
New guidelines and definitions for type I photodynamic therapy
Mingle
Li†
 *a,
Jianhua
Xiong†
ab,
Yingying
Zhang†
ab,
Le
Yu†
c,
Lizhou
Yue
ab,
Changyu
Yoon
d,
Yujin
Kim
d,
Yang
Zhou
e,
Xiaoqiang
Chen
*a,
Yunjie
Xu
*d,
Xiaojun
Peng
*a and
Jong Seung
Kim
*d
*a,
Jianhua
Xiong†
ab,
Yingying
Zhang†
ab,
Le
Yu†
c,
Lizhou
Yue
ab,
Changyu
Yoon
d,
Yujin
Kim
d,
Yang
Zhou
e,
Xiaoqiang
Chen
*a,
Yunjie
Xu
*d,
Xiaojun
Peng
*a and
Jong Seung
Kim
*d
aCollege of Materials Science and Engineering, Shenzhen University, Shenzhen 518060, China. E-mail: limingle@szu.edu.cn; chenxq@szu.edu.cn; pengxj@dlut.edu.cn
bCollege of Physics and Optoelectronic Engineering, Shenzhen University, Shenzhen 518060, China
cCollege of Chemical Science and Technology, Yunnan University, Kunming 650091, China
dDepartment of Chemistry, Korea University, Seoul 02841, Korea. E-mail: xuyunjie87@korea.ac.kr; jongskim@korea.ac.kr
eKey Laboratory of Advanced Materials of Tropical Island Resources of Ministry of Education and School of Chemistry and Chemical Engineering, Hainan University, Haikou, Hainan 570228, China
First published on 20th June 2025
Abstract
The advent of photochemical technologies has revolutionized biology and medicine, offering groundbreaking innovations in cancer treatment and beyond. Among these, photodynamic therapy (PDT) has emerged as a promising approach to cancer therapy, leveraging cytotoxic reactive oxygen species (ROS) to eliminate cancer cells. While traditional type II PDT relies on high oxygen levels and consumes substantial amounts of oxygen, type I PDT requires less oxygen and holds great potential in addressing the hypoxic microenvironments characteristic of solid tumors. Over the last six years, our research team has made pioneering contributions to this field, with a particular focus on type I photosensitizers (PSs) and their diverse applications, including oxygen-sparing PDT, mitochondrial respiration inhibitors, modulation of cellular self-protection pathways, targeted cancer cell destruction, regulation of cellular signaling pathways, immune activation via nanomedicines, and intracellular oxygen-independent artificial photoredox catalysis. Notably, in 2018, our research proposed a “partial oxygen-recyclable mechanism” mediated by O2˙−, successfully revealing why the type I mechanism can be used for overcoming PDT hypoxia resistance. This revitalized interest in type I PDT and inspired numerous research groups worldwide to develop a plethora of new O2˙− photogenerators. However, inconsistencies in mechanistic interpretations, detection methodologies, and application strategies have arisen due to fragmented communication within the field of photoscience and ambiguity in some key definitions. Given our research team's significant contributions and expertise in the type I PDT domain, we believe it is imperative to present a comprehensive review to establish standardized definitions, mechanisms, molecular designs, detection techniques, and clinical applications of type I PDT in cancer diagnosis and treatment. Our goal is to provide a clear and authoritative resource for both specialists and non-specialists, fostering a deeper understanding of type I PDT and inspiring future innovations to advance more effective and clinically relevant therapies for cancer treatment.
 From left to right: Lizhou Yue, Yang Zhou, Xiaoqiang Chen, Xiaojun Peng, Mingle Li, Yingying Zhang, Jianhua Xiong | Mingle Li (PhD, Dalian Univ. of Tech., 2019) is a full professor at Shenzhen University, specializing in type I PDT and O2-independent photocatalytic biomolecular regulation in living settings. Jianhua Xiong (PhD, Wuhan Univ., 2024) is a postdoc under Prof Xiaojun Peng at Shenzhen University, specializing in the design and synthesis of organic small-molecule prodrugs and their antitumor mechanisms. Yingying Zhang (PhD, Nanjing Forestry Univ., 2024) is a postdoc under Prof Xiaojun Peng at Shenzhen University, specializing in photosensitive dyes for type I PDT and photocatalytic therapy. Lizhou Yue (PhD, Guangxi Univ., 2024) is a postdoc under Prof Xiaojun Peng at Shenzhen University, specializing in molecular probes, biomedical imaging, and type I photosensitizers. Yang Zhou (PhD, Kent State Univ., 2017) is a full Professor at Hainan University, specializing in organic functional NO donors for biomedical applications. Xiaoqiang Chen (PhD, Dalian Univ. of Tech., 2007) is a full professor at Shenzhen University, specializing in organic/supramolecular photochemistry, biological sensors, and mRNA delivery materials. Xiaojun Peng (PhD, Dalian Univ. of Tech., 1990) is an Academician of the Chinese Academy of Sciences, specializing in functional dyes for fluorescent bioimaging/labeling and digital printing/recording. |
 Le Yu | Le Yu (PhD, Korea Univ., 2024) is currently a lecturer at Yunnan University, with research interests in fluorescence probes, photoacoustic imaging, photodynamic therapy, and photothermal therapy. |
 From left to right: Changyu Yoon, Jong Seung Kim, Yunjie Xu, Yujin Kim | Changyu Yoon (BS, Hallym Univ., 2022) is pursuing his PhD degree under Prof Jong Seung Kim at Korea University, focusing on designing disease-selective small-molecule photosensitizers with enhanced photochemical reactivity for PDT. Yujin Kim (BS, Korea Univ., 2023) is pursuing her PhD degree under Prof Jong Seung Kim at Korea University, focusing on the development of drug delivery systems in cancer therapy. Yunjie Xu (PhD, Jilin Univ., 2018) is currently pursuing postdoctoral training under the supervision of Prof Jong Seung Kim at Korea University, focusing on the antitumor mechanism (e.g., pyroptosis, ferroptosis, and autophagy) of prodrugs and small-molecular photoactive agents. Jong Seung Kim (PhD, Texas Tech Univ., 1993) is an Academician of the Korea Academy of Sciences and a distinguished Professor at Korea University in Seoul, focusing on drug delivery systems, in vivo imaging of pathologies, and phototherapeutic chemistry. He has been a highly cited researcher since 2014 and the author of more than 700 papers with an h-index of 127. |
Key learning points(1) The historical perspective and definitions of type I photodynamic therapy.(2) The photophysical and photochemical processes of type I photosensitizers. (3) Detection techniques for type I ROS in chemistry and biology. (4) A palette of type I photosensitizers in cancer treatment. (5) Challenges and opportunities in advancing type I PS research. |
1. Introduction
Innovations in phototheranostic technologies have profoundly impacted life sciences, enabling precise exploration and manipulation of complex biochemical processes.1–5 Among these, photodynamic therapy (PDT) has emerged as a groundbreaking approach in tumor treatment, celebrated for its unparalleled temporal and spatial precision, minimal invasiveness, and high therapeutic selectivity.6,7 PDT relies on three essential components—photosensitizers (PSs), light sources, and sufficient oxygen—for its efficacy.8 By leveraging the metabolic activity of tumor cells, PSs preferentially accumulate in tumor tissue, where they can be activated by specific laser irradiation to generate reactive oxygen species (ROS), which can specifically ablate malignant cells and neovascularization with remarkable precision.9,10 Such a dual targeting mechanism—combining preferential PS accumulation with precise light delivery—establishes PDT as a uniquely selective therapeutic modality with minimized collateral damage.1PDT is broadly classified into type I and type II mechanisms based on the mode of ROS generation.11,12 The well-established type II PDT involves PS excitation upon light irradiation, transitioning to a triplet excited state through intersystem crossing (ISC).13 The triplet PS transfers energy to molecular oxygen, generating singlet oxygen (1O2), a potent oxidizing agent capable of inducing oxidative damage and cell death.14 However, the reliance of type II PDT on molecular oxygen presents a critical limitation in hypoxic tumor microenvironments (pO2 < 5 mmHg),15 a hallmark of solid tumors that significantly reduces its therapeutic efficacy. Current strategies to overcome hypoxia include hyperbaric O2 therapy, nanotechnology-based O2 delivery and in situ O2 production, but still face several concerns, such as the risk of oxygen poisoning and tumor-specific accumulation and controlled release.16–19
In contrast, type I PDT has gained considerable attention due to its reduced O2 dependency and a unique “partial oxygen-recyclable” anti-hypoxia mechanism (elaborated in Section 3.2.1).20 Since our seminal discovery in 2018 of thio-Nile blue (ENBS) as a novel generator of superoxide anions (O2˙−),21 interest in type I PDT has surged, leading to substantial research advancements globally. However, the diversity in detection methods and ROS identification mechanisms has introduced challenges, often complicating the accurate understanding and optimization of type I PDT.
Recognizing this need, we leverage our expertise and recent breakthroughs in tumor phototherapy to provide a comprehensive guide addressing these challenges. In this review, we redefine type I PDT, systematically outlining molecular design principles, photochemical mechanisms, and methodologies for ROS detection. Beginning with an in-depth exploration of type I photochemistry, this review consolidates cutting-edge advancements and offers practical insights into molecular PS design for type I PDT. In particular, we introduce, for the first time, fundamental chemical principles regarding “New Guidelines and Definitions for type I PDT”, aiming to inspire the development of more effective, innovative therapeutic strategies.
2 Status quo of type I PDT
Since its identification in 1991, type I PDT has undergone rapid development, transitioning into a new era after more than three decades of progress.22 Initially focused on understanding the electron transfer-based type I photosensitized oxidation and its biological application (i.e., type I PDT photochemical mechanism and therapeutic efficacy), research has now advanced to the systematic design of innovative type I PSs. These include not only organic small molecular dyes23,24 but also inorganic (hybrid) nanomaterials25 for achieving superb therapeutic outcomes.The development of type I PDT can be roughly partitioned into two stages: (1) the first stage is from approximately 1991 to 2012. During this period, researchers unveiled the fundamental photochemical mechanism of type I PDT (i.e., electron transfer process)26 and identified some PSs enabling the generation of O2˙− (but not specific), including organic molecular dyes (e.g., methylene blue and cationic boron-dipyrromethene),27 water-soluble bacteriochlorophyll derivatives (e.g., WTS09 and WTS11)28,29 and metal oxide nanoparticles (e.g., TiO2, ZnO, Al2O3, CeO2, and Fe2O3 nanoparticles).30 (2) The second stage is in the last decade. To address the limitations of traditional type I PDT including (1) limited tissue penetration depth of excitation light; (2) non-specific O2˙− generation hampers anti-hypoxia efficiency in O2-deficient environments (e.g., tumor microenvironment); (3) the lack of targeted specificity of PSs, leading to oxidative damage in normal tissues, researchers have developed and modified numerous type I PSs. For instance, Lin et al. introduced upconversion-based TiO2 nanomaterials for near-infrared (NIR) light excitation, enhancing tissue penetration depth;31 Li and co-workers developed the first exclusive NIR O2˙− photogenerator (ENBS-B)21 and later designed a binary photodynamic O2-economizer (SORgenTAM)32 to mitigate tumor hypoxia by inhibiting intracellular O2 consumption, thereby improving therapeutic efficacy; additionally, Li et al. further proposed a structure-inherent targeting (SIT) strategy using Förster resonance energy transfer (FRET) theory to improve the tumor-targeting ability of type I PSs.33
Despite these advancements, significant challenges remain in fully understanding the relationship between the therapeutic efficacy of type I PDT and its underlying biological mechanism. Recent studies have classified O2-independent photoredox catalysis within cells as type I PDT.34,35 However, according to the definition of PDT, light, oxygen, and PSs are the three essential elements required.26 Without oxygen, these reactions should be categorized as photocatalytic therapy rather than PDT, as they merely borrow from the mechanisms of type I reactions without aligning with PDT's strict definition. Originally, type I PDT was believed to primarily induce apoptosis to achieve therapeutic effects. Recent findings, however, suggest that pyroptosis may also serve as a “potential contributor” to cell death,36,37 although the dominance of apoptosis versus pyroptosis is likely influenced by multiple factors, including PS localization, intracellular ROS flux, light dose, and cellular context (e.g., caspase expression levels).38 This raises critical questions: Are there other undiscovered therapeutic mechanisms underlying type I PDT? How can existing approaches be integrated to develop a “one for all” strategy that addresses type I PDT limitations? These challenges are driving scientists to conduct more comprehensive and in-depth research, paving the way for future advancements in clinical treatment.
3. New definition of type I PDT
In the evolving landscape of PDT, inconsistencies in terminology and definitions regarding type I photosensitized oxidation reactions have hindered progress. Discrepancies in reaction mechanisms, detection techniques, and application fields, often stemming from insufficient communication among professionals in photoscience, have created unnecessary misunderstandings and confusion. To foster a unified and accurate understanding of type I mechanisms, it is essential to bridge these gaps and establish a coherent framework. This section introduces an updated and comprehensive definition of type I PSs, highlighting their mechanisms of action and detection methodologies. By tracing the historical development of type I PDT, we provide a detailed exploration of its photophysical and photochemical processes, such as the photoinduced electron transfer mechanism in organic molecular dyes and the photon-generated electron–hole pair mechanism in inorganic nanomaterials. In particular, to present a holistic perspective of this dynamic field, we summarize the chemistry and biology of type I related ROS, including O2˙−, hydroxyl radicals (˙OH), and hydrogen peroxide (H2O2). Drawing upon our research group's unique insights, we delve into the current status and challenges of type I PDT, offering a clear and forward-looking analysis of this transformative therapy. They are detailed in the following sections:3.1. Photophysical and photochemical processes
Since its initial conceptualization by von Tappeiner and A. Jodlbauer in 1907,39,40 PDT has undergone extensive development, demonstrating significant potential in the treatment of solid tumors. The pivotal distinction between type I and type II PDT mechanisms was first tentatively proposed by Foote in 1991,22 significantly advancing the field (Fig. 1). Subsequent discoveries have further elucidated type I PDT. For example, in 2002, methylene blue was identified as capable of generating O2˙− at aqueous micelle interfaces,27 and by 2015, metal oxides were also implicated in type I PDT mechanisms.31 Jean Cadet and colleagues made a landmark contribution in 2017 by formally proposing “ten tips for defining type I and type II photosensitized oxidation reactions”, providing a comprehensive framework for distinguishing these two pathways.41 This work has charted a forward-looking path of guidance for subsequent research into type I PDT properties of both organic molecules and inorganic compounds. Despite these advancements, however, the fundamental question of why type I PDT exhibits a lower oxygen dependence compared to type II PDT remains unresolved. In 2018, our research group made a pivotal discovery with sulfur (S)-substituted Nile blue analogs (ENBS), which uniquely function as selective generators of O2˙− for PDT.21 Within cancer cells, it was revealed that photogenerated O2˙− could engage in superoxide dismutase (SOD)-mediated disproportionation reactions to form H2O2, then participate in Haber–Weiss or Fenton reaction to produce highly toxic ˙OH radicals. This mechanism permits an O2-recycling cascade. Hence, even in a severely O2-poor microenvironment, photoinduced O2˙− can be sustained at a therapeutically effective level.21 This breakthrough drew significant attention and spurred numerous subsequent studies, advancing the field of type I PDT and expanding its applications.20,37,42,43 Currently, type I PDT has transcended traditional boundaries, finding relevance in supramolecular systems,44–46 bacterial cell membrane disruption47–49 and even environmental pollutant degradation.50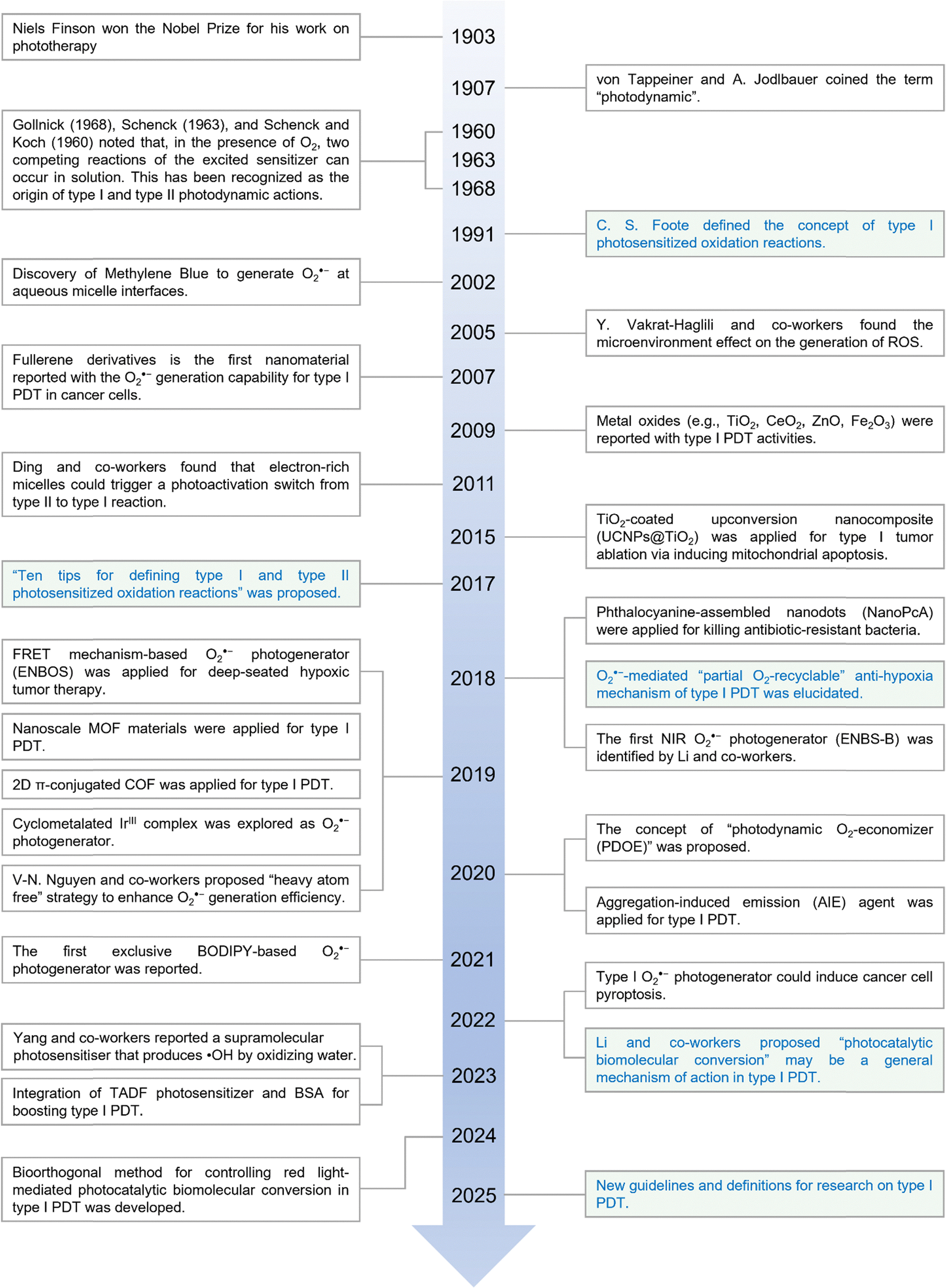 | ||
| Fig. 1 A concise timeline of key milestones in type I PDT research. | ||
In type I PDT, PSs play a pivotal role in driving the therapeutic process. So far, a diverse array of PSs have been developed, which can be broadly categorized into two categories: (1) organic molecular dyes and (2) inorganic (hybrid) nanoparticles. At the core of type I PDT lies a fundamental photochemical reaction in which electrons are transferred from PSs to oxygen or other substrates, generating radical cations or anions. Notably, such an electron transfer mechanism differs significantly between organic molecules and inorganic nanoparticles, reflecting their distinct photophysical and photochemical properties.
As depicted in Fig. 2a, upon photoirradiation, type I organic molecular PSs absorb photons and transition to a short-lived excited singlet state (i.e., 1[PS*]). From this state, PSs can return to their ground state (i.e., 0[PS]) via fluorescence emission or a nonradiative decay process (e.g., vibrational relaxation). Alternatively, 1[PS*] can undergo intersystem crossing (ISC), a nonradiative transition to a long-lived triplet state (i.e., 3[PS*]), which is either isoenergetic or slightly lower in energy. Conventionally, 3[PS*] is understood to directly transfer electrons to surrounding O2 (and water), resulting in the formation of O2˙− and ˙OH radicals. These radicals can initiate cascaded reactions that disrupt cellular components, such as membranes and lipids, ultimately leading to cell death (Fig. 2b).51 This mechanism represents the most well-characterized and widely studied type I pathway. However, an often-overlooked alternative pathway involves electron transfer between 3[PS*] and biomolecules (Fig. 2c). Cells contain a variety of electron-rich biomolecules, e.g., amino acids, proteins, RNA, DNA, and NAD(P)H. Upon photoexcitation, these biomolecules can transfer electrons to the excited PSs, forming anionic PS species. These intermediates subsequently transfer one electron to O2 to generate O2˙−, highlighting the ability of 3[PS*] to engage oxygen indirectly.52–54 This mechanism, in theory, is similar to photoredox catalysis, thus it has gradually evolved into a newly recognized type I mechanism of action termed “photocatalytic biomolecular conversion” (discussed further in Section 6).55
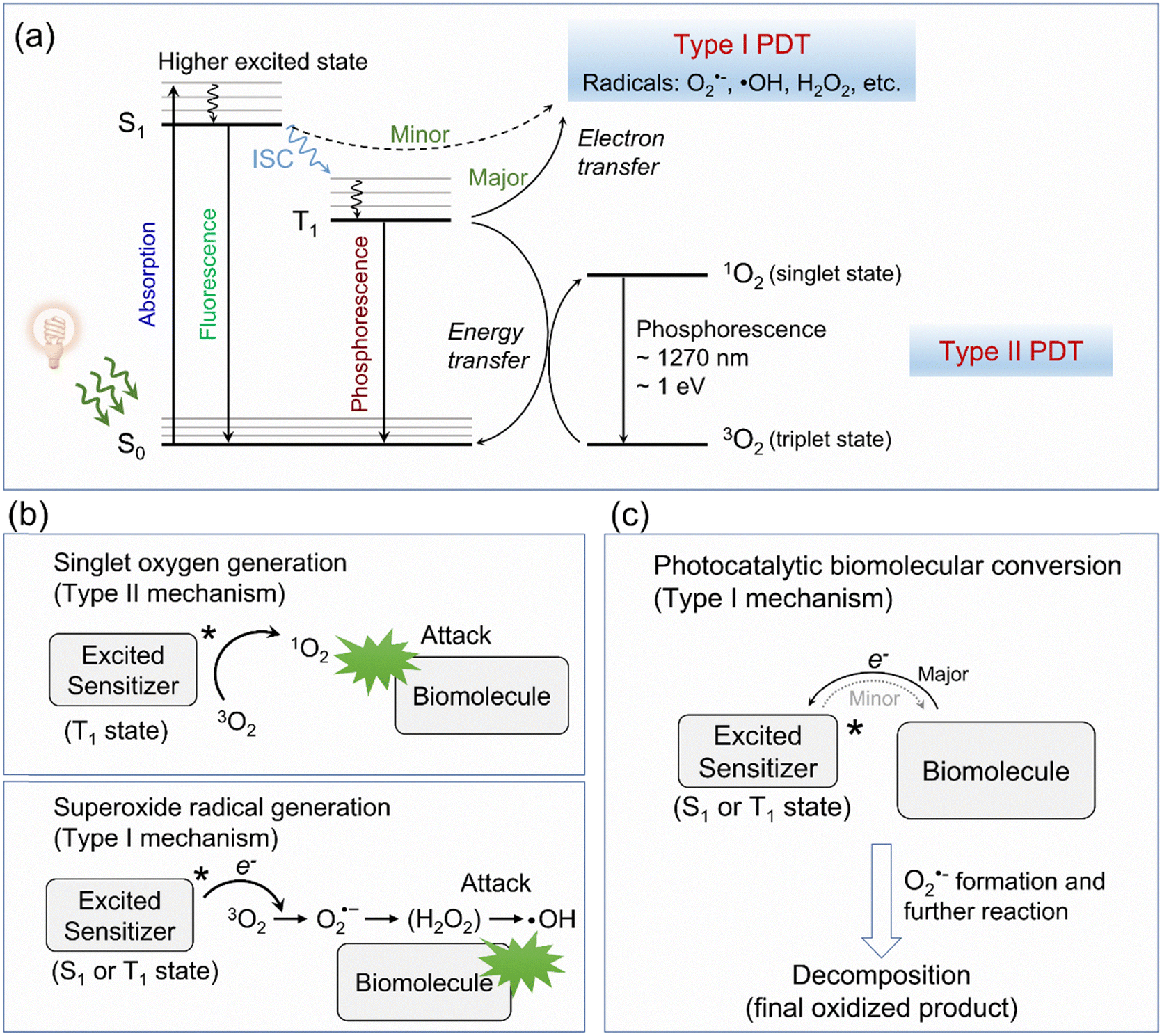 | ||
| Fig. 2 Type I PDT mechanism in organic molecules. (a) Jablonski energy diagram of photochemistry in PDT. (b) Schematic illustration showing how the ROS are generated in conventional type II and type I mechanisms of action. (c) Schematic illustration of photocatalytic biomolecular conversion as a new type I PDT mode of action. | ||
It is worth noting that theoretically, singlet state PSs (i.e., 1[PS*]) are also capable of initiating electron transfer pathways to finish type I photochemical reactions. However, due to the extremely short lifetime of 1[PS*], this pathway is less significant and is not considered a dominant mechanism in type I PDT, often being overlooked in mechanistic studies.
Different from type I organic molecular PSs, inorganic (hybrid) nanomaterials displayed a “photogenerated electron holes” mechanism (as depicted in Fig. 3a).25,56 Upon photoirradiation, the energy of the light sources must exceed the bandgap of the nanomaterials. This excitation promotes electrons from the valence band (VB) to the conduction band (CB), creating a charge-separated state characterized by an electron–hole pair. In this process, electrons in the CB can transfer to molecular oxygen, generating O2˙− for type I PDT. Simultaneously, holes in the VB react with water molecules, forming ˙OH for a photochemotherapy (PCT) process that is independent of O2 and distinct from PDT. Thus, type I PDT in inorganic (hybrid) nanomaterials is often accompanied by PCT. A prominent example is modified titanium dioxide (TiO2), an ideal type I inorganic (hybrid) PS due to its efficient photogenerated charge separation.56 Upon illumination, photogenerated electrons produce O2˙−, and photogenerated holes react with water to form ˙OH radicals, representing an O2-independent PCT process. Similar mechanisms have been observed in diverse inorganic (hybrid) nanomaterials (Fig. 3b), including covalent organic frameworks (COFs),57,58 metal–organic frameworks (MOFs),59 porous organic polymers (POPs),60 and carbon dots (CDs).61,62
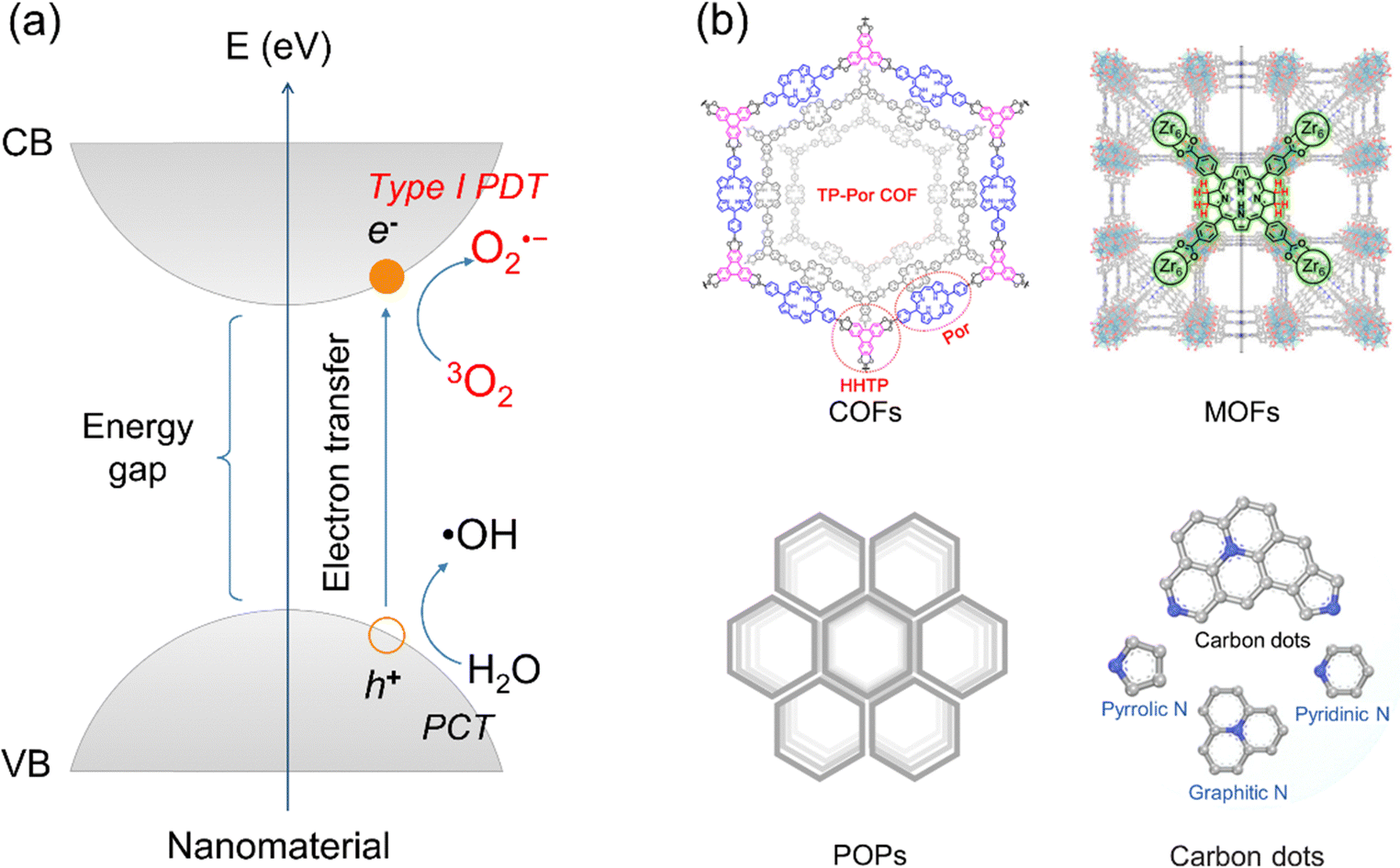 | ||
| Fig. 3 (a) Schematic diagram of the typical excitation process in inorganic nanomaterials, illustrating the transition of electrons from the valence band (VB) to the conduction band (CB) upon photoirradiation, followed by type I PDT and PCT processes. (b) Examples of diverse inorganic and hybrid nanomaterials employed in type I PDT, such as covalent organic frameworks (COFs), metal–organic frameworks (MOFs), porous organic polymers (POPs), and carbon dots (CDs). | ||
3.2. Type I ROS chemistry and biology
ROS refers to a group of chemically reactive molecules derived from the incomplete reduction of oxygen or water, mainly including 1O2, O2˙−, ˙OH, and H2O2.63 These molecules are integral to the growth and development of living organisms, playing a pivotal role in regulating various physiological functions.64 However, excessive ROS production can disrupt cellular homeostasis, leading to oxidative stress. This condition is implicated in numerous diseases, including chronic inflammation,65 cancer,66 neurodegenerative diseases,67etc. Over the last century, significant advances have been made in understanding the intricate redox chemistry of ROS, which has, in turn, accelerated the development of ROS-based therapeutics, especially PDT.68 This section focuses on the photophysical and photochemical properties of O2˙−, ˙OH, and H2O2, the key ROS species involved in type I PDT (Fig. 4).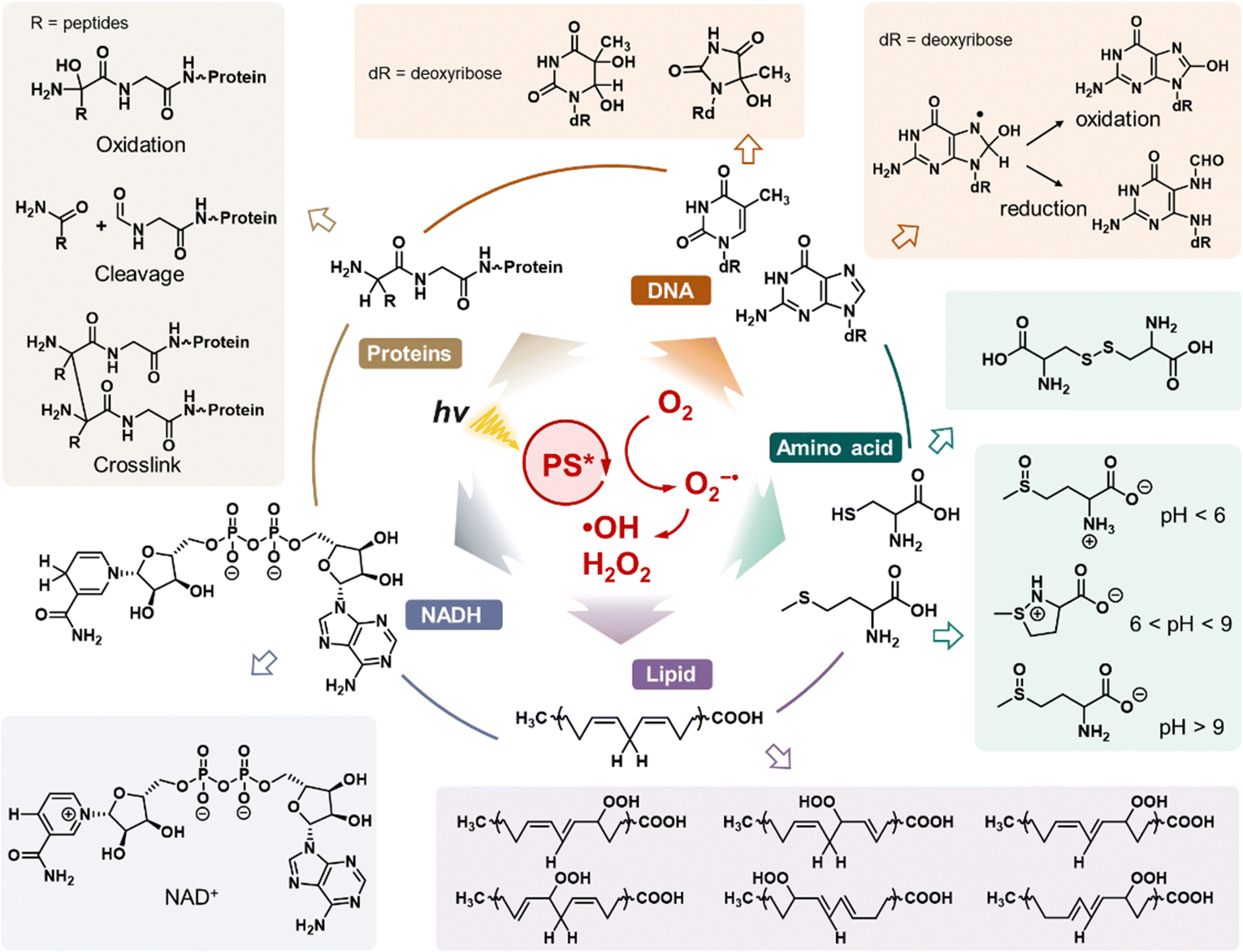 | ||
| Fig. 4 Biomolecule oxidation pathways mediated by ROS generated during type I PDT. The diagram illustrates key oxidative reactions induced by ROS, including ˙OH, O2˙−, and associated cascade reactions. | ||
Despite its short lifespan and limited diffusion, O2˙− is highly effective in initiating cascade biochemical reactions that lead to target cell lethality, such as in cancer therapy.21
1. Disproportionation reaction
O2˙− is catalyzed by intracellular SOD (i.e., superoxide dismutase) to form H2O2 and molecular oxygen via a rapid disproportionation reaction with a rate constant of 6.4 × 109 M−1 s−1 (reaction (1)).71
2. Fenton and Haber–Weiss reactions
H2O2 produced in the disproportionation participates in the Fenton reaction, reacting with Fe2+ to generate ˙OH, a highly toxic species. Concurrently, remaining O2˙− can react with Fe3+ to regenerate oxygen (reaction (2)).72 O2˙− also modulates H2O2 reduction through the Haber–Weiss reaction, producing ˙OH and oxygen (reaction (3)).
These highly reactive ˙OH radicals cause irreversible oxidative damage to target cells, effectively eliminating them. Furthermore, the oxygen generated during these processes alleviates hypoxia-resistance in cancer cells, enhancing the therapeutic efficacy of type I PDT and endowing it with a “partial oxygen-recyclable” anti-hypoxia mechanism (as proposed by Li et al. in 2018).
Reactions:
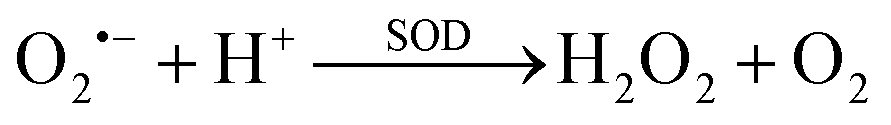 | (1) |
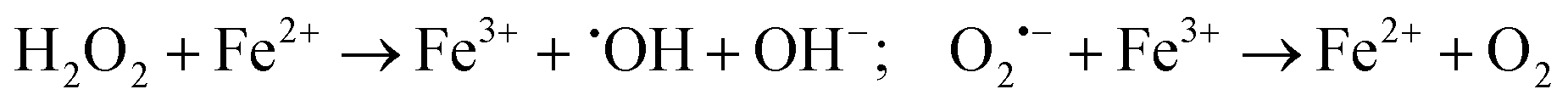 | (2) |
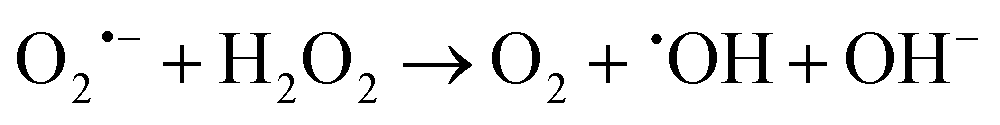 | (3) |
Significantly, ˙OH is the most lethal ROS produced during the type I PDT process due to its extraordinary oxidative properties. The oxidative potential of the ˙OH/H2O pair is as high as +2.30 eV, enabling ˙OH to abstract electrons from almost all nearby molecules at extremely high reaction rates (reaction rate constants between 109 and 1010 M−1 s−1).75 For instance, ˙OH readily participates in hydrogen-abstraction reactions, producing water and substrate radicals (R˙).76 Additionally, ˙OH can react with low-valence metal cations through an electron transfer process, resulting in the formation of high-valence metal cations and OH−.75 Furthermore, ˙OH interacts with the pentyl group of unsaturated fatty acids, initiating lipid peroxidation. This process generates incorporated ROS, such as peroxyl radicals (ROO˙), which further propagate oxidative damage.77 Due to its potent oxidative properties, ˙OH generated in type I PDT inflicts significant damage on a variety of intracellular components, including proteins, DNA, amino acids, lipids, and NAD(P)H, ultimately leading to the destruction of targeted cells.72 Given its reliable and robust oxidative capacity, enhancing ˙OH production in type I PDT has emerged as a prominent research hotspot in recent years.
It is noteworthy that type I PDT does not generate H2O2 directly but relies on the dismutation of O2˙−, as discussed in Section 2.2.1. Biologically, the cumulation of intracellular H2O2 also primarily arises from O2˙− dismutation catalyzed by SOD (reaction (1)).79 In addition to SOD, various enzymes, including peroxidases, catalases (CAT), and peroxiredoxins, play critical roles in H2O2 metabolism. Ascorbate peroxidase, a specific type of peroxidase, catalyzes H2O2 decomposition through a ping-pong mechanism, producing water and ascorbate radicals. This mechanism represents one of the primary pathways for intracellular H2O2 scavenging.80 In addition, CAT also plays an important role in the catalysis of H2O2. Of note, CAT-mediated H2O2 decomposition yields molecular oxygen, distinguishing it from the ascorbate peroxidase pathway.81 This oxygen production is particularly significant in mitigating tumor hypoxia, thereby having been widely used for enhancing the effectiveness of PDT.
4. Methods to identify type I PDT
Type I PDT has rapidly emerged as a powerful tool for cancer and anti-infection therapy. However, there remains a lack of clear guidelines for identifying type I PSs. Accurate characterization of ROS is crucial for distinguishing between type I and type II PDT modalities. This section outlines fundamental chemical principles and methodologies for precisely analyzing ROS during PDT processes, both in solution tests and in cellular experiments.4.1. Detection methods for O2˙−
The detection of O2˙−, a key ROS in type I PDT, can be achieved through three primary approaches: (1) optical absorption in ultraviolet and visible (UV) and infrared (IR) regions (Section 4.1.1); (2) fluorescence assay (Section 4.1.2); and (3) spin-trapping electron spin resonance (ESR, Section 4.1.3).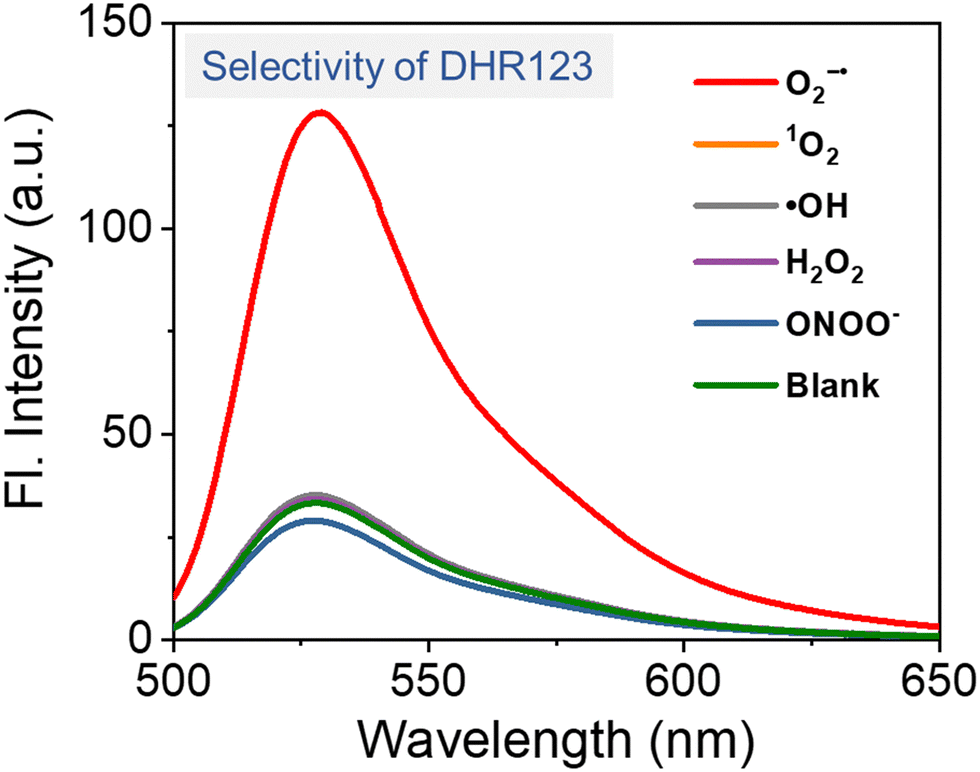 | ||
| Fig. 5 Fluorescence intensity of DHR123 for the detection of various ROS and ONOO− in aqueous solution, confirming that DHR123 exhibits selective response to O2˙−. | ||
While multiple fluorescence probes have been widely used for O2˙− detection, their limitations must be carefully considered. For DHR123, although its specificity for O2˙− in solution is confirmed (Fig. 5), its reactivity with H2O2 in the presence of peroxidases restricts its utility in cellular environments where endogenous peroxidases are abundant. DHE and MitoSOX Red, while membrane-permeable and suitable for intracellular O2˙− detection, require integration into DNA for fluorescence emission. This DNA dependency limits their application in non-nuclear or extracellular contexts (e.g., mitochondrial matrix and cytoplasmic vesicles). Furthermore, DHE exhibits non-specific oxidation by other ROS (e.g., ONOO−, HOCl) and redox-active metal ions in cells, leading to potential false-positive signals. To mitigate this, parallel control experiments using O2˙− scavengers (e.g., superoxide dismutase, SOD) is essential for validation. Based on their ease of use in experimental setups and the reliability of the results, DHR123 and DHE are considered the preferred probes for detecting O2˙− in solution and within cells, respectively (Table 1).
| Probes | Analyte sensitivity | False-positive risks | Preferred scope of application | Validation protocols |
|---|---|---|---|---|
| Note: +high; −low | ||||
| DHR123 | + + + (O2˙−) | − − − (solution test) | Solution test | (1) Mixing DHR123 with PSs in an aqueous solution, followed by light irradiation. |
| − − − (other ROS) | + + + (in cell test) | (2) Measuring the fluorescence spectra to confirm O2˙− generation. | ||
| DHE | + + + (O2˙−) | + + + (solution test) | In-cell test | (1) Adding 10 μM DHE to PS-treated cells and incubating for 30 mins at 37 °C, then subjecting the cells to light irradiation. |
| + + + (other ROS) | − − − (in cell test) | (2) Using confocal microscopy to observe the red fluorescence in the nucleus. | ||
4.2. Detection methods for ˙OH
Due to its extremely high reactivity, ˙OH is often regarded as the most toxic ROS in type I PDT. The primary detection methods for ˙OH include (1) fluorescence assay (Section 4.2.1) and (2) spin-trapping ESR (Section 4.2.2).4.3. Detection methods for H2O2
Among the ROS involved in type I PDT, H2O2 stands out as the only stable molecule. This stability allows for its separate detection after the decay of other ROS (i.e., O2˙− and ˙OH). To date, two primary methods have been widely employed for H2O2 detection: (1) direct measurement of optical absorption in the UV region (Sections 4.3.1) and (2) fluorescence assays (Section 4.3.2).| Probes | Analyte | λ abs (nm)a | λ em (nm)a | Scope of application |
|---|---|---|---|---|
| a In aqueous solution. | ||||
| DHR123 | O2˙− | 509 | 529 | Solution test |
| DHE | O2˙− | 518 | 606 | In-cell test |
| MitoSOX | O2˙− | 510 | 580 | In-cell test |
| HKSOX-1 | O2˙− | 509 | 534 | Solution and in-cell tests |
| HPF | ˙OH | 490 | 515 | Solution and in-cell tests |
| APF | ˙OH | 490 | 515 | Solution and in-cell tests |
| HPA | H2O2 | 320 | 420 | Solution and in-cell tests |
| Amplex Red | H2O2 | 572 | 583 | Solution and in-cell tests |
| DMPO | ˙OH or O2˙− | — | — | Solution test |
| BMPO | ˙OH or O2˙− | — | — | Solution test |
| DEPMPO | O2˙− | — | — | Solution test |
| CPYPMPO | ˙OH | — | — | Solution test |
4.4. Validation via ROS scavengers
The identification of dominant ROS in type I PDT necessitates the use of selective scavengers as critical analytical tools. A panel of well-characterized scavengers was shown here, each targeting specific ROS with distinct mechanisms. For example, sodium pyruvate mediates non-enzymatic decarboxylation, specifically neutralizing H2O2 without affecting other ROS species.99 Superoxide dismutase (SOD), a cell-impermeable enzyme, can only catalyze the dismutation of extracellular O2˙−, while Tiron (4,5-dihydroxy-1,3-benzenedisulfonic acid) can penetrate cell membranes to quench intracellular O2˙− and modulate electron transfer pathways.88 D-mannitol acts as an efficient ˙OH scavenger through radical hydrogen abstraction.100 Co-treatment of PS-loaded systems with these scavengers under illumination, followed by comparative cytotoxicity or ROS quantification assays, provides direct evidence for ROS specificity and their relative contributions to PDT (Table 3).32| ROS species | Scavenger | Mechanism | Scope of application |
|---|---|---|---|
| O2˙− | SOD | Catalyzes O2˙− dismutation to H2O2 and O2 | Solution test |
| O2˙− | Tiron | Scavenging O2˙− | Solution and in-cell tests |
| ˙OH | D-Mannitol | Hydroxyl radical quenching | Solution and in-cell tests |
| H2O2 | Sodium pyruvate | Non-enzymatic H2O2 decomposition | Solution and in-cell tests |
5. A palette of type I PSs
This section presents a fresh perspective on the design mechanism of type I PSs, particularly from the viewpoint of small molecular chemistry. Despite significant advancements, systematic and rational approaches to designing exclusive type I PSs remain rare and elusive. To address this, we summarize state-of-the-art breakthroughs from the last seven years, focusing on molecular design, photochemical mechanisms, and their implications for cancer imaging, diagnosis, and therapeutics. Additionally, we explore methods to enhance type I photochemistry, providing a foundation for improved therapeutic efficacy. Detailed discussions are organized in the following subsections.5.1. Phenothiazine derivatives
Efficient therapy in PDT requires careful selection of the underlying mechanism. The dynamics of substances generated during PDT differ significantly between type I and type II mechanisms. For instance, type II PDT often fails in a hypoxia environment due to oxygen depletion, making mechanism selection a critical factor in successful treatment design. Type I PDT offers a promising alternative in such environments, but its efficacy relies heavily on the PS molecules. A practical molecule design tailored to type I mechanisms often sparks debate due to the challenges in achieving specificity and efficacy. Continuous validation and refinement are crucial to ensuring therapeutic success. This section highlights key studies, particularly on ENBS, offering valuable insights and directions for future research.In 2018, we reported the first-generation specific O2˙− generator, ENBS, as a landmark discovery stemming from an in-depth exploration of Nile blue dye modifications with chalcogen elements (Fig. 6a).21 ENBS is characterized by its amphiphilic properties and versatile chemical reaction sites, enabling functionalization and optimization for diverse applications (Fig. 6b). Over the last seven years, extensive research has demonstrated its utility in targeted cancer therapy, cell signaling regulation, immune activation, phage engineering, and nanomedicine.32,33,48,49,101–110 For example, ENBS conjugated with targeting biomolecules (e.g., ascorbate analog,111 antibodies,102 and biotin21) selectively accumulates in cancer cells, achieving specific ablation (Fig. 6a and c). Additionally, the molecular design optimizes intersystem crossing to favor type I PDT, as evidenced by imbalanced optical absorbance and fluorescence changes (Fig. 6d).33 This study gives an impressive perception of the molecular design for O2˙− generators. When combined with immunotherapy, ENBS exhibits synergistic effects. For instance, overexpressed ROS from PDT can induce pyroptosis and stimulate immune responses, especially when paired with 1-MT to inhibit IDO (indoleamine 2,3-dioxygenase, an immune suppressor), resulting in enhanced ROS release and tandem immunotherapy effects (Fig. 6e).112 Some other studies from our group also expand the family of ENBS, such as targeting droplets for ferroptosis induction,104 activating iterative revolutions of cancer immunity for photoimmunotherapy,113 electrostatically attracting siRNA for synergistic gene therapy,109 engineering photo-PROTAC augmenter for pyroptosis activation,107 and self-reporting photodynamic nanobody for sustainable large-volume tumor eradication.110 These studies have established ENBS as a benchmark for type I PDT research, solidifying our group's position in the field both domestically and internationally.
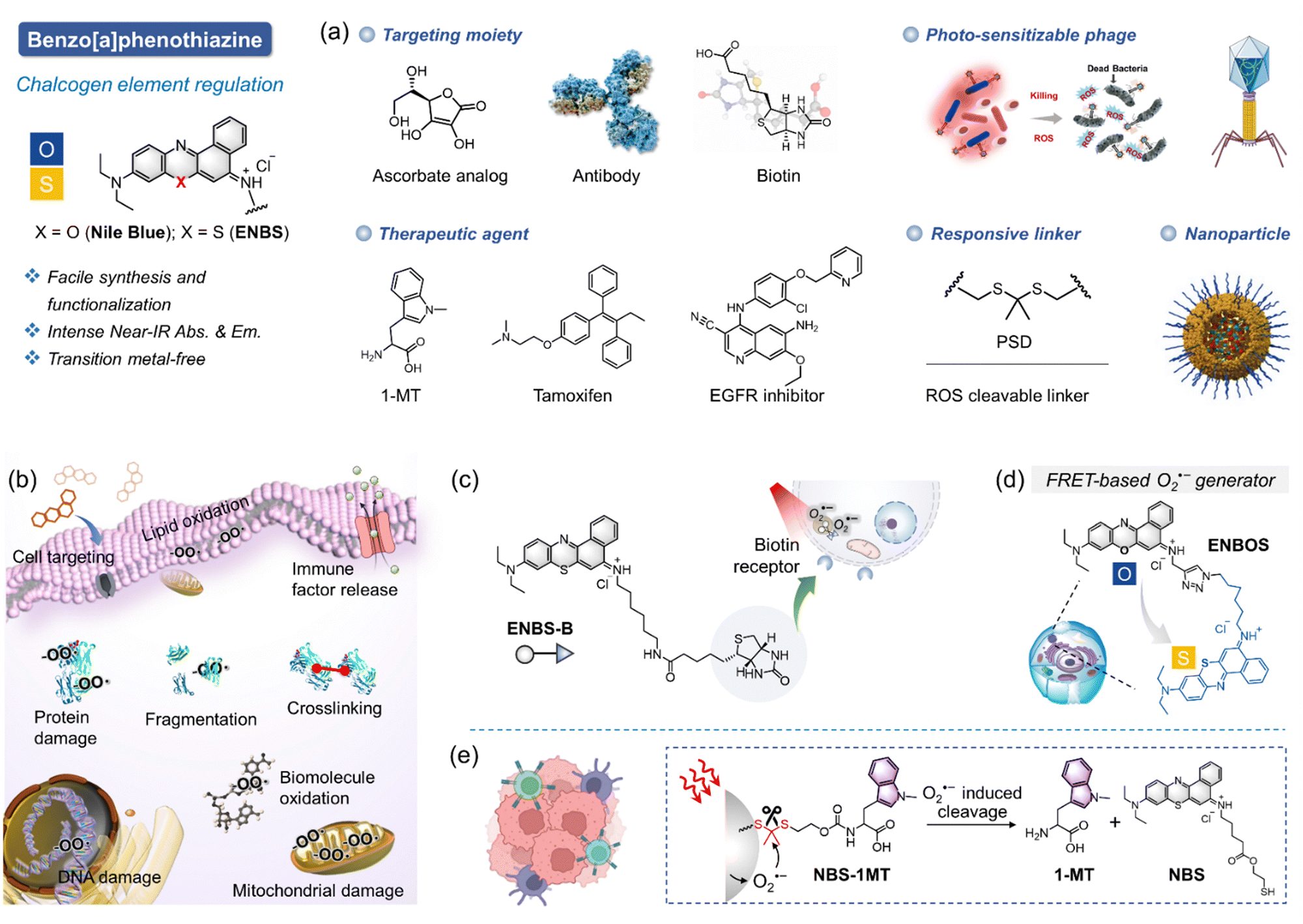 | ||
| Fig. 6 First-generation exclusive type I O2˙− photogenerator (ENBS) utilizing a chalcogen element regulation strategy to modify Nile blue molecule. (a) ENBS-based emerging applications, such as photoimmunotherapy, synergetic photochemotherapy, photo-controlled drug release, photosensitizable phage, and nanomedicine. (b) Cellular mechanisms underlying type I PDT. (c) Schematic illustration of a biotin-targeted type I O2˙− photogenerator. (d) Schematic illustration of a FRET-based type I O2˙− photogenerator. (e) Schematic illustration of photo-controlled immune drug 1-MT release for synergistic type I photoimmunotherapy. Figure created with Biorender.com. | ||
The excellent properties of ENBS explored in our research have inspired other groups, further advancing molecular design and broadening application scope. For example, ENBS can be integrated into nanocarriers to enable multifunctional applications, such as drug delivery, PDT, and MR imaging (Fig. 7).114 Besides, PSs with endoplasmic reticulum targeting abilities play a key role in enhancing the efficacy of PDT. NBS-ER115 was designed to target the ER and generate O2˙− under hypoxic conditions (Fig. 8a). This ability effectively triggers ER stress and induces apoptosis, showing significant PDT efficacy and good biocompatibility in 4T1 tumor-bearing mice. Some approaches utilizing O2˙− indirectly for therapeutic purposes were also reported (e.g., BPT in Fig. 8b).116 Peroxynitrite (ONOO−), a cytotoxic agent with potent anticancer activity, can overcome drug resistance through the in situ reaction of O2˙− and NO˙. By regulating O2˙− production via light control, precise ONOO− production is achieved, overcoming challenges like low selectivity and short biological half-life (Fig. 8b). Insights from antibacterial studies also contribute to understanding PDT's selectivity in cancer treatment. For example, replacing oxygen in Nile blue derivatives with chalcogen elements alters ROS generation and antibacterial effects (Fig. 8c).117 It shows a different pattern depending on which chalcogen element (i.e., S or Se) replaces the oxygen site in the basic skeleton of Nile blue. The O2˙− generated by NBS-N shows high effectiveness in targeting Gram-positive bacteria. In contrast, NBSe-N primarily produces 1O2, which damages cell membranes non-selectively due to its high reactivity. This highlights the importance of selectivity in type I PDT, especially for bacteria treatment, where precision in targeting is essential. Aggregation of PSs in aqueous media is a well-known challenge as it can result in quenching of ROS production and conversion of absorbed light energy into heat, reducing the overall efficacy of PDT. To address this issue, recent research has shown that assembling immune interferon gene stimulants into polymers containing NBS (PNBS/diABZI)42 can form nanoagonists (Fig. 8d). These nanoagonists facilitate H-aggregation and intersystem crossing, enhancing the production of O2˙− and 1O2 by 3-fold. Additionally, combining ENBS (M-TPO)37 with mitochondrial targeting drugs enables interference with signal pathway crosstalk, providing precise spatiotemporal control over inflammatory responses (Fig. 8e). This approach holds promise for designing strategies to mediate pyroptosis in cancer cells expressing GSDME, which could be a powerful therapeutic avenue. Besides, the design of ROS-activatable ENBS (NTP)118 for anticancer drug delivery further expands potential applications of type I PSs in cancer therapy (Fig. 8f). These findings not only demonstrate the effectiveness of ENBS as a robust O2˙− generator but also offer valuable insights for scientists exploring new strategies in cancer biomedicine.
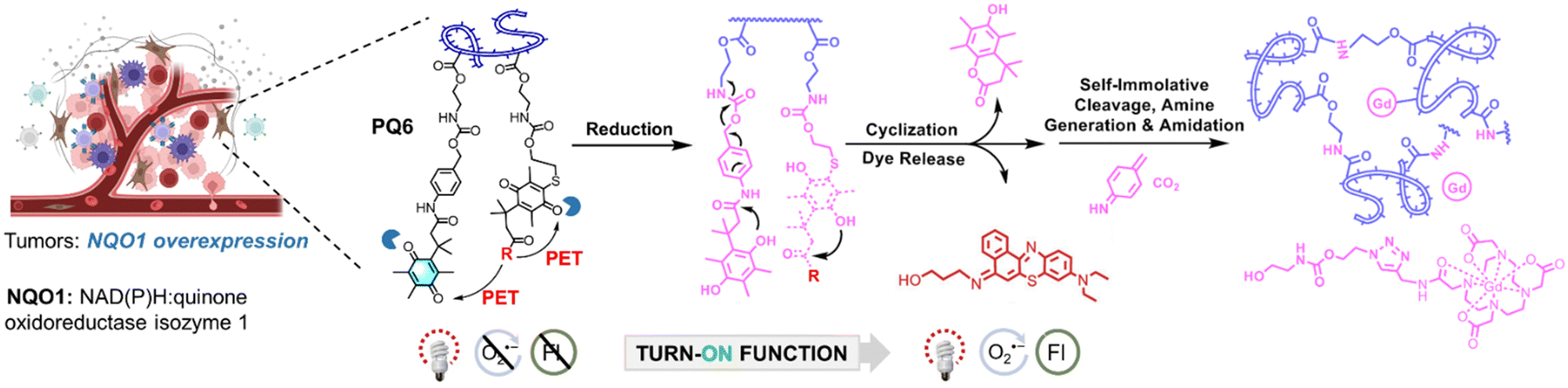 | ||
| Fig. 7 ENBS molecule mediated self-assembled vesicles of amphiphilic block copolymers for activated NIR fluorescence imaging and tissue-specific type I PDT under cellular and in vivo conditions. Figure created with Biorender.com. | ||
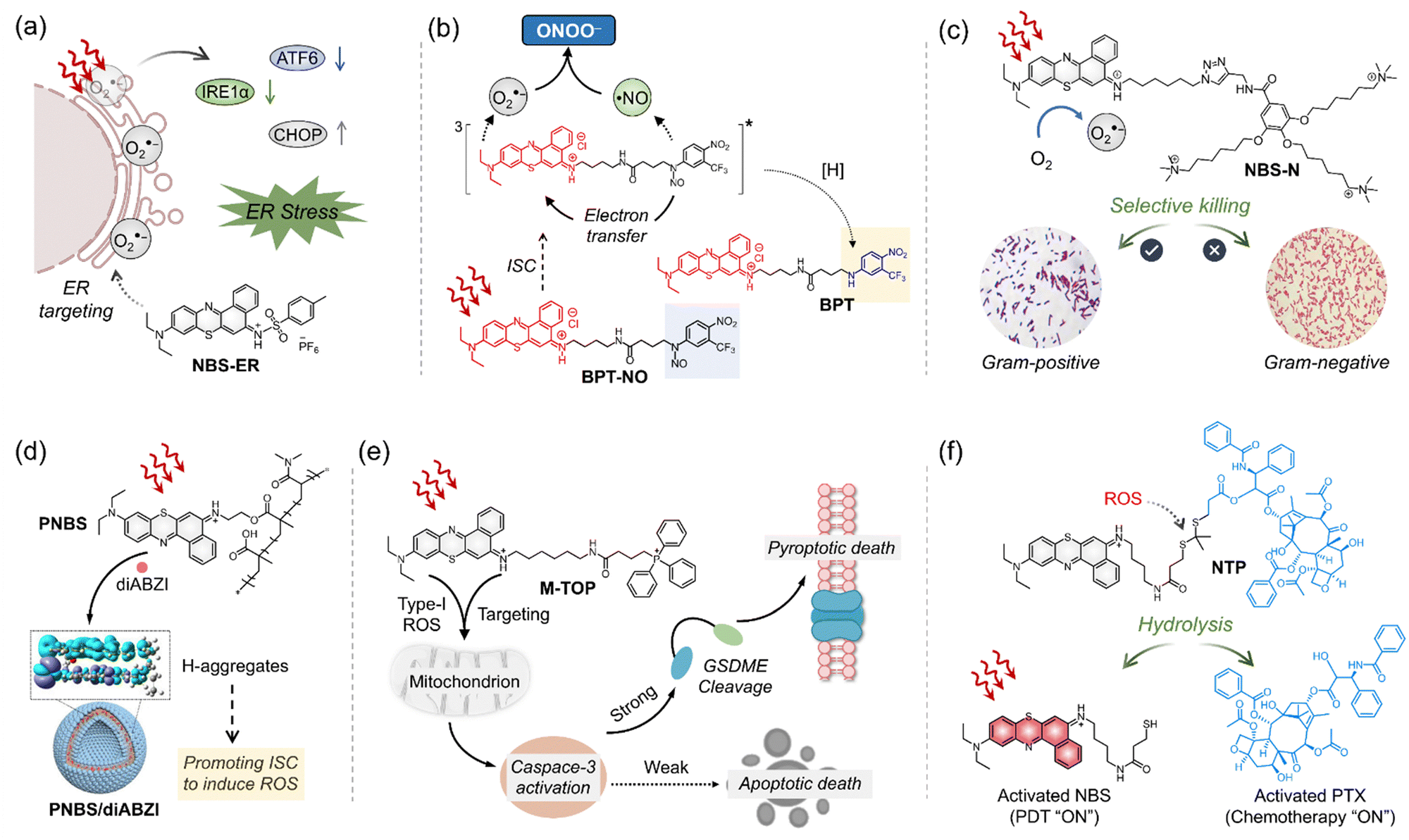 | ||
| Fig. 8 Schematic illustration of chemical structures and mechanisms of ENBS analog type I PSs. (a) Type I PS (NBS-ER) designed for endoplasmic reticulum targeting research. (b) Proposed molecular mechanism leading to the generation of ONOO− and the stable photoproduct BPT upon red light excitation of the molecular hybrid BPT-NO. (c) Schematic illustration of O2˙− generated by NBS-N, along with the selective bactericidal effect on Gram-positive bacteria and the healing of Gram-positive bacterial infections in murine wound models using NBS-N. (d) Development of a nanoagonist (PNBS/diABZI) through the combination of stimulator of interferon genes (STING) and ENBS, aimed at enhancing tumor response. (e) ENBS PS (M-TOP) with mitochondrial targeting for optimized immunotherapy drug delivery. (f) ROS-activatable ENBS PS (NTP) for targeted anticancer drug delivery. Figure created with Biorender.com. | ||
5.2. Porphyrin/chlorin/phthalocyanine-based photosensitizers
Light-induced radical generation is a hallmark of a photo-derived therapeutic approach. Since the pioneering introduction of porphyrin/phthalocyanine derivatives as PSs, these compounds have found wide applications across various fields, often incorporated into composite materials.119,120 These PSs serve as a foundation for the development of type I PS through the following strategic approaches. (1) Electron-donating strategy: when a PS is incorporated with an electron-rich polymer, its type I efficiency is enhanced. (2) Self-assembly strategy: the effectiveness of type I photosensitization increases when the PS self-assembles into electron-rich nanoparticles, particularly in aqueous environments. These strategies have been explored in several studies, illustrating their impact on the photoactivation processes of PSs. For example, modulation of type I and II photoactivation mechanisms of PSs has been investigated by assessing how the carrier microenvironment affects the photophysical properties of the PS, subsequently influencing its therapeutic efficacy.121 As illustrated in Fig. 9a, incorporating 5,10,15,20-tetrakis(meso-hydroxyphenyl)porphyrin (mTHPP) to electron-rich polymer, (2-(diisopropylamino)ethyl methacrylate) (PDPA) micelles, significantly increased the generation of O2˙− over 1O2, transitioning the photoactivation process of mTHPP from type II to type I mode. The use of PDPA micelles (PEO-b-PDPA) enhanced phototoxicity in multiple cancer cell lines compared to the electron-deficient poly(D,L-lactide) control. These data suggest that micelle carriers not only improve the bioavailability of PS but also modulate their photochemical properties, enhancing PDT efficacy. In another study, the self-assembly of a novel phthalocyanine molecule into nanodot (NanoPcA) demonstrated highly efficient O2˙− generation through the type I mechanism (Fig. 9b).122 This innovative approach underscores the potential of self-assembled nanoparticles for advancing the effectiveness of type I PDT.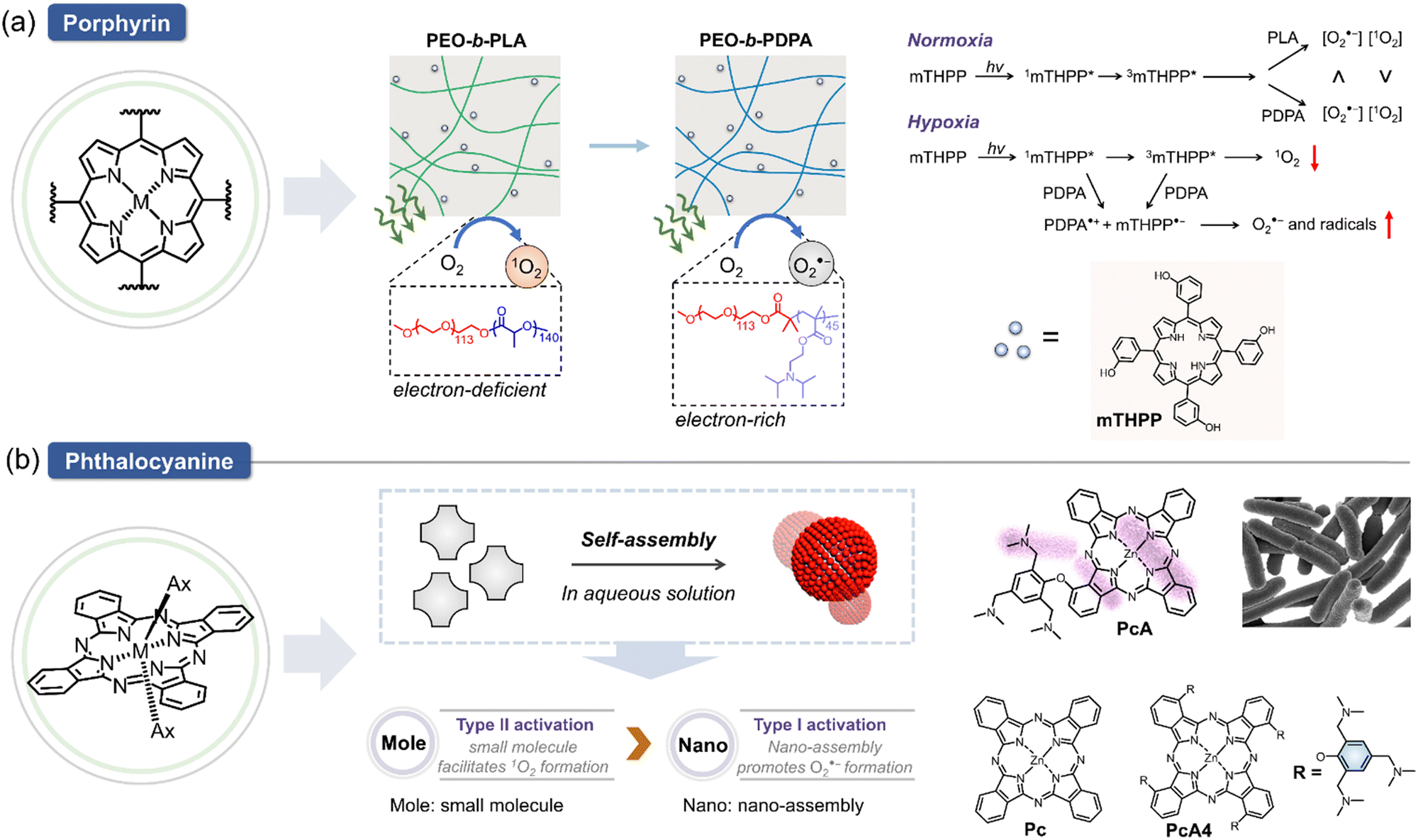 | ||
| Fig. 9 Chemical structures of porphyrin/phthalocyanine derived type I PSs. (a) Schematic showing porphyrin-derived PSs (mTHPP) incorporated into electron-rich micelles can trigger a transition of PDT mechanism of action from type II to type I mode. The ROS generation mechanism of mTHPP under hypoxia and normoxia is shown. (b) Schematic illustration of phthalocyanine-derived nanodots can enhance type I PDT efficiency; the chemical structure of PcA and control molecules are shown. | ||
In addition, a variety of type I PS based on chlorin derivatives have been developed due to their high yield of exogenous O2˙− generation, excellent therapeutic efficacy, as well as a straightforward preparation method. In 2009, two novel observations were made regarding light-induced ROS generation in aqueous solutions using water-soluble derivatives of the photosynthetic pigment bacteriochlorophyll a (Bchl), WST09 and WST11 (Fig. 10a).29 These studies revealed that WST11 exclusively generates O2˙− and ˙OH, with no detectable formation of 1O2 in aqueous solutions. Furthermore, WST11 forms a noncovalent complex with human serum albumin (HSA), which functions as a photocatalytic oxidoreductase, enabling approximately 15 cycles of electron transfer from the HSA protein to molecular oxygen in the solution. These findings challenge the conventional paradigm of porphyrin- and chlorin-based PDT, which is traditionally considered to be primarily mediated by the formation of 1O2, particularly in vascular-targeted photodynamic therapy (VTP). In another study, Yang et al. measured the PDT activities for dicyano-bacteriochlorins, including the parent compound (BC), dicyano derivative (NC)2BC, and corresponding zinc (NC)2BC-Zn and palladium chelate (NC)2BC-Pd, against cancer cells (Fig. 10b).123 Their results demonstrated that the PS with the shortest triplet lifetime, (NC)2BC-Pd, exhibited the highest activity. This outcome suggests that O2˙− and ˙OH, well-known precursors of important pathophysiological processes, can be leveraged for effective tumor eradication, leading to new design paradigms for type I PS undergoing photoactivation.
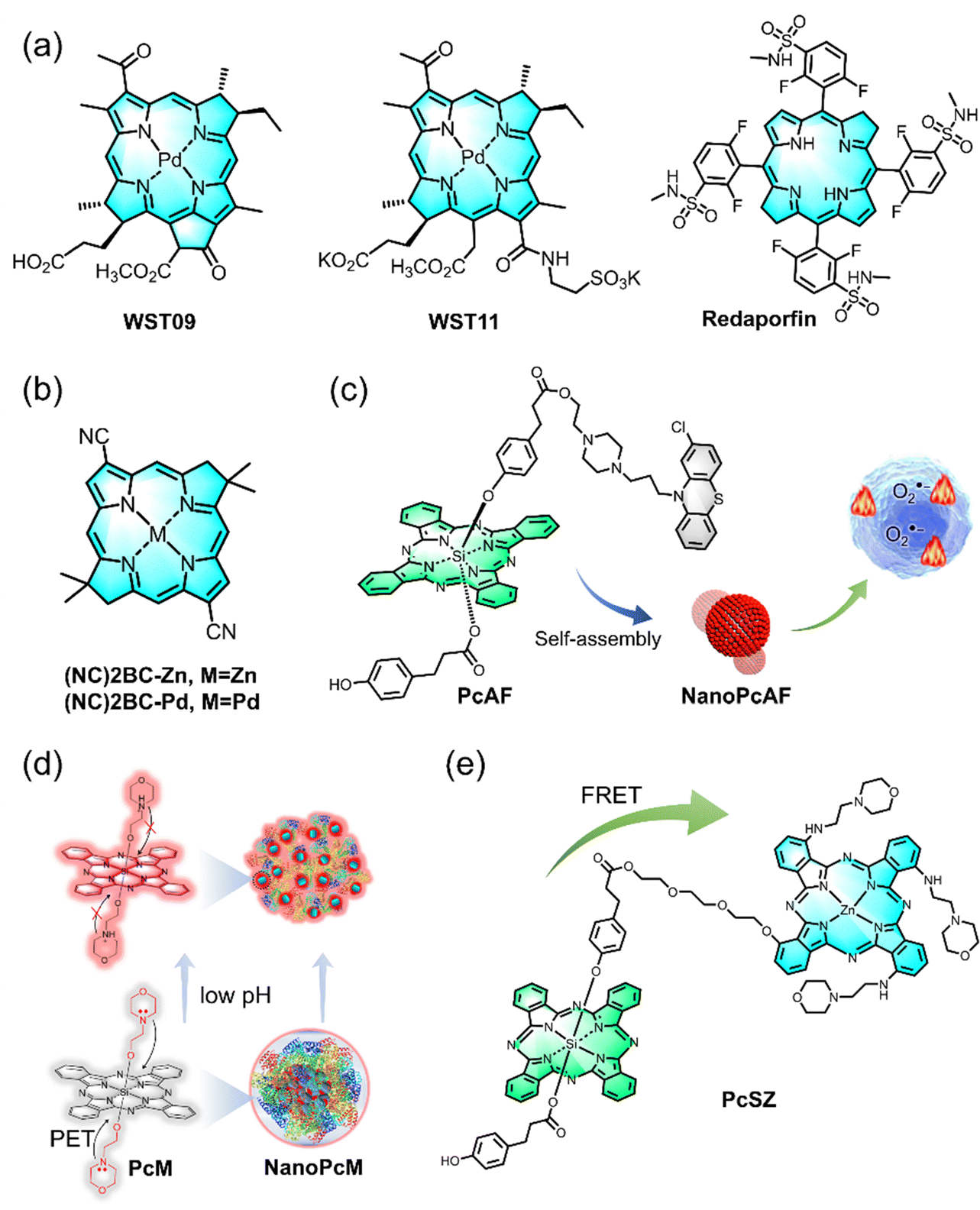 | ||
| Fig. 10 Schematic illustration of porphyrin and phthalocyanine derived PS for enhanced O2˙− generation yield. (a) Chemical structure of [Pd] bacteriopheophorbide derivate, WST09, WST11, and Redaporfin. (b) Chemical structure of phthalocyanine ((NC)2BC-Zn and (NC)2BC-pd). (c) Chemical structure of phthalocyanine (PcAF) and its assemblies NanoPcAF used for type I PDT. (d) Chemical structure of silicon phthalocyanine (PcM) and its assemblies NanoPcM designed for turn-on imaging and PDT-based immunotherapy. (e) Chemical structure of silicon phthalocyanine derivates (PcSZ) for enhanced O2˙− generation via the FRET effect. | ||
While porphyrin and chlorin-based type I PSs have been extensively studied, the development of type I PSs using phthalocyanine has also been actively pursued. A novel design was introduced to shift the photophysical and photochemical properties of traditional phthalocyanine-based PSs from type II photoreaction to efficient type I photoreaction and vibrational relaxation-induced photothermal conversion for photothermal therapy (PTT) as well as multimodal imaging (Fig. 10c).124 One such innovation, NanoPcAF, is a nanostructured PS based on the self-assembly of silicon(IV) phthalocyanine axially monosubstituted with perphenazine. NanoPcAF demonstrated substantial O2˙− generation through the type I mechanism and excellent photothermal performance, overcoming tumor hypoxia in PDT. As a result, NanoPcAF exhibited a synergistic effect combining type I photoreaction and photothermal action, effectively inhibiting tumor growth. Furthermore, there is an increasing demand in clinical applications for titanium phthalocyanine-based PSs with high-definition fluorescence imaging capabilities, alongside outstanding therapeutic effects. A noteworthy example is the combination of morpholine-modified silicon phthalocyanine (PcM) with serum albumin (SA) through supramolecular self-assembly technology. This strategy not only reduces background noise but also enhances tumor-targeting fluorescence imaging, addressing key clinical demands (Fig. 10d).125 In the latest research, the group explored the use of silicon(IV) phthalocyanine analogs (PcS) as energy donors and successfully paired them with zinc(II) phthalocyanine derivatives (serving as energy acceptors, denoted as PcZ). Using fluorescence resonance energy transfer (FRET) technology, they constructed an efficient O2˙− generator, PcSZ.126 This innovation not only enhances energy conversion efficiency but also improves the safety of the system. These findings offer valuable insights for the future development of type I PSs using phthalocyanine derivatives, with the potential to drive further advancements in related fields (Fig. 10e).
5.3. BODIPY dyes
With the promising therapeutic outcomes of PDT, the development of advanced PS continues to gain momentum. Among these, boron dipyrromethene (BODIPY) stands out as one of the most actively studied PSs due to its high triplet quantum yield and efficient ROS generation. In general, BODIPY sensitizers are classified as type II PSs, capable of producing 1O2 with ultrahigh quantum yield.127,128 However, through strategic molecular engineering, its derivatives offer notable advantages, including low oxygen-dependent type I mechanisms for enhanced hypoxic cancer treatment. Like porphyrin/phthalocyanine, BODIPY serves as an excellent starting point for designing type I PSs, utilizing strategies such as: (1) promoting electron donating efficiency via the introduction of electron-rich polymer (Fig. 11a); (2) triplet state formation from an effective excited-state relaxation by introducing α,β-linked BODIPY dimer/trimer (Fig. 11b); and (3) converting type II to type I PSs via host–guest strategies (Fig. 11c).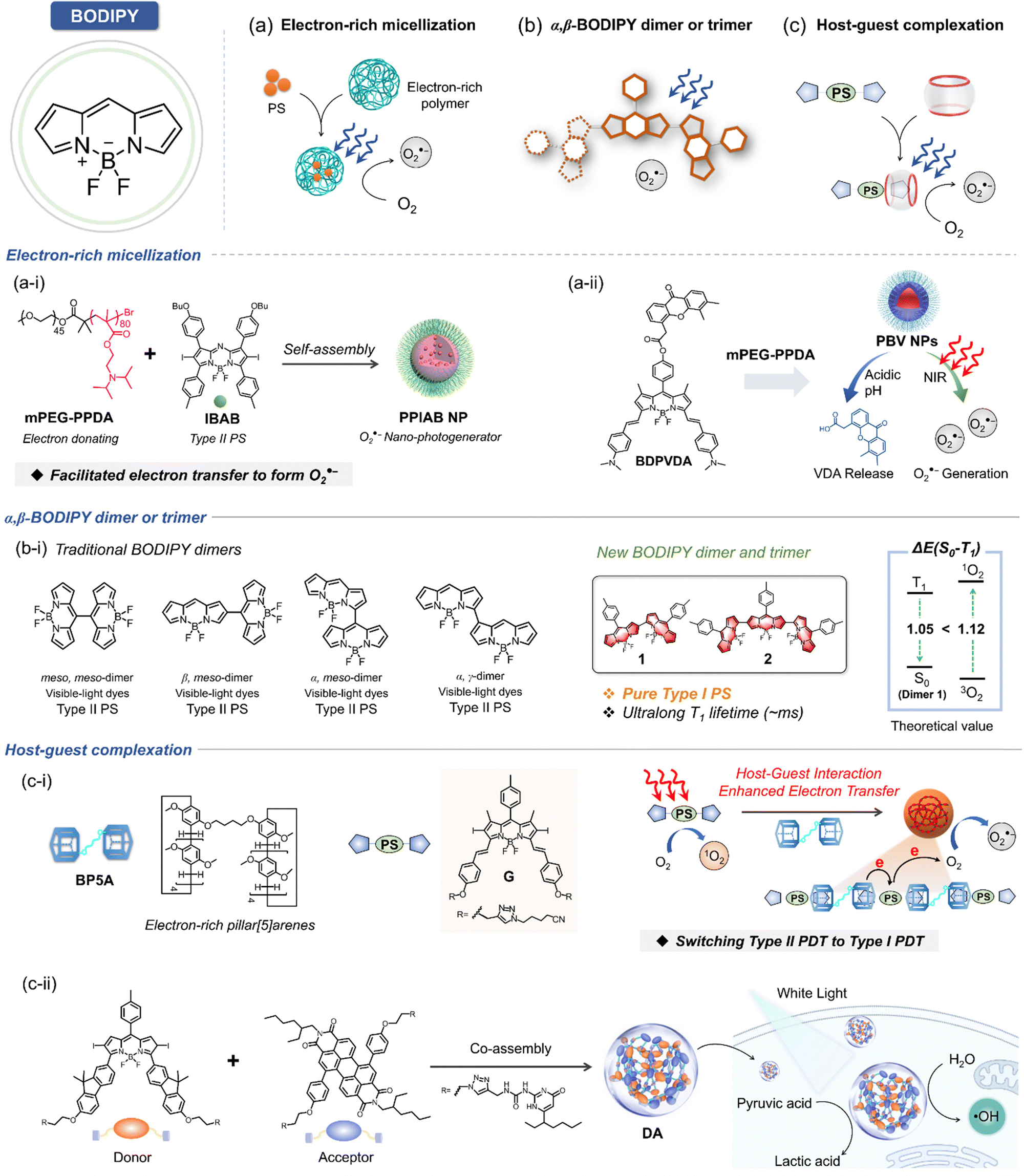 | ||
| Fig. 11 Schematic illustration of chemical structures of BODIPY derived type I PSs. (a) Scheme of electron-rich polymer nanoparticles (NPs). (a-i) Chemical structure of BODIPY derived type I PS IBAB and its assemblies PPIABNP. (a-ii) Chemical structure of BODIPY derived type I PS BDPVDA and its assemblies PBVNPs. (b) Scheme of BODIPY dimers and trimers. (b-i) Chemical structures of traditional and newly developed BODIPY dimers. (c) Scheme of host–guest complexation. (c-i) BODIPY derivates as guests, pllar[5]arenes as hosts, forming nanoparticle assemblies to shift PDT from type II to type I PDT. (c-ii) BODIPY-derivated PS (DA) capable of directly catalyzing the oxidation of water to release highly cytotoxic ˙OH. | ||
To address the limitations of traditional BODIPY-based PSs under hypoxic conditions, PPIABNPs were developed.129 This aza-BODIPY-based nanoplatform incorporates a heavy atom to promote ISC and is encapsulated by the electron-rich polymer mPEG-PPDA for improved electron-donating efficiency (Fig. 11a–i).129 This approach boosted O2˙− photogeneration, effectively overcoming hypoxia-induced challenges in PDT. Upon irradiation, aza-BODIPY rapidly transitions to its singlet state and subsequently changes to a charge-separated state facilitated by the electron-rich environment (i.e., electron-rich polymer, mPEG-PPDA), promoting O2˙− photogeneration. Building on this, PBVNPs were developed by encapsulating the BODIPY-based conjugate in an electron-rich amphiphilic mPEG-PPDA polymer (Fig. 11a-ii).130 These NPs exhibited excellent core–shell intermolecular electron transfer, efficiently converting O2 into O2˙− under NIR laser irradiation, even under hypoxic conditions. Moreover, in vitro and in vivo studies showed that PBVNPs could release vascular-disrupting agents (VDAs) at the tumor site, disrupting tumor vascular vasculature and hindering metastasis pathways.
Yang's research group reported BODIPY-based type I PSs without heavy atoms to mitigate dark toxicity.131 They constructed a series of α, β-linked BODIPY dimers and trimers to tune electron interaction between BODIPY moieties, enhancing ISC efficiency through connections between the electron-deficient α site and the electron-rich β site (Fig. 11b-i). This strategy leveraged effective excited state relaxation, where the triplet state (T1) was populated by internal conversion (IC) from the intermediate triplet (T2) state. Using this strategy, trimer 2 demonstrated significantly enhanced O2˙− generation due to its low reduction potential and an ultralong T1 state lifetime. This was achieved through intermolecular charge transfer to molecular oxygen (Fig. 11b-i). Notably, the generation of 1O2 was effectively suppressed. This suppression occurred because the energy gap between the T1–S0 states was reduced to a level smaller than the gap between 3O2 and 1O2. Consequently, the T1 energy states were insufficient to enable 1O2 formation via excitation energy transfer, thereby inhibiting type II ROS generation.
Building on these findings, the same research group introduced a series of strategies to develop type I exclusive PSs.45,132 To overcome the competition between triplet–triplet energy transfer and type II process, they utilized a host–guest complexation approach to convert conventional type II PS into type I PSs. Electron-rich pillar [5] arenes served as the hosts, while iodide BODIPY-based electron deficient type II PSs acted as the guests (Fig. 11c-i).132 Upon irradiation, host–guest complexation promoted intermolecular electron transfer from the host to the guest, which was subsequently delivered to molecular oxygen via the type I mechanism, resulting in efficient O2˙− generation. Antitumor experiments validated this type I-exclusive photosensitization strategy, demonstrating its high PDT efficacy even under hypoxic conditions.
In a recently launched in-depth study, Yang et al. successfully designed and synthesized a BODIPY PS featuring fluorene as an electron-donating group. This PS was synergistically assembled with perylene diimide, an efficient electron acceptor, to construct a supramolecular photodynamic system based on quadruple hydrogen bonding (Fig. 11c-ii).45 The resulting innovative PS (DA) exhibits remarkable intermolecular electron transfer and charge separation capabilities under light excitation, efficiently facilitating the generation of radical ion pairs. Notably, under strictly anaerobic conditions, DA demonstrated the ability to directly catalyze water oxidation, releasing highly cytotoxic ˙OH. Concurrently, it transferred captured electrons to pyruvate molecules, enabling a catalytic cycle mechanism tailored for tumor systems. This dual-action functionality highlights the potential of this advanced PS design for enhancing PDT efficacy in challenging hypoxic tumor environments.
5.4. Transition metal complexes
Transition metal-based PSs (TMPs) are increasingly gaining attention in PDT, especially for their efficacy in addressing hypoxic tumors.133–136 The unique photophysical and chemical properties of TMPs make them promising candidates for enhancing the type I PDT pathway. Key advantages of TMPs in improving PDT outcomes include their supportive electronic configurations, water solubility, remarkable endocytosis efficiency, organelle targeting capabilities, high molar absorption coefficients, and deep tissue penetration due to absorbance in the NIR region.135 These PSs also exhibit excellent photostability and efficiently generate ROS by interacting with biomolecules such as amino acids, peptides, and proteins.52 The ability of TMPs to activate the type I pathway and generate ROS stems from their partially filled d-orbitals, which facilitate electron transfer to nearby biomolecules. Moreover, the absorbance wavelengths of TMPs can be finely tuned by modifying the central metal ion and electronic environment for their chelating ligands. Through metal–ligand charge transfer (MLCT), the central metal in its reduced form can participate in multiple redox cycles, transferring electrons to oxygen and nearby molecules to generate O2˙−, ˙OH, H2O2, and other ROS.137,138 Beyond the type I pathway, the heavy atom effect inherent in TMPs extends the triplet state lifetime, enabling more efficient energy transfer to molecular oxygen for the generation of 1O2. This dual capability enhances PDT efficacy by combining type I and type II pathways synergistically. TMPs are predominantly hydrophilic, facilitating their excretion from normal cells and reducing dark toxicity.139 Moreover, certain transition metals can coordinate with serum proteins, extending their circulation time and promoting effective accumulation at the tumor site.139 While their relatively large size can increase the risk of accumulation in organs and potential long-term toxicity, the high molar absorption coefficient and selective tumor targeting of TMPs allow for reduced dosing, mitigating toxicity concerns. Recent advancements have highlighted several examples of TMPs, particularly those based on ruthenium (Ru), iridium (Ir), osmium (Os), platinum (Pt), and rhenium (Rh), demonstrating efficient tumor therapy through the type I PDT pathway.The success of a PS in type I PDT depends on its ability to efficiently transfer electrons to molecular oxygen and neighboring biomolecules, facilitating ROS generation and cancer cell eradication. In this section, we explore recent breakthroughs in TMP-based PDT, emphasizing studies that characterize type I pathways using spectroscopic and validate their efficacy in both in vitro and in vivo tumor models.
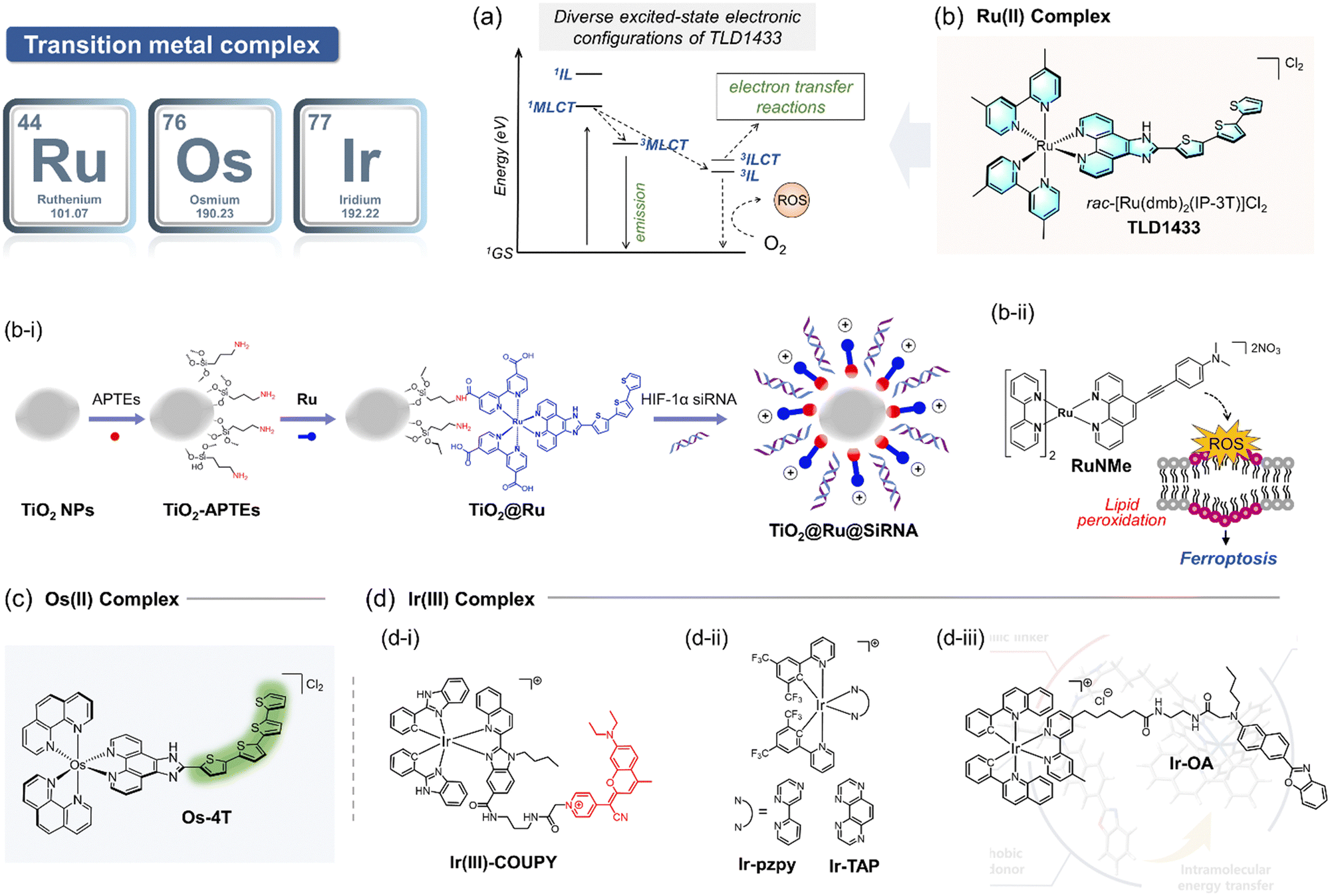 | ||
| Fig. 12 Schematic illustration of representative examples of transition metal-based PSs studied for type I pathway of PDT. (a) Exited state configuration of the ruthenium complex TLD1433, currently in phase II clinical trials. (b) Molecular structure of TLD1433. (b-i) Systematic presentation of the preparation process for the titanium-modified Ru complex TiO2@Ru@SiRNA. (b-ii) Chemical structures of Ru(II) complexes RuNMe and illustration of the photoinduced ferroptosis triggered by RuNMe upon irradiation. (c) Chemical structure of Os-4T. (d-i) Chemical structures of Ir(III)–COUPY. (d-ii) Chemical structures of Ir-pzpy and Ir-TAP. (d-iii) Molecular structure of Ir-OA. | ||
Encouraged by the early success of Ru complexes in PDT, recent studies have focused on developing Ru-based PSs that specifically and effectively activate the type I PDT pathway. For example, a hypoxia-compatible nanocomposite, TiO2@Ru@siRNA, was developed by coating TiO2-nanoparticles with a Ru PS and loading them with hypoxia-inducible factor-1α (HIF-1α) siRNA (Fig. 12b-i).140 This nanocomposite was shown to localize successfully in lysosomes and, under visible light irradiation (525 nm), generate both O2˙− and 1O2, while escaping the HIF-1α signaling pathway. TiO2@Ru@siRNA demonstrated effective tumor growth inhibition by reducing hypoxia, increasing ROS generation via the type I pathway, and enhancing the cancer immune response in an immune-deficient patient-derived xenograft (PDX) model. Additionally, Ru complexes with finely tuned conjugated ligands have shown impressive results in type I PDT. For instance, a series of Ru(II) polypyridine complexes were designed to generate a high quantum yield of O2˙− upon visible light irradiation.142 Among these, the RuNMe derivative (Fig. 12b-ii) demonstrated the most effective inhibition of tumor cells due to its superior cell uptake compared to other derivatives. Recent advances highlight the potential of Ru complexes as exceptional PS candidates for exclusive type I PDT pathways. To maximize their effectiveness, key design considerations should include achieving high quantum yields for electron transfer to biomolecules rather than oxygen, strong targeting and localization abilities, and enhanced biocompatibility. These features are crucial for ensuring efficient and selective PDT via the type I pathway.
Building on the intrinsic strengths of Os, McFarland et al. reported a series of Os(II) complexes conjugated with varying numbers of thiophene rings for application in PDT against hypoxic tumors. These complexes are structurally analogous to the ruthenium-based TLD1433,145 which is currently in clinical trials. The Os(II) complexes exhibited broad absorption in the visible to NIR region, extending up to 740 nm, with photocytotoxicity increasing alongside the number of thiophene rings. These complexes were found to have good biocompatibility and exhibited comparatively low cytotoxicity to normal cells. Notably, the Os-4T complex (Fig. 12c)145 is the first Os-based PS to demonstrate photocytotoxicity in a 1% oxygen environment, achieving the highest phototherapeutic index reported to date. This pivotal study highlighted the significant potential of osmium complexes in the type I pathway of PDT. Recent research has further demonstrated that Os complexes offer several advantages over other PSs such as a broader absorption spectrum, stronger spin–orbital coupling, faster ISC, and prolonged lifetime of triplet excited state. These properties make them highly promising candidates for the development of PSs specifically tailored for type I PDT applications.
Further advancements include two Ir-based complexes, Ir-pzpy and Ir-TAP (Fig. 12d-ii).148 The 1,4,5,8-tetraazaphenanthrene (TAP) ligand-based complex Ir-TAP showed effective binding with the neighboring biomolecules and exhibited stronger cytotoxicity under hypoxic conditions, despite generating a lower quantum yield of 1O2. These findings underscore the importance of optimizing and tuning chelating ligands to increase the production of other ROS, thereby enhancing the efficacy of type I PDT under hypoxic conditions. Another notable example is the iridium PS Ir-OA, which demonstrated accelerated ROS production within mitochondria (Fig. 12d-iii).149 This complex also exhibited the unique ability to monitor mitochondrial microenvironments, including polarity, viscosity, and morphology, further expanding its functionality in PDT applications. Designing TMPS specifically for the type I pathway of PDT requires careful consideration of the chelating ligand structure and its interaction with the central metal.
Recent studies highlight that the efficiency of TMPS can be significantly enhanced through strategic modifications, such as optimization of the structure of chelating ligands by incorporating donor groups, selection of a central metal, and accumulation of chromophores to enhance absorption ranges. TMPs offer immense potential to generate diverse ROS species, enabling highly efficient and selective type I PDT. Despite concerns about toxicity, these can often be mitigated by employing lower doses, thanks to the high molar extinction coefficient of these complexes. With their ability to deliver superior photophysical properties, stability under physiological conditions, and tumor selectivity, Ir complexes present versatile opportunities for designing PSs tailored to specific cancer types and therapeutic needs.
5.5. AIE gens
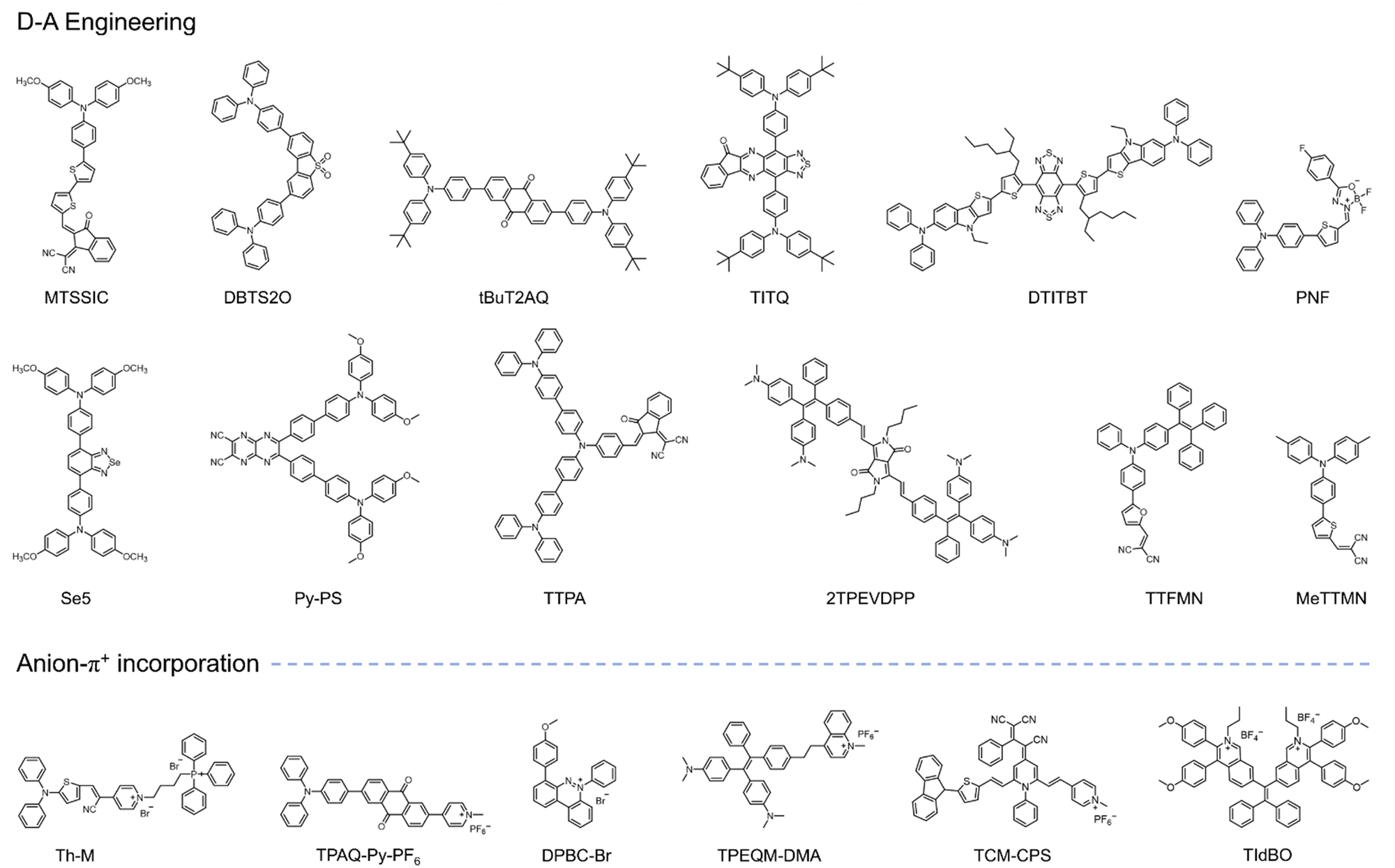
The concept of aggregate-induced emission (AIE) was first proposed in 2001.150 AIE-active fluorophores, known as AIEgens, exhibit minimal fluorescence in solution but emit intensely in the aggregated state, effectively avoiding the aggregation-caused quenching (ACQ) effects. This unique property has made AIEgens a promising tool for various applications, including PDT. In PDT, aggregation is one of the strategies proposed to enhance ISC, thereby increasing ROS generation. However, traditional PSs (porphyrins, phthalocyanine indocyanine, etc.) being hydrophobic, often aggregate in physiological environments, resulting in ACQ effects. This strong π–π interaction between aggregates reduces their ROS generation efficiency. In contrast, PSs with AIE characteristics exhibit weak fluorescence and limited ROS generation in solution due to energy dissipation through intramolecular motion. Upon aggregation, this non-radiative heat dissipation is significantly reduced due to restricted intramolecular motion (RIM). This promotes fluorescence and enhances the ISC process, leading to increased ROS generation.151 Additionally, the twisted conformation of AIE PSs reduces intermolecular π–π stacking, contributing to both their AIE properties and aggregation-induced generation of ROS (AIG-ROS) characteristics.Given these advantages, significant efforts have been devoted to developing innovative AIE-based PSs with enhanced ROS generation capabilities and improved therapeutic outcomes. The ISC process, which initiates ROS generation, is critical for the efficiency of AIE PSs. To optimize photosensitization, it is essential to achieve a higher ISC rate, ensuring adequate triplet-state production. Various studies have proposed strategies to enhance the ISC process in AIE PSs, including spin–orbit coupling (SOC) enhancement, donor–acceptor (D–A) molecular engineering,152 and other approaches. Although substantial progress has been made in designing type I AIE PSs, challenges remain in preferentially generating type I free radical species over type II ROS. This section summarizes the current strategies for developing type I AIE PSs, categorized into three main approaches: (1) D–A engineering, (2) anion–π+ incorporation, and (3) acceptor-shielding strategy, as shown in Fig. 13.
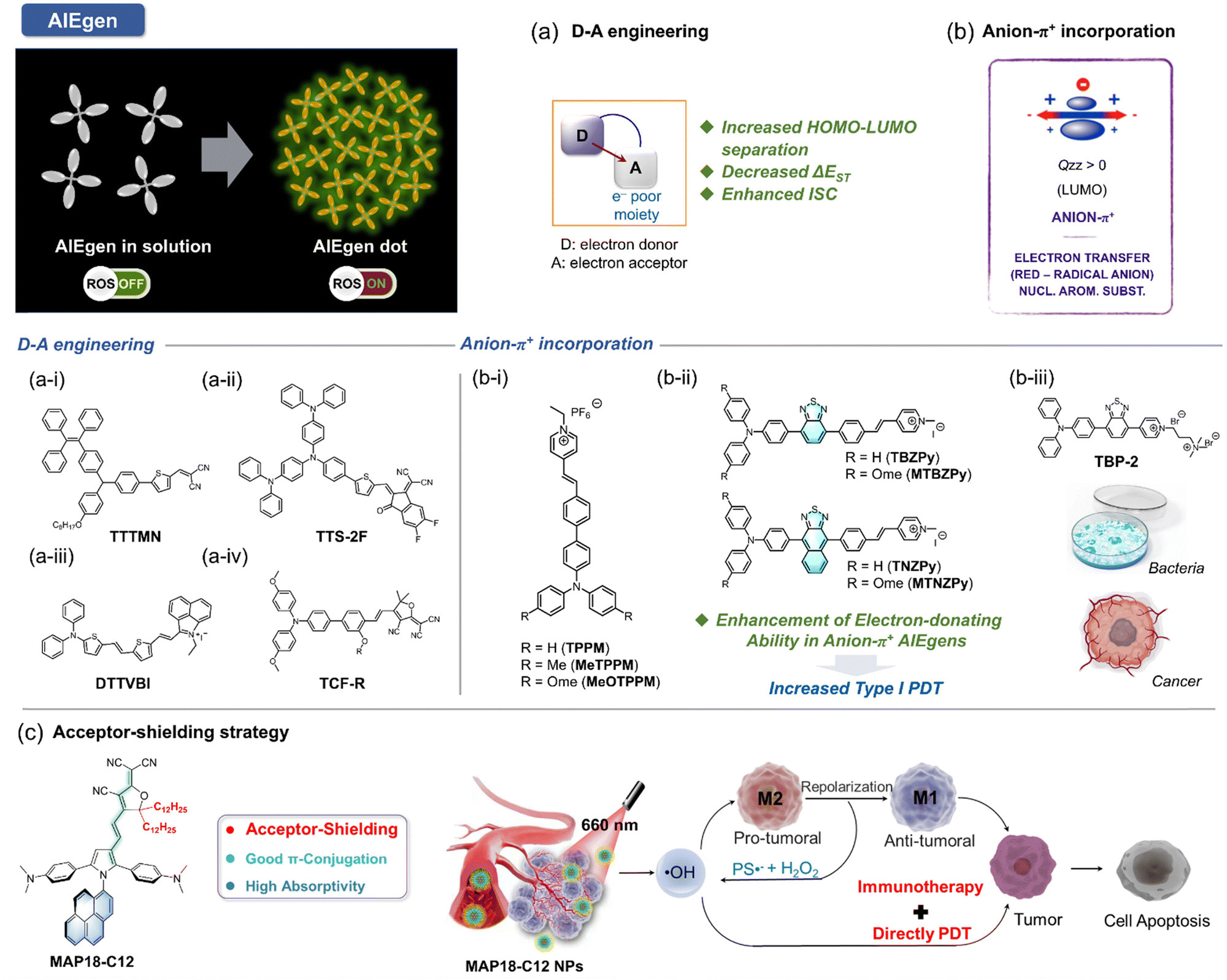 | ||
| Fig. 13 Schematic illustration of AIE-based PSs in solution and aggregated states and their ROS generation behaviors. Illustration of (a) D–A and (b) anion–π+ strategies and associated photochemical factors facilitating type I ROS generation. Chemical structures of AIE-based type I PSs employing (a-i to a-iii) D–A and (b-i to b-iii) anion–π+ strategies respectively. (c) Chemical structure of AIE-based type PS designed with acceptor–shielding strategy, coupled with a schematic illustration of type I PDT and immunotherapy mechanisms. Figure b-iii created with Biorender.com. | ||
Numerous efforts have been directed toward developing efficient type I AIE PSs using D–A strategies, focusing on enhanced O2˙− production capabilities and broad biomedical applications (Fig. 14).156–165 Notably, molecular frameworks incorporating D–A designs, such as TTS-2F, DTTVBI, and TCF-R, provide additional benefits such as absorption in the NIR region. PSs with absorption and emission improve tissue penetration while minimizing auto-fluorescence interference. Furthermore, their broad absorption profiles enable the use of white light sources for ROS generation, thereby simplifying the PDT process. The structural tunability of type I AIE PSs for preferred photochemical mechanisms allows for dual-modal therapies, such as combined type I PDT and PTT, within a single molecular framework. For instance, the AIE PS TTS-2F (Fig. 13a-ii) demonstrates the potential for synergistic type I PDT and PTT.154 PTT induces localized heating, enhancing O2 supply in tumor tissues through improved blood flow, which, in turn, boosts the efficacy of type I PDT. The improved PDT then reinforces PTT efficacy, creating a positive feedback loop. Given the competitiveness of energy dissipation processes, careful design of molecular scaffold is crucial to regulate energy dissipation equilibrium through controlled intramolecular rotations. This strategic approach provides a foundation for advancing multifunctional AIE type I PS for NIR-II fluorescence-guided type I PDT and synergistic PTT therapies.
 | ||
| Fig. 14 Schematic illustration of molecular structures of AIE-based type I PSs developed through D–A engineering or anion–π+ incorporation strategies. | ||
5.6. Other examples
This section highlights a few promising PSs selected based on their innovative approaches and significant interest in the field. These examples represent diverse strategies for achieving type I PDT, aiming to inspire further research and deepen understanding in this area.A notable example is the use of semiconducting materials with an acceptor–donor–acceptor (A–D–A) configuration to develop a π-conjugated molecular system exhibiting both type I & II capabilities, as well as NIR-II emission properties. One such system, COi6-4Cl (Fig. 15a), incorporates electron-rich oxygen atoms in the donor core to enhance electron-donating ability, linked to dicyanovinylindanone as the acceptor moiety with two chloride functional groups.179 This structure results in a strong intramolecular charge transfer effect and a low band gap (1.27 eV). Further, COi6-4Cl demonstrates the ability to produce both 1O2 and ˙OH radicals due to small ΔEST (0.27 eV) and improved kISC, confirmed by solution tests. Theoretical studies revealed that the bridged molecular structure suppresses non-radiative decay by reducing molecular vibrations, while bulky side alkyl chains minimize π–π stacking, avoiding ACQ effects. For enhanced biocompatibility, COi6-4Cl was encapsulated into polystyrene5000-b-poly(ethylene glycol)5000 (PS-b-PEG), forming spherical nanoparticles with significant colloidal stability (up to 50 days). These nanoparticles exhibited concentration-dependent cytotoxicity against cancer cells under both hypoxic and normoxic conditions. The NIR-II imaging capability of COi6-4Cl nanoparticles enables deep tissue penetration and accurate discrimination between normal and cancerous tissues in tumor-bearing mouse models. As compared to chlorin e6 (Ce6) as a clinical PDT standard (660 nm laser irradiation), COi6-4Cl NPs (808 nm laser irradiation) showed significant anti-tumor efficacy.
 | ||
| Fig. 15 Schematic illustration of chemical structures of representative semiconducting materials serving as type I PSs for PDT. (a) Chemical structure of the π-conjugated molecule COi6-4Cl. (b) Chemical structure of PTS, PTSe, and PTTe used as NIR-II type I PDT/PTT PSs. | ||
In recent years, interest in NIR-II-based PDT/PTT agents has surged due to their advantages, such as deep tissue penetration (>1 cm) and higher maximum permissible exposure limits (1.0 W cm−2 for 1064 nm and 0.33 W cm−2 for 808 nm). These properties hold promise for treating large, deeply infiltrated, or surgically inoperable malignant tumors while sparing patients from invasive procedures or radiotherapy. However, developing type I-based NIR-II PSs remains a challenge. Key hurdles include improving the ISC rate constant to generate triplet excitons efficiently and enabling electron transfer from molecular oxygen to generate O2˙− regardless of reducing intermediates. Additionally, achieving low Gibbs free energy (ΔG < 0) is crucial for producing H2O2 and ˙OH through superoxide disproportionation, while type II PDT requires that the T1 state energy exceeds the oxygen sensitization threshold (0.98 eV). In general, type II processes tend to proceed at a faster rate compared to type I processes. To address this, a D–A strategy was employed to design three narrow-bandgap semiconducting materials, namely, PTS, PTSe, and PTTe (Fig. 15b).180 These materials consist of thienoisoindigo (as an electron donor) and thiophene, selenophene, or tellurophene (as an electron donor) respectively. This approach enabled the achievement of NIR-II absorption and enhanced KISC to generate the triplet excitons (heavy atom effect, Se/Te). Additionally, these structural adjustments facilitated the type I process over the type II process by tunning the Gibbs free energy value between PTS, PTSe, PTTe, and 3O2 to values below zero. This optimization enhanced the efficiency of type I PDT. The biocompatible NPs of PTTe NPs prepared using the nanoprecipitation method were validated for NIR-II (1064 nm) type I PDT/PTT applications. These validations included in vitro and in vivo models under both normoxic and hypoxic conditions, showcasing the therapeutic potential of PTTeNPs. The results, depicted in Fig. 15, emphasize the advantage of combining type I PDT and PTT capabilities in a single system for effective cancer treatment.
Another paradigm of type I PSs stems from the design of cyanine- and squaraine-based dyes. For instance, Li et al. developed an anionic pentamethine cyanine PS (CST-Pco)181 through a counterion engineering strategy to address the limitations of ACQ and low ROS efficiency in traditional cyanine dyes (Fig. 16). By incorporating a triphenylphosphonium cation (Pco) with oligoethylene glycol chains as the counterion, the authors enhanced dye–counterion interactions and optimized amphiphilicity, effectively suppressing excessive aggregation while preserving molecular symmetry and photophysical properties. In the aggregated state, CST-Pco exhibited strong NIR-II fluorescence emission (quantum yield up to 3.8%) and efficient type I ROS (e.g., O2˙−) generation. Dual excitation wavelength modulation (760 nm for ROS production vs. 808 nm for fluorescence imaging) enabled dynamic theranostic switching. Formulated nanoparticles demonstrated enhanced tumor-targeting capability, achieving >6 mm imaging depth in vivo with a tumor-to-background signal ratio 7.6-fold higher than indocyanine green (ICG). Three PDT treatments yielded a 93.5% tumor suppression rate without systemic toxicity.
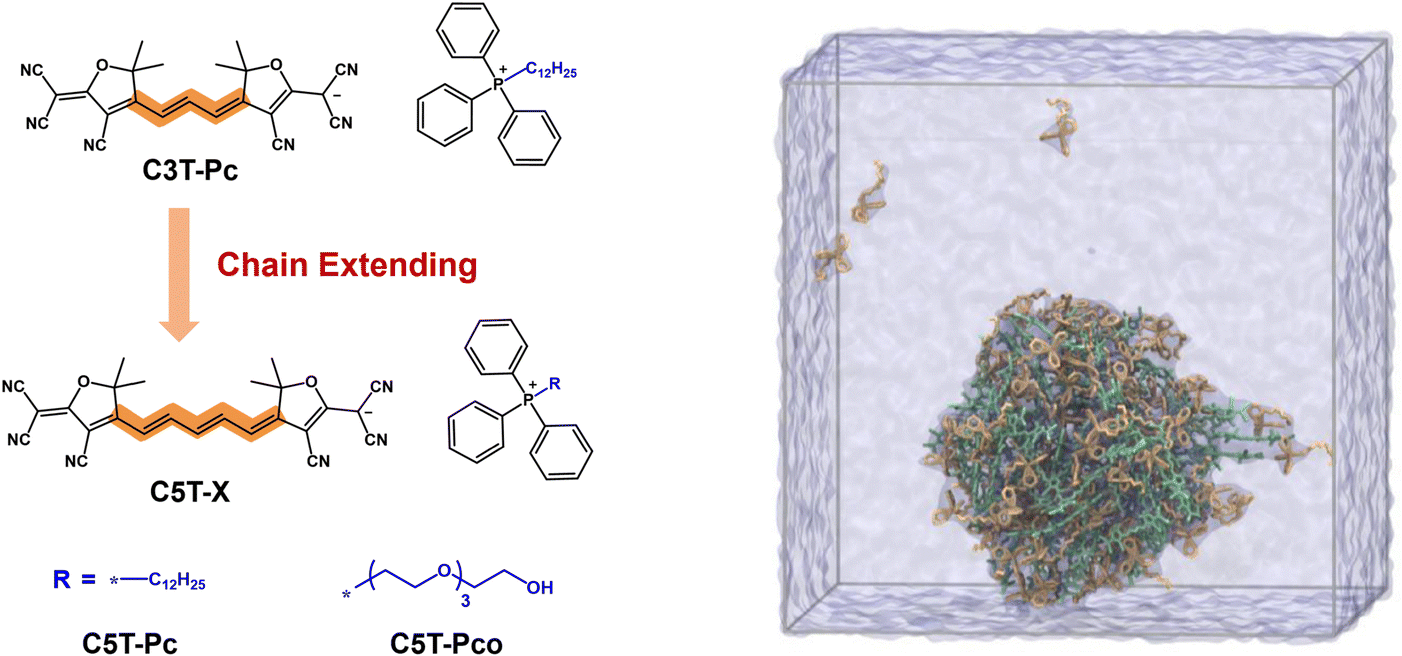 | ||
| Fig. 16 Schematic illustration of chemical structures of CST-Pco and calculated aggregation behavior of C5T-Pco under nanoconfinement conditions based on MD simulations.181 | ||
In parallel, to challenge the conventional view that H-aggregation quenches photosensitization, a squaraine (SQ)-based supramolecular photosensitizer (TPE-SQ7)182 with ordered H-aggregates was designed (Fig. 17). By integrating tetraphenylethylene (TPE) and sulfonate groups, the H-aggregates promoted ISC and narrowed the singlet–triplet energy gap (ΔEST), leading to enhanced type I ˙OH generation while maintaining a high photothermal conversion efficiency of 54.2%. TPE-SQ7 NPs demonstrated potent cytotoxicity under both normoxia and hypoxia (2% O2), achieving a 94.4% tumor inhibition rate after three type I PDT/PTT treatments. Additionally, a broad-spectrum antibacterial efficacy was also seen in TPE-SQ7 NPs, exhibiting >99% antibacterial efficacy against Staphylococcus aureus and Escherichia coli. This study redefines the role of H-aggregates in photodynamic processes, offering a dual-functional platform for hypoxia-tolerant cancer therapy and antimicrobial applications.
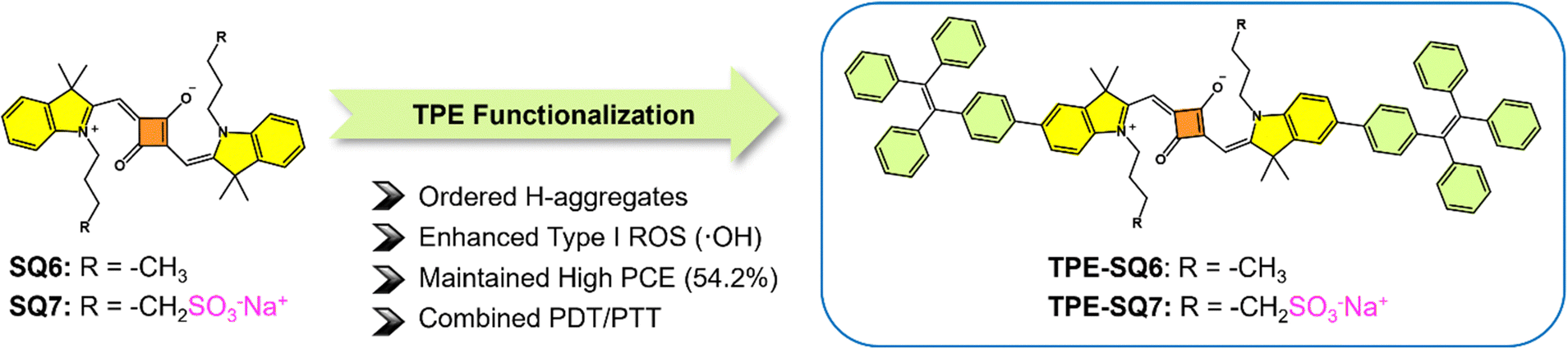 | ||
| Fig. 17 Schematic illustration of molecular engineering of SQ-based PSs. | ||
Interestingly, Peng et al. recently introduced a new type I PDT modality by integrating PDT with photocatalysis – offering a promising solution to the persistent challenge of drug resistance in tumor therapy. In this system, a julolidine-modified crystal violet (CVJ) was developed as a type I PS.183 This molecular modification effectively mitigated the issue of intramolecular rotation-induced non-radiative decay, a key limitation that hampers ROS generation in conventional CV. In CVJ, the enhanced torsional strain reduces nonradiative transitions in highly viscous environments, thereby boosting type I ROS production. Further, Langlois’ reagent (CF3SO2Na) was employed, which under photocatalytic activation produces ˙CF3 and SO2. The ˙CF3 radicals exhibit superior oxidative potential compared to conventional ROS, while SO2 serves to deplete intracellular GSH, effectively disrupting redox homeostasis and inhibiting PS efflux from cancer cells (Fig. 18). Incorporation of this multifunctional system into biocompatible NPs demonstrated potent anticancer activity in both in vitro and in vivo models of drug-resistant tumors. This pioneering study not only underscores the therapeutic potential of ˙CF3-based oxidative stress in cancer therapy but also introduces photocatalysis-induced ferroptosis as an innovative and mechanistically distinct strategy for overcoming multidrug resistance.
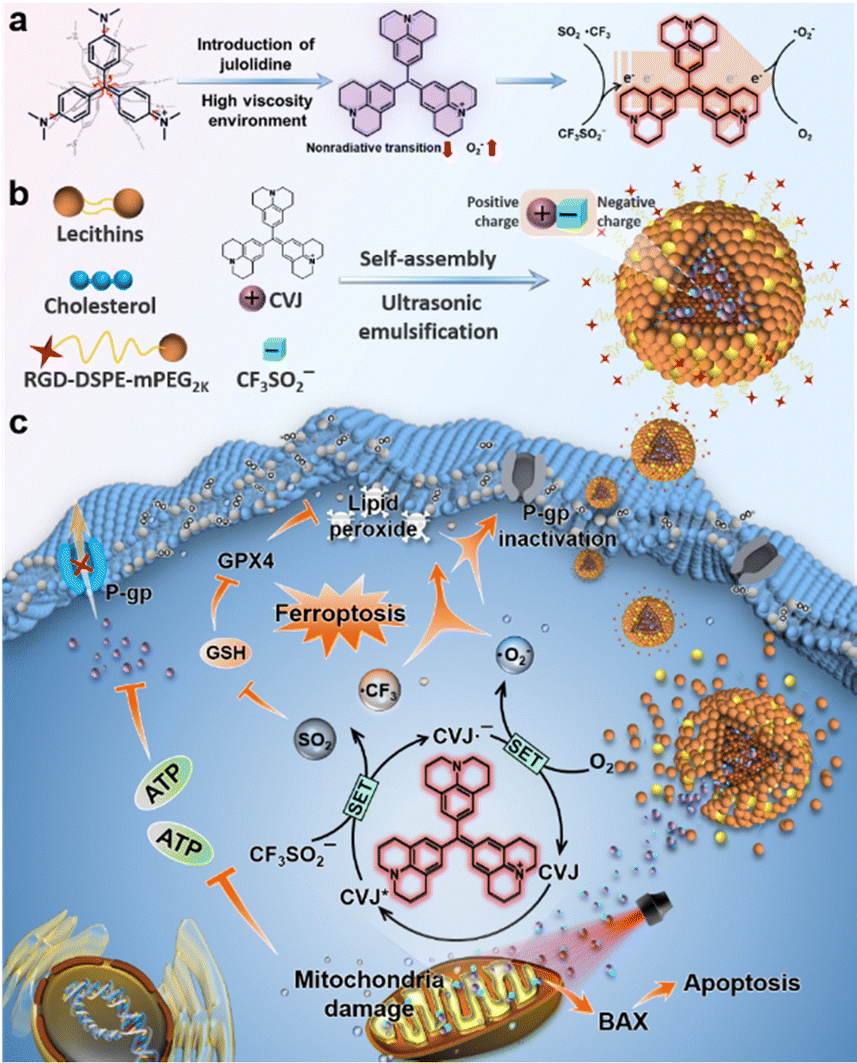 | ||
| Fig. 18 Schematic illustration of CF3-CVJ NPs preparation and underlying photocatalysis-based ferroptosis mechanism for drug-resistant cancer treatment. (a) Chemical structures of crystal violet, julolidine-based crystal violet, and its photocatalytic process. (b) Preparation of CF3-CVJ NPs by the reverse evaporation method. (c) Mechanism of ferroptosis-based cell death triggered by CF3-CVJ NPs under irradiation. Adapted with permission from ref. 183. Copyright 2022 Elsevier B.V. | ||
While organic small molecular-based PSs typically function as type II PDT agents, their supramolecular assemblies can transition to type I PSs with appropriate structural modifications. For instance, the self-assembly of phthalocyanines bearing electron-donating amine groups favors type I ROS generation over type II activity.122 The electron-donating amine groups in proximity to the triplet state enable rapid reductive quenching, forming radical anions, which then transfer electrons to molecular oxygen and nearby substrates. However, introducing electron-donating substituents into organic scaffolds can be challenging. Kida et al. addressed this by developing supramolecular PS by using a charge-separated state (CS) of monomeric PS FI-C2 and FI-C18 (Fig. 19).184 Under the CS state, electron transfer is stabilized over the triplet state, effectively suppressing type II ROS generation and favoring type I ROS production. Fluorescein, a conventional type II PS in its monomeric state, demonstrated photosensitization switching to type I activity upon self-assembly. Electrochemical and spectroscopic studies confirmed the CS state under irradiation, and ROS indicators (DHR123 and ABDA) alongside ESR experiments validated type I ROS generation. In cancer cells, the NPs demonstrated light intensity dependent type I PDT activity. This investigation offers a novel strategy for developing type I PSs in monomeric organic systems and supramolecular assemblies, expanding the toolkit for photodynamic cancer therapies.
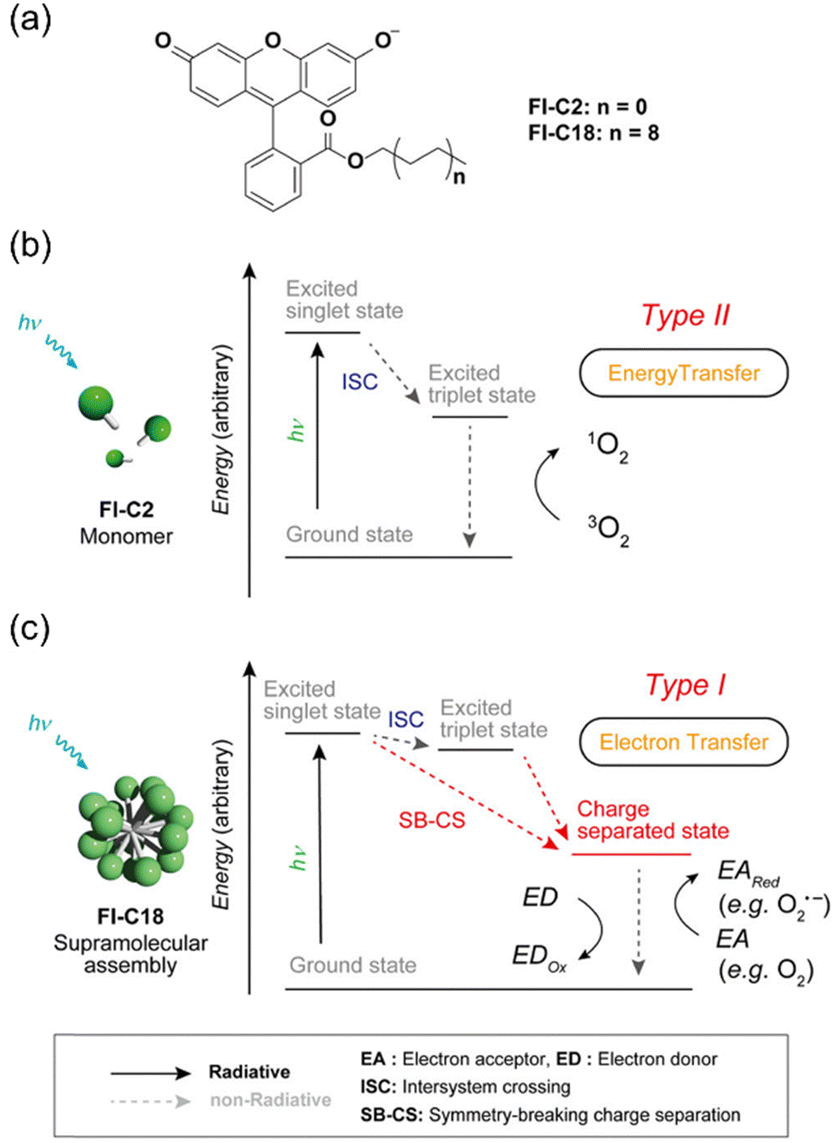 | ||
| Fig. 19 Schematic illustration of the assembly process of PSs. (a) Chemical structures of FI-C2 and FI-C18 PSs. (b) Schematic illustration of FI-C2 mediated type II and (c) FI-C18 mediated type I photosensitization mechanisms, respectively. Please note that the radiative decay forms of the excited states are omitted for better clarity. | ||
Despite the remarkable strides made in type I PDT research, its therapeutic efficacy still faces significant challenges. One major limitation lies in the reliance of most reported type I nanosensitizers on the passive targeting mechanisms, which often result in suboptimal drug accumulation within tumor tissues, falling short of therapeutic expectations. To address this, researchers have explored advanced delivery strategies, such as tumor-selective antibodies, ligand modifications, and tumor-associated factor activation. However, these promising active targeting techniques have yet to see widespread implementation in type I nanosensitizer systems. In the latest research, Song et al. synthesized a novel amphiphilic PS (PS-02)185 that combines thermally activated delayed fluorescence (TADF) with enhanced type I photosensitization capabilities (Fig. 20a). This design leverages a piperazine unit (6-NS) as an electron donor, covalently attached to the PS to create a localized “electron-rich environment”. The electron-deficient ligand 6-NS acts as an “electron transfer cage”, effectively suppressing the unwanted electron transfer (ET) process. Upon precise targeting and binding to the carbonic anhydrase IX (CAIX), the inherent electron-rich property of CAIX “unlocks” the ET effect, dramatically boosting type I photosensitization. Remarkably, this system achieves a 10.7-fold increase in the production of O2˙− compared to unmodified PS NPs. Further advancing the field, the team also developed PS@BSA, a novel type I PS integrating a fluorescein derivative with TADF properties into bovine serum albumin (BSA) (Fig. 20b).186 This ingenious system harnesses the fluorescein derivative as an “electron pump” and BSA as an “electron reservoir”, thanks to its electron-rich amino acid composition. The PS “electron pump” draws electrons from BSA and transfers them to 3O2, while BSA continuously replenishes electrons, driving efficient O2 generation. This synergistic mechanism not only accelerates type I PDT processes but also provides new perspectives and a robust theoretical foundation for designing next-generation type I PSs.
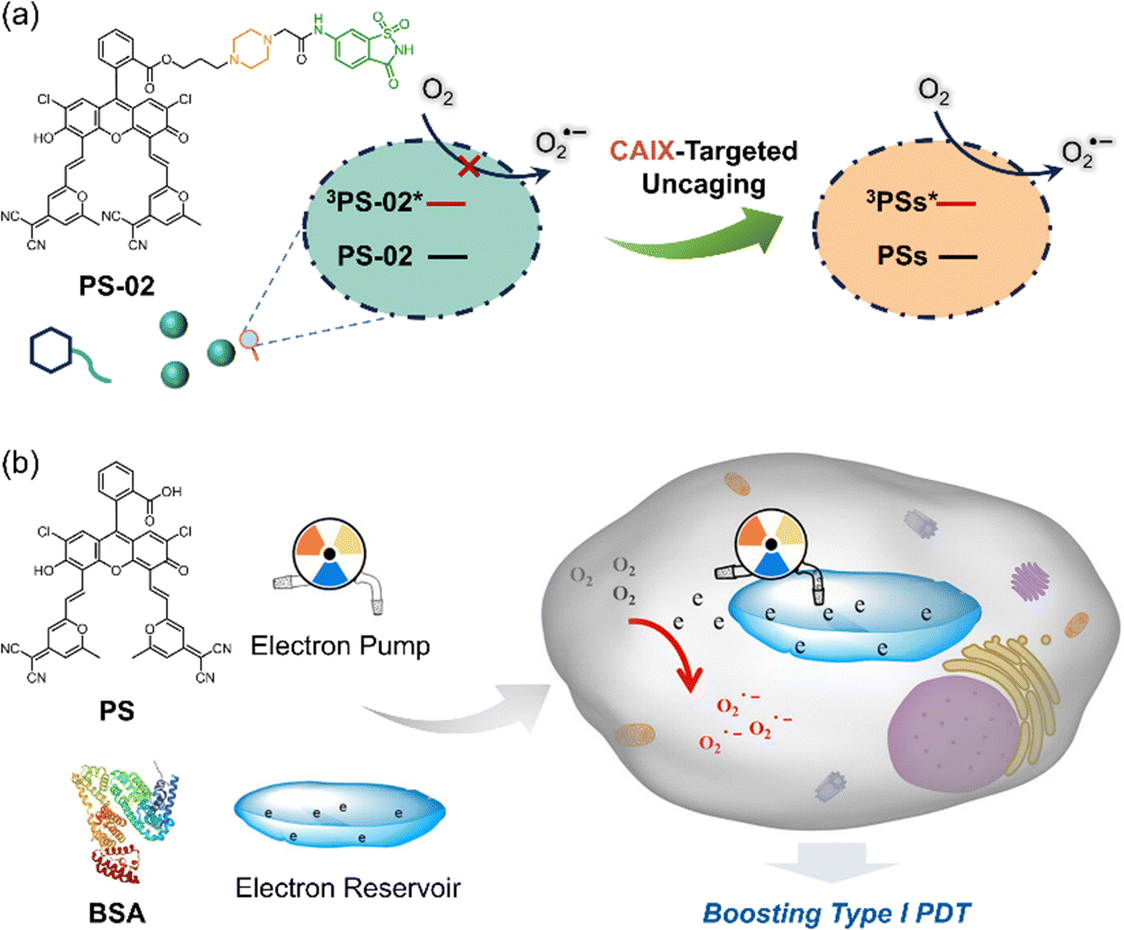 | ||
| Fig. 20 Schematic illustration of fluorescein derivatives thermally activated delayed fluorescence (TADF) type I PS. (a) Fluorescein derivatives PS-02 for promoting type I ROS. (b) Fluorescein derivatives PS PS@BSA as an “electron pump” and BSA as an “electron reservoir” for promoting type I photosensitive process. | ||
6. Is O2-independent photoredox catalysis a type I PDT?
Recent developments in type I PDT have introduced exciting new paradigms, such as the “photocatalytic oxidation of NADH in cells”, which has inspired the development of a variety of novel agents by research groups worldwide.52–55,187–189 In this section, we aim to highlight these significant advancements and emerging subdisciplines, to use cross-disciplinary insights to further accelerate the evolution of type I PDT.In conventional type I PDT, photosensitizers (PSs) absorb light, which triggers an electron transfer process. In this process, the excited-state PSs (i.e., [PS*]) typically donate an electron to oxygen, leading to the generation of O2˙−. However, this electron transfer is generally irreversible, meaning that PSs used in type I PDT are non-recyclable, resulting in the loss of PSs over time and diminishing the therapeutic efficacy of PDT. Fortunately, within living cells, numerous electron-rich substrates—such as proteins, nucleic acids, and small molecules (e.g., NADH, Fig. 21)—are readily available to donate electrons to exited PSs, even under mild reaction conditions (e.g., physiological environments). This interaction allows type I PSs to function like photocatalysts in the electron transfer process, making them recyclable. This catalytic nature opens up new avenues for developing alternative mechanisms of action in cancer phototherapy.
 | ||
| Fig. 21 Schematic illustration of the principle of PSs for photoredox catalysis of NADH in cells. | ||
The process of utilizing transition metal complexes for photocatalytic oxidation of NADH is increasingly becoming a research focus. For example, modification to Ir(III) complexes with quinoline has led to the development of photocatalysts (QAIC) that can monitor real-time dynamic changes in NADH and trigger PDT through NADH depletion (Fig. 22a).190 Furthermore, metal complexes substituted with Ru(II), such as chloromethyl-modified Ru(II) complexes (RuNH2), have also been employed in photocatalytic NADH conversion, targeting mitochondria (Fig. 22b).191 These innovative designs enable the photocatalytic conversion of biomolecules while simultaneously generating O2˙− and regenerating the PSs (acting as photocatalysts), suggesting that the photocatalytic redox imbalance of biomolecules also serves as a potential antitumor mechanism of action in type I PDT.
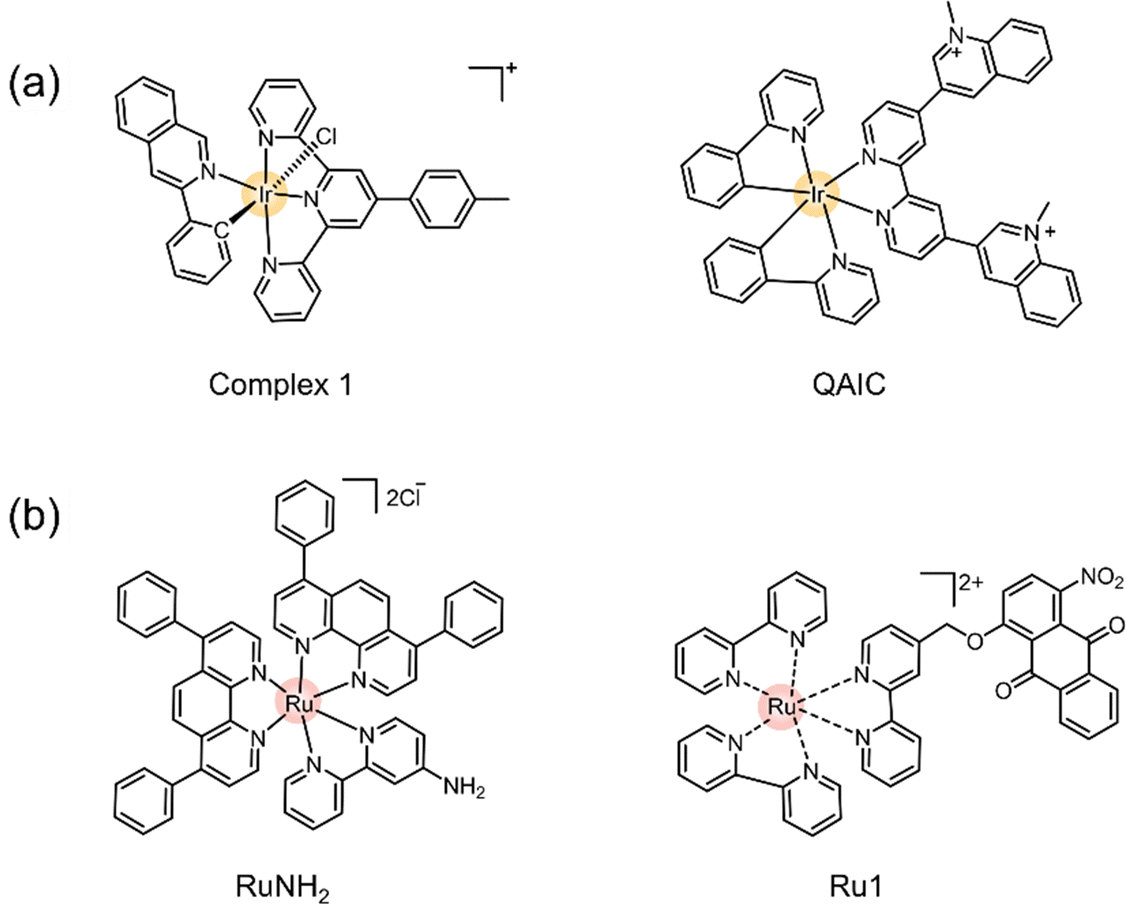 | ||
| Fig. 22 Schematic illustration of the chemical structure of transition metal complexes: (a) Ir and (b) Ru. | ||
In contrast to the above-mentioned paradigms, a particularly impressive mechanism of action, namely O2-independent photoredox catalysis in cells, has emerged recently. In 2019, Sadler and his colleagues made a groundbreaking contribution by reporting an intracellular photoredox catalysis mechanism with an O2-independent mechanism of action, which could effectively ablate cancer cells under hypoxic conditions.52 Under light excitation, the transition metal catalyst, [Ir(ttpy)(pq)Cl]PF6 (i.e., complex 1, Fig. 22a), not only catalyzed the oxidation of the cellular coenzyme NADH to form NAD+ but also drove the reduction of cytochrome C (Cyt c, Fe3+) to Cyt c (Fe2+) in an O2-independent manner. It is well-established that NADH and Cyt c (Fe3+) are critical components in the electron transport chain (ETC) in mitochondria. Therefore, the photocatalytic redox imbalance of NADH/NAD+ and Cyt c (Fe3+)/Cyt c (Fe2+) disrupt energy metabolism and cause an imbalance of redox homeostasis in cancer cells. Motivated by these findings, our group further proposed a conditionally activatable photoredox catalysis (ConAPC) concept in 2021, wherein the inherent photocatalytic properties could be selectively activated by enzymes in cells.187 In our work, we showed that a selenium-substituted Nile blue analog (Se–NH2), a metal-free agent, is an effective NIR triplet state photocatalyst that could reach photooxidation of NADH to NAD+ and further induce the reduction of Cyt c (Fe3+), as shown in Fig. 23. By introducing a nitroreductase-responsive group, Se–NH2 was further expanded into Se–NO2, which is capable of both enzyme-responsive and light-activated. This system enabled high spatiotemporal resolution for selective fluorescence labeling of hypoxic tumor cells and cascade regulation of electron flow in the mitochondrial ETC.
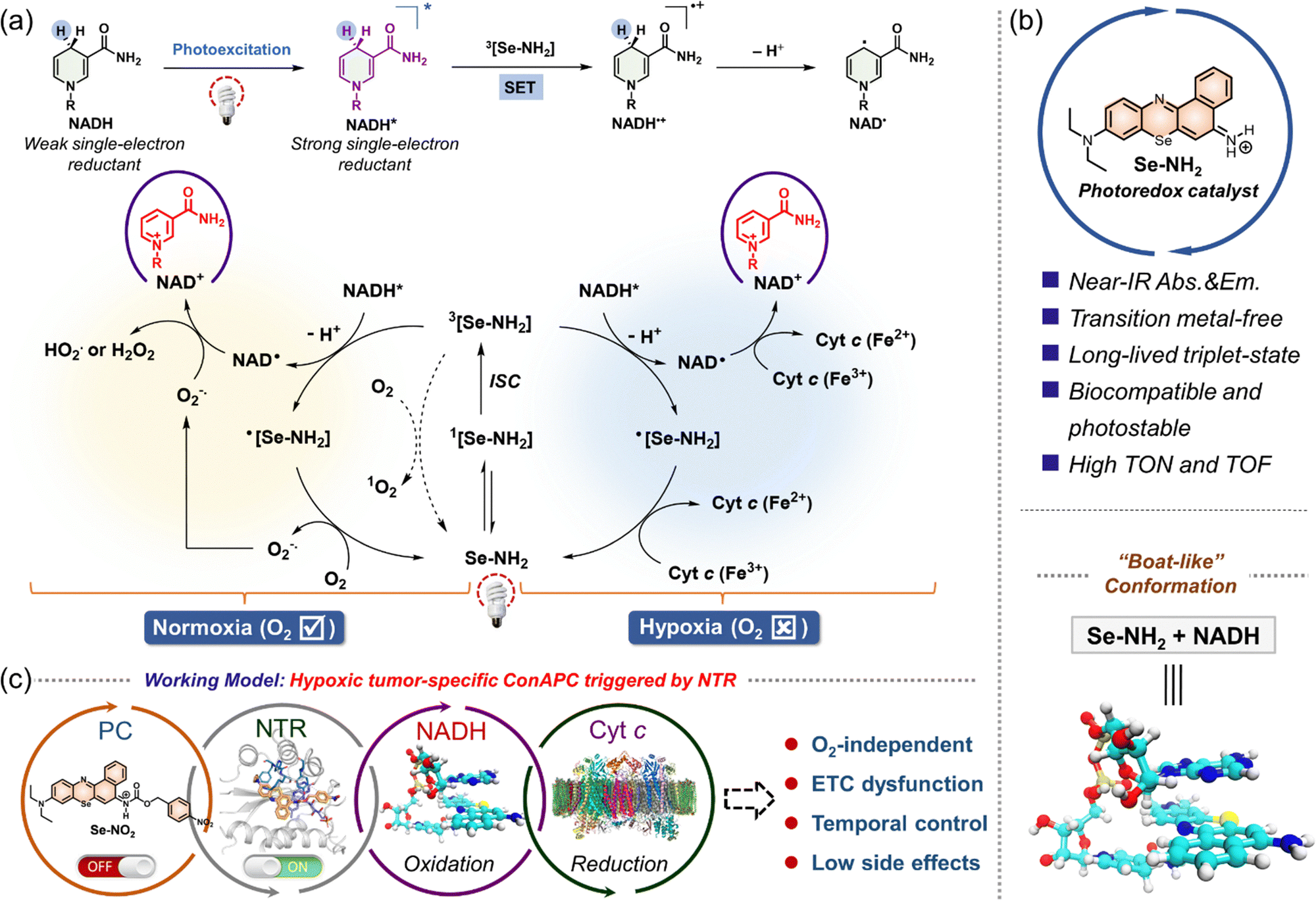 | ||
| Fig. 23 Schematic illustration of O2-independent photoredox catalysis process. (a) Plausible tandem photocatalytic conversion of cellular electron source NADH and hemoprotein Cyt c via an O2-independent pathway. (b) Chemical structure of newly developed NIR photoredox catalyst Se–NH2 and its advantages. The optimized molecular conformation between Se–NH2 and NADH is also shown. (c) Conditional activatable photoredox catalysis (ConAPC) for spatial-temporal control of photocatalytic activities in living systems. Abbreviations: TOF, turnover frequency; TON, turnover number; SET, singlet electron transfer. Adapted with permission from ref. 187. Copyright 2021 American Chemical Society. | ||
Inspired by these pioneering works, more and more researchers are focusing on exploring O2-independent modalities within the domain of PDT, such as Yao et al., who proposed O2-independent RNA degradation via excitation energy transfer between Se-substituted Nile blue PSs and RNA molecules (also defined as type III PDT).192 However, this raises a pivotal question: can O2-independent photoredox catalysis truly be classified as a form of PDT? In recent years, several definitions and research findings have contributed to the growing body of literature in this area. Through extensive discussions with prominent photochemists worldwide, a consensus has gradually emerged: photoredox catalysis, although it shares certain similarities with PDT, should not be strictly classified as PDT. PDT inherently involves three key components—O2, PS, and light. Without the participation of O2 in the process, it is unreasonable to categorize any phototherapeutic modality as PDT. This perspective fundamentally contradicts the essence of PDT in terms of its definition. Instead, a more accurate term for this approach would be “photocatalytic therapy”, which better captures the unique characteristics and mechanistic details.
7. Conclusion
The remarkable advancements in PDT over the last decade, particularly in the domain of type I PDT, have ushered in significant breakthroughs for the treatment of malignant tumors. Innovations in developing novel PSs to overcome the challenges posed by hypoxic tumor microenvironments, as well as emerging strategies targeting cellular organelles to enhance therapeutic efficacy, have not only challenged longstanding assumptions in biology and medicine but have also provided fresh perspectives and methodologies for tumor treatment. Researchers globally have been tackling the issue of tumor hypoxia by developing various PSs for type I PDT. However, despite these promising advancements, clear guidelines for designing effective type I PSs remain elusive.Drawing from our research group's extensive experience in the field, we aim to offer a coherent design framework for both current and future researchers working on type I PDT. This review aspires to serve as a comprehensive “guidebook”, systematically organizing and defining key aspects such as the definition, mechanisms, detection methods, and applications of type I PDT. The ultimate goal is to provide researchers with an accessible and practical resource that will further propel advancements in PDT and encourage the innovative design and application of type I PSs.
However, several urgent challenges remain that need to be addressed:
(1) Designing exclusive type I PSs from a molecular perspective remains a complex task. Despite significant research, the scientific community has yet to establish a clear and general structure–activity relationship for type I PSs, making it difficult to predict the photoreactions based solely on the molecular structure and modifications of specific scaffolds. This complexity arises from the need to understand the multidimensional interplay between factors such as molecular architecture (e.g., electron donor–acceptor strength), excited-state dynamics, and the surrounding microenvironment. However, emerging computational approaches—particularly time-dependent density functional theory (TD-DFT) and machine learning algorithms—show promise in deciphering these relationships, enabling predictive modeling of electron transfer pathways and facilitating the targeted design of type I-dominant PSs.
(2) Standardizing strategies for calculating the yield of O2˙− is crucial. Current detection methods, such as ESR or fluorescent probes, provide qualitative assessments of type I ROS generation but lack quantitative precision.
(3) Oxygen dependence continues to be an issue, despite the partial oxygen cycling capabilities of many type I PSs. While some PSs can reduce their oxygen dependency, oxygen is still a vital component in photosensitive reactions.
(4) Translating in vitro and in vivo findings into clinical applications is challenging. The absence of a clear dose–response relationship remains a major obstacle to the clinical adoption and widespread use of type I PDT.
(5) Light source optimization plays a pivotal role in enhancing PDT outcomes, given its centrality in biomedical optical technology. Developing a light source that allows precise spatiotemporal control while minimizing side effects is essential for improving PDT's therapeutic efficacy. One promising avenue could be constructing a light transmission system using functionalized LED beads. This could represent an integrated approach to optimize future research and applications. Additionally, creating small, flexible alternating current electroluminescent (ACEL) devices that combine PSs and light sources into therapeutic patches capable of simultaneously performing photodiagnosis and PDT could further boost the effectiveness of type I PDT.
(6) Accurately assessing the therapeutic effects of type I PDT and optimizing treatment plans is another key challenge. This requires a holistic evaluation of factors such as tumor type, stage, size, and PS characteristics, alongside the parameters of the light source. A personalized treatment plan must integrate these variables for maximum impact. Although type I PDT can be combined with other therapeutic modalities (e.g., radiotherapy and chemotherapy) to enhance efficacy, determining the most effective combination and sequence of treatments, as well as evaluating the synergistic effects, still demands further investigation.
(7) Establishing clear design principles for PSs compatible with clinical workflows is important. Several critical factors should be prioritized in type I PS development: (i) biocompatibility and rapid clearance: emphasizing PSs with reduced systemic toxicity and short half-lives to minimize photosensitivity side effects; (ii) tumor microenvironment responsiveness: engineering PSs that respond to specific tumor hallmarks—such as hypoxia, acidosis, or elevated enzyme levels. This can significantly enhance therapeutic selectivity and minimize off-target effects; (iii) scalable synthesis: prioritizing cost-effective, reproducible synthesis routes to facilitate large-scale production for clinical use; (iv) multimodal integration: designing PSs compatible with imaging modalities (e.g., fluorescence-guided surgery) to streamline clinical workflows.
In conclusion, type I PDT has significantly expanded the spectrum of available therapeutic options within the complex physiological context of cancer treatment. Ongoing research and the development of both current and future PSs hold great promise, offering the potential to deepen our understanding of biological systems. This deeper insight not only paves the way for a more thorough understanding of cancer treatment mechanisms but also drives the development of more efficient and targeted PSs, especially those capable of precise tumor targeting and self-degradation. As technological innovation continues to advance at a rapid pace, there is a strong belief that type I PDT is poised for a bright future in providing precise, safe, and effective treatments.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. L. wishes to thank the support of the National Natural Science Foundation of China (Grant No. 22308220), Shenzhen University Third-Phase Project of Constructing High-Level University (Grant No. 000001032104), the Research Team Cultivation Program of Shenzhen University (Grant No. 2023QNT005), and the Guangdong Province Key Areas Special Project for Regular Colleges and Universities (Grant No. 2024ZDZX2018). X. C. is thankful for the support of the National Natural Science Foundation of China (Grant No. 22090011), Shenzhen University 2035 Program for Excellent Research (Grant No. 00000208), Guangdong Basic and Applied Basic Research Foundation (Grant No. 2023B1515120001), and the Shenzhen Science and Technology Program (Grant No. RCBS20231211090515015). X. P. wants to thank the support of the Shenzhen University 2035 Program for Excellent Research (Grant No. 00000225). J. S. K is thankful for the National Research Foundation of Korea (2018R1A3B1052702). This work was also supported by the National Research Foundation of Korea (Grant No. 2018R1A3B1052702, J. S. K.; RS-2023-00241100, Y. X.). The authors also gratefully thank the support from the National Natural Science Foundation of China (Grant No. 82203050, Y. X.).References
- X. Li, J. F. Lovell, J. Yoon and X. Chen, Nat. Rev. Clin. Oncol., 2020, 17, 657–674 CrossRef PubMed.
- B. M. Vickerman, E. M. Zywot, T. K. Tarrant and D. S. Lawrence, Nat. Rev. Chem., 2021, 5, 816–834 CrossRef PubMed.
- L. Wu, J. Huang, K. Pu and T. D. James, Nat. Rev. Chem., 2021, 5, 406–421 CrossRef CAS PubMed.
- H. Xiong, Y. Xu, B. Kim, H. Rha, B. Zhang, M. Li, G.-F. Yang and J. S. Kim, Chem, 2023, 9, 29–64 CAS.
- A. Sharma, P. Verwilst, M. Li, D. Ma, N. Singh, J. Yoo, Y. Kim, Y. Yang, J.-H. Zhu, H. Huang, X.-L. Hu, X.-P. He, L. Zeng, T. D. James, X. Peng, J. L. Sessler and J. S. Kim, Chem. Rev., 2024, 124, 2699–2804 CrossRef CAS PubMed.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- G. Obaid, J. P. Celli, M. Broekgaarden, A.-L. Bulin, P. Uusimaa, B. Pogue, T. Hasan and H.-C. Huang, Nat. Rev. Bioeng., 2024, 2, 752–769 CrossRef CAS PubMed.
- E. Nestoros, A. Sharma, E. Kim, J. S. Kim and M. Vendrell, Nat. Rev. Chem., 2025, 9, 46–60 CrossRef CAS PubMed.
- V.-N. Nguyen, Z. Zhao, B. Z. Tang and J. Yoon, Chem. Soc. Rev., 2022, 51, 3324–3340 RSC.
- T. C. Pham, V.-N. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 121, 13454–13619 CrossRef CAS PubMed.
- S. Kwiatkowski, B. Knap, D. Przystupski, J. Saczko, E. Kędzierska, K. Knap-Czop, J. Kotlińska, O. Michel, K. Kotowski and J. Kulbacka, Biomed. Pharmacother., 2018, 106, 1098–1107 CrossRef PubMed.
- J. Yuan, H. Yang, W. Huang, S. Liu, H. Zhang, X. Zhang and X. Peng, Chem. Soc. Rev., 2025, 54, 341–366 RSC.
- G. Feng, G.-Q. Zhang and D. Ding, Chem. Soc. Rev., 2020, 49, 8179–8234 RSC.
- M. Przygoda, D. Bartusik-Aebisher, K. Dynarowicz, G. Cieślar, A. Kawczyk-Krupka and D. Aebisher, Int. J. Mol. Sci., 2023, 24, 16890 CrossRef CAS PubMed.
- V. Petrova, M. Annicchiarico-Petruzzelli, G. Melino and I. Amelio, Oncogenesis, 2018, 7, 10 CrossRef PubMed.
- Y. Liu, Y. Jiang, M. Zhang, Z. Tang, M. He and W. Bu, Acc. Chem. Res., 2018, 51, 2502–2511 CrossRef CAS PubMed.
- Y. Wan, L.-H. Fu, C. Li, J. Lin and P. Huang, Adv. Mater., 2021, 33, 2103978 CrossRef CAS PubMed.
- J. Xie, Y. Wang, W. Choi, P. Jangili, Y. Ge, Y. Xu, J. Kang, L. Liu, B. Zhang, Z. Xie, J. He, N. Xie, G. Nie, H. Zhang and J. S. Kim, Chem. Soc. Rev., 2021, 50, 9152–9201 RSC.
- Y. Zhang, Y. Zhou, Y. Jia, T. Wang and D. Meng, Front. Med., 2023, 10, 160774 Search PubMed.
- M. Li, Y. Xu, X. Peng and J. S. Kim, Acc. Chem. Res., 2022, 55, 3253–3264 CrossRef CAS PubMed.
- M. Li, J. Xia, R. Tian, J. Wang, J. Fan, J. Du, S. Long, X. Song, J. W. Foley and X. Peng, J. Am. Chem. Soc., 2018, 140, 14851–14859 CrossRef CAS PubMed.
- C. S. Foote, Photochem. Photobiol., 1991, 54, 659 CrossRef CAS PubMed.
- D. Chen, Q. Xu, W. Wang, J. Shao, W. Huang and X. Dong, Small, 2021, 17, 2006742 CrossRef CAS PubMed.
- J. Li, Z. Zhuang, Z. Zhao and B. Z. Tang, View, 2022, 3, 20200121 CrossRef CAS.
- Y.-Y. Wang, Y.-C. Liu, H. Sun and D.-S. Guo, Coord. Chem. Rev., 2019, 395, 46–62 CrossRef CAS.
- K. Plaetzer, B. Krammer, J. Berlanda, F. Berr and T. Kiesslich, Lasers Med. Sci., 2009, 24, 259–268 CrossRef CAS PubMed.
- H. C. Junqueira, D. Severino, L. G. Dias, M. S. Gugliotti and M. S. Baptista, Phys. Chem. Chem. Phys., 2002, 4, 2320–2328 RSC.
- Y. Vakrat-Haglili, L. Weiner, V. Brumfeld, A. Brandis, Y. Salomon, B. McLlroy, B. C. Wilson, A. Pawlak, M. Rozanowska, T. Sarna and A. Scherz, J. Am. Chem. Soc., 2005, 127, 6487–6497 CrossRef CAS PubMed.
- I. Ashur, R. Goldschmidt, I. Pinkas, Y. Salomon, G. Szewczyk, T. Sarna and A. Scherz, J. Phys. Chem. A, 2009, 113, 8027–8037 CrossRef CAS PubMed.
- Y. Li, W. Zhang, J. Niu and Y. Chen, ACS Nano, 2012, 6, 5164–5173 CrossRef CAS PubMed.
- Z. Hou, Y. Zhang, K. Deng, Y. Chen, X. Li, X. Deng, Z. Cheng, H. Lian, C. Li and J. Lin, ACS Nano, 2015, 9, 2584–2599 CrossRef CAS PubMed.
- M. Li, Y. Shao, J. H. Kim, Z. Pu, X. Zhao, H. Huang, T. Xiong, Y. Kang, G. Li, K. Shao, J. Fan, J. W. Foley, J. S. Kim and X. Peng, J. Am. Chem. Soc., 2020, 142, 5380–5388 CrossRef CAS PubMed.
- M. Li, T. Xiong, J. Du, R. Tian, M. Xiao, L. Guo, S. Long, J. Fan, W. Sun, K. Shao, X. Song, J. W. Foley and X. Peng, J. Am. Chem. Soc., 2019, 141, 2695–2702 CrossRef CAS PubMed.
- Y. Tang, Y. Li, B. Li, W. Song, G. Qi, J. Tian, W. Huang, Q. Fan and B. Liu, Nat. Commun., 2024, 15, 2530 CrossRef CAS PubMed.
- Y. Yin, X. Ge, J. Ouyang and N. Na, Nat. Commun., 2024, 15, 2954 CrossRef CAS PubMed.
- L. Yu, Y. Xu, Z. Pu, H. Kang, M. Li, J. L. Sessler and J. S. Kim, J. Am. Chem. Soc., 2022, 144, 11326–11337 CrossRef CAS PubMed.
- Z. Yi, X. Qin, L. Zhang, H. Chen, T. Song, Z. Luo, T. Wang, J. Lau, Y. Wu, T. B. Toh, C.-S. Lee, W. Bu and X. Liu, J. Am. Chem. Soc., 2024, 146, 9413–9421 CrossRef CAS PubMed.
- M. Li, J. Kim, H. Rha, S. Son, M. S. Levine, Y. Xu, J. L. Sessler and J. S. Kim, J. Am. Chem. Soc., 2023, 145, 6007–6023 CrossRef CAS PubMed.
- R. Ackroyd, C. Kelty, N. Brown and M. Reed, Photochem. Photobiol., 2001, 74, 656–669 CrossRef CAS PubMed.
- M. D. Daniell and J. S. Hill, ANZ. J. Surg., 2008, 61, 340–348 Search PubMed.
- M. S. Baptista, J. Cadet, P. Di Mascio, A. A. Ghogare, A. Greer, M. R. Hamblin, C. Lorente, S. C. Nunez, M. S. Ribeiro, A. H. Thomas, M. Vignoni and T. M. Yoshimura, Photochem. Photobiol., 2017, 93, 912–919 CrossRef CAS PubMed.
- Z. Wang, W. Ma, Z. Yang, D. O. Kiesewetter, Y. Wu, L. Lang, G. Zhang, S. Nakuchima, J. Chen, Y. Su, S. Han, L.-G. Wu, A. J. Jin and W. Huang, J. Am. Chem. Soc., 2024, 146, 28973–28984 CrossRef CAS PubMed.
- G. Li, L. Gu, C. Yang, X. Kong, Y. Qin and L. Wu, ACS Mater. Lett., 2024, 6, 1820–1830 CrossRef CAS.
- Z. Fan, K.-X. Teng, Y.-Y. Xu, L.-Y. Niu and Q.-Z. Yang, Angew. Chem., Int. Ed., 2025, 64, e202413595 CrossRef CAS PubMed.
- K.-X. Teng, L.-Y. Niu and Q.-Z. Yang, J. Am. Chem. Soc., 2023, 145, 4081–4087 CrossRef CAS PubMed.
- K.-X. Teng, L.-Y. Niu, N. Xie and Q.-Z. Yang, Nat. Commun., 2022, 13, 6179 CrossRef CAS PubMed.
- J. Jiang, X. Lv, H. Cheng, D. Yang, W. Xu, Y. Hu, Y. Song and G. Zeng, Acta Biomater., 2024, 177, 1–19 CrossRef CAS PubMed.
- B. Ran, Y. Yuan, W. Xia, M. Li, Q. Yao, Z. Wang, L. Wang, X. Li, Y. Xu and X. Peng, Chem. Sci., 2021, 12, 1054–1061 RSC.
- T. Xiong, F. Ning, Y. Chen, M. Gu, M. Li, X. Chen, L. Wang, J. Fan and X. Peng, ACS Nano, 2025, 19, 2822–2833 CrossRef CAS PubMed.
- Y. Hou, P. Zhou, F. Liu, K. Tong, Y. Lu, Z. Li, J. Liang and M. Tong, Nat. Commun., 2024, 15, 7350 Search PubMed.
- K. Jakubczyk, K. Dec, J. Kałduńska, D. Kawczuga, J. Kochman and K. Janda, Pol. Merkuriusz Lek., 2020, 48, 124–127 Search PubMed.
- H. Huang, S. Banerjee, K. Qiu, P. Zhang, O. Blacque, T. Malcomson, M. J. Paterson, G. J. Clarkson, M. Staniforth, V. G. Stavros, G. Gasser, H. Chao and P. J. Sadler, Nat. Chem., 2019, 11, 1041–1048 CrossRef CAS PubMed.
- Y. Xu, C. V. Chau, J. Lee, A. C. Sedgwick, L. Yu, M. Li, X. Peng, J. S. Kim and J. L. Sessler, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2314620121 CrossRef CAS PubMed.
- J. Kim, Y. Xu, J. H. Lim, J. Y. Lee, M. Li, J. M. Fox, M. Vendrell and J. S. Kim, J. Am. Chem. Soc., 2025, 147, 701–712 Search PubMed.
- M. Li, Y. Xu, Z. Pu, T. Xiong, H. Huang, S. Long, S. Son, L. Yu, N. Singh, Y. Tong, J. L. Sessler, X. Peng and J. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2210504119 Search PubMed.
- S. Park, Y. Keum and J. Park, Chem. Commun., 2022, 58, 607–618 Search PubMed.
- L. Zhang, S. Wang, Y. Zhou, C. Wang, X.-Z. Zhang and H. Deng, Angew. Chem., Int. Ed., 2019, 58, 14213–14218 CrossRef CAS PubMed.
- Y. Liu, L. Yuan, W. Chi, W.-K. Han, J. Zhang, H. Pang, Z. Wang and Z.-G. Gu, Nat. Commun., 2024, 15, 7150 Search PubMed.
- G. Lan, K. Ni, S. S. Veroneau, X. Feng, G. T. Nash, T. Luo, Z. Xu and W. Lin, J. Am. Chem. Soc., 2019, 141, 4204–4208 Search PubMed.
- Y. Zhu, P. Xu, X. Zhang and D. Wu, Chem. Soc. Rev., 2022, 51, 1377–1414 Search PubMed.
- Y. Zhang, Q. Jia, F. Nan, J. Wang, K. Liang, J. Li, X. Xue, H. Ren, W. Liu, J. Ge and P. Wang, Biomaterials, 2023, 293, 121953 Search PubMed.
- X. Zhang, L. Li, B. Wang, Z. Cai, B. Zhang, F. Chen, G. Xing, K. Li and S. Qu, Angew. Chem., Int. Ed., 2024, 63, e202410522 CrossRef CAS PubMed.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 Search PubMed.
- C. C. Winterbourn, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS PubMed.
- P. Fraisl, J. Aragonés and P. Carmeliet, Nat. Rev. Drug Discovery, 2009, 8, 139–152 Search PubMed.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed.
- K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205–214 CrossRef CAS PubMed.
- B. Yang, Y. Chen and J. Shi, Chem. Rev., 2019, 119, 4881–4985 Search PubMed.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029–3085 Search PubMed.
- H. Mattila, S. Khorobrykh, V. Havurinne and E. Tyystjärvi, J. Photochem. Photobiol., B, 2015, 152, 176–214 CrossRef CAS PubMed.
- B. Gray and A. J. Carmichael, Biochem. J., 1992, 281, 795–802 CrossRef CAS PubMed.
- P. a Ma, H. Xiao, C. Yu, J. Liu, Z. Cheng, H. Song, X. Zhang, C. Li, J. Wang, Z. Gu and J. Lin, Nano Lett., 2017, 17, 928–937 CrossRef CAS PubMed.
- H. Sies, Eur. J. Biochem., 1993, 215, 213–219 Search PubMed.
- E. Codorniu-Hernández and P. G. Kusalik, J. Am. Chem. Soc., 2012, 134, 532–538 Search PubMed.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- G. N. R. Tripathi, J. Am. Chem. Soc., 1998, 120, 4161–4166 CrossRef CAS.
- W. H. Koppenol and J. Butler, Adv. Free Radical Biol. Med., 1985, 1, 91–131 Search PubMed.
- C. C. Winterbourn and D. Metodiewa, Free Radical Biol. Med., 1999, 27, 322–328 CrossRef CAS PubMed.
- I. Batinić-Haberle, J. S. Rebouças and I. Spasojević, Antioxid. Redox Signaling, 2010, 13, 877–918 Search PubMed.
- C. Miyake, F. Michihata and K. Asada, Plant Cell Physiol., 1991, 32, 33–43 Search PubMed.
- S. Z. F. Phua, G. Yang, W. Q. Lim, A. Verma, H. Chen, T. Thanabalu and Y. Zhao, ACS Nano, 2019, 13, 4742–4751 Search PubMed.
- B. H. J. Bielski, D. E. Cabelli, R. L. Arudi and A. B. Ross, J. Phys. Chem. Ref. Data, 1985, 14, 1041–1100 Search PubMed.
- Y. Gong, M. Zhou and L. Andrews, Chem. Rev., 2009, 109, 6765–6808 CrossRef CAS PubMed.
- J. S. Nam, M.-G. Kang, J. Kang, S.-Y. Park, S. J. C. Lee, H.-T. Kim, J. K. Seo, O.-H. Kwon, M. H. Lim, H.-W. Rhee and T.-H. Kwon, J. Am. Chem. Soc., 2016, 138, 10968–10977 CrossRef CAS PubMed.
- L. M. Henderson and J. B. Chappell, Eur. J. Biochem., 1993, 217, 973–980 Search PubMed.
- Y. Qin, M. Lu and X. Gong, Cell Biol. Int., 2008, 32, 224–228 CrossRef CAS PubMed.
- J. A. Royall and H. Ischiropoulos, Arch. Biochem. Biophys., 1993, 302, 348–355 Search PubMed.
- L. Burnaugh, K. Sabeur and B. A. Ball, Theriogenology, 2007, 67, 580–589 CrossRef CAS PubMed.
- J. Zielonka and B. Kalyanaraman, Free Radical Biol. Med., 2010, 48, 983–1001 Search PubMed.
- J. J. Hu, N.-K. Wong, S. Ye, X. Chen, M.-Y. Lu, A. Q. Zhao, Y. Guo, A. C.-H. Ma, A. Y.-H. Leung, J. Shen and D. Yang, J. Am. Chem. Soc., 2015, 137, 6837–6843 Search PubMed.
- C. L. Hawkins and M. J. Davies, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 708–721 Search PubMed.
- H. Zhao, J. Joseph, H. Zhang, H. Karoui and B. Kalyanaraman, Free Radical Biol. Med., 2001, 31, 599–606 Search PubMed.
- K. Saito, M. Takahashi, M. Kamibayashi, T. Ozawa and M. Kohno, Free Radical Res., 2009, 43, 668–676 CrossRef CAS PubMed.
- K.-i Setsukinai, Y. Urano, K. Kakinuma, H. J. Majima and T. Nagano, J. Biol. Chem., 2003, 278, 3170–3175 CrossRef CAS PubMed.
- M. Saita, K. Kobatashi, F. Yoshino, H. Hase, T. Nonami, K. Kimoto and M.-C.-I. Leed, Dent. Mater. J., 2012, 31, 458–464 CrossRef CAS PubMed.
- V. Brezová, D. Dvoranová and A. Staško, Res. Chem. Intermed., 2007, 33, 251–268 Search PubMed.
- H. Zhang, L.-H. Guo, L. Zhao, B. Wan and Y. Yang, J. Phys. Chem. Lett., 2015, 6, 958–963 CrossRef CAS PubMed.
- B. Zhao, F. A. Summers and R. P. Mason, Free Radical Biol. Med., 2012, 53, 1080–1087 CrossRef CAS PubMed.
- R. T. Mallet, J. E. Squires, S. Bhatia and J. Sun, J. Mol. Cell. Cardiol., 2002, 34, 1173–1184 Search PubMed.
- J. M. Desesso, A. R. Scialli and G. C. Goeringer, Teratology, 1994, 49, 248–259 CrossRef CAS PubMed.
- T. Xiong, Y. Chen, Q. Peng, M. Li, S. Lu, X. Chen, J. Fan, L. Wang and X. Peng, J. Am. Chem. Soc., 2024, 146, 24158–24166 CrossRef CAS PubMed.
- T. Xiong, Q. Peng, Y. Chen, M. Li, J. Du, J. Fan, L. Jia and X. Peng, Adv. Funct. Mater., 2021, 31, 2103629 CrossRef CAS.
- D. Huang, H. Huang, M. Li, J. Fan, W. Sun, J. Du, S. Long and X. Peng, Adv. Funct. Mater., 2022, 32, 2208105 CrossRef CAS.
- T. Xiong, Y. Chen, Q. Peng, S. Lu, S. Long, M. Li, H. Wang, S. Lu, X. Chen, J. Fan, L. Wang and X. Peng, Adv. Mater., 2024, 36, 2309711 Search PubMed.
- T. Xiong, Y. Chen, Q. Peng, X. Zhou, M. Li, S. Lu, X. Chen, J. Fan, L. Wang and X. Peng, Adv. Mater., 2025, 37, 2410992 Search PubMed.
- T. Xiong, M. Li, Y. Chen, J. Du, J. Fan and X. Peng, Chem. Sci., 2021, 12, 2515–2520 RSC.
- D. Huang, Y. Zou, H. Huang, J. Yin, S. Long, W. Sun, J. Du, J. Fan, X. Chen and X. Peng, Adv. Mater., 2024, 36, 2313460 CrossRef CAS PubMed.
- C. Shi, X. Zhou, Q. Zhao, Z. Zhang, H. Ma, Y. Lu, Z. Huang, W. Sun, J. Du, J. Fan and X. Peng, CCS Chem., 2022, 4, 2662–2673 CrossRef CAS.
- Y. Yang, H. Ning, T. Xia, J. Du, W. Sun, J. Fan and X. Peng, Adv. Mater., 2023, 35, 2301409 CrossRef CAS PubMed.
- Y. Chen, T. Xiong, Q. Peng, J. Du, W. Sun, J. Fan and X. Peng, Nat. Commun., 2024, 15, 6935 Search PubMed.
- S. Koo, M.-G. Lee, A. Sharma, M. Li, X. Zhang, K. Pu, S.-G. Chi and J. S. Kim, Angew. Chem., Int. Ed., 2022, 61, e202110832 Search PubMed.
- Y. Lu, F. Xu, Y. Wang, C. Shi, Y. Sha, G. He, Q. Yao, K. Shao, W. Sun, J. Du, J. Fan and X. Peng, Biomaterials, 2021, 278, 121167 CrossRef CAS PubMed.
- J. Ding, Y. Lu, X. Zhao, S. Long, J. Du, W. Sun, J. Fan and X. Peng, Adv. Mater., 2024, 36, 2400196 Search PubMed.
- C. Yao, Y. Li, Z. Wang, C. Song, X. Hu and S. Liu, ACS Nano, 2020, 14, 1919–1935 Search PubMed.
- S. Li, Y. Chen, Y. Wu, S. Yao, H. Yuan, Y. Tan, F. Qi, W. He and Z. Guo, Chem. – Eur. J., 2022, 28, e202202680 CrossRef CAS PubMed.
- C. Parisi, M. Failla, A. Fraix, L. Menilli, F. Moret, E. Reddi, B. Rolando, F. Spyrakis, L. Lazzarato, R. Fruttero, A. Gasco and S. Sortino, Chem. Sci., 2021, 12, 4740–4746 Search PubMed.
- X. Wu, M. Yang, J. S. Kim, R. Wang, G. Kim, J. Ha, H. Kim, Y. Cho, K. T. Nam and J. Yoon, Angew. Chem., Int. Ed., 2022, 61, e202200808 CrossRef CAS PubMed.
- S. Jiang, B. Gurram, J. Zhu, S. Lei, Y. Zhang, T. He, O. Tagit, H. Fang, P. Huang and J. Lin, Adv. Sci., 2024, 12, 2409960 Search PubMed.
- K. Lu, C. He, N. Guo, C. Chan, K. Ni, R. R. Weichselbaum and W. Lin, J. Am. Chem. Soc., 2016, 138, 12502–12510 CrossRef CAS PubMed.
- J. Liu, D. W. Kang, Y. Fan, G. T. Nash, X. Jiang, R. R. Weichselbaum and W. Lin, J. Am. Chem. Soc., 2024, 146, 849–857 CrossRef CAS PubMed.
- H. Ding, H. Yu, Y. Dong, R. Tian, G. Huang, D. A. Boothman, B. D. Sumer and J. Gao, J. Controlled Release, 2011, 156, 276–280 CrossRef CAS PubMed.
- X. Li, D. Lee, J.-D. Huang and J. Yoon, Angew. Chem., Int. Ed., 2018, 57, 9885–9890 CrossRef CAS PubMed.
- E. Yang, J. R. Diers, Y.-Y. Huang, M. R. Hamblin, J. S. Lindsey, D. F. Bocian and D. Holten, Photochem. Photobiol., 2013, 89, 605–618 CrossRef CAS PubMed.
- Y.-Y. Zhao, L. Zhang, Z. Chen, B.-Y. Zheng, M. Ke, X. Li and J.-D. Huang, J. Am. Chem. Soc., 2021, 143, 13980–13989 CrossRef CAS PubMed.
- R. Wang, K.-H. Kim, J. Yoo, X. Li, N. Kwon, Y.-H. Jeon, S.-K. Shin, S. S. Han, D.-S. Lee and J. Yoon, ACS Nano, 2022, 16, 3045–3058 Search PubMed.
- Y.-Y. Zhao, X. Zhang, Y. Xu, Z. Chen, B. Hwang, H. Kim, H. Liu, X. Li and J. Yoon, Angew. Chem., Int. Ed., 2024, 63, e202411514 CrossRef CAS PubMed.
- M. Li, S. Long, Y. Kang, L. Guo, J. Wang, J. Fan, J. Du and X. Peng, J. Am. Chem. Soc., 2018, 140, 15820–15826 CrossRef CAS PubMed.
- A. Turksoy, D. Yildiz and E. U. Akkaya, Coord. Chem. Rev., 2019, 379, 47–64 CrossRef CAS.
- D. Chen, Z. Wang, H. Dai, X. Lv, Q. Ma, D.-P. Yang, J. Shao, Z. Xu and X. Dong, Small Methods, 2020, 4, 2000013 CrossRef CAS.
- D. Chen, Q. Yu, X. Huang, H. Dai, T. Luo, J. Shao, P. Chen, J. Chen, W. Huang and X. Dong, Small, 2020, 16, 2001059 Search PubMed.
- K.-X. Teng, W.-K. Chen, L.-Y. Niu, W.-H. Fang, G. Cui and Q.-Z. Yang, Angew. Chem., Int. Ed., 2021, 60, 19912–19920 CrossRef CAS PubMed.
- K.-X. Teng, L.-Y. Niu and Q.-Z. Yang, Chem. Sci., 2022, 13, 5951–5956 Search PubMed.
- M. Parasram and V. Gevorgyan, Chem. Soc. Rev., 2017, 46, 6227–6240 Search PubMed.
- L. C.-C. Lee and K. K.-W. Lo, J. Am. Chem. Soc., 2022, 144, 14420–14440 CrossRef CAS PubMed.
- C. Imberti, P. Zhang, H. Huang and P. J. Sadler, Angew. Chem., Int. Ed., 2020, 59, 61–73 CrossRef CAS PubMed.
- Q. Fu, C. Wei and M. Wang, ACS Nano, 2024, 18, 12049–12095 Search PubMed.
- Y. Wu, S. Li, Y. Chen, W. He and Z. Guo, Chem. Sci., 2022, 13, 5085–5106 RSC.
- L. Gourdon, K. Cariou and G. Gasser, Chem. Soc. Rev., 2022, 51, 1167–1195 RSC.
- Y. Song, Q. You and X. Chen, Adv. Mater., 2023, 35, 2212102 CrossRef CAS PubMed.
- J.-Y. Zhou, W.-J. Wang, C.-Y. Zhang, Y.-Y. Ling, X.-J. Hong, Q. Su, W.-G. Li, Z.-W. Mao, B. Cheng, C.-P. Tan and T. Wu, Biomaterials, 2022, 289, 121757 CrossRef CAS PubMed.
- S. Monro, K. L. Colón, H. Yin, J. Roque, III, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed.
- F. Qi, H. Yuan, Y. Chen, X.-X. Peng, Y. Wu, W. He and Z. Guo, CCS Chem., 2022, 5, 1583–1591 CrossRef.
- A. Mani, T. Feng, A. Gandioso, R. Vinck, A. Notaro, L. Gourdon, P. Burckel, B. Saubaméa, O. Blacque, K. Cariou, J.-E. Belgaied, H. Chao and G. Gasser, Angew. Chem., Int. Ed., 2023, 62, e202218347 CrossRef CAS PubMed.
- J. Karges, R. W. Stokes and S. M. Cohen, Trends Chem., 2021, 3, 523–534 CrossRef CAS PubMed.
- J. A. Roque, P. C. Barrett, H. D. Cole, L. M. Lifshits, G. Shi, S. Monro, D. von Dohlen, S. Kim, N. Russo, G. Deep, C. G. Cameron, M. E. Alberto and S. A. McFarland, Chem. Sci., 2020, 11, 9784–9806 RSC.
- S. Jing, X. Wu, D. Niu, J. Wang, C.-H. Leung and W. Wang, Molecules, 2024, 29, 256 CrossRef CAS PubMed.
- V. Novohradsky, A. Rovira, C. Hally, A. Galindo, G. Vigueras, A. Gandioso, M. Svitelova, R. Bresolí-Obach, H. Kostrhunova, L. Markova, J. Kasparkova, S. Nonell, J. Ruiz, V. Brabec and V. Marchán, Angew. Chem., Int. Ed., 2019, 58, 6311–6315 Search PubMed.
- R. Bevernaegie, B. Doix, E. Bastien, A. Diman, A. Decottignies, O. Feron and B. Elias, J. Am. Chem. Soc., 2019, 141, 18486–18491 Search PubMed.
- C. Lee, J. S. Nam, C. G. Lee, M. Park, C.-M. Yoo, H.-W. Rhee, J. K. Seo and T.-H. Kwon, Nat. Commun., 2021, 12, 26 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 Search PubMed.
- S. Cui, S. Dai, N. Lin, X. Wu, J. Shi, B. Tong, P. Liu, Z. Cai and Y. Dong, Small, 2022, 18, 2203825 CrossRef CAS PubMed.
- W. Zhang, M. Kang, X. Li, Y. Pan, Z. Li, Y. Zhang, C. Liao, G. Xu, Z. Zhang, B. Z. Tang, Z. Xu and D. Wang, Adv. Mater., 2024, 36, 2410142 Search PubMed.
- S. Zhen, Z. Xu, M. Suo, T. Zhang, M. Lyu, T. Li, T. Zhang, M. Li, Z. Zhao and B. Z. Tang, Adv. Mater., 2025, 37, 2411133 Search PubMed.
- P. Xiao, W. Xie, J. Zhang, Q. Wu, Z. Shen, C. Guo, Y. Wu, F. Wang, B. Z. Tang and D. Wang, J. Am. Chem. Soc., 2023, 145, 334–344 CrossRef CAS PubMed.
- C. Xiang, Y. Liu, Q. Ding, T. Jiang, C. Li, J. Xiang, X. Yang, T. Yang, Y. Wang, Y. Tan, L. Mei, Z. Lu, J. S. Kim and P. Gong, Adv. Funct. Mater., 2025, 35, 2417979 Search PubMed.
- J. Gong, X. Wang, W. Zhang, Y. Wu, K. Li, R. Sha, L. Liu, C. Li, L. Feng, G. Jiang, J. Wang and B. Z. Tang, Chem. Sci., 2024, 15, 13001–13010 Search PubMed.
- H. Wang, T. Qin, W. Wang, X. Zhou, F. Lin, G. Liang, Z. Yang, Z. Chi and B. Z. Tang, Adv. Sci., 2023, 10, 2301902 CrossRef CAS PubMed.
- R. Lin, J. Liu, W. Xu, Z. Liu, X. He, C. Zheng, M. Kang, X. Li, Z. Zhang, H.-T. Feng, J. W. Y. Lam, D. Wang, M. Chen and B. Z. Tang, Adv. Mater., 2023, 35, 2303212 CrossRef CAS PubMed.
- W. Zhang, X. Li, M. Kang, Z. Zhang, Y. Pei, M. Fan, D. Yan, Y. Zhang, C. Yang, G. Xu, D. Wang, Z. Xu and B. Z. Tang, ACS Mater. Lett., 2024, 6, 2174–2185 CrossRef CAS.
- D. Li, X. Chen, W. Dai, Q. Jin, D. Wang, J. Ji and B. Z. Tang, Adv. Mater., 2024, 36, 2306476 Search PubMed.
- J. Liu, X. Ou, K. Wang, K. Wang, L. Gui, F. Song, C. Chen, J. W. Y. Lam, Z. Yuan and B. Z. Tang, Adv. Funct. Mater., 2024, 34, 2410202 Search PubMed.
- M. M. S. Lee, D. M. Lin, J. H. C. Chau, E. Y. Yu, D. Ding, R. T. K. Kwok, D. Wang and B. Z. Tang, ACS Nano, 2023, 17, 11039–11053 Search PubMed.
- M. Kang, Z. Zhang, W. Xu, H. Wen, W. Zhu, Q. Wu, H. Wu, J. Gong, Z. Wang, D. Wang and B. Z. Tang, Adv. Sci., 2021, 8, 2100524 Search PubMed.
- L. Feng, C. Li, L. Liu, Z. Wang, Z. Chen, J. Yu, W. Ji, G. Jiang, P. Zhang, J. Wang and B. Z. Tang, ACS Nano, 2022, 16, 4162–4174 CrossRef CAS PubMed.
- J. Wang, X. Gu, P. Zhang, X. Huang, X. Zheng, M. Chen, H. Feng, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16974–16979 CrossRef CAS PubMed.
- Y. Zhao, Y. Cotelle, L. Liu, J. López-Andarias, A.-B. Bornhof, M. Akamatsu, N. Sakai and S. Matile, Acc. Chem. Res., 2018, 51, 2255–2263 CrossRef CAS PubMed.
- X. Zhao, Y. Dai, F. Ma, S. Misal, K. Hasrat, H. Zhu and Z. Qi, Chem. Eng. J., 2021, 410, 128133 Search PubMed.
- Q. Wan, R. Zhang, Z. Zhuang, Y. Li, Y. Huang, Z. Wang, W. Zhang, J. Hou and B. Z. Tang, Adv. Funct. Mater., 2020, 30, 2002057 Search PubMed.
- S. Ning, M. Lyu, D. Zhu, J. W. Y. Lam, Q. Huang, T. Zhang and B. Z. Tang, ACS Nano, 2023, 17, 10206–10217 Search PubMed.
- Y. Li, F. Liu, J. Zhang, X. Liu, P. Xiao, H. Bai, S. Chen, D. Wang, S. H. P. Sung, R. T. K. Kwok, J. Shen, K. Zhu and B. Z. Tang, Adv. Sci., 2021, 8, 2001750 CrossRef CAS PubMed.
- D. Zhu, J. Zhang, G. Luo, Y. Duo and B. Z. Tang, Adv. Sci., 2021, 8, 2004769 CrossRef CAS PubMed.
- Y. Li, D. Zhang, Y. Yu, L. Zhang, L. Li, L. Shi, G. Feng and B. Z. Tang, ACS Nano, 2023, 17, 16993–17003 CrossRef CAS PubMed.
- J. Zhuang, B. Wang, H. Chen, K. Zhang, N. Li, N. Zhao and B. Z. Tang, ACS Nano, 2023, 17, 9110–9125 CrossRef CAS PubMed.
- Z. Liu, Q. Wang, W. Qiu, Y. Lyu, Z. Zhu, X. Zhao and W.-H. Zhu, Chem. Sci., 2022, 13, 3599–3608 RSC.
- K. Chen, P. He, Z. Wang and B. Z. Tang, ACS Nano, 2021, 15, 7735–7743 CrossRef CAS PubMed.
- L. Liu, C. Li, J. Gong, Y. Zhang, W. Ji, L. Feng, G. Jiang, J. Wang and B. Z. Tang, Angew. Chem., Int. Ed., 2023, 62, e202307776 CrossRef CAS PubMed.
- J. Qu, Y. Zhang, Z. Cai, B. Tong, H. Xie, Y. Dong and J. Shi, Nanoscale, 2022, 14, 14064–14072 RSC.
- L. Li, C. Shao, T. Liu, Z. Chao, H. Chen, F. Xiao, H. He, Z. Wei, Y. Zhu, H. Wang, X. Zhang, Y. Wen, B. Yang, F. He and L. Tian, Adv. Mater., 2020, 32, 2003471 CrossRef CAS PubMed.
- K. Wen, H. Tan, Q. Peng, H. Chen, H. Ma, L. Wang, A. Peng, Q. Shi, X. Cai and H. Huang, Adv. Mater., 2022, 34, 2108146 CrossRef CAS PubMed.
- Y. Li, F. Qu, F. Wan, C. Zhong, J. Rao, Y. Liu, Z. Li, J. Zhu and Z. A. Li, Nat. Commun., 2025, 16, 762 CrossRef CAS PubMed.
- W. Qiao, T. Ma, G. Xie, J. Xu, Z.-R. Yang, C. Zhong, H. Jiang, J. Xia, L. Zhang, J. Zhu and Z. A. Li, ACS Nano, 2024, 18, 25671–25684 CrossRef CAS PubMed.
- J. Zheng, J. Du, H. Ge, N. Xu, Q. Yao, S. Long, J. Fan and X. Peng, Chem. Eng. J., 2022, 449, 136565 CrossRef CAS.
- H. Shigemitsu, K. Ohkubo, K. Sato, A. Bunno, T. Mori, Y. Osakada, M. Fujitsuka and T. Kida, JACS Au, 2022, 2, 1472–1478 CrossRef CAS PubMed.
- W. Chen, Z. Wang, G. Hong, J. Du, F. Song and X. Peng, Chem. Sci., 2024, 15, 10945–10953 RSC.
- W. Chen, Z. Wang, M. Tian, G. Hong, Y. Wu, M. Sui, M. Chen, J. An, F. Song and X. Peng, J. Am. Chem. Soc., 2023, 145, 8130–8140 CrossRef CAS PubMed.
- M. Li, K. H. Gebremedhin, D. Ma, Z. Pu, T. Xiong, Y. Xu, J. S. Kim and X. Peng, J. Am. Chem. Soc., 2022, 144, 163–173 CrossRef CAS PubMed.
- L. Yu, K.-W. Lee, Y.-Q. Zhao, Y. Xu, Y. Zhou, M. Li and J. S. Kim, Inorg. Chem., 2023, 62, 18767–18778 CrossRef CAS PubMed.
- J. H. Kim, J. Lee, K.-W. Lee, H. Xiong, M. Li and J. S. Kim, JACS Au, 2024, 4, 3657–3667 CrossRef CAS PubMed.
- S. Shamjith, M. M. Joseph, V. P. Murali, G. S. Remya, J. B. Nair, C. H. Suresh and K. K. Maiti, Biosens. Bioelectron., 2022, 204, 114087 CrossRef CAS PubMed.
- J. Liu, A. W. Prentice, G. J. Clarkson, J. M. Woolley, V. G. Stavros, M. J. Paterson and P. J. Sadler, Adv. Mater., 2023, 35, 2210363 CrossRef CAS PubMed.
- Q. Yao, J. Fan, S. Long, X. Zhao, H. Li, J. Du, K. Shao and X. Peng, Chem, 2022, 8, 197–209 CAS.
Footnote |
| † Equally contributed. |
| This journal is © The Royal Society of Chemistry 2025 |