MXenes enhance electrocatalytic water electrolysis of NiFe layered double hydroxides through bifunctional heterostructuring†
Received
28th March 2025
, Accepted 23rd May 2025
First published on 30th May 2025
Abstract
Transition metal-based layered double hydroxides (TM-LDHs) are among the most promising catalytic materials for the electrochemical reactions involved in energy conversion and storage technology. We systematically investigate NiFe-LDH-based electrocatalysts toward application in water electrolysis. We start with the highly accurate advanced density functional theory description of NiFe-LDH's fundamental properties, and demonstrate that coupling a spin-polarized p-band or d-band center model with the Gibbs free energy calculations explains NiFe-LDH's oxygen evolution reaction (OER) mechanism. By involving the related transient states, a reversible oxygen vacancy assisted reaction mechanism has been directly observed and motivated by the high spin transition metal impurity which is further confirmed by the time-consuming hybrid functional method. To further facilitate the electrocatalytic activity of NiFe-LDH, we study NiFe-LDH/MXene heterostructures where the essential semiconductor-to-metallic transition takes place by the additional Ti-3d orbitals and the interfacial non-covalent interaction between the two catalysts. On the basis of calculated results, we propose a link between microscopic properties and macroscopic electrocatalytic kinetics of heterogenous electrocatalysts. Accurately describing the electronic and magnetic structures of electrocatalysts leads us to a step-by-step process for tailoring desired electrocatalytic properties, especially for the high spin state contained TM-LDHs. A descriptor based on combination of the calculated d-band center of transition metal and p-band center of oxygen is the key to predicting electrochemical activity and stability of oxide electrocatalysts. From our results, we establish a design strategy for NiFe-LDH-based bifunctional electrocatalyst fabrication.
Introduction
Hydrogen energy is an important aspect for renewable energy; it involves using hydrogen or hydrogen-containing compounds to produce energy. The energy industry requires new technologies that are not only highly efficient, but also offer environmental and social benefits. Energy efficiency and environmental sustainability requirements are why scientists seek new alternatives to fossil fuels. Hydrogen is an ideal candidate due to its zero emission of carbonaceous species during utilization. Hydrogen can be harvested by water splitting; its only by-product, oxygen, is also a valuable industrial gas. In the last twenty years, water splitting via photocatalytic approaches has attracted a lot of attention since it directly uses solar energy.1,2 More recently, water electrolysis has been an adaptable technique that efficiently produces hydrogen using electrochemical energy devices.3,4 Generally, water electrolysis includes two electrodes, one cathode for the hydrogen evolution reaction (HER) and one anode for the oxygen evolution reaction (OER). The overall electrochemical reaction comprises two complementary half-reactions, the HER and OER. To substantially increase water electrolysis efficiency, highly efficient, low cost and environment-friendly electrocatalysts for the HER and OER are required.
Over the past decade, electrocatalysts have been studied extensively in energy conversion and storage technologies.5–7 Among them, layered double hydroxides (LDHs) are new, highly active OER electrocatalysts in an alkaline environment that can be used directly in water electrolysis.8,9 A crystal structure the same as the mineral hydrotalcite (Mg6Al2CO3(OH)16·4(H2O)) is often denoted as a layered double hydroxide (LDH), with nickel iron layered double hydroxides (NiFe-LDHs) being investigated widely.10–14 The geometric structure of NiFe-LDH can be conveniently understood by incorporating iron (Fe) atoms in nickel hydroxides. Among the four phases of nickel hydroxides, the β-Ni(OH)2 phase is the most studied. In this phase, the atomic layers are held together by weak van der Waals forces. Within the layers of the hexagonal lattice, nickel (Ni) atoms are in their most common +2 oxidation state. Replacing Ni2+ with Fe3+ ions results in positively charged layers which can be further compensated by intercalated anions like carbonate (CO3−2). Water molecules can also be intercalated in their interlayer spacing, forming hydrogen bonds (H-bonds).11 Owing to NiFe-LDHs’ complex and variable geometric structures combined with the high cost of computing large magnetic systems, only their simplified single layer structure has been used in previous reports. Even though, diversities appear in electrochemical reaction mechanisms because calculated energetic parameters are highly dependent on accurate reaction environment predictions or say accurate electronic and magnetic structures of reaction active centers. It is necessary to carefully analyze NiFe-LDH's geometric and electronic properties and multiple reaction pathways using a precise theoretical approach. Besides, 3d transition metal can be modified to be highly active for specific catalytic reactions via varying their coordination environment where high-throughput computational screening can be applied. But the complexity of the electrochemical reactions triggered the divergences of the activities depending on different chemical environments and theoretical study methods which indicate that more reliable clear-out reference data are needed.
Lately, multifunctional electrocatalysts such as trifunctional electrocatalysts for the HER/OER/ORR and bifunctional electrocatalysts for overall water splitting are topical.15–17 Most recently, the work by Kim et al. caught our attention, as they proposed that doping specific MXene surfaces with single atoms can be an effective strategy to modulate their bifunctional or multifunctional catalytic behavior, attributed to the intrinsic stability and metallic nature of MXenes.18,19 On the other hand, multifunctional heterostructured electrocatalysts can achieve higher efficiencies than separate single functional catalysts owing to working condition divergences.20 Although current experimental strategies for producing multifunctional catalysts have improved quickly, it still remains challenging due to complicated fabrication procedures that involve multiple atomic doping or hybridizing steps. The resulting complex atomic structures of catalysts may make it difficult to understand the actual catalytic mechanism of the whole system.16 Thus, developing new multifunctional electrocatalysts via a simple synthesis approach is still needed.
This work proposes a common strategy for developing LDH-based bifunctional electrocatalysts by synthesizing LDH with suitable metallic MXenes. Our systematic investigation starts by precisely describing NiFe-LDH's geometric, electronic, magnetic, and electrochemical properties. These calculations lead us to the real OER active phase of NiFe-LDH and the corresponding reaction mechanism. The synergistic effect between Fe–O–Ni and Ni–O–Ni moieties allows NiFe-LDH to become an efficient and durable electrocatalyst material. We proposed a detailed reaction mechanism involving oxygen vacancy formation and related transient states through both the advanced DFT method (PBE + U) and the Hartree–Fock/DFT hybrid functional method (B3LYP). The oxygen-rich layered structure of NiFe-LDH offers sufficient geometric features for the lattice oxygen vacancy assisted OER mechanism. Tailoring a NiFe-LDH system to have better electrocatalytic properties, meanwhile fabricating a bifunctional NiFe-LDH-based heterocatalyst, for this purpose we studied several functional MXenes. We analyzed the correlation between their HER activities and electronic structures and found that the oxygen p-band center describes reaction activity and MXene surface specificity. Eventually, NiFe-LDH/MXene heterostructures were established. The homogenous and fully applicable reaction active sites on both sides allow the NiFe-LDH/MXene heterostructure to be utilized as a benchmark bifunctional electrocatalyst for overall water splitting. Moreover, the heterostructure's interfacial architecture allows easy access for electron transfer, which may further enhance electrocatalytic activities on both sides. The screening procedure introduced in this work offers a new principle for tailoring electrocatalytic materials to their activity.
Calculation methods
To study the geometric, electronic, magnetic, and electrochemical properties of our structures, we carried out first-principles calculations based on the density functional theory (DFT) approach as implemented in the Vienna ab initio simulation package (VASP).21 For all calculations, we used the projector augmented wave (PAW) pseudopotentials within 600 eV energy cutoff for the plane wave basis set.22,23 The exchange–correlation energy was modelled using the Perdew–Burke–Ernzerhof (PBE) type generalized gradient approximation (GGA) functional.24 To accurately describe Ni and Fe localized d-electrons, the PBE + U method with effective U parameter (Ueff) values of 6.50 eV for Ni and 4.50 eV for Fe were used. Ueff = U − J, U and J are the effective on-site Coulomb and exchange corrections.25 These Ueff values were carefully tested as described below. To further confirm the effectiveness of the PBE + U method, the expensive unscreened hybrid functional B3LYP method is also used and compared with the PBE + U method.26 Ni and Fe atoms’ initial magnetic moments were set at 2.00μB and 5.00μB, respectively. The atomic ratio between Ni and Fe is 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The van der Waals density functional method (vdW-DF) within the optB86b functional was included to describe dispersion interactions in the interlayer spacing.27,28 For structure optimization, the energy and force criterions were set at 10−6 eV and 0.01 eV Å−1, respectively. A 15 Å vacuum layer was used for the surface structure. The charge transfer calculation was performed using the Bader charge analysis.29 Spin-polarization was included in all calculations.
:1. The van der Waals density functional method (vdW-DF) within the optB86b functional was included to describe dispersion interactions in the interlayer spacing.27,28 For structure optimization, the energy and force criterions were set at 10−6 eV and 0.01 eV Å−1, respectively. A 15 Å vacuum layer was used for the surface structure. The charge transfer calculation was performed using the Bader charge analysis.29 Spin-polarization was included in all calculations.
Here, we determined the HER procedure using an approach developed by Nørskov et al.:30
where * denotes an active site for H
+ adsorption, and H* is the intermediate state on the catalytic surface. The intermediate state's Gibbs free energy was calculated as:
| | | ΔGH* = ΔEH* + ΔZPE − TΔS | (3) |
where Δ
EH* is the hydrogen adsorption energy, ΔZPE is the zero-point energy difference between the intermediate state and the gas phase hydrogen, and Δ
S is the vibrational entropy difference between these two hydrogen states at temperature
T. The hydrogen adsorption energy was evaluated as:
| |  | (4) |
where
EH@sur is the total energy of the intermediate adsorbed surface,
Esur is the total energy of the pristine surface, and
EH2 is the total energy of the gas phase hydrogen molecule.
In alkaline solution, the OER procedure can be described via these elementary steps:6,31
| | | OH* + OH− → O* + H2O + e− | (6) |
| | | OOH* + OH− → O2 + * + H2O + e− | (8) |
where * indicates the reaction active site for intermediate OH*, O*, and OOH*. The Gibbs free energy of each step was calculated as:
| | | ΔG = ΔE + ΔZPE − TΔS − neUR + ΔG(pH) | (9) |
where Δ
E is the adsorption energy of each intermediate, and ΔZPE and Δ
S are the differences in zero-point energy and entropy at temperature
T caused by the reaction. −
neUR is the bias correction where
UR is the electrode potential relative to RHE. Δ
G(pH) = −
kTln[H
+] is the free energy correction induced by the hydrogen ion concentration. The chemisorption energy of intermediate OH*, O*, and OOH* is:
| |  | (10) |
| | | EO* = EO@sur − Esur − EH2O + EH2 | (11) |
| |  | (12) |
where
EOH@sur,
EO@sur, and
EOOH@sur are the total energy of intermediate state surfaces,
Esur is the total energy of a pristine surface, and
EH2O and
EH2 are the total energy of water and hydrogen molecules in the gas phase. All energy terms were calculated using DFT calculations, while gas phase entropy data were taken from experimental standard tables.
6
In a conventional d-band center model, a metal d-band center is computed as the centroid of the projected density of states (pDOS) of the metal-d orbitals.32–34 Hence, in our work, for spin-polarized systems, the oxygen p-band center or metal d-band center was calculated as:35
| |  | (13) |
where
E′ =
E −
EF is the energy corrected relative to the Fermi level, and
Dσ(
E′) is the
pDOS of the O-2p or metal-3d orbitals for different spin components (σ = ↑,↓). All data were extracted from DFT calculations.
In this work, the structural thermodynamic stability of the NiFe-LDH/MXene heterostructures was evaluated based on binding energy, which was calculated as:
| | | Eb = Ehetero − ELDH − EMXenes | (14) |
where
Ehetero,
ELDH and
EMXenes are the total energy of NiFe-LDH/MXene heterostructures, isolated NiFe-LDH and MXene slabs, respectively.
Results and discussion
Geometric, electronic, and electrocatalytic properties of NiFe-LDH
Hubbard Ueff correction testing.
To determine the Hubbard Ueff correction for NiFe-LDH PBE + U calculation, we first performed geometric and electronic structure testing of the corresponding host structures. We used brucite-like β-Ni(OH)2 and lepidocrocite γ-FeOOH crystal structures for seeking Ni and Fe's Ueff values, respectively. Both structures are hydroxide compounds consisting of octahedral sheets with Ni2+ or Fe3+ ions occupying the octahedral interstices. The sheets are stacked by weak non-covalent interactions. We used antiferromagnetic configurations for both β-Ni(OH)2 and γ-FeOOH, which are the most energetically favorable structures according to our calculations and previous experimental reports.36–42 The optimized β-Ni(OH)2 and γ-FeOOH atomic structures with their antiferromagnetic ordering are shown in Fig. S1a and S2a (ESI†). The Ueff testing results are shown in these figures as well. We analyzed the calculated energy band gap (Eg) and magnetic moment (μM) of metal (Ni/Fe) atoms to evaluate Ueff values (Fig. S1b and S2b, ESI†). By comparing these results with experimental data, we chose the Ueff value at 6.50 eV for Ni and 4.50 eV for Fe for further calculations. Ueff value testing is performed rather than using a linear response calculation because our study involves a high spin state Fe3+ with five unpaired d-electrons; a linear response approach may endure errors in the self-consistent response step.43,44
NiFe-LDH property analysis.
The optimized geometric structure of NiFe-LDH is shown in Fig. 1a. Calculated lattice constants of NiFe-LDH are a = 12.33 Å, b = 6.17 Å, and c = 22.60 Å. This results in an interlayer spacing of 7.53 Å. Within the interlayer spacing, the intercalated species CO2 and H2O interact with the hydroxide terminated surfaces of Ni0.75Fe0.25(OH)2 layers through H-bonds (marked in Fig. 1a). Calculated density of states (DOS) and projected density of states (pDOS) of NiFe-LDH using the PBE+U method are presented in Fig. 1b and c, which indicate a semiconductor characteristic with a band gap of ∼2.00 eV. Compared to the host structure β-Ni(OH)2 (Fig. S1c, ESI†), the Fe orbitals of NiFe-LDH hybridize with the O orbitals and act as impurity states in the energy band gap between the Ni–O hybridized states in the valence band and conduction band (from −0.12 eV to 4.00 eV). The appearance of these impurity states reduces the band gap above the Fermi level, thus enhancing NiFe-LDH's electrical conductivity. The detailed pDOS (Fig. S3, ESI†) show that these impurity states are mainly attributed to Fe–d orbitals within spin-down electrons, implying that the NiFe-LDH Fe atoms are in a +3 oxidation state with a high spin d5 electron configuration. The electron configuration schemes for Ni2+ and Fe3+ are shown in Fig. 1d. In the trigonal antiprismatic (D3d) ligand field (the octahedron compressed along C3), Ni2+ has a d8 electron configuration within fully occupied e′ (dx2−y2, dxy) and a1 (dz2) orbitals and singly occupied e′′ (dyz, dxz) orbitals. In the weak ligand field, Fe3+ prefers a high spin d5 electron configuration in which e′, a1 and e′′ orbitals are all half occupied, which causes the impurity state to rise. This is also confirmed by the calculated magnetic moment of Ni and Fe atoms, 1.78μB and 4.23μB, respectively. This result demonstrates that our initial magnetic moment setting effectively acts as a seed for establishing the correct magnetic ground state structure of NiFe-LDH, thereby enhancing the overall efficiency of the calculation. To further verify the PBE+U calculation results of NiFe-LDH, the B3LYP hybrid functional calculations are performed. As shown in Fig. S4 (ESI†), the B3LYP calculations exhibit similar atomic orbital behavior to PBE+U results with slightly more localized states. So, our calculation approach accurately describes NiFe-LDH's electronic properties and metal site oxidation states.
 |
| | Fig. 1 (a) Geometric structure of the NiFe-LDH unit cell, left side is the top view and right side is the side view. The crystal cell is represented by a gray box and H-bonds are marked with gray dashed lines. The lattice constants are also shown in the figure. (b) Density of states (DOS) and (c) projected density of states (pDOS) of NiFe-LDH using the PBE+U method. The Fermi level is shifted to zero. (d) Scheme of ligand field splitting in trigonal antiprismatic (D3d) geometries of Ni2+ and Fe3+ electron configurations. | |
These just-above-the-Fermi-level impurity states suggest that the doped Fe atoms boost NiFe-LDH's electrochemical activity. Therefore, we further investigated NiFe-LDH's catalytic properties with regard to the OER procedure. As shown in Fig. 2, we studied the potential OER pathways on the oxygen terminated NiFe-LDH (001) surface, also called the reaction active phase. This is primarily due to previous experimental studies demonstrating that the intrinsic catalytic activity of LDH does not stem from the bulk structure itself, but rather from distinct active surface phases that emerge during the OER process.45–48 Because this reaction active surface is oxygen terminated, the calculated OER process starts from the O* intermediate state to form OOH* (eqn (7)) as shown in Fig. 2a and b, rather than the formation of the OH* intermediate state in the conventional process (eqn (5)). Two possible reaction active centers are denoted as Fe–O–Ni (path1) and Ni–O–Ni (path2). Both are three metal coordinated oxygen, one with two Ni–O bonds and one Fe–O bond, while the other with only Ni–O bonds. In the NiFe-LDH lattice, each Fe atom is surrounded by six Ni atoms, thus the ratio of Fe–O–Ni to Ni–O–Ni is 3:1. Schematic illustrations of both OER pathways are shown in Fig. 2 with their corresponding free energy diagrams. As shown in Fig. 2c, the reaction rate determining step (RDS) for both paths is the deprotonation process with an energy barrier value of 2.20 eV (path1) and 2.49 eV (path2). This deprotonation process is further facilitated by the complex structured H-bond network of NiFe-LDH, as indicated by Fig. 1a that the H-bonds formed between the outmost hydrogen atoms on the Ni0.75Fe0.25(OH)2 layers and the oxygen atoms of the intercalated species boost OH group deprotonation via strong dipole–dipole attraction; while the H-bonds between CO32− ions and H2O molecules may assist in deprotonation and the proton transfer through the Grotthuss mechanism. Most interestingly, Fig. 2a and b also demonstrate that both reaction paths involve the formation of oxygen vacancies. However, the energy barrier of path1 (1.63 eV) is lower than that of path2 (1.85 eV). As mentioned above, the number of Fe–O–Ni moieties is three times Ni–O–Ni moieties. Therefore, we conclude that path1 (the Fe–O–Ni moiety) dominates NiFe-LDH's OER process. Herein, like the above electronic structure calculations, we used the B3LYP method to confirm the catalytic property calculations. The B3LYP results (Fig. S5a, ESI†) show a similar reaction trend to the PBE+U method with a slightly higher (0.11–0.16 eV) energy barrier of RDS, which again proved the accuracy of our calculation.
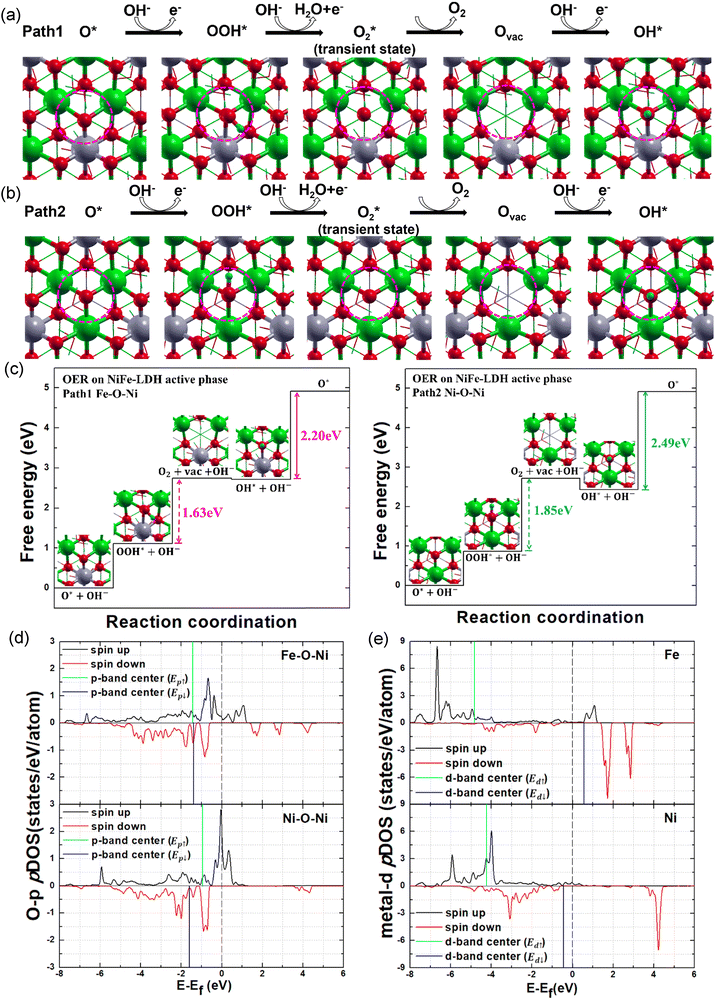 |
| | Fig. 2 Scheme of NiFe-LDH active phase OER pathways (a) path1: Fe–O–Ni moiety and (b) path2: Ni–O–Ni moiety as reaction active centers (marked with pink circles). Ni, Fe, O, C and H atoms in green, silver, red, yellow and cyan colors. The topmost layer is shown in ball-stick mode and other layers are shown in thin stick mode. (c) Gibbs free energy diagrams of NiFe-LDH OER pathways using the PBE+U method. The reaction energy barriers are shown with arrows. pDOS of NiFe-LDH (d) O-2p orbitals and (e) metal-3d orbitals using the PBE+U method. The p-band and d-band centers of spin-up (cyan line) and spin-down (blue line) states are shown in the figure. | |
To further examine the reaction mechanism, we calculated the O p-band center and metal d-band center of the NiFe-LDH active phase. Instead of applying the conventional d-band center model, we defined two band centers for spin-polarized NiFe-LDH, one for spin up and one for spin down electronic states. Fig. 2d shows that the Fe–O–Ni moiety's spin up p-band center (Ep↑) shifts away from the Fermi level compared to the Ni–O–Ni moiety, while the spin down component (Ep↓) moves slightly upward, resulting in weaker binding with the reaction intermediates. Thus, the RDS energy barrier on the Fe–O–Ni site is reduced. To clarify O vacancy formation via Ni–O–Ni and Fe–O–Ni moieties, we compared the calculated Ni and Fe d-band centers (Fig. 2e). Interaction between O and Fe d-band centers for down spins (Ed↓) is attractive because Fe Ed↓ is unoccupied while interaction with the Fe d-band center for up spins (Ed↑) is repulsive because Fe Ed↑ is occupied. These two competing effects loosen the O–Fe bond resulting in easier O vacancy formation at the Fe–O–Ni site. The split between Fe Ed↓ and Ed↑ is large, which also confirms that Fe is in its high spin state. These results are also supported by the B3LYP calculations shown in Fig. S5b (ESI†). Therefore, we conclude that the high spin Fe impurity boosts NiFe-LDH's OER activity by weakening the metal–O bonds to facilitate O vacancy formation, meanwhile reducing interactions between the reaction intermediates and the catalytic surface to accelerate the reaction's initial stage. Contrarily, the host Ni–O–Ni moiety with stronger metal–O bonds maintains NiFe-LDH lattice stability, which is also essential for ensuring the long-term electrochemical durability of a promising electrocatalyst.
NiFe-LDH/MXene bifunctional heterostructures
As mentioned above, on-site reaction activity and electron transfer ability are the two crucial factors for new electrocatalyst design. For NiFe-LDH-based catalysts, impure Fe atoms introduce several discontinuous energy states just above the Fermi level, resulting in a smaller energy band gap (∼0.8 eV) than the host structure. However, this semiconductor property still limits the catalyst's electrical conductivity. Therefore, to further improve the catalyst, we built a bifunctional heterogenous catalyst by combining functionalized MXenes onto NiFe-LDH which may be achieved via a simple electrochemical transfer approach.
MXenes’ electronic properties and electrocatalytic activity.
Five functionalized MXenes, Ti4C3O2, Ti3C2O2, Ti4N3O2, Mo3C2O2, and Ti2MoC2O2, were studied. They all demonstrate good metallic features as indicated by their electronic structures (Fig. 3a and Fig. S6, ESI†). All of their geometric structures were obtained using the crystal structures of their corresponding MAX phase. The in-plane lattice mismatch between them and NiFe-LDH is in the range from 3.56% (Ti4N3O2) to 0.23% (Ti4C3O2). We investigated the HER catalytic activity of MXenes as bifunctional heterocatalysts with NiFe-LDH. Fig. 3b indicates that Ti4C3O2 exhibits the best HER activity with ΔGH* = −0.01 eV, followed by Ti3C2O2 and Ti2MoC2O2. Ti4N3O2 and Mo3C2O2 binding with H* were too weak and too strong, respectively. To explain this, we calculated the O p-band center for each case and compared them in Fig. 3c. Among the five MXenes, TixCx−1O2 (x = 3,4) shows a moderate O p-band center relative to the Fermi level, which is consistent with its nearly zero free energy value. The closer the O p-band center to the Fermi level, the stronger the hydrogen adsorbed to the MXene. In addition to the p-band center of all O-2p electronic states (cyan line), the p-band center of only occupied states (green line) is also displayed in Fig. 3c. This can be used for comparison with X-ray emission spectroscopy (XES) data. Each MXene's detailed pDOS and their reaction intermediate adsorbed structures are included in the ESI† (Fig. S6 and S7, ESI†).
 |
| | Fig. 3 (a) Geometric structure (top view) and DOS of Ti4N3O2, Ti3C2O2, Ti4C3O2, Mo3C2O2 and Ti2MoC2O2. (b) Gibbs free energy diagrams of the five MXenes’ HER processes. (c) The pDOS of each MXene's O-2p orbitals. The p-band center of all electronic states (cyan line) and only occupied electronic states (green line) are shown in the figure. | |
NiFe-LDH/MXene bifunctional heterostructures.
Following our NiFe-LDH OER activity and MXene HER activity investigations, we built a bifunctional heterocatalyst by fabricating the MXene layers onto the NiFe-LDH. The lateral unit cell size of this heterostructure corresponds to the 1 × 2 supercell of NiFe-LDH (a = 12.33 Å, b = 6.17 Å), where the MXene supercell was adjusted. The smallest lattice mismatch is considered here, and we thus focus only on the NiFe-LDH/Ti4C3O2 heterostructure. Other MXenes could be used with other LDHs in the future. We tested different interlayer stackings (Fig. S8, ESI†) and obtained NiFe-LDH/Ti4C3O2 (“AA stacking”), the most energetically favorable structure. NiFe-LDH/Ti4C3O2's geometric and electronic structures are shown in Fig. 4. The binding energy of the NiFe-LDH/Ti4C3O2 heterostructure is −1.49 eV per cell which is sufficiently low to warrant the thermodynamic stability of the heterostructure. The averaged interfacial distance between NiFe-LDH and Ti4C3O2 layers is 1.87 Å, which indicates strong non-covalent interactions (H-bonds) in the interfacial spacing (Fig. 4a). Compared to the pristine NiFe-LDH (Fig. 1c), the NiFe-LDH/Ti4C3O2 heterostructure's pDOS (Fig. 4b) at the Fermi level is enhanced and continued, resulting in good metallic behavior. The detailed pDOS near the Fermi level of the heterostructure (Fig. 4c) demonstrates that the heterostructure's excellent electronic properties are caused not only by the rise of Ti-3d orbitals from the additional Ti4C3O2 layer, but also by the interfacial effect between them. The H-bonds formed in the NiFe-LDH/Ti4C3O2 interfacial spacing shift the Fermi level into the O-2p valence band, causing the Fermi level to be dominated by the O-2p states. The charge density calculation also confirms that the Ti4C3O2 layer gain of 0.73 e per cell is from NiFe-LDH via the interfacial structure (Fig. 4d). Fig. S9 (ESI†) illustrates that the interfacial effect between NiFe-LDH and Ti4C3O2 layers can be further improved by reducing the interlayer distance. As the interlayer spacing decreases by 10%, 20%, and 30% of the initial distance, the interfacial charge transfer increases to 0.78 e per cell, 0.86 e per cell, and 1.03 e per cell, respectively. The charge density difference and DOS are compared in Fig. S9 (ESI†). The charge density difference clearly shows the expansion of the charge transfer at the interface while the DOS exhibits Fermi level enhancement. This demonstrates how the heterostructure's electric conductivity may be improved. And the interlayer spacing engineering can be realized prior to electrocatalytic application through controlled synthesis strategies, such as hydrothermal synthesis, solvothermal treatments, or annealing under inert or reductive atmospheres, which simulate high-temperature/high-pressure environments at the material preparation stage. This pre-catalysis structural tuning strategy ensures the practical relevance of the design and aligns with standard experimental capabilities in water electrolysis research.
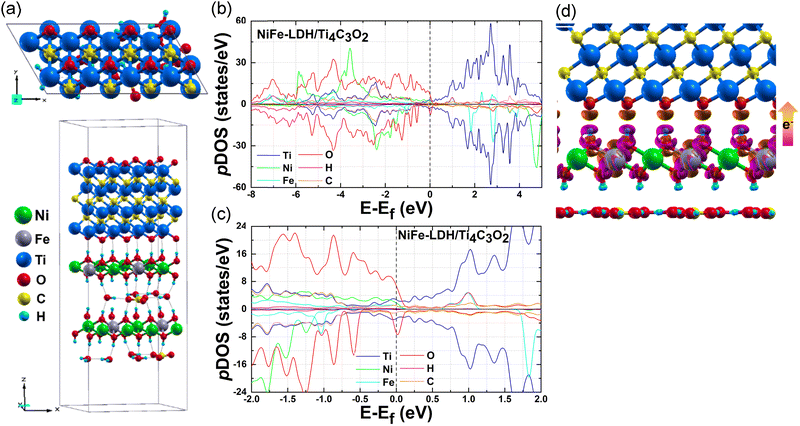 |
| | Fig. 4 (a) Geometric structures of the NiFe-LDH/Ti4C3O2 heterostructure, top view (up) and side view (down). The crystal cell is represented by a gray box and H-bonds are marked with gray dashed lines. NiFe-LDH/Ti4C3O2 heterostructure's (b) total pDOS and (c) zoomed up view of pDOS near the Fermi level. (d) Charge density difference distribution of the NiFe-LDH/Ti4C3O2 interface. The pink color indicates electron loss, while the orange color represents electron gain with an isosurface level of 0.02 e Å−3. | |
Conclusions
Based on a series of systematic DFT calculations, we demonstrated that NiFe-LDH/MXene heterostructures work as a new family of easily accessible, highly efficient, and durable bifunctional electrocatalysts for overall water splitting. We presented a fundamental explanation of the geometric, electronic, and magnetic properties of large NiFe-LDH systems from a molecular orbital perspective. Using NiFe-LDH calculated OER free energy diagrid, we identified synergistic effects between Fe–O–Ni and Ni–O–Ni moieties. Together, these two types of moieties, Fe–O–Ni for catalytic efficiency and Ni–O–Ni for structural integrity, are both crucial for the performance of NiFe-LDH based electrocatalysts. Furthermore, we believe that the synergy between the adjacent atomic-scale sites as well as between the large-scope heterostructures is significant for electrocatalysts. Therefore, to increase NiFe-LDH electrical conductivity and achieve a semiconductor-to-metallic transition, as well as compose an OER and HER benchmark bifunctional electrocatalyst, highly catalytic active and metallic functionalized MXenes should be applied to NiFe-LDH. Correlations between catalytic activity and their electronic structures were revealed for both NiFe-LDH/MXene heterostructure electrocatalytic surfaces. The calculated O p-band center is a good descriptor for screening oxide electrocatalyst electrochemical activity and stability. Combining this descriptor with machine learning methods could create a new way to systematically screen nanomaterials with specific catalytic properties. This work proposes a new design principle for constructing three-dimensional (3D) bifunctional heterocatalysts by composing single functional electrocatalytic material like LDHs for the OER and MXenes for the HER using a simple electrochemical transfer approach. The deliberate design of 3D heterostructured electrocatalysts serves not only to enhance electrical conductivity but also to improve interfacial charge transfer kinetics, tailor the local electronic environment, and optimize the overall electrocatalytic efficiency. As a uniquely structured 3D OER electrode synthesized via electrochemical integration, it enables accelerated OER kinetics and improved mass transport, thereby achieving an ultra-low overpotential.49,50 Tailoring heteromaterials’ electronic structures and electrocatalytic activity with excellent properties based on theoretical calculations offers a perspective on how to develop a general strategy for synthesizing new heterocatalysts.
Author contributions
Nannan Li: idea, conceptualization, investigation, data analysis, writing – original draft. Xiaotong Han: idea, conceptualization, writing – review & editing. Ho Seok Park: conceptualization, writing – review & editing. Jin Yong Lee: conceptualization, writing – review & editing, supervision, project administration.
Data availability
The software packages used for the calculations are provided in references. More details of the data obtained and analyzed in the current study will be made available from the corresponding author upon request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
N. Li was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. NRF-2022R1A2C1092044). J. Y. Lee acknowledges financial support from the National Research Foundation (NRF) grant funded by the Korean government (2022R1A4A1032832).
References
- T. Jafari, E. Moharreri, A. S. Amin, R. Miao, W. Song and S. L. Suib, Molecules, 2016, 21, 900, DOI:10.3390/molecules21070900.
- J. N. Tiwari, A. N. Singh, S. Sultan and K. S. Kim, Adv. Energy Mater., 2020, 10, 2000280, DOI:10.1002/aenm.202000280.
- S. Shiva Kumar and V. Himabindu, Mater. Sci. Energy Technol., 2019, 2, 442–454, DOI:10.1016/j.mset.2019.03.002.
- M. A. Khan, H. Zhao, W. Zou, Z. Chen, W. Cao, J. Fang, J. Xu, L. Zhang and J. Zhang, Electrochem. Energy Rev., 2018, 1, 483–530, DOI:10.1007/s41918-018-0014-z.
- J. N. Tiwari, S. Sultan, C. W. Myung, T. Yoon, N. Li, M. Ha, A. M. Harzandi, H. J. Park, D. Y. Kim, S. S. Chandrasekaran, W. G. Lee, V. Vij, H. Kang, T. J. Shin, H. S. Shin, G. Lee, Z. Lee and K. S. Kim, Nat. Energy, 2018, 3, 773–782, DOI:10.1038/s41560-018-0209-x.
- N. Li, Y. Nam and J. Y. Lee, Appl. Surf. Sci., 2020, 514, 146073, DOI:10.1016/j.apsusc.2020.146073.
- X. Han, N. Li, P. Xiong, M. G. Jung, Y. Kang, Q. Dou, Q. Liu, J. Y. Lee and H. S. Park, InfoMat, 2021, 1–11, DOI:10.1002/inf2.12226.
- S. Nayak and K. M. Parida, Sci. Rep., 2019, 9, 2458, DOI:10.1038/s41598-019-39009-4.
- D. Tonelli, I. Gualandi, E. Musella and E. Scavetta, Nanomaterials, 2021, 11, 725, DOI:10.3390/nano11030725.
- J. Mohammed-Ibrahim, J. Power Sources, 2020, 448, 227375, DOI:10.1016/j.jpowsour.2019.227375.
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 6, 1600621, DOI:10.1002/aenm.201600621.
- F. Ning, M. Shao, S. Xu, Y. Fu, R. Zhang, M. Wei, D. G. Evans and X. Duan, Energy Environ. Sci., 2016, 9, 2633–2643, 10.1039/C6EE01092J.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan, A. Bergmann, J. Drnec, J. F. d Araujo, M. Gliech, D. Teschner, J. Zhu, W.-X. Li, J. Greeley, B. R. Cuenya and P. Strasser, Nat. Commun., 2020, 11, 2522, DOI:10.1038/s41467-020-16237-1.
- Q. Xiang, F. Li, W. Chen, Y. Ma, Y. Wu, X. Gu, Y. Qin, P. Tao, C. Song, W. Shang, H. Zhu, T. Deng and J. Wu, ACS Energy Lett., 2018, 3, 2357–2365, DOI:10.1021/acsenergylett.8b01466.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452, DOI:10.1038/nnano.2015.48.
- Y. Jia, L. Zhang, A. Du, G. Gao, J. Chen, X. Yan, C. L. Brown and X. Yao, Adv. Mater., 2016, 28, 9532–9538, DOI:10.1002/adma.201602912.
- X. Han, N. Li, Y. B. Kang, Q. Dou, P. Xiong, Q. Liu, J. Y. Lee, L. Dai and H. S. Park, ACS Energy Lett., 2021, 6, 2460–2468, DOI:10.1021/acsenergylett.1c00936.
- R. Anand, A. S. Nissimagoudar, M. Umer, M. Ha, M. Zafari, S. Umer, G. Lee and K. S. Kim, Adv. Energy Mater., 2021, 11, 2102388, DOI:10.1002/aenm.202102388.
- R. Anand, B. Ram, M. Umer, M. Zafari, S. Umer, G. Lee and K. S. Kim, J. Mater. Chem. A, 2022, 10, 22500, 10.1039/D2TA06297F.
- H. Wang, H.-W. Lee, Y. Deng, Z. Lu, P.-C. Hsu, Y. Liu, D. Lin and Y. Cui, Nat. Commun., 2015, 6, 7261, DOI:10.1038/ncomms8261.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186, DOI:10.1103/PhysRevB.54.11169.
- P. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953, DOI:10.1103/PhysRevB.50.17953.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758, DOI:10.1103/PhysRevB.59.1758.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865, DOI:10.1103/PhysRevLett.77.3865.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509, DOI:10.1103/PhysRevB.57.1505.
- P. J. Stephens, F. J. Devlin, F. J. Devlin and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627, DOI:10.1021/j100096a001.
- J. Klimeš, D. R. Bowler and A. Michaelides, J. Phys.: Condens. Matter, 2009, 22, 022201, DOI:10.1088/0953-8984/22/2/022201.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131, DOI:10.1103/PhysRevB.83.195131.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360, DOI:10.1016/j.commatsci.2005.04.010.
- J. Nørskov, T. Bligaard, A. Logadottir, J. Kitchin, J. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23–J26, DOI:10.1149/1.1856988.
- J. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. Kitchin and T. Bligaard, J. Phys. Chem. B, 2004, 108, 17886–17892, DOI:10.1021/jp047349j.
- B. Hammer and J. K. Nørskov, Nature, 1995, 376, 238–240, DOI:10.1038/376238a0.
- J. K. Nørskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943, DOI:10.1073/pnas.1006652108.
- R. Jacobs, J. Hwang, Y. Shao-Horn and D. Morgan, Chem. Mater., 2019, 31, 785–797, DOI:10.1021/acs.chemmater.8b03840.
- S. Bhattacharjee, U. V. Waghmare and S.-C. Lee, Sci. Rep., 2016, 6, 35916, DOI:10.1038/srep35916.
- M. Fracchia, A. Visibile, E. Ahlberg, A. Vertova, A. Minguzzi, P. Ghigna and S. Rondinini, ACS Appl. Energy Mater., 2018, 1, 1716–1725, DOI:10.1021/acsaem.8b00209.
- Y. Sakamoto, Y. Noda, K. Ohno, K. Koike, K. Fujii, T. M. Suzuki, T. Morikawa and S. Nakamura, Phys. Chem. Chem. Phys., 2019, 21, 18486–18494, 10.1039/C9CP00157C.
- D. J. Pope, A. E. Clark, K. M. Rosso and M. P. Prange, J. Appl. Phys., 2020, 128, 103906, DOI:10.1063/5.0009300.
- H. Zhang, M. Bayne, S. Fernando, B. Legg, M. Zhu, R. L. Penn and J. F. Banfield, J. Phys. Chem. C, 2011, 115, 17704–17710, DOI:10.1021/jp205192a.
- P. Hermet, L. Gourrier, J.-L. Bantignies, D. Ravot, T. Michel, S. Deabate, P. Boulet and F. Henn, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 235211, DOI:10.1103/PhysRevB.84.235211.
- D. S. Hall, D. J. Lockwood, C. Bock and B. R. MacDougall, Proc. R. Soc. A, 2015, 471, 20140792, DOI:10.1098/rspa.2014.0792.
- H. Wang and C. Song, Eur. Phys. J. B, 2019, 92, 37, DOI:10.1140/epjb/e2019-90369-6.
- M. Cococcioni and S. de Gironcoli, Am. Phys. Soc., 2005, 71, 035105, DOI:10.1103/PhysRevB.71.035105.
- E. B. Linscott, D. J. Cole, M. C. Payne and D. D. O’Regan, Phys. Rev. B, 2018, 98, 235157, DOI:10.1103/PhysRevB.98.235157.
- S. A. Chala, M.-C. Tsai, B. W. Olbasa, K. Lakshmanan, W.-H. Huang, W.-N. Su, Y.-F. Liao, J.-F. Lee, H. Dai and B. J. Hwang, ACS Nano, 2021, 15, 14996–15006, DOI:10.1021/acsnano.1c05250.
- Z. Qiu, C.-W. Tai, G. A. Niklasson and T. Edvinsson, Energy Environ. Sci., 2019, 12, 572–581, 10.1039/C8EE03282C.
- D. Tyndall, M. J. Craig, L. Gannon, C. McGuinness, N. McEvoy, A. Roy, M. García-Melchor, M. P. Browne and V. Nicolosi, J. Mater. Chem. A, 2023, 11, 4067–4077, 10.1039/D2TA07261K.
- R. Chen, Y. Yang, W. Wu, S. Chen, Z. Wang, Y. Zhu and N. Cheng, Chem. Eng. J., 2024, 480, 148100, DOI:10.1016/j.cej.2023.148100.
- P. Thangavel, G. Kim and K. S. Kim, J. Mater. Chem. A, 2021, 9, 14043, 10.1039/D1TA02883A.
- P. Thangavel, M. Ha, S. Kumaraguru, A. Meena, A. N. Singh, A. M. Harzandia and K. S. Kim, Energy Environ. Sci., 2020, 13, 3447, 10.1039/D0EE00877J.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary Fig. S1–S9. See DOI: https://doi.org/10.1039/d5cp01202c |
| ‡ Nannan Li and Xiaotong Han contributed equally to this work. |
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Xiaotong
Han‡
b,
Ho Seok
Park
a,
Xiaotong
Han‡
b,
Ho Seok
Park







