Unveiling the effect of choline chloride on hydrophobic association of methane†
Received
21st May 2025
, Accepted 4th August 2025
First published on 5th August 2025
Abstract
Elucidating how osmolytes influence hydrophobic interactions in small nonpolar solutes helps to explain their role in biology. This is crucial for understanding protein stability, enzyme–substrate binding, and membrane self-assembly. Motivated by this, we employ equilibrium simulations and umbrella sampling to explore the molecular basis of the osmolyte-induced association of methane molecules. Our study reveals a remarkable enhancement in methane association driven by choline chloride (ChCl). Radial distribution functions (RDFs) and solvent accessible surface area show increased methane aggregation with the addition of ChCl compared to pure water. Detailed cluster analysis highlights the formation of larger and more compact methane clusters at higher ChCl concentrations. The addition of ChCl induces dewetting of the methane surface, enhancing methane association, as confirmed by preferential interaction parameter calculations for water. Simultaneously, choline molecules cluster around methane, creating a hydrophobic environment that further promotes association. Our microscopic structural investigation shows that the addition of ChCl disrupts the water hydrogen-bond network around methane. Hydrogen bond analysis reveals an increase in choline–choline bonds and a decrease in both water–water and choline–water hydrogen bonds near methane. PMF and association constants obtained from umbrella sampling further confirm the enhanced hydrophobic association between methane molecules in the presence of ChCl compared to pure water. In summary, our findings highlight the crucial role of osmolytes in stabilizing hydrophobic interactions among nonpolar solutes, which has broad implications for various biological processes.
1 Introduction
Hydrophobic interactions1–4 are key driving forces in many biomolecular processes. These include protein folding, enzyme–substrate binding, and membrane–protein self-assembly.5–7 These interactions are determined by a delicate balance of direct and water-mediated interactions between nonpolar solutes, and their strength depends on solute size and nature of the attractive intermolecular interactions.5,8,9 For example, many phenomena related to the hydrophobic effect are observed due to expulsion of nonpolar solutes from aqueous environments due to the attraction between water molecules being greater than the attraction of solute–water. The hydrophobic effect can be broadly categorized into two aspects: hydrophobic interaction and hydrophobic hydration. Hydrophobic interaction refers to the direct interaction between nonpolar solutes in an aqueous environment, while hydrophobic hydration describes the indirect effects involving microscopic structural and dynamical changes in water during the dissolution of nonpolar solutes. The immediate consequence of the hydrophobic effect is the minimization of the solvent-exposed surface area of nonpolar solute leading to self-aggregation or folding into compact states.4,9 Water is an integral component of the cellular environment and plays a vital role in regulating numerous biological processes.10–13 The structure of water near nonpolar solutes significantly influences the extent of water-mediated interactions between them. These water-structure changes near nonpolar solutes are strongly dependent on solute size. For small hydrophobic solutes, water molecules can accommodate them within cavities formed by the hydrogen-bond network, resulting in minimal or no loss of hydrogen bonds.14 In contrast, larger hydrophobic solutes cannot fit into the cavities formed by the hydrogen bond network of surrounding water. This leads to the breaking of hydrogen bonds near the solute.3,15 A crossover between these two regimes occurs at a solute size of approximately 1 nm, as reported by Chandler.3
Several studies have shown that hydrophobic interactions among biomolecular components during protein folding, enzyme–substrate binding, and bilayer self-assembly are strongly influenced by environmental conditions.16–19 For instance, proteins tend to unfold under harsh conditions like high temperature,20 high salt concentrations,17 or high pressure.16 Chemical environments also play a critical role; for example, proteins unfold in the presence of 8 M urea.21 Conversely, a range of small organic molecules known as protecting osmolytes help to stabilize proteins in their folded state under stress. Examples include sugars (e.g., trehalose22), methylamines (e.g., TMAO, ChCl23,24), and amino acids (e.g., glycine betaine25). Among these, ChCl, a methylamine-class osmolyte, is widely used to protect protein structure under stress and has gained attention for its diverse applications in the chemical and food industries.26–32 The presence of such osmolytes helps maintain the delicate balance between hydrophobic interactions and the folding–unfolding equilibrium of proteins.16–19,33 However, the molecular mechanisms by which osmolytes stabilize the folded state is not fully understood. Due to the complexity of larger biomolecules, small nonpolar solutes serve as effective models to study osmolyte-induced effects on hydrophobic interactions at the fundamental level. Methane, a simple nonpolar solute, is often used to mimic hydrophobic residues34 within proteins and thus reduces the complexity of the system. Hence, in this study, we focus on methane molecules in the presence of ChCl to investigate how protecting osmolytes influence hydrophobic interactions, an insight critical to understanding broader biological phenomena. While experimental techniques such as IR spectroscopy, neutron scattering, and NMR have been used to study hydrophobic effects and the influence of osmolyte, they often lack atomic-level resolution. In this context, all-atom molecular dynamics simulations provide a powerful computational approach to uncover the underlying microscopic mechanisms.
The structural and thermodynamic behavior of hydrophobic surfaces ranging from small nonpolar solutes to larger macromolecules in water and osmolyte mixtures have been extensively studied.35–37 For example, it has been shown that while urea stabilizes the association of methane molecules, it destabilizes the association of larger nonpolar solutes.35–37 Multiple studies have attempted to quantify the strength of hydrophobic interactions by comparing the association or dissociation tendencies between pairs of nonpolar solutes or hydrophobic polymers.34,38,39 Further investigations into the effects of urea and TMAO using both all-atom and united-atom descriptions of methane have revealed that urea enhances hydrophobic association, whereas TMAO has a negligible effect.34 Other works have reported that the addition of urea promotes methane clustering due to the preferential binding of methane with water.40–42 Garde et al. demonstrated that TMAO does not alter hydrophobic interactions between methane molecules because the enthalpic and entropic contributions nearly compensate each other. They found that contact minima between two methanes are entropically favored, while solvent-separated minima are stabilized by enthalpy.43 Beyond TMAO, other osmolytes such as trehalose and glycine betaine have also been explored.44,45 For instance, Dixit et al. studied the hydrophobic association and solvation of methane in aqueous urea and glycine betaine solutions and found that both osmolytes enhance hydrophobic association, primarily due to favorable entropic contributions.44 A related study explored the effects of urea and taurine on the hydrophobic association and solvation of methane and neopentane, revealing that methane association is entropy-driven, while for neopentane, both enthalpy and entropy contribute favorably.46 In contrast, Paul et al. used a simulation approach to report that the addition of trehalose reduces hydrophobic interactions among neopentane molecules.45 The effect of increasing pressure on the hydration structure of neopentane molecules has shown a reduction in hydrophobic association but stabilization of the solvent-separated minimum.47 Moreover, Sharma et al. studied caffeine self-association in water from 275 K to 350 K and observed a reduction in association with increasing temperature.48 Additionally, the introduction of ChCl has been found to disrupt the tetrahedral structure and orientation of water around methane.49 From a dynamical perspective, ChCl appears to push water molecules toward methane, forming water cages that hinder the translational motion of methane.50 A study by Ramachandran et al. investigated the impact of amino acids on methane hydrate formation. They observed that at lower concentrations, phenylalanine and tryptophan promote the nucleation and growth of methane hydrates, while higher concentrations of these amino acids inhibit these processes.51 In terms of hydrophobic effect, it has been reported that hydrophobic polymers adopt more compact conformations in ChCl solutions compared to pure water.52 Other studies have also reported the influence of ChCl-based deep eutectic solvents on the self-assembly behavior of ionic surfactants and amphiphilic star-block copolymers.53–55 Similarly, Mondal et al. showed the stabilization of collapsed conformations in model Lennard-Jones and synthetic polystyrene polymers through both simulations and experiments.56,57 Nayar et al. found that low concentrations of TMAO (less than 2 M) can stabilize the collapsed state of hydrophobic polymers.58 Most of the above-mentioned works reported the role of various osmolytes on the hydrophobic interactions considering nonpolar solutes of different length scales. Because of the different combinations of the osmolytes present in the biological systems, understanding how the hydrophobic interactions are influencing in the presence of all these osmolytes is necessary. Although osmolytes like TMAO, glycine betaine, and trehalose have been widely studied for their effects on the hydrophobic interaction of simple nonpolar solutes, we are not aware of any systematic investigation addressing the effect of ChCl on hydrophobic interactions. This gap motivates our study, where we use molecular dynamics simulations to explore the influence of varying concentrations of ChCl on the association behavior of methane molecules.
In the present work, we investigate the role of ChCl in hydrophobic interactions between methane molecules using molecular dynamics simulations. Our primary focus is to provide a structural and molecular-level understanding of the hydrophobic effect in the presence of this protecting osmolyte. Our findings demonstrate that the hydrophobic association between methane molecules is enhanced with the progressive addition of ChCl. The aggregation of choline molecules contributes to creating a more hydrophobic environment, thereby promoting stronger associations among methane molecules. Furthermore, our study reveals detailed molecular insights into the interactions among methane, water, and ChCl that drive this enhanced association. These observations are further supported by free energy analyses.
The manuscript is organized as follows: the next section describes the simulation methodology. The results and discussion section is divided into four subsections. The first subsection deals with the initial signature of the osmolyte-driven hydrophobic association of methane. The subsequent subsections outline the evidence of methane clustering, a molecular-level picture of the arrangement of solvent and cosolvent around methane, and free energy analysis of methane association. A summary of our findings from the current study is provided in the final section.
2 Methods
2.1 System and force field
We consider an all-atom model of 100 methane molecules placed randomly in a cubic simulation box with periodic boundary conditions employed in all three directions. The dimensions and space between methane molecules and the edge of the simulation box are chosen carefully to avoid self-interactions across the periodic images. Then we add the required number of choline, chloride ions, and water to generate the four different systems that we consider in this study. The initial configuration of all considered systems is generated using Packmol software.59 We simulate the following four systems namely, S0, S1, S2, and S3. S0: 100 methane molecules immersed in 2000 water molecules, S1: 100 methane molecules in a mixture of 100 ChCl and 2000 water molecules, S2: 100 methane molecules in a mixture of 250 ChCl and 2000 water molecules, and S3: 100 methane molecules in a mixture of 500 ChCl and 2000 water molecules. Hydrophobic moieties have the intrinsic characteristic of avoiding contact between their exposed surfaces and aqueous environments. To explicitly discard the contribution of this intrinsic behavior in aqueous environments, we kept the number of water molecules constant in all the systems. Hence, the box dimensions are not the same for all the systems and range from ∼4 to ∼5.5 nm. Additionally, we simulate neat water to investigate the role of methane and ChCl in the structural rearrangement of water. A snapshot of the simulation box showing a representative configuration of the S1 system and structure of methane, choline chloride, and water molecules is provided in Fig. 1a and b. The OPLS-AA60 force field is used to define topological and interaction parameters for methane and ChCl. The OPLS-AA force field is intended for use in liquid phase simulations and it has been extensively used for studying nonpolar solutes in solvent–cosolvent mixtures.44,61 We use the SPC/E water model, which is one of the successful models to reproduce the structural and dynamical properties of bulk water.62 Earlier, we validated this combination of OPLS-AA force field and SPC/E water model by computing diffusion coefficients and free energy of solvation values for methane in bulk water and compared them with experimentally reported values.49 Our observations are in good agreement with the experimental data.63,64 We performed two replicas of the simulations for all systems and provided the analyses of the generated output in the present manuscript. To confirm the convergence of the two replicas of the simulated trajectories we compute the kinetic, potential, and total energy of the system as well as the density of the system and the results are shown in Fig. S1 and S2 of the Supplementary Information (SI).
 |
| | Fig. 1 (a) A representative configuration of a system containing methane molecules (blue color) in water (red color), choline (violet color), and chloride (green color). Methane is shown in VDW representation, ChCl in stick representation, and water in solvent representation for visualization purposes. (b) Structures of methane, choline chloride, and water in ball and stick representation. | |
2.2 Equilibrium simulations
We performed equilibrium classical all-atom molecular dynamics simulations of the abovementioned systems using GROMACS65 version 2021. The initial configuration of each system is initially subjected to the 5000 steps of energy minimization using the steepest descent algorithm.66 This energy-minimized configuration is further equilibrated under NVT ensemble for 1 ns using a velocity rescale thermostat.67 Subsequently, NPT equilibration is carried out for 5 ns using a Berendsen barostat.68 This will achieve a steady temperature of 300 K and pressure of 1 atm for all systems. The final configuration obtained from NPT equilibration is used as a starting structure for the production run of 500 ns in the NPT ensemble. The equation of motion is solved with an integration time step of 2 fs and trajectories are recorded at every 10 ps interval. All bonds are constrained to their equilibrium bond length using the LINCS algorithm.69 All the water molecules were simulated as rigid using the SETTLE algorithm.70 We compute electrostatic interactions using the particle mesh Ewald method71 with a cutoff of 1.0 nm and grid spacing of 0.12 nm. Van der Waals interactions are treated using the minimum image convention72 with a spherical cutoff of 1.0 nm. To study the role of osmolytes on hydrophobic interactions among nonpolar solutes, in-built modules of GROMACS65-2021, some in-house scripts and codes are used. For visualization, VMD 1.9.373 and OVITO74 are used.
2.3 Umbrella sampling simulations
The free energy landscape for methane association or dissociation along the reaction coordinate in varying concentrations of ChCl is obtained by performing umbrella sampling simulations via PLUMED75 patched with GROMACS. We choose the distance between the center of mass (COM) of methane molecules as a reaction coordinate. The four systems are composed of only two methane molecules in each system. The number of ChCl and water molecules is the same as that used for equilibrium simulations to compare the results from both unbiased and biased simulations. We generate a total of 25 windows for COM distances ranging from 0.3 to 1.5 nm at a spacing of 0.05 nm between adjacent windows. A harmonic potential with a force constant of 836.8 kJ mol−1 nm−2 is used to harmonically restrain the COM distance to the desired value between two methane molecules. In each window, we first perform 2 ns equilibration under the NPT ensemble using a Nosé–Hoover thermostat76 and Parrinello-Rahman barostat77 to maintain the temperature and pressure throughout at constant values of 300 K and 1 atm, respectively. Furthermore, the production run of 10 ns is carried out in each window under the NPT ensemble. Finally, the weighted histogram analysis method (WHAM)78 is used to calculate the PMF. There will be an entropic contribution to the PMF due to the rotation of the nonpolar solutes. This entropic correction is done by adding the term 2kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(r) to the PMF obtained from WHAM, where r represents the COM distance between the methane molecules. Error bars in the PMF are computed using the block analysis method. The resulting PMF is scaled to zero at larger COM distances.
ln(r) to the PMF obtained from WHAM, where r represents the COM distance between the methane molecules. Error bars in the PMF are computed using the block analysis method. The resulting PMF is scaled to zero at larger COM distances.
3 Results and discussion
First, we qualitatively investigate the effect of increasing the concentration of ChCl on the hydrophobic interactions of methane. A reasonable approach is to investigate the distribution or arrangement of methane molecules with the addition of ChCl. For this purpose, we compute the RDFs and solvent-accessible surface area (SASA) of methane in the following sections. Moreover, cluster analysis will provide an overall idea of the influence of ChCl on the hydrophobic association of methane. The underlying microscopic arrangement of water and choline surrounding methane is confirmed by looking into their local structure around methane in terms of RDFs and cumulative coordination numbers (CNs), as outlined in the subsequent sections.
3.1 Qualitative picture of osmolyte-driven methane association
To gain a molecular-level understanding of the effect of varying concentrations of ChCl on hydrophobic interactions of methane, we calculate RDFs based on the COM of methane. Fig. 2a represents methane–methane RDFs involving their COM for all systems considered for this study. The first peak of the methane–methane RDF appears at ∼0.39 nm and the second peak is found at ∼0.7 nm. Similar peak positions for methane–methane RDFs in water and aqueous solutions of glycine betaine or taurine were reported earlier.44,46 These peak positions remain unaltered for all four systems, S0 to S3. It is interesting to note that the first peak of RDF becomes more intense while progressing from S0 to S3 showing the initial signature of the aggregation of methane molecules with increasing concentration of ChCl. For example, the peak height for the methane–methane RDF in the S0 system is ∼9.05, and this peak height gradually increases from ∼13.7 to ∼31.3 for systems S1 to S3. Furthermore, we also compute the pair potential of mean force (PMF) from the methane–methane RDF using the following equation,| | | W(r) = −kBTlngMe–Me(r) | (1) |
where kB is the Boltzmann constant, T is the temperature (300 K) and gMe–Me(r) is the RDF based on the COM of methane molecules. The relative trend in the PMF between methane molecules for all systems studied is shown in Fig. S3 and Table S1 of the SI. The first minimum in the PMF represents the contact minimum (CM) hence, hydrophobic association, and the subsequent minimum is the solvent-separated minimum (SSM) i.e., hydrophobic solvation of methane molecules depending on the concentration of ChCl. Fig. S3 illustrates that the population of CM is significantly larger than that of the SSM, indicating the stronger hydrophobic association between methane molecules compared with its hydrophobic solvation. With increasing ChCl concentration from system S0 to S3, the depth of the contact minimum (CM) increases from ∼−5.51 to ∼−8.61 kJ mol−1 (Table S1), while the position of the CM remains constant at ∼0.39 nm across all systems, indicating enhanced hydrophobic association of methane in the presence of ChCl. This appearance of the CM at ∼0.39 nm is previously reported for methane in different cosolvent mixtures.44,46
 |
| | Fig. 2 (a) RDFs of methane around methane calculated from the COM of methane molecules. (b) Probability distribution of solvent accessible surface area, P(SASA) of methane for all the systems studied. | |
Next, to confirm the enhancement in methane association with the addition of ChCl, we calculate the SASA of methane and present its probability distribution in Fig. 2b. SASA describes the extent of the surface area over which contact between solute and solvent can occur. It can be seen from Fig. 2b that the SASA of all methane molecules in systems S0 to S3 extends from ∼40 to ∼75 nm2. The peak of the SASA distribution shifts to lower values with increasing ChCl concentration from S0 to S3. For instance, the peak of P(SASA) for S0 is at ∼59 nm2 and it is shifted to ∼55 nm2, ∼50 nm2, and ∼47 nm2 for systems S1 to S3. The appearance of a small peak for P(SASA) at slightly higher values in the S3 system is the consequence of the presence of some free methane molecules outside the cluster whose surface area is exposed to the surrounding solvent molecules. This decrease in SASA values gives evidence of the enhanced aggregation tendency of methane molecules in the presence of ChCl (S1–S3) compared to pure water (S0). The average SASA per methane calculated from the two replicas of the simulations shows a qualitatively similar trend of enhanced aggregation. The average SASA per methane from two replicas of the simulations and their uncertainty is tabulated in Table 1. The values obtained from the two replicas of the simulations are in close agreement with each other, with very minimal uncertainties.
Table 1 Average SASA per methane molecule from two replicas of the simulations for all different systems considered. All values are in nm2. Errors corresponding to the standard deviation are also given in the last column
| System |
Average SASA per methane (replica-1) |
Average SASA per methane (replica-2) |
Uncertainty |
| S0 |
0.59 |
0.60 |
0.00707 |
| S1 |
0.55 |
0.53 |
0.01414 |
| S2 |
0.50 |
0.52 |
0.01414 |
| S3 |
0.48 |
0.49 |
0.00707 |
The analyses of the RDFs and SASA reveal the enhanced aggregation of methane with the progressive addition of ChCl. These findings provide initial evidence for the stronger hydrophobic association of methane in ChCl compared than in water. The next section describes the detailed cluster analysis performed to determine the extent of the hydrophobic association between methane molecules depending on the concentration of ChCl.
3.2 Cluster analysis
Cluster analysis is a useful tool as it provides quantitative insight into how the hydrophobic association of methane changes with changes in the concentration of ChCl. We find that the methane molecules are coming closer to each other with increasing ChCl concentrations from RDFs and SASA as discussed above in Section 3.1. We estimate the clusters of the methane in different systems using the clustering algorithm: we considered an aggregate of methane molecules to be a cluster if one methane molecule is within 0.6 nm distance of a neighboring methane molecule, as shown in Fig. 3a. This distance cutoff is taken from the first minima of the methane–methane RDF. For all four systems, we estimate the probability distribution of the number (NCluster) and average size (SCluster) of the cluster which is represented as violin plots in Fig. 3b and c. This calculation is performed for the last 100 ns of the trajectories for each of the systems. As shown in Fig. 3b, the formation of higher-order methane clusters in all systems is quite evident owing to the presence of an aqueous environment. However, the extent of the clustering is different depending on the concentration of ChCl. For the S0 system, the violin plot shows a broad vertical spread across the entire range of NCluster values, indicating that the formation of varying numbers of clusters is probable. A slightly higher probability is observed around 15–17 clusters, as reflected by the increased width of the violin plot in this region. This vertical spread gradually narrows, and the width of the violin plot becomes increasingly concentrated around lower cluster numbers from S1 to S3. This trend indicates a reduction in the number of clusters with increasing ChCl concentration. Notably, the S3 system exhibits two distinct subpopulations, evident from the bimodal nature of its distribution. This can be attributed to the presence of other methane molecules forming multiple smaller clusters. This observation is consistent with the appearance of an additional small peak at slightly higher SASA values in the P(SASA) distribution for the S3 system (Section 3.1). The narrowing width of the violin plot around lower NCluster values suggests that fewer but more stable clusters are formed at higher ChCl concentrations. Fig. 3c presents the probability distribution of the average cluster size for systems S0–S3. The average size of a cluster is defined by the number of methane molecules it contains. The violin plots corresponding to SCluster reveal three distinct modes, indicating the presence of three subpopulations of cluster sizes for each system. However, the width of each mode varies across the four systems. For systems S0 and S1, the two lower subpopulations exhibit broader distributions compared to those in S2 and S3, suggesting that smaller average cluster sizes are more probable in pure water (S0) and at lower ChCl concentrations (S1). As we transition to S2 and S3, the widths of these lower subpopulations decrease, while the upper subpopulation becomes broader. This indicates that the formation of clusters containing 80–85 methane molecules becomes more frequent in S2 and S3, due to the tendency to form larger clusters at higher ChCl concentrations. Furthermore, these larger clusters appear to be more stable, as evidenced by reduced fluctuations and a more consistent trend in the time evolution of NCluster and SLargestcluster, as shown in Fig. S4 and S5 of the SI. Overall, the cluster analysis provides both qualitative and quantitative insights into the formation of fewer, larger, and more stable clusters with increasing ChCl concentration.
 |
| | Fig. 3 (a) A clustering algorithm identifying methane molecules as part of the same cluster if they are within 0.6 nm of a neighboring methane molecule. (b) A violin plot showing the distribution of cluster number and size, NCluster and SCluster respectively, for all the systems studied. The white dot at the middle of each distribution represents the mean number and size of the clusters. | |
As discussed earlier, the formation of higher-order clusters is evident across all systems. To further substantiate this observation, we calculated the size of the largest clusters and presented the corresponding histograms in Fig. 4a. The size of the largest cluster is defined by the average number of methane molecules in the largest cluster. As shown in Fig. 4a, the S0 system exhibits frequent occurrences of clusters comprising approximately 80 methane molecules. There is an increase in the occurrence of larger cluster events from S1 to S3.
 |
| | Fig. 4 (a) Histogram showing the size of the largest cluster. (b) Clustering degree, ϕ, of the methane molecules, and (c) Compaction degree, χ, of the clusters formed for all the systems studied. The errors corresponding to the standard deviation are also plotted with the data in the plot (b) and (c). | |
All the above discussions suggest that the clustering tendency of methane depends strongly on the surrounding aqueous environment. In addition, we would like to emphasize that the concentration of methane in aqueous solution also influences the degree of clustering. In our system, the methane concentration exceeds its experimental solubility, resulting in a supersaturated initial condition. As reported by Grabowska et al., higher concentrations of methane can lead to faster nucleation of methane hydrates.79 Therefore, it is reasonable to hypothesize that such a high methane concentration could promote spontaneous aggregation and potentially result in phase separation, where methane molecules aggregate into a single large cluster, separating from the aqueous phase. To examine whether high methane concentration under ambient conditions leads to phase separation over longer timescales, we extend the simulation of the S0 system from 500 ns to 1 μs. To assess the influence of methane concentration over this extended duration, we analyze the methane–methane RDFs based on their COM, the SASA of methane, NCluster, SCluster, and SLargestcluster. The detailed analysis and corresponding plots are provided in Fig. S6(a), (b) and S7(a)–(c) of the SI. We compare the results obtained from both the original 500 ns and extended 1 μs trajectories (Table S2 in SI). The observations from the extended 1 μs simulation are in close agreement with those from the 500 ns simulation. This clearly indicates that the initial supersaturated concentration of methane, under ambient temperature and pressure, does not solely lead to phase separation over a timescale of up to 1 μs. While extreme thermodynamic conditions, such as high pressure and low temperature, may facilitate phase separation when the initial concentration of a nonpolar solute is high. Our study focuses on hydrophobic association under ambient temperature and pressure therefore, we do not observe any indication of phase separation. It is important to note that the objective of our work is not to characterize the complete kinetic evolution of the system, but rather to compare the effect of choline chloride (ChCl) on methane aggregation under ambient conditions and within accessible simulation timescales. Several other studies have also explored systems with high initial concentrations of nonpolar solutes in aqueous solutions, including in the presence of osmolytes, to examine hydrophobic association. Their observations are consistent with our results and support our conclusions.44,51
We quantify the aggregation propensity of methane and stability of the clusters at all concentrations of ChCl by calculating the clustering degree (ϕ) and compaction degree (χ) as follows,
| |  | (2) |
| | 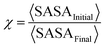 | (3) |
where 〈
NLargestcluster〉 is the average number of methane molecules in the largest cluster formed,
NTotal is the total number of methane molecules in each system, and 〈SASA
Initial〉 and 〈SASA
Final〉 refer to the average SASA of all methane molecules averaged over the initial 10 ns and final 10 ns of the total 500 ns trajectory, respectively. These quantities provide a quantitative framework to evaluate the clustering propensity, stability, and structural characteristics of the formed clusters. The increased values of both parameters,
ϕ and
χ, clearly reflect a higher clustering propensity.
Fig. 4b and c present the average clustering and compaction degrees obtained from three independent replicas of the trajectory for all studied systems. A detailed examination of
Fig. 4b reveals that the clustering degree,
ϕ, increases with increasing ChCl concentration. This indicates that the average number of methane molecules participating in the largest cluster increases with ChCl addition, which is consistent with the distribution of the largest cluster sizes shown in
Fig. 4a. Similarly, the compaction degree,
χ, also increases from system S0 to S3, reflecting a reduction in SASA for the final-state configurations due to enhanced clustering at higher ChCl concentrations. The higher values of
χ in systems containing ChCl further support the formation of tighter and more compact clusters. This observation is further supported by the snapshots of methane clusters extracted from the final simulation frames, as shown in
Fig. 5a–d. Visual inspection of these snapshots shows that in the S0 system, methane molecules are dispersed throughout the simulation box, indicating minimal clustering in pure water. In contrast, with increasing ChCl concentration from S1 to S3, the formation of larger and more compact clusters becomes increasingly evident, consistent with the trends observed in the cluster analysis. These compact clusters appear to be stable as indicated by the time evolution of the
NCluster and
SLargestcluster at higher ChCl concentrations, as shown in Fig. S4 and S5 of the SI. To determine the effect of simulation box size on the stability of clusters formed in each system, we considered additional simulations of both pure water (S0) and aqueous choline chloride (S3) systems, each constructed at three different system sizes while maintaining consistent methane-to-solvent ratios (Table S3). As presented in the SI (Fig. S8a–c and S9a–c), methane clustering in S0 systems is prominent in smaller boxes but is suppressed in the largest box, where methane remained uniformly dispersed. These observations are consistent with the conclusions drawn by Weijs
et al., where they mentioned larger box sizes or dilute conditions allow more diffusion of the molecules, hence preventing aggregation or nucleation.
80 In contrast, all S3 systems exhibited persistent clustering regardless of box size, indicating that choline chloride promotes aggregation even under more dilute or expanded conditions.
 |
| | Fig. 5 Representative snapshots of the (a) S0, (b) S1, (c) S2, and (d) S3 systems recovered from the final time frame of the simulation. Methane is shown in VDW representation, and carbon and hydrogen atoms of methane are shown in blue and white colors respectively. ChCl concentration is increased by adding the required number of ChCl while keeping the number of methane and water molecules conserved across all the systems. As a result, box dimensions are changing and each edge length of the box shown is in nm. Water and ChCl molecules present in the simulation box are not shown for visual clarity. Snapshots show the enhanced clustering and formation of the compact clusters at higher ChCl concentration. | |
In summary, the results from detailed cluster analyses reveal the formation of fewer but larger and more stable methane clusters with increasing ChCl concentration. These clusters become progressively more compact and stable, demonstrating that ChCl enhances the clustering tendency of methane and promotes hydrophobic association between methane molecules.
3.3 Molecular picture of the arrangement of water and choline around methane
3.3.1 Distribution of solvent and cosolvent around methane.
To gain insights into the microscopic arrangement of constituents surrounding the methane, we compute the RDFs of water oxygen (Ow), the COM of choline, and the hydrophobic part of choline (Cc) around methane COM as shown in Fig. 6a–d. The corresponding cumulative CNs of the above-mentioned constituents are calculated as,| | 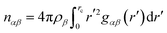 | (4) |
where nαβ is the number of atoms of type β around an atom of type α in a spherical shell extending from 0 to rc. ρβ and rc are the number density of β type atoms in the system and the distance of the first minimum in the corresponding RDFs respectively. The values of rc for water oxygen, choline, and hydrophobic part of choline are 0.55, 0.7, and 0.5 nm respectively. The cumulative CNs are plotted together with the RDFs and represented in Fig. 6a–d. The average first shell cumulative CNs of water oxygen, choline, and Cc obtained by integrating the respective RDFs up to the first minimum is tabulated in Table 2. As evident from Fig. 6a, choline is an unsymmetric molecule with a major hydrophobic domain abbreviated as Cc, comprised of three methyl carbon atoms (C1, C2, and C3) attached to the quaternary nitrogen atom. For computing RDFs between methane and water, we consider the carbon atom of methane and oxygen atom of water (Ow). Similarly, for methane–choline and methane–Cc RDFs, we consider the carbon atom of methane and COM of choline or methyl carbons of choline respectively. For all three RDFs, water, choline, and Cc around methane the first peak heights are less than 1, indicating a lower fraction of these constituents in the vicinity of methane compared to the bulk. The first peak of the methane–water RDF appears at ∼0.37 nm, suggesting that the first solvation shell of water around methane lies at this distance and extends up to ∼0.55 nm. This peak position remains unchanged across all four systems (Fig. 6b). We observe that the water density around methane is highest in the absence of ChCl (S0), as evident from the maximum peak height of the RDF (∼0.73), which decreases upon the gradual addition of ChCl (systems S1–S3). This reduction in peak height indicates the exclusion of water molecules from the methane vicinity, a phenomenon previously reported in studies exploring the hydrophobic association of nonpolar solutes in aqueous environments.48 This notable depletion of water density around methane correlates with the enhanced aggregation tendency of methane in ChCl-containing systems, as discussed earlier in Sections 3.1 and 3.2. The qualitative trend in the cumulative CNs is similar to that of the RDFs. With increasing ChCl concentration from S0 to S3, the cumulative coordination number (CN) of water around methane decreases, further supporting the dewetting of the methane surface due to increased hydrophobic association. The average first-shell cumulative CNs for water and choline are provided in Table 2. It is important to note that the ChCl concentration was increased while keeping the number of water molecules constant across all systems. This results in an increase in box size, as water molecules were not replaced to maintain ChCl concentration. Consequently, a slight reduction in water or choline's cumulative CNs is expected due to the increased box volume. To account for this, we normalize the cumulative CN values of water and choline with respect to system S0 (for water) and S1 (for choline). These normalized values, representing expected cumulative CNs by taking the reduced water and choline density into account, are shown in parentheses in Table 2. The increasing difference between actual and normalized (expected) cumulative CN values for water in the first solvation shell with ChCl concentration indicates a real depletion of water molecules from the vicinity of methane. In contrast, the relatively small difference in cumulative CNs suggests that the change in box size has minimal impact on the actual cumulative CN. The decreasing CNs (both cumulative and average first shell cumulative CNs) and RDF peak height of water from S0 to S3 further supports enhanced methane aggregation in ChCl-containing systems, as discussed previously in Sections 3.1 and 3.2. Similar dewetting of hydrophobic surfaces has also been reported for self-aggregation of nonpolar DTBM (di-t-butyl-methane) molecules in water.81 Examining the RDFs of choline around methane considering their COMs (Fig. 6c), the peak position remains fixed at ∼0.55 nm across all systems. Although choline's local density around methane is lower than its bulk concentration, its accumulation near methane increases with ChCl concentration. Comparing the RDFs of water and choline (Fig. 6b and c) shows that choline is more enriched near methane than water, as indicated by the higher peak intensities for RDFs of choline: ∼0.55, ∼0.71, and ∼0.83 for S1 to S3, respectively. This suggests that choline contributes to creating a hydrophobic environment around methane, facilitating clustering. To investigate this in more detail, we compute the RDF of Cc (the hydrophobic part of choline) around methane COM. Fig. 6d shows that the solvation shell for Cc is centered at ∼0.4 nm and the RDF peak heights increase from S1 to S3. This indicates that the hydrophobic moiety of choline preferentially orients toward methane, thereby enhancing the hydrophobic environment and promoting self-aggregation of methane. The same trend is observed for the cumulative CNs of both choline and Cc (Fig. 6c and d). Moreover, both actual and normalized first-shell cumulative CNs of choline and Cc increases with ChCl concentration, with the differences between them becoming more pronounced from S1 to S3. This suggests the accumulation of choline and its hydrophobic domain in the solvation shell of methane.
 |
| | Fig. 6 (a) A pictorial representation of the hydrophobic domain of choline with atoms labelled. RDFs of (b) water oxygen atoms (Ow), (c) choline around methane considering the COM of choline, and (d) the hydrophobic domain of choline (Cc) around the methane COM for all the systems studied. Cumulative CNs of the respective constituents obtained by integrating RDFs are also plotted in the same plots as dashed lines. | |
Table 2 Average first shell cumulative CNs for water (Ow), COM of choline, and the hydrophobic part of choline (Cc) around methane COM for all systems considered. The numbers in parentheses are described in the text
| System |
CN (water) |
CN (choline) |
CN (Cc) |
| S0 |
9.94 |
— |
— |
| S1 |
7.63 (7.67) |
0.55 |
0.37 |
| S2 |
5.32 (5.71) |
1.26 (1.03) |
0.89 (0.70) |
| S3 |
3.47 (3.94) |
1.98 (1.42) |
1.49 (0.96) |
Collectively, the analysis of water and choline RDFs around methane reveals two complementary behaviors: dewetting of the methane surface due to water exclusion and accumulation of choline in its vicinity. This is further supported by a decrease in the local density of water and an increase in the local density of choline as shown in Fig. S10 of the SI. Both phenomena play a crucial role in enhancing methane–methane aggregation in the presence of ChCl.
To confirm whether the arrangement of water and choline in the methane solvation shell as observed from their RDFs represents the actual underlying molecular picture, we compute the preferential interaction parameters between methane–water and methane–choline, relative to methane–methane interactions. These preferential interaction parameters, shown in Fig. 7a–c, quantify the tendency of methane to preferentially associate with itself over water or choline in the surrounding environment. The parameter is calculated as follows,
where, in the case of Γ
MeMe–W, the indices
i and
j represent methane and water, respectively, while for Γ
MeMe–Ch,
i and
j correspond to methane and choline. The term
ρi denotes the number density of methane in the given system. Moreover, the preferential interaction parameter for the solute methane in the solvent mixture of water and ChCl is calculated as,
| | | ΓMeW–Ch = ρW(GMe–W − GMe–Ch) | (6) |
 |
| | Fig. 7 Preferential interaction parameter for methane considering (a) methane–water, (b) methane–choline and (c) water–choline binary mixtures for all the systems studied. | |
In the above two equations, Gi−j is the Kirkwood–Buff integral and is defined as,
| | 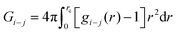 | (7) |
where
rc is the distance at which the integral approaches zero. According to the above expressions, a positive value of Γ
ii−j (Γ
ii−j > 0) indicates preferential accumulation of species
i around itself over species
j, whereas a negative value (Γ
ii−j < 0) suggests depletion. As shown in
Fig. 7a and b, both Γ
MeMe–W and Γ
MeMe–Ch are greater than zero for all four systems, indicating that methane preferentially interacts with itself over water and choline. This preferential accumulation of methane around itself supports the self-association tendency of methane, as discussed in the previous Sections 3.1, 3.2 and 3.3.1. Furthermore, as the number of choline molecules increases from S0 to S3, both Γ
MeMe–W and Γ
MeMe–Ch become more positive. This trend highlights the enhanced self-association of methane with increasing ChCl concentration, correlating with the observed increase in clustering and compaction degree of methane clusters discussed previously (Section 3.2). Similar preferential accumulation behavior has been reported previously, for instance, DTBM molecules preferentially associating with themselves over water and caffeine.
81 On the other hand,
Fig. 7c shows the preferential interaction parameter Γ
MeW–Ch, which is negative, indicating that methane prefers to interact with choline rather than with water. This observation aligns with the increased peak intensity for choline in the methane–choline and methane–Cc RDFs (Section 3.3.1).
Overall, the microscopic arrangement of water and choline around methane, and the extent of their interactions reveal that methane preferentially interacts with itself over water and choline. Additionally, when comparing methane's interactions with water and choline, it shows a stronger affinity for choline. The hydrophobic solvation provided by choline's hydrophobic moiety facilitates stronger hydrophobic associations among methane molecules. A similar stabilizing effect has been reported for hydrophobic polymers solvated by the three methyl groups of TMAO.56
3.3.2 Analysis of water and choline structure around methane.
It is well established that alterations in the structure of the hydrogen-bonded network of water surrounding a nonpolar solute is length-scale dependent.3 However, the addition of cosolvents can further alter the water structure. To examine the effect of introducing ChCl on the structure of water, we evaluate the RDFs between oxygen atoms of water (Ow–Ow RDF) and between hydrogen and oxygen atoms of water (Ow–Hw RDF). Fig. 8a and b present the Ow–Ow and Ow–Hw RDFs, respectively, with the peak regions magnified and shown in the lower panels for all systems. For comparison, the RDFs for Ow–Ow in bulk water (without methane) is also included to evaluate the influence of the nonpolar solute on water structure. Additionally, the cumulative CNs are plotted in the same figure. The actual and normalized cumulative CNs for the first solvation shell, obtained by integrating the Ow–Ow RDFs up to a first minimum (rc = 0.34 nm), along with the corresponding peak intensities for the first and second solvation shells, are provided in Table 3. From Fig. 8a, the first and second peaks of the Ow–Ow RDF appear at approximately ∼0.27 nm and ∼0.45 nm, respectively. These distances correspond to the hydrogen-bonded first neighbors and the tetrahedrally positioned second neighbors, and are consistent with previously reported values for similar systems involving nonpolar solutes in water–cosolvent mixtures.45,48 A slight shift in these peak positions toward shorter distances is observed with increasing ChCl concentration from S0 to S3. Additionally, the increasing ChCl concentration leads to an enhancement in the height of the first peak, while the second peak becomes less pronounced compared to in bulk water and the S0 system. This suggests that water becomes locally more structured near the solute, but the second hydration shell collapses upon the introduction of ChCl. Such behavior reflects the disruption of the extended hydrogen-bonded network of water by ChCl, as also reported in earlier studies.49 This structural change is reflected in the cumulative CN plots shown in Fig. 8a. Both the actual and normalized first-shell cumulative CNs decrease with increasing ChCl concentration, indicating a reduction in the number of neighboring water molecules. We also present Ow–Hw RDFs in Fig. 8b for different ChCl concentrations, along with their corresponding cumulative CNs. First-shell cumulative CNs (actual and normalized) are listed in Table 4. These values provide insight into the water–water hydrogen bonding behavior. For the Ow–Hw RDFs, both the first and second peak intensities increase with ChCl concentration, indicating a more localized arrangement of hydrogen atoms around the oxygen atoms of water. However, the corresponding cumulative CNs decrease. To capture more structural details, we provide the number of hydrogen atoms (Hw) around oxygen atoms (Ow), and vice versa, along with the total cumulative CNs. The decrease in the total cumulative CNs is supported by the reduction in the water–water hydrogen bonds as discussed in the next section. The observed decrease in first-shell cumulative CNs, and the marked difference between actual and normalized cumulative CNs, arise from the occupation of hydrogen-bonding sites by choline, which disrupts the native water hydrogen-bonded network.
 |
| | Fig. 8 (a) RDFs involving (a) water oxygens (Ow–Ow) (b) hydrogen atoms around oxygen atoms of the water (Ow–Hw) for all the systems investigated. The peak positions of both the RDFs, Ow–Ow and Ow–Hw are magnified and provided in the bottom panel of the respective plots. RDFs obtained from the simulation of the bulk water are also provided for comparison purposes and represented in black color. Cumulative CNs of the respective constituents obtained by integrating RDFs are also plotted in the same plots with the dashed lines. | |
Table 3 Peak heights corresponding to the first and second peaks in the Ow–Ow RDFs and average first shell cumulative CN for water (Ow) around water (Ow) for all systems considered. The numbers in parentheses are described in the text
| System |
First peak heights |
Second peak heights |
CN (Ow–Ow) |
| S0 |
3.42 |
1.18 |
4.55 |
| S1 |
3.92 |
1.17 |
3.77 (3.51) |
| S2 |
4.48 |
1.11 |
2.90 (2.61) |
| S3 |
5.01 |
1.02 |
2.01 (1.80) |
Table 4 Average first shell cumulative CN for water hydrogen around water oxygen (Ow–Hw), water oxygen around water hydrogen (Hw–Ow), and total cumulative CN (Ow–Hw + Hw–Ow) for all systems considered. The numbers in parentheses are described in the text. The cumulative CN calculations were performed using a cutoff distance (rc) of 0.25 nm
| System |
CN (Ow–Hw) |
CN (Hw–Ow) |
Total CN (Ow–Hw + Hw–Ow) |
| S0 |
0.96 |
0.96 |
1.92 |
| S1 |
0.81 (0.74) |
0.81 (0.74) |
1.62 (1.48) |
| S2 |
0.64 (0.55) |
0.64 (0.55) |
1.28 (1.10) |
| S3 |
0.46 (0.38) |
0.46 (0.38) |
0.92 (0.76) |
To explore how the addition of ChCl affects the tetrahedral arrangement of water, we compute the probability distribution of tetrahedral order parameter (TOP), P(qtet) and subtended angle, P(θ). As seen from Fig. 9b and c, results from neat water are also presented for comparison purposes and to compare the effect of the presence of methane and ChCl on the microscopic structure of the water. Also, peak values of P(qtet) and P(θ) are included in Table 5. TOP is evaluated using the following expression,82,83
| | 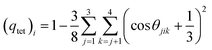 | (8) |
where,
θjik is the angle formed between the lines connecting central water's oxygen (
i) to oxygens of its two nearest partners (
j,
k) of the four immediate water molecules (
Fig. 9a). We include electronegative atoms of both water and choline as partners for forming a hydrogen bond network with central water's Ow.
qtet = 1 signifies a perfectly tetrahedral structure whereas,
qtet = 0 designates a completely random orientation. In a perfectly tetrahedral ice-like structure, if a water molecule is located at the center of a regular tetrahedron formed by its four nearest neighbors occupying four vertices, the angle between any two nearest neighboring water oxygens satisfies cos
θjik = −1/3 which corresponds to the ideal tetrahedral angle of approximately ∼109.5°. Under these conditions,
qtet reaches its maximum value of 1, indicating a perfectly tetrahedral arrangement. In contrast, in a completely random system, such as an ideal gas, the spatial arrangement of molecules is random, and the angles between neighboring molecules are uncorrelated. In this case, the six angles associated with a central molecule vary independently, and the average value of the tetrahedral order parameter approaches zero as
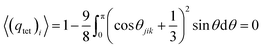
.
82,83
 |
| | Fig. 9 (a) Schematic showing the tetrahedral geometry of water with the angle formed between the lines connecting the central water's oxygen (i) to the oxygens of its two nearest partners (j, k) of the four immediate water molecules. Probability distribution of the tetrahedral order parameter, P(qtet) and angle subtended by two partners with the central water's Ow, P(θ) for (b) solvation shell water and (c) bulk water molecules for all the systems studied. Probability distributions for P(qtet) and P(θ) obtained from the simulation of the neat water are also provided for comparison purposes represented by the black color. | |
Table 5 Peak positions of the probability distribution of tetrahedral order parameter, P(qtet), and angle θ1 (between two neighbors and central water oxygen), for solvation shell and bulk water molecules across all systems. Peak positions of qtet and angle θ1 obtained from the simulation of the neat water are also provided for comparison purposes
| System |
q
tet
|
θ
1 (degree) |
| Solvation shell water |
Bulk water |
Solvation shell water |
Bulk water |
| Neat water |
— |
0.75 |
— |
55.0 |
| S0 |
0.49 |
0.73 |
52.8 |
54.3 |
| S1 |
0.45 |
0.47 |
51.1 |
50.6 |
| S2 |
0.44 |
0.45 |
48.89 |
49.25 |
| S3 |
0.40 |
0.43 |
46.13 |
45.72 |
The perfectly tetrahedral geometry of the water hydrogen bond network is influenced by the presence of solute or cosolvents. Therefore, we compute the P(qtet), P(θ) considering neat water, and present the values in Fig. 9b and c for comparison purposes. The qtet distribution for neat water has a main peak at higher and a shoulder at lower qtet values, indicating the presence of a dominant population of structured water molecules, along with a smaller population of disordered water molecules respectively. We compute P(qtet) and P(θ) separately for two categories of water molecules: those within 0.55 nm from the methane COM (Fig. 9b), referring to them as “solvation shell water”, and those beyond 1 nm from the methane COM, referring to them as “bulk water” (Fig. 9c). For solvation shell water molecules, we observe a main peak at lower and shoulder at higher qtet values suggesting a larger population of disordered water and smaller population of tetrahedrally ordered water. The higher population of disordered water is attributed to the disruption due to the presence of methane molecules. It is evident from Fig. 9b that the peak of the distribution for P(qtet) shifts to a lower value of qtet as we increase the number of ChCl molecules in the system. This increased deviation of the peak from 1 in P(qtet) indicates that ChCl introduces an additional disruption in the tetrahedral arrangement of the water and structural disorder in the methane solvation shell. The decreasing intensity of the shoulder at higher qtet values from S1 to S3 further supports the observation that ChCl contributes to this disruption beyond what is caused by methane alone. For example, the peak position of P(qtet) for BW is 0.75, and it gradually deviates, taking values from 0.49 to 0.40 for systems S0 to S3. In this context, the increased deviation in the water arrangement from tetrahedral geometry with increasing ChCl concentration contradicts the increased first peak height of the Ow–Ow RDF (Fig. 8a). This contradiction arises because choline induces crowding and imposes closer packing on water molecules, hence, the local structure of water becomes more evident. However, due to the presence of the nonpolar solute, water molecules near the solute adopt a tangential orientation to avoid unfavorable polar interactions with the water and thus lack the appropriate orientation to preserve the tetra-coordinated hydrogen bond network. This tangential alignment of water molecules near the methane surface in the presence of ChCl has been reported in previous studies.49 Additionally, some of the coordination sites in the water hydrogen-bonded network are occupied by the choline molecules, which are also potential hydrogen bond-forming partners, resulting in the deviation of the tetrahedral arrangement of the water molecules. This is further supported by the collapse of the second-shell water molecules in the Ow–Ow RDFs as discussed above (Section 3.3.2) due to the presence of choline at those distances.
It is evident that the qtet values are strongly influenced by the distribution of θjik, the O–O–O angle among the nearest oxygen atoms of water or choline. The trend obtained for P(qtet) can be supported by the deviation of θjik. Fig. 9b shows the probability distribution of θjik, and this distribution has two characteristic peaks that correspond to the two different alignments of the water molecules. These two peaks correspond to the angles θ1 and θ2. The disorder in the structure of the water is dependent on the position and intensity of θ1, and these values are tabulated in Table 5. The peak at ∼100° (θ2) is due to the presence of water molecules with the appropriate tetrahedral arrangement. This angle accounts for the experimentally observed local tetrahedral angle by the water oxygen. As we increase the ChCl concentration, we find flattening of the distribution with peaks near ∼100°, whereas the distribution with peaks near ∼40–60° starts to become sharper and shifts to lower values of θ1. These peaks corresponding to angle θ1 indicate the presence of interstitial water molecules that are not directly hydrogen-bonded to the central water molecules. Additionally, some of the vertices in the tetra-coordinated geometry of the water network are occupied by choline, leading to structural disruption.49 Hence, increasing the concentration of ChCl decreases the population of the θ2 angle while increasing the population of interstitial water, which has a disordered geometry. This further confirms the loss in the tetrahedrality of the water at higher ChCl concentrations for solvation shell water molecules.
When analyzing the probability distribution of qtet for bulk water molecules (Fig. 9c), the profiles for neat water and S0 appear to be nearly the same, both showing a prominent peak at higher qtet values and a shoulder at lower values. This indicates that the majority of bulk water remains structurally ordered and is less affected by the presence of methane aggregates. However, for systems S1 to S3, the main peak shifts toward lower qtet values, signifying increasing structural disorder introduced by ChCl. This qualitative trend of increasing disorder in water structures is also reflected in the distribution of θjik for bulk water. Overall, this observation supports the fact that the increased aggregation tendency of methane leads to the release of water molecules into the bulk. Also, water molecules acquire a disrupted structural arrangement, which leads to weaker interactions between water and methane. Hence, methane preferentially accumulates around itself, leading to enhanced clustering as the concentration of ChCl increases.
Next, we compute the number of hydrogen bonds between water–water (W–W), choline–choline (Ch–Ch), and choline–water (Ch–W) to support the observations obtained from their RDFs. This analysis is carried out using the GROMACS “gmx hbond” tool considering the last equilibrated section of the trajectories. Hydrogen bonds are identified using standard geometric criteria, with a donor–acceptor distance ≤ 0.35 nm and a hydrogen–donor–acceptor angle < 30°. The histograms of the normalized number of hydrogen bonds among the aforementioned constituents are shown in Fig. 10a–c. The number of water–water hydrogen bonds is normalized with respect to the total number of water molecules, while choline–choline and choline–water hydrogen bonds are normalized with respect to the total number of choline molecules in each respective system. The average values of these normalized hydrogen bonds are also listed in Table 6. Choline possesses hydrogen bond donor and acceptor sites, allowing it to form hydrogen bonds both with water and with other choline molecules. These interactions influence the surrounding water structure and promote choline self-aggregation, ultimately affecting choline's role in facilitating the hydrophobic association of methane. This self-aggregation of the choline is further confirmed by investigating the RDFs and cumulative CNs of choline (Ch) around choline considering their COMs, and the hydrophobic domain (Cc) around the hydrophobic domain of tagged choline (Fig. S11a and b and Table S4). The appearance of the Ch–Ch and Cc–Cc, g(r) peak positions at lower distances with increasing ChCl concentrations shows the tendency of choline for self-aggregation and subsequently the enhanced hydrophobic environment preferable for methane association. We observe that, upon the addition of ChCl, the number of Ch–Ch hydrogen bonds (per choline) increases, while the number of W–W (per water) and Ch–W (per choline) hydrogen bonds decreases. This reduction in W–W and Ch–W hydrogen bonding arises from the collapse of the second solvation shell of water around both water and choline, as well as a decrease in the first-shell cumulative CN of water around these species, as discussed in the previous Section 3.3.2. The reduction in the Ch–W hydrogen bond is also supported by the reduced cumulative CNs of water in the solvation shell of choline (Fig. S11c and Table S4). Notably, the increase in Ch–Ch hydrogen bonds supports the self-aggregation of choline, which plays a critical role in enhancing the clustering of methane by providing a more hydrophobic solvation environment. A similar reduction in W–W hydrogen bonds around methane and neopentane has been reported previously.45
 |
| | Fig. 10 Histogram showing a normalized number of hydrogen bonds between (a) water–water (W–W) (b) choline–choline (Ch–Ch) and (c) choline–water (Ch–W) for all the systems studied. W–W hydrogen bonds are normalized with respect to the total number of water molecules and Ch–Ch and Ch–W hydrogen bonds are normalized with respect to the total number of choline molecules. | |
Table 6 Average number of normalized hydrogen bonds between water–water (W–W), choline–choline (Ch–Ch), and choline–water (Ch–W) for all S0–S3 systems. The number of W–W hydrogen bonds is normalized with respect to the total number of water molecules, while Ch–Ch and Ch–W hydrogen bonds are normalized with respect to the total number of choline molecules in each respective system
| System |
W–W |
Ch–Ch |
Ch–W |
| S0 |
1.78 |
— |
— |
| S1 |
1.51 |
0.016 |
1.65 |
| S2 |
1.21 |
0.032 |
1.26 |
| S3 |
0.89 |
0.040 |
0.84 |
Collectively, the RDFs, cumulative CNs, and hydrogen bond analyses indicate the exclusion of water from the vicinity of methane and choline, alongside enhanced choline–choline interactions. While the removal of water from the methane solvation shell via hydrogen bonding contributes to the typical salting-out effect, choline plays an additional and distinct role beyond this mechanism. Unlike other osmolytes, choline is an asymmetric molecule with two distinct domains: a hydrophilic hydroxyl group (Oc) capable of forming hydrogen bonding with water, and a comparatively large hydrophobic region (three methyl groups attached to quaternary nitrogen). This character introduces a unique solvation environment around methane molecules that is not simply attributed to a general osmolyte-induced salting-out effect. The hydrophobic portion of choline actively contributes to the solvation structure around methane, promoting aggregation through direct hydrophobic interactions, which is a unique, molecule-specific contribution. This is also reported in our earlier study in which we showed that the hydrophobic domain of choline prefers to face methane and its hydrophilic end makes the hydrogen bonds with water present in the methane solvation shell.49 This combination establishes a favorable environment for the hydrophobic association of methane molecules.
3.4 Unraveling the energetics of methane association
To investigate the role of ChCl on the pairwise hydrophobic interaction between methane molecules and to validate the observations obtained from the unbiased trajectories, we compute the potential of mean force (PMF) using the COM distance between two methane molecules as the collective variable. The unbiased PMF is defined as,| | | W(r) = −kBTlnP(dMe–Me) | (9) |
The unbiased probability, P(dMe–Me), is calculated using WHAM and is provided in Fig. S12 of the SI. As evident from Fig. 11, the free energy landscape features two basins corresponding to negative free energies. The first basin appears at around ∼0.37–0.39 nm depending on the system and represents the contact minimum (CM) where two methane molecules are in direct contact. The second basin, known as the solvent-separated minimum (SSM), appears at ∼0.7 nm and corresponds to a configuration in which the pair of methane molecules is separated by a layer of solvent molecules. The representative configurations corresponding to the CM and SSM are shown in Fig. 11b. A desolvation barrier is present between the CM and SSM. For the S0 system (methane in water only), we find the CM is at ∼0.387 nm with a free energy of ∼−2.44 kJ mol−1 (Fig. 11c). This CM depth (∼−2.44 kJ mol−1) and the desolvation barrier of ∼3.78 kJ mol−1 (Table 7) are in good agreement with previous studies that also investigated the PMF of a methane pair in pure water at 300 K.44,46 These CM and SSM indicate hydrophobic association and solvation, respectively. Focusing on the S1 system, the free energies at the CM and the desolvation barrier are ∼−3.02 kJ mol−1 and ∼4.29 kJ mol−1 (Table 7), respectively, with their positions at ∼0.378 nm and ∼0.57 nm. For the S2 and S3 systems, the positions of the CM are at ∼0.375 nm and ∼0.372 nm, with corresponding free energies of ∼−3.56 kJ mol−1 and ∼−5.00 kJ mol−1. The desolvation barrier-free energies are ∼4.89 kJ mol−1 and ∼5.39 kJ mol−1, respectively. This indicates that with increasing ChCl concentration from S0 to S3, the CM shifts to shorter distances and becomes more stabilized, as seen from the increasing depth (more negative free energy) in Fig. 11c. The increasing desolvation barrier (Table 7), calculated with respect to the CM, also suggests enhanced stability of the contact pair configuration over the SSM configuration. Fig. 11c reveals that the CM position decreases while the free energy at the CM becomes more negative, supporting the enhanced hydrophobic association of methane. In contrast, the SSM shows minimal depth and increasing barrier-free energy, indicating less stability for the SSM configuration and, therefore, reduced hydrophobic solvation of methane. To gain deeper insight into how ChCl modulates methane hydrophobic interactions, we evaluate the corresponding free energy differences at the CM by subtracting the free energy in the water-only system (S0) from that in the ChCl-containing systems as: ΔG = Gmin(CS+W) − Gmin(W), where CS denotes the cosolvent (ChCl) and W denotes water. These ΔG values are presented in Fig. 11d. With increasing ChCl concentration from S1 to S3, ΔG becomes more negative. For example, S1 has a ΔG of ∼−0.58 kJ mol−1, S2 of ∼−1.12 kJ mol−1, and S3 of ∼−2.56 kJ mol−1. This trend indicates that the addition of ChCl promotes the stabilization of the CM configuration and hence enhances the hydrophobic association between methane molecules. Similar observations in PMF analyses of methane in other cosolvent–solvent mixtures have been reported, and our results are consistent with those findings.44,46
 |
| | Fig. 11 (a) Free energy profile (PMF) between two methane molecules considering the COM distance between them as a reaction coordinate for all the systems studied. The error bars corresponding to standard deviation are also plotted (b) different methane-pair representative configurations corresponding to the contact minimum (CM) and solvent-separated minimum (SSM). (c) The position of CM and free energies corresponding to the CM for all systems S0 to S3 and (d) the difference in free energies, ΔG = Gmin(CS+W) − Gmin(W) (CS = cosolvent, W = water) at the CM for all systems. | |
Table 7 Position of the desolvation barrier, free energy values at desolvation barrier (GBarrier), and difference in the free energies at the contact minimum (CM) and desolvation barrier i.e. barrier heights (GBarrier − GMin) obtained from the PMF profile for all studied systems
| System |
Position of barrier (nm) |
G
Barrier (kJ mol−1) |
G
Barrier − GMin (kJ mol−1) |
| S0 |
0.57 |
1.34 |
3.78 |
| S1 |
0.57 |
1.27 |
4.29 |
| S2 |
0.57 |
1.33 |
4.89 |
| S3 |
0.57 |
0.39 |
5.39 |
Additionally, to understand the extent of the association of methane in water as well as ChCl, we calculate the association constant, Ka, by integrating the PMF up to the desolvation barrier, which marks the outer limit for the methane–methane contact configuration. The association constant, Ka is defined using the following expression,34
| | 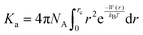 | (10) |
Here, rc is the position of the desolvation barrier in the respective PMF profile and NA is the Avogadro's number. We note that greater hydrophobic association is related to the higher values of Ka. The values of Ka for different systems are tabulated in Table 8. From this table, we observe that the value of Ka is increasing from system S0 to S3, favoring the associated state of the methane at higher ChCl concentrations. Moreover, the binding efficiency of the methane with itself can be quantified in terms of the binding energy evaluated from the PMFs as,81
| | 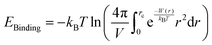 | (11) |
| | 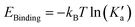 | (12) |
where
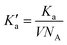
and
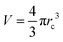
is the volume occupied by the interacting methane pair. A more negative value of
EBinding indicates a stronger attractive interaction between the methane molecules. The calculated values of
EBinding are also presented in
Table 8. Notably, the
EBinding values become increasingly negative from the S0 to S3 systems, suggesting a stronger binding affinity between methane molecules in the presence of ChCl compared to pure water.
Table 8 Association constants, Ka, and binding energies (EBinding) obtained from the PMFs for methane in all systems
| System |
K
a (M−1) |
E
Binding (kJ mol−1) |
| S0 |
0.55 |
−0.41 |
| S1 |
0.69 |
−0.96 |
| S2 |
0.87 |
−1.56 |
| S3 |
1.18 |
−2.32 |
Altogether, the trends in the PMFs, association constant, and binding energy provide compelling evidence of the enhanced hydrophobic association between methane molecules with increasing ChCl concentration. These findings are consistent with the results obtained from the unbiased trajectories, reinforcing the role of ChCl in promoting methane clustering. The energetic analysis confirms the stabilizing effect of ChCl on the methane hydrophobic association, further supporting the idea that the addition of ChCl leads to a more favorable environment for methane aggregation compared to pure water.
4 Summary
In this work, we performed molecular dynamics simulations of multiple methane molecules in pure water and in aqueous solutions with varying concentrations of ChCl to investigate the impact of ChCl on hydrophobic interactions. Our results show that methane aggregation increases with higher ChCl concentration through methane–methane RDFs and SASA. Cluster analysis indicates a greater tendency to form larger, more compact clusters, while the total number of clusters decreases. Potential of mean force (PMF) calculations reveals that the contact minimum shifts to lower free energy with increasing ChCl, indicating stronger stabilization of methane contact pairs. The increasing desolvation barrier further suggests a higher energetic cost to separate methane molecules. These findings consistently demonstrate that ChCl enhances hydrophobic association, promoting tighter and more stable methane clustering compared to pure water.
Further insights from RDFs, CNs, and preferential interaction parameters reveals dewetting of the methane surface and accumulation of choline, especially its hydrophobic regions, near methane. This creates a locally more hydrophobic environment that promotes methane aggregation. While methane generally prefers self-association, in water–ChCl mixtures it shows stronger preferential interaction with choline over water. Structural analysis indicates that choline disrupts the water network by collapsing the second solvation shell and reducing water density around methane. The tetrahedral order parameter (TOP) further shows increasing disorder in the local water structure due to both choline and methane. RDFs between choline molecules indicate enhanced choline self-association with increasing ChCl concentration, along with reduced water coordination number around choline. Hydrogen bond analysis confirms decreased water–water and choline–water hydrogen bonds, and increased choline–choline hydrogen bonding. Altogether, these results suggest that choline clustering around itself and around methane enhances local hydrophobicity and drives methane aggregation at higher ChCl concentrations.
In summary, this study highlights the significant role of ChCl in enhancing hydrophobic interactions between methane molecules. Looking forward, it would be interesting to extend this study by investigating nonpolar solutes of larger molecular size to better mimic hydrophobic domains in proteins. A comparative study involving various osmolytes and nonpolar solutes of different sizes and complexities could provide deeper insights into the mechanisms underlying biomolecular stability, folding, and self-assembly processes in cellular environments in the presence of stabilizing osmolytes.
Author contributions
Pooja Nanavare: conceptualization, formal analysis, simulations, methodology, writing – original draft, and writing – review and editing. Rajarshi Chakrabarti: conceptualization, supervision, and writing – review and editing.
Conflicts of interest
There are no conflicts to declare.
Data availability
Supplementary information is available: Convergence analysis between independent simulation replicas; Time evolution of the simulated densities of the considered systems; Methane–methane pair potential of mean force profile using unbiased trajectories; Time profile of the number and size of the largest cluster; No signs of macroscopic phase separation: cluster analysis of extended trajectories; system size dependence of methane aggregation; Local solvation environment of methane; Microscopic arrangement of constituents around choline; Methane–methane center of mass distribution recovered from umbrella sampling simulations. See DOI: https://doi.org/10.1039/d5cp01919b
The datasets generated during the current study are included in the main manuscript and the SI. The data are available from the authors upon reasonable request.
Acknowledgements
P. N. thanks UGC for a fellowship. P. N. acknowledges Dr. Ligesh Theeyancheri for helpful discussions. We acknowledge the SpaceTime-2 supercomputing facility at IIT Bombay for the computing time. We dedicate this article to Prof. N. Sathyamurthy.
References
- L. R. Pratt and A. Pohorille, Chem. Rev., 2002, 102, 2671–2692 CrossRef CAS PubMed.
- D. Chandler, Nature, 2007, 445, 831–832 CrossRef CAS PubMed.
- D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS PubMed.
- D. Ben-Amotz, Annu. Rev. Phys. Chem., 2016, 67, 617–638 CrossRef CAS PubMed.
- K. A. Dill, Biochemistry, 1990, 29, 7133–7155 CrossRef CAS PubMed.
-
C. Tanford, The hydrophobic effect: formation of micelles and biological membranes, J. Wiley & Sons, 1980, vol. 233 Search PubMed.
- J. W. Brady, L. Tavagnacco, L. Ehrlich, M. Chen, U. Schnupf, M. E. Himmel, M.-L. Saboungi and A. Cesàro, Eur. Biophys. J., 2012, 41, 369–377 CrossRef CAS PubMed.
- L. R. Pratt, Annu. Rev. Phys. Chem., 2002, 53, 409–436 CrossRef CAS PubMed.
-
A. Y. Ben-Naim, Hydrophobic interactions, Springer Science & Business Media, 2012 Search PubMed.
-
B. Bagchi, Water in biological and chemical processes: from structure and dynamics to function, Cambridge University Press, 2013 Search PubMed.
- P. Ball, Chem. Rev., 2008, 108, 74–108 CrossRef CAS PubMed.
- B. Bagchi, Chem. Rev., 2005, 105, 3197–3219 CrossRef CAS PubMed.
- N. Nandi, K. Bhattacharyya and B. Bagchi, Chem. Rev., 2000, 100, 2013–2046 CrossRef CAS PubMed.
- F. H. Stillinger, Science, 1980, 209, 451–457 CrossRef CAS PubMed.
- R. Zangi and B. Berne, J. Phys. Chem. B, 2008, 112, 8634–8644 CrossRef CAS PubMed.
- J. R. Grigera and A. N. McCarthy, Biophys. J., 2010, 98, 1626–1631 CrossRef CAS PubMed.
- C. L. Dias and H. S. Chan, J. Phys. Chem. B, 2014, 118, 7488–7509 CrossRef CAS PubMed.
- E. P. O'Brien, R. I. Dima, B. Brooks and D. Thirumalai, J. Am. Chem. Soc., 2007, 129, 7346–7353 CrossRef.
- S. Roy and B. Bagchi, J. Phys. Chem. B, 2014, 118, 5691–5697 CrossRef CAS PubMed.
- R. Gazi, S. Maity and M. Jana, ACS Omega, 2023, 8, 2832–2843 CrossRef CAS PubMed.
- M.-E. Lee and N. F. van der Vegt, J. Am. Chem. Soc., 2006, 128, 4948–4949 CrossRef CAS PubMed.
- S. Paul and S. Paul, J. Phys. Chem. B, 2015, 119, 10975–10988 CrossRef CAS PubMed.
- S. Sarkar, S. Ghosh and R. Chakrabarti, RSC Adv., 2017, 7, 52888–52906 RSC.
- S. S. Cho, G. Reddy, J. E. Straub and D. Thirumalai, J. Phys. Chem. B, 2011, 115, 13401–13407 CrossRef CAS PubMed.
- M. Mukherjee and J. Mondal, J. Phys. Chem. B, 2020, 124, 6565–6574 CrossRef CAS.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Green Chem., 2016, 18, 2736–2744 RSC.
- C. Li, D. Li, S. Zou, Z. Li, J. Yin, A. Wang, Y. Cui, Z. Yao and Q. Zhao, Green Chem., 2013, 15, 2793–2799 RSC.
- A. P. Abbott, R. C. Harris and K. S. Ryder, J. Phys. Chem. B, 2007, 111, 4910–4913 CrossRef CAS PubMed.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- D. Tolmachev, N. Lukasheva, R. Ramazanov, V. Nazarychev, N. Borzdun, I. Volgin, M. Andreeva, A. Glova, S. Melnikova and A. Dobrovskiy,
et al.
, Int. J. Mol. Sci., 2022, 23, 645 CrossRef CAS PubMed.
- S. Supriyati, I. G. M. Budiarsana, L. Praharani, R. Krisnan and I. K. Sutama, J. Anim. Sci. Technol., 2016, 58, 1–12 CrossRef.
- F. Khorrami and M. H. Kowsari, J. Phys. Chem. B, 2020, 124, 3770–3783 CrossRef CAS PubMed.
- P. Ganguly, J. Polák, N. F. van der Vegt, J. Heyda and J.-E. Shea, J. Phys. Chem. B, 2020, 124, 6181–6197 CrossRef CAS.
- R. Sarma and S. Paul, J. Phys. Chem. B, 2012, 116, 2831–2841 CrossRef CAS PubMed.
- S. Shimizu and H. S. Chan, J. Am. Chem. Soc., 2001, 123, 2083–2084 CrossRef CAS.
- S. Shimizu and H. S. Chan, Proteins: Struct., Funct., Bioinf., 2002, 48, 15–30 CrossRef CAS PubMed.
- S. Shimizu and H. S. Chan, Proteins: Struct., Funct., Bioinf., 2002, 49, 560–566 CrossRef CAS.
- R. Sahu and D. Nayar, J. Chem. Phys., 2021, 155, 024903 CrossRef CAS PubMed.
- S. Bharadwaj, D. Nayar, C. Dalgicdir and N. F. van der Vegt, J. Chem. Phys., 2021, 154, 134903 CrossRef CAS PubMed.
- D. Trzesniak, N. F. van der Vegt and W. F. van Gunsteren, Phys. Chem. Chem. Phys., 2004, 6, 697–702 RSC.
- M. Ikeguchi, S. Nakamura and K. Shimizu, J. Am. Chem. Soc., 2001, 123, 677–682 CrossRef CAS PubMed.
- C. Oostenbrink and W. F. van Gunsteren, Phys. Chem. Chem. Phys., 2005, 7, 53–58 RSC.
- M. V. Athawale, S. Sarupria and S. Garde, J. Phys. Chem. B, 2008, 112, 5661–5670 CrossRef CAS PubMed.
- M. K. Dixit, A. A. Siddique and B. Tembe, J. Phys. Chem. B, 2015, 119, 10941–10953 CrossRef CAS PubMed.
- S. Paul and S. Paul, J. Chem. Phys., 2013, 139, 044508 CrossRef PubMed.
- M. Dixit, T. Hajari and B. Tembe, J. Mol. Liq., 2016, 223, 660–671 CrossRef CAS.
- R. Sarma and S. Paul, J. Chem. Phys., 2012, 136, 114510 CrossRef PubMed.
- B. Sharma and S. Paul, J. Phys. Chem. B, 2015, 119, 6421–6432 CrossRef CAS PubMed.
- P. Nanavare, A. R. Choudhury, S. Sarkar, A. Maity and R. Chakrabarti, ChemPhysChem, 2022, 23, e202200446 CrossRef CAS PubMed.
- P. Nanavare, L. Theeyancheri, S. Sarkar and R. Chakrabarti, Chem. Phys. Impact, 2023, 6, 100223 CrossRef.
- Aashu, S. Rawat and C. Ramachandran, Phys. Chem. Chem. Phys., 2025, 27, 10591–10605 RSC.
- P. Nanavare, S. Sarkar, A. B. Jena and R. Chakrabarti, Phys. Chem. Chem. Phys., 2024, 26, 24021–24040 RSC.
- I. V. Voroshylova, E. S. Ferreira and M. N. D. Cordeiro, Molecules, 2025, 30, 574 CrossRef CAS PubMed.
- Y. Vora, O. Sethi, S. N. Bariya, S. L. Gawali, S. S. Soni, T. S. Kang, P. A. Hassan and K. Kuperkar, Phys. Chem. Chem. Phys., 2025, 27, 1171–1186 RSC.
- P. Patidar, B. Kanoje, A. Bahadur, K. Kuperkar, D. Ray, V. K. Aswal, M. Wang, L.-J. Chen and P. Bahadur, Colloid Polym. Sci., 2021, 299, 117–128 CrossRef CAS.
- J. Mondal, G. Stirnemann and B. Berne, J. Phys. Chem. B, 2013, 117, 8723–8732 CrossRef CAS PubMed.
- J. Mondal, D. Halverson, I. T. Li, G. Stirnemann, G. C. Walker and B. J. Berne, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9270–9275 CrossRef CAS PubMed.
- D. Nayar and N. F. van der Vegt, J. Phys. Chem. B, 2018, 122, 3587–3595 CrossRef CAS PubMed.
- L. Martnez, R. Andrade, E. G. Birgin and J. M. Martnez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- B. Doherty and O. Acevedo, J. Phys. Chem. B, 2018, 122, 9982–9993 CrossRef CAS PubMed.
- R. Zangi, R. Zhou and B. Berne, J. Am. Chem. Soc., 2009, 131, 1535–1541 CrossRef CAS PubMed.
- S. Chatterjee, P. G. Debenedetti, F. H. Stillinger and R. M. Lynden-Bell, J. Chem. Phys., 2008, 128, 124511 CrossRef PubMed.
- A. Ben-Naim and Y. Marcus, J. Chem. Phys., 1984, 81, 2016–2027 CrossRef CAS.
- P. Witherspoon and D. Saraf, J. Phys. Chem., 1965, 69, 3752–3755 CrossRef CAS.
- B. Hess, C. Kutzner, D. Van Der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- S. S. Petrova and A. D. Solov'ev, Hist. Math., 1997, 24, 361–375 CrossRef.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- H. J. Berendsen, J. V. Postma, W. F. Van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- M. P. Allen, Mol. Phys., 2019, 117, 2391–2417 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- A. Stukowski, Modell. Simul. Mater. Sci. Eng., 2009, 18, 015012 Search PubMed.
- M. Bonomi, D. Branduardi, G. Bussi, C. Camilloni, D. Provasi, P. Raiteri, D. Donadio, F. Marinelli, F. Pietrucci and R. A. Broglia,
et al.
, Comput. Phys. Commun., 2009, 180, 1961–1972 Search PubMed.
- D. J. Evans and B. L. Holian, J. Chem. Phys., 1985, 83, 4069–4074 CrossRef CAS.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 Search PubMed.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- J. Grabowska, S. Blazquez, E. Sanz, I. M. Zerón, J. Algaba, J. M. Mguez, F. J. Blas and C. Vega, J. Phys. Chem. B, 2022, 126, 8553–8570 CrossRef CAS PubMed.
- J. H. Weijs, J. R. Seddon and D. Lohse, ChemPhysChem, 2012, 13, 2197–2204 CrossRef CAS PubMed.
- B. Sharma and S. Paul, J. Mol. Liq., 2016, 224, 930–939 CrossRef CAS.
- P.-L. Chau and A. Hardwick, Mol. Phys., 1998, 93, 511–518 CrossRef CAS.
- J. R. Errington and P. G. Debenedetti, Nature, 2001, 409, 318–321 Search PubMed.
Footnote |
| † We dedicate this article to Prof. N. Sathyamurthy. |
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Rajarshi
Chakrabarti
and
Rajarshi
Chakrabarti













 .82,83
.82,83






 and
and  is the volume occupied by the interacting methane pair. A more negative value of EBinding indicates a stronger attractive interaction between the methane molecules. The calculated values of EBinding are also presented in Table 8. Notably, the EBinding values become increasingly negative from the S0 to S3 systems, suggesting a stronger binding affinity between methane molecules in the presence of ChCl compared to pure water.
is the volume occupied by the interacting methane pair. A more negative value of EBinding indicates a stronger attractive interaction between the methane molecules. The calculated values of EBinding are also presented in Table 8. Notably, the EBinding values become increasingly negative from the S0 to S3 systems, suggesting a stronger binding affinity between methane molecules in the presence of ChCl compared to pure water.