
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnergy partitioning in H2 formation on interstellar carbonaceous grains. Insights from ab initio molecular dynamics simulations†
Léana
Jubert
 a,
Berta
Martínez-Bachs
a,
Gerard
Pareras
a and
Albert
Rimola
*ab
a,
Berta
Martínez-Bachs
a,
Gerard
Pareras
a and
Albert
Rimola
*ab
aDepartament de Química, Universitat Autònoma de Barcelona, Bellaterra 08193, Catalonia, Spain. E-mail: albert.rimola@uab.cat
bAccademia delle Scienze di Torino, Via Maria Vittoria, 3, 10123 Torino, Italy
First published on 26th June 2025
Abstract
Molecular hydrogen (H2) stands as the most abundant molecule within the interstellar medium (ISM), primarily originating from the coupling of two H atoms on the surfaces of dust grains. The role of dust grains during the H2 formation is of third bodies, dissipating the nascent reaction energy and thereby stabilizing the newly formed molecule and preventing it from dissociating back. Whether the formed H2 remains adsorbed or not on the surface (in this latter case undergoing chemical desorption, CD) largely depends on the type of grain and its capability to absorb the reaction energy excess. In diffuse interstellar clouds, dust grains are typically bare and are composed primarily of silicates or carbonaceous materials, while in denser regions they are covered in ices mostly of water. While water-ice-covered grains have been elucidated to be efficient third bodies, the behavior of carbonaceous grains is still unknown. In this study, ab initio molecular dynamics (AIMD) simulations are employed to analyze how the reaction energy is distributed between the newly formed H2 and a large graphene slab, as a model of carbonaceous grains in diffuse clouds, and assess the feasibility of CD. The results indicate that only a fraction of the reaction energy is absorbed by the surface, leaving the newly formed H2 with sufficient internal energy for CD to occur.
1 Introduction
Molecular hydrogen (H2) is the most abundant molecule in the Universe and plays a fundamental role in the evolution of the interstellar medium (ISM). It is a major contributor to the cooling of interstellar clouds, a fundamental process that triggers star formation and drives galaxy evolution.1,2 Additionally, H2 serves as a key intermediate in the formation of larger molecules due to its ability to generate the transient H3+ species, an exceptional protonation source that initiates complex ion–molecule reactions, ultimately leading to the synthesis of numerous interstellar molecules.3,4The efficiency of H2 formation depends on the physical and chemical conditions of the ISM, which vary across different regions, including diffuse and dense molecular clouds, each with distinct characteristics. Diffuse clouds are characterized to present low atomic densities (10–500 atom cm−3) and gas temperatures between 50–100 K.5 Due to the low density, ultraviolet (UV) photons and cosmic rays can penetrate the cloud, destroying any chemical compound of more than three atoms. Thus, in diffuse clouds, the gas–grain interaction can be envisioned as bare grains, primarily composed of silicates6 and carbonaceous materials,7 with atoms and diatomic species adsorbed on them. In contrast, molecular clouds are denser (104–105 atom cm−3) and with lower temperatures (5–10 K).5 Here, the higher density largely prevents UV and cosmic rays from penetrating the cloud, and hence, relatively more complex chemical species can survive. Thus, the refractory material of the grains is covered in ices of volatile species due to their accretion and in situ formation on their surfaces, forming a core-mantle grain structure. The dominant species of the ices is H2 O (the most abundant solid-state species in the ISM), along with CO, CO2, NH3, CH3 OH, CH4, among others.
Interstellar grains are essential in astrochemistry as they serve as solid-state supports for chemical reactions.8–13 Their role can be divided into four main functions: (i) as reactant concentrators,14,15 trapping molecules on their surfaces to facilitate reactions; (ii) as reactant suppliers,16–18 directly participating themselves in reactions; (iii) as chemical catalysts,19–22 providing alternative reaction pathways with lower activation energy; and (iv) as third bodies,23–26 absorbing the excess of energy released by largely exoergic chemical reactions, this way stabilizing the newly formed products.
H2 formation can result from the coupling of two hydrogen atoms. This H + H association reaction is thermodynamically largely favorable, with a reaction energy of ≈−440 kJ mol−1 (≈−4.50 eV). However, to ensure the stability of the newly formed molecule, this energy excess must be dissipated; otherwise, H2 would dissociate back to reactants. In the gas phase, direct association of two H atoms is extremely inefficient due to the need for radiative stabilization via quadrupole transitions with lifetimes on the order of 100 years. This renders such processes practically irrelevant under interstellar conditions. Radiative association processes (e.g., H + H+ or H + H−) are also insufficient, as their rate constants are 6 orders of magnitude lower than required to explain the observed abundance of H2 in the ISM. Therefore, it is widely accepted that H2 primarily forms on the surfaces of interstellar dust grains.27,28
Acting as third bodies, grains do not participate directly in the reaction, but they facilitate the formation of H2 by absorbing the energy released during the reaction. The third-body effect is essential not only for stabilizing the newly formed molecule but also for promoting its chemical desorption (CD). CD is a chemically driven process in which the newly formed molecule is ejected from the surface into the gas phase due to the energy released by the reaction. This excess energy can lead to localized heating of the surface,29 or it may be retained as internal energy in the product (e.g., translation or rotation).30 Experimental and theoretical studies31–38 have highlighted the importance of CD in astrochemistry, particularly in cold ISM environments where thermal desorption is ineffective, thus explaining the unexpected gas-phase abundance of certain molecules.
Previous studies23,24,26 have investigated the third-body effect of water-ice-covered grains39–41 in various chemical reactions, like the formation of HCO,23 NH3,24 PH,25 as well as H2via H + H association.26 These studies focused on dense molecular clouds, where water ice mantles were found to be particularly efficient in dissipating reaction energies. This efficiency was attributed to an effective vibrational coupling between the newly formed products and the water molecules of the ice. Nevertheless, within diffuse clouds, the capability of the grain refractory materials as third bodies has never been explored, which is here hypothesized to be different due to their different heat capacity compared to water ice and the different nature of the molecule/surface interactions. Investigating their role is, therefore, critical to understanding formation in such environments. To this aim, in the present work, we address the H2 formation on graphene (as a paradigmatic case of interstellar carbonaceous material) to assess its function as third body and the likelihood of CD of the newly formed H2 molecule.
H2 formation on surfaces has been postulated to occur via three mechanisms: Langmuir–Hinshelwood (LH), Eley–Rideal (ER), and hot-atom (HA). In the LH mechanism, two adsorbed H atoms diffuse and react to form H2, a process supported by both experimental42–46 and theoretical studies.47–49
Adsorption of hydrogen atoms on carbonaceous surfaces can occur via physisorption and chemisorption, as demonstrated in numerous experimental and theoretical studies.46,50–56 In the physisorbed state, H atoms are weakly bound through van der Waals interactions and preferentially located above the centre of the carbon hexagons, with an equilibrium distance of approximately 3.0 Å from the surface.57 In contrast, chemisorption involves the formation of stronger covalent bonds directly atop carbon atoms, occurring at shorter equilibrium distances, around 1.8 Å. This bonding requires lattice reconstruction, where the carbon atom involved rehybridizes from sp2 to sp3 and puckers out of the graphene plane by ≈0.36 Å.58 Theoretical studies support that H atoms, once adsorbed, can diffuse/recombine through quantum tunnelling or thermal hopping.55,59–62 A comprehensive review63 has examined H2 formation on various surfaces, including carbonaceous materials, highlighting key processes such as adsorption, diffusion, energy dissipation, and CD. The associative desorption of H2 and D2 from chemisorbed hydrogen on highly oriented pyrolytic graphite (HOPG) surfaces at 2100 K has been studied experimentally.64 These investigations reveal that a significant portion of the total reaction energy (≈328 kJ mol−1 or 3.4 eV) is converted into translational energy with distributions peaking near 125 kJ mol−1 (1.3 eV). Quantum mechanical simulations further indicate that 40–50% of the total energy from H2 formation via the ER mechanism is carried away as translational energy of H2, the remaining energy being distributed between the internal energy of the product and transferred to the surface.65 Several studies have investigated the vibrational excitation of nascent H2. Some report low excitation levels with most molecules desorbing in the vibrational ground state (ν = 0).44,65,66 Others, however, have found significant excitation with a substantial fraction of H2 emerging in higher vibrational states such as ν = 4 and ν = 5.49,67,68
Despite significant progress in understanding H2 formation on surfaces, the mechanisms of energy dissipation and the role of grains as third bodies remain partly unexplored. A particularly powerful approach for studying the third-body effect is by means ab initio molecular dynamics (AIMD) simulations. These simulations incorporate electronic structure while treating nuclei classically, accounting for bond formation, breaking, and product behavior. In particular, microcanonical (NVE) AIMD simulations, where total energy remains constant, help analyze energy distribution of a chemical reaction, namely, in the form of potential energy and kinetic energy, and for the latter term, whether retained by the newly formed molecule or transferred to the surface.
A recent study69 employing a similar ab initio molecular dynamics approach investigated H2 formation on C60 fullerene, primarily focusing on the ER mechanism following hydrogen atom impact. While they also explored LH reactions across a broad temperature range (10–4000 K), representing thermal shock, their emphasis remained on structural aspects of bond formation and surface reactivity. In contrast, our work focuses on the energy partitioning during H2 formation, specifically tracking how the reaction energy is distributed between the product and the surface. Additionally, we provide detailed insight into the vibrational excitation state of the nascent H2 molecule and the energy dissipation dynamics within the carbonaceous grain.
In this work, we employ AIMD simulations grounded on the density functional theory (DFT) to investigate energy dissipation and the feasibility of CD during H2 formation on graphene. The formation of H2 through the coupling of H atoms is assumed to proceed via an LH mechanism, where adsorbed H atoms diffuse and react on the graphene surface. AIMD calculations, combined with graphene models, provide an ideal framework to capture temperature effects and surface conditions relevant to diffuse interstellar clouds, allowing us to examine their impact on energy partitioning and the feasibility of CD processes.
2 Methodology
2.1 Atomistic slab model for graphene
Carbonaceous materials cover diverse systems, including amorphous carbon with aliphatic H-rich and aromatic H-poor components, graphite, silicon carbide, and polycyclic aromatic hydrocarbons (PAHs). In this study, we use a graphene surface as an atomistic model to represent carbonaceous materials. Graphene effectively captures the properties of graphite, PAHs, and partially aromatic H-poor carbonaceous materials in the context of H2 formation.The graphene structure was modeled using a periodic approach, starting from its primitive unit cell,70 which was expanded into a (15 × 15) supercell resulting in a single-layer graphene sheet composed of 512 carbon atoms (see Fig. 1). This extended structure provides a sufficiently large surface to minimize finite-size effects. Upon optimization, the lattice parameters of the unit cell are a = 39.47 Å, b = 39.47 Å, and (α = β = 90°) and (γ = 120°). A 40 Å vacuum gap was introduced along the out-of-plane (c = 40 Å) to prevent interactions between simulation boxes.
 | ||
| Fig. 1 Top view of the super cell unit of the graphene slab model used in this work. | ||
2.2 Computational details
Both static calculations and AIMD simulations were performed with the CP2K package,71 specifically employing the Quickstep module.71The exchange–correlation energy was described using the generalized gradient approximation (GGA) PBEsol functional,72 which offers a good balance between computational efficiency and accuracy, and is particularly well-suited for solid-state systems as supported by previous benchmarking studies.73,74 Additionally, we also performed preliminary calculations for the H2 formation in the gas phase (i.e., in absence of the graphene surface) and found that the reaction energies calculated at PBEsol and CCSD(T)-F12 are in excellent agreement (results available in the ESI†). To account for dispersion interactions, the D3 correction with Becke–Johnson damping (D3(BJ)) was applied.75 Although H2 lacks a net electric dipole moment, it possesses a significant electric quadrupole moment, which leads to only weak interactions. As a result, dispersive forces arising from quadrupole moment interactions (like those present in the H2/graphene system) play a crucial role. Previous studies have shown that neglecting these interactions can significantly underestimate binding energies (e.g., ref. 76).
Core electrons were described using Goedecker–Teter–Hetter pseudopotentials,77 while a hybrid approach combining Gaussian and plane waves was used to describe valence electrons.78 A contracted Gaussian double-ζ MOLOPT basis set79 with a single polarization function (DZVP) was employed, along with plane waves with an energy cutoff set at 500 Ry.
The bare surface was first optimized by relaxing atomic positions and cell parameters. The optimized lattice parameters were then fixed for both static calculations and AIMD simulations, allowing only atomic positions to relax. Static calculations were performed to characterize the potential energy surfaces (PESs) of the H2 formation reaction by locating the stationary points corresponding to reactants, products, and transition states (TSs). The reactant geometries were first optimized in the triplet electronic state to prevent spontaneous H2 formation due to the Pauli exclusion principle. This optimized triplet structure was then used for a second optimization in the open–shell singlet biradical system. To locate TSs, we used the climbing image nudged elastic band (CI-NEB) method in CP2K71 and vibrational analysis was performed to confirm their structures. All calculations were carried out adopting the broken symmetry approach,80 which is essential when dealing with systems involving open–shell singlet biradical systems.
AIMD simulations were run within the (NVE) microcanonical ensemble, which maintains a fixed number of particles (N), constant volume (V), and constant total energy (E). These simulations were run for two picoseconds with a timestep of 0.2 femtoseconds. Simulations were performed at varying initial temperatures (10, 30, and 50 K) to model different conditions typical of interstellar clouds. The dynamics of the system were propagated in time to observe the formation of the new H2 molecule and analyze its subsequent behavior. Throughout the simulations, the energy released by the reactions was tracked by calculating the kinetic energy from the particle velocities along the trajectories. This approach allowed us to decompose the kinetic energy into contributions from the newly formed H2 molecule and the graphene surface. Moving averages were calculated over 50 steps of the molecular dynamics trajectory to estimate observables. In cases where due to the random initial atomic velocities assigned in CP2K, we were not able to obtain H2 product to form, we applied the cascade method. This method provides an additional 2.5 kJ mol−1 of energy to each H atom in the correct direction, helping to drive the reaction forward while minimally perturbing the overall system dynamics.
To determine the vibrational states of the H2 molecule during AIMD simulations, we constructed the H–H PES by scanning the H2 bond length from 0.3 to 3.0 Å in 0.01 Å steps. The resulting energy profile was fitted to a Morse potential, providing the equilibrium bond length, dissociation energy, and force constant. These parameters determined the vibrational energy levels, turning points, and equilibrium position, which were then compared with AIMD trajectories to assign the vibrational state at each oscillation.
To study how energy transfers from the excited H2 molecule to phonon modes of graphene, we used the trajectory analyzer and visualizer (TRAVIS),81,82 a program designed for analyzing and visualizing trajectories from molecular dynamics simulation, including AIMD simulations. In particular, TRAVIS was here employed to calculate the vibrational density of states (VDOS) using the Fourier transform of the velocity autocorrelation function from the simulation trajectories. The VDOS helped identify surface vibrational modes involved in energy dissipation, providing insight into the interaction between graphene phonons and excited H2 and the overall energy relaxation process.
2.3 Binding energy calculations
The binding energy (BE) of H2 on the graphene surface was calculated to assess their interaction strength. The adsorption energy, ΔEads, was first calculated as:| ΔEads = ECPLX − (Egraphene + EH2) | (1) |
To correct for Basis Set Superposition Error (BSSE),83 which arises when interacting molecules artificially overstabilize their energy due to basis functions overlap, the counterpoise correction84 was applied. This estimates BSSE by recalculating energies with ghost orbitals and subtracting the error:
| ΔECPads = ΔEads − BSSE | (2) |
Finally, the BE was determined as ΔECPads in opposite sign:
| BE = −ΔECPads | (3) |
3 Results and discussion
3.1 Potential energy surface of the H2 formation on graphene
Previous to the AIMD simulations, we first calculated the PES of the H2 formation on the graphene slab model to determine if the process presents an energy barrier. While the reaction is barrierless in the gas phase due to spin–spin coupling, an energy barrier could emerge due to H/graphene interactions, as seen in radical–radical couplings on water ice surfaces.85 Thus, different PESs were explored adopting an Lh mechanism and assuming that diffusion of the H atoms has already occurred, placing them in proximity to directly simulate the reaction. Three different initial H positions were considered, representing different adsorption regimes (i.e., physisorption and chemisorption), allowing us to explore the impact of the initial H adsorption states on the energy dissipation. These configurations, illustrated in Fig. 2 (see the reactant structures), are described as follows:• PP structure: both H atoms are physisorbed above the centre of two adjacent hexagons in the graphene lattice, sharing a common edge defined by two interconnected carbon atoms. The optimized structure (triplet electronic state) shows an H–H separation of 3.02 Å and H–C distances of approximately 2.86 Å, placing the H atoms about 3.0 Å above the basal plane. These values are consistent with prior studies on physisorption of H atoms on graphene.49,57
• PC structure: one H atom is chemisorbed on a carbon atom in the graphene lattice, while the second is physisorbed above the centre of the same hexagon. In the optimized triplet state, the H–H distance is 2.19 Å. The chemisorbed H atom forms a covalent bond with an H–C distance of 1.13 Å and induces a noticeable out-of-plane puckering of the bonded carbon atom. The physisorbed H atom is positioned approximately 2.79 Å above the surface.
• CC structure: both H atoms are chemisorbed on carbon atoms belonging to two adjacent hexagons. The optimized singlet open–shell structure is characterized by an H–H distance of 2.91 Å and H–C bond lengths of 1.13 Å. As in the PC case, clear puckering of the carbon atoms bonded to hydrogen takes place. This structural distortion is consistent with earlier reports.58,65
Fig. 2 also represents the stationary points of the reactions starting from each initial structure. For PP and PC, reactants are optimized in the triplet electronic state, as the singlet open–shell biradical collapses spontaneously into the H2 product, indicating a barrierless reaction.
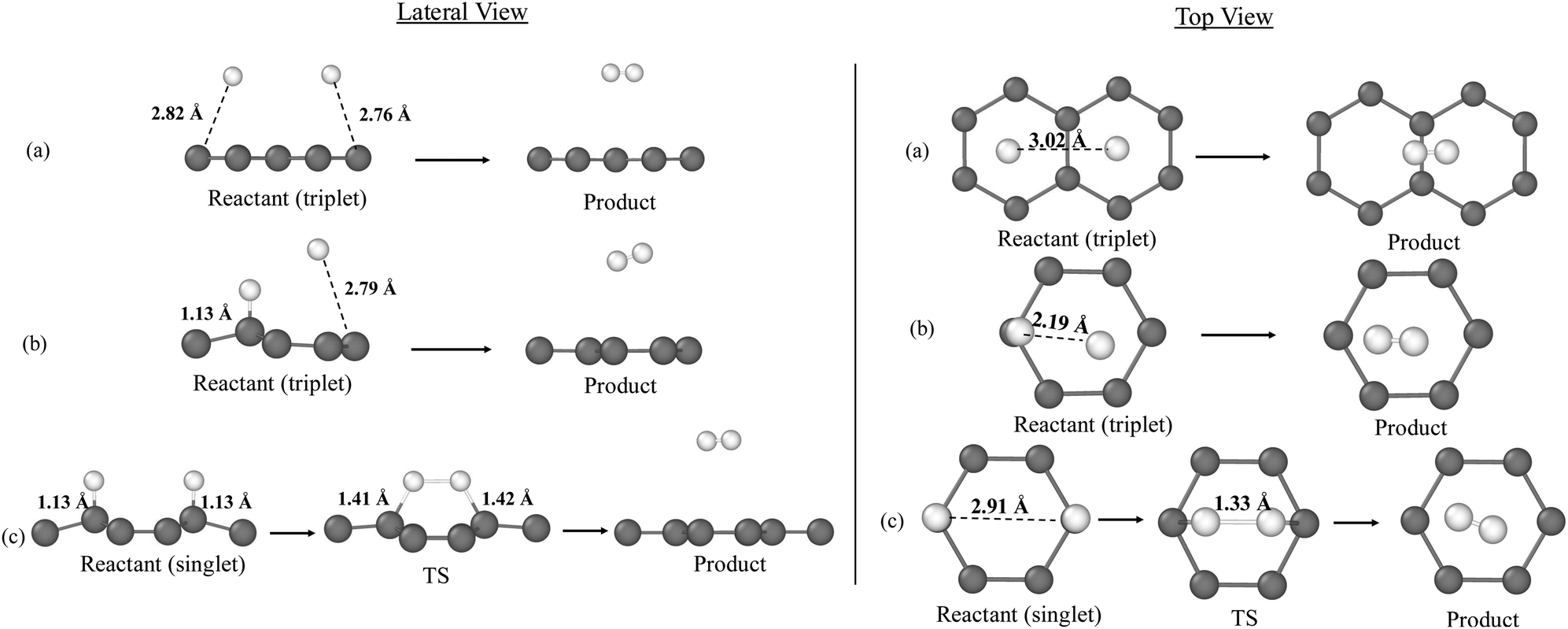 | ||
| Fig. 2 PBEsol-D3(BJ)-optimized geometries of the stationary points for the H2 formation on graphene according to the initial structures: (a) PP, (b) PC, (c) CC. For each configuration, the lateral view is displayed on the left and the top view on the right. Colour-coding: white, H atoms; grey, C atoms. | ||
In contrast, for CC, the reaction encounters a substantial potential energy barrier of 119.6 kJ mol−1 due to the H–C bonds, which must be broken before H2 can form. These bonds stabilize the CC reactant system at an energy minimum, thus reducing the energy released to −158.9 kJ mol−1 (see Table 1), significantly lower than in the gas phase.
| Initial structure | ΔErx (kJ mol−1) | ΔE‡ (kJ mol−1) |
|---|---|---|
| PP | −443.2 | Barrierless |
| PC | −356.8 | Barrierless |
| CC | − 158.9 | 119.6 |
For PP, both H atoms are physisorbed, resulting in a larger energy release of −443.2 kJ mol−1, closer to the gas-phase value. PC represents an intermediate case, where one H atom is chemisorbed, and the other is physisorbed, leading to a smaller energy release than PP but larger than CC (−356.8 kJ mol−1).
3.2 Ab initio molecular dynamics simulations of H2 formation on graphene
The results concerning the energy evolution and partitioning during the H2 formation on the graphene layer obtained from the AIMD simulations are described here. For the barrierless reactions (namely, from structures PP and PC), the optimized geometries of the reactants were used as the starting points of the AIMD simulations. For the reaction presenting a potential energy barrier (that from structure CC), preliminary AIMD simulations from the open–shell singlet reactant were performed to check if CC spontaneously evolves towards H2 formation caused by dynamic effects. As expected, given the substantial barrier, no spontaneous reaction was observed. Consequently, to facilitate the reaction, the TS was chosen as the starting structure for the AIMD simulations applying the cascade method (explained in Section 2, Methodology). Choosing the TS as the initial structure is justified by the potential role of quantum tunneling, which is not accounted for in AIMD simulations, as quantum phenomena such as tunneling cannot be captured. Tunneling is particularly significant in reactions involving hydrogen atoms and at very low temperatures. However, this approach overestimates the reaction energy to be considered in the analysis and accordingly, results for this specific case can be taken as insights and the values semi-qualitative. | ||
| Fig. 3 Evolution of the averaged energy components (in kJ mol−1) over 400 fs displayed in plots (a)–(i) depending on the initial temperature (10, 30 and 50 K): in green, the potential energy (Vtot); in orange, the total kinetic energy (Ttot); in red, the H2 kinetic energy (TH2); in blue, the graphene kinetic energy (Tgraphene); in grey, the total energy (Etot). The first data point has been subtracted from each series, normalizing all values to start at 0 kJ mol−1 except for graphene kinetic energy. | ||
The evolution of Ttot and Vtot exhibit a symmetric behavior in accordance with the principle of energy conservation. During H2 formation, the system releases energy as the H–H bond forms, leading to a sharp rise in Ttot and a corresponding drop in Vtot.
The formation of H2 is characterized by the rapid rise in TH2, followed by an increase in TGraphene, indicating an energy transfer from the energy released during H2 formation to the graphene surface.
Analyses of the TH2 and TGraphene evolutions (shown in Table 2) reveal that the amount of the reaction energy that is transferred from H2 to the surface depends on the initial temperature and the initial structure. In general, the larger the initial temperature, the larger the amount of energy transferred to the surface. While at an initial temperature of 10 K the fraction of kinetic energy retained by H2 is 0.46–0.68 (meaning an absorption of 32–54% of the energy released during the H2 formation by the graphene surface), at 50 K the fraction decreases to 0.28–0.40 (meaning an increment of the absorption of the released energy by graphene to 60–72%, see Table 2). Thus, it seems that higher temperatures improve the vibrational coupling between H2 and graphene, this way favoring the energy transfer between the two partners. However, we observe an exception in PP at 10 K, where the energy transfer is higher than at 30 K. This coincides with the transient formation of a C–H bond along the trajectory of PP at 10 K, which favors a direct, enhanced energy transfer.
| Structure | Temperature | t des |
|
|
trans-T | trans-Tz | BE | Vibrational state |
|---|---|---|---|---|---|---|---|---|
| PP | 10 | 172.4 | 0.38 | 0.62 | 200.6 | 11.4 | 4.6–4.9 | 14 → 10 |
| 30 | 117.6 | 0.32 | 0.68 | 203.3 | 21.1 | 4.6–4.9 | 14 → 14 | |
| 50 | 116.4 | 0.60 | 0.40 | 209.7 | 28.5 | 4.6–4.9 | 14 → 12 | |
| PC | 10 | 129.4 | 0.32 | 0.68 | 106.7 | 48.4 | 4.6–4.9 | 10 → 9 |
| 30 | 131.2 | 0.55 | 0.45 | 105.2 | 43.1 | 4.6–4.9 | 9 → 9 | |
| 50 | 132.2 | 0.67 | 0.33 | 105.2 | 39.9 | 4.6–4.9 | 9 → 9 | |
| CC | 10 | 59.6 | 0.54 | 0.46 | 100.1 | 86.7 | 4.6–4.9 | 3 → 3 |
| 30 | 58.8 | 0.71 | 0.29 | 100.4 | 87.7 | 4.6–4.9 | 3 → 2 | |
| 50 | 58.2 | 0.72 | 0.28 | 101.1 | 89.0 | 4.6–4.9 | 3 → 2 |
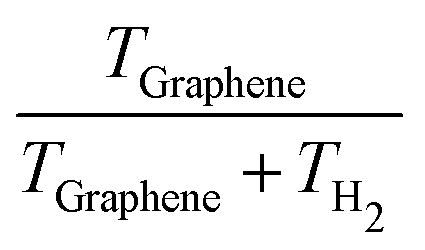

Clearly, once formed, the H2 molecule retains a significant amount of the reaction energy in the form of internal kinetic energy, which is distributed among its degrees of freedom, that is, among the vibrational frequency (which becomes excited in the moment of the H2 formation) and the rotational and translational components. Translational kinetic energy (trans-T) of H2 along the x and y directions contributes to the diffusion of H2 across the graphene surface. However, the z component of trans-T (perpendicular to graphene) is crucial for desorption, as it drives the possibility of CD. If the z component of the translational kinetic energy (trans-Tz) of H2 exceeds the binding energy (BE) of H2 on graphene, the molecule has enough internal energy to overcome the interaction with the surface as to be desorbed and ejected into the gas phase. This is what is observed in the simulations, which is explained as follows.
The BEs calculated for the distinct positions of H2 on graphene span the 4.6–4.9 kJ mol−1 range, which agree with BE values reported in previous studies.86,87 CD of H2 takes place within 58.2 to 172.4 fs after its formations (see Table 2). In these cases, graphene absorbs a small fraction of the energy released during H2 formation, meaning that the rest is retained by the molecule. Accordingly, trans-Tz values for all the trajectories (between 11.4 and 89.0 kJ mol−1, depending on the initial temperatures and the positions, see Table 2) are significantly larger than the H2 BEs such that CD of the newly formed H2 molecule thus takes place.
In the interstellar medium (ISM), the vibrational state of newly formed H2 molecules is influenced by the likelihood of these molecules leaving the grain surface before being “thermalized” through interactions with the grain surfaces.88 The level of vibrational excitation in these H2 molecules has significant implications for two key astrophysical areas: (i) facilitating gas-phase reactions with high activation energy barriers, which are crucial for initiating the chemistry in molecular clouds;89 and (ii) enabling the potential detection of newly formed H2via emission resulting from its vibrational relaxation49,65 through weak quadrupole transitions that can be detected with e.g., the NIRSpec and MIRI instruments of JWST, especially in warm molecular gas (T > 100 K) such as in shocked regions, photodissociation regions, star-forming regions and circumstellar disks, thereby allowing for the measurement of formation rates in molecular clouds and providing constraints for theoretical astrochemical models. Because of that, we analyse the level of vibrational excitations of H2 once it is formed and its degree of relaxation on the surface and, to be the case, upon CD.
By adopting the procedure explained in Section 2 (Methodology), we identify that, in almost all the cases, H2 is immediately excited once it is formed. Table 2 shows the vibrational states for each initial structure. For PP, H2 initially forms in highly excited vibrational states, specifically at ν = 14, indicating that a significant amount of energy is stored in bond oscillations. At 30 K, there is no observed vibrational relaxation immediately after formation. However, at 10 K and 50 K, the final vibrational state decreases to ν = 10 and ν = 12 respectively, suggesting that some vibrational energy relaxation occurs over time. Remarkably, largest vibrational relaxation occurs at 10 K, which is consistent with the largest energy transfer due to forming the transient C–H chemical bond (see above). In contrast, for CC, H2 forms in a much lower vibrational state (ν = 3), which is consistent with the lower reaction energy, with only a slight relaxation observed after desorption at 30 K and 50 K, lowering the vibrational state to ν = 2. For PC, H2 is initially produced in a mid-to-high vibrational state at ν = 9–10. After desorption at 10 K, a small vibrational relaxation occurs, reducing the vibrational state from ν = 10 to ν = 9. However, at higher temperatures of 30 K and 50 K, no vibrational relaxation is observed.
Our simulations also highlight a correlation between the initial binding configuration of surface H atoms and the vibrational energy retained by the desorbing H2 molecule. While this may appear intuitive, our analysis quantifies this effect and emphasizes how specific binding sites influence the internal energy budget of the product. For CC, where chemisorbed hydrogen atoms recombine, the lower reaction energy results in H2 formed in a lower vibrational state. Conversely, for PP, a significant amount of energy is released upon recombination, leading to the formation of H2 in highly excited vibrational states. PC represents an intermediate case, with moderate energy release and an initial vibrational state between the one of CC and PP. Additionally, our results show little to no vibrational relaxation after desorption, suggesting that energy dissipation through vibrational relaxation is minimal in these processes.
Previous experimental work67 highlighted that recombination of (apparently physisorbed) H and D atoms on a highly oriented pyrolytic graphite (HOPG) surface at 15 K forms HD in ν = 5–7 vibrational states. Theoretically,68 the H2 formation on HOPG has been studied, in this case adopting an ER mechanism in which a chemisorbed H atom directly reacts with an incoming, gaseous H atom. To compare with the experimental data, authors performed a set of approximations to determine the vibrational states of a newly formed HD, showing a similar ν = 5–7 vibrational state distribution. This later study would be consistent with our PC initial structures, in which H2 forms and keeps at ca. ν = 9. Although the results of these studies do not entirely agree, it is important to note that we are here considering H2 formation, while the others focus on HD formation. Because HD has a larger reduced mass, its vibrational energy levels are more closely spaced than those of H2. As a result, if the total energy transferred into the vibrational degree of freedom is similar for both molecules, vibrational state distribution of HD will center around a higher ν quantum number. However, assuming the energy transfer occurs on similar timescales, HD relaxation involves more vibrational states due to their smaller energy gaps, leading to a lower final vibrational state compared to H2. This can help reconcile the differences between the studies.
To further investigate the energy transfer process, we computed the VDOS power spectrum (which highlights the surface vibrational modes during the simulations) considering only the graphene surface, both in its isolated state and during the reaction but neglecting the atoms of H2. The difference between these two VDOS spectra (ΔVDOS) reveals the changes in the vibrational mode populations of graphene along the trajectories, thereby capturing the changes in the population of the surface vibrations due to the reaction. Thus, by examining the increase in the population of the graphene vibrational modes (reflected by an increase of the ΔVDOS) we can determine the magnitude of the vibrational coupling between the H2 molecule and the graphene surface, gaining insight into the mechanism that governs the energy dissipation. Fig. 4 shows the calculated ΔVDOS for the H2 formation on the different structures and temperatures.
 | ||
| Fig. 4 Difference of the simulated VDOS power spectrum (ΔVDOS) displayed in plots (a)–(i) depending on the initial temperature (10, 30 and 50 K), between the computed trajectories for the graphene surface when the reaction takes place and for the isolated layer. | ||
Graphene presents a low-frequency region (ν = 0–1000 cm−1) assigned to out-of-plane bending vibrations, and a higher frequency region (ν = 1000–1800 cm−1) assigned to in-plane bending and stretching vibrations. According to ΔVDOS, in all the structures, these frequency regions become more populated, indicating that the main mechanism responsible for the dissipation of the large excess of the reaction energy is the intermolecular vibrational coupling between the vibrational mode of the newly born H2 and the graphene phonon modes. As expected, in CC (that with the highest energy transfer), the graphene vibrations become more populated than in PP and PC, suggesting a stronger coupling effect in CC. Additionally, in PC at 10 K, we observe a population increase similar to CC, consistent with the transient C–H bond formation in this case, which likely contributes to the high energy transfer observed.
However, the vibrational coupling between H2 and graphene is of limited efficiency. By comparing these results with those of Ferrero et al.24 in the formation of NH3 on amorphous water ice surface, the increase of the population of the vibrational modes of the water ice (particularly the low-frequency libration modes) is significantly greater than that occurring in the graphene surface. The difference is due to the fact that the interaction of NH3 with water ice surface takes place mainly through hydrogen bonds, which establish a direct path towards favorable a vibrational coupling between the two components, while the interaction between H2 and graphene is essentially through dispersion forces, giving rise a less efficient vibrational coupling.
Our results, thus, confirm the energy dissipation mechanism occurring during the H2 formation on graphene. Upon H2 formation, the molecule enters a highly excited vibrational state due to the significant energy released during the reaction, which is momentarily stored in its vibrational degree of freedom. Over time, this energy is transferred to the graphene surface, resulting in a progressive decrease in the vibrational state of the molecule. This energy transfer manifests as an increase in TGraphene and in the population of its vibrational modes. The energy transfer is driven by the coupling between the H2 highly excited vibrational mode and the graphene lattice phonon modes, these latter serving as key elements for absorbing and redistributing the energy released during the reaction.
Finally, to further investigate the spatial distribution of the energy dissipation throughout the graphene surface, we analyzed the transfer of energy from the reaction site to the surrounding environment. This analysis involved examining the distribution of kinetic energy per carbon atom across four concentric shells centered on the H2 formation reaction site. The spheres were defined with radii of 5, 10, 15, and 20 Å, with each shell containing atoms not included in the smaller shells. Here, for the sake of brevity and in view that the trends are general, we only show the results for PP at the different temperatures (Fig. 5), while those for the other structures are available in the ESI.†
 | ||
| Fig. 5 Evolution of the averaged TGraphene (normalized per carbon atom) over 400 fs (in kJ mol−1) displayed in plots (a)–(c) depending on the initial temperature (10, 30 and 50 K), in concentric shells centered at the H2 formation reaction site in the PP structure at the different temperatures. | ||
Notable variations in the energy distribution across the shells of carbon atoms can be distinguished. Close to the reaction site, the first shell exhibits a sharp, highly localized peak. This energy then propagates outward like a wave, with the second shell experiencing a smaller, slightly delayed peak. This delay increases with each successive shell as the energy moves through the lattice. The highest peak occurs in the first shell, confirming that the reaction energy is initially concentrated at the reaction site. The energy dissipates rapidly as graphene efficiently distributes the energy across its structure until it becomes evenly spread over the surface. Ultimately, TGraphene stabilizes, becoming uniformly distributed across the graphene sheet and preventing any localized energy concentration. However, as temperature increases, both the peak value in the second shell and the baseline level of the shell become higher. This indicates that at elevated temperatures, the ability of graphene to absorb and retain energy is enhanced.
In Pantaleone's work, different initial structures were used, which differ on the surface morphology of the H adsorption sites. However, in all the cases, the reactive H atoms were physisorbed on the water ice surfaces and with an H to H distance of about 4 Å. Due to that, all the tested reactions were found to be barrierless, by means of both static calculations (aiming to characterize the PES) and AIMD simulations. This is similar to what happens on graphene when at least one H atom is physisorbed. But when both atoms are chemisorbed, an unfeasible situation on ices, the reaction has a potential energy barrier, which is associated with the H–C chemical bond cleavage to form H2.
Another interesting aspect to compare is the capability of the surfaces to absorb and dissipate the energy released by the reaction. On both surfaces, the newly formed H2 molecule relaxes, meaning that there is an energy transfer from the molecule to the surfaces. However, the fraction of energy transferred to the surface is different: on water ices, the surfaces absorbed about 45–65% of the released energy, while on graphene between 32–54%. From this data clearly emerges the fact that water ices are more efficient third bodies than graphene, as already mentioned above when comparing the vibrational population of a newly formed NH3 molecule on water ice. The reason of this difference is the major capacity of water ice to vibrationally couple with the adsorbates, due to being a material presenting local dipole moments, at variance with graphene.
Interestingly, such a smaller capacity to absorb the reaction energy by graphene compared to water ice is important as regards CD. Indeed, on graphene, CD of the newly H2 formed molecule is observed in almost all the cases, precisely because the H2 retains a notable fraction of the released energy in the form of kinetic energy, which has not been transferred to the surface. In contrast, on water ice surfaces, CD is observed in only one case. In Pantaleone et al., the analysis of the trans-Tz was not done, and CD was predicted to occur on the basis that the total trans-T was greater than 10 times the H2 BE. Furthermore, CD was observed to occur when the formed H2 molecule diffuses across the surface and stumbles over a surface irregularity (in that case, a dangling H atom of an outermost H2 O molecule of the ice), in which all the trans-T is channeled towards the desorption direction. This is different from the H2 CD on the graphene layer, as it is an ideal surface without defects. Accordingly, on graphene, the CD is purely driven by the amount of trans-Tz contained by H2 upon formation.
The different capability of the surfaces to dissipate the reaction energy excess and the chances to have CD is also reflected in the vibrational states of the emerging H2 molecule. On water ices, the H2 molecule also forms in a vibrationally excited state (between ν = 4–6), which lowers to ν = 1–2 along the simulation. On graphene, however, we observe a strong dependence of the initial excited vibrational state of H2 with the initial H positions, which in turn is regulated by the reaction energy. When the two H atoms are physisorbed (PP structure, presenting the most favorable reaction energy), the H2 molecule forms in highly excited vibrational states (ν = 14); when the two H atoms are chemisorbed (CC structure, presenting the less favorable reaction energies) the H2 molecule forms in low vibrational states (ν = 3); when one H atom is physisorbed and the other chemisorbed (PC structure, presenting an intermediate reaction energy) the H2 molecule forms in moderately high vibrational states (ν = 9–10).
4 Conclusions
This study investigates the role of graphene, as a test case of cosmic carbonaceous material, as a third body in energy dissipation during interstellar H2 formation on its surface. Using (NVE) AIMD simulations, the partitioning of the reaction energy excess at three interstellar temperatures and considering different initial H adsorption states has been examined, revealing how energy is distributed between graphene and the newly formed H2 molecule.Our results show that between 32% and 72% of the reaction energy is transferred to the graphene surface, with higher temperatures enhancing energy transfer. Such values infer that in most of the cases, H2 chemical desorption takes place due to the highly exothermic reaction providing enough energy to overcome the H2 binding energy on graphene.
Energy dissipates through vibrational coupling, populating the vibrational modes of graphene. While graphene is less efficient at absorbing energy compared to water ice due to its stiffness and the weaker H2/graphene intermolecular interactions (based on dispersion forces), the energy transfer occurs quickly, dissipating and reaching an uniform distribution across the surface. Notably, sites where H atoms were initially chemisorbed exhibit larger vibrational population, leading to greater energy transfer. As a result, the H2 molecule formed from this initial adsorption state is in low vibrational states, as it is able to transfer their excess energy more effectively to the surface. In contrast, when H atoms are physisorbed, the vibrational population of the graphene modes is smaller, meaning a lower energy transfer to the surface. Consequently, for these cases, newly formed H2 molecules are released into the gas in medium-high vibrational states.
Our analysis primarily focuses on the vibrational excitation of nascent H2. Although rotational excitation is known to play a significant role in surface recombination reactions,65,66 this aspect is not included in the present study. Clearly, incorporating rotational degrees of freedom would provide a more complete picture of energy partitioning during H2 formation.
One key limitation of AIMD is neglecting the zero-point energy correction, which can lead to an underestimation of the actual energy available in the system. Additionally, AIMD treats energy transfer classically, while in reality, vibrational relaxation occurs in discrete quantum steps. Studying this process through ring polymer molecular dynamics simulations in the future would help to better understand the vibrational relaxation dynamics of H2 on graphene.
In view of the present results, we consider that exploring other molecules and surfaces is essential to generalizing (or not) these findings in astrophysical environments. Extending this approach to different chemical species and grain surfaces of different nature, such as amorphous carbon, silicates, or mixed-composition grains, would provide a broader understanding of energy dissipation mechanisms and chemical desorption phenomenon.
Author contributions
Léana Jubert: data curation (equal); formal analysis (equal); investigation (equal); methodology (equal); software (equal); validation (equal); visualization (equal); writing – original draft (lead). Berta Martínez-Bachs: data curation (equal); formal analysis (equal); investigation (equal); methodology (equal); software (equal); supervising (equal); validation (equal); visualization (equal); writing – review & editing (supporting) Gerard Pareras: data curation (equal); formal analysis (equal); investigation (equal); methodology (equal); software (equal); supervising (equal); validation (equal); visualization (equal); writing – review & editing (supporting) Albert Rimola: conceptualization (lead); funding acquisition (lead); investigation (equal); project administration (lead); resources (lead); supervision (equal); validation (equal); writing – review & editing (lead).Conflicts of interest
There are no conflicts to declare.Data availability
The data underlying this article are freely available in Zenodo at https://doi.org/10.5281/zenodo.15005628.Acknowledgements
This project has received funding within the European Unions Horizon 2020 research and innovation program from the European Research Council (ERC) for the projects “Quantum Chemistry on Interstellar Grains” (QUANTUMGRAIN), grant agreement no. 865657, and the European Unions Horizon Europe research and innovation program under the Marie Sklodowska-Curie grant agreement no. 101105235 for the funding of CHAOS project. The Spanish MICINN is also acknowledged for funding the projects PID2021-126427NB-I00 and CNS2023-144902. The authors thankfully acknowledge the supercomputational facilities provided by CSUC. A. R. acknowledges Accademia delle Scienze di Torino for supporting the project “In silico interstellar grain-surface chemistry”. A. R. gratefully acknowledges support through 2023 ICREA Award.References
- S. Schlemmer, Angew. Chem., Int. Ed., 2011, 50, 2214–2215 CrossRef CAS PubMed.
- J. Le Bourlot, G. Pineau des Forêts and D. R. Flower, Mon. Not. R. Astron. Soc., 1999, 305, 802–810 CrossRef CAS.
- T. R. Geballe, Philos. Trans. R. Soc., A, 2000, 358, 2503–2513 CrossRef CAS.
- T. Oka, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12235–12242 CrossRef CAS PubMed.
- T. P. Snow and B. J. McCall, Annu. Rev. Astron. Astrophys., 2006, 44, 367–414 CrossRef CAS.
- T. Henning, Annu. Rev. Astron. Astrophys., 2010, 48, 21–46 CrossRef CAS.
- B. Draine, Annu. Rev. Astron. Astrophys., 2003, 41, 241–289 CrossRef.
- W. D. Watson and E. E. Salpeter, Astrophys. J., 1972, 174, 321 CrossRef CAS.
- W. Duley and D. Williams, Interstellar Chemistry, Academic Press, 1984 Search PubMed.
- D. A. Williams, Astrochemistry, 1987, pp. 531–536 Search PubMed.
- E. Herbst, Chem. Soc. Rev., 2001, 30, 168–176 RSC.
- E. Herbst, Q. Chang and H. M. Cuppen, J. Phys.: Conf. Ser., 2005, 6, 18 CrossRef CAS.
- T. Suhasaria and V. Mennella, Front. Astron. Space Sci., 2021, 8, 655883 CrossRef.
- N. Watanabe and A. Kouchi, Astrophys. J., 2002, 571, L173 CrossRef CAS.
- A. Rimola, V. Taquet, P. Ugliengo, N. Balucani and C. Ceccarelli, Astron. Astrophys., 2014, 572, A70 CrossRef.
- A. Rimola, D. Skouteris, N. Balucani, C. Ceccarelli, J. Enrique-Romero, V. Taquet and P. Ugliengo, ACS Earth Space Chem., 2018, 2, 720–734 CrossRef CAS.
- G. Molpeceres and J. Kästner, Astrophys. J., 2021, 910, 55 CrossRef CAS.
- J. Perrero, J. Enrique-Romero, B. Martínez-Bachs, C. Ceccarelli, N. Balucani, P. Ugliengo and A. Rimola, ACS Earth Space Chem., 2022, 6, 496–511 CrossRef CAS PubMed.
- B. Martinez-Bachs, A. Anguera-Gonzalez, G. Pareras and A. Rimola, ChemPhysChem, 2024, 25, e202400272 CrossRef CAS PubMed.
- G. Pareras, V. Cabedo, M. McCoustra and A. Rimola, Astron. Astrophys., 2024, 687, A230 CrossRef CAS.
- V. Cabedo, G. Pareras, J. Allitt, A. Rimola, J. Llorca, H. H. P. Yiu and M. R. S. McCoustra, Mon. Not. R. Astron. Soc., 2024, 535, 2714–2723 CrossRef CAS.
- G. Pareras, V. Cabedo, M. McCoustra and A. Rimola, Astron. Astrophys., 2023, 680, A57 CrossRef CAS.
- S. Pantaleone, J. Enrique-Romero, C. Ceccarelli, P. Ugliengo, N. Balucani and A. Rimola, Astrophys. J., 2020, 897, 56 CrossRef CAS.
- S. Ferrero, S. Pantaleone, C. Ceccarelli, P. Ugliengo, M. Sodupe and A. Rimola, Astrophys. J., 2023, 944, 142 CrossRef.
- G. Molpeceres, V. Zaverkin, K. Furuya, Y. Aikawa and J. Kästner, Astron. Astrophys., 2023, 673, A51 CrossRef CAS.
- S. Pantaleone, J. Enrique-Romero, C. Ceccarelli, S. Ferrero, N. Balucani, A. Rimola and P. Ugliengo, Astrophys. J., 2021, 917, 49 CrossRef CAS.
- D. Hollenbach and E. E. Salpeter, Astrophys. J., 1971, 163, 155 CrossRef CAS.
- R. J. Gould and E. E. Salpeter, Astrophys. J., 1963, 138, 393 CrossRef CAS.
- J. Takahashi and D. A. Williams, Mon. Not. R. Astron. Soc., 2000, 314, 273–278 CrossRef CAS.
- A. Fredon, A. K. Radchenko and H. M. Cuppen, Acc. Chem. Res., 2021, 54, 745–753 CrossRef CAS PubMed.
- F. Dulieu, E. Congiu, J. Noble, S. Baouche, H. Chaabouni, A. Moudens, M. Minissale and S. Cazaux, Sci. Rep., 2013, 3, 1338 CrossRef PubMed.
- M. Minissale, F. Dulieu, S. Cazaux and S. Hocuk, Astron. Astrophys., 2016, 585, A24 CrossRef.
- M. Minissale and F. Dulieu, J. Chem. Phys., 2014, 141, 014304 CrossRef CAS PubMed.
- E. Congiu, E. Matar, L. E. Kristensen, F. Dulieu and J. L. Lemaire, Mon. Not. R. Astron. Soc.: Lett., 2009, 397, L96–L100 CrossRef.
- R. T. Garrod, V. Wakelam and E. Herbst, Astron. Astrophys., 2007, 467, 1103–1115 CrossRef CAS.
- A. I. Vasyunin and E. Herbst, Astrophys. J., 2013, 769, 34 CrossRef.
- S. Cazaux, V. Cobut, M. Marseille, M. Spaans and P. Caselli, Astron. Astrophys., 2010, 522, A74 CrossRef.
- S. Cazaux, M. Minissale, F. Dulieu and S. Hocuk, Astron. Astrophys., 2016, 585, A55 CrossRef.
- R. G. Smith, K. Sellgren and A. T. Tokunaga, Astrophys. J., 1988, 334, 209 CrossRef CAS.
- T. J. Millar and D. A. Williams, Dust and chemistry in astronomy, 1993 Search PubMed.
- A. A. Boogert, P. A. Gerakines and D. C. Whittet, Annu. Rev. Astron. Astrophys., 2015, 53, 541–581 CrossRef CAS.
- V. Pirronello, C. Liu, L. Shen and G. Vidali, Astrophys. J., 1997, 475, L69 CrossRef CAS.
- V. Pirronello, C. Liu, J. E. Roser and G. Vidali, Astron. Astrophys., 1999, 344, 681–686 CAS.
- J. S. Perry, J. M. Gingell, K. A. Newson, J. To, N. Watanabe and S. D. Price, Meas. Sci. Technol., 2002, 13, 1414 CrossRef CAS.
- G. Vidali, J. Roser, G. Manicò and V. Pirronello, Adv. Space Res., 2004, 33, 6–13 CrossRef CAS.
- N. Katz, I. Furman, O. Biham, V. Pirronello and G. Vidali, Astrophys. J., 1999, 522, 305 CrossRef CAS.
- H. M. Cuppen and E. Herbst, Mon. Not. R. Astron. Soc., 2005, 361, 565–576 CrossRef CAS.
- S. Morisset, F. Aguillon, M. Sizun and V. Sidis, J. Chem. Phys., 2004, 121, 6493–6501 CrossRef CAS PubMed.
- B. Kerkeni and D. C. Clary, Chem. Phys., 2007, 338, 1–10 CrossRef CAS.
- Z. Medina and B. Jackson, J. Chem. Phys., 2008, 128, 114704 CrossRef PubMed.
- S. Cazaux, S. Morisset, M. Spaans and A. Allouche, Astron. Astrophys., 2011, 535, A27 CrossRef.
- M. Bonfanti, R. Martinazzo, G. F. Tantardini and A. Ponti, J. Phys. Chem. C, 2007, 111, 5825–5829 CrossRef CAS.
- S. Morisset and A. Allouche, J. Chem. Phys., 2008, 129, 024509 CrossRef CAS PubMed.
- L. Hornekær, i c v Šljivančanin, W. Xu, R. Otero, E. Rauls, I. Stensgaard, E. Lægsgaard, B. Hammer and F. Besenbacher, Phys. Rev. Lett., 2006, 96, 156104 CrossRef PubMed.
- L. Hornekær, E. Rauls, W. Xu, i c v Šljivančanin, R. Otero, I. Stensgaard, E. Lægsgaard, B. Hammer and F. Besenbacher, Phys. Rev. Lett., 2006, 97, 186102 CrossRef PubMed.
- D. Bossion, A. Sarangi, S. Aalto, C. Esmerian, S. R. Hashemi, K. K. Knudsen, W. Vlemmings and G. Nyman, Astron. Astrophys., 2024, 692, A249 CrossRef CAS.
- L. Jeloaica and V. Sidis, Chem. Phys. Lett., 1999, 300, 157–162 CrossRef CAS.
- X. Sha, B. Jackson, D. Lemoine and B. Lepetit, J. Chem. Phys., 2004, 122, 014709 CrossRef PubMed.
- E. Han, W. Fang, M. Stamatakis, J. O. Richardson and J. Chen, J. Phys. Chem. Lett., 2022, 13, 3173–3181 CrossRef CAS PubMed.
- O. Biham, I. Furman, N. Katz, V. Pirronello and G. Vidali, Mon. Not. R. Astron. Soc., 1998, 296, 869–872 CrossRef CAS.
- S. Cazaux and A. G. G. M. Tielens, Astrophys. J., 2004, 604, 222 CrossRef CAS.
- T. P. M. Goumans and J. Kästner, Angew. Chem., Int. Ed., 2010, 49, 7350–7352 CrossRef CAS PubMed.
- V. Wakelam, E. Bron, S. Cazaux, F. Dulieu, C. Gry, P. Guillard, E. Habart, L. Hornekær, S. Morisset, G. Nyman, V. Pirronello, S. Price, V. Valdivia, G. Vidali and N. Watanabe, Mol. Astrophys., 2017, 9, 1–36 CrossRef.
- S. Baouche, G. Gamborg, V. V. Petrunin, A. C. Luntz, A. Baurichter and L. Hornekær, J. Chem. Phys., 2006, 125, 084712 CrossRef CAS PubMed.
- A. J. H. M. Meijer, A. Farebrother, D. C. Clary and A. J. Fisher, J. Phys. Chem. A, 2001, 105, 2173–2182 CrossRef CAS.
- S. C. Creighan, J. S. A. Perry and S. D. Price, J. Chem. Phys., 2006, 124, 114701 CrossRef PubMed.
- E. R. Latimer, F. Islam and S. D. Price, Chem. Phys. Lett., 2008, 455, 174–177 CrossRef CAS.
- S. Casolo, G. F. Tantardini and R. Martinazzo, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6674 CrossRef CAS PubMed.
- Y. Guo and D. R. McKenzie, Commun. Chem., 2025, 8, 97 CrossRef CAS PubMed.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- T. D. Kühne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Glöβ, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- M. De La Pierre, R. Orlando, L. Maschio, K. Doll, P. Ugliengo and R. Dovesi, J. Comput. Chem., 2011, 32, 1775–1784 CrossRef CAS PubMed.
- L. Maschio, M. Ferrabone, A. Meyer, J. Garza and R. Dovesi, Chem. Phys. Lett., 2011, 501, 612–618 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Ferrero, L. Zamirri, C. Ceccarelli, A. Witzel, A. Rimola and P. Ugliengo, Astrophys. J., 2020, 904, 11 CrossRef CAS.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS PubMed.
- G. Lippert, J. Hutter and M. Parrinello, Mol. Phys., 1997, 92, 477–487 CrossRef CAS.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2003, 118, 4365–4369 CrossRef CAS.
- F. Neese, J. Phys. Chem. Solids, 2004, 65, 781–785 CrossRef CAS.
- M. Brehm and B. Kirchner, J. Chem. Inf. Model., 2011, 51, 2007–2023 CrossRef CAS PubMed.
- M. Brehm, M. Thomas, S. Gehrke and B. Kirchner, J. Chem. Phys., 2020, 152, 164105 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- E. R. Davidson and D. Feller, Chem. Rev., 1986, 86, 681–696 CrossRef CAS.
- J. Enrique-Romero, A. Rimola, C. Ceccarelli, P. Ugliengo, N. Balucani and D. Skouteris, Astrophys. J., Suppl. Ser., 2022, 259, 39 CrossRef CAS.
- W. Tian, Y. Zhang, Y. Wang, T. Liu and H. Cui, Int. J. Hydrogen Energy, 2020, 45, 12376–12383 CrossRef CAS.
- J. Petucci, C. LeBlond, M. Karimi and G. Vidali, J. Chem. Phys., 2013, 139, 044706 CrossRef PubMed.
- N. Watanabe, Y. Kimura, A. Kouchi, T. Chigai, T. Hama and V. Pirronello, Astrophys. J., Lett., 2010, 714, L233 CrossRef CAS.
- M. Agúndez, J. R. Goicoechea, J. Cernicharo, A. Faure and E. Roueff, Astrophys. J., 2010, 713, 662 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp01585e |
| This journal is © the Owner Societies 2025 |