
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstrate charge transfer drives the adsorption site of metal-phthalocyanines and porphyrins on coinage metal surfaces†
Silvia
Carlotto
 ab,
Iulia
Cojocariu
cd,
Vitaliy
Feyer
ef,
Luca
Schio
g,
Luca
Floreano
g and
Maurizio
Casarin
*a
ab,
Iulia
Cojocariu
cd,
Vitaliy
Feyer
ef,
Luca
Schio
g,
Luca
Floreano
g and
Maurizio
Casarin
*a
aDipartimento di Scienze Chimiche, Università degli Studi di Padova, Via Francesco Marzolo 1, 35131 Padova, Italy. E-mail: maurizio.casarin@unipd.it
bCNR – ICMATE, Via Francesco Marzolo 1, 35131 Padova, Italy
cDipartimento di Fisica, Università degli Studi di Trieste, Via Alfonso Valerio 2, 34127 Trieste, Italy
dElettra-Sincrotrone, S.C.p.A., Basovizza S.S. 14 – km 163.5, 34149 Trieste, Italy
eForschungszentrum Jülich GmbH, Peter Grünberg Institute (PGI-6), Leo-Brandt-Strabe, 52428 Jülich, Germany
fDuisburg-Essen University, Department of Physics and Center for Nanointegration Duisburg-Essen (CENIDE), 47048 Duisburg, Germany
gIOM – CNR, Lab. TASC, Basovizza S.S. 14, km 163.5, 34149 Trieste, Italy
First published on 16th May 2025
Abstract
The Frontier electronic structure of tetraphenylporphyrinato (TPP2−) and phthalocyaninato (Pc2−) square planar transition metal complexes (MTPP and MPc; M = V, Cr, Mn, Fe, Co, Ni, Cu, and Zn) has been revisited through DFT calculations. The different ground state symmetry and spin multiplicity between MPc and MTPP of the same M is shown to originate from the different Pc2− and TPP2− ligand field, stronger in the former ligand than in the latter. The corresponding spatial localization and symmetry of the unoccupied molecular orbitals postulate unescapable geometric constraints to their overlap with the electron cloud of a crystalline metal surface. From comparison with literature experimental evidence, we show that the adsorption geometry (atomic site and azimuthal orientation) of MTPPs and MPcs on the low index crystal planes of coinage metals (CM = Au, Ag, Cu) may be predicted when two conditions are satisfied: (i) evidence of a surface → adsorbate charge transfer, (ii) absence of significant distortion of the macrocycle upon adsorption. In this regard, the overall susceptibility to charge transfer is determined by the strength of the molecular ligand field (i.e., charge transfer to MPc is more favoured than to MTPP) and inversely linked to the electronegativity of the surface atoms (being Au the most inert CM substrate thanks to its highest electronegativity).
1. Introduction
The control of the interfacial structure between organic/metalorganic molecular layers and the underlying substrate is crucial to reproducing the functional properties of the organic/metalorganic component in surface-supported devices. Even tiny alterations of the interface structure may severely modify the behavior of the overlayer both at the supramolecular and the single-molecule level, thus generating significant changes independently of the investigated property (mechanical,1 electronic,2–4 optical,5,6 magnetic7,8) or the application field (sensing, catalysis, light-to-energy conversion, molecular electronics, nonlinear optics, spintronics). Among surface-supported layers, those made up of transition-metal complexes of porphyrin-related molecules (see the upper panel of Fig. 1)‡ are particularly relevant because of the pivotal role played by porphyrins not only in fundamental biological processes such as oxygen transport and storage, the photosynthesis, and the electron transport during cellular respiration and photosynthesis,10,11 but also in technological fields ranging from electronics12 to solar cells,13 and sensors,14 thus justifying the cross-disciplinary interest towards them and motivation to develop new porphyrin-like species, whose electronic and optical properties may be tuned through molecular engineering.15–23 | ||
| Fig. 1 Idealized representation of D4h MTPP and MPc isolated molecules. White, gray, blue, and yellow spheres are representative of H, C, N, and M atoms, respectively. In the adopted framework, M lies in the origin O of the coordinate axes, M–NPy bonds are aligned with OX and OY axes, and the σh plane corresponds to the XY plane. The atom numbering adopted for the planar, aromatic macrocycle core (pmc) is that recommended by IUPAC. | ||
Unlike porphyrins, neither the H2Pc free-base nor MPc complexes (see the lower panel of Fig. 1) are present in Nature. Nonetheless, they have been attracting a growing interdisciplinary interest20,24–30 since their synthesis by serendipity at the end of the twenties of the last century31–39 because their technological potential spans a huge range of applications including catalysts,26 dyestuffs for textiles and inks, intrinsic semiconductors, chemical sensors, organic light-emitting diodes, organic photovoltaic cells, thin-film transistors, materials for nonlinear optics, spintronics, and laser recording as well.40–43
From a structural point of view, isolated MTPPs are usually characterized by a planar, aromatic macrocycle core (hereafter, pmc) consisting of four Py-like rings held together by four methine groups in m positions (see the upper panel of Fig. 1 where the adopted atom numbering is that recommended by IUPAC and m sites correspond to the 5, 10, 15, and 20 positions). Additionally, 1, 4, 6, 9, 11, 14, 16, and 19 (2, 3, 7, 8, 12, 13, 17, and 18) positions are commonly referred to as α-positions (β-positions). Similarly to MTPPs, isolated MPcs consist of four i-Ind (a benzene ring fused with Py) fragments held together by four aza-nitrogen atoms occupying the pmc m positions (see the lower panel of Fig. 1 where the adopted atom numbering is again the one recommended by IUPAC).
Upon moving from the isolated to the adsorbed species, the coordinative arrangements of the on-surface stabilized arrays of M ions may be exploited to boost free molecules' peculiar properties,44,45 to stabilize M in unusual spin and oxidation states46–53 or, in wider terms, to control and tune their chemical reactivity.54–58 As such, a crucial issue is the pmc adsorption site, usually determined by combining the outcomes of STM measurements with the results of expensive and time-consuming quantum mechanical calculations, commonly based on DFT.20,23 However, DFT-based numerical experiments have often been restricted to the generation of phenomenological descriptions rather than exploring in detail the anchoring configurations of the adsorbate–substrate pair.20,23
Li et al.25 tackled the single-molecule chemistry of MPcs on the (111) surface of group 11 elements (CM) by combining first-principles simulations with STM, which, besides information about the local adsorption geometry and the electronic properties of chemisorbed MPcs, was exploited to carry out the so-called single-molecule surgery to control the Kondo effect59 at the MPc/CM(111) interface. Nevertheless, the DFT numerical experiments carried out by Li et al.25 did not provide any atomistic modeling of the MPc–CM(111) anchoring configuration to explain, for instance, why the chemisorption site of the same MPc may be different on the (111) surface of diverse CM or why the chemisorption site of diverse MPcs may be different on the (111) surface of the same group 11 element. In this regard, we recently proposed convincing modeling of the NiTPP anchoring to the Cu(100) surface, as well as an atomistic view of the Cu(100) → NiTPP charge transfer taking place at the interface.60 Starting from the experimentally reported NiII (d8) → NiI (d9) reduction at the interface,61 the NiTPP chemisorption site and the molecular orientation on Cu(100) can be unequivocally assigned as confirmed by the structural outcomes of PED measurements.60 The charge transfer from specific substrate atoms to specific adsorbate atoms follows the spatial superposition of the symmetry-allowed substrate and molecular orbitals, according to the matching of the intramolecular atomic structure with the substrate lattice spacing. More specifically, symmetry and geometry arguments made it practically unessential to carry out costly numerical experiments to assess the chemisorption configuration. Our analysis relied on identifying the NiTPP lowest-lying unoccupied MOs and the SALCs of the substrate topmost atoms (SCu) 4s AOs involved in the charge transfer process. The latter point implies that each SCu participates in the adsorbate/substrate interaction, with its single electron occupying its 4s AO, while the electrons of the completely occupied 3d shell are simple bystanders.
The use of MTPP and MPc symmetry, orbitals, and spectra62 coupled with a descriptor able to provide information about the different nobleness of group 11 elements turned out to be Hobson's choice to set up a protocol able to provide a semiquantitative modeling of the SCM → MTPP/MPc charge transfer when MTPP/MPc complexes are chemisorbed on CM (100), (110), and (111) surfaces. In this regard, it is notable that Kepp thoroughly investigated the chemical causes of M nobleness a few years ago.63 More specifically, he considered all the group 10 and 11 elements as well as the heaviest element of group 12 (Hg), and he tested 12 different descriptors, including, by the way, the d-band center energy of the solid, the first IE and the EA, the Pauling electronegativity χ,64 the bulk polycrystalline M work function, the relativistic s-shell contraction, the oxophilicity of M, and the cohesive free energy (free energy of atomization) of the bulk M state, to conclude that the M's χ explains best the M nobleness.63 The noblest metal is then Au, whose high χ (2.54 Pauling's units, the highest among metals)65,66 is determined by concurrent effects such as the high effective nuclear charge and the contraction of the half-filled 6s AO.67 As such, Pyykkö67–69 pointed out that the ratio of relativistic and nonrelativistic 6s shell radii in the atomic GS of the elements with the atomic number Z ranging from 55 to 100 has a marked minimum for Z = 79, and there is often talk of the “gold maximum” of relativistic effects in group 11, whose elements are all characterized by the electronic configuration d10s1.65
Even though the high Auχ65,66 provides a rationale for understanding the inertness of gold surfaces independently of Miller indices and chemisorption sites’ local symmetry, genuine unpublished results and literature data will be compared with the provisions of the proposed approach in the forthcoming discussion. In more detail: (i) results on the Frontier electronic structure of isolated MTPP and MPc (M = V, Cr, Mn, Fe, Co, Ni, Cu, Zn), critically revisited and compared with literature experimental and theoretical evidence in the ESI,† are summarized in Paragraph 3.1; (ii) clean, bulk-terminated CM (100), (110), and (111) surfaces have been considered in Paragraph 3.2 by focusing on the local symmetry properties of their most common chemisorption sites; (iii) the proposed approach is applied in Paragraph 3.3 to selected MTPP/CM and MPc/CM interfaces (sub-Paragraphs 3.3.1–3.3.5) representing as many case studies.
2. Computational details
The GS electronic configurations of MPcs (ZM = 25–30) have been thoroughly investigated by multireference electronic structure methods in the past.70 Nonetheless, the GS structural and electronic properties of free MTPP and MPc complexes (ZM = 23–30) have been re-investigated herein at the DFT level by exploiting the ADF package.71 ADF numerical experiments have been carried out within the assumption of an idealized D4h symmetry72 (see Fig. 1) and by running nonrelativistic, spin-polarized calculations with generalized gradient corrections self-consistently included through the BP86 formula.73,74 A triple-ζ with a polarization function Slater-type basis set has been adopted for all the atoms; moreover, the (1s–2p)M, 1sN, and 1sC cores have been kept frozen throughout the calculations.§¶ IEs and EAs of valence MOs have been estimated through spin-polarized TS calculations.75 The adopted set-up allowed the comparison of ADF results with homogeneous theoretical outcomes pertinent to CoTPP,51,52,56,76 NiTPP,52,53,60 CuTPP,77,78 MPc (M = V,79–81 Cr,79 Mn,79 Fe,79 and Cu81–84). In a few selected cases (vide infra), further numerical experiments have been carried out by employing the same ADF package, the same basis sets, and a hybrid functional (B3LYP),85 incorporating a portion of exact exchange (20%) from Hartree–Fock theory.3. Results and discussion
3.1 Isolated MTPP and MPc
The square planar arrangement of MII ions in both MTPP and MPc lifts the five-fold degeneracy of the M 3d AOs to generate, within the assumption of a local D4h symmetry72 and the framework adopted in Fig. 1, five spin-up orbitals (SO↑) of symmetry a1g (z2), b1g (x2–y2), b2g (xy), eg (xz, yz) and as many SO↓ of the same symmetry. These SOs may be grouped in parallel (‖) and perpendicular (⊥) πt2g-like (π‖b2g + πeg) SOs and σeg-like SOs (σa1g + σb1g) by exploiting the parenthood between D4h and Oh complexes.72 It is noteworthy that the M σeg-like σb↑1g SO, antibonding in nature with respect to the M–NPy σ interaction, is certainly occupied in the 3d5 HS MnTPP,86 3d9 CuTPP78,87,88 and CuPc,81–84,89–91 and 3d10 ZnTPP and ZnPc complexes;90–92 moreover, only the closed-shell ZnTPP and ZnPc have no vacancy in the Zn 3d-based SOs. Additionally, the Pc2− ligand field is experimentally and theoretically found slightly stronger than the TPP2− one78,86,93 (see Section ESI.1 of the ESI†). This last evidence scarcely affects MTPP and MPc complexes whose πt↑2g-/πt↓2g-like SOs are filled, and the 2A1g (CoII, 3d7), 1A1g (NiII, 3d8), 2B1g (CuII, 3d9), and 1A1g (ZnII, 3d10) GSs uniquely determined;|| however, it could be relevant in lighter complexes, whose GS electronic terms are still debated79,94 and could be different upon moving from MTPP to MPc.78,86,93As mentioned in the Introduction, the frontier electronic structure of MTPP and MPc isolated complexes has been critically revisited and thoroughly compared with literature data in the ESI† (Section ESI.2). The relevant results of such a review will be briefly discussed by referring to Tables 1, 2, and Fig. 2.
| σa1g | σb1g | πb2g | πeg | a1u↓ | a2u↓ | |
|---|---|---|---|---|---|---|
| a HS MnTPP TSIE calculations have been run by adopting the B3LYP functional. | ||||||
| H2TPP | — | — | — | — | 6.77 (10au) | 6.48 (13b1u) |
| H2Pc | — | — | — | — | 6.57 (4au) | 7.74 (7b1u) |
| VTPP | 6.29↑ | — | 6.37↑ | 5.69 ↑ | 6.72 | 6.50 |
| VPc | 6.74↑ | — | 6.59↑ | 6.17 ↑ | 6.49 | 7.98 |
| CrTPP | 7.28↑ | — | 7.69↑ | 6.63↑ | 6.69 | 6.54 |
| CrPc | 7.73↑ | — | 7.44↑ | 7.03↑ | 6.50 | 7.99 |
| MnTPP | 9.61↑ | 5.88 ↑ | 9.49↑ | 10.53↑ | 6.55 | 6.36 |
| MnPc | 8.40↑ | — | 9.33↑ | 5.99 ↓ | 6.57 | 8.00 |
| FeTPP | 6.18 ↓ | — | 6.45↓ | 7.08↑ | 6.70 | 6.57 |
| FePc | 6.65↓ | — | 6.77↓ | 8.63↑ | 6.51 | 8.01 |
| CoTPP | 8.94↑ | — | 7.60↓ | 6.85↓ | 6.69 | 6.60 |
| CoPc | 9.14↑ | — | 7.62↓ | 7.32↓ | 6.54 | 8.01 |
| NiTPP | 7.65↓ | — | 8.63↓ | 7.03↓ | 6.70 | 6.62 |
| NiPc | 8.36↓ | — | 8.03↓ | 7.47↓ | 6.55 | 8.03 |
| CuTPP | 9.72↓ | 6.95↑ | 9.39↓ | 9.64↓ | 6.72 | 6.55 |
| CuPc | 9.17↓ | 7.28↑ | 8.08↓ | 7.77↓ | 6.56 | 8.01 |
| ZnTPP | 6.72 | 6.54 | ||||
| ZnPc | 6.55 | 7.98 | ||||
| σa1g | σb1g | πb2g | πeg | pmceg | pmcb1u | |
|---|---|---|---|---|---|---|
| a HS MnTPP TSEA calculations have been run by adopting the B3LYP functional. b TSEA positive values correspond to the IE of the MTPP− (MPc−) species; negative values indicate an unfavorable (costly) electron capture. | ||||||
| VTPP | 0.66↓ | 0.31↑ | 0.01↓ | 2.23 ↑ | 1.81↓ | 0.43↑ |
| VPc | 1.18↓ | −0.95↑ | 0.53↓ | 2.94 ↑ | 2.50↓ | 0.99↑ |
| CrTPP | 0.39↓ | 0.32↑ | 0.13↓ | −0.37↓ | 1.76 ↓ | 0.38↑ |
| CrPc | 1.13↓ | −0.59↑ | 0.87↓ | 0.96↓ | 2.47 ↓ | 0.97↑ |
| MnTPP | −0.20↓ | −3.59↓ | −1.17↓ | −1.57↓ | 1.33 ↓ | −0.09↑ |
| MnPc | 2.16↓ | 1.14↑ | 2.25↓ | 1.69↓ | 2.84 ↓ | 0.93↑ |
| FeTPP | — | 0.41↑ | — | 0.71↓ | 1.85 ↓ | 0.35↑ |
| FePc | — | 0.30↑ | — | 1.44↓ | 2.61 ↓ | 0.95↑ |
| CoTPP | 2.13 ↓ | 1.06↑ | — | — | 1.52↑ | 0.26↑ |
| CoPc | 3.00 ↓ | 1.45↑ | — | — | 2.17↑ | 0.90↑ |
| NiTPP | — | 1.47↑ | — | — | 1.53 ↑ | 0.27↑ |
| NiPc | — | 1.88↑ | — | — | 2.19 ↑ | 0.91↑ |
| CuTPP | — | 1.35↓ | — | — | 1.61 ↑ | 0.33↑ |
| CuPc | — | 1.78↓ | — | — | 2.25 ↑ | 0.94↑ |
| ZnTPP | — | — | — | — | 1.64 ↑ | 0.38↑ |
| ZnPc | — | — | — | — | 2.26 ↑ | 0.96↑ |
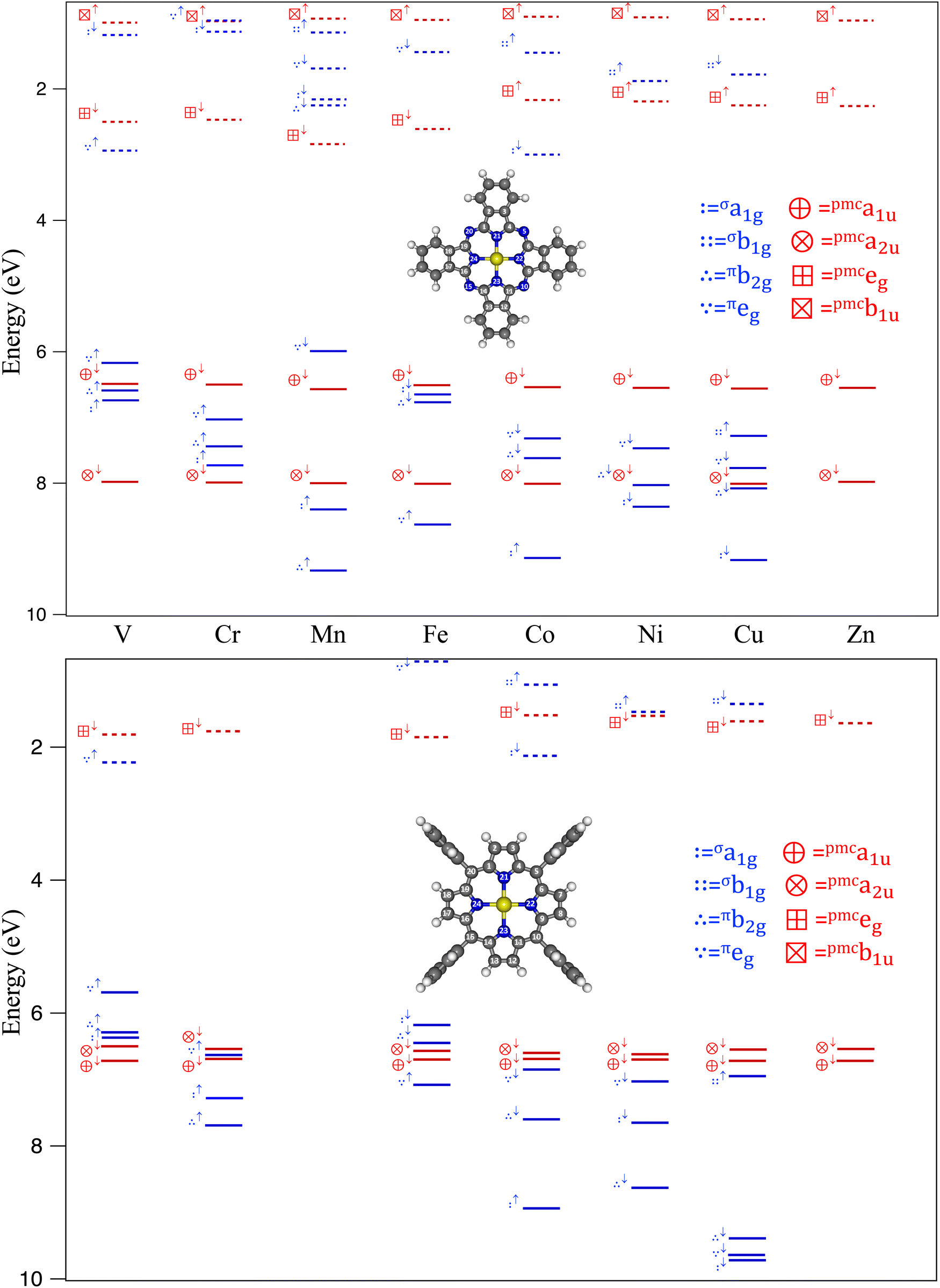 | ||
| Fig. 2 TSIEs (solid lines) and TSEAs (dotted lines) of MPc and MTPP M 3d-based (blue lines) and pmcπ-based (red lines) spin orbitals. MnTPP TSIEs and TSEAs are not included in the figure, having been estimated by adopting the B3LYP73,74,85 functional rather than the BP86 one73,74 (see the main text). | ||
Starting from the occupied FMOs and, more specifically, from the pmcπ ones; i.e., the H2TPP 13b1u and 10au FMOs, the H2Pc 7b1u and 4au FMOs, the MTPP 12a2u and 2a1u FMOs, and the MPc 6a2u and 2a1u FMOs, the following statements hold: (i) TSIEs of the 12a2u (13b1u in H2TPP)** FMO are very similar along the whole TPP series (the IE range they cover is less than 0.2 eV wide; see Table 1 and Fig. 2, where the MnTPP 12a2u FMO TSIE is not considered because evaluated by running B3LYP-based TS calculations (see Section ESI.2, ESI†)); (ii) TSIEs of the 6a2u (7b1u in H2Pc)** FMO are very similar along the whole Pc series and vary between 7.74 and 8.01 eV (see Table 1 and Fig. 2); (iii) nodal properties of the 2a1u MO (the 10au FMO in H2TPP and the 4au FMO in H2Pc)** make corresponding TSIEs very similar to each other; the |ΔTSIE| between the 12a2u and 2a1u (10au and 13b1u in H2TPP)** FMOs is tiny (∼0.2 eV) in MTPP; in addition, the ΔTSIE between the 6a2u and the 2a1u (7b1u and 4au in H2Pc)** FMOs is rather constant (∼1.5 eV) in MPc. Moving to the M 3d-based occupied FMOs, photoemission processes involving an M 3d-based MO and lying at the lowest IE are limited to VTPP, FeTPP (TSIEs of MnTPP FMOs have been computed by running B3LYP-based TS calculations; see Section ESI.2, ESI†), VPc, and MnPc.
Data on unoccupied MTPP and MPc FMOs are, for this contribution, much more interesting to be jointly considered. Let us start from the pmcπ* FMOs; i.e., the quasi degenerate 12b2g/12b3g and the 11au FMOs in H2TPP, the quasi degenerate 6b2g/63g and the 5au FMOs in H2Pc, and the eg and b1u FMOs in MTPP and MPc (see Table 2 and Fig. 2). Similarly to the free H2TPP and H2Pc species, the pmceg and pmcb1u MPc TSEAs lye deeper in energy than the MTPP ones; moreover, the MPc pmcb1u TSEAs (see Table 2 and Fig. 2) and, separately, the MTPP pmcb1u TSEAs are scarcely affected by the presence of different M along the two series because of the symmetry forbidden participation of the M AOs to the b1u MOs. Moving to the M 3d-based unoccupied FMOs, two things are particularly relevant: (i) both in MTPP and MPc, the largest EA corresponds to an M 3d-based unoccupied FMO only in V (πe↑g) and Co (σa↓1g) complexes (see Table 2 and Fig. 2); (ii) among the MTPP complexes, CoTPP is the only one with both σeg-like FMOs (the σa↓1g and σb↑1g SOs) able to be involved in a charge transfer processes (see Table 2 and Fig. 2).
3.2 Bulk-terminated CM surfaces
Copper, silver, and gold share the same space group 225 (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) and the cubic-close-packed (ccp) crystal structure; corresponding cell parameters are a = b = c (Cua = 3.61496 Å; Aga = 4.0853 Å; Aua = 4.0782 Å) and α = β = γ = 90°.95 A schematic representation of the two outermost layers of the bulk terminated CM(100) and CM(110) surfaces is displayed in Fig. 3,†† which also includes a top view of the three CM(111) unreconstructed topmost layers (high-symmetry chemisorption sites are also pinpointed in the figure).
m) and the cubic-close-packed (ccp) crystal structure; corresponding cell parameters are a = b = c (Cua = 3.61496 Å; Aga = 4.0853 Å; Aua = 4.0782 Å) and α = β = γ = 90°.95 A schematic representation of the two outermost layers of the bulk terminated CM(100) and CM(110) surfaces is displayed in Fig. 3,†† which also includes a top view of the three CM(111) unreconstructed topmost layers (high-symmetry chemisorption sites are also pinpointed in the figure).
 | ||
| Fig. 3 Schematic representation of the high symmetry chemisorption sites (in yellow) on the CM(100), CM(110), and CM(111) bulk-terminated surfaces. T, B, and H stand for top, bridge, and hole, respectively. The topmost layer lies in the XY plane; moreover, the shades of blue correspond to different layers, with the most intense color corresponding to the deepest one. | ||
The inspection of Fig. 3 reveals that local symmetry properties of these sites are quite different. More specifically, both CM(100) T and H sites are characterized by a local fourfold symmetry (C4), reduced to a local C2 symmetry when the B site is considered; (ii) no CM(110) chemisorption site (T, H, SB, and LB) exceeds a local C2 symmetry; (iii) CM(111) T, H, and B sites have a local C6, C3, and C2 symmetry, respectively. Incidentally, details of possible surface reconstructions, such as those affecting all the Au low-index planes, are neglected herein.‡‡ More generally, an interacting adsorbate (i.e., subject to charge transfer) will locally disrupt the collective (periodic) surface properties, and the frontier orbitals of the SCM atoms underneath the adsorbate will recover their atomic-like symmetry.
Coming back to the topmost layer of the bulk terminated CM surfaces, the single ns AO (n = 4, 5, and 6 for Cu, Ag, and Au, respectively) localized on a SCM T site (STCM), independently of the local symmetry, will be a basis for the totally symmetric ir (a, in the Schoenflies notation).72 The situation is a bit more complicated when SALCs of the ns AOs centered on chemisorption sites’ neighbours are considered: (i) the four SALCs of the ns AOs centered on the STCM(100) nnn and SHCM(100) nn (STCMnnn100 and SHCMnn100, respectively) are bases for the irs a, b, and e (see Fig. S13 and S14 of the ESI†) of the C4 point group,72 while those of the ns AOs centered on SBCMnn100 transform as the irs a and b (see Fig. S15 of the ESI†) of the C2 point group,72 and simply correspond to the in-phase and out-of-phase linear combinations of the two ns AOs; (ii) SALCs of the ns AOs centered on SBCMnn110 and SHCMnn110 transform as the irs a and b (see above); (iii) SALCs of the ns AOs centered on STCMnnn111 are bases for the irs a, b, and e (see Fig. S16 of the ESI†); SALCs of the ns AOs centered on SHCMnn111 transform as the irs a and e (see Fig. S17 of the ESI†); SALCs of the ns AOs centered on SBCMnn111 are of symmetry a and b (see Fig. S15 of the ESI†).§§
3.3 MTPP and MPc chemisorbed on CM surfaces
The core of the present paper consists of providing a convincing molecular picture of the MTPP and MPc grafting to CM surfaces and then a local point of view of the substrate → adsorbate charge transfer, if present, by combining elementary symmetry and geometry arguments with readily accessible information about the unoccupied frontier electronic structure of the free adsorbates. Far from attempting a systematic analysis of the adsorption of all the MTPP and MPc herein considered on the different chemisorption sites present on CM(100), CM(110), and CM(111) surfaces, we will limit ourselves to focus on selected case studies for which (i) the adsorbate → substrate charge transfer is established and (ii) the structural perturbations undergone by the pmc upon adsorption are tiny.A few years ago, Mabrouk and Majewski101 theoretically investigated the stability and the electronic and magnetic properties of VPc grafted to Au(111) by exploiting VASP.102 The authors considered all the possible high-symmetry chemisorption sites reported in the bottom panel of Fig. 3 as well as different molecular orientations to conclude that the VPc chemisorption on Au(111) is weakly dependent on the adsorption site (the ΔE between the most (Hfcc) and least (T) stable sites amounts to 120 meV) and the substrate → adsorbate charge transfer is weak and quantifiable in 0.62 electrons.101 Even without supercell periodic calculations and STM measurements, let us see how VPc ADF results combined with symmetry and geometrical considerations may provide useful insights into the grafting of VPc to Ag(111).
VPc TSEAs of the V 3d-based SOs are all but one positive (see Table 2 and Fig. 2); moreover, both the πe↑g and pmce↓g TSEAs are very high (2.94 and 2.50 eV, respectively). In addition, the experimentally revealed role played by the VPc σeg-like σa1g SO in the substrate → adsorbate charge transfer100 necessarily implies the interaction of the V 3dz2 AO with 5s SAg SALCs transforming as the ir a in the chemisorption site local symmetry. As such, it can be useful to remember that the σa↓1g TSEA (1.18 eV; see Table 2 and Fig. 2) is significantly smaller than the πe↑g and pmce↓g TSEAs (see above). Schematic representations of the 5s SAg SALCs transforming as the ir a for the T, B, and H¶¶ sites are superimposed to the optimized pmcVPc placed at 2.8 Å|||| above the bulk terminated Ag(111) in Fig. 4 (STAgnn111–V, SBAgnn111–V, and SHAgnn111–V internuclear distances are 2.80, 3.15, and 3.26 Å, respectively).
 | ||
Fig. 4 Schematic representation of the STAgnn111 (a), SBAgnn111 (b), and SHAgnn111 (c) 5s AOs SALCs of symmetry a on the Ag(111) bulk-terminated surface superimposed to the optimized pmcVPc placed at 2.8 Å above the substrate and oriented with V–NPy bonds aligned along the [2![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif) ] and [01] directions. Only Ag(111) topmost layer atoms are displayed for clarity. Large red spheres represent SAgnn 5s AOs. ] and [01] directions. Only Ag(111) topmost layer atoms are displayed for clarity. Large red spheres represent SAgnn 5s AOs. | ||
Elementary symmetry, geometrical, and overlap considerations allow the following statements: (i) the participation of the VPc πe↑g SOs to the STAgnn111 → VPc charge transfer is symmetry forbidden at the T site (see the 3D CPs displayed in Fig. 5); (ii) the contribution of the VPc πe↑g SOs to the STAgnnn111 → VPc charge transfer should be very weak as the STAgnnn111–V internuclear distance is 4.02 Å; (iii) similarly weak should be the contribution of both the VPc πe↑g and pmce↓g SOs (see Fig. 5) to the SHAgnn111 → VPc charge transfer (the local C3 symmetry of the H site is incompatible with the adsorbate local C4 symmetry); (iv) both the VPc 3d-based σeg-like σa↓1g (see Fig. 4) and πt2g-like πe↑g (see Fig. 5) may effectively participate to the SBAgnn111 → VPc charge transfer; (v) the VPc pmce↑g (see Fig. 5) is well tailored to participate to the SBAgnn111/SBAgnnn111 → VPc charge transfer (with VPc sitting at the B site, placed at 2.8 Å above the bulk-terminated Ag(111), and oriented as in Fig. 5, the SBAgnn111–NPy and SBAgnnn111–Nm internuclear distances are almost identical (2.85 and 2.84 Å, respectively)). The VPc chemisorption B site, with V–NPy bonds oriented along the [01] and [2] directions, seems then inescapable. This conclusion is only apparently in contrast with Mabrouk and Majewski101 results because one must consider on one side the lower nobleness of Ag compared to Au (Agχ and Auχ are 1.94 and 2.54 Pauling's units65,66) and, on the other side, the weak dependence on the adsorption site of the VPc chemisorption on Au(111). Results so far presented are useful to get information about the faint VPc species generated in situ on Ag(111),100 and can be straightforwardly extended to other interfaces.
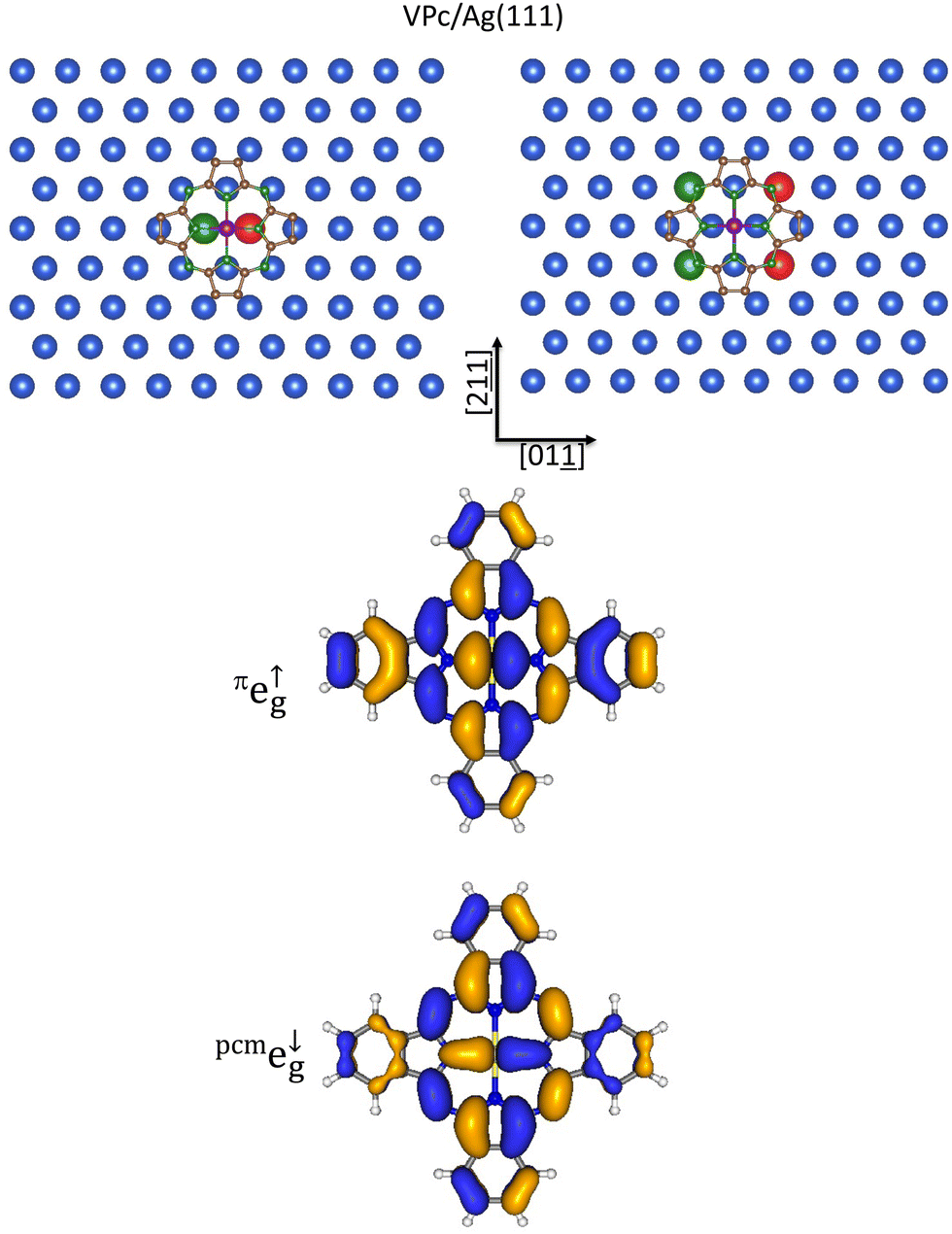 | ||
| Fig. 5 Schematic representation of the SBAgnn111 (upper left panel) and SBAgnnn111 (upper right panel) 5s AOs SALC of symmetry b on the Ag(111) bulk-terminated surface superimposed to the optimized pmcVPc placed at 2.8 Å above the substrate and oriented with V–NPy bonds aligned along the [2] and [0] directions. Only Ag(111) topmost layer atoms are displayed for clarity. Large spheres represent the SAgnn and SAgnnn 5s AOs, while different colors account for their different phases. 3D CPs of one component of the VII πt2g-like πe↑g SOs (middle panel) and one component of the pmce↓g π* SOs (lower panel). Displayed isosurfaces correspond to ±0.02 e½ Å−3/2 values. | ||
As already stressed, the inspection of Table 2 and Fig. 2 indicates that: (i) the TSEAs of M 3d-based SOs exceed those of the pmcπ* ones only in the V and Co complexes; (ii) the CoPc σeg-like σa↓1g TSEA has the highest value (3.00 eV) among those reported in the table and displayed in the figure. Moreover, Fig. 4 reveals at glance that STAgnn111 allows the best overlap between the CoII 3dz2 AO and the SAg SALCs transforming as the ir a. The huge CoPc EA also makes possible the Au(111) → CoPc charge transfer (see above) despite the high Auχ (2.54 Pauling's units65,66), while the different behavior of CoPc59 (T site, large charge transfer) and VPc101 (B site, weak charge transfer) on Au(111) may be reasonably traced back to (i) the fulfillment of CoPc πt↑2g-/t↓2g-like SOs and (ii) the different TSEA of the σeg-like σa↓1g in VPc and CoPc (see Table 2 and Fig. 2).
Results about the CoPc/Au(111) interface may be now exploited to shed new light into the CoPc/Au(110)107 and CoPc/Au(100)108,109 ones and for which the CoPc chemisorption site is still unknown. As such, it is experimentally established that the CoPc molecular plane lies parallel to the surface and results firmly anchored to the substrate both in CoPc/Au(110) and CoPc/Au(100);107,108 moreover, NEXAFS at the Co L2,3-edges and valence band photoemission suggested a reduction of the spin magnetic moment for CoPc/Au(110).107 Photoemission studies of CoPc on Au(100) also demonstrate that the strong Au(100) → CoPc charge transfer determines the CoII (d7) → CoI (d8) surface reduction, most likely involving the Co-based σeg-like σa↓1g SO, and then the quenching of the adsorbate magnetic moment (S = 0).109 The giant CoPc first EA level (see Table 2 and Fig. 2) and the significant charge transfer from the Au(110) and Au(100) surfaces into the Co 3d-based SOs prompt us to indicate the T site as the most favorite not only for the CoPc chemisorption on Au(111) but also on Au(110) and Au(100). Concerning the azimuthal orientation of CoPc on Au(110), the inspection of Fig. 3 and 6 reveals that an effective substrate → adsorbate charge transfer involving the lowest-lying pmcπ* orbitals (the CoPc pmce↑g SOs TSEA = 2.17 eV; see Table 2 and Fig. 2) should imply the out-of-phase linear combination of the STAunnn110 6s AOs (see the upper panel of Fig. 6) and the CoPc pmce↑g SO having a node localized on the NPy atoms aligned to the [001] direction (see the lower panel of Fig. 6). Incidentally, with the planar CoPc placed at 3 Å above the bulk-terminated Au(110) surface, the Co–STAunnn110 and NPy–STAunnn110 internuclear distances are 4.16 and 3.15 Å, respectively.
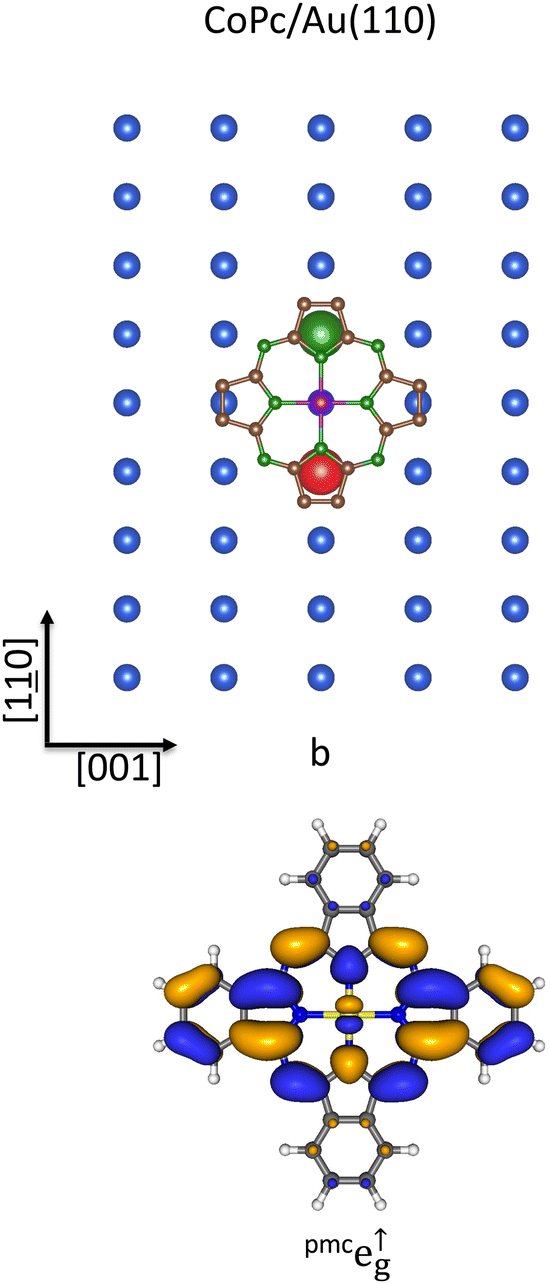 | ||
| Fig. 6 Schematic representation of the STAunnn110 6s AOs SALC of symmetry b on the Au(110) bulk-terminated surfaces superimposed to the optimized pmcCoPc placed at 3.0 Å above the substrate and oriented with Co–NPy bonds aligned along the [10] and [001] directions. Only Au(110) topmost layer atoms are displayed for clarity. Large spheres represent STAunnn110 6s AOs, while different colors account for their different phases (upper panel). 3D CP of one component of the pmce↑g SOs (lower panel). Displayed isosurfaces correspond to ±0.02 e½ Å−3/2 values. | ||
For the Au(100) surface, the experimental evidence reported by Lindner et al.,109 combined with the CoPc and CM(100) local fourfold symmetry, is also consistent with CoPc occupying a T site and the Co–NPy bonds oriented along the [01] and [011] directions (see Fig. 3 and 7). As such, it is noteworthy that the chemisorption T site and the proposed orientation are best suited to favor the charge transfer from the STAunn100 6s AO into the σeg-like σa↓1g SO through a direct Co–STAunn100 through-space interaction, which determines the experimentally detected CoII (d7) → CoI (d8) surface reduction.109 Incidentally, the CoPc chemisorption at the Ag(100) H site, as proposed by Mugarza et al.,110 is contradictory with the simultaneously reported charge transfer of one electron into the molecule; as such, it can be discarded.
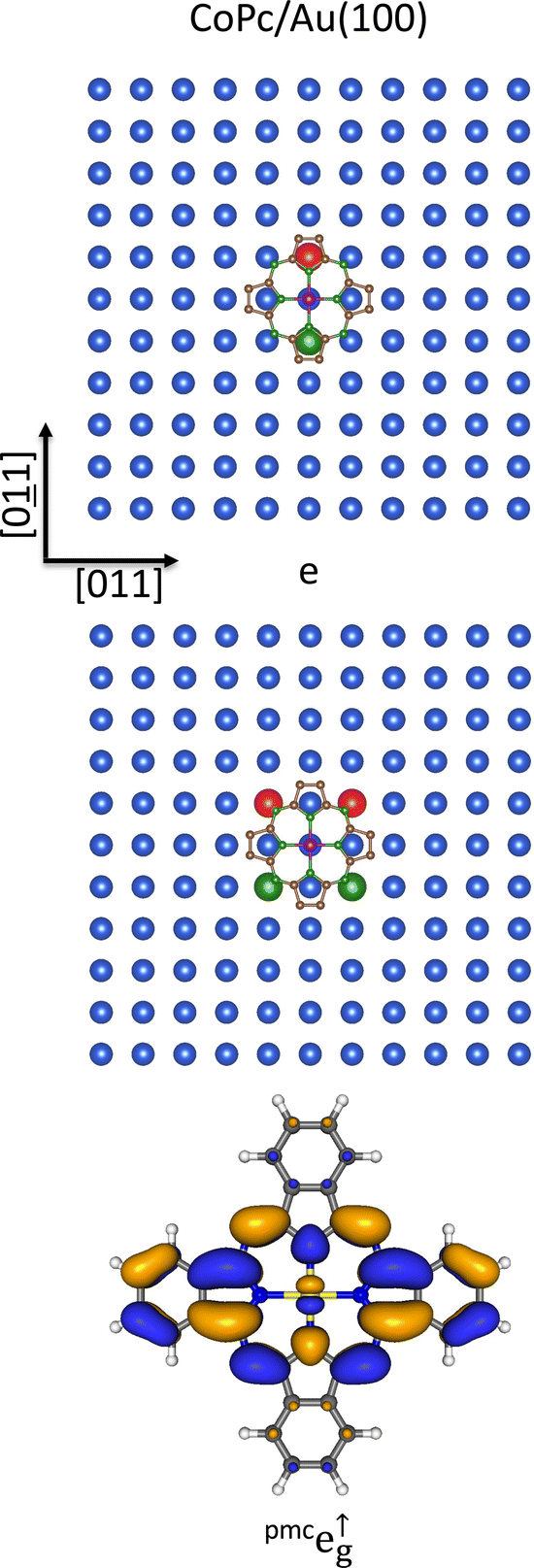 | ||
| Fig. 7 Schematic representation of one component of the STAunnn100 (upper panel), STAunnnn100 (middle panel) 6s AOs SALC of symmetry e on the Au(100) bulk-terminated surface superimposed to the optimized pmcCoPc placed at 3.0 Å above the substrate and oriented with Co–NPy bonds aligned to the [01] and [011] directions. Only Au(100) topmost layer atoms are displayed for clarity. Large spheres represent STAunnn100 and STAunnnn100 6s AOs, while different colors account for their different phases. 3D CP of one component of the pmce↑g SOs (lower panel). Displayed isosurfaces correspond to ±0.02 e½ Å−3/2 values. | ||
The overlap between the SALCs of Au 6s AOs localized on the STAunnn100 and STAunnnn100, transforming as the ir e in the local C4 symmetry, and the CoPc pmce↑g SO (see Fig. 7), provides a theoretical rationale for the tight anchoring of the adsorbate to the substrate. By the way, within the assumption of the CoPc molecular plane at 3 Å from the surface and the Co–NPy bonds oriented along the [01] and [011] directions (see Fig. 7), the STAunnn100–NPy and STAunnnn100–Nm internuclear distances are 3.15 and 3.08 Å, respectively.
The FePc 3A2g IS GS determined by the [σa↑↓π1g‖b↑↓π2g⊥e↑↑g] configuration (see Table S14 of the ESI†) would prevent the chemisorption site T because the antibonding component of the interaction between the completely occupied FePc σeg-like σa↑↓1g MO (the FeII 3dz2-based MO) and the STAunn100 6s AO would result partially occupied. Nevertheless, the minute ΔE between the FePc 3Eg(1) excited state generated by the [σa↑π1g‖b↑↓π2g⊥e↑↑↓g] configuration and the 3A2g GS (47 meV; see Table S14 of the ESI†) must be kept in mind because tiny ligand field perturbations, for instance, those induced by chemisorption, could generate different occupation numbers of the FeII 3d-based SOs. As such, the TSEAs of the 3Eg(1) πt2g-like (π⊥e↓g) 12eg LUMOs (3.04 eV) and σeg-like (σa↓1g) 21a1g LUMO+2 (2.70 eV), much higher than those reported in Table 2 for the low-lying empty SOs of the isolated molecule in its 3A2g GS, are consistent with a higher FePc electron-withdrawing capability in the 3Eg(1) state than in the 3A2g one. The π⊥e↓g and σa↓1g SOs bareness and their high TSEAs make them very well suited to actively participate to the substrate → adsorbate charge transfer and then to the anchoring of FePc to both Au(110) and Au(100).
Besides the abnormal gold nobleness65,66 and the 3Eg(1)π⊥e↓g SOs TSEA (3.04 eV), higher than the 3Eg(1) σa↓1g one (2.70 eV), we may further account for the symmetry and geometry constraints to determine the FePc chemisorption site on Au(110). Starting with symmetry considerations: (i) the participation of the FeII πt2g-like π⊥e↓g SOs to the substrate → adsorbate charge transfer is symmetry forbidden for FePc chemisorbed at a T site of the Au(110) and Au(100) surfaces but symmetry allowed for FePc chemisorbed at the SB site in the former case and the B one in the latter (see Fig. 8);*** (ii) the participation of the FeII σeg-like σa↓1g SO to the anchoring of FePc to both Au(110) and Au(100) is symmetry allowed for FePc chemisorbed at the SB site in the former case and the B one in the latter (see Fig. 8);*** (iii) the FePc pmceg π* SOs (TSEA = 2.03 eV) may actively participate to the anchoring of FePc to both Au(110) and Au(100) when chemisorbed at the SB site in the former case and the B one in the latter. With specific reference to the third point, it has to be noted that the substrate → adsorbate charge transfer will involve different pmc atoms upon moving from the FePc/Au(110) interface to the FePc/Au(100) one (see Fig. 9).
 | ||
| Fig. 8 Schematic representation of the SSBAunn110 6s AOs SALCs of symmetry b (upper, left panel) and a (upper, right panel) on the Au(110) bulk-terminated surface superimposed to the optimized pmcFePc placed at 2.85 Å above the substrate and oriented with Fe–NPy bonds aligned to the [10] and [001] directions. Only Au(110) topmost layer atoms are displayed for clarity. Large spheres represent SSBAunn110 6s AOs, while different colors account for their different phases. Schematic representation of the SBAunn100 6s AOs SALCs of symmetry b (middle, left panel) and a (middle, right panel) on the Au(100) bulk-terminated surface superimposed to the optimized pmcFePc placed at 2.85 Å above the substrate and oriented with Fe–NPy bonds aligned to the [10] and [011] directions. Only Au(100) topmost layer atoms are displayed for clarity. Large spheres represent SBAunn100 6s AOs, while different colors account for their different phases. 3D CPs of one component of the partially occupied FePc 3Eg(1) πt2g-like πe↓g SOs (lower, left panel), unoccupied FePc 3Eg(1) pmce↑g π* SOs (lower, middle panel), and unoccupied FePc 3Eg(1) σa↓1g SO (lower, right panel). Displayed isosurfaces correspond to ±0.02 e½ Å−3/2 values. | ||
 | ||
| Fig. 9 Schematic representation of the SSBAunnn110 and SBAunnn100 6s AOs SALCs of symmetry b on the Au(110) (upper panel) and Au(100) (lower panel) bulk-terminated surfaces superimposed to the optimized pmcFePc placed at 2.85 Å above the substrate and oriented with Fe–NPy bonds aligned to the [10] and [001] directions. Only Au(110) and Au(100) topmost layer atoms are displayed for clarity. Large spheres represent SSBAunnn110 and SBAunnn100 6s AOs, while different colors account for their different phases. | ||
From a geometry perspective, starting with FePc positioned 2.85 Å111 above the bulk terminated Au(110) at the SB site*** (see Fig. 8 and 9), the SSBAunn110–Fe, SSBAunn110–NPy, and SSBAunnn110–Cβ internuclear distances are 3.19, 2.89, and 2.94 Å, respectively; moreover, with FePc positioned 2.85 Å111 above the bulk terminated Au(100) at the B site (see Fig. 8 and 9), the SBAunn100–Fe, SBAunn100–NPy, SBAunnn100–Cα, and SBAunnn100–Nm internuclear distances are 3.19, 2.89, 2.87, and 3.04 Å, respectively.
SALCs displayed in Fig. 9 and the 3D CP of the FePc pmce↑g π* SOs (not herein reported because indistinguishable from the 3D CP of the CoPc pmce↑g π* SOs reported in Fig. 7) allow us to assess that, for FePc at 2.85 Å111 from the bulk terminated Au(110) and positioned at the SB site, the substrate → adsorbate charge transfer involving the pmce↑g π* SOs will mainly concern the NPy and Cβ 2pz AOs. For FePc at the same distance from the bulk terminated Au(100) and positioned at the B site, the substrate → adsorbate charge transfer involving the pmce↑g π* SOs will mainly concern the NPy, Nm, and Cα 2pz AOs. The most stable chemisorption site of FePc on Au(110) and Au(100) should be then the SB and B one, respectively.
Strictly related to the just considered FePc/Au(110) interface, is the FePc/Ag(110) system; an interface able to catalyze the four-electron oxygen reduction reaction to form H2O from O246,112 and closely related to the oxygen-binding active center of hemoglobin. Some of us have thoroughly investigated the adsorbate–substrate interactions taking place at the FePc/Ag(110) interface through the combined use of high-resolution STM measurements and DFT supercell periodic calculations including semiempirical dispersion interactions.111 Experimental measurements unveiled that FePc molecules result parallel to the Ag(110) surface and arranged in rows running along the [001] direction (see the central panel of Fig. 3); moreover, two closely related phases (c(10 × 4) and p(10 × 4) superstructures) observed after depositing FePc at RT and persisting after extensive annealing at 473 K, were revealed at the sub-ML regime.
DFT calculations, indicated that: (i) chemisorption sites of the c(10 × 4) and p(10 × 4) superstructures are different, they correspond to T and SB sites, respectively, with a height of FeII with respect to the Ag(110) topmost layer of 2.85 Å in the c(10 × 4) phase and 2.54 Å in the p(10 × 4) one; (ii) in the c(10 × 4) phase, all the FePc molecules are oriented with their  axes (those aligned with Fe–NPy bonds) forming a 45° angle with the [10] direction of the Ag substrate; (iii) in the p(10 × 4) phase, FePc molecules are arranged in an alternated sequence of two linear arrays, both aligned along the [001] direction of the Ag substrate and differing for the angle formed by their
axes (those aligned with Fe–NPy bonds) forming a 45° angle with the [10] direction of the Ag substrate; (iii) in the p(10 × 4) phase, FePc molecules are arranged in an alternated sequence of two linear arrays, both aligned along the [001] direction of the Ag substrate and differing for the angle formed by their  axes and the [10] direction ((30 ± 2)° and (−30 ± 2)°, respectively).111 Finally, even though the FePc IS state was mentioned, the authors provided no information about its symmetry.111 All this evidence may be straightforwardly rationalized by referring, besides the symmetry and the geometry arguments invoked to assess the chemisorption site of FePc on Au(110), to the different Auχ and Agχ.65,66 The Agχ value, significantly lower than the Auχ one, and the high TSEA (2.70 eV) of the 3Eg(1) σeg-like σa↓1g SO, concur to favor a direct Fe–Ag bonding interaction between the FeII σeg-like σa↓1g and the STAgnn110 5s AO and then a T chemisorption site. The FePc sitting at the T site does not prevent the possibility of an alternative chemisorption site, the SB one, with both the FeII πt2g-like π⊥e↓g and σeg-like σa↓1g SOs contributing to the adsorbate–substrate grafting. We remark that the Fe atom optimized eight of 2.54 Å characterizing the p(10 × 4) phase111 implies a SSBAgnn110–Fe internuclear distance of 2.92 Å, very similar to the STAgnn110–Fe one (2.85 Å). Finally, the results herein reported for the FePc/Ag(110) interface can be straightforwardly transferred to the FePc/Cu(110) one.113
axes and the [10] direction ((30 ± 2)° and (−30 ± 2)°, respectively).111 Finally, even though the FePc IS state was mentioned, the authors provided no information about its symmetry.111 All this evidence may be straightforwardly rationalized by referring, besides the symmetry and the geometry arguments invoked to assess the chemisorption site of FePc on Au(110), to the different Auχ and Agχ.65,66 The Agχ value, significantly lower than the Auχ one, and the high TSEA (2.70 eV) of the 3Eg(1) σeg-like σa↓1g SO, concur to favor a direct Fe–Ag bonding interaction between the FeII σeg-like σa↓1g and the STAgnn110 5s AO and then a T chemisorption site. The FePc sitting at the T site does not prevent the possibility of an alternative chemisorption site, the SB one, with both the FeII πt2g-like π⊥e↓g and σeg-like σa↓1g SOs contributing to the adsorbate–substrate grafting. We remark that the Fe atom optimized eight of 2.54 Å characterizing the p(10 × 4) phase111 implies a SSBAgnn110–Fe internuclear distance of 2.92 Å, very similar to the STAgnn110–Fe one (2.85 Å). Finally, the results herein reported for the FePc/Ag(110) interface can be straightforwardly transferred to the FePc/Cu(110) one.113
Experimentally, Schwarz et al.126 ultimately stated the SB adsorption geometry and the adsorption height (2.25 ± 0.04 Å) of CoP on Cu(111) by combining STM, high-resolution XPS, XSW measurements, and DFT calculations. However, when TPPs are concerned, intermolecular interactions allowed by the rotational degrees of freedom of Ph fragment decorating the pmc may overcome those of M and pmc with the substrate. Some of us revealed by PED the coexistence of Hhpc and Hfcc sites for a saturated CoTPP layer on Ag(111), as driven by the Ph intercalation among adjacent molecules (the so-called T-type interaction).127
At first sight, the experimental and theoretical results collected by Schwarz et al.126 are liable to affect the proposed approach; nevertheless, a thorough analysis of data reported in Table 2 and displayed in Fig. 2 demonstrates the opposite. The CoTPP highest TSEA (2.13 eV) is the largest among MTPP complexes (alike to CoPc among MPcs); however, it is noteworthy that the CoPc first affinity level (3.00 eV) significantly exceeds the CoTPP one. In addition, the TSEA of the CoTPP σeg-like 3dx2–y2-based 13b↓1g SO (1.06 eV), is the highest among lighter MTPP (ΔTSEA between CoTPP σeg-like SOs = 1.07 eV). Incidentally, the ΔTSEA between the CoP σeg-like SOs amounts to 1.00 eV.††† Now, the participation of the σeg-like 3dx2–y2-based SO to the substrate → adsorbate charge transfer is prevented by symmetry when the adsorbate is chemisorbed at the T site, while the contribution of both σeg-like MOs is symmetry allowed when the adsorbate occupies a B site (SB for the CM(110) surface). This is ultimately stated both experimentally and theoretically126 for the least electronegative CM;65,66 thus, it sounds reasonable that the chemisorption site of CoTPP on CM surfaces is, in the presence of a relevant substrate → adsorbate charge transfer involving the CoII (d7) → CoI (d8) reduction, the B one (SB for the CM(110)).
We recently addressed the adsorption configuration of NiTPP on Cu(100) by symmetry arguments as follows. The isolated NiTPP has a closed-shell nature and a 1A1g GS determined by the πt2g6-σeg2-like configuration; moreover, the empty NiII 3d-based σeg-like MO corresponds to the GS 12b1g LUMO (see Fig. 10) whose TSEA (1.47 eV, see Table 2 and Fig. 2) is closely spaced to that of the pmcπ* 13eg MOs (1.53 eV, see Table 2 and Fig. 2) and significantly higher than the TSEA of 9b1upmcπ* MO (the forth low-lying NiTPP empty orbital; see Table 2, Fig. 2, and Fig. 10).
 | ||
| Fig. 10 3D CPs of low-lying NiTPP empty MOs (only one component of the 13eg orbital is reported); displayed isosurfaces correspond to ±0.02 e½ Å−3/2 values. | ||
Experimental and theoretical outcomes pertinent to the NiTPP/Cu(100) interface revealed that: (i) both the substrate and the adsorbate have a local C4 symmetry;128 (ii) the huge Cu(100) → NiTPP charge transfer taking place at the interface determines the partial occupation of all four low-lying unoccupied NiTPP MOs (the σeg-like 12b1g, as well as the 13eg and 9b1upmcπ* MOs; see Fig. 10);128 (iii) the pmc of the chemisorbed NiTPP has a flat geometry,128 it lies at 1.93 Å from the topmost layer of the surface,60 and is characterized by the presence of the highly reactive 3d9 NiI species,61 which excludes asorption at a T site by symmetry constraints.60 With specific reference to the last point, the NiII (3d8) → NiI (3d9) surface reduction necessarily involves the only NiII 3d empty AO (3dx2–y2), basis for the ir b in the local C4 symmetry, whose overlap with the STCunn100 4s AO (basis for the ir a in the same fourfold symmetry) is identically zero. Thus, the substrate → adsorbed charge transfer rules the NiTPP adsorption on a specific chemisorption site; i.e., the fourfold hollow site characterizing the Cu(100) surface (see the top panel of Fig. 3).60 Incidentally, STM and STS results recently collected by Okuyama et al. indicate that CuPc (a single vacancy in the CuII 3dx2–y2) on Cu(100) maintains its flat geometry and occupies a C4 H site.129
As far as the NiTPP/Cu(110) interface is concerned, MOT130 results indicated a relevant adsorbate–substrate interaction characterized by a significant Cu(110) → NiTPP charge transfer and involving both the 13eg and 9b1upmcπ* MOs of the isolated NiTPP. Zamborlini117 also emphasized that, similarly to NiTPP/Cu(100), no contribution from the σeg-like 12b1g MO was revealed in the NiTPP/Cu(110) μ-ARPES maps. Incidentally, the absence of such a contribution in the NiTPP/Cu(100) μ-ARPES maps, successively revealed by NEXAFS measurements at the Ni L3-edge,61 was tentatively ascribed to the fact that the σeg-like 12b1g MO momentum map shows only four narrow lobes, presumably located outside the experimentally probed k-space range.117
To the best of the authors’ knowledge, NEXAFS measurements at the Ni L3-edge of the NiTPP/Cu(110) interface able to unequivocally determine the Ni oxidation state are not present in the literature. Nevertheless, it has been claimed that the Cu(110) → NiTPP charge transfer taking place at the NiTPP/Cu(110) interface is similar to that characterizing the NiTPP/Cu(100).117 Thus the frontier electronic structure of the free NiTPP prevents a T chemisorption site and favors a SB site in agreement with periodic DFT calculations.117 In fact, the empty Ni 3d-based MO reminiscent of the σeg-like 12b1g orbital may interact, in the local C2 symmetry, with the SSBCunn110 4s AOs SALC of symmetry a (see the left panel of Fig. 11). The inspection of Fig. 11, where the SALCs of symmetry b and a of the SSBCunnn110 4s AOs are displayed in the central and right panels, makes evident that they are very well suited for transferring electronic charge into the pmcπ* MOs reminiscent of the free NiTPP 13eg and 9b1u orbitals, respectively.
 | ||
| Fig. 11 Schematic representation of the SSBCunn110 4s AOs SALC of symmetry a in the local C2 symmetry of the Cu(110) bulk-terminate surface (left panel), SSBCunnn110 4s AOs SALC of symmetry b in the local C2 symmetry of the Cu(110) bulk-terminate surface (central panel), SSBCunnn110 4s AOs SALC of symmetry a in the local C2 symmetry of the Cu(110) bulk-terminate surface (right panel) superimposed to the optimized pmcNiTPP placed at 2 Å above the substrate and oriented with Ni–NPy bonds aligned along the [10] and [001] directions. Only Cu(110) topmost layer atoms are displayed for clarity. Large spheres represent SSBCunn110 and SSBCunnn110 4s AOs, while different colors account for their different phases. | ||
The crystal structure shared by Cu and Ag, their very similar χ values (1.90 and 1.94 Pauling's units, respectively),65,66 and the unoccupied frontier electronic structure of NiTPP would prompt to foresee the same SB chemisorption site for NiTPP/Cu(110) and NiTPP/Ag(110) interfaces, eventually imaging a weaker adsorbate–substrate interaction in the latter case determined by the larger size of Ag atoms (rAg = 1.60 Å) compared to that of the Cu ones (rCu = 1.35 Å).65
] and [2] directions.
The last substrate to be considered is the CM(110) one. Again, the inspection of Fig. 3 (middle panel) is enlightening. T and SB sites must be rejected because they are unfitted to minimize the repulsive interaction between ZnII πt2g-like/σeg-like completely occupied MOs and the underneath SCM atoms. As far as the CM(110) H and LB sites are concerned, a schematic representation of the Ag(110) with the optimized pmcZnTPP placed at 2.8 Å132 above the substrate and oriented with Zn–NPy bonds aligned along the [10] and [001] directions is reported in Fig. 12. The inspection of the figure reveals at once that the overlap between the SHAgnn110 5s SALC of symmetry b and the 13egpmcπ* MOs (see Fig. 10; NiTPP and ZnTPP 3D plots of the 13eg MOs are indistinguishable) is poorer for the pmcZnTPP at the H site than for the pmcZnTPP at the LB one. In this regard, it must be mentioned that, with the pmcZnTPP at 2.8 Å from the unreconstructed surface,132 the shortest (longest) SHAgnn110–NPy internuclear distance is 3.15 Å (3.52 Å), while the SHAgnn110–Cm and SHAgnn110–Cα are 3.00 and 2.94 Å, respectively. At variance, NPy atoms lying along the [001] direction are perfectly on top SLBAgnn110 (SLBAgnn110–NPy internuclear distance is 2.80 Å); moreover, the SLBAgnnn110–Cm and SLBAgnnn110–Cα are 2.86 and 2.95 Å, respectively. In the presence of a significant substrate → adsorbate charge transfer, which, as already mentioned implies the maximization of the overlap between low-lying pmcπ* MOs and SCM ns SALCs of suitable symmetry, the favored CM(110) chemisorption site should be the LB one.
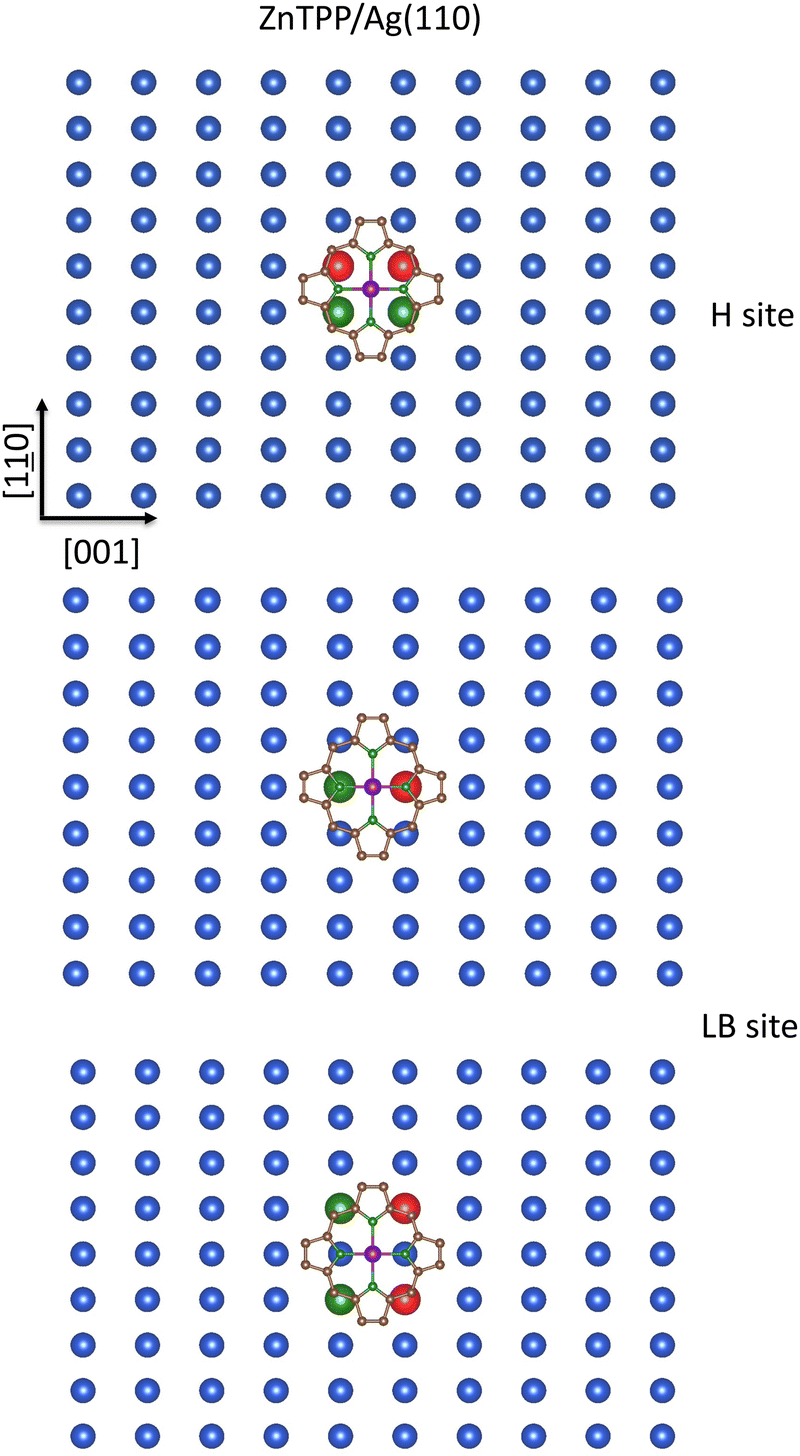 | ||
| Fig. 12 Schematic representation of the SHAgnn110 (upper panel), SLBAgnn110 (middle panel), and SLBAgnnn110 (bottom panel) 5s AOs SALCs transforming as the ir b in the local C2 symmetry of the Ag(110) bulk-terminate surface superimposed to the optimized pmcZnTPP placed at 2.8 Å above the substrate and oriented with Ni–NPy bonds aligned along the [10] and [001] directions. Only Ag(110) topmost layer atoms are displayed for clarity. Large spheres represent SAgnn and SAgnnn 5s AOs, while different colors account for their different phases. | ||
Available experimental evidence well agrees with the above-reported considerations. Baklanov et al.132 investigated the adsorption configuration of ZnP on Cu(111) and Ag(111) by combining XSW, XPS, STM, bond-resolved AFM, LICAD, and DFT-based supercell periodic calculations (limited to the ZnP/Cu(111) interface) to conclude that the ZnP species chemisorbed on Cu(111) occupies the B site with two Zn–NPy bonds oriented along the primary axis (see Scheme 1 of ref. 132). In addition, De Luca et al.,133 based on STM and PES measurements on the ZnTPP/Au(111) interface, proposed the same B chemisorption site, superseding former claims of adsorption on T site.134 Finally, Amsalem et al.135 investigated the electronic and vibrational properties of the ZnPc/Ag(110) interface by LEED, STM, and HREELS measurements, whose outcomes were consistent with a quite strong substrate → adsorbate charge transfer, implying the partial filling of the ZnPc π* LUMO. Amsalem et al.135 also proposed a tentative model for the geometrical structure of 1 ML ZnPc/Ag(110), although arbitrarily placing the ZnII ions on T and SB sites instead of the LB one.135
4. Conclusions
Structural and electronic reasons determining a crystal field stronger in the Pc2− ligand than in the TPP2− one have been identified, and the impact of this difference on the frontier electronic structure of free MTPP and MPc (ZM = 23–30; the largest series so far ever considered) has been investigated by DFT calculations with the ADF package. Easily accessible information about group 11 elements (crystal structure,95 nobleness,63 surface geometry136) combined with a thorough description of the free MTPP and MPc frontier electronic structure offer a cost-effective approach to foresee the chemisorption site of MTPP and MPc on low-index Cu, Ag, and Au surfaces, provided that two requirements are fulfilled: an established substrate → adsorbate charge transfer; the absence of significant distorsion of the macrocycle upon adsorption. A thorough comparison of the electronic structures of isolated MTPP and MPc and their building blocks indicates a more efficient substrate → adsorbate charge transfer in MPc than in MTPP. The stronger MPc ligand field, determined by the shrinking of the coordinative pocket and the higher π acceptor capability of MPc (see the ESI†), yields an overall electronegativity larger in MPc than in MTPP, which brings the MPc LUMOs closer in energy to the SCM SALCs. Because of their very similar electronegativities,65,66 Cu and Ag surfaces show similar behaviors and predictable chemisorption sites, while the Au nobleness63 prevents weighty charge transfer and only the chemisorption site of FePc (in its 3Eg(1) state) and CoPc on Au surfaces59,137,138 may be shortly, easily, and cheaply foreseen. This information also provides an useful input to avoid time-expensive trial-and-error computations, when the full interfacial structural and electronic details details must be retrieved through costly numerical experiments. The predictive capability of the semi-quantitative approach herein proposed reinforces the idea that the adsorbate–substrate interaction, if strong, is a phenomenon mainly local in character139 where the accuracy of the local description of the potential is more relevant than the inclusion of long-range effects and then it can be suitably treated through the molecular cluster approximation.139–141Acronyms
| ADF | Amsterdam density functional |
| AFM | Atomic force microscopy |
| AO | Atomic orbital |
| ARPES | Angular resolved photoelectron spectroscopy |
| ASPCS | Atomic subshell photoionization cross-sections |
| B | Bridge chemisorption site |
| B3LYP | Becke, 3-parameter, Lee–Yang–Parr |
| BP86 | Becke–Perdew 86 |
| CASPTn | Multi reference complete active space perturbation theory |
| CASSCF | Complete active space self-consistent field |
| CM | Coinage metal |
| C n | Clockwise rotation through  radiants radiants |
| CP | Contour plot |
| DFT | Density functional theory |
| DMRG | Density matrix renormalization group |
| DV-Xα | Discrete variational Xα |
| EA | Electron affinity |
| ESI | Electronic supplementary material |
| fcc | Face centered cubic |
| FMO | Frontier molecular orbital |
| GED | Gas phase electron diffraction |
| GGA | Generalized gradient approximation |
| GS | Ground state |
| H | Hollow chemisorption site |
| H2P | Porphine |
| H2Pc | Phthalocyanine |
| H2Pz | Porphyrazine |
| H2TPP | meso-Tetraphenylporphyrin |
| hcp | Hexagonal close packed |
| HOMO | Highest occupied molecular orbital |
| HREELS | High resolution electron energy loss spectroscopy |
| HS | High spin |
| i-Ind | Isoindole |
| IE | Ionization energy |
| IETS | Inelastic electron tunneling spectroscopy |
| IPS | Inverse photoemission spectroscopy |
| ir | Irreducible representation |
| IR | InfraRed |
| IS | Intermediate spin |
| IUPAC | Internation union of pure and applied chemistry |
| LB | Long bridge |
| LEED | Low energy electron diffraction |
| LICAD | Ligand-induced central atom displacement |
| LS | Low spin |
| LUMO | Lowest unoccupied molecular orbital |
| m | meso |
| M | Transition metal |
| ML | Monolayer |
| MO | Molecular orbital |
| MOT | Molecular orbital tomography |
| MP | Porphinato transition metal complex |
| MPc | Phthalocyaninato transition metal complex |
| MTPP | Tetraphenylporphirinato transition metal complex |
| n | Principal quantum number |
| NEXAFS | Near edge X-ray absorption fine structure |
| nn | Nearest neighbour |
| nnn | Next nearest neighbour |
| OEP2− | 2,3,7,8,12,13,17,18-Octaethylporphyrinato ion |
| OMTS | Orbital-mediated tunneling spectroscopy |
| P2− | Porphinato ion |
| Pc2− | Phthalocyaninato ion |
| PED | Photoelectron diffraction |
| PES | Photoelectron spectroscopy |
| Ph | Phenyl group |
| pmc | Planar aromatic macro cycle core |
| PPhMe2 | Dimethylphenyl phosphine |
| Py | Pyrrole |
| RASPT2 | Multiconfigurational second-order perturbation theory restricted active space |
| RT | Room temperature |
| S | Total spin quantum number |
| SALC | Symmetry adapted linear combination |
| SB | Short bridge |
| SCM | Surface coinage metal |
| SO | Spin orbital |
| STM | Scanning tunneling microscopy |
| STS | Scanning tunneling specroscopy |
| T | Top chemisorption site |
| TS | Transition state |
| TSEA | Transition state electron affinity |
| TSIE | Transition state ionization energy |
| THF | Tetrahydrofuran |
| TPP2− | meso-Tetraphenyl porphirinato ion |
| TTP2− | meso-Tetra-p-tolylporphyrinato ion |
| UV | Ultraviolet |
| VASP | Vienna ab initio Simulation Package |
| VdW | van der Waals |
| XC | Exchange–correlation |
| XMCD | X-Ray magnetic circular dichroism |
| XPS | X-Ray photoelectron spectroscopy |
| XSW | X-Ray standing wave |
| Z | Atomic number |
| χ | Electronegativity |
| μ | Magnetic moment |
| ν | Frequency |
| 3D | Three dimensional |
Author contributions
S. C.: investigation, data curation, writing – review & editing; I. C.: investigation, data curation, writing – review & editing; V. F.: investigation, data curation, writing – review & editing; L. S.: investigation, data curation, writing – review & editing; L. F.: investigation, data curation, writing – review & editing; M. C.: conceptualization, investigation, methodology, resources, supervision, writing original draft, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- J. J. Davis, G. A. Orlowski, H. Rahman and P. D. Beer, Chem. Commun., 2010, 46, 54–63 RSC.
- B. Schmaltz, T. Weil and K. Müllen, Adv. Mater., 2009, 21, 1067–1078 CrossRef CAS.
- W. Auwärter, K. Seufert, F. Bischoff, D. Ecija, S. Vijayaraghavan, S. Joshi, F. Klappenberger, N. Samudrala and J. V. Barth, Nat. Nanotechnol., 2012, 7, 41–46 CrossRef PubMed.
- D. G. De Oteyza, J. M. García-Lastra, M. Corso, B. P. Doyle, L. Floreano, A. Morgante, Y. Wakayama, A. Rubio and J. E. Ortega, Adv. Funct. Mater., 2009, 19, 3567–3573 CrossRef CAS.
- W. R. Browne and B. L. Feringa, Ann. Rev. Phys. Chem., 2009, 60, 407–428 CrossRef CAS PubMed.
- J. Dintinger, S. Klein and T. W. Ebbesen, Adv. Mater., 2006, 18, 1267–1270 CrossRef CAS.
- P. Gambardella, S. Stepanow, A. Dmitriev, J. Honolka, F. M. F. de Groot, M. Lingenfelder, S. S. Gupta, D. D. Sarma, P. Bencok, S. Stanescu, S. Clair, S. Pons, N. Lin, A. P. Seitsonen, H. Brune, J. V. Barth and K. Kern, Nat. Mater., 2009, 8, 189–193 CrossRef CAS PubMed.
- D. Gatteschi, A. Cornia, M. Mannini and R. Sessoli, Inorg. Chem., 2009, 48, 3408–3419 CrossRef CAS PubMed.
- P. V. Bernhardt and G. A. Lawrance, Comprehensive Coordination Chemistry II: Transition Metal Groups 9–12, ed. D. E. Fenton, 2003, vol. 6 Search PubMed.
- D. L. Nelson, M. M. Cox and A. A. Hoskins, Lehninger Principles of Biochemistry, Freeman, W.H. Macmillan Learning, New York, NY, USA, 8th edn, 2021 Search PubMed.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, W. H. Freeman, New York, NY, USA, 5th edn, 2002 Search PubMed.
- A. Tsuda and A. Osuka, Science, 2001, 293, 79–82 CrossRef CAS PubMed.
- M. Planells, A. Forneli, E. Martínez-Ferrero, A. Sánchez-Díaz, M. A. Sarmentero, P. Ballester, E. Palomares and B. C. O’Regan, Appl. Phys. Lett., 2008, 92, 153506 CrossRef.
- N. A. Rakow and K. S. Suslick, Nature, 2000, 406, 710–713 CrossRef CAS PubMed.
- The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, NY, USA, 2000 Search PubMed.
- J. Mack and M. J. Stillman, The Porphyrin Handbook, 2003, vol. 16, p. 43 Search PubMed.
- M. K. Engel, The Porphyrin Handbook, 2003, vol. 20, pp. 1–246 Search PubMed.
- C. Di Natale, D. Monti and R. Paolesse, Mater. Today, 2010, 13, 46–52 CrossRef CAS.
- A. W. Hains, Z. Liang, M. A. Woodhouse and B. A. Gregg, Chem. Rev., 2010, 110, 6689–6735 CrossRef CAS PubMed.
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259–379 CrossRef CAS.
- J. M. Gottfried, On-Surface Synthesis, Springer, 2016 Search PubMed.
- Y.-f Geng, P. Li, J.-z Li, X.-m Zhang, Q.-d Zeng and C. Wang, Coord. Chem. Rev., 2017, 337, 145–177 CrossRef CAS.
- J. J. Ortiz-Garcia and R. C. R. C. Quardokus, J. Vac. Sci. Technol., A, 2023, 41, 030801 CrossRef CAS.
- D. Dini and M. Hanack, J. Porphyrins Phthalocyanines, 2004, 8, 915–933 CrossRef CAS.
- Z. Li, B. Li, J. Yang and J. G. Hou, Acc. Chem. Res., 2010, 43, 954–962 CrossRef CAS PubMed.
- A. B. Sorokin, Chem. Rev., 2013, 113, 8152–8191 CrossRef CAS PubMed.
- Q. Zhou, Z.-F. Liu, T. J. Marks and P. Darancet, J. Phys. Chem. A, 2021, 125, 4055–4061 CrossRef CAS PubMed.
- M. Urbani, M.-E. Ragoussi, M. K. Nazeeruddin and T. Torres, Coord. Chem. Rev., 2019, 381, 1–64 CrossRef CAS.
- S. Keshipour and A. Asghari, Int. J. Hydrogen Energy, 2022, 47, 12865–12881 CrossRef CAS.
- A. Galstyan, Chem. – Eur. J., 2020, 27, 1903–1920 CrossRef PubMed.
- H. de Diesbach and E. von der Weid, Helv. Chim. Acta, 1927, 10, 886–888 CrossRef CAS.
- I. M. Heilbron, F. Irving, R. P. Linstead and J. F. Thorpe (to Imperial Chemical Industries, Ltd.) British Patent 410814 (May 16, 1934).
- R. B. Linstead, J. Chem. Soc., 1934, 1016–1017 RSC.
- C. E. Dent and R. B. Linstead, J. Chem. Soc., 1934, 1027–1031 RSC.
- R. B. Linstead and A. R. Lowe, J. Chem. Soc., 1934, 1031–1033 RSC.
- C. E. Dent, R. B. Linstead and A. R. Lowe, J. Chem. Soc., 1934, 1033–1039 RSC.
- P. A. Barrett, C. E. Dent and R. P. Linstead, J. Chem. Soc., 1936, 1719–1736 RSC.
- R. P. Linstead and J. M. Robertson, J. Chem. Soc., 1936, 1736–1738 RSC.
- M. A. Dahlen, Ind. Eng. Chem., 1939, 31, 839–847 CrossRef CAS.
- M. D. Angione, R. Pilolli, S. Cotrone, M. Magliulo, A. Mallardi, G. Palazzo, L. Sabbatini, D. Fine, A. Dodabalapur, N. Cioffi and L. Torsi, Mater. Today, 2011, 14, 424–433 CrossRef CAS.
- S. H. Jang and A. K. Y. Jen, Structured Organic Non-Linear Optics, Academic Press, Amsterdam, 2011 Search PubMed.
- T. Nguyen, Surf. Coat. Technol., 2011, 206, 742–752 CrossRef CAS.
- W. Cao and J. Xue, Energy Environ. Sci., 2014, 7, 2123–2144 RSC.
- B. Hulsken, R. Van Hameren, J. W. Gerritsen, T. Khoury, P. Thordarson, M. J. Crossley, A. E. Rowan, R. J. M. Nolte, J. A. A. W. Elemans and S. Speller, Nat. Nanotechnol., 2007, 2, 285–289 CrossRef CAS PubMed.
- K. S. Mali, N. Pearce, S. De Feyter and N. R. Champness, Chem. Soc. Rev., 2017, 46, 2520–2542 RSC.
- F. Sedona, M. Di Marino, D. Forrer, A. Vittadini, M. Casarin, A. Cossaro, L. Floreano, A. Verdini and M. Sambi, Nat. Mater., 2012, 11, 970–977 CrossRef CAS PubMed.
- S. Carlotto, M. Sambi, F. Sedona, A. Vittadini, J. Bartolomé, F. Bartolomé and M. Casarin, Phys. Chem. Chem. Phys., 2016, 18, 28110–28116 RSC.
- E. Bartolomé, J. Bartolomé, F. Sedona, J. Lobo-Checa, D. Forrer, J. Herrero-Albillos, M. Piantek, J. Herrero-Martín, D. Betto, E. Velez-Fort, L. M. García, M. Panighel, A. Mugarza, M. Sambi and F. Bartolomé, J. Phys. Chem. C, 2020, 124, 13993–14006 CrossRef.
- I. Cojocariu, S. Carlotto, H. M. Sturmeit, G. Zamborlini, M. Cinchetti, A. Cossaro, A. Verdini, L. Floreano, M. Jugovac, P. Puschnig, C. Piamonteze, M. Casarin, V. Feyer and C. M. Schneider, Chem. – Eur. J., 2021, 27, 3526–3535 CrossRef CAS PubMed.
- H. M. Sturmeit, I. Cojocariu, A. Windischbacher, P. Puschnig, C. Piamonteze, M. Jugovac, A. Sala, C. Africh, G. Comelli, A. Cossaro, A. Verdini, L. Floreano, M. Stredansky, E. Vesselli, C. Hohner, M. Kettner, J. Libuda, C. M. Schneider, G. Zamborlini, M. Cinchetti and V. Feyer, Small, 2021, 17, 2104779 CrossRef CAS PubMed.
- I. Cojocariu, S. Carlotto, G. Zamborlini, M. Jugovac, L. Schio, L. Floreano, M. Casarin, V. Feyer and C. M. Schneider, J. Mater. Chem. C, 2021, 9, 12559–12565 RSC.
- I. Cojocariu, S. Carlotto, M. Jugovac, L. Floreano, M. Casarin, V. Feyer and C. M. Schneider, J. Mater. Chem. C, 2022, 10, 9748–9757 RSC.
- I. Cojocariu, S. Carlotto, D. Baranowski, M. Jugovac, J. Dreiser, L. Schio, L. Floreano, M. Casarin, V. Feyer and C. M. Schneider, J. Mater. Chem. C, 2023, 11, 15521–15530 RSC.
- P. Knecht, J. Reichert, P. S. Deimel, P. Feulner, F. Haag, F. Allegretti, M. Garnica, M. Schwarz, W. Auwärter, P. T. P. Ryan, T.-L. Lee, D. A. Duncan, A. P. Seitsonen, J. V. Barth and A. C. Papageorgiou, Angew. Chem., Int. Ed., 2021, 60, 16561–16567 CrossRef CAS PubMed.
- A. Basagni, L. Colazzo, F. Sedona, M. Di Marino, T. Carofiglio, E. Lubian, D. Forrer, A. Vittadini, M. Casarin, A. Verdini, A. Cossaro, L. Floreano and M. Sambi, Chem. – Eur. J., 2014, 20, 14296–14304 CrossRef CAS PubMed.
- I. Cojocariu, S. Carlotto, D. Baranowski, M. Jugovac, L. Schio, L. Floreano, M. Casarin, V. Feyer and C. M. Schneider, Inorg. Chim. Acta, 2023, 556, 121657 CrossRef CAS.
- T. Furuyama, K. Satoh, T. Kushiya and N. Kobayashi, J. Am. Chem. Soc., 2014, 136, 765–776 CrossRef CAS PubMed.
- G. Raposo-Hernández, E. Sánchez Marcos, R. R. Pappalardo and J. M. Martínez, J. Chem. Phys., 2023, 159, 064110 CrossRef PubMed.
- A. Zhao, Q. Li, L. Chen, H. Xiang, W. Wang, S. Pan, B. Wang, X. Xiao, J. Yang, J. G. Hou and Q. Zhu, Science, 2005, 309, 1542–1544 CrossRef CAS PubMed.
- S. Carlotto, A. Verdini, G. Zamborlini, I. Cojocariu, V. Feyer, L. Floreano and M. Casarin, Phys. Chem. Chem. Phys., 2023, 25, 26779–26786 RSC.
- G. Zamborlini, M. Jugovac, A. Cossaro, A. Verdini, L. Floreano, D. Lüftner, P. Puschnig, V. Feyer and C. M. Schneider, Chem. Commun., 2018, 54, 13423–13426 RSC.
- M. Orchin and H. H. Jaffé, Symmetry, Orbitals, and Spectra (S.O.S.), Wiley-Interscience, New York, 1971 Search PubMed.
- K. P. Kepp, ChemPhysChem, 2020, 21, 360–369 CrossRef CAS PubMed.
- L. Pauling, The nature of the chemical bond, Cornel University, Ithaca, NY, 3rd edn, 1960, p. 88 Search PubMed.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Butterworth-Heinemann, Cambridge, 2nd edn, 1984, p. 1176 Search PubMed.
- https://www.webelements.com/periodicity/eneg_pauling/ reports Cuχ = 1.90, Agχ = 1.93, and Auχ = 2.54 Pauling's units.
- P. Pyykkö and J.-P. Descalux, Acc. Chem. Res., 1979, 12, 276–281 CrossRef.
- P. Pyykkö, Chem. Rev., 1988, 88, 563–594 CrossRef.
- P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef PubMed.
- A. J. Wallace, B. E. Williamson and D. L. Crittenden, Can. J. Chem., 2016, 94, 1163 CrossRef CAS.
- ADF2014, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, https://www.scm.com.
- B. E. Douglas and C. A. Hollingsworth, Symmetry in Bonding and Spectra, an Introduction, Academic Press, Orlando, FL, USA, 1985 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- J. C. Slater, Quantum Theory of Molecules and Solids. The Self-Consistent-Field for Molecules and Solids, McGraw-Hill, New York, 1974, vol. 4 Search PubMed.
- S. Carlotto, I. Cojocariu, V. Feyer, L. Floreano and M. Casarin, Nanomaterials, 2022, 12, 218 CrossRef CAS PubMed.
- G. Mangione, S. Carlotto, M. Sambi, G. Ligorio, M. Timpel, A. Vittadini, M. V. Nardi and M. Casarin, Phys. Chem. Chem. Phys., 2016, 18, 18727–18738 RSC.
- G. Mangione, M. Sambi, S. Carlotto, A. Vittadini, G. Ligorio, M. Timpel, L. Pasquali, A. Giglia, M. V. Nardi and M. Casarin, Phys. Chem. Chem. Phys., 2016, 18, 24890–24904 RSC.
- S. Carlotto, M. Sambi, F. Sedona, A. Vittadini and M. Casarin, Nanomaterials, 2021, 11, 54 CrossRef CAS PubMed.
- S. Carlotto, M. Sambi, M. Rancan and M. Casarin, Inorg. Chem., 2018, 57, 1859–1869 CrossRef CAS PubMed.
- M. Casarin and S. Carlotto, Eur. J. Inorg. Chem., 2018, 3145 CrossRef CAS.
- M. V. Nardi, F. Detto, L. Aversa, R. Verucchi, G. Salviati, S. Iannotta and M. Casarin, Phys. Chem. Chem. Phys., 2013, 15, 12864–12881 RSC.
- G. Mangione, M. Sambi, M. V. Nardi and M. Casarin, Phys. Chem. Chem. Phys., 2014, 16, 19852–19855 RSC.
- G. Mangione, L. Pandolfo, M. Sambi, G. Ligorio, M. V. Nardi, A. Cossaro, L. Floreano and M. Casarin, Eur. J. Inorg. Chem., 2015, 2709–2713 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. A. Reed, J. K. Kouba, C. J. Grimes and S. K. Cheung, Inorg. Chem., 1978, 17, 2666–2670 CrossRef CAS.
- M.-S. Liao and S. Scheiner, J. Chem. Phys., 2002, 117, 205–219 CrossRef CAS.
- M.-S. Liao, J. D. Watts and M.-J. Huang, J. Phys. Chem. A, 2007, 111, 5927–5935 CrossRef CAS PubMed.
- A. B. P. Lever, J. Chem. Soc., 1965, 1821–1829 RSC.
- M.-S. Liao and S. Scheiner, J. Comput. Chem., 2002, 23, 1391–1403 CrossRef CAS PubMed.
- M.-S. Liao and S. Scheiner, J. Chem. Phys., 2001, 114, 9780–9791 CrossRef CAS.
- K. A. Nguyen and R. Pachter, J. Chem. Phys., 2001, 114, 10757–10767 CrossRef CAS.
- J. F. Kirner, W. Dow and W. R. Scheidt, Inorg. Chem., 1976, 15, 1685–1690 CrossRef CAS.
- D. Nachtigallová, A. Antalík, R. Lo, R. Sedlák, D. Manna, J. Tucek, J. Ugolotti, L. Veis, O. Legeza, J. Pittner, R. Zboril and P. Hobza, Chem. – Eur. J., 2018, 24, 13413–13417 CrossRef PubMed.
- https://www.webelements.com/ .
- S. B. Darling, A. W. Rosenbaum, Y. Wang and S. J. Sibener, Langmuir, 2002, 18, 7462–7468 CrossRef CAS.
- D. Gibbs, B. M. Ocko, D. M. Zehner and S. G. J. Mochrie, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 7330–7344 CrossRef CAS PubMed.
- D. L. Abernathy, D. Gibbs, G. Griibel, K. G. Huang, S. G. J. Mochrie, A. R. Sandy and D. M. Zehner, Surf. Sci., 1993, 283, 260–276 CrossRef CAS.
- L. Floreano, A. Cossaro, R. Gotter, A. Verdini, G. Bavdek, F. Evangelista, A. Ruocco, A. Morgante and D. Cvetko, J. Phys. Chem. C, 2008, 112, 10794–10802 CrossRef CAS.
- K. Eguchi, T. Nakagawa, Y. Takagi and T. Yokoyama, J. Phys. Chem. C, 2015, 119, 9805–9815 CrossRef CAS.
- M. Mabrouk and J. A. Majewski, Comput. Theor. Chem., 2021, 1202, 1133189 Search PubMed.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- W. H. Blades, A. C. Reber, S. N. Khanna, L. Lopez-Sosa, P. Calaminici and A. M. Köster, J. Phys. Chem. A, 2017, 121, 2990–2999 CrossRef CAS PubMed.
- J. D. Baran, J. A. Larsson, R. A. J. Woolley, Y. Cong, P. J. Moriarty, A. A. Cafolla, K. Schulte and V. R. Dhanak, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 075413 CrossRef.
- J. D. Baran and J. A. Larsson, J. Phys. Chem. C, 2013, 117, 23887–23898 CrossRef CAS.
- M. Schmid, A. Kaftan, H.-P. Steinrück and J. M. Gottfried, Surf. Sci., 2012, 606, 945–949 CrossRef CAS.
- M. G. Betti, P. Gargiani, R. Frisenda, R. Biagi, A. Cossaro, A. Verdini, L. Floreano and C. Mariani, J. Phys. Chem. C, 2010, 114, 21638–21644 CrossRef CAS.
- O. V. Molodtsova, M. Knupfer, Yu. A. Ossipyan and V. Yu Aristov, J. Appl. Phys., 2008, 104, 083704 CrossRef.
- S. Lindner, U. Treske, M. Grobosch and M. Knupfer, Appl. Phys. A: Mater. Sci. Process., 2011, 105, 921–925 CrossRef CAS.
- A. Mugarza, R. Roble, C. Krull, R. Korytar, N. Lorente and P. Gambardella, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155437 CrossRef.
- M. Casarin, M. Di Marino, D. Forrer, M. Sambi, F. Sedona, E. Tondello, A. Vittadini, V. Barone and M. Pavone, J. Phys. Chem. C, 2010, 114, 2144–2153 CrossRef CAS.
- J. H. Zagal, Coord. Chem. Rev., 1992, 119, 89–136 CrossRef CAS.
- R. A. Rehman, C. Y. Liang, Z. H. Jie, W. Ke, D. W. Dong, L. H. Yang, H. P. Mo and B. S. Ning, Chin. Phys. B, 2013, 22, 063101 CrossRef.
- T. Houwaart, T. Le Bahers, P. Sautet, W. Auwärter, K. Seufert, J. V. Barth and M. L. Bocquet, Surf. Sci., 2015, 635, 108–114 CrossRef CAS.
- A. Weber-Bargioni, W. Auwärter, F. Klappenberger, J. Reichert, S. Lefrançois, T. Strunskus, C. Wöll, A. Schiffrin, Y. Pennec and J. V. Barth, ChemPhysChem, 2008, 9, 89–94 CrossRef CAS PubMed.
- P. Donovan, A. Robin, M. S. Dyer, M. Persson and R. Raval, Chem. – Eur. J., 2010, 16, 11641–11652 CrossRef CAS PubMed.
- G. Zamborlini, Organic-Metal Hybrid Interfaces at the Mesoscopic Scale, Forschungszentrum Jülich GmbH, Zentralbibliothek, Verlag, 2018 Search PubMed.
- J. M. Gottfried and H. Marbach, Z. Phys. Chem., 2009, 223, 53–74 CrossRef CAS.
- W. Hieringer, K. Flechtner, A. Kretschmann, K. Seufert, W. Auwärter, J. V. Barth, A. Görling, H. P. Steinrück and J. M. Gottfried, J. Am. Chem. Soc., 2011, 133, 6206–6222 CrossRef CAS PubMed.
- T. Lukasczyk, K. Flechtner, L. R. Merte, N. Jux, F. Maier, J. M. Gottfried and H. P. Steinrück, J. Phys. Chem. C, 2007, 111, 3090–3098 CrossRef CAS.
- D. Wechsler, M. Franke, Q. Tariq, L. Zhang, T. L. Lee, P. K. Thakur, N. Tsud, S. Bercha, K. C. Prince, H. P. Steinrück and O. Lytken, J. Phys. Chem. C, 2017, 121, 5667–5674 CrossRef CAS.
- W. Auwärter, K. Seufert, F. Klappenberger, J. Reichert, A. Weber-Bargioni, A. Verdini, D. Cvetko, M. Dell’Angela, L. Floreano, A. Cossaro, G. Bavdek, A. Morgante, A. P. Seitsonen and J. V. Barth, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 245403 CrossRef.
- L. Scudiero, D. E. Barlow and K. W. Hipps, J. Phys. Chem. B, 2000, 104, 11899–11905 CrossRef CAS.
- K. W. Hipps and U. Mazur, J. Porphyrins Phthalocyanines, 2012, 16, 1–9 CrossRef.
- D. E. Barlow, L. Scudiero and K. W. Hipps, Langmuir, 2004, 20, 4413–4421 CrossRef CAS PubMed.
- M. Schwarz, M. Garnica, D. A. Duncan, A. P. Paz, J. Ducke, P. S. Deimel, P. K. Thakur, T.-L. Lee, A. Rubio, J. V. Barth, F. Allegretti and W. Auwa
![[r with combining umlaut]](https://www.rsc.org/images/entities/char_0072_0308.gif) ter, J. Phys. Chem. C, 2018, 122, 5452–5461 CrossRef CAS.
ter, J. Phys. Chem. C, 2018, 122, 5452–5461 CrossRef CAS. - W. Auwärter, K. Seufert, F. Klappenberger, J. Reichert, A. Weber-Bargioni, A. Verdini, D. Cvetko, M. Dell’Angela, L. Floreano, A. Cossaro, G. Bavdek, A. Morgante, A. P. Seitsonen and J. V. Barth, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 245403 CrossRef.
- G. Zamborlini, D. Lüftner, Z. Feng, B. Kollmann, P. Puschnig, C. Dri, M. Panighel, G. Di Santo, A. Goldoni, G. Comelli, M. Jugovac, V. Feyer and C. M. Schneider, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed.
- H. Okuyama, S. Kuwayama, Y. Nakazawa, S. Hatta and T. Aruga, Surf. Sci., 2022, 723, 122126 CrossRef CAS.
- P. Puschnig, S. Berkebile, A. J. Fleming, G. Koller, K. Emtsev, T. Seyller, J. D. Riley, C. Ambrosch-Draxl, F. P. Netzer and M. G. Ramsey, Science, 2009, 326, 702–706 CrossRef CAS PubMed.
- https://doi.org/10.1351/goldbook.T06456 .
- A. Baklanov, J. T. Küchle, D. A. Duncan, P. T. P. Ryan, R. J. Maurer, M. Schwarz, E. C. Rascon, I. Piquero-Zulaica, H. T. Ngo, A. Riss, F. Allegretti and W. Auwärter, J. Phys. Chem. C, 2023, 127, 7501–7512 CrossRef CAS.
- O. De Luca, T. Caruso, I. Grimaldi, A. Policicchio, V. Formoso, J. Fujii, I. VoborniK, D. Pacilé, M. Papagno and R. G. Agostino, Nanotechnology, 2020, 31, 365603 CrossRef CAS PubMed.
- C. Ruggieri, S. Rangan, R. A. Bartynski and E. Galoppini, J. Phys. Chem. C, 2015, 119, 6101–6110 CrossRef CAS.
- P. Amsalem, L. Giovanelli, J. M. Themlin and T. Angot, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 235426 CrossRef.
- A. Zangwill, Physics at Surfaces, Cambridge University Press, Cambridge, 1996 Search PubMed.
- L. Massimi, M. Angelucci, P. Gargiani, M. G. Betti, S. Montoro and C. Mariani, J. Chem. Phys., 2014, 140, 244704 CrossRef PubMed.
- M. G. Betti, P. Gargiani, R. Frisenda, R. Biagi, A. Cossaro, A. Verdini, L. Floreano and C. Mariani, J. Phys. Chem. C, 2010, 114, 21638–21644 CrossRef CAS.
- M. Casarin, C. Maccato and A. Vittadini, J. Phys. Chem. B, 1998, 102, 10745–10752 CrossRef CAS.
- M. Casarin, G. Granozzi, M. Sambi, E. Tondello and A. Vittadini, Surf. Sci., 1994, 307–309, 95–100 CrossRef CAS.
- M. Casarin, C. Maccato and A. Vittadini, J. Chem. Soc., Faraday Trans., 1998, 94, 797–804 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Frontier electronic structure of MTPP and MPc (M = V, Cr, Mn, Fe, Co, Ni, Cu, and Zn) isolated complexes. Optimized coordinates of Py, i-Ind, H2P, H2Pz, H2TPP, H2Pc, P2−, Pz2−, TPP2−, and Pc2− in Tables S1–S10; optimized coordinates of MTPP and MPc in Tables S13, S16–S24, S27, S28, S30, S31, S33, and S34; optimized coordinates of CoPc+, NiPc+, CuPc+, and ZnPc+ in Tables S25, S29, S32, and S35; H2TPP, H2Pc, MTPP, and MPc TSIEs in Table S11; MTPP and MPc optimized/experimental M–NPy bond lengths in Table S12; MTPP and MPc GS terms and low-lying excited state terms sharing the same GS spin multiplicity in Table S14; MTPP and MPc TSEAs in Table S15; theoretical/experimental ν values of the IR-active 22eu vibrational mode of MPc and MPc+ (M = Co, Ni, Cu, and Zn) in Table S25; selected internuclear distances (Å) in: D2h H2P and H2Pz (Fig. S1); D2h H2TPP and H2Pc (Fig. S2); D4h P2− and Pz2− (Fig. S3); D4h TPP2− and Pc2− (Fig. S4); energy position of H2P, H2TPP, H2Pc, and H2Pz GS FMOs (Fig. S5); 3D CPs of the Py and i-Ind HOMOs (Fig. S6); 3D CP of the i-IND 1a2 π MO (Fig. S7); 3D CPs of H2P FMOs generated by the 1a2 Py HOMO (Fig. S8); 3D CPs of low-lying H2P π MOs (Fig. S9); parenthood between pmcπ and pmcπ* Frontier MOs upon D2h → D4h switching (Fig. S10); 3D CPs of low-lying TPP2− and Pc2− π* MOs (Fig. S11); superposition of the CoPc (2A1g) and CoPc+ (3A1u), NiPc (1A1g) and NiPc+ (2A1u), CuPc (2B1g) and CuPc+ (3B1u), ZnPc (1A1g) and ZnPc+ (2A1u) otpimized structures in Fig. S12; schematic representation of the SCM ns AOs SALCs in Fig. S13–S17. See DOI: https://doi.org/10.1039/d5cp01576f |
| ‡ Porphine (H2P) is an aromatic (4n + 2; n = 4 for the shortest cyclic path), heterocyclic, organic compound consisting of four pyrrole fragments held together by as many methylidene bridges, which makes it the simplest tetrapyrrole (C20H14N4). Because of the H2P low solubility, the interest for the pristine molecule is mainly theoretical. Substituted porphines correspond to porphyrins; meso-tetraphenylporphyrin (H2TPP) is the most common.9 The acronym herein used for M complexes of H2TPP is MTPP. |
| § ADF outcomes about isolated MTPP and MPc are compared with the results of the literature in the ESI.† |
| ¶ Throughout the paper, the MO numbering corresponds to all-electron calculations independently of adopting the frozen core approximation. |
| || Both CoII (3d7) and NiII (3d8) complexes are characterized by a low-spin (LS) state with S = ½ and S = 0, respectively (see Section ESI.2, ESI†). |
** Upon the D2h → D4h switching, the following correlations hold between irreducible representations (ir): (B2g + B3g) → Eg;  ; ;  ; ;  (see Appendix 3 of ref. 72). (see Appendix 3 of ref. 72). |
| †† The internuclear distance between SCMnn is the same, independently of the surface Miller indexes: 2.556, 2.889, and 2.884 Å in Cu, Ag, and Au, respectively.95 |
| ‡‡ The complex herringbone reconstruction of Au(111) corresponds to a compressed atomic layer with an average lattice spacing of 2.76 Å along the [110] direction,96 instead of the corresponding bulk plane value of 2.88 Å, despite the hexagonal symmetry is locally preserved. The Au(100) surface also bears a large cell reconstruction, resulting in a surface layer with an atomic density 25% larger than in the (100) bulk plane. The corresponding quasi-hexagonal surface layer displays an average interatomic spacing (∼2.78 Å) very close to that of the (111)-herringbone reconstruction.97,98 Finally, the Au(110) surface displays the renown (1 × 2) missing row reconstruction, which exhibits local (111) microfacets. The missing row reconstruction may be lifted by the adsorption of large heteroaromatic molecules like CuPc, which locally deconstructs the surface beneath the molecular scaffold.99 |
| §§ The schematic representations of the CM(111) STCMnn and SHCMnnns AOs SALCs sketched in Fig. S16 and S17 (ESI†) do not consider the ns AOs normalization coefficients; the radii of the ns AOs localized on different SCMnn are then the same. The interested reader may refer to Chapter 7 of ref. 72. |
| ¶¶ Besides CM(111) T and B sites, two types of H sites characterize the surface: the Hhcp and the Hfcc in Fig. 3. If the attention is limited to the outermost CM(111) layer, Hhcp and Hfcc have the same environment. |
| |||| Blades et al. have thoroughly investigated the electronic and molecular properties of VCux+, VAgx+, and VAux+ (x = 3, 14) clusters. The V–Ag bond distance varies between 2.76 Å (VAg3+) and 2.95 Å (VAg14+) with a mean value of 2.8 Å.103 |
| *** The Au(110) LB chemisorption site has not being herein considered because, with FePc positioned 2.85 Å above the bulk-terminated surface,111 the Fe–SLBAgnn internuclear distance amounts to 3.504 Å. |
| ††† The TSEA of the CoP 3dz2-based σa↓1g and 3dx2–y2-based σb↑1g SOs are 1.73 eV and 0.73 eV, respectively. |
| This journal is © the Owner Societies 2025 |