
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Density functional benchmark for quadruple hydrogen bonds†
Usman
Ahmed
 a,
Mikael P.
Johansson
ab,
Susi
Lehtola
a and
Dage
Sundholm
*a
a,
Mikael P.
Johansson
ab,
Susi
Lehtola
a and
Dage
Sundholm
*a
aDepartment of Chemistry, University of Helsinki, P.O. Box 55, A. I. Virtasen aukio 1, FI-00014, Finland. E-mail: susi.lehtola@helsinki.fi; sundholm@chem.helsinki.fi
bCSC-IT Center for Science Ltd., P.O. Box 405, FI-02101 Espoo, Finland. E-mail: mikael.johansson@csc.fi
First published on 3rd April 2025
Abstract
Hydrogen bonding is an important non-covalent interaction that plays a major role in molecular self-organization and supramolecular structures. It can be described accurately with ab initio quantum chemical wave function methods, which become computationally expensive for large molecular assemblies. Density functional theory (DFT) offers a better balance between accuracy and computational cost, and can be routinely applied to large systems. A large number of density functional approximations (DFAs) has been developed, but their accuracy depend on the application, necessitating benchmark studies to guide their selection for use in applications. Some of us have recently determined highly accurate hydrogen bonding energies of 14 quadruply hydrogen-bonded dimers by extrapolating coupled-cluster energies to the complete basis set limit as well as extrapolating electron correlation contributions with a continued-fraction approach [U. Ahmed et al., Phys. Chem. Chem. Phys., 2024, 26, 24470–24476]. In this work, we study the reproduction of these bonding energies at the DFT level using 152 DFAs. The top ten density functional approximations are composed of eight variants of the Berkeley functionals both with and without dispersion corrections, and two Minnesota 2011 functionals augmented with a further dispersion correction. We find the B97M-V functional with the non-local correlation functional replaced by an empirical D3BJ dispersion correction to be the best DFA, while changes to the dispersion part in other Berkeley functionals lead to poorer performance in our study.
1 Introduction
Molecular self-assembly is a spontaneous process where disordered molecules organize into ordered structures without external influence.2,3 The process leads to large supramolecular architectures, which are complexed through non-covalent interactions that guide the self-assembly. Understanding the complex self-assembly mechanism is necessary for the design of large supramolecular systems.Hydrogen bonding is one of the most important non-covalent interactions governing the self-assembly due to its strength, selectivity, and directionality.4,5 The significance of hydrogen bonding for stabilizing molecules and biomolecules is well known from, e.g., its role in the structure of deoxyribonucleic acid (DNA). The strength of hydrogen bonding depends strongly on the number of bonds, each hydrogen bond increasing the strength of the bonding. Molecules with four hydrogen bonds per molecule are examples of multiple hydrogen bonding in self-organized systems. Self-complementarity enables identical molecules to interact and form larger and uniform supramolecular structures. Self-complementary arrays of four hydrogen bonds can bind together in either a DDAA–AADD or a DADA–ADAD bonding motif, where D and A denote hydrogen bond donors and acceptors, respectively. These motifs are particularly consequential for designing stable assemblies with a given structure.
The quadruply hydrogen bonded systems having self-complementary DDAA–AADD or DADA–ADAD motifs have been of scientific interest during the past few decades.6–18 According to the traditional view, dimers with the DDAA–AADD motif have four attractive secondary A⋯D interactions leading to stronger hydrogen bonding than in dimers with the DADA–ADAD motif, where all secondary interactions have been considered to be repulsive.19 However, some of us showed recently that dimers with the DDAA–AADD motif have five attractive secondary interactions (A⋯D and A⋯A) and those with the DADA–ADAD motifs have three attractive secondary interactions, because the secondary acceptor–acceptor (A⋯A) interactions are also attractive.1,20
Accurate computational analysis of the strength and stability of supramolecular systems with hydrogen bonding is challenging, because the molecules are large. While ab initio correlated methods do offer an accurate description of hydrogen bonding, the rapidly increasing computational costs hamper computational studies of large molecules. Kohn–Sham density functional theory (DFT)21 has become the method of choice for studying such systems.22 DFT is widely used due to its cost effectiveness and availability in all modern quantum chemical software packages. The exact exchange–correlation functional is not known, and more than 600 density functional approximations (DFAs) have so far been proposed and implemented.23 Since the accuracy of the DFAs varies depending on the application, it is not obvious which functional should be used for studying molecules with multiple hydrogen bonds.
The recent work by Montero de Hijes et al.24 on the density isobars of water and the melting temperature of ice based on machine learned density functional potential energy surfaces underlines the need for reinvestigating DFT models of hydrogen bonds. Their study found small changes in the semi-empirical dispersion correction in DFT calculations to result in large changes in the bulk properties of water and ice. As multiple hydrogen bonding carries a significant role for the structure of water, identifying functionals (which include dispersion corrections) that work especially well for modeling highly hydrogen bonded systems is useful to guide future studies of complex liquids and other systems where hydrogen-bonding interactions play a major role.
The accuracy of various DFAs for modeling hydrogen bonds has been evaluated in a number of works.25–29 More recent DFA benchmark studies of hydrogen-bonding strengths have employed the S22, S66 and S66x8 datasets.30–45 The pronounced relevance of hydrogen bonding in chemistry and their DFT modeling is underlined by the fact that hydrogen bond strengths have been included in the fitting of the Minnesota functionals since their first versions,29,46,47 for example.
In this work, we study how well various DFAs reproduce the hydrogen bonding in 14 quadruply hydrogen-bonded dimers with N⋯H⋯O interactions. Half of the molecules exhibit the DDAA–AADD hydrogen-bonding motif and the other half has the DADA–ADAD motif. We consider 152 DFAs including the Hartree–Fock (HF) method. The employed dispersion correction schemes include Grimme's empirical dispersion corrections (DFT-D3,48 DFT-D449) and a non-local (NL) correlation functional (VV10).50
Some of us have recently reported accurate reference values for the molecules studied in this work, which were calculated with domain-based local pair natural orbital coupled-cluster theory at the singles, doubles and perturbative triples DLPNO-CCSD(T) level extrapolated to the complete basis set limit.1 Ahmed et al.1 also extrapolated the reference energies towards the Schrödinger limit following Goodson,51 producing the data that we use as reference in this work.
This work is outlined as follows. The computational details are given in Section 2 along with the list of the studied DFAs. The obtained results are presented in Section 3: basis-set convergence is discussed in Section 3.1, and the results are analyzed in Section 3.2. The work is summarized and its main conclusions are drawn in Section 4.
2 Computational methods
The benchmark considers the 14 dimers of the study of Ahmed et al.1 that determined hydrogen-bonding energies for TPSSh-D3/def2-TZVPP optimized geometries at the DLPNO-CCSD(T) level of theory extrapolated to the complete basis set limit, as well as towards the Schrödinger limit using the continued-fraction (cf) scheme of Goodson.51 We denote the reference level of theory as CCSD(T)-cf. The reference energies and molecular structures of the dimers reported in the ESI† are taken from Ahmed et al.1 The molecular structures of the monomers with the DADA and DDAA motifs are shown in Fig. 1 and 2, respectively. | ||
| Fig. 1 The monomers of the quadruple hydrogen-bonded dimers with DADA–ADAD hydrogen-bonding pattern. The previously studied DADA071 is omitted because the CCSD(T)-cf reference data are not available. The figures are made with Jmol.52 | ||
 | ||
| Fig. 2 The monomers of the quadruple hydrogen-bonded dimers with the DDAA–AADD hydrogen-bonding pattern. The previously studied DDAA011 is omitted because the CCSD(T)-cf reference data are not available. The figures are made with Jmol.52 | ||
The hydrogen-bonding energies were calculated in this work at the DFT level of theory with versions 1.7 and 1.9 of the Psi4 program,53 employing the fixed TPSSh-D3/def2-TZVPP geometries of Ahmed et al.1 We used the systematic Karlsruhe family of basis sets to study the convergence of the hydrogen-bonding energies towards the complete basis set limit. We used split-valence polarized (def2-SVP), triple-ζ valence polarized (def2-TZVP) or double valence polarized (def2-TZVPP), as well as quadruple-ζ valence with a double set of polarization functions (def2-QZVPP) basis sets in this study.54 We note here that since the considered molecules only contain the H, C, N, O, and S atoms, the def2-QZVP basis is identical to def2-QZVPP and only the latter is therefore considered, while the difference in def2-TZVP and def2-TZVPP is limited to a larger basis set for H in def2-TZVPP. The effect of diffuse basis functions was also studied with the corresponding basis sets (def2-SVPD, def2-TZVPD, def2-TZVPPD, and def2-QZVPPD).55
The hydrogen-bonding energies were corrected for basis set superposition errors (BSSE) calculated using the counterpoise correction method.56,57 The BSSE was obtained by comparing the energies of the two monomers of the dimer structure calculated using the dimer basis set to the monomer energies calculated using the monomer basis sets and the same molecular structure. The BSSE correction was added to the hydrogen-bonding energy calculated for fully optimized monomers using the basis set of the monomers.
The calculations were performed using the default (75,302) exchange–correlation grid in Psi4, which uses 75 radial grid points placed following Treutler and Ahlrichs,58 and an angular Lebedev grid with 302 points.59 The VV10 non-local correlation functional50 was likewise evaluated on the corresponding (50,146) default grid. The resolution-of-the-identity approximation was used in all calculations with the universal Hartree–Fock auxiliary basis sets.60
The 75 base DFAs considered in this work, which have been listed in Table 1, comprise 14 semi-local generalized gradient approximation (GGA) functionals, 21 global hybrid (GH) GGA functionals, 13 range-separated (RS) GGA functionals, 13 semi-local meta-GGA (mGGA) functionals, 8 GH mGGA functionals, and 5 RS mGGA functionals, as well as HF (a GH functional without a semi-local exchange–correlation part). All of the density functionals were evaluated with Libxc,23 as usual in Psi4. Versions 6.0.0 and 6.2.2 of the library were employed. We note that some functionals such as B3LYP, HSE03, and HSE06 have various definitions in the literature,61 which is avoided through the use of the standard implementations in Libxc. Table 1 also shows the empirical (DFT-D3,48 DFT-D449) dispersion corrections or non-local correlation (VV1050) additionally considered with some of the functionals. Various combinations of these dispersion models (or the lack thereof) with the 75 base DFAs yield the 152 DFAs investigated in this study.
| Functional | Type | D3 | D4 | NL | Ref. | Functional | Type | D3 | D4 | NL | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Empirical dispersion with Becke–Johnson145 3-body damping. b The functional was parametrized with Grimme's original dispersion correction, which is not available in Psi4, so the calculations with this functional do not include dispersion. c Empirical dispersion with Becke–Johnson145 2-body damping. | |||||||||||
| B97-D3 | GGA | 62 | 62 | ωB97 | RS GGA | 63 | |||||
| BLYP | GGA | 48 | 49 | 64 | 65–67 | ωB97X | RS GGA | 49 | 63 | ||
| BOP | GGA | 48 | 68 | ωB97X-Db | RS GGA | 69 | |||||
| BP86 | GGA | 35 | 70 | 65 and 71 | ωB97X-V | RS GGA | 72 | 73 | |||
| CHACHIYO | GGA | 74 and 75 | ωPBE | RS GGA | 48 | 76 and 77 | |||||
| KT1 | GGA | 78 | CAM-B3LYP | RS GGA | 48 | 79 | |||||
| KT2 | GGA | 78 | CAMh-B3LYP | RS GGA | 80 | ||||||
| KT3 | GGA | 81 | CAM-QTP-00 | RS GGA | 82 | ||||||
| N12 | GGA | 83 | 84 | CAM-QTP-01 | RS GGA | 85 | |||||
| PBE | GGA | 48 | 49 | 70 | 86 and 87 | CAM-QTP-02 | RS GGA | 88 | |||
| revPBE | GGA | 48 | 49 | 64 | 89 | HSE03 | RS GGA | 48 | 49 | 90 and 91 | |
| rPBE | GGA | 48 | 49 | 92 | HSE06 | RS GGA | 48 | 49 | 90, 91 and 93 | ||
| PW91 | GGA | 94 | 49 | 95 and 96 | N12-SX | RS GGA | 83 | 97 | |||
| XLYP | GGA | 48 | 49 | 98 | B97M-V | mGGA | 72 | 99 | |||
| HF | GH | 48 | 49 | M06-L | mGGA | 100 | |||||
| B1LYP | GH GGA | 48 | 49 | 101 | revM06-L | mGGA | 102 | ||||
| B3LYP | GH GGA | 48 | 49 | 64 | 103 | M11-L | mGGA | 83 | 104 | ||
| revB3LYP | GH GGA | 105 | MN12-L | mGGA | 83 | 106 | |||||
| B3PW91 | GH GGA | 48 | 64 | 107 | MN15-L | mGGA | 108 | ||||
| B3P86 | GH GGA | 35 | 107 | MVS | mGGA | 109 and 110 | |||||
| B97 | GH GGA | 48 | 111 | SCAN | mGGA | 48 | 49 | 112 | |||
| B97-1 | GH GGA | 35 | 113 | rSCAN | mGGA | 114 | |||||
| B97-2 | GH GGA | 35 | 115 | r2SCAN | mGGA | 48 | 116 | 117 and 118 | |||
| B97-3 | GH GGA | 119 | TASK | mGGA | 120 and 121 | ||||||
| BHandH | GH GGA | 122 | TPSS | mGGA | 48 | 49 | 64 | 123 and 124 | |||
| BHandHLYP | GH GGA | 65 and 122 | revTPSS | mGGA | 48 | 49 | 125 | 110 and 126 | |||
| BMK | GH GGA | 48 | 127 | M06 | GH mGGA | 31 | |||||
| HCTH120 | GH GGA | 48 | 128 | revM06 | GH mGGA | 129 | |||||
| HCTH407 | GH GGA | 48 | 130 | M06-2X | GH mGGA | 31 | |||||
| MPW1PW | GH GGA | 48 | 49 | 131 | M08-HX | GH mGGA | 132 | ||||
| O3LYP | GH GGA | 48 | 49 | 133 and 134 | M08-SO | GH mGGA | 132 | ||||
| PBE0 | GH GGA | 48 | 49 | 70 | 135 and 136 | MN15 | GH mGGA | 48 | 137 | ||
| revPBE0 | GH GGA | 48 | 49 | 64 | 86, 87 and 89 | TPSSh | GH mGGA | 48 | 49 | 27 | |
| QTP17 | GH GGA | 138 | revTPSSh | GH mGGA | 48 | 49 | 139 | ||||
| SOGGA11-X | GH GGA | 83 | 140 | ωB97M-V | RS mGGA | 72 | 141 | ||||
| X3LYP | GH GGA | 48 | 49 | 98 | M06-SX | RS mGGA | 142 | ||||
| M11 | RS mGGA | 48 | 143 | ||||||||
| revM11 | RS mGGA | 144 | |||||||||
| MN12-SX | RS mGGA | 83 | 97 | ||||||||
The study by Ahmed et al.1 showed that solvent effects increase the hydrogen-bonding energy of the tautomers DADA08 and DDAA06 by only 4–5%. We have not considered any solvent effects in this work because they are relatively small. Solvent effects are not expected to significantly affect the ranking of the DFAs.
3 Results
3.1 Basis-set convergence
We start the analysis by determining the basis-set convergence of the hydrogen bonding energies. We use the B97M-D3 functional and use the largest basis set considered (def2-QZVPPD) to determine reference values for this part of the study. The resulting basis-set truncation errors in the hydrogen-bonding energies are shown in Fig. 3 as a function of basis set.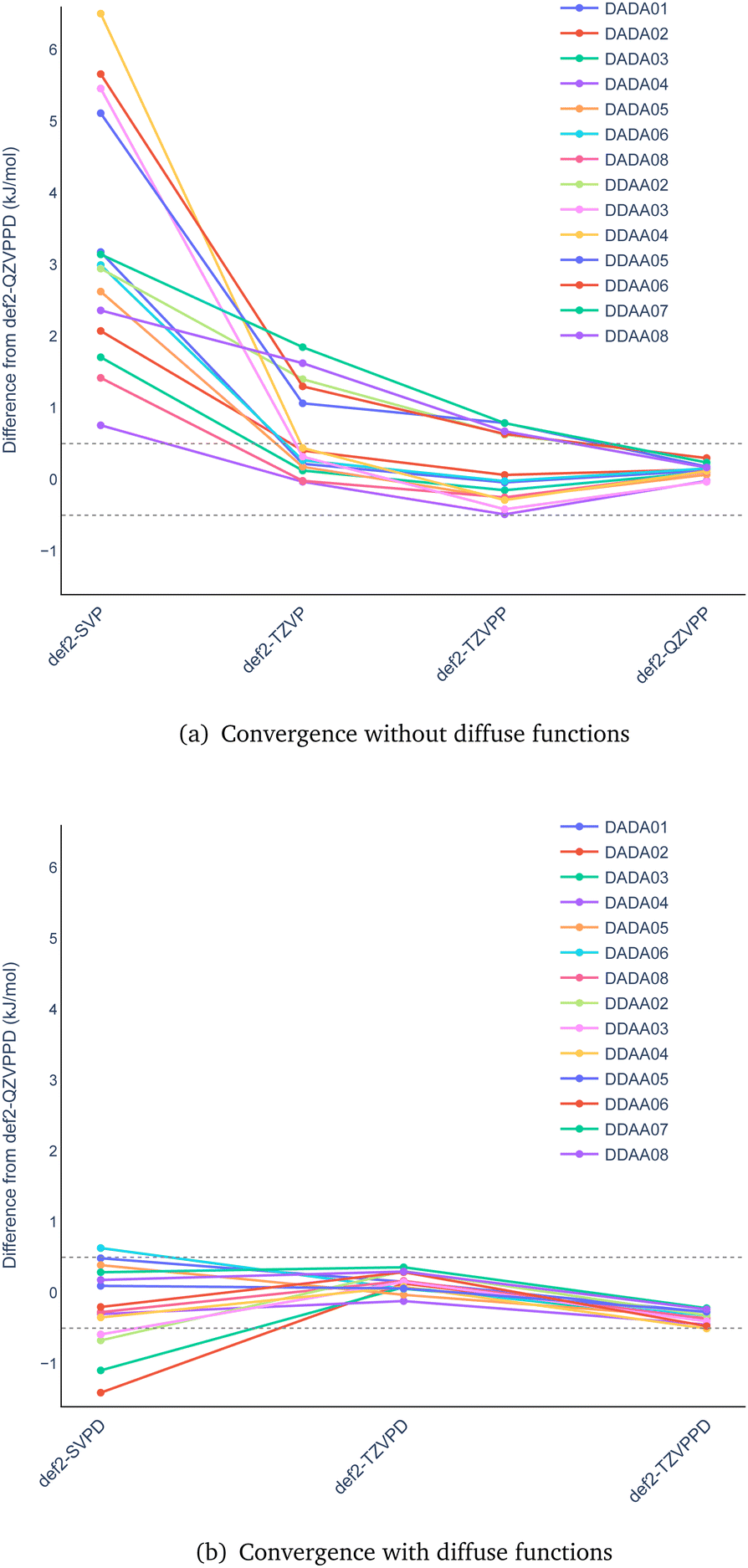 | ||
| Fig. 3 Basis-set convergence plot of the hydrogen-bonding energies relative to B97M-D3/def2-QZVPPD values calculated (a) without and (b) with diffuse basis functions. The figures show an energy window of ±0.5 kJ mol−1 as horizontal dashed lines. | ||
Fig. 3(a) shows the convergence for the basis sets without diffuse functions. The truncation errors decrease systematically, as the basis set is increased from split-valence with polarization to polarized valence triple- and quadruple-ζ. The convergence is relatively slow with the cardinal number, and not all triple-ζ results are not yet converged to ±0.5 kJ mol−1 precision.
Fig. 3(b) shows the analogous results including diffuse functions. The range of truncation errors for the polarized split-valence basis sets drops significantly from ≈0.8–7 kJ mol−1 with def2-SVP in Fig. 3(a) to ≈−1.5–0.5 kJ mol−1 with def2-SVPD; this highlights the need to include diffuse functions for describing non-covalent bonding, as in the present hydrogen bonded systems. A convergence to within ±0.5 kJ mol−1 precision with respect to the basis set is already achieved with the def2-TZVPD basis set. It is thus clear from the data in Fig. 3 that the def2-QZVPPD basis set should afford benchmark accuracy hydrogen-bond energies suitable for the present study, and we will use the def2-QZVPPD basis set for the rest of the study.
3.2 The best density functional approximations
The density functional approximations were ranked based on their mean absolute errors (MAE). The ranking is shown in Tables 2 and 3 together with the corresponding MAEs and maximum absolute errors (MAX). The lowest MAE is obtained with the B97M-D3 functional, which is the B97M-V functional where the VV10 nonlocal correlation functional (indicated by -V in the functional's name) has been replaced with an empirical D3 dispersion correction; the original B97M-V functional is ranked 4th. The second-lowest MAE is achieved with the ωB97M-V functional, which is a RS GGA functional again with VV10 dispersion. Replacing VV10 with the D3 dispersion correction drops the ranking to the 7th place.| Rank | Functional | MAE | MAX | Rank | Functional | MAE | MAX |
|---|---|---|---|---|---|---|---|
| 1 | B97M-D3 | 1.82 | 5.24 | 39 | BLYP-D4 | 6.36 | 14.27 |
| 2 | ωB97M-V | 2.06 | 5.45 | 40 | TPSS-D3 | 6.37 | 12.98 |
| 3 | M11-L-D3 | 2.13 | 5.22 | 41 | M08-HX | 6.43 | 10.81 |
| 4 | B97M-V | 2.17 | 6.71 | 42 | ωB97X-D4 | 6.67 | 11.55 |
| 5 | ωB97 | 2.21 | 5.86 | 43 | revTPSS-NL | 6.73 | 12.11 |
| 6 | ωB97X-V | 2.43 | 6.09 | 44 | r2SCAN-D4 | 7.07 | 13.55 |
| 7 | ωB97M-D3 | 2.48 | 5.24 | 45 | TPSSh-D3 | 7.15 | 12.97 |
| 8 | ωB97X-D3 | 3.09 | 6.25 | 46 | r2SCAN-D3 | 7.29 | 13.63 |
| 9 | ωB97X | 3.11 | 7.98 | 47 | HF-D4 | 7.31 | 14.14 |
| 10 | M11-D3 | 3.17 | 7.27 | 48 | PBE-D3 | 7.65 | 14.64 |
| 11 | revTPSSh-D3 | 3.20 | 7.55 | 49 | PW91-D3 | 7.70 | 14.23 |
| 12 | SCAN | 3.43 | 8.91 | 50 | revM11 | 7.98 | 13.51 |
| 13 | revTPSS-D3 | 3.48 | 9.21 | 51 | TPSS-D4 | 7.99 | 15.50 |
| 14 | revPBE-NL | 3.64 | 8.94 | 52 | B1LYP-D3 | 8.12 | 12.75 |
| 15 | revTPSS-D4 | 3.65 | 9.64 | 53 | M06-L | 8.26 | 13.41 |
| 16 | revTPSSh-D4 | 3.99 | 8.98 | 54 | N12-SX | 8.29 | 25.27 |
| 17 | XLYP-D3 | 4.21 | 10.57 | 55 | MN12-SX-D3 | 8.45 | 14.75 |
| 18 | ωPBE-D3 | 4.24 | 12.62 | 56 | BMK-D3 | 8.49 | 14.50 |
| 19 | rPBE-D4 | 4.30 | 10.82 | 57 | CAM-QTP-01 | 8.54 | 20.80 |
| 20 | B97-D3 | 4.45 | 12.54 | 58 | BLYP-NL | 8.66 | 13.94 |
| 21 | BLYP-D3 | 4.58 | 11.96 | 59 | TPSSh-D4 | 8.82 | 14.81 |
| 22 | XLYP-D4 | 4.72 | 11.76 | 60 | PBE-D4 | 9.26 | 16.42 |
| 23 | BOP-D3 | 4.73 | 12.09 | 61 | HSE03 | 9.35 | 29.92 |
| 24 | HCTH407-D3 | 4.78 | 15.06 | 62 | SCAN-D4 | 9.75 | 16.54 |
| 25 | revPBE-D4 | 4.83 | 12.10 | 63 | M06 | 9.85 | 16.87 |
| 26 | HCTH120-D3 | 4.84 | 14.53 | 64 | B3PW91-D3 | 9.92 | 16.30 |
| 27 | rPBE-D3 | 5.03 | 16.16 | 65 | M11 | 9.98 | 14.92 |
| 28 | revPBE0-D3 | 5.05 | 8.89 | 66 | B3LYP-D3 | 10.05 | 14.89 |
| 29 | M06-2X | 5.12 | 9.73 | 67 | B1LYP-D4 | 10.09 | 14.61 |
| 30 | revPBE-D3 | 5.37 | 15.66 | 68 | MPW1PW-D3 | 10.14 | 16.95 |
| 31 | revPBE0-NL | 5.42 | 11.61 | 69 | MN12-L-D3 | 10.25 | 17.57 |
| 32 | CAM-QTP-00 | 5.53 | 15.23 | 70 | PW91-D4 | 10.48 | 17.06 |
| 33 | rSCAN | 5.56 | 16.54 | 71 | PBE-NL | 10.65 | 16.64 |
| 34 | B97-1-D3 | 5.59 | 10.42 | 72 | MPW1PW-D4 | 10.74 | 17.73 |
| 35 | MVS | 5.66 | 13.53 | 73 | HSE06 | 10.74 | 31.54 |
| 36 | B97-2-D3 | 5.89 | 10.47 | 74 | PBE0-D3 | 10.82 | 17.40 |
| 37 | r2SCAN | 5.93 | 15.69 | 75 | SCAN-D3 | 10.88 | 17.64 |
| 38 | revPBE0-D4 | 6.01 | 11.03 | 76 | TPSS-NL | 10.88 | 16.88 |
| Rank | Functional | MAE | MAX | Rank | Functional | MAE | MAX |
|---|---|---|---|---|---|---|---|
| 77 | B3LYP-D4 | 11.05 | 15.90 | 115 | BMK | 19.84 | 31.71 |
| 78 | BP86-D3 | 11.10 | 19.82 | 116 | X3LYP | 19.96 | 40.69 |
| 79 | revM06 | 11.20 | 15.90 | 117 | N12 | 20.22 | 48.75 |
| 80 | X3LYP-D3 | 11.26 | 16.17 | 118 | CAMh-B3LYP | 20.44 | 41.43 |
| 81 | HSE06-D3 | 11.35 | 17.78 | 119 | ωB97X-D | 20.59 | 42.63 |
| 82 | B3PW91-NL | 11.60 | 18.99 | 120 | MN12-L | 20.79 | 27.11 |
| 83 | X3LYP-D4 | 11.68 | 16.45 | 121 | MPW1PW | 21.09 | 47.74 |
| 84 | CAM-B3LYP-D3 | 11.78 | 17.57 | 122 | TPSSh | 23.77 | 50.62 |
| 85 | PBE0-D4 | 11.90 | 18.31 | 123 | KT3 | 23.86 | 51.05 |
| 86 | CAM-B3LYP | 12.10 | 29.56 | 124 | ωPBE | 23.91 | 48.60 |
| 87 | M08-SO | 12.13 | 19.50 | 125 | revTPSSh | 24.32 | 46.15 |
| 88 | SOGGA11-X-D3 | 12.13 | 17.82 | 126 | TPSS | 25.91 | 53.91 |
| 89 | B3LYP-NL | 12.18 | 17.31 | 127 | B3LYP | 26.54 | 50.90 |
| 90 | PBE0-NL | 12.22 | 19.09 | 128 | revTPSS | 26.68 | 49.15 |
| 91 | HSE03-D3 | 12.53 | 19.42 | 129 | BP86 | 27.33 | 58.38 |
| 92 | MN15-D3 | 12.64 | 18.31 | 130 | B1LYP | 28.11 | 51.93 |
| 93 | PW91 | 12.80 | 35.00 | 131 | B97-0 | 28.81 | 53.34 |
| 94 | PBE0 | 13.04 | 34.52 | 132 | O3LYP-D4 | 29.21 | 51.57 |
| 95 | HSE06-D4 | 13.12 | 19.58 | 133 | O3LYP-D3 | 29.77 | 52.14 |
| 96 | MN15 | 13.33 | 19.31 | 134 | B3PW91 | 29.82 | 61.28 |
| 97 | MN15-L | 13.33 | 19.31 | 135 | QTP-17 | 30.03 | 47.11 |
| 98 | CAM-QTP-02 | 13.66 | 26.81 | 136 | B97-3 | 30.95 | 53.53 |
| 99 | M06-SX | 13.71 | 19.06 | 137 | KT1 | 31.33 | 39.56 |
| 100 | B3P86-D3 | 14.21 | 20.56 | 138 | B97-2 | 33.77 | 64.10 |
| 101 | HSE03-D4 | 14.26 | 20.90 | 139 | HCTH120 | 36.29 | 65.40 |
| 102 | BHANDHLYP | 15.21 | 33.09 | 140 | revPBE0 | 37.87 | 69.50 |
| 103 | SOGGA11-X | 15.59 | 31.93 | 141 | XLYP | 38.30 | 65.73 |
| 104 | M11-L | 15.69 | 26.51 | 142 | BLYP | 39.54 | 69.55 |
| 105 | BP86-NL | 16.09 | 23.11 | 143 | KT2 | 39.62 | 49.78 |
| 106 | N12-SX-D3 | 16.34 | 21.96 | 144 | BHANDH | 44.53 | 61.65 |
| 107 | HF-D3 | 16.63 | 25.87 | 145 | HF | 46.07 | 71.35 |
| 108 | PBE | 17.27 | 40.79 | 146 | HCTH407 | 47.72 | 79.14 |
| 109 | revM06-L | 17.67 | 26.51 | 147 | revPBE | 50.92 | 88.16 |
| 110 | B3P86 | 17.84 | 43.49 | 148 | rPBE | 51.03 | 85.81 |
| 111 | B97-1 | 17.96 | 36.89 | 149 | O3LYP | 61.36 | 102.66 |
| 112 | N12-D3 | 18.19 | 25.07 | 150 | BOP | 63.69 | 105.40 |
| 113 | revB3LYP | 19.61 | 41.55 | 151 | CHACHIYO | 63.76 | 111.80 |
| 114 | MN12-SX | 19.79 | 25.07 | 152 | TASK | 148.78 | 216.80 |
The Minnesota M11-L-D3 holds 3rd place on the list with the added D3 dispersion corrections, while the original functional parametrized without the empirical dispersion term (M11-L) holds the 104th place.
The DFAs ranked 5th to 9th are all Berkeley ωB97 (RS GGA) functionals with different dispersion correction terms. ωB97X-D3 is the ωB97X-V73 functional with a D3 Becke–Johnson48,145 dispersion correction instead of VV10,72 and it should not to be confused with the ωB97X-D3 functional of Lin et al.146 which was not studied in this work.
The Minnesota M11 functional with an additional D3 correction is on the 10th place, while the original functional without the dispersion term ranks 65th.
The ωB97X-D4 functional is ranked 42nd, which is 34 places below the rank of ωB97X-D3. This is likely because ωB97X-D4 is based on a different base functional than ωB97X-D3: it is built on top of an older functional, ωB97X instead of ωB97X-V.49 While a dispersion correction was included in the fitting of the ωB97X-V functional through the VV10 functional, ωB97X was fit without dispersion terms.
Various mGGA functionals and one revised GGA functional are ranked 11th to 16th.
Five of the six studied Berkeley functionals are ranked among the 10 best performing DFAs. mGGA and global hybrid (GH) GGA functionals also appear among the 20 most accurate functionals for hydrogen-bonding energies. The best DFAs at the GGA level are ranked 17th (XLYP-D3), 19th (rPBE-D4), 21st (BLYP-D3) and 22nd (XLYP-D4).
The error distributions for the hydrogen-bonding energies of the 36 top DFAs are shown as violin plots in Fig. 4, while violin plots for the remaining DFAs are included in the ESI.† The average errors in the hydrogen-bonding energies for the top DFAs are close to zero, except for M11-D3, whose distribution is almost completely on the negative side, implying that it systematically underestimates the hydrogen bonding energies. Similar plots are shown for the DFAs ranked 13th–24th in Fig. 4(b), where the distributions are broader, and the average is larger than zero for many DFAs, which implies that the hydrogen-bonding energies are overestimated. The error distributions for the DFAs ranked between 25th and 36th shown in Fig. 4(c) are broader and more scattered than for the higher-ranked DFAs. The violin plots for the rest of the DFAs are given in the ESI.†
 | ||
| Fig. 4 Violin plots showing the error distributions in the hydrogen-bonding energies (in kJ mol−1) for the investigated DFAs. The violin plots for the DFAs ranked 1st–12th are shown in (a). Violin plots for those ranked 13th–24th and 25th–36th are shown in (b) and (c), respectively. The reference hydrogen-bonding energies are extrapolated CCSD(T)-cf values from the literature.1 | ||
Hydrogen-bonding energies have been previously calculated in the basis-set limit at the second-order Møller–Plesset perturbation theory (MP2) level.20 Since all deviations have the same sign, MP2 systematically overestimates the hydrogen-bonding energies, whereas the deviations of the hydrogen-bonding energies calculated using the top DFAs are both positive and negative. The hydrogen-bonding energies at the MP2 level correlate linearly with the reference energies. The angular coefficient of the linear fit of the MP2 energies is 1.022 ± 0.02 with an offset of −5.4 ± 3.3 kJ mol−1. The MAE and MAX values at the MP2 level are 8.3 kJ mol−1 and 14.6 kJ mol−1, respectively, corresponding to the 55th place in the ranking list.
3.3 Meta-GGA functionals
The best performing DFAs at the mGGA level are B97M-D3 (1st), M11-L-D3 (3rd) and B97M-V (4th). SCAN and revTPSS-D3 are mGGA functionals that also perform well, holding rank 12 and 13, respectively. SCAN exhibits worse accuracy when paired with the D3 (ranked 75th) or D4 (ranked 62th) dispersion, suggesting overbinding. In contrast, the revTPSS functional improves its performance significantly by adding the empirical dispersion correction term to the functional, the rank increasing from 125th to 13th (D3) or 15th (D4), which suggests that the hydrogen bonds are too weak at the revTPSS level, which is rectified by the dispersion correction. The rest of the mGGA functionals among the first third part of the ranking list includes rSCAN (33rd), MVS (35th) and r2SCAN (37th). However, by considering the dispersion interaction using the D3 correction term, the performance of r2SCAN drops from 37th to 46th and to 44th with D4. Common mGGA functionals such as M06-L (53rd) and MN15-L (97th) are in the second third part of the ranking list, whereas M11-L without dispersion (104th), revM06-L (109th), MN12-L (120th), TPSS (126th), revTPSS (128th) and TASK (152nd) are in last third of the ranking list. The performance of the TPSS, revTPSS, MN12-L, and MN12-L functionals improves significantly when adding the empirical dispersion term. The accuracy is in a few cases improved by using D4 instead of D3, whereas using the VV10 dispersion correction leads to a lower accuracy than obtained with the D3 correction.3.4 Range-separated hybrid functionals
The Berkeley range-separated functionals perform well. The ωB97M-V is the best RS mGGA functional in the 2nd place and the Minnesota M11 functional combined with the D3 correlation term is ranked 10th. The best of the rest of the RS mGGA functionals is revM11, which is ranked 50th. Three of the five RS GGA functionals from Berkeley are ranked among the 10 best DFAs, while adding a dispersion correction to ωB97X leads to worse performace and 42nd rank. The ωB97X-D functional performs poorly in our study (119th), but this is likely caused by lack of dispersion in our calculations with this functional; see Table 1.The best RS GGA functional from Gainesville (QTP) is 32nd, whereas the rest of the RS GGA functionals are ranked between 54th and 135th. The short-range-only HSE03 and HSE06 functionals rank 61st and 73rd, respectively. Adding the dispersion D3 term to CAM-B3LYP has a very small effect on its MAE, whereas D3 improves its performance by decreasing the MAX value by 40%.
3.5 Global hybrid functionals
We have investigated the performance of 21 global hybrid GGA functionals and added the D3 dispersion terms to 16 of them. Their rank without the D3 term is in general 100th and larger. The only exceptions is PBE0 (94th). GH GGA functionals perform better when adding the D3 dispersion term. The best GH GGA functionals with the D3 correction are B97 (20th), HCTH407 (24th), HCTH120 (26th), revPBE0 (28th), B97-1 (34th) and B97-2 (36th). The ranking of the other GH GGA-D3 functionals is 52nd and larger. The use of D4 or NL dispersion corrections leads in general to a slightly less accurate functional than with D3.The best global hybrid mGGA functional without additional dispersion term is M06-2X in the 29th place. The M08-HX functional is ranked 41st, whereas the rest of the GH mGGA functionals is ranked between 63rd and 125th. The performance of the TPSSh functional improves from 122nd to 45th (D3) and 59th (D4) when adding the empirical dispersion term to the functional. Adding an empirical dispersion correction to the revTPSSh functional improves its performance significantly: the ranking of revTPSSh increases from 125th to 11th (revTPSSh-D3) or 16th (revTPSSh-D4).
MAX values generally increase with increasing MAE value. However, revPBE0-D3 and M06-2X have much smaller MAX values than some of the higher ranked DFAs. Judged from the MAX value, revPBE0-D3 and M06-2X would be ranked 12th and 17th instead of 28th and 29th based on the MAE value, respectively.
3.6 GGA functionals
The revPBE-NL and XLYP-D3 functionals are the best performing DFAs among the GGAs. Their performance drops from the 14th place to 124th, and 17th rank to 141th without dispersion corrections, respectively. The rPBE-D4 and BLYP-D3 functionals are the next ones on the GGA ranking list ranked 19th and 21st, respectively. The BOP functional places 23rd. Other GGA functionals among the first 50 on the ranking list are PBE-D3 and PW91-D3, which are ranked 48th and 49th, respectively. The rest of the GGA functionals including dispersion effects appear among the second third on the ranking lists. The ranking of HF is 145th, which improves to 46th and 107th with D3 and D4 dispersion corrections, respectively.4 Summary and conclusions
We calculated the hydrogen-bonding strength for 14 dimers using 152 density functional approximations (DFAs). The accuracy of the calculated bond strengths with various DFAs was assessed by comparison to the literature CCSD(T)-cf energies of Ahmed et al.,1 which they extrapolated to the complete basis set limit, as well as to the Schrödinger limit using the continued-fraction methodology of Goodson.51We began the work by validating the numerical reliability of our computational method by assessing the convergence of the hydrogen-bonding strengths across the Karlsruhe basis set family for the B97M-D3 functional. Taking the def2-QZVPPD values as reference, we observed that diffuse functions are important to obtain converged interaction energies, and we therefore recommend their use in modeling hydrogen bonds. As the def2-TZVPD and def2-TZVPPD values are within 0.5 kJ mol−1 of the def2-QZVPPD data, we can safely assume that the quadruple-ζ reference calculations are indeed of benchmark quality. The def2-QZVPPD basis set is often used for benchmarking in the literature, and has been employed in the fitting of the ωB97M-V functional,141 for example.
Having validated the numerical methodology, we continued with the benchmark. We found the top of the list to be dominated by the Berkeley functionals, either with or without dispersion. The most accurate DFA for hydrogen-bonding strengths was found to be B97M-D3, which is the B97M-V functional whose VV10 dispersion correction has been replaced by the empirical D3 dispersion correction. We found the accuracy of mGGA functionals to be surprisingly sensitive to the description of the dispersion energy, tentatively supporting the recent findings of Montero de Hijes et al.24 Other DFAs that accurately predicted hydrogen-bonding strengths included the range-separated ωB97M-V mGGA functional, the ωB97, ωB97X, ωB97X-V range-separated GGA functionals, as well as the semi-local M11-L-D3 mGGA and the range-separated M11-D3 mGGA functional.
Data availability
The data supporting this article have been included as part of the ESI.† The employed codes Psi4, gimp 2.10.36, and Jmol version 14 can be found at https://psicode.org, https://www.gimp.org, an https://jmol.sourceforge.net. All programs are freely available.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Academy of Finland through project numbers 314821, 340583, 350282, and 353749. It has also been financially supported by the Magnus Ehrnrooth Foundation, the Oskar Öflund Foundation, and by the Swedish Cultural Foundation in Finland. We acknowledge computational resources from CSC – IT Center for Science, Finland.Notes and references
- U. Ahmed, C. D. Daub, D. Sundholm and M. P. Johansson, Attractive acceptor–acceptor interactions in self-complementary quadruple hydrogen bonds for molecular self-assembly, Phys. Chem. Chem. Phys., 2024, 26, 24470–24476, 10.1039/D4CP02361G.
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Molecular Self-Assembly and Nanochemistry: a Chemical Strategy for the Synthesis of Nanostructures, Science, 1991, 254, 1312–1319, DOI:10.1126/science.1962191.
- D. Pochan and O. Scherman, Introduction: Molecular Self-Assembly, Chem. Rev., 2021, 121, 13699–13700, DOI:10.1021/acs.chemrev.1c00884.
- G. Cooke and V. M. Rotello, Methods of modulating hydrogen bonded interactions in synthetic host-guest systems, Chem. Soc. Rev., 2002, 31, 275–286, 10.1039/B103906G.
- A. Buckingham, J. Del Bene and S. McDowell, The hydrogen bond, Chem. Phys. Lett., 2008, 463, 1–10, DOI:10.1016/j.cplett.2008.06.060.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, Strong Dimerization of Ureidopyrimidones via Quadruple Hydrogen Bonding, J. Am. Chem. Soc., 1998, 120, 6761–6769, DOI:10.1021/ja974112a.
- V. G. H. Lafitte, A. E. Aliev, H. C. Hailes, K. Bala and P. Golding, Ureidopyrimidinones Incorporating a Functionalizable p-Aminophenyl Electron-Donating Group at C-6, J. Org. Chem., 2005, 70, 2701–2707, DOI:10.1021/jo048223l.
- P. S. Corbin and S. C. Zimmerman, Complexation-Induced Unfolding of Heterocyclic Ureas: A Hydrogen-Bonded Sheetlike Heterodimer, J. Am. Chem. Soc., 2000, 122, 3779–3780, DOI:10.1021/ja992830m.
- T. Moriuchi, T. Tamura and T. Hirao, Self-Assembly of Dipeptidyl Ureas: A New Class of Hydrogen-Bonded Molecular Duplexes, J. Am. Chem. Soc., 2002, 124, 9356–9357, DOI:10.1021/ja020098c.
- A. R. Sanford, K. Yamato, X. Yang, L. Yuan, Y. Han and B. Gong, Well-defined secondary structures, Eur. J. Biochem., 2004, 271, 1416–1425, DOI:10.1111/j.1432-1033.2004.04062.x.
- X. Zhao, X.-Z. Wang, X.-K. Jiang, Y.-Q. Chen, Z.-T. Li and G.-J. Chen, Hydrazide-Based Quadruply Hydrogen-Bonded Heterodimers. Structure, Assembling Selectivity, and Supramolecular Substitution, J. Am. Chem. Soc., 2003, 125, 15128–15139, DOI:10.1021/ja037312x.
- P. K. Baruah, R. Gonnade, U. D. Phalgune and G. J. Sanjayan, Self-Assembly with Degenerate Prototropy, J. Org. Chem., 2005, 70, 6461–6467, DOI:10.1021/jo0508705.
- P. Otte, J. Taubitz and U. Lüning, Lipophilicity Enhancing Substituents for ADDA Recognition Domains of DAAD·ADDA Heterodimers with Quadruple Hydrogen Bonds, Eur. J. Org. Chem., 2013, 2130–2139, DOI:10.1002/ejoc.201201450.
- B. Ośmiałowski, E. Kolehmainen, S. Ikonen, A. Valkonen, A. Kwiatkowski, I. Grela and E. Haapaniemi, 2-Acylamino- and 2,4-Bis(acylamino)pyrimidines as Supramolecular Synthons Analyzed by Multiple Noncovalent Interactions. DFT, X-ray Diffraction, and NMR Spectral Studies, J. Org. Chem., 2012, 77, 9609–9619, DOI:10.1021/jo301643z.
- V. G. H. Lafitte, A. E. Aliev, E. Greco, K. Bala, P. Golding and H. C. Hailes, Quadruple hydrogen bonded cytosine modules: N-1 functionalised arrays, New J. Chem., 2011, 35, 1522–1527, 10.1039/C1NJ20162J.
- E. Greco, A. E. Aliev, V. G. H. Lafitte, K. Bala, D. Duncan, L. Pilon, P. Golding and H. C. Hailes, Cytosine modules in quadruple hydrogen bonded arrays, New J. Chem., 2010, 34, 2634–2642, 10.1039/C0NJ00197J.
- A. M. Martin, R. S. Butler, I. Ghiviriga, R. E. Giessert, K. A. Abboud and R. K. Castellano, Self-complementary purines by quadruple hydrogen bonding, Chem. Commun., 2006, 4413–4415, 10.1039/B610239E.
- S. Kheria, S. Rayavarapu, A. S. Kotmale, R. G. Gonnade and G. J. Sanjayan, Triazine-Based Highly Stable AADD-Type Self-Complementary Quadruple Hydrogen-Bonded Systems Devoid of Prototropy, Chem. – Eur. J., 2017, 23, 783–787, DOI:10.1002/chem.201605208.
- P. K. Baruah and S. Khan, Self-complementary quadruple hydrogen bonding motifs: from design to function, RSC Adv., 2013, 3, 21202–21217, 10.1039/C3RA43814G.
- U. Ahmed, D. Sundholm and M. P. Johansson, The effect of hydrogen bonding on the π depletion and the π–π stacking interaction, Phys. Chem. Chem. Phys., 2024, 26, 27431–27438, 10.1039/D4CP02889A.
- W. Kohn and L. J. Sham, Self-Consistent Equations Including Exchange and Correlation Effects, Phys. Rev., 1965, 140, A1133–A1138, DOI:10.1103/PhysRev.140.A1133.
- A. D. Becke, Perspective: Fifty years of density-functional theory in chemical physics, J. Chem. Phys., 2014, 140, 18A301, DOI:10.1063/1.4869598.
- S. Lehtola, C. Steigemann, M. J. T. Oliveira and M. A. L. Marques, Recent developments in LIBXC—a comprehensive library of functionals for density functional theory, SoftwareX, 2018, 7, 1–5, DOI:10.1016/j.softx.2017.11.002.
- P. Montero de Hijes, C. Dellago, R. Jinnouchi and G. Kresse, Density isobar of water and melting temperature of ice: Assessing common density functionals, J. Chem. Phys., 2024, 161, 131102, DOI:10.1063/5.0227514.
- C. Tuma, A. D. Boese and N. C. Handy, Predicting the binding energies of H-bonded complexes: A comparative DFT study, Phys. Chem. Chem. Phys., 1999, 1, 3939–3947, 10.1039/A904357H.
- A. Rabuck and G. Scuseria, Performance of recently developed kinetic energy density functionals for the calculation of hydrogen binding strengths and hydrogen-bonded structures, Theo. Chem. Acc., 2000, 104, 439–444, DOI:10.1007/s002140000163.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes, J. Chem. Phys., 2003, 119, 12129–12137, DOI:10.1063/1.1626543.
- X. Xu and W. A. Goddard, Bonding Properties of the Water Dimer: A Comparative Study of Density Functional Theories, J. Phys. Chem. A, 2004, 108, 2305–2313, DOI:10.1021/jp035869t.
- Y. Zhao and D. G. Truhlar, Benchmark Databases for Nonbonded Interactions and Their Use To Test Density Functional Theory, J. Chem. Theory Comput., 2005, 1, 415–432, DOI:10.1021/ct049851d.
- Y. Zhao and D. G. Truhlar, Density Functionals for Noncovalent Interaction Energies of Biological Importance, J. Chem. Theory Comput., 2007, 3, 289–300, DOI:10.1021/ct6002719.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other function, Theor. Chem. Acc., 2008, 120, 215–241, DOI:10.1007/s00214-007-0310-x.
- P. Jurečka, J. Šponer, J. Černý and P. Hobza, Benchmark database of accurate (MP2 and CCSD(T) complete basis set limit) interaction energies of small model complexes, DNA base pairs, and amino acid pairs, Phys. Chem. Chem. Phys., 2006, 8, 1985–1993, 10.1039/B600027D.
- Y. Zhao and D. G. Truhlar, Density Functionals with Broad Applicability in Chemistry, Acc. Chem. Res., 2008, 41, 157–167, DOI:10.1021/ar700111a.
- L. Goerigk, H. Kruse and S. Grimme, Benchmarking Density Functional Methods against the S66 and S66x8 Datasets for Non-Covalent Interactions, ChemPhysChem, 2011, 12, 3421–3433, DOI:10.1002/cphc.201100826.
- L. Goerigk, A. Hansen, C. Bauer, S. Ehrlich, A. Najibi and S. Grimme, A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions, Phys. Chem. Chem. Phys., 2017, 19, 32184–32215, 10.1039/C7CP04913G.
- L. A. Burns, Á. Mayagoitia-Vázquez, B. G. Sumpter and C. D. Sherrill, Density-functional approaches to noncovalent interactions: A comparison of dispersion corrections (DFT-D), exchange-hole dipole moment (XDM) theory, and specialized functionals, J. Chem. Phys., 2011, 134, 084107, DOI:10.1063/1.3545971.
- J. Aragó, E. Ortí and J. C. Sancho-García, Nonlocal van der Waals Approach Merged with Double-Hybrid Density Functionals: Toward the Accurate Treatment of Noncovalent Interactions, J. Chem. Theory Comput., 2013, 9, 3437–3443, DOI:10.1021/ct4003527.
- G. A. DiLabio, E. R. Johnson and A. Otero-de-la Roza, Performance of conventional and dispersion-corrected density-functional theory methods for hydrogen bonding interaction energies, Phys. Chem. Chem. Phys., 2013, 15, 12821–12828, 10.1039/C3CP51559A.
- L. M. Roch and K. K. Baldridge, Dispersion-Corrected Spin-Component-Scaled Double-Hybrid Density Functional Theory: Implementation and Performance for Non-covalent Interactions, J. Chem. Theory Comput., 2017, 13, 2650–2666, DOI:10.1021/acs.jctc.7b00220.
- K. Remya and C. H. Suresh, Which density functional is close to CCSD accuracy to describe geometry and interaction energy of small noncovalent dimers? A benchmark study using Gaussian09, J. Comput. Chem., 2013, 34, 1341–1353, DOI:10.1002/jcc.23263.
- P. Jurečka, J. Černý, P. Hobza and D. R. Salahub, Density functional theory augmented with an empirical dispersion term. Interaction energies and geometries of 80 noncovalent complexes compared with ab initio quantum mechanics calculations, J. Comput. Chem., 2007, 28, 555–569, DOI:10.1002/jcc.20570.
- B. Brauer, M. K. Kesharwani, S. Kozuch and J. M. L. Martin, The S66x8 benchmark for noncovalent interactions revisited: explicitly correlated ab initio methods and density functional theory, Phys. Chem. Chem. Phys., 2016, 18, 20905–20925, 10.1039/C6CP00688D.
- G. Santra, E. Semidalas, N. Mehta, A. Karton and J. M. L. Martin, S66x8 noncovalent interactions revisited: new benchmark and performance of composite localized coupled-cluster methods, Phys. Chem. Chem. Phys., 2022, 24, 25555–25570, 10.1039/D2CP03938A.
- J. Řezáč, Non-Covalent Interactions Atlas Benchmark Data Sets: Hydrogen Bonding, J. Chem. Theory Comput., 2020, 16, 2355–2368, DOI:10.1021/acs.jctc.9b01265.
- J. Řezáč, Non-Covalent Interactions Atlas Benchmark Data Sets 2: Hydrogen Bonding in an Extended Chemical Space, J. Chem. Theory Comput., 2020, 16, 6305–6316, DOI:10.1021/acs.jctc.0c00715.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, Exchange-correlation functional with broad accuracy for metallic and nonmetallic compounds, kinetics, and noncovalent interactions, J. Chem. Phys., 2005, 123, 161103, DOI:10.1063/1.2126975.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions, J. Chem. Theory Comput., 2006, 2, 364–382, DOI:10.1021/ct0502763.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465, DOI:10.1002/jcc.21759.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, A generally applicable atomic-charge dependent London dispersion correction, J. Chem. Phys., 2019, 150, 154122, DOI:10.1063/1.5090222.
- O. A. Vydrov and T. Van Voorhis, Nonlocal van der Waals density functional: the simpler the better, J. Chem. Phys., 2010, 133, 244103, DOI:10.1063/1.3521275.
- D. Z. Goodson, Extrapolating the coupled-cluster sequence toward the full configuration-interaction limit, J. Chem. Phys., 2002, 116, 6948–6956, DOI:10.1063/1.1462620.
- Jmol: an open-source Java viewer for chemical structures in 3D, https://www.jmol.org.
- D. G. A. Smith, L. A. Burns, A. C. Simmonett, R. M. Parrish, M. C. Schieber, R. Galvelis, P. Kraus, H. Kruse, R. Di Remigio, A. Alenaizan, A. M. James, S. Lehtola, J. P. Misiewicz, M. Scheurer, R. A. Shaw, J. B. Schriber, Y. Xie, Z. L. Glick, D. A. Sirianni, J. S. O'Brien, J. M. Waldrop, A. Kumar, E. G. Hohenstein, B. P. Pritchard, B. R. Brooks, H. F. Schaefer, A. Y. Sokolov, K. Patkowski, A. E. DePrince, U. Bozkaya, R. A. King, F. A. Evangelista, J. M. Turney, T. D. Crawford and C. D. Sherrill, Psi4 1.4: Open-source software for high-throughput quantum chemistry, J. Chem. Phys., 2020, 152, 184108, DOI:10.1063/5.0006002.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. Rappoport and F. Furche, Property-optimized Gaussian basis sets for molecular response calculations, J. Chem. Phys., 2010, 133, 134105, DOI:10.1063/1.3484283.
- H. Jansen and P. Ros, Non-empirical molecular orbital calculations on the protonation of carbon monoxide, Chem. Phys. Lett., 1969, 3, 140–143, DOI:10.1016/0009-2614(69)80118-1.
- S. F. Boys and F. Bernardi, The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors, Mol. Phys., 1970, 19, 553–566, DOI:10.1080/00268977000101561.
- O. Treutler and R. Ahlrichs, Efficient molecular numerical integration schemes, J. Chem. Phys., 1995, 102, 346–354, DOI:10.1063/1.469408.
- V. I. Lebedev, A quadrature formula for the sphere of 59th algebraic order of accuracy, Russ. Acad. Sci. Dokl. Math., 1995, 50, 283–286 Search PubMed.
- F. Weigend, Hartree–Fock exchange fitting basis sets for H to Rn, J. Comput. Chem., 2008, 29, 167–175, DOI:10.1002/jcc.20702.
- S. Lehtola and M. A. L. Marques, Reproducibility of density functional approximations: How new functionals should be reported, J. Chem. Phys., 2023, 159, 114116, DOI:10.1063/5.0167763.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799, DOI:10.1002/jcc.20495.
- J.-D. Chai and M. Head-Gordon, Systematic optimization of long-range corrected hybrid density functionals, J. Chem. Phys., 2008, 128, 084106, DOI:10.1063/1.2834918.
- W. Hujo and S. Grimme, Performance of the van der Waals Density Functional VV10 and (hybrid)GGA Variants for Thermochemistry and Noncovalent Interactions, J. Chem. Theory Comput., 2011, 7, 3866–3871, DOI:10.1021/ct200644w.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100, DOI:10.1103/PhysRevA.38.3098.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 37, 785–789, DOI:10.1103/PhysRevB.37.785.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr, Chem. Phys. Lett., 1989, 157, 200–206, DOI:10.1016/0009-2614(89)87234-3.
- T. Tsuneda, T. Suzumura and K. Hirao, A new one-parameter progressive Colle-Salvetti-type correlation functional, J. Chem. Phys., 1999, 110, 10664–10678, DOI:10.1063/1.479012.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620, 10.1039/b810189b.
- M. K. Kesharwani, A. Karton and J. M. L. Martin, Benchmark ab Initio Conformational Energies for the Proteinogenic Amino Acids through Explicitly Correlated Methods. Assessment of Density Functional Methods, J. Chem. Theory Comput., 2016, 12, 444–454, DOI:10.1021/acs.jctc.5b01066.
- J. P. Perdew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B:Condens. Matter Mater. Phys., 1986, 33, 8822–8824, DOI:10.1103/PhysRevB.33.8822.
- A. Najibi and L. Goerigk, The Nonlocal Kernel in van der Waals Density Functionals as an Additive Correction: An Extensive Analysis with Special Emphasis on the B97M-V and ωB97M-V Approaches, J. Chem. Theory Comput., 2018, 14, 5725–5738, DOI:10.1021/acs.jctc.8b00842.
- N. Mardirossian and M. Head-Gordon, ωB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy, Phys. Chem. Chem. Phys., 2014, 16, 9904–9924, 10.1039/c3cp54374a.
- T. Chachiyo and H. Chachiyo, Simple and Accurate Exchange Energy for Density Functional Theory, Molecules, 2020, 25, 3485, DOI:10.3390/molecules25153485.
- T. Chachiyo and H. Chachiyo, Understanding electron correlation energy through density functional theory, Comput. Theor. Chem., 2020, 1172, 112669, DOI:10.1016/j.comptc.2019.112669.
- T. M. Henderson, B. G. Janesko and G. E. Scuseria, Generalized gradient approximation model exchange holes for range-separated hybrids, J. Chem. Phys., 2008, 128, 194105, DOI:10.1063/1.2921797.
- E. Weintraub, T. M. Henderson and G. E. Scuseria, Long-Range-Corrected Hybrids Based on a New Model Exchange Hole, J. Chem. Theory Comput., 2009, 5, 754–762, DOI:10.1021/ct800530u.
- T. W. Keal and D. J. Tozer, The exchange-correlation potential in Kohn–Sham nuclear magnetic resonance shielding calculations, J. Chem. Phys., 2003, 119, 3015–3024, DOI:10.1063/1.1590634.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57, DOI:10.1016/j.cplett.2004.06.011.
- Y. Shao, Y. Mei, D. Sundholm and V. R. I. Kaila, Benchmarking the Performance of Time-Dependent Density Functional Theory Methods on Biochromophores, J. Chem. Theory Comput., 2020, 16, 587–600, DOI:10.1021/acs.jctc.9b00823.
- T. W. Keal and D. J. Tozer, A semiempirical generalized gradient approximation exchange-correlation functional, J. Chem. Phys., 2004, 121, 5654–5660, DOI:10.1063/1.1784777.
- P. Verma and R. J. Bartlett, Increasing the applicability of density functional theory. IV. Consequences of ionization-potential improved exchange-correlation potentials, J. Chem. Phys., 2014, 140, 18A534, DOI:10.1063/1.4871409.
- L. Goerigk, Treating London-Dispersion Effects with the Latest Minnesota Density Functionals: Problems and Possible Solutions, J. Phys. Chem. Lett., 2015, 6, 3891–3896, DOI:10.1021/acs.jpclett.5b01591.
- R. Peverati and D. G. Truhlar, Exchange–Correlation Functional with Good Accuracy for Both Structural and Energetic Properties while Depending Only on the Density and Its Gradient, J. Chem. Theory Comput., 2012, 8, 2310–2319, DOI:10.1021/ct3002656.
- Y. Jin and R. J. Bartlett, The QTP family of consistent functionals and potentials in Kohn–Sham density functional theory, J. Chem. Phys., 2016, 145, 034107, DOI:10.1063/1.4955497.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868, DOI:10.1103/PhysRevLett.77.3865.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)], Phys. Rev. Lett., 1997, 78, 1396, DOI:10.1103/PhysRevLett.78.1396.
- R. L. A. Haiduke and R. J. Bartlett, Non-empirical exchange-correlation parameterizations based on exact conditions from correlated orbital theory, J. Chem. Phys., 2018, 148, 184106, DOI:10.1063/1.5025723.
- Y. Zhang and W. Yang, Comment on “Generalized Gradient Approximation Made Simple”, Phys. Rev. Lett., 1998, 80, 890, DOI:10.1103/PhysRevLett.80.890.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Hybrid functionals based on a screened Coulomb potential, J. Chem. Phys., 2003, 118, 8207–8215, DOI:10.1063/1.1564060.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Erratum: “Hybrid functionals based on a screened Coulomb potential” [J. Chem. Phys. 118, 8207 (2003)], J. Chem. Phys., 2006, 124, 219906, DOI:10.1063/1.2204597.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Improved adsorption energetics within density-functional theory using revised Perdew–Burke–Ernzerhof functionals, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 7413–7421, DOI:10.1103/PhysRevB.59.7413.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, Influence of the exchange screening parameter on the performance of screened hybrid functionals, J. Chem. Phys., 2006, 125, 224106, DOI:10.1063/1.2404663.
- J. R. Reimers, D. Panduwinata, J. Visser, Y. Chin, C. Tang, L. Goerigk, M. J. Ford, M. Sintic, T.-J. Sum, M. J. J. Coenen, B. L. M. Hendriksen, J. A. A. W. Elemans, N. S. Hush and M. J. Crossley, A priori calculations of the free energy of formation from solution of polymorphic self-assembled monolayers, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E6101, DOI:10.1073/pnas.1516984112.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation, Phys. Rev. B:Condens. Matter Mater. Phys., 1992, 46, 6671–6687, DOI:10.1103/PhysRevB.46.6671.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Erratum: Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 48, 4978, DOI:10.1103/PhysRevB.48.4978.2.
- R. Peverati and D. G. Truhlar, Screened-exchange density functionals with broad accuracy for chemistry and solid-state physics, Phys. Chem. Chem. Phys., 2012, 14, 16187–16191, 10.1039/c2cp42576a.
- X. Xu and W. A. Goddard, From The Cover: The X3LYP extended density functional for accurate descriptions of nonbond interactions, spin states, and thermochemical properties, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 2673–2677, DOI:10.1073/pnas.0308730100.
- N. Mardirossian and M. Head-Gordon, Mapping the genome of meta-generalized gradient approximation density functionals: The search for B97M-V, J. Chem. Phys., 2015, 142, 074111, DOI:10.1063/1.4907719.
- Y. Zhao and D. G. Truhlar, A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions, J. Chem. Phys., 2006, 125, 194101, DOI:10.1063/1.2370993.
- C. Adamo and V. Barone, Toward reliable adiabatic connection models free from adjustable parameters, Chem. Phys. Lett., 1997, 274, 242–250, DOI:10.1016/S0009-2614(97)00651-9.
- Y. Wang, X. Jin, H. S. Yu, D. G. Truhlar and X. He, Revised M06-L functional for improved accuracy on chemical reaction barrier heights, noncovalent interactions, and solid-state physics, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8487–8492, DOI:10.1073/pnas.1705670114.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 1994, 98, 11623–11627, DOI:10.1021/j100096a001.
- R. Peverati and D. G. Truhlar, M11-L: A Local Density Functional That Provides Improved Accuracy for Electronic Structure Calculations in Chemistry and Physics, J. Phys. Chem. Lett., 2012, 3, 117–124, DOI:10.1021/jz201525m.
- L. Lu, H. Hu, H. Hou and B. Wang, An improved B3LYP method in the calculation of organic thermochemistry and reactivity, Comput. Theor. Chem., 2013, 1015, 64–71, DOI:10.1016/j.comptc.2013.04.009.
- R. Peverati and D. G. Truhlar, An improved and broadly accurate local approximation to the exchange-correlation density functional: The MN12-L functional for electronic structure calculations in chemistry and physics, Phys. Chem. Chem. Phys., 2012, 14, 13171, 10.1039/c2cp42025b.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652, DOI:10.1063/1.464913.
- H. S. Yu, X. He and D. G. Truhlar, MN15-L: A New Local Exchange-Correlation Functional for Kohn–Sham Density Functional Theory with Broad Accuracy for Atoms, Molecules, and Solids, J. Chem. Theory Comput., 2016, 12, 1280–1293, DOI:10.1021/acs.jctc.5b01082.
- J. Sun, J. P. Perdew and A. Ruzsinszky, Semilocal density functional obeying a strongly tightened bound for exchange, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 685–689, DOI:10.1073/pnas.1423145112.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, L. A. Constantin and J. Sun, Workhorse Semilocal Density Functional for Condensed Matter Physics and Quantum Chemistry, Phys. Rev. Lett., 2009, 103, 026403, DOI:10.1103/PhysRevLett.103.026403.
- A. D. Becke, Density-functional thermochemistry. V. Systematic optimization of exchange-correlation functionals, J. Chem. Phys., 1997, 107, 8554–8560, DOI:10.1063/1.475007.
- J. Sun, A. Ruzsinszky and J. P. Perdew, Strongly Constrained and Appropriately Normed Semilocal Density Functional, Phys. Rev. Lett., 2015, 115, 036402, DOI:10.1103/PhysRevLett.115.036402.
- F. A. Hamprecht, A. J. Cohen, D. J. Tozer and N. C. Handy, Development and assessment of new exchange-correlation functionals, J. Chem. Phys., 1998, 109, 6264, DOI:10.1063/1.477267.
- A. P. Bartók and J. R. Yates, Regularized SCAN functional, J. Chem. Phys., 2019, 150, 161101, DOI:10.1063/1.5094646.
- P. J. Wilson, T. J. Bradley and D. J. Tozer, Hybrid exchange-correlation functional determined from thermochemical data and ab initio potentials, J. Chem. Phys., 2001, 115, 9233, DOI:10.1063/1.1412605.
- S. Ehlert, U. Huniar, J. Ning, J. W. Furness, J. Sun, A. D. Kaplan, J. P. Perdew and J. G. Brandenburg, r2SCAN-D4: Dispersion corrected meta-generalized gradient approximation for general chemical applications, J. Chem. Phys., 2021, 154, 061101, DOI:10.1063/5.0041008.
- J. W. Furness, A. D. Kaplan, J. Ning, J. P. Perdew and J. Sun, Accurate and Numerically Efficient r2SCAN Meta-Generalized Gradient Approximation, J. Phys. Chem. Lett., 2020, 11, 8208–8215, DOI:10.1021/acs.jpclett.0c02405.
- J. W. Furness, A. D. Kaplan, J. Ning, J. P. Perdew and J. Sun, Correction to “Accurate and Numerically Efficient r2SCAN Meta-Generalized Gradient Approximation”, J. Phys. Chem. Lett., 2020, 11, 9248, DOI:10.1021/acs.jpclett.0c03077.
- T. W. Keal and D. J. Tozer, Semiempirical hybrid functional with improved performance in an extensive chemical assessment, J. Chem. Phys., 2005, 123, 121103, DOI:10.1063/1.2061227.
- T. Aschebrock and S. Kümmel, Ultranonlocality and accurate band gaps from a meta-generalized gradient approximation, Phys. Rev. Res., 2019, 1, 033082, DOI:10.1103/PhysRevResearch.1.033082.
- J. P. Perdew and Y. Wang, Accurate and simple analytic representation of the electron-gas correlation energy, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249, DOI:10.1103/PhysRevB.45.13244.
- A. D. Becke, A new mixing of Hartree–Fock and local density-functional theories, J. Chem. Phys., 1993, 98, 1372–1377, DOI:10.1063/1.464304.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Climbing the Density Functional Ladder: Nonempirical Meta-Generalized Gradient Approximation Designed for Molecules and Solids, Phys. Rev. Lett., 2003, 91, 146401, DOI:10.1103/PhysRevLett.91.146401.
- J. P. Perdew, J. Tao, V. N. Staroverov and G. E. Scuseria, Meta-generalized gradient approximation: Explanation of a realistic nonempirical density functional, J. Chem. Phys., 2004, 120, 6898–6911, DOI:10.1063/1.1665298.
- H. Kruse, P. Banáš and J. Šponer, Investigations of Stacked DNA Base-Pair Steps: Highly Accurate Stacking Interaction Energies, Energy Decomposition, and Many-Body Stacking Effects, J. Chem. Theory Comput., 2018, 15, 95–115, DOI:10.1021/acs.jctc.8b00643.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, L. A. Constantin and J. Sun, Erratum: Workhorse Semilocal Density Functional for Condensed Matter Physics and Quantum Chemistry [Phys. Rev. Lett. 103, 026403 (2009)], Phys. Rev. Lett., 2011, 106, 179902, DOI:10.1103/PhysRevLett.106.179902.
- A. D. Boese and J. M. L. Martin, Development of density functionals for thermochemical kinetics, J. Chem. Phys., 2004, 121, 3405, DOI:10.1063/1.1774975.
- A. D. Boese, N. L. Doltsinis, N. C. Handy and M. Sprik, New generalized gradient approximation functionals, J. Chem. Phys., 2000, 112, 1670, DOI:10.1063/1.480732.
- Y. Wang, P. Verma, X. Jin, D. G. Truhlar and X. He, Revised M06 density functional for main-group and transition-metal chemistry, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10257–10262, DOI:10.1073/pnas.1810421115.
- A. D. Boese and N. C. Handy, A new parametrization of exchange–correlation generalized gradient approximation functionals, J. Chem. Phys., 2001, 114, 5497, DOI:10.1063/1.1347371.
- C. Adamo and V. Barone, Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models, J. Chem. Phys., 1998, 108, 664–675, DOI:10.1063/1.475428.
- Y. Zhao and D. G. Truhlar, Exploring the Limit of Accuracy of the Global Hybrid Meta Density Functional for Main-Group Thermochemistry, Kinetics, and Noncovalent Interactions, J. Chem. Theory Comput., 2008, 4, 1849–1868, DOI:10.1021/ct800246v.
- W.-M. Hoe, A. J. Cohen and N. C. Handy, Assessment of a new local exchange functional OPTX, Chem. Phys. Lett., 2001, 341, 319–328, DOI:10.1016/S0009-2614(01)00581-4.
- A. J. Cohen and N. C. Handy, Dynamic correlation, Mol. Phys., 2001, 99, 607–615, DOI:10.1080/00268970010023435.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170, DOI:10.1063/1.478522.
- M. Ernzerhof and G. E. Scuseria, Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional, J. Chem. Phys., 1999, 110, 5029–5036, DOI:10.1063/1.478401.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions, Chem. Sci., 2016, 7, 5032–5051, 10.1039/C6SC00705H.
- Y. Jin and R. J. Bartlett, Accurate computation of X-ray absorption spectra with ionization potential optimized global hybrid functional, J. Chem. Phys., 2018, 149, 064111, DOI:10.1063/1.5038434.
- G. I. Csonka, J. P. Perdew and A. Ruzsinszky, Global Hybrid Functionals: A Look at the Engine under the Hood, J. Chem. Theory Comput., 2010, 6, 3688–3703, DOI:10.1021/ct100488v.
- R. Peverati and D. G. Truhlar, Communication: A global hybrid generalized gradient approximation to the exchange-correlation functional that satisfies the second-order density-gradient constraint and has broad applicability in chemistry, J. Chem. Phys., 2011, 135, 191102, DOI:10.1063/1.3663871.
- N. Mardirossian and M. Head-Gordon, ωB97M-V: A combinatorially optimized, range-separated hybrid, meta-GGA density functional with VV10 nonlocal correlation, J. Chem. Phys., 2016, 144, 214110, DOI:10.1063/1.4952647.
- Y. Wang, P. Verma, L. Zhang, Y. Li, Z. Liu, D. G. Truhlar and X. He, M06-SX screened-exchange density functional for chemistry and solid-state physics, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2294–2301, DOI:10.1073/pnas.1913699117.
- R. Peverati and D. G. Truhlar, Improving the Accuracy of Hybrid Meta-GGA Density Functionals by Range Separation, J. Phys. Chem. Lett., 2011, 2, 2810–2817, DOI:10.1021/jz201170d.
- P. Verma, Y. Wang, S. Ghosh, X. He and D. G. Truhlar, Revised M11 Exchange-Correlation Functional for Electronic Excitation Energies and Ground-State Properties, J. Phys. Chem. A, 2019, 123, 2966–2990, DOI:10.1021/acs.jpca.8b11499.
- E. R. Johnson and A. D. Becke, A post-Hartree–Fock model of intermolecular interactions, J. Chem. Phys., 2005, 123, 024101, DOI:10.1063/1.1949201.
- Y.-S. Lin, G.-D. Li, S.-P. Mao and J.-D. Chai, Long-Range Corrected Hybrid Density Functionals with Improved Dispersion Corrections, J. Chem. Theory Comput., 2013, 9, 263–272, DOI:10.1021/ct300715s.
Footnote |
| † Electronic supplementary information (ESI) available: The Libxc identifiers of the employed functionals, violin plots for the density functional approximations ranked between 37 and 152, as well as reproduced data from Ahmed et al.:1 the employed TPSSh-D3/def2-TZVPP optimized Cartesian coordinates of the studied molecules along with visualizations of the molecular structures, as well as the coupled-cluster reference energies. See DOI: https://doi.org/10.1039/d5cp00836k |
| This journal is © the Owner Societies 2025 |