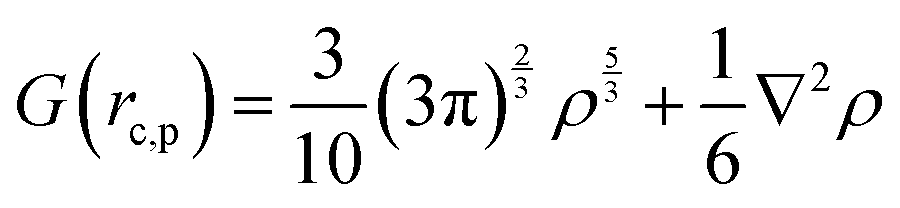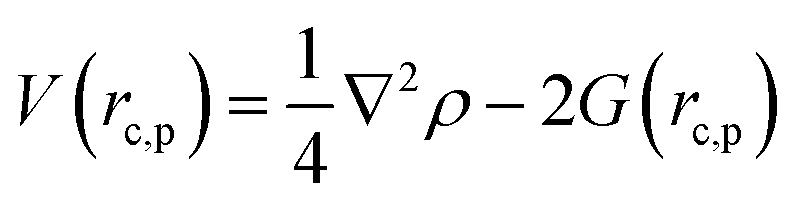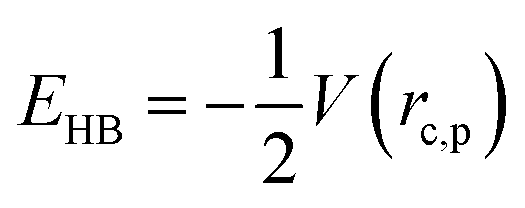Importance of intermolecular –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e002.gif) C–H⋯X–C (X = F, O, N) and –Y–H⋯F–C (Y = O, N) hydrogen bonds in crystal structures
C–H⋯X–C (X = F, O, N) and –Y–H⋯F–C (Y = O, N) hydrogen bonds in crystal structures
Received
9th October 2025
, Accepted 3rd November 2025
First published on 4th November 2025
Introduction
Intermolecular interactions play a significant role in building crystalline architecture.1 Strong hydrogen bonds like –O–H⋯O, –O–H⋯N, –N–H⋯O, –N–H⋯N, etc, that guide and alter crystal structures efficiently through the formation of various supramolecular motifs like dimers, trimers, tetramers, chains, ladders, etc.2 Their pivotal role in crystal engineering has been well documented.3 While the stronger hydrogen bonds behave predictively,4 the weaker ones are significantly unpredictable.5 Several weak hydrogen bonds like C–H⋯O, C–H⋯N, C–H⋯S, etc have been documented in the literature.6 Many weak hydrogen bonds involving less electronegative donors like the C–H group have been shown to alter the crystal packing of molecules,7 which also contain strong hydrogen bonds. Still, their dominance is inhibited by several weaker hydrogen bonds, thereby leading to a different crystal packing of a series of isomeric molecules. Weak hydrogen bonds involving the aromatic –C–H group as donors with various electronegative acceptors (F, Cl, O, N, etc) have been studied extensively.8–11 It has been demonstrated that aromatic C–H groups (pKa > 30) act as hydrogen bond donors and form various supramolecular motifs in crystal structures, thereby contributing to the stabilization of such structures.12,13 Although pKa is measured in solution, as the crystals of organic compounds are mostly grown from solutions of the compound, the acidity of hydrogens plays a significant role in the formation of various crystal lattices. Therefore, the interactions that are observed in the solid state as a result of predominant intermolecular interactions that prevail in solution, resulting in nucleation and growth of a crystalline architecture. We have been involved in the study of weak interactions involving “organic fluorine” for the last couple of decades and elucidated the significance of intermolecular –C–H⋯F–C hydrogen bonds and –C–F⋯F–C interactions, specifically the C atoms being part of an aromatic ring.14–20 It was demonstrated that intermolecular –C–H⋯F–C hydrogen bonds are capable of producing head-to-head and head-to-tail dimers, molecular chains and ribbons, etc, just like strong hydrogen bonds offered by –COOH and –CHNH– groups. It was also elucidated that weak –C–F⋯F–C interactions can generate molecular chains and dimers.21,22 The acetylenic hydrogen (pKa ≈25), being more acidic than aromatic hydrogens, is expected to act as a better hydrogen bond donor and form stronger hydrogen bonds with highly electronegative elements like F, O, and N, bonded to C, compared to the aromatic hydrogens. Previously, Böese and co-workers reported molecular complexes of acetylene with various small organic molecules using in situ crystallization experiments.23–25 Acetylenic groups have been reported to act as weak π acceptors by many groups through both experimental and computational methods.26–28 Crystal structures of many metal complexes have been found to have weak –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H⋯O–C hydrogen bonds involving acetylene C–H group as a donor and the O atom (H2O, –COO−, M
C–H⋯O–C hydrogen bonds involving acetylene C–H group as a donor and the O atom (H2O, –COO−, M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CO, PO etc) as acceptor in the Cambridge Structural Database.29–34 It is noteworthy that the number of structures found to have hydrogen bonds of the type –CC–H⋯F–C is significantly less in the database compared to –C(sp2/sp3)–H⋯F–C hydrogen bonds (Table 1). Interestingly, one of the early reports demonstrated that a molecule preferred to have –C(sp3)–H⋯F–C hydrogen bonds with F–C(sp2) group, wherein a terminal –CC–H group was available to form a hydrogen bond with the >CO group and the F–C(sp2) groups present in the molecule.35 This observation triggered this investigation to explore the capability of the –CC–H group as a hydrogen bond donor and its significance in crystal engineering. To understand the significance of –CC–H⋯F–C hydrogen bonds in crystal packing, we studied the structures reported in the Cambridge Structural Database (CSD),29 calculated the stabilization energy of the dimers formed by such interaction using Gaussian 1636 and compared the same with the dimers formed by –CC–H⋯X–C (X = O, N) and –Y–H⋯F–C (Y = O, N) hydrogen bonds. Further, we intended to compare the topological properties of these interacting molecular pairs using Bader's Atoms in Molecules (AIM)37 approach using AIM200038 package. The salient features of our results are reported in this article.
O, CO, PO etc) as acceptor in the Cambridge Structural Database.29–34 It is noteworthy that the number of structures found to have hydrogen bonds of the type –CC–H⋯F–C is significantly less in the database compared to –C(sp2/sp3)–H⋯F–C hydrogen bonds (Table 1). Interestingly, one of the early reports demonstrated that a molecule preferred to have –C(sp3)–H⋯F–C hydrogen bonds with F–C(sp2) group, wherein a terminal –CC–H group was available to form a hydrogen bond with the >CO group and the F–C(sp2) groups present in the molecule.35 This observation triggered this investigation to explore the capability of the –CC–H group as a hydrogen bond donor and its significance in crystal engineering. To understand the significance of –CC–H⋯F–C hydrogen bonds in crystal packing, we studied the structures reported in the Cambridge Structural Database (CSD),29 calculated the stabilization energy of the dimers formed by such interaction using Gaussian 1636 and compared the same with the dimers formed by –CC–H⋯X–C (X = O, N) and –Y–H⋯F–C (Y = O, N) hydrogen bonds. Further, we intended to compare the topological properties of these interacting molecular pairs using Bader's Atoms in Molecules (AIM)37 approach using AIM200038 package. The salient features of our results are reported in this article.
Table 1 CSD search results on C–H⋯F–C hydrogen bonds with special emphasis on –CC–H⋯F–C
| Sl. No. |
Type of interaction |
Number of hits |
| Search 1 |
–CC–H⋯F–C |
40 |
| Search 2 |
–CC–H⋯F–C(Ar) |
32 |
| Search 3 |
–CC–H⋯F–C(sp2) |
3 |
| Search 4 |
–CC–H⋯F–CC* |
1 |
| Search 5 |
(Ar)C–H⋯F–C |
8291 |
| Search 6 |
(sp2)C–H⋯F–C |
1126 |
| Search 7 |
C–H⋯F–C |
14620 |
| Search 8 |
−CC–H⋯O–C |
250 |
| Search 9 |
−CC–H⋯N–C |
37 |
| Search 10 |
N–H⋯F–C |
509 |
| Search 11 |
O–H⋯F–C |
330 |
Methodology
Cambridge structural database search
An extensive search was conducted using the structures reported in the 2024 edition of the Cambridge Structural Database, using the program Conquest version 2024.1.0 for C–H⋯F–C hydrogen bonds with a special emphasis on –CC–H⋯F–C hydrogen bonds. The search was conducted using H⋯F distance ranging between 1 and 2.67 Å and the ∠C–H⋯F between 90° and 180°. The hits were restricted to structures with an R factor less than 5%, only organic molecules, no disordered molecules, and no powder structures were considered. The –CC–H⋯F–C hydrogen bonds were classified into 4 different subgroups where the hybridization of the C atom of the F–C acceptor was specified (no restriction, aromatic, sp2, and sp). It was observed that the –CC–F group is reported only once (REFCODE NEDMIV) in CSD, and the molecule displayed the desired hydrogen bond (dH⋯F = 2.668 Å and ∠C–H⋯F = 132.7°). Details of these searches are listed in Table 1. To compare the occurrence and characteristics of X–H⋯F–C (Y = N, O) hydrogen bonds (where the pKa of N–H and O–H groups are >7 and 4–18, respectively) with our target hydrogen bond (–CC–H⋯X–C, X = F, O, and N), additional searches were conducted using the same conditions. These results are also listed in Table 1. It is observed that the number of molecules having an acetylenic group is much less in the CSD (total number 4950). Among them, only 379 molecules contain one or more F–C groups. Among these 379 molecules, the occurrence of the desired hydrogen bond falling within our search criteria is significantly less (Table 1). The pKa of O–H and N–H groups are significantly less than that of aromatic and acetylenic C–H groups. Therefore, in principle, they (O–H and N–H groups) should act as better donors for a hydrogen bond. Hence, we also searched for hydrogen bonding interactions wherein the donor atom was O or N (N–H⋯F–C and O–H⋯F–C). These results are listed in Table 1. Some of the molecules from their respective searches have been shown in Fig. S1–S5.
Computational study
Based on the results of the CSD search and analysis of the search results, the stabilization energy of the dimers formed by –CC–H⋯X–C (X = F, N, O) (i.e. Searches 1, 2 3, 4, 8, and 9), and Y–H⋯F–C (Y = N, O) (Searches 10, and 11) among selected molecules was calculated using Gaussian 16. Among the hits from the database search, the smallest molecules with a lower number of non-hydrogen atoms were (for computational ease) identified for computational study such that the pool of molecules represented the entire range of H⋯X distance ranging between 1 and 2.67 Å and the ∠C–H⋯F/∠Y–H⋯F between 90° and 180°. The energy of the monomer and the energy of the dimers were calculated using the density functional theory (DFT) implementing B3LYP functional and 6-31+g(d) basis set with the basis set superposition error (BSSE) correction for the dimers. It was also made sure that the selected molecules did not have any other intermolecular interactions between the pair of molecules that were linked by the desired hydrogen bond. The wavefunctions for the molecular pairs were also generated using Gaussian 16 for atoms in molecules (AIM) calculations. AIM2000 was used to obtain the topological parameters of the pair of molecules connected by the desired hydrogen bond. From AIM analysis, the electron density at the bond critical points (ρ(r)cp, e Å−3) of the desired interactions, the corresponding Laplacian (∇2ρ(r)cp, e Å−3), and the length of the bond path (Rij, Å) were extracted. The Gcp (kJ mol−1 Bohr−3), Vcp (kJ mol−1 Bohr−3), and Hcp (kJ mol−1 Bohr−3) (Kinetic, potential, and total energy density respectively) were also calculated.
Results and discussion
The CSD search results indicate that the number of reported crystal structures of molecules having both –CC–H and C–F groups is very low in the database. Therefore, the number of hits having the desired hydrogen bond (Searches 1–4, and 8–11) is also low compared to the other related hydrogen bonds (Searches 5–7). This does not indicate that the –CC–H group acts as a poor donor towards the organic fluorine and other electronegative elements. The stabilization energy for these dimers is calculated using the formula:
EStabilization![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) energy = EDimer − (EMonomer1 + EMonomer2) energy = EDimer − (EMonomer1 + EMonomer2) |
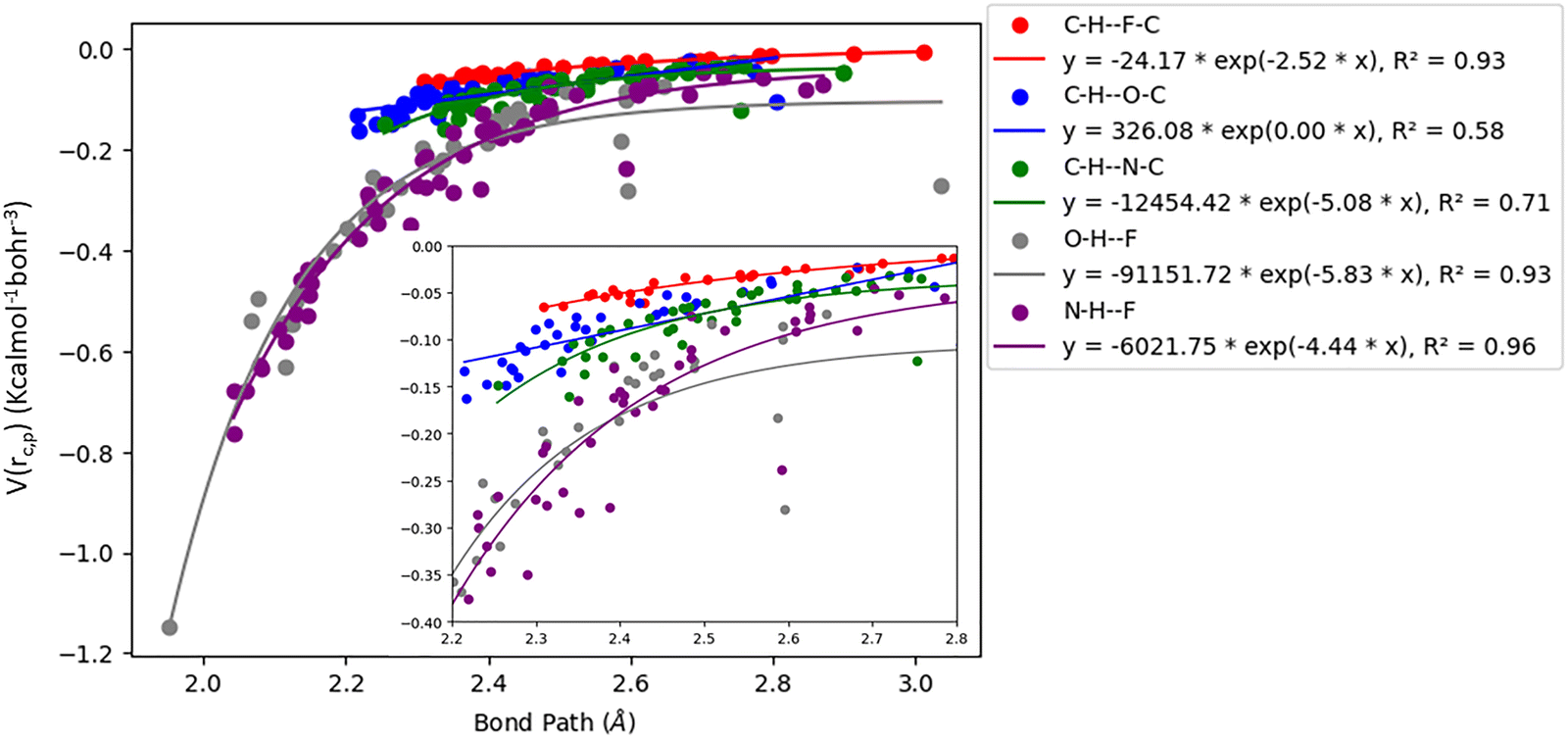
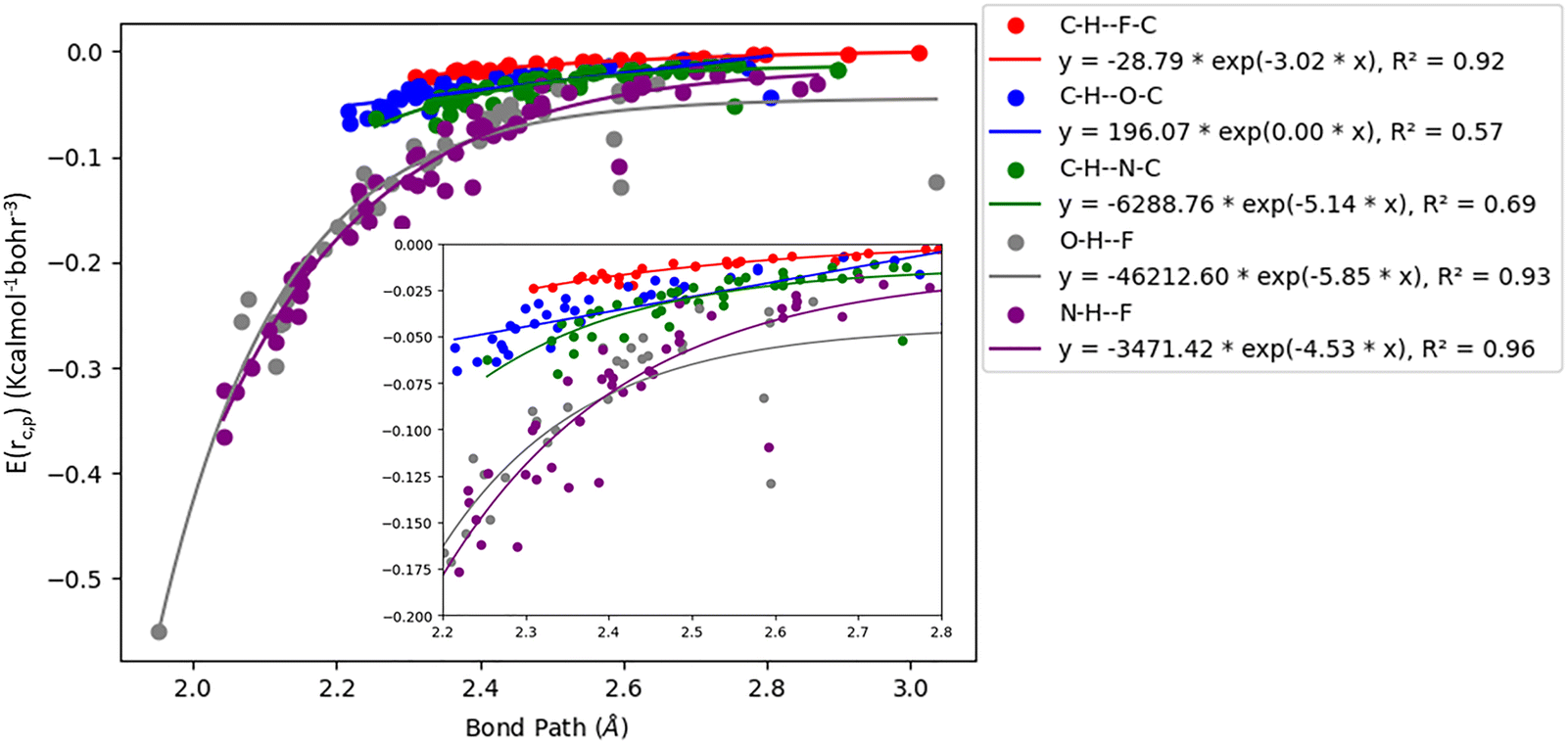
Gaussian 16 was used to generate wavefunctions for the selected dimers, and AIM2000 was used to calculate the topological parameters of the pair of molecules interacting by the –CC–H⋯X–C (where X = F, N, O) and Y–H⋯F–C (where Y = N, O) hydrogen bonds. AIM analysis provided the electron density (ρ(r)cp, e Å−3), and Laplacian (∇2ρ(r)cp, e Å−5) at the bond critical point (BCP) and the bond path (Rij, Å). Additionally, the kinetic (G(rc,p)), potential (V(rc,p)), hydrogen bond energy (EHB), and total energy densities (E(rc,p)) at the bond critical point were also calculated using the following equations37
| E(rc,p) = G(rc,p) + V(rc,p) |
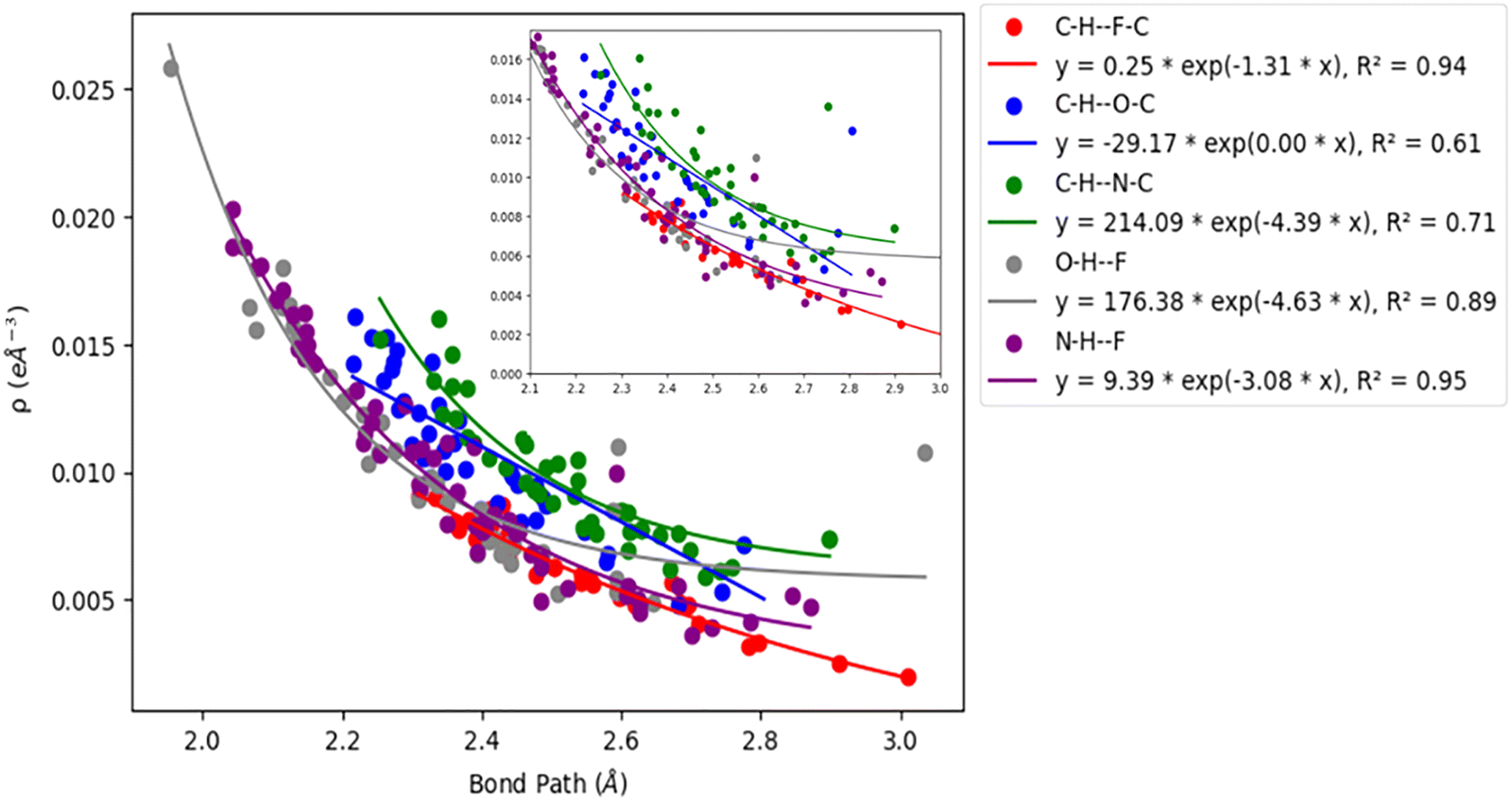
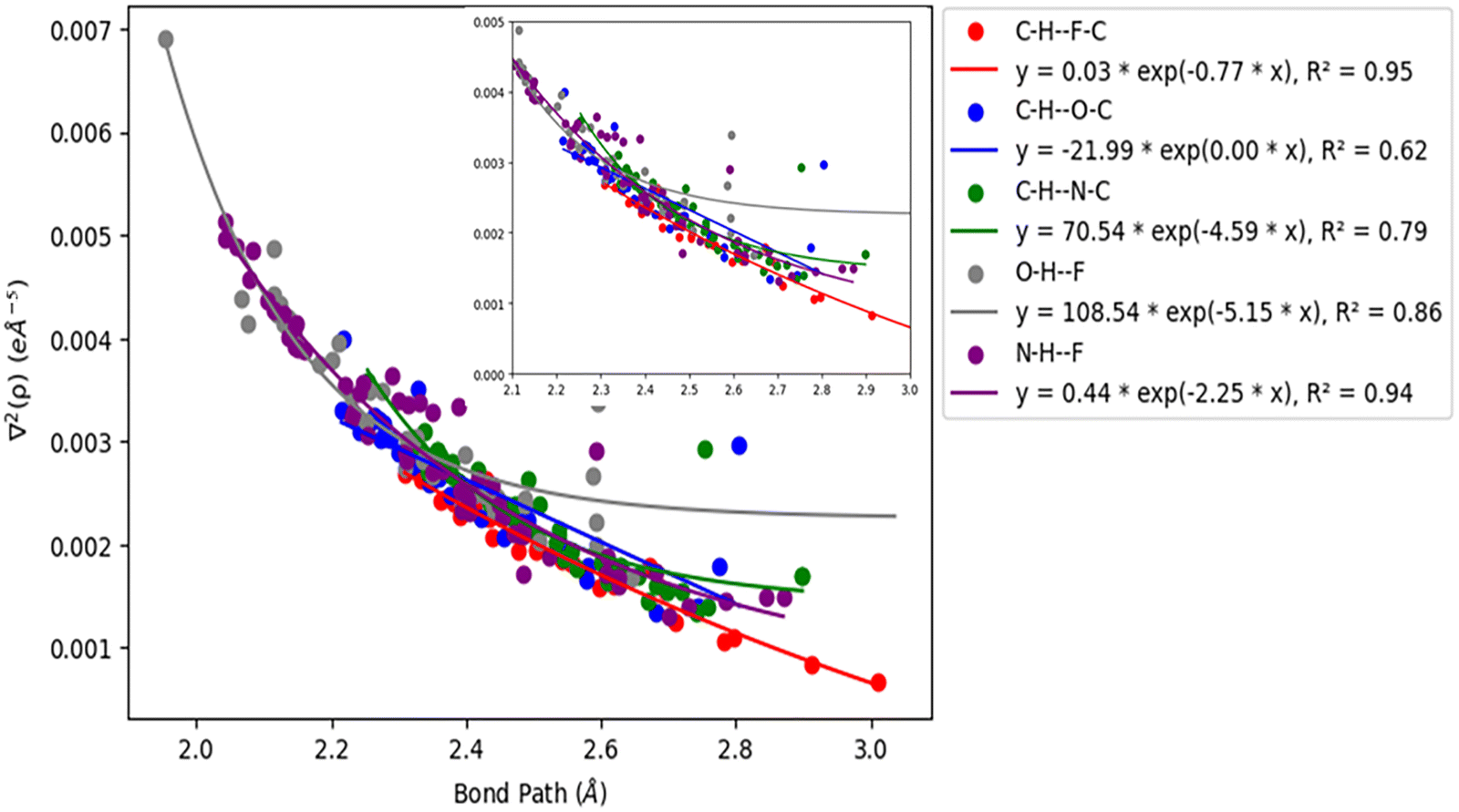
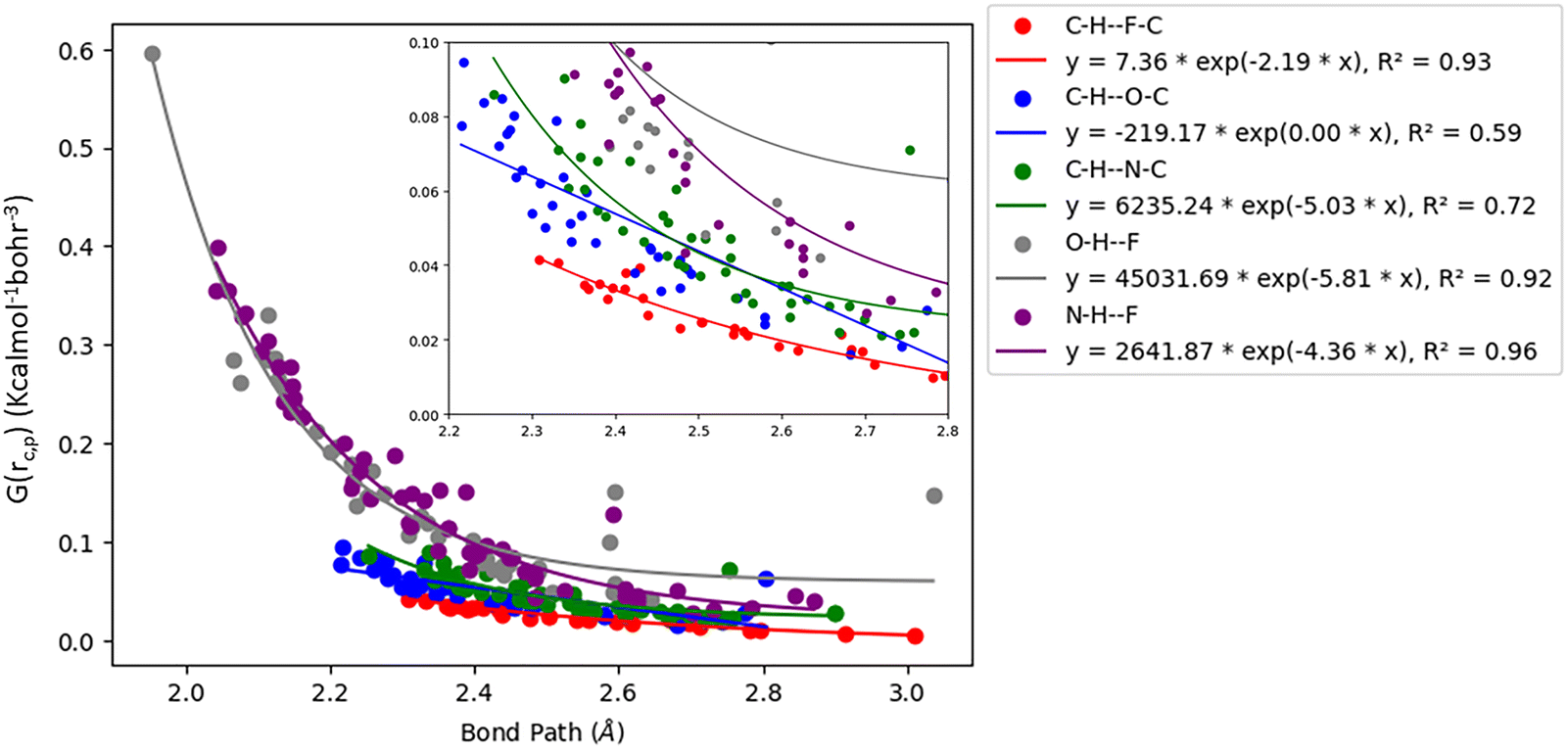
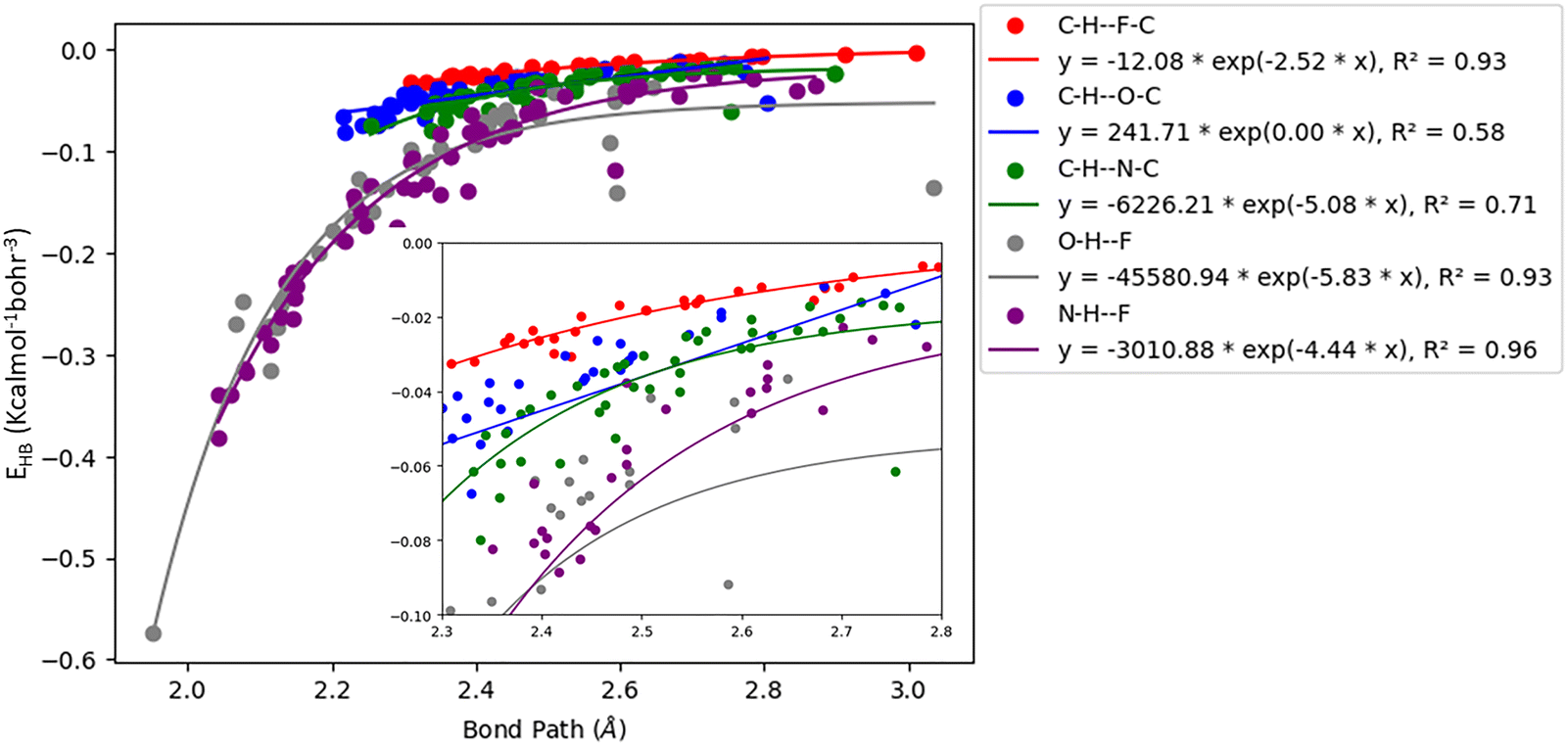
Acetylenic hydrogen (pKa ≈ 25) is considered to be more acidic than aromatic hydrogens (pKa ≈ 30), thus making it a better hydrogen bond donor. Hence it is expected that –CC–H group maybe capable of forming stronger hydrogen bonds with highly electronegative elements such as F, O, and N bonded to carbon compared to aromatic hydrogen atoms. Our AIM 2000 analysis reveals that the electron density and the Laplacian at the critical point (3, −1) are lowest in dimers formed via –CC–H⋯F–C hydrogen bonds. To comprehend the data reported in Tables S1–S5, we plotted (a) ρ(e Å) vs. Rij(Å) (Fig. 1), (b) ∇2ρ(e Å−5) vs. Rij(Å) (Fig. 2), (c) G(rc,p), vs. (Rij) (Fig. 3), (d) V(rc,p) vs. (Rij) (Fig. 4), (e) E(rc,p) vs. (Rij) (Fig. 5), and (f) EHBvs. (Rij) (Fig. 6) with appropriate color-coding for different types of interactions using the data reported in Tables S1–S5. Although the electron densities at the BCPs and the corresponding Laplacian for the dimers formed by –CC–H⋯X–C (X = O, N) and –Y–H⋯F–C (Y = O, N) hydrogen bonds are similar, dimers with –CC–H⋯F–C hydrogen bonds generally resulted in marginally lower values of electron density and Laplacian at the BCP for the studied dimers (Fig. 1 and 2).
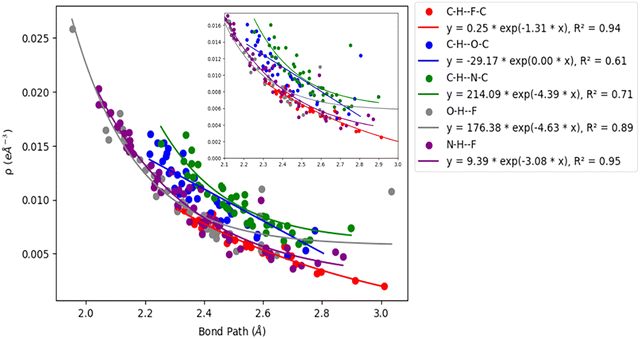 |
| | Fig. 1 Correlation plot of electron density (ρ(e Å)) at the BCP and bond path. | |
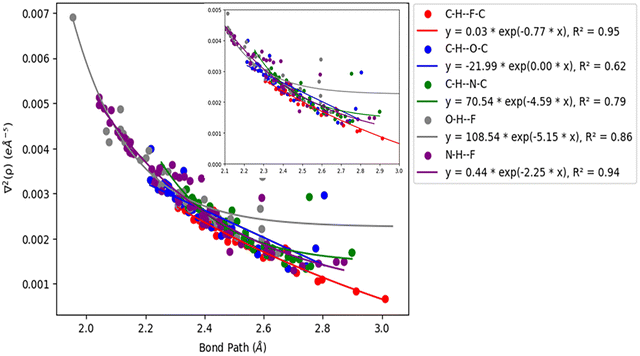 |
| | Fig. 2 Correlation plot of Laplacian of the electron density (∇2ρ(e Å−5)) and bond path. | |
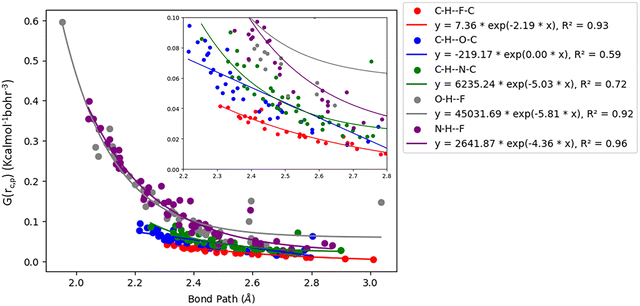 |
| | Fig. 3 Variation of kinetic energy density (G(rc,p)) with Bond Path for different interactions. | |
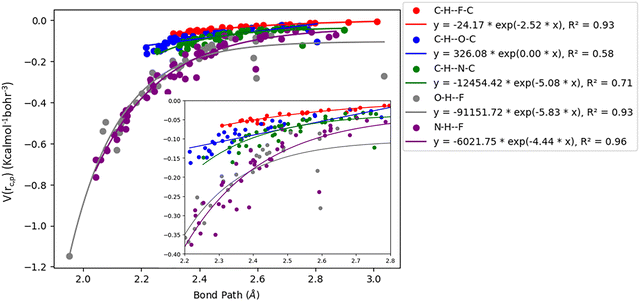 |
| | Fig. 4 Correlation plot of potential energy density (V(rc,p)) with bond path for different interactions. | |
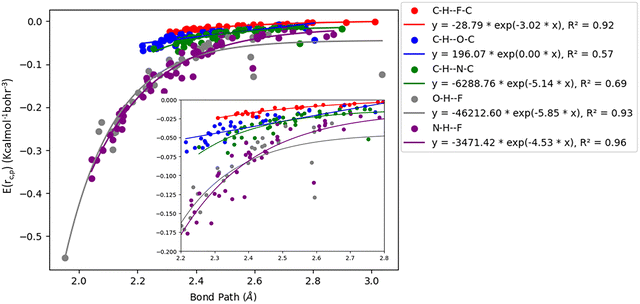 |
| | Fig. 5 Correlation plot of the total energy density (E(rc,p)) with bond path for different interactions. | |
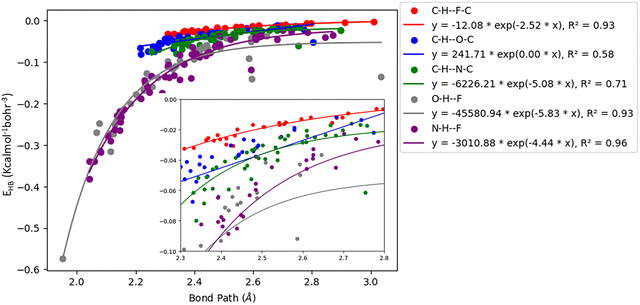 |
| | Fig. 6 Correlation plot of the hydrogen bond energy density (EHB) with bond path for different interactions. | |
The plots of G(rc,p), against bond path (Rij) show that the values for –CC–H⋯F–C interactions are consistently lower (less positive) compared to the other sets of interactions (Fig. 3). Similarly, the plots of V(rc,p), E(rc,p), and EHB against Rij demonstrate that the values for –CC–H⋯F–C interactions are consistently higher (less negative) than those for the other interactions (Fig. 4–6). Although the differences in the values of G(rc,p), V(rc,p), E(rc,p), and EHB are minimal and lie within a comparable range with the other interactions, the variations observed indicate that all the dimers are stable and highlight the importance of –CC–H as a hydrogen bond donor and its futuristic applications in crystal engineering.
Conclusions
The pKa value of the acetylenic hydrogen in the ethynyl group (–CC–H) indicates that it exhibits greater acidity than hydrogen atoms attached to aromatic carbons (sp-hybridized versus sp2-hybridized carbons). Our current comprehensive analysis of the crystal structures using computational methods reveals that the ethynyl group functions as a hydrogen bond donor, contributing to the stabilization of crystal structures. Furthermore, the pKa values of hydroxyl (O–H) and amino (N–H) groups demonstrate that these functionalities possess an even greater acidic character than the acetylenic C–H bond. Computational energy assessments and topological parameter evaluations suggest that the acetylenic group acts as an equally effective donor as oxygen and nitrogen atoms in hydrogen bonding interactions. This again underlines the importance of the intermolecular hydrogen bonding interactions involving −CC–H group in stabilizing the crystal structures. The study provides useful insights on –CC–H⋯F–C hydrogen bonds and their ability to act as a significant attractive interaction in the crystal lattice. It is noteworthy that the scarcity of –CC–H⋯F–C hydrogen bonds compared to the –C–H⋯F–C hydrogen bonds in CSD is solely due to the unavailability of crystal structures of molecules containing both –CC–H and F–C groups in the Cambridge Structural Database but their role in crystal packing cannot be refuted or ignored, rather can be explored in detail with more targeted synthesis of molecules for futuristic applications.
Author contributions
Agantuk Saha and Sakshi conducted the database search and analysis, computations, analysis of computational data, and drafted the manuscript and SI. Angshuman Roy Choudhury conceptualised the problem, supervised the computational study and associated data analysis, modified the draft manuscript, scrutinised the presented data, and finalised the manuscript.
Conflicts of interest
There are no conflicts to declare.
Data availability
All the data files, including the input and output files used for the computations reported in this manuscript, are available with us and will be produced if the reviewers or readers require the files for their analysis and understanding.
Supplementary information is available. See DOI: https://doi.org/10.1039/d5cp03886c.
Acknowledgements
The authors thank IISER Mohali for all the research facilities and infrastructure, especially for the licenses of CSD, Gaussian16, and AIM2000 packages. Sakshi thanks IISER Mohali for SRF. We thank Prof. N. Sathyamurthy, founder director of IISER Mohali, for his continuous support and encouragement. This manuscript is dedicated to Prof. N. Sathyamurthy.
References
- A. V. Vologzhanina, Intermolecular Interactions in Functional Crystalline Materials: From Data to Knowledge, Crystals, 2019, 9(9), 478 CrossRef CAS.
- C. B. Aakeroy and K. R. Seddon, The Hydrogen Bond and Crystal Engineering, Chem. Soc. Rev., 1993, 22, 397–407 RSC.
- G. R. Desiraju, Reflections on the Hydrogen Bond in Crystal Engineering, Cryst. Growth Des., 2011, 11(0034), 896–898 CrossRef CAS.
- K. C. K. Swamy, S. Kumaraswamy and P. Kommana, Very Strong C−H⋯O, N−H⋯O, and O−H⋯O Hydrogen Bonds Involving a Cyclic Phosphate, J. Am. Chem. Soc., 2001, 123(50), 12642–12649 CrossRef CAS PubMed.
-
G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, Oxford, 1999 Search PubMed.
- M. Domagała and S. J. Grabowski, C−H⋯N and C−H⋯S Hydrogen Bonds- Influence of Hybridization on Their Strength, J. Phys. Chem. A, 2005, 109, 5683–5688 CrossRef PubMed.
- G. Kaur, S. Singh, A. Sreekumar and A. R. Choudhury, The evaluation of the role of C–H⋯F hydrogen bonds in crystal altering the packing modes in the presence of strong hydrogen bond, J. Mol. Struct., 2016, 1106, 154–169 CrossRef CAS.
- G. Kaur and A. R. Choudhury, A comprehensive understanding of the synthons involving C–H⋯F–C hydrogen bond (s) from structural and computational analyses, CrystEngComm, 2015, 17, 2949–2963 RSC.
- S. Ghosh, P. Chopra and S. Wategaonkar, C−H⋯S Interaction Exhibits all the Characteristics of Conventional Hydrogen Bonds, Phys. Chem. Chem. Phys., 2020, 22, 17482–17493 RSC.
- C. B. Aakeröy, T. A. Evans, K. R. Seddon and I. Palink, The C−H⋯Cl hydrogen bond: does it exist?, New J. Chem., 1999, 23, 145–152 RSC.
- C. L. D. Gibb, E. D. Stevens and B. C. Gibb, C−H⋯X−R (X = Cl, Br, and I) Hydrogen Bonds Drive the Complexation Properties of a Nanoscale Molecular Basket, J. Am. Chem. Soc., 2001, 123(24), 5849–5850 CrossRef CAS PubMed.
- H. R. Yadav and A. R. Choudhury, Can C−H⋯F−C hydrogen bonds alter crystal packing features in the presence of NH⋯OC hydrogen bond?, J. Mol. Struct., 2017, 1150, 469–480 CrossRef CAS.
- A. R. Choudhury, U. K. Urs, T. N. Guru Row and K. Nagarajan, Weak interactions involving organic fluorine: a comparative study of the crystal packing in substituted isoquinolines, J. Mol. Struct., 2002, 605, 71–77 CrossRef CAS.
- A. R. Choudhury and T. N. Guru Row, How Realistic Are Interactions Involving Organic Fluorine in Crystal Engineering? Insights from Packing Features in Substituted Isoquinolines, Cryst. Growth Des., 2004, 4, 47–52 CrossRef CAS.
- A. R. Choudhury and T. N. Guru Row, Organic fluorine as crystal engineering tool: Evidence from packing features in fluorine substituted isoquinolines, CrystEngComm, 2006, 3, 265–274 RSC.
- G. Kaur, P. Panini, D. Chopra and A. R. Choudhury, “Structural investigation of weak intermolecular interactions in fluorine substituted isomeric N benzylideneanilines”, Cryst. Growth Des., 2012, 12, 5096–5110 CrossRef CAS.
- S. Dhingra, D. J. Barman, H. R. Yadav, J. Eyyathiyil, P. Bhowmik, P. Kaur, D. Adhikari and A. R. Choudhury, Structural and computational understanding of weak interactions in “bridge-flipped” isomeric tetrafluoro-bis-benzylideneanilines, CrystEngComm, 2018, 20, 716–727 RSC.
- L. Singla, H. R. Yadav and A. R. Choudhury, Evaluation of Fluorine-Mediated Intermolecular Interactions in Tetrafluorinated Tetrahydroisoquinoline Derivatives: Synthesis and Computational Studies, Acta Crystallogr., 2020, B76, 604–617 Search PubMed.
- L. Singla, H. R. Yadav and A. R. Choudhury, Structural and Computational Analysis of Organic Fluorine-Mediated Interactions in Controlling the Crystal Packing of Tetrafluorinated Secondary Amides in the Presence of Weak C−H⋯O−C Hydrogen Bonds, Cryst. Growth Des., 2022, 22(3), 1604–1622 CrossRef CAS.
- L. Singla, A. Kumar, C. M. Robertson, P. Munshi and A. R. Choudhury, Investigation of C−F⋯F−C
Interactions Using Experimental and Theoretical Charge Density Analyses, Cryst. Growth Des., 2023, 23, 853–861 CrossRef CAS.
- R. J. Baker, P. E. Colavita, D. M. Murphy, J. A. Platts and J. D. Wallis, Fluorine-Fluorine Interactions in the Solid State: An Experimental and Theoretical Study, J. Phys. Chem. A, 2012, 116, 1435–1444 CrossRef CAS PubMed.
- S. K. Nayak, M. K. Reddy, T. N. Guru Row and D. Chopra, Role of Hetero-Halogen (F⋯X, X = Cl, Br, and I) or Homo-Halogen (X⋯X, X = F, Cl, Br, and I) Interactions in Substituted Benzanilides, Cryst. Growth Des., 2011, 11, 1578–1596 CrossRef CAS.
- R. Boese, D. Blaser and G. Jansen, Synthesis and Theoretical Characterization of an Acetylene-Ammonia Cocrystal, J. Am. Chem. Soc., 2009, 131, 2104–2106 CrossRef CAS PubMed.
- M. T. Kirchner, D. Das, R. Boese, A. Gehrke and D. Blaser, Co-crystallization with acetylene. Part III. Molecular complexes with aromatic azacycles, CrystEngComm, 2004, 6(63), 360–366 RSC.
- M. T. Kirchner, D. Das and R. Boese, Cocrystallization with Acetylene: Molecular Complex with Methanol, Cryst. Growth Des., 2008, 8(3), 763–765 CrossRef CAS.
- K. Verma, K. Dave and K. S. Viswanathan, Hydrogen-Bonded Complexes of Phenylacetylene−Acetylene: Who is the Proton Donor?, J. Phys. Chem. A, 2015, 119, 12656–12664 CrossRef CAS PubMed.
- G. Karir and K. S. Viswanathan, Phenylacetylene-water complex: Is it n⋯σ or H⋯π in the matrix?, J. Mol. Struct., 2016, 1107, 145–156 CrossRef CAS.
- G. Karir, M. Fatima and K. S. Viswanathan, The elusive ≡C-H⋯O complex in the hydrogen bonded systems of Phenylacetylene: A Matrix Isolation Infrared and Ab Initio Study, J. Phys. Chem. A, 2017, 121, 5797–5808 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Cryst., 2016, B72, 171–179 Search PubMed.
- W.-Y. Wong, K.-Y. Ho and K.-H. Choi, New ferrocenyl heterometallic complexes of 2,7-diethynylfluoren-9-one, J. Organomet. Chem., 2003, 670, 17–26 CrossRef CAS.
- D. J. Davidson, A. P. McKay, D. B. Cordes, J. D. Woollins and N. J. Westwood, The Covalent Linking of Organophosphorus Heterocycles to Date Palm Wood-Derived Lignin: Hunting for New Materials with Flame-Retardant Potential, Molecules, 2023, 28(23), 7885 CrossRef CAS PubMed.
- Y. Asawa, S. Hatsuzawa, A. Yoshimori, K. Yamada, A. Katoh, H. Kouji and H. Nakamura, Comprehensive exploration of chemical space using trisubstituted carboranes, Sci. Rep., 2021, 11, 24101 CrossRef CAS PubMed.
- L. Wu, W. Zhong, B. Xu, Z. Wei and X. Liu, Synthesis and characterization of copper(ii) complexes with multidentate ligands as catalysts for the direct hydroxylation of benzene to phenol, Dalton Trans., 2015, 44, 8013 RSC.
- C. Balakrishnan and M. A. Neelakantan, Crystal structure and bio-catalytic potential of oxovanadium (IV) Schiff base complexes derived from 2-hydroxy-4-(prop-2-yn-1-yloxy) benzaldehyde and alicyclic/aromatic diamines, Inorg. Chim. Acta, 2018, 469, 503–514 CrossRef CAS.
- A. R. Choudhury, R. Bhat, T. N. Guru Row and S. Chandrasekaran, Weak Csp3−H⋯F−C Interaction Overshadows the Strong C ≡ C−H⋯O = C Hydrogen Bond: Structure of Pentafluorophenyl Prop-2-ynyl Carbonate, Cryst. Growth Des., 2007, 7(5), 844–846 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
-
R. F. W. Bader, Atoms in Molecules: A Quantum Theory, International Series of Monographs on Chemistry 22, Oxford Univ. Press, 1990 Search PubMed.
- F. Biegler-König, J. Schönbohmand and D. Bayles, AIM2000 - A Program to Analyze and Visualize Atoms in Molecules, J. Comput. Chem., 2001, 22, 545–559 CrossRef.
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*