Angle-dependent electrocatalytic activity of twisted bilayer graphene for the hydrogen evolution reaction†
Received
15th April 2025
, Accepted 7th July 2025
First published on 8th July 2025
Abstract
Two-dimensional (2D) materials are attractive for their unique electronic structures and electrocatalytic properties. In this work, we propose to use the twist angle as a knob to tune the electrocatalytic properties of 2D materials. As proof of concept, we investigate the effects of twist angle (>10°) on the electrocatalytic properties of twisted bilayer graphene (tBLG). We predict the activity of tBLG with the twist angle of 13.174° and 21.787° for the hydrogen evolution reaction (HER) using a density functional theory (DFT) calculation and computational hydrogen electrode (CHE) approach. We calculate the hydrogen adsorption energy (ΔGH*) at various sites on tBLG and examine their angle-dependency. By comparing the ΔGH* for different active sites of untwisted bilayer graphene (BLG) and tBLG, we find that the ΔGH* decreases with the increase of the twist angle. As a result, the thermodynamic limiting potential for the HER increases with the twist angle. Furthermore, the ΔGH* shows a correlation with the layer distance (![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) ) and the site location on the 2D plane. Detailed analysis reveals that the twist of bilayer graphene could increase the z height (dz) of the active sites as a function of their distance to the symmetry centers, alter the local geometry of the active sites, and therefore modify the ΔGH*. These results indicate that the twist angle can be effectively used as a knob to fine-tune the electrocatalytic properties of 2D materials.
) and the site location on the 2D plane. Detailed analysis reveals that the twist of bilayer graphene could increase the z height (dz) of the active sites as a function of their distance to the symmetry centers, alter the local geometry of the active sites, and therefore modify the ΔGH*. These results indicate that the twist angle can be effectively used as a knob to fine-tune the electrocatalytic properties of 2D materials.
1. Introduction
Two-dimensional (2D) materials are promising electrocatalysts with their advantages of ultra-thin layered structure, large specific surface area, high density of surface active sites, and fast charge transmission.1–9 Among the 2D materials, graphene,10–12 nitrogen-doped carbon,13–16 metal halogens,17,18 and layered double hydroxides19,20 have received great attention for their unique electrocatalytic properties. Various structural and compositional strategies, such as elemental doping, defect engineering, ligand modification, spin state tunning, and van der Waals (vdW) heterostructures, have been developed to improve their electrocatalytic activity.4,13,21–27 Recently, physicists revealed that twisted 2D materials possess exceptional traits like unconventional superconductivity,28,29 and orbital magnetism.28,30 This exciting finding leads us to speculate whether the twisted 2D materials may have unique chemical and thermodynamic properties that differ from their untwisted counterparts. The twist angles of layered 2D materials may alter the interaction between the reactants and the catalytic sites on 2D materials at different degrees depending on the twist angle of adjacent layers, eventually leading to angle-dependent electrocatalytic properties. Thus, the twist angles of layered 2D materials can potentially be used as a novel knob to tune their catalytic properties. This new strategy is worth exploring, as it may lead to surprising discoveries.
Twisted bilayer graphene (tBLG) exhibits remarkably diverse electronic properties that evolve non-monotonically with rotation angle. This angle-dependent behavior manifests in three distinct regimes: at small twist angles (θ < 3°), long-range moiré superlattices dominate, generating flat electronic bands and correlated quantum states.29 In the intermediate regime (3° ≤ θ ≤ 10°), expanding AA-stacking domains create heterogeneous electronic environments with localized states and stacking-dependent interlayer distances.31 At large twist angles (θ > 10°), unique symmetry features are retained.
Herein, we use the hydrogen evolution reaction (HER) as a model reaction to investigate the origin of the angle-dependent electrocatalytic activity of twisted bilayer graphene (tBLG) at twist angle >10°. The HER could be considered as the simplest electrochemical reaction and the foundation of multiple electron-proton transfer reactions.32,33 The hydrogen adsorption free energy (ΔGH*) at all on-top sites of untwisted bilayer graphene (BLG) and tBLG with twist angles of θ = 13.174° and 21.787° is meticulously calculated and examined for their angle-dependency. The results demonstrate that the ΔGH* decreases with the increase of the twist angle, and the UL for the HER increases with the twist angle. And the ΔGH* varies as a function of the site position parameters on the 2D plane, including the z-distance to the bottom layer and distance to the high-symmetry centers. Consequently, the twist of bilayer graphene alters the local geometry of the active sites, thereby modifying the ΔGH*. This work provides a helpful footstone for further broadening the application of twisted 2D materials for catalysis.
2. Models and methods
2.1. Models of twisted bilayer graphene
There are three types of high-symmetry stacking structures for bilayer graphene, namely AB-, AA-, and saddle-point (SP-) stacking configurations, as shown in Fig. 1a.34 Each configuration has two high-symmetry twist centers, 1 and 2. We construct the models of tBLG by twisting one constituent lattice in the AB stacking configuration around center 1 (Fig. 1b). The unit cell of a high-symmetry tBLG has two basis vectors A1 = ma1 + na2 and A2 = −na1 + (m + n)a2, where a1 and a2 are the basis vectors of the untwisted BLG lattice, and m and n are coprime indices.35 Table S1 (ESI†) summarizes the coprime indices, θ, and the number of atoms in the primitive cells of high-symmetry tBLG. Considering the computational cost of the density functional theory (DFT) calculation and the complexity/specificity of models and electronic structures, we first focus on the tBLG at twist angle >10°, specifically θ = 13.174° and θ = 21.787°, namely tBLG (13.174°) and tBLG (21.787°).
 |
| | Fig. 1 (a) Three types of high-symmetry bilayer graphene (BLG) in AB-, AA-, and saddle-point (SP-) stacking configurations. (b) Twist angle (θ) is produced by twisting one constituent lattice of BLG in the AB-stacking configuration. | |
For comparison, we redefine two unit cells of the BLG lattice (θ = 0°) to include the same number of C atoms as their tBLG counterparts, namely BLG-28(0°) and BLG-76(0°). Using the unit cells of tBLG and BLG of the same C atom numbers ensures the modeling of H adsorption at the same H-coverage. A vacuum space of 20 Å along the z direction was added to avoid interactions between adjacent slabs.
2.2. Methods
Structure optimization and energy calculation were performed by the Vienna ab initio simulation package (VASP 5.4.4).36,37 The core-electron and exchange–correlation interactions were described by the Perdew–Burke–Ernzerhof (GGA-PBE) functional38 with the projector-augmented-wave (PAW)39,40 pseudopotential. To consider the van der Waals interaction, Grimme's DFT-D3 was utilized.41 A cut-off energy of 400 eV was set for the plane wave basis. The Monkhorst–Pack k-point mesh 3 × 3 × 1 was employed for all BLG and tBLG models. The structure optimization was completed until the forces and energies were less than 0.02 eV Å−1 and 10−5 eV per atom, respectively.
The calculation of the H adsorption free energy was performed by using the computational hydrogen electrode (CHE) approach.42 In this framework, the free energy of the electron–proton pair (H+ + e−) can be referenced to the chemical potential of gaseous H2 at equilibrium (0 V vs. standard hydrogen electrode). The H adsorption energy can be computed using the following eqn (1) and (2):
| | | ΔEH* = EH* − E* − 1/2EH2(g) | (1) |
| | | ΔGH* = ΔEH* + ΔEZPE + ΔU(T) − TΔS | (2) |
where
EH* is the electronic energy after H adsorption;
E* is the electronic energy of the clean surface;
EH2(g) denotes the electronic energy of gaseous H
2; Δ
EH* is the electronic energy difference after and before adsorption of H; Δ
EZPE is the change of zero-point energy; Δ
U(
T) is the change of internal energy;
T is temperature (
T = 298.15 K), and Δ
S is the entropy change (
T = 298.15 K).
The thermodynamic limiting potential is calculated using eqn (3):
3. Results and discussion
Fig. 2 shows optimized structures for untwisted bilayer graphene (BLG-76 and BLG-28) and twisted bilayer graphene (tBLG) with θ of 13.174° and 21.787°. After twisting the upper layer by θ = 13.174° and 21.787°, the average distance in the z-direction between the top and bottom layer atoms () increases from 3.492 Å (0°) to 3.564 Å (13.174°) by 2.06% and 3.523 Å (21.787°) by 0.89%, indicating that the twist destabilizes the bilayer structure slightly. The surface structure of the BLG and tBLG differs significantly and the on-top adsorption sites can be categorized into various types based on their local geometry and symmetry. Two types of on-top H-adsorption sites are identified on the BLG surface (Fig. 2a and c) while more types of unique sites form at the tBLG surfaces due to the twist of the upper layer (Fig. 2b and d). Both tBLG(13.174°) and tBLG(21.787°) have a space group P3 symmetry, including two types of symmetry centers C1 and C2 (Fig. S1, ESI†). To clarify the adsorption site classification and spatial distribution around the C1/C2 centers, high-symmetry adsorption sites are defined relative to C1/C2. C1-centered sites: directly atop the moiré hollow (AA-stacked regions); C2-centered sites: near stacking-domain boundaries (AB/BA transitions). And the H-adsorption sites are arranged in three-phases and repeated every 120° with the three-fold rotational axis shown as triangles. After twist of the upper surface, the number of types of sites increases from two to seven (θ = 13.174°) and three (θ = 21.787°), respectively. The z-distance (dz) of the active site relative to the bottom layer is calculated by:| | dz = zi-top − ![[z with combining macron]](https://www.rsc.org/images/entities/i_char_007a_0304.gif) bottom bottom | (4) |
where zi-top is the z-height of the active site in the top layer, and bottom is the average z-height of the bottom layer atoms. The dz for each type of site varies on the 2D plane as a function of their relative position to the C1 and C2 symmetry centers, as shown in Fig. 2b and d. The sites are colored in different saturation levels to show the change of dz, indicating a change in the geometric structure of the sites. The dz increases slightly as the active site locates further away from the C1 to C2 center. For tBLG with θ = 13.174° and 21.787°, the dz increases from 3.555 to 3.574 Å and from 3.521 to 3.525 Å, respectively. These active sites with varied local geometry could lead to diverse performances for the hydrogen evolution reaction.
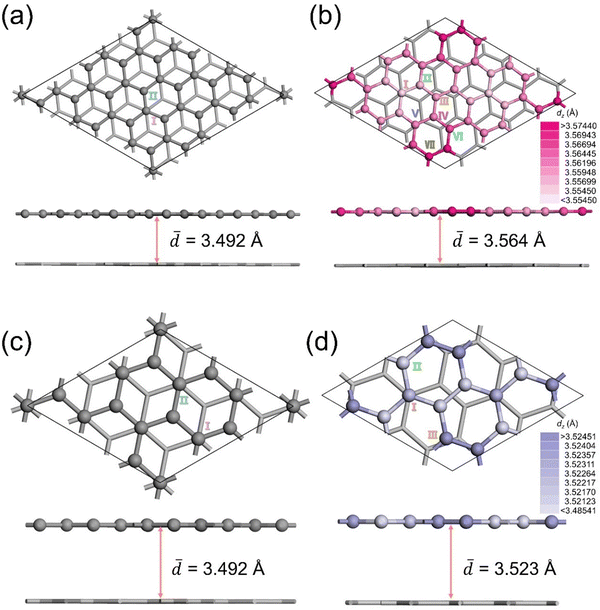 |
| | Fig. 2 Optimized structures and the active sites of (a) BLG-76(0°), (b) tBLG(13.174°), (c) BLG-28(0°), and (d) tBLG(21.787°) and the mapping of dz. Color bar stands for dz values. stands for the difference between average z-distance of the top and bottom layer atoms. | |
The adsorption energy of hydrogen atoms at the active site (ΔGH*) has been proven to be a suitable activity descriptor for a broad range of HER catalysts, including both metal-based and metal-free materials.42,43 We modeled the structures for H adsorbed at the two sites of BLG and the ten sites of tBLG. Fig. 3a–d show the color rendered mapping of ΔGH* on tBLG and BLG (detailed structures and values are shown in Fig. S2 and S3, ESI†). The color from dark to bright represents the ΔGH* from low to high, corresponding to H-binding from strong to weak. For tBLG(21.787°) and tBLG(13.174°), site II adjacent to the C1 center exhibits the highest ΔGH*. The ΔGH of the sites except that on the C1 center decreases as the site is further away from the C1 center and closer to the C2 center. (The difference in ΔGH* for various sites is significant and larger than the precision from the DFT calculation with a STD of 0.001 eV in Table S2 (ESI†).) Fig. 4a and b show the linear correlation between the bilayer distance of each site and ΔGH*. As dz increases, the corresponding sites ΔGH* decrease. Fig. 4c and d show the location-dependency of ΔGH* and dz of the sites in the 2D plane of tBLG, in which the atoms closer to the C1 center have higher ΔGH* and shorter dz. Moreover, compared with BLG, the in tBLG increases with twist angle (Fig. 2 and 3), corresponding to the decreased ΔGH* (average value). Therefore, increased z-distance (e.g., at AA-stacked regions) reduces the interlayer orbital overlap, weakens sp2–sp2 hybridization, and enhances pz-orbital availability for H adsorption. Furthermore, Fig. S4 (ESI†) compares the ΔGH* for the single monolayer graphene (MLG), BLG, and tBLG. The ΔGH* increases monotonically from MLG to BLG but decreases in tBLG. These results indicate that interlayer coupling in BLG weakens the binding strength, while the introduction of a twist angle further modulates the coupling, ultimately enhancing the binding strength.
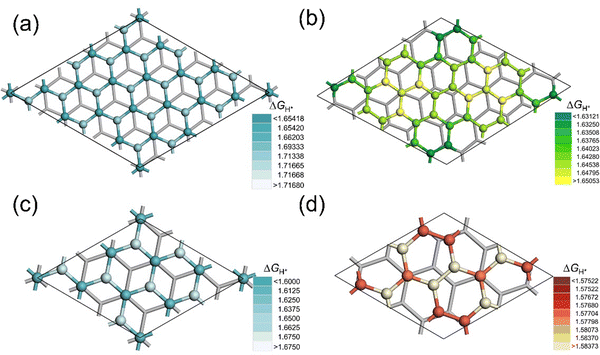 |
| | Fig. 3 The energy-site relationship mapping for (a) BLG-76 (0°), (b) tBLG(13.174°), (c) BLG-28(0°), and (d) tBLG(21.787°). Color bar stands for H adsorption free energy value. | |
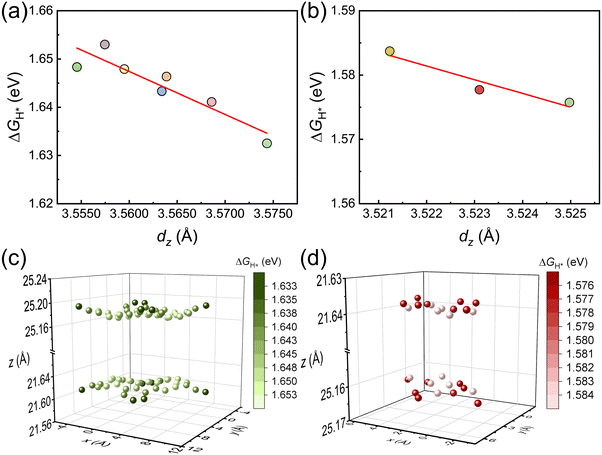 |
| | Fig. 4 Correlation between the dz and ΔGH* for (a) tBLG(13.174°) and (b) tBLG (21.787°). Points with different colors represent various active sites. 3D scatter plots of the relationship between the atom position and ΔGH* for (c) tBLG(13.174°) and (d) tBLG(21.787°). The color gradient represents ΔGH*. | |
Fig. 5a shows the relationship between twist angle and ΔGH*. The average of ΔGH* was presented by the saturated point. From θ = 0° to 13.174° and θ = 0° to 21.787°, ΔGH* has obviously decreased. The ΔΔGH* is used to further quantify the change of ΔGH* with twist angle:
| | | ΔΔGH* = ΔGH*(θ) − ΔGH*(0°) | (5) |
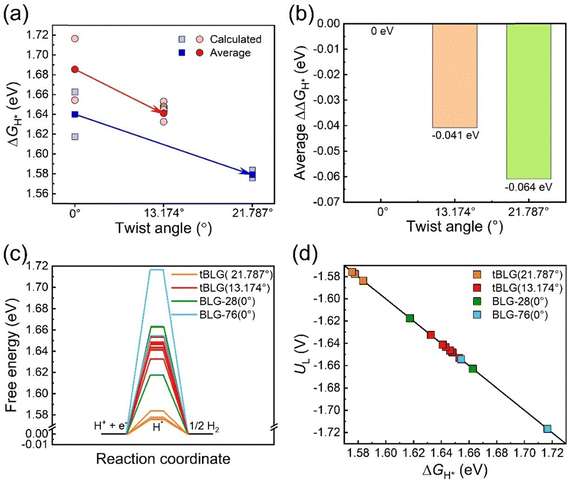 |
| | Fig. 5 (a) Dependence of ΔGH* on the twist angle. Saturated points represent the average values of ΔGH*; unsaturated points show individual calculated ΔGH* values. Red data points correspond to the ΔGH* values for tBLG(13.174°); blue data points represent ΔGH* values for tBLG(21.787°). (b) Average ΔΔGH* as a function of the twist angle. (c) Free energy diagram of the HER at various sites of the tBLG and BLG surface. (d) Thermodynamic limiting potentials (UL) of the HER as a function of ΔGH*. | |
The average ΔΔGH* decreases from −0.041 eV to −0.061 eV when θ increases from 13.174° to 21.787° (Fig. 5b), indicating that the larger twisted angle leads to a larger change of ΔGH*. Fig. 5c shows the reaction free energy diagram for the HER at different sites. Based on the Sabatier principle, the binding of atomic H to an effective HER catalyst should neither be too strong nor too weak. Consequently, smaller |ΔGH*| values represent a lower thermodynamic barrier for the HER. For an ideal HER catalyst, the ΔGH* should be 0 eV. The positive values of ΔGH* represents the under-binding of the H* intermediate and are the origin of the thermodynamic overpotential. The lower ΔGH* values on tBLG than on BLG indicate that twist angle promotes the HER activity. The thermodynamic limiting potential (UL) is calculated using eqn (3). Correspondingly, the UL for the HER also increases with the twist angle (Fig. 5d). We also considered the electronic structure effect on the HER (Fig. S5 and S6, ESI†), but found that the charge density difference almost has no difference between BLG and tBLG surfaces. This might be caused by the weak interaction in bilayer graphene. Notably, no significant correlation emerged between the Bader charge of the active sites and ΔGH* as demonstrated in Fig. S7a and b (ESI†). Additional insights were gained from projected density of states (PDOS) calculations comparing the BLG and tBLG configurations (Fig. S7c and d, ESI†). The twist induced a distinct change in electronic structure, manifested by a reduction in the number of peaks at the Fermi level from three to one following twisting. But the corresponding p band center of site-PDOS lacks clear correlation with adsorption energy trends across different configurations (Fig. S7e and f, ESI†). Therefore, correlation analyses reveal that geometric parameters outperform local electronic descriptors (Bader charge or PDOS) in predicting site-adsorption energies. The twist angle generates various active site structures and increases the bilayer distance, further enhancing the HER activity.
4. Conclusion
We scrutinized the angle-dependent electrocatalytic activity of the twisted bilayer graphene (tBLG) with twist angles greater than 10° for the HER using DFT calculations and the CHE approach. The results show that the twisting of BLG leads to an increase in the bilayer distance, which follows a decreasing trend as the site is located further away from the C1 to C2 symmetry center. Meanwhile, the intensity of hydrogen adsorption also varies as a function of site position in the 2D plane, and the ΔGH* decreases as the dz of the active site increases. Both dz and ΔGH* correlate within the 2D plane of tBLG, which is attributed to the local geometry in the tBLG structures generated by the twist. This effect is further enhanced with the increase of the the twist angle. As a result, twisting the BLG results in the 2D position-dependency of various active sites with unique local structures and fine-tuned HER activities. This study suggests that twisting can be a novel strategy to design 2D materials with precisely controlled catalytic activities.
Author contributions
Lifang Chen: investigation, methodology, writing – original draft, visualization. Jin Li: investigation. Xi Yin: conceptualization, investigation, writing – review & editing, supervision, funding acquisition.
Conflicts of interest
The authors declare no competing financial interest.
Data availability
All the data are available in the main text and the ESI.†
Acknowledgements
Financial support from the State Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences is greatly appreciated. This study was financially supported by Shanxi Province Youth Natural Science Foundation Grant No. 202103021223458, and Shanxi Province Grant No. 2023SHB003. The authors would like to thank Dr Xiaodong Wen and Dr Xingchen Liu at ICC CAS for their support for calculations during the project.
References
- S. Yu, C. Zhang and H. Yang, Chem. Rev., 2023, 123, 3443–3492 CrossRef CAS PubMed.
- K. Wan, T. Chu, B. Li, P. Ming and C. Zhang, Adv. Sci., 2023, 10, e2203391 CrossRef PubMed.
- X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 16064 CrossRef CAS.
- Q. Zhu, Y. Qu, D. Liu, K. W. Ng and H. Pan, ACS Appl. Nano Mater., 2020, 3, 6270–6296 CrossRef CAS.
- A. VahidMohammadi, J. Rosen and Y. Gogotsi, Science, 2021, 372, eabf1581 CrossRef CAS PubMed.
- Y. Wang, H. Su, Y. He, L. Li, S. Zhu, H. Shen, P. Xie, X. Fu, G. Zhou, C. Feng, D. Zhao, F. Xiao, X. Zhu, Y. Zeng, M. Shao, S. Chen, G. Wu, J. Zeng and C. Wang, Chem. Rev., 2020, 120, 12217–12314 CrossRef CAS PubMed.
- X. Yin, M. Shi, K. S. Kwok, H. Zhao, D. L. Gray, J. A. Bertke and H. Yang, Nano Res., 2018, 11, 3442–3452 CrossRef CAS.
- Y. T. Pan, X. Yin, K. S. Kwok and H. Yang, Nano Lett., 2014, 14, 5953–5959 CrossRef CAS PubMed.
- X. Yin, X. Liu, Y.-T. Pan, K. A. Walsh and H. Yang, Nano Lett., 2014, 14, 7188–7194 CrossRef CAS PubMed.
- Q. Wan, H. Guo and S. Lin, ACS Catal., 2022, 12, 14601–14608 CrossRef CAS.
- Y. Qu, Y. Ke, Y. Shao, W. Chen, C. T. Kwok, X. Shi and H. Pan, J. Phys. Chem. C, 2018, 122, 25331–25338 CrossRef CAS.
- R. Cepitis, N. Kongi, J. Rossmeisl and V. Ivanistsev, ACS Energy Lett., 2023, 8, 1330–1335 CrossRef CAS PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS PubMed.
- H. Yang, N. Lu, J. Zhang, R. Wang, S. Tian, M. Wang, Z. Wang, K. Tao, F. Ma and S. Peng, Carbon Energy, 2023, 5, e337 CrossRef CAS.
- P. Cui, L. Zhao, Y. Long, L. Dai and C. Hu, Angew. Chem., Int. Ed., 2023, 135, e202218269 CrossRef.
- X. Guo, S. Zheng, Y. Luo and H. Pang, Chem. Eng. J., 2020, 401, 126005 CrossRef CAS.
- M. Liu, J. A. Wang, W. Klysubun, G. G. Wang, S. Sattayaporn, F. Li, Y. W. Cai, F. Zhang, J. Yu and Y. Yang, Nat. Commun., 2021, 12, 5260 CrossRef CAS PubMed.
- K. Wang, K. Yu, S. Xu, S. Yuan, L. Xiang, B. Pang, J. Zheng and N. Li, Appl. Catal., B, 2023, 328, 122445 CrossRef CAS.
- H. Lei, Q. Wan, S. Tan, Z. Wang and W. Mai, Adv. Mater., 2023, 35, e2208209 CrossRef PubMed.
- H. Yi, S. Liu, C. Lai, G. Zeng, M. Li, X. Liu, B. Li, X. Huo, L. Qin, L. Li, M. Zhang, Y. Fu, Z. An and L. Chen, Adv. Energy Mater., 2021, 11, 2002863 CrossRef CAS.
- S. Yao, X. Zhang, A. Chen, Z. Zhang, M. Jiao and Z. Zhou, J. Mater. Chem. A, 2019, 7, 19290–19296 RSC.
- R. Ma, J. Wang, Y. Tang and J. Wang, J. Phys. Chem. Lett., 2022, 13, 168–174 CrossRef CAS PubMed.
- K. Yuan, D. Lutzenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. H. Leung, Y. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS PubMed.
- X. Wei, X. Luo, H. Wang, W. Gu, W. Cai, Y. Lin and C. Zhu, Appl. Catal., B, 2020, 263, 118347 CrossRef CAS.
- R. Lu, C. Quan, C. Zhang, Q. He, X. Liao, Z. Wang and Y. Zhao, Nano Res., 2022, 15, 6067–6075 CrossRef CAS.
- Z. Zhang, P. Liu, Y. Song, Y. Hou, B. Xu, T. Liao, H. Zhang, J. Guo and Z. Sun, Adv. Sci., 2022, 9, e2204297 CrossRef PubMed.
- U. I. Kramm, J. Herranz, N. Larouche, T. M. Arruda, M. Lefevre, F. Jaouen, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach, S. Mukerjee and J. P. Dodelet, Phys. Chem. Chem. Phys., 2012, 14, 11673–11688 RSC.
- X. Lu, P. Stepanov, W. Yang, M. Xie, M. A. Aamir, I. Das, C. Urgell, K. Watanabe, T. Taniguchi, G. Zhang, A. Bachtold, A. H. MacDonald and D. K. Efetov, Nature, 2019, 574, 653–657 CrossRef CAS PubMed.
- Y. Cao, V. Fatemi, S. Fang, K. Watanabe, T. Taniguchi, E. Kaxiras and P. Jarillo-Herrero, Nature, 2018, 556, 43–50 CrossRef CAS PubMed.
- A. L. Sharpe, E. J. Fox, A. W. Barnard, J. Finney, K. Watanabe, T. Taniguchi, M. A. Kastner and D. Goldhaber-Gordon, Science, 2019, 365, 605–608 CrossRef CAS PubMed.
- Y. Choi, H. Kim, C. Lewandowski, Y. Peng, A. Thomson, R. Polski, Y. Zhang, K. Watanabe, T. Taniguchi, J. Alicea and S. Nadj-Perge, Nat. Phys., 2021, 17, 1375–1381 Search PubMed.
- B. H. Solis and S. Hammes-Schiffer, Inorg. Chem., 2014, 53, 6427–6443 CrossRef CAS PubMed.
- F. Sun, Q. Tang and D.-E. Jiang, ACS Catal., 2022, 12, 8404–8433 CrossRef CAS.
- Y. Yu, K. Zhang, H. Parks, M. Babar, S. Carr, I. M. Craig, M. Van Winkle, A. Lyssenko, T. Taniguchi, K. Watanabe, V. Viswanathan and D. K. Bediako, Nat. Chem., 2022, 14, 267–273 CrossRef CAS PubMed.
- K. Uchida, S. Furuya, J.-I. Iwata and A. Oshiyama, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 155451 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Jin
Li
a,
Jin
Li




