Boosting hydrazine electrooxidation on Ru-coordinated heteronuclear double metal atom catalysts
Received
9th August 2025
, Accepted 25th September 2025
First published on 25th September 2025
Abstract
The hydrazine oxidation reaction (HzOR) is considered as an efficient alternative anodic reaction to the oxygen evolution reaction for low-energy hydrogen production. Consequently, developing highly efficient electrocatalysts for the HzOR is important. By using density functional theory (DFT) calculations, we evaluate the HzOR activity of dual-metal atom catalysts (DACs), specifically Ru coordinated with 3d–5d transition metals, anchored on nitrogen-doped graphene (RuM@N6C, where M = Ti–Cu, Zr–Mo, Ru–Pd, W, Ir and Pt). Among these DACs, RuCo@N6C and RuCu@N6C exhibit high catalytic activity with low limiting potential values of −0.13 and 0.00 V, respectively. The electron transfer, crystal orbital Hamiltonian population and electron localization function are further analyzed to prove that the moderate metal coordination favored the reduction of the strong adsorption of the Ru site to the *N2H3 intermediate. In addition, the excellent thermodynamic stability of RuCo@N6C and RuCu@N6C was also identified. These findings underscore the crucial role of electron transfer in the HzOR and highlight the potential of Ru-coordinated heteronuclear DACs in bridging the gap between the sustainable hydrogen production and ecosystem governance technologies.
1. Introduction
Hydrazine (N2H4) is a versatile liquid energy carrier that has gained significant attention due to its excellent energy density, ease of storage and transport and favourable thermodynamic properties.1 As a clean and efficient fuel, N2H4 has become a focus of research in energy technologies. In recent years, renewable hydrogen production via direct water splitting has achieved a series of advancements.2–5 In traditional electrochemical water splitting for hydrogen production, the high thermodynamic barrier of 1.23 V (vs. RHE) for the anodic oxygen evolution reaction (OER) imposes a huge energy consumption, whereas the hydrazine oxidation reaction (HzOR) is considered as an efficient alternative pathway to the OER due to its 0.33 V theoretical potential.6,7 However, the catalytic activity of the HzOR catalysts remains unsatisfactory, highlighting the need for the development of highly efficient materials. In addition, theoretical insights into the underlying mechanisms of new materials are essential for advancing materials chemistry.
As for HzOR electrocatalysis, single-atom catalysts (SACs) show commendable performances.8–12 Jiao et al. investigated a series of 14 SACs (single transition metal atoms anchored on the C2N monolayer, TM@C2N) using the first-principles calculations and identified Ru@C2N as the best catalyst with the lowest limiting potential of −0.24 V.13 Dual-atom catalysts (DACs) have atomically dispersed active sites and exhibit similar advantages to SACs in terms of maximum utilization efficiency of metal sites. Moreover, DACs possess two adjacent metal atoms as the active center, potentially resulting in a new catalytic mechanism and greatly improved structural adjustability compared with SACs.14–16 Theoretical studies of DACs have already demonstrated their application potential in numerous fields, such as the carbon dioxide reduction reaction (CO2RR)17,18 and the nitrogen reduction reaction (NRR).19 Furthermore, the cooperative interaction between the two adjacent metal atoms results in a unique electronic structure of both metal atoms that is different from the corresponding single-atom counterparts, which potentially optimizes HzOR performance by adjusting intermediate adsorption.20,21 On the other hand, nitrogen-doped graphene has attracted widespread attention for its ability to provide anchor sites for metals due to its high carrier density and mobility.22 In particular, FeZn@N6C,23 CoCu@N6C,24 and RuCo@N6C25 have been successfully synthesized, where RuCo@N6C was used as a bifunctional oxygen electrocatalyst for oxygen electrocatalysis, demonstrating the potential of nitrogen-doped graphene as an ideal support for DACs. Given the excellent HzOR catalytic performance of the Ru-containing catalysts discussed above, constructing Ru-based DACs on nitrogen-doped graphene is promising for obtaining high catalytic activity, and the corresponding reaction mechanism deserves further investigation.
In this study, we employed density functional theory (DFT) based first-principles calculations to explore the performance and mechanisms of Ru paired with transition metals (TM) supported on nitrogen-doped graphene for the HzOR. The investigated catalysts, denoted as RuM@N6C DACs (containing TMs including Ti–Cu, Zr–Mo, Ru–Pd, W, Ir and Pt) had different adsorption sites for the activation of N2H4. Furthermore, we identified an optimized volcano-shaped correlation between the adsorption free energy of the N2H4 molecule and the limiting potential (UL) for hydrazine oxidation to nitrogen synthesis across all heteronuclear RuM@N6C DACs. Notably, RuCo@N6C and RuCu@N6C exhibited excellent HzOR catalytic activity, with limiting potentials of −0.13 V and 0.00 V, respectively. Additionally, we use the electron transfer (Q) and the crystal orbital Hamilton population (COHP) to demonstrate the mechanism of how TMs with moderate chemical activity atoms (e.g. Co, Cu) coordinated with Ru in DACs can reduce UL by effectively modulating the adsorption capacity of the reaction intermediates. These results give an insight into how the HzOR selectivity can be modulated by the dual metal coordination composition and further provide theoretical guidance for designing catalysts for hydrazine assisted hydrogen production.
2. Computational details
Spin-polarized first-principles calculations have been performed based on DFT implemented in the Vienna Ab initio Simulation Package (VASP),26,27 and the data were post-processed using VASPKIT.28 The interaction between the valence electrons and the ionic core is treated using the projector augmented wave (PAW) method29 and the exchange–correlation effect is accounted by the Perdew–Burke–Ernzerhof (PBE) functional30 with the DFT-D3 method to correct the van der Waals interaction.31 The plane wave energy cutoff was set to be 450 eV. The convergence criteria for Hellmann–Feynman force and energy are set to be 0.02 eV Å−1 and 10−5 eV, respectively. A 4 × 4 graphene supercell with a vacuum space of ∼15 Å is adopted to build the atomic model of RuM@N6C, for which the Monkhorst–Pack (MP) grids of 3 × 3 × 1 and 6 × 6 × 1 are adopted to perform structural optimization and the calculation of densities of states (DOS),31 respectively. The COHP was calculated with the Local Orbital Basis Suite Towards Electronic-Structure Reconstruction (LOBSTER) program.32 Bader charge analysis was performed to quantitate the electron transfer.33Ab initio molecular dynamics (AIMD) simulation was adopted to evaluate the thermal stability of the proposed catalysts, for which the simulation was performed under a constant volume and temperature (NVT) ensemble. The total simulation time we adopted is 6 ps with a time step of 2 fs.34 During the AIMD simulations, the temperature is controlled using a Nosé–Hoover thermostat. The total electronic energies of various HzOR intermediates (NxHy) and adsorption energy (ΔEads(*NxHy)) for NxHy on the catalyst are defined as:| | | E(NxHy) = E(N2H4) − [(4 − y)/2E(H2)] | (1) |
| | | ΔEads(*NxHy) = E(*NxHy) − E(*) − E(NxHy) | (2) |
where x = 2, y = 0, 1, 2, 3 and 4 and the E(N2H4) and E(H2) are the total electronic energies of the isolated N2H4 and H2 molecules in a vacuum, respectively, and ΔE(*NxHy) and E(*) are the total electronic energies of the catalyst with and without intermediates, respectively. The computational hydrogen electrode (CHE) model35 is used to calculate the free energy change of the reaction elementary step based on the following formula:where ΔE was calculated based on the DFT total energy, and ΔEZPE and TΔS are the contributions of zero-point energy and entropy difference of the reactants and products (298.15 K, 1 atm.), respectively. EZPE and TS of the free molecules (Table S1, SI) and the adsorbates (Table S2, SI) were obtained by calculating the vibrational frequencies. The reaction free energies of each step during the HzOR process are denoted in Table S3 (SI). The UL was calculated with:where ΔGmax is the free energy change of the potential determining step (PDS), which is the most endothermic one among all elementary steps along the lowest-energy pathway.
3. Results and discussion
3.1. Structure and stability of RuM@N6C DACs
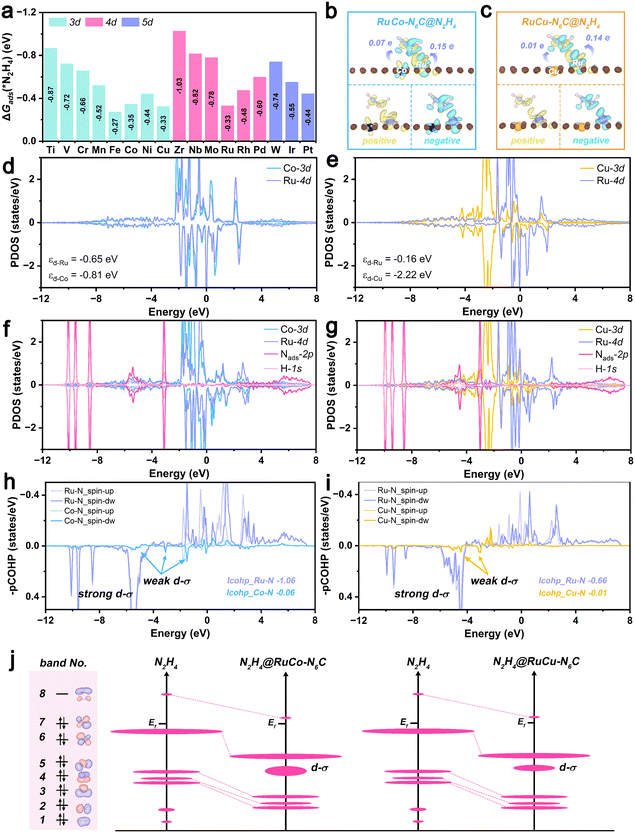
The structure model of RuM@N6C DACs consists of two parts, namely, nitrogen-doped graphene and the Ru-metal dimer. For the optimized structure of the substrate nitrogen-doped graphene, which contains six pyridine N atoms, the lattice parameters are given in Fig. S1 (SI). The active centre consists of the Ru-metal dimer as shown in Fig. 1a, where a Ru atom is paired with a TM atom, and each TM is coordinated with 3 N atoms. The TM atoms selected to coordinate with Ru are shown in Fig. 1b, and considering the strength of adsorption, not too strong (Sc, Ti, Y and Zr) and not too weak (Zn, Ag, Cd, Au and Hg), combined with the commonness of TMs (except Tc, Ta, Re and Os), the middle TMs with moderate chemical activity have been considered to constitute heteronuclear RuM@N6C DACs. The optimized geometric configuration is presented in Fig. S2 (SI). To evaluate the stability of RuM@N6C DACs, we first use the formation energy (Ef) as the first screening factor. The value of Ef and the energy of TM atoms in their bulk (ETM-bulk) are listed in Table S4 (SI). All RuM@N6C DACs exhibit Ef < 0, indicating their thermodynamic stability.36 The structure parameters are given in Table S5 (SI). Both the largest bond lengths between TM atoms and their neighbouring N atoms and the Ru–M bond lengths are smaller than sum of their corresponding covalent radii. The stability of the RuM@N6C DACs is further confirmed by the calculated total density of states (TDOS) and COHP. As shown in Fig. S3 (SI) and Fig. 1e, f, there is significant electronic state hybridization spanning a large energy range for the bonded atoms, confirming the formation of strong chemical bonds.
 |
| | Fig. 1 (a) Schematic diagram of RuM@N6C DACs, showing the Ru atom (purple sphere) paired with a transition metal (green sphere), each metal atom coordinated with three nitrogen atoms (grey spheres), and nitrogen-doped graphene (brown spheres). (b) The TM selected to coordinate with Ru. (c) Formation energy and binding energy of the studied RuM@N6C DACs. The COHP of (e) RuCo@N6C and (f) RuCu@N6C. | |
3.2. Adsorption and activation of N2H4
Due to the special spatial configuration of the N2H4 molecule, we investigated the role of different metal sites in the adsorption and activation of N2H4, and the calculated adsorption free energies and optimized adsorption configuration for *N2H4 are shown in Table S6 and Fig. S4–S6 (SI), respectively. The corresponding energies of the optimal adsorption sites are shown in Fig. 2a. Interestingly, the adsorption energies have a periodic pattern, and the TM located on the left side of the periodic table of elements paired with Ru made RuM@N6C DACs have a strong adsorption energy for *N2H4, which also means that it is difficult for further HzOR dehydrogenation to occur. Thus, we speculate that the RuM@N6C DACs, where TM on the right side of the periodic table is paired with Ru as the optimal adsorption site, show high catalytic activity for the HzOR.
 |
| | Fig. 2 (a) The energies of the optimal adsorption sites. The CDD of (b) RuCo@N6C and (c) RuCu@N6C. The PDOS of (d) RuCo@N6C and (e) RuCu@N6C. The PDOS of (f) RuCo@N6C and (g) RuCu@N6C with N2H4. The COHP of (h) RuCo@N6C and (i) RuCu@N6C with N2H4. (j) Schematic diagram of the activation mechanism of N2H4. | |
To gain fundamental insight into the N2H4 adsorption and activation mechanism, the charge density difference (CDD), DOS and COHP were investigated. Considering the moderate chemical activity, RuCo@N6C and RuCu@N6C are chosen as representatives. As shown in Fig. 2b and c, N2H4 adsorption and activation should follow the typical Blyholder model.37 The electron depletion (cyan region) can be seen on the Ru atom and σ* orbitals of N2H4, which indicates that Ru site back-donates electrons to the σ* orbitals of N2H4 and the σ orbitals of N2H4 donate electrons to the empty d states of the Ru site. Overall, the N2H4 molecule gains 0.15e and 0.14e from the active sites of RuCo@N6C and RuCu@N6C, respectively. The CDD diagram of N2H4 on Ru2@N6C is shown in Fig. S7 (SI), the N2H4 molecule gains 0.17e from the active site of Ru2@N6C. Compared to the above moderate chemical system, the Ru2@N6C system has a stronger bond between the active site and the N2H4 molecule, which may be detrimental to the next step of the dehydrogenation reaction. In order to explore the adsorption and bonding strength between the active site and the N2H4 molecule, we further evaluated the partial densities of states (PDOS) and COHP. The calculated PDOS diagrams of the Ru–M d orbital of RuM@N6C DACs are shown in Fig. S8 (SI), and the PDOS of selected RuCo@N6C and RuCu@N6C are presented in Fig. 2d and e, respectively. For the RuCo@N6C system, significant orbital overlap was observed between Co-3d and Ru-4d, indicating that strong hybridization in RuCo@N6C arises between Co-3d and Ru-4d. For the RuCu@N6C system, due to the characteristics of the electronic structure of the Cu atom, the overlap between Cu-3d and Ru-4d is not as large as the former, but there is still a significant overlap in the 2 eV to −2 eV energy region. Meanwhile, the d-band center (εd) of the Ru atom for RuCo@N6C is −0.64 eV, which is higher than that of the Co atom (εd = −0.81 eV). The d-band center of the Ru atom in RuCu@N6C follows a similar trend compared with RuCo@N6C, where the εd values of Ru and Cu are −0.16 and −2.22 eV, respectively. According to the d-band center theory, the higher d-band center will contribute to the stronger adsorption. The d-band center of different metal atoms shows a consistent trend with adsorption free energies at different adsorption sites.
The PDOS diagrams for N2H4 adsorbed on RuCo@N6C, RuCu@N6C and free N2H4 are shown in Fig. 2f, g and Fig. S9 (SI), respectively. The electronic state hybridization between the molecular orbitals of N2H4 and the d orbitals of Ru and other metal atoms can be observed at ∼5 eV. The COHP analysis could give more quantitative information on the chemical bond between N2H4 and the dominant metal active sites, and the details of more negative integrated COHP (ICOHP) values are shown in Tables S9–S11 (SI). As shown in Fig. 2h and i, the ICOHP values (−1.06 eV) of Ru–N for the RuCo@N6C system indicate that Ru is the dominant adsorption metal active site for adsorbing N2H4, proving the generation of a d–σ bond during adsorption. For the RuCu@N6C system, the ICOHP values (−0.66 and −0.01 eV for Ru–N and Cu–N) are lower than those of RuCo@N6C system, which confirms the stronger adsorption of N2H4 on RuCo@N6C. The PDOS and COHP of the Ru2@N6C system are shown in Fig. S10 and S11 (SI), respectively. For the Ru2@N6C system, the stronger d–σ bond than those of RuCo@N6C and RuCu@N6C leads to difficulties in the first dehydrogenation step to generate *N2H3, resulting in a higher energy barrier for the RDS. The ICOHP values (−0.14 and −1.26 eV for Ru1–N and Ru2–N) further prove the excessive N2H4 adsorption ability of the Ru2@N6C system. Overall, a schematic diagram of the activation mechanism of N2H4 is shown in Fig. 2j. The adsorption process causes the empty orbital (band no. 8) of N2H4 to shift downwards, approaching the Fermi energy level (Ef), which is conducive to electron transfer from the metal active site, thus causing molecule activation, while the orbital splits out the d–σ bond, and N2H4 of the RuCo@N6C system has a larger orbital width and overlap than that of RuCu@N6C system, indicating a higher activation intensity.
3.3. HzOR catalytic mechanism
To clarify the HzOR mechanism, we calculated ΔG profiles for all intermediates on RuM@N6C DACs. In addition to the previously mentioned optimized adsorption geometry configuration for N2H4 (Fig. S5 and S6, SI), the configuration of the rest of the reaction intermediates (*N2H3–*N2) is shown in Fig. S12—S15 (SI). As evidenced in Fig. 3a and Table S7 (SI), the rate-determining step (RDS) of RuM@N6C DACs is mainly the step2 (N2H4–N2H3, ΔG2) and step3 (N2H3–N2H2, ΔG3), which may be due to selective modulation of the *N2H3 intermediates by the moderate RuM@N6C DACs, such as RuCo@N6C and RuCu@N6C, indicating that the inclusion of Cu and Co atoms as the TMs to coordinate with Ru significantly improves the adsorption of the intermediates. In addition, the UL values for RuCo@N6C (−0.13 V) and RuCu@N6C (0.00 V) are less negative than those of the other RuM@N6C and Ru2@N6C (−0.41 V) catalysts, indicating the thermodynamic advantage of these two materials. We further explored the relationship between ΔGads(*N2H4) and ΔG2 and ΔG3, respectively. As shown in Fig. 3b and c, linear correlations can be observed, thus ΔGads(*N2H4) can be selected as a descriptor. A volcano plot of limiting potential on RuM@N6C DACs is shown in Fig. 3d, where RuCo@N6C and RuCu@N6C exactly stand near the top of the volcano, confirming that the moderate adsorption of N2H4 is pivotal for the HzOR. Since the electrochemical HzOR occurs in an aqueous solution, we used the VASPsol implicit solvation model to include by default the effects of solvation on catalytic activity, which was validated for the key DACs (RuCo@N6C, RuCu@N6C and Ru2@N6C),38 as shown in Fig. S16 and Table S8, with respective overpotentials (UL) of −0.14, 0.07 and −0.40 V. These values are nearly consistent with those obtained without taking into account of solvation effects. Meanwhile, ΔGads(*N2H4) of RuCo@N6C and RuCu@N6C is stable under different U values, indicating the exchange–correlation functional using PBE is sufficient to explain the relationship of volcano plot for HzOR, as shown in Fig. S17 and S18 (SI).
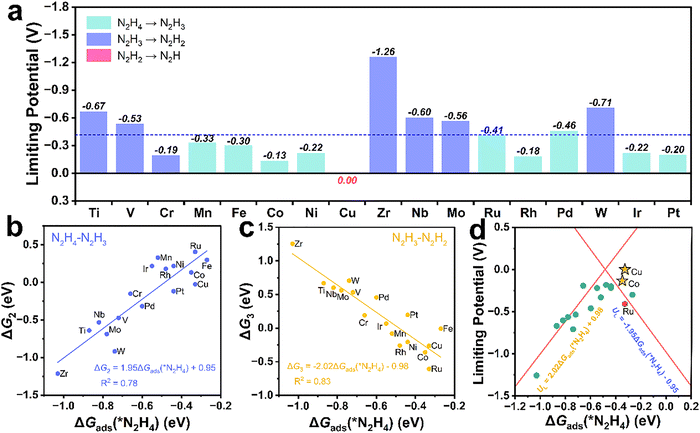 |
| | Fig. 3 (a) Summary of the limiting potentials of the HzOR on RuM@N6C DACs. (b) The relationship between ΔGads(*N2H4) and ΔG2 (N2H4–N2H3). (c) The relationship between ΔGads(*N2H4) and ΔG3 (N2H3–N2H2). (d) HzOR volcano plot of RuM@N6C DACs with a descriptor (ΔGads(*N2H4)), where the red hexagon represents Ru2@N6C, the outline of the volcano plot are composed of two function lines in red gained by Fig. 3(b) and (c). | |
3.4. HzOR performance of RuCo@N6C and RuCu@N6C
As plotted in Fig. 4a and b, the detailed free energy diagrams of the HzOR on RuCo@N6C and RuCu@N6C are further analyzed to validate their hydrazine-to-nitrogen performance. The top views of the corresponding structures are shown in Fig. 4c and d. The free energy diagram of the HzOR on Ru2@N6C is shown in Fig. S19 (SI) for comparison. N2H4 can be stably adsorbed with a Ru–N bond and then goes through the first dehydrogenation step to form *N2H3 with energy changes of 0.13 eV for RuCo@N6C and −0.13 eV for RuCu@N6C. In the subsequent steps (*N2H3–*N2H2–*N2H–*N2), stepwise dehydrogenation occurs and the corresponding energies drop by 0.36, 0.17 and 0.38 eV for RuCo@N6C and 0.26, 0.00 and 0.40 eV for RuCu@N6C, respectively. Eventually, *N2 species desorb from RuM@N6C, and the energies of of RuCo@N6C and RuCu@N6C are −0.19 and −0.30 eV, respectively.
 |
| | Fig. 4 Free energy diagrams of (a) RuCo@N6C and (b) RuCu@N6C, respectively. The top view of the optimized geometry adsorption configuration of (c) RuCo@N6C and (d) RuCu@N6C, respectively. The charge variation between the metal atoms and the intermediates of (e) RuCo@N6C and (f) RuCu@N6C, respectively. | |
To quantify the electron transfer contribution of the active center Ru–M metal pair to the intermediates, charge variations of metal atoms during the HzOR on RuCo@N6C, RuCu@N6C and Ru2@N6C are shown in Fig. 4e, f and Fig. S20 (SI). It is found that the tendency of charge variations on RuCo@N6C and RuCu@N6C is analogous. Specially, in the first step the Ru atom is the dominant metal active site for both systems, which is consistent with the previous discussion. For the RuCu@N6C system, the electronic contribution of the Cu atom to the intermediates increases gradually during the steps 1–6 (*N2H4–*N2H3–*N2H2–*N2H–*N2), indicating that the inclusion of the Cu atom as the TM to coordinate with Ru significantly improved the adsorption of the intermediates. For the RuCo@N6C system, the harmonious coordination of Co and Ru atoms leads to an increase in the electron transfer number of the Ru–Co pair compared with the Ru2@N6C system, resulting in the activation of the intermediates, thus the energy barrier can be efficiently decreased.
3.5. Origin of the enhanced HzOR activity of RuCo@N6C and RuCu@N6C
The CDD diagrams of N2H3 on RuCo@N6C, RuCu@N6C and Ru2@N6C are shown in Fig. 5a–c. Pronounced charge redistribution between Co, Cu and Ru and N2H3 indicates a strong orbital interaction between Ru–M metal pairs and N2H3. To evaluate the N2H3 adsorption stability, we also calculate the COHP, further demonstrating the bonding strength between N of N2H3 and the metal d orbital. As shown in Fig. 5d–f, for RuCo@N6C, RuCu@N6C and Ru2@N6C, significant orbital overlap was observed among Co-3d, Cu-3d, Ru-4d and N-2p, further indicating a strong interaction between the intermediates and active sites. The values of ICOHP are −2.77, −2.25 and −2.57 eV for RuCo@N6C, RuCu@N6C and Ru2@N6C, respectively (Tables S12–S14, SI). The RuCu@N6C system has a more negative ICOHP value, representing a more stable adsorption configuration, thus the next dehydrogenation step of N2H3 can be accelerated. The electron localization function (ELF) was analyzed to further prove the bonding characteristics. The results showed active interactions between the active sites (Ru and TM) and the N of N2H3. Charge transfer predominantly occurred from Ru or TM atoms to the nitrogen atoms, forming the stronger bonds (Ru–N or TM–N), which enhanced the stability of the adsorption of N2H3, shown in Fig. 5-i and Fig. S21–S23 (SI). This illustrates that the TM atom (moderate active site) of RuCo@N6C and RuCu@N6C can more effectively adjust the adsorption of the key intermediate (N2H3) than that of Ru2@N6C, resulting in the enhancement of the HzOR activity.
 |
| | Fig. 5 The charge density difference of (a) RuCo@N6C, (b) RuCu@N6C and (c) Ru2@N6C, respectively. The COHP of TM–N (between N of N2H3 and the metal d orbital) on (d) RuCo@N6C, (e) RuCu@N6C and (f) Ru2@N6C, respectively. The electron localization function (ELF) diagrams of (g) RuCo@N6C, (h) RuCu@N6C and (i) Ru2@N6C, respectively. | |
3.6. Electrochemical stability of RuCo@N6C and RuCu@N6C
To further confirm the thermodynamic stability of RuCo@N6C and RuCu@N6C, AIMD simulations were performed and the results are shown in Fig. 6a and b, respectively. RuCo@N6C and RuCu@N6C maintained their complete structures during the simulations at 300 K for 6 ps. In addition, the energies of RuCo@N6C and RuCu@N6C remained relatively stable, indicating their good thermodynamic stability. The excellent thermal stabilities of RuCo@N6C and RuCu@N6C suggest that they could serve as durable electrocatalysts for the HzOR.
 |
| | Fig. 6 Temperature and energy evolution during the AIMD simulation of (a) RuCo@N6C and (b) RuCu@N6C, respectively. Insets show the top and side views of the snapshots after 6 ps simulation. | |
4. Conclusions
In summary, we systematically investigated Ru-coordinated 3d–5d TM atoms, anchored on the nitrogen-doped graphene DACs for the HzOR. RuCo@N6C and RuCu@N6C showed significantly enhanced catalytic performance due to their good stability, moderate adsorption ability, and significant electron transfer for N2H4. Through the cooperative interaction of TM atoms with Ru, RuCo@N6C and RuCu@N6C effectively modulate the adsorption of the key *N2H3 intermediate, and exhibit the limiting potentials of −0.13 and 0.00 V, which are much lower than that of Ru2@N6C (−0.41 V). The excellent thermal stability of RuCo@N6C and RuCu@N6C was further verified through AIMD simulations. These findings underscore the potential of DACs containing Ru coordinated with a moderate TM atom and are expected to inspire further experimental and theoretical research in this area.
Conflicts of interest
The authors declare no conflicts of interest.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: the optimized structure and related data are shown in SI. See DOI: https://doi.org/10.1039/d5cp03046c.
Acknowledgements
The authors appreciate the support from the National Natural Science Foundation of China (No. 52371161, 52371232, 52271152).
References
- T. Sakamoto, A. Serov, T. Masuda, M. Kamakura, K. Yoshimoto, T. Omata, H. Kishi, S. Yamaguchi, A. Hori, Y. Horiuchi, T. Terada, K. Artyushkova, P. Atanassov and H. Tanaka, Highly durable direct hydrazine hydrate anion exchange membrane fuel cell, J. Power Sources, 2018, 375, 291–299 CrossRef CAS
 .
.
- K. Jiang, Z. Liu, Y. Lu, M. Wang, D. Chen, L. Cai, T. Chan, P. Liu, A. Pan and Y. Tan, Rapid Melt-Quenching Enables General Synthesis of High-Loading Single-Atom Catalysts with Bicontinuous Nanoporous Structure, Adv. Mater., 2023, 35, 2207850 CrossRef CAS PubMed .
- L. Cai, H. Bai, C. Kao, K. Jiang, H. Pan, Y. Lu and Y. Tan, Platinum–Ruthenium Dual-Atomic Sites Dispersed in Nanoporous Ni0.85 Se Enabling Ampere-Level Current Density Hydrogen Production, Small, 2024, 20, 2311178 CrossRef CAS PubMed .
- K. Jiang, Z. Liu, Z. Wang, F. Xie, X. Yuan and Y. Tan, Manipulating Interfacial Water Via Metallic Pt1 Co6 Sites on Self-Adaptive Metal Phosphides to Enhance Water Electrolysis, Adv. Mater., 2025, 37, 2419644 CrossRef CAS PubMed .
- J. Peng, Z. Wang, K. Jiang, M. Peng, N. Palaniyandy, J. Ren and Y. Tan, Phosphorus dopants triggered single-atom platinum catalysis for efficient hydrogen evolution in proton exchange membrane electrolyzers, J. Mater. Chem. A, 2024, 12, 17395–17403 RSC .
- C. Mo, J. Jian, J. Li, Z. Fang, Z. Zhao, Z. Yuan, M. Yang, Y. Zhang, L. Dai and D. Yu, Boosting water oxidation on metal-free carbon nanotubes via directional interfacial charge-transfer induced by an adsorbed polyelectrolyte, Energy Environ. Sci., 2018, 11, 3334–3341 RSC .
- K. Zeng and D. Zhang, Recent progress in alkaline water electrolysis for hydrogen production and applications, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS .
- C. Zhang, W. Yuan, Q. Wang, X. Peng, X. Liu and J. Luo, Single Cu Atoms as Catalysts for Efficient Hydrazine Oxidation Reaction, ChemNanoMat, 2020, 6, 1474–1478 CrossRef CAS .
- J. Zhang, Y. Wang, C. Yang, S. Chen, Z. Li, Y. Cheng, H. Wang, Y. Xiang, S. Lu and S. Wang, Elucidating the electro-catalytic oxidation of hydrazine over carbon nanotube-based transition metal single atom catalysts, Nano Res., 2021, 14, 4650–4657 CrossRef CAS .
- S. Zhou, Y. Zhao, R. Shi, Y. Wang, A. Ashok, F. Héraly, T. Zhang and J. Yuan, Vacancy-Rich MXene-Immobilized Ni Single Atoms as a High-Performance Electrocatalyst for the Hydrazine Oxidation Reaction, Adv. Mater., 2022, 34, 2204388 CrossRef CAS PubMed .
- R. G. Kadam, T. Zhang, D. Zaoralová, M. Medveď, A. Bakandritsos, O. Tomanec, M. Petr, J. Zhu Chen, J. T. Miller, M. Otyepka, R. Zbořil, T. Asefa and M. B. Gawande, Single Co-Atoms as Electrocatalysts for Efficient Hydrazine Oxidation Reaction, Small, 2021, 17, 2006477 CrossRef CAS PubMed .
- F. Luo, S. Pan, Y. Xie, C. Li, Y. Yu, H. Bao and Z. Yang, Hydrazine-Assisted Acidic Water Splitting Driven by Iridium Single Atoms, Adv. Sci., 2023, 10, 2305058 CrossRef CAS PubMed .
- D. Jiao, Y. Tian, H. Wang, Q. Cai and J. Zhao, Single transition metal atoms anchored on a C2 N monolayer
as efficient catalysts for hydrazine electrooxidation, Phys. Chem. Chem. Phys., 2020, 22, 16691–16700 RSC .
- J. Xu, S. Lai, D. Qi, M. Hu, X. Peng, Y. Liu, W. Liu, G. Hu, H. Xu, F. Li, C. Li, J. He, L. Zhuo, J. Sun, Y. Qiu, S. Zhang, J. Luo and X. Liu, Atomic Fe–Zn dual-metal sites for high-efficiency pH-universal oxygen reduction catalysis, Nano Res., 2021, 14, 1374–1381 CrossRef CAS .
- D. Yu, Y. Ma, F. Hu, C. Lin, L. Li, H. Chen, X. Han and S. Peng, Dual-Sites Coordination Engineering of Single Atom Catalysts for Flexible Metal–Air Batteries, Adv. Energy Mater., 2021, 11, 2101242 CrossRef CAS .
- Z. Wang, X. Jin, C. Zhu, Y. Liu, H. Tan, R. Ku, Y. Zhang, L. Zhou, Z. Liu, S. Hwang and H. J. Fan, Atomically Dispersed Co2–N6 and Fe–N4 Costructures Boost Oxygen Reduction Reaction in Both Alkaline and Acidic Media, Adv. Mater., 2021, 33, 2104718 CrossRef CAS PubMed .
- F.-Y. Ma, P. Huang, J. Zhou, H.-W. Zeng, J.-W. Zhang, H. Zhao, Y.-M. Dong, Y.-F. Zhu and Y. Wang, In situ revealing C–C coupling behavior for CO2 electroreduction on tensile strain Ptδ+–Cuδ+ dual sites, Rare Met., 2024, 43, 6436–6446 CrossRef CAS .
- Q. Li, L.-G. Wang and J.-B. Wu, Recent advances in dual-atom catalysts for energy catalysis, Rare Met., 2025, 44, 841–867 CrossRef CAS .
- H. Li, Z. Zhao, Q. Cai, L. Yin and J. Zhao, Nitrogen electroreduction performance of transition metal dimers embedded into N-doped graphene: a theoretical prediction, J. Mater. Chem. A, 2020, 8, 4533–4543 RSC .
- X. Zhang, Y. Li, M. Jiang, J. Wei, X. Ding, C. Zhu, H. He, H. Lai and J. Shi, Engineering the coordination environment in atomic Fe/Ni dual-sites for efficient oxygen electrocatalysis in Zn–air and Mg–air batteries, Chem. Eng. J., 2021, 426, 130758 CrossRef CAS .
- Y. He, X. Yang, Y. Li, L. Liu, S. Guo, C. Shu, F. Liu, Y. Liu, Q. Tan and G. Wu, Atomically Dispersed Fe–Co Dual Metal Sites as Bifunctional Oxygen Electrocatalysts for Rechargeable and Flexible Zn–Air Batteries, ACS Catal., 2022, 12, 1216–1227 CrossRef CAS .
- Z. Zhu, M. Chen, M. Sun, J. Wang, Y. Zhou, X. Li and H. Tao, Mixture screening strategy of efficient transition metal heteronuclear dual-atom electrocatalysts toward nitrogen fixation, Phys. Chem. Chem. Phys., 2022, 24, 26776–26784 RSC .
- L. Zhang, G. Fan, W. Xu, M. Yu, L. Wang, Z. Yan and F. Cheng, Isolated diatomic Zn–Fe in N-doped carbon for electrocatalytic nitrogen reduction to ammonia, Chem. Commun., 2020, 56, 11957–11960 RSC .
- Y. Chen, J. Lin, Q. Pan, X. Liu, T. Ma and X. Wang, Inter-Metal Interaction of Dual-Atom Catalysts in Heterogeneous Catalysis, Angew. Chem., Int. Ed., 2023, 62, e202306469 CrossRef CAS PubMed .
- L. Zhang, J. Yao, J. Zhang, W. He, Y. Li, L. Liang, C. Liu, H. Liu and Q. Hao, Engineering Co and Ru dual-metal atoms on nitrogen-doped carbon as highly efficient bifunctional oxygen electrocatalysts, Catal. Sci. Technol., 2022, 12, 5435–5441 RSC .
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS .
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed .
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS .
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS .
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed .
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed .
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, Crystal Orbital Hamilton Population (COHP) Analysis As Projected from Plane-Wave Basis Sets, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef CAS PubMed .
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef .
- G. J. Martyna, M. L. Klein and M. Tuckerman, Nosé–Hoover chains: The canonical ensemble via continuous dynamics, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef .
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef PubMed .
- C. N. Sun, Z. L. Wang, X. Lang, Z. Wen and Q. Jiang, Synergistic Effect of Active Sites of Double-Atom Catalysts for Nitrogen Reduction Reaction, ChemSusChem, 2021, 14, 4593–4600 CrossRef CAS PubMed .
- G. Blyholder, Molecular Orbital View of Chemisorbed Carbon Monoxide, J. Phys. Chem., 1964, 68, 2772–2777 CrossRef CAS .
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed .
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 abc,
Hui
Jiang
abc,
Hui
Jiang





