
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of substitution pattern on the dynamics of internal conversion and intersystem crossing in thiopyridone isomers†
Douglas
Garratt‡
 a,
Sambit K.
Das‡
b,
Kacie J.
Nelson
c,
Jessica
Harich
d,
Antonia
Freibert
d,
Camila
Bacellar
e,
Claudio
Cirelli
e,
Philip J. M.
Johnson
e,
Rebeca G.
Castillo
f,
Marija R.
Zoric
c,
Ru-Pan
Wang
d,
Hyeongtaek
Lim
c,
Amy A.
Cordones
c,
Nils
Huse
d,
Michael
Odelius
*b and
Kelly
Gaffney
*c
a,
Sambit K.
Das‡
b,
Kacie J.
Nelson
c,
Jessica
Harich
d,
Antonia
Freibert
d,
Camila
Bacellar
e,
Claudio
Cirelli
e,
Philip J. M.
Johnson
e,
Rebeca G.
Castillo
f,
Marija R.
Zoric
c,
Ru-Pan
Wang
d,
Hyeongtaek
Lim
c,
Amy A.
Cordones
c,
Nils
Huse
d,
Michael
Odelius
*b and
Kelly
Gaffney
*c
aLinac Coherent Light Source (LCLS), SLAC National Accelerator Laboratory, 2575 Sand Hill Road, Menlo Park, CA 94025, USA
bDepartment of Physics, Stockholm University, AlbaNova University Center, SE-106 91 Stockholm, Sweden
cStanford PULSE Institute, SLAC National Accelerator Laboratory, Stanford University, Menlo Park, California 94025, USA
dDepartment of Physics, University of Hamburg and Center for Free-Electron Laser Science, Hamburg, Germany
ePaul Scherrer Institute, CH-5232 Villigen PSI, Switzerland
fLaboratory of Ultrafast Spectroscopy, École Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland
First published on 10th July 2025
Abstract
We report a combined experimental and theoretical investigation of the ultrafast internal conversion (IC) and intersystem crossing (ISC) dynamics of two thiopyridone (TP) isomers in solution. Our study used ultrafast transient X-ray absorption spectroscopy (XAS) at the sulfur K-edge, in conjunction with electronic excited state surface hopping molecular dynamics and simulations of the excited state XAS, to investigate the impact of the functional group substitution pattern and solvent on the dynamics of IC and ISC. The combination of the localized X-ray probe and the simulation results enables, in part, the differentiation between ππ* and nπ* character excited states, as well as singlet and triplet states. Access to nπ* character excitations has particular value since they often prove challenging to assess with optical spectroscopy. For 2-TP, the photoexcited S2 (ππ*) state rapidly undergoes IC to the S1 (nπ*) state below the instrument response time, followed by ISC to the T1 (ππ*) state on a timescale of 600 fs in acetonitrile. For 4-TP, the timescale of S2 to S1 IC increases to 330 fs and the timescale of ISC increases to more than 10 ps. The differences between isomers are rationalized by considering the key role of the, nπ* intermediates in mediating the intersystem crossing of these systems. Varying the substitution pattern of the molecule can stabilize or destabilize these intermediates leading to the increase in ISC rate in the ortho isomer as compared to the para isomer, while changing the solvent from acetonitrile to water had minimal effect on the electronic excited state relaxation mechanism.
1 Introduction
The interplay of internal conversion and intersystem crossing is crucial in determining the excited state properties and reactivity of organic molecules. Many organic chromophores undergo rapid internal conversion to the ground state following photoexcitation making them photo-stable and non-reactive, important properties for the photoprotection of DNA from ultraviolet (UV) light.1,2 Intersystem crossing generates population in triplet and higher lying spin states which have long radiative lifetimes.3 For triplet states with slow non-radiative decay, these long-lived electronic excited states often prove photochemically active, making them useful in organic photosensitizers and in organic photocatalysis.4–6 Understanding the chemical and environmental factors which determine the relative rates and efficiency of ISC and IC therefore offers a route towards optimizing the excited state properties of a molecular system for a particular application. The rate and efficiency of these processes ultimately depend on the character of the electronic excited states,7–9 the shape of the corresponding excited state potential energy surfaces9,10 and the dynamics at points where these surfaces are close in energy or intersect. In organic heteroaromatic compounds internal conversion and intersystem crossing are often mediated by excited states with nπ* character. These states have low absorption cross section from the ground state meaning that they are populated indirectly via internal conversion from bright ππ* character states following photoexcitation. In a single-electron picture, nπ* states correspond to excitation of a lone pair electron which is localized at the heteroatom and in the plane of the π-conjugated molecule to an out-of-plane, delocalized π* orbital. The strong localization of the n hole and the fact that the orbital is perpendicular to the π molecular orbital enables efficient intersystem crossing between states of nπ* and ππ* character in accordance with El-Sayed's rules.11,12 Therefore the relative energetics and coupling between the nπ* and ππ* character states are important in determining the rate of intersystem crossing in these systems. In turn the energetics of these states can be strongly influenced by both solvent environment and chemical modifications.The class of thiocarbonyl compounds form a striking example of how substitution of an oxygen atom with a sulfur can dramatically impact the optical properties and excited state dynamics of a system. Thiocarbonyls typically undergo efficient intersystem crossing to form long-lived triplet excited states with rich photochemistry.13 Of particular recent interest are sulfur substituted nucleobases (thiobases) which absorb in the UV A–B region and have close to unity triplet yield, leading to potential applications in photodynamic therapy.14–16 This behavior is unlike that of the canonical nucleobases which absorb below 250 nm and undergo rapid and efficient IC to the ground state.17,18 Previous time resolved studies on these systems19–21 have suggested that the efficient triplet state population in thiobases is primarily due to the stabilizing effect of the sulfur atom on the heteroatom-centered nπ* and ππ* excited states. This lowers the energy of these states relative to the conical intersections responsible for the fast non-radiative decay of canonical nucleobases to the ground state and therefore blocks these relaxation channels in thiobases. The efficient ISC is then due to a combination of the strong localization of the n and π orbitals on the sulfur atom and the energetic proximity of the sulfur centered nπ* and ππ* states.
Directly probing these processes with optical spectroscopy is challenging due to their ultrafast nature and the fact that nπ* states typically have low optical cross section,22 meaning that their role is often inferred indirectly. Time resolved X-ray spectroscopy offers a new tool in this regard due to the distinct X-ray transitions which, in an orbital picture, depend on the dipole overlap between core orbitals localized at the heteroatom and the unoccupied valence orbitals of the molecule.23–25 This has been utilized in pioneering X-ray absorption studies to directly probe the role of dark nπ* excitations in the internal conversion and electronic relaxation of organic chromophores26–29 using either high-harmonic generation or X-ray free electron laser (XFEL) based sources in the soft X-ray range. Extending time resolved heteroatom K-edge absorption spectroscopy to biologically and chemically relevant sulfur containing compounds requires probing in the tender X-ray range (2–5 keV). Time resolved studies at synchrotron facilities have demonstrated the chemical sensitivity of S K-edge X-ray spectroscopy to photochemical reaction products,30–33 and more recently developed XFEL sources and beamlines in the tender X-ray spectral range are opening up time-resolved S K-edge spectroscopy on femtosecond timescales.34,35
The focus of this work is thiopyridone (TP), the sulfur substituted analogue of hydroxypyridine, which in turn is one of the simplest model systems for the photophysics and tautomerization of DNA base-pairs. Photo-excitation of thiopyridone leads to rapid and efficient intersystem crossing to a long-lived reactive triplet state.36,37 Previous X-ray and optical studies on 2-thiopyridone38,39 have focused on the photochemistry of the triplet state, in particular the possibility of it undergoing excited-state proton transfer to form the thiol form 2-mercaptopyridine (2-MP). However, the process of triplet state formation has not been resolved due to the ultrafast timescales involved. Theoretical calculations for 2-TP40–42 have predicted rich intersystem crossing dynamics which proceed via a dark singlet nπ* character state and a triplet excited state of mixed nπ*, ππ* character to form the lowest triplet state within 1 ps.
Here, we directly probe the role of the intermediate dark nπ* states in the IC and ISC of thiopyridone with time resolved sulfur K-edge X-ray absorption spectroscopy, electronic excited state molecular dynamics with surface hopping to describe non-adiabatic changes in electronic state, and quantum chemical simulations of the sulfur K-edge X-ray absorption spectra. To understand the impact of chemical structure and solvent environment on the excited state dynamics, we compare 2-TP (ortho isomer) with 4-TP (para isomer) dissolved in acetonitrile and water. This combination of experimental and theoretical methods provides a firm foundation for interpreting the isomer dependence of the electronic excited state dynamics of solution phase thiopyridone.
1.1 Methods
We pumped 2-TP/4-TP in acetonitrile and 2-TP in water at a wavelength of 357 nm (3.47 eV) at a fluence of 5.9 mJ cm−2 giving an excitation fraction of approximately 8% for 2-TP and 30% for 4-TP. The pulse intensity was chosen so that the pulse energy dependence of the transient bleach signal at 5 ps was in the linear regime (see Fig. S4, ESI†). We recorded the majority of the data with the pump polarization parallel to the X-ray polarization. For 2-TP in acetonitrile, we also recorded a full dataset of kinetic and spectral traces with perpendicular pump and probe polarization in order to resolve the polarization anisotropy of the XA signal. For all samples, we recorded energy scans for a series of quasi-logarithmically spaced time delays between 0.2 and 200 ps and time delay scans to ∼100 ps (sample dependent) were recorded at the main absorption features. The temporal instrument response function was estimated to be 90 fs from fits to the kinetic traces. To account for temporal drifts in the laser X-ray arrival time, reference kinetic traces at the bleach feature were recorded regularly and the time axis of subsequent or prior scans were aligned to the nearest reference scan. Note that this means that there is a systematic uncertainty in the exact position of time zero in the experimental kinetic traces. We also subtracted the below-edge absorption from the static spectra and normalized the difference spectra to the static absorption above the edge.
ESMD simulations of the two constitutional isomers were executed using the surface hopping52 approach, as implemented in the SHARC-3.0 code.53–55 Simulations were performed on the 2-TPacetoimpl and 4-TPacetoimpl models, with forces, energies, and wavefunction overlaps from TDDFT calculations in ORCA using the same settings as in the static calculations. Initial conditions with geometries and associated velocities were generated from a discrete sampling of the Wigner distribution of vibrational normal modes. From analysis of the corresponding UV-visible spectra, we concluded that photoexcitations associated with  excitations are deemed essential for the current study. Accordingly, out of the 300 initial conditions in the Wigner sampling, we simulated 150 trajectories from the ESMD-S2 set for the 2-TP and 4-TP models. The default parameters in SHARC were used for determining the non-adiabatic coupling and spin–orbit coupling (SOC) in the ESMD simulations.56–63
excitations are deemed essential for the current study. Accordingly, out of the 300 initial conditions in the Wigner sampling, we simulated 150 trajectories from the ESMD-S2 set for the 2-TP and 4-TP models. The default parameters in SHARC were used for determining the non-adiabatic coupling and spin–orbit coupling (SOC) in the ESMD simulations.56–63
We simulated sulfur K-edge XA spectra for 2-TP and 4-TP with a multi reference approach to assign features in the transient spectra. Initially, we compared the valence excitation results from TDDFT with those obtained using multistate complete active space perturbation theory (MS-CASPT2)64–68 with a CAS(12, 10) active space in OpenMolcas v.23.06.69 To accommodate the core excitations, the active space was expanded using restricted active space perturbation theory (RASPT2).65,70 The sulfur 1s core orbital (with two electrons) was incorporated into the active space alongside at most one hole in the core orbital. The resulting RAS(14,1,0;1,10,0) active space, combined with the highly excited state scheme,71,72 allowed us to determine the core excited states and derive XA spectra for ground and valence excited states. The transition dipole moments of the valence and core excitations were calculated in the integrated restricted active space state interaction (RASSI) framework73,74 in OpenMolcas.
2 Results
2.1 Valence electronic excited states
To understand the character of the valence electronic excited state structure, we first examine the energy and orbital character of the excited states of 2-TP and 4-TP at the optimized geometry of the ground state (Franck–Condon point).Fig. 1A and B presents the vertical excitation energies and energetic ordering of the low lying singlet and triplet states in 2-TP and 4-TP calculated via TDDFT in an implicit acetonitrile solvent medium. The full datasets at the TDDFT and CASPT2 levels of theory are tabulated in Tables S12, S15 (2-TP) and S13, S16 (4-TP) in the ESI.† The frontier molecular orbitals contributing to the low-lying singlet and triplet states are shown in Fig. 1D. The complete active space implemented in the multi reference approach is illustrated in Fig. S13 in the ESI.† As noted in previous studies, the n and π orbitals both have significant sulfur 3p character and are distinguished by the orientation of the orbital with respect to the plane of the molecule. The n orbitals are in the molecular plane and the π orbitals are out of plane. Transient S K-edge X-ray absorption spectroscopy (XAS) directly probes the S 3p character of the unoccupied molecular orbitals and provides a powerful means of characterizing the orbital character of electronic excited states.
 | ||
| Fig. 1 (A) Qualitative depiction of the singlet (Sn) and triplet (Tn) excited state energies of 2-TP at the Franck–Condon point calculated at the TDDFT level (energies summarized in Tables S12 and S13 in the ESI†). The dominant expected IC and ISC pathway is depicted on the diagram. (B) Equivalent diagram for 4-TP, ISC from S1 to T2 in this isomer is expected to be less efficient due to the same orbital character of these states at the Franck–Condon point as illustrated with the dashed orange line. (C) Experimental UV-vis spectra of 2-TP and 4-TP in water and acetonitrile. The inset in the top right shows a zoom-in on the 2.2 eV to 3.5 eV region of 2-TP and 4-TP in acetonitrile. (D) Frontier molecular orbitals which describe the low lying singlet and triplet states of 2-TP and 4-TP. | ||
Experimentally measured UV-visible spectra for both isomers in acetonitrile and water are shown in Fig. 1C. The UV-visible spectrum of 2-TP shows two absorption bands at approximately 3.4 and 4.2 eV in acetonitrile. This finding is in qualitative agreement with the computed UV-vis spectra in Fig. S14 (ESI†). Based on the TDDFT and CASPT2 calculations, and consistent with previous studies,75,76 we assign the lower lying absorption band to the  state, while the higher lying band is assigned to
state, while the higher lying band is assigned to  and higher singlet state excitations. The UV-visible spectrum of 4-TP is dominated by a single absorption at around 3.55 eV which is assigned primarily to the
and higher singlet state excitations. The UV-visible spectrum of 4-TP is dominated by a single absorption at around 3.55 eV which is assigned primarily to the  state. Higher lying singlet states in this isomer absorb above 5 eV. For both isomers, photo-excitation at 3.5 eV populates the
state. Higher lying singlet states in this isomer absorb above 5 eV. For both isomers, photo-excitation at 3.5 eV populates the 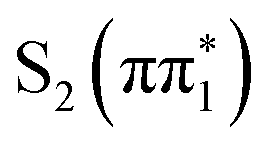 state.
state.
The TDDFT calculations suggest that the S1 state of 2-TP has 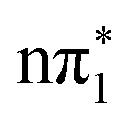 character and is very close in energy to the
character and is very close in energy to the  state (separated by less than 200 meV). Indeed, the CASPT2 calculation in this study of 2-TP gives an inverted energetic ordering
state (separated by less than 200 meV). Indeed, the CASPT2 calculation in this study of 2-TP gives an inverted energetic ordering  and
and 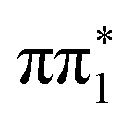 character singlet states but also place them very close in energy. The lowest lying triplet state of 2-TP has
character singlet states but also place them very close in energy. The lowest lying triplet state of 2-TP has 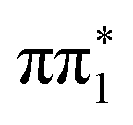 character and is separated by more than 1 eV from
character and is separated by more than 1 eV from  , indicating that direct intersystem crossing from
, indicating that direct intersystem crossing from  is unlikely to be efficient. However, in between these states are two higher lying triplet states of
is unlikely to be efficient. However, in between these states are two higher lying triplet states of 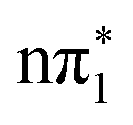 and
and  characters which are not only close in energy to each other but also to the
characters which are not only close in energy to each other but also to the  state at the Franck–Condon point. The small energy separation between these states gives a potential ISC pathway from
state at the Franck–Condon point. The small energy separation between these states gives a potential ISC pathway from  to
to  through these states.
through these states.
For 4-TP, TDDFT calculations at the electronic ground state geometry give the same energetic ordering of the singlet and triplet states as 2-TP but predict a stabilization of the  ,
, 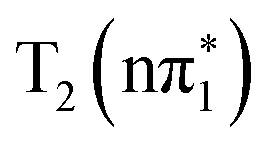 and
and  states relative to the bright
states relative to the bright  state and the higher lying
state and the higher lying  state. We see evidence for the stabilization of the
state. We see evidence for the stabilization of the 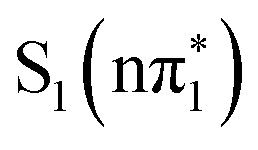 state of 4-TP in the UV-vis spectra, which has a very weak low energy shoulder on the 3.5 eV absorption band in acetonitrile due to the
state of 4-TP in the UV-vis spectra, which has a very weak low energy shoulder on the 3.5 eV absorption band in acetonitrile due to the 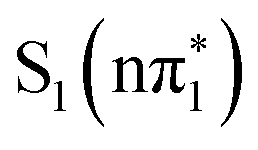 state (see Fig. S5, ESI†). The strength of this absorption feature is approximately 0.5% of the main ππ* band. The equivalent lowest energy band in 2-TP does not exhibit a low energy shoulder, consistent with the weak absorption of the S1 state overlapping in energy with the bright state transitions at the Franck–Condon point. Therefore, the energy separation between the S1 state and the bright S2 state calculated to be larger in 4-TP than in 2-TP appears consistent with the experimental spectra.
state (see Fig. S5, ESI†). The strength of this absorption feature is approximately 0.5% of the main ππ* band. The equivalent lowest energy band in 2-TP does not exhibit a low energy shoulder, consistent with the weak absorption of the S1 state overlapping in energy with the bright state transitions at the Franck–Condon point. Therefore, the energy separation between the S1 state and the bright S2 state calculated to be larger in 4-TP than in 2-TP appears consistent with the experimental spectra.
2.2 ESMD calculations
Fig. 2 summarizes the results from the excited state molecular dynamics simulations performed for 2-TP and 4-TP in an implicit acetonitrile solvent. Panels A and B show the time-dependent adiabatic state populations over the 1 ps simulation time for 2-TP and 4-TP respectively. For additional details, see Section S5.2.2 of the ESI.† In general and consistent with the qualitative interpretation of the excited state ordering in the ground state geometry, we see a much faster build-up of the T1 state population in 2-TP than in 4-TP. For 2-TP, T1 exhibits a dominant state population among all the adiabatic states within 1 ps. In contrast, in 4-TP only a minimal triplet state population appears within the first picosecond, with a significant population accumulating in the S1 state. | ||
| Fig. 2 Dynamical simulations of 2-TP and 4-TP molecules in acetonitrile solution (implicit solvation model). The population is presented in the adiabatic representation. Panels (A) and (B) show population dynamics of the ESMD-S2 set of trajectories for 2-TP and 4-TP respectively. | ||
For 2-TP, the simulations initially show a very rapid IC from the bright S2 state into the dark S1 state, followed by a subsequent rise of triplet states population. A transient T2 population builds up over the first 200 fs, which eventually decays into T1. To identify the ISC pathway, we follow the transition matrix shown in Table 1, which collects the number of hops between states in the dynamical simulation. This shows a large number of S2 → S1 transitions consistent with the fast initial IC to S1. The dominant ISC transition occurs through S1 → T2, followed by the S1 → T3 channel, corresponding to triplet  and
and  electronic state configurations in the Franck–Condon region, respectively. The S1 → T3 pathway does not lead to significant T3 population within the state dynamics, largely due to a fast T3 → T2 internal conversion. Subsequently, an efficient IC from T2 to T1 results in a considerable T1 population within 1 ps. The ESMD simulation thus suggests that the dominant photorelaxation pathway to T1 in 2-TP is as follows:
electronic state configurations in the Franck–Condon region, respectively. The S1 → T3 pathway does not lead to significant T3 population within the state dynamics, largely due to a fast T3 → T2 internal conversion. Subsequently, an efficient IC from T2 to T1 results in a considerable T1 population within 1 ps. The ESMD simulation thus suggests that the dominant photorelaxation pathway to T1 in 2-TP is as follows:  . Excitations to higher lying singlet states are insignificant and do not affect the ISC dynamics due to rapid internal conversion to the S1 state within the first 50 fs.
. Excitations to higher lying singlet states are insignificant and do not affect the ISC dynamics due to rapid internal conversion to the S1 state within the first 50 fs.
| S0 | S1 | S2 | S3 | S4 | S5 | T1 | T2 | T3 | |
|---|---|---|---|---|---|---|---|---|---|
| 2-TP | |||||||||
| S0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| S1 | 0 | 0 | 140 | 0 | −1 | 0 | 0 | −83 | −30 |
| S2 | 0 | −140 | 0 | 0 | 1 | 0 | 0 | 8 | −17 |
| S3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| S4 | 0 | 1 | −1 | 0 | 0 | 0 | 0 | 0 | 0 |
| S5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| T1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 102 | −11 |
| T2 | 0 | 83 | −8 | 0 | 0 | 0 | −102 | 0 | 53 |
| T3 | 0 | 30 | 17 | 0 | 0 | 0 | 11 | −53 | 0 |
| 4-TP | |||||||||
| S0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| S1 | 0 | 0 | 134 | −4 | 0 | 0 | 1 | −20 | −2 |
| S2 | 0 | −134 | 0 | 3 | 1 | 0 | 0 | −1 | −1 |
| S3 | 0 | 4 | −3 | 0 | −1 | 0 | 0 | 0 | 0 |
| S4 | 0 | 0 | −1 | 1 | 0 | 0 | 0 | 0 | 0 |
| S5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| T1 | 0 | −1 | 0 | 0 | 0 | 0 | 0 | 18 | 0 |
| T2 | 0 | 20 | 1 | 0 | 0 | 0 | −18 | 0 | 2 |
| T3 | 0 | 2 | 1 | 0 | 0 | 0 | 0 | −2 | 0 |
For 4-TP, the ESMD simulations show a much slower rate of internal conversion from S2 to S1 with population transfer between these states taking place over a time scale of ∼200 fs. This is consistent with an increased energy separation between the S1 state and the higher lying singlet states in 4-TP as compared to 2-TP. ISC from S1 is also much slower in this isomer and primarily proceeds via S1 → T2 ISC followed by T2 → T1 IC, as shown in Table 1. Therefore despite the S1 → T2 pathway being the predominant route for singlet-to-triplet transitions in both isomers, the rate of intersystem crossing is significantly faster in the ortho than in the para isomer. This can be explained by considering the underlying electronic characters of these states. While the S1 state is  in both 2-TP and 4-TP, the character of the T2 state is
in both 2-TP and 4-TP, the character of the T2 state is 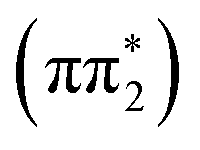 in 2-TP and
in 2-TP and  in 4-TP. Consequently, the dominant ISC pathway is El-Sayed11,12,77 allowed in 2-TP, but El-Sayed forbidden in 4-TP. For 4-TP, the corresponding El-Sayed allowed pathway is an S1 → T3 transition, which is inefficient due to the large energy gap between the S1 and T3 states.
in 4-TP. Consequently, the dominant ISC pathway is El-Sayed11,12,77 allowed in 2-TP, but El-Sayed forbidden in 4-TP. For 4-TP, the corresponding El-Sayed allowed pathway is an S1 → T3 transition, which is inefficient due to the large energy gap between the S1 and T3 states.
We also note oscillatory population dynamics along the S1, S2, T2, and T3 states of 2-TP in the ESMD. These oscillations are due to minimal energy separation between the two singlet states, which leads to frequent re-ordering of nπ* and ππ* states. The same is true for the two triplet states. This results in recurrent state transitions between the electronic states. However, the oscillatory behavior vanishes when the population dynamics is followed along the character of these states, instead of the adiabatic state representation. Such oscillatory behavior is absent in the T1 population of 2-TP and all electronic states in 4-TP, suggesting that the states are energetically distinct. Additionally, photo-induced stretching vibrations have been observed in the dynamical simulation. These vibrations tend to induce motion in the aromatic ring, which is governed by the geometrical differences between the relaxed geometries of S1 and S2. Thus, the recurrent surface crossings along these modes can give rise to the oscillatory population transfer between S1 and S2. An analysis was performed on the evolution of the vibrational normal modes of the ground state in the simulation, which allowed for the identification of some of the associated vibrations. We present a detailed discussion of these vibrational dynamics in Section S5.2.3 of the ESI,† including Fig. S17–S19, and Table S18.
The theoretical predictions point towards the key importance of the energy and character of the triplet intermediates in the intersystem crossing dynamics of thiopyridone. The simulations show a slower ISC rate in 4-TP than in 2-TP, in agreement with picosecond optical transient absorption measurements by Alam et al.,36 which reported a delayed rise time of 144 ps of the triplet–triplet absorption band in 4-TP and an instrument response limited, sub 100 ps ISC timescale in 2-TP. However, optical spectroscopy is not strongly sensitive to nπ* character states and the time resolution of these measurements was not sufficient to resolve the formation of the T1 state in 2-TP.
2.3 Time-resolved X-ray absorption spectroscopy
To probe the IC and ISC dynamics of these isomers, we employ time resolved X-ray absorption spectroscopy at the sulfur K-edge. This method probes the sulfur 3p character of the unoccupied valence orbitals of the molecule and is therefore expected to be sensitive to both nπ* and ππ* excited states due to the significant sulfur 3p character of the n and π orbitals holes created by the valence excited states.The static S K-edge XA spectra of 2-TP and 4-TP in water and acetonitrile (Fig. 3) show three main absorption features around the absorption edge. The lowest peak at around 2471 eV corresponds to a S1s → π* transition associated with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond and the two higher lying peaks at 2473.3 eV and 2475 eV correspond to S1s → σ* transitions. The energy of the S1s → π* peak is 0.4 eV lower in energy in 4-TP compared to 2-TP and shows a solvatochromic shift in going from acetonitrile to water. Comparison with the calculated ground state XA spectra shown in Fig. 5 indicates that this peak has contributions from transitions to the
S bond and the two higher lying peaks at 2473.3 eV and 2475 eV correspond to S1s → σ* transitions. The energy of the S1s → π* peak is 0.4 eV lower in energy in 4-TP compared to 2-TP and shows a solvatochromic shift in going from acetonitrile to water. Comparison with the calculated ground state XA spectra shown in Fig. 5 indicates that this peak has contributions from transitions to the 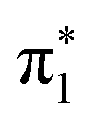 and
and  orbitals in 2-TP, while in 4-TP it corresponds to a transition to the
orbitals in 2-TP, while in 4-TP it corresponds to a transition to the  orbital, reflecting the lack of amplitude on the sulfur atom in the
orbital, reflecting the lack of amplitude on the sulfur atom in the  orbital in this isomer (see Fig. 1D). The peak at 2475 eV, visible in the acetonitrile spectra of both isomers, also shows a strong sensitivity to hydrogen bonding and is significantly broadened and shifted to higher energy in water.
orbital in this isomer (see Fig. 1D). The peak at 2475 eV, visible in the acetonitrile spectra of both isomers, also shows a strong sensitivity to hydrogen bonding and is significantly broadened and shifted to higher energy in water.
 | ||
| Fig. 3 (A) Static XA spectra of 2-TP and 4-TP in acetonitrile and water. (B) Illustration of the sulfur K-edge X-ray transient absorption spectroscopy methodology, where the UV pulse in orange opens a new channel for the subsequent X-ray pulse in blue. The plot shows static and pumped XA spectra recorded for 2-TP in acetonitrile at 2 ps. The orbital character of the XA features are labeled on the plot. The inset shows a qualitative illustration of the time resolved X-ray absorption methodology. An optical pump pulse photo-excites the system and the ensuing dynamics are probed with a time delayed X-ray pulse at the sulfur K-edge. The strong new feature below the edge in the pumped spectrum is due to a transition into the vacancy in the π or n orbital. | ||
For the time resolved measurements, we first focus on a comparison between 2-TP and 4-TP in acetonitrile. The key spectral changes observed in 2-TP and 4-TP over the first 4 ps following photo-excitation are depicted in Fig. 4. The difference XA spectra of 2-TP (Fig. 4A) show a strong below-edge transient absorption at positive time delays corresponding to a transition into the vacant n or π orbital. At short time delays this absorption feature is accompanied by an increase in absorption at around 2475 eV. Within the first picosecond, the below-edge transient absorption peak blue-shifts in energy by (0.37 ± 0.01) eV from (2467.78 ± 0.01) eV to (2468.15 ± 0.01) eV with a time constant of (0.58 ± 0.04) ps. The fit of the spectral data indicates that this shift in energy is best described by a change in amplitude of two discrete peaks, corresponding to distinct electronic states, rather than a continuous energy shift of a single peak. A detailed discussion of this analysis can be found in Section S4 of the ESI.† The shift in the below-edge absorption is accompanied by the decay of the 2475 eV feature, also on a sub-picosecond timescale. After this point in time, the energy of the spectral features remains constant, but the relative amplitudes of the peaks continue to evolve on a timescale of 5 ps, with the magnitude of the below-edge transient absorption and S1s → π* bleach increasing with time and the magnitude of the S1s → σ* bleach decreasing with time.
 | ||
| Fig. 4 Summary of the experimental transient XA spectra of 2-TP and 4-TP in acetonitrile. (A) Full transient XA spectra of 2-TP recorded at 0.2, 0.5 and 2 ps time delay. The inset shows a zoom in to the below edge transient absorption feature. (B) Kinetic traces of 2-TP at select photon energies over the first 4 ps. The energetic positions of the traces correspond to the color coded lines in panel (A). The data points show the raw data and associated uncertainty while the solid lines show the result of a global fit to the traces detailed in the text. (C) and (D) Equivalent plots for 4-TP in acetonitrile. | ||
This spectral evolution is reflected in the kinetic traces and corresponding fits, as shown in Fig. 4B. We fit the kinetics with a global sequential kinetic model described in detail in Section S2.2 of the ESI,† where the final species in the model is assumed to be long-lived on the timescale of the experimental data and corresponds to a triplet electronic excited state. The number of kinetic species required was determined by comparing the reduced χ2 of the fit for 2, 3 and 4 species models as discussed in the ESI,† Section S3. For 2-TP, three kinetic species gave the best description of the data. The time constants obtained from the model using three kinetic species are collected in Table 2 and the result of the fit can be found in Fig. 4B. The first kinetic species, which decays with a (0.58 ± 0.04) ps time constant describes the spectral shift in the below-edge feature and the decay of the 2475 eV absorption, while the second spectral evolution associated with a (4.8 ± 0.5) ps time constant describes changes in peak amplitudes associated with a decay in the anisotropy of the transient XA signal as discussed in the ESI,† Section S3.1.3. Therefore, we assign the first time constant to ISC to the long-lived triplet state, while we assign the slower time constant to rotational diffusion effects. Our analysis indicates our measurement lacks the time resolution to resolve the S2 → S1 IC consistent with the sub-50 fs IC dynamics seen in the ESMD for 2-TP.
| Parameter | 2-TP acetonitrile | 4-TP acetonitrile |
|---|---|---|
| E 1 (eV) | (2467.78 ± 0.01) eV | (2467.2 ± 0.2) eV |
| E 2 (eV) | (2468.15 ± 0.01) eV | (2467.7 ± 0.1) eV |
| E 3 (eV) | N/A | (2467.93 ± 0.07) eV |
| τ 1 (ps) | (0.58 ± 0.04) ps | (0.33 ± 0.03) ps |
| τ 2 (ps) | (4.8 ± 0.5) ps | (15 ± 6) ps |
| τ 3 (ps) | N/A | (46 ± 1) ps |
To assign the spectral features, we compare the experimental spectra with RASPT2 calculations of the S K-edge XA spectra for 2-TP in its low lying singlet and triplet excited states shown in Fig. 5A. Note that the state associated with a particular character may vary with molecular geometry and computation method. Moreover, in the ESMD simulation, the dynamics primarily involves singlet or triplet states associated with  character orbital. Therefore, we focus on states exhibiting the desired characteristics (
character orbital. Therefore, we focus on states exhibiting the desired characteristics ( and
and  ) and spin multiplicity in the X-ray spectral calculations, rather than their precise excited state ordering. When referring specifically to the calculated XA spectra we therefore drop the subscript denoting the adiabatic ordering of the state and label them
) and spin multiplicity in the X-ray spectral calculations, rather than their precise excited state ordering. When referring specifically to the calculated XA spectra we therefore drop the subscript denoting the adiabatic ordering of the state and label them  ,
,  ,
,  and
and  . We reintroduce the subscript when making assignments of spectra to adiabatic states in the ESMD calculations. Sulfur K-edge XA spectra were calculated for the ground state geometry and for the relaxed geometries of the excited states to investigate the effect of vibrational relaxation. The calculated spectra shown in Fig. 5 correspond to the excited state geometry. Additional discussion of the computed spectral features can be found in Section S5.3.1 of the ESI,† including Fig. S20 and Tables S19 and S20. Consistent with the experimental data, the calculated XA spectra for all the excited states considered show a strong absorption feature below the edge and an additional absorption feature above the S1s → σ* transition. An analysis of the features based on the natural transition orbitals (NTO) and transition dipole moments is shown in Fig. S8 and discussed in Section S5.3.2 of the ESI.† In essence, the below-edge feature corresponds to a transition into the vacant n orbital in the case of
. We reintroduce the subscript when making assignments of spectra to adiabatic states in the ESMD calculations. Sulfur K-edge XA spectra were calculated for the ground state geometry and for the relaxed geometries of the excited states to investigate the effect of vibrational relaxation. The calculated spectra shown in Fig. 5 correspond to the excited state geometry. Additional discussion of the computed spectral features can be found in Section S5.3.1 of the ESI,† including Fig. S20 and Tables S19 and S20. Consistent with the experimental data, the calculated XA spectra for all the excited states considered show a strong absorption feature below the edge and an additional absorption feature above the S1s → σ* transition. An analysis of the features based on the natural transition orbitals (NTO) and transition dipole moments is shown in Fig. S8 and discussed in Section S5.3.2 of the ESI.† In essence, the below-edge feature corresponds to a transition into the vacant n orbital in the case of  and
and  states or the vacant π orbital for the
states or the vacant π orbital for the 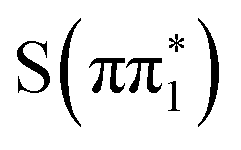 and
and  states while the higher lying feature is due to a change in energy of the S1s → σ* transition in the excited state.
states while the higher lying feature is due to a change in energy of the S1s → σ* transition in the excited state.
 | ||
| Fig. 5 S K-edge XA spectra of (A) 2-TPacetoimpl and (B) 4-TPacetoimpl models for the S1, S2, T1 and T2 states. Spectra are calculated at the optimized geometry of the respective valence excited state. The experimental spectra at 0.2 ps, 2 ps are shown for comparison. The dashed lines on the experimental spectra show the peak position obtained from a multi-peak fit to the spectra summarized in Table 2. The energy axis of the theoretical spectra is shifted by 4.45 eV to align the ground state π* resonance with the experiment. Ground state experimental and RASPT2-computed spectra are compared in the top panel (black and gray curves respectively). | ||
While the singlet and triplet states with 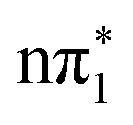 character have essentially identical XA spectra at their relaxed geometries, there are clear differences between these and the calculated XA spectra of the ππ* character states
character have essentially identical XA spectra at their relaxed geometries, there are clear differences between these and the calculated XA spectra of the ππ* character states 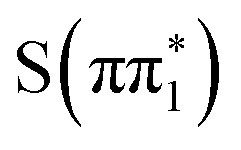 and
and 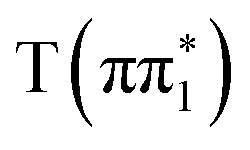 . The spectral position of the below-edge transient absorption feature is within 10 meV for the
. The spectral position of the below-edge transient absorption feature is within 10 meV for the  ,
,  and
and  states, but is significantly blue-shifted in the
states, but is significantly blue-shifted in the 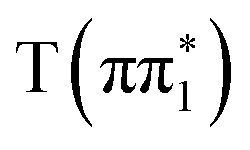 state, consistent with differences between the experimental spectra at 0.2 ps and 2 ps, as shown in Fig. 5. The calculated energies of the below-edge peak for each state are summarized in Table S21 of the ESI.† The absolute energetics and magnitude of the predicted shift of the
state, consistent with differences between the experimental spectra at 0.2 ps and 2 ps, as shown in Fig. 5. The calculated energies of the below-edge peak for each state are summarized in Table S21 of the ESI.† The absolute energetics and magnitude of the predicted shift of the  state relative to
state relative to  is in excellent agreement with the experimentally measured value of (0.37 ± 0.01) eV. Furthermore, the experimentally measured timescale for the shift in the energy of the transient absorption of (0.58 ± 0.04) ps shows excellent agreement with the ESMD prediction of the timescale of the
is in excellent agreement with the experimentally measured value of (0.37 ± 0.01) eV. Furthermore, the experimentally measured timescale for the shift in the energy of the transient absorption of (0.58 ± 0.04) ps shows excellent agreement with the ESMD prediction of the timescale of the  state formation in 2-TP.
state formation in 2-TP.
We also considered the effect of vibrational relaxation on the excited state sulfur K-edge spectra by calculating the spectra for both the ground and excited state geometries. As discussed in the ESI,† Section S5.3.1, structural relaxation blue-shifts the energy of the below-edge feature in the singlet states by 400 meV to 500 meV but does not significantly affect the triplet spectra. However, the energy of the below-edge XA feature in the ground state geometry in either the  or
or 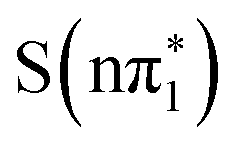 state is calculated to be much lower than what is observed experimentally at 200 fs and therefore ISC to
state is calculated to be much lower than what is observed experimentally at 200 fs and therefore ISC to  from a higher lying state gives a better description of the measured data than vibrational relaxation of the
from a higher lying state gives a better description of the measured data than vibrational relaxation of the  state. In addition, vibrational relaxation would lead to a continuous shift in energy of the below-edge feature with time delay. Fitting to the available experimental spectra (Fig. S10 described in Section S4 of the ESI†) shows that a transition between two discrete peaks better represents the experimental observations than a single shifting peak.
state. In addition, vibrational relaxation would lead to a continuous shift in energy of the below-edge feature with time delay. Fitting to the available experimental spectra (Fig. S10 described in Section S4 of the ESI†) shows that a transition between two discrete peaks better represents the experimental observations than a single shifting peak.
Based on the below-edge absorption feature alone, the time-resolved experimental difference spectra of 2-TP are consistent with an electronic transition from either 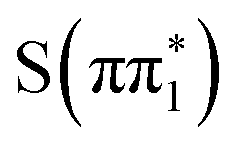 ,
,  , or
, or  to the long-lived
to the long-lived  state. We also considered the high-lying absorption feature at ∼2475 eV which shows differences in energy and amplitude between the calculated
state. We also considered the high-lying absorption feature at ∼2475 eV which shows differences in energy and amplitude between the calculated  ,
,  ,
,  , and
, and  XA spectra. The feature is predicted to be strongest and at highest energy in the nπ* character states,
XA spectra. The feature is predicted to be strongest and at highest energy in the nπ* character states,  ,
, 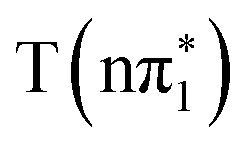 . It is somewhat red shifted and reduced in amplitude in
. It is somewhat red shifted and reduced in amplitude in  and further red shifted and reduced in amplitude for
and further red shifted and reduced in amplitude for  . Therefore the experimental data, which shows a decay of the feature to a small residual shoulder of the S1s → σ* transition with the same time constant as the blue-shift of the lower energy excited state absorption, is in better agreement with an nπ* to ππ* transition (either
. Therefore the experimental data, which shows a decay of the feature to a small residual shoulder of the S1s → σ* transition with the same time constant as the blue-shift of the lower energy excited state absorption, is in better agreement with an nπ* to ππ* transition (either 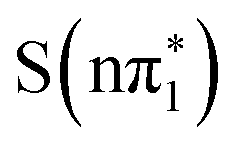 , or
, or  to
to  ) rather than a direct
) rather than a direct  to
to  transition, which would give an increase in the magnitude of this feature with time. The assignment of the photo-induced absorption at 2475 eV based on the calculated spectra is less robust than the analysis of the excited-state absorption at 2468 eV because of the limitations for higher energy transitions in the active space used for the RASPT2 calculations.
transition, which would give an increase in the magnitude of this feature with time. The assignment of the photo-induced absorption at 2475 eV based on the calculated spectra is less robust than the analysis of the excited-state absorption at 2468 eV because of the limitations for higher energy transitions in the active space used for the RASPT2 calculations.
2.4 4-TP
Fig. 4C and D summarizes the measured S K-edge time-resolved XA spectra of 4-TP in acetonitrile. We see similar features in this isomer as in 2-TP, including a strong below-edge transient absorption feature and a weaker increase in absorption above the S1s → σ* transition accompanied by bleaching of the static XA features. However, there are some notable differences with respect to the 2-TP spectra. Focusing on the kinetic traces shown in Fig. 4D, there is a faster decay (0.33 ± 0.03 ps time constant) in the lowest energy trace recorded at 2467.19 eV (red kinetic trace in Fig. 4D). Secondly, the amplitude of the difference signal at very early time delays is reduced relative to the signal strength at 2467.89 eV after 1 ps (purple kinetic trace). This is reflected in the full spectra shown in Fig. 4 panel C, which shows only a weak shoulder on the low energy side of the below-edge peak at 200 fs. Then, between 200 and 500 fs, there is an increase in the amplitude of the below-edge peak. The kinetic traces and the evolution associated spectra obtained from a global fit to these traces shown in Fig. S6 of the ESI† indicate that this shoulder is due to a reduced amplitude feature which has mostly decayed by 200 fs. Note that we did not record spectra at shorter time delays due to uncertainty in the position of time zero. As detailed in Section S4 of the ESI,† we fit the below-edge absorption feature recorded at select time delays with three Lorentzian peaks. In this fit, we constrain the energy of the peaks to be constant with time delay and the width of the peaks to be the same. The energy of the fitted peaks is summarized in Table 2. The lowest energy fitted peak at (2467.2 ± 0.2) eV only has amplitude in the earliest time delay spectrum, indicating that it corresponds to the fast decay component seen in the 2467.19 eV kinetic trace.
Fig. 5B shows the calculated sulfur K-edge spectra of 4-TP for the two lowest lying singlet and triplet excited states. The states with nπ* character,  and
and 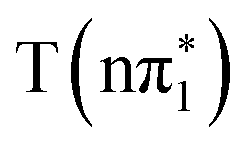 , are again predicted to have identical below-edge absorption in 4-TP. The
, are again predicted to have identical below-edge absorption in 4-TP. The  state is also predicted to give a very similar below-edge feature with slightly reduced amplitude and shifted energy. Therefore transitions between these states do not explain the reduced amplitude of the below-edge feature at early time delays observed in the experiment. This means that intersystem crossing from
state is also predicted to give a very similar below-edge feature with slightly reduced amplitude and shifted energy. Therefore transitions between these states do not explain the reduced amplitude of the below-edge feature at early time delays observed in the experiment. This means that intersystem crossing from  would not explain the experimental observations in 4-TP on sub-picosecond timescale. In contrast, the calculated
would not explain the experimental observations in 4-TP on sub-picosecond timescale. In contrast, the calculated 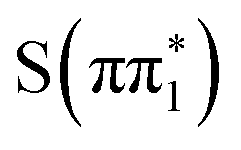 state spectrum shows reduced below-edge absorption cross section and is red shifted with respect to the other states. The calculated energy of this feature in the
state spectrum shows reduced below-edge absorption cross section and is red shifted with respect to the other states. The calculated energy of this feature in the  state of 2467.28 eV shows good agreement with the experimental value of (2467.2 ± 0.2) eV obtained from the fit. Therefore, we conclude the experimental spectra are most consistent with the (0.33 ± 0.03) ps time constant describing the relaxation of the
state of 2467.28 eV shows good agreement with the experimental value of (2467.2 ± 0.2) eV obtained from the fit. Therefore, we conclude the experimental spectra are most consistent with the (0.33 ± 0.03) ps time constant describing the relaxation of the  state. In principle, this could be due to either internal conversion to
state. In principle, this could be due to either internal conversion to  or ISC to one of the triplet states. However, the measured time constant is also in excellent agreement with the predicted timescale of
or ISC to one of the triplet states. However, the measured time constant is also in excellent agreement with the predicted timescale of 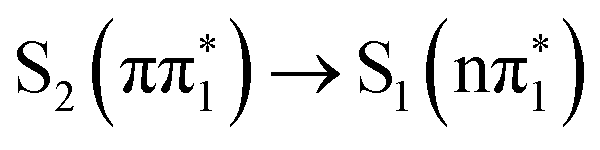 internal conversion from the ESMD calculations. Therefore, while we cannot rule out alternative ISC or IC channels in 4-TP on the basis of the experimental spectra alone, the good theoretical agreement between both the XA spectra and the observed timescales indicates S2 (ππ*) → S1 (nπ*) internal conversion with a time constant of (0.33 ± 0.03) ps provides the best description of the data recorded at short time delays.
internal conversion from the ESMD calculations. Therefore, while we cannot rule out alternative ISC or IC channels in 4-TP on the basis of the experimental spectra alone, the good theoretical agreement between both the XA spectra and the observed timescales indicates S2 (ππ*) → S1 (nπ*) internal conversion with a time constant of (0.33 ± 0.03) ps provides the best description of the data recorded at short time delays.
Unlike 2-TP, which does not show significant spectral evolution beyond the first picosecond, we observe subtle changes in the spectral position of the below-edge absorption feature on longer timescales in 4-TP. Between 0.2 ps and 20 ps there is a small shift in the central energy of the below-edge absorption feature as well as a decay of the low energy shoulder assigned to the  state. Within the three peak spectral model described above, this corresponds to a transition between a peak centered at (2467.7 ± 0.1) eV, which has dominant amplitude at 0.2 ps to a peak centered at (2467.93 ± 0.07) eV which grows in amplitude and dominates the spectrum at 20 ps (see Fig. S4 in Section S4 of the ESI†). Further, in the para isomer, the ESMD simulations and previous optical studies by Alam et al.36 suggest that the triplet state will be formed on longer timescales. The calculated spectra show a small difference in the energy of the below-edge absorption feature between the singlet and triplet nπ* states at around 2467.66 eV and the
state. Within the three peak spectral model described above, this corresponds to a transition between a peak centered at (2467.7 ± 0.1) eV, which has dominant amplitude at 0.2 ps to a peak centered at (2467.93 ± 0.07) eV which grows in amplitude and dominates the spectrum at 20 ps (see Fig. S4 in Section S4 of the ESI†). Further, in the para isomer, the ESMD simulations and previous optical studies by Alam et al.36 suggest that the triplet state will be formed on longer timescales. The calculated spectra show a small difference in the energy of the below-edge absorption feature between the singlet and triplet nπ* states at around 2467.66 eV and the  state at 2467.83 eV. These energies are in good agreement with the experimentally fitted values indicating that intersystem crossing from
state at 2467.83 eV. These energies are in good agreement with the experimentally fitted values indicating that intersystem crossing from  to
to  may play a role in the spectral changes observed on a ∼10 ps timescale. With this in mind, we fit the kinetic traces in 4-TP to a 4 component sequential kinetic model, which gives two time constants describing the longer timescale evolution summarized in Table 2. We tentatively assign the faster (15 ± 6) ps component to rotational diffusion, and the (46 ± 1) ps component to the formation of the triplet state (see Section S4 of the ESI† for detailed discussion). However, due to the limited number of kinetic traces recorded and the lack of distinct spectral changes, it is difficult to definitively distinguish rotational dynamics from potential electronic state changes in this isomer. We also note that Alam et al. concluded ISC occurred with a significantly longer time constant of 144 ps.
may play a role in the spectral changes observed on a ∼10 ps timescale. With this in mind, we fit the kinetic traces in 4-TP to a 4 component sequential kinetic model, which gives two time constants describing the longer timescale evolution summarized in Table 2. We tentatively assign the faster (15 ± 6) ps component to rotational diffusion, and the (46 ± 1) ps component to the formation of the triplet state (see Section S4 of the ESI† for detailed discussion). However, due to the limited number of kinetic traces recorded and the lack of distinct spectral changes, it is difficult to definitively distinguish rotational dynamics from potential electronic state changes in this isomer. We also note that Alam et al. concluded ISC occurred with a significantly longer time constant of 144 ps.
3 Discussion and closing remarks
We have used time resolved X-ray absorption spectroscopy at the sulfur K-edge to access the IC and ISC dynamics of 2-TP and 4-TP in acetonitrile and water. The experimental measurements are in excellent agreement with key predictions from ESMD simulations of the excited state dynamics and calculated excited-state S K-edge spectra. For 2-TP, the sulfur K-edge time-resolved XAS primarily probes the timescale of triplet state formation which is measured to be (0.58 ± 0.04) ps in acetonitrile and (0.68 ± 0.04) ps in water. This observed timescale is in good agreement with the overall triplet formation time from the ESMD calculations. The energetic shift in the below-edge absorption feature of (0.37 ± 0.01) eV is also in excellent agreement with the calculated energy difference between this feature for the state and the higher lying excited states. However, the definitive assignment of the short timescale experimental spectra based on the RASPT2 calculations of the XA spectra is precluded by the small predicted energy shift between the
state and the higher lying excited states. However, the definitive assignment of the short timescale experimental spectra based on the RASPT2 calculations of the XA spectra is precluded by the small predicted energy shift between the  ,
, 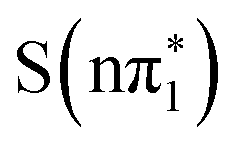 , and
, and  states. The similarity of the XA spectra of these states in part reflects the theoretical prediction that they are very close in energy and mixed in character in 2-TP. Despite this limitation, a comparison of the calculated ESMD and the S K-edge XAS for the electronic excited state difference spectra with the experimental spectral dynamics leads us to conclude that the population dynamics are dominated by the decay of the
states. The similarity of the XA spectra of these states in part reflects the theoretical prediction that they are very close in energy and mixed in character in 2-TP. Despite this limitation, a comparison of the calculated ESMD and the S K-edge XAS for the electronic excited state difference spectra with the experimental spectral dynamics leads us to conclude that the population dynamics are dominated by the decay of the  excited state and the formation of the long-lived
excited state and the formation of the long-lived  character excited state. The simulation of 2-TP dynamics indicates the experiment lacks the necessary time-resolution to observe the S2 → S1 IC.
character excited state. The simulation of 2-TP dynamics indicates the experiment lacks the necessary time-resolution to observe the S2 → S1 IC.
In contrast both IC and ISC are much slower in 4-TP. We assign the sub-ps population dynamics in the experiment to the S2 → S1 internal conversion on a (0.33 ± 0.03) ps timescale in acetonitrile and (0.30 ± 0.08) ps in water. This adds evidence in support of the hypothesis that a stabilization of the S1 (nπ*) state in 4-TP is responsible for the increased IC timescale in this isomer. We also observe subtle spectral changes on a timescale of (46 ± 1) ps in 4-TP which we tentatively assign to ISC to the T1 triplet state. This is again consistent with the theoretical prediction that ISC is less efficient in this isomer due to the energetically favored pathway  being nominally El-Sayed forbidden and the El-Sayed allowed pathway through
being nominally El-Sayed forbidden and the El-Sayed allowed pathway through  being inefficient due to the higher energy of the
being inefficient due to the higher energy of the 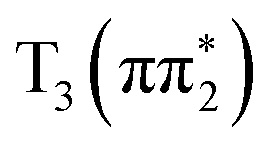 state relative to S1. We note that the determination of the ISC pathway proves less experimentally conclusive for 4-TP than 2-TP, due to challenges in separating ISC on >10 ps timescale from rotational effects. Therefore, the conclusions drawn for 4-TP regarding the timescale ISC relies on the good agreement between the fast time constant extracted from the experimental data and timescale of IC predicted in the ESMD calculations, as well as the good agreement between the calculated XA spectra and the peak positions extracted from the available experimental spectra.
state relative to S1. We note that the determination of the ISC pathway proves less experimentally conclusive for 4-TP than 2-TP, due to challenges in separating ISC on >10 ps timescale from rotational effects. Therefore, the conclusions drawn for 4-TP regarding the timescale ISC relies on the good agreement between the fast time constant extracted from the experimental data and timescale of IC predicted in the ESMD calculations, as well as the good agreement between the calculated XA spectra and the peak positions extracted from the available experimental spectra.
Due to the large contribution of sulfur 3p character in the π and n frontier orbitals occupied in the electronic ground state and depopulated in the lowest energy valence electronic excited states of 2-TP and 4-TP, time-resolved sulfur K-edge absorption proves to be sensitive to both nπ* and ππ* character excited states with either singlet and triplet spin multiplicities. While time resolved S K-edge XAS provides a universal probe of the low lying electronic excited states in these systems, distinguishing nπ* and ππ* states, which are close in energy, is challenging due to the ∼0.7 eV lifetime broadening of the absorption features and the similar S1s → π and S1s → n cross-sections. We have augmented our measurements with electronic excited state molecular dynamics simulations and RASPT2 simulations of the S K-edge difference spectra for distinct electronic excited state configurations. These theoretical findings prove essential to extract robust mechanistic information from the experimental results. Future studies targeting time resolved S K-edge RIXS could improve the spectral resolution beyond the 1s core hole lifetime broadening and potentially distinguish singlet from triplet excitations based on the anti-Stokes scattering signal.78 In general, polarization-dependent ultrafast XAS will assist in identifying the character of the electronic excited states. Unfortunately for 2-TP and 4-TP, excitation of the S-1s electron into the n or the π vacancy created in nπ* and ππ* character valence excited states have polarizations nearly orthogonal to the S2 (ππ*) valence excited state and, consequently, exhibit the same polarization dependence. Also combining S K-edge spectroscopy with either X-ray measurements at a complimentary absorption edge or optical spectroscopy could further disentangle nπ* and ππ* states which are close in energy. For example in thiopyridone, the nπ* states are predicted to have reduced electron density on the nitrogen atom meaning that nitrogen or carbon K-edge spectroscopy could selectively probe the role of ππ* states in the photophysics of these molecules.
In the theoretical investigations, we assessed the accuracy of the implicit solution models in comparison with other gas phase and micro-solvated models, referred to as 2-TPgas, 2-TPwaterexpl, 4-TPgas, and 4-TPwaterexpl. To summarize briefly, the results indicate consistency between the overall non-adiabatic dynamics of the explicit aqueous (micro-solvated) solvation models and those observed in the implicit acetonitrile solvation models. These similarities are primarily noticed in the evolution of electronic states, while structural variations exist between the two solvent models. In contrast, simulations of gas phase models at the TDDFT and CASSCF levels, including the previously published investigation,41 suggest large variations in the electronic structure of the gas phase and the solvated thiopyridone systems. Such alterations are attributed to the difference in the valence excited states and their underlying characteristics, which may modify the nature of the dynamics. Similar observations are also made in the computed sulfur K-edge absorption spectra of 2-TP. For instance, as presented in Table S21 (ESI†), while the computed spectral shifts of the transient absorption features of 2-TP are consistent among the solvated models, the largest deviations are noted for the gas phase calculations. Nevertheless, these intricate details of the theoretical aspects are outside the scope of the present paper and will be the subject of an independent study.79
Finally, previous studies have suggested that 2-TP may undergo excited-state proton transfer on ultrafast timescales76 prior to triplet state formation. We did not see evidence for this process in the isomers and solvents addressed here. Our results are consistent with previous studies which found that the photochemistry of thiopyridones originates from the long-lived triplet state rather than the singlets which are rapidly depopulated in <1 ps in the case of 2-TP. We also performed comparative measurements on the triplet state photochemistry between the isomers and solvents discussed in this work. These measurements indicate that the formation of excimers is the dominant deactivation mechanism of the triplet state. The dynamics of excimer formation will be discussed in a future publication.80
Author contributions
DG: formal analysis, investigation, project administration. SKD: formal analysis, software. KJN: investigation, formal analysis. JH: investigation. AF: investigation. CB: investigation. CC: investigation, software. PJMJ: investigation. RGC: investigation. MRZ: investigation. RPW: investigation. HL: investigation. AAC: supervision, resources. NH: supervision, resources. MO: conceptualization, supervision, funding acquisition. KG: conceptualization, supervision, funding acquisition. DG, SKD, MO and KG wrote the manuscript with comments from all of the authors.Conflicts of interest
There are no conflicts to declare.Data availability
Data for this article, including underlying datasets for each figure are available at supporting data: influence of substitution pattern on the dynamics of internal conversion and intersystem crossing in thiopyridone isomers at https://doi.org/10.17605/OSF.IO/CTJ7A. Additional supporting data has been included as part of the ESI.†Acknowledgements
We acknowledge the Paul Scherrer Institute, Villigen, Switzerland for provision of free-electron laser beamtime at the Alvra instrument of the SwissFEL ARAMIS branch. We thank Emma V. Beale and Dardan Gashi for sample delivery support in the experiments. K. G., D. G., M. Z., H. L., K. J. N. and A. A. C. were supported by the Department of Energy, Laboratory Directed Research and Development program at SLAC National Accelerator Laboratory, under contract DE-AC02-76SF00515. M. O. acknowledges funding from Swedish Research Council grant agreement no. 2021-04521. S. K. D. and M. O. acknowledge support from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 860553 (ITN Network SMART-X) and valuable discussions with Michael R. Coates, who also prepared a series of structures for the explicit aqueous solvation model. The computations were enabled by resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) and the Swedish National Infrastructure for Computing (SNIC) at NSC and PDC partially funded by the Swedish Research Council through grant agreement no. 2022-06725 and no. 2018-05973. J. H., A. F. and N. H. acknowledge financial support from the Cluster of Excellence ‘CUI: Advanced Imaging of Matter’ of the Deutsche Forschungs-gemeinschaft (DFG) – EXC 2056 – project ID 390715994.Notes and references
- W. J. Schreier, P. Gilch and W. Zinth, Annu. Rev. Phys. Chem., 2015, 66, 497–519 CrossRef CAS PubMed.
- A. Pinnola and R. Bassi, Biochem. Soc. Trans., 2018, 46, 467–482 CrossRef CAS PubMed.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC.
- X. Zhang, Z. Wang, Y. Hou, Y. Yan, J. Zhao and B. Dick, J. Mater. Chem. C, 2021, 9, 11944–11973 RSC.
- M. A. Bryden and E. Zysman-Colman, Chem. Soc. Rev., 2021, 50, 7587–7680 RSC.
- T. Bortolato, S. Cuadros, G. Simionato and L. Dell’Amico, Chem. Commun., 2022, 58, 1263–1283 RSC.
- M. C. Daza, M. Doerr, S. Salzmann, C. M. Marian and W. Thiel, Phys. Chem. Chem. Phys., 2009, 11, 1688–1696 RSC.
- M. Baba, J. Phys. Chem. A, 2011, 115, 9514–9519 CrossRef CAS PubMed.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
- C. M. Marian, Annu. Rev. Phys. Chem., 2021, 72, 617–640 CrossRef CAS PubMed.
- M. A. El-Sayed, Acc. Chem. Res., 1968, 1, 8–16 CrossRef CAS.
- S. Lower and M. El-Sayed, Chem. Rev., 1966, 66, 199–241 CrossRef CAS.
- J. D. Coyle, Tetrahedron, 1985, 41, 5393–5425 CrossRef CAS.
- T. C. Pham, V.-N. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 121, 13454–13619 CrossRef CAS PubMed.
- V.-N. Nguyen, Y. Yan, J. Zhao and J. Yoon, Acc. Chem. Res., 2020, 54, 207–220 CrossRef PubMed.
- J. Tang, L. Wang, A. Loredo, C. Cole and H. Xiao, Chem. Sci., 2020, 11, 6701–6708 RSC.
- R. Borrego-Varillas, A. Nenov, P. Kabaciński, I. Conti, L. Ganzer, A. Oriana, V. K. Jaiswal, I. Delfino, O. Weingart and C. Manzoni, et al. , Nat. Commun., 2021, 12, 7285 CrossRef CAS PubMed.
- V. I. Prokhorenko, A. Picchiotti, M. Pola, A. G. Dijkstra and R. D. Miller, J. Phys. Chem. Lett., 2016, 7, 4445–4450 CrossRef CAS PubMed.
- S. Mai, M. Pollum, L. Martínez-Fernández, N. Dunn, P. Marquetand, I. Corral, C. E. Crespo-Hernández and L. González, Nat. Commun., 2016, 7, 13077 CrossRef CAS PubMed.
- R. Borrego-Varillas, D. C. Teles-Ferreira, A. Nenov, I. Conti, L. Ganzer, C. Manzoni, M. Garavelli, A. Maria de Paula and G. Cerullo, J. Am. Chem. Soc., 2018, 140, 16087–16093 CrossRef CAS PubMed.
- D. Mayer, F. Lever, D. Picconi, J. Metje, S. Alisauskas, F. Calegari, S. Düsterer, C. Ehlert, R. Feifel and M. Niebuhr, et al. , Nat. Commun., 2022, 13, 198 CrossRef CAS PubMed.
- D. C. Teles-Ferreira, I. Conti, R. Borrego-Varillas, A. Nenov, I. H. Van Stokkum, L. Ganzer, C. Manzoni, A. M. de Paula, G. Cerullo and M. Garavelli, Chem. – Eur. J., 2020, 26, 336–343 CrossRef CAS PubMed.
- R. M. Jay, K. Kunnus, P. Wernet and K. J. Gaffney, Annu. Rev. Phys. Chem., 2022, 73, 187–208 CrossRef CAS PubMed.
- D. Garratt, M. Matthews and J. Marangos, Struct. Dyn., 2024, 11, 010901 CrossRef CAS PubMed.
- U. Bergmann, J. Kern, R. W. Schoenlein, P. Wernet, V. K. Yachandra and J. Yano, Nat. Rev. Phys., 2021, 3, 264–282 CrossRef CAS PubMed.
- T. Wolf, R. H. Myhre, J. Cryan, S. Coriani, R. Squibb, A. Battistoni, N. Berrah, C. Bostedt, P. Bucksbaum and G. Coslovich, et al. , Nat. Commun., 2017, 8, 29 CrossRef CAS PubMed.
- V. Scutelnic, S. Tsuru, M. Pápai, Z. Yang, M. Epshtein, T. Xue, E. Haugen, Y. Kobayashi, A. I. Krylov and K. B. Møller, et al. , Nat. Commun., 2021, 12, 5003 CrossRef CAS PubMed.
- Y.-P. Chang, T. Balciunas, Z. Yin, M. Sapunar, B. N. Tenorio, A. C. Paul, S. Tsuru, H. Koch, J.-P. Wolf and S. Coriani, et al. , Nat. Phys., 2024, 1–9 Search PubMed.
- A. Freibert, D. Mendive-Tapia, N. Huse and O. Vendrell, J. Phys. B: At., Mol. Opt. Phys., 2022, 54, 244003 CrossRef.
- M. Ochmann, I. Von Ahnen, A. A. Cordones, A. Hussain, J. H. Lee, K. Hong, K. Adamczyk, O. Vendrell, T. K. Kim and R. W. Schoenlein, et al. , J. Am. Chem. Soc., 2017, 139, 4797–4804 CrossRef CAS PubMed.
- M. Ochmann, A. Hussain, I. Von Ahnen, A. A. Cordones, K. Hong, J. H. Lee, R. Ma, K. Adamczyk, T. K. Kim and R. W. Schoenlein, et al. , J. Am. Chem. Soc., 2018, 140, 6554–6561 CrossRef CAS PubMed.
- K. Ledbetter, C. B. Larsen, H. Lim, M. R. Zoric, S. Koroidov, C. D. Pemmaraju, K. J. Gaffney and A. A. Cordones, Inorg. Chem., 2022, 61, 9868–9876 CrossRef CAS PubMed.
- A. A. Cordones, J. H. Lee, K. Hong, H. Cho, K. Garg, M. Boggio-Pasqua, J. J. Rack, N. Huse, R. W. Schoenlein and T. K. Kim, Nat. Commun., 2018, 9, 1989 CrossRef PubMed.
- M. Ochmann, J. Harich, R. Ma, A. Freibert, Y. Kim, M. Gopannagari, D. H. Hong, D. Nam, S. Kim and M. Kim, et al. , Nat. Commun., 2024, 15, 8838 CrossRef CAS PubMed.
- Y. Kim, D. Nam, R. Ma, S. Kim, M.-J. Kim, J. Kim, I. Eom, J. H. Lee and T. K. Kim, J. Synchrotron Radiat., 2022, 29, 194–201 CrossRef CAS PubMed.
- M. Alam, et al. , J. Chem. Soc., Perkin Trans. 2, 1998, 817–824 RSC.
- R. Du, C. Liu, Y. Zhao, K.-M. Pei, H.-G. Wang, X. Zheng, M. Li, J.-D. Xue and D. L. Phillips, J. Phys. Chem. B, 2011, 115, 8266–8277 CrossRef CAS PubMed.
- B. E. Van Kuiken, M. R. Ross, M. L. Strader, A. A. Cordones, H. Cho, J. H. Lee, R. W. Schoenlein and M. Khalil, Struct. Dyn., 2017, 4, 044021 CrossRef PubMed.
- S. Eckert, J. Norell, R. M. Jay, M. Fondell, R. Mitzner, M. Odelius and A. Föhlisch, Chem. – Eur. J., 2019, 25, 1733–1739 CrossRef CAS PubMed.
- J. Norell, S. Eckert, B. E. Van Kuiken, A. Föhlisch and M. Odelius, J. Chem. Phys., 2019, 151, 114117 CrossRef CAS PubMed.
- J. Norell, M. Odelius and M. Vacher, Struct. Dyn., 2020, 7, 024101 CrossRef CAS PubMed.
- J. Norell, A. Ljungdahl and M. Odelius, J. Phys. Chem. B, 2019, 123, 5555–5567 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian∼16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- R. Cammi, B. Mennucci and J. Tomasi, J. Phys. Chem. A, 2000, 104, 5631–5637 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- J. C. Tully, J. Chem. Phys., 1990, 93, 1061–1071 CrossRef CAS.
- M. Richter, P. Marquetand, J. González-Vázquez, I. Sola and L. González, J. Chem. Theory Comput., 2011, 7, 1253–1258 CrossRef CAS PubMed.
- S. Mai, P. Marquetand and L. González, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1370 Search PubMed.
- S. Mai, P. Marquetand and L. González, Int. J. Quantum Chem., 2015, 115, 1215–1231 CrossRef CAS.
- G. Granucci, M. Persico and A. Toniolo, J. Chem. Phys., 2001, 114, 10608–10615 CrossRef CAS.
- F. Plasser, G. Granucci, J. Pittner, M. Barbatti, M. Persico and H. Lischka, J. Chem. Phys., 2012, 137, 22A514 CrossRef PubMed.
- F. Plasser, M. Ruckenbauer, S. Mai, M. Oppel, P. Marquetand and L. González, J. Chem. Theory Comput., 2016, 12, 1207–1219 CrossRef CAS PubMed.
- D. R. Yarkony, Nonadiabatic Derivative Couplings, John Wiley & Sons, Ltd, 2002 Search PubMed.
- B. H. Lengsfield III and D. R. Yarkony, J. Chem. Phys., 1986, 84, 348–353 CrossRef.
- B. De Souza, G. Farias, F. Neese and R. Izsak, J. Chem. Theory Comput., 2019, 15, 1896–1904 CrossRef CAS PubMed.
- G. Granucci and M. Persico, J. Chem. Phys., 2007, 126, 134114 CrossRef PubMed.
- G. Granucci, M. Persico and A. Zoccante, J. Chem. Phys., 2010, 133, 134111 CrossRef PubMed.
- B. O. Roos, P. R. Taylor and P. E. Siegbahn, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- P. Å. Malmqvist, A. Rendell and B. O. Roos, J. Chem. Phys., 1990, 94, 5477–5482 CrossRef CAS.
- B. O. Roos, et al. , Adv. Chem. Phys., 2007, 69, 399–445 CrossRef.
- K. Andersson, P.-Å. Malmqvist and B. O. Roos, J. Chem. Phys., 1992, 96, 1218–1226 CrossRef CAS.
- J. Finley, P.-Å. Malmqvist, B. O. Roos and L. Serrano-Andrés, Chem. Phys. Lett., 1998, 288, 299–306 CrossRef CAS.
- G. Li Manni, I. F. Galván, A. Alavi, F. Aleotti, F. Aquilante, J. Autschbach, D. Avagliano, A. Baiardi, J. J. Bao and S. Battaglia, et al. , J. Chem. Theory Comput., 2023, 19, 6933–6991 CrossRef CAS PubMed.
- V. Sauri, L. Serrano-Andrés, A. R. M. Shahi, L. Gagliardi, S. Vancoillie and K. Pierloot, J. Chem. Theory Comput., 2011, 7, 153–168 CrossRef CAS PubMed.
- M. Guo, L. K. Sørensen, M. G. Delcey, R. V. Pinjari and M. Lundberg, Phys. Chem. Chem. Phys., 2016, 18, 3250–3259 RSC.
- M. G. Delcey, L. K. Sørensen, M. Vacher, R. C. Couto and M. Lundberg, J. Comput. Chem., 2019, 40, 1789–1799 CrossRef CAS PubMed.
- P. Å. Malmqvist and B. O. Roos, Chem. Phys. Lett., 1989, 155, 189–194 CrossRef CAS.
- P. Å. Malmqvist, B. O. Roos and B. Schimmelpfennig, Chem. Phys. Lett., 2002, 357, 230–240 CrossRef CAS.
- S. Eckert, P. Miedema, W. Quevedo, B. O’Cinneide, M. Fondell, M. Beye, A. Pietzsch, M. Ross, M. Khalil and A. Föhlisch, Chem. Phys. Lett., 2016, 647, 103–106 CrossRef CAS.
- S. Eckert, J. Norell, P. S. Miedema, M. Beye, M. Fondell, W. Quevedo, B. Kennedy, M. Hantschmann, A. Pietzsch and B. E. Van Kuiken, et al. , Angew. Chem., Int. Ed., 2017, 56, 6088–6092 CrossRef CAS PubMed.
- M. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- K. Kunnus, I. Josefsson, I. Rajkovic, S. Schreck, W. Quevedo, M. Beye, S. Grübel, M. Scholz, D. Nordlund and W. Zhang, et al. , New J. Phys., 2016, 18, 103011 CrossRef.
- S. K. Das, D. Garratt, K. Gaffney and M. Odelius, Frontier Orbital Changes Induce Slow-down of Excited State Population Dynamics in Thiopyridone Isomers, https://chemrxiv.org/engage/chemrxiv/article-details/67c180a6fa469535b94b0725 Search PubMed.
- S. K. Das, D. Garratt, K. J. Nelson, J. Harich, A. Freibert, C. Bacellar, C. Cirelli, P. J. M. Johnson, M. Zoric, R.-P. Wang, H. Lim, A. A. Cordones, N. Huse, K. Gaffney and M. Odelius, Probing Excimer Dynamics in Aromatic Thiones with Sulfur K-edge Absorption Spectroscopy (Unpublished).
Footnotes |
| † Electronic supplementary information (ESI) available: Supplemental experimental datasets for 2-TP, 4-TP in water. Full experimental data-sets to 100 ps time delay. Power titration scans performed for each sample. Anisotropy of the XAS signal in 2-TP and a discussion of rotational effects. Detailed description of the data analysis and fitting procedures. Computational details, optimized molecular structures, Wigner sampling, UV-vis spectra, excited state dynamics dynamics trajectories, polarization dependence of calculated XA spectra, comparison of theoretical solvation models. See DOI: https://doi.org/10.1039/d5cp01456e |
| ‡ These authors contributed equally. |
| This journal is © the Owner Societies 2025 |