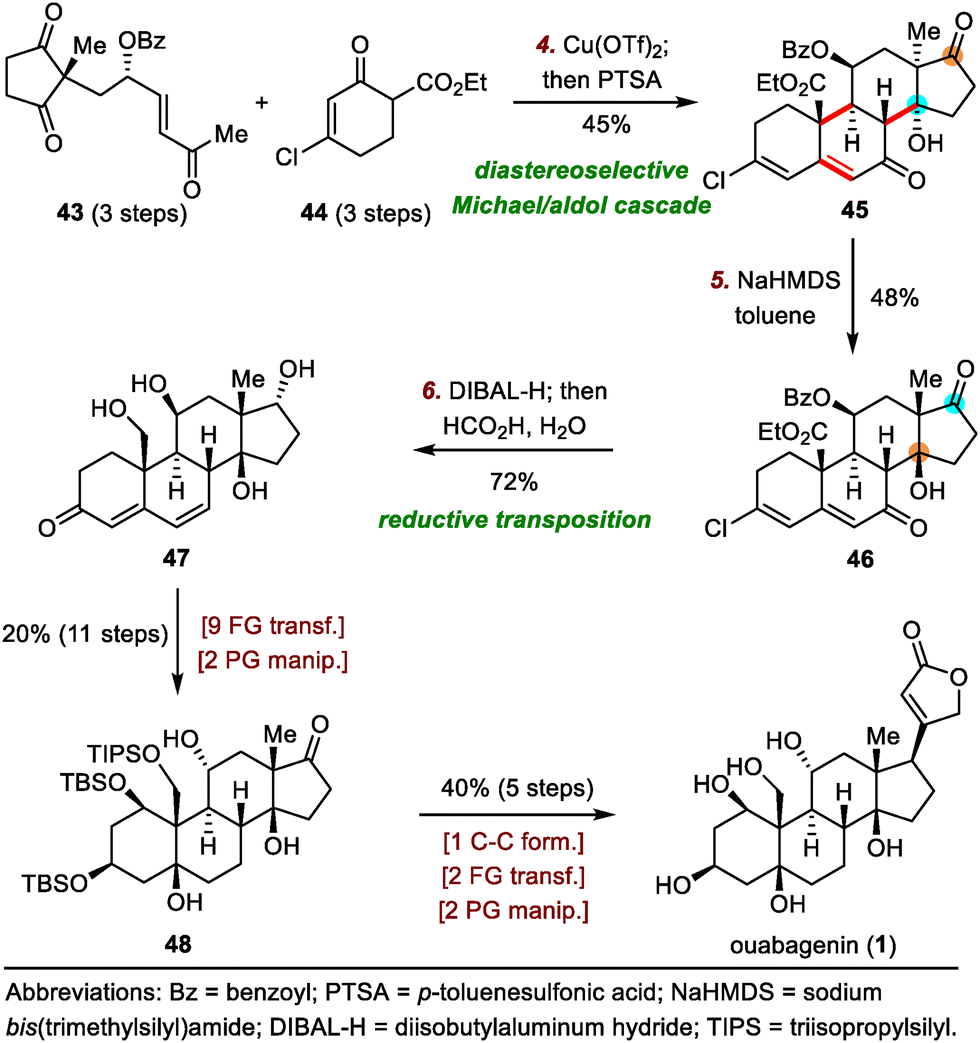 | ||
| Scheme 8 Nagorny's synthesis of ouabagenin (1). | ||
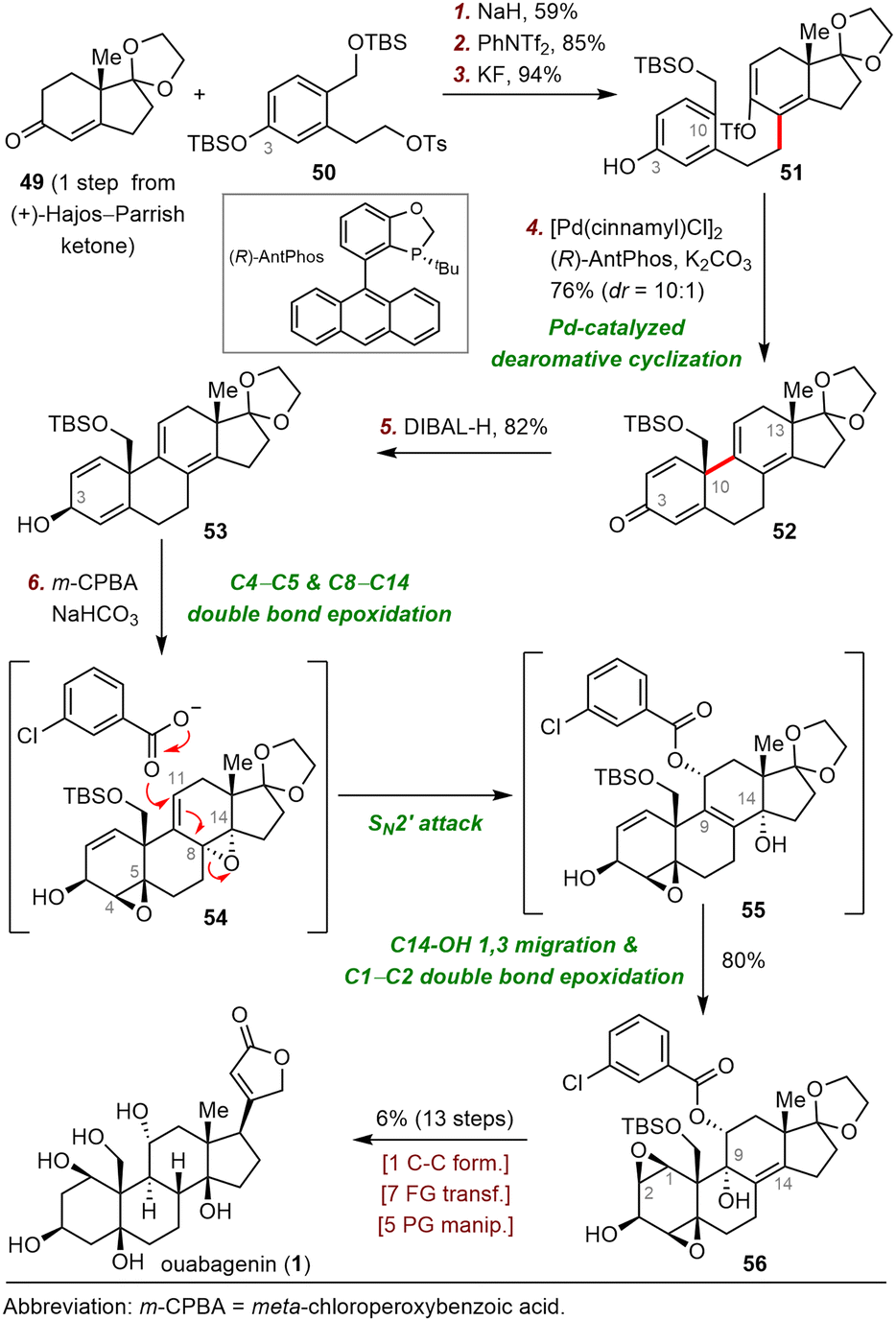 | ||
| Scheme 9 Tang's synthesis of ouabagenin (1). | ||
The synthesis began with an SN2 reaction of enone 49 with tosylate 50 to afford a coupling product (not shown) that was subjected to triflation and selective removal of the C3 tert-butyl(dimethyl)silyl (TBS) group to yield phenol 51, which was the precursor for the dearomative cyclization reaction. Pleasingly, palladium-catalyzed enantioselective dearomative cyclization of 51 in the presence of (R)-AntPhos as a ligand smoothly gave dienone 52 in 76% yield with a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio (dr) at C10. In contrast, replacing (R)-AntPhos with (rac)-AntPhos led to a poor dr (1.5:1), indicating that the stereochemistry at C13 had little influence on the stereochemistry at C10.
:1 diastereomeric ratio (dr) at C10. In contrast, replacing (R)-AntPhos with (rac)-AntPhos led to a poor dr (1.5:1), indicating that the stereochemistry at C13 had little influence on the stereochemistry at C10.
With compound 52, which possesses the desired tetracyclic framework, in hand, Tang and colleagues had reached the stage for installing multiple hydroxyl groups onto the skeleton. To this end, reduction of the C3-ketone of 52 with DIBAL-H delivered alcohol 53, which was epoxidized with m-chloroperoxybenzoic acid (m-CPBA) to produce diepoxide 56 in 80% yield. After characterizing two key intermediates during the above-described process, Tang et al. proposed the following plausible pathway: (1) diastereoselective epoxidation of the C4–C5 and C8–C14 double bonds gives 54, (2) SN2′ attack at C11 of 54 produces allylic alcohol 55, and (3) 1,3-migration of the allylic alcohol from C14 to C9 and subsequent C3-OH-directed epoxidation of the C1–C2 double bond generate 56. Ouabagenin (1) was synthesized from 56 in thirteen steps.
Wortmannin belongs to the furanosteroid subclass of steroids and was originally isolated from Penicillium wortmannii.44 This natural product possesses a highly reactive [6,6,5]-furanocyclohexadienone lactone moiety and two all-carbon quaternary stereogenic centers, one at C10 and another at C13. The highly oxidized skeleton of wortmannin, coupled with its biological importance, has gained tremendous attention from the synthetic organic community, which culminated in two synthetic routes by Shibasaki45 and one route by Luo.46 In 1996, Shibasaki and co-workers accomplished a semisynthesis of wortmannin from hydrocortisone.45a In 2002, they developed a second strategy which involved the use of an intramolecular Heck reaction to construct the B ring and the synthetically challenging C10 quaternary stereogenic center.45b Using the second strategy, these investigators accomplished a formal enantioselective synthesis of wortmannin in 2005.45c
Luo's 2017 synthesis featured convergent assembly of the B ring via a palladium-catalyzed cascade reaction and an intramolecular Friedel–Crafts alkylation to forge the C7–C8 and C5–C10 bonds, respectively (Scheme 10).46 The synthesis began with a coupling reaction between carboxylic acid 57 and propargyl carbonate 58 in the presence of Pd2(dba)3 (dba, dibenzylideneacetone) via a π-propargyl-palladium species to afford furan 59. In contrast, direct alkylation of (+)-Hajos–Parrish ketone with a halofuran electrophile resulted in double alkylation and required a lengthy preparation of the electrophilic reagent. Furan 59 was transformed into epoxide 60 in six steps, setting the stage for the second key bond-forming reaction. Specifically, the use of hexafluoro-2-propanol as a solvent and Ph4PBF4 as an additive facilitated the desired Friedel–Crafts alkylation at the β-position of the furan with the epoxide to give a tetracyclic intermediate (not shown), which then underwent desilylation to afford 61 in 47% yield over two steps. Compound 61 was elaborated to wortmannin (2) in nine steps.
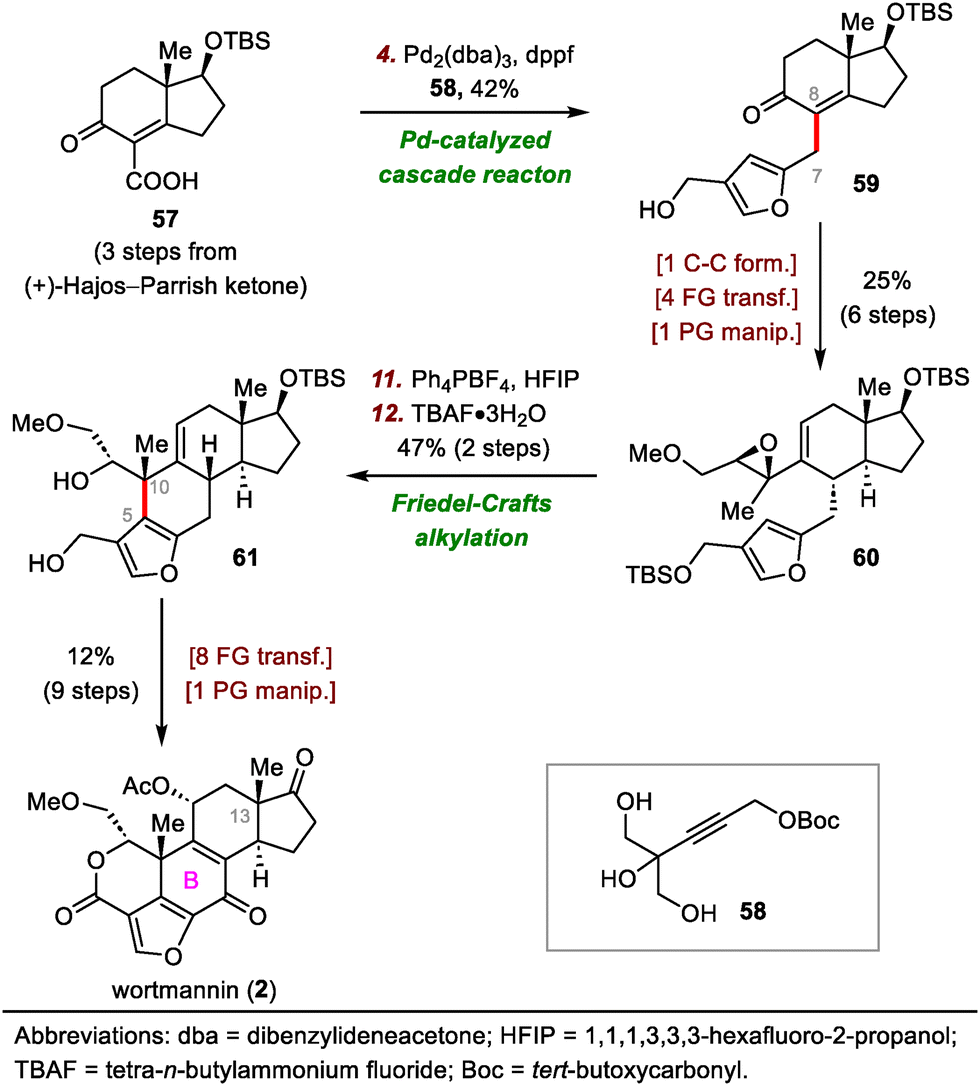 | ||
| Scheme 10 Luo's synthesis of wortmannin (2). | ||
To date, three impressive total syntheses of viridin and viridiol have been reported, one each by the groups of Sorensen,52 Guerrero,53 and Gao.54 In 2004, Sorensen and co-workers disclosed the first asymmetric total syntheses of viridin and viridiol, which were accomplished by means of a rhodium-catalyzed cyclotrimerization to form the tetrasubstituted phenyl ring (C ring) and a thermal electrocyclic rearrangement to establish the B ring.
Guerrero's synthetic strategy was based mainly on bond disconnection at the central B ring (Scheme 11).53 The synthesis commenced with a Liebeskind stannane-thioester coupling reaction between furan 62 and indanone 63, furnishing diketone 64 in 83% yield. Products resulting from oxidative addition of the aryl triflate were not observed. Ketone 64 underwent a highly enantioselective palladium-catalyzed intramolecular Heck reaction to forge the B ring, giving rise to alkene 65 in 75% yield with high enantioselectivity (>99% enantiomeric excess).55 Overall, these key C–C bond forming reactions allowed for quick construction of the pentacyclic skeleton of viridin and viridiol in only two steps from simple building blocks. Elaboration of 65 to viridiol (3) required seven steps. Finally, oxidation of viridiol (3) with catalysis by (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) furnished viridin (67) in 46% yield.
 | ||
| Scheme 11 Guerrero's syntheses of viridin (67) and viridiol (3). | ||
:1 dr. A two-step sequence transformed 69 to ester 70. This compound was then converted to the corresponding Weinreb amide, which underwent nucleophilic addition with a lithium reagent derived from bromide 71 and tert-butyllithium (tBuLi) to furnish ketone 72 in 71% yield over two steps.
 | ||
| Scheme 12 Gao's syntheses of viridin (67) and viridiol (3). | ||
Next, treatment of alkene 72 with Co(Salen)Cl (73) in the presence of phenylsilane (PhSiH3) resulted in a radical cyclization reaction that furnished desired tetracyclic ketone 75.56 The cyclization may have proceeded via radical intermediate 74, which led to desired stereochemistry at C10 via conformational control. Ketone 75 was then transformed to viridin (67) in seven steps, and selective reduction of the C3-ketone of viridin with sodium borohydride (NaBH4) uneventfully provided viridiol (3) in 68% yield.
In 2022, Heretsch reported the first syntheses of dankasterones A and B and periconiastone A, which were accomplished by means of a radical framework reconstruction strategy.60 Shortly after Heretsch's semisynthesis, Ma and co-workers reported their total syntheses of these abeo-steroids, wherein radical cyclization mediated by metal-hydride hydrogen atom transfer was employed to construct the cis-decalin framework (Scheme 13).61
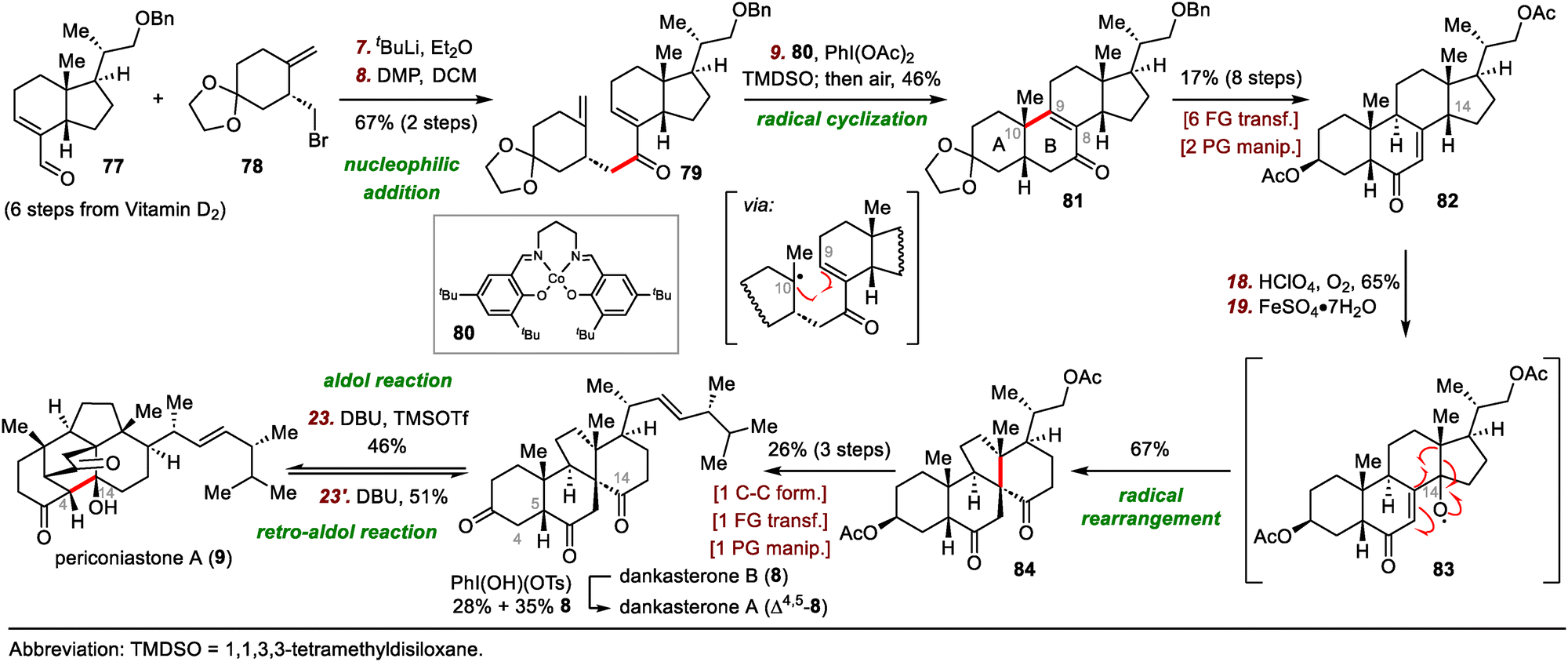 | ||
| Scheme 13 Ma's syntheses of dankasterones A (Δ4,5-8) and B (8) and periconiastone A (9). | ||
Ma's syntheses began with the union of chiral fragments 77 and 78via nucleophilic addition and Dess–Martin periodinane oxidation of the resulting secondary alcohol, generating enone 79 in 67% yield over two steps. After extensive optimization of various reaction parameters, tetracyclic compound 81, which has the desired cis-fused A/B ring system, was obtained in 46% yield by cycloisomerization of 79. A cobalt catalyst was found to be superior to iron and manganese catalysts:62 Fe(dpm)3 (dpm, 2,2,6,6-tetramethylheptane-3,5-dionate) delivered the desired Giese-type cyclization product (not shown) but in only 33% yield with reduction of the C8–C9 bond, and all attempts with manganese catalysts failed to give any useful products.
Compound 81 was transformed to enone 82 in eight steps, setting the stage for the pivotal radical rearrangement to build the 13(14 → 8)abeo-steroid skeleton. Specifically, treatment of enone 82 with O2 in the presence of perchloric acid (HClO4) led to the formation of a C14 hydroperoxide intermediate, which underwent radical rearrangement to afford desired product 84 in 67% yield, possibly via radical intermediate 83. Finally, spirocycle 84 was converted to dankasterone B (8) in three steps. Selective dehydrogenation of 8 at C4–C5 delivered dankasterone A (Δ4,5-8). Furthermore, biomimetic interconversion of 8 and periconiastone A (9) through intramolecular aldol and retro-aldol reactions was achieved with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU).
 | ||
| Scheme 14 Our group's syntheses of clionastatins A (91) and B (4). | ||
Our group's syntheses started with the preparation of donor 86 for the acyl radical conjugate addition reaction. To this end, an Ireland–Claisen rearrangement of chiral allylic acetate 85 with KHMDS and trimethylsilyl chloride afforded an 80% yield of a carboxylic acid intermediate, which was converted to acyl telluride 86 in 79% yield. Upon treatment with triethyl borane (Et3B) and air, 86 smoothly underwent an acyl radical conjugate addition reaction with enone 87,66 affording a triketone intermediate that was transformed to enol triflate 88 in 50% yield over two steps. Unfortunately, direct Heck cyclization67 of 88 failed to provide any cyclized product; triketone 89 was the only product (75% yield). Formation of 89 could be attributed to the extremely high acidity of C14–H, which is located at the α-position relative to the ketone at C15. This problem was avoided by subjecting 88 to a Luche reduction to give a mixture of diols, which underwent the desired Heck cyclization reaction to furnish enone 90 (46% yield over two steps) after in situ oxidation of the cyclized diols with Dess–Martin periodinane (DMP).
With tetracyclic enone 90 in hand, we adjusted the oxidation states and installed the alkyl chloride and vinyl chloride functional groups. Specifically, clionastatin A (91) was obtained from 90 in nine steps. Selective chlorination of 91 at C16 furnished clionastatin B (4).
2.3. Convergent assembly of the C ring
Like disconnection at the B ring (Scheme 6), disconnection at the C ring leads to two fragments of comparable structural complexity (the A/B ring system and the D ring), and this disconnection has also been widely recognized as an efficient strategy for steroid synthesis (Scheme 15). In this subsection, representative recent synthetic work on building the C ring and associated stereogenic centers via various bond-forming reactions will be highlighted. | ||
| Scheme 15 Retrosynthetic disconnections at the C ring. | ||
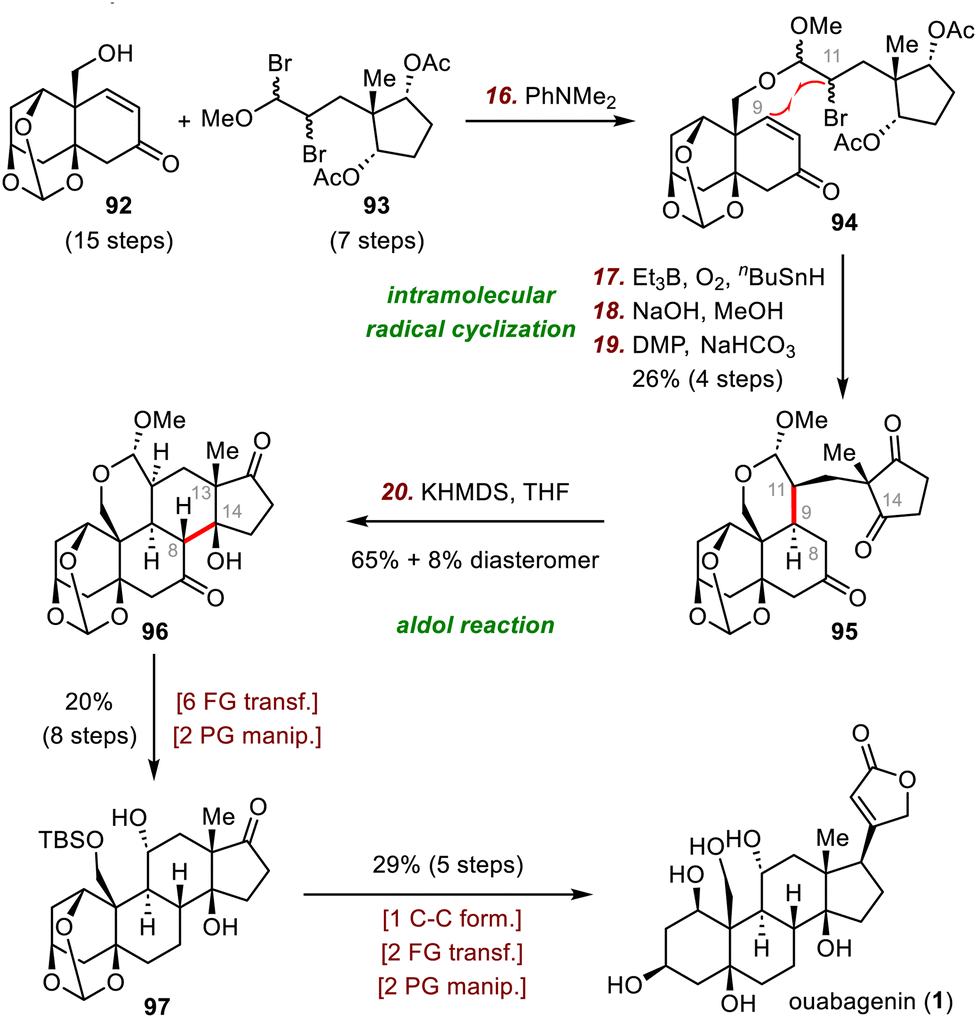 | ||
| Scheme 16 Inoue's synthesis of ouabagenin (1). | ||
The Inoue group's synthesis of ouabagenin began with the joining of fragments 92 and 93via nucleophilic substitution to furnish acetal 94. Upon treatment of 94 with Et3B and tributyltin hydride (n-Bu3SnH) in the presence of O2, a secondary radical generated from the bromide engaged in the desired 6-exo radical cyclization to form the key C9–C11 bond. Subsequent hydrolysis of the acetates and DMP oxidation of the resulting alcohols gave triketone 95 in 26% yield over four steps. Next, an intramolecular aldol reaction between C8 and C14 of 95 in the presence of KHMDS as the base afforded 96 (65% yield), which has the desired tetracyclic skeleton, along with an 8% yield of the diastereomer with the opposite configurations at C13 and C14. Further elaboration (eight steps) afforded diol 97, which was transformed to ouabagenin (1) in five steps.
In 1972, Imhof and co-workers reported a semisynthesis of batrachotoxinin A,72 and the first total synthesis of this molecule was achieved by the Kishi group in 1998.73 Subsequent total syntheses of batrachotoxin and batrachotoxinin A were reported by the groups of Du Bois,74 Luo,75 and Inoue.76
Du Bois's syntheses of batrachotoxinin A and batrachotoxin commenced with a nucleophilic addition reaction between enone 99 and the vinyl lithium species (generated by reaction of vinyl bromide 98 with tBuLi) to furnish enyne 100 (65% yield), which has the key C8–C14 bond (Scheme 17). Enyne 100 was transformed to 101 in two steps, setting the stage for a radical cascade cyclization to install the C ring and the all-carbon quaternary stereocenter at C13. To this end, treatment of 101 with Et3B and n-Bu3SnH in the presence of O2 produced a vinyl radical intermediate (not shown), which underwent successive 6-endo-trig and 5-exo-dig radical cyclizations followed by 1,4-hydrogen atom transfer of vinyl radical intermediate 102 to afford 103 in 75% yield. This unprecedented enyne radical cyclization afforded the tetracyclic skeleton of batrachotoxin in only four steps from fragments 98 and 99.
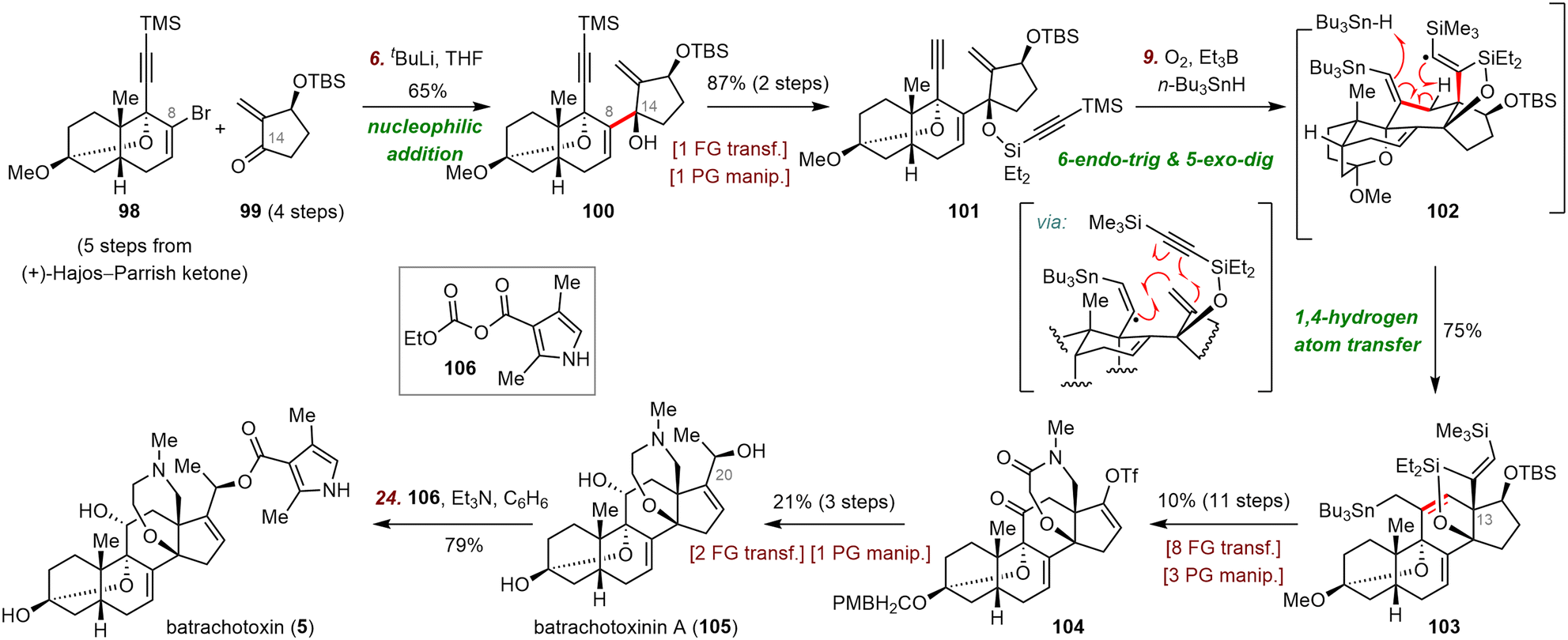 | ||
| Scheme 17 Du Bois's syntheses of batrachotoxinin A (105) and batrachotoxin (5). | ||
Subsequently, a series of redox manipulations furnished batrachotoxin and batrachotoxinin A. Specifically, elaboration of 103 led to vinyl triflate 104, which was converted to batrachotoxinin A (105) in three steps. Esterification of batrachotoxinin A with mixed anhydride 106 furnished batrachotoxin (5) in 79% yield.
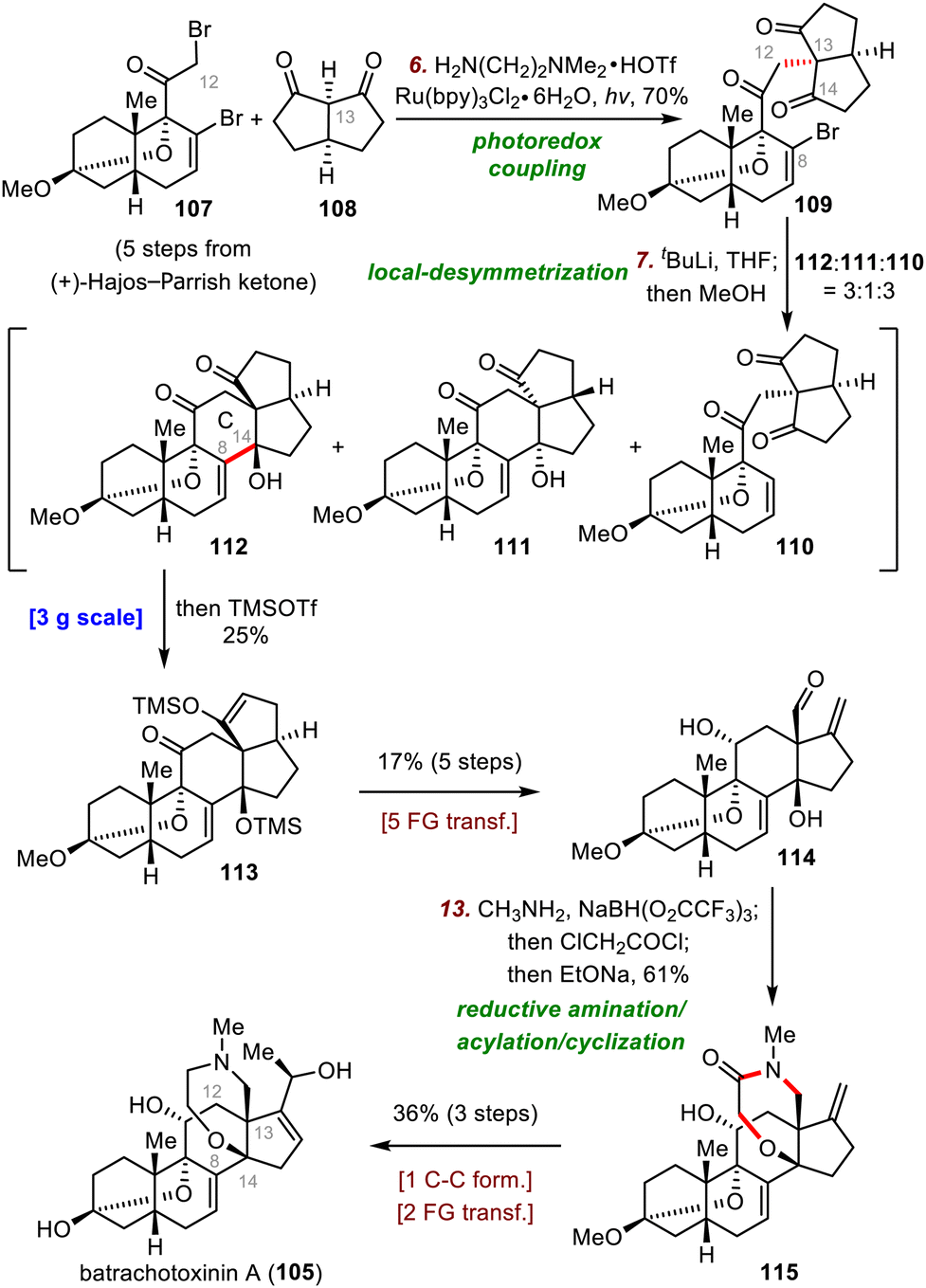 | ||
| Scheme 18 Luo's synthesis of batrachotoxinin A (105). | ||
A powerful photoredox alkylation reaction that was first developed by the MacMillan group for asymmetric photoalkylation of aldehydes77 and later improved by the Luo group for asymmetric α-photoalkylation of β-ketocarbonyls78 was found to be exceptionally useful for photoredox coupling of bromide 107 and diketone 108 to furnish vinyl bromide 109 in 70% yield. In contrast, all attempts to construct the C12–C13 bond via traditional SN2 reactions failed. Subsequently, closure of the C ring was pursued by means of a local-desymmetrization nucleophilic addition reaction between C8 and C14. Specifically, treatment of 109 with tBuLi at −98 °C gave a 3:1:3 mixture of desired alcohol 112, diastereomer 111, and debromination product 110, and in situ silylation of desired product 112 gave silyl enol ether 113 in 25% yield. Although the yield of the desired cyclized product was modest, the reaction could be easily performed on a 3 g scale, providing enough material for the following transformations. 113 was converted to aldehyde 114 in five steps, and then a one-pot reductive amination/acylation/cyclization of 114 was employed to build the homomorpholinamide ring: 115 was obtained in 61% yield, thereby forming the required ring system. Finally, 115 was converted to batrachotoxinin A (105) in three steps.
 | ||
| Scheme 19 Inoue's syntheses of batrachotoxinin A (105) and batrachotoxin (5). | ||
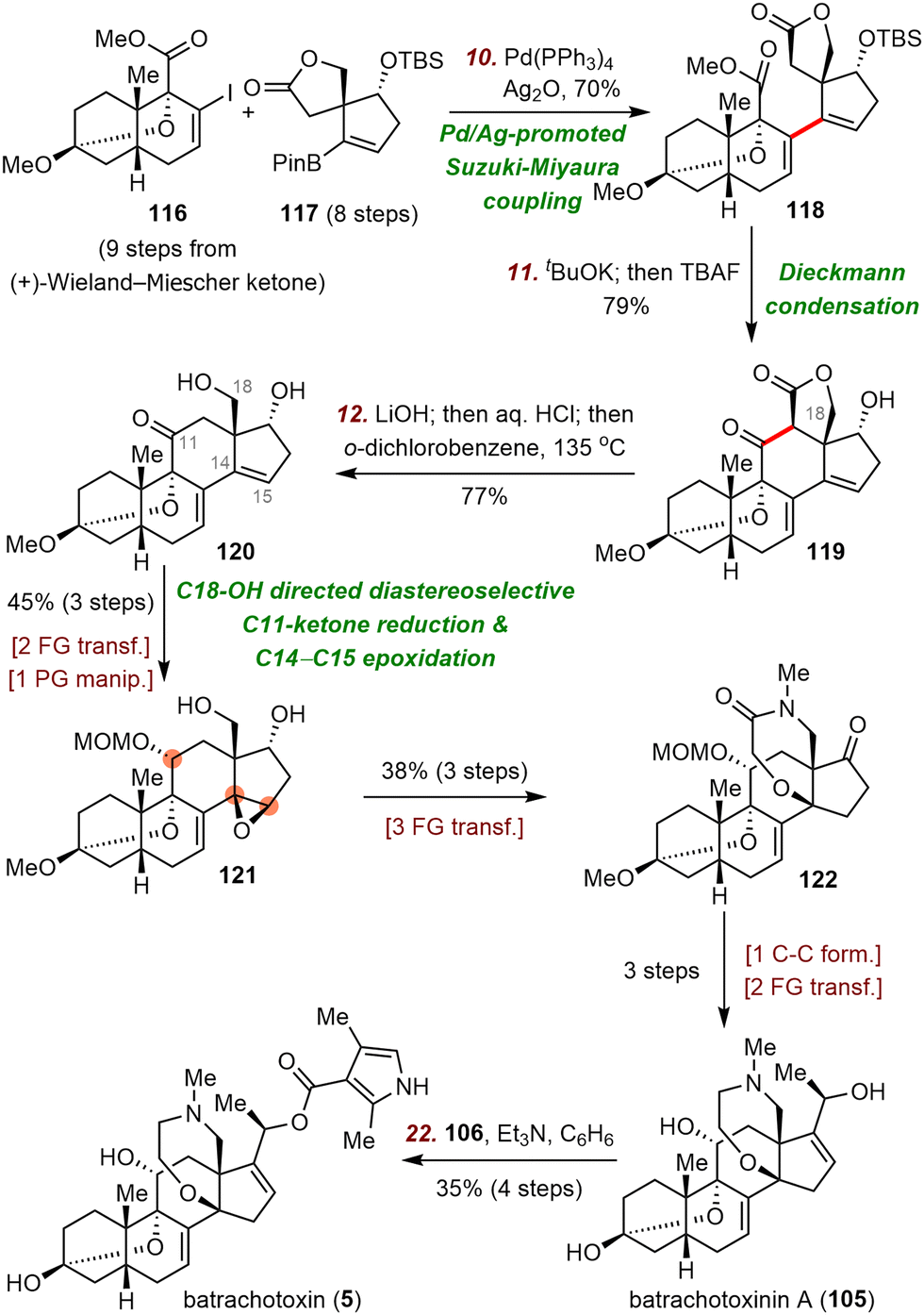
First, Inoue et al. used a Suzuki–Miyaura coupling reaction79 to connect two chiral fragments, vinyl iodide 116 and pinacol boronate 117, delivering ester 118 in 70% yield. Subsequent potassium tert-butoxide (tBuOK)-promoted intramolecular Dieckmann condensation and one-pot desilylation led to lactone 119 in 79% yield. Saponification and subsequent decarboxylation at high temperature released the C18-alcohol, giving rise to diol 120. This compound underwent C18-alcohol-directed diastereoselective C11-ketone reduction and C14–C15 epoxidation to deliver 121 in three steps. Alcohol 121 was transformed to batrachotoxinin A (105) in six steps, and esterification of batrachotoxinin A gave batrachotoxin (5).
 | ||
| Scheme 20 Luo's syntheses of Veratrum alkaloids. | ||
First, chiral furan 123, accessed from (+)-Hajos–Parrish ketone in six steps, was oxidized by 1O2, and pyridine-catalyzed isomerization of the resulting electron-deficient cis olefin and Pinnick oxidation of the aldehyde intermediate furnished carboxylic acid 124 in 87% yield over two steps.84 Conversion of 124 into the corresponding anhydride was followed by amidation with free amine 126 (prepared from 125 in six steps) to afford amide 127. The amide was heated to reflux in THF to promote the intramolecular Diels–Alder reaction, which furnished bromide 128 in 92% yield. Moving forward, the crucial C8–C14 bond was established by means of an intramolecular radical cyclization, affording cyanide 129 in 89% yield after in situ reduction of the C11 ketone. In this way, the hexacyclic carbon framework was constructed in only ten steps. With sufficient quantities of 129 in hand, Luo et al. divergently synthesized Veratrum alkaloids zygadenine (12), the alkamine of veramadine A (130), germine (131), and protoverine (132) through judicious application of a series of redox manipulations.85
The synthesis of cyclopamine by Gao and co-workers began with preparation of compound 135, the precursor for a photoinduced excited-state Nazarov cyclization (Scheme 21).93 To this end, nucleophilic addition of an aryl lithium species (generated by a lithium/bromide exchange reaction between 133 and tBuLi) to aldehyde 134 gave a secondary alcohol, which was oxidized to deliver enone 135 in 52% yield over two steps. The subsequent Nazarov cyclization proceeded smoothly upon irradiation of 135 with UV light (366 nm),96 generating tetracyclic intermediate 137 in 67% yield, possibly via disrotatory electrocyclization of 136. Intermediate 137 was transformed into veratramine (13) in two steps. Veratramine served as the starting material for a relay synthesis of cyclopamine (14). Specifically, a four-step sequence converted 13 to epoxide 138, which underwent acid-induced cyclization to afford tertiary alcohol 139. Finally, 139 was elaborated to cyclopamine (14) via dehydration and deprotection in two steps.
 | ||
| Scheme 21 Gao's syntheses of veratramine (13) and cyclopamine (14). | ||
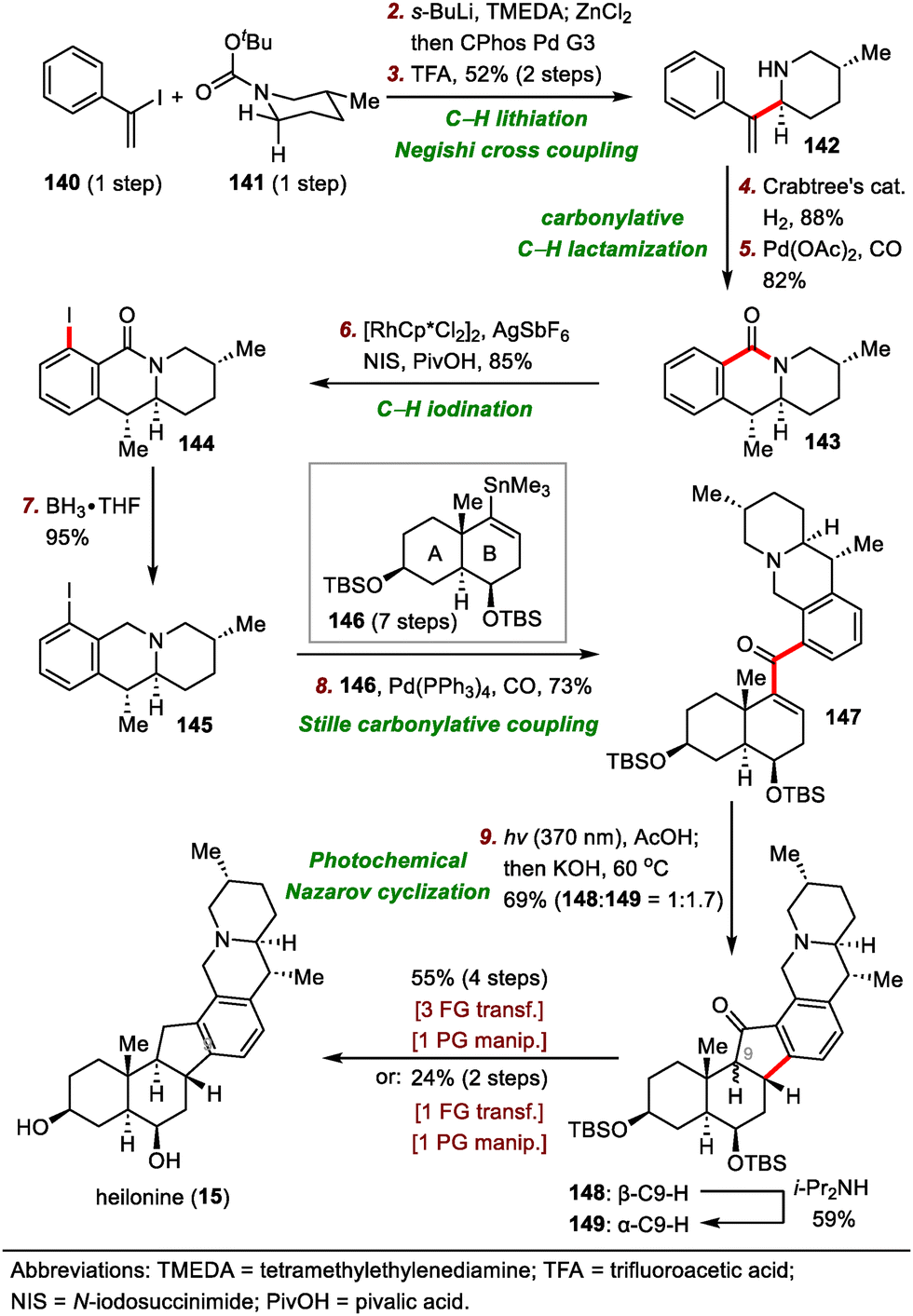
The synthesis began with a tert-butyloxycarbonyl (Boc)–directed C–H lithiation of 141 followed by transmetalation with ZnCl2 to generate an organozinc reagent (Scheme 22), which was coupled with vinyl iodide 140via a Negishi cross-coupling to furnish chiral piperidine 142 in two steps after removal of the Boc group. Hydrogenation of the exo-methylene group of 142 using Crabtree's catalyst was followed by free amine–directed carbonylative C–H lactamization to generate lactam 143. The newly formed lactam moiety then served as a directing group for a rhodium-catalyzed C–H iodination of 143 to install an ortho iodide atom, giving iodide 144 in 85% yield. Reduction of the lactam moiety of 144 produced piperidine 145 in 95% yield, which underwent palladium-catalyzed Stille carbonylative coupling with A/B ring system 146 (prepared in seven steps from Wieland–Miescher ketone) to deliver ketone 147 in 73% yield, setting the stage for the Nazarov cyclization to build the hexacyclic framework. Of note, reduction of the carbonyl group of the lactam to the methylene group before cross-coupling was essential to get a high yield, probably because of the unfavorable steric effects of the carbonyl group.
 | ||
| Scheme 22 Dai's synthesis of heilonine (15). | ||
After extensive optimization of reaction conditions for the Nazarov cyclization, irradiation of 147 with UV light (370 nm) in the presence of AcOH was found to lead to a mixture of diastereomers that could be epimerized at C9 in situ with KOH to produce a 1.7:1 mixture of desired cyclized product 149 and undesired product 148 in a total yield of 69%. Notably, 148 could be isolated and resubjected to the epimerization conditions to furnish 149 in 59% yield. Finally, 149 was converted to heilonine (15) in either two or four steps.
2.4. Convergent assembly of the D ring
Retrosynthetic bond disconnection at the D ring has not been explored as thoroughly as disconnection at the B and C rings (Scheme 23). This subsection will cover Rawal's synthesis of heilonine (15) by means of an intramolecular [2+2+2] cycloisomerization to install the central tetrasubstituted benzene moiety98 and Baran's synthesis of cyclopamine (14) via a nucleophilic addition and late-stage ring-closing metathesis (RCM) reaction to build the central cyclohexene motif.92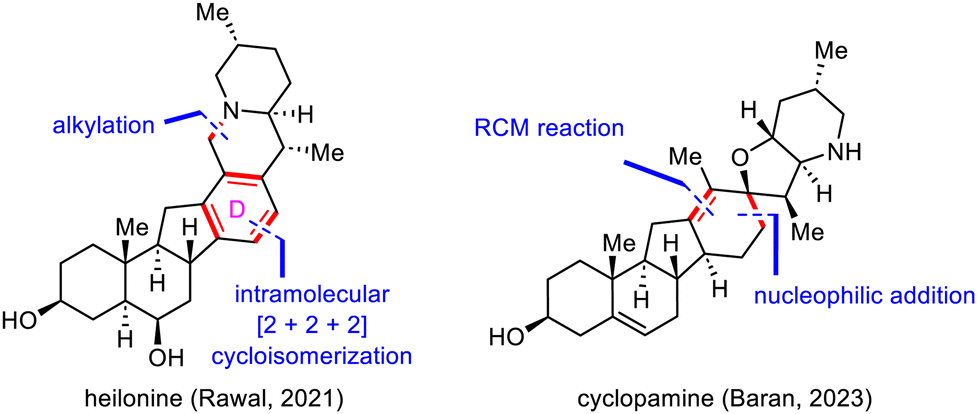 | ||
| Scheme 23 Retrosynthetic disconnection at the D ring. | ||
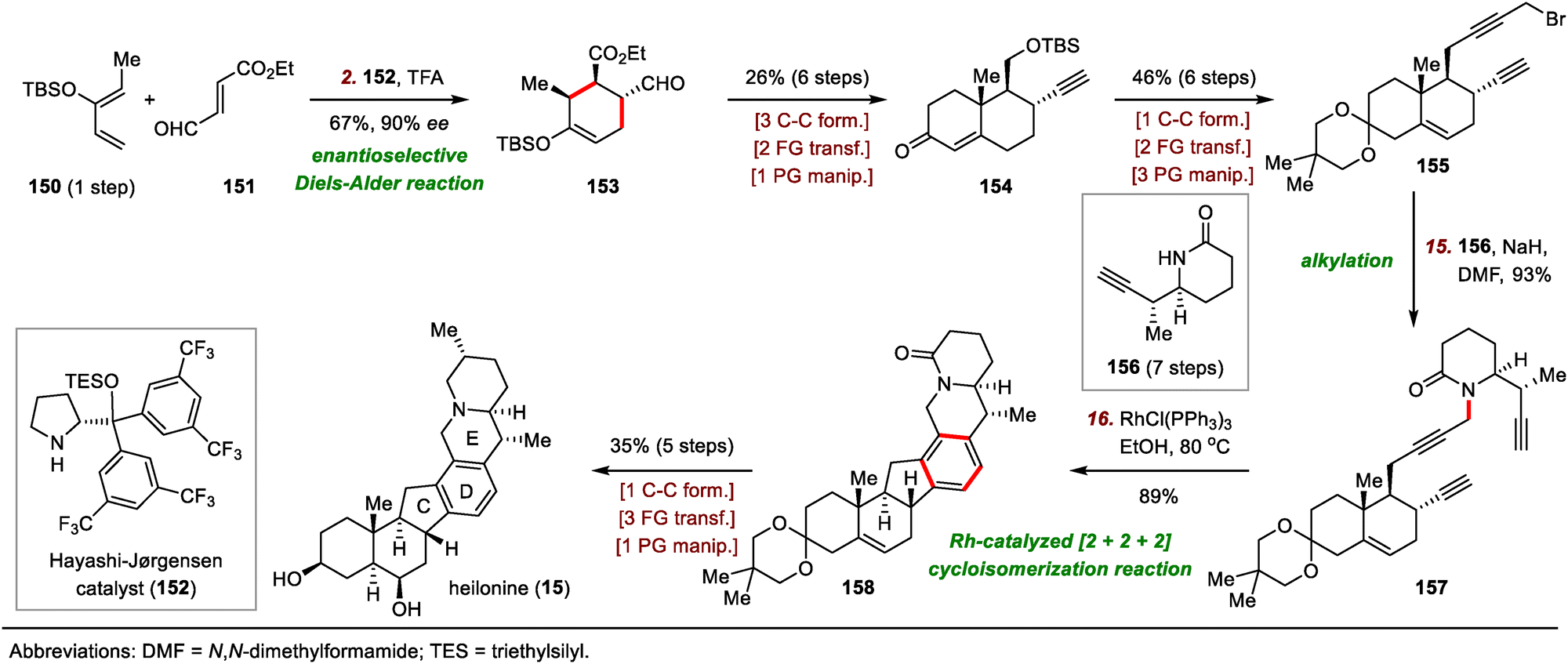 | ||
| Scheme 24 Rawal's synthesis of heilonine (15). | ||
 | ||
| Scheme 25 Baran's synthesis of cyclopamine (14). | ||
3. Synthesis of SNPs via semisynthetic strategies
Because convergent synthesis of SNPs offers flexibility in bond disconnection and easy preinstallation of various functional groups on the coupling fragments, convergent strategies have been widely pursued, as exemplified above. However, semisynthetic strategies, which generally utilize inexpensive commercially available steroids as starting materials, have their own advantages: (1) allowing for concise syntheses by making full use of the existing 6/6/6/5 tetracyclic ring system and the stereogenic centers of the starting steroids, (2) facilitating the discovery of novel synthetic transformations of the rigid framework of steroids, and (3) providing experimental evidence for proposed pathways for SNP biosynthesis. This section focuses mainly on efficient syntheses of SNPs by semisynthetic strategies. In general, these strategies pose two key challenges: (1) site-selective oxidation of the C(sp3)–H bonds of the steroid skeletons to install the desired functional groups at specific positions and (2) skeletal reorganization of the tetracyclic skeletons of classical steroids to access the core frameworks of rearranged steroids. Accordingly, this section is divided into two subsections, one covering the redox-relay strategy and one covering the skeletal reorganization strategy.3.1. Redox-relay strategy
Redox and stereochemical relays (Scheme 26), defined by Baran and co-workers as the transfer of redox and stereochemical information from one site to another within a molecular framework, are powerful tools for the semisynthesis of highly oxidized SNPs.32 This strategy relies mainly on the directing effects of existing hydroxyl groups and on conformational control to achieve the desired regio- and stereoselectivities. In this subsection, the syntheses of ouabagenin by Baran et al.32 and clionastatins A and B by Zhang et al.65 are used to demonstrate how the oxidation states of steroids can be efficiently installed by means of a redox-relay strategy.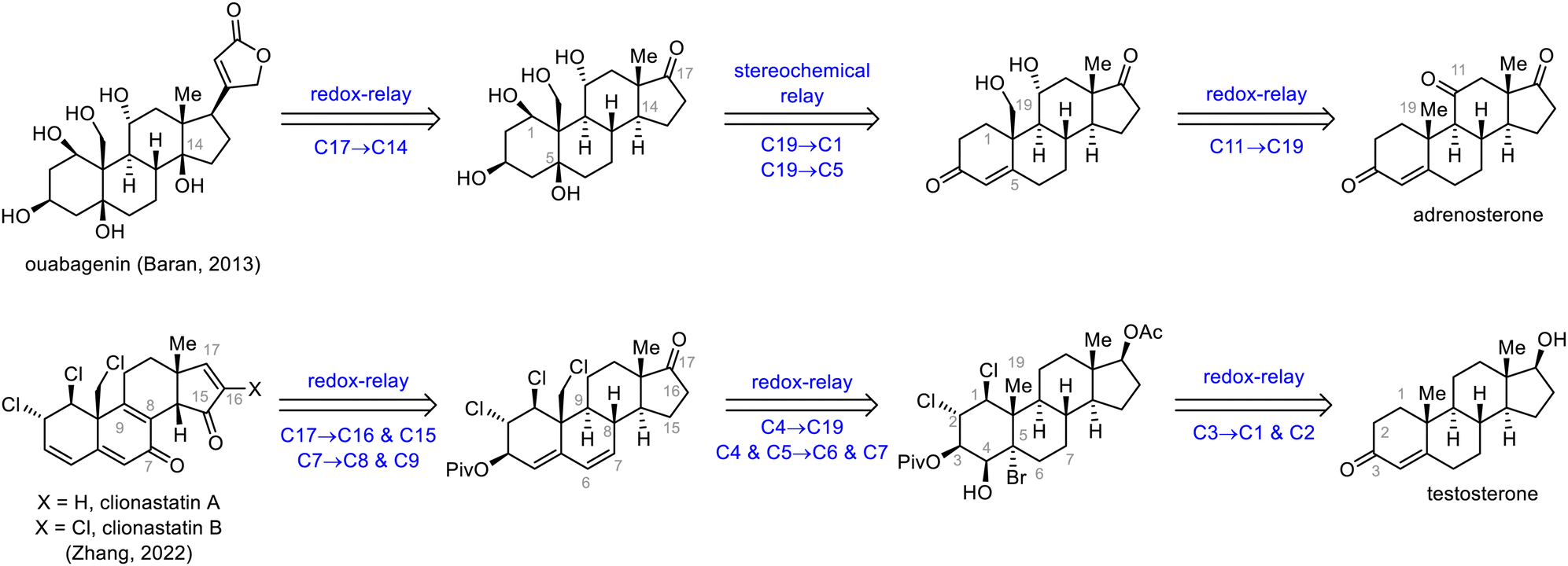 | ||
| Scheme 26 Redox-relay strategies for the synthesis of highly oxidized SNPs. | ||
 | ||
| Scheme 27 Baran's synthesis of ouabagenin (1). | ||
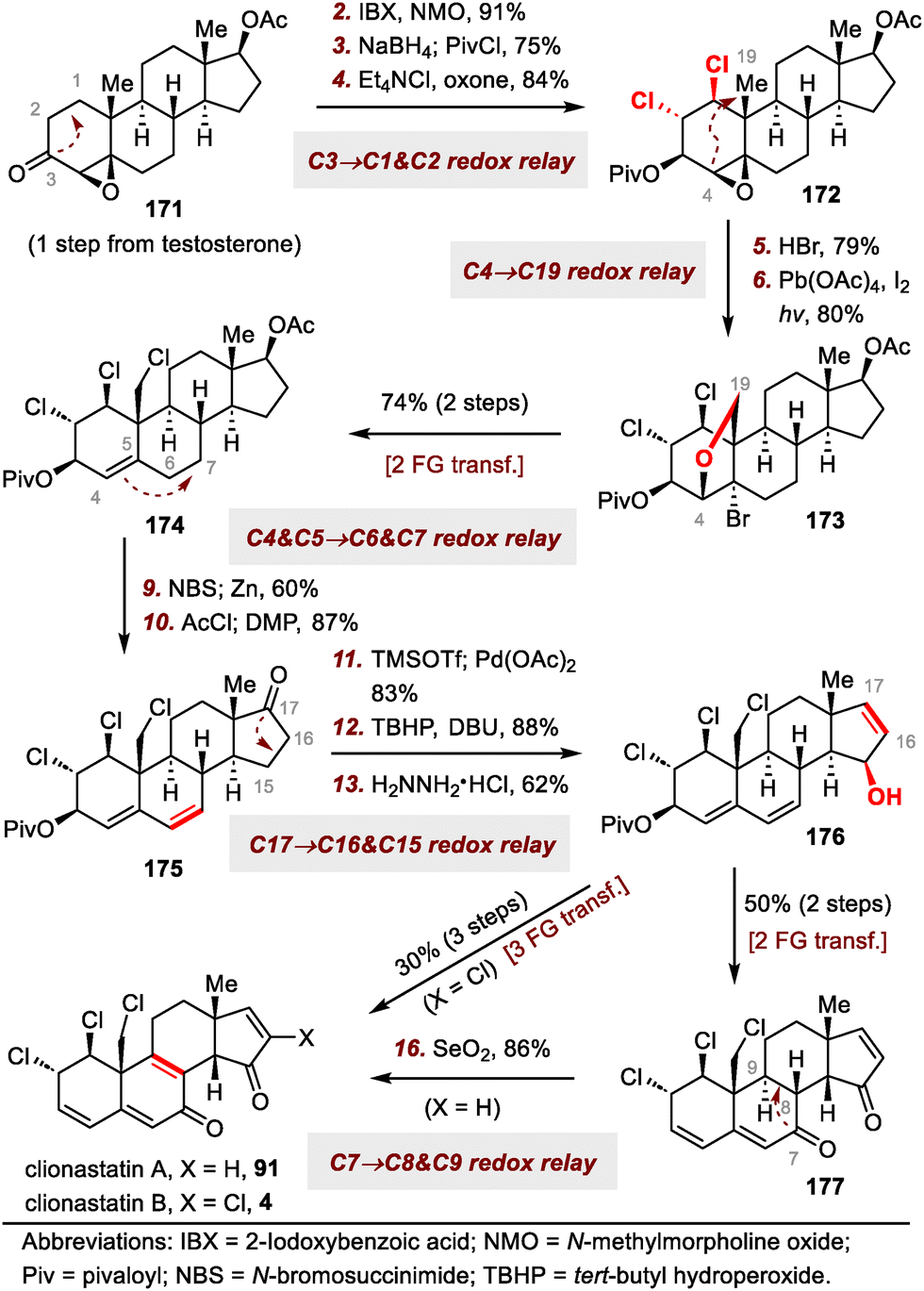 | ||
| Scheme 28 Zhang's syntheses of clionastatins A (91) and B (4). | ||
3.2. Skeletal reorganization strategies
Skeletal reorganization of classical steroids, which can be viewed as migration of one or more C–C bonds from one carbon to another within the tetracyclic steroidal skeleton, has emerged as an effective strategy for rapidly accessing the core frameworks of rearranged SNPs, enabling concise, scalable syntheses of these molecules.17d,20 In this subsection, four molecules have been selected to showcase how the complex core frameworks of rearranged steroids can be rapidly accessed from the tetracyclic skeletons of inexpensive commercially available steroids via skeletal reorganization (Scheme 29). In most cases, the proposed biosynthetic pathway of the rearranged steroid proved to be a critical inspiration in realizing the desired skeletal reorganization.103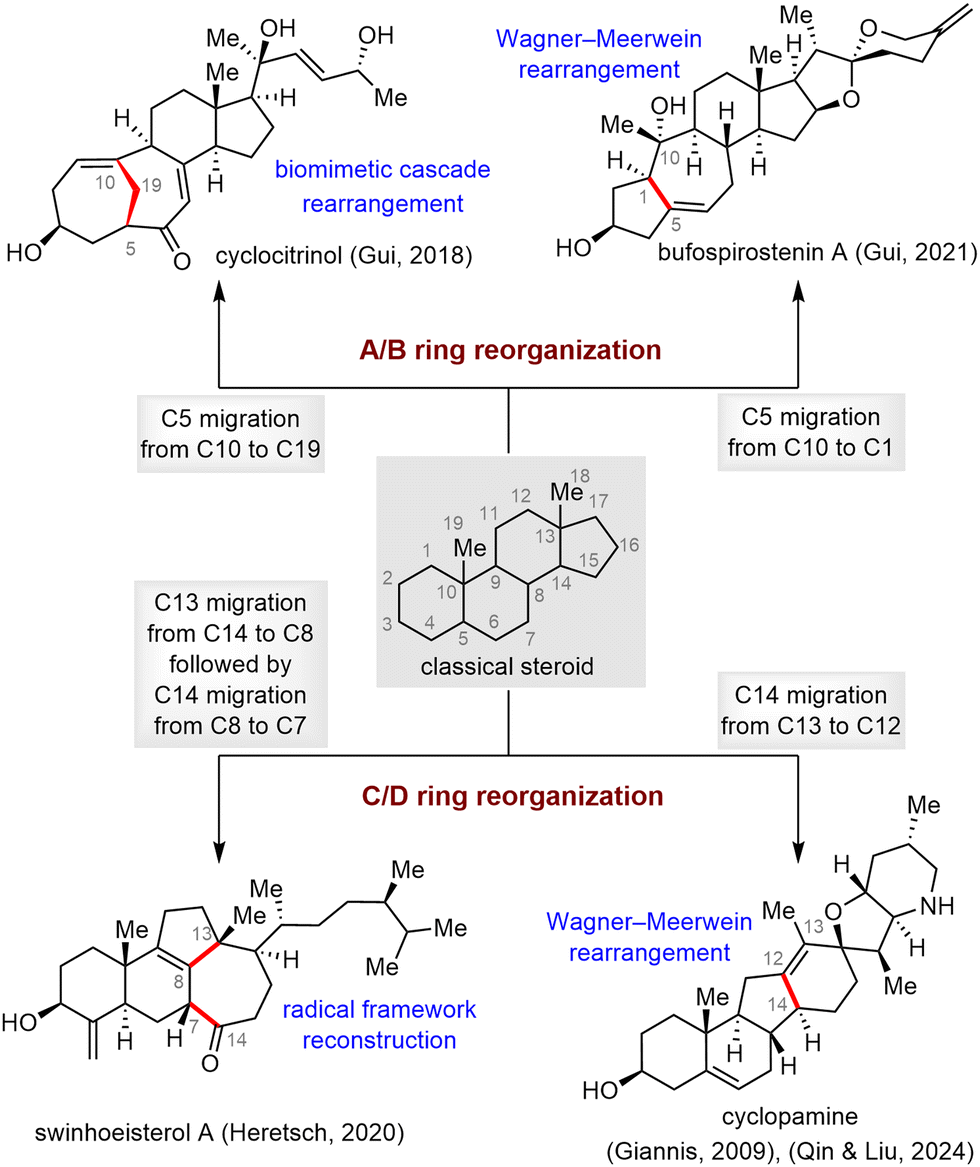 | ||
| Scheme 29 Skeletal reorganization strategies for synthesis of rearranged steroids. | ||
As shown in Scheme 30, heating a solution of allylic sulfoxide 178 in toluene in the presence of O-methylquinine as a base effected a syn-elimination reaction to give diene 179. This intermediate underwent cyclopropanation, and a subsequent ring scission reaction of cyclopropylcarbinyl cation 180 generated triene 182 and cycloheptatriene 181, possibly through regioselective deprotonations at C1 and C9, respectively. This biomimetic cascade rearrangement allowed the construction of the synthetically challenging bridged A/B ring system in only eight steps from commercially available pregnenolone. Moving forward, triene 182, which has the desired C1–C10 double bond, was elaborated to cyclocitrinol (11) in two steps.
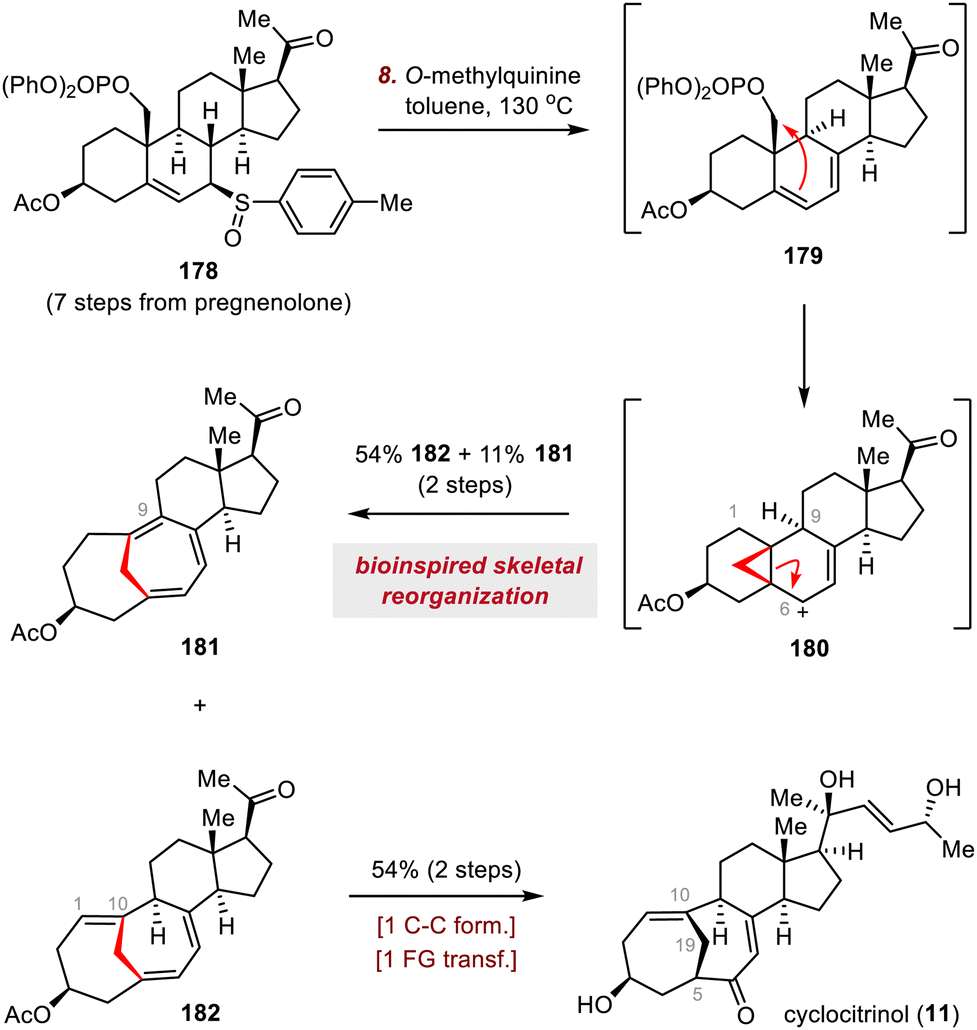 | ||
| Scheme 30 Our group's synthesis of cyclocitrinol (11). | ||
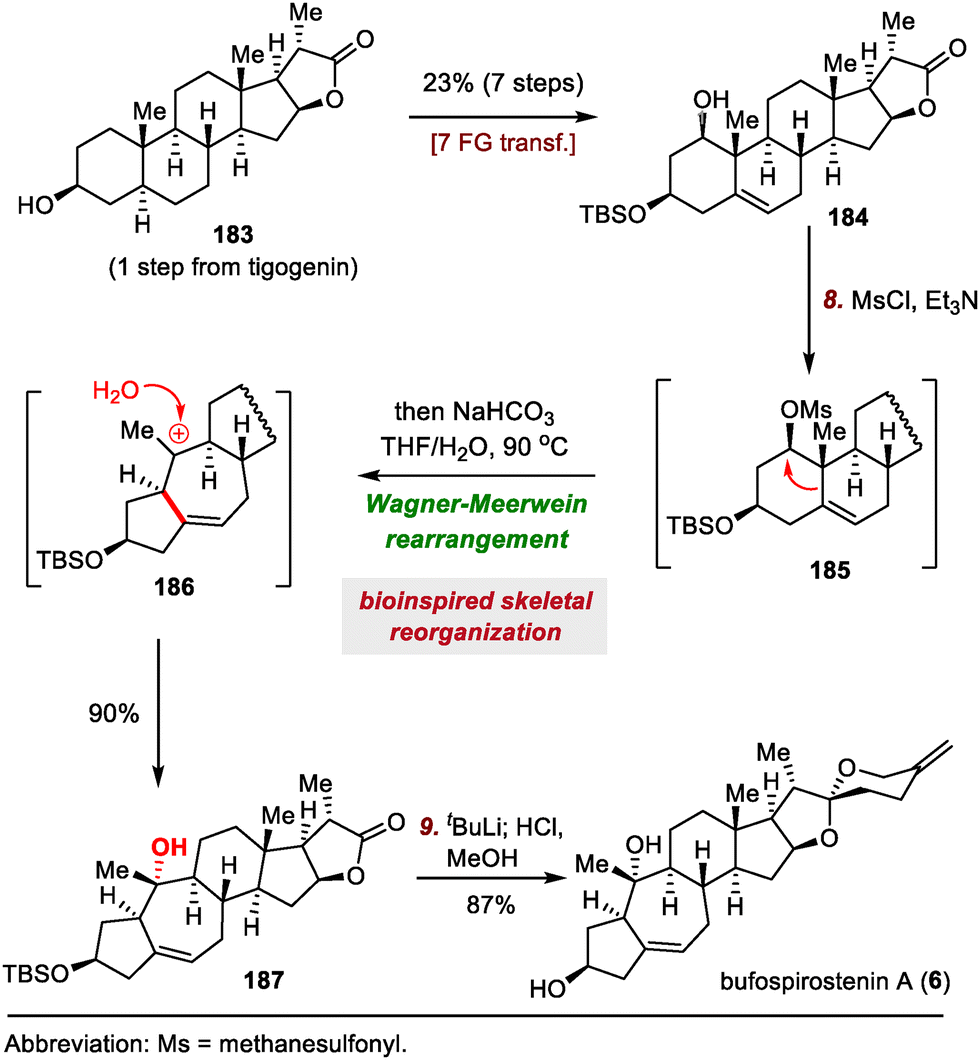 | ||
| Scheme 31 Our group's synthesis of bufospirostenin A (6). | ||
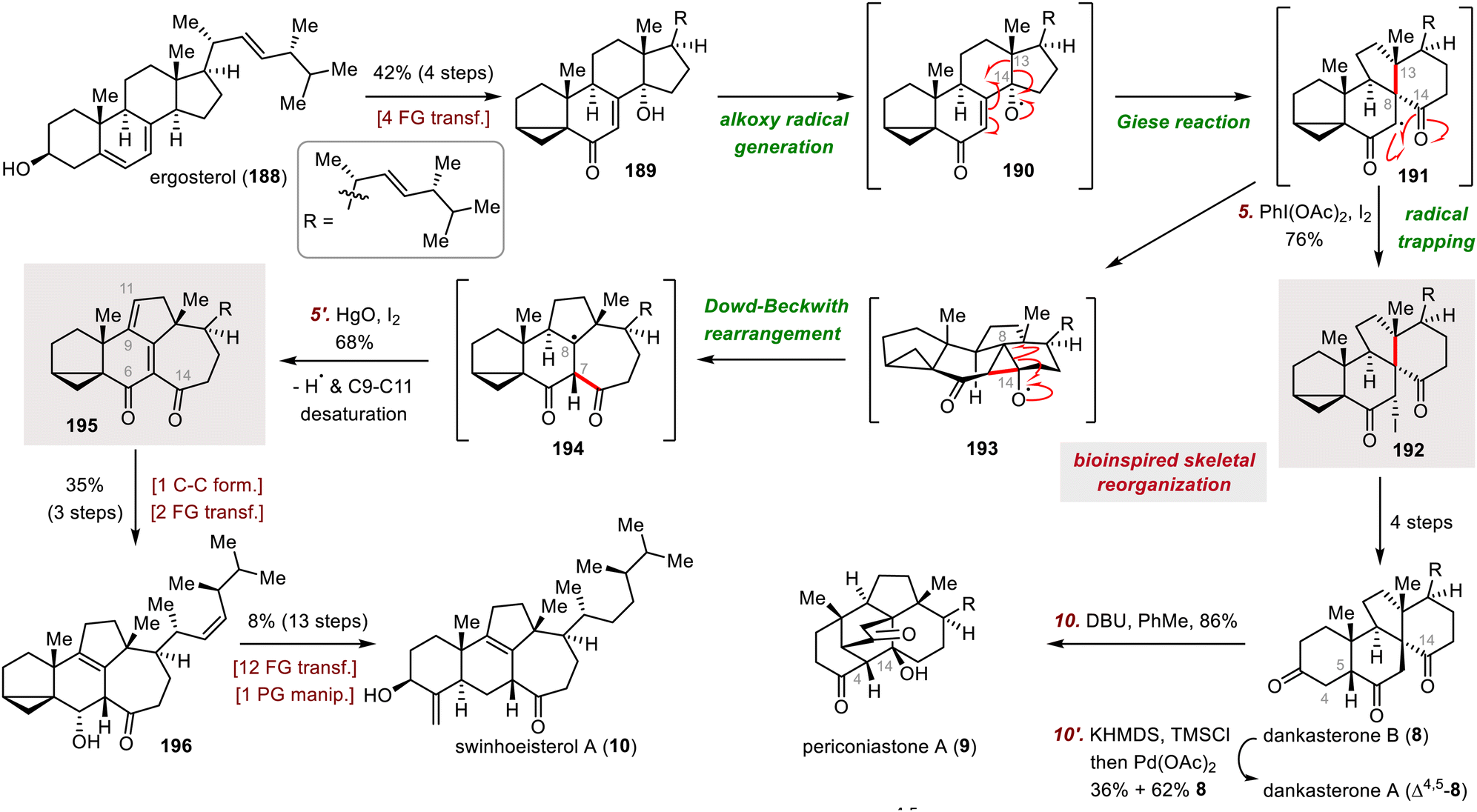 | ||
| Scheme 32 Heretsch's syntheses of swinhoeisterol A (10), dankasterones A (Δ4,5-8) and B (8), and periconiastone A (9). | ||
A four-step sequence was used to convert iodide 192 to dankasterone B (8), from which dankasterone A (Δ4,5-8) was accessed via a Saegusa–Ito oxidation to desaturate the C4–C5 bond. Alternatively, treatment of dankasterone B with DBU triggered an intramolecular aldol condensation between C4 and C14, affording periconiastone A (9) in 86% yield. Swinhoeisterol A (10) was synthesized from diketone 195 in seventeen steps, including a series of redox manipulations and installation of the side chain. It is worth noting that, during the revision of this manuscript, our group reported the divergent total syntheses of swinhoeisterols A–C from the Wieland–Miescher ketone. The synthetic strategy involves a tandem Negishi/Heck cross-coupling followed by a Baran reductive olefin coupling to construct the characteristic 5/7-fused ring system.110
:3 ratio), with desired exo alkene 200 being the major isomer. Alkene 200 was converted to 201 in five steps, and the synthesis of cyclopamine (14) was completed in six steps from 201.
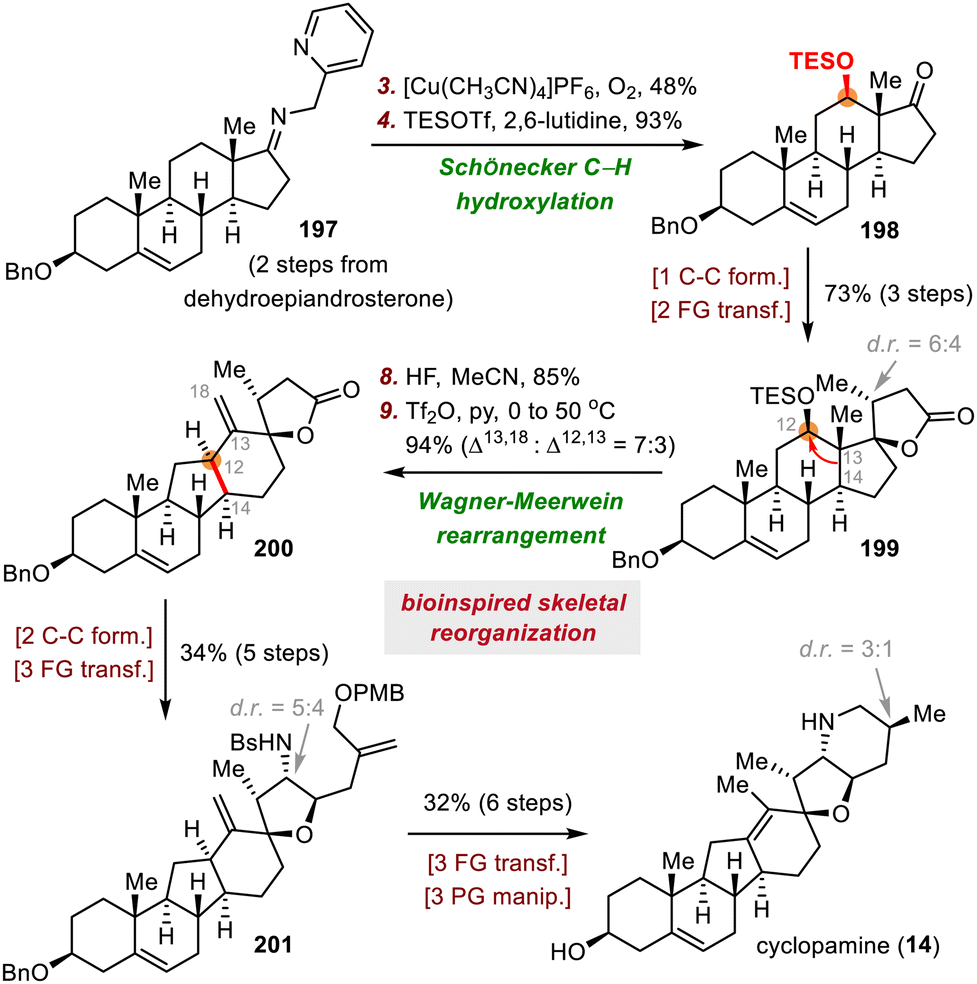 | ||
| Scheme 33 Giannis's synthesis of cyclopamine (14). | ||
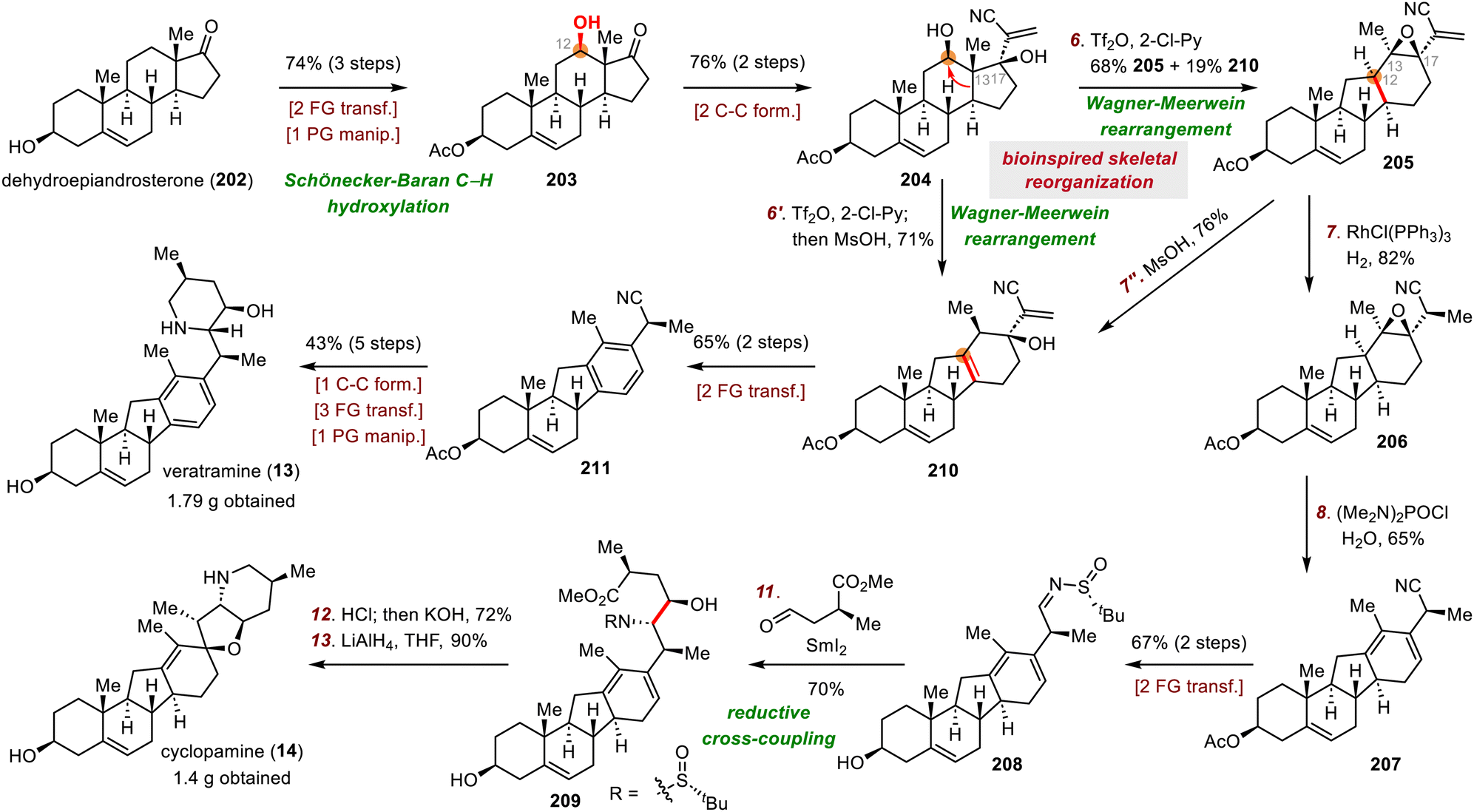 | ||
| Scheme 34 Qin and Liu's syntheses of cyclopamine (14) and veratramine (13). | ||
First, treatment of diol 204 with triflic anhydride and 2-chloropyridine initiated the desired Wagner–Meerwein rearrangement, and the resulting C13 carbocation was captured by the adjacent C17 hydroxyl group to give epoxide 205 in 68% yield, along with a 19% yield of alkene 210. Directed hydrogenation of epoxide 205 using Wilkinson's catalyst (RhCl(PPh3)3) afforded 206 with a dr of 33:1. The epoxide was converted to a diene via treatment of 206 with (Me2N)2POCl/H2O to give conjugated diene 207 in 65% yield. The cyano group of 207 was converted to a sulfinyl imine (208) in two steps, and reductive cross-coupling of 208 with a chiral aldehyde furnished aminoalcohol 209 in 70% yield. Bis-cyclization of 209 using HCl and reduction of the resulting amide with lithium aluminium hydride (LiAlH4) gave cyclopamine (14) in two steps.
Second, in situ treatment of epoxide 205 with methanesulfonic acid (MsOH) gave thermodynamically stable tetrasubstituted alkene 210, probably through acid-promoted epoxide opening followed by a 1,2-H shift and deprotonation. Veratramine (13) was prepared from 210 in only seven steps. The routes to both 13 and 14 resulted in gram quantities of the target natural products.
4. Conclusion and outlook
The unique and highly functionalized frameworks of SNPs and their intriguing biological activities have rendered these molecules popular targets for both synthetic and medicinal chemists, and significant synthetic and methodological breakthroughs have been achieved. This review has highlighted representative efficient syntheses of SNPs accomplished during the past two decades, grouping the convergent and semisynthetic syntheses on the basis of the key C–C bond forming reactions.Convergent strategies for SNP synthesis necessitate the rational design of coupling partners, and the coupling and cyclization methods must be judiciously chosen to establish the target polycyclic frameworks. Functionalization of the coupling partners with the maximum number of functional groups minimizes the number of post-coupling redox manipulations. Intramolecular cyclization reactions can be used to build ring systems and install associated stereogenic centers, and synthetic chemists have thus been motivated to invent robust new methodologies. In comparison, semisynthetic strategies for SNP synthesis use known steroids as starting materials, and these strategies typically necessitate a series of redox and stereochemical relays to functionalize the existing C–H bonds, along with skeletal reorganizations to build the rearranged skeletons. For the latter task, biogenetic hypotheses play an important inspirational role in the design of the key cascade transformations. Clearly, both convergent and semisynthetic strategies have their own advantages and disadvantages, and the choice of an appropriate strategy for specific targets largely depends on the synthetic efficiency of each approach, as well as on the opportunities it provides to discover new chemistry.
In recent years, artificial intelligence (AI) techniques have been increasingly applied in the field of chemistry,113 demonstrating remarkable potential in accelerating the synthesis of complex molecules.114 Traditionally, the total synthesis of natural products has relied heavily on chemists’ specialized knowledge and extensive experimental experience to design retrosynthetic routes—a process that is often time-consuming, labor-intensive, and expensive. In contrast, AI can automatically integrate vast amounts of chemical literature and reaction data into structured databases, enabling the prediction of reaction feasibility and the recommendation of optimal conditions (e.g., temperature, solvents, and catalysts). Clearly, this modern approach streamlines the synthetic planning process by minimizing the need for extensive trial-and-error experimentation, thereby significantly reducing experimental costs and enhancing overall efficiency.114c One of the most formidable challenges in the synthesis of SNPs lies in the construction of highly oxidized, polycyclic frameworks. By embracing AI-assisted strategies, chemists are now better equipped to address such challenges, making the efficient and scalable synthesis of these structurally intricate molecules increasingly achievable.115
In addition, recent years have seen remarkable breakthroughs in enzymatic C–H oxygenation reactions, which exhibit high chemo- and stereoselectivities.116 Consequently, chemoenzymatic strategies, that is, strategies that combine chemical synthesis with enzymatic C–H oxidation, have become an effective alternative for concise syntheses of bioactive SNPs.117
Last but not least, it is important to note that, despite the significant progress made in the synthesis of SNPs, the application of SNP analogs or modified advanced intermediates for biological studies, which is crucial for elucidating detailed structure–activity relationships (SAR) and accelerating drug discovery, still remains in its early stages.118 This limitation may stem from insufficient interdisciplinary integration. The pursuit of greater efficiency in this exciting field will undoubtedly continue, and we hope this review will further promote the exploration of novel methods and strategies, as well as the development of new steroid drugs in the near future.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the National Key Research and Development Program of China (2021YFF0502400 and 2022YFC2303100), the Talent Plan of Shanghai Branch, Chinese Academy of Sciences (CASSHB-QNPD-2023-009), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB1060000) and the CAS Project for Young Scientists in Basic Research (YSBR-095). This work has been supported by the New Cornerstone Science Foundation through the XPLORER PRIZE (J. G.).Notes and references
- (a) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed; (b) K. C. Nicolaou, D. Vourloumis, N. Winssinger and P. S. Baran, Angew. Chem., Int. Ed., 2000, 39, 44–122 CrossRef CAS; (c) S. B. Bharate and C. W. Lindsley, J. Med. Chem., 2024, 67, 20723–20730 CrossRef CAS PubMed.
- (a) D. A. Dias, S. Urban and U. Roessner, Metabolites, 2012, 2, 303–336 CrossRef CAS PubMed; (b) A. G. Atanasov, S. B. Zotchev, V. M. Dirsch, the International Natural Product Sciences Taskforce and C. T. Supuran, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed.
- P. S. Baran, J. Am. Chem. Soc., 2018, 140, 4751–4755 CrossRef CAS PubMed.
- L. W. Hernandez and D. Sarlah, Chem. – Eur. J., 2019, 25, 13248–13270 CrossRef CAS PubMed.
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
- A. Chaudhuri and D. Anand, Biomed. Spectrosc. Imaging, 2017, 6, 1–24 CAS.
- K. C. Nicolaou and T. Montagnon, Molecules That Changed the World, Wiley-VCH, Weinheim, Germany, 2008, pp. 79–90 Search PubMed.
- (a) W. S. Johnson, Acc. Chem. Res., 1968, 1, 1–8 CrossRef CAS; (b) W. S. Johnson, Bioorg. Chem., 1976, 5, 51–98 CrossRef CAS; (c) W. S. Johnson, Angew. Chem., Int. Ed. Engl., 1976, 15, 9–17 CrossRef CAS PubMed; (d) R. A. Yoder and J. N. Johnston, Chem. Rev., 2005, 105, 4730–4756 CrossRef CAS PubMed.
- F. Noack, R. C. Heinze and P. Heretsch, Synthesis, 2019, 2039–2057 CAS.
- S. Prévost, N. Dupré, M. Leutzsch, Q. Wang, V. Wakchaure and B. List, Angew. Chem., Int. Ed., 2014, 53, 8770–8773 CrossRef PubMed.
- W. Che, L. Wojitas, C. Shan and J. M. Lopchuk, Sci. Adv., 2024, 10, eadp9375 CrossRef CAS PubMed.
- K. Cen, J. Bao, X. Wang, H. Tian, Y. Wang and J. Gui, J. Am. Chem. Soc., 2024, 146, 6481–6486 CrossRef CAS PubMed.
- X. Wang, G. Huang, Y. Wang and J. Gui, J. Am. Chem. Soc., 2023, 145, 9354–9363 CrossRef CAS PubMed.
- J.-H. Fan, J.-J. Wang, F. Li, G. Wang, Q. Guo, L. W. Chung and C.-C. Li, CCS Chem., 2021, 3, 348–357 CrossRef CAS.
- (a) J. Xie, X. Liu, N. Zhang, S. Choi and G. Dong, J. Am. Chem. Soc., 2021, 143, 19311–19316 CrossRef CAS PubMed; (b) J. Xie, Z. Zheng, X. Liu, N. Zhang, S. Choi, C. He and G. Dong, J. Am. Chem. Soc., 2023, 145, 4828–4852 CrossRef CAS PubMed.
- A. R. Bucknam and G. C. Micalizio, J. Am. Chem. Soc., 2022, 144, 8493–8497 CrossRef CAS PubMed.
- (a) F. L. Duecker, F. Reuß and P. Heretsch, Org. Biomol. Chem., 2019, 17, 1624–1633 RSC; (b) L. Min, L.-P. Zhong and C.-C. Li, Acc. Chem. Res., 2023, 56, 2378–2390 CrossRef CAS PubMed; (c) M. Alekseychuk and P. Heretsch, Chem. Commun., 2023, 59, 6811–6826 RSC; (d) Y. Wang and J. Gui, Acc. Chem. Res., 2024, 57, 568–579 CAS; (e) H. R. Khatri, N. Carney, R. Rutkoski, B. Bhattarai and P. Nagorny, Eur. J. Org. Chem., 2020, 755–776 CrossRef CAS; (f) M. T. C. Pessôa, L. A. Barbosa and J. A. F. P. Villar, Stud. Nat. Prod. Chem., 2018, 57, 79–113 Search PubMed; (g) P. Gupta and G. Panda, Eur. J. Org. Chem., 2014, 8004–8019 CrossRef CAS; (h) M. Kotora, F. Hessler and B. Eignerová, Eur. J. Org. Chem., 2012, 29–42 CrossRef CAS; (i) R. Skoda-Földes and L. Kollár, Chem. Rev., 2003, 103, 4095–4130 CrossRef PubMed; (j) J. R. Hanson, Nat. Prod. Rep., 2010, 27, 887–899 RSC; (k) D. F. Covey, Steroids, 2009, 74, 577–585 CrossRef CAS PubMed; (l) M. Ibrahim-Ouali and L. Rocheblave, Steroids, 2008, 73, 375–407 CrossRef CAS PubMed; (m) M. Ibrahim-Ouali and M. Santelli, Steroids, 2006, 71, 1025–1044 CrossRef CAS PubMed; (n) M. Ibrahim-Ouali, Steroids, 2007, 72, 475–508 CrossRef CAS PubMed; (o) J.-F. Biellmann, Chem. Rev., 2003, 103, 2019–2034 CrossRef CAS PubMed; (p) A.-S. Chapelon, D. Moraléda, R. Rodriguez, C. Ollivier and M. Santelli, Tetrahedron, 2007, 63, 11511–11616 CrossRef CAS.
- (a) D. Urabe, T. Asaba and M. Inoue, Chem. Rev., 2015, 115, 9207–9231 CrossRef CAS PubMed; (b) Y. Gao and D. Ma, Acc. Chem. Res., 2021, 54, 569–582 CrossRef CAS PubMed.
- Z. Wang and C. Hui, Org. Biomol. Chem., 2021, 19, 3791–3812 RSC.
- Z. Zhang, X. Qian, Y. Gu and J. Gui, Nat. Prod. Rep., 2024, 41, 251–272 RSC.
- Z. G. Brill, M. L. Condakes, C. P. Ting and T. J. Maimone, Chem. Rev., 2017, 117, 11753–11795 CrossRef CAS PubMed.
- M.-J. Cheng, L.-P. Zhong, C.-C. Gu, X.-J. Zhu, B. Chen, J.-S. Liu, L. Wang, W.-C. Ye and C.-C. Li, J. Am. Chem. Soc., 2020, 142, 12602–12607 CrossRef CAS PubMed.
- L.-P. Zhong, R. Feng, J.-J. Wang and C.-C. Li, J. Am. Chem. Soc., 2023, 145, 2098–2103 CrossRef CAS PubMed.
- H.-Y. Tian, L.-J. Ruan, T. Yu, Q.-F. Zheng, N.-H. Chen, R.-B. Wu, X.-Q. Zhang, L. Wang, R.-W. Jiang and W.-C. Ye, J. Nat. Prod., 2017, 80, 1182–1186 CrossRef CAS PubMed.
- (a) Y. Wang, H. Tian and J. Gui, J. Am. Chem. Soc., 2021, 143, 19576–19586 CrossRef CAS PubMed; (b) P. Yang, Y.-Y. Li, H. Tian, G.-L. Qian, Y. Wang, X. Hong and J. Gui, J. Am. Chem. Soc., 2022, 144, 17769–17775 CrossRef CAS PubMed.
- J. Huang, T. Cao, Z. Zhang and Z. Yang, J. Am. Chem. Soc., 2022, 144, 2479–2483 CrossRef CAS PubMed.
- (a) J. Blanco-Urgoiti, L. Añorbe, L. Pérez-Serrano, G. Domínguez and J. Pérez-Castells, Chem. Soc. Rev., 2004, 33, 32–42 RSC; (b) Z. Yang, Acc. Chem. Res., 2021, 54, 556–568 CrossRef CAS PubMed.
- H.-Y. Tian, L. Wang, X.-Q. Zhang, Y. Wang, D.-M. Zhang, R.-W. Jiang, Z. Liu, J.-S. Liu, Y.-L. Li and W.-C. Ye, Chem. – Eur. J., 2010, 16, 10989–10993 CrossRef CAS PubMed.
- (a) P. A. Wender and D. Sperandio, J. Org. Chem., 1998, 63, 4164–4165 CrossRef CAS; (b) B. M. Trost, F. D. Toste and H. Shen, J. Am. Chem. Soc., 2000, 122, 2379–2380 CrossRef CAS.
- H. R. El-Seedi, S. A. M. Khalifa, E. A. Taher, M. A. Farag, A. Saeed, M. Gamal, M.-E. F. Hegazy, D. Youssef, S. G. Musharraf, M. M. Alajlani, J. Xiao and T. Efferth, Pharmacol. Res., 2019, 141, 123–175 CrossRef CAS PubMed.
- C. Mannich and G. Siewert, Ber. Dtsch. Chem. Ges. A, 1942, 75, 737–750 CrossRef.
- (a) H. Renata, Q. Zhou and P. S. Baran, Science, 2013, 339, 59–63 CrossRef CAS PubMed; (b) H. Renata, Q. Zhou, G. Dünstl, J. Felding, R. R. Merchant, C.-H. Yeh and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 1330–1340 CrossRef CAS PubMed.
- H. Zhang, M. Sridhar Reddy, S. Phoenix and P. Deslongchamps, Angew. Chem., Int. Ed., 2008, 47, 1272–1275 CrossRef CAS PubMed.
- K. Mukai, S. Kasuya, Y. Nakagawa, D. Urabe and M. Inoue, Chem. Sci., 2015, 6, 3383–3387 RSC.
- H. R. Khatri, B. Bhattarai, W. Kaplan, Z. Li, M. J. Curtis Long, Y. Aye and P. Nagorny, J. Am. Chem. Soc., 2019, 141, 4849–4860 CrossRef CAS PubMed.
- J. Sun, Y. Chen, S. S. Ragab, W. Gu, Z. Tang, Y. Tang and W. Tang, Angew. Chem., Int. Ed., 2023, 62, e202303639 CrossRef CAS PubMed.
- (a) J.-F. Lavallée and P. Deslongchamps, Tetrahedron Lett., 1988, 29, 6033–6036 CrossRef; (b) S. Trudeau and P. Deslongchamps, J. Org. Chem., 2004, 69, 832–838 CrossRef CAS PubMed.
- N. R. Cichowicz, W. Kaplan, Y. Khomutnyk, B. Bhattarai, Z. Sun and P. Nagorny, J. Am. Chem. Soc., 2015, 137, 14341–14348 CrossRef CAS PubMed.
- W. Kaplan, H. R. Khatri and P. Nagorny, J. Am. Chem. Soc., 2016, 138, 7194–7198 CrossRef CAS PubMed.
- B. Bhattarai and P. Nagorny, Org. Lett., 2018, 20, 154–157 CrossRef CAS PubMed.
- K. Du, P. Guo, Y. Chen, Z. Cao, Z. Wang and W. Tang, Angew. Chem., Int. Ed., 2015, 54, 3033–3037 CrossRef CAS PubMed.
- (a) P. Liu, H. Cheng, T. M. Roberts and J. J. Zhao, Nat. Rev. Drug Discovery, 2009, 8, 627–644 CrossRef CAS PubMed; (b) J. Yang, J. Nie, X. Ma, Y. Wei, Y. Peng and X. Wei, Mol. Cancer, 2019, 18, 26 CrossRef PubMed.
- (a) F. Marra, M. Pinzani, R. DeFranco, G. Laffi and P. Gentilini, FEBS Lett., 1995, 376, 141–145 CrossRef CAS PubMed; (b) G. Powis, R. Bonjouklian, M. M. Berggren, A. Gallegos, R. Abraham, C. Ashendel, L. Zalkow, W. F. Matter, J. Dodge, G. Grindey and C. J. Vlahos, Cancer Res., 1994, 54, 2419–2423 CAS; (c) P. Wipf and R. J. Halter, Org. Biomol. Chem., 2005, 3, 2053–2061 RSC.
- (a) P. W. Brian, P. J. Curtis, H. G. Hemming and G. F. L. Norris, Trans. Br. Mycol. Soc., 1957, 40, 365–368 CrossRef CAS; (b) T. J. Petcher, H. P. Weber and Z. Kis, J. Chem. Soc., Chem. Commun., 1972, 1061–1062 RSC; (c) J. MacMillan, T. J. Simpson and S. K. Yeboah, J. Chem. Soc., Chem. Commun., 1972, 1063 RSC.
- (a) S. Sato, M. Nakada and M. Shibasaki, Tetrahedron Lett., 1996, 37, 6141–6144 CrossRef CAS; (b) T. Mizutani, S. Honzawa, S.-y Tosaki and M. Shibasaki, Angew. Chem., Int. Ed., 2002, 41, 4680–4682 CrossRef CAS PubMed; (c) H. Shigehisa, T. Mizutani, S.-y Tosaki, T. Ohshima and M. Shibasaki, Tetrahedron, 2005, 61, 5057–5065 CrossRef CAS.
- Y. Guo, T. Quan, Y. Lu and T. Luo, J. Am. Chem. Soc., 2017, 139, 6815–6818 CrossRef CAS PubMed.
- (a) P. W. Brian and J. G. McGowan, Nature, 1945, 156, 144–145 CrossRef CAS; (b) J. F. Grove, P. McCloskey and J. S. Moffatt, J. Chem. Soc. C, 1966, 743–747 RSC.
- J. S. Moffatt, J. D. Bu’Lock and T. H. Yuen, J. Chem. Soc. D, 1969, 839a RSC.
- (a) R. W. Jones and J. G. Hancock, Can. J. Microbiol., 1987, 33, 963–966 CrossRef CAS PubMed; (b) G.-A. Pakora, S. Mann, D. M. Kone and D. Buisson, Bioorg. Chem., 2021, 112, 104959 CrossRef CAS PubMed.
- G.-Q. Wang, G.-D. Chen, S.-Y. Qin, D. Hu, T. Awakawa, S.-Y. Li, J.-M. Lv, C.-X. Wang, X.-S. Yao, I. Abe and H. Gao, Nat. Commun., 2018, 9, 1838 CrossRef PubMed.
- J. R. Hanson, Nat. Prod. Rep., 1995, 12, 381–384 RSC.
- E. A. Anderson, E. J. Alexanian and E. J. Sorensen, Angew. Chem., Int. Ed., 2004, 43, 1998–2001 CrossRef CAS PubMed.
- M. Del Bel, A. R. Abela, J. D. Ng and C. A. Guerrero, J. Am. Chem. Soc., 2017, 139, 6819–6822 CrossRef CAS PubMed.
- Y. Ji, Z. Xin, H. He and S. Gao, J. Am. Chem. Soc., 2019, 141, 16208–16212 CrossRef CAS PubMed.
- A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945–2964 CrossRef CAS PubMed.
- (a) S. W. M. Crossley, F. Barabé and R. A. Shenvi, J. Am. Chem. Soc., 2014, 136, 16788–16791 CrossRef CAS PubMed; (b) S. A. Green, S. W. M. Crossley, J. L. M. Matos, S. Vásquez-Céspedes, S. L. Shevick and R. A. Shenvi, Acc. Chem. Res., 2018, 51, 2628–2640 CrossRef CAS PubMed; (c) J. M. Smith, S. J. Harwood and P. S. Baran, Acc. Chem. Res., 2018, 51, 1807–1817 CrossRef CAS PubMed.
- T. Amagata, M. Doi, M. Tohgo, K. Minoura and A. Numata, Chem. Commun., 1999, 1321–1322 RSC.
- T. Amagata, M. Tanaka, T. Yamada, M. Doi, K. Minoura, H. Ohishi, T. Yamori and A. Numata, J. Nat. Prod., 2007, 70, 1731–1740 CrossRef CAS PubMed.
- W. Gao, C. Chai, Y. He, F. Li, X. Hao, F. Cao, L. Gu, J. Liu, Z. Hu and Y. Zhang, Org. Lett., 2019, 21, 8469–8472 CrossRef CAS PubMed.
- (a) F. L. Duecker, R. C. Heinze and P. Heretsch, J. Am. Chem. Soc., 2020, 142, 104–108 CrossRef CAS PubMed; (b) F. L. Duecker, R. C. Heinze, S. Steinhauer and P. Heretsch, Chem. – Eur. J., 2020, 26, 9971–9981 CrossRef CAS PubMed.
- (a) P. Chen, C. Wang, R. Yang, H. Xu, J. Wu, H. Jiang, K. Chen and Z. Ma, Angew. Chem., Int. Ed., 2021, 60, 5512–5518 CrossRef CAS PubMed; (b) S. Deng, H. Xu, H. Jiang and Z. Ma, Org. Chem. Front., 2022, 9, 3961–3965 RSC.
- S. W. M. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Chem. Rev., 2016, 116, 8912–9000 CrossRef CAS PubMed.
- E. Fattorusso, O. Taglialatela-Scafati, F. Petrucci, G. Bavestrello, B. Calcinai, C. Cerrano, P. Di Meglio and A. Ianaro, Org. Lett., 2004, 6, 1633–1635 CrossRef CAS PubMed.
- W. Ju, X. Wang, H. Tian and J. Gui, J. Am. Chem. Soc., 2021, 143, 13016–13021 CrossRef CAS PubMed.
- H. Cui, Y. Shen, Y. Chen, R. Wang, H. Wei, P. Fu, X. Lei, H. Wang, R. Bi and Y. Zhang, J. Am. Chem. Soc., 2022, 144, 8938–8944 CrossRef CAS PubMed.
- (a) K. Masuda, M. Nagatomo and M. Inoue, Nat. Chem., 2017, 9, 207–212 CrossRef CAS PubMed; (b) D. Kuwana, M. Nagatomo and M. Inoue, Org. Lett., 2019, 21, 7619–7623 CrossRef CAS PubMed; (c) H. Fujino, T. Fukuda, M. Nagatomo and M. Inoue, J. Am. Chem. Soc., 2020, 142, 13227–13234 CrossRef CAS PubMed.
- S. Laschat, F. Narjes and L. E. Overman, Tetrahedron, 1994, 50, 347–358 CrossRef CAS.
- K. Mukai, D. Urabe, S. Kasuya, N. Aoki and M. Inoue, Angew. Chem., Int. Ed., 2013, 52, 5300–5304 CrossRef CAS PubMed.
- T. Tokuyama, J. Daly and B. Witkop, J. Am. Chem. Soc., 1969, 91, 3931–3938 CrossRef CAS PubMed.
- (a) N. J. Linford, A. R. Cantrell, Y. Qu, T. Scheuer and W. A. Catterall, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 13947–13952 CrossRef CAS PubMed; (b) S.-Y. Wang, J. Mitchell, D. B. Tikhonov, B. S. Zhorov and G. K. Wang, Mol. Pharmacol., 2006, 69, 788–795 CrossRef CAS PubMed.
- (a) E. X. Albuquerque, J. W. Daly and B. Witkop, Science, 1971, 172, 995–1002 CrossRef CAS PubMed; (b) G. B. Brown, Int. Rev. Neurobiol., 1988, 29, 77–116 CrossRef CAS PubMed.
- (a) R. Imhof, M. E. Gössinger, W. Graf, H. Berner, M. L. Berner-Fenz and H. Wehrli, Helv. Chim. Acta, 1972, 55, 1151–1153 CrossRef CAS PubMed; (b) R. Imhof, E. Gössinger, W. Graf, L. Berner-Fenz, H. Berner, R. Schaufelberger and H. Wehrli, Helv. Chim. Acta, 1973, 56, 139–162 CrossRef CAS PubMed.
- M. Kurosu, L. R. Marcin, T. J. Grinsteiner and Y. Kishi, J. Am. Chem. Soc., 1998, 120, 6627–6628 CrossRef CAS.
- M. M. Logan, T. Toma, R. Thomas-Tran and J. Du Bois, Science, 2016, 354, 865–869 CrossRef CAS PubMed.
- Y. Guo, Z. Guo, J.-T. Lu, R. Fang, S.-C. Chen and T. Luo, J. Am. Chem. Soc., 2020, 142, 3675–3679 CrossRef CAS PubMed.
- (a) Y. Watanabe, H. Morozumi, H. Mutoh, K. Hagiwara and M. Inoue, Angew. Chem., Int. Ed., 2023, 62, e202309688 CrossRef CAS PubMed; (b) Y. Watanabe, K. Sakata, D. Urabe, K. Hagiwara and M. Inoue, J. Org. Chem., 2023, 88, 17479–17484 CrossRef CAS PubMed.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- (a) Y. Zhu, L. Zhang and S. Luo, J. Am. Chem. Soc., 2014, 136, 14642–14645 CrossRef CAS PubMed; (b) W. Zhang, Y. Zhu, L. Zhang and S. Luo, Chin. J. Chem., 2018, 36, 716–722 CrossRef CAS.
- (a) J. Uenishi, J. M. Beau, R. W. Armstrong and Y. Kishi, J. Am. Chem. Soc., 1987, 109, 4756–4758 CrossRef CAS; (b) D. Imao, B. W. Glasspoole, V. S. Laberge and C. M. Crudden, J. Am. Chem. Soc., 2009, 131, 5024–5025 CrossRef CAS PubMed.
- (a) S. M. Kupchan, J. Am. Chem. Soc., 1956, 78, 3546–3547 CrossRef CAS; (b) T. J. Gilbertson, Phytochemistry, 1973, 12, 2079–2080 CrossRef CAS.
- S. M. Kupchan and C. R. Narayanan, J. Am. Chem. Soc., 1959, 81, 1913–1921 CrossRef CAS.
- S. M. Kupchan, C. I. Ayres, M. Neeman, R. H. Hensler, T. Masamune and S. Rajagopalan, J. Am. Chem. Soc., 1960, 82, 2242–2251 CrossRef CAS.
- (a) H.-J. Li, Y. Jiang and P. Li, Nat. Prod. Rep., 2006, 23, 735–752 RSC; (b) M. L. Dirks, J. T. Seale, J. M. Collins and O. M. McDougal, Molecules, 2021, 26, 5934 CrossRef CAS PubMed.
- Y. Guo, J.-T. Lu, R. Fang, Y. Jiao, J. Liu and T. Luo, J. Am. Chem. Soc., 2023, 145, 20202–20207 CrossRef CAS PubMed.
- Y. Guo, R. Fang, Y. Jiao, J. Liu, J.-T. Lu and T. Luo, Nat. Commun., 2024, 15, 7639 CrossRef CAS PubMed.
- K. Saito, Bull. Chem. Soc. Jpn., 2006, 15, 22–27 CrossRef.
- R. F. Keller, Phytochemistry, 1968, 7, 303–306 CrossRef.
- H.-Y. Kim, S.-W. Lee, S.-K. Choi, J. Ashim, W. Kim, S.-M. Beak, J.-K. Park, J. E. Han, G.-J. Cho, Z. Y. Ryoo, J. Jeong, Y.-H. Lee, H. Jeong, W. Yu and S. Park, Am. J. Chin. Med., 2023, 51, 1309–1333 CrossRef CAS PubMed.
- M. K. Cooper, J. A. Porter, K. E. Young and P. A. Beachy, Science, 1998, 280, 1603–1607 CrossRef CAS PubMed.
- W. S. Johnson, H. A. P. DeJongh, C. E. Coverdale, J. W. Scott and U. Burckhardt, J. Am. Chem. Soc., 1967, 89, 4523–4524 CrossRef CAS.
- (a) A. Giannis, P. Heretsch, V. Sarli and A. Stößel, Angew. Chem., Int. Ed., 2009, 48, 7911–7914 CrossRef CAS PubMed; (b) P. Heretsch, S. Rabe and A. Giannis, J. Am. Chem. Soc., 2010, 132, 9968–9969 CrossRef CAS PubMed.
- M. Sofiadis, D. Xu, A. J. Rodriguez, B. Nissl, S. Clementson, N. N. Petersen and P. S. Baran, J. Am. Chem. Soc., 2023, 145, 21760–21765 CrossRef CAS PubMed.
- H. Shao, W. Liu, M. Liu, H. He, Q.-L. Zhou, S.-F. Zhu and S. Gao, J. Am. Chem. Soc., 2023, 145, 25086–25092 CrossRef CAS PubMed.
- W. Hou, H. Lin, Y. Wu, C. Li, J. Chen, X.-Y. Liu and Y. Qin, Nat. Commun., 2024, 15, 5332 CrossRef CAS PubMed.
- M. D. Zott, D. W. Zuschlag and D. H. Trauner, J. Am. Chem. Soc., 2025, 147, 3010–3016 CrossRef CAS PubMed.
- (a) J. K. Crandall and R. P. Haseltine, J. Am. Chem. Soc., 1968, 90, 6251–6253 CrossRef CAS; (b) A. B. Smith and W. C. Agosta, J. Am. Chem. Soc., 1973, 95, 1961–1968 CrossRef CAS; (c) S. Cai, Z. Xiao, Y. Shi and S. Gao, Chem. – Eur. J., 2014, 20, 8677–8681 CrossRef CAS PubMed.
- Y. Kitamura, M. Nishizawa, K. Kaneko, M. Shiro, Y.-P. Chen and H.-Y. Hsu, Tetrahedron, 1989, 45, 7281–7286 CrossRef CAS.
- K. J. Cassaidy and V. H. Rawal, J. Am. Chem. Soc., 2021, 143, 16394–16400 CrossRef CAS PubMed.
- Y. Jin, S. Hok, J. Bacsa and M. Dai, J. Am. Chem. Soc., 2024, 146, 1825–1831 CrossRef CAS PubMed.
- (a) H. Gotoh and Y. Hayashi, Org. Lett., 2007, 9, 2859–2862 CrossRef CAS PubMed; (b) L. You, X.-T. Liang, L.-M. Xu, Y.-F. Wang, J.-J. Zhang, Q. Su, Y.-H. Li, B. Zhang, S.-L. Yang, J.-H. Chen and Z. Yang, J. Am. Chem. Soc., 2015, 137, 10120–10123 CrossRef CAS PubMed.
- (a) R. Grigg, R. Scott and P. Stevenson, Tetrahedron Lett., 1982, 23, 2691–2692 CrossRef CAS; (b) S. J. Neeson and P. J. Stevenson, Tetrahedron Lett., 1988, 29, 813–814 CrossRef CAS.
- B. M. Trost and J. E. Schultz, Synthesis, 2019, 1–30 CAS.
- (a) R. Bao, H. Zhang and Y. Tang, Acc. Chem. Res., 2021, 54, 3720–3733 CrossRef CAS PubMed; (b) L. Chen, P. Chen and Y. Jia, Acc. Chem. Res., 2024, 57, 3524–3540 CrossRef CAS PubMed.
- A. G. Kozlovsky, V. P. Zhelifonova, V. M. Adanin, S. M. Ozerskaya and U. Grafe, Pharmazie, 2000, 55, 470–471 CAS.
- Z.-H. He, C.-L. Xie, T. Wu, Y.-T. Yue, C.-F. Wang, L. Xu, M.-M. Xie, Y. Zhang, Y.-J. Hao, R. Xu and X.-W. Yang, J. Nat. Prod., 2023, 86, 157–165 CrossRef CAS PubMed.
- L. Du, T. Zhu, Y. Fang, Q. Gu and W. Zhu, J. Nat. Prod., 2008, 71, 1343–1351 CrossRef CAS PubMed.
- J. Liu, J. Wu, J.-H. Fan, X. Yan, G. Mei and C.-C. Li, J. Am. Chem. Soc., 2018, 140, 5365–5369 CrossRef CAS PubMed.
- Y. Wang, W. Ju, H. Tian, W. Tian and J. Gui, J. Am. Chem. Soc., 2018, 140, 9413–9416 CrossRef CAS PubMed.
- Y. Wang, W. Ju, H. Tian, S. Sun, X. Li, W. Tian and J. Gui, J. Am. Chem. Soc., 2019, 141, 5021–5033 CrossRef CAS PubMed.
- G. Huang, X. Zhang, Y.-C. Gu and J. Gui, J. Am. Chem. Soc., 2025, 147 DOI:10.1021/jacs.5c07053.
- B. Schönecker, T. Zheldakova, Y. Liu, M. Kötteritzsch, W. Günther and H. Görls, Angew. Chem., Int. Ed., 2003, 42, 3240–3244 CrossRef PubMed.
- (a) Y. Y. See, A. T. Herrmann, Y. Aihara and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 13776–13779 CrossRef CAS PubMed; (b) R. Trammell, Y. Y. See, A. T. Herrmann, N. Xie, D. E. Díaz, M. A. Siegler, P. S. Baran and I. Garcia-Bosch, J. Org. Chem., 2017, 82, 7887–7904 CrossRef CAS PubMed.
- Z. J. Baum, X. Yu, P. Y. Ayala, Y. Zhao, S. P. Watkins and Q. Zhou, J. Chem. Inf. Model., 2021, 61, 3197–3212 CrossRef CAS PubMed.
- (a) Y. Wang, C. Pang, Y. Wang, J. Jin, J. Zhang, X. Zeng, R. Su, Q. Zou and L. Wei, Nat. Commun., 2023, 14, 6155 CrossRef CAS PubMed; (b) Y. Jiang, Y. Yu, M. Kong, Y. Mei, L. Yuan, Z. Huang, K. Kuang, Z. Wang, H. Yao, J. Zou, C. W. Coley and Y. Wei, Engineering, 2023, 25, 32–50 CrossRef; (c) R. S. Aal, E. Ali, J. Meng, M. E. I. Khan and X. Jiang, Artif. Intell. Chem., 2024, 2, 100049 CrossRef; (d) F. Strieth-Kalthoff, S. Szymkuć, K. Molga, A. Aspuru-Guzik, F. Glorius and B. A. Grzybowski, J. Am. Chem. Soc., 2024, 146, 11005–11017 CAS; (e) D. Meijer, M. A. Beniddir, C. W. Coley, Y. M. Mejri, M. Öztürk, J. J. J. van der Hooft, M. H. Medema and A. Skiredj, Nat. Prod. Rep., 2025, 42, 654–662 RSC; (f) Y. Wei, L. Shan, T. Qiu, D. Lu and Z. Liu, Chin. J. Chem. Eng., 2025, 77, 273–292 CrossRef CAS; (g) A. Gangwal and A. Lavecchia, J. Med. Chem., 2025, 68, 3948–3969 CrossRef CAS PubMed.
- (a) Y. Lin, R. Zhang, D. Wang and T. Cernak, Science, 2023, 379, 453–457 CrossRef CAS PubMed; (b) P. Zhang, J. Eun, M. Elkin, Y. Zhao, R. L. Cantrell and T. R. Newhouse, Nat. Synth., 2023, 2, 527–534 CrossRef CAS; (c) C. Li and R. A. Shenvi, Nature, 2025, 638, 980–986 CrossRef PubMed.
- (a) J. Dong, E. Fernández-Fueyo, F. Hollmann, C. E. Paul, M. Pesic, S. Schmidt, Y. Wang, S. Younes and W. Zhang, Angew. Chem., Int. Ed., 2018, 57, 9238–9261 CrossRef CAS PubMed; (b) E. King-Smith, C. R. Zwick, III and H. Renata, Biochemistry, 2018, 57, 403–412 CrossRef CAS PubMed; (c) S. Chakrabarty, Y. Wang, J. C. Perkins and A. R. H. Narayan, Chem. Soc. Rev., 2020, 49, 8137–8155 RSC; (d) J. He, K. Yokoi, B. Wixted, B. Zhang, Y. Kawamata, H. Renata and P. S. Baran, Science, 2024, 386, 1421–1427 CrossRef CAS PubMed.
- (a) Y. Peng, C. Gao, Z. Zhang, S. Wu, J. Zhao and A. Li, ACS Catal., 2022, 12, 2907–2914 CrossRef CAS; (b) M. Zheng, Z. Lin, S. Lin and X. Qu, Eur. J. Org. Chem., 2024, e202301066 CrossRef CAS; (c) H. Song, Z. Zhang, C. Cao, Z. Tang, J. Gui and W. Liu, Angew. Chem., Int. Ed., 2024, 63, e202319624 CrossRef CAS PubMed.
- (a) M. E. Maier, Org. Biomol. Chem., 2015, 13, 5302–5343 RSC; (b) B. Hong, T. Luo and X. Lei, ACS Cent. Sci., 2020, 6, 622–635 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |