
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organoantimony: a versatile main-group platform for pnictogen-bonding and redox catalysis
Elisa
Chakraborty
and
Robin
Weiss
 *
*
Institut de Chimie Analytique et Réactivité Moléculaire en Normandie – CARMeN, Université de Caen Normandie, ENSICAEN, INSA Rouen Normandie, Université de Rouen Normandie, CNRS, UMR 6064, F-14050 Caen, France. E-mail: robin.weiss@unicaen.fr
First published on 17th October 2025
Abstract
The chemistry of main-group elements has seen remarkable advances in organic synthesis and catalysis, redefining the boundaries of redox and acid–base reactivity traditionally dominated by transition metals. Among these developments, organoantimony chemistry has remained largely overlooked. Yet, recent discoveries have unveiled its multifaceted reactivity, spanning redox processes and Lewis acidity through the so-called pnictogen bond, positioning it as a promising underexplored platform for organic synthesis and catalysis. This Tutorial Review delves into the fundamental principles underlying organoantimony reactivity, drawing comparisons with other main-group elements while highlighting its distinctive behaviour. We examine its emerging roles in non-covalent interactions, organometallic transformations, and redox catalysis, offering a comprehensive perspective on its synthetic potential.
 Elisa Chakraborty | Elisa Chakraborty obtained her MSc degree in chemistry from the Indian Institute of Technology Bombay, Mumbai, India. She is pursuing her doctoral studies under the guidance of Pr. Robin Weiss, at Institut de Chimie Analytique et Réactivité Moléculaire en Normandie – CARMeN (France, 2024). Her present research explores heavy main-group elements for organic synthesis and catalysis. |
 Robin Weiss | Robin Weiss obtained his PhD in organic chemistry under the supervision of Dr Mamane and Pr. Pale at the University of Strasbourg (France, 2021). After a postdoctoral stint with Prof. Fürstner in the Max-Planck Institut für Kohlenforschung (Germany, 2022), he joined Pr. Jierry as a postdoctoral fellow at the Institut Charles Sadron (France, 2023). He began his independent career at the University of Caen Normandie (France, 2024), as a Junior Professor, where he holds a Chair of Organic Chemistry. His research interests cover non-covalent interactions, main-group chemistry and asymmetric homogeneous catalysis. In recognition of his achievements, he has received the 2022 René Dabard Prize for significant progress in the area of organocatalysis. |
Key learning points(1) Exploring the multifaceted reactivity of organoantimony (Lewis base, Lewis acid, and metal-like reactivity).(2) Understanding the unorthodox nature of organoantimony to establish interaction with a Lewis base via the so-called pnictogen bond. (3) Learning how organoantimony reactivity can be harnessed with appropriate ligand functionalization. (4) Gaining insight into different applications of organoantimony in catalysis and organic synthesis. (5) Identifying future perspectives and challenges in catalysis with organoantimony chemistry. |
1. Introduction
The last decades have witnessed major discoveries in the chemistry of main-group elements.1 This series of concerted efforts from the synthetic community in main-group chemistry has now undoubtedly challenged the primacy of d block elements in terms of redox and acid reactivity. One can mention several breakthroughs such as the frustrated Lewis pair concept,2,3 the use of main group elements as transition-metal-like species (B, Si, P, Sn, and Ge)4,5 and very recently bismuth,6 and the use of halogen and chalcogen as Lewis acids with the contribution of σ- and π-holes.7–9Despite the wealth of developments in the redox reactivity of main-group elements, their application in catalytic processes, as well as the exploitation of heavier p-block elements, is still in its infancy. The main difficulty stems from the substantial energy difference between frontier orbitals; consequently, redox events are often unidirectional and mainly thermodynamically driven.1 For example, low valent elements of groups 13 and 14 preferentially undergo oxidative additions, while elements from groups 16 and 17 more likely exhibit reductive elimination.10
Based on these observations, elements of group 15, the pnictogens (Pn), located halfway within p-block elements, present interesting intermediate reactivity which was exploited in numerous redox couples: PIII/PV,11 AsIII/AsV,12,13 BiI/BiIII and BiIII/BiV.14 Furthermore, it is worth mentioning the extensive development of their Lewis acid properties, which they have undergone in parallel.15–18 In comparison, organoantimony chemistry has received very little attention and its reactivity has long been relegated to the field of Lewis bases and to highly reactive Lewis acids with reference to archetypal antimony halides such as SbCl3 or SbF5.19,20
In light of the recent advances, antimony chemistry is now attracting renewed interest. Metal-like behaviour and Lewis acid properties enabling the building of strong interaction, namely the pnictogen bond, were lately pinpointed, promoting de facto organoantimony compounds to the rank of a promising platform for organic synthesis and ultimately catalysis (Fig. 1).21–23
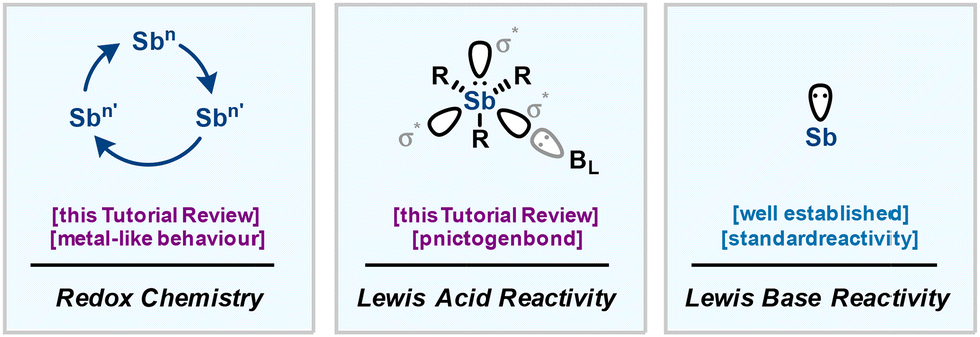 | ||
| Fig. 1 Multifaceted reactivity of organoantimony divided into three categories: redox chemistry, Lewis acid reactivity and Lewis base reactivity. | ||
This Tutorial Review aims to familiarize readers with recent advances in the burgeoning field of organoantimony, highlighting its non-orthodox reactivity based on concepts including pnictogen bonding and redox chemistry. The major goals are to (1) establish the origins of organoantimony multifaceted reactivity, (2) expose singular and similar reactivity regarding other elements, and (3) provide a comprehensive understanding and rationale of organoantimony for redox chemistry and pnictogen bonding. Towards these objectives, the present Tutorial Review is articulated in three main parts covering (i) pnictogen bonding for organic synthesis and non-covalent catalysis, (ii) organoantimony in organometallic chemistry, and (iii) organoantimony redox chemistry for organic synthesis and catalysis. Finally, and most importantly, we hope to convey not only an insight into organoantimony synthetic capabilities, but also to instigate additional effort in this fascinating field of research from synthetic organic and inorganic communities.
2. Synthesis of organoantimony compounds
While the synthetic landscape of organoantimony compounds encompasses a range of methodologies tailored to the desired oxidation state, substitution pattern, and electronic environment around the antimony center, this section focuses on the most general applicable strategies for accessing commonly encountered species, rather than providing an exhaustive and detailed account of all methods.Much of the synthetic chemistry of antimony starts with antimony halides (SbX3 and SbX5), which can be used to design the targeted structure around the Sb center. Organoantimony(III) derivatives (SbAr3) are most commonly synthesized via nucleophilic substitution of stibine halides (SbX3) with organolithium or Grignard reagents, affording stable and isolable species used as precursors in further functionalization or oxidation reactions. Synthesis of stibines SbArAr′2 flanked with different moieties can be achieved with the same procedure starting from halo-aryl-stibines SbArX2 (Fig. 2(A)). SbArX2 and SbAr2X are synthesized from SbAr3 and SbX3 through a disproportionation reaction leading to SbArX2 or SbAr2X, depending on the stoichiometry (Fig. 2(B)). The same methodology, including nucleophilic substitution with organometallic reagents, can be applied to obtain pentavalent organoantimony SbAr5 starting from SbX5. Organoantimony(V) derivatives including SbAr3X2 are typically generated via oxidation of stibines (SbAr3) using mild oxidants, such as Br2, Cl2, or H2O2. Of particular interest, 1,2-quinone derivatives can both serve as an oxidant for the Sb(III) center and, once reduced in catecholate motifs, as efficient stabilizing ligands around the antimony center. The resulting triaryl-catecholatostiboranes can alternatively be generated via substitution of triaryl-dihalo-stiboranes (SbAr3X2) with catechol derivatives. The stability of the pentavalent state is strongly influenced by the steric and electronic nature of the organic group conditioning; therefore, the purification is achieved mainly via recrystallization and, in rare cases, on gel silica chromatography. Cationic organoantimony(V) species are generally accessed through alkylation or Lewis acid mediated-abstraction of halides on stibines (SbAr3) and organoantimony derivatives (SbAr3X2 and SbAr4X) respectively (Fig. 2(C)).
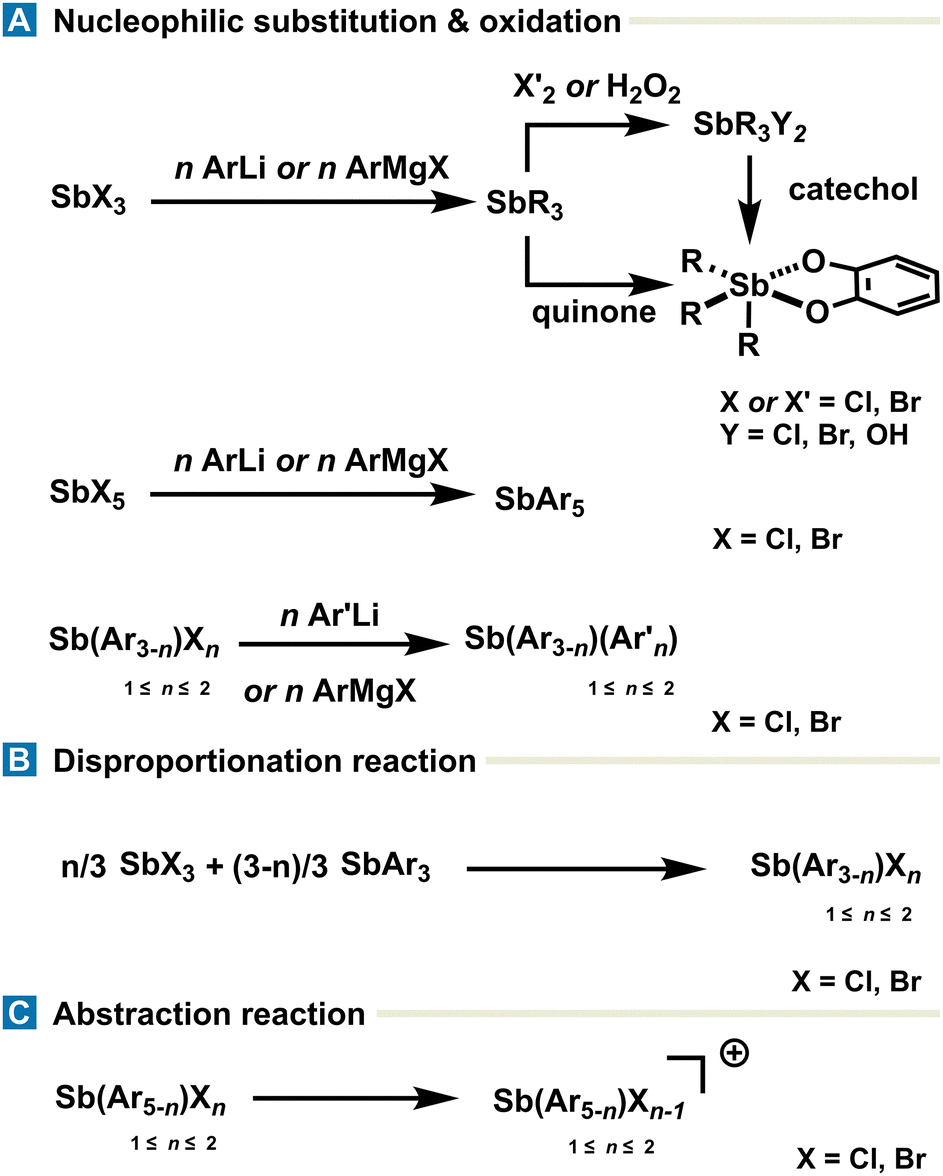 | ||
| Fig. 2 Overview of a few key syntheses of common organoantimony derivatives obtained by (A) nucleophilic substitution or oxidation, (B) disproportionation or (C) abstraction reaction. | ||
3. Pnictogen bonding for organic synthesis and noncovalent catalysis
Historically, the Lewis acidity of antimony has been assessed in Gutmann's early work24 denoting the higher affinity of SbCl5 for halides compared to BCl3. Although this property has been elegantly harnessed in superacid chemistry by Olah,25 limited effort has been put into the development of antimony-based Lewis acids until the twenty-first century. This domain is now expanding concomitantly with an improved grasp provided by the pnictogen bond model.3.1. Pnictogen bonding: features and properties
Formation of weak to strong adducts between Lewis bases and organoantimony derivatives is indicative of a certain acidity for the latter. However, the distinctive absence of a nonbonding vacant orbital in organoantimony derivatives prevents us from explaining the origin of its acidity, as in the case of a classical Lewis acid (Fig. 3). Instead, this pnictogen acidity can be rationalized by means of the σ-hole model. A σ-hole is defined as a region of depleted electron density along the extension of a covalent bond Sb–R (typically Sb–C or Sb–X), creating a site of positive electrostatic potential. This polarized region arises from the anisotropic distribution of electron density around antimony and can interact with a Lewis base to give rise to what is referred to as a pnictogen bond. Pnictogen bonding (PnB) is described as an attractive and directional interaction between an electron-rich acceptor (a pnictogen bond acceptor), such as a Lewis base (A), and the positively charged electrostatic surface, namely a σ-hole, of a pnictogen (a pnictogen bond donor) (Fig. 4(A)). In addition to the pnictogen bond definition provided by IUPAC,26 fundamental theoretical investigations have unambiguously contributed to its classification.27 Very importantly, it is worth noting that the σ-hole, located at the antipode of the σSb–R covalent bond, overlaps intrinsically with the σ*Sb–R/X antibonding orbital which can therefore also interact with the pnictogen bond acceptor via a charge transfer. Thus, the pnictogen bond can be described as a contribution of electrostatic interactions through the σ-hole model and charge transfer through orbital interactions (Fig. 3).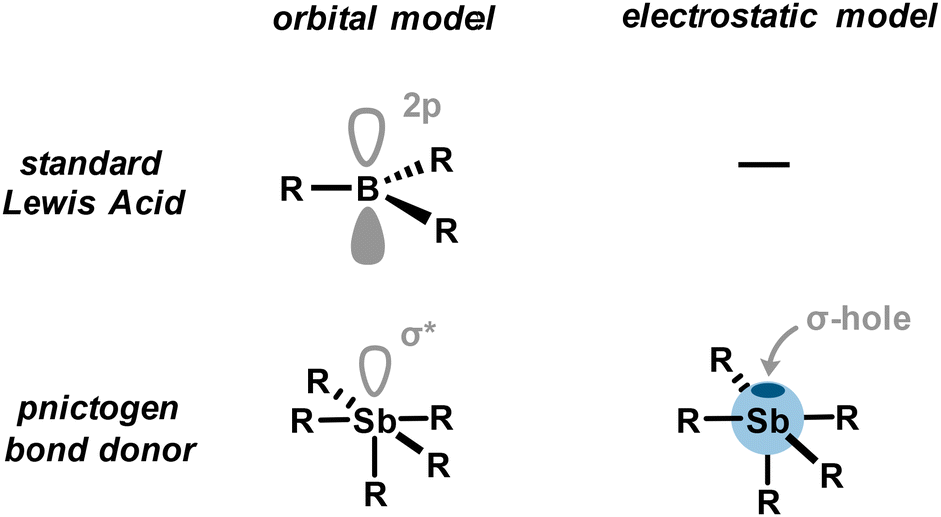 | ||
| Fig. 3 Comparison of models employed for a standard Lewis acid and pnictogen bond donor. | ||
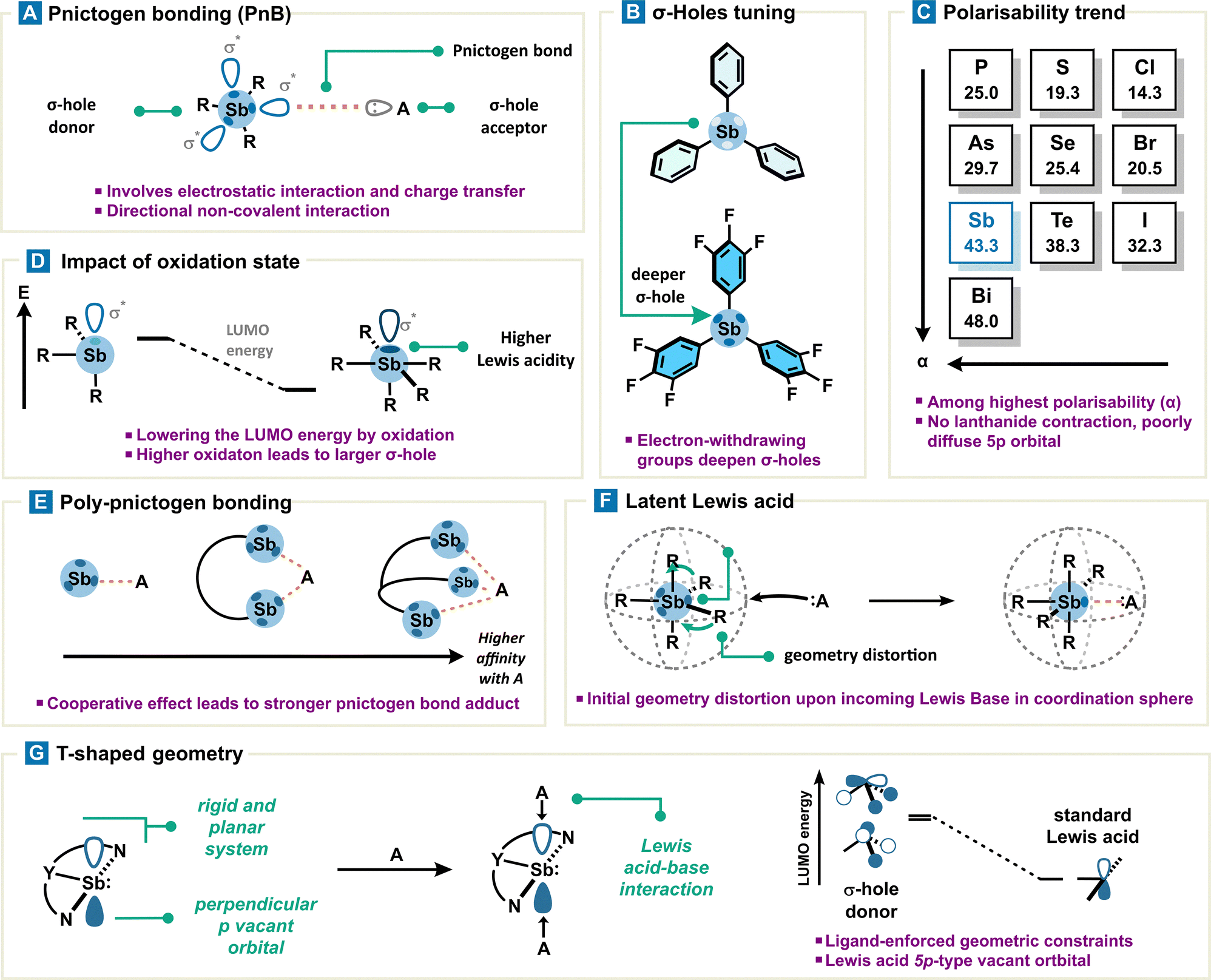 | ||
| Fig. 4 (A) Description of a pnictogen bond between R3Sb and a Lewis base (A) and (B) increase of σ-hole depth (light to dark blue) by introduction of an electron-withdrawing group (EWG), (C) polarizability trend in the periodic table, (D) effect of oxidation state at the Sb center on σ-hole and the LUMO leading overall to an higher Lewis acidity, (E) schematic representation of poly-pnictogen bonding, (F) distortion of geometry induced by a Lewis base leading to the formation of a pnictogen bond, and (G) properties of antimony as a function of its geometry: evolution of a pnictogen bond donor (pyramidal geometry) towards a genuine antimony-based Lewis acid (planar geometry). | ||
Consequently, pnictogen bond donors may be regarded as a particular class of Lewis acids. Like conventional Lewis acids, they are capable of accepting an electron pair; however, the accepting orbital is an antibonding σ* molecular orbital rather than a nonbonding atomic orbital. This distinction implies that the balance of interactions differs, combining a well-defined electrostatic potential (the σ-hole) with a characteristic orbital framework that sets them apart from classical Lewis acids. These differences give rise to unique features such as weaker and reversible PnB which are, however, generally more robust compared to other non-covalent interactions (e.g., hydrogen bond, π-stacking, and dispersion forces). Thus, for the sake of clarity and due to close similarities, PnB can be considered as the non-covalent counterpart of a classical Lewis acid. By extension to the latter and keeping in mind these differences but also similarities, it is therefore frequent to refer to the Lewis acidity of a pnictogen bond donor.
Overall, PnB is defined as a non-covalent interaction whose bonding energy ranges from 10 to 60 kcal mol−1. PnB involving antimony is also distinguished by a superior covalency degree in comparison with other elements of group 15, 16 and 17, due to better charge transfer contributions. Indeed, the latter can contribute significantly to the pnictogen bond which can result in an interaction energy twice as great. To this extent, PnB may be rather considered as a coordination bond in some cases.
Antimony presents inherent features that distinguish its uniqueness. The knowledge and proper application of these properties should allow the design of an effective pnictogen bond donor, which can be summarized as follows:
• Lewis acidity is proportional to the electro-attractivity of the rest of the molecule; therefore, the presence of an electron-withdrawing group (EWG) strengthens PnB by depleting electron density around antimony (Fig. 4(B)). This has the beneficial effect of further deepening the σ-hole. Additionally, the introduction of a positive charge on the organoantimony scaffold or antimony center substantially increases its Lewis acidity.28
• As a general rule, σ-hole based non-covalent interaction strengthens with heavier atoms, in concert with σ-hole donor atomic polarizability (α). A diffuse electronic cloud also allows an easier electronic density relocation around the σ-hole donor. In consequence, the position of Sb in the periodic table (αSb = 43.3) makes it a natural choice regarding other pnictogens (αAs = 29.7, αP = 25.0) and other columns (αTe = 38.3, αSe = 25.4, αS = 19.3, and αI = 32.3, αBr = 20.5, αCl = 14.3) (Fig. 4(C)).
• The exception to the preceding principle is often observed with organobismuth, which forms a weaker pnictogen bond with respect to organoantimony, despite a higher polarizability (αBi = 48.0). Antimony orbital considerations allow a simple rationalization by comparing the 5p valence orbitals which are less diffused than the 6p valence orbitals of Bi. Therefore, Bi-based PnB suffers from stronger Pauli repulsion which cannot be compensated. This discrepancy within the pnictogen family arises from the lanthanide contraction leading to only a small variation (0.09 Å) in covalent radii from Sb to Bi (1.39 vs. 1.48 Å). This smaller-than-expected size variation results in shorter-than-expected covalent bonds with Bi, which electronically repel any incoming Lewis base more easily (Pauli repulsion) and in fine weaken the Lewis acidity of Bi compared to Sb.29 In addition, Sb-based PnB preeminence over Bi-based PnB with a hard Lewis base (O, N) also results from size compatibility of orbitals with regard to the hard–soft acid–base theory.30
• A higher oxidation state leads to stronger σ-holes and energy-lowered σ*Sb–R orbitals at the metalloid center, propelling antimony V to the rank of a better pnictogen bond donor in comparison with antimony III.31 More precisely, oxidation of the antimony center increases the interaction energy of PnB by ca. 30 to 50% (Fig. 4(D)).
• A tethering effect might occur if a Lewis base or a bulky group is present on the organoantimony scaffold: this results in hindering of the pnictogen bond formation with an external Lewis base. Usually, this effect can be observed with aryl-antimony flanked in the ortho position with a pnictogen bond acceptor or bulky substituent.32,33
• Use of a multivalent pnictogen bond donor enhances substrate binding via a cooperative effect involving more than one pnictogen bond (Fig. 4(E)).34–36
• Pentavalent organoantimony adopting a trigonal bipyramid type geometry can be considered as a latent pnictogen bond donor due to its sterically hindered σ-holes. Nevertheless, PnB formation is still possible: upon approach of a Lewis base, the Sb coordination sphere distorts its initial geometry into an octahedral complex, revealing in fine an accessible σ-hole (Fig. 4(F)). The strain energy associated with organoantimony deformation is even lower when initial geometrical constraints are already present.37
• Constraining antimony geometry from a pyramidal to a planar form with adequate ligands leads to a complete molecular orbital rearrangement and in fine a different reactivity. Non-planar antimony derivatives are usually characterized by both high nucleophilicity and electrophilicity; most importantly, the lowest unoccupied molecular orbital (LUMO) consists mainly of a vacant p orbital at the Sb center. In this peculiar case, the interaction based on a vacant p orbital and a Lewis base can’t be considered as a pnictogen bond since no σ* orbitals are involved.
3.2. Organoantimony derivatives as Lewis bases and acid coordination sites
Organoantimony chemistry displays several reactivities. Hence, before applications of PnB are detailed below, other reactivities more related to the field of covalent chemistry such as basicity and coordination at the antimony center will be succinctly presented to avoid confusion in this section.By extension to lighter pnictogens, organoantimony features a Lewis base character through its electron lone pair. This facet was historically identified with electron-rich stibines being able to react with various acids.38 This property was also clearly underlined in metallic complexes including triarylstibine and stibinidene, whose antimony free electron pairs are delocalized towards metal centers such as palladium or {W(CO)5} fragments, acting thus as L-type ligands (Scheme 1).39,40
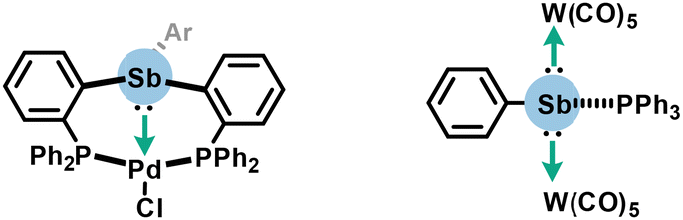 | ||
| Scheme 1 Complexes featuring the Lewis base character of organoantimony. | ||
In contrast, organoantimony derivatives also act as strong acid sites, which, in the presence of Lewis bases, could lead to the formation of covalent bonds. Interestingly, their formation involves a σ-hole. The transition state leading to their formation of these robust adducts involves a pnictogen bond which is fleetingly formed before evolving into a covalent bond. As a consequence, the formation of the new covalent bond leads to the subsequent cleavage of another Sb–R bond or the formation of an ate complex (Fig. 5(A)). Such a non-isolable pnictogen bond is called a transient PnB. A typical example of a transient pnictogen bond driving the formation of an ate complex is the binding of fluoride or chloride for sensing or anion transport applications.41 Another relevant example of transient PnB is the complexation of formaldehyde with phosphino-stiborane (Fig. 5(B)),42 which may suggest new potential applications due to a reactivity reminiscent of frustrated Lewis pairs. While the formation of a covalent bond may involve transient PnB, this aspect will not be addressed in the next discussion which highlights instead non-covalent catalytic processes driven by PnB.
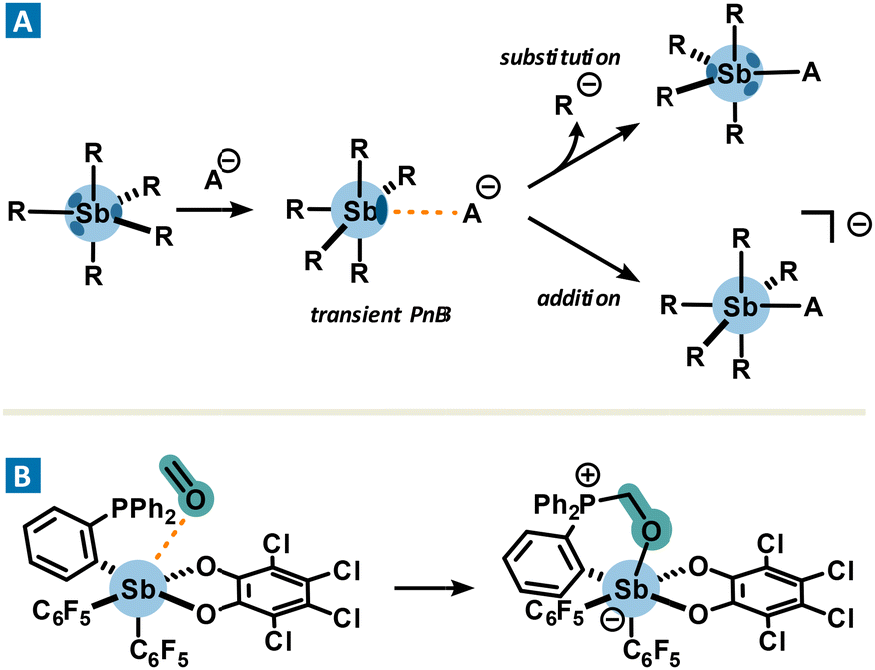 | ||
| Fig. 5 Formation of a transient pnictogen bond resulting in a substitution (A) or an addition reaction (A) and (B). | ||
3.3. Pnictogen bonding for organic synthesis and catalysis
For the practical use of organoantimony reagents in organic synthesis, stability and ease of handling are critical. The sensitivity of antimony-based pnictogen bond donors toward oxygen and moisture strongly depends on both the oxidation state of antimony and the supporting ligands. Electron-deficient trivalent stibines are prone to hydrolysis and degradation in the presence of air and water, whereas electron-rich stibines can often be stored on the bench without special precautions. More elaborate cationic complexes bearing weakly coordinating anions are generally more sensitive, requiring rigorously dried solvents and inert-atmosphere techniques. Yet, several recent studies have shown that certain acidic stibonium salts remain air-stable and catalytically active under open-flask conditions, thereby broadening their synthetic utility.43,44 In contrast, many Sb(V) derivatives, most notably pentavalent catecholatostiboranes, exhibit remarkable robustness, tolerating air and moisture over extended periods without decomposition.41,45 Overall, while highly electrophilic Sb(III) species demand careful exclusion of oxygen and water, the most developed Sb(V) family, catecholatostiboranes, can be regarded as bench-stable reagents, thus offering practicing chemists a series of chemicals ranging from sensitive to water- and air-tolerant systems.Lewis acid properties of organoantimony in catalysis have been reported since the early 2000s, despite the absence of any model at that time. In 2010, the Wong group introduced an azastibocine perfluoroalkylsulfonate whose stability was attributed to an intramolecular pnictogen bond which simultaneously rigidifies the molecular scaffold (Scheme 2).46 This catalyst was successfully used in a highly diastereoselective Mannich reaction, leading to beta-amino ketones in sustainable solvents such as water. The catalytic activity was unequivocally attributed to the Lewis acidity of the antimony center, deepened by the perfluoroalkylsulfonate moiety. Importantly, no selectivity was observed with the organobismuth analogue, despite its catalytic efficiency.47
 | ||
| Scheme 2 Mannich reaction catalysed with azastibocine derivatives. | ||
As established in a systematic study performed by Gabbaï's group in 2014, tetraarylstibonium salts are potent pnictogen bond donors and have a propensity to activate oxygen-based Lewis bases such as tetrahydrofuran. Capitalizing on these results, the same group reported activation and hydrosilylation of benzaldehyde derivatives with a bifunctional pnictogen bond donor bearing two triarylmethylstibonium cations. Unambiguously, the authors demonstrated a mechanism occurring via carbonyl activation, with a stronger catalytic effect ascribed to a double electrophilic activation due to the ditopic nature of distibonium salts (Scheme 3).
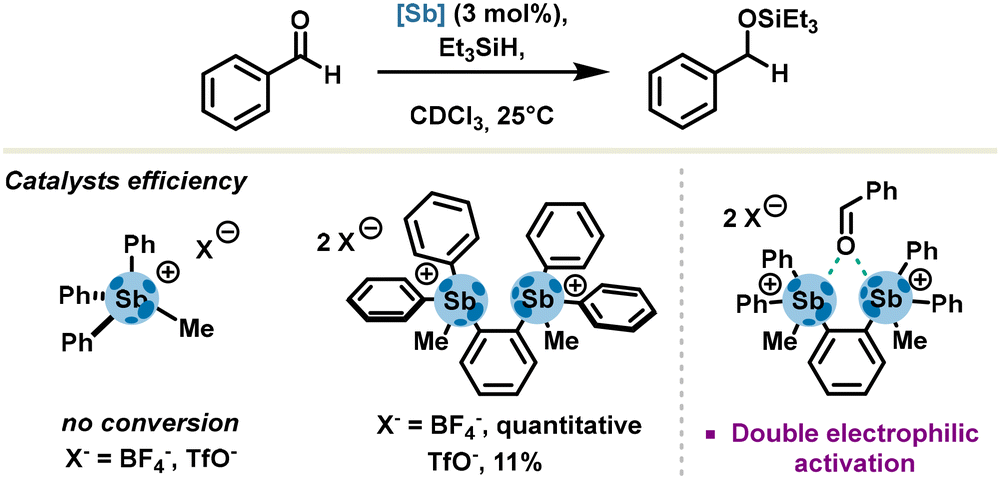 | ||
| Scheme 3 Hydrosilylation of an aldehyde with stibonium salts. | ||
Concomitantly, the Hudnall group also took advantage of these features, using stibonium salts in acetalization reactions with catalyst loadings as low as 0.1 mol%.48 In all these examples, high catalytic activity of stibonium salts is denoted with a very low catalyst loading, similar to those usually employed with metals.
The pnictogen bond concept in catalysis was first introduced by the Matile group in 2018,49 in a comprehensive study establishing the direct relationship between catalytic activity, σ-hole depth, and polarizability of σ-hole donors. Due to a stronger polarizability, antimony-based σ-hole donors have demonstrated the best results in benchmark reactions such as Reissert-type substitution of isoquinolines and substitution of 1-chloroisochromane in the presence of silyl enol ether (Scheme 4). High yields (up to 91%) were obtained with 20 mol% of catalyst loadings. Due to the propensity of σ-hole donors to form strong adducts in the presence of halides, the asserted mechanism involves chloride abstraction, releasing a highly reactive onium ion which subsequently undergoes nucleophilic addition in the presence of the silyl enol ether. PnB surpassed halogen bonding and chalcogen bonding, affording a reaction rate increase from 3 to 80-fold. As expected, substitution of stibine with highly electron-withdrawing pentafluorophenyl motifs led to the best catalytic activity, with a σ-hole deepening of 39% in comparison with triphenylstibine.
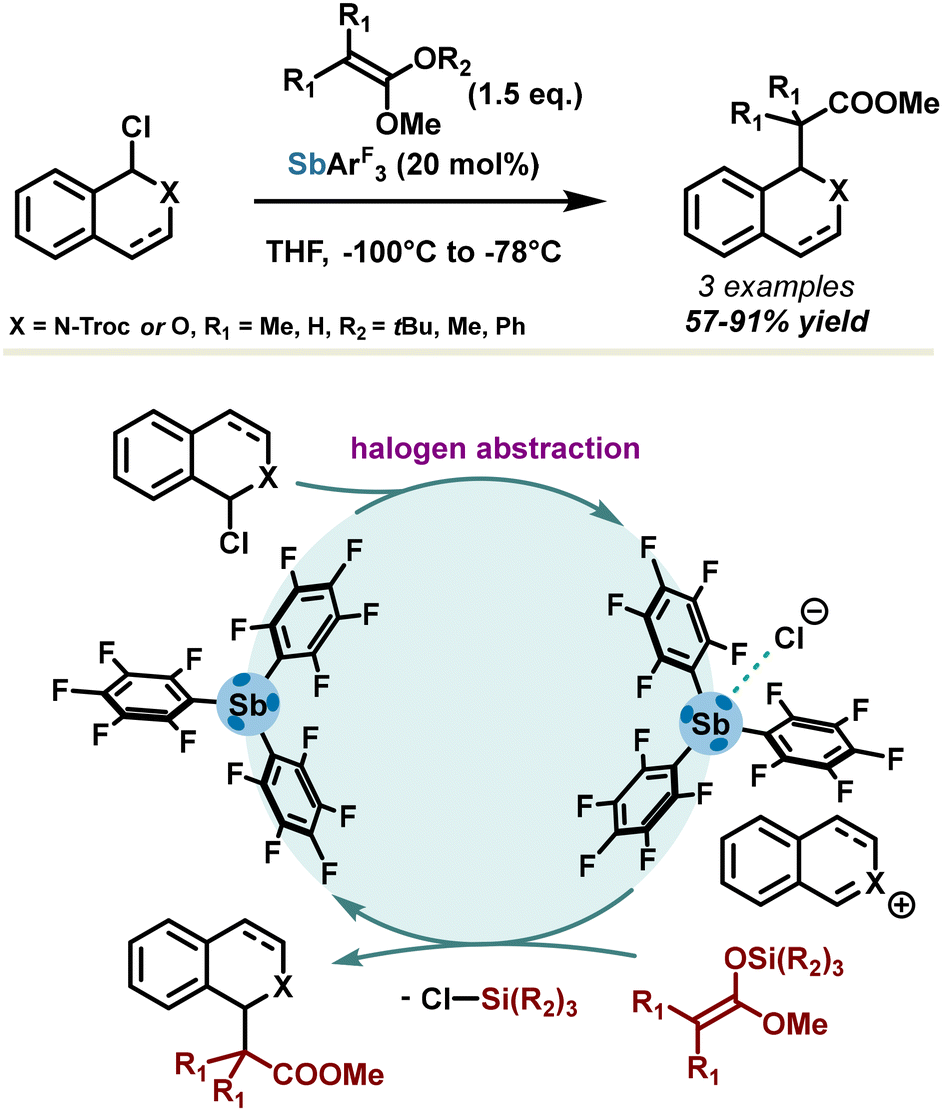 | ||
| Scheme 4 First proof-of-concept of halogen abstraction catalysis invoking the PnB concept. | ||
In continuation of these pioneering groundworks, an additional series of antimony-based catalysts were developed on the basis of the PnB rationale. In view of strengthening σ-holes, features such as positive charge at the antimony center, a strong electron-withdrawing motif, a high oxidation state (+V) and bidentate motifs were targeted. In order to evaluate new pnictogen bond donors, transfer hydrogenation of quinolines with Hantzsch ester was recurrently selected as a standard benchmark reaction due to its proven responsiveness towards σ-hole-directed catalysis (Scheme 5).50
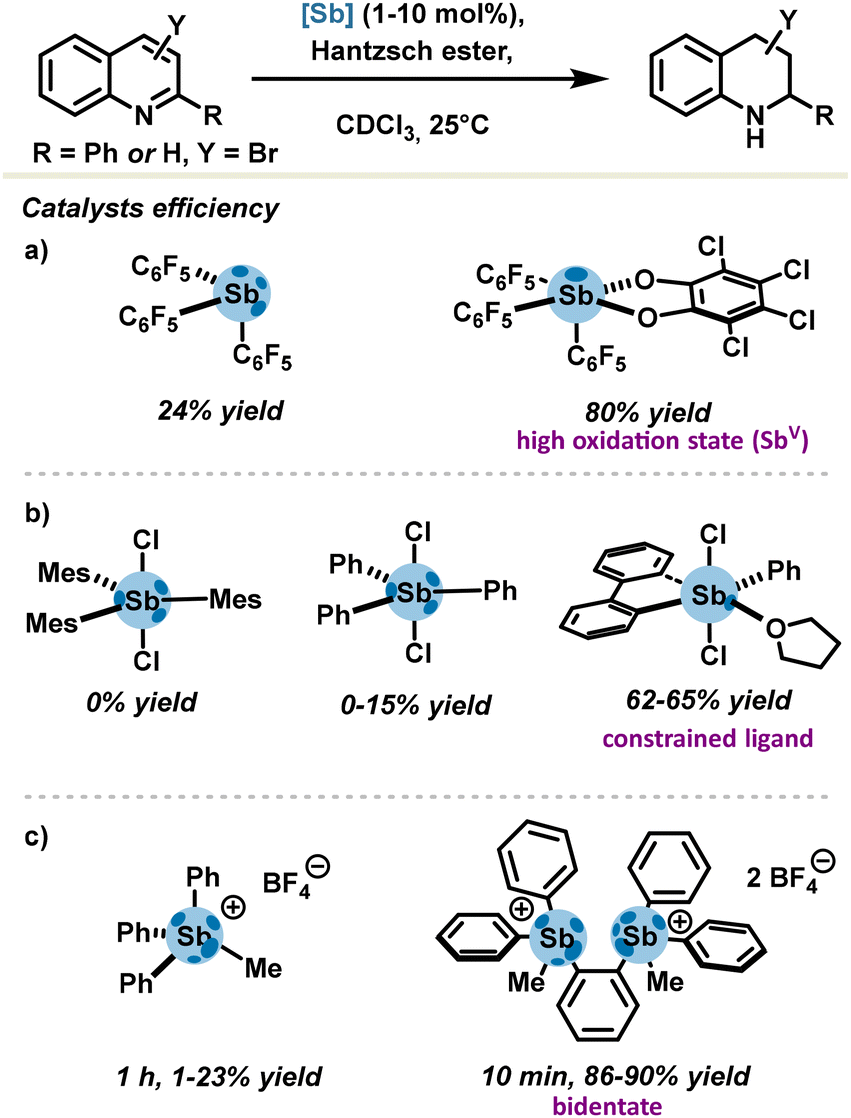 | ||
| Scheme 5 Transfer hydrogenation of quinolines catalysed by pnictogen bonding with (a) fluorinated organoantimony, (b) sterically hindered and accessible stiboranes, (c) and stibonium salts. | ||
The tris(pentafluorophenyl)stibine gave poor conversion in comparison with the corresponding stiborane (Scheme 5(a)) (24% yield versus 80%). Although the stiborane is fitted with only one σ-hole, it presents a higher Lewis acidity which arises from the high oxidation state of antimony V (Fig. 4(D)).31 Notably, pre-organization around the metalloid center is of great importance and drastically impacts the outcome of a reaction. Gabbaï's group compared the catalytic activities of several dichlorostiboranes (Scheme 5(b)). Dichloromesitylstiborane was completely inactive, very certainly due to the important tethering effect from mesityl motifs, precluding any access to σ-holes. Reduction of steric hindrance in dichlorophenylstiborane did not lead to considerable improvements; up to 15% yield was solely observed.37 This result may be explained by an excessive energy cost for geometry rearrangement when transitioning from the ground state to the PnB complex. In order to bypass this activation strain, the dichlorostiborane can be replaced by the 5-phenyl-5,5-dichloro-λ5-dibenzostibole whose antimony center is subjected to a greater strain in the 5-membered heterocycle. The latter thus requires a lower strain energy to access the prerequisite octahedral geometry for PnB complex formation and is in fine more Lewis acidic by virtue of its pre-templated geometry. The new pnictogen bond donor was also isolated as an adduct via an Sb⋯O pnictogen bond with the solvent (in this case, tetrahydrofuran) which is depicted in Scheme 5(b), thus confirming higher σ-hole availability and Lewis acidity. In addition, good catalytic activity was observed with catalyst loadings as low as 1 mol%.
In 2019, Gabbaï's group also demonstrated in the same transfer hydrogenation of quinolines the excellent catalytic activity of a bis-stibonium salt (Scheme 5(c)), which was attributed to a double activation mode of a substrate through a doubly coordinated bimolecular complex involving two Sb⋯N interactions, one of which was clearly identified as a PnB.51 The bis-stibonium salt achieved up to 90% yield with 5 mol% of catalyst loadings. In comparison, the control mono-stibonium salt delivered a 24% yield at best, underlining the importance of pre-organization effects rather than those featured in bidentate catalysts.
Even though stibonium salts do not suffer much strain energy re-organization, ligand motifs sometimes lead to a reduced catalytic activity due to the tethering effect. In another systematic study, Gabbaï's group examined the influence of steric hindrance around the Sb center within a series of tetraarylstibonium triflates (Scheme 6).52 Unhindered tetraphenylstibonium triflate, with 10 mol% catalyst loading, achieved a good yield (53%) in a model cycloaddition reaction between oxiranes and isocyanates affording oxazolidinones as a mixture of two regioisomers. Steric tethering of σ-holes with methyl motifs led to a slight deterioration of the yield (46%). In contrast, the presence of σ-hole stopper motifs, namely intramolecular σ-hole acceptor motifs, drastically diminished yields up to 6% by preventing any possibility of establishing an external pnictogen bond.
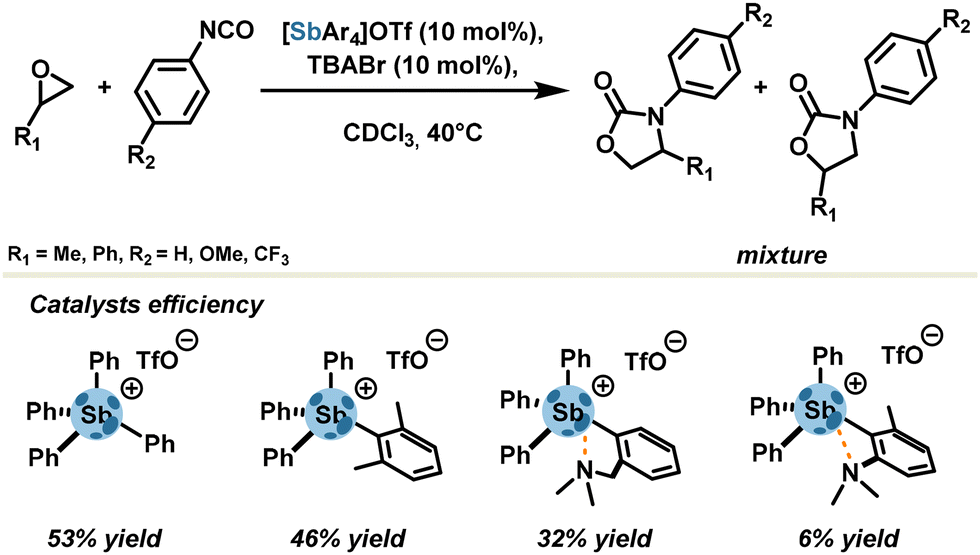 | ||
| Scheme 6 Synthesis of oxazolidinone catalysed by tetraarylstibonium cations. | ||
In parallel, it should be noted that the Takagi group has also used stibonium salts in cationic polymerization of styrene derivatives. In conjunction with σ-hole activation rules, the stibonium cation flanked with the most electron withdrawing groups offered the highest catalytic activity (with a monomer conversion of 91% in 8 h).53
In the context of asymmetric catalysis, the Zhang group achieved a significant foray in 2021, reporting the first stable stibonium stiborate salt in enantiocontrolled transfer hydrogenation reactions.54 A variety of substrates such as quinolines, benzoxazines and benzoxazinones were reduced in excellent yields, with catalyst loadings as low as 0.05 mol%. Enantioselectivity up to 98% ee could be achieved via an asymmetric-counter-ion-directed-catalysis mechanism due to a weakly coordinating chiral stiborate anion (Scheme 7). This is the first example of chiral induction with antimony-based PnB, which paves the way towards further development of chiral PnB catalysts. One major challenge remains to be the introduction of chirality directly on the σ-hole donor, providing access to a putative class of neutral chiral pnictogen bond donors.
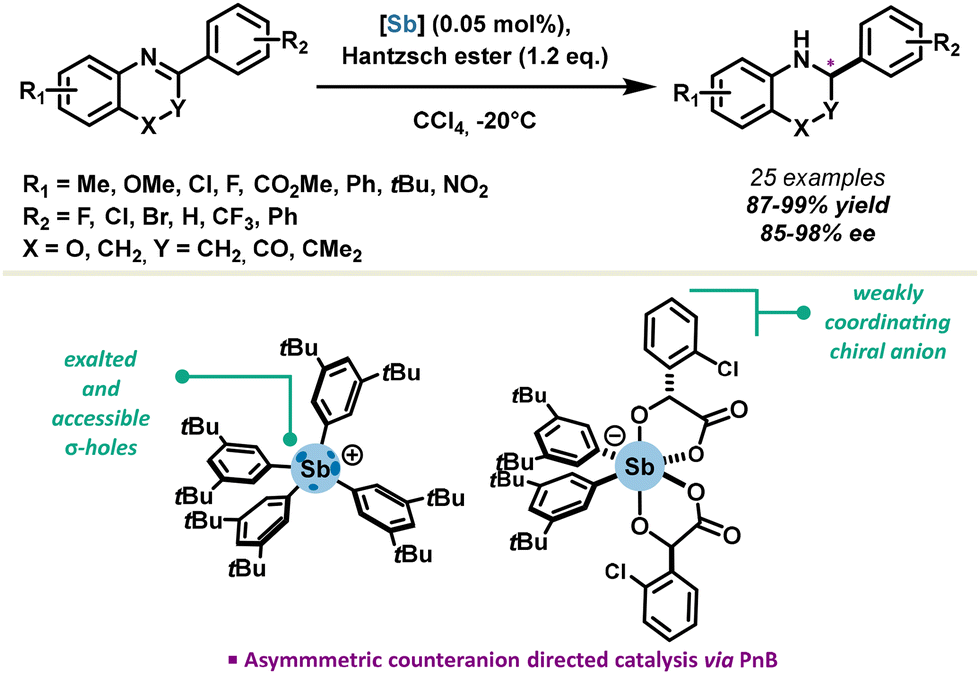 | ||
| Scheme 7 Asymmetric pnictogen bonding catalysis. | ||
As extensively exemplified by the Matile group, the emergence and importance of organoantimony in the field of catalysis might also be attributed to its unique features, which are certainly more than just extensions of other σ-hole donors. As a first incursion, epoxide-opening polyether cascade cyclisations were systematically studied and used as a responsive tool for supramolecular catalysis assessment.33 PnB catalysis distinctively provided 6-endo-tet anti-Baldwin products, with important variations between stibines and stiboranes as PnB catalysts regarding regio- and stereoselectivity. A cyclisation reaction example is depicted with a simple monoepoxide reagent in Scheme 8(a), catalyzed by several Lewis acids (Scheme 8(b)). Covalent Lewis acid catalysis reasonably led to the Baldwin product, with Lewis acids such as BF3 or SbCl3 as well as other σ-hole donors based on bismuth, tin or germanium.32 This peculiar reactivity may be attributed to multi-supramolecular coordination enabled by hydrogen bonding, PnB and lonepair–π interactions in the presence of polyepoxide substrates. By contrast, the previously cited interactions, combined with the highly acidic Sb center and the hydrophobic platform of stiboranes, lead to a possible catalyst–substrate complex giving rise to this unorthodox selectivity (Scheme 8(c)). This underlying mechanism was investigated and proposed later on.55 These lessons on antimony catalysis led authors to describe PnB catalysis as the non-covalent supramolecular counterpart of classical covalent Lewis acid catalysis.
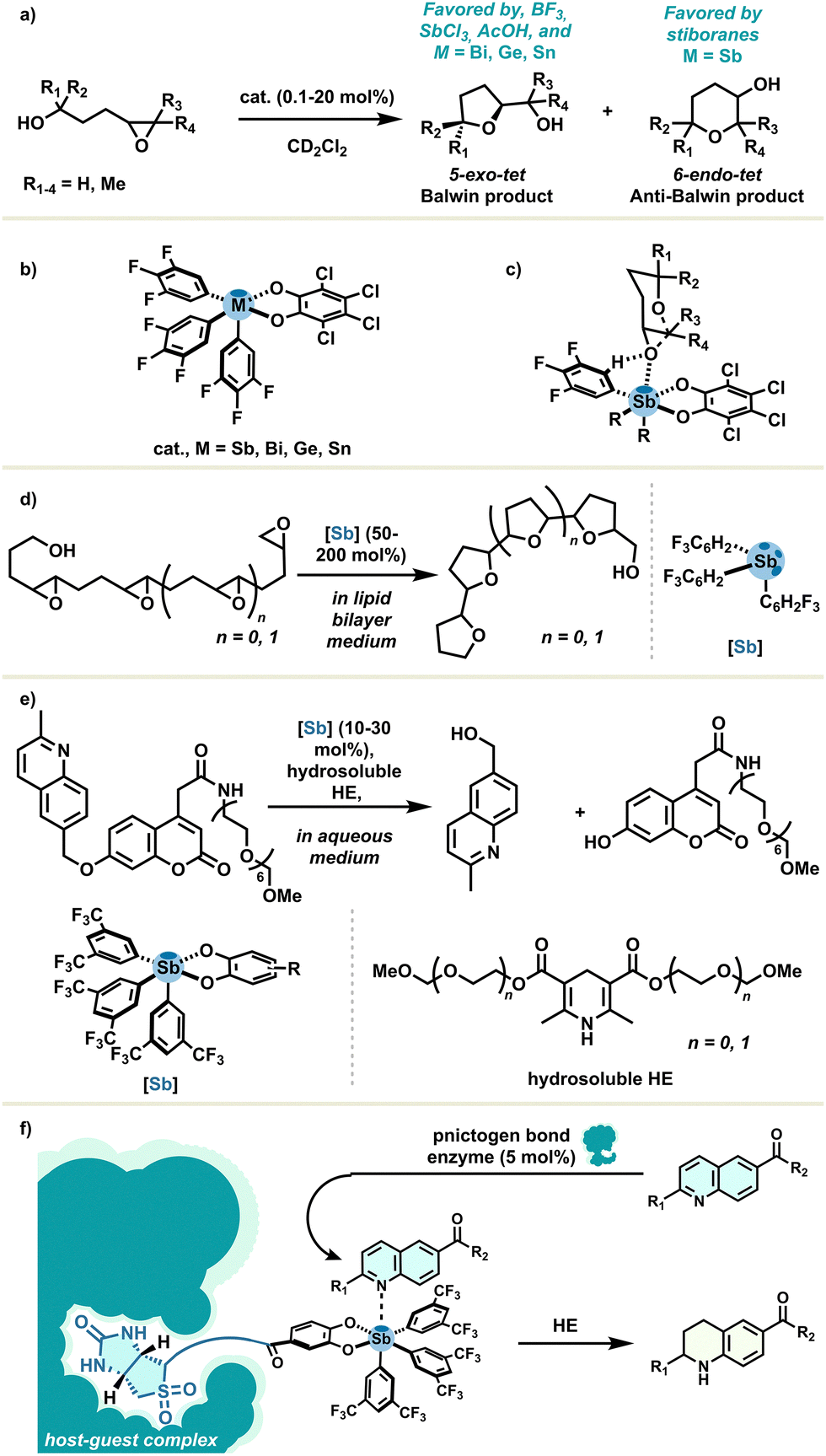 | ||
| Scheme 8 (a) Epoxide-opening cyclisation with (b) several σ-hole donors, (c) potential catalyst–substrate transition state, (d) oligoepoxide-opening cyclisation in a lipid bilayer membrane, (e) transfer hydrogenation reaction with hydrosoluble starting materials using standard stiboranes, and (f) transfer hydrogenation reaction catalysed by pnictogen bond enzyme. | ||
The propensity of organoantimony to engage in transmembrane transport has received a lot of attention.41 In 2021, the same group reported this notion in combination with PnB catalysis, which was found to occur with a rate enhancement beyond one million in oligoepoxide-opening cyclization in lipid bilayer membranes (Scheme 8(d)), whereas no conversion was observed in solution.56
Capitalising on the strong catalytic activity of pnictogen bond donors in non-trivial environments, the authors established, afterwards, a transfer hydrogenation reaction with hydrosoluble fluorogenic quinolines and Hantszch ester in aqueous medium, catalysed with stiboranes (Scheme 8(e)). This remarkable result underlines the ability of non-polar σ-holes to catalyse in aqueous systems, allowing us to move one step further towards biological conditions and promising valuable contributions for sustainable antimony chemistry.57 Building on this momentum, the Matile group proposed host–guest complexes between wild-type streptavidin enzymes and biotinylated stiboranes for catalysing the hydrogenation transfer reaction (Scheme 8(f)).58 Remarkably, σ-hole donor–artificial enzyme complexes were found to operate clearly with PnB at the micromolar level. In addition, the chiral environment provided by enzymes has improved the diastereoselectivity outcome very modestly.
Trivalent antimony also has its place in pnictogen bond catalysis: for illustration, the Venugopal group reported in 2021 the efficient reduction of oxide phosphine into corresponding phosphine under mild conditions with silane as the hydrogen source. The catalyst, a stibenium triflate, was used with 10 mol% loading and achieved up to 92% yield. Remarkably, stibenium salt proved to be more effective with respect to bismuthenium salt (Scheme 9).59 This observation was correlated with computational calculations revealing a LUMO lowered in energy for antimony derivatives.
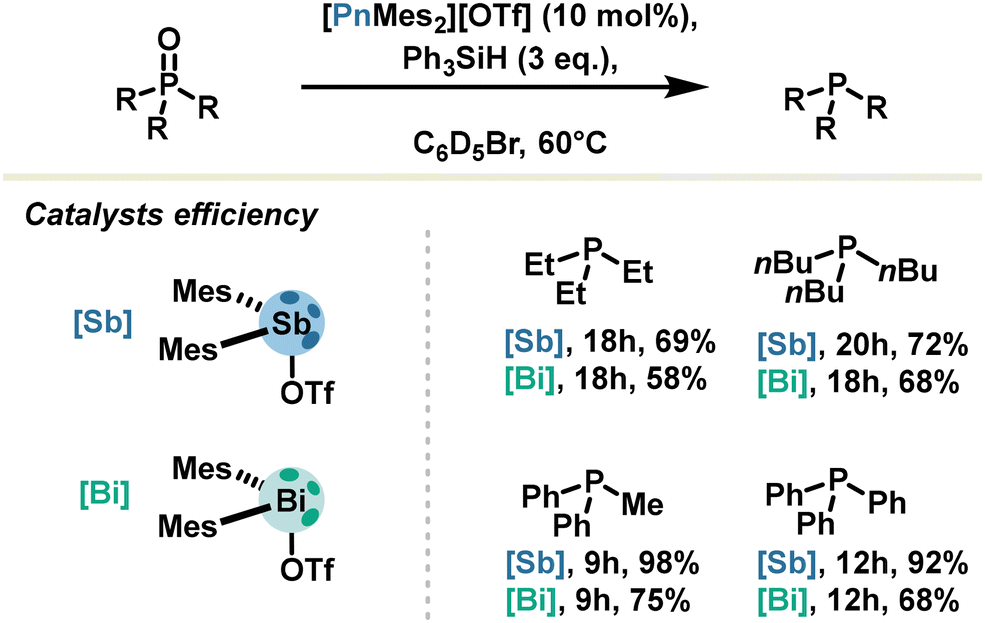 | ||
| Scheme 9 Oxide phosphine reduction catalysed by bismuthenium- and stibenium salts. | ||
The same group went one step further in the stibenium salt design by flanking the catalyst scaffold with a Lewis base in order to generate an intramolecular stabilizing Sb⋯N pnictogen bond. Aluminate anion was used as an extremely weakly coordinating counteranion for unleashing the Lewis acid potential. The resulting stibenium salt generated in situ proved to be highly acidic and was used respectively in a cross-, a ring opening- and a ring closing carbonyl olefin metathesis with moderate to good yields and 10 mol% of catalyst loadings (Scheme 10(a)).60
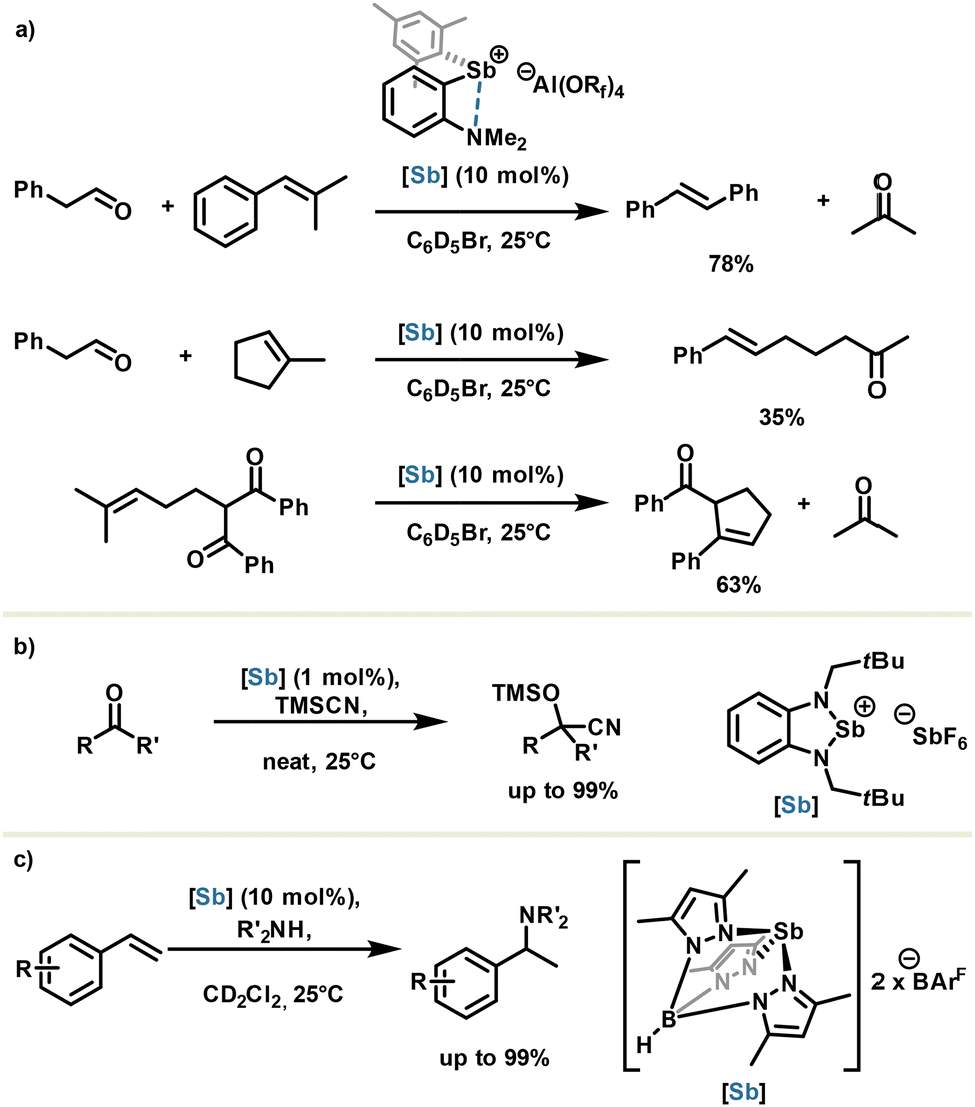 | ||
| Scheme 10 (a) Carbonyl olefin metathesis, (b) cyanosilylation and (c) hydroamination reactions catalyzed by cationic antimony derivatives. | ||
Other stibenium salts were also described as being highly active in the cyanosilylation reaction (Scheme 10(b)),61,62 and very recently, in 2025, the Venugopal group reported the metal-free hydroamination of styrene using a highly constrained stibenium with a trispyrazolylborate moiety (Scheme 10(c)).63 This highlights the importance of ligand design for organoantimony reactivity.
4. Organoantimony derivatives for organometallic chemistry
4.1. Organoantimony, a not-so-innocent ligand
Antimony-based ligands are also used in metallo-organoantimony complexes due to their free electron pairs. For example, 2,2′-bis(diarylstibino)-1,1′-binaphthyl (BINASb) was reported as an efficient Ligand in rhodium complexes for ketone hydrosilylation. Noteworthily, partial enantioselectivity was obtained with BINASb complexes, up to 34% ee; whereas almost no chiral induction was observed with inferior homologue complexes based on 2,2′-bis(diarylphosphino)-1,1′-binaphthyl (BINAP) (Fig. 6(a)).64 In a comprehensive work of organometallic synthesis and antimony-based ligand investigation, the Gabbaï group reported a large palette of antimony–metal complexes including late transition metals such as iridium, rhodium, platinum, palladium, gold and nickel.40,65 According to the organoantimony nature, the Sb⋯M coordination type may differ. First, stibines which are good L-ligands enable donor–acceptor interaction Sb → M from the antimony as the donor towards the metal as the acceptor. This peculiar interaction greatly influences the transition metal behaviour by polarizing electron density in the Sb⋯M bond. It is worth noting that the Lewis acidic antimony center may concurrently engage in a secondary interaction with an external Lewis base at the antipode of the Sb–M bond, via the σ*Sb–M molecular orbital (Fig. 6(b)). The overall effect results in a weaker Sb⋯M interaction. | ||
| Fig. 6 (a) Application of ligand antimony in the hydrogenation reaction, (b) ligand-non-innocence and (c) redox-non-innocence in an antimony derivative, and (d) hydroamination of alkynes with a non-innocent antimony–gold complex. | ||
This effect, termed by the Gabbaï group as coordination-non-innocence, offers the possibility to fine-tune metal reactivity. In the second place, two-electron oxidation of heterobimetallic Sb(III)–metal complexes may occur partially or completely at the redox-non-innocent antimony center. Resulting Sb(V)–metal complexes display strong acidity at the Sb center which in turn reverts Sb⋯M interaction into a new donor–acceptor interaction M → Sb from the metal as the donor towards antimony as the acceptor (Fig. 6(c)). Taking advantage of both ligand- and redox-non-innocence of ambiphilic antimony ligands, the Gabbaï group achieved synthesis of Sb–metal complexes with exacerbated reactivity at the metal center, which were nicely exemplified in catalysis.
One of the most striking examples is the hydroamination of acetylenes with antimony–gold complexes in which a high oxidation state Sb(V) was revealed to be vital for catalytic activity (Fig. 6(d)).40 To this end, the authors selected a neutral trifluorostiborane moiety as a Z-ligand for activating the metal center.66 Later on, the authors demonstrated the benefits of monocationic and dicationic stibonium as Z-ligands, respectively, in cycloisomerisation of 1,6-enyne and polymerisation and hydroamination of styrene.67,68
4.2. Metal–antimony exchange in cross-coupling reactions
The C–Sb bond has nearly half the bond dissociation energy of a C–C bond (ESb–C = 215 kJ mol−1).69 For this reason, organoantimony derivatives might be considered as transmetalation agents which can be synergistically coupled in metal-catalysed reactions. As a common feature, antimony-based cross-coupling usually requires mild conditions without additives such as bases. Notably, organoantimony compounds are known to be effective in carbon–carbon bond formation such as Heck-, Hiyama-, Stille- and Suzuki-type cross-coupling reactions.70,71 A representative example is a pallado-catalysed oxidative cross-coupling of aryl trifluoroborates and aryl stibines, reported in 2018 by Chaplin and Hooper (Scheme 11(a)). The use of aryl stibines affords very good selectivity in favor of cross-coupling versus undesired homo-coupling, with respect to other non-antimony-based aryl transfer reagents such as BiPh3, PbPh4, SnPh4 and BPh3. This methodology is of certain interest owing to the ready availability of stibines in only one step and good to excellent yields obtained with both electron-rich and electron-poor stibines.72 Application of less toxic and stable organoantimony has been described by the Yasuike group using difluorostiborane in the synthesis of 4-arylcoumarins via an oxidative Heck-type arylation followed by a cyclization (Scheme 11(b)).73 One can notice that additive-free and aerobic conditions yielded the desired products in good to excellent yields. The organobismuth analogue failed to provide corresponding 4-arylcoumarins in appreciable yield (35% in 24 h).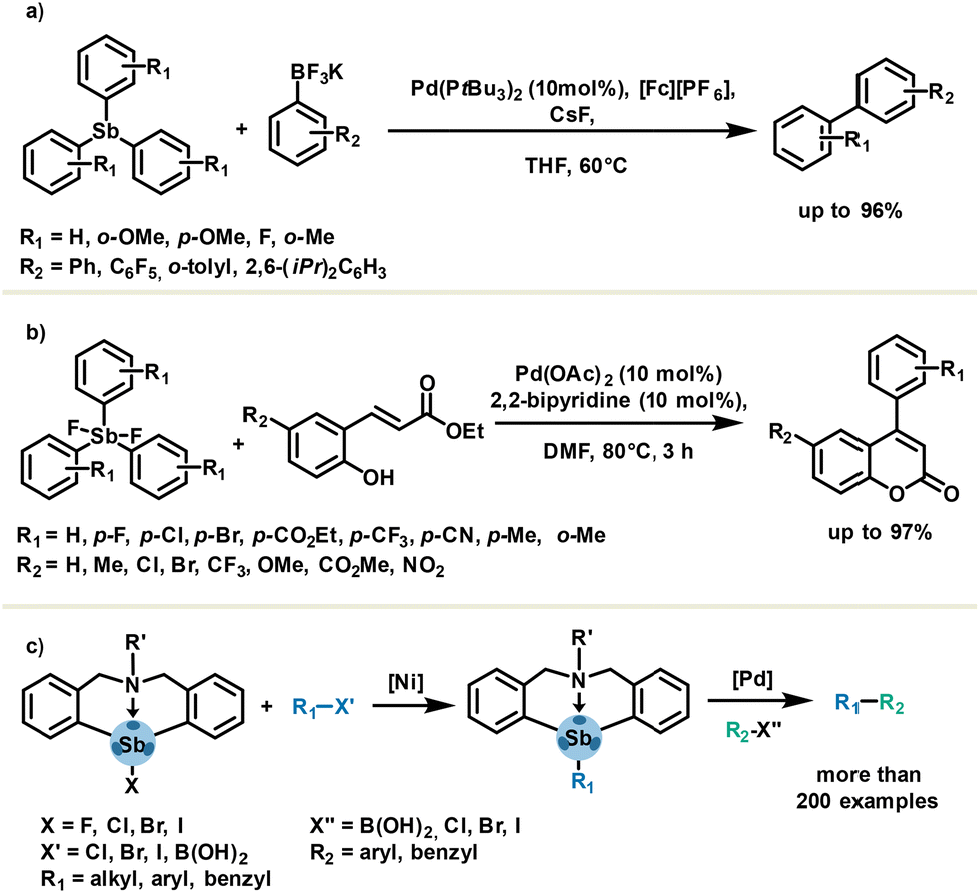 | ||
| Scheme 11 Metal-catalysed cross-coupling in the presence of (a) stibines, (b) difluorostiboranes, (c) and azastibocine derivatives as aryl transfer agents. | ||
Another class of organoantimony such as azastibocines features an intramolecular PnB Sb⋯N, which is responsible for enhanced reactivity in cross-coupling reactions. Taking advantage of this feature, the Qiu group investigated and developed several nickel- and palladium-based cross-coupling methodologies (Scheme 11(c)). This impressive work enabled fully operational synthetic routes for the coupling of a wide range of building blocks in a two-step reaction protocol.74–76
5. Organoantimony redox chemistry in catalysis and organic synthesis
5.1. Elementary steps: on the footsteps of d-block elements
The previous section describes the behaviour of organoantimony as a ligand or at best a coupling reagent in transmetalation processes catalysed by transition metals and its use in organometallic chemistry. Accordingly, in this section, we will focus on redox properties of organoantimony compounds and their reactivity as such, by providing an overview with non-exhaustive but relevant examples. In addition to redox reactions reported in the literature, the legitimacy of redox properties probably stems from a number of clues which are drawn as follows:• Antimony is located in the middle range of the pnictogen family; therefore, one can expect relatively mild conditions for Sb reduction (compared to P and As which require strong reducing agents) and oxidation (compared to bismuth which requires highly oxidizing agents). Additionally, low valent antimony (SbI) exhibits strong nucleophilicity, a consequence of its p-orbital lone pair being highly prone to oxidation (Fig. 7 and 8(A)).
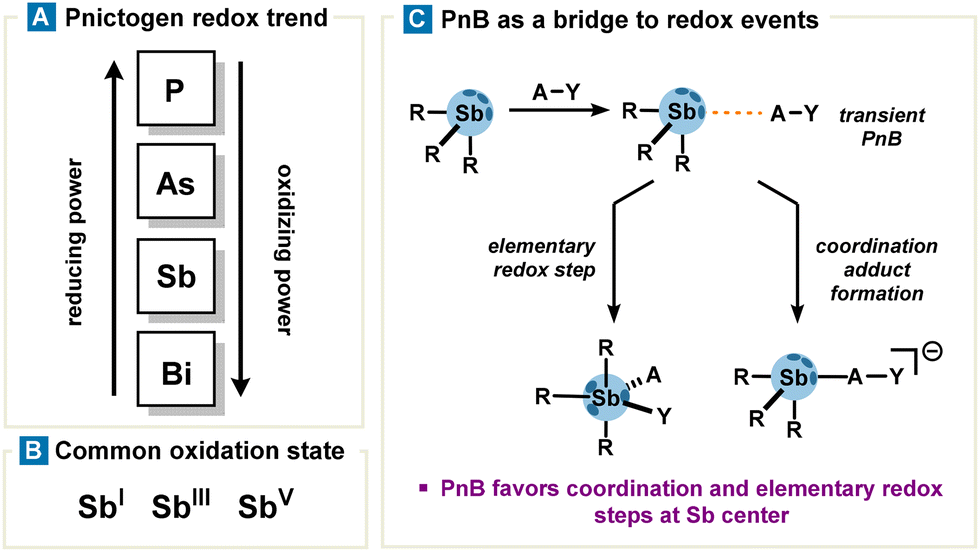 | ||
| Fig. 7 (A) Vertical periodic redox trend of the pnictogen family, (B) common oxidation state of antimony, and (C) transient PnB tunnelling redox events such as formation of a coordination bond and an elementary redox step consisting of an oxidative addition. | ||
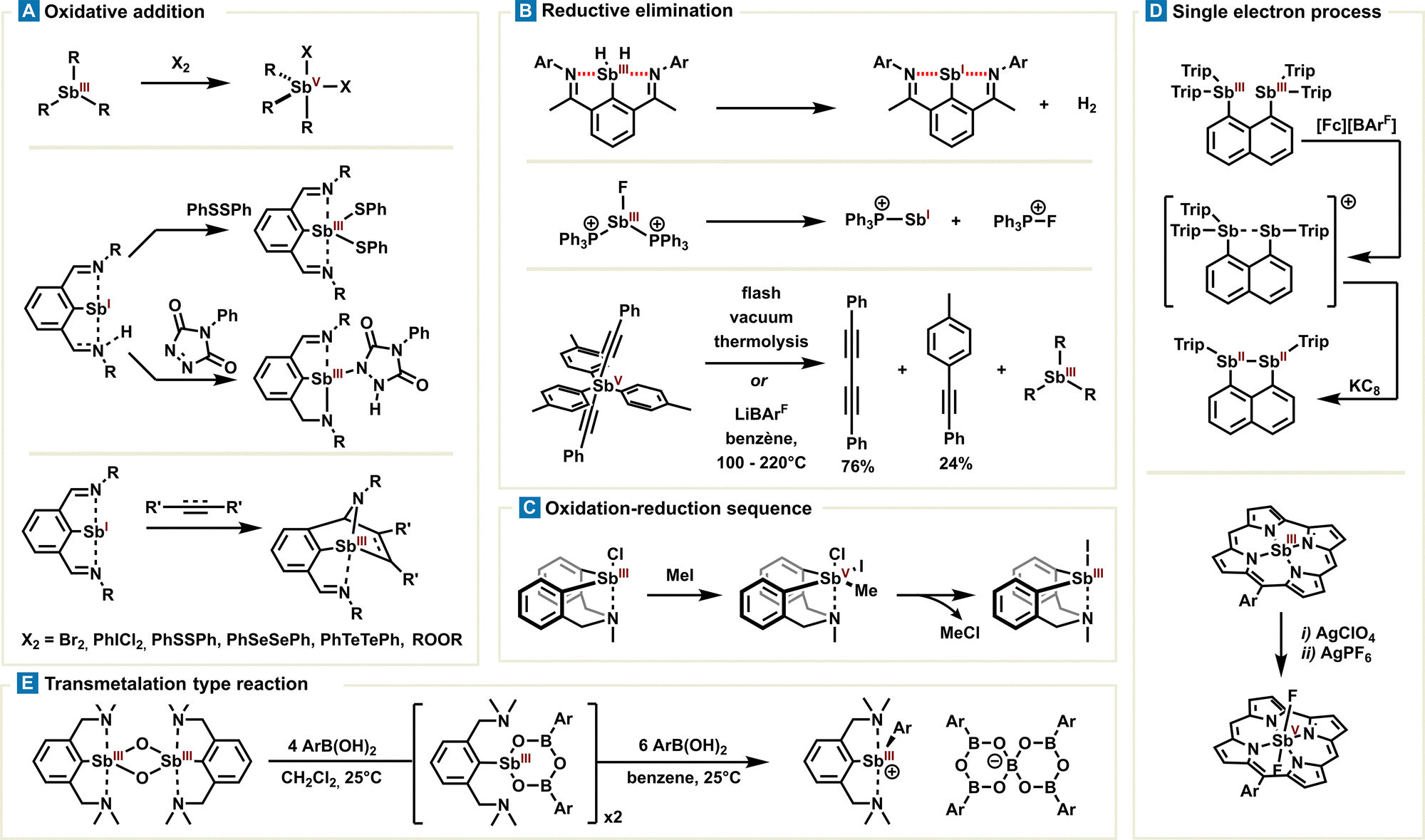 | ||
| Fig. 8 Overview of a typical organometallic-type reaction encountered in organoantimony chemistry: (A) oxidative additions, (B) reductive elimination, (C) sequential oxidation-reduction, (D) single electron oxidation or reduction, (D) transmetallation type reaction. | ||
• The valence electronic ns2np3 configuration of antimony allows several oxidation states ranging from −III to +V. Noteworthily, antimony exhibits several stable oxidation states constituting two main redox couples SbV/SbIII and SbIII/SbI (Fig. 7(B)).10
• Processes of rupture or formation of Sb–H or Sb–C bonds are feasible in terms of required energy (ESb–H = 255 kJ mol−1, ESb–C = 215 kJ mol−1).69
• Antimony geometry can be distorted into a planar form by means of tailored ligands, while requiring a low barrier energy of 10 kJ mol−1.77,78 The planarization enables a reduction of the large energy gap between frontier orbitals and induces the formation of an empty p orbital at the antimony center, rendering it more electrophilic. Moreover, planar antimony derivatives combine at the Sb center both (i) an empty p molecular orbital and (ii) an s molecular orbital containing a free electron pair, which is a net advantage for mimicking transition metal reactivity (Fig. 4(G)). Overall, these features should favour putative redox processes.
• Ligand moieties around the Sb center are as essential as the ligands for metals. Strongly electron-withdrawing substituents stabilize oxidized states and also enable lowering of the LUMO energy (Fig. 4(D)).
• Pre-association via PnB can orient and polarize substrates with the Sb center, effectively lowering the activation barrier for a simple coordination step as well as for elementary redox steps (Fig. 7(C)).
Several oxidative additions of stibines(III) were reported using oxidizing agents such as peroxides, bromine, selenium- or tellurium-based diphenyldichalcogenides or iodobenzene dichloride (Fig. 8(A)).79–82 Stabilised stibinidenes(I) also undergo oxidative addition of diphenyldisulfide, as reported by the Dostál group. The 2,6-bis(ketimine)phenyl or 2,6-(methylamine)(ketimine)phenyl ligands impose both a trigonal planar geometry and a steric shielding of Sb(I) via intramolecular Sb⋯N PnB. Planarization results in the formation of the Sb center of both an s-type lone pair and an empty p molecular orbital, being partly responsible for the redox reactivity. Antimony may also benefit from a ligand-cooperativity effect as observed in the oxidation reaction with alkyne or alkene cyclisation, providing the corresponding stibines(III) (Fig. 8(A)). In parallel, reductive eliminations of poorly stable stibines were observed by elimination of H2 or a phosphonium cation.78,83 More blatantly, the Akiba group reported in 2015 a systematic study of ligand exchange and ligand-coupling reactions with pentavalent antimony derivatives,84 based on their anterior observations (Fig. 8(B)).85 The authors confirmed reductive elimination on antimony Sb(V), leading to diyne and biaryl products, in the presence of a catalytic amount of lithium tetrakis(pentafluorophenyl)borate under strong heating. The reductive elimination also proceeds without a catalyst by means of flash vacuum thermolysis; nevertheless, the activation mode of the lithium cation is relatively unclear and still remains to be utterly clarified (Fig. 8(B)).
Noteworthily, a stoichiometric two-electron redox process has been described with the azostibocine scaffold in the presence of methyl iodide. Although the mechanism has not been proven, one can assume oxidative addition on the antimony center via the stiborane intermediate, followed by methyl chloride elimination (Fig. 8(C)).86 Alternatively, 1-electron-sequence oxidation or reduction is also possible. In this context, Schulz and Haberhauer reported remarkable stepwise single-electron oxidation and reduction reactions of peri-substituted bisstibino-naphthalene(III) into corresponding distibane (formally: II) (Fig. 8(D)).87 Later, the Bröring group reported sequential oxidation of antimony(III) corroles towards antimony(V) species with silver salts. Interestingly, the authors observed no physical oxidation state +IV after the first oxidation but instead corrole oxidation due to non-innocent behaviour of the latter (Fig. 8(D)).88 These sequential steps allow for the selective adjustment of the antimony oxidation state and a subsequent study of the intermediates which are of great importance for the development of new redox processes.
One can mention a canonical transmetalation involving a highly strained cyclic lithium borate and stibine(III) reported by the Gabbaï group.89 However, the underlying mechanism has not been determined and the driving force of the reaction is more likely to be associated with a strain-release mechanism, making it a case apart. In this line, the Dostál group reported later the transmetalation of boronic acid on a stibaheteroboroxine derivative. Whereas the transmetalation conditions require 10 equivalents of boronic acid, this reaction is the first of its kind and is conceptually highly significant in antimony chemistry (Fig. 8(E)).90 Since the transmetalation is a recurring step in organometallic chemistry, this naturally raises the important question of antimony's ability to undertake other or similar related redox reaction steps.
Stibine oxide tends to polymerise in most cases; however, to overcome this problem, the Johnstone group has reported a sterically hindered monomeric stibine oxide based on a 2,6-diisopropylphenyl ligand, opening therefore a new avenue for oxide stibine chemistry.91 The hindered monomeric stibine oxide undergoes reduction with silanes under mild conditions, as well as oxide transfer involving the cleavage of the Sb–O bond, ultimately leading to the corresponding difluorostiborane (Scheme 12(a)). The same group also highlighted the peculiar reactivity of stibine oxides, which promotes canonical C–F and Si–F bond cleavage (Scheme 12(b)).92 The ambiphilic nature of stibine oxide might serve as a starting point for small molecule activation through isohypsic processes, similar to metals in catalysis.
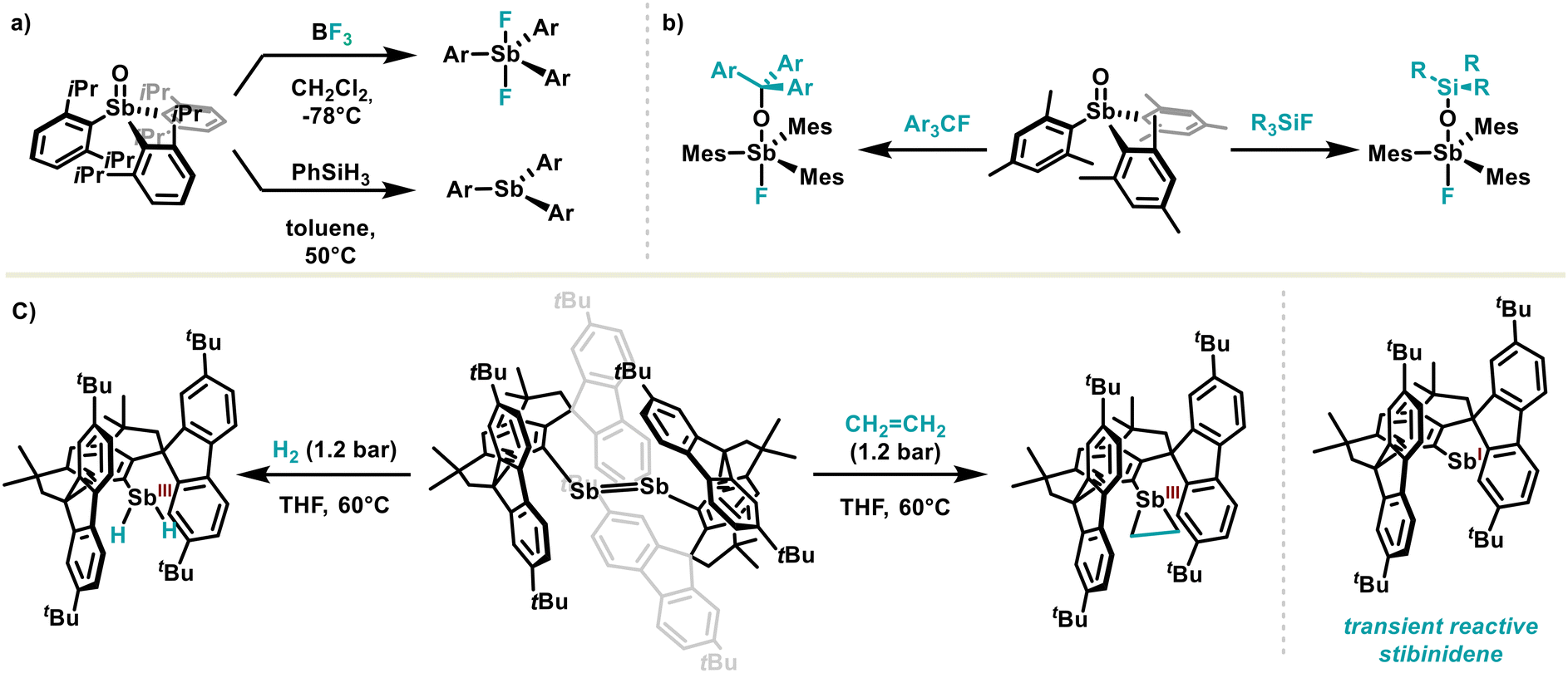 | ||
| Scheme 12 (a) Hindered oxide stibine reactivity involving isohypsic oxide transfer, reduction, (b) canonical insertion into the C–F and Si–F bond, and (c) small molecule activation with distibene via an oxidative addition. | ||
Taking small molecule activation one step further, the Cornella group superbly achieved in 2023 hydrogen and ethylene activation with sterically distorted distibenes, under very mild conditions (Scheme 12(c)).93 By using bulky hydrindacene ligands, the authors obtained distibenes featuring unusual long Sb![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Sb bond lengths. This property leads distibene to dissociate in solution, providing transient reactive monomeric stibinidene(I), which reacts with hydrogen and ethylene. In stibinidene(I), metal-like reactivity is due to dual orbital interaction between organoantimony and the substrate. Oxidative addition of small molecules is certainly of high importance and there is plenty of room for improvement by better understanding of both the metal-like reactivity of Sb(I) and potential Sb redox catalysis.10,94
Sb bond lengths. This property leads distibene to dissociate in solution, providing transient reactive monomeric stibinidene(I), which reacts with hydrogen and ethylene. In stibinidene(I), metal-like reactivity is due to dual orbital interaction between organoantimony and the substrate. Oxidative addition of small molecules is certainly of high importance and there is plenty of room for improvement by better understanding of both the metal-like reactivity of Sb(I) and potential Sb redox catalysis.10,94
Other elementary redox steps in antimony redox chemistry have also been unlocked, such as C–H activation, which is, par excellence, the hallmark of transition metals. The Periana group was the first to use stiboranes(V) in the C–H activation of small molecules such as methane or ethane. The antimony complex was shown to activate small molecules through the formation of a polarized C–Sb bond, leading to a pentavalent complex after ligand decoordination (Scheme 13). The high oxidation state of the latter favors the reductive elimination, releasing both the stibine(III) and the C–H activated product.95 The authors also demonstrated quantitative C–H activation with high selectivity by replacing trifluoroacetic acid with sulfuric acid and using a pentamethoxy antimony(V) derivative; noteworthily, a C–H activation mechanism in a similar fashion was confirmed.96 This proof of concept of C–H activation of light alkanes adds another metal-like feature to the antimony toolbox. Moreover, the pentavalent antimony scaffold employed in this study is relatively simple, offering considerable opportunities for system improvement through the introduction of more complex ligands. It is also worth noting that other C–H activation reactions have been reported on larger molecules, such as the phenolate anion, using dichlorostibine(III).97
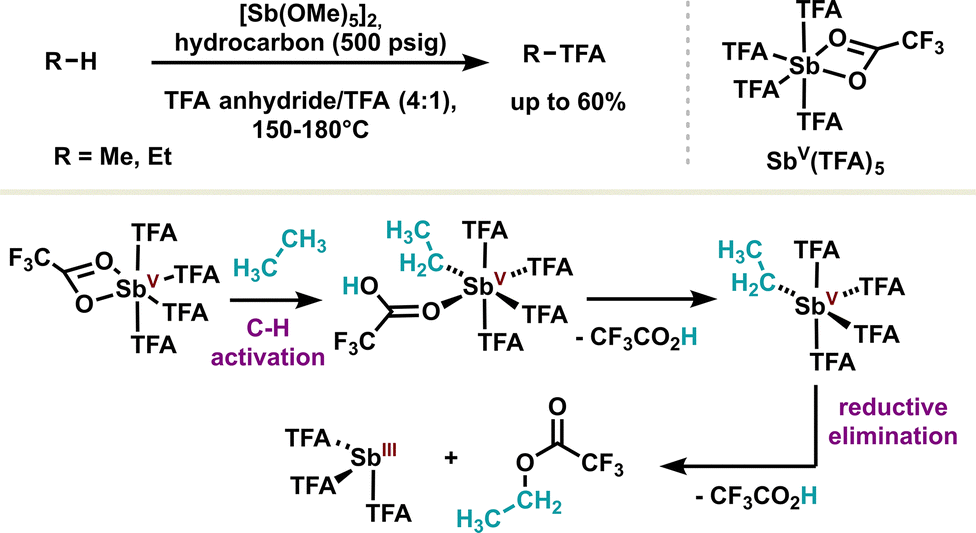 | ||
| Scheme 13 Pentavalent antimony(V)-mediated C–H activation of hydrocarbons. | ||
5.2. Low-valent organoantimony SbIII/SbII and SbIII/SbI in catalysis and organic synthesis
The use of low-valent organoantimony in catalysis and organic synthesis is particularly rare; nevertheless, one can cite a few examples that harness antimony reactivity, benefiting from geometry-constraining effects or the non-innocent nature of the ligands.Capitalizing on the ambiphilic properties of stibinidene(I), the Zhou group used these features for Sb(III)/Sb(I) redox catalysis with 10 mol% loading for hydroboration of disulfides. The propensity of stibinidene(I) to undergo oxidative addition was used to initiate the catalytic cycle with diaryldisulfides ArSSAr. The resulting stibine(III) performs a hydrogen transfer with pinacolborane (HBpin), leading to a sulfidoborate ArSBpin and a fleeting hydridostibine(III) intermediate which regenerates spontaneously into the catalyst through a reductive elimination (Scheme 14).98 The basic character of the stibinidene(I) catalyst was subsequently put to contribution for in situ valorization of the so-obtained sulfidoborate and thiophenol products into β-sulfido carbonyl compounds through a Michael reaction. The one-pot access to β-sulfido carbonyl compounds through low-valent antimony redox catalysis, coupled synergistically with antimony Lewis base catalysis, is unprecedented and is expected to pave the way for using the many facets of antimony in catalysis. Moreover, similar antimony reactivity with respect to organobismuth should legitimate further investigations in this area.
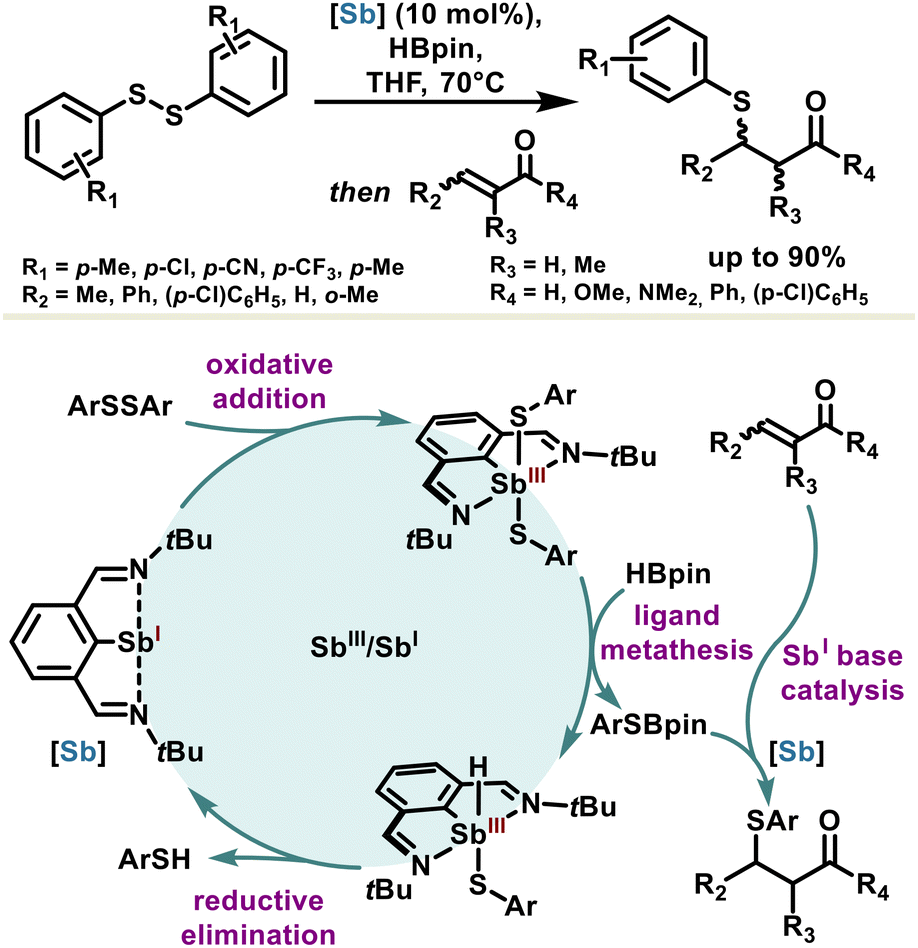 | ||
| Scheme 14 Synergic catalysis based on redox and basic properties of antimony for the one-pot synthesis of β-sulfido carbonyl compounds. | ||
Antimony redox properties were also harnessed for hydrogen production in electrocatalytic processes. The challenge lies rather in determining the mechanistic aspects of the hydrogen evolution reaction which have been extensively studied with transition metals, but poorly explored with main-group-element-based electrocatalysts. Brudvig, Batista and Crabtree group reported the hydrogen evolution reaction with an Sb(III) porphyrin active catalyst in which antimony is the catalytic center for proton reduction. In this electrocatalytic reaction, the antimony catalyst navigates through three formal oxidation states (V/IV/III) whose redox potentials are directly modulated by an antimony non-innocent polypyrrole ligand. This flexibility is a key advantage of the system, unlike metal-based hydrogen evolution reaction catalysts, where the redox-active site and the substrate-binding site are inherently linked, restricting the independent tuning of electronic properties.99
Alternatively, the catalyst can also be supported directly in an electrode as reported by the Jiang and Vogiatzis group which developed an antimony(III) salen heterogenized in a glassy carbon electrode.100 The reaction proceeds via an antimony-assisted and ligand-centered pathway involving sequential two-electron reduction and protonation steps to produce H2 electrocatalytically. A simplified redox catalytic cycle is depicted in Scheme 15, in which the antimony(III) salen results in a stibinidene(I) after two sequential electron reduction–protonation steps. The latter undergoes a formal oxidative addition leading to a hydridostibine(III) which, after hydrogen elimination, regenerates the stibine catalyst. Put in perspective, these results provide an in-depth analysis of the underlying redox mechanism, opening new avenues in heterogeneous redox antimony electrocatalysis.
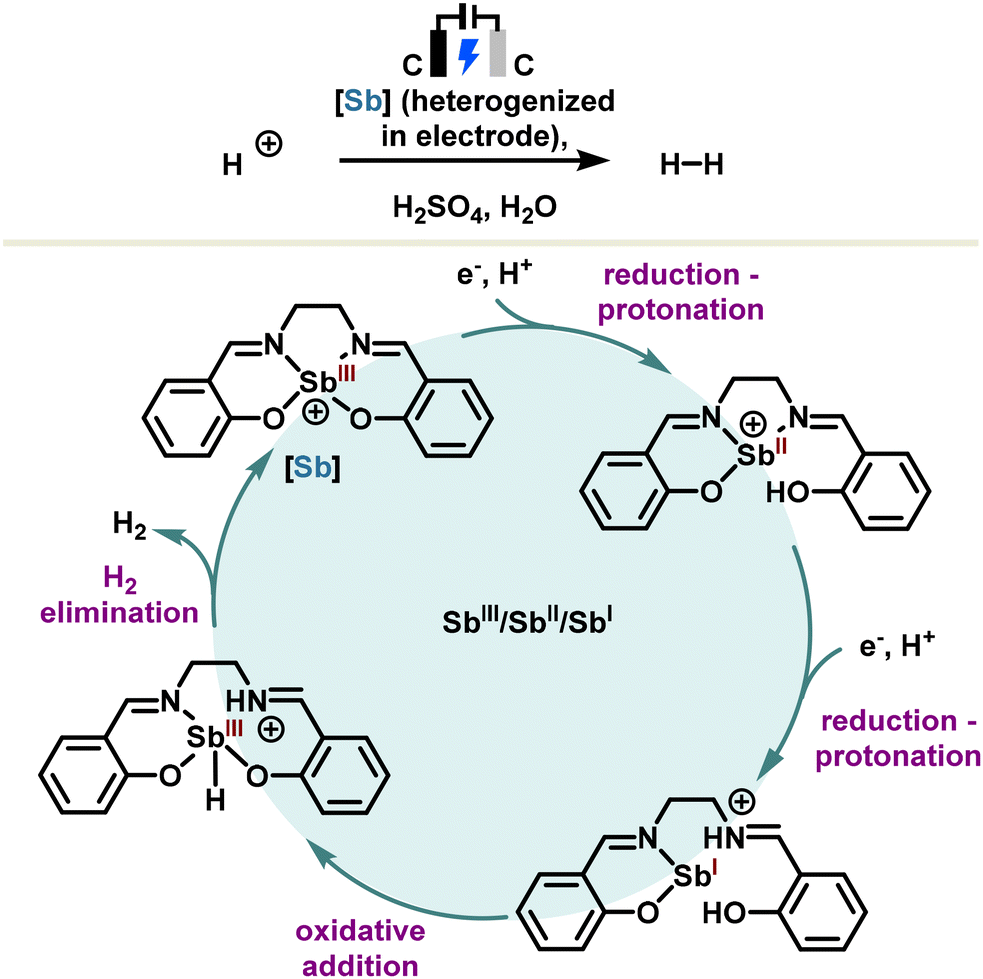 | ||
| Scheme 15 Electrocatalytic hydrogen evolution reaction with an antimony(III) salen. | ||
Hydrometalation of alkenes and alkynes is a key elementary reaction that offers a direct route to alkyl- or alkenyl-metal intermediates which are highly valuable in organic synthesis applications. To this extent, the Chitinis group reported uncatalyzed hydrostibination reactions of alkene and alkyne bonds, as a new class of elementary hydrometallation reactions requiring neither a radical initiator nor additives such as metals or Lewis acids (Scheme 16).101 With the intention to design a highly reactive hydrostibination reagent whose LUMO is sufficiently low to engage with unsaturated substrates, the authors prepared a hydridostibine(III) with a rigid naphthalenediamine scaffold. Increase of the Sb–N bond polarity with diamine ligands enables LUMO localization on the Sb center and enhances its Lewis acidity (with respect to Sb–C bonds); these features are required conditions for hydrometallation and somehow reminiscent of borane by analogy. Most importantly, the planar nature of the naphthalenediamine ligand, flanked by a bulky triethylsilyl group, imposes a distorted geometry around the antimony center, which maintains its acidity by preventing any back donation of nitrogen lone pair. In addition, the silyl groups ensure stability of the reagent against H2 elimination and promote higher reactivity of the Sb–H bond via a geometry-permitted β-silicon effect (σN–Si→ σSb–H). The meticulously designed reagent performed hydrostibination reactions in excellent yields with alkynes and alkenes but also with azobenzene and activated aldehydes. Strikingly, the hydrostibination reaction from terminal arylacetylenes yields selectively to anti-Markovnikov Z-olefins, which is in contradiction with anti-Markovnikov E-olefins usually observed via syn-addition in the hydroboration reaction. The unpredicted selectivity emerges from a homolytic mechanism initiated by the generation of a stibinyl(II) radical, resulting originally from hydrogen transfer between hydridostibine(III) and arylacetylenes (Scheme 16). The stibinyl(II) radical participates then in a cycle featuring two oxidation states Sb(II) and Sb(III), leading after a final hydrogen transfer to the hydrostibination product.102 The so-obtained Z-olefins are rather difficult to obtain and usually require metal complexes with tailored ligands, which renders hydrostibination stereoselectivity both unorthodox and of great interest for synthetic methodology.
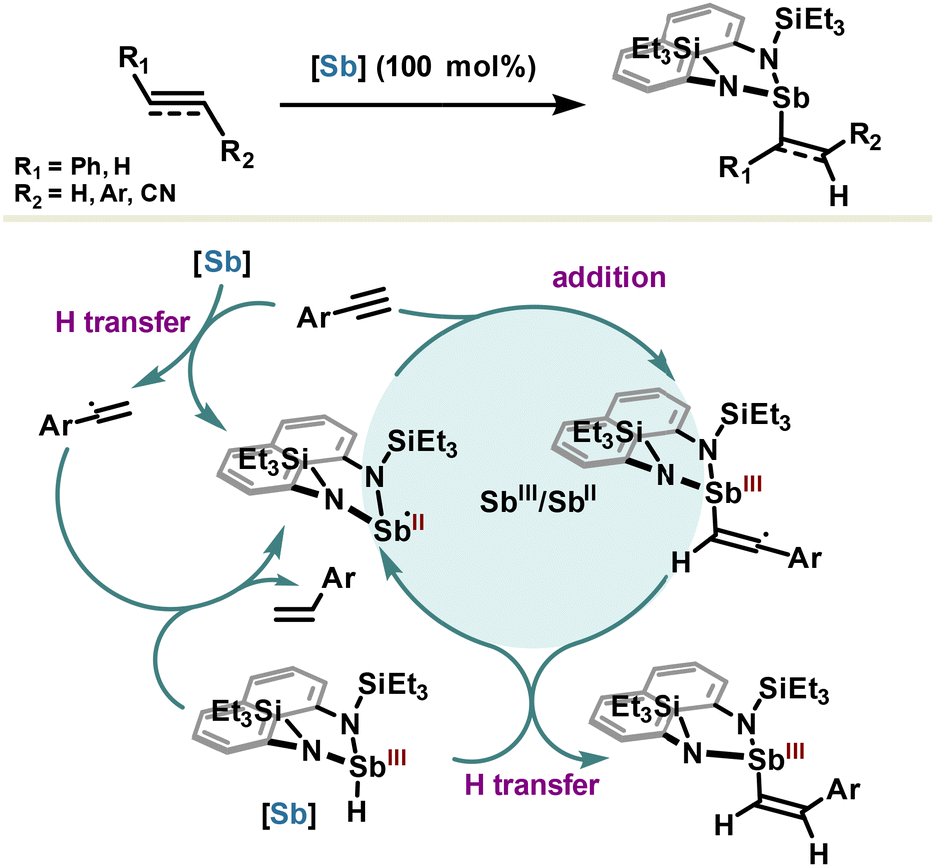 | ||
| Scheme 16 Hydrostibination reaction for the selective synthesis of Z-olefins. | ||
5.3. High-valent organoantimony SbV/SbIV/SbIII in catalysis
The first reported catalytic redox system using the redox couple Sb(V)/Sb(III) was reported in 1985 by the Akiba group for α-diketone synthesis (Scheme 17).103 The active catalyst, dibromophenylstiborane(V), has been exploited for its ability to chelate α-hydroxy ketones under their enolic form with a prior ligand exchange. Although the exact mechanism remains to be explored, a rational pathway tends rather towards the reductive elimination of the resulting complex, leading to the α-diketone and triphenyl stibine(III). The latter can be reconverted into dibromophenylstiborane(V) in the presence of a stoichiometric quantity of a dibrominated co-oxidant. Later, milder and more sustainable conditions were developed, notably by using a polymeric oxide triphenylstibine as the catalyst. The mechanism is assumed to be the same and the authors avoided stoichiometric use of co-oxidant, capitalizing on the oxidizing properties of oxygen to close the catalytic cycle (Scheme 17).104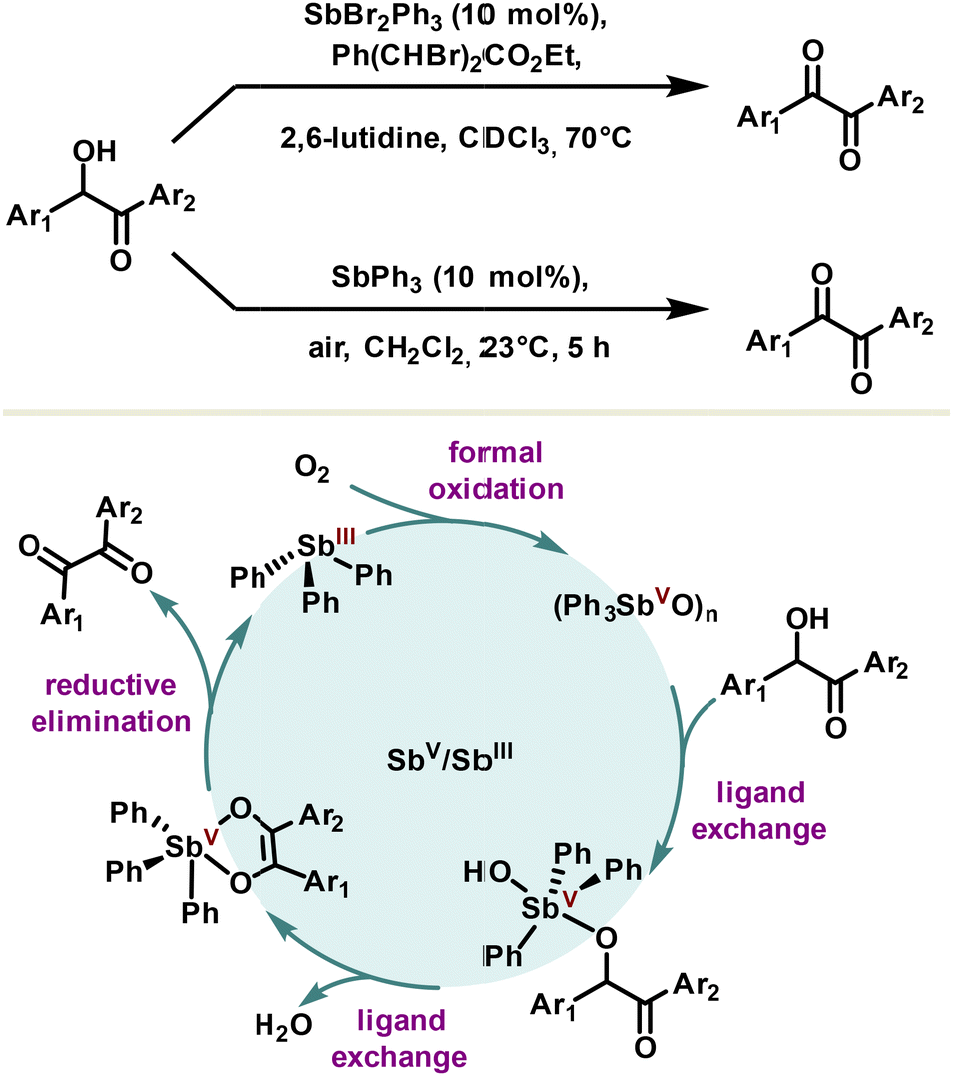 | ||
| Scheme 17 Catalytic oxidation of α-hydroxyketones with stiborane and polymeric stibine oxide. | ||
The use of other homologues such as triphenylarsine or triphenylbismuth is not effective in this reaction, reflecting thus the complementary importance of antimony in catalysis.10
Cross-dehydrogenative coupling is an efficient tool for the creation of carbon–carbon bonds; however, the developed methodologies are largely based on transition metals. An alternative system using N-hydroxyphthalimide and a redox active antimonate salt, NaSbCl6, as a non-innocent counteranion co-catalyst was reported by the Kobayashi group for the aerobic cross-dehydrogenative coupling of N-tetrahydroisoquinolines (Scheme 18).105 Although the mechanism is not established, it is assumed that the single electron oxidation of tetrahydroisoquinolines is catalyzed by a hexachloroantimonate(V) anion providing an aminium radical cation. Aerobic condition leads to the reoxidation of the antimony(IV) species into an antimonate(V) anion catalyst and the formation of an oxygen radical anion. The latter abstracts subsequently the hydrogen of the N-hydroxyphthalimide, delivering the phthalimide N-oxide radical and the peroxide anion. Lastly, the aminium radical cation undergoes hydrogen radical abstraction to yield the iminium cation which readily reacts with nucleophiles to furnish the final cross-products. Overall, the antimony-based co-catalysis of cross-dehydrogenative coupling does not offer significant advantages compared to other metal-based systems. However, from a fundamental point of view, the implementation of redox-active antimonate counteranions in catalysis might lead to further investigation in this area. It should be noted, however, that a main challenge lies in the assessment of the catalytically active species, since halide antimony derivatives tend to disproportionate.
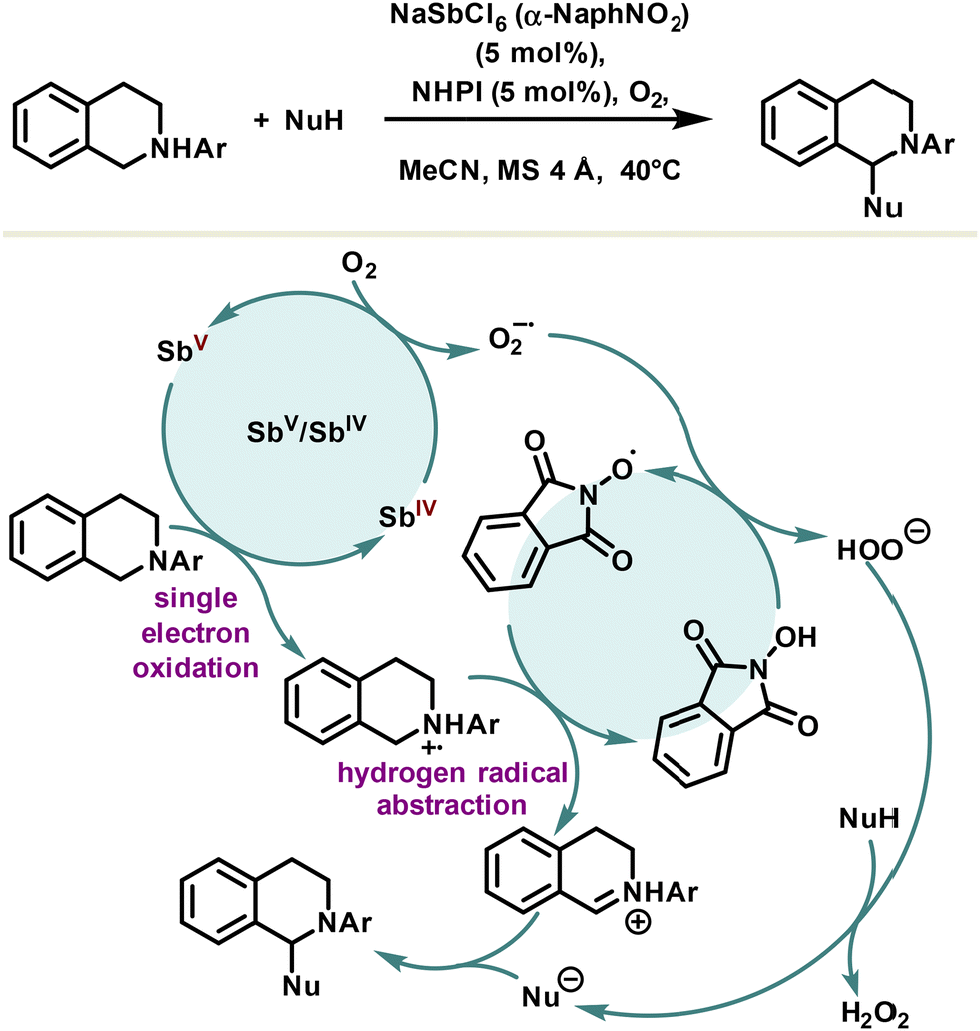 | ||
| Scheme 18 Cross-dehydrogenative coupling reaction using a redox-active antimony salt as a co-catalyst. | ||
In the same way as non-noble transition metals, the redox properties of antimony derivatives prompt them to be oxidized under aerobic conditions, which makes them a potent platform for the two-electron reduction of O2. Inspired by these features, the Gabbaï group designed a bifunctional platform to achieve the four-electron reduction of O2, which is often considered a more difficult task.106 For this purpose, the authors took advantage of a bis-diphenylstibine based on a xanthene or dihydroacridine planar scaffold as depicted in Scheme 19. In the presence of an electron-rich ortho-quinone, a formal oxidation proceeds on both stibines(III), going to the +V oxidation state, in parallel to the four-electron reduction of oxygen. The resulting stable α,α,β,β-tetraolate distiborane can be triggered sequentially via a formal ligand exchange in acidic medium, followed by a reductive elimination mediated by an arylthiol reducing agent, regenerating in fine the starting bis diphenylstibine(III). Overall, the authors introduced an innovative strategy based on an antimony platform for the metal-free reduction of dioxygen. Despite the obvious difficulty in achieving a catalytic reaction, sequential reaction conditions were implemented to regenerate the initial distibine, paving the way for further catalytic development.
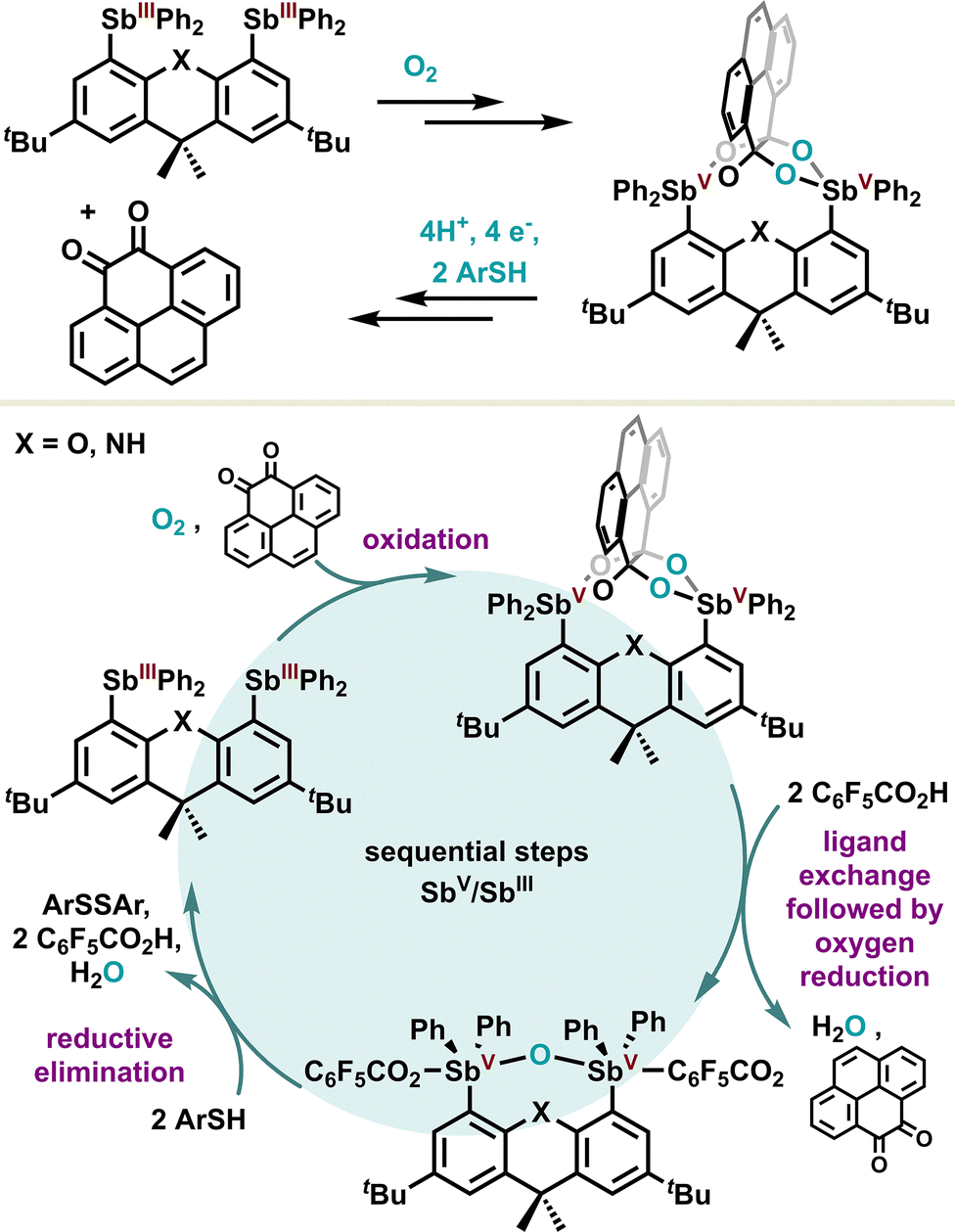 | ||
| Scheme 19 Four-electron reduction of dioxygen with distibines. | ||
5.4. Redox neutral catalysed and mediated reaction of SbIII
Redox neutral catalysis can be described as a series of elementary steps (analogous to those observed with transition metals) part of a catalytic cycle, preserving the oxidation state of the metal or metalloid center. Several examples based on organobismuth or organophosphorus compounds have already been reported.10,14 However, only one example using antimony has been published based on redox neutral catalysis definition. It was reported in 2022 by the Coles group for the catalytic hydrophosphination of phenyl isocyanate leading non-selectively to the products of mono- and di-insertion (Scheme 20).107 An antimony(III) phosphanide catalyst was developed capitalizing on a bis(amidodimethyl)disiloxane ligand, offering similar advantages with respect to the hydrostibination catalyst developed by the Chitnis group (Scheme 16), namely higher stability via steric hindrance, and a reactive Sb–P bond owing to constrained geometry and β-silicon effect. The mechanism may certainly involve an adduct between antimony(III) phosphanide and phenyl isocyanate via a Sb⋯N or Sb⋯O PnB promoting the first insertion step. The resulting phosphanylcarboxamidate antimony(III) complex undergoes then a second insertion reaction or directly releases the phosphanylcarboxamide product via a hydrogen transfer step which formally refers to a ligand exchange. While the selectivity still remains to be controlled, this redox neutral hydrophosphination reaction is a first proof of concept in antimony chemistry.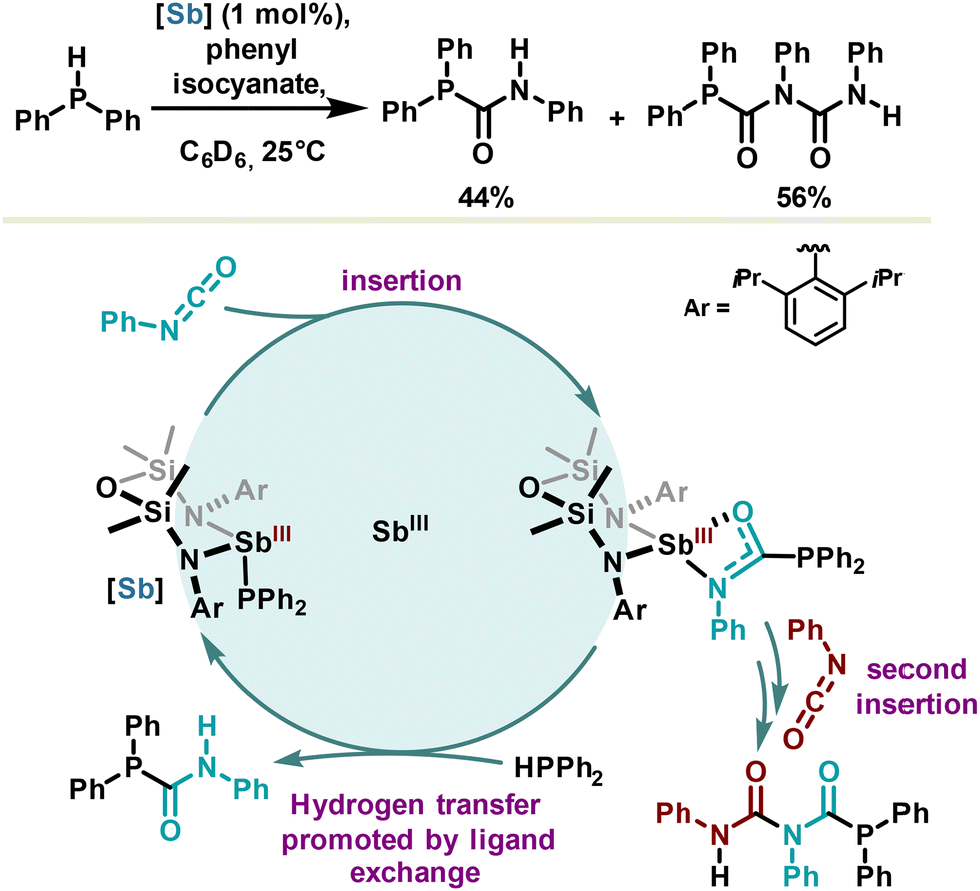 | ||
| Scheme 20 Hydrophosphination of phenyl isocyanate in redox neutral antimony catalysis. | ||
6. Conclusions
Over the past few years, the fields of organic chemistry and catalysis have experienced major upheavals due to the pursuit of metal-free alternatives and the aspiration to discover new reactivities. In this context, rapid advancements in organoantimony chemistry have positioned it as a compelling platform for organic synthesis and catalysis, bridging the gap between traditional main-group chemistry and transition metal reactivity. This attention was greatly fostered by the combination of redox versatility and the emerging role of pnictogen bonding, which arises from the Lewis acidity of organoantimony. This latter has greatly contributed to broadening the scope of non-covalent interactions and their applications in catalysis. However, despite the significant progress made in recent years, several challenges remain. A major limitation is the relatively narrow understanding of structure–redox reactivity relationships until now. While promising applications have been demonstrated in redox transformations and small-molecule activation, the full potential of organoantimony in redox catalysis has yet to be fully realized.Looking ahead, key areas of future research include not only the design of more robust and highly selective organoantimony catalysts, but also the seamless integration of chiral pnictogen bond donors into asymmetric catalysis, and the exploration of novel reactivity modes inspired by transition metals. It is important to note that σ-hole-based asymmetric catalysis is a current topic of high interest and is still in its infancy with respect to pnictogen bond. Furthermore, interdisciplinary approaches combining computational chemistry, mechanistic studies, and innovative ligand design will be crucial in refining and expanding the practical applications of organoantimony chemistry.
Conflicts of interest
There are no conflicts to declare.Data availability
All data discussed in this Tutorial Reviews are available in the cited literature. No new data were generated.Acknowledgements
Generous financial support for this work by the French Agency of Research (ANR-23-CPJ1-0018-01) and Région Normandie is gratefully acknowledged. RW and EC acknowledge the University of Caen Normandie and ENSICAEN for essential research infrastructure and logistical support. Special appreciation is extended to the University of Caen Normandie for its continuous support within the framework of the RW tenure-track position.Notes and references
- P. P. Power, Nature, 2010, 463(7278), 171–177 CrossRef CAS PubMed.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314(5802), 1124–1126 CrossRef CAS PubMed.
- F. G. Fontaine and D. W. Stephan, Philos. Trans. R. Soc., A, 2017, 375, 20170004 CrossRef PubMed.
- C. Hansell, Nat. Chem., 2015, 7, 88 CrossRef CAS PubMed.
- L. Greb, F. Ebner, Y. Ginzburg and L. M. Sigmund, Eur. J. Inorg. Chem., 2020, 3030–3047 CrossRef CAS.
- O. Planas, F. Wang, M. Leutzsch and J. Cornella, Science, 2020, 367(6475), 313–317 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- R. Weiss, E. Aubert, P. Pale and V. Mamane, Angew. Chem., Int. Ed., 2021, 60, 19281–19286 CrossRef CAS PubMed.
- D. Jovanovic, M. Poliyodath Mohanan and S. M. Huber, Angew. Chem., Int. Ed., 2024, 63, e202404823 CrossRef CAS PubMed.
- J. M. Lipshultz, G. Li and A. T. Radosevich, J. Am. Chem. Soc., 2021, 143, 1699–1721 CrossRef CAS PubMed.
- C. Xie, A. J. Smaligo, X.-R. Song and O. Kwon, ACS Cent. Sci., 2021, 7, 536–558 CrossRef CAS PubMed.
- P. Cao, C.-Y. Li, Y.-B. Kang, Z. Xie, X.-L. Sun and Y. Tang, J. Org. Chem., 2007, 72, 6628–6630 CrossRef CAS PubMed.
- R. Inaba, I. Kawashima, T. Fujii, T. Yumura, H. Imoto and K. Naka, Chem. – Eur. J., 2020, 26, 13400–13407 CrossRef CAS PubMed.
- H. W. Moon and J. Cornella, ACS Catal., 2022, 12, 1382–1393 CrossRef CAS PubMed.
- J. M. Bothwell, S. W. Krabbe and R. S. Mohan, Chem. Soc. Rev., 2011, 40, 4649–4707 RSC.
- A. Pogoreltsev, Y. Tulchinsky, N. Fridman and M. Gandelman, J. Am. Chem. Soc., 2017, 139, 4062–4067 CrossRef CAS PubMed.
- J. M. Bayne and D. W. Stephan, Chem. Soc. Rev., 2016, 45, 765–774 RSC.
- J. Zhou, L. L. Liu, L. L. Cao and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 5407–5412 CrossRef CAS PubMed.
- Y. Matano, Antimony and Bismuth in Organic Synthesis, in Main Group Metals in Organic Synthesis, John Wiley & Sons, Ltd, 2004, pp. 753–811 Search PubMed.
- G. G. Yakobson and G. G. Furin, Synthesis, 1980, 345–364 CrossRef CAS.
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- A. Frontera and A. Bauza, Int. J. Mol. Sci., 2021, 22, 12550 CrossRef CAS PubMed.
- While this review was in preparation, additional examples appeared: (a) S. Fernando, Y. C. Chan, S. Fernandez, E. Sabater, G. Tizzard, S. J. Coles, D. M. Andrada and O. Planas, Angew. Chem., Int. Ed., 2025, 64, e202505697 CrossRef CAS PubMed; (b) S. G. Kim, J. Oh, D. Kim, D. E. Choi and S. J. Hwang, JACS Au, 2025, 5, 2779–2791 CrossRef CAS PubMed; (c) Z. Zhang, K. Li, M. Huang, T. Chen and J. Zhou, J. Am. Chem. Soc., 2025, 147, 10066–10072 CrossRef CAS PubMed; (d) F. Şit, N. H. Hunter, L. T. Maltz and F. P. Gabbaï, Angew. Chem., Int. Ed., 2025, 64, e202505893 CrossRef PubMed.
- V. Gutmann, H. Hubacek and A. Steininger, Monasth. Chem., 1964, 95, 678–686 CrossRef CAS.
- G. A. Olah, J. Org. Chem., 2001, 66, 5943–5957 CrossRef CAS PubMed.
- G. Resnati, D. L. Bryce, G. R. Desiraju, A. Frontera, I. Krossing, A. C. Legon, P. Metrangolo, F. Nicotra, K. Rissanen, S. Scheiner and G. Terraneo, Pure Appl. Chem., 2024, 96, 135–145 CrossRef CAS.
- L. de A. Santos, T. A. Hamlin, T. C. Ramalho and F. M. Bickelhaupt, Phys. Chem. Chem. Phys., 2021, 23, 13842–13852 RSC.
- A. P. M. Robertson, N. Burford, R. McDonald and M. J. Ferguson, Angew. Chem., Int. Ed., 2014, 53, 3480–3483 CrossRef CAS PubMed.
- L. T. Maltz and F. P. Gabbaï, Inorg. Chem., 2023, 62, 13566–13572 CrossRef CAS PubMed.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 643 CrossRef CAS.
- M. Yang, D. Tofan, C.-H. Chen, K. M. Jack and F. P. Gabbaï, Angew. Chem., Int. Ed., 2018, 57, 13868–13872 CrossRef CAS PubMed.
- H. Chen, A. Frontera, M. Ángeles Gutiérrez López, N. Sakai and S. Matile, Helv. Chim. Acta, 2022, 105, e202200119 CrossRef CAS.
- A. Gini, M. Paraja, B. Galmés, C. Besnard, A. I. Poblador-Bahamonde, N. Sakai, A. Frontera and S. Matile, Chem. Sci., 2020, 11, 7086–7091 RSC.
- D. You, B. Zhou, M. Hirai and F. P. Gabbaï, Org. Biomol. Chem., 2021, 19, 4949–4957 RSC.
- J. L. Beckmann, J. Krieft, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2023, 62, e202310439 CrossRef CAS PubMed.
- J. L. Beckmann, J. Krieft, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem. Sci., 2023, 14, 13551–13559 RSC.
- J. E. Smith and F. P. Gabbaï, Organometallics, 2023, 42, 240–245 CrossRef CAS PubMed.
- Y. Matano, Main Group Metals in Organic Synthesis, John Wiley & Sons, Ltd, 2004, pp. 753–811 Search PubMed.
- B. Sigwarth, U. Weber, L. Zsolnai and G. Huttner, Chem. Ber., 1985, 118, 3114–3126 CrossRef CAS.
- J. S. Jones and F. P. Gabbaï, Acc. Chem. Res., 2016, 49, 857–867 CrossRef CAS PubMed.
- B. L. Murphy and F. P. Gabbaï, J. Am. Chem. Soc., 2023, 145, 19458–19477 CrossRef CAS PubMed.
- D. Tofan and F. P. Gabbaï, Chem. Sci., 2016, 7, 6768–6778 RSC.
- B. Pan and F. P. Gabbaï, J. Am. Chem. Soc., 2014, 136, 9564–9567 CrossRef CAS PubMed.
- N. Li, R. Qiu, X. Zhang, Y. Chen, S.-F. Yin and X. Xu, Tetrahedron, 2015, 71, 4275–4281 CrossRef CAS.
- M. Hall and D. B. Sowerby, J. Am. Chem. Soc., 1980, 102, 628–632 CrossRef CAS.
- J. Xia, R. Qiu, S. Yin, X. Zhang, S. Luo, C.-T. Au, K. Xia and W.-Y. Wong, J. Organomet. Chem., 2010, 695, 1487–1492 CrossRef CAS.
- X. Zhang, S. Yin, R. Qiu, J. Xia, W. Dai, Z. Yu, C.-T. Au and W.-Y. Wong, J. Organomet. Chem., 2009, 694, 3559–3564 CrossRef CAS.
- R. A. Ugarte and T. W. Hudnall, Green Chem., 2017, 19, 1990–1998 RSC.
- S. Benz, A. I. Poblador-Bahamonde, N. Low-Ders and S. Matile, Angew. Chem., Int. Ed., 2018, 57, 5408–5412 CrossRef CAS PubMed.
- M. Breugst and J. J. Koenig, Eur. J. Org. Chem., 2020, 5473–5487 CrossRef CAS.
- M. Yang, M. Hirai and F. P. Gabbaï, Dalton Trans., 2019, 48, 6685–6689 RSC.
- M. Yang, N. Pati, G. Bélanger-Chabot, M. Hirai and F. P. Gabbaï, Dalton Trans., 2018, 47, 11843–11850 RSC.
- T. Hasegawa and K. Takagi, J. Polym. Sci., 2023, 61, 2655–2664 CrossRef CAS.
- J. Zhang, J. Wei, W.-Y. Ding, S. Li, S.-H. Xiang and B. Tan, J. Am. Chem. Soc., 2021, 143, 6382–6387 CrossRef CAS PubMed.
- M. Paraja, A. Gini, N. Sakai and S. Matile, Chem. – Eur. J., 2020, 26, 15471–15476 CrossRef CAS PubMed.
- H. V. Humeniuk, A. Gini, X. Hao, F. Coelho, N. Sakai and S. Matile, JACS Au, 2021, 1, 1588–1593 CrossRef CAS PubMed.
- G. Renno, Q.-X. Zhang, A. Frontera, N. Sakai and S. Matile, Helv. Chim. Acta, 2024, 107, e202400015 CrossRef CAS.
- G. Renno, D. Chen, Q.-X. Zhang, R. M. Gomila, A. Frontera, N. Sakai, T. R. Ward and S. Matile, Angew. Chem., Int. Ed., 2024, 63, e202411347 CrossRef CAS PubMed.
- D. Sharma, S. Balasubramaniam, S. Kumar, E. D. Jemmis and A. Venugopal, Chem. Commun., 2021, 57, 8889–8892 RSC.
- D. Sharma, A. Benny, R. Gupta, E. D. Jemmis and A. Venugopal, Chem. Commun., 2022, 58, 11009–11012 RSC.
- N. Sen, P. Gothe, P. Sarkar, S. Das, S. Tothadi, S. K. Pati and S. Khan, Chem. Commun., 2022, 58, 10380–10383 RSC.
- X. Lan, X. Zhang, Y. Mei, C. Hu and L. L. Liu, Dalton Trans., 2023, 52, 15660–15664 RSC.
- D. Sharma, A. Benny, A. P. Andrews, T. Rajeshkumar, L. Maron and A. Venugopal, Nat. Synth., 2025, 1–9 Search PubMed.
- S. Yasuike, S. Okajima, K. Yamaguchi, H. Seki and J. Kurita, Tetrahedron, 2003, 59, 4959–4966 CrossRef CAS.
- N. K. Srungavruksham, Y.-H. Liu, M.-K. Tsai and C.-W. Chiu, Inorg. Chem., 2020, 59, 4468–4474 CrossRef CAS PubMed.
- H. Yang and F. P. Gabbaï, J. Am. Chem. Soc., 2015, 137, 13425–13432 CrossRef CAS PubMed.
- D. You, H. Yang, S. Sen and F. P. Gabbaï, J. Am. Chem. Soc., 2018, 140, 9644–9651 CrossRef CAS PubMed.
- Y.-H. Lo and F. P. Gabbaï, Angew. Chem., Int. Ed., 2019, 58, 10194–10197 CrossRef CAS PubMed.
- C. J. Carmalt and N. C. Norman, Chemistry of Arsenic, Antimony, and Bismuth, Blackie Academic and Professional, London, 1998 Search PubMed.
- S. Yasuike, W. Qin, Y. Sugawara and J. Kurita, Tetrahedron Lett., 2007, 48, 721–724 CrossRef CAS.
- B. Marciniec, C. Pietraszuk, P. Pawluć and H. Maciejewski, Chem. Rev., 2022, 122, 3996–4090 CrossRef CAS PubMed.
- Q. Simpson, M. J. G. Sinclair, D. W. Lupton, A. B. Chaplin and J. F. Hooper, Org. Lett., 2018, 20, 5537–5540 CrossRef CAS PubMed.
- Y. Kitamura, M. Matsumura, Y. Kato, Y. Murata and S. Yasuike, Eur. J. Org. Chem., 2020, 1652–1657 CrossRef CAS.
- D. Zhang, Z. Xu, T. Tang, L. Le, C. Wang, N. Kambe and R. Qiu, Org. Lett., 2022, 24, 3155–3160 CrossRef CAS PubMed.
- D. Zhang, L. Le, R. Qiu, W.-Y. Wong and N. Kambe, Angew. Chem., Int. Ed., 2021, 60, 3104–3114 CrossRef CAS PubMed.
- D. Zhang, T. Tang, Z. Zhang, L. Le, Z. Xu, H. Lu, Z. Tong, D. Zeng, W.-Y. Wong, S.-F. Yin, A. Ghaderi, N. Kambe and R. Qiu, ACS Catal., 2022, 12, 854–867 CrossRef CAS.
- K. M. Marczenko, J. A. Zurakowski, M. B. Kindervater, S. Jee, T. Hynes, N. Roberts, S. Park, U. Werner-Zwanziger, M. Lumsden, D. N. Langelaan and S. S. Chitnis, Chem. – Eur. J., 2019, 25, 16414–16424 CrossRef CAS PubMed.
- P. Šimon, F. de Proft, R. Jambor, A. Růžička and L. Dostál, Angew. Chem., Int. Ed., 2010, 49(32), 5468–5471 CrossRef PubMed.
- W. Malisch, H.-A. Kaul, E. Groß and U. Thewalt, Angew. Chem., Int. Ed. Engl., 1982, 21(S7), 1281–1288 CrossRef.
- W. Malisch and P. Panster, Angew. Chem., Int. Ed. Engl., 1974, 13, 670–672 CrossRef.
- P. Šimon, R. Jambor, A. Růžička and L. Dostál, Organometallics, 2013, 32, 239–248 CrossRef.
- S. Sahu and F. P. Gabbaï, J. Am. Chem. Soc., 2017, 139(14), 5035–5038 CrossRef CAS PubMed.
- S. S. Chitnis, A. P. M. Robertson, N. Burford, J. J. Weigand and R. Fischer, Chem. Sci., 2015, 6(4), 2559–2574 RSC.
- G. Schröder, T. Okinaka, Y. Mimura, M. Watanabe, T. Matsuzaki, A. Hasuoka, Y. Yamamoto, S. Matsukawa and K. Akiba, Chem. – Eur. J., 2007, 13, 2517–2529 CrossRef PubMed.
- K. Akiba, T. Okinaka, M. Nakatani and Y. Yamamoto, Tetrahedron Lett., 1987, 28, 3367–3368 CrossRef CAS.
- K. Ohkata, S. Takemoto, M. Ohnishi and K. Akiba, Tetrahedron Lett., 1989, 30(36), 4841–4844 CrossRef CAS.
- A. Gehlhaar, H. M. Weinert, C. Wölper, N. Semleit, G. Haberhauer and S. Schulz, Chem. Commun., 2022, 58, 6682–6685 RSC.
- S. Eulberg, N. Schulze, J. Krumsieck, N. Klein and M. Bröring, Angew. Chem., Int. Ed., 2023, 62, e202306598 CrossRef CAS PubMed.
- C. R. Wade, M. R. Saber and F. P. Gabbaï, Heteroat. Chem., 2011, 22(3–4), 500–505 CrossRef CAS.
- L. Dostál, R. Jambor, A. Růžička, R. Jirásko, A. Lyčka, J. Beckmann and S. Ketkov, Inorg. Chem., 2015, 54, 6010–6019 CrossRef PubMed.
- J. S. Wenger, M. Weng, G. N. George and T. C. Johnstone, Nat. Chem., 2023, 1–8 Search PubMed.
- J. S. Wenger and T. C. Johnstone, J. Am. Chem. Soc., 2024, 146, 19350–19359 CrossRef CAS PubMed.
- Y. Pang, M. Leutzsch, N. Nöthling and J. Cornella, Angew. Chem., Int. Ed., 2023, 62, e202302071 CrossRef CAS PubMed.
- L. Dostál, Coord. Chem. Rev., 2017, 353, 142–158 CrossRef.
- A. Koppaka, S. H. Park, B. G. Hashiguchi, N. J. Gunsalus, C. R. King, M. M. Konnick, D. H. Ess and R. A. Periana, Angew. Chem., Int. Ed., 2019, 58, 2241–2245 CrossRef CAS PubMed.
- S.-S. Chen, A. Koppaka, R. A. Periana and D. H. Ess, J. Am. Chem. Soc., 2021, 143, 18242–18250 CrossRef CAS PubMed.
- G. Duneş, M. Cordier, S. Kahlal, A. Pöllnitz, J.-Y. Saillard, C. Silvestru and Y. Sarazin, Dalton Trans., 2024, 53, 15427–15440 RSC.
- M. Huang, K. Li, Z. Zhang and J. Zhou, J. Am. Chem. Soc., 2024, 146, 20432–20438 CrossRef CAS PubMed.
- J. Jiang, K. L. Materna, S. Hedström, K. R. Yang, R. H. Crabtree, V. S. Batista and G. W. Brudvig, Angew. Chem., Int. Ed., 2017, 56, 9111–9115 CrossRef CAS PubMed.
- C. K. Williams, G. A. McCarver, A. Chaturvedi, S. Sinha, M. Ang, K. D. Vogiatzis and J. “Jimmy” Jiang, Chem. – Eur. J., 2022, 28, e202201323 CrossRef CAS PubMed.
- K. M. Marczenko, J. A. Zurakowski, K. L. Bamford, J. W. M. MacMillan and S. S. Chitnis, Angew. Chem., Int. Ed., 2019, 58, 18096–18101 CrossRef CAS PubMed.
- J. W. M. MacMillan, K. M. Marczenko, E. R. Johnson and S. S. Chitnis, Chem. – Eur. J., 2020, 26, 17134–17142 CrossRef CAS PubMed.
- K. Akiba, H. Ohnari and K. Ohkata, Chem. Lett., 1985,(10), 1577–1580 CrossRef CAS.
- S. Yasuike, Y. Kishi, S. Kawara and J. Kurita, Chem. Pharm. Bull., 2005, 53, 425–427 CrossRef CAS PubMed.
- A. Tanoue, W.-J. Yoo and S. Kobayashi, Adv. Synth. Catal., 2013, 355, 269–273 CrossRef CAS.
- B. Zhou and F. P. Gabbaï, J. Am. Chem. Soc., 2023, 145, 13758–13767 CrossRef CAS PubMed.
- R. J. Schwamm and M. P. Coles, Eur. J. Inorg. Chem., 2022, e202200064 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |