
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of thermal treatments on high surface area anatase TiO2†
Chiara
Nannuzzi‡
 *a,
Ton V. W.
Janssens
b and
Gloria
Berlier
*a
*a,
Ton V. W.
Janssens
b and
Gloria
Berlier
*a
aDepartment of Chemistry, NIS and INSTM Centers, Università di Torino, via P. Giuria 7, 10125 Turin, Italy. E-mail: chiara.nannuzzi2@unibo.it; gloria.berlier@unito.it
bUmicore ApS, kogle Allè 1, 2970 Hørsholm, Denmark
First published on 1st July 2025
Abstract
Titanium dioxide (TiO2) is widely employed in catalysis, energy conversion, and environmental applications, where high surface area and crystallinity play crucial roles in enhancing performance. However, achieving thermal stability while maintaining these properties remains challenging. This study investigates the structural and textural evolution of a high-surface-area anatase TiO2 subjected to various thermal treatments, including vacuum, static, and flow conditions. Through N2 adsorption, X-ray diffraction, electron microscopy, and in situ infrared spectroscopy, we elucidate the interplay between surface area, crystal growth, surface sites and exposed crystallographic surfaces. Thermal treatments in vacuum preserve the surface area up to 450 °C, while static calcination leads to significant reduction and scarce reproducibility. Controlled flow conditions improve stability, though humidity accelerates sintering. Spectroscopic analyses reveal a strong interaction with water, influencing hydroxylation and dehydroxylation dynamics. Our findings provide insights into optimizing TiO2 processing conditions for applications requiring high thermal stability and catalytic efficiency.
Introduction
Titanium dioxide (TiO2) is a versatile material with a wide array of applications ranging from photocatalysis, solar energy harvesting, and environmental remediation to its roles in sensors, catalysis, and biomedicine. Its exceptional electrical, optical, and photocatalytic properties arise from its strong oxidation and reduction potential when exposed to photon energy.1,2 These characteristics make TiO2 a critical material for addressing global challenges in sustainable energy and environmental protection.TiO2 is one of the most studied materials, and many synthetic approaches have been developed in the years to obtain materials with different crystal phase, particle dimension and morphology.1,3–5
A particularly important property of TiO2's functionality is its surface area, which significantly influences its efficiency in applications such as heterogeneous catalysis, photocatalysis, and dye-sensitized solar cells (DSSCs).4,6–8 High-surface-area TiO2 enhances interaction with reactants, facilitates charge transfer processes, and increases the density of active sites, which are essential for improved catalytic and photoactive performance.9,10 Despite these advantages, challenges remain in synthesizing TiO2 presenting at the same time high crystallinity and large surface area, as traditional methods often require trade-offs between these attributes. Approaches such as hydrothermal treatments and templated synthesis have shown promise in addressing these challenges by enabling controlled manipulation of morphology and crystalline phases.11–13
High surface area TiO2 materials are typically characterized by low thermal stability, resulting in a loss of surface area as a result of particle agglomeration and consequential sintering, often accompanied by the transition of anatase to the most stable rutile phase.1,3,14–16 One example of application is the use of TiO2 as support for VOx based catalysts for the NH3-based Selective Catalytic Reduction (NH3-SCR), which is used for the removal of NOx in the exhaust gas treatment of light-duty and heavy-duty diesel vehicles.17–20 Notwithstanding their low cost and better tolerance to SO2 poisoning, VOx/TiO2 catalysts display lower activity with respect to Cu-exchanged zeolites in the low temperature range (between 150 and 300 °C).19,21–23 One approach to improve the catalyst activity is to increase the surface area of the support, which turns in increases the number of active sites.24 A thermal treatment is necessary during the synthesis of VOx/TiO2 catalysts, to decompose the precursor of the VOx active phase (ammonium metavanadate or other vanadium salts). It is clear that a high surface area TiO2 support can be used to improve VOx/TiO2 activity provided that it is stable during the synthesis and in reaction conditions. This means that the use of a high surface area TiO2 requires a preliminary investigation of the changes induced by thermal treatments on morphology, crystal phase, surface area, and porosity for all the applications which requires preliminary thermal treatments and/or high temperature operational conditions.
This work focuses on the characterization of high-surface-area anatase TiO2, exploring the interplay between surface area, crystallinity, and surface sites distribution as a function of different thermal treatments. The aim is to provide insights into the relationship between atomic surface structure and structural/textural parameters, and to optimize the experimental conditions to obtain a stable high-surface-area TiO2 with enhanced performance and applicability across a broad spectrum of scientific and industrial domains.
Experimental
Materials and methods
The TiO2 materials studied in this work were supplied by Umicore ApS Denmark.24The specific surface areas (SSA) of TiO2 as such and thermally treated in different conditions was determined from the N2 adsorption isotherms at liquid nitrogen temperature with ASAP 2020 Micromeritics instrument, and according to the Brunauer–Emmett–Teller (BET) equation. The pore dimensions were determined with the Barrett–Joyner–Halenda (BJH) equation on the adsorption curve. In some case, pore size distribution was calculated by the DFT tool available in the Microactive software (Micromeritics), using the classical type, cylinder geometry, and the N2 Harkins and Jura thickness curve. Before the N2 adsorption measurements, the samples were outgassed at room temperature (RT), until a residual pressure of 1 × 10−1 mbar was reached and heated in vacuum for 2 h at different temperatures.
A Nabertherm LT muffle was used for calcination in static conditions, in a temperature range between 100 and 500 °C, for 1 or 5 h. A tubular Carbolite furnace was used for flow conditions treatments at 350 °C in dry and wet air (synthetic air bottle was used, with the addition of water in the case of wet condition). In this case a total flow of 150 mL min−1 was used. TGA measurements were performed with thermogravimetric analyzer TA-Q600, with 100 mL min−1 flow of air. About 20 mg of each sample was analyzed in Al2O3 pans. Equilibration at 30 °C for 30 minutes was set before heating up to 900 °C, with a ramp of 5 °C min−1. Weights were normalized to 100% after removal of physisorbed water at 150 °C.
X-ray diffraction patterns were collected with a PANalytical X’Pert Pro powder diffractometer, equipped with a X’Celerator detector, using Cu Kα (1.5418 Å) radiation generated at 45 kV and 40 mA. The patterns were measured in the 2θ range from 20–85°, with a step size of 0.017° and a counting time of 120 seconds per step; Bragg–Brentano geometry was used with ¼ slits. Before the measurements, all samples were soft-milled and positioned in the sample holder carefully to avoid preferential orientations. The crystal domains (d) along [101], [001], and [100] directions were calculated with Scherrer equation, where K is a shape factor (in this study 0.9), λ is the X-ray wavelength, Bexp is the experimental peak width at half maximum (FWHM) at the given 2θ, Bins is the instrumental broadening (measured on Si reference in the same analysis condition), and θ is the measured Bragg angle in radians. In the calculation of the FWHM, used in the Scherrer equation:  , the broadening in the 35–40 2θ range corresponding to the (004) reflection is associated with a significant error in the estimation of the crystallite dimension along the c-axis, due to the overlapping with the (103) and (112) reflections. To obtain a more accurate value for the width of the (004) reflection, this range was fitted with three pseudo-Voigt profiles located at 37.03° (103), 37.89° (004), and 38.66° (112); the angle positions were taken from the anatase TiO2 PDF reference (ICDD PDF 00-021-1272).
, the broadening in the 35–40 2θ range corresponding to the (004) reflection is associated with a significant error in the estimation of the crystallite dimension along the c-axis, due to the overlapping with the (103) and (112) reflections. To obtain a more accurate value for the width of the (004) reflection, this range was fitted with three pseudo-Voigt profiles located at 37.03° (103), 37.89° (004), and 38.66° (112); the angle positions were taken from the anatase TiO2 PDF reference (ICDD PDF 00-021-1272).
Information on the morphology of the TiO2 particles was obtained by electron microscopy. High-resolution transmission electron microscopy (HR-TEM) was performed on a JEOL 3010-UHR microscope operated at 300 kV, using samples of TiO2 dispersed on Cu grids covered in lacey carbon. Samples were suspended in water and sonicated for 30 minutes before performing the measurements.
Fourier transform infrared (IR) experiments were carried out on samples pressed into thin self-supporting pellets for transmission measurements. The pellets were fixed in a gold frame, and placed inside an in situ IR cell, which allows carrying out thermal treatments in vacuum, performing low-temperature measurements and controlling the gas atmosphere. The spectra were measured in transmission mode with 4 cm−1 resolution on a Bruker N Invenio spectrophotometer equipped with a mercury cadmium telluride (MCT) cryodetector. Backgrounds and sample spectra were recorded measuring 128 interferogram scans to achieve a good signal-to-noise ratio. All the reported spectra were normalized by using the optical density of prepared pellets (weight of pellet divided by area), to correct for possible variations in the thickness of the pellets.
The nature and distribution of surface sites were investigated using CO as probe molecule. Before introducing CO, the samples were treated at 500 °C for 3 h under dynamic vacuum to maintain a residual pressure of 5 × 10−4 mbar, before exposure to 80 mbar of O2. After 10 min in O2 the samples were cooled to 150 °C, where O2 was removed, and then further cooled to room temperature. Then the sample was moved to the spectrophotometer, cooled down with liquid nitrogen and 40 mbar of CO were introduced into the cell. To study the CO stretching region (νCO), the spectra recorded after CO adsorption were processed by subtracting the spectra collected prior to the exposure. The relative amount of different surface sites was evaluated by fitting the corresponding components with Lorentz/Gaussian curves with the OPUS software. The calculated areas were normalized to the optical densities. The results are compared with those obtained on an anatase TiO2 with similar morphology and lower surface area (ls-TiO2, 89 m2 g−1).24
In situ IR experiments during thermal treatments in gas flow were performed with the Aabspec cell. A total flow of 50 mL min−1 (synthetic air N2 + 20% O2) was used for these experiments and the temperature ramp was set at 5 °C min−1.
Results and discussion
Raw material (r-TiO2)
The textural properties of r-TiO2 material, that is surface area (SSA) and pore size and volume, were obtained from the N2 adsorption measurements. Before performing the measure, r-TiO2 was outgassed at room temperature, to avoid possible changes induced by thermal treatments. The isotherm and pore distribution curves of r-TiO2 are reported in Fig. 1. The isotherm can be classified as type IV with a broad hysteresis loop starting around p/p0 = 0.6, with SSA of 345 m2 g−1 and pore volume of 0.35 cm3 g−1 (Table 1). A narrow pore size distribution centered around 20 Å is observed, a value close to the limit for the validity of the BJH method, and confirmed by DFT analysis. In other words, the raw material has pores at the borderline between micro and mesopores. | ||
| Fig. 1 N2 sorption isotherm of r-TiO2 outgassed at room temperature and corresponding pore size distribution, calculated by BJH and DFT (left and right, respectively). | ||
| Sample | Temperature (°C) | SSA (m2 g−1) | Pore size (Å) | Pore volume (cm3 g−1) |
|---|---|---|---|---|
| r-TiO2 | RT | 345 | ∼20 | 0.35 |
| 150v-TiO2 | 150 | 390 | ∼20 | 0.36 |
| 200v-TiO2 | 200 | 385 | ∼20 | 0.37 |
| 350v-TiO2 | 350 | 355 | ∼20 | 0.32 |
| 450v-TiO2 | 450 | 312 | ∼20 | 0.37 |
The X-ray diffraction pattern of r-TiO2, reported in black in Fig. 2, only shows the anatase phase, with a non-complete crystallinity, indicated by the broadening of the peaks, together with the non-flat baseline. In particular, the three reflections in the 35–40 2θ range are convoluted in a single broad peak. The average size of crystal domains (d) along the [101], [001] and [100] directions were determined using the Scherrer equation, from the (101), (004) and (200) anatase reflections, resulting in values of 6, 3, and 8 nm along b, c and a directions, respectively. These dimensions can be ascribed to an elongated shape along the b and a axes, suggesting a platelet-like morphology.1,25–27
 | ||
| Fig. 2 XRD patterns in 20–85 2θ range of r-TiO2 as such and after calcination in static conditions at different temperatures, for 5 and 1 h (full and dotted curves, respectively). Patterns are normalized for the (101) diffraction peak of anatase TiO2 labeled with * and vertically translated for clarity. | ||
Transmission electron microscopy (TEM) was used to investigate the morphology of r-TiO2 material (Fig. 3). The sample was dispersed in H2O and sonicated, but this treatment was not sufficient to avoid a high degree of agglomeration. Due to agglomeration, it is very difficult to distinguish single particles in order to obtain information on the exposed crystal planes. On the whole, a qualitative analysis- not detailed in the manuscript- was performed using Gatan DigitalMicrograph software to obtain the interplanar distances of the families of planes giving diffraction, which were then compared to those of anatase TiO2 (ICDD PDF file 00-021-1272). This analysis revealed a high relative amount of the (101) plane, followed by (001) and (100), respectively. Nevertheless, it was possible to estimate the particles’ dimensions, around 5 nm, from high magnification images (right panel of Fig. 3), in good agreement with the crystallite dimensions obtained with the Scherrer analysis.
 | ||
| Fig. 3 HR-TEM images at low and high magnification, left and right respectively, of r-TiO2. | ||
Effect of thermal treatments
High surface area TiO2 materials are typically characterized by low thermal stability, resulting in a loss of surface area as a result of particle agglomeration and consequential sintering, often accompanied by the transition of anatase to the most stable rutile phase.1,3,14,15 Since many applications require temperatures higher than room temperature, we have studied the effect of thermal treatments in different conditions on crystallinity, textural properties and surface sites distribution.Thermal treatments were carried out in vacuum (directly in the cell used for N2 adsorption measurements, without exposure to atmosphere), in static muffle and in flow conditions. The resulting samples are labeled as xy-TiO2, where x is the temperature used in the treatment and y is the condition (v as vacuum, s as static). Dry- and wet-TiO2 refer to samples thermally treated in flow conditions at 350 °C (see below).
Vacuum conditions
The r-TiO2 material was studied after pre-treatments performed in vacuum before the N2 adsorption measurements, directly in the measurement cell. The textural properties resulting from treatments at different temperatures are listed in Table 1. The corresponding isotherms are reported in Fig. S1 in the ESI.†The results show an increase of SSA after mild thermal treatments (150 and 200 °C), with respect to r-TiO2 outgassed at room temperature. This could be explained by the desorption of physisorbed water that was not removed by room temperature outgassing. The SSA starts to decrease with respect to 150v- and 200v-TiO2 after thermal treatments at 350 and 450 °C. However, thermal treatments in vacuum did not affect the porosity, both in terms of pores distribution and pore volume.
Static conditions
The results described in the previous section show that thermal treatments in vacuum only slightly affect the textural properties of the high surface area material up to 450 °C. However, this kind of treatment is not suitable for large scale applications, also considering the fact that thermal treatments are one of the main bottlenecks for companies in terms of costs and energy consumption. Thus, calcination in a static oven was performed at different temperatures, for 1 or 5 h.For each temperature, the length of the treatment does not particularly affect the shape of the XRD patterns, in terms of peak intensity and widths. Crystallinity is increasing with increasing temperature, already with the treatment at 200 °C, thus, the gradual increase in the size of the crystalline domains in all the samples (Table 2). The two peaks in the 51–57 2θ interval starts to be resolved at 300 °C, while the three peaks between 35–40 2θ, become resolved only after the treatment at 450 °C. The observed preferential growth of the crystallites along a and b directions, observed for r-TiO2, is maintained after the thermal treatments. The anatase structure is preserved up to 500 °C, without the formation of extra-phases in appreciable amounts. Particle size after thermal treatments at 450/500 °C is comparable to the anatase particles in the benchmark P25 TiO2 sample (26 nm).27
| Sample | Length (h) | d 101 (nm) | d 004 (nm) | d 200 (nm) |
|---|---|---|---|---|
| 100s-TiO2 | 1 | 7 | 4 | 9 |
| 5 | 7 | 5 | 9 | |
| 200s-TiO2 | 1 | 10 | 7 | 10 |
| 5 | 10 | 7 | 10 | |
| 300s-TiO2 | 1 | 13 | 6 | 14 |
| 5 | 14 | 7 | 15 | |
| 350s-TiO2 | 5 | 17 | 8 | 18 |
| 450s-TiO2 | 1 | 17 | 9 | 18 |
| 5 | 21 | 15 | 24 | |
| 500s-TiO2 | 5 | 23 | 15 | 24 |
While the length of the thermal treatment only slightly affects the diffraction pattern and particle size, this parameter seems to have more effect on the textural properties. The N2 sorption isotherms and pore size distribution of a selection of samples are reported in Fig. 4. The isotherms measured on samples treated in the 300–450 °C temperature range show differences particularly at high p/p0 values, that is in the range corresponding to capillary condensation in mesopores. In fact, the considered samples show distinct pore size distributions, as evidenced in the right panel of Fig. 4. The measured surface areas and porosity parameters are resumed in Table 3. Both temperature and length of the treatment affect SSA and pore size, but some discrepancies are observed. The surface area increases from 300 to 350 °C after 1 h treatment, from 152 to 172 cm2 g−1, while it does not significantly change when the treatment was carried out for 5 h. The treatment at 350 °C for 5 h was repeated, leading to a different surface area. Generally, the thermal treatments in this temperature range cause a clear change in the pore size, which becomes larger and less homogeneous, particularly in the case of the sample treated at 450 °C for 5 h. This change does not sensibly affect the pore volume with respect to pristine r-TiO2, which was characterized by a narrow distribution of pores around 20 Å. This could be related to agglomeration and sintering of TiO2 particles during thermal treatments, where a large number of small pores condense in a lower number of larger ones. To explore this aspect, pore size distribution was calculated by DFT (Fig. S2, ESI†). The samples treated at 350 and 450 °C for 5 h show a bimodal distribution, with a fraction of pores around 25 Å and a broader distribution of bigger pores.
 | ||
| Fig. 4 N2 sorption isotherms and corresponding BJH pore size distribution (left and right panels, respectively), after calcination in static conditions at different temperature and time lengths. For sake of clarity only the adsorption curves have been plotted. | ||
| Sample | Length (h) | SSA (m2 g−1) | Pore size (Å) | Pore volume (cm3 g−1) |
|---|---|---|---|---|
| 300s-TiO2 | 1 | 152 | 55 | 0.35 |
| 5 | 140 | 62 | 0.36 | |
| 350s-TiO2 | 1 | 172 | 50 | 0.36 |
| 5 | 138 | 23, 80 | 0.37 | |
| 5 | 116 | 27, 80 | 0.34 | |
| 450s-TiO2 | 5 | 97 | 23, 120 | 0.31 |
In general, these results suggest a low control and reproducibility of textural properties when the thermal treatment is carried out in static conditions, as will be further discussed in the following.
Flow conditions
The discrepancies and low reproducibility observed after treatments in static conditions could be related to variations in the ambient conditions, specifically regarding air humidity. To verify this hypothesis, thermal treatments were carried out in controlled atmosphere in flow conditions. Specifically, dry and wet (3% H2O) air flows were used for the treatment of r-TiO2 at 350 °C for 1 h. The choice of the parameters was made with the aim to minimize the loss in surface area caused by the thermal treatment, which appears to be more consistent at higher temperature. Moreover, this temperature was found to be sufficient for the decomposition of the ammonium metavanadate salt used as a precursor of VOx in the synthesis of VOx/TiO2 catalysts and relevant for the application of these catalysts in the NH3-SCR reaction.24Surface area, porosity, and crystalline domain size measured after the treatments are reported in Table 4. The corresponding experimental data are shown in Fig. S3 and S4 (ESI†). To check for reproducibility, the treatment was performed three times for each condition. Values of surface area and porosity were found to be consistent, with variations well within the experimental error (5% for BET surface area), indicating a good reproducibility for this method, at variance with what was obtained in static conditions.
| Sample | SSA (m2 g−1) | Pore size (Å) | Pore volume (cm3 g−1) | d 101 (nm) | d 004 (nm) | d 200 (nm) |
|---|---|---|---|---|---|---|
| Dry-TiO2 | 182 ± 1 | 48 ± 1 | 0.36 ± 0.01 | 12 | 7 | 12 |
| Wet-TiO2 | 148 ± 3 | 62 ± 2 | 0.36 ± 0.01 | 13 | 7 | 13 |
The surface area changes with the flow composition: in particular, the presence of H2O leads to a higher decrease in the surface area than the dry flow. However, the pore volume does not significantly change in the different conditions. The decrease of surface area with the wet flow is associated to an increase of the pore size, which moves from around 48 to 62 nm, with a small broadening of the size distribution (Fig. S3, right panel, ESI†). Calcination in dry air flow was also carried out at 300 °C for 1 h (not reported), resulting in a similar surface area (183 m2 g−1).
As discussed above, the FWHM of (101), (004), and (200) diffraction peaks (XRD patterns reported in Fig. S4, ESI†) were used in the Scherrer equation to obtain the crystal domains along b, c, and a axes, respectively (Table 4), and similar dimensions were obtained in the two cases.
In summary, the data show that the presence of H2O vapor during the thermal treatment negatively affects surface area, the decrease of which has been related to the sintering of the nanoparticles,1 also confirmed by the increase in the crystallite dimensions along a and b axes (see Table 4).
This supports the hypothesis that the low reproducibility of the results obtained after thermal treatments in static conditions without environmental control are related to variations of humidity, and could explain why the treatments in vacuum have a lower impact on the material textural properties.
Surface sites
The results discussed so far indicates that the thermal treatments applied to the r-TiO2 sample yield different outcomes in terms of textural properties and crystallite domains. Typically, a high surface area correlates with high defectivity represented by OH groups and undercoordinated Ti4+ sites located on steps, edges, and corners of the particles.28–30 Therefore, it is crucial to examine the surface sites of the r-TiO2 material. The use of CO molecules as probe followed by IR spectroscopy is one of the most commonly employed technique for this purpose.27,28,30–33To obtain reliable results, ensuring a bare and clean surface of the material before sending the CO probe is essential, as this allows for direct interaction between the CO molecules and the surface sites. This implies that a thermal treatment is required to remove the physisorbed water, followed by oxidation at high temperatures to eliminate any organic matter present on the titanium oxide surface.26,27,29,30,34
In this study, we investigated the OH surface groups of r-TiO2 during thermal treatments using in situ IR in flow conditions, and probed the different Ti4+ sites with CO molecules after a thermal treatment in vacuum.
The r-TiO2 was heated in air flow, from room temperature to 500 °C, with a 1 h hold at the final temperature. During the heating process, IR spectra were recorded every minute in order to track changes in the OH population. The collected spectra, displayed in Fig. 5, show the νOH region (3800–2400 cm−1) and the bending (δ) H–O–H mode of physisorbed water (1800–1500 cm−1)35 in the left and right panel, respectively.
 | ||
| Fig. 5 In situ IR spectra of r-TiO2 during thermal treatment in air flow, from 25 °C to 500 °C. Total flow 50 mL min−1, ramp 5 °C min−1. | ||
At 100 °C (dark green spectrum), the IR spectra exhibit a broad and intense band (out of scale) centered at 3200 cm−1, characteristic of hydrogen-bonding interactions among water molecules and between water molecules and surface OH groups.36 As the temperature increases, the absorption gradually decreases, revealing the typical features of surface Ti–OH groups. By 150 °C, three distinct peaks at 3630, 3655, and 3679 cm−1 emerged. These features merged into two less-resolved components (light green spectra form 300 °C) showing a maximum at 3662 cm−1 and a shoulder at 3681 cm−1. By further increasing the temperature, the shoulder decreased in intensity, and a new component appeared at higher frequencies (3700 cm−1) at 450 °C. Beyond this temperature, only a decrease in the whole intensity was observed.
The three peaks observed at 150 °C can be attributed to weakly/strongly H-bonded OH groups which dehydroxylate at increasing temperatures, releasing water molecules. This leads to the appearance of free OH groups that vibrate at higher frequencies, 3700 and 3681 cm−1, located on edges, vertices and corners, and extended surfaces of the TiO2 particles.37,38 Notice that the positions of these bands is slightly different with respect to what measured at low temperature (see below). In the right panel, the intensity of the δ H–O–H mode peak at 1626 cm−1 indicates the presence of H2O on the surface of r-TiO2. As the temperature increased, the intensity of this peak decreased due to water desorption, with the complete disappearance of the signal at 450 °C, coinciding with the emergence of isolated OH groups.
Next, we studied the Ti4+ surface sites by CO adsorption monitored by IR spectroscopy, after a pretreatment in vacuum at 500 °C for 3 h, followed by oxidation in 80 mbar of O2 for 10 minutes (Fig. 6). This procedure was optimized after several trials (not reported), to remove strongly adsorbed water molecules. Following this treatment, the surface area of r-TiO2 decreased to 227 m2 g−1, representing a 34/42% reduction from the initial SSA value measured after RT/150 °C vacuum treatment, respectively (see Table 1). For comparison, we report the spectra obtained in the same conditions on an anatase TiO2 with similar morphology and surface area of 89 m2 g−1 (labelled as low surface area TiO2, ls-TiO2). The main properties of this material are summarized in Table S1 and Fig. S5 (ESI†).
 | ||
| Fig. 6 Normalized IR spectra of CO adsorption (40 mbar) on r-TiO2 in the νOH and νCO regions, left and right panels, respectively (bottom, blue). For comparison the top curves (red) show the corresponding spectra measured on a low surface area TiO2 (ls-TiO2) with similar properties (see text). The left panel also reports the spectra of both TiO2 before CO adsorption, after thermal treatment in vacuum in vacuum at 500 °C for 3 h followed by oxidation. These spectra were used as a reference to obtain the background subtracted spectra reported in the right panel. Spectra normalized with respect to the pellet thickness. | ||
The spectroscopic fingerprints of the two TiO2 in the νOH region are reported in the left panel of Fig. 6. Two main features are located at 3720 and 3671 cm−1, corresponding to isolated Ti–OH in the vertices and edges, and bridged Ti–OH–Ti sites on extended surfaces, respectively.37,39–41 On r-TiO2 two clear shoulders are observed at higher and lower frequency with respect to the main peaks, at 3740 and 3642 cm−1. The former is attributed to OH groups on the (001) surface, formed by a dissociative adsorption of H2O.42–44 The latter, also detected in the in situ experiment (see Fig. 5), indicates that some H2O molecules are adsorbed on the (101) surface, in accordance with Arrouvel et al., suggesting a density ≤10.8 H2O/nm2.42 This is also in agreement with the weak signal at 1620 cm−1 related to the bending mode of H2O36 persisted in r-TiO2 after the pretreatment (not reported), indicating the difficulty in the removal all the physisorbed H2O in this sample.
When CO is admitted, the OH groups weakly interact with it, forming OH⋯CO adducts, observed as a broad band centered at 3560 cm−1. However, while bridged Ti–OH–Ti are almost completely involved in the interaction, isolated Ti–OH ones are less affected, in line with a lower acidity.39
Moving to the νCO region (right panel of Fig. 6), the main peak at 2180 cm−1 is assigned to CO interacting with 5-fold-coordinated Ti4+ ions located on the extended (101) facet, while at 2165 cm−1 the interaction between CO and Ti4+ on the (100) facets is observed.28,31,33,45,46 The peak at 2153 cm−1 corresponds to CO interacting with OH groups, while the peak at 2139 cm−1 is attributed to liquid-like form of adsorbed CO, which does not imply specific interactions with TiO2.32,33,47 The signal related to CO interaction with defective Ti4+ sites is located at relatively high frequency (2207 cm−1) and is associated with 4-fold or 3-fold Ti4+ sites located on steps, edges, and corners on high-index crystal planes.27,48 No information can be obtained on the presence and extension of the (001) surface since the interaction of CO with the corresponding Ti4+ sites can be observed only at lower temperature.25,31
More interesting is the comparison of the relative concentration of surface sites in the two samples with similar morphology and different surface areas. Focusing on the νCO bands, the area of the different components related to Ti4+⋯CO on different surfaces or defects, described above, has been calculated after curve fitting (Table S2, ESI†). Based on this, we calculated the ratio of the different surface sites in the two samples (Table 5).
| Surface sites | Ratio r-TiO2/ls-TiO2 |
|---|---|
| Defects | 2.4 |
| (101) | 1.4 |
| (100) | 4.7 |
| Total Ti4+ | 1.6 |
| OH | 3.4 |
Looking into the single components, the ratio between the estimated amount of defective sites in the two samples reflects the ratio between surface areas (2.4 vs. 2.6), agreeing with the association between this textural property and the defectivity of the material. This does not hold for the probed Ti4+ sites on the (101) facets (2180 cm−1). This can be in part explained by the persistence of some adsorbed water molecules in the high surface area material (see above, band at 3642 cm−1) and also indicates that this surface, which is the most stable surface on anatase,1 is less extended in the irregular particles of r-TiO2. Indeed, we observe a higher relative concentration of Ti4+ on (100) (band at 2165 cm−1), which supports the hypothesis that the small particles of the high surface area materials exposes different preferential surfaces, even if both samples show the same preferential elongation along the c-axis (Table 2 and Table S1, ESI†). This hypothesis is further supported by the ratio between the total probed Ti4+ sites (1.6). Since in the tested conditions it is not possible to monitor the Ti4+ sites on the (001) surface, it is reasonable to assume that this surface has a large extension on ls-TiO2, in agreement with the observation of the νOH component at 3740 cm−1 attributed to OH groups from a dissociative adsorption of H2O on the (001) surface.42–44 Finally, the ratio between the integrated areas of OH groups in the two samples greatly exceeds the ratio between surface areas (Table 5). However, this value could be overestimated, due to the presence in r-TiO2 of the two components at 3740 and 3642 cm−1 related to H2O adsorption.
In this work, we investigated the changes in the textural properties and surface sites of an high surface area anatase TiO2 after thermal treatments in different conditions. Our findings demonstrate that the surface area of r-TiO2 is highly dependent on the specific treatment protocol and conditions, that is using a static oven, a tubular flow furnace (under dry or wet air), or vacuum treatments (e.g. in the BET cell or in the IR procedure).
Thermal treatments under vacuum had a minimal impact on the surface area and porosity of r-TiO2 up to 450 °C, with the highest surface area (390 m2 g−1) measured after outgassing at 150 °C, suggesting that heating in vacuum tends to preserve the material's surface area. However, a significant decrease in the surface area (227 m2 g−1) was observed after the vacuum treatment at 500 °C, necessary for IR studies. This is important for understanding parameters that influence aggregation and sintering, as well as for semiquantitative considerations in IR spectra analysis. Notice that this was not observed for the reference ls-TiO2 material, which preserved its surface area after the same treatment (89 m2 g−1).
In contrast, treatments in oven under static conditions posed challenges in terms of reproducibility and sensitivity to treatment duration. Calcination at 300 °C resulted in a notable decrease in surface area, with values of 152 and 140 m2 g−1 for 1 and 5 h, respectively, representing a 61 and 64% loss compared to the initial value of 390 m2 g−1. Moreover, 1 h of calcination at 350 °C led to a higher surface area of 172 m2 g−1 overcoming the measure error of 5%. After 450 °C and for longer calcination, a further decrease up to 97 m2 g−1 was detected. The decrease in surface area is associated with an increase in crystal size domains, as calculated by the Scherrer equation. Some of the data measured after thermal treatments around 350 °C indicate scarce control and reproducibility under static conditions. These inconsistencies may stem on variations in humidity. To address this, calcination in a tubular oven in dry flow was carried out at the fixed temperature of 350 °C. By applying these conditions, higher reproducibility was obtained. Additionally, the presence of H2O was found to promote TiO2 sintering, as evidenced by a slight increase in crystal domain size and a decrease in surface area to 145 m2 g−1, corresponding to a 20% reduction compared to dry conditions (181 m2 g−1).
Scherrer analysis provided crucial insights into the morphology of the r-TiO2 samples, which cannot be analyzed in depth by TEM, due to the high agglomeration of the particles. By analyzing the growth along different crystallographic axes, the elongated shapes were attributed to a platelet-like morphology, in analogy with what observed for ls-TiO2.24
The in situ IR study of r-TiO2 during heating in air flow revealed a strong interaction between water and surface sites, which, considering the morphology, stands with the presence of extended (001) surfaces that strongly interact with water.25,31,49 This surface is also responsible for dissociating H2O into the corresponding OH groups, which are stable up to 450 °C (shoulder at 3642 cm−1).
Furthermore, CO adsorption at low temperature was followed by IR to analyze the material surface sites. The surface sites probed by CO adsorption, as shown in Fig. 6, were similar in both materials, with the main difference related to their intensity (Table 5). A high surface area generally correlates with an increased number of defective sites and OH groups. The former is confirmed by the semiquantitative analysis performed by the integration of the corresponding IR peaks (2207 cm−1) in r-TiO2 and in a benchmark anatase TiO2 sample (ls-TiO2) with a SSA of 89 m2 g−1 (see Table 5). As for the latter, the measured amount is markedly higher with respect to what expected on the basis of surface area, but also includes contributions from dissociated and strongly adsorbed H2O molecules. Moreover, the lower relative amount of Ti4+ sites on the main anatase (101) surface (2180 cm−1) coupled to the higher relative amount of Ti4+ sites on (100) (2165 cm−1) and the indirect indication about a significant extension of the (001) surface, testified by Scherrer analysis performed on XRD pattern and the presence of OH groups vibrating at higher frequencies, points to a higher contribution of typically less abundant facets in r-TiO2 material with respect to what expected on anatase1,31,40 and observed on a ‘standard’ ls-TiO2 sample. This feature can be related to the smaller particle size of r-TiO2 (≈5 nm).
TiO2 were analyzed by thermogravimetric analysis in the 150–900 °C range (Fig. 7). r-TiO2 shows a large weight loss between 30 and 150 °C, accounting for ca. 15% of dry weight, vs. ca. 1.3% in ls-TiO2 (Fig. S6, ESI†). This could suggest a higher hydrophilicity of r-TiO2, since the amount of physisorbed water is not proportional to surface area.
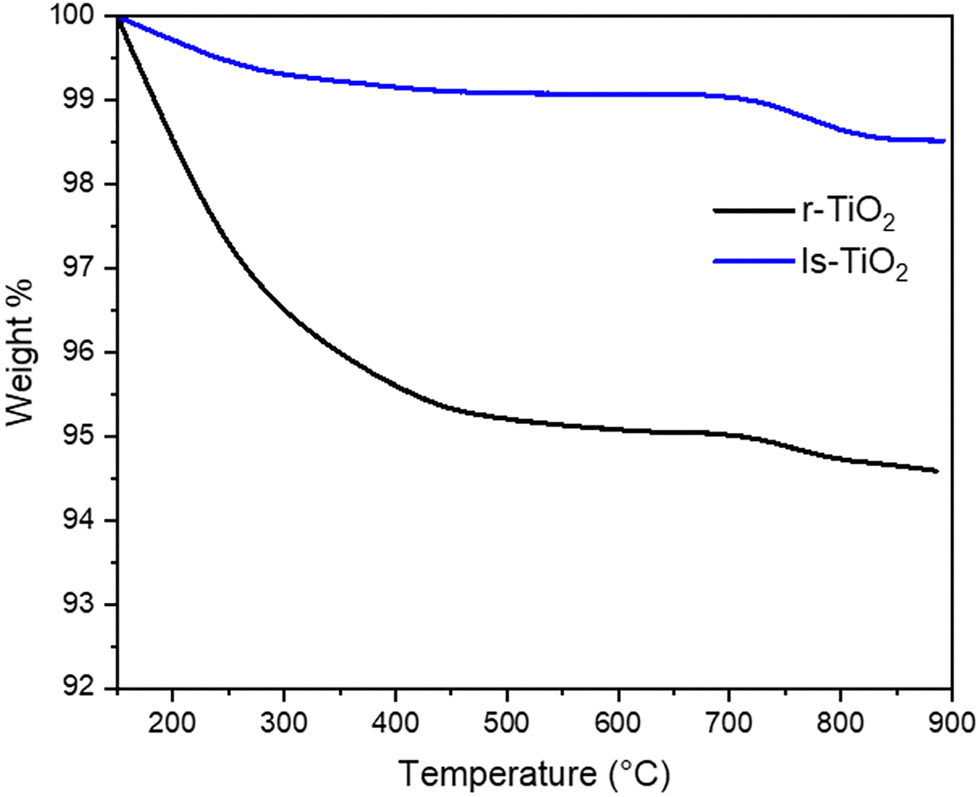 | ||
| Fig. 7 TGA curves of ls-TiO2 (blue) and r-TiO2 (black) heated in air flow. Weights were normalized to 100% to the dry weight of the material, after the loss of physisorbed water between 30 and 150 °C. Heating rate 5 °C min−1. | ||
The data reported in Fig. 7 were thus normalized with respect to the dry weight measured after heating at 150 °C.
After water desorption, ls-TiO2 shows a ≈ 1.8% weight loss up to 900 °C, which can be related to dehydroxylation, that is water through a condensation reaction between surface Ti–OH groups.
The extent of dehydroxylation is higher in r-TiO2, resulting in a total weight loss of ca. 7.6%. This result closely aligns with the surface area ratio of 4.4 (390 vs. 89 m2 g−1), indicating that the amount of OH groups on the surface is directly proportional to the surface area.
For both materials, most of the dehydroxylation takes place up to 500 °C, which is the temperature used in the pretreatment for the in situ IR study. Again, this is more evident for r-TiO2, and suggests that the decrease in surface area observed by high temperature treatments is related to dehydroxylation involving vicinal particles, ruling the aggregation and sintering process.
Conclusions
The findings of this study highlight the significant influence of thermal treatments on the textural properties of an high surface area anatase TiO2, labeled as r-TiO2. Increasing the treatment temperature consistently led to a gradual reduction in surface area, while extending the treatment duration from 1 to 5 h under static conditions caused an even greater decrease. Vacuum pre-treatment before BET measurements resulted in a smaller surface area reduction compared to other methods, particularly those involving a static oven, which produced lower surface area values and scarce reproducibility of the results. In contrast, thermal treatments under flow conditions, more suitable for industrial applications, offered better reproducibility and generally higher surface areas with respect to treatments in static oven. The presence of H2O in the flow was found to promote sintering, leading to a slight increase in crystal domain size and a more pronounced surface area reduction, further corroborating results obtained from static calcination.Although the high degree of agglomeration hindered an in depth analysis of r-TiO2 morphology, combining Scherrer analysis with TEM suggested an elongated, platelet-like morphology, in analogy with a benchmark anatase TiO2 sample with lower surface area (ls-TiO2). In situ IR spectroscopy during thermal heating provided valuable insights into the behavior of OH groups as temperature increased, with three distinct H-bonded OH groups converting into free OH groups located on edges, vertices, corners and extended surfaces after water desorption at 450 °C. The high temperature indicates a strong water interaction with surface sites, particularly on the (101) and (001) facets.
CO adsorption followed by IR allowed us to provide a qualitative description of TiO2 surface sites in the high surface area material, and to compare the concentration of defects (4-fold and 3-fold Ti4+ sites at steps, edges, and corners) and the extension of preferential surfaces with respect to the benchmark material. The results point to a higher contribution of typically less abundant facets, that is (100) and (001), in the r-TiO2 material. Particularly, the presence of a significant amount of the (001) is indicated by the strong resistance of the OH groups formed by dissociative adsorption of H2O observed in the material (band at 3740 cm−1).
Finally, TGA analysis confirmed that the amount of OH groups on r-TiO2 is proportional to surface area, by comparison to the benchmark ls-TiO2 sample. Under heating in flow in TGA, ls-TiO2 undergoes a severe dehydroxylation, reaching a concentration of OH groups similar to the low surface area material around 500 °C, as confirmed by IR spectroscopy. The severe dehydroxylation can be related to the thermal instability of the material, probably occurring during the aggregation and sintering processes leading to a decrease in surface area.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
C. Nannuzzi and G. Berlier acknowledge support from the Project CH4.0 under the MUR program “Dipartimenti di Eccellenza 2023–2027” (CUP: D13C22003520001).References
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- T. Peng, D. Zhao, K. Dai, W. Shi and K. Hirao, J. Phys. Chem. B, 2005, 109, 4947–4952 CrossRef CAS.
- S. Reghunath, D. Pinheiro and S. D. KR, Appl. Surf. Sci. Adv., 2021, 3, 100063 CrossRef.
- R. Zhang, A. A. Elzatahry, S. S. Al-Deyab and D. Zhao, Nano Today, 2012, 7, 344–366 CrossRef CAS.
- M. Bankmann, R. Brand, B. H. Engler and J. Ohmer, Catal. Today, 1992, 14, 225–242 CrossRef CAS.
- H. J. Snaith and L. Schmidt-Mende, Adv. Mater., 2007, 19, 3187–3200 CrossRef CAS.
- V. Dhas, S. Muduli, S. Agarkar, A. Rana, B. Hannoyer, R. Banerjee and S. Ogale, Sol. Energy, 2011, 85, 1213–1219 CrossRef CAS.
- Y. V. Kolen’ko, B. R. Churagulov, M. Kunst, L. Mazerolles and C. Colbeau-Justin, Appl. Catal., B, 2004, 54, 51–58 CrossRef.
- J. Bisquert, D. Cahen, G. Hodes, S. Ruhle and A. Zaban, J. Phys. Chem. B, 2004, 108, 8106–8118 CrossRef CAS.
- T. Fröschl, U. Hörmann, P. Kubiak, G. Kucerová, M. Pfanzelt, C. K. Weiss, R. J. Behm, N. Hüsing, U. Kaiser, K. Landfester and M. Wohlfahrt-Mehrens, Chem. Soc. Rev., 2012, 41, 5313–5360 RSC.
- D. S. Kim and S. Y. Kwak, Appl. Catal., A, 2007, 323, 110–118 CrossRef CAS.
- G. Colón, M. C. Hidalgo and J. A. Navío, Catal. Today, 2002, 76, 91–101 CrossRef.
- Y. Dai, S. Shen, Z. Ma, L. Ma, Z. Sun, J. Yu, C. Wan, S. Han, D. Mao and G. Lu, J. Nanosci. Nanotechnol., 2017, 17, 3772–3778 CrossRef CAS.
- J. Krýsa, M. Keppert, J. Jirkovský, V. Štengl and J. Šubrt, Mater. Chem. Phys., 2004, 86, 333–339 CrossRef.
- T. V. Nguyen, H. C. Lee and O. B. Yang, Sol. Energy Mater. Sol. Cells, 2006, 90, 967–981 CrossRef CAS.
- V. N. Koparde and P. T. Cummings, ACS Nano, 2008, 2, 1620–1624 CrossRef CAS.
- E. Tronconi and I. Nova, Urea-SCR Technology for deNOx After Treatment of Diesel Exhausts, Springer International Publishing, 2014 Search PubMed.
- K. Skalska, J. S. Miller and S. Ledakowicz, Sci. Total Environ., 2010, 408, 3976–3989 CrossRef CAS.
- L. Han, S. Cai, M. Gao, J. Hasegawa, P. Wang, J. Zhang, L. Shi and D. Zhang, Chem. Rev., 2019, 119, 10916–10976 CrossRef CAS.
- Z. Shi, Q. Peng, E. J. iaqiang, B. Xie, J. Wei, R. Yin and G. Fu, Fuel, 2023, 331, 125885 CrossRef CAS.
- A. J. Shih, I. Khurana, H. Li, J. González, A. Kumar, C. Paolucci, T. M. Lardinois, C. B. Jones, J. D. Albarracin Caballero, K. Kamasamudram, A. Yezerets, W. N. Delgass, J. T. Miller, A. L. Villa, W. F. Schneider, R. Gounder and F. H. Ribeiro, Appl. Catal., A, 2019, 574, 122–131 CrossRef CAS.
- J. P. Dunn, H. G. Stenger and I. E. Wachs, Catal. Today, 1999, 51, 301–318 CrossRef CAS.
- M. Qing, S. Su, L. Wang, L. Liu, K. Xu, L. He, X. Jun, S. Hu, Y. Wang and J. Xiang, Chem. Eng. J., 2019, 361, 1215–1224 CrossRef CAS.
- C. Nannuzzi, L. Mino, S. Bordiga, A. H. Pedersen, J. M. Houghton, P. N. R. Vennestrøm, T. V. W. Janssens and G. Berlier, J. Catal., 2023, 421, 228–239 CrossRef CAS.
- K. Bourikas, C. Kordulis and A. Lycourghiotis, Chem. Rev., 2014, 114, 9754–9823 CrossRef CAS.
- L. Mino, F. Pellegrino, S. Rades, J. Radnik, V. Hodoroaba, G. Spoto, V. Maurino and G. Martra, ACS Appl. Nano Mater., 2018, 1, 5355–5365 CrossRef CAS.
- L. Mino, G. Spoto, S. Bordiga and A. Zecchina, J. Phys. Chem. C, 2012, 116, 17008–17018 CrossRef CAS.
- K. I. Hadjiivanov and D. G. Klissurski, Chem. Soc. Rev., 1996, 25, 61–69 RSC.
- L. Mino, A. M. Ferrari, V. Lacivita, G. Spoto, S. Bordiga and A. Zecchina, J. Phys. Chem. C, 2011, 115, 7694–7700 CrossRef CAS.
- A. Zecchina, D. Scarano, S. Bordiga, G. Ricchiardi, G. Spoto and F. Geobaldo, Catal. Today, 1996, 27, 403–435 CrossRef CAS.
- L. Mino, A. M. Ferrari, V. Lacivita, G. Spoto, S. Bordiga and A. Zecchina, J. Phys. Chem. C, 2011, 115, 7694–7700 CrossRef CAS.
- K. Hadjiivanov, J. Lamotte and J. C. Lavalley, Langmuir, 1997, 13, 3374–3381 CrossRef CAS.
- K. I. Hadjiivanov and G. N. Vayssilov, Adv. Catal., 2002, 47, 307–511 CAS.
- C. Deiana, M. Minella, G. Tabacchi, V. Maurino, E. Fois and G. Martra, Phys. Chem. Chem. Phys., 2013, 15, 307–315 RSC.
- M. Takeuchi, L. Bertinetti, G. Martra, S. Coluccia and M. Anpo, Appl. Catal., A, 2006, 307, 13–20 CrossRef CAS.
- N. B. Colthup, L. H. Daly and S. E. Wiberly, Introduction to Infrared and Raman Spectroscopy, New York, 2nd edn, 1975 Search PubMed.
- A. Mahdavi-Shakib, J. M. Arce-Ramos, R. N. Austin, T. J. Schwartz, L. C. Grabow and B. G. Frederick, J. Phys. Chem. C, 2019, 123, 24533–24548 CrossRef CAS.
- A. A. Tsyganenko and V. N. Filimonov, J. Mol. Struct., 1973, 19, 579–589 CrossRef CAS.
- L. Mino, Á. Morales-García, S. T. Bromley and F. Illas, Nanoscale, 2021, 13, 6577–6585 RSC.
- S. Dzwigaj, C. Arrouvel, M. Breysse, C. Geantet, S. Inoue, H. Toulhoat and P. Raybaud, J. Catal., 2005, 236, 245–250 CrossRef CAS.
- K. Hadjiivanov, Identification and Characterization of Surface Hydroxyl Groups by Infrared Spectroscopy, Elsevier Inc., 1st edn, 2014, vol. 57 Search PubMed.
- C. Arrouvel, M. Digne, M. Breysse, H. Toulhoat and P. Raybaud, J. Catal., 2004, 222, 152–166 CrossRef CAS.
- A. Travert, O. V. Manoilova, A. A. Tsyganenko, F. Maugé and J. C. Lavalley, J. Phys. Chem. B, 2002, 106, 1350–1362 CrossRef CAS.
- C. Morterra, J. Chem. Soc., 1988, 84, 1617–1637 CAS.
- S. Pantaleone, F. Pellegrino, V. Maurino, M. Corno, P. Ugliengo and L. Mino, J. Mater. Chem. A, 2024, 12, 4325–4332 RSC.
- C. Morterra, E. Garrone, V. Bolis and B. Fubini, Spectrochim. Acta, Part A, 1987, 43, 1577–1581 CrossRef.
- A. A. Tsyganenko, L. A. Denisenko, S. M. Zverev and V. N. Filimonov, J. Catal., 1985, 94, 10–15 CrossRef CAS.
- H. Knozinger and S. Huber, Faraday Trans., 1998, 94(15), 2047–2059 RSC.
- B. Zhang, F. Wei, Q. Wu, L. Piao, M. Liu and Z. Jin, J. Phys. Chem. C, 2015, 119, 6094–6100 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp01731a |
| ‡ Current address: CIRI-FRAME, University of Bologna, viale Ciro Menotti 48, 48122 Marina di Ravenna, Italy. |
| This journal is © the Owner Societies 2025 |