
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePartial atomic charge of oxygen and hydrogen-bonding ability: insights from mass-selective IR spectroscopy of jet-cooled hydrogen-bonded complexes†
Akshay Kumar
Sahu
ab,
Anant Ram
Satpathi
ab,
Saiprakash
Rout
 ab,
Radharaman
Samanta
ab,
Laxmipriya
Dash
ab and
Himansu S.
Biswal
*ab
ab,
Radharaman
Samanta
ab,
Laxmipriya
Dash
ab and
Himansu S.
Biswal
*ab
aSchool of Chemical Sciences, National Institute of Science Education and Research (NISER), PO-Bhimpur-Padanpur, Via-Jatni, District-Khurda, PIN - 752050, Bhubaneswar, India. E-mail: himansu@niser.ac.in
bHomi Bhabha National Institute, Training School Complex, Anushakti Nagar, Mumbai 400094, India
First published on 30th September 2025
Abstract
The hydrogen bond is a fundamental non-covalent interaction that underpins the structure and function of chemical and biological systems. While the nature of conventional hydrogen bonding is predominantly electrostatic, accurately describing the atomic charge distribution within molecules remains a significant challenge, as atomic charges are not physically observable quantities. In this work, we present a systematic experimental investigation of hydrogen bonding in the gas phase using mass-selective IR spectroscopy of jet-cooled p-cresol–acceptor dimers. The redshift of the p-cresol OH stretching frequency (ΔνOH) serves as a direct measure of hydrogen-bond strength. We analysed two series of acceptors, acyclic alcohols and ethers, which demonstrate increasing inductive effects, and cyclic ethers, which reveal the influence of resonance. The gas-phase spectroscopy results provide a dataset that serves as a benchmark for validating computational chemistry models used for atomic charge calculation. It was demonstrated that while standard hydrogen-bonding descriptors correlate well with the experimental data, many popular partial atomic charge models fail to reproduce the observed chemical trends. This failure could be due to the over-reliance of the models on the electronegativity of directly bonded atoms. By highlighting this discrepancy, this work serves as a valuable cautionary example regarding the use of atomic charges and underscores the need for better theoretical frameworks for atomic charge estimation.
Introduction
The hydrogen bond (H-bond) is one of the most important non-covalent interactions in chemistry and biology.1,2 Its strength and directionality govern molecular recognition, drive protein folding, and dictate the structure of liquid water.3–5 The nature of conventional H-bonds, such as O–H⋯O, O–H⋯N, is predominantly electrostatic,6,7 meaning that their strengths are highly dependent on the charge distribution of the interacting molecules.8,9 For this reason, computational models of H-bonded systems, such as force fields for molecular dynamics simulations, heavily rely on the concept of partial atomic charges (PAC).10–12 While indispensable for modelling, PACs are not physically observable.13 Their calculation from a quantum-mechanical wavefunction is a fundamentally ambiguous process, and over 50 different methodologies have been proposed since Mulliken's pioneering work in 1955.14–18 This proliferation of methods highlights the lack of a universal, physically rigorous approach. For computational chemists, this raises a crucial question: which method best reproduces experimental reality? Extensive efforts have been made to validate PAC models using theoretical parameters.19–24 Direct experimental benchmarks remain rare. One notable example is the work of R. J. Lovelock in 2018.25 Lovelock employed X-ray photoelectron spectroscopy (XPS) and near-edge X-ray absorption fine structure (NEXAFS) spectroscopy to investigate atomic charges on nitrogen atoms in nine ionic liquids. However, the relationship between computed charges and experimentally measurable properties remains poorly established.Gas-phase spectroscopy of isolated, jet-cooled complexes provides an ideal experimental method to address this question. The absence of a solvent enables a direct study of the intrinsic properties of a H-bond, free from external influence.26–29 In this study, we use resonance-ion-dip infrared (RIDIR) spectroscopy to measure the OH stretching frequency redshift (ΔνOH) of H-bonded p-cresol (pCR) dimers. The magnitude of this redshift serves as a direct, sensitive probe of the H-bond strength, which is itself proportional to the electron-donating ability (or the electron density) of the acceptor molecule, making this interaction an ideal testbed for evaluating PAC models.30–32
Herein, this experimental technique has been used to investigate two distinct series of hydrogen-bond acceptors: (I) cyclic ethers: this series includes furan (FRN), 2,3-dihydrofuran (2,3-DHF), 2,5-dihydrofuran (2,5-DHF), and tetrahydrofuran (THF). These molecules allow us to investigate the role of resonance and conjugation on the electron density of the acceptor oxygen within a five-membered ring structure. (II) Acyclic alcohols and ethers: This series includes water (H2O), methanol (MeOH), ethanol (EtOH), iso-propanol (iPrOH), t-butanol (tBuOH), dimethyl ether (DME), diethyl ether (DEE), and di-isopropyl ether (DIE). We chose these molecules to systematically probe how increasing alkyl substitution, which is a well-documented source of inductive electron-donating effects, influences electron density at the oxygen and thus the H-bond strength.33,34 By combining these two experimental series, we build a comprehensive dataset that serves as a benchmark for evaluating computational PAC models.
In this work, we systematically evaluate 18 widely used charge assignment schemes, including Hirshfeld,35 Voronoi deformation density (VDD),36 Mulliken,14–16 Löwdin,37 Modified Mulliken by Ros & Schuit (SCPA),38 Modified Mulliken by Stout & Politzer (SP),39 Modified Mulliken by Bickelhaupt (Bickel),40 Becke,41 Atomic Dipole Corrected Hirshfeld (ADCH),42 CHELPG,43 Merz–Kollmann (M–K),44 Charge Model-5 (CM5),45 RESP,46 electronegativity equalization method (EEM),47 partial equalization of orbital electronegativity (PEOE),48,49 minimal basis iterative stockholder (MBIS),50 natural population analysis (NPA),51 and Bader's atoms in molecules (AIM).52 While the experimental data provide clear, chemically intuitive trends, it can be shown that many widely used computational charge models fail to reproduce these same trends. This work does not aim to propose a new charge model; instead, it uses the experimental data to clearly demonstrate the limitations of existing charge methodologies and serves as a cautionary example for non-experts.
Results and discussion
Experimental trends in hydrogen-bonding strength
To probe the strength of H-bonding, we employed supersonic jet-cooled, mass-selective IR (RIDIR) spectroscopy. RIDIR spectra were obtained by introducing IR photons 50–100 ns prior to UV excitation and ionisation. The UV excitation frequency was fixed at 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 331 cm−1 for the pCR monomer, while for the pCR dimers with cyclic ethers, the frequencies were 35119 cm−1 (pCR–FRN), 34925 cm−1 (pCR–2,3-DHF), 34756 cm−1 (pCR–2,5-DHF), and 34849 cm−1 (pCR–THF). For the alcohol dimers, the corresponding excitation frequencies were 34974, 34906, 34909, 34952, and 34s935 cm−1 for H2O, MeOH, EtOH, iPrOH, and tBuOH, respectively. For the acyclic ethers, the excitation frequencies were 34871 cm−1 (DME), 34880 cm−1 (DEE), and 34839 cm−1 (DIE). The ionisation frequency for the 2C-R2PI spectra was fixed at 37700 cm−1. The corresponding mass-selective REMPI spectra are shown in Fig. S1–S3.
331 cm−1 for the pCR monomer, while for the pCR dimers with cyclic ethers, the frequencies were 35119 cm−1 (pCR–FRN), 34925 cm−1 (pCR–2,3-DHF), 34756 cm−1 (pCR–2,5-DHF), and 34849 cm−1 (pCR–THF). For the alcohol dimers, the corresponding excitation frequencies were 34974, 34906, 34909, 34952, and 34s935 cm−1 for H2O, MeOH, EtOH, iPrOH, and tBuOH, respectively. For the acyclic ethers, the excitation frequencies were 34871 cm−1 (DME), 34880 cm−1 (DEE), and 34839 cm−1 (DIE). The ionisation frequency for the 2C-R2PI spectra was fixed at 37700 cm−1. The corresponding mass-selective REMPI spectra are shown in Fig. S1–S3.
The redshift of the pCR OH stretching frequency (ΔνOH) from its monomer value of 3658 cm−1 provides a reliable measure of H-bond strength. For the cyclic five-membered ethers (Fig. 1a–e), the observed order of H-bond strength, based on ΔνOH, is chemically intuitive: FRN (−78 cm−1) < 2,3-DHF (−188 cm−1) < 2,5-DHF (−282 cm−1) < THF (−318 cm−1). This trend directly reflects the degree of conjugation of the oxygen lone pair. In FRN, the oxygen lone pair is fully conjugated with two double bonds, which significantly reduces its electron density and H-bonding ability. In 2,3-DHF, the conjugation with a single double bond weakens this effect, while in 2,5-DHF, the double bond is not conjugated with the oxygen, leaving a higher electron density on the oxygen. Finally, in THF, the oxygen lone pair is fully localised and available for bonding, making it the strongest acceptor.
 | ||
| Fig. 1 Mass-selected vibrational spectra (RIDIR spectra) of Left Panel: H-bonded dimers of pCR with cyclic ethers, including (b) pCR–FRN, (c) pCR–2,3-DHF, (d) pCR–2,5-DHF, and (e) pCR–THF; Middle Panel: H-bonded dimers of pCR with alcohols, including (g)–(k) pCR with H2O, MeOH, EtOH, iPrOH, and tBuOH, respectively; Right Panel: H-bonded dimers of pCR with acyclic ethers, including (m)–(p) pCR with H2O, DME, DEE, and DIE, respectively. For comparison, the RIDIR spectrum of the pCR monomer is included at the top of each panel. The RIDIR spectra for the pCR–H2O, pCR–MeOH, pCR–EtOH, and pCR–DEE dimers have been previously reported in the literature; the experiments were reproduced here, yielding similar results.53–55 | ||
For the acyclic alcohols and ethers, the H-bond strength also follows a well-established trend that is consistent with the increasing electron-donating inductive effect of the alkyl groups. The corresponding ΔνOH values are −127, −191, −217, −241, and −257 cm−1 for the alcohol series (H2O, MeOH, EtOH, iPrOH, and tBuOH, respectively), as shown in Fig. 1f–k. For the ether series, the redshifts are −127, −242, −280, and −303 cm−1 for the corresponding dimers with H2O, DME, DEE, and DIE (Fig. 1l–p). This data confirms that the H-bond strength increases with the electron-donating ability of the substituents. It is important to note that previous studies by some of us have reported the pCR–H2O,53pCR–MeOH,54pCR–EtOH,54 and pCR–DEE dimers,55 and our current results are in excellent agreement with those works, validating our experimental setup.
Computational analysis of H-bonding strength and atomic charges
The validation of the experimental data was carried out by comparing it to established theoretical descriptors of H-bond strength. Quantum-chemical calculations were performed to evaluate the OH⋯O bond length, binding energy, QTAIM electron density at the bond critical point (ρ(BCP)), and NBO donor–acceptor interaction energy (E(2)). The values are given in Table S1.For cyclic ethers, all parameters, i.e., the OH⋯O bond length, binding energy, BCP electron density, and E(2), show excellent linear correlations with ΔνOH, with R2 values of 0.963, 0.982, 0.994, and 0.990, respectively (Fig. 2a–d). This confirms that the experimental frequency shift is a reliable indicator of H-bond strength. For the alcohols and acyclic ethers, similar correlations were observed, except in the case of the binding energy (R2 = 0.700); the other parameters gave R2 values of 0.916 (bond length), 0.904 (BCP electron density), and 0.816 (E(2)) (Fig. 2g–j). Again, these results validate ΔνOH as a consistent probe of H-bonding strength. The molecular graph generated from QTAIM analysis and the donor–acceptor natural orbital generated from NBO analysis are shown in Fig. S4 and S5, respectively.
 | ||
| Fig. 2 Correlation plots between the redshift of the pCR OH stretching frequency (ΔνOH) with various H-bond descriptors such as OH⋯O bond length, binding energy, ρ(BCP), NBO interaction energy (E(2)), electrostatic energy contribution, and MESP on oxygen for Top panel: (a)–(f) H-bonded complexes with cyclic ethers and Bottom panel: (g)–(l) H-bonded complexes with alcohols and acyclic ethers. | ||
To probe the nature of the interactions, SAPT2 energy decomposition analysis was performed, which confirmed that the OH⋯O hydrogen bonds were predominantly electrostatic in nature (Fig. S6). The values of the different components of SAPT2 analysis are given in Table S2. The strong correlation between the electrostatic contribution and ΔνOH, with R2 values of 0.987 for cyclic ethers (Fig. 2e) and 0.955 for alcohols and acyclic ethers (Fig. 2k), further reinforces the idea that our ΔνOH values are a direct measure of the electron density on the oxygen atom.
The molecular electrostatic potential (MESP) at the oxygen atom was calculated and compared with the ΔνOH. The MESP values are given in Table S1. The MESP maps of the molecules are shown in Fig. S7. For the cyclic ethers, which share a common molecular backbone, the MESP values correlated strongly with the shifts (R2 = 0.988; Fig. 2f). However, a perplexing anomaly emerged for the acyclic systems: the alcohols exhibited higher MESP values (more negative potential) on the oxygen than the ethers. This trend is counterintuitive given the expected stronger inductive effect from the alkyl groups in ethers (Fig. 2l). The computed PACs on the oxygen atom for cyclic and acyclic systems, obtained using 18 different population analysis schemes, are given in Tables 1 and 2, respectively. Because different methods yield different charge trends, we first screened these methods using the cyclic ether data, which showed consistent H-bonding trends. Among the tested methods, the ADCH, CM5, and PEOE charges exhibited the most consistent correlations with H-bond strength (Table S3) and were therefore chosen for further analysis owing to their robustness and widespread applicability.56–58
| FRN | 2,3-DHF | 2,5-DHF | THF | |
|---|---|---|---|---|
| Hirshfeld | −0.142 | −0.207 | −0.226 | −0.228 |
| VDD | −0.166 | −0.252 | −0.273 | −0.275 |
| Mulliken | −0.715 | −0.897 | −0.929 | −0.911 |
| Löwdin | 0.075 | 0.020 | 0.007 | 0.000 |
| SCPA | −0.985 | −0.685 | −0.860 | −1.232 |
| SP | −0.919 | −1.039 | −0.941 | −0.892 |
| Bickel | −0.760 | −0.837 | −0.943 | −0.844 |
| Becke | −0.338 | −0.389 | −0.476 | −0.476 |
| ADCH | −0.164 | −0.246 | −0.309 | −0.324 |
| CHELPG | −0.153 | −0.311 | −0.585 | −0.466 |
| MK | −0.114 | −0.268 | −0.546 | −0.425 |
| CM5 | −0.220 | −0.291 | −0.316 | −0.323 |
| RESP | −0.114 | −0.268 | −0.543 | −0.423 |
| PEOE | −0.326 | −0.354 | −0.373 | −0.381 |
| MIBS | −0.254 | −0.400 | −0.466 | −0.455 |
| EEM | −0.550 | −0.539 | −0.522 | −0.528 |
| NPA | −0.580 | −0.667 | −0.689 | −0.688 |
| AIM | −1.072 | −1.055 | −1.023 | −1.038 |
| H2O | MeOH | EtOH | iPrOH | tBuOH | DME | DEE | DIE | |
|---|---|---|---|---|---|---|---|---|
| Hirshfeld | −0.312 | −0.267 | −0.270 | −0.271 | −0.268 | −0.215 | −0.211 | −0.214 |
| VDD | −0.286 | −0.292 | −0.292 | −0.296 | −0.288 | −0.273 | −0.264 | −0.267 |
| Mulliken | −0.315 | −0.611 | −0.687 | −0.748 | −0.679 | −0.830 | −0.924 | −1.052 |
| Löwdin | 0.169 | 0.071 | 0.097 | 0.118 | 0.136 | −0.049 | 0.006 | 0.044 |
| SCPA | −0.383 | −0.953 | −0.891 | −0.708 | −0.154 | −1.507 | −1.444 | −0.787 |
| SP | −0.621 | −0.769 | −0.880 | −0.899 | −1.207 | −0.858 | −0.683 | −0.782 |
| Bickel | −0.338 | −0.600 | −0.696 | −0.742 | −1.125 | −0.785 | −0.758 | −1.298 |
| Becke | −0.707 | −0.603 | −0.647 | −0.689 | −0.726 | −0.416 | −0.525 | −0.632 |
| ADCH | −0.707 | −0.519 | −0.515 | −0.505 | −0.487 | −0.317 | −0.291 | −0.289 |
| CHELPG | −0.725 | −0.644 | −0.694 | −0.719 | −0.772 | −0.379 | −0.491 | −0.618 |
| MK | −0.720 | −0.632 | −0.689 | −0.713 | −0.748 | −0.314 | −0.474 | −0.581 |
| CM5 | −0.642 | −0.486 | −0.485 | −0.482 | −0.476 | −0.324 | −0.313 | −0.307 |
| RESP | −0.719 | −0.631 | −0.687 | −0.710 | −0.746 | −0.314 | −0.468 | −0.573 |
| PEOE | −0.411 | −0.400 | −0.397 | −0.394 | −0.391 | −0.388 | −0.382 | −0.376 |
| MIBS | −0.897 | −0.662 | −0.683 | −0.693 | −0.688 | −0.414 | −0.438 | −0.467 |
| EEM | −0.642 | −0.591 | −0.602 | −0.608 | −0.613 | −0.526 | −0.538 | −0.544 |
| NPA | −0.963 | −0.808 | −0.812 | −0.814 | −0.823 | −0.686 | −0.701 | −0.709 |
| AIM | −1.203 | −1.120 | −1.124 | −1.122 | −1.122 | −1.063 | −1.070 | −1.064 |
The computed charges were plotted against the experimental ΔνOH values. As shown in Fig. 3, the cyclic ethers exhibit strong linear correlations between the IR shifts and the PACs of oxygen obtained from ADCH, CM5, and PEOE (Fig. 3a–c). In these systems, a more negative oxygen charge corresponds to a larger IR redshift, which is expected. This provided an initial indication that these methods could correctly capture the charge trends. However, when we applied these same charge models to the acyclic alcohols and ethers, the correlations failed. Despite the clear experimental trend of increasing H-bond strength with increasing alkyl substitution, the computed oxygen charges often became less negative; the ethers consistently display the least-negative charges, followed by alcohols, with water exhibiting the most-negative oxygen charge (Fig. 3d–f). This trend is in stark contradiction to the experimental data, in which the H-bond strength increases from water to alcohols to ethers. This result serves as a cautionary example. It demonstrates that many widely used computational charge models, while mathematically sound, can produce charges that are inconsistent with fundamental, experimentally validated trends. This is particularly problematic for non-experts who might use these models without fully understanding their limitations.
 | ||
| Fig. 3 Correlation plots between the atomic charges and the redshift of the pCR OH stretching frequency (ΔνOH). Top panel: (a)–(c) ΔνOHvs. uncorrected ADCH, CM5, and PEOE charges for H-bonded dimers with cyclic ethers (FRN, 2,3-DHF, 2,5-DHF, and THF); Bottom panel: (d)–(f) ΔνOHvs. uncorrected ADCH, CM5, and PEOE charges for H-bonded dimers with alcohols and acyclic ethers. | ||
Inclusion of electronegativity corrections
The ADCH and CM5 charges show different slopes for different alkyl groups, whereas PEOE yields nearly identical slopes for all three types of alkyl group (Fig. 3d–f). This suggests that the electronegativity of the atoms bonded to the oxygen atoms plays some role, as the PEOE methods calculate atomic charges by partially equalising the orbital electronegativity of the atoms.48,49 This is again supported by the fact that the average electronegativity difference (ΔEN) between oxygen and its bonded atoms is approximately 1.24 for water (O–H bond), 1.06 for alcohols (O–C and O–H), and 0.89 for ethers (O–C bonds).As presented in Table 2, larger ΔEN values consistently correspond to more-negative computed oxygen charges, which explains why water consistently shows the most negative charge across different models, despite being the weakest H-bond acceptor in this series.
To further illustrate the dominant influence of electronegativity, empirical adjustments were introduced to the computed charges. A correction was first applied based on the electronegativity difference ΔEN1 between the central oxygen and the atoms directly bonded to it (eqn (2)–(4)).
 | (1) |
 | (2) |
 | (3) |
 | (4) |
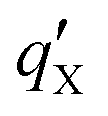 is the correction parameter for X = ADCH, CM5 and PEOE.
is the correction parameter for X = ADCH, CM5 and PEOE.
This adjustment significantly improved the correlations with ΔνOH (Fig. 4a–c); the only significant deviations were observed for MeOH, DME, and iPrOH, DIE and t-BuOH, which may be attributed to the fact that in these molecules, the central atom is bonded to a –CH3, –C3H7 and –C4H9 group, respectively, whereas EtOH and DEE have a –C2H5 group, similar to FRN, 2,3-DHF, 2,5-DHF, and THF. To address these cases, we refined the correction by incorporating the average electronegativity of more distal substituents (ENav; Eqn5).
| QCorrX = QX − qX | (5) |
| qADCH = −1.24ΔEN1 − 0.03(ENC − ENav) + 1.11 | (6) |
| qCM5 = −1.04ΔEN1 − 0.01(ENC − ENav) + 0.92 | (7) |
| qPEOE = −0.14ΔEN1 − 0.01(ENC − ENav) + 0.12 | (8) |
 | ||
Fig. 4 Correlation plots between corrected atomic charges and redshift of the pCR OH stretching frequency (ΔνOH) of H-bonded dimers. Top panel: (a)–(c) ΔνOHvs. ΔEN1-corrected charges obtained from ADCH  , CM5 , CM5  , and PEOE , and PEOE  , Bottom panel: (d)–(f) ΔνOHvs. ΔENav-corrected charges obtained from ADCH (QCorrADCH), CM5 (QCorrCM5), and PEOE (QCorrPEOE). , Bottom panel: (d)–(f) ΔνOHvs. ΔENav-corrected charges obtained from ADCH (QCorrADCH), CM5 (QCorrCM5), and PEOE (QCorrPEOE). | ||
The variable qX in eqn 5 represents the modified charge, where X = ADCH, CM5 and PEOE, ENC refers to the electronegativity of the carbon atom (2.55), and ENav is the average electronegativity of the other atoms at all positions other than position one. The values of EN1 and ENav are provided in Table S4. Electronegativity (EN) calculations for various molecules were carried out using the molecular structures shown in Fig. S9. The molecule DEE was chosen as the reference, as FRN, 2,3-DHF, 2,5-DHF, and THF all share the same backbone. This molecule contains three distinct positions. At position 1, the central oxygen atom is bonded to two carbon atoms, giving EN1 = 2.55. At position 2, six atoms (two carbons and four hydrogens) are present, resulting in EN2 = 2.32. At position 3, six hydrogen atoms are present, giving EN3 = 2.20 (Fig. S9a). The average electronegativity is therefore ENav = 2.26. A more detailed explanation of the other molecules, the determination of these quantities, and the procedure used to derive the equations (eqn 2–4 and 6–8) are provided in the SI.
This refined, multi-parameter adjustment incorporating both ΔEN1 and the average electronegativity of the surrounding atoms (ENav) yielded a marked improvement in the correlation between the computed atomic charges and the experimental ΔνOH shifts (Fig. 3d–f), achieving R2 values of 0.976, 0.943, and 0.949 for ADCH, CM5, and PEOE, respectively. The unadjusted, ΔEN1-adjusted, and ENav-adjusted charges for ADCH, CM5, and PEOE are summarised in Tables S5–S7. Among these, the ADCH method shows the strongest linear correlation after adjustment.
To demonstrate the generality of this effect, we extended the analysis to nitrogen-containing systems (NH3, diethylamine (DEA), triethylamine (TEA)) for which the gas-phase IR spectra were available.59 Experimental gas-phase basicity trends and ΔνOH shifts for the H-bonded complexes of these compounds with phenol reveal that TEA is a stronger hydrogen-bond acceptor and more basic than DEA, while NH3 is the weakest.60 However, the unadjusted ADCH charges predicted the opposite trend. When the influence of ΔEN was incorporated using the empirical relation given in eqn (S5), the corrected ADCH charges agreed with the experimental observations (Fig. S8), reinforcing the consistent impact of electronegativity across different classes of H-bond acceptors. This result is also consistent with the observations of Snurr et al.,23 who found that the PAC model is more dependent on directly bonded atoms, with a secondary yet significant contribution from distal atoms.
The ability to “fix” the models by accounting for a simple, fundamental property, such as electronegativity, highlights that the underlying algorithms of these models are not correctly capturing the electronic distribution in a chemically intuitive manner.
Conclusions
In this work, a comprehensive experimental dataset of H-bonding in the gas phase has been presented, which provides a benchmark for evaluating quantum chemical models. The systematic investigation of both cyclic and acyclic acceptors reveals clear, intuitive H-bonding strength trends. However, comparing these data with 18 popular charge calculation methods reveals a poor correlation between the computed atomic charges of the H-bond acceptors and the corresponding H-bond strengths, particularly for systems such as the OH⋯O H-bonding examples studied in the present work, which involve various substituents. These findings serve as a critical warning: many widely used charge models are inherently biased by the electronegativity of bonded atoms, leading them to produce results that contradict well-established chemical principles and experimental observations. This highlights the need for caution when using these models and underscores the importance of experimental validation. This manuscript offers a high-quality dataset that can be used to develop and test new theoretical frameworks, which will ultimately contribute to the creation of more physically meaningful atomic charges, which will be essential for accurately modelling a vast range of chemical phenomena.Author contributions
Conceptualization: HSB, methodology: HSB, AKS, ARS, investigation: AKS, ARS, SR, RS, LD, HSB, visualization: AKS, ARS, SR, RS, LD, HSB, funding acquisition: HSB, project administration: HSB, supervision: HSB, writing: original draft: AKS, HSB, writing: review & editing: AKS, ARS, SR, RS, LD, HSBConflicts of interest
There are no conflicts to declare.Data availability
The data supporting this work are uploaded as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cp03250d.Acknowledgements
A. K. S., A. R. S, S. R., R. S., L. D., and H. S. B. acknowledge financial support from the Department of Atomic Energy, Anusandhan National Research Foundation (ANRF) (Project File No: CRG/2022/001096), Govt. of India.Notes and references
- G. A. Jeffrey, An introduction to hydrogen bonding, Oxford university pressNew York, 1997 Search PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg and P. Hobza, Pure Appl. Chem., 2011, 83, 1637–1641 CrossRef.
- L. Pauling and R. B. Corey, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 251–256 CAS.
- C. N. Pace, H. Fu, K. Lee Fryar, J. Landua, S. R. Trevino, D. Schell, R. L. Thurlkill, S. Imura, J. M. Scholtz and K. Gajiwala, Prot. Sci., 2014, 23, 652–661 CAS.
- F. H. Stillinger, Science, 1980, 209, 451–457 CAS.
- S. Scheiner, Hydrogen bonding: a theoretical perspective, Oxford University Press, 1997 Search PubMed.
- K. Morokuma and K. Kitaura, Chemical Applications of Atomic and Molecular Electrostatic Potentials: Reactivity, Structure, Scattering, and Energetics of Organic, Inorganic, and Biological Systems, Springer, 1981, pp. 215–242 Search PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CAS.
- G. R. Desiraju and T. Steiner, The weak hydrogen bond: in structural chemistry and biology, International Union of Crystal, 2001 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CAS.
- S. J. Weiner, P. A. Kollman, D. A. Case, U. C. Singh, C. Ghio, G. Alagona, S. Profeta and P. Weiner, J. Am. Chem. Soc., 1984, 106, 765–784 CAS.
- A. D. MacKerell Jr, J. Comput. Chem., 2004, 25, 1584–1604 Search PubMed.
- J. Meister and W. H. E. Schwarz, J. Phys. Chem., 1994, 98, 8245–8252 CrossRef CAS.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef CAS.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1841–1846 CrossRef CAS.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 2338–2342 CrossRef CAS.
- T. Lu and Q. Chen, Exploring Chemical Concepts Through Theory and Computation, 2024, pp. 161–188 Search PubMed.
- T. Lu and F.-W. Chen, Acta Phys. – Chim. Sin, 2012, 28, 1–18 CAS.
- B. Wang, S. L. Li and D. G. Truhlar, J. Chem. Theory Comput., 2014, 10, 5640–5650 CrossRef CAS PubMed.
- T. A. Manz and D. S. Sholl, J. Chem. Theory Comput., 2010, 6, 2455–2468 CrossRef CAS PubMed.
- J. Zhao, Z.-W. Zhu, D.-X. Zhao and Z.-Z. Yang, Phys. Chem. Chem. Phys., 2023, 25, 9020–9030 RSC.
- K. B. Wiberg and P. R. Rablen, J. Org. Chem., 2018, 83, 15463–15469 CAS.
- S. Kancharlapalli, A. Gopalan, M. Haranczyk and R. Q. Snurr, J. Chem. Theory Comput., 2021, 17, 3052–3064 CAS.
- K. B. Wiberg and P. R. Rablen, J. Comput. Chem., 1993, 14, 1504–1518 CAS.
- R. M. Fogarty, R. P. Matthews, C. R. Ashworth, A. Brandt-Talbot, R. G. Palgrave, R. A. Bourne, T. Vander Hoogerstraete, P. A. Hunt and K. R. J. Lovelock, J. Chem. Phys., 2018, 148, 193817 Search PubMed.
- Y. Yamada, T. Ebata, M. Kayano and N. Mikami, J. Chem. Phys., 2004, 120, 7400–7409 CAS.
- T. Ebata, A. Iwasaki and N. Mikami, J. Phys. Chem. A, 2000, 104, 7974–7979 CAS.
- S. Kumar, P. Biswas, I. Kaul and A. Das, J. Phys. Chem. A, 2011, 115, 7461–7472 CAS.
- S. Ghosh, P. Chopra and S. Wategaonkar, Phys. Chem. Chem. Phys., 2020, 22, 17482–17493 CAS.
- D. Cheshmedzhieva, S. Ilieva, B. Hadjieva and B. Galabov, J. Phys. Org. Chem., 2021, 34, e4258 CAS.
- B. Galabov, V. A. Popov, D. Cheshmedzhieva, S. Ilieva and H. F. Schaefer Iii, Chem. Phys. Lett., 2022, 791, 139378 CAS.
- A. K. Sahu, A. R. Satpathi, S. Rout, P. Mohanty, L. Dash and H. S. Biswal, J. Phys. Chem. Lett., 2024, 15, 11445–11453 CAS.
- J. Clayden, Organic Chemistry, 2nd edn, Oxford University Press, Oxford, 2012 Search PubMed.
- R. A. Benkeser, R. A. Hickner and D. I. Hoke, J. Am. Chem. Soc., 1958, 80, 2279–2282 Search PubMed.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef.
- C. Fonseca Guerra, J.-W. Handgraaf, E. J. Baerends and F. M. Bickelhaupt, J. Comput. Chem., 2004, 25, 189–210 CrossRef PubMed.
- P. O. Löwdin, J. Chem. Phys., 1950, 18, 365–375 CrossRef.
- P. Ros and G. C. A. Schuit, Theor. Chim. Acta, 1966, 4, 1–12 CrossRef.
- E. W. Stout and P. Politzer, Theor. Chim. Acta, 1968, 12, 379–386 CrossRef.
- F. M. Bickelhaupt, N. J. R. van Eikema Hommes, C. Fonseca Guerra and E. J. Baerends, Organometallics, 1996, 15, 2923–2931 CrossRef.
- A. D. Becke, J. Chem. Phys., 1988, 88, 2547–2553 CrossRef.
- T. Lu and F. Chen, J. Theor. Comput. Chem., 2012, 11, 163–183 CrossRef.
- C. M. Breneman and K. B. Wiberg, J. Comput. Chem., 1990, 11, 361–373 CrossRef.
- B. H. Besler, K. M. Merz Jr and P. A. Kollman, J. Comput. Chem., 1990, 11, 431–439 CrossRef.
- A. V. Marenich, S. V. Jerome, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2012, 8, 527–541 CrossRef PubMed.
- J. Wang, P. Cieplak and P. A. Kollman, J. Comput. Chem., 2000, 21, 1049–1074 CrossRef CAS.
- W. J. Mortier, K. Van Genechten and J. Gasteiger, J. Am. Chem. Soc., 1985, 107, 829–835 CrossRef CAS.
- J. Gasteiger and M. Marsili, Tetrahedron Lett., 1978, 19, 3181–3184 CrossRef.
- J. Gasteiger and M. Marsili, Tetrahedron, 1980, 36, 3219–3228 CrossRef CAS.
- T. Verstraelen, S. Vandenbrande, F. Heidar-Zadeh and L. Vanduy, J. Chem. Theory Comput., 2016, 12, 3894–3912 CrossRef CAS PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- R. F. W. Bader, Atoms In Molecules: A Quantum Theory, Clarendon Press, Oxford, UK, 1990 Search PubMed.
- H. S. Biswal, P. R. Shirhatti and S. Wategaonkar, J. Phys. Chem. A, 2009, 113, 5633–5643 CrossRef CAS PubMed.
- H. S. Biswal, P. R. Shirhatti and S. Wategaonkar, J. Phys. Chem. A, 2010, 114, 6944–6955 CrossRef CAS PubMed.
- H. S. Biswal and S. Wategaonkar, J. Phys. Chem. A, 2010, 114, 5947–5957 CrossRef CAS PubMed.
- Y. Fu, H. Liu, B. Z. Tang and Z. Zhao, Nat. Commun., 2023, 14, 2019 CrossRef CAS PubMed.
- Y. Yuan, L. Zhou, H. Robatjazi, J. L. Bao, J. Zhou, A. Bayles, L. Yuan, M. Lou, M. Lou, S. Khatiwada, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2022, 378, 889–893 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- A. Iwasaki, A. Fujii, T. Watanabe, T. Ebata and N. Mikami, J. Phys. Chem., 1996, 100, 16053–16057 CrossRef CAS.
- J. I. Brauman, J. M. Riveros and L. K. Blair, J. Am. Chem. Soc., 1971, 93, 3914–3916 CrossRef CAS.
Footnote |
| † Dedicated to Professor Resnati, celebrating a career in fluorine and noncovalent chemistry on the occasion of his 70th birthday |
| This journal is © the Owner Societies 2025 |