Spin: an essential factor in advancing the oxygen evolution reaction on 2D Fe-MOF†
Received
26th March 2025
, Accepted 7th May 2025
First published on 9th May 2025
Abstract
The oxygen evolution reaction (OER) is a crucial component in oxygen-involving reactions and plays a vital role in developing sustainable energy conversion technologies. However, it still requires developing efficient catalysts that can overcome the sluggish reaction kinetics. Recent studies on oxygen electrocatalysis predominantly focussed on the thermodynamic viewpoint of oxygen adsorption, while the catalytic role of spin remains greatly elusive. In this work, we investigated the impact of spin on the OER performance of a two-dimensional iron-based metal–organic framework (2D Fe-MOF) using spin-polarized first-principles calculations. Our results reveal that the pristine Fe-MOF in the high spin state exhibits electronic properties suitable for an OER electrocatalyst. Even after adsorption, the Fe-MOF preserves its high spin state; such magnetic stability ensures the consistent application of the OER. Moreover, adsorption on a 2D Fe-MOF is spin-dependent. It validates that the spin states can regulate the adsorption strength for the OER. Remarkably, the spin-sensitive 2D Fe-MOF yields a significantly low overpotential of 0.49 V, comparable to precious catalysts. Furthermore, the spin-related charge transfer and orbital interaction originate from the overlapping between the O pz of the oxygen intermediates and the Fe dz2 of the Fe active site. This reveals that the OER on the Fe-MOF is dependent on the selective spin-orbital. Overall, the spin is inevitable in enhancing the OER process, making our work valuable in the development of MOF catalysts. Our finding enriches the atomistic understanding of the OER in the development of noble-metal-free MOF catalysts.
Introduction
Spin is a fundamental quantum characteristic of electrons, possessing two possible states: spin-up or spin-down. Organic molecules generally have spin-up and spin-down electrons equally distributed, resulting in a paired spin or singlet state. In contrast to this generalization, molecular oxygen (O2) in the ground state has two unpaired electrons or a triplet state (S = 1), which is ∼1 eV energetically more favorable than its singlet state (S = 0). Spin configurations of O2 suggest that the spin can play a crucial role in oxygen-involved catalytic reactions.1–6 In particular, the oxygen evolution reaction (OER) necessitates a spin transition, as singlet H2O and OH must transform into triplet O2 as a final product (Scheme S1 in the ESI†). Such a singlet-to-triplet process causes the spin-forbidden transition that contributes to the inherently sluggish reaction kinetics with high overpotential in the OER.2,3,7 Thus, spin-dependent oxygen reactions can proceed more efficiently in the presence of appropriate catalysts that overcome the barrier associated with spin transitions.1,2 Despite its fundamental importance, the spin impact has been largely overlooked in traditional catalyst designs. Addressing this challenge requires new strategies incorporating the spin as an additional degree of freedom into catalyst development, as supported by previous studies of organometallic materials, molecular complexes, and metal-based oxides.8–14 In this context, spin-polarized surfaces can regulate the spin states of reactants and intermediates, accelerating slow spin-transition kinetics and enhancing OER efficiency.1,4,15
Transition metal (TM) containing catalysts have been suited as spin-polarized catalysts to improve oxygen reactions because their d-states interact with the unpaired electron and the spin configuration from oxygen intermediates.3,4 In particular, the high spin of Fe is likely to promote the OER for pairing the spin-oriented electrons from triplet O2.16 In addition, Fe is an abundant and cost-effective metal in the earth's crust and exhibits significant characteristics in the OER due to its chemical environment.17–21 Significant efforts have been made in graphene-based single-atomic-catalysts (SACs) for oxygen reactions.22–26 Namely, FeN4-embedded SACs have been widely investigated as promising catalysts for the OER due to their suitable binding strength for oxygen intermediates.24,25 However, SACs in fabrication still face challenges such as low extrinsic metal loading and undesirable atom aggregation, which hinder not only the homogeneous dispersion of metal active sites but also the precise relationship between spin-state regulation and catalytic activity.27,28
While burgeoning the development of SACs, two-dimensional metal–organic frameworks (2D MOFs) have been investigated due to their planar structure, hierarchical porosity, high stability, good electrical conductivity, and catalytic activity.29–40 Importantly, 2D MOF materials have been facilitated by a self-assembled structure built from organic linkers and uniformly distributed intrinsic metal nodes, creating planar periodic networks with π-conjugated ligand environments and isolated single-metal sites.
Such intrinsic metal site in the 2D MOF allows the systematic modulation of high-, intermediate-, and low-spin configurations, which can serve as an ideal platform for regulating the spin state.41,42 Nevertheless, the progress in addressing the spin impact on 2D MOFs remains limited. In particular, the underlying relationships between the geometry, spin, and electronic and catalytic properties have not been fully understood.
In this work, we emphasize the significance of the spin impact by regulating the spin state of the Fe site in the 2D Fe-MOF to define its optimal spin-polarized configuration for the OER. Upon adsorption of oxygen intermediates, the Fe-MOF satisfies the favourable free energy criteria required for the OER, thereby facilitating the reaction. This study not only deepens the understanding of spin-dependent OER electrocatalysis on the Fe-MOF but also paves the way for future research by modulating a spin-based design strategy to develop an efficient MOF electrocatalyst and enhance OER performance.
Results and discussion
Structure and electronic properties of a pristine monolayer
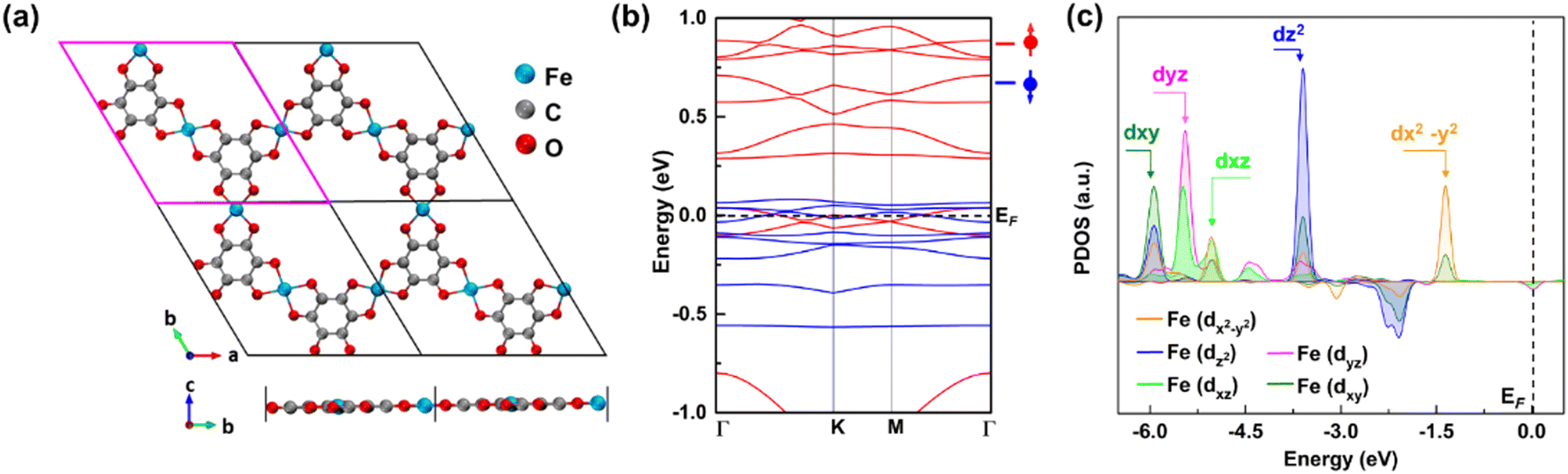
We selected the 2D Fe-MOF with Fe-metal nodes and benzene organic linkers since it is a compact representative of 2D MOFs as a case study. Fig. 1a shows the periodic structural view of a 2D Fe-MOF monolayer built from the Fe3(C6O6)2-unit cell, where a Fe atom is surrounded by four O ligands, creating planar coordination as a FeO4 complex that is connected with the neighbouring carbon rings. The optimized lattice parameter is a = b = 13.377 Å, consistent with the previous result.43 The nearest Fe–Fe distance in the monolayer is 6.69 Å, thus too far to be directly coupled. We compared the electronic configurations for the 2D Fe-MOF at various spin states (Fig. S1, ESI†); the high spin-polarized electronic configuration is preferable in the present system. Thus, Fig. 1b shows the spin-polarized band structure near the Fermi level (EF) from −1 to 1 eV, where a dashed line indicates the position of EF at 0 eV. Foremost, the Fe-MOF shows a metallic character with uneven spin channels. In particular, the energy states near the EF exhibit a notable absence of a band gap, resulting in its behaviour as a metallic conductor. This inherent conductivity plays a crucial role in facilitating rapid electron transfer during chemical reactions, particularly aiding in the efficient generation from singlet H2O/OH− to triplet O2. Fig. 1c presents the projected density of states (PDOS) of the Fe site to further discern the electronic feature of the Fe-MOF. It shows that the Fe-d orbital splits into energetically different states of dx2–y2, dz2, dyz, dxz, and dxy, due to the crystal field (Fig. S2, ESI†). The sub-energy levels further facilitate the capture of the O molecule during the reaction. In addition, we estimated that the Fe-MOF bears the high spin state of Fe with +3 valence (Table S1, ESI†). Thus, the Fe-MOF exhibits the high spin-polarized electronic characteristics that make it well-suited for use as an electrocatalyst.
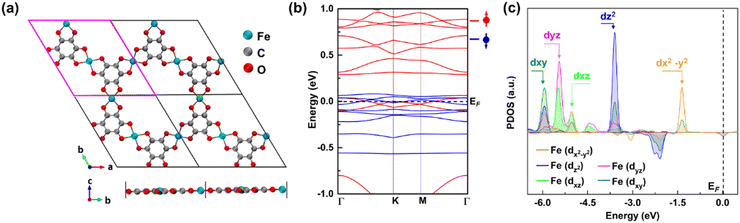 |
| | Fig. 1 Structural top and side views of the (a) periodic supercell. (b) High-spin-polarized band structure of the Fe-MOF monolayer, where red (blue) lines represent the spin-up (down) bands. (c) Orbital-projected density of states (PDOS) of the Fe atom in a high spin state. | |
Adsorption on the Fe-MOF monolayer
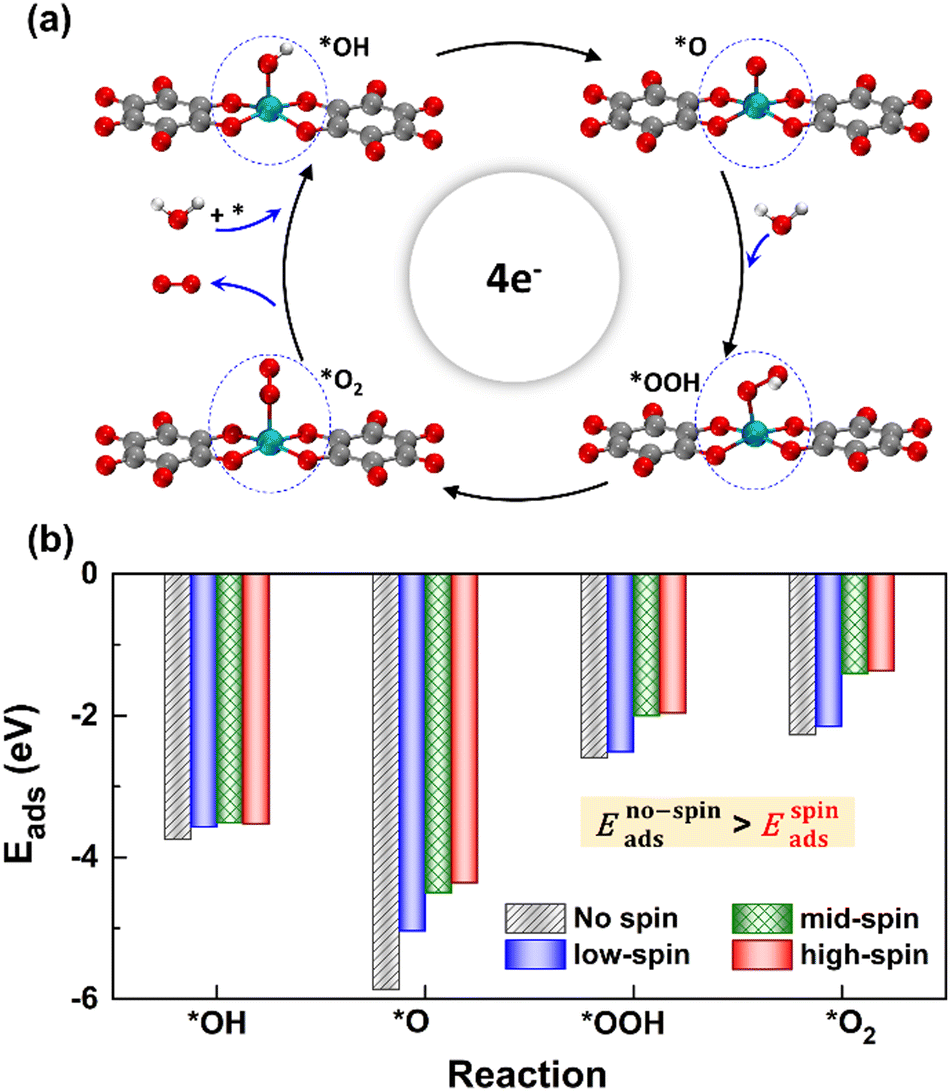
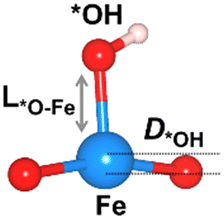
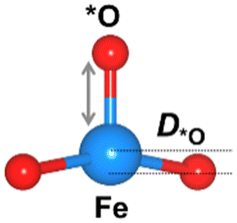
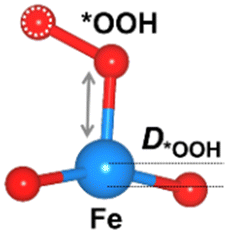
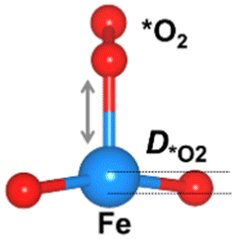
We considered four crucial reaction steps for the OER to define the catalytic feature of the Fe-MOF (Fig. 2a). The oxygen intermediates prefer to adsorb on the central Fe site of FeO4 (dashed blue circle). The resultant configurations are marked as *OH, *O, *OOH, and *O2, respectively. To initiate the OER process on the Fe-MOF, we calculated the adsorption energy (Eads) of oxygen intermediates at various spin states. Eads for each case shows a negative value (Fig. 2b and Table S2, ESI†), which is also consistent with a tendency of integrated crystal orbital Hamiltonian population (Fig. S3, ESI†). It implies that the O species can be spontaneously adsorbed on the Fe site. Moreover, the adsorption on the Fe-MOF is spin-dependent; Eads excluding spin provides a large value, while Eads including spin endows a relatively low value. It indicates that the spin states can be used to regulate the adsorption strength and further facilitate the oxygen reaction process.
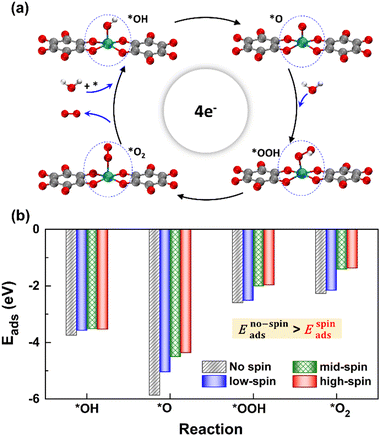 |
| | Fig. 2 (a) Structural perspective view of oxygen intermediates of the OER on the 2D Fe-MOF. (b) Eads values for the 2D Fe-MOF at various spin states; the black bar represents Eads in the absence of spin, while the blue, green, and red bars indicate Eads that belongs to the low-, mid-, and high-spin states, respectively. | |
The Eads value also differs at each oxygen intermediate and follows the trend of *O > *OH > *OOH > *O2, causing the features of the various oxygen species. Importantly, our result satisfies the condition for an effective catalyst (*OH > *OOH) to facilitate continuous product release.5 Herein, while the first oxygen intermediate (*OH) adsorbs at the Fe active site, the oxygen product after *OOH is released, attainable in a next OER cycle. Subsequently, the Eads value of *O2 is significantly lower than others. Such weak adsorption of *O2 is the key to enhancing the OER. Hence, the present Fe-MOF at the high spin demonstrates promising catalytic activation and holds potential as an effective OER catalyst.
Structural distortion
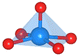
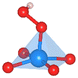
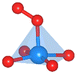
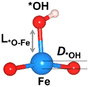
The oxygen intermediate disturbs the original structure of the Fe-MOF when the oxygen reaction occurs, creating a pyramidal geometry (insets in Table 1). To be precise, we compare the structural distortion that occurs in the environment of FeO4, in both the absence and presence of oxygen intermediates (details are provided in Tables S3 and S4, ESI†). Herein, the oxygen intermediate evidently binds to the Fe site, forming the *O–Fe bond with its bond length L*O–Fe. A longer bond yields weaker adsorption, as validated by the less negative value of overall ICOHP and vice versa (Fig. S4a, ESI†).
Table 1 Bond lengths (L*O–Fe) for a *O–Fe pair and displacement (DFe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) site) of the Fe site along the z-axis, where * represents the FeO4 environment
site) of the Fe site along the z-axis, where * represents the FeO4 environment
|
|
|
* |
*OH |
*O |
*OOH |
*O2 |
|
|
Perspective view |

|
|
|
|
|
| Distortion |
Side view |

|
|
|
|
|
| *L (Å) |
— |
1.811 |
1.675 |
1.890 |
2.094 |
|
D
Fesite (Å) |
— |
0.307 |
0.297 |
0.257 |
0.261 |
Since O2 is a decisive step, but also the bottleneck for the OER process, the O2 desorption should be feasible to overcome and proceed with the reaction. Our result shows that the bond index of *O2–Fe in the high spin state is significantly lower than the non-spin case, suggesting that the O2 desorption can be more favourable in this state (Fig. S4b, ESI†). Thus, the high-spin-polarized configuration is considered for further calculations. We illustrate another notable change created in FeO4 after the adsorption, where the Fe site moves upward relative to the basal plane along the z-direction. Such a local displacement of the Fe site (DFesite) disturbs the formal orbitals of Fe. Thus, the structural distortions during the entire OER process can lead to substantial changes in the electronic and magnetic properties of the MOF, which in turn affects the catalytic performance of the MOF.6
Electronic and magnetic properties of the Fe-MOF after the adsorption
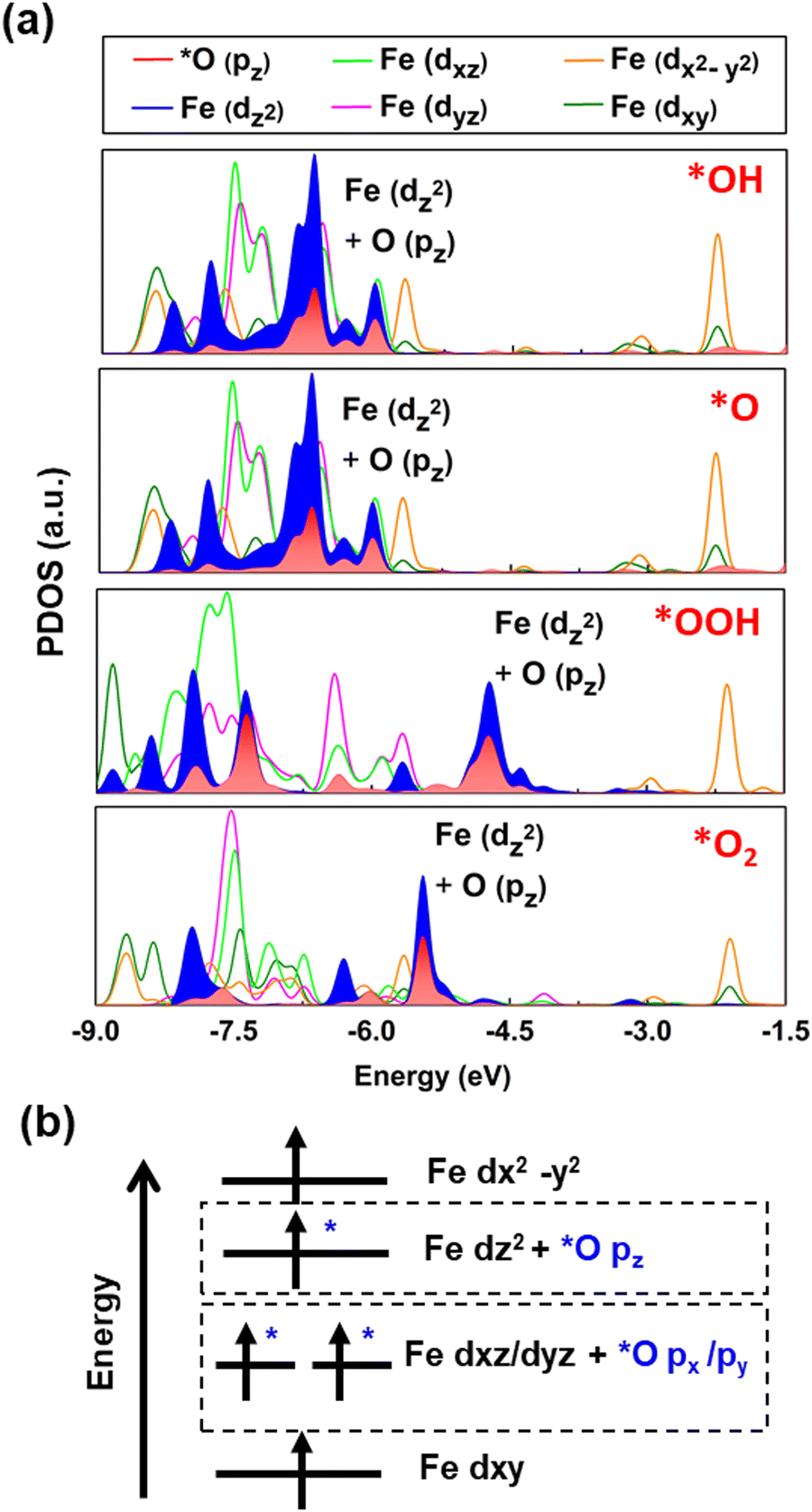
We calculated the projected density of states (PDOS) for the oxygen intermediates and the Fe site to gain deeper insights into the changes in the electronic structure following adsorption. Fig. 3a demonstrates that *O pz of the absorbed intermediates hybridizes Fe dz2. Meanwhile, *O px and *O py merely overlap Fe dxz or dyz, respectively (see Fig. S5 and S6, ESI†). However, there is no obvious overlapping with Fe dx2–y2 and Fe dxy. Hence, it is indeed a selective orbital-dependent interaction.44 Notably, the PDOS after adsorption of oxygen intermediates shows a generic trend; the orbital diagram of the Fe-MOF is summarized in a single scheme (Fig. 3b). Herein, dz2, dyz and dxz orbitals hybridize with the oxygen intermediates; however, the dx2–y2 and dxy orbitals interact less with the intermediate. This situation infers that the electrons predominantly transfer between certain orbitals of the Fe atom and the oxygen intermediate. As seen in the PDOS, the hybridization of Fe dz2 and *O pz of the absorbed oxygen intermediates steadily persists near EF. This result implies that the dz2-orbital predominantly participates in the reaction process, conditioning a charge transfer from the monolayer to the adsorbed oxygen intermediates along the z-direction. In short, spin-resolved DOS analysis reveals that the Fe-MOF with the discrete Fe-d states exhibit a strong spatial orbital overlap and orbital-dependent interaction with the adsorbed intermediates.
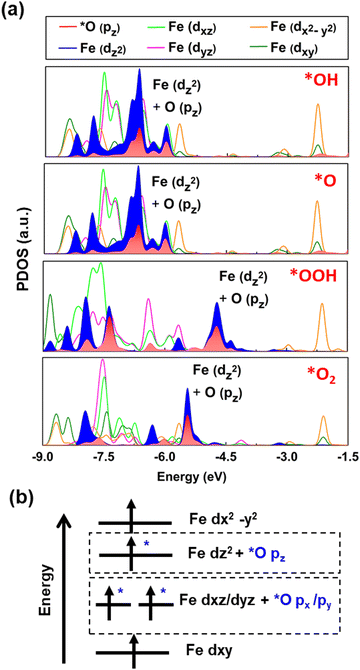 |
| | Fig. 3 (a) The spin-resolved PDOS of the Fe site and O intermediates after the adsorption, highlighting the hybridized orbitals of Fe dz2 and *O pz along the spin-up channel, for a clarity. (b) The schematic orbital diagram of the Fe-MOF at the high spin after the adsorption of the oxygen species, based on our solid evidence. | |
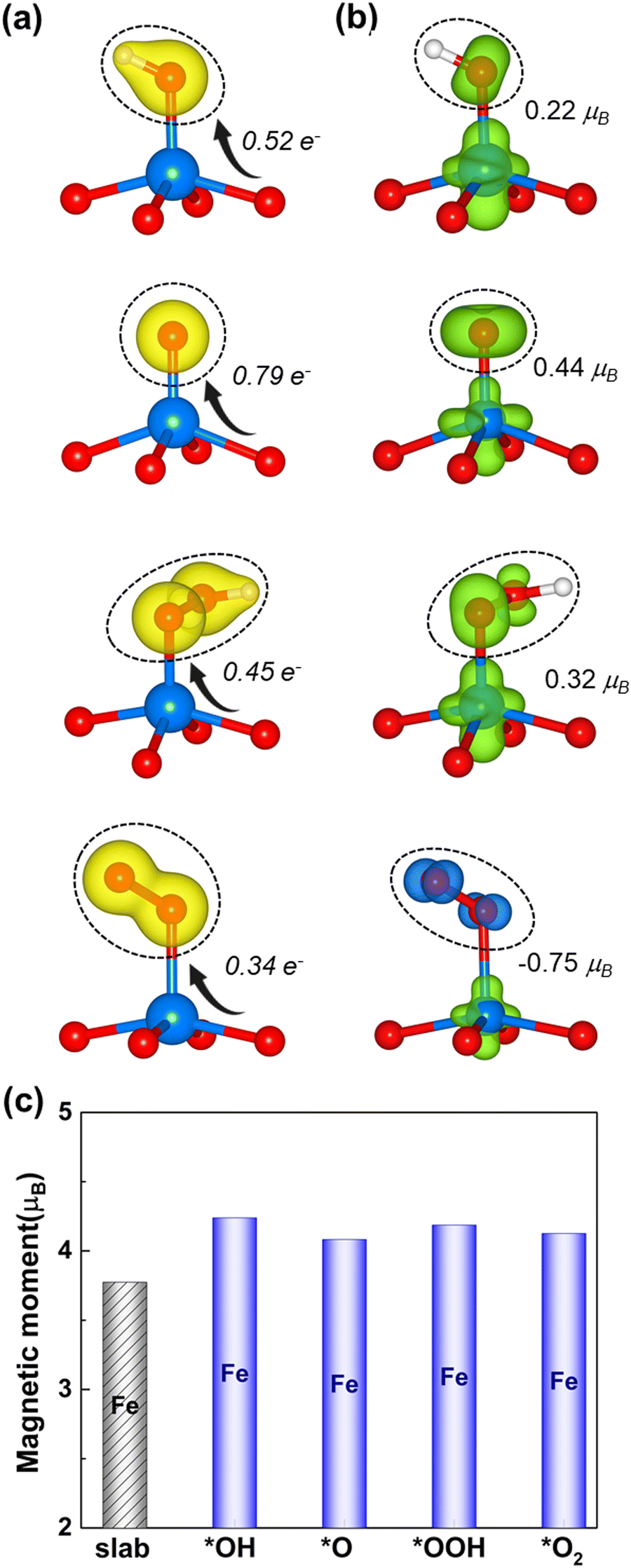
Fig. 4 displays the charge density difference (Δρ) and the spin redistribution of O intermediates adsorbed on the Fe site. As seen in Fig. 4a, Δρ is mostly found on the O-intermediates and neutral on the rest of the architecture, where the yellow iso-surface indicates the charge accumulation. This situation indicates that the monolayer with the high spin donates the charge to the attached O-atom and also affects the end O-/H-atom. The Bader charge analysis also supports that the O-intermediates gain 0.52, 0.79, 0.45, and 0.34e− charges from the monolayer with the high spin, indicating the electron transfer between the monolayer and the intermediates. It also shows the trend of *O > *OH > *OOH > *O2 which is consistent with that of ICOHP. Given this observed trend, it becomes apparent that a higher (lower) electron transfer to the adsorbed oxygen intermediates implies a strong (moderate) degree of adsorption (the ICOHP value in Fig. S3, ESI†)
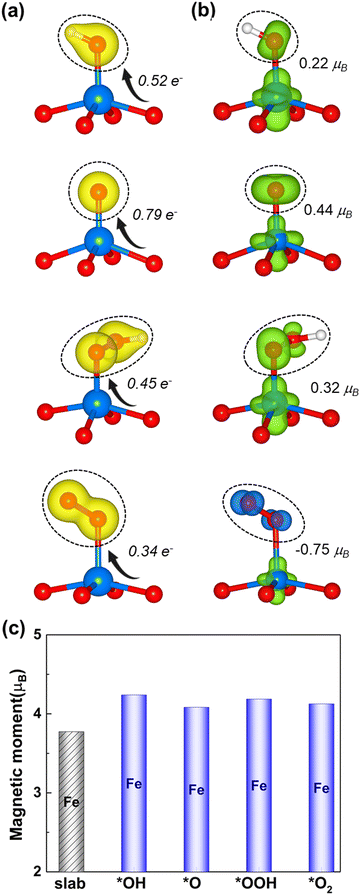 |
| | Fig. 4 (a) The charge density difference (left) of the O intermediates on the high spin Fe site, together with the Bader charge. The yellow isosurface represents the positive charge density. (b) The spin density difference (right) and the green (blue) isosurface represents the positive (negative) charges polarized in the up (down) spins. The black circle highlights the corresponding O intermediate. (c) The spin magnetic moment of Fe before (black column) and after (blue column) the adsorption. | |
Furthermore, it is worth considering the induced magnetic moment of oxygen intermediates (Fig. 4b). The *OH, *O, and *OOH intermediates yield small positive values of 0.22, 0.44, and 0.32μB, respectively, which originate from their weak diamagnetic response to the Fe active site of the high spin-polarized 2D Fe-MOF. In contrast, the *O2 intermediate shows a negatively induced magnetic moment that is 2 times larger than those of the *OH and *OOH intermediates. This observation suggests that the paramagnetic properties of *O2 quickly respond to the Fe active site of the high spin-polarized 2D Fe-MOF. Meanwhile, the spin magnetic moment of the Fe ion slightly changes from 3.77μB to 4.1μB, indicating that the Fe ion preserves its high-spin state after the adsorption (Fig. 4c). Such magnetic moment stability allows Fe to proceed with consistent bonding–antibonding interactions with the oxygen intermediates, which is a crucial factor for catalytic performance.
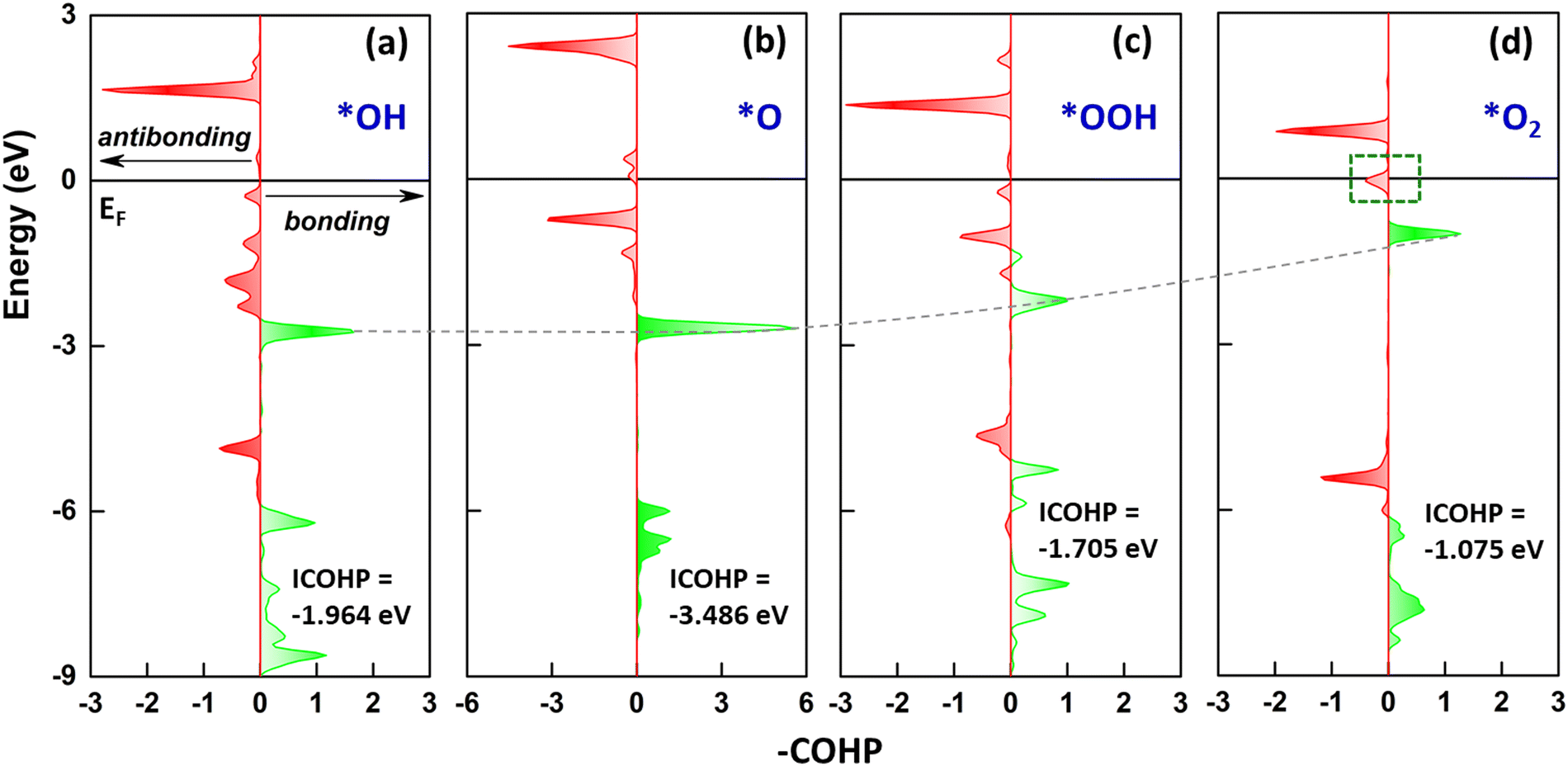
We have conducted the projected crystal orbital Hamiltonian population (pCOHP) analysis to evaluate the catalytic properties. As presented above in PDOS, we found that the oxygen intermediates strongly interact with the Fe site through the hybridization of their O pz and Fe dz2 orbitals. Therefore, the antibonding-bonding properties of the O–Fe pair at certain orbitals are worth considering. In this regard, Fig. 5 represents the corresponding profile for the coupling of Fe dz2 and O pz. The bonding orbital states are below the EF, further attributing to the bond strength between the oxygen intermediate and the Fe site. For the *OH and *O, the Fe–O pair shows significant bonding states over the antibonding states with a more negative ICOHP, indicating a stronger chemisorption. Starting with the *OOH, the bond states yield less negative ICOHP, indicating the bond strength weakening between the Fe and the intermediate, which is the essential catalytic property necessary for OER completion. Meanwhile, the antibonding states above EF reflect an interaction feature between the Fe–O pair. For the *OH, the antibonding states (<2.5 eV) indicate that the first reaction step is firmly attached to the surface, conditioning well for the next reaction step. The antibonding states for *O remain within the relevant energy range, enabling the reaction to proceed consistently. Starting with the *OOH, the antibonding states shift to the vicinity of EF, further facilitating the reaction. For the final decisive step of the OER, the downshift of antibonding states becomes significant. Notably, a small but non-negligible anti-bonding state crossing EF is crucial for the O2 release, beneficial for the completion of the reaction cycle.
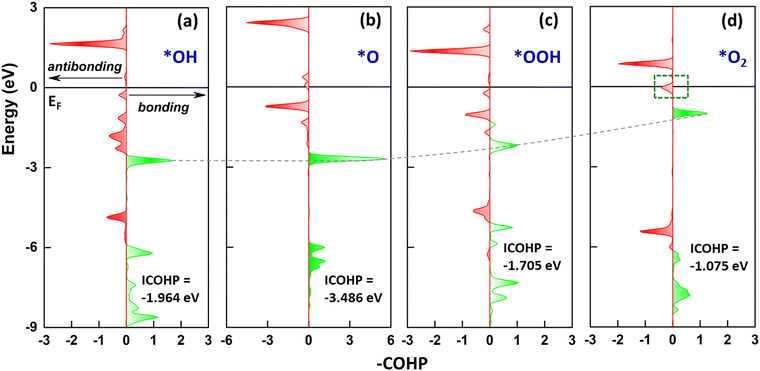 |
| | Fig. 5 The projected crystal orbital Hamilton population (pCOHP) analysis of (a) *OH, (b) *O, (c) *OOH, and (d) *O2 absorbed on the Fe site, representing the pair of Fe dz2 and O pz, where the bonding orbital state is a positive value (right arrow) and the antibonding orbital state is a negative value (left arrow). | |
Catalytic performance
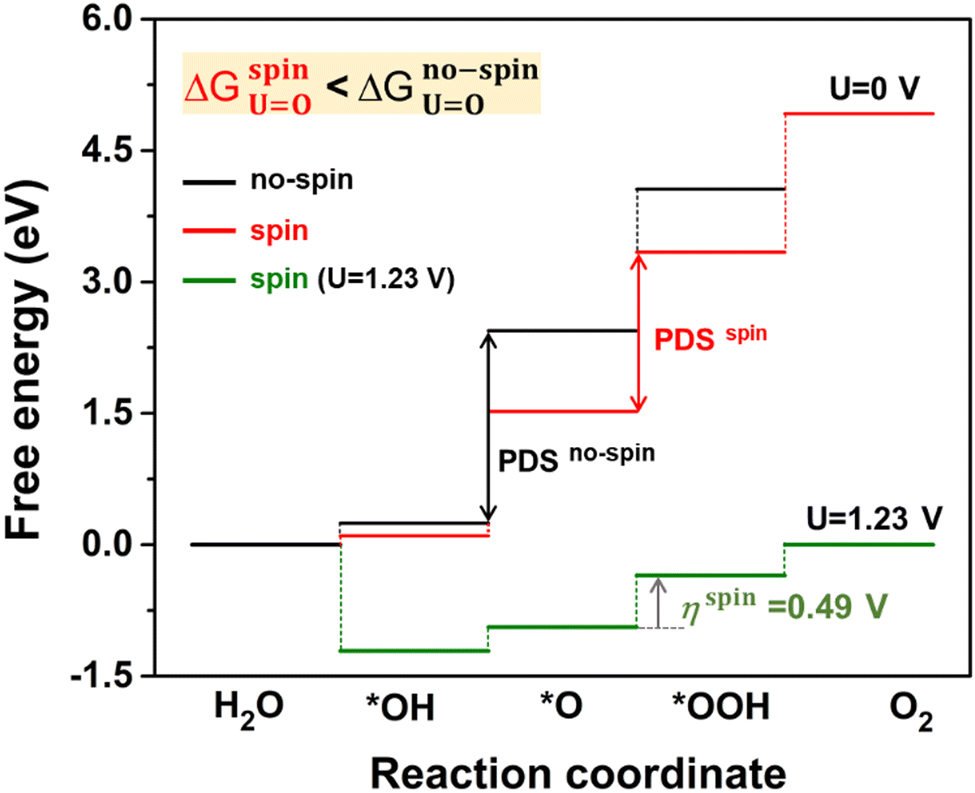
Fig. 6 displays the Gibbs free energy change (ΔG) of reaction steps at two different potentials, including a reference potential of 0 V and an equilibrium potential of 1.23 V (details in Table S5, ESI†). Foremost, we compared the spin-polarized and unpolarized energy changes of the OER at U = 0 V. All the reaction steps for the spin-polarized configuration show the lower ΔG values rather than that of the non-spin-polarized one. Such energy difference indicates that the spin-polarized 2D Fe-MOF is energetically favourable and evidently beneficial to the OER. At U = 1.23 V, the ΔG decreases to the negative (exothermic) value, satisfying the criterion of ΔG ≤ 0 eV. It indicates that the product becomes thermodynamically stable. Furthermore, the overpotential for the spin-polarized configuration reduced to 0.49 V (0.97 V excluding spin), which is comparable to those of well-known OER catalysts.45,46 This confirms that the spin impact is inevitably significant to the catalytic performance of the Fe-MOF.
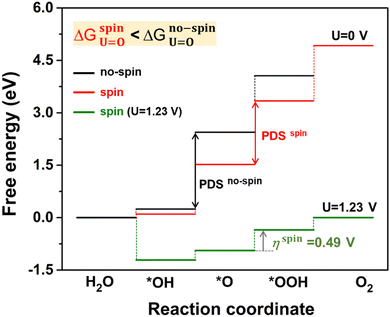 |
| | Fig. 6 Gibbs free energy changes (in eV) for O intermediates on the Fe-MOF during the OER. The free energies are calculated at two different potentials. The potential-determining step (PDS) is *OOH, while the overpotential η is found to be 0.49 V at the equilibrium potential, including a spin. | |
Conclusions
In this work, we focus on how spin affects the OER at the atomic scale using spin-polarized first-principles DFT calculations. The structural, electronic, and magnetic properties of a 2D Fe-MOF have been investigated at various spin states during the OER process. The electronic structure of the Fe-MOF in the high spin state behaves as a metallic conductor, making it a suitable OER electrocatalyst. Once the adsorption occurs, the spin-dependent Eads endows a favourable value that can facilitate the oxygen reaction process. The spin-resolved PDOS results reveal that the Fe site splits into the evenly distributed discrete levels, promoting the electron transfer and enhancing the OER. When the spin is considered in the free energy calculations, the free energy changes and the overpotential of the 2D Fe-MOF in the high spin state is significantly reduced. It indicates that the spin-polarized 2D Fe-MOF is energetically favourable for the OER. Overall, our finding not only provides insights into advancing OER mechanisms on the noble-metal-free MOF catalyst but also is used to rationally design a new OER catalyst with exceptional performance by tuning its spin state.
Computational details
First-principles calculations based on density functional theory (DFT) were performed using the Vienna Ab Initio Simulation Package (VASP, v.5.4.4).47 All calculations were carried out within the framework of projector augmented wave (PAW) pseudopotentials and plane-wave basis set with a cut-off energy of 520 eV. The exchange–correlation function was treated with a spin-polarized PBE+U method (U = 4), including Grimme's D3 corrections. All calculations in this work were included SOC for the iron-based MOF to estimate the spin-related properties. The Γ-centred grid of 8 × 8 × 1 k-points for the sampling of Brillouin zone (BZ) was applied in the calculations. In the structure, a vacuum region higher than 15 Å is added to the perpendicular direction to the basal plane, in order to simulate an isolated layer without spurious interactions between layers in the adjacent periodic cells. All structures throughout DFT calculations were fully optimized until the convergence thresholds of the total energy and the atomic forces were below 0.1 μeV and 1 meV Å−1, respectively. The geometry, spin density, and charge density differences were visualized using VESTA software.48 The crystal orbital Hamilton population (COHP) and crystal orbital bond index (COBI) were implemented using the LOBSTER code to analyse the electronic bonding characteristics.49
The adsorption energy (Eads) of oxygen species on the 2D Fe-MOF is estimated using the following equation:
| Eads = Esystem − Epristine − EOspecies |
where
Esystem,
Epristine, and
EOspecies are the total energies of the O-adsorbed system, the pristine monolayer, and the free oxygen species, respectively. The negative value of
Eads indicates exothermic adsorption.
The spin-resolved charge density difference (Δρ) is obtained as follows:
| Δρ = ρsystem − ρpristine − ρOspecies |
where
ρsystem,
ρpristine and
ρOspecies represent the charge density distribution of the O-adsorbed system and the pristine monolayer as well as an O species, respectively. In addition, Bader charge analysis is performed to show the electron-gaining/withdrawing of oxygenated intermediates.
50
The OER in the acidic medium (pH = 0) is described as follows:51
Four elementary reaction steps can be expressed as follows:
| H2O + * → *OH + (H+ + e−) |
| H2O + *O → *OOH + (H+ + e−) |
| *OOH → * + O2 + (H+ + e−) |
More precisely, H2O reaches the catalyst (denoted as *), forming an OH intermediate attached to the surface. This first elementary configuration is hereafter marked as *OH. After separating the (H++ e−) pair, the O intermediate remains on the surface, generating the configuration marked as *O. Thereafter, another H2O interacts with the *O, forming the next configuration of *OOH. By releasing the final (H+ + e−) pair, the O2 is formed from the *OOH.
The Gibbs free energy is expressed as follows:52
| ΔG = ΔE + ΔZPE − TΔS + ΔGU |
where Δ
E, ΔZPE, and Δ
S are the reaction energy found by the DFT calculation, difference in zero-point vibrational energy and entropy during the reaction process, respectively.
T is the temperature of 298.15 K. In addition, Δ
GU = −
eU, where
U is the potential electrode. One can calculate the free energy changes for each step (Δ
GOH*, Δ
GO*, Δ
GOOH*, and Δ
GO2) accordingly. The main criterion to facilitate the overall reaction is that the free energy of each elementary reaction must be less than or equal to zero (Δ
G ≤ 0 eV). To define the optimal potential required to facilitate the OER fairly well, we used the strategy to stepwise increase the potential (
U ≥ 0 V).
The difference between two free energies is calculated as follows:53
An overpotential in the free energy profile of the OER is defined as follows:
where max[PDS] is the maximum free energy change in the four-step OER (max [Δ
G1, Δ
G2, Δ
G3, and Δ
G4]), referred to as the potential-determining step, PDS. Besides,
e is the electronic charge and 1.23 V is the oxidation potential needed for an ideal OER catalyst. A low overpotential is preferable for the OER.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful to the Center of Atomic Initiative for New Materials (AI-Mat), National Taiwan University (project no. 114L900803, 111-2113-M-002-022 and 113-2639-M-002-005-ASP) for funding support. B. S. acknowledges the financial support from the National Science and Technology Council (project no. 113-2113-M-002-022-MY2) in Taiwan. The high-performance computing facility supported by the Computer & Information Networking Center of National Taiwan University is also acknowledged.
Notes and references
- X. Ren, T. Wu and Y. Sun,
et al.
, Nat. Commun., 2021, 12, 2608 CrossRef CAS PubMed.
- L. Jisi,
et al.
, Adv. Energy Sustainable Res., 2021, 2, 2100034 CrossRef.
- Z. Gang,
et al.
, Nat. Commun., 2021, 12, 4827 CrossRef PubMed.
- L. Kang,
et al.
, Nat. Commun., 2022, 13, 2075 Search PubMed.
- Z. Zhirong,
et al.
, Angew. Chem., Int. Ed., 2023, 62, e202216837 CrossRef PubMed.
- L. Tong,
et al.
, ACS Nano, 2024, 18, 28433–28443 CrossRef PubMed.
- B. Haoyun and F. Jinxian,
et al.
, Small, 2023, 19, 2205638 CrossRef PubMed.
- D. Schröder,
et al.
, Acc. Chem. Res., 2000, 33, 139–145 Search PubMed.
- Y. Y. Sun,
et al.
, J. Am. Chem. Soc., 2007, 129, 12606–12607 CrossRef CAS PubMed.
- Y. Sun,
et al.
, J. Am. Chem. Soc., 2016, 138, 3715–3730 CrossRef CAS PubMed.
- A. J. Pohlman,
et al.
, PCCP, 2020, 22, 16641–16647 Search PubMed.
- A. Mirzanejad,
et al.
, PCCP, 2022, 24, 20721–20727 RSC.
- T. Wu,
et al.
, PNAS, 2024, 121, e2318652121 CrossRef CAS PubMed.
- P. He,
et al.
, Chem. – Eur. J., 2024, 30, e202403437 Search PubMed.
- S. Shubin,
et al.
, Adv. Mater., 2024, 36, 2312524 Search PubMed.
- S. Yuanmiao,
et al.
, Adv. Mater., 2020, 32, 2003297 CrossRef PubMed.
- N. Shuai,
et al.
, J. Am. Chem. Soc., 2019, 141, 7005–7013 CrossRef PubMed.
- W. Jinlei,
et al.
, Dalton Trans., 2020, 49, 14290 RSC.
- X. Meiling,
et al.
, Adv. Mater., 2020, 32, 2004900 CrossRef PubMed.
- F. Xing,
et al.
, ACS Energy Lett., 2022, 7, 343–348 Search PubMed.
- C. Baisong,
et al.
, Nanoscale Horiz., 2022, 7, 1340 Search PubMed.
- L. Zejun,
et al.
, ACS Nano, 2021, 15, 7105–7113 CrossRef PubMed.
- C. Shanyong,
et al.
, J. Am. Chem. Soc., 2022, 144, 14505–14516 CrossRef PubMed.
- W. Yanan,
et al.
, J. Phys. Chem. Lett., 2022, 13, 5969–5976 CrossRef PubMed.
- W. Daoxiong,
et al.
, J. Mater. Chem. A, 2023, 11, 13502–13509 RSC.
- Y. Mingyuan,
et al.
, ACS Catal., 2024, 14, 6816–6826 CrossRef.
- B. Dinesh,
et al.
, ChemElectroChem, 2024, 11, e202400034 CrossRef.
- Y. Xue and H. Ethan,
et al.
, iScience, 2023, 26, 107275 CrossRef PubMed.
- J. Park,
et al.
, J. Am. Chem. Soc., 2018, 140, 14533–14537 CrossRef CAS PubMed.
- W. Junru,
et al.
, J. Phys. Chem. C, 2020, 124, 9350–9359 CrossRef.
- P. Jihye,
et al.
, ACS Appl. Mater. Interfaces, 2020, 12, 39074–39081 CrossRef PubMed.
- Z. Juan,
et al.
, ACS Sustainable Chem. Eng., 2020, 8, 7472–7479 CrossRef.
- L. Tianchun,
et al.
, J. Mater. Chem. A, 2021, 9, 24887–24894 RSC.
- X. Yi,
et al.
, Electrochim. Acta, 2021, 378, 138028 CrossRef.
- Z. Man-Rong,
et al.
, ACS Appl. Mater. Interfaces, 2021, 13, 26109–26122 CrossRef PubMed.
- F. Zhen,
et al.
, J. Mater. Chem. A, 2022, 10, 4731–4738 RSC.
- W. Anyang,
et al.
, J. Mater. Chem. A, 2022, 10, 13005–13012 RSC.
- G. Zhixiao,
et al.
, Appl. Surf. Sci., 2022, 601, 154187 Search PubMed.
- S. Xiaowen,
et al.
, J. Phys. Chem. C, 2022, 126, 16606–16614 CrossRef.
- W. Feifan,
et al.
, J. Mater. Chem. A, 2024, 12, 28764–28770 RSC.
- L. Liu and Z. Qiu,
et al.
, Chem, 2022, 8, 1822–1854 Search PubMed.
- L. Jingjuan,
et al.
, Acc. Chem. Res., 2024, 57, 1032–1045 CrossRef PubMed.
- M. Bohayra,
et al.
, Appl. Mater. Today, 2019, 15, 405–415 CrossRef.
- H. Chen,
et al.
, J. Am. Chem. Soc., 2024, 146, 12395–12400 CrossRef PubMed.
- J. K. Nørskov,
et al.
, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef PubMed.
- M. C. Isabela,
et al.
, ChemCatChem, 2011, 3, 1159–1165 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- M. Koichi and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653–658 CrossRef.
- D. L. Volker,
et al.
, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- M. Koper, J. Electroanal. Chem., 2011, 660, 254–260 CrossRef CAS.
- J. Rossmeisl,
et al.
, Chem. Phys., 2005, 319, 178–184 CrossRef CAS.
- X. Haoxiang,
et al.
, Nat. Catal., 2024, 7, 207–218 CrossRef.
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Michitoshi
Hayashi
ab,
Michitoshi
Hayashi