
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO oxidation on bimetallic Re–Pt clusters: unraveling the role of oxygen coverage†
Andrés
Álvarez-García
 a,
Ignacio L.
Garzón
ab and
Luis M.
Molina
*b
a,
Ignacio L.
Garzón
ab and
Luis M.
Molina
*b
aInstituto de Física, Universidad Nacional Autónoma de México, Apartado Postal 20-364, Ciudad de México 01000, Mexico
bDepartamento de Física Teórica, Atómica y Optica, Universidad de Valladolid, E-47011 Valladolid, Spain. E-mail: lmolina@uva.es
First published on 6th May 2025
Abstract
ReOx and PtOx clusters are able to catalyze CO oxidation under mild pressure and temperature conditions. One possible way of designing novel catalysts with enhanced activity is to form bimetallic alloys. Here, we present a systematic theoretical study of the oxidation of RexPty clusters (x + y = 5) and their performance as catalysts for CO oxidation. Re-rich clusters reach high oxygen saturation levels, with up to 16 adsorbed oxygen atoms under standard reaction conditions, with a tendency to form interconnected ReO3 and ReO4 units. In contrast, Pt-rich clusters maintain some Pt–Pt bonds, with the amount of oxygen adsorbed decreasing steadily with increasing Pt concentration. Comparing reaction mechanisms for oxidized and unoxidized clusters, a superior catalytic performance for CO oxidation is found for the oxidized clusters, due to both a weaker interaction with CO and to a less stable nature of the adsorbed CO2 reaction intermediate. Finally, strong changes in reactivity are found with varying Re:Pt stoichiometry. The most active cluster is Re4Pt1O12, which can catalyze the reaction even at low temperatures, while the Re3Pt2O12 and Re2Pt3O12 clusters require higher temperatures for similar performance. These results provide insight into the oxidation of metal clusters on a subnanometer scale and their potential use as catalysts for the CO oxidation reaction.
1 Introduction
Metal oxidation is a process that is often accomplished under ambient conditions. The first step of oxidation involves oxygen dissociation by coordinatively unsaturated metal atoms on the surface.1 In the process, several phenomena occur, such as the chemisorption of atomic oxygen, the oxygen coverage on the surface, the formation of buried oxides and oxygen atoms within the subsurface, and even surface reconstruction.2,3 The final result can be a protective oxide layer or bulk oxide, depending on the properties of the metal and existing reaction conditions.4The oxidation process at the subnanometer and nanometer scale has received increasing attention.5–8 The stepwise adsorption of atomic oxygen is the initial step in this process, which results in the redistribution of charge, the formation of metal–oxygen bonds, and the loss of electrons from the metal atoms.9,10 Trinchero et al. explored the oxidation of subnanometer clusters for several transition metals. The results showed that the energetic behavior of these clusters is in concordance with the periodic trends of oxidation of bulk metals. This indicates that the stability of the clusters may be determined by the nature of local bonding.11 Subsequent studies have concentrated on the oxidation of Cu5 clusters through the successive adsorption of O2 molecules. They found that the Cu5 clusters are resistant to irreversible oxidation, providing evidence of their thermal stability.12,13 Apart from this, some computational research has also explored the oxidation of sub-nanometer Pt clusters, though not with oxygen saturation.14–16 Investigating the lowest energy isomers and oxygen coverage for larger metal clusters, such as Cu55 or Ni13Ag42, can be quite challenging.17,18 Therefore, studying the thermal stability and oxygen saturation of sub-nanometer clusters is crucial.19
The degree of oxidation in these small metal clusters has a strong influence on their reactivity for a wide range of chemical reactions.19–23 Among them, one prototypical case is the oxidation of CO. Several studies have analyzed the structure, stability and reactivity for CO2 production in PtOx clusters under realistic pressure and temperature conditions;14–16,24 these clusters and Pt2+ ions are active for CO oxidation even at low temperatures.25 The importance of the Pt–O–Pt motif in activating oxidation reactions has also been analysed.26 An interesting way of enhancing the catalytic activity of platinum is to alloy it with rhenium.27,28 It has been shown that the synergy between a Pt metal surface and neighboring ReOx clusters enhances the catalytic performance for the preferential oxidation of CO.29 In bimetallic Re–Pt clusters, the ratio of Re:Pt influences the catalytic activity of the system, where the lower affinity of Re atoms for CO compared to Pt atoms helps avoid catalyst poisoning.30,31 However, to our knowledge, the effect of Re:Pt composition on the oxidation of bimetallic clusters and its role in the CO oxidation reaction have not been explored in a systematic way.
This has motivated us to investigate the structure, stability, and reactivity of oxidized RexPty (x + y = 5) clusters using density functional theory (DFT) calculations, first-principles thermodynamics, and the nudged elastic band (NEB) method for the search of transition states. First, we investigate the structures and oxygen binding energies of oxidized Re–Pt clusters up to saturation. Then, an ab initio thermodynamics approach32 is employed to study these oxidized clusters under different pressure and temperature conditions. Following this, CO is adsorbed onto the clusters, considering the effect of oxygen coverage. We finally study the reaction mechanisms for CO oxidation, comparing two scenarios with the clusters initially either oxidized or unoxidized, and taking into account thermal effects for each energy barrier. The results show that varying the Re:Pt relative stoichiometry has drastic effects in the reaction energetics leading to CO2 production. Also, they highlight the importance of studying the reaction under realistic conditions with the clusters previously oxidized, since only in these cases meaningful conclusions can be derived about their intrinsic reactivity. Overall, the results indicate that for some compositions, binary Re–Pt clusters have the potential to act as very reactive catalysts for CO oxidation.
2 Computational setup
Density functional theory (DFT) calculations were performed using the VASP33 code with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional.34 A plane-wave basis with a 400 eV cutoff energy was utilized. The projector augmented wave (PAW) method was adopted to describe the core–valence electron interactions.35,36 Geometry optimizations were conducted with a convergence criterion of 0.01 eV Å−1 for forces and an electronic convergence tolerance of 10−8 eV. The K-point sampling of the Brillouin zone was performed at the gamma point, and the electronic occupation was handled with the Methfessel–Paxton scheme, with a smearing width of 0.01 eV.37Gas-phase Re–Pt clusters were modeled within a cubic cell measuring 15 × 15 × 15 Å. The oxidation of Re–Pt clusters was studied through the sequential adsorption of oxygen atoms. The potential energy surface (PES) was thoroughly investigated to ascertain the most stable isomers of oxidized clusters. Subsequently, the projected density of states (DOS) for each atom was calculated to understand the electronic structure and bonding of the systems. Moreover, phonon calculations were performed using a finite differences approach, wherein the dynamical matrix was constructed and diagonalized to obtain the frequencies.38
The reaction mechanisms for CO oxidation on unoxidized and oxidized Re–Pt clusters were calculated. Unoxidized clusters refer to metal clusters with a single O2 molecule dissociated, while oxidized clusters correspond to metal clusters with the most stable oxygen coverage at 300 K and 1 bar. The transition states (TS) were identified using the climb image nudged-elastic-band (CI-NEB) method.39 Each TS corresponds to a maximum in the PES characterized by a single imaginary frequency. Furthermore, the thermal correction for the CO oxidation mechanism was evaluated using first-principles atomistic thermodynamics.32,40 This approach facilitates a better understanding of the reaction under realistic temperature and pressure conditions.
3 Results and discussion
3.1 Oxidation of RexPty clusters
Prior to the reactivity studies, an exhaustive isomer search for the most stable conformations of the bare RexPty clusters must be performed. For each of the six compositions under study (that is, Re5, Re4Pt1, Re3Pt2, Re2Pt3, Re1Pt4 and Pt5), this has been already done in previous works,41,42 using the same DFT setup. Then, starting from such geometries (shown in Fig. 1), we have sequentially dissociated several oxygen molecules on each RexPty cluster. Fig. 1 shows the relaxed structures for the most stable conformations found during this oxidation process. For each additional oxygen molecule, we have calculated its dissociative binding energy as| Eb = E[(n − 2)O/RexPty)] + E[O2] − E[nO/RexPty] |
 | ||
| Fig. 1 Most stable structures and dissociative binding energies (in eV) for the oxidized RexPty clusters. Teal spheres: Re atoms. Silver spheres: Pt atoms. Red spheres: oxygen atoms. | ||
The isomer search for stable structures of nO–RexPty was performed sequentially, testing a large number of alternatives with the oxygen atoms adsorbed at various sites around the cluster. Once the most stable conformation was determined for a given oxygen coverage, it was used as the starting point for the adsorption of two more oxygen atoms, testing again various alternate structures. During this process, the clusters were found to be fluxional, undergoing important distortions as the oxygen coverage increased; therefore, for each case, other alternate RexPty conformations were tried. In the ESI† (Fig. S1–S6), the most relevant conformations for each nO–RexPty cluster are shown, together with the relative energies with respect to the global minimum.
In each case, we have added oxygen atoms to the cluster until its saturation coverage is found, according to the thermodynamic model which will be discussed below. In general, the high dissociation energies suggest that molecular O2 will easily dissociate on these clusters, with small or moderate activation barriers. Previous work by the authors on bare RexPty clusters shows that, indeed, the activation barriers for O2 dissociation at these clusters are small (usually below 0.5 eV) or even non-existent.43 This indicates that thermal effects will only play a minor role, with the clusters reaching oxygen saturation coverages under standard reaction conditions, according to the results of the thermodynamic modeling discussed below.
In the case of pure Re5, the high intrinsic reactivity of the Re atoms towards oxidation results in very high dissociation energies for each additional oxygen molecule. Since molecular adsorption of O2 results in binding energies of the order of 3–4 eV (see Fig. 4 in ref. 43), by dissociating O2 into adsorbed O atoms, there is an energy gain of more than 3 eV. Therefore, with relatively small barriers, even at small temperatures, kinetic effects will not prevent the expected complete oxidation of the Re5 cluster. The dissociation energies for Re5 remain very high up to 8 oxygen atoms, decreasing a little bit between 10 and 16 oxygen atoms. It is interesting to note that, as the oxygen coverage increases, strong changes take place in the structure of the Re5 cluster. Up to 10 oxygen atoms (corresponding to a ReO2 oxide stoichiometry), the Re–Re bond distances are similar to the ones for the clean Re5 cluster. However, the addition of more oxygen results in a sharp increase of the Re–Re bond distances; the cluster, instead of being a Re5 central unit surrounded by O adatoms, transforms into a collection of interconnected tetrahedral ReO4 units. W. Chen and co-workers calculated the oxidation of a single rhenium atom and determined that the most stable structures for ReO3 and ReO4 are trigonal and distorted tetrahedral, respectively.44 Another study found that the most stable structure of Re3O11 consists of tetrahedral and trigonal bipyramidal coordination for the Re atoms.45 In contrast, Re5Ox clusters with a high oxygen coverage (up to 10) only exhibit tetrahedral units.
For Re4Pt1, similar results are found, with incremental dissociation energies above 6 eV for up to 8 oxygen atoms (corresponding to the ReO2 stoichiometry) and slightly smaller (4–5 eV) or larger oxygen coverages. An analogous behavior is found for clusters with increasing Pt content; addition of oxygen atoms beyond the ReO2 ratio results in a decrease of dissociation energies below 6 eV per O2 molecule. In the case of clusters with several Pt atoms, the higher reactivity of the Re sites causes the oxygen atoms to preferentially bind at those sites, until ReO3 or ReO4 tetrahedral units are formed. Depending on the relative amount of platinum in the cluster, these units can be linked together, or completely isolated from each other, as observed in Re3Pt2–12O and in all high oxygen content cases of Re2Pt3 and Re1Pt4.
For Pt-rich clusters (Re2Pt3 and Re1Pt4), after Re sites have been completely saturated with oxygen, additional oxygen atoms bind exclusively to the Pt sites. However, the intrinsic lower reactivity of Pt causes the dissociative binding energies to fall to around 2–3 eV per O2 molecule. In the case of pure Pt5, saturation coverage is found for only 6 oxygen atoms, with a sharp decrease above that number. Similar results are found for Re2Pt3 and Re1Pt4 where, after each Re atom adsorbs 3–4 oxygen atoms, it is only possible to bind a maximum of 1 oxygen atom per Pt site. Interestingly, for Re2Pt3 at its saturation coverage (12 oxygen atoms), the last O2 molecule adsorbs molecularly, highlighting the much weaker bonding of oxygen at Pt sites. The structures found for the Pt5Ox clusters are similar to those reported for other oxidized Pt clusters.14,15,24,46 Pt atoms in the Pt5O10 cluster have a planar square coordination as the PtO bulk.47 In oxygen saturation, Pt–Pt bonds preserve more than Re–Re bonds since Re atoms are more prone to oxidation. Some Pt5Ox clusters exhibit a symmetrical structure. For instance, the Pt5O8 and Pt5O10 clusters have C2v and D5h symmetry groups, respectively.
In the process of gradual oxidation of the RexPty clusters, we have also performed a detailed analysis of the electronic structure of these nO–RexPty clusters. The complete results are shown in the ESI† (Fig. S15–S22). Fig. 2 shows a summary of the density of electronic states (DOS) for situations where the clusters are partially or completely covered by adsorbed oxygen atoms. In each case, the figures show the projected DOS into platinum, rhenium and oxygen states (black, green and red curves, respectively). The results show several interesting effects. First, it must be noted that for the Pt–Re mixed clusters, the DOS is formed by a superposition of Re d-states, which lie above and below the Fermi level, and Pt d-states, which are located just below the Fermi level. As oxygen atoms start to bind to the cluster, the oxygen sp states preferentially hybridize with Re states at the bottom of the valence d-band, with the Pt states being left unhybridized until oxygen accumulates on the cluster. Second, the accumulation of oxygen results in a broadening of the valence band and also in a shift of the Fermi level to lower binding energies (where we take the vacuum level as the energy reference). Finally, we note that as the amount of oxygen adatoms reaches concentrations corresponding to saturation coverages, the electronic structure resembles the one corresponding to a metallic oxide, with a sizable gap separating the valence and conduction bands of the oxide. However, as the relative concentration of Pt and Re changes, sizable variations take place, involving both the size of the HOMO–LUMO gap and the location of the Fermi level relative to this energy gap. For pure Re, some isolated states appear in the middle of the gap, with the Fermi level being located approximately at the bottom of the conduction band. For Pt, the situation is reversed, and the Fermi level lies at the top of the conduction band. Re4Pt1 is an interesting case, with a marked tendency, for almost every oxygen concentration, towards the opening of HOMO–LUMO gaps with the Fermi level at their center.
 | ||
| Fig. 2 Densities of electronic states (DOS) for some selected nO–RexPty oxidized clusters, with various degrees of oxidation. The black curves show the contribution from Pt states, the green curves show the contribution from Re states, while the red curves show the projection of the DOS on oxygen s and p orbitals. Dashed blue lines highlight the location of the Fermi level in each case. When the cluster has a non-zero magnetic moment, both spin-up and spin-down contributions are shown separately. | ||
3.2 (p,T)-phase diagrams of oxidized nO–RexPty clusters
Next, the relative stability of oxidized Re–Pt clusters was determined by calculating the Gibbs free energy of formation.| ΔG(p,T) = GRexPtyOn(p,T) − GRexPty(p,T) − n·μO(p,T) |
where kB is the Boltzmann constant and p0 is the standard pressure (1 bar). The first term represents the temperature dependency of chemical potential at p0, while the second term is the pressure dependency through the ratio p/p0. The oxygen atoms in the Re–Pt clusters are in equilibrium with the gas-phase O2. Consequently, the molecular oxygen potential can be related to the atomic oxygen potential through the following equation:


To ensure high accuracy, μO2(p0,T) was sourced from the NIST-JANAF thermochemical tables.50 The most stable phase is defined as the one that minimizes the Gibbs free energy of formation.51 Based on this principle, (p,T)-phase diagrams were used to determine the most stable oxygen coverage of the Re–Pt cluster for a given temperature and oxygen pressure. A range of pressures, from ultra-high vacuum (UHV) to standard pressure, was examined, while temperatures were varied from 200 K to 1000 K. This methodology for calculating the (p,T)-phase diagrams is analogous to other theoretical reports on oxidized metal clusters.12,15,16,52
For the most stable nO–RexPty structures (Fig. 1), we have calculated the (p,T) phase diagram using the first principles atomistic thermodynamics. The results are shown in Fig. 3. The selected temperature and the oxygen partial pressure range have been chosen to circumvent crossing the phase boundaries of O2. These parameters also comprise conditions relevant to industrial catalytic oxidation processes and ensure that the ideal gas assumption is valid. (p,T)-Phase diagrams indicate that Re–Pt clusters are susceptible to oxidation over a wide range of temperatures and pressures. The Pt5 cluster constitutes the sole exception, manifesting as a metallic phase exclusively under low pressures (near UHV) and elevated temperatures (above 900 K). Conversely, the Re5 cluster exhibits remarkable stability at oxygen saturation, even under very low O2 pressure. All Re–Pt clusters reach their maximum oxygen coverage under standard pressure and temperature conditions. As it should be expected, increasing the Pt content results in a steady decrease of the amount of oxygen atoms adsorbed under standard conditions, from 16 oxygen atoms in the case of Re5 to only six in the case of Pt5. These results reflect the fact, discussed above, that a sharp drop in oxygen binding energy takes place when rhenium atoms are saturated with 3–4 oxygen adatoms each.
 | ||
| Fig. 3 Pressure–temperature (p,T) phase diagrams for oxidized RexPty clusters: (a) Re5On, (b) Re4Pt1On, (c) Re3Pt2On, (d) Re2Pt3On, (e) Re1Pt4On, (f) Pt5On. Each colored area represents a stable phase in thermodynamic equilibrium. The oxygen pressure ratio (p/p0) is plotted on a logarithmic scale. | ||
For Re-rich clusters (Re4Pt3 and Re3Pt2), the same phases are obtained (containing 10, 12, and 14 oxygen atoms) but in different proportions. At low O2 pressure, the Re4Pt1O14 cluster requires a higher temperature to lose oxygen atoms compared to the Re3Pt2O14 cluster. Additionally, the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 oxygen/metal ratio is more representative in the cluster with the lower Re content (Re3Pt2). In the case of Re2Pt3 and Re1Pt4 (Pt-rich clusters), the most stable phases under standard pressure and temperature conditions are represented by Re2Pt3O12 and Re1Pt4O8, respectively. The oxidation of subnanometer Re–Pt clusters is affected by the presence of a single Re atom. For example, the Re1Pt4 cluster does not present a metallic phase due to the strong affinity of rhenium for oxygen.
:1 oxygen/metal ratio is more representative in the cluster with the lower Re content (Re3Pt2). In the case of Re2Pt3 and Re1Pt4 (Pt-rich clusters), the most stable phases under standard pressure and temperature conditions are represented by Re2Pt3O12 and Re1Pt4O8, respectively. The oxidation of subnanometer Re–Pt clusters is affected by the presence of a single Re atom. For example, the Re1Pt4 cluster does not present a metallic phase due to the strong affinity of rhenium for oxygen.
3.3 CO adsorption at oxidized nO–RexPty clusters
After obtaining the most stable conformations for oxidized RexPty clusters with a varying amount of dissociated oxygen atoms, the next step in the CO oxidation reaction involves the study of the interaction of CO with these oxygen-covered clusters. For each RexPty cluster with a varying amount of oxygen adatoms (up to saturation coverage), we have systematically studied the adsorption of CO at each possible binding site. Fig. 4 shows the most stable adsorption conformation of CO for each cluster under study, with varying amounts of dissociated O2 molecules, representing situations where the cluster is partially or fully oxidized. In the ESI† (Fig. S7–S12) we report, for each case, the results of the extensive search for the most stable conformers, together with the relative energies.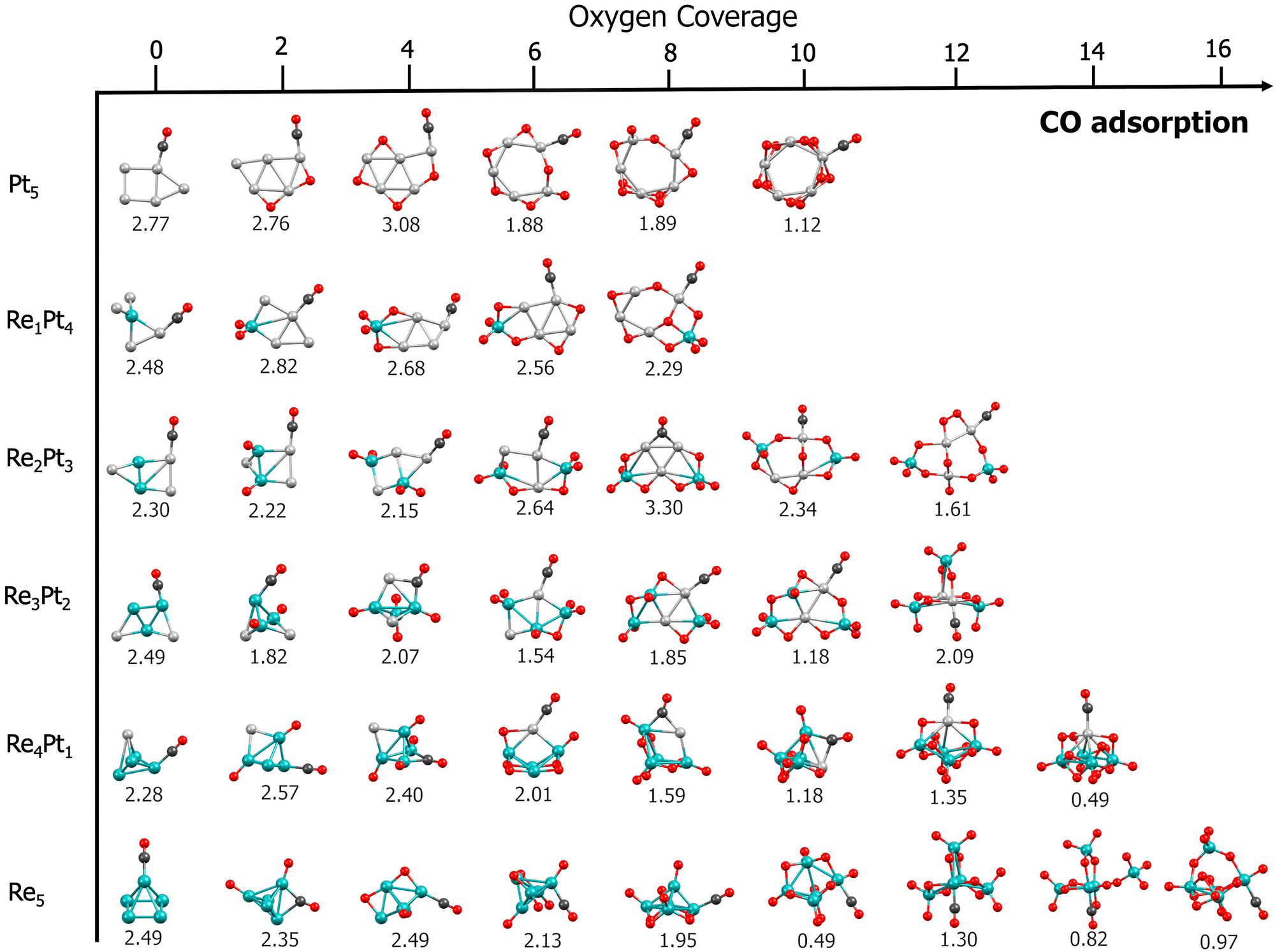 | ||
| Fig. 4 Structures and binding energies (in eV) for the adsorption of a CO molecule at oxidized RexPty clusters, with varying amounts of adsorbed oxygen. Teal spheres: Re atoms. Silver spheres: Pt atoms. Red spheres: oxygen atoms. Black spheres: carbon atoms. | ||
For all the RexPty compositions, CO interacts strongly with the bare unoxidized clusters, with binding energies of the order of 2.3–2.7 eV. In the case of Re-rich clusters, saturating the cluster with oxygen adatoms has a strong influence on the strength of the CO–metal bonds. When the amount of oxygen on the cluster is below the stoichiometry corresponding to the ReO2 composition, relatively high (1.5–2.0 eV) CO binding energies are found, only slightly smaller than the values found for the clean clusters without oxygen. However, a further increase in oxygen coverage generally results in a sharp drop in CO binding to values of the order of 1 eV or even smaller. It is interesting to note that when the cluster contains platinum, CO will always preferentially bind at Pt sites. Contrary to the case of Re-rich clusters, for Pt-rich clusters, the oxidation of the cluster does not cause a systematic decrease in CO binding energies. In many instances, the CO adsorption at a partially oxidized Pt site leads to a significant increase in the CO binding energy, which may sometimes exceed 3 eV.
Finally, we also considered the effect of multiple binding of CO molecules at the bare RexPty clusters. Results for adsorption of up to three CO molecules are shown in the ESI† (Fig. S13 and S14). In general, these clusters can adsorb several CO molecules without a noticeable drop in the binding energies (Re1Pt4 is the sole exception, which can nevertheless be attributed to isomerization effects). Since the difference in binding energies for CO and atomic oxygen is very high for Re-rich clusters, it can be concluded that CO accumulation at these types of cluster is very unlikely, as dissociating oxygen molecules will easily displace CO. This situation is reversed for pure Pt5, where CO binding energies compete with dissociative oxygen binding energies. Then, it is possible that for this cluster, CO will accumulate under standard reaction conditions, preventing oxygen dissociation, and thus resulting in a reduced activity for CO oxidation.
3.4 CO2 formation at unoxidized and oxidized RexPty clusters
After identifying the most stable initial conformations for CO adsorbed at oxidized nO–RexPty clusters, we have studied the reaction between CO and a coadsorbed oxygen atom. This part of the reaction involves two steps. First, the carbon atom of CO reacts with a neighboring oxygen atom, forming a CO2 molecule bonded to the Pt–Re cluster. Second, this bonded CO2 entity needs to be desorbed to the gas phase, by breaking all C–metal and O–metal bonds. To gain a better understanding on the effect caused by the partial or total oxidation of the cluster, we have compared the reaction pathways leading to CO2 formation in two situations. First, we have calculated the reaction barriers for clusters with a single O2 molecule dissociated at the cluster. Second, we have studied the same process for CO2 formation at nO–RexPty clusters with oxygen coverages corresponding to standard reaction conditions, obtained from the results in Fig. 3.Fig. 5 shows the energy profiles for the case of clusters with only a single O2 molecule dissociated on them. The highest barrier is always the first step, which involves forming a new C–O bond. This barrier is quite high, nearly 2 eV for Re-rich clusters, while for Pt-rich clusters, it decreases a little to values of the order of 1 eV. The formation of CO2 involves the displacement of both the adsorbed CO and a nearby oxygen atom from their positions. In the intermediate I, the O–C⋯O distance varies from 3.13 Å (on Pt5O2) to 4.23 Å (on Re1Pt4O2). Interestingly, the largest displacement along the reaction coordinate corresponds to the lowest energy barrier. For the transition states (TS1), the new C–O bond distance ranges from 1.6 Å to 1.9 Å, with the longest distance observed in the Re1Pt4O2 cluster. It suggests that the energetic cost of CO2 formation is not directly related to the adsorbate displacement along the reaction coordinate.
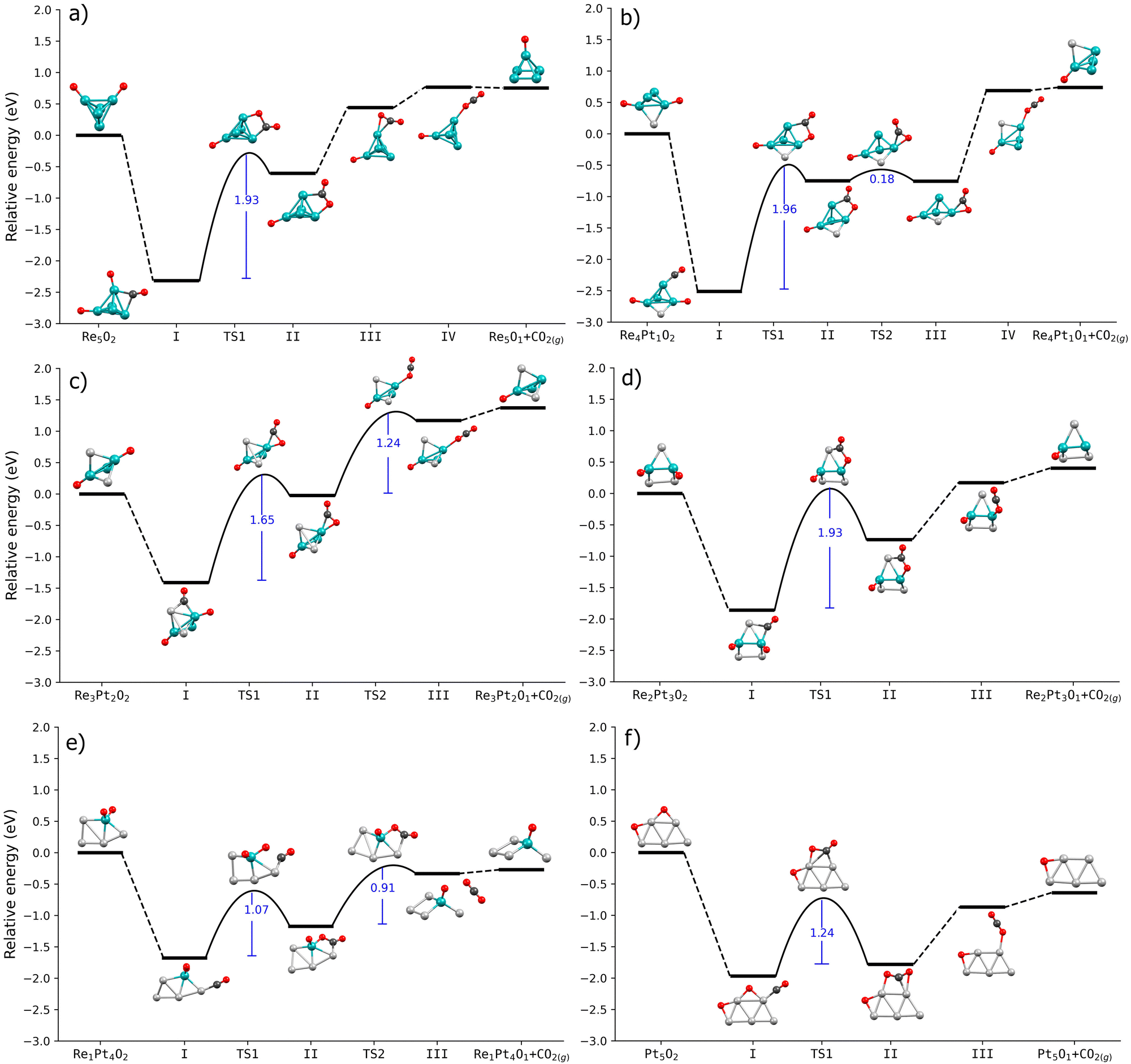 | ||
| Fig. 5 Energy profiles of CO oxidation on RexPtyO2 clusters via the Langmuir–Hinshelwood (LH) mechanism: (a) Re5O2, (b) Re4Pt1O2, (c) Re3Pt2O2, (d) Re2Pt3O2, (e) Re1Pt4O2, (f) Pt5O2. The energy barrier values are shown in blue, with the first barrier corresponding to the formation of CO2. Teal spheres: Re atoms. Silver spheres: Pt atoms. Red spheres: oxygen atoms. Black spheres: carbon atoms. | ||
Comparing the results for all the RexPty compositions, we observe that the stability of the initial state (with CO and O coadsorbed, labeled as I in the figures) is quite high with respect to the relative energy of the final state, with CO2 detached from the cluster. This means that we can expect a high energetic cost to both form CO2 and to detach it from the cluster, something that will result in a very low activity for CO oxidation. For Re-rich clusters, this effect is more pronounced, with the total cost for the formation of C–O bonds (step II) and the final CO2 desorption is of the order of 3 eV. For Pt-rich clusters, the intermediate states in the reaction are comparatively less stable, although the energetic cost for CO2 formation is still relatively high (around 1.5 eV). These results can easily be interpreted in terms of the differences in binding energies of CO and O between Re-rich and Pt-rich clusters. Oxygen atoms bind very strongly to Re, and if we add the binding energy of CO (more than 2 eV), the sum of both values is much larger than the enthalpy for the CO + 1/2O2 reaction, meaning that it will be very costly to detach the CO2 molecule adsorbed at the cluster. In the case of platinum, the binding energy of oxygen decreases, reducing slightly the stability of the intermediate states.
Then, we have extended the study of reaction mechanisms to the case of RexPty covered with oxygen atoms, at coverages consistent with standard reaction conditions, according to the calculated phase diagrams. For each cluster with a given oxygen coverage, we have studied the reaction mechanisms using the following procedure: first, starting with the most stable configuration for CO adsorption, we have calculated the stability of the adsorbed CO2 intermediate species, for several cases where CO reacts with neighboring oxygen adatoms. Then, after identifying which oxygen atom is more reactive towards the production of CO2, we have calculated for such situation the activation barriers for CO2 formation and subsequent CO2 detachment from the cluster. These results are displayed in Fig. 6.
 | ||
| Fig. 6 Energy profiles of CO oxidation on oxidized RexPtyOn clusters via the Langmuir–Hinshelwood (LH) mechanism: (a) Re5O16, (b) Re4Pt1O12, (c) Re3Pt2O12, (d) Re2Pt3O12, (e) Re1Pt4O8, and (f) Pt5O6. The energy barrier values are shown in blue, with the first barrier corresponding to the formation of CO2. Teal spheres: Re atoms. Silver spheres: Pt atoms. Red spheres: oxygen atoms. Black spheres: carbon atoms. | ||
We observe significant differences in the reaction pathways of a single dissociated O2 molecule on RexPty clusters compared to clusters that have been oxidized at their saturation coverage. First of all, for both Re-rich and Pt-rich compositions, now the relative energies of states I and II (adsorbed CO, and CO2 intermediate) are comparable with the relative energy of CO2 desorbed into the gas phase. This means that once CO2 is formed, it is not necessary to overcome large barriers to detach the reaction product from the cluster. In one case (Re2Pt3O12), this reaction intermediate is actually less stable than CO2 in the gas phase. We can attribute this behavior to the weaker O–metal bonding that occurs for clusters with increasing amount of adsorbed oxygen, as compared to the clusters with only a dissociated O2 molecule on them. Also, for many compositions (specially for Re-rich clusters), CO binding energies are also smaller at increasing oxygen coverages. This indicates that since the reactants are less strongly bound, the CO2 intermediate will also be less stable compared to the gas-phase molecule, making its desorption easier. The reduced CO binding energy has also some important consequences for the overall reactivity of the clusters. For example, in the case of Re5O16, the CO binding energy (0.97 eV) is smaller than the activation energy for the formation of absorbed CO2 (1.22 eV). Consequently, it is energetically more favorable for CO to desorb back into the gas phase rather than forming CO2. Therefore, the oxidized Re5 is expected to exhibit low catalytic activity.
In the oxidized clusters, the energy barriers for the reaction between CO and oxygen adatoms show drastic changes with the Pt–Re stoichiometry. They change in a non regular fashion, with very high values (around 1.90 eV) for Re3Pt2 and Re1Pt4, moderately high values (1.2–1.3 eV) for Re2Pt3, Re5 and Pt5, and a very small 0.46 eV barrier for Re4Pt1. The case of the Re4Pt1 with 12 preadsorbed oxygen atoms is particularly interesting. Our findings indicate that the reaction pathway for this cluster combines all the favorable factors necessary for high reactivity toward CO2 production. First, CO binding at the oxidized Re4Pt1 cluster is reasonably strong, with 1.35 eV. Second, there is only a very small barrier of 0.46 eV for the formation of adsorbed CO2. Finally, the desorption of CO2 is energetically unexpensive and involves only a moderate barrier of 0.69 eV. Contrary to the rest of cases, the rate-limiting step is the breaking of the CO2–cluster bonds, and not the CO + O bond formation.
We have also evaluated the effect of temperature on the barriers of CO oxidation on the oxidized RexPty clusters (Fig. 7). The Gibbs free energy at 1 bar pressure was calculated using first-principles thermodynamics. The energy barrier of CO2 formation decreases with temperature, particularly in Re4Pt1O12, Re3Pt2O12, Re2Pt3O12 and Pt5O6 clusters. At high temperatures, these systems exhibit a energy barrier of less than 1 eV (Fig. 7a). The effect of temperature for the range studied (from 200 to 1000 K) is not the same in all clusters. For instance, the energy barrier of the Re5O16 cluster decreases by about 0.2 eV, while the Re3Pt2O12 cluster exhibits a more significant reduction of around 0.9 eV. This variation is attributed to entropic effects resulting from conformational changes in CO2 formation.
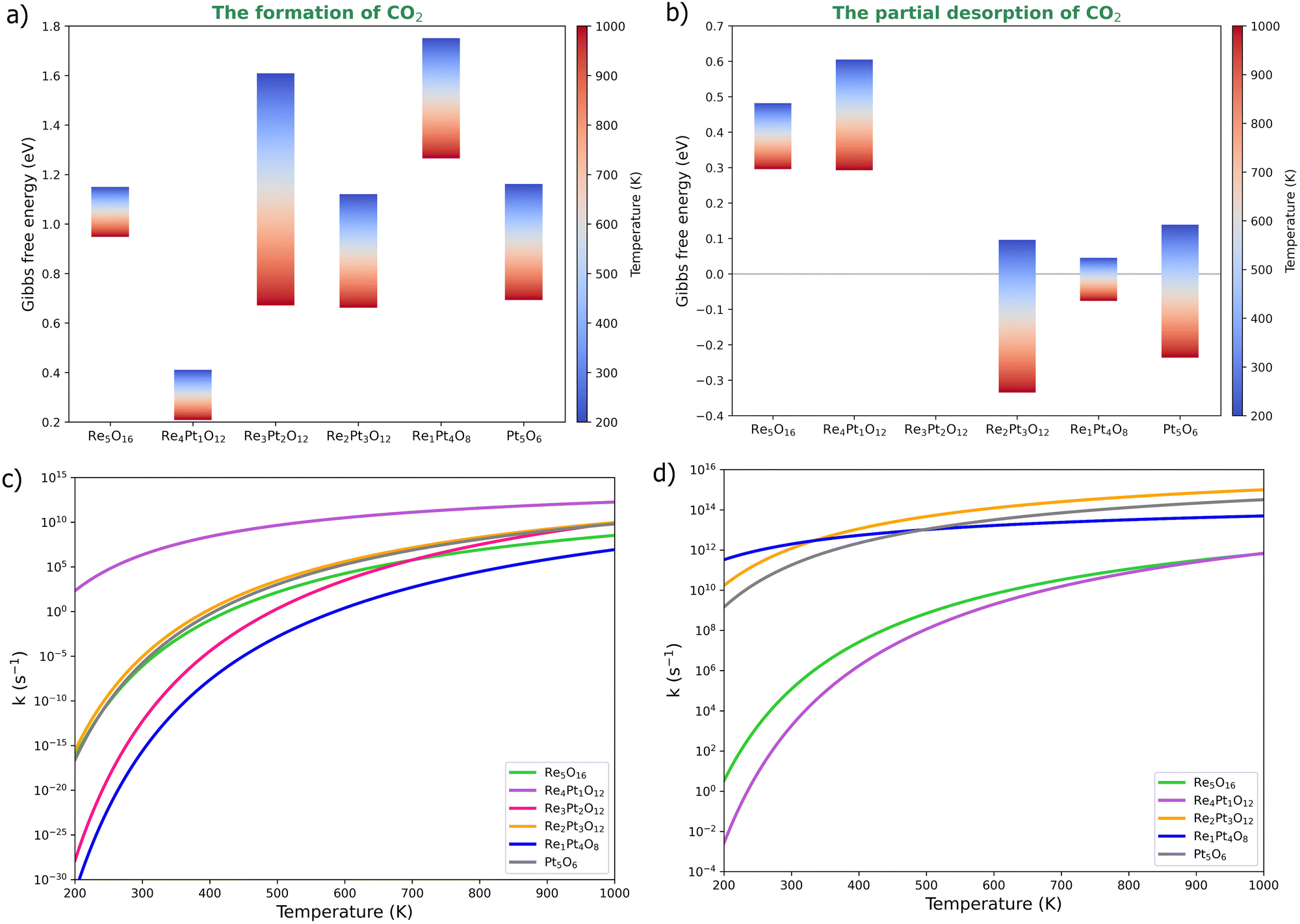 | ||
| Fig. 7 Effect of temperature on the CO oxidation barriers at the oxidized RexPty clusters. (a) and (c) Gibbs free energies and rate constants for the formation of CO2. (b) and (d) Gibbs free energies and rate constants for the partial desorption of CO2. | ||
As mentioned, CO2 detachment on oxidized clusters occurs with low energy requirements, except for the Pt5O6 cluster, which requires approximately 1.5 eV to detach the product. CO2 desorption was performed in steps, where partial desorption involves breaking a bond between CO2 and the cluster, usually the metal–carbon bond. This elementary reaction involves an isomeric change in CO2 formed on the cluster. The energy barriers for this step decrease with temperature, even disappearing in the Re2Pt3O12, Re1Pt4O8, and Pt5O6 clusters (Fig. 7b). Re-rich clusters (Re5O16 and Re4Pt1O12) exhibit partial desorption energy barriers with a moderate height (less than 0.7 eV), which decrease to 0.3 eV with increasing temperature. Finally, total detachment involves the breaking of the last metal–oxygen bond holding CO2 to the cluster, where no energy barrier was found for this final step.
Subsequently, the rate constants for the elementary reaction steps were calculated using the Arrhenius equation53

4 Conclusions
In summary, a systematic study of the oxidation of subnanometer RexPty (x + y = 5) clusters has been carried out, determining the most stable structures for the dissociative adsorption of several oxygen molecules. Oxygen saturation is associated with a sharp decrease in the dissociate binding energy; this takes place between six (for pure Pt5) and sixteen (for pure Re5) oxygen adatoms, depending on the cluster stoichiometry. The higher intrinsic reactivity of Re atoms towards oxygen results in a steady increase of the oxygen saturation coverage as the clusters become richer in rhenium. Also, it results in a preferential binding of oxygen at Re sites, until ReO3 or ReO4 units are formed. Then, some oxygen atoms can also bind at Pt sites. When such ReO4 units are formed, Re-rich clusters tend to elongate and eventually rupture their Re–Re bonds, while Pt-rich clusters preserve some of their Pt–Pt bonds. For Re5, at the saturation coverage, the structure resembles that of the Re2O7 rhenium oxide, with interconnected tetrahedral ReO4 units. In the case of Re4 Pt, the cluster effectively becomes a platinum single-atom catalyst (SAC) adsorbed on a small rhenium oxide support. As the oxygen molecules adsorb and dissociate, the electronic structure is characterized by the hybridization of oxygen sp states with rhenium d-states, a broadening of the valence band, and shifts in the Fermi level. When the clusters become completely oxidized, the densities of states resemble the ones for metallic oxides, although they show very complex variations with the Re/Pt composition. In some cases (14O–Re4 Pt, for example), large HOMO–LUMO gaps are found; in others (14O–Re5), some isolated electronic states appear in the middle of the HOMO–LUMO gap.The conclusions obtained after analyzing the sequential oxygen binding energies have been further confirmed by the results of the ab initio thermodynamics approach and the calculated (p,T) phase diagrams. Only the pure Pt5 cluster has a stable metallic phase at high temperatures and low pressures. All the clusters containing Re are stable only in their oxidized phase at any given temperature and pressure, a fact which is a consequence of the strong oxophilic nature of this metal. As mentioned above, the degree of oxidation of the Re–Pt clusters is determined by their composition, ranging from 6 oxygen atoms in the Pt5 cluster to 16 oxygen atoms in the Re5 cluster under standard pressure and temperature conditions. Thus, the oxygen coverage for Re-rich clusters can exceed the 1:2 metal/oxygen ratio, indicating that rhenium can easily attain high oxidation states even at low temperatures.
The reactivity of the oxidized clusters as catalysts of the CO oxidation reaction has been also studied in detail. Reaction pathways and energy barriers were calculated for RexPty clusters with either a single dissociated O2 molecule or a fully saturated molecule with oxygen under standard pressure and temperature conditions. Unoxidized clusters exhibit high CO adsorption energy, which is a factor that can lead to catalyst poisoning and loss of active sites. This situation changes when the clusters become fully oxidized; while Pt-rich clusters still have a relatively high affinity for CO (even when oxygen is coadsorbed at Pt sites), the oxidation of Re-rich clusters causes a sharp decrease in CO binding energies, as carbon monoxide cannot bind at Re sites where three or more oxygen atoms have already been adsorbed.
The comparison between unoxidized clusters and fully oxidized ones also shows that the overall reaction energetics undergo drastic changes with varying oxygen coverage. For clusters with a single dissociated O2 molecule, a very strong adsorbed CO2 reaction intermediate is formed. This is partially due to a high CO binding energy, but also to the high dissociative binding energy for the first two oxygen atoms. This results in a very difficult desorption of the CO2 molecule to gas phase, rendering the clusters inactive at such low oxygen coverages. In contrast, fully oxidized clusters require much less energy for CO2 desorption. Comparing various stoichiometries, we found large variations in the activation barriers, which will result in huge variations of reactivity with composition. The stoichiometry with the best performance was found to be Re4Pt1; when oxidized to 12O–Re4Pt1, CO can still easily adsorb at the Pt site, reacting with neighboring oxygen adatoms with very small activation barriers, forming a loosely bound CO2 intermediate which can easily desorb from the cluster. Effectively, this case can be interpreted in terms of a favorable synergy caused by the presence of a reactive Pt species, which binds CO, and neighboring ReOx species which provide reactive oxygen species. Such a cooperative effect between Re and Pt has been also reported in the experimental work of Ishida et al.,29 where the interplay between Pt and ReOx results in high activity for the preferential oxidation of CO.
Finally, we calculated the effect of temperature on CO oxidation using ab initio thermodynamics. The Re4Pt1O12 cluster serves as a CO catalyst at low temperatures, while the Re2Pt3O12 and Re3Pt2O12 clusters may only catalyze the reaction at high temperatures (near 1000 K). The structure of oxidized Re4Pt1 can be considered as a prototype of a platinum single-atom-catalyst (SAC) supported on a small rhenium oxide support (Pt1/Re4O12). The Pt atom has been used as the SAC, as it can be easily dispersed as stable supported entities on reducible metal oxides, such as CeOx or FeOx.54–56 These highly localized active sites reduce CO poisoning and promote CO2 desorption more effectively than conventional catalysts.57 In the case of Pt1/Re4O12, the Pt atom serves as an active site for CO oxidation at low temperatures in an oxidizing atmosphere.
Overall, our results contribute to a better understanding of the oxidation of metal clusters at the subnanometer scale. Also, we find that these small binary Pt–Re clusters can be active for the CO oxidation reaction even at low temperatures, with strong variations of activity with composition and oxidation state. Thus, these DFT calculations highlight the importance of studying the reaction mechanisms under realistic reaction conditions, which in the present case involve highly oxidized clusters.
Author contributions
Conceptualisation: AAG, ILG, and LMM; formal analysis: AAG, ILG, and LMM; funding acquisition: ILG and LMM; supervision: ILG and LMM; visualisation: AAG; writing – original draft: AAG; writing – review & editing: ILG and LMM.Data availability
The data supporting this article have been included as part of the ESI.† Further information and details are available from the corresponding author upon reasonable request.Conflicts of interest
The authors have no conflicts to disclose.Acknowledgements
Andrés Álvarez-García gratefully thanks CONAHCyT-Mexico for the PhD scholarship No. 957574. Luis M. Molina acknowledges the support from Ministerio de Ciencia e Innovación of Spain (Grant PID2022-138340OB-I00 funded by MCIN/AEI/10.13039/501100011033 and FSE+) and from the University of Valladolid (GIR Nanostructure Physics). Ignacio L. Garzón was supported by PASPA-DGAPA-UNAM through a sabbatical fellowship at the Universidad de Valladolid, Spain. ILG also acknowledges the hospitality of the Departamento de Física Teórica, Atómica y Óptica de la Facultad de Ciencias de la Universidad de Valladolid, Spain, during a sabbatical year. Calculations were performed at the DGTIC-UNAM Supercomputing Center under Project LANCAD-UNAM-DGTIC-049. We also acknowledge networking within the COST Action CA21101 “Confined molecular systems: from a new generation of materials to the stars” (COSY) supported by COST (European Cooperation in Science and Technology).References
- H. Over and A. Seitsonen, Science, 2002, 297, 2003–2005 CrossRef CAS PubMed.
- J. C. Goloboy and W. G. Klemperer, Angew. Chem., Int. Ed., 2009, 48, 3562–3564 CrossRef CAS PubMed.
- W. X. Li, L. Österlund, E. K. Vestergaard, R. T. Vang, J. Matthiesen, T. M. Pedersen, E. Lægsgaard, B. Hammer and F. Besenbacher, Phys. Rev. Lett., 2004, 93, 146104 CrossRef CAS PubMed.
- H. Kan and J. Weaver, Surf. Sci., 2009, 603, 2671–2682 CrossRef CAS.
- M. B. Torres, E. M. Fernández and L. C. Balbás, J. Phys. Chem. A, 2008, 112, 6678–6689 CrossRef CAS PubMed.
- E. M. Fernández and L. C. Balbás, J. Chem. Phys., 2016, 144, 224308 CrossRef PubMed.
- L. X. Chen, Z. Wen, Z. W. Chen, C. V. Singh and Q. Jiang, J. Mater. Chem. A, 2021, 9, 11726–11733 RSC.
- E. Fernández, M. Boronat and A. Corma, Phys. Chem. Chem. Phys., 2022, 24, 4504–4514 RSC.
- A. Sumer, J. Phys. Chem. A, 2021, 125, 5201–5211 CrossRef CAS PubMed.
- Z. Luo, A. W. Castleman and S. N. Khanna, Chem. Rev., 2016, 116, 14456–14492 CrossRef CAS PubMed.
- A. Trinchero, S. Klacar, L. O. Paz-Borbón, A. Hellman and H. Grönbeck, J. Phys. Chem. C, 2015, 119, 10797–10803 CrossRef CAS.
- D. Buceta, S. Huseyinova, M. Cuerva, H. Lozano, L. J. Giovanetti, J. M. Ramallo-López, P. López-Caballero, A. Zanchet, A. O. Mitrushchenkov, A. W. Hauser, G. Barone, C. Huck-Iriart, C. Escudero, J. C. Hernández-Garrido, J. J. Calvino, M. López-Haro, M. P. de Lara-Castells, F. G. Requejo and M. A. López-Quintela, Chem. – Eur. J., 2023, 29, e202301517 CrossRef CAS PubMed.
- J. Garrido-Aldea and M. P. de Lara-Castells, Phys. Chem. Chem. Phys., 2022, 24, 24810–24822 RSC.
- C. Kerpal, D. Harding, A. Hermes, G. Meijer, S. Mackenzie and A. Fielicke, J. Phys. Chem. A, 2013, 117, 1233–1239 CrossRef CAS PubMed.
- M. Taleblou, M. Camellone, S. Fabris and S. Piccinin, J. Phys. Chem. C, 2022, 126, 10880–10888 CrossRef CAS.
- L. Xu, G. Henkelman, C. T. Campbell and H. Jónsson, Surf. Sci., 2006, 600, 1351–1362 CrossRef CAS.
- L. Ma and J. Akola, Phys. Chem. Chem. Phys., 2019, 21, 11351–11358 RSC.
- R. H. Aguilera-Del-Toro, F. Aguilera-Granja and A. Vega, Phys. Chem. Chem. Phys., 2023, 26, 3117–3125 RSC.
- M. Schmidt, P. Cahuzac, C. Bréchignac and H.-P. Cheng, J. Chem. Phys., 2003, 118, 10956–10962 CrossRef CAS.
- C. C. Chang, Y. C. Chen, K. C. Wu, H. N. Priyadarshini, L. Y. Lee, J. L. Chen, C. R. Lee, C. W. Pao and D. Y. Wang, ChemCatChem, 2024, e202400596 CrossRef CAS.
- S. Yamazoe, K. Koyasu and T. Tsukuda, Acc. Chem. Res., 2014, 47, 816–824 CrossRef CAS PubMed.
- M. D’Andria, F. Krumeich, Z. Yao, F. R. Wang and A. T. Güntner, Adv. Sci., 2024, 11, 2308224 CrossRef PubMed.
- X. N. Li, L. N. Wang, L. H. Mou and S. G. He, J. Phys. Chem. A, 2019, 123, 9257–9267 CrossRef CAS PubMed.
- J. E. Q. Domínguez, K. M. Neyman and A. Bruix, J. Chem. Phys., 2022, 157, 094709 CrossRef PubMed.
- E. A. Lashina, E. M. Slavinskaya, O. A. Stonkus, A. I. Stadnichenko, A. V. Romanenko and A. I. Boronin, Chem. Eng. Sci., 2023, 267, 118328 CrossRef CAS.
- D. Vasilchenko, T. Asanova, B. Kolesov, A. Tsygankova, A. Stadnichenko, E. Slavinskaya, E. Gerasimov, K. Lomachenko, A. Boronin and S. Korenev, ChemCatChem, 2020, 12, 1413–1428 CrossRef CAS.
- R. P. Galhenage, K. Xie, H. Yan, G. S. Seuser and D. A. Chen, J. Phys. Chem. C, 2016, 120, 10866–10878 CrossRef CAS.
- A. S. Duke, R. P. Galhenage, S. A. Tenney, P. Sutter and D. A. Chen, J. Phys. Chem. C, 2015, 119, 381–391 CrossRef CAS.
- Y. Ishida, T. Ebashi, S. I. Ito, T. Kubota, K. Kunimori and K. Tomishige, Chem. Commun., 2009, 5308–5310 RSC.
- K. Azzam, I. Babich, K. Seshan and L. Lefferts, Appl. Catal., B, 2008, 80, 129–140 CrossRef CAS.
- K. G. Azzam, I. V. Babich, K. Seshan, B. L. Mojet and L. Lefferts, ChemCatChem, 2013, 5, 557–564 CrossRef CAS.
- K. Reuter, C. Stampf and M. Scheffler, in AB Initio Atomistic Thermodynamics and Statistical Mechanics of Surface Properties and Functions, ed. S. Yip, Springer, Netherlands, Dordrecht, 2005, pp. 149–194 Search PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- M. Methfessel and A. T. Paxton, Phys. Rev. B:Condens. Matter Mater. Phys., 1989, 40, 3616–3621 CrossRef CAS PubMed.
- X. Wu, D. Vanderbilt and D. R. Hamann, Phys. Rev. B:Condens. Matter Mater. Phys., 2005, 72, 035105 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- K. Reuter and M. Scheffler, Phys. Rev. B:Condens. Matter Mater. Phys., 2002, 65, 1–11 CrossRef.
- L. E. Gálvez-González, L. O. Paz-Borbón and A. Posada-Amarillas, Surf. Sci., 2022, 725, 122157 CrossRef.
- A. Álvarez-García, J. C. Luque-Ceballos, L. O. Paz-Borbón and I. L. Garzón, Comput. Mater. Sci., 2022, 214, 111697 CrossRef.
- A. Álvarez García, L. M. Molina and I. L. Garzón, Phys. Chem. Chem. Phys., 2024, 26, 15902–15915 RSC.
- W.-J. Chen, H.-J. Zhai, X. Huang and L.-S. Wang, Chem. Phys. Lett., 2011, 512, 49–53 CrossRef CAS.
- Y. X. Zhao, X. L. Ding, Y. P. Ma, Z. C. Wang and S. G. He, Theor. Chem. Acc., 2010, 127, 449–465 Search PubMed.
- Y. Nakajima, M. A. Latif, T. Nagata, K. Ohshimo and F. Misaizu, J. Phys. Chem. A, 2023, 127, 3570–3576 CrossRef CAS PubMed.
- J. R. McBride, G. W. Graham, C. R. Peters and W. H. Weber, J. Appl. Phys., 1991, 69, 1596–1604 CrossRef CAS.
- A. Hjorth Larsen, J. Jørgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dulak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. Bergmann Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys. Condens. Matter., 2017, 29, 273002 CrossRef PubMed.
- K. Reuter, Catal. Lett., 2016, 146, 541–563 CrossRef CAS.
- M. Chase, NIST-JANAF Thermochemical Tables, 4th edition, American Institute of Physics, vol. 1, 1998 Search PubMed.
- W. Zhang, J. R. Smith and X. G. Wang, Phys. Rev. B:Condens. Matter Mater. Phys., 2004, 70, 024103 CrossRef.
- S. Bhattacharya, S. V. Levchenko, L. M. Ghiringhelli and M. Scheffler, Phys. Rev. Lett., 2013, 111, 084704 CrossRef PubMed.
- J. T. Hirvi, T. J. J. Kinnunen, M. Suvanto, T. A. Pakkanen and J. K. Nørskov, J. Chem. Phys., 2010, 133, 084704 CrossRef PubMed.
- D. Jiang, Y. Yao, T. Li, G. Wan, X. I. Pereira-Hernández, Y. Lu, J. Tian, K. Khivantsev, M. H. Engelhard, C. Sun, C. E. García-Vargas, A. S. Hoffman, S. R. Bare, A. K. Datye, L. Hu and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 26054–26062 CrossRef CAS PubMed.
- Y. Tang, Y. G. Wang and J. Li, J. Phys. Chem. C, 2017, 121, 11281–11289 CrossRef CAS.
- Z. Zhang, J. Tian, Y. Lu, S. Yang, D. Jiang, W. Huang, Y. Li, J. Hong, A. S. Hoffman, S. R. Bare, M. H. Engelhard, A. K. Datye and Y. Wang, Nat. Commun., 2023, 14, 2664 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S6 present the most stable isomers for multiple oxygen adsorption on RexPty clusters. Fig. S7–S12 show the most stable isomers for CO adsorption on oxidized RexPty clusters. Fig. S13 and S14 show the most stable isomers for multiple CO adsorption on RexPty clusters. Fig. S15–S22 present the projected densities of states for oxidized RexPty clusters. See DOI: https://doi.org/10.1039/d5cp00995b |
| This journal is © the Owner Societies 2025 |