
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the binding free energy landscape of intrinsically disordered protein–protein interactions: insights into the AF9–BCOR complex implicated in leukemia†
Shilpa
Sharma
 and
Arjun
Saha
*
and
Arjun
Saha
*
Department of Chemistry and Biochemistry, University of Wisconsin-Milwaukee, Milwaukee, Wisconsin 53211, USA. E-mail: sharma69@uwm.edu; saha6@uwm.edu
First published on 23rd April 2025
Abstract
Chromosomal rearrangements involving the mixed-lineage leukemia (MLL) gene are implicated in acute leukemias with poor prognosis. In MLL-rearranged leukemias, the aberrant recruitment of transcriptional and epigenetic modifier complexes is driven primarily by the MLL–AF9 fusion protein. AF9 typically inhibits transcription by recruiting BCL-6 corepressor (BCOR); however, the direct fusion of AF9 with MLL results in a loss of context dependence in AF9 recruitment and causes oncogenic transformation of hematopoietic cells. Notably, the E531R mutation in AF9, which disrupts the binding between the MLL–AF9 fusion protein and BCOR, abrogates the leukemogenic potential in a mouse model, underscoring its significance as a therapeutic target in leukemia. AF9 and BCOR interact through their intrinsically disordered regions (IDRs), which undergo conformational folding upon complex formation. Understanding this conformational transition is critical for guiding drug discovery efforts but interactions mediated by IDRs remain challenging to study due to their dynamic nature. We propose a hybrid method by combining conventional and replica exchange molecular dynamics (REMD) simulations, to investigate the binding free energy landscape (BFEL) of wild type (WT) and mutant (MT) AF9–BCOR complexes. REMD simulations of WT AF9 alone revealed a significant loss of β-sheets and mutation accelerated the rate of β-sheet disappearance due to the formation of non-native contacts. BFEL of WT AF9–BCOR complex exhibited several local minima, highlighting C-terminal BCOR interactions as potential target for therapeutic intervention. Mutation disrupted the native interactions in AF9–BCOR complex and showed poor binding affinity. Our study uncovers the interaction dynamics of AF9–BCOR and introduces an innovative approach for mapping protein–protein interaction energy landscapes, offering valuable insights to advance targeted drug design.
Introduction
Proteins are the fundamental building blocks of cells, orchestrating nearly all biological processes through their diverse functions. Many of these functions, including vital processes such as cell signaling, transcriptional regulation, and immune response, are mediated by protein–protein interactions (PPIs).1 These interactions are crucial for maintaining cellular homeostasis, and their dysregulation has been implicated in a wide range of diseases, including neurodegenerative disorders, cardiovascular diseases, and cancers.2,3 It is essential to study PPIs to unravel the molecular mechanisms underlying these processes and understand how their perturbation leads to diseases. Proteins participating in PPIs span a structural continuum from fully ordered to entirely disordered, with many exhibiting a combination of both. Among these, intrinsically disordered proteins (IDPs), which lack a stable three-dimensional structure in their unbound/bound state, play a pivotal role. IDPs are uniquely characterized by their structural flexibility, enabling them to adopt specific conformations upon binding to their interaction partners—a phenomenon often referred to as “folding upon binding”.4,5 This dynamic adaptability makes IDPs highly versatile, allowing a single IDP to interact with multiple partners and perform distinct roles in different cellular contexts. Consequently, intrinsically disordered proteins (IDPs) containing intrinsically disordered regions (IDRs) longer than 30 residues are highly prevalent in eukaryotic organisms, comprising approximately 44% of the human proteome.6 Studying these flexible proteins and their interactions not only enhances our understanding of cellular mechanisms but also provides valuable insights for developing therapeutic interventions targeting diseases caused by aberrant PPI.7,8 Investigating protein–protein interactions involving IDPs presents significant experimental and computational challenges. Due to their dynamic and flexible nature, IDPs often produce diffuse and overlapping signals in nuclear magnetic resonance (NMR) spectroscopy, making structural characterization difficult. Additionally, IDPs are prone to aggregation under suboptimal experimental conditions, further complicating analysis. Their inherent structural flexibility also hinders crystallization, resulting in a limited number of documented IDP structures in the RCSB Protein Data Bank. Traditional methods such as X-ray crystallography, SAXS, and FRET often capture only averaged structural features, failing to resolve diverse conformations, crucial for understanding IDPs.9 Despite these challenges, both experimental and computational advancements have contributed to the progress in this field. Techniques like non-uniform sampling (NUS) and fast NMR methods have improved data acquisition and resolution for IDP studies.10 On the computational front, the development of IDP-specific force fields9 and enhanced sampling techniques has enabled more comprehensive exploration of their conformational landscapes.11 Despite significant advancements, there remains an urgent need for more innovative approaches to fully unravel the complexity of protein–protein interactions, especially those involving intrinsically disordered regions. Static representations of protein structures are insufficient to capture the dynamic nature of macromolecular motion. Investigating protein dynamics and conformational heterogeneity is essential for deepening our understanding of biomolecular systems and developing effective therapeutic strategies for diseases.To this end, we developed a computational protocol by combining enhanced sampling simulations with equilibrium (conventional) MD simulations to characterize the binding free energy landscape (BFEL) of a protein–protein interaction mediated by two intrinsically disordered regions. We call it Mix & Match (M&M) method. The BFEL serves as a predictive map, identifying the most dominant and thermodynamically favorable binding pose for the protein–protein or protein–ligand complexes.12 The M&M method is a hybrid method which combines the power of enhanced sampling technique (replica exchange MD in this case) and computational flexibility of equilibrium simulations. It operates on the principle that by having a diverse set of conformations for both the binding partners, one can combine these conformations in all conceivable ways to generate a complete set of probable binding poses for the complex. In the present study, we applied the M&M method to investigate the AF9–BCOR complex formation, linked to leukemia. Mixed lineage leukemia (MLL) gene regulates the transcription of many genes and is also required for H3K4 trimethylation and transcriptional initiation at a subset of HOX promoters.13 Chromosomal translocations involving the MLL gene are responsible for a subset of acute leukemias characterized by poor prognosis and early relapse.14 N-Terminal domain of MLL gene can fuse with one of over 60 translocation partners.15 ANC1 Homology Domain (AHD) of AF9 fuses with N-terminal part of MLL and these fused proteins then interact with different transcription elongation factors like AF4,16,17 DOT1L18,19 or transcription repressors like BCOR and CBX820 (Fig. 1). Direct fusion of AF9 and MLL results in loss of context dependence of AF9 recruitment. MLL fusion protein cause oncogenic transformation of hematopoietic cells by constitutive recruitment of elongation factors to HOX promoters, resulting in over-expression of target genes. Direct BCOR recruitment to MLL–AF9 is essential for leukemia.20 The importance of the BCOR interaction for MLL–AF9 leukemia has implications for the development of efficacious targeted therapies for this devastating disease, particularly in light of the limited efficacy seen in initial clinical testing of enzymatic inhibitors of DOT1L.20
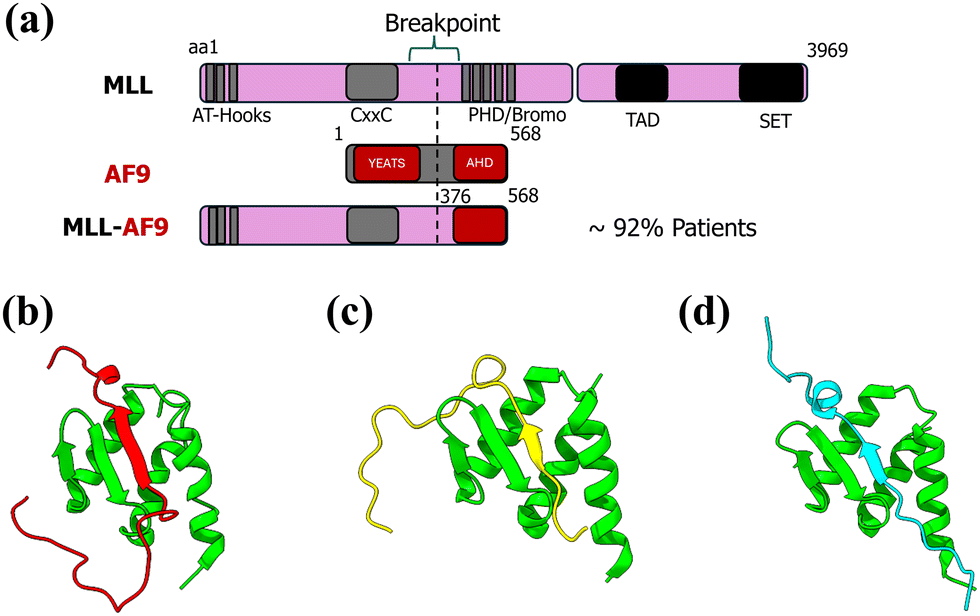 | ||
| Fig. 1 (a) Depiction of translocation of MLL and AF9 gene to form fusion MLL–AF9 gene, AF9 complex with transcription factors like (b) BCOR, (c) DOT1L, and (d) CBX8. | ||
Bushweller and colleagues17,18,20 have significantly advanced our understanding of AF9 and its interactions through over a decade of meticulous research. They have provided NMR structures of AF9 in complex with various partners, including DOT1L (PDB: 2MV718), BCOR (PDB: 6B7G20), AF4 (PDB: 2LM017), and CBX8 (PDB: 2N4Q20). These studies revealed that AF9 employs a folding-upon-binding mechanism across all these complexes, sharing a common binding interface. These interactions are mutually exclusive, highlighting the complex's dynamic nature. The AF9–BCOR complex structure reveals a critical interaction site where a β-strand of alternating hydrophobic BCOR residues forms an antiparallel β-sheet with a β-hairpin in AF9. The remaining AF9 residues fold into three helices, which wrap around the binding site, making additional contacts with the BCOR peptide and burying hydrophobic side chains. Their NMR relaxation measurements indicated that AF9 retains significant dynamic behavior even in complex, potentially facilitating exchange between disordered partners.17 The AF9–BCOR interaction plays a critical role in leukemogenesis by modulating EYA1, SIX, and MYC gene activities. Loss of direct BCOR binding to MLL–AF9 causes partial differentiation, increased proliferation, and abrogates the leukemogenic potential of the fusion protein in mouse models. Notably, the E531R mutation in AF9 selectively disrupts its binding with BCOR and was unable to induce leukemia in vivo. The mutation specifically downregulates EYA1, the most affected direct target gene.20 These findings underscore the therapeutic potential of targeting the AF9–BCOR interaction for the treatment of leukemia. Schmidt et al. reported that extended BCOR (residues 1175–1226) binds more effectively with AF9 as compared to short BCOR (residues 1175–1207, PDB: 6B7G) with a dissociation constant (KD) 32 ± 20 nM. However, due to experimental challenges, its structure could not be resolved through NMR.20 They mentioned the importance of extended C-terminal region in designing inhibitors for AF9, therefore, it is important to model the interaction of AF9 with extended BCOR (BCORext) computationally and to analyze the effect of mutation on this complex.
In this study, we employ our advanced M&M method to analyze the AF9–BCOR complex, generating a comprehensive free energy landscape for the binding of these intrinsically disordered proteins. This approach enables us to address previously unresolved questions regarding the AF9–BCOR interaction: (1) we investigate the unbound structure and dynamics of AF9, specifically examining the impact of the E531R mutation on its behavior. (2) By constructing detailed binding free-energy landscapes, we explore the conformational ensembles of both the wild-type (WT) and mutant (MT) AF9–BCOR complexes, identifying the most probable binding poses and revealing key structural insights. (3) We further analyze the WT and MT AF9 complexes in conjunction with extended BCOR, uncovering critical interactions and dynamics.
The binding free energy landscapes generated through the M&M method reveal multiple local minima, with a few predicted conformations demonstrating stronger binding affinities than the experimentally determined structure. These findings provide unprecedented insights into the AF9–BCOR interaction, uncovering essential contributing interactions that can guide the rational design of small molecule or peptide inhibitors, paving the way for targeted therapeutic interventions.
Methods
Preparation of systems
The starting structure of the AF9 (AHD)–BCOR complex was obtained from RCSB (PDB entry: 6B7G20) and the individual proteins were obtained from the complex structure by extracting the coordinates of only AF9 (AHD) or BCOR. For convenience, AF9 (AHD) will be referred to as AF9. The E531R AF9 mutant was prepared using CHARMM-GUI.21 All the simulation files for WT and MT AF9 were generated through CHARMM-GUI.For preparing the AF9 complex with extended BCOR, the residues 1208–1226 of BCOR were modelled using ModLoop tool of MODELLER.22,23
Preparation of AF9–BCOR artificial complexes
Two protocols under M&M method were used to generate all the artificial complexes. In the first protocol, the conformation of BCOR was kept exactly same as it was observed in the experimental complex structure (PDB: 6B7G) and in the second protocol, random conformations of BCOR were used to prepare artificial complexes. Schematic representations of the two approaches are shown in Fig. 2 and 3. The details of two protocols are as follows: | ||
| Fig. 2 Schematic representation of Protocol 1 for the preparation of AF9–BCOR artificial complexes. | ||
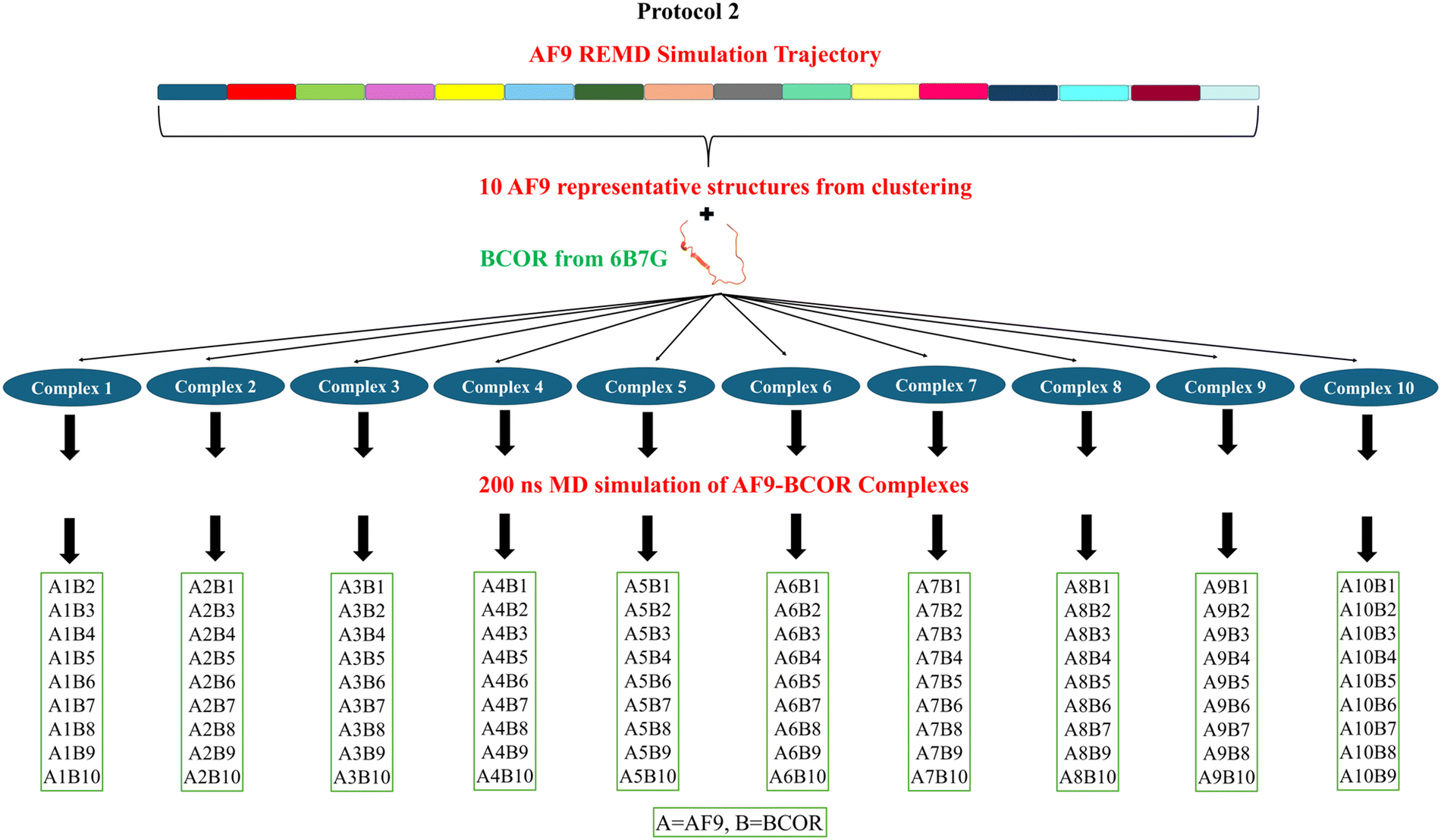 | ||
| Fig. 3 Schematic representation of Protocol 2 for the preparation of AF9–BCOR artificial complexes. | ||
Protocol 1: first, Cα based RMSD clustering was performed on WT AF9 alone REMD simulation trajectory at every 5 ns. The representative structure of AF9 was picked from the most populated cluster and aligned with the experimentally determined structure (PDB: 6B7G) to prepare the artificial complexes. The structure of BCOR conformation was kept the same. In some complexes minor transformations in the structures were done to resolve any bad contacts (clashing of atoms). A total of 40 artificial complexes were prepared using this method; their structures are represented in Fig. S1 (ESI†).
Likewise, the artificial complexes of MT AF9–BCOR were prepared by extracting the MT AF9 structures from the REMD simulation trajectory at every 25 ns interval through clustering analysis. By keeping the BCOR conformation same, 8 artificial MT complexes were prepared by aligning them with WT NMR complex.
Protocol 2: by following the same clustering approach in Protocol 1, the AF9 trajectory was clustered at 10 ns intervals providing 20 representative structures of AF9. Out of these 20, 10 structures were picked that are significantly different. These structures were used to prepare the complexes with BCOR as mentioned in Protocol 1, and the prepared complexes were simulated using equilibrium molecular dynamics for 200 ns. The complexes were named as complex 1, complex 2, … and so on, which consisted of AF9 1-BCOR 1, AF9 2-BCOR 2, … and so on. Now additional complexes were prepared out of these ten simulated complexes again using mix and match (M&M) method. Here, the artificial complexes were prepared by combining AF9 1 with BCOR 2, 3, …, 10, AF9 2 with BCOR 1, 3, …, 10, AF9 3 with BCOR 1, 2, 4, …, 10 and so on. The complexes were prepared by aligning them with WT AF9–BCOR complex NMR structure (PDB: 6B7G), structural transformations were performed to remove clashes. The second approach yielded 100 AF9–BCOR artificial complexes. This protocol assigns relevant flexibility to the BCOR structure as well by extracting their random conformations.
All the prepared artificial complexes along with WT (NMR) AF9–BCOR complex were used as initial structures to perform equilibrium molecular dynamics simulations.
Simulation details
Replica Exchange molecular dynamics (REMD) simulation to generate the representative structures of unfolded, unbound AF9
The simulation files for REMD simulations were prepared using CHARMM-GUI21 and simulations were performed using GROMACS-2023.24 For obtaining the bonded and non-bonded parameters for AF9 protein, CHARMM36m force field25 was chosen. The CHARMM36m force field has been shown to perform well in simulations of intrinsically disordered proteins (IDPs), accurately reproducing both structured and unstructured regions of the protein.6,26,27 The protonation states of amino acids were adjusted according to physiological pH (pH = 7.4). The protein was placed at the center of a cubic box ensuring a 1.0 nm distance between protein atoms and the box edge. The box was solvated using the TIP3P water model28 which added approximately 7700 water molecules. The charge on the protein was neutralized by adding 0.15 M sodium chloride. After neutralization, energy minimization was carried out using 5000 steps of the steepest descent method to remove any unfavorable contacts from the protein system. Minimized structure was equilibrated in two steps: first, under NVT ensemble for 500 ps to maintain a constant temperature (303.15 K), followed by equilibration under NPT ensemble for 20 ns to maintain a constant pressure (1 bar). The v-rescale method29 was used for temperature maintenance and c-rescale method30 was used for pressure coupling. The equilibrated structure was simulated using replica exchange molecular dynamics (REMD) method at different temperatures in the range 303.15–400 K. Each replica was simulated for 200 ns at its respective temperature. The exchange probability was kept at 0.2 which resulted in 44 replicas distributed in the given temperature range. Exchange of replicas between adjacent replicas was attempted at every 100 steps. CHARMM-GUI uses an algorithm developed by Patriksson and van der Spoel31 to generate equally distributed replicas within a given temperature range to ensure efficient sampling and optimal exchange probabilities during replica exchange molecular dynamics (REMD) simulations. The algorithm requires information about the lower and upper temperature limits, exchange probability, and the number of atoms in the system. It also assumes the energy distribution to be Gaussian. Particle Mesh Ewald (PME) method32 was used for long-range electrostatics and a cut-off of 1.2 nm was selected for both electrostatics and van der Waals interactions. For constraining the bonds involving H-atoms LINCS method33 was utilized, and periodic boundary conditions (PBC) were applied in all the three directions. 2 fs timestep was used for the simulations. After the simulation, replica at 303.15 K was chosen for further analysis. We have also run the REMD simulation by varying the temperature range 303.15–450 K and 303.15–600 K. Additionally, we examined the effect of varying the acceptance ratio from 0.2 to 0.4 and effect of force field AMBER99SB-Disp with TIP4P-D water model.34 REMD simulation of MT AF9 was carried out similar to WT AF9.Equilibrium molecular dynamics simulations
The equilibrium or conventional all-atom molecular dynamics (cMD) were performed using GROMACS-202324 package. CHARMM36m force field was used for protein parameters. All AF9–BCOR complexes were prepared using the same protocol as implemented in the REMD simulations. All other simulation parameters were kept consistent with those used in the REMD setup. After monitoring the convergence of temperature, pressure, density etc., the complexes were simulated for 200 ns under NPT ensemble. 2 fs timestep was used for the simulations. MT complexes were simulated for 500 ns and WT/MT complexes with extended BCOR were simulated for 400 ns.Analysis of trajectories
Molecular mechanics-generalized born surface area (MM-GBSA) method for binding free energy calculations
MM-GBSA method was employed for calculating the binding free energies of all the simulated AF9–BCOR complexes. MM-GBSA is an end-point method to calculate the binding free energy from the single trajectory of solvated protein–ligand/protein complex.35 MM-GBSA method is very useful for ranking large number of complexes as it is computationally less expensive. There are more accurate methods available for the absolute binding free energy calculations, but their usability becomes questionable for larger systems like protein–protein, protein–RNA complexes and when there are hundreds of complexes to screen. To calculate the binding free energy of complexes gmx_MMPBSA software36 was used which uses AMBER's cpptraj and MMPBSA.py tools. Cpptraj first converts the files from GROMACS format to AMBER format and then MMPBSA.py script calculates the binding free energy according to the input parameters. Binding free energy was calculated according to the thermodynamic cycle given in Scheme S1 (ESI†).For binding free energy calculations, 100 frames were selected from the last 50 ns of the trajectory, contribution of each residue of complex was also calculated for the last 50 ns. The average value and standard deviation across these frames were reported. Prior to running calculations with gmx_MMPBSA, periodic boundary conditions (PBC) were removed from the GROMACS output trajectory. The binding free energy of a complex is be given as:
| ΔGbind = Gcomplex − (Greceptor − Gligand) | (1) |
| ΔGbind = ΔEMM + ΔGsolvation− TΔS | (2) |
The CHARMM36m25 force field was used to compute the molecular mechanics energy term (ΔEMM), which is the combination of bonded (bonds, angles, dihedrals) and non-bonded (van der Waals and electrostatic) energy terms in the gas phase:
| ΔEMM = (ΔEbonds + ΔEangle + ΔEdihedral) + ΔEvdW + ΔEelectrostatic | (3) |
| ΔGsolvation = ΔGpolar + ΔGnon-polar | (4) |
The polar component of the solvation free energy (ΔGpolar) was estimated using the GB/OBC (Generalized Born/Optimized Born Chain) (igb = 5),37 while the non-polar solvation free energy (ΔGnon-polar) was calculated using the following equation:
| ΔGSA = γ·ΔSASA + β | (5) |
Here, ΔSASA denotes the change in solvent-accessible surface area upon complex formation, with empirical constants γ = 0.0072 kcal Å−2 mol−1 and β = 0, as defined for the GB models.37 Entropic contributions were not included in this study, as we are comparing relative binding free energies within the same protein–protein system. Accordingly, the total binding free energy can be expressed as the sum of the individual energy components:
| ΔGbind = (ΔEbonds + ΔEangle + ΔEdihedral) + ΔEvdW + ΔEelectrostatic + ΔGpolar + ΔGnon-polar | (6) |
Construction of binding free energy landscapes (BFEL)
Initially we had 141 WT complexes including the NMR predicted structure and 8 complexes for the MT AF9–BCOR. Simulation of some of the artificial complexes of WT resulted in a structure very different from the experimental structure and for the same reason those complexes were discarded. After filtering out the bad complexes, 107 WT complexes, and 8 MT complexes were chosen for the construction of binding free energy landscape (BFEL). The total and βAF9–βBCOR binding free energies for all the prepared complexes were obtained from MM-GBSA method as mentioned earlier. For βAF9, residues 541–548 of AF9 and for βBCOR, residues 1197–1204 of BCOR were selected. We selected root mean square deviation (RMSD) and radius of gyration (Rg) as reaction coordinates for constructing the protein–protein binding free energy landscape, as they are robust and widely accepted structural descriptors that effectively capture essential aspects of conformational dynamics.38,39 RMSD quantifies the overall deviation of a given conformation from a reference structure, providing insight into global structural transitions, while Rg reflects the compactness or expansion of the complex, highlighting folding or unfolding events. By using these coordinates, we were able to project the conformational ensemble sampled during MD simulations onto three and two-dimensional space that meaningfully represents the structural heterogeneity of the binding interface. RMSD and Rg values were calculated with respect to the experimental structure. Their average values were calculated for the same number of frames, i.e. 100 frames from last 50 ns, for which binding free energy were calculated. A three-dimensional (3D) binding free energy landscape was constructed as a function of average Cα RMSD and complex Rg values. Two dimensional (2D) free energy diagram of WT and MT AF9–BCOR complexes was plotted as a function of RMSD and Rg by using the Boltzmann eqn (7) to calculate the free energy values, using a python script.ΔG = −kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(Pi/Pref) ln(Pi/Pref) | (7) |
Calculation of structural properties
To evaluate structural changes, various structure-based properties were calculated, and interaction-related properties were analyzed to monitor the interplay between AF9 and BCOR. Root mean square deviation (RMSD) was used to quantify structural changes over time relative to a reference structure. For this, the Cα atoms of the protein in each frame were aligned to the Cα atoms of the first frame. To calculate the residue wise fluctuations, root mean square fluctuations (RMSF) were calculated for the last 25 ns trajectory. Radius of gyration (Rg) provides a means to measure the protein compactness, it was calculated for all the atoms in protein. Hydrogen bonds (H-bonds) were identified based on distance and angle criteria, where the donor–acceptor distance was ≤0.35 nm, and the acceptor–donor–hydrogen bond angle was ≤30°. Clustering analysis was performed using the linkage method, which grouped structures based on RMSD. Structures with an RMSD ≤ 0.15 nm were assigned to the same cluster, and the mid-structure of the most populated cluster was used to prepare artificial complexes. Secondary structure analysis was conducted using the DSSP (Dictionary of Secondary Structure in Proteins) algorithm implemented in GROMACS. For distance analysis, the center-of-mass distance between selected residues was calculated. Native contacts were calculated with gmx mindist command in GROMACS. Visualization of protein structures was done using UCSF ChimeraX,40 while data plotting and binding free energy landscape construction were carried out using Origin 202441 and Matplotlib.42 To smoothen the free energy landscapes, the 3D smoother tool in Origin was utilized.Results and discussion
The structure of the AF9 (AHD) in its unbound state remains unresolved due to experimental limitations. To address this, we hypothesize that starting from the bound state structure will enable us to derive a conformation closely resembling its unbound state. To achieve this, we employed replica exchange molecular dynamics (REMD) simulations, which enhance the sampling of conformational states by simulating multiple copies of the system at varying temperatures and allowing frequent exchanges between conformations. The success of this approach depends on several critical factors, including the selection of an appropriate temperature range, the number of replicas, exchange probability, exchange frequency, and force field parameters. To identify the optimal temperature range, REMD simulations of wild-type AF9 were performed across three temperature ranges: (1) 303.15–400 K, (2) 303.15–450 K, and (3) 303.15–600 K. Additionally, for determining the ideal exchange probability, simulations were performed with 0.2 and 0.4 exchange probabilities within the 303.15–400 K temperature range. The reliability of different force fields, specifically CHARMM36m and AMBER99SB-DISP, was also assessed for comparison.Despite varying parameters such as temperature, exchange probability, and force field, the sampled conformations remained consistent. Consequently, to investigate the structural behavior of WT and MT AF9, we selected the temperature range of 303.15–400 K, a 0.2 exchange probability, and utilized the CHARMM36m force field, as per standard practice. The complete overlap of the probability density profiles for the potential energies of all simulated replicas of both WT and MT AF9 demonstrates the successful convergence of the REMD simulations (Fig. S2, ESI†).
Replica exchange molecular dynamics (REMD) simulations of the AF9 (AHD) highlight the varying stability of its secondary structure and effect of mutation on its stability
The structural changes in WT and MT AF9 were monitored by calculating root mean square deviation (RMSD) of Cα atoms throughout the simulation (Fig. 4(a)). The RMSD of Cα atoms in WT AF9 exhibited an initial rise, followed by fluctuations between 25–50 ns, then a gradual decrease around 75 ns, continuing to decline until 125 ns. A subsequent increase in RMSD at 125 ns was followed by a slight decrease at 175 ns, indicating significant structural rearrangements in WT AF9. For the E531R mutant (MT) AF9, the RMSD increased gradually until 60 ns, followed by a sharp rise between 60–75 ns, before stabilizing for the remainder of the trajectory. Notably, MT AF9 exhibited a consistently higher RMSD compared to the WT variant, highlighting the distinctive structural dynamics of the mutant form.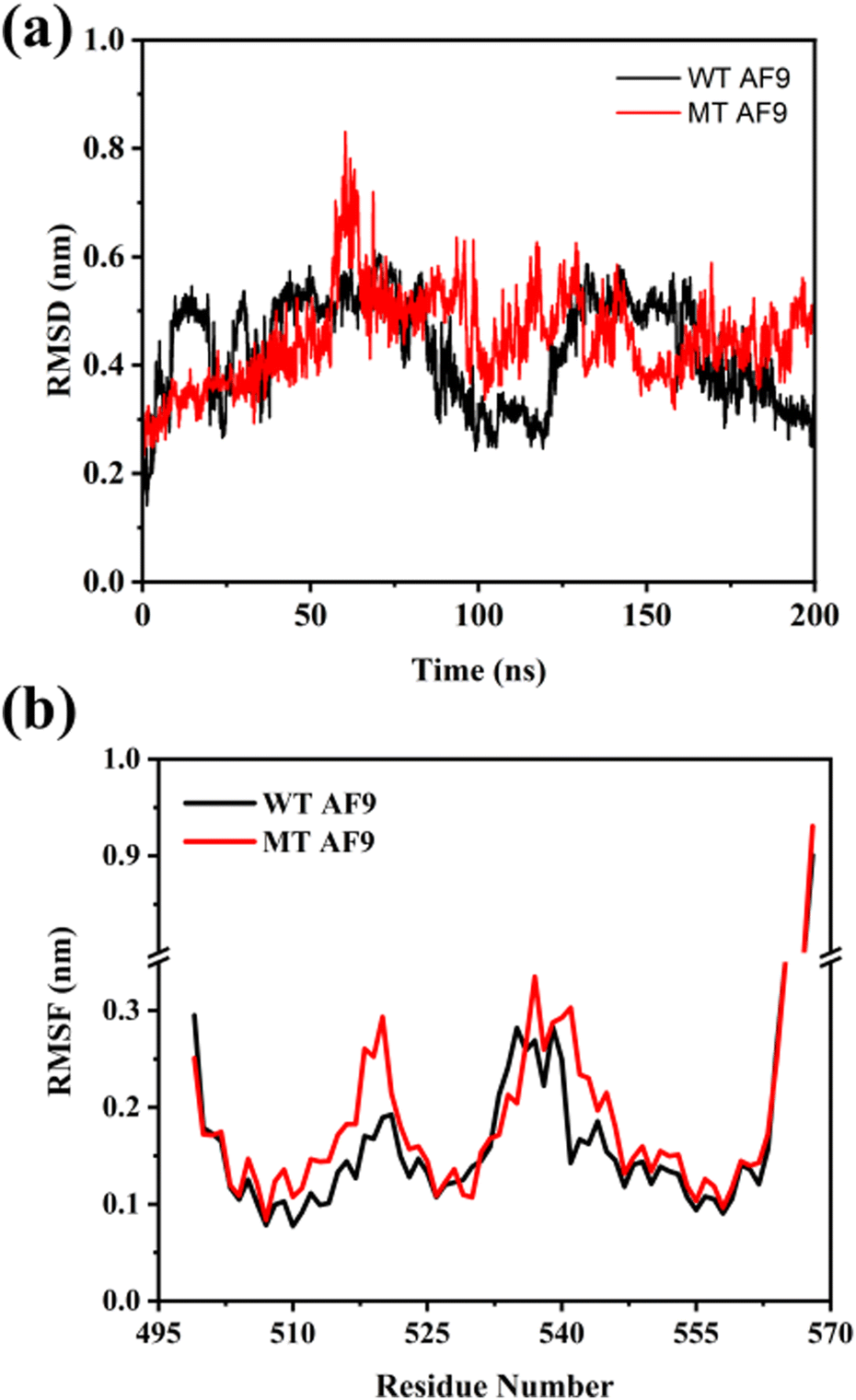 | ||
| Fig. 4 Comparison of (a) Cα root mean square deviation (RMSD) over entire simulation time and (b) Cα root mean square fluctuation (RMSF) for the last 25 ns of WT and MT AF9 simulations. | ||
Residue-wise fluctuations in WT and MT AF9 were analyzed using root mean square fluctuation (RMSF) plots for the last 25 ns of the trajectory (Fig. 4(b)). Fig. S3 (ESI†) shows the labeled secondary structure components of AF9 and BCOR complex. Both WT and MT AF9 displayed flexibility in the regions 507–525 and 535–545, along with pronounced fluctuations at the N- and C-termini. Importantly, residues 507–514 form the α2 helix, while residues 537–539 and 542–546 form β1 and β2 sheets, respectively, in the bound conformation of AF9. Enhanced fluctuations in these secondary structure elements, particularly α2 and β2, were observed in MT AF9, suggesting its destabilizing effect.
To further examine the evolution of secondary structure of WT and MT AF9, the DSSP algorithm was used. For WT AF9, the β-sheets (β1 and β2) dissolved into random coils around 30 ns, while the four major α-helices remained stable throughout the simulation (Fig. S4(a), ESI†). In contrast, MT AF9 exhibited an accelerated dissolution of β-sheets, occurring around 15 ns, nearly twice as fast as in WT AF9 (Fig. S4(b), ESI†). Additionally, perturbations were observed in α3, the helix carrying the E531R mutation. Quantitative analysis of secondary structure content revealed a faster reduction in β-sheet content for MT AF9 (Fig. 5(a)). Both WT and MT AF9 displayed a transient decrease in α-helix content between 40–75 ns, with no significant changes exhibited thereafter (Fig. 5(b)). However, MT AF9 exhibited a slight overall reduction in α-helix content compared to WT, consistent with observations in DSSP plot (Fig. S4, ESI†). The disappearance of β-sheets upon BCOR removal underscores their intrinsically disordered nature, suggesting AF9 adopts an ordered conformation only when bound to BCOR. Conversely, the stability of the α-helices highlights their rigidity, which may provide structural support during functional transitions. The intrinsic disorder of AF9 likely facilitates its ability to adapt and interact with multiple partners, while the rigidity of the α-helices ensures a stable framework for these structural rearrangements. The E531R mutation not only accelerates β-sheet dissolution but also induces subtle perturbations in α3 helix, potentially affecting the functional dynamics of AF9.
 | ||
| Fig. 5 Comparison of secondary structure content (a) β-sheet and (b) α-helix formation with time in WT and MT AF9. | ||
To further investigate the structural heterogeneity of WT and MT AF9, clustering analysis was performed at 10 ns intervals (Fig. S5(a), ESI†). WT AF9 exhibited increased cluster formation between 40–50 ns, 70–90 ns, and 160–170 ns, corresponding to the increased RMSD fluctuations observed in these intervals. In contrast, MT AF9 showed higher cluster formation during 40–70 ns, 90–120 ns, and 160–170 ns. Overall, both WT and MT AF9 demonstrated structural heterogeneity, with a general trend indicating that a higher number of clusters at a given time interval corresponds to a lower population in the most dominant cluster (Fig. S5(b), ESI†).
To examine more closely the tertiary structure of WT and MT AF9 in the absence of BCOR, the most representative structures at 10 ns intervals were extracted through clustering analysis and visualized (Fig. 6). In WT AF9, the secondary structure remained intact up to 10 ns, after which the β-sheets began to dissolve, completely disappearing by 40 ns. This dissolution aligns with RMSD fluctuations observed between 25–50 ns. The resulting loop, formed from the dissolved β-sheets, adopted diverse conformations, while the C-terminal residues exhibited significant fluctuations throughout the simulation. These RMSD changes are primarily attributed to perturbations in the β-sheet-turned-loop region and the C-terminal residues. In MT AF9, β-sheet dissolution occurred earlier, between 10–20 ns, followed by major conformational changes in the resulting loop. After 80 ns, the α3 helix displayed partial unfolding, and the C-terminal residues showed pronounced perturbations throughout the simulation. Consistent with earlier analyses, visualization of the tertiary structures confirmed that in its unbound state, AF9 adopts a disordered conformation in regions involved in interface formation. The mutation not only accelerated the transition to random conformations but also induced specific structural changes in the α3 helix. The comparison of the aligned representative structures of WT and MT AF9, from the last 5 ns, indicated a noticeable difference in the loop formed due to disappearance of β-sheets and also in α3,4 helices (Fig. S3, ESI†). Thus, mutation causes a change in the secondary and tertiary structures of AF9 by affecting the loop and alpha helices. Leach et al. conducted CD experiments to evaluate the secondary structure of AF9, revealing that, in isolation, it is predominantly a random coil with only a minor β-structure component predicted by K2D.17 However, our simulations did not fully replicate this behavior, suggesting potential limitations in experimental predictions, differences in timescales, or the influence of environmental factors not captured in the simulations.
 | ||
| Fig. 6 Conformations of WT and MT AF9 obtained from the clustering analysis of REMD simulations performed at 303.15–400 K. | ||
To validate the reliability of our simulations, we compared the results from multiple REMD simulations performed under varying conditions. Simulations across broader temperature ranges (303.15–450 K and 303.15–600 K) and with an increased exchange probability of 0.4, all yielded similar results (Fig. S6 and S7, ESI†). This confirms the lability of the AF9 β-sheets in the absence of their binding partner, while the α-helices remained rigid under all conditions. Based on these findings, we conclude that the rigidity of the α-helices in AF9 (AHD) provides structural support and facilitates interactions with different partners as required by its function, whereas the flexibility of the β-sheets enables rapid partner switching.
Non-native contacts formed by mutated AF9 lock the protein into a specific conformation
While comparing the 3D structures of WT and MT AF9, a noticeable change in the loop – resulted from the dissolution of β hairpin – was observed, which might be the result of altered interactions due to mutation (Fig. S8(a), ESI†). Therefore, to determine the interaction between residue Glu531 in WT or Arg531 in MT and neighboring residues, the distance between residue 531 and center-of-mass of residues 535–541 was monitored, as shown in Fig. 7. In WT AF9, the distance remained constant throughout the simulation length, with average distance ∼0.68 nm. Unlike WT AF9, the observed distance in MT AF9 decreased to ∼0.50 nm after 60 ns, which suggested the formation of intramolecular interaction between Arg531, and neighboring residues present in loop. | ||
| Fig. 7 Distance between residue 531 and centre-of-mass of residues 535–541 in WT and MT AF9. | ||
To look more closely on the type of interaction present, we calculated the hydrogen bonds between residue 531 and residues 535–541 (Fig. S9, ESI†).
Residue 531 showed hydrogen bonding with residues 536 and 537 in WT and MT AF9, however, the frequency of hydrogen bond formation is very less in WT AF9. Additionally, one more hydrogen bond was present between residue 531 and residue 540 in MT AF9, which remained absent in WT AF9 (Fig. S8(b), ESI†).
The interaction analyses suggest that the underlying reason of difference in the tertiary structures of WT and MT AF9 can be attributed to interaction between the mutated residue and loop residues. These additional interactions have the potential to lock the MT AF9 structure in a particular conformation, rendering it incapable of forming native interactions with BCOR. It also explains the early dissolution of β-sheets in MT AF9.
Binding free energy landscape (BFEL) of WT AF9–BCOR complex shows multiple thermodynamically stable structures
As detailed in the methodology section, the binding free energy landscape (BFEL) of the WT AF9–BCOR complex was constructed based on the total binding free energy and the βAF9–βBCOR interaction energy (Fig. 8 and Fig. S10, ESI†). Fig. S11 (ESI†) presents the energy landscapes with raw data points shown. A total of 140 artificial AF9–BCOR complexes, generated using Protocols 1 and 2, were simulated alongside the experimentally determined NMR complex. After the simulation, we visually inspected the complexes and excluded any that looked very different from the WT NMR complex, as they seemed overly artificial. This refinement process resulted in 107 complexes being retained for BFEL analysis. Table S1 (ESI†) lists the average binding energies of selected complexes along with their respective RMSD and Rg values. The BFEL plot for total energy (Fig. 8(a)) showed that about one-third of the complexes had poor binding energies between −55 to −26 kcal mol−1 (in red), with noticeably higher RMSD and Rg values compared to the NMR structure, suggesting more extended structures. Most complexes had binding energies between −70 to −55 kcal mol−1 (in green), while very few had greater binding energies lower than −80 kcal mol−1 (in blue and purple). | ||
| Fig. 8 Binding free energy landscape (BFEL) of the wild-type (WT) AF9–BCOR complex, constructed based on the total binding free energy calculations. (a) 3D BFEL and (b) 2D BFEL. The Z-axis and color scale represent the binding free energy values (calculated via MM-GBSA) for all the structural complexes between WT-AF9 and WT-BCOR. These complexes are denoted as RAF9n·RBCORn, where n = 1 to 107. Here, RAF9n and RBCORn refer to the coordinates generated through Protocols 1 and 2 (see Methods). The X- and Y-axes represent the radius of gyration (Rg) and root-mean-square deviation (RMSD) of each complex. Arrows indicate the secondary structures of four minimum-energy conformations (including the experimental structure), identified through clustering of the final 25 ns of simulation. | ||
The WT complex BFEL revealed three key energy minima. The first minimum was located at RMSD 1–1.2 nm and Rg 1.6–1.7 nm, while the second and third minima appeared between RMSD 1.3–1.5 nm and Rg 1.8–2.0 nm. These minima corresponded to complexes with better binding free energy than the experimentally determined structure. The BFEL also showed several local minima with significant energy barriers, highlighting the complexity of the energy landscape. The most thermodynamically stable structures, Conf 1, Conf 3, and Conf 4, were marked on the 2D BFEL plot (Fig. 8(b)) for further analysis. Conf 1 suggested compact structures similar to the experimental structure (Conf 2), while Conf 3 and Conf 4 displayed higher RMSD and Rg values, with structural differences in their bound conformations.
To elucidate the molecular basis of the enhanced interactions observed in Conf 1, Conf 3, and Conf 4 compared to the NMR-derived structure (Conf 2), we calculated the per-residue contributions to the binding free energy (Fig. 9). The analysis revealed that in Conf 1, residue Ser549 of AF9 and residues Asn1177, Ser1180, and Glu1184 of BCOR contributed significantly more to the binding affinity than in Conf 2, leading to an overall increase in binding energy. Similarly, in Conf 3, Asp546 and Leu547 of AF9, along with Ser1180, Glu1184, Leu1191, Val1196, and Ile1198 of BCOR, demonstrated enhanced contributions. In Conf 4, Asp546, Leu547, and Cys548 of AF9, together with Leu1191, Asn1193, Leu1194, Lys1195, Val1195, Cys1197, Ile1198, Glu1199, Gly1202, His1204, and Pro1205 of BCOR, exhibited energetically more favorable interactions compared to Conf 2. A key structural feature in Conf 3 and Conf 4 was the reduction of the β-hairpin of AF9 to a single β-sheet, which facilitated more effective interactions with BCOR. This structural reorganization was reflected in the increased contributions from residues 546–548 of AF9 and multiple C-terminal residues of BCOR. These findings provide critical insights for rational inhibitor design.
 | ||
| Fig. 9 Per-residue contribution to the total binding free energy of AF9–BCOR complex in minimum energy structures (Conf 1–Conf 4) obtained from BFEL. Additional interactions formed in Conf 4 are highlighted by circle. | ||
To further investigate the binding interactions between the two primary interacting chains, we generated the binding free energy landscape (BFEL) for the βAF9–βBCOR interaction energy (Fig. S10, ESI†). The BFEL revealed a single dominant energy minimum, corresponding to Conf 1, which closely resembled Conf 2 (the NMR structure). Notably, most complexes exhibited poor binding energies (red regions), indicating that many structures with moderate interaction energies in the full interaction BFEL were likely engaged in non-native interactions. This suggests that only a subset of conformations exhibit optimal binding stability. The BFEL highlights the critical role of the AF9 interaction with C-terminal region of BCOR in efficient complex formation. These regions should be strategically targeted to design high-affinity AF9 binders, potentially leading to more potent inhibitors that effectively disrupt the AF9–BCOR complex.
Mutant complex binding free energy diagram shows decreased binding between AF9 and BCOR
To understand the impact of E531R mutation on AF9–BCOR complex formation, the binding energies of 8 artificial mutant complexes were calculated and compared with that of WT AF9–BCOR complex (Fig. 10). The MT complexes showed decreased binding energies as compared to WT complexes; no binding was observed when the interaction of βAF9–βBCOR was calculated (Table S2, ESI†). This observation corroborates well with the experimental findings20 where it was observed that E531R mutation impede the AF9 interaction with BCOR. | ||
| Fig. 10 2D-Binding free energy diagram of MT AF9–BCOR interaction constructed by calculating the total binding free energy (black) and βAF9–βBCOR interaction energy (red) of artificial complexes of MT AF9–BCOR. | ||
The contribution of each residue to the βAF9–βBCOR binding was also calculated for WT (NMR) and MT (complex 8, prepared with MT AF9 structure between 175–200 ns) AF9–BCOR complex (Fig. S12, ESI†). The energy contribution of each residue to the energy was very low in MT complex as compared to the WT complex which confirms the destabilizing effect of mutation on the complex formation.
To shed more light on the structural and dynamical differences between the wild-type (WT, NMR structure) and MT AF9–BCOR complexes, we analyzed key structural properties over time. The comparison of root mean square deviation (RMSD) and radius of gyration (Rg) (Fig. S13(a) and (b), ESI†) revealed greater fluctuations in the MT complex, suggesting increased structural instability. Particularly, a sudden decrease in RMSD and Rg values between 75–100 ns indicated a collapse of the MT complex structure, likely due to altered intermolecular interactions. Further analysis revealed that the MT complex exhibited a significantly lower number of native contacts between AF9 and BCOR, along with a reduced formation of hydrogen bonds within the βAF9–βBCOR interface compared to the WT complex (Fig. S13(c) and (d), ESI†). These findings suggest that the E531R mutation weakens the AF9–BCOR interaction, likely by disrupting native stabilizing interactions and impairing the formation of key hydrogen bonds.
We also generated the 2D free energy landscape of WT and MT complexes with respect to RMSD and Rg values (Fig. S14, ESI†) and visualized the minimum energy structures. The WT complex exhibited a broad distribution of lower energy conformations with very small energy barrier, visualization of the most stable structure suggested a well-defined binding conformation (Fig. S14(a), ESI†). The FEL of the MT complex (E531R mutation) displayed more dispersed and higher-energy regions compared to WT, indicating greater structural fluctuations and less stability (Fig. S14(b), ESI†). The structure of the MT complex revealed that mutant AF9 was unable to bind to BCOR in the same manner as was observed in the WT complex. This disruption was caused by the destabilizing effect of the mutation, leading to the formation of a non-native complex.
Extended BCOR forms additional contacts with the AF9 α-helix
The extended C-terminal region of BCOR was found to play a crucial role in the AF9–BCOR interaction, and the E531R mutation appeared to hinder their binding. To further investigate this effect, we focused on analyzing the interaction between residue 531 and the extended BCOR C-terminal region (residues 1208–1226) in both the WT and MT complexes. First, we examined the hydrogen bond (H-bond) formation between residue 531 and the extended BCOR C-tail (Fig. S15(a), ESI†). Initially, no H-bonds were observed in either the WT or MT complexes. However, around 75 ns, transient H-bond formation was detected in the WT complex, which became consistent after 135 ns. In contrast, no such H-bonding was observed in the MT complex, indicating that the E531R mutation disrupts potential stabilizing interactions. To assess other distance–dependent interactions such as electrostatic and hydrophobic interactions, we measured the distance between residue 531 and the center of mass of the extended BCOR C-tail (Fig. S15(b), ESI†). Until 50 ns, the distance remained stable in both the complexes. However, in the MT complex, a slight decrease in distance was observed, stabilizing around 1 nm, suggesting the absence of significant interactions. Conversely, in the WT complex, a sudden decrease in distance was evident, reaching a stable value of approximately 0.25 nm after 300 ns, indicating strong interactions between residue 531 and the extended BCOR C-tail. Further, we calculated the distances between residue 531 and individual residues 1200–1226 in both WT and MT complexes. In the WT complex, residue 1206 exhibited close interaction with residue 531, whereas in the MT complex, no such interaction was observed (Fig. S15(c), ESI†). This suggests that the E531R mutation disrupts critical residue-specific interactions necessary for stable AF9–BCOR binding.To further investigate the orientation of the extended C-tail of BCOR, we visualized the final frame of the simulation (Fig. 11). Starting from the same initial complex structure (Fig. 11(a)), in the WT AF9–BCORext complex, the extended C-tail of BCOR was observed to wrap around the α2 helix of AF9, facilitated by favorable electrostatic interactions between oppositely charged residues (Fig. 11(b)). In contrast, in the MT AF9–BCORext complex, the extended BCOR C-tail adopted a different orientation, moving away from AF9 due to electrostatic repulsion between the mutated Arg531 and the charged residues of the C-tail (Fig. 11(c)). These findings align well with experimental observations, highlighting the critical role of the BCOR C-terminal tail in stabilizing AF9–BCOR complex formation.
 | ||
| Fig. 11 Visualization of interaction between WT/MT AF9 and extended BCOR before and after the MD simulation. (a) Initial structure (t = 0 ns) of WT/MT AF9–BCORext complex, (b) final structure (t = 400 ns) structure of WT AF9–BCORext complex, and (c) final structure (t = 400 ns) structure of MT AF9–BCORext complex. The mutated residue 531 is shown in orange, in WT complex residue K1206 (shown in magenta) forms H-bond with E531. The extended part of BCOR i.e. residues 1208–1226 are highlighted in cyan and all other residues are highlighted in red. WT AF9 is colored in yellow and MT AF9 is colored in green. | ||
Conclusions
Understanding the complex interactions between protein and protein/peptide, especially involving intrinsically disordered region, is crucial for advancing drug discovery and therapeutic development. To address this, we developed a useful computational approach, “Mix and Match” (M&M), designed to capture the full binding free energy landscape (BFEL) of protein–protein and protein–peptide interactions. This approach explicitly accounts for the intrinsic flexibility of interacting partners, enabling a more comprehensive mapping of their binding energy profiles. We applied the M&M method to study the AF9–BCOR complex, which is linked to leukemia, and examined the effect of the E531R mutation on complex formation. We explored the structural dynamics of WT and MT AF9 with replica exchange molecular dynamics (REMD) simulations. Our findings revealed that while AF9's β-sheets exhibited significant flexibility, its α-helices remained rigid within the studied timescales. This behavior persisted even when we varied temperature ranges, exchange probabilities, and force field–water model combinations (AMBER99SB-Disp/TIP4P-D). However, we acknowledge the possibility of different behaviors emerging over much longer timescales. Mutation E531R further accelerated the dissolution of β-sheets into random structured loop. Further investigation revealed that this mutation introduced non-native contacts between Arg531 and residues Phe536, His537, and Asn540, stabilizing an alternative conformation of AF9. Using our M&M method, we constructed the binding free energy landscape (BFEL) of WT AF9–BCOR complexes by calculating both the total binding energy and βAF9–βBCOR interaction energies. The BFEL revealed four distinct lower energy binding conformations (local minima), including the experimentally determined structure (Conf 2). Conf 3 and Conf 4 emerged as the most stable structures, primarily stabilized by interactions between β1 of AF9 and the C-terminal region of BCOR, a key target previously proposed in experimental studies.20 This finding indicates that disrupting these interactions could be a promising approach for designing inhibitors of AF9–BCOR binding. The E531R mutation significantly weakened AF9–BCOR binding, leading to the formation of only non-native complexes—as indicated by a significant reduction in native contacts and hydrogen bonds. We further examined WT and MT AF9 binding to an extended BCOR structure. In the WT complex, Glu531 played a crucial stabilizing role, forming hydrogen bonds and electrostatic interactions with BCOR's C-terminal charged residues like Lys1206. In contrast, Arg531 in the MT complex repelled BCOR's extended C-terminal region due to charge repulsion, further disrupting complex formation—again corroborating well with the experimental findings. Overall, our research provides a useful method for studying protein–protein and protein–peptide interactions. The method was successfully applied to study the binding free energy landscape of AF9–BCOR complex implicated in leukemia. We propose to target the interactions created by the C-terminal of BCOR for a successful inhibition of AF9–BCOR complex formation.Author contributions
A. S. and S. S. conceived the idea. A. S. supervised the project. S. S. performed all the simulations and analyzed the data. S. S. wrote the manuscript and A. S. edited the final version for publication.Data availability
The free software tools used in this study, including CHARMM-GUI (https://www.charmm-gui.org), UCSF ChimeraX (https://www.cgl.ucsf.edu/chimerax/), and OriginPro2024 trial version (https://www.originlab.com) are freely available at their Web sites. The gmx_MMPBSA (https://valdes-tresanco-ms.github.io/gmx_MMPBSA/dev/installation/) is available to use with GROMACS 2023. All the input files required to reproduce the results and the initial as well as final structures are available for free at GitHub (https://github.com/SahaLabGitHub/AF9_BCOR).Conflicts of interest
There is no conflict of interest.Acknowledgements
The authors thank the UWM startup Fund and the UWMRF Catalyst Grant (Award number: MIL121666). The authors also thank UWM Research Computing and the UWM Department of Chemistry & Biochemistry for technical and facility support. The authors declare no competing financial interest.References
- M. P. H. Stumpf, T. Thorne, E. de Silva, R. Stewart, H. Jun An, M. Lappe and C. Wiuf, Estimating the size of the human interactome, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6959–6964 CrossRef PubMed.
- D. Ryan and J. Matthews, Protein–protein interactions in human disease, Curr. Opin. Struct. Biol., 2005, 15, 441–446 CrossRef.
- G. Kar, A. Gursoy and O. Keskin, Human Cancer Protein–Protein Interaction Network: A Structural Perspective, PLoS Comput. Biol., 2009, 5, e1000601 CrossRef PubMed.
- V. Receveur-Bréchot, J.-M. Bourhis, V. N. Uversky, B. Canard and S. Longhi, Assessing protein disorder and induced folding, Proteins: Struct., Funct., Bioinf., 2006, 62, 24–45 CrossRef PubMed.
- C. J. Oldfield, Y. Cheng, M. S. Cortese, P. Romero, V. N. Uversky and A. K. Dunker, Coupled Folding and Binding with α-Helix-Forming Molecular Recognition Elements, Biochemistry, 2005, 44, 12454–12470 CrossRef PubMed.
- M. Arai, S. Suetaka and K. Ooka, Dynamics and interactions of intrinsically disordered proteins, Curr. Opin. Struct. Biol., 2024, 84, 102734 CrossRef PubMed.
- D. E. Scott, A. R. Bayly, C. Abell and J. Skidmore, Small molecules, big targets: drug discovery faces the protein–protein interaction challenge, Nat. Rev. Drug Discovery, 2016, 15, 533–550 CrossRef CAS PubMed.
- J. A. Wells and C. L. McClendon, Reaching for high-hanging fruit in drug discovery at protein–protein interfaces, Nature, 2007, 450, 1001–1009 CrossRef CAS.
- J. Mu, H. Liu, J. Zhang, R. Luo and H.-F. Chen, Recent Force Field Strategies for Intrinsically Disordered Proteins, J. Chem. Inf. Model., 2021, 61, 1037–1047 CrossRef CAS PubMed.
- S. Maiti, A. Singh, T. Maji, N. V. Saibo and S. De, Experimental methods to study the structure and dynamics of intrinsically disordered regions in proteins, Curr. Res. Struct. Biol., 2024, 7, 100138 CrossRef CAS.
- U. R. Shrestha, J. C. Smith and L. Petridis, Full structural ensembles of intrinsically disordered proteins from unbiased molecular dynamics simulations, Commun. Biol., 2021, 4, 243 CrossRef CAS PubMed.
- F. Bai, Y. Xu, J. Chen, Q. Liu, J. Gu, X. Wang, J. Ma, H. Li, J. N. Onuchic and H. Jiang, Free energy landscape for the binding process of Huperzine A to acetylcholinesterase, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4273–4278 CrossRef.
- M. G. Guenther, R. G. Jenner, B. Chevalier, T. Nakamura, C. M. Croce, E. Canaani and R. A. Young, Global and Hox-specific roles for the MLL1 methyltransferase, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8603–8608 CrossRef PubMed.
- C. Meyer, B. Schneider, S. Jakob, S. Strehl, A. Attarbaschi, S. Schnittger, C. Schoch, M. W. J. C. Jansen, J. J. M. van Dongen, M. L. den Boer, R. Pieters, M. G. Ennas, E. Angelucci, U. Koehl, J. Greil, F. Griesinger, U. zur Stadt, C. Eckert, T. Szczepański, F. K. Niggli, B. W. Schäfer, H. Kempski, H. J. M. Brady, J. Zuna, J. Trka, L. L. Nigro, A. Biondi, E. Delabesse, E. Macintyre, M. Stanulla, M. Schrappe, O. A. Haas, T. Burmeister, T. Dingermann, T. Klingebiel and R. Marschalek, The MLL recombinome of acute leukemias, Leukemia, 2006, 20, 777–784 CrossRef CAS.
- C. Meyer, E. Kowarz, J. Hofmann, A. Renneville, J. Zuna, J. Trka, R. Ben Abdelali, E. Macintyre, E. De Braekeleer and M. De Braekeleer, New insights to the MLL recombinome of acute leukemias, Leukemia, 2009, 23, 1490–1499 CrossRef CAS.
- A. Yokoyama, M. Lin, A. Naresh, I. Kitabayashi and M. L. Cleary, A Higher-Order Complex Containing AF4 and ENL Family Proteins with P-TEFb Facilitates Oncogenic and Physiologic MLL-Dependent Transcription, Cancer Cell, 2010, 17, 198–212 CrossRef.
- B. I. Leach, A. Kuntimaddi, C. R. Schmidt, T. Cierpicki, S. A. Johnson and J. H. Bushweller, Leukemia Fusion Target AF9 Is an Intrinsically Disordered Transcriptional Regulator that Recruits Multiple Partners via Coupled Folding and Binding, Structure, 2013, 21, 176–183 CrossRef PubMed.
- A. Kuntimaddi, N. J. Achille, J. Thorpe, A. A. Lokken, R. Singh, C. S. Hemenway, M. Adli, N. J. Zeleznik-Le and J. H. Bushweller, Degree of Recruitment of DOT1L to MLL–AF9 Defines Level of H3K79 Di- and Tri-methylation on Target Genes and Transformation Potential, Cell Rep., 2015, 11, 808–820 CrossRef PubMed.
- Y. Li, H. Wen, Y. Xi, K. Tanaka, H. Wang, D. Peng, Y. Ren, Q. Jin, S. Y. R. Dent, W. Li, H. Li and X. Shi, AF9 YEATS Domain Links Histone Acetylation to DOT1L-Mediated H3K79 Methylation, Cell, 2014, 159, 558–571 CrossRef.
- C. R. Schmidt, N. J. Achille, A. Kuntimaddi, A. M. Boulton, B. I. Leach, S. Zhang, N. J. Zeleznik-Le and J. H. Bushweller, BCOR Binding to MLL–AF9 Is Essential for Leukemia via Altered EYA1, SIX, and MYC Activity, Blood Cancer Discovery, 2020, 1, 162–177 CrossRef PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, CHARMM-GUI: a web-based graphical user interface for CHARMM, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef PubMed.
- A. Fiser and A. Sali, ModLoop: automated modeling of loops in protein structures, Bioinformatics, 2003, 19, 2500–2501 CrossRef.
- A. Fiser, R. K. G. Do and A. Šali, Modeling of loops in protein structures, Protein Sci., 2000, 9, 1753–1773 CrossRef.
- P. Bauer, B. Hess and E. Lindahl, GROMACS 2022.6 Manual, 2023 Search PubMed.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmüller and A. D. MacKerell, CHARMM36m: an improved force field for folded and intrinsically disordered proteins, Nat. Methods, 2017, 14, 71–73 CrossRef PubMed.
- P. R. Banerjee, A. S. Holehouse, R. Kriwacki, P. Robustelli, H. Jiang, A. I. Sobolevsky, J. M. Hurley and J. T. Mendell, Dissecting the biophysics and biology of intrinsically disordered proteins, Trends Biochem. Sci., 2024, 49, 101–104 CrossRef PubMed.
- H. Jeong, S. Clark, A. Goehring, S. Dehghani-Ghahnaviyeh, A. Rasouli, E. Tajkhorshid and E. Gouaux, Structures of the TMC-1 complex illuminate mechanosensory transduction, Nature, 2022, 610, 796–803 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79, 926–935 CrossRef.
- G. Bussi, D. Donadio and M. Parrinello, Canonical sampling through velocity rescaling, J. Chem. Phys., 2007, 126, 014101 CrossRef.
- M. Bernetti and G. Bussi, Pressure control using stochastic cell rescaling, J. Chem. Phys., 2020, 153, 114107 CrossRef.
- A. Patriksson and D. van der Spoel, A temperature predictor for parallel tempering simulations, Phys. Chem. Chem. Phys., 2008, 10, 2073–2077 RSC.
- H. G. Petersen, Accuracy and efficiency of the particle mesh Ewald method, J. Chem. Phys., 1995, 103, 3668–3679 CrossRef.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, LINCS: a linear constraint solver for molecular simulations, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- P. Robustelli, S. Piana and D. E. Shaw, Developing a molecular dynamics force field for both folded and disordered protein states, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4758–E4766 CrossRef CAS.
- S. Genheden and U. Ryde, The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS PubMed.
- M. S. Valdés-Tresanco, M. E. Valdés-Tresanco, P. A. Valiente and E. Moreno, gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS, J. Chem. Theory Comput., 2021, 17, 6281–6291 CrossRef PubMed.
- A. V. Onufriev and D. A. Case, Generalized Born Implicit Solvent Models for Biomolecules, Annu. Rev. Biophys., 2019, 48, 275–296 CrossRef CAS PubMed.
- V. Dalal, P. Dhankhar, V. Singh, G. Rakhaminov, D. Golemi-Kotra and P. Kumar, Structure-Based Identification of Potential Drugs Against FmtA of Staphylococcus aureus: Virtual Screening, Molecular Dynamics, MM-GBSA, and QM/MM, Protein J., 2021, 40, 148–165 CrossRef CAS.
- Q. Yu, W. Ye, W. Wang and H.-F. Chen, Global Conformational Selection and Local Induced Fit for the Recognition between Intrinsic Disordered p53 and CBP, PLoS One, 2013, 8, e59627 CrossRef CAS.
- E. C. Meng, T. D. Goddard, E. F. Pettersen, G. S. Couch, Z. J. Pearson, J. H. Morris and T. E. Ferrin, UCSF ChimeraX: tools for structure building and analysis, Protein Sci., 2023, 32, e4792 CrossRef CAS.
- OriginPro, Version 2024, OriginLab Corporation, Northampton, MA, USA Search PubMed.
- J. D. Hunter, Matplotlib: A 2D Graphics Environment, Comput. Sci. Eng., 2007, 9, 90–95 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp01009h |
| This journal is © the Owner Societies 2025 |