
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning of aromaticity and reactivity in gold-substituted cyclopropenyl cations†
Erick
Cerpa
 *a,
Alba
Vargas-Caamal
a,
Luz
Diego
b,
David
Arias-Olivares
c,
Diego V.
Moreno
d,
Jorge I.
Martínez-Araya
e,
Rafael
Islas
*e and
Gabriel
Merino
*f
*a,
Alba
Vargas-Caamal
a,
Luz
Diego
b,
David
Arias-Olivares
c,
Diego V.
Moreno
d,
Jorge I.
Martínez-Araya
e,
Rafael
Islas
*e and
Gabriel
Merino
*f
aDepartamento de Formación Básica Disciplinaria, Unidad Profesional Interdisciplinaria de Ingeniería Campus Guanajuato, Instituto Politécnico Nacional, C.P. 36275, Silao de la Victoria, Gto, Mexico. E-mail: jcerpa@ipn.mx
bDoctorado en Fisicoquímica Molecular, Facultad de Ciencias Exactas, Universidad Andres Bello, Av. República 275, Santiago 8370146, Chile
cCenter of Applied Nanoscience (CANS), Facultad de Ciencias Exactas, Universidad Andres Bello, Av. República 275, Santiago 8370146, Chile
dLaboratorio de Química Computacional, Programa de Química, Universidad de Ciencias Aplicadas y Ambientales (U.D.C.A.), Calle 222 #55-37, Bogotá 111166, Colombia
eDepartamento de Ciencias Químicas, Facultad de Ciencias Exactas, Universidad Andres Bello, Av. República 275, Santiago 8370146, Chile. E-mail: rafael.islas@unab.cl
fDepartamento de Física Aplicada, Centro de Investigación y de Estudios, Avanzados, Mérida Km. 6 Antigua carretera a Progreso Apdo. Postal 73, Cordemex, Yuc., Mérida, Mexico. E-mail: gmerino@cinvestav.mx
First published on 12th May 2025
Abstract
This study investigate the structural, electronic, and reactivity properties of the aurocarbon C3H2Au+, C3HAu2+, and C3Au3+ clusters. A potential energy surface exploration indicates that the most stable isomers adopt cyclic structures, supporting the gold–hydrogen analogy in these cationic systems. These cyclic forms preserve the characteristic geometry of the cyclopropenyl cation, with covalent C–Au interactions confirmed by multiple bonding analyses. All three systems exhibit a delocalized 3c–2e π-bond across the carbon ring, consistent with an aromatic behavior. The substitution of hydrogen by gold alters local reactivity patterns.
Introduction
Gold clusters and their compounds have attracted attention in nanoscience and nanotechnology due to their applications in catalysis, medicine, and electronics.1–5 Among these, aurocarbons, a class of gold clusters containing carbon atoms, have emerged as promising candidates for facilitating diverse chemical transformations. For instance, they play a key role in the environmentally relevant conversion of carbon monoxide to carbon dioxide.6 Density functional theory (DFT) studies by Tiwari revealed that small aurocarbons, such as C4H3Au and C4Au4, exhibit enhanced catalytic activity for CO oxidation, particularly when CO pre-adsorbed on all gold sites in C4Au4.7 These findings suggest that carbon-intercalated gold clusters may serve as efficient catalysts. Beyond CO oxidation, aurocarbons have also shown potential for SO2 reduction with CO and for acetylene hydracyanation.8,9 Their stability is often attributed to aurophilic interactions (strong mutual attractions between gold atoms). This phenomenon was explored in detail by Zaleski-Ejgierd and Pyykkö, who compared the bonding characteristics of aurocarbons with their carbon-only counterparts.10Naumkin has also extensively investigated the formation, reactivity, and properties of carbon–gold nanoclusters. In 2006, he reported the C5Au12 system and its charged derivatives (C5Au12− and C5Au12+). These core–shell aurocarbons consist to a tetrahedral C5 radical (carbon core) encapsulated by a 12-atom gold shell, mimicking the bonding environment of the C5H12 molecule. Using DFT calculations, the study evaluated the structural stability, electronic properties, and bonding interactions of neutral and ionic forms. Two main classes of isomers were identified: an icosahedral-like (Ih) isomer and an octahedral-like (Oh) isomer, both resembling similar structures found in the Au13 cluster and C5H12.11 Building on this approach, Naumkin later explored core-doping in the C10Au18 system. He reported a structural analogue of di-tert-butyl-acetylene C10H18 with near-C3 symmetry. A key finding was that such clusters allow flexible tuning of catalytic properties: the C10 dopant can selectively alter the electron-donating capacity of the gold cluster while maintaining its electron-acceptor efficiency.12
Ramachandran and Naumkin further investigated the structure, stability, and electronic properties of C4Aun clusters, where n = 4, 6, and 8. They classified the isomers into four categories: hydrocarbon analogues, carbon chains, carbon rings adsorbed on gold surface, and carbon cores encapsulated by gold atoms. The most stable structures typically featured linear or surface-bound carbon moieties rather than encapsulated cores. For example, the lowest-energy isomers of C4Au4 resembles a gold analogue of 1,2,3-butatriene, while the cyclobutadiene-like structure (C4Au4) lies 36.9 kcal mol−1 higher in energy. Despite their lower thermodynamic stability, encapsulated-carbon isomers exhibit variations in ionization energies, electron affinities, and HOMO–LUMO gaps, properties that make them attractive for the rational design of tunable nanocatalyst.13
The structural characterization of aurocarbons clusters has been widely explored. In 2015, Meng et al. determined the structures of four AuC3H− isomers using emission spectroscopy and ab initio calculations.14 Ticknor reported the formation of AuCn+ species via laser vaporization and mass spectrometry15 techniques that also yielded AuCn+ (n = 1–11, 18), Au2Cn+ (n = 1–16), Au3Cn+ (n = 1–10), and AuC3H− clusters, which are single-gold analogues of the C3H2 carbene.14,16 Theoretical and experimental efforts have further examined gold carbide clusters and the synthesis of gold–acetylenes complexes (Au–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Au).8,17,18
C–Au).8,17,18
Beyond structural aspects, several studies have proposed an analogy between gold and hydrogen in these clusters. This so-called gold–hydrogen analogy suggests that hydrogen can mimic the behavior of a gold atom within a cluster.19–21 Zheng et al. investigated this concept by analyzing auro-acetylide species (AuC2H, AuC2Au, and Au2C2H) using photoelectron spectroscopy and DFT calculations, finding that AuC2H and AuC2Au share similar electronic and bonding properties.18 Wang et al. expanded this idea, exploring the formation of aurosilicon, auroboron, and aurocarbon clusters, all characterized by strong Au–E covalent bonds (E = Si, B, C).18,21,22 In addition, the combination of photoelectron imaging spectroscopy and DFT calculations has been used to elucidate the structures of four AuC3H− isomers.14 These techniques were subsequently applied to study AuC4−, AuC6−, and AuCnH− (n = 2, 4, and 6) clusters, leading to the discovery of auropolyynes (Au(C2)nAu−, n = 1–3) via high-resolution photoelectron imaging and ab initio calculations.23
Further insight into bonding in gold–carbon systems comes from Armentrout's study of the gold carbene cation (AuCH2+), which used molecular orbital analysis to reveal covalent C–Au bonding.24 Megha et al. revisited the gold–hydrogen analogy by studying CO and O2 adsorption on pristine and hydrogen-doped gold clusters (Au7H), confirming the analogy in the most stable planar isomer, although deviations were obtained in higher-energy structures.19 Tang et al. synthesized and characterized phenylgold (C6H5Au) and its anion (C6H5Au−) using laser vaporization, photoelectron spectroscopy, and DFT calculations.25 Tsipis and Gkarbounis investigated aromatic aurocarbons (C6Au6), the benzene analogue, predicting their stability and analyzing their magnetic properties. Their calculations indicated that coinage-metal-substituted benzenes exhibit reduced aromaticity compared to benzene.26 More recently, Lu examined M5C6 systems (M = Cu, Ag and Au), concluding that Au5C6− exhibits σ-aromaticity.27
In this work, we investigate the sequential H-to-Au substitution in C3H3+,28 leading to the fully substituted species C3Au3+. Cyclic structures analogous to C3H3+ emerge as the lowest-energy configurations, displaying marked electron delocalization and tunable reactivity. To characterize the bonding and electronic properties of C3H2Au+, C3HAu2+ and C3Au3+, we employed Natural Population Analysis (NPA),29 Wiberg bond Indices (WBI),30 the Electron Localization Function (ELF),31 and Adaptive Natural Density Partitioning (AdNDP).32 Aromaticity was further assessed via magnetically induced current density (MICD) and induced magnetic field (Bind) analysis,33 both confirming the presence of diatropic ring currents. Finally, reactivity trends were examined using local hyper-softness (LHS),34–39 revealing that even a single H-to-Au substitution alters local reactivity patterns.
Computational details
The potential energy surfaces (PESs) of the clusters were explored using a modified genetic algorithm, as implemented in GLOMOS,40–42 which has been previously validated in related studies.43–48 Initial geometries were optimized at both the PBE0/def2-TZVP and PBE0-D349/def2-TZVP50,51 levels. The same isomers were located with both levels, with relative energy differences of less than 2 kcal mol−1 (see Fig. S1–S4, ESI†). These results indicate that, for the systems examined here, dispersion interactions do not alter the overall topology of the PES, although they do introduce slight shifts in the relative energies of the isomers. The structure were further refined at the MP2/def2-TZVP level,50 incorporating effective core potentials for gold.52 Harmonic frequency analysis confirmed that all structures correspond to local minima. All calculations were carried out with Gaussian 16.53 To analyze stability, bonding characteristics, and electron delocalization, we computed NPA charges,29 WBI,30 ELF,31 and AdNDP32 analysis using Multiwfn.54MICD55 was computed via the response function56 and the perturbing operator for the magnetic field using DIRAC 19.57 The wavefunction was obtained with the four-component Dirac–Coulomb Hamiltonian using unrestricted kinetic balance,57 employing the PBE0 functional51 and the cc-pVDZ58 basis set for carbon and hydrogen atoms. Gold atoms were treated with the Dyall double-ζ basis set.59 Shielding tensors required for the calculation of the induced magnetic field (Bind)33 were computed with ORCA60 using the PBE0 functional and the def2-QZVP52,61 basis set. Isolines maps and isosurfaces were visualized using VisIt.62
Conceptual density functional theory (c-DFT)63 was used to evaluate local reactivity using LHS,34–39 a descriptor that is more accurate and size-consistent than local softness, dual descriptors, or fukui functions.64 LHS65,66 values were computed at the MP2/def2-QZVP level using Gaussian 16. To simplify the calculations, we adopted the frontier molecular orbital approximation (FMOA) rather than the finite difference approximation (FDA), ensuring a reliable reactivity description even in systems with orbital degeneracy.67 The robustness of the dual descriptor, which underlies LHS, has been validated in systems as diiodine, where relativistic effects are significant, confirming that FMOA-based dual descriptors provide a reliable qualitative description.68
Results and discussion
A PES exploration identified global minima and low-energy isomers for the singlet and triplet states of the C3HAu2+, C3H2Au+ and C3Au3+ systems. In each case, the most stable isomer preserves the three-carbon cyclic geometry of the cyclopropenyl cation (c-C3H3+), with energy differences of less than 33 kcal mol−1 relative to linear isomers resembling the propargyl cation (l-C3H3+) (Fig. 1, additional isomers in Fig. S1–S4, ESI†). In other words, these results support the gold–hydrogen analogy in cationic systems.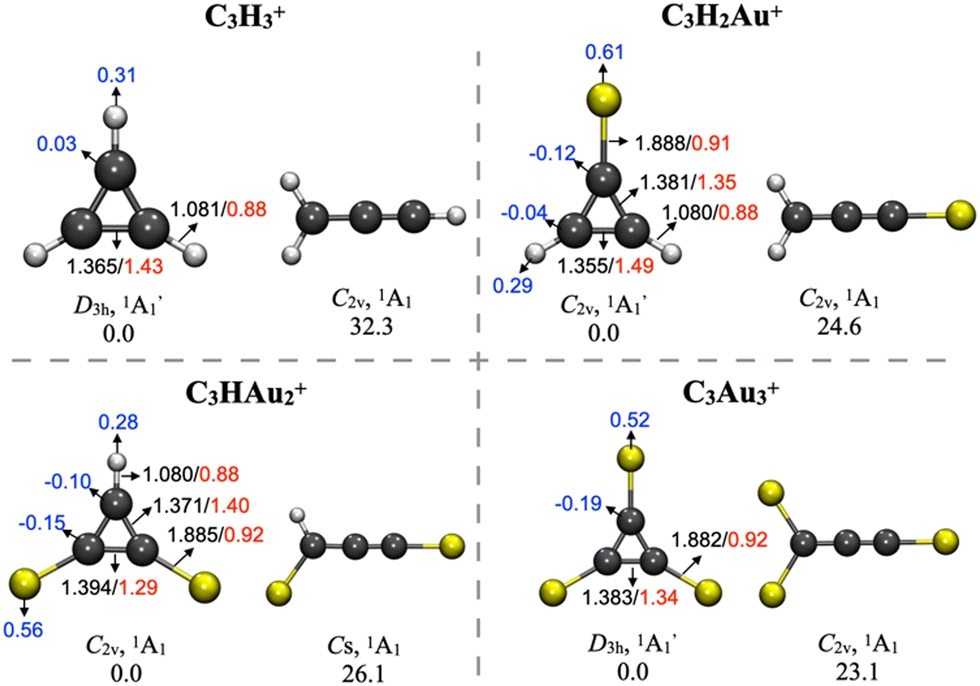 | ||
| Fig. 1 Relative energies (kcal mol−1) of the low-energy isomers of C3H3+, C3H2Au+, C3HAu2+, and C3Au3+, calculated at the MP2/def2-TZVP level. Bond lengths (Å, black), natural population analysis charges (|e|, blue), and Wiberg bond indices (red) are indicated. | ||
In the most stable isomers, the C3 ring remains intact, maintaining constant C–H bond lengths, while the C–C bond lengths increase slightly from 1.37 to 1.39 Å upon H-to-Au substitution (Fig. 1). The resulting C–Au bonds (∼1.88 Å) are consistent with reported Au–CO bond lengths (1.84–2.03 Å);.69 WBI confirm covalent C–Au interactions (WBIC–Au = 0.92) and reveal a decrease in WBIC–C values from 1.43 in C3H3+ to 1.34 in C3Au3+. Furthermore, replacing H with Au increases charge accumulation on the carbon skeleton, with NPA charges indicating a shift toward more positive on the Au atoms and more negative values on the C atoms.
ELF values close to 1 confirm covalent bonding across the systems (Fig. 2). Among these, C–Au bonds exhibit the lowest ELF values (0.80–0.90), deviating from ideal covalent values yet remaining within the covalent range. In Au-substituted systems, C–C bonds display slightly reduced ELF values, consistent with their elongation. These trend in ELF agree with the WBI data, collectively confirming the presence of strong C–H and C–Au covalent bonds and a weakening of C–C bond orders upon Au substitution.
 | ||
| Fig. 2 Color-filled map ELF maps of C3H3+, C3H2Au+, C3HAu2+, and C3Au3+. | ||
AdNDP analysis of C3Au3+ (Fig. 3) identifies three 2c–2e σ-bonds between carbon atoms, three C–Au σ-bonds, and a delocalized 3c–2e π-bond across the carbon ring, satisfying Hückel's rule and confirming aromaticity. Each Au atom contributes five 5d orbitals corresponding to lone pairs, resulting in a total of fifteen. These orbitals exhibit slightly reduced occupation numbers (ONs) between 1.96 and 1.91 |e|, indicating partial charge transfer to the carbon atoms but no participation in π-delocalization. This bonding pattern, including the presence of a 3c–2e π-bond, is also present in the related complexes C3H2Au+ and C3HAu2+ (Fig. S5 and S6, ESI†).
 | ||
| Fig. 3 Adaptive natural density partitioning plots of C3Au3+ at the PBE0-D3/def2-TZVP level, using pseudopotentials for Au atom. | ||
These results are consistent with the NPA charge analysis, which shows that gold substitution increases the negative charge on the carbon atoms and the positive charge on the gold atoms. Accordingly, the AdNDP analysis confirms that the C3 ring in C3Au3+ retains an aromatic character similar to that of the prototypical C3H3+ system (Fig. S7, ESI†), with a delocalized 3c–2e π-bond localized over the carbon atoms.
During the review process, one of the reviewers inquired about the effect of adding an electron to C3H3+ and C3Au3+, a particular insightful question. The contrasting behavior of the resulting C3H3 and C3Au3 clusters arises from fundamental differences in electron delocalization and orbital symmetry, which govern their geometric and electronic structures. A key aspect lies in the nature of the molecular orbital populated upon reduction of the corresponding cation.
For C3H3+, the added electron occupies a degenerate π-antibonding orbital (Fig. S8, ESI†). This electron configuration violates Hückel's 4n + 2 rule for aromaticity, and the presence of an unpaired electron in a π*-orbital destabilizes the system. This electronic degeneracy triggers a Jahn–Teller distortion, lowering the symmetry from D3h to C2v, disrupting π-delocalization and eliminating aromatic stabilization. Geometry optimizations at the PBE0-D3/def2-TZVP level yields two imaginary frequencies at the D3h geometry. Upon relaxation, the molecule adopts a lower-symmetry C2v geometry, characterized by bond length alternation (1.38 Å vs. 1.42 Å), consistent with π-antibonding interactions and the loss of aromatic character. In contrast, the addition of an electron to C3Au3+ populates a σ-bonding orbital (Fig. S8, ESI†). So, occupation of the σ-orbital does not induce the destabilization observed in the hydrocarbon analogue. Consequently, the neutral C3Au3 retains its D3h symmetry and shows no imaginary frequencies, consistent with a true local minimum.
To further evaluate aromaticity across the aurocarbon series, MICD analysis was performed. All systems exhibit diatropic ring currents (Fig. 4), supporting their aromatic character. The strength of the current density was integrated in the xy plane (orange line in Fig. 4), which bisects a representative C–C bond in each system. These values are shown in Fig. 4 and further support the aromatic behavior of the molecules. Interestingly, the addition of Au results in a gradual decrease in current density strength. The diatropic response around the Au atoms overlaps with that of the C3 core, forming a net diatropic current. This effect is particularly evident in C3H2Au+ and C3HAu2+ (see Fig. S9a, ESI†), where current density plots ≥2 Bohr above the molecular plane reveal this combined delocalization pattern. Notably, C3Au3+ shows a weaker diatropic current density (7.9 nA T−1) compared to C3H3+ (9.1 nA T−1), consistent with the trend in Fig. S9a and b (ESI†). In contrast, both C4H4 and C4Au4 exhibit paratropic current densities, consistent with their antiaromatic character. For C4H4, the integrated current density strength was calculated as −22.8 nA T−1, in good agreement with the previously reported value of −19.9 nA T−1.70 For C4Au4, a paratropic current density of approximately −14.7 nA T−1 was obtained. The corresponding current density plots for both species are shown in Fig. S9b (ESI†).
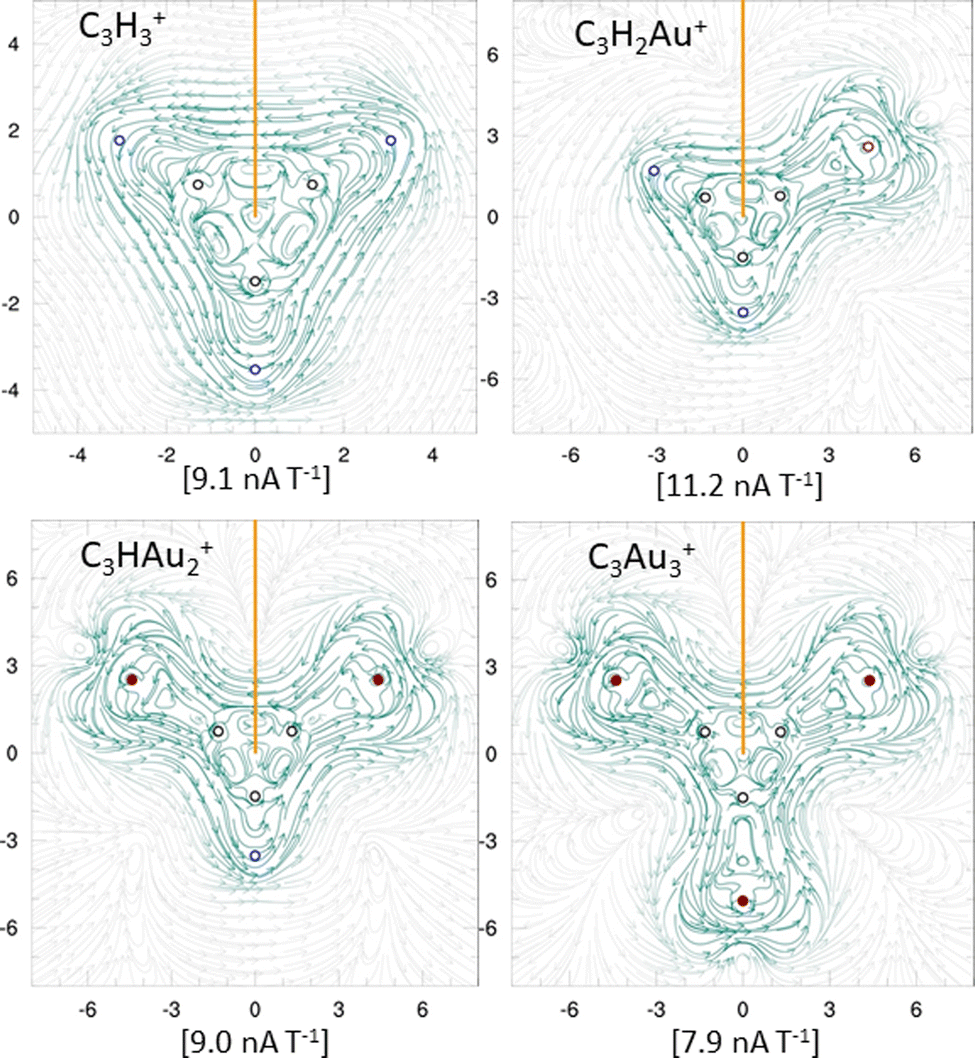 | ||
| Fig. 4 Magnetically induced current density maps for: (A) C3H3+, (B) C3H2Au+, (C) C3HAu2+, and (D) C3Au3+. Counterclockwise currents correspond to diatropic behavior. Black, blue, and red spheres represent C, H, and Au atoms, respectively. Numbers in brackets indicate current strength (nA T−1), and the orange line denotes the integration plane. | ||
Isolines maps of the z-component of the induced magnetic field (Bindz, equivalent to NICSzz)71 also reveal diatropic ring currents in all systems (Fig. 5). Substitution of protons by Au atoms has minimal impact on the electron delocalization within the C3 ring. (Fig. 5). However, due to Au's large atomic radius, localized diatropic regions appear around the heavy atoms, likely due to contributions from the 5d electrons. As more protons are replaced by gold, these diatropic regions become increasingly pronounced. Fig. S10 (ESI†) shows Bindz isosurfaces for C3H3+, C3H2Au+, C3HAu2+, and C3Au3+, revealing a broader magnetic response in C3Au3+. Finally, Fig. S11 (ESI†) shows the Bindz isosurfaces for C4H4 and C4Au4, confirming that their antiaromatic character, evidenced by a paratropic magnetic response, persists even after hydrogen-to-gold substitution.
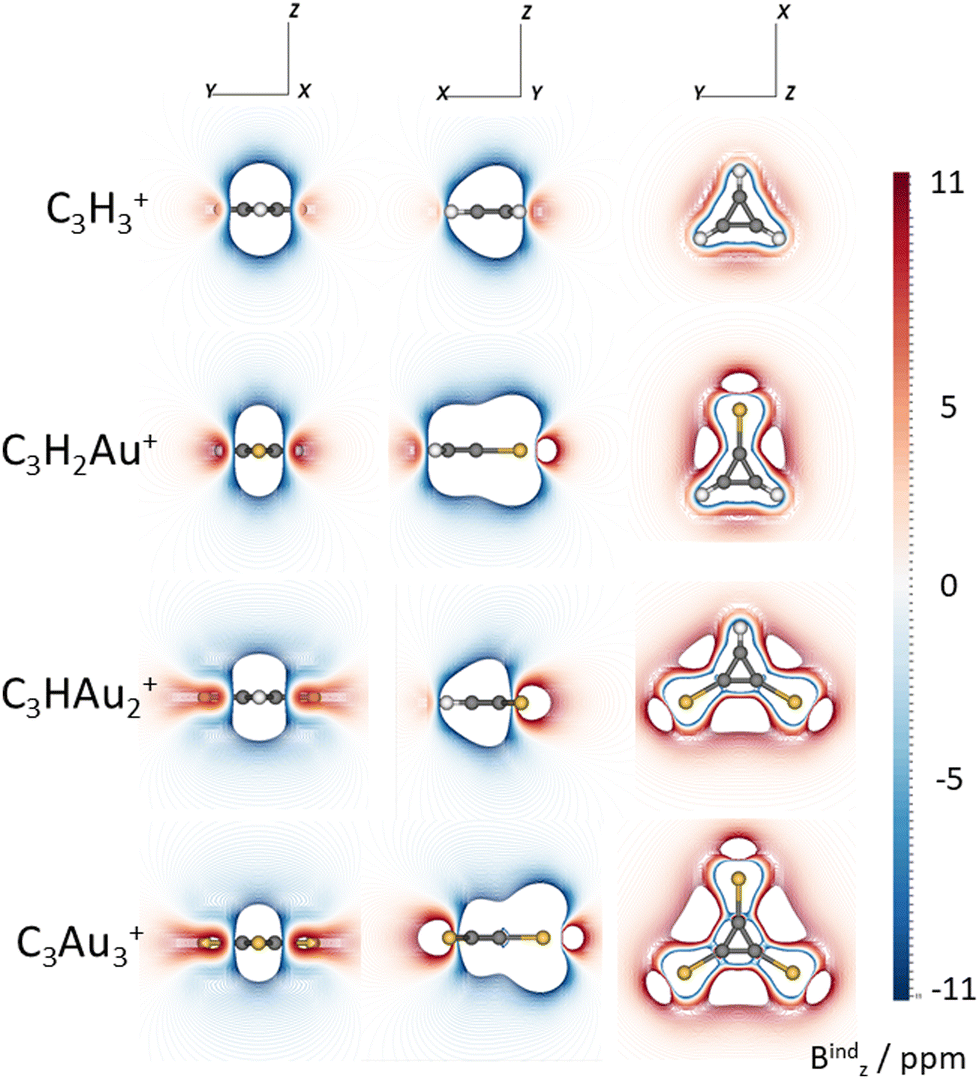 | ||
| Fig. 5 Isolines maps of the z-component of the induced magnetic field (Bindz) for all systems. Units are ppm. Carbon, hydrogen, and gold atoms are represented as black, white, and yellow spheres, respectively. | ||
Do the geometric and electronic changes induced by H-to-Au substitution affect the reactivity of aurocarbons? To address this question, we performed LHS analysis to identify covalent nucleophilic and electrophilic sites. Fig. 6 shows the LHS isosurfaces, which aligns with the symmetric irreducible representations of their corresponding point groups: D3h for C3H3+ and C3Au3+, and C2v for C3H2Au+ and C3HAu2+. The cyclopropenyl cation (Fig. 6(A)) acts as an electrophile, accepting electron density above and below the molecular plane. Replacing H with Au localizes the electrophilicity character at the gold atoms while enhancing nucleophilic behavior in their vicinity (Fig. 6(B)–(D)). This dual reactivity pattern persists throughout the substitution series: The Au-decorated vertices retain pronounced electrophilic character, while nucleophilic regions emerge around the Au atom. Thus, H-to-Au substitution shifts the electrophilic behavior from a delocalized, planar nature to a more localized, vertex-centered pattern.
 | ||
| Fig. 6 Isosurfaces of local hyper-softness (LHS) at ±0.00075 |e|3 Hartree−2 Bohr−3: (A) C3H3+, (B) C3H2Au+, (C) C3HAu2+, (D) C3Au3+. Upper panels show front views; lower panels show side views. Dark lobes correspond to positive LHS values (+0.00075), and white lobes to negative values (−0.00075). Additional isosurface cuts are provided in the ESI† (Fig. S12). | ||
To further quantify this trend, we computed the electrophilicity index (ω),72 which estimates the energy stabilization upon electron saturation of an electrophile. As shown in Table S1 (ESI†), C3H3+ exhibits the highest ω value, serving as the reference. The following trend is found:
| ω(C3H3+) > ω(C3Au3+) > ω(C3H2Au+) > ω(C3HAu2+) |
We also evaluated the electron-donating (−ω−) and electron-accepting (ω+) powers.73 C3H2Au+ emerges as the most electro-donating species, whereas C3H3+, consistent with its electrophilic nature, shows minimal electron-donating capability. These trends are further summarized using the net electrophilicity index (Δω±):74
| Δω±(C3H3+) > Δω±(C3Au3+) > Δω±(C3H2Au+) > Δω±(C3HAu2+) |
Finally, the softness index (S)75 shows the opposite trend:
| S(C3HAu+) > S(C3Au3+) > S(C3H2Au+) > S(C3H3+) |
Altogether, these results confirm that while C3H3+ is a prototypical electrophile, its Au-substituted analogues display a richer dual character that becomes more pronounced with increasing Au content.
Conclusion
Sequential H-to-Au substitution in C3H3+ preserves the cyclic geometry of the parent cyclopropenyl cation, as confirmed by potential energy surface analyses. The structural similarity between cyclic and linear isomers reinforces the gold–hydrogen analogy in these cationic systems. Substitution leads to elongated C–C bonds (1.37–1.39 Å) and the formation of covalent C–Au bonds (∼1.88 Å), supported by Wiberg bond indices (WBI > 0.9) and charge transfer from Au to C. Electron Localization Function analysis further validates the covalent nature of the C–Au interactions, albeit with minor deviations attributed to the higher electronegativity of gold.Aromaticity is preserved across all substituted clusters. This is evidenced by AdNDP, magnetically induced current density, and induced magnetic field analyses, all of which confirm the presence of diatropic ring currents. Gold's large atomic radius induces localized diatropic regions, particularly in C3Au3+, thereby altering the electron delocalization pattern while maintaining aromatic behavior.
So, H-to-Au substitution reshapes reactivity. The Au atoms act as electrophilic sites, while the adjacent carbon atoms exhibit enhanced nucleophilic character. This dual behavior becomes more pronounced with increasing substitution, gradually transforming the prototypical nucleophilic C3H3+ into a system with vertex-centered, tunable reactivity. These adaptable electronic features make aurocarbons promising candidates for catalytic applications that demand site-specific reactivity.
Author contributions
Erick Cerpa: conceptualization, writing – review & editing, investigation; Alba Vargas-Caamal: writing – original draft, investigation; Luz Diego: writing – review & editing, investigation; David Arias-Olivares: writing – review & editing, investigation; Diego V. Moreno: software, editing, investigation; Jorge I. Martínez-Araya: writing – review & editing, investigation; Rafael Islas: writing – review & editing, investigation, formal analysis; Gabriel Merino: writing – review & editing, supervision.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. D. acknowledges National Agency for Research and Development (ANID)/Scholarship Program/becas doctorado nacional/2023-21231670 (L-D). J. I. M-A. acknowledges the financial support provided by FONDECYT grant no 1181504.Notes and references
- Y. Zhang, X. Cui, F. Shi and Y. Deng, Chem. Rev., 2012, 112, 2467–2505 CrossRef CAS PubMed.
- G. Schmid and U. Simon, Chem. Commun., 2005, 697–710 RSC.
- C. F. Shaw, Chem. Rev., 1999, 99, 2589–2600 CrossRef CAS PubMed.
- S. Taghizadeh, V. Alimardani, P. L. Roudbali, Y. Ghasemi and E. Kaviani, Photodiagn. Photodyn. Ther., 2019, 25, 389–400 CrossRef CAS PubMed.
- D.-e Jiang, K. Nobusada, W. Luo and R. L. Whetten, ACS Nano, 2009, 3, 2351–2357 CrossRef CAS PubMed.
- J. Mathews and L. Watters, J. Am. Chem. Soc., 1900, 22, 108–111 CrossRef.
- M. Tiwari and C. N. Ramachandran, Chem. Phys. Impact, 2021, 2, 100023 CrossRef.
- M. Tiwari and C. N. Ramachandran, Chem. Phys. Lett., 2020, 761, 138059 CrossRef CAS.
- M. Tiwari, Vinit and C. N. Ramachandran, Chem. Phys. Lett., 2019, 726, 111–116 CrossRef CAS.
- P. Zaleski-Ejgierd and P. Pyykkö, Can. J. Chem., 2009, 87, 798–801 CrossRef CAS.
- F. Naumkin, Phys. Chem. Chem. Phys., 2006, 8, 2539–2545 RSC.
- F. Y. Naumkin, Chem. Phys. Lett., 2008, 466, 44–49 CrossRef CAS.
- C. N. Ramachandran and F. Y. Naumkin, J. Phys. Chem. A, 2013, 117, 6803–6808 CrossRef CAS PubMed.
- J.-H. Meng, Q.-Y. Liu and S.-G. He, J. Phys. Chem. A, 2015, 119, 11265–11270 CrossRef CAS PubMed.
- B. W. Ticknor, B. Bandyopadhyay and M. A. Duncan, J. Phys. Chem. A, 2008, 112, 12355–12366 CrossRef CAS PubMed.
- J. Havel, E. M. Peña-Méndez, F. Amato, N. R. Panyala and V. Buršíková, Resour. Chem. Mater., 2014, 28, 297–304 CAS.
- M. Tiwari and C. N. Ramachandran, Chem. Phys. Lett., 2022, 793, 139387 CrossRef CAS.
- P. Wang, W. Zhang, X.-L. Xu, J. Yuan, H.-G. Xu and W. Zheng, J. Chem. Phys., 2017, 146(19), 194303 CrossRef PubMed.
- Megha, C. Kamal, K. Mondal, T. K. Ghanty and A. Banerjee, J. Phys. Chem. A, 2019, 123, 1973–1982 CrossRef CAS PubMed.
- B. Kiran, X. Li, H.-J. Zhai, L.-F. Cui and L.-S. Wang, Angew. Chem., Int. Ed., 2004, 43, 2125–2129 CrossRef CAS PubMed.
- X. Li, B. Kiran and L.-S. Wang, J. Phys. Chem. A, 2005, 109, 4366–4374 CrossRef CAS PubMed.
- L.-S. Wang, Phys. Chem. Chem. Phys., 2010, 12, 8694–8705 RSC.
- I. León, F. Ruipérez, J. M. Ugalde and L.-S. Wang, J. Chem. Phys., 2018, 149, 144307 CrossRef PubMed.
- P. B. Armentrout, B. C. Stevenson, F. Yang, F. J. Wensink, O. V. Lushchikova and J. M. Bakker, J. Phys. Chem. A, 2019, 123, 8932–8941 CrossRef CAS PubMed.
- S. Sun, X. Xing, H. Liu and Z. Tang, J. Phys. Chem. A, 2005, 109, 11742–11751 CrossRef CAS PubMed.
- A. C. Tsipis, Organometallics, 2012, 31, 7206–7212 CrossRef CAS.
- S.-J. Lu, Chem. Phys. Lett., 2024, 849, 141425 CrossRef CAS.
- D. Zhao, K. D. Doney and H. Linnartz, Astrophys. J., Lett., 2014, 791, L28 CrossRef.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC.
- R. Islas, T. Heine and G. Merino, Acc. Chem. Res., 2012, 45, 215–228 CrossRef CAS PubMed.
- C. Cárdenas, N. Rabi, P. W. Ayers, C. Morell, P. Jaramillo and P. Fuentealba, J. Phys. Chem. A, 2009, 113, 8660–8667 CrossRef PubMed.
- J. I. Martínez-Araya, J. Phys. Chem. C, 2013, 117, 24773–24786 CrossRef.
- J. I. Martínez-Araya and D. Glossman-Mitnik, Chem. Phys. Lett., 2015, 618, 162–167 CrossRef.
- J. I. Martínez-Araya, A. Grand and D. Glossman-Mitnik, Phys. Chem. Chem. Phys., 2015, 17, 29764–29775 RSC.
- J. I. Martínez-Araya and D. Glossman-Mitnik, J. Mol. Model., 2018, 24, 42 CrossRef PubMed.
- J. I. Martínez-Araya and R. Islas, J. Comput. Chem., 2022, 43, 29–42 CrossRef PubMed.
- F. Ortiz-Chi and G. Merino, GLOMOS, CINVESTAV, Mérida, México, 2020 Search PubMed.
- A. Ramírez-Manzanares, J. Peña, J. M. Azpiroz and G. Merino, Comput. Chem., 2015, 36, 1456–1466 CrossRef PubMed.
- R. Grande-Aztatzi, P. R. Martínez-Alanis, J. L. Cabellos, E. Osorio, A. Martínez and G. Merino, J. Comput. Chem., 2014, 35, 2288–2296 CrossRef CAS PubMed.
- A. Vargas-Caamal, J. L. Cabellos, F. Ortiz-Chi, H. S. Rzepa, A. Restrepo and G. Merino, Chem. – Eur. J., 2016, 22, 2812–2818 CrossRef CAS PubMed.
- A. C. Castro, E. Osorio, J. L. Cabellos, E. Cerpa, E. Matito, M. Solà, M. Swart and G. Merino, Chem. – Eur. J., 2014, 20, 4583–4590 CrossRef CAS PubMed.
- A. Vargas-Caamal, S. Pan, F. Ortiz-Chi, J. L. Cabellos, R. A. Boto, J. Contreras-Garcia, A. Restrepo, P. K. Chattaraj and G. Merino, Phys. Chem. Chem. Phys., 2016, 18, 550–556 RSC.
- A. Vargas-Caamal, F. Ortiz-Chi, D. Moreno, A. Restrepo, G. Merino and J. L. Cabellos, Theor. Chem. Acc., 2015, 134, 16 Search PubMed.
- F. Murillo, A. Vargas-Caamal, S. Pan, J. L. Cabellos, M. J. Mora-Fonz, A. Muñoz-Castro, A. Restrepo and G. Merino, Phys. Chem. Chem. Phys., 2017, 19, 17088–17093 RSC.
- A. Vargas-Caamal, E. Dzib, F. Ortiz-Chi, A. Restrepo and G. Merino, ChemPhysChem, 2023, 24, e202200582 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. B.01, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. A. Elvidge and L. M. Jackman, J. Chem. Soc., 1961, 1, 859–866 RSC.
- T. Saue and H. J. A. Jensen, J. Chem. Phys., 2003, 118, 522–536 CrossRef CAS.
- T. Saue, R. Bast, A. S. P. Gomes, H. J. A. Jensen, L. Visscher, I. A. Aucar, R. Di Remigio, K. G. Dyall, E. Eliav, E. Fasshauer, T. Fleig, L. Halbert, E. D. Hedegård, B. Helmich-Paris, M. Iliaš, C. R. Jacob, S. Knecht, J. K. Laerdahl, M. L. Vidal, M. K. Nayak, M. Olejniczak, J. M. H. Olsen, M. Pernpointner, B. Senjean, A. Shee, A. Sunaga and J. N. P. van Stralen, J. Chem. Phys., 2020, 152, 204104 CrossRef CAS PubMed.
- T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- K. G. Dyall and A. S. P. Gomes, Theor. Chem. Acc., 2010, 125, 97–100 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- F. Weigend, F. Furche and R. Ahlrichs, J. Chem. Phys., 2003, 119, 12753–12762 CrossRef CAS.
- H. Childs, E. Brugger, B. Whitlock, J. Meredith, S. Ahern, D. Pugmire, K. Biagas, M. Miller, C. Harrison and G. H. Weber, Int. J. High Perform. Comput. Appl., 2012, 34(6), 676–691 CrossRef.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1874 CrossRef CAS PubMed.
- J. I. Martínez-Araya, J. Math. Chem., 2024, 62, 461–475 CrossRef.
- C. Morell, A. Grand and A. Toro-Labbé, J. Phys. Chem. A, 2005, 109, 205–212 CrossRef CAS PubMed.
- C. Morell, A. Grand and A. Toro-Labbé, Chem. Phys. Lett., 2006, 425, 342–346 CrossRef CAS.
- J. Martínez, Chem. Phys. Lett., 2009, 478, 310–322 CrossRef.
- J. I. Martínez-Araya, Front. Chem., 2022, 10, 869110 CrossRef PubMed.
- P. Schwerdtfeger, M. Lein, R. P. Krawczyk and C. R. Jacob, J. Chem. Phys., 2008, 128, 124302 CrossRef PubMed.
- H. Fliegl, D. Sundholm, S. Taubert, J. Jusélius and W. Klopper, J. Phys. Chem. A, 2009, 113(30), 8668–8676 CrossRef CAS PubMed.
- P. v R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- R. G. Parr, L. v Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- J. L. Gázquez, A. Cedillo and A. Vela, J. Phys. Chem. A, 2007, 111, 1966–1970 CrossRef PubMed.
- P. K. Chattaraj, A. Chakraborty and S. Giri, J. Phys. Chem. A, 2009, 113, 10068–10074 CrossRef CAS PubMed.
- W. Yang and R. G. Parr, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 6723–6726 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp01112d |
| This journal is © the Owner Societies 2025 |