
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular dynamics simulations reveal a strong binding capacity of colossolactone H to the EGFR inactive conformation†
Duc Toan
Truong
 ab,
Kiet
Ho
c,
Chinh Tam
Thai
d,
Dung
Do Thi Mai
d,
Nguyen Minh
Tam
e,
Viet Bac T.
Phung
f and
Minh Tho
Nguyen
*ab
ab,
Kiet
Ho
c,
Chinh Tam
Thai
d,
Dung
Do Thi Mai
d,
Nguyen Minh
Tam
e,
Viet Bac T.
Phung
f and
Minh Tho
Nguyen
*ab
aLaboratory for Chemical Computation and Modeling, Institute for Computational Science and Artificial Intelligence, Van Lang University, Ho Chi Minh City, Vietnam. E-mail: toan.truongduc@vlu.edu.vn; minhtho.nguyen@vlu.edu.vn
bFaculty of Applied Technology, School of Technology, Van Lang University, Ho Chi Minh City, Vietnam
cInstitute for Computational Science and Technology (ICST), Ho Chi Minh City, Vietnam
dHanoi University of Pharmacy, 13-15 Le Thanh Tong, Hoan Kiem, Ha Noi, Vietnam
eFaculty of Basic Sciences, University of Phan Thiet, 225 Nguyen Thong, Phan Thiet City, Binh Thuan Province, Vietnam
fCenter for Environmental Intelligence and College of Engineering and Computer Science, Vin University, Gia Lam District, Hanoi, Vietnam
First published on 9th June 2025
Abstract
The major side effects of in-use drugs, such as gefitinib, erlotinib and osimetinib, have led to inherent limitations and considerable concerns regarding the use of tyrosine kinase inhibitors (TKIs) in cancer treatment. Natural compounds can effectively inhibit the expression of epidermal growth factor receptor (EGFR) tyrosine kinase protein; therefore, they are considered not only a functional product but also a substitute in cancer therapy. Based on the experimental findings of the bioactivities of natural compounds extracted from Ganoderma lucidum belonging to the family Ganodermataceae, which are commonly known as lingzhi and have been used since ancient times in Asian traditional medicine, we performed a theoretical study on the anti-tumor abilities of these colossolactone derivatives. Our work aims to understand the molecular interactions between a lactone compound and an EGFR intracellular protein, a common target in the development of TKI cancer drugs. A series of 16 colossolactone derivatives were placed in either the ATP-competition region or the allosteric active and inactive sites to explore their binding mechanism and rank their binding affinities. The latter was determined using steered molecular dynamics simulations and the umbrella sampling method. Of the 16 natural derivatives, colossolactone H was found to bind strongly to the allosteric pocket of EGFR-TKI and did not compete with the first-generation TKIs, which prefer to interact with the ATP region of the EGFR active state. The binding affinity of this lactone was 16 kcal mol−1. Our calculated results offer a rational explanation for previous experimental (in vivo) tests and promote the use of colossolactone H as a natural compound, providing an efficient synergistic drug combination for various cancer treatments.
1. Introduction
It has long been known that the epidermal growth factor receptor (EGFR) plays a crucial role in various biological processes, such as cell proliferation, differentiation and survival.1 Unfortunately, overexpression of this receptor is associated with the growth of several types of cancer.2 In the field of EGFR-targeting treatments, two main approaches have been proposed—the use of monoclonal antibodies (mAbs) as well as small compounds known as tyrosine kinase inhibitors (TKIs).3,4 mAbs such as cetuximab and panitumumab are utilized to specifically target the extracellular domain of EGFR.5 In contrast, low molecular weight TKIs are used to target the intracellular tyrosine kinase (TK) domain of the receptor.6 Although the use of TKIs has been successful in saving numerous human lives, this treatment still poses several risks, notably the development of drug resistance over time and side effects such as skin rash and diarrhea.3,7 In addition, the efficacy of these treatments can vary significantly among patients, emphasizing the need for precision medicine.8 Therefore, numerous scientific studies have been conducted, focusing on the interactions between small ligands and EGFR to overcome these inherent limitations and improve patient conditions.In 2002, Stamos et al.9 reported the first structural information on the interactions between erlotinib and the EGFR-TK domain. Their finding suggested that erlotinib can take on an active conformation of the receptor without the activation of the phosphorylation loop.9 This provided an insight into the binding mode of erlotinib and the mechanism of activation by receptor dimerization. Two years later, Wood et al.10 reported a second interactive structure of the EGFR-TK domain with the lapatinib drug, suggesting that it seemingly represents an authentic inactive form of the enzyme.10 The inactive state is caused by an outward rotation of the C-helix, which interrupts the salt-bridge connecting the active site of Lys745 and Glu762 in the C-helix. This rotation also stabilizes the structure with a helical turn, which is part of the activation loop.11 The activation of the EGFR kinase can arise from either a ligand-induced dimerization or an oncogenic mutation. When the ligand binds to the extracellular region of the receptor, the intracellular kinase domain undergoes asymmetric dimerization in which the C-lobe of one molecule binds to the N-lobe of the other. Such an interaction essentially pushes the C-helix inward, thereby kinetically establishing an active state. Another way to activate EGFR kinase is through oncogenic mutations. The N-terminal segment of the activation loop forms a narrow helix around Leu858, which stabilizes the inactive state of the EGFR-TK domain. Thus, the L858R mutation releases the C-helix, leading to the formation of the active state of the EGFR-TK domain.12,13
Clinical studies conducted over the past two decades have demonstrated the efficacy of targeted EGFR therapy using tyrosine kinase inhibitors (TKIs). It effectively inhibits tumor growth and increases the survival rates among patients.14 For now, four well-known generations of EGFR-TKIs have been developed for the treatment of non-small cell lung cancer (NSCLC) in which the EGFR acts as a primary driver gene.15 The first generation of EGFR-TKIs includes the Gefitinib drug16–18 (which was approved by the US Food and Drug Administration in 2003), Erlotinib,19 Lapatinib,20 and Icotinib.21 The second generation of EGFR-TKIs possesses a side chain that can form an irreversible bond with Cys797,22 such as the Afatinib23 and Dacomitinib24 drugs. The third generation was created using a new aminopyrimidine framework that can selectively act on mutations and irreversibly bind to the TK domain.15 Prominent representatives of this generation include the Osimertinib,25 Rociletinib,26 Olmutinib27 and Avitinib28,29 drugs. Extensive investigations are currently conducted into the fourth-generation medications whose main purpose is to address various resistant mutations, such as the C797S, T790M and L858R.30 These drugs function as EGFR allosteric inhibitors (EAI) instead of inhibiting the ATP competition of the EGFR-TK domain, such as in the case of earlier generations.31,32 In 2019, the EAI-045 was reported as the first EAI.33
However, natural products have long been a plentiful resource in the pharmaceutical industry, especially for drug innovation and upgrading. Several previous reports have pointed out that some natural derivatives can inhibit strong cancer mutations following the activation of the EGFR signaling pathways.34–36 In the context that synthetic compounds can cause side effects and drug resistance in the EGFR-TK domain, the discovery and development of EGFR-TKI compounds derived from natural sources are of great importance, especially for developing countries possessing a rich inventory of natural products.
Medicinal mushroom species belonging to the genus Ganoderma in the family Ganodermataceae have been used since ancient times in Asian traditional medicine, such as Ganoderma lucidum, commonly known as lingzhi. The health benefits of chemical derivatives from Ganoderma species were investigated, of which the lanostane triterpenoids emerged as a significant metabolite category.37 Within the Ganoderma family, the Ganoderma colossum, which stands out as a distinctive member, is characterized by yellow basidiocarps on its surface38 and possesses unique biological properties, such as antibacterial,39 anti-cancer,40 and anti-HIV41 activities. Earlier investigations conducted by Isaka and colleagues42,43 revealed that Ganoderma colossum is a rich source of distinctive triterpenoids containing a diversity of structures and physiological activities. More interestingly, the fruiting bodies of this species have been known to produce a unique lanostane-type triterpene, which is characterized by the presence of six-membered, even seven-membered, lactone rings, named colossolactones.44 The chemical structures of colossolactones are characterized by a steroidal framework, which can include variations, such as oxa-A-homo-steroidal or oxa-A,B-dihomo-steroidal structures. Until now, the structures of a series of 16 colossolactons in Ganoderma colossum have been revealed, including colossolactones A–H,44,45 colossolactones I–VIII41,46 and the newest one, namely the colossolactone J.43 The chemical structures and some related information of these 16 lactones are presented in Table 1. These compounds are further diversified through modifications in functional groups, including dihydroxy alkenyl and lactone residues, which significantly enhance their biological activities. In what follows, for the sake of simplification, the term lactone stands for colossolactone.
| Colossolactone I |

|
Colossolactone A |

|
| C30H46O3 | C32H52O5 | ||
| ID: 139584889 | ID: 10118664 | ||
| Colossolactone II |

|
Colossolactone B |

|
| C30H46O4 | C32H48O5 | ||
| ID: 139585541 | ID: 10164616 | ||
| Colossolactone III |

|
Colossolactone C |
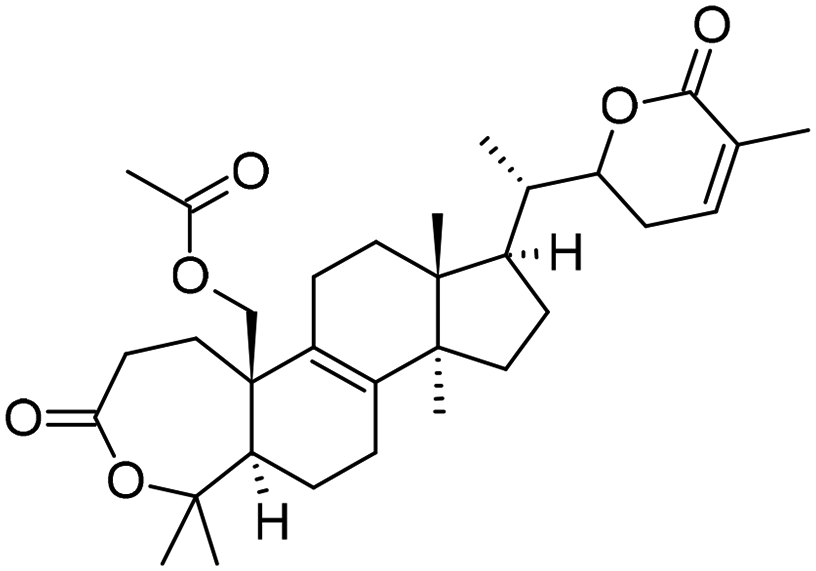
|
| C31H46O4 | C32H46O6 | ||
| ID: 139588615 | ID: 10301878 | ||
| Colossolactone IV |

|
Colossolactone D |

|
| C30H44O5 | C30H40O5 | ||
| ID: 139587125 | 10277517 | ||
| Colossolactone V |

|
Colossolactone E |

|
| C35H54O9 | C32H42O6 | ||
| ID: 24898463 | ID: 44560569 | ||
| Colossolactone VI |

|
Colossolactone F |
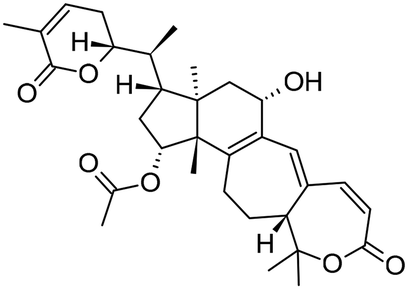
|
| C35H52O9 | C32H42O7 | ||
| ID: 24898245 | ID: 10280390 | ||
| Colossolactone VII |

|
Colossolactone G |

|
| C33H50O7 | C32H42O7 | ||
| ID: 24898464 | ID: 44560614 | ||
| Colossolactone VIII |

|
Colossolactone H |

|
| C32H42O7 | ID: C32H42O6 | ||
| ID: 24898686 |
The lactones V, VI and E (cf.Table 1) were established to exhibit inhibitory activities against the HIV-1 protease with IC50 values of 9.0, >100 and 8.0 μg mL−1, respectively.41 Although lactone E and 23-hydroxy-colossolactone E perform activities against the Bacillus subtilis and Pseudomonas syringae diseases, the B does not show any antibacterial activity.39 The E shows an antimalarial activity against Plasmodium falciparum with IC50 values of 10.0 μM,47 while the IV exerts anticancer activities against breast (MCF-7), cervix (HeLa), colorectal (HCT-116) and liver (HepG2) cancer cells with IC50 values ranging from 4.9 to 64.2 μM, and with an R-fraction of less than 47.5%.48
The lactone structures shown in Table 1 are renowned for their distinctive ability to enhance efficacy when combined with established chemotherapeutic agents. The presence of lactone G strengthens the anticancer effects of 5-fluorouracil (5-FU) and gemcitabine (GCB) in colorectal cancer cells. The combination of G with the latter drugs results in a synergistic effect, improving the cell cycle arrest and apoptosis in cancer cells.49 Beyond the treatment of colorectal cancer, lactones (G–H) have been found to reinforce the capacities of other anticancer agents, such as the gefitinib and sorafenib drugs, in the fight against non-small cell lung cancer and hepatocellular carcinoma, respectively.45,50 These findings demonstrate the possibilities of implementing these natural lactone compounds as complementary agents in various cancer therapies. They also emphasize the need for a better understanding of their chemical mechanisms and clinical activities.
Over the past decade, advances in computational methods have allowed us to quantitatively explore the interactions between small compounds and the EGFR intracellular domain51,52 and thereby obtain appropriate information, leading to relevant treatment. Numerous promising compounds were identified in various experimental studies, and their molecular level interaction dynamics were revealed through computational simulations. For instance, Nasser et al.53 discovered a novel class of pyrimidine-5-carbonitrile derivatives that inhibit the TK domain of EGFR wild-type and EGFR T790M mutations. Truong et al.54 combined umbrella sampling and steered molecular dynamics simulations to explain the EGFR inhibition mechanism of new imidazole[1,5-a]pyridine derivatives. Different teams55–57 used docking computations and molecular dynamics simulations to model the activities of new quinazoline derivatives targeted at EGFR.
In another approach, chemical derivatives isolated from natural sources were identified by computations as effective EGFR-TKIs, such as natural flavonoids, that can inhibit EGFR-TK.58 In addition, machine learning models were also applied along with docking and molecular dynamics simulations to the traditional Chinese medicine database to determine the potential candidates for EGFR-TKIs.59 Colossolactone compounds were also considered in previous computational studies by Rangsinth et al.60 and Jana et al.61 that targeted the SARS-CoV-2 main protease. Notably, assay experiments have shown that these derivatives can effectively reduce the size of cancerous tumors when they are used in combination with the conventional drug gefitinib.45 However, the atomic-level inhibition mechanism related to the properties of lactone compounds has not yet been elucidated.
In this context, motivated by the reasons mentioned above, we set out to carry out a theoretical investigation on the molecular interactions between all available colossolactone compounds, identified to our best knowledge (cf.Table 1), with the EGFR intracellular protein using computational approaches. In this study, we aim to leverage computational analyses, for the first time, on the binding capacities of a series of 16 colossolactone derivatives targeting three specific regions of the EGFR, namely, (a) the ATP-binding region of the EGFR active state, (b) the ATP-binding region of the EGFR inactive state, and (c) the allosteric region of the EGFR inactive state. In each of the 48 ligand–protein complexes considered, we follow a computational strategy in carrying out the following steps: (i) generation of initial conformation by molecular docking simulations, (ii) sampling of ligand–protein conformations by molecular dynamics (MD) simulations, (iii) evaluation of binding affinities by the steered molecular dynamic simulations, and finally (iv) computation of the absolute binding free energy using the umbrella sampling MD method.
The computed results help us open an avenue for utilizing lactone derivatives as potential scaffolds in the development of new drugs (targeting allosteric sites). More impressively, of the 16 natural derivatives displayed in Table 1, lactone H binds strongly to the allosteric pocket and does not compete with tyrosine kinase inhibitors (TKIs), which prefer only the ATP region of an EGFR active state. Overall, we offer a rational explanation for the results of previous experimental (in vivo) testing and promote the colossolactone H as a natural compound, leading to an effective use in synergistic drug combinations.45,49,50
2. Materials and methods
2.1. Receptor and ligand preparation
The EGFR tyrosine kinase domain is formed by five sub-regions: the N-terminal lobe, the C-terminal lobe, the hinge region, the ATP binding site and the allosteric pocket.62 Particularly, the N-terminal lobe contains the C-terminal helix, which is reported to play an essential role in the activation of the EGFR protein conformation change from an inactive to an active state.11 Both N-terminal and C-terminal lobes are connected by a thin hinge region where the ATP-binding site is located.63 Thr790 is known as a gatekeeper of the ATP-binding site and is associated with resistance to EGFR-TKIs, as demonstrated by T790M, one of the most common mutations.64 The allosteric pocket adjacent to the ATP binding site is present only when the C-helix loop moves outward and the EGFR protein is in an inactive conformation.13 The Lys745–Glu762 salt bridge and DFG-motif (including D855, F856 and G857) are expected to play a key role in sealing the allosteric pocket and stabilizing the active state of the EGFR.65The 3D conformations of the EGFR-TK domain are downloaded from the RCSB Protein Data Bank (RCSB PDB) under the codes 2GS6 and 2GS7, representing the active and inactive states, respectively. The missing residues are constructed using the MODELLER server66,67 after the removal of all water molecules. The geometric structures of the 16 lactone compounds considered are taken from the PubChem database,68 namely lactones I, II, III, IV, V, VI, VII, VIII, A, B, C, D, E, F, G and H, and all their corresponding 2D structures, as shown in detail in Table 1. The restricted molecular electrostatic potential69 (RESP) method is utilized to fit the atomic net charges in each compound, based on density functional theory (DFT) calculations performed using Gaussian 16 (G16) software,70 using the hybrid B3LYP functional in conjunction with the 6-31+G(d,p) basis set. AMBER Tools71 with an antechamber module is then used to convert the G16 results, including the geometries of the compounds considered, into Gromacs input files. All frontier orbitals (HOMO and LUMO) of the ligands are plotted, as illustrated in Fig. S1 of the ESI.†
Three-dimensional structures of EGFR in both active and inactive states, along with the binding positions of colossolactone H, are illustrated in Fig. 1 to highlight the conformational differences and potential interaction sites between the ligand and the protein.
 | ||
| Fig. 1 EGFR backbone is illustrated as a cyan cartoon in both the active (A) and inactive (B) and (C) conformations. The αC-helix is highlighted in red, adopting the “in” conformation in the active state (A) and “out” conformation in the inactive state (B) and (C). Key amino acid residues involved in the ligand binding region are depicted as yellow sticks. Colossolactone H is shown as a magenta stick, docked into the ATP-binding site (A) and (B) and allosteric site (C) of EGFR. | ||
2.2. Molecular docking
The receptors and ligands are prepared for docking simulations using the AutoDockTools (ADT) package implemented in the MGLTools software suite.72 AutoDock Vina script version 1.2.373 is then used to initially assess the ligand-binding pose and affinity. All lactone compounds considered are docked into the ATP binding site and allosteric pocket of the TK domain with a box size of 80 × 80 × 80 Å3, centered at the Thr790 residue for ATP-site docking and at the Asp855 residue for allosteric-site docking. The exhaustiveness and space grid amount to 200 and 0.375, respectively.2.3. Molecular dynamics simulations
GROMACS software74 version 2023.1 is now used to simulate all conformational changes in the complexes formed upon the interaction of EGFR-TK and lactone ligands. The topology parameters of the receptor are generated using the Amber99SB-ILDN force field,75 in combination with the TIP3P water model.76 The solvated protein–ligand complex and ions are placed in a rectangular box under periodic boundary conditions with a size of 10.0 × 10.0 × 10.0 nm3. The resulting system, consisting of more than 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms, is subjected to an energy minimization and convergence process to ensure that no atom experiences a maximum force exceeding 100 kJ mol−1 nm−1. To relax the system to equilibrium, 100 ps of NVT, including a constant number of particles, volume, and temperature, and 100 ps of NPT (including a constant number of particles, pressure and temperature) simulations are carried out. Finally, starting from the end of the NPT phase, 100 ns MD trajectories are produced for each complex of ligand–protein. The Langevin thermostat and the Berendsen barostat are used to restrain the temperature and pressure of the systems at 300 K and 1 atm, respectively.77,78 All hydrogen bonds are constrained using the LINCS algorithm.79 The short-range electrostatic calculations using a cut-off of 1.0 nm and the long-range electrostatic ones were treated with the Particle Mesh Ewald (PME) algorithm.80 All simulations are carried out with a time step of 2 fs, and snapshots are saved every 10 fs.
000 atoms, is subjected to an energy minimization and convergence process to ensure that no atom experiences a maximum force exceeding 100 kJ mol−1 nm−1. To relax the system to equilibrium, 100 ps of NVT, including a constant number of particles, volume, and temperature, and 100 ps of NPT (including a constant number of particles, pressure and temperature) simulations are carried out. Finally, starting from the end of the NPT phase, 100 ns MD trajectories are produced for each complex of ligand–protein. The Langevin thermostat and the Berendsen barostat are used to restrain the temperature and pressure of the systems at 300 K and 1 atm, respectively.77,78 All hydrogen bonds are constrained using the LINCS algorithm.79 The short-range electrostatic calculations using a cut-off of 1.0 nm and the long-range electrostatic ones were treated with the Particle Mesh Ewald (PME) algorithm.80 All simulations are carried out with a time step of 2 fs, and snapshots are saved every 10 fs.
To visualize the outcome data of the MD simulations method, the number of hydrogen bonds, numcount and interaction energy between the ligand and receptor were computed as performed in our previous studies.54 The free energy surface (FES) is crucial information for understanding the thermodynamics and kinetic mechanism of a bio-macromolecular complex. In this study, all FES maps are constructed by two components: the ligand's solvent accessible surface areas (SASA) and the number of contacts between the ligand and protein. These quantities are computed under the support of Gromacs packages: gmx hbond, gmx gmx sham, gmx sasa, and gmx mindist.
2.4. Steered molecular dynamics simulations
In the present work, we employ a non-equilibrium technique, known as steered molecular dynamics (SMD) simulation, to classify the relative affinity between ligand and protein. This involves the application of an external force to the center of mass (COM) of the ligand with a constant pulling speed of 1 m s−1 and a spring coefficient k of 600 kJ mol−1 nm−1. The pulling direction is determined and aligned with the z-direction of the simulation box using the CAVER program.81 All Cα atoms of the protein are constrained by a harmonic potential at k = 100 kJ mol−1 nm−1. The time-dependent force and time-dependent displacement are recorded every 10 fs. For each protein–ligand complex involved, 50 independent trajectories are fully simulated using the GROMACS package, with a simulation time of the trajectory set to 3 ns.2.5. Umbrella sampling (US) and potential of mean force (PMF) calculations
In this task, our US performance implies that several protein–ligand conformations are involved, called US windows, in which a ligand moves by 0.025 nm from adjacent windows. Although this is a reliable method for calculating equilibrium free energy, the US is not able to generate protein–ligand dissociation, and thus energy, by itself. These setups are implemented using preliminary SMD simulations at a very slow speed of 0.1 m s−1. In the case of a ligand moving every 0.025 nm, more than 120 windows are extracted. The NVT and NPT equilibrium phases are ordinarily executed before performing a 10-ns MD simulation to record the center of mass of the ligand. The free energy curve derived from the potential mean force (PMF) step is then constructed using the GROMACS package.3. Results and discussion
3.1. Initial protein–ligand binding modes generated by docking algorithms
All initial conformations of the complexes generated are obtained by docking 16 lactone derivatives, as listed in Table 1, into both the ATP region and allosteric binding region of an EGFR wide type, as illustrated in Fig. S2–S4 in the ESI.† Based on these initial structures, the images describe how 16 lactone derivatives interact with key residues located either in the ATP binding site in both active state and inactive state, or in the allosteric pocket of the EGFR intracellular domain. Two parameters, hydrogen bonds and non-bonding contacts, are identified.3.2. Colossolactone derivatives in the ATP-binding site of the EGFR active state
As depicted in Fig. S2 (ESI†), with the exception of III, C, D and E, the remaining lactones exhibit at least one hydrogen bond with the receptor when binding to the ATP region. In more detail, II, IV, VII and VIII form hydrogen bonds with Asp855, Ser720, Cys797 and Thr854, respectively. In particular, the docking results show many hydrogen bonds formed following interaction between A and five residues of EGFR, including Leu718, Glu762, Phe795, Thr854 and Asp855. Among these, two residues are frequently involved in the formation of hydrogen bonds in most cases: when colossolatone derivatives are docked in either the ATP pocket of both active and inactive states or the allosteric pocket of inactive states. The first is Thr854 with a hydrogen bond formed at the oxygen atom, and the second is Asp855 with a hydrogen bond at the nitrogen atom. In the only case of colossolactone H, one hydrogen bond is formed with the oxygen atom of Asp855. All hydrogen bonds of lactones are formed at the oxygen atom, and the range of the bond length varies from 2.77 to 3.34 Å. The non-bonding contacts, visualized using the LigPlot+ software with default parameters, show that some contacts attain rather long distances ranging from 2.90 to 3.90 Å. In addition, the docking results also point out that Leu718, Phe723, Val726, Ala743, Lys745, Met766, Thr790, Gly796, Cys797, Asp800 and Leu844 are in regular contact with each lactone.The docking energy of the lactones considered is provided in Tables 2–4 based on an analysis of the initial docked conformations. The best conformation with the lowest docking energy is subsequently extracted and further analyzed. When the EGFR is located in its active state, I, III, VII and F hold interaction energies below −9.0 kcal mol−1. Next, lactone VI has a docking energy smaller than −6.6 kcal mol−1. Despite having the largest number of hydrogen bonds, the docking energy of A (E ∼ −7.6 kcal mol−1) is larger than that of the compounds that do not form any hydrogen bond with the receptor, such as III (E = −9.4 kcal mol−1), C (E = −8.1 kcal mol−1), D (E = −8.7 kcal mol−1) and E (E = −8.1 kcal mol−1). This clearly demonstrates that no relationship between the number of hydrogen bonds and the docking energy can be established, and the same conclusion is reached in the case of non-bonding contacts.
| ATP binding site (active state) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| N 0 | Index | Docking energy (kcal mol−1) | Mindist to T790 (nm) | Number of contact | Number of hydrogen bonds | Coulomb potential (kcal mol−1) | VDW potential (kcal mol−1) | SASA (nm2) | Interaction energy (kcal mol−1) |
| Note: computed docking energies and averaged numerical data from the last 20 ns MD simulations: minimum distances between the ligands and local residue T790–Mindist (nm), number of contacts between the ligands and protein, number of hydrogen bonds formed between the ligands and protein, electrostatic potential energies, Coulomb potential (kcal mol−1), Lennard-Jones potential energies, VDW potential (kcal mol−1), solvent accessible surface areas (SASA, nm2), and interaction energy (kcal mol−1). The average value of the minimum distance between the ligand and T790 was calculated when the ligand fluctuated in the ATP region of the EGFR mutant. All 16 derivatives were stable in the EGFR binding site after 100 ns MD simulations. | |||||||||
| 1 | I | −9.1 | 0.23 | 2496 | 0.1 | −2.51 | −36.3 | 7.06 | −38.8 |
| 2 | II | −8.7 | 0.22 | 2508 | 0.52 | −1.71 | −38.1 | 7.17 | −39.8 |
| 3 | III | −9.4 | 0.22 | 2506 | 0.37 | −3.77 | −38.3 | 7.39 | −42.1 |
| 4 | IV | −8.5 | 0.31 | 2481 | 0.73 | −4.53 | −36.9 | 7.43 | −41.4 |
| 5 | V | −7.4 | 0.44 | 2477 | 0.74 | −3.06 | −37.5 | 9.28 | −40.6 |
| 6 | VI | −6.6 | 0.3 | 2449 | 1.21 | −5.11 | −32.2 | 9.37 | −37.3 |
| 7 | VII | −9.2 | 0.28 | 2478 | 0.57 | −8.47 | −37.1 | 8.52 | −45.6 |
| 8 | VIII | −7.3 | 0.24 | 2507 | 0.44 | −1.6 | −42.3 | 7.98 | −43.9 |
| 9 | A | −7.6 | 0.24 | 2533 | 0.35 | −8.06 | −42.3 | 7.9 | −50.4 |
| 10 | B | −8.1 | 0.24 | 2509 | 0.1 | −3.71 | −39.1 | 7.73 | −42.8 |
| 11 | C | −8.1 | 0.61 | 2427 | 0.47 | −1.2 | −28.5 | 7.78 | −29.7 |
| 12 | D | −8.7 | 0.26 | 2471 | 0.08 | −6.88 | −34 | 7.34 | −40.9 |
| 13 | E | −8.1 | 0.26 | 2516 | 0.05 | −1.83 | −43.1 | 7.9 | −44.9 |
| 14 | F | −9.2 | 0.48 | 2473 | 2.28 | −18.2 | −36.7 | 7.8 | −54.9 |
| 15 | G | −8.2 | 0.25 | 2483 | 0.07 | −4.31 | −37.8 | 7.87 | −42.1 |
| 16 | H | −8.5 | 0.24 | 2531 | 0.53 | −2.89 | −45.5 | 8 | −48.4 |
| ATP binding site (inactive state) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| N 0 | Index | Docking energy (kcal mol−1) | Mindist to T790 (nm) | Number of contact | Number of hydrogen bonds | Coulomb potential (kcal mol−1) | VDW potential (kcal mol−1) | SASA (nm2) | Interaction energy (kcal mol−1) |
| Note: computed docking energies and averaged numerical data from the last 20 ns MD simulations: minimum distances between the ligands and local residue T790–Mindist (nm), number of contacts between the ligands and protein, number of hydrogen bonds between the ligands and protein, electrostatic potential energies, Coulomb potential (kcal mol−1), Lennard-Jones potential energies, VDW potential (kcal mol−1), solvent accessible surface areas (SASA, nm2), and interaction energy (kcal mol−1). The average value of the minimum distance between the ligand and T790 was calculated when the ligand fluctuated in the ATP binding region of the inactive EGFR mutant. All 16 derivatives were stable in the EGFR binding site after 100 ns MD simulations. | |||||||||
| 1 | I | −10.2 | 0.25 | 2833 | 1.63 | −43.08 | −43.1 | 7.06 | −86.2 |
| 2 | II | −10.1 | 0.25 | 2422 | 0.04 | −36.76 | −36.8 | 7.04 | −73.5 |
| 3 | III | −10.2 | 0.23 | 3396 | 0.01 | −51.85 | −51.8 | 7.23 | −103.7 |
| 4 | IV | −9.2 | 1.00 | 1671 | 1.64 | −28.74 | −28.7 | 7.25 | −57.5 |
| 5 | V | −7.7 | 0.95 | 1876 | 1.04 | −34.62 | −34.6 | 8.12 | −69.3 |
| 6 | VI | −6.8 | 0.77 | 2055 | 0.95 | −34.95 | −35.0 | 7.90 | −69.9 |
| 7 | VII | −7.5 | 0.30 | 2571 | 1.23 | −40.56 | −40.6 | 8.26 | −81.1 |
| 8 | VIII | −9.0 | 0.24 | 2995 | 1.31 | −45.44 | −45.4 | 7.70 | −90.9 |
| 9 | A | −8.2 | 0.85 | 1894 | 1.39 | −29.42 | −29.4 | 7.34 | −58.8 |
| 10 | B | −9.7 | 0.67 | 2548 | 1.77 | −40.00 | −40.0 | 7.60 | −80.0 |
| 11 | C | −8.0 | 0.23 | 3118 | 0.00 | −49.03 | −49.0 | 7.66 | −98.1 |
| 12 | D | −10.2 | 0.24 | 2324 | 1.26 | −37.44 | −37.4 | 7.24 | −74.9 |
| 13 | E | −10.4 | 1.25 | 944 | 0.61 | −17.85 | −17.8 | 7.63 | −35.7 |
| 14 | F | −9.0 | 0.58 | 2255 | 0.62 | −40.42 | −40.4 | 7.91 | −80.8 |
| 15 | G | −10.2 | 0.80 | 1712 | 0.25 | −28.87 | −28.9 | 7.79 | −57.7 |
| 16 | H | −9.9 | 0.38 | 2286 | 1.15 | −36.79 | −36.8 | 7.88 | −73.6 |
| Allosteric pocket (inactive state) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| N 0 | Index | Docking energy (kcal mol−1) | Mindist to T790 (nm) | Number of contact | Number of hydrogen bonds | Coulomb potential (kcal mol−1) | VDW potential (kcal mol−1) | SASA (nm2) | Interaction energy (kcal mol−1) |
| Note: computed docking energies and averaged numerical data from the last 20 ns MD simulations: minimum distances between the ligands and local residue K745–Mindist (nm), number of contacts between ligands and protein, number of hydrogen bonds between ligands and protein, electrostatic potential energies, Coulomb potential (kcal mol−1), Lennard-Jones potential energies, VDW potential (kcal mol−1), solvent accessible surface areas (SASA, nm2), and interaction energy (kcal mol−1). The average value of the minimum distance between the ligand and T790 was calculated when the ligand fluctuated in the allosteric pocket of the EGFR mutant. All 16 derivatives considered were stable in the EGFR allosteric area after 100 ns MD simulations. | |||||||||
| 1 | I | −9.0 | 0.28 | 3282 | 0.83 | −7.47 | −50.9 | 7.13 | −58.4 |
| 2 | II | −8.3 | 0.24 | 3147 | 0.94 | −10 | −46.9 | 7.2 | −56.9 |
| 3 | III | −8.9 | 0.33 | 3196 | 0.72 | −6.88 | −51.2 | 7.38 | −58.1 |
| 4 | IV | −8.9 | 0.27 | 3138 | 0.35 | −4.55 | −49 | 7.32 | −53.6 |
| 5 | V | −6.9 | 0.71 | 2452 | 1.25 | −8.23 | −39.3 | 9.12 | −47.5 |
| 6 | VI | −7.5 | 0.59 | 2475 | 0.5 | −7.1 | −39.6 | 9.21 | −46.7 |
| 7 | VII | −7.3 | 0.81 | 2359 | 0.1 | −3.75 | −37.7 | 8.26 | −41.5 |
| 8 | VIII | −9.0 | 0.24 | 3061 | 0.11 | −7.11 | −50.2 | 7.77 | −57.3 |
| 9 | A | −7.9 | 0.56 | 3025 | 2.15 | −18.5 | −43.5 | 7.68 | −62.0 |
| 10 | B | −8.1 | 0.25 | 3234 | 0.71 | −7.35 | −50.2 | 7.82 | −57.6 |
| 11 | C | −8.4 | 0.29 | 3393 | 0.92 | −11.3 | −53.8 | 7.71 | −65.1 |
| 12 | D | −9.8 | 0.25 | 3060 | 0.51 | −5.26 | −48.7 | 7.24 | −54.0 |
| 13 | E | −9.1 | 0.22 | 3069 | 0.18 | −6.03 | −49.5 | 7.8 | −55.5 |
| 14 | F | −8.0 | 0.22 | 3109 | 0.87 | −8.3 | −50.8 | 7.92 | −59.1 |
| 15 | G | −8.6 | 0.23 | 3201 | 0.37 | −7.9 | −52 | 7.84 | −59.9 |
| 16 | H | −8.4 | 0.23 | 3270 | 0.004 | −5.31 | −53.5 | 7.88 | −58.8 |
3.3. Colossolactone derivatives in the ATP-binding site of the EGFR inactive state
Docking results obtained when EGFR is situated in its inactive state are presented in Table 3 and Fig. S3 (ESI†). At this EGFR state, the lactones involve docking energy values ranging from −6.8 to −10.4 kcal mol−1, with I, II, III, D, E and G exhibiting docking energies smaller than −10 kcal mol−1. Meanwhile, V, VI and VII attain the highest docking energy values, ranging from −6.8 to −7.7 kcal mol−1. Except for III, IV, VIII, B, C, D and F, which do not form any hydrogen bonds with EGFR, the remaining complexes are all stabilized by hydrogen bond interactions with intermolecular distances ranging from 2.74 to 3.28 Å. Similar to the case of EGFR in the active state, V and A still form the largest number of hydrogen bonds with EGFR (4 hydrogen bonds), while their docking energies are not the lowest, but they are relatively high. This again reaffirms the lack of correlation between the number of hydrogen bonds in the complex and the corresponding docking energy.3.4. Colossolactone derivatives in the allosteric pocket of the EGFR inactive state
Because the allosteric binding site now becomes a potential inhibition target to develop the fourth generation of EGFR-TKIs, we set out to focus on investigating the interactions of the 16 lactones examined (cf.Table 1) in this region. The docking energies of the lactones in the allosteric pocket of the EGFR-TK domain are ranked, as displayed in Table 4. The computed docking energies range from −6.9 to −9.8 kcal mol−1. Compared with the ATP binding site, these compounds involve fewer hydrogen bonds; most of them have only one hydrogen bond (see details in Fig. S4, ESI†). Although compounds II, IV and D each have 2 bonds, only A enjoys 3 bonds. Species I and H do not form any hydrogen bond. Phe856 is the preferred residue to form a hydrogen bond with ligands besides residue Lys745 and Thr790. All 16 compounds considered share contact with some residues, such as Lys745, Leu747, Ile759, Glu762, Met766, Leu777, Leu788, Thr854, Asp855 and Leu858. These results help us identify that this coordinate in the protein can be a potential binding site for our ligands.3.5. Molecular dynamics simulations
Receptor dynamics constitute a crucial component in the understanding of the behavior of protein–ligand complexes. Conventional docking methods, however, often disregard this aspect by treating the receptor as a rigid structure, thereby overlooking the intrinsic flexibility of the binding site during ligand recognition and accommodation. Despite this limitation, docking is still a fast and simple step in generating the complex configurations to be used for subsequent steps. We perform 100 ns MD simulations for complexes of the series of 16 lactones considered engaging with the EGFR-TK domain at two binding sites, including the ATP binding sites in both active and inactive states, and the allosteric binding site based on the docking configuration. The results obtained after MD simulations are recorded and averaged in Tables 2–4.All derivatives appear to be stabilized within the receptor binding site in view of the small average distances to the local residues, as illustrated in Table S3 (ESI†). At the ATP binding site in the active state, the number of contacts between lactones and the EGFR-TK domain amounts to over 2000, and the largest averaged number of hydrogen bonds is about 3. With EGFR in the inactive state, except for E, which involves only 944 contacts, each of the remaining lactones induces more than 1600 contacts. Regarding the number of hydrogen bonds formed, C did not form any. However, several lactones exhibit more than 1 hydrogen bond, such as colossolactones I, IV, V, VII, VIII, A, B, D and H.
Meanwhile, the number of contacts initiated by those ligands and residues in the allosteric region of the receptor goes mostly over 3000 contacts, but the number of hydrogen bonds remains not larger than 1, except for V and A, which have averages of 1.25 and 2.15 bonds, respectively. In part, the larger number of contacts suggests that the ligand is very hydrophobic. Tables 2–4 also list the SASA values of lactones within the complexes, ranging from 7000 to 9000 (nm2) at both binding sites.
The Coulombic interaction energy and Lennard-Jones energy are two well-known parameters that can be used to quantitatively evaluate the interactions between chemical objects, and they can be extracted from MD simulations. Basically, the more negative they are, the stronger the interactions between the objects, or in other words, the more they prefer to pair with each other. The last two columns of Tables 2–4 show the average values of Coulombic interaction energy and Lennard-Jones energy between the EGFR-TK domain and the lactones at the two binding sites, and all of them are actually negative values for both the ATP and allosteric sites. When the interactions of ligands occur at the ATP binding sites (both for active and inactive states) and the allosteric site, the correlation coefficients between the number of contacts and Lennard-Jones energy are up to 0.93, 0.98 and 0.94, respectively.
However, when paying attention to the minimum distances (cf. mindists) of lactones with the key residues in binding sites, we find that no ligand has a mindist value to any critical residue greater than 1.0 nm, except for the distance of 1.04 nm between lactone C and residue Leu859. Information on the distances for the series of 16 lactones at the ATP binding site is shown in Table S1 (ESI,† active state) and Table S2 (ESI,† inactive state). In particular, the mindist values of residues Gly721, Lys745, Thr790, Gln791, Leu792, Met793, Cys797 and Arg841 for the lactones are all smaller than 0.5 nm. Calculated results indicate the proximity of the residue Ala722 to lactones IV, V, VI, and VII, with mindist <0.44 nm. During the simulation, C consistently maintains a distance greater than 0.5 nm from the key residues. This implies that C may not be capable of inhibiting this protein by competing with ATP. In the inactive state, as shown in Table S2 (ESI†), most lactones maintain proximity to key residues, with mindist values being predominantly smaller than 0.5 nm. Exceptions arise for residue Leu858, where most of the lactones show distances exceeding 1.0 nm. Lactones I, III, VII and D consistently maintain tight interactions with residues Thr790, Leu792, Met793, Cys797 and Arg841, all with mindist values smaller than 0.3 nm. However, E exhibits particularly large distances to residues Thr790, Gln791, Leu792, Met793 and Leu858, ranging from 0.9 nm to over 1.5 nm, which suggests a weaker inhibitory potential in this state.
As illustrated in Table S3 (ESI†), the minimum distance between the lactones and some key residues in the allosteric region can be observed. Lys745, Ile759, Glu762 and Leu788 emerge as the preferred residues of all 16 lactones due to their mindist values of around 0.3 nm. On closer inspection, the mindist parameters of V, VI, VII and A reveal estimated values exceeding 0.5 nm. Some of the examples include the distances between lactone V and Cys775, lactone V and Thr790, and lactone V and Thr854 of 0.87, 0.72 and 0.69 nm, respectively. Both VI and A are 0.7 nm away from Cys775. Remarkably, VII always maintains quite large distances from some key residues, such as Ala763 (0.7 nm), Cys775 (1.11 nm), Leu777 (0.74 nm), Thr790 and Phe856 (0.81 nm) and Thr854 (0.82 nm).
3.6. Representative configurations
During the MD simulation processes, several snapshots are generated over time. Depending on the selected parameters, 5000 snapshots over time are then extracted to represent the dynamics of the system. Here, we use the last 50 ns of each 100 ns MD trajectory to extract representative configurations denoted by zero Gibbs (free) energy change, implying that these configurations appear most frequently at equilibrium. Fig. 2 depicts the free energy landscape of a representative structure of H; this is established from the number of contacts and SASA, as well as the interaction plot of representative snapshots. Overall, Lys745 and Thr790 are two residues that frequently appear in the interaction of ligands at both binding sites of the EGFR-TK domain. In addition, Leu718, Val726, Ala743, Cys797, Arg841 and Leu844 are the preferred residues in the ligand interactions at the ATP binding site, while the favorite amino acids of ligands in allosteric include Phe723, Ile759, Glu762, Met766, Leu777, Leu788, Asp855, Phe856, Leu858 and Ala859. The number of contacts, which is counted from representative structures in the case of ligand binding to the ATP binding site (in both active and inactive states), tends to decrease compared to the docking configurations. | ||
| Fig. 2 Free energy landscapes and representative structures are obtained from 100 ns MD simulation of colossolactone H interacting with the ATP binding region of (a) the active EGFR (upper case), (b) the inactive EGFR (mediate case) and (c) the allosteric binding region of inactive EGFR (lower case). Distances are presented in angstroms. | ||
Meanwhile, in the allosteric site, some lactones remain almost unchanged or have the contact number increased, including IV, B, C, D, E, F and G. However, the number of hydrogen bonds formed by lactone derivatives and EGFR in representative structures is also recorded. In comparison to the corresponding docking values, while the hydrogen bond in the case of ligands located at the ATP binding site is rather arbitrary, that in the allosteric region is not significantly changed. Several preferred residues induce hydrogen bond formation with ligands, such as Lys745, Met793, Arg803, and Thr854 in the ATP binding site, and Lys745, Asp855, Phe856 and Leu858 in the allosteric site.
3.7. Binding affinities of 16 colossolactones
We now evaluate the binding affinities of the 16 lactone derivatives considered using both SMD and umbrella sampling MD approaches. An external force is applied to explore the non-bonding interaction between the ligand and the receptor. To rank the binding affinity of 16 lactones to the EGFR-TK domain, including in both ATP and allosteric areas, 50 independent trajectories are carried out, and various non-equilibrium quantities are extracted. The calculated values are listed in Table 5. In simulations, the external force applied to the ligand increases with time and quickly reaches a maximum value Fmax to break the association between ligand and receptor. This causes the ligand to move far away from the EGFR binding site. A ruptured force with a higher value indicates a larger binding affinity. Commonly, the SMD approach is used as a powerful tool in drug screening.82 All of the time-dependent force, pulling work and non-equilibrium free energy data are available in the ESI,† repository.| Ligand | ATP binding region (active state) | ATP binding region (inactive state) | Allosteric binding pocket (inactive state) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N 0 | Index | F max (pN) | W pull (kcal mol−1) | ΔG‡unbind (kcal mol−1) | F max (pN) | W pull (kcal mol−1) | ΔG‡unbind (kcal mol−1) | F max (pN) | W pull (kcal mol−1) | ΔG‡unbind (kcal mol−1) |
| 1 | I | 883 | 73.9 | 56.5 | 673.1 | 58.6 | 27.4 | 494.6 | 40.9 | 19.5 |
| 2 | II | 850 | 70.1 | 51.3 | 405.8 | 25.9 | 14.7 | 523.9 | 46.3 | 19.8 |
| 3 | III | 910 | 82.6 | 58.4 | 873.2 | 79 | 55.6 | 476.0 | 42.7 | 16.1 |
| 4 | IV | 670 | 68.5 | 33.9 | 403.4 | 31.6 | 11.5 | 443.0 | 37.7 | 14.5 |
| 5 | V | 744 | 77.5 | 27.4 | 561.2 | 46.9 | 20.9 | 435.2 | 36.0 | 13.8 |
| 6 | VI | 992 | 113.0 | 69.1 | 437.2 | 31.5 | 15 | 487.0 | 46.8 | 15.5 |
| 7 | VII | 694 | 72.1 | 29.1 | 540.8 | 48.9 | 20.9 | 444.2 | 41.4 | 13.3 |
| 8 | VIII | 1130 | 119.0 | 89.9 | 531.5 | 45.9 | 20 | 754.0 | 77.4 | 41.3 |
| 9 | A | 1000 | 95.1 | 72.5 | 569 | 48.7 | 22.4 | 386.5 | 28.8 | 10.9 |
| 10 | B | 1070 | 101.0 | 82.1 | 587 | 42 | 25.9 | 523.1 | 42.4 | 22.0 |
| 11 | C | 626 | 41.6 | 28.5 | 710.3 | 63.6 | 35.8 | 478.7 | 39.3 | 18.4 |
| 12 | D | 932 | 88.6 | 60.2 | 455.7 | 36.6 | 16.6 | 472.1 | 41.3 | 16.0 |
| 13 | E | 1090 | 103.0 | 83.3 | 388.5 | 28.6 | 10.6 | 819.8 | 78.3 | 46.8 |
| 14 | F | 1190 | 120.0 | 99.0 | 500.9 | 38 | 19.1 | 729.2 | 72.9 | 36.4 |
| 15 | G | 596 | 47.1 | 25.8 | 421 | 31 | 13.6 | 798.1 | 73.6 | 47.9 |
| 16 | H | 509 | 44.7 | 16.9 | 985.5 | 92.6 | 68.3 | 917.6 | 86.8 | 61.2 |
The highest value of rupture force (cf.Table 5), in the case of interaction with the ATP binding site (active state) of the EGFR-TK domain, belongs to lactone F, with the value of Fmax = 1190 pN. In addition, some lactones with great rupture force values emerge, including VIII (Fmax = 1130 pN), E (Fmax = 1090 pN), B (Fmax = 1070 pN), and A (Fmax = 1000 pN). At the bottom of the ranking list are lactone G with a rupture force of 596 pN and lactone H with a rupture force of 509 pN.
We also calculate the pulling work, Wpull, based on the force–displacement profile. The top two pulling work values are associated with F (120 kcal mol−1) and VIII (119 kcal mol−1). Three other high values include VI (113 kcal mol−1), E (103 kcal mol−1) and B (101 kcal mol−1). Lactones G, H and C rank at the bottom of the list with the pulling work values of 47, 45 and 42 kcal mol−1, respectively. Simulated results point out that some pairs of Wpull values, such as F and VII (120 and 119 kcal mol−1), B and E (101 and 103 kcal mol−1), II and VII (70 and 72 kcal mol−1), and lactones I and VII (74 and 72 kcal mol−1), are almost indistinguishable from each other owing to the small differences when accounting for the expected error margins.
The unbinding barrier, ΔG‡unbind, calculated from Jarzynski's equation with the extension of Hummer and Szabo83 allow us to determine the phase transition point from binding to unbinding of lactones. A large range of this energy barrier value is found, ranging from 17 to 99 kcal mol−1. Similar to the case of pulling work, some unbinding barrier values overlap with each other; for example, I and III have values of 56.5 and 58.4 kcal mol−1, respectively; V, VII, and C have values of 27.4, 29.1, and 28.5 kcal mol−1, respectively; and B and E have values of 82.1 and 83.3 kcal mol−1, respectively. This overlap turns out to be a disadvantage when comparing dynamics and selecting objects. However, in the context of the present study, the overlap of the pulling work or the unbinding barrier can be interpreted because of the behavior of these compounds, which are quite similar to each other when interacting with the ATP binding region of the EGFR-TK domain.
When analyzing the results of SMD simulations for the ATP binding site (inactive state), a lower range of rupture force Fmax values are observed, spanning from 388.5 pN (lactone E) to 985.5 pN (lactone H). Notably, lactone H exhibits the highest Fmax value, thus highlighting its strong binding affinity in the inactive conformation of the ATP binding site. In terms of pulling work Wpull, its values range from 26 (II) to 93 kcal mol−1 (H). Similarly, H is characterized by the highest Wpull value, further reinforcing its stability in this binding state.
The unbinding barriers ΔG‡unbind follow a similar trend, with H leading the ranking at 68 kcal mol−1, while E has the lowest value of 11 kcal mol−1. These results suggest that lactone H enjoys the strongest interaction with the inactive ATP binding site, while other lactones, such as E and II, achieve weaker binding. The variations and overlaps among some unbinding barriers indicate similar binding behaviors for certain pairs of ligands in this conformation.
In parallel, 50 independent SMD trajectories are applied, and some dynamic parameters of 16 lactones are considered in the interaction with the allosteric binding site of the EGFR-TK domain are computed. The corresponding results are listed in Table 5. In general, the calculated values are smaller than those in the cases at the ATP binding site. The Fmax value varies from 386.5 to 917.6 pN. Surprisingly, H occupies a top position in the ranking of rupture force. Additionally, the first position in the pulling work and unbinding barrier involves H, with 87 and 61 kcal mol−1, respectively. This implies that lactone H can be regarded as an effective candidate for an allosteric inhibition of the EGFR-TK domain, instead of competition with ATP at the ATP binding site.
Following H, lactones E, G, VIII and F emerge with rather large rupture force values of 820, 798, 754 and 729 kcal mol−1, respectively. The remaining values are substantially smaller, ranging from 300 to 600 kcal mol−1. The rupture force of A is found to be the smallest value. The range of pulling work and unbinding barriers varies from 29 to 87 kcal mol−1 and from 11 to 61 kcal mol−1, respectively. Again, we encounter an overlap between some pairs of values when considering the expected error margins. This demonstrates that some lactone pairs are almost identical in their interactions with EGFR at the active sites.
The SMD simulation method is effective only when comparing the binding affinities in cases where multiple ligands consistently bind to the same region. Here, the system includes two ATP regions and one allosteric region. Hence, this makes the SMD approach useless when comparing the behaviour of a ligand, such as lactone H, when binding to different regions of EGFR. Fortunately, this inherent limitation can be overcome using the umbrella sampling (US) method when an equilibrium dissociation process can be generated. To generate reliable data, however, the US method consumes a great quantity of computational resources, as well as real simulation time, making it difficult to carry out US computations for all systems considered. Therefore, we focus only on exploring the equilibrium free energy of lactone H in both the ATP regions (active and inactive state) and the allosteric region. More than 120 windows with 10 ns each are simulated to ensure overlap between two adjacent windows in such a way that we can view the histograms plotted in Fig. 3. Free energy profiles are also constructed.
 | ||
| Fig. 3 (A) Free energy profile obtained when colossolactone H is in an interacting complex with the ATP region of the EGFR active state (in red), ATP region of the EGFR inactive state (in green) and allosteric region of EGFR inactive state (in blue). Diagrams of the umbrella sampling method are also shown in the interaction of the ligand with the ATP active state (B), ATP inactive state (C), and allosteric inactive state (D). | ||
The US method is known to be an appropriate method for calculating the free energy of non-covalent ligand–protein binding. The free energy difference between the bound state when the ligand is located in the ATP binding region, and the unbound state when the ligand is rejected far away from the EGFR can be related to the strength of the ligand–protein binding affinity. In the case of lactone H binding in the ATP binding site at the active state of EGFR, the free energy profile shows a small value of −5 kcal mol−1. This value is approximately 3 times as small as the value of −16 kcal mol−1 in the case of this derivative binding in the allosteric pocket. The inspiring value of −16 kcal mol−1 obtained using the PMF method points out the ability of H to inhibit the EGFR allosteric pocket. In addition, the low binding free energy value of −5 kcal mol−1, corresponding to that of H in the ATP region, allows us to predict that lactone H is not involved in the binding mode to the EGFR active structure. Consequently, it does not compete with the ATP molecule or other TKIs (such as gefitinib or osimertinib) in the ATP region of the EGFR active state.
In the last case, when H binds to the ATP region of the inactive state, a quite good binding free energy of −13 kcal mol−1 is computed. This numerical value emphasizes that lactone H prefers to interact with the EGFR inactive conformation rather than with its active counterpart.
3.8. Interactions of colossolactone H with 40 residues located in the allosteric region of the EGFR inactive state
Let us now consider in more detail the lactone H, which enjoys the largest interaction energy. It undergoes contact with the EGFR protein primarily involving the van der Waals (VDW) interaction energy rather than the electrostatic interaction energy because the total VDW energy dominated the total Coulomb energy (−52 vs. −6 kcal mol−1). Table S4 (ESI†) presents the interaction between H and 40 residues situated in the allosteric pocket, which forms at least one contact with H in the last 20 ns of the simulations. Similarly, the probability of distribution is evaluated for three separated fragments contributing to the interaction between the ligand and the key residues of EGFR. The distribution map is presented in Fig. 4, and all related data are given in Table S5 (ESI†). Accordingly, the lactone H frequently undergoes interactions with 12 residues, including Lys745, Leu747, Ile759, Glu762, Ala763, Met766, Leu777, Leu788, Thr790, Asp855, Leu858 and Ala859, with the highest average number of contact (larger than 100 contacts formed per frame) being 223, 264, 304, 220, 118, 259, 231, 311, 125, 212, 103 and 153, respectively. It is noteworthy that Leu747 tends to favor interaction with the G1 and G2 fragments of H (Fig. 4), as it makes 165 and 113 contacts per frame, respectively (as also shown in Table S5 and ESI†).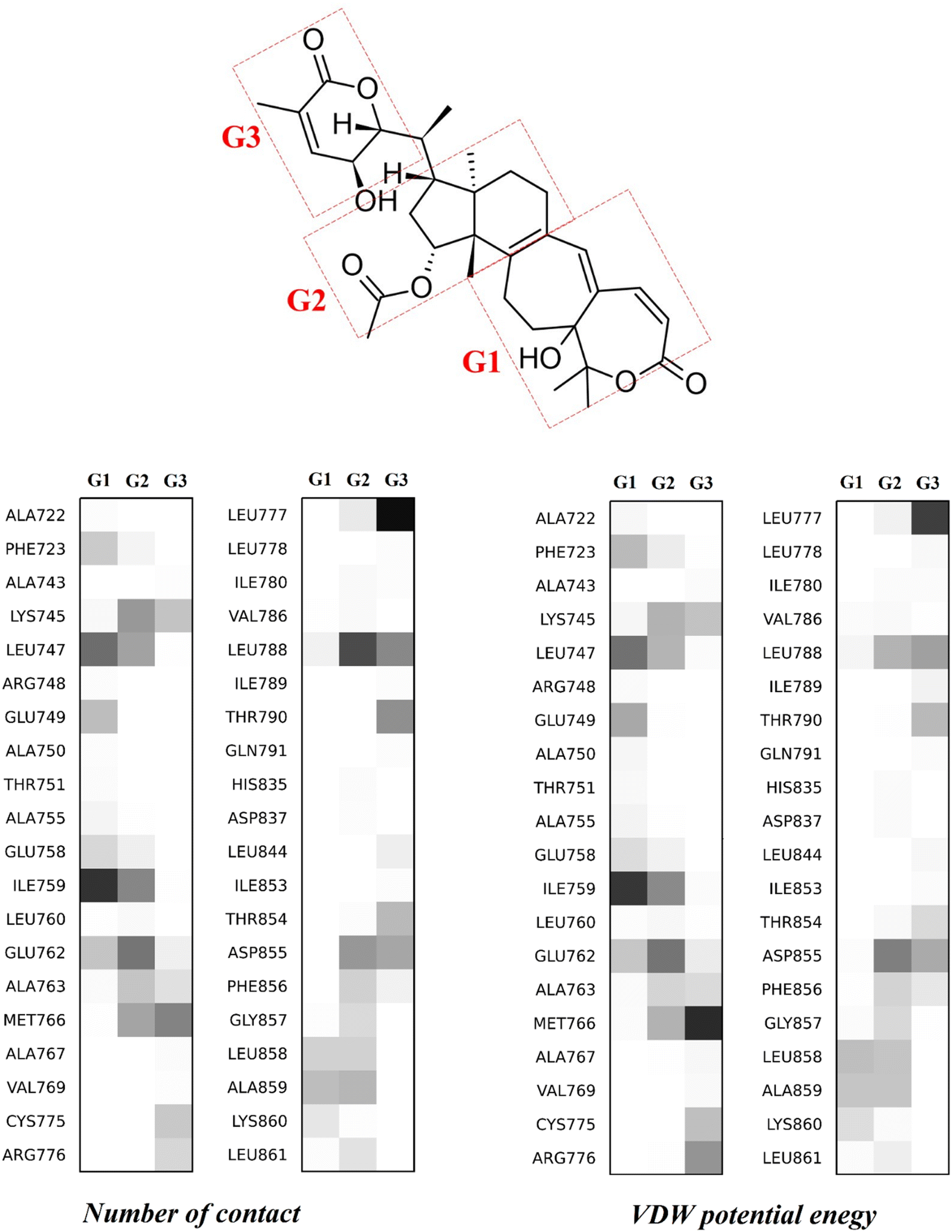 | ||
| Fig. 4 Distribution map of the number of contacts (lower left) and the non-polar potential energy (lower right) between three separated fragments of lactone H (upper panel), identified as the G1–G2–G3 group, and 40 residues of the EGFR tyrosine domain. | ||
The largest number of contacts belongs to residue Leu788, with 165 and 120 contacts with the G2 and G3 fragments, respectively (cf.Fig. 4). Remarkably, in the top 5 largest numbers of contacts, the amino acid Leucine appears three times. Except for Leu788, Leu747 and Leu777 situated in the top 5, Leu 760, Leu778, Leu844, Leu858 and Leu861 are also involved, contributing up to 982 contacts to the total of 3270 contacts (accounting for up to 30%) between lactone H and EGFR.
Despite having a high contact frequency, the eight Leucine amino acids interact with H through a weak electrostatic interaction with an energy of only −0.9 kcal mol−1. These data show that lactone H is stabilized in the allosteric region of EGFR under the dominance of the van der Waals interaction, whose energy amounts to −14 kcal mol−1, in the case that all eight Leucine amino acids are added together. Besides, Ile759, Met766 and Asp855 are three residues possessing the strongest VDW interactions, with calculated potential values of −4.2, −4.3 and −4.5 kcal mol−1, respectively. For insight into the three separated structures of lactone H, the G1, G2 and G3 fragments (Fig. 4) contribute the VDW potential energies of −14, −19 and −19 kcal mol−1, respectively, which are at least 4 times higher than the relevant amounts of Coulomb potential energies with the values of −2.4, −0.1 and −4.1 kcal mol−1, respectively. Although not all Coulomb interaction pairs are considered, four pairs are found, G1-Ile759, G3-Met766, G3-Leu777 and G2-Asp855, which contribute some marked results of −2.8, −2.9, −3.2 and −2.4 kcal mol−1, respectively, to the interactions.
4. Concluding remarks
In the context of acquired drug resistance complicating drug therapies in cancer treatment, the use of natural compounds provides us with much hope. This prompted us to explore further to obtain new insights into the interaction dynamics of a series of 16 colossolactones at the binding active sites of the EGFR tyrosine kinase domain, including both ATP and allosteric regions. The binding affinities of 16 colossolatones extracted from the Ganoderma colossum are ranked via molecular dynamics (MD) simulations. The computed results of the present theoretical study provide us with strong support for related experimental studies and contribute to an explanation for the enhancement by the presence of lactone H in the use of the gefitinib drug in anti-tumor treatment. The lactone H does not compete with the TKIs of the ATP region of the EGFR active state.The umbrella sampling MD, which is a reliable simulation method, emphasizes the high binding free energies of lactone H. Based on these absolute binding free energies, we inspiringly predict that colossolactone H extracted from lingzhi, a well-known Asian natural product, can be used as a good treatment drug because it strongly binds to the inactive conformation of EGFR, either in the ATP or the allosteric region. This leads to the recommended implementation of a synergistic drug combination in cancer treatment involving colossolactone H and gefitinib, which is a well-established drug. This result opens an avenue for the use of colossolactone derivatives as effective scaffolds to develop new anti-cancer drugs.
Conflicts of interest
The authors declare no conflict of interest.Data availability
(1) The molecular dynamics program GROMACS has been used.74 (2) Plots of the frontier orbitals (HOMO–LUMO) of 16 colossolactones are presented; analysed results of SMD simulations based on the interactions between the 16 colossolactones and EGFR at different positions are provided. (3) Some of the supporting data for this study can be accessed via a shared Google drive folder. All files are organized to facilitate reuse and independent verification of the study's findings: https://drive.google.com/drive/folders/1X1eBQ_GDf5QyWN4anDW0oJGF007qrty3?usp=drive_link. This folder includes the following: INPUT files for steered molecular dynamics (SMD): these contain the pre-aligned pulling directions along the Z-axis for each ligand–protein complex, allowing users to easily reproduce the simulations and visualize the trajectory using PyMOL. Simulation output data: time-dependent force profiles, pulling work, and non-equilibrium energies for all ligands. Umbrella sampling data: free energy profiles, transition state estimations, and corresponding RMSD plots are included to complement the MM-PBSA results and highlight differences in methodological outcomes.Acknowledgements
The authors acknowledge the support provided by the VinUniversity Center for Environmental Intelligence under Flagship Project VUNI.CEI.FS_0007. We thank the Polish high performance computing infrastructure PLGrid for computing times within Grant no. PLG/2024.017274 to Dr Mateusz Chwastyk at the Polish Academy of Science.References
- C. Yewale, D. Baradia, I. Vhora, S. Patil and A. Misra, Biomaterials, 2013, 34, 8690–8707 CrossRef CAS PubMed.
- Y. Ohsaki, S. Tanno, Y. Fujita, E. Toyoshima, S. Fujiuchi, Y. Nishigaki, S. Ishida, A. Nagase, N. Miyokawa, S. Hirata and K. Kikuchi, Oncol. Rep., 2000, 7, 603–607 CAS.
- O. Dassonville, A. Bozec, J. L. Fischel and G. Milano, Crit. Rev. Oncol. Hematol., 2007, 62, 53–61 CrossRef PubMed.
- S. E. Abdullah, M. Haigentz, Jr. and B. Piperdi, Chemother. Res. Pract., 2012, 2012, 351210 Search PubMed.
- K. Kyriakopoulou, E. Kefali, Z. Piperigkou, H. Bassiony and N. K. Karamanos, Cell. Signalling, 2018, 51, 99–109 CrossRef CAS PubMed.
- R. Roskoski, Jr., Pharmacol. Res., 2019, 139, 395–411 CrossRef.
- R. S. Herbst, P. M. LoRusso, M. Purdom and D. Ward, Clin. Lung Cancer, 2003, 4, 366–369 CrossRef CAS PubMed.
- C. K. Lee, L. Davies, Y. L. Wu, T. Mitsudomi, A. Inoue, R. Rosell, C. Zhou, K. Nakagawa, S. Thongprasert, M. Fukuoka, S. Lord, I. Marschner, Y. K. Tu, R. J. Gralla, V. Gebski, T. Mok and J. C. Yang, J. Natl. Cancer Inst., 2017, 109, dwj279 Search PubMed.
- J. Stamos, M. X. Sliwkowski and C. Eigenbrot, J. Biol. Chem., 2002, 277, 46265–46272 CrossRef CAS.
- E. R. Wood, A. T. Truesdale, O. B. McDonald, D. Yuan, A. Hassell, S. H. Dickerson, B. Ellis, C. Pennisi, E. Horne, K. Lackey, K. J. Alligood, D. W. Rusnak, T. M. Gilmer and L. Shewchuk, Cancer Res., 2004, 64, 6652–6659 CrossRef CAS.
- M. J. Eck and C. H. Yun, Biochim. Biophys. Acta, 2010, 1804, 559–566 CrossRef CAS.
- C. H. Yun, T. J. Boggon, Y. Li, M. S. Woo, H. Greulich, M. Meyerson and M. J. Eck, Cancer Cell, 2007, 11, 217–227 CrossRef CAS.
- X. Zhang, J. Gureasko, K. Shen, P. A. Cole and J. Kuriyan, Cell, 2006, 125, 1137–1149 CrossRef CAS PubMed.
- X. Tian, T. Gu, M. H. Lee and Z. Dong, Biochim. Biophys. Acta, Rev. Cancer, 2022, 1877, 188645 CrossRef CAS.
- W. Pao and J. Chmielecki, Nat. Rev. Cancer, 2010, 10, 760–774 CrossRef CAS.
- F. Blackhall, M. Ranson and N. Thatcher, Lancet Oncol., 2006, 7, 499–507 CrossRef CAS.
- M. H. Cohen, G. A. Williams, R. Sridhara, G. Chen, W. D. McGuinn, Jr., D. Morse, S. Abraham, A. Rahman, C. Liang, R. Lostritto, A. Baird and R. Pazdur, Clin. Cancer Res., 2004, 10, 1212–1218 CrossRef CAS.
- A. E. Wakeling, S. P. Guy, J. R. Woodburn, S. E. Ashton, B. J. Curry, A. J. Barker and K. H. Gibson, Cancer Res., 2002, 62, 5749–5754 CAS.
- J. C. Kim, M. A. Ali, A. Nandi, P. Mukhopadhyay, H. Choy, C. Cao and D. Saha, Indian J. Biochem. Biophys., 2005, 42, 358–365 CAS.
- S. R. Johnston and A. Leary, Drugs Today, 2006, 42, 441–453 CrossRef CAS.
- F. Tan, X. Shen, D. Wang, G. Xie, X. Zhang, L. Ding, Y. Hu, W. He, Y. Wang and Y. Wang, Lung Cancer, 2012, 76, 177–182 CrossRef PubMed.
- X. Song, L. Cao, B. Ni, J. Wang, X. Qin, X. Sun, B. Xu, X. Wang and J. Li, Front. Pharmacol., 2023, 14, 1090500 CrossRef CAS.
- P. Giordano, A. Manzo, A. Montanino, R. Costanzo, C. Sandomenico, M. C. Piccirillo, G. Daniele, N. Normanno, G. Carillio, G. Rocco, R. Bianco, F. Perrone and A. Morabito, Crit. Rev. Oncol. Hematol., 2016, 97, 143–151 CrossRef.
- C. Brzezniak, C. A. Carter and G. Giaccone, Expert Opin. Pharmacother., 2013, 14, 247–253 CrossRef CAS PubMed.
- J. Remon, C. E. Steuer, S. S. Ramalingam and E. Felip, Ann. Oncol., 2018, 29, i20–i27 CrossRef CAS.
- L. V. Sequist, J. C. Soria, J. W. Goldman, H. A. Wakelee, S. M. Gadgeel, A. Varga, V. Papadimitrakopoulou, B. J. Solomon, G. R. Oxnard, R. Dziadziuszko, D. L. Aisner, R. C. Doebele, C. Galasso, E. B. Garon, R. S. Heist, J. Logan, J. W. Neal, M. A. Mendenhall, S. Nichols, Z. Piotrowska, A. J. Wozniak, M. Raponi, C. A. Karlovich, S. Jaw-Tsai, J. Isaacson, D. Despain, S. L. Matheny, L. Rolfe, A. R. Allen and D. R. Camidge, N. Engl. J. Med., 2015, 372, 1700–1709 CrossRef PubMed.
- E. S. Kim, Drugs, 2016, 76, 1153–1157 CrossRef CAS.
- H. Wang, L. Zhang, P. Hu, X. Zheng, X. Si, X. Zhang and M. Wang, Lung Cancer, 2018, 122, 1–6 CrossRef.
- X. Xu, Chin. J. Cancer, 2015, 34, 285–287 CAS.
- Y. Zhang, L. Tong, F. Yan, P. Huang, C. L. Zhu and C. Pan, Bioorg. Chem., 2024, 147, 107394 CrossRef CAS PubMed.
- Y. C. Zhang, Q. Zhou and Y. L. Wu, Cancer Lett., 2019, 459, 240–247 CrossRef CAS.
- H. Patel, R. Pawara, A. Ansari and S. Surana, Eur. J. Med. Chem., 2017, 142, 32–47 CrossRef CAS.
- T. S. Beyett, C. To, D. E. Heppner, J. K. Rana, A. M. Schmoker, J. Jang, D. J. H. De Clercq, G. Gomez, D. A. Scott, N. S. Gray, P. A. Janne and M. J. Eck, Nat. Commun., 2022, 13, 2530 CrossRef CAS.
- B. Yin, D. M. Fang, X. L. Zhou and F. Gao, Eur. J. Med. Chem., 2019, 182, 111664 CrossRef CAS PubMed.
- Z. Wang, S. Lin, D. Wang and L. Huang, Eur. J. Integr. Med., 2014, 6, 565–570 CrossRef.
- Y. Yu, S. M. Fan, Y. C. Ye, S. Tashiro, S. Onodera and T. Ikejima, Free Radical Res., 2012, 46, 1393–1405 CrossRef CAS PubMed.
- S. Baby, A. J. Johnson and B. Govindan, Phytochemistry, 2015, 114, 66–101 CrossRef CAS PubMed.
- M. Isaka, P. Chinthanom, R. Choeyklin, T. Thummarukcharoen, P. Rachtawee, M. Sappan, K. Srichomthong, R. Fujii, K. Kawashima and S. Mori, J. Nat. Prod., 2020, 83, 2066–2075 CrossRef CAS PubMed.
- L. Ofodile, N. Uma, R. Grayer, O. Ogundipe and M. Simmonds, Phytother. Res., 2012, 26, 748–751 CrossRef CAS PubMed.
- C.-J. Weng, P.-S. Fang, D.-H. Chen, K.-D. Chen and G.-C. Yen, J. Agric. Food Chem., 2010, 58, 7657–7663 CrossRef CAS PubMed.
- R. S. El Dine, A. M. El Halawany, C.-M. Ma and M. Hattori, J. Nat. Prod., 2008, 71, 1022–1026 CrossRef CAS.
- M. Isaka, P. Chinthanom, V. Vichai, S. Sommai and R. Choeyklin, J. Nat. Prod., 2020, 83, 3404–3412 CrossRef CAS.
- P. Chinthanom, M. Sappan, K. Srichomthong, T. Boonpratuang and M. Isaka, Nat. Prod. Res., 2023, 37, 2639–2646 CrossRef CAS PubMed.
- P. Kleinwächter, N. Anh, T. T. Kiet, B. Schlegel, H.-M. Dahse, A. Härtl and U. Gräfe, J. Nat. Prod., 2001, 64, 236–239 CrossRef PubMed.
- S. Y. Chen, C. L. Chang, T. H. Chen, Y. W. Chang and S. B. Lin, Fitoterapia, 2016, 114, 81–91 CrossRef CAS PubMed.
- R. S. El Dine, A. M. El Halawany, C. M. Ma and M. Hattori, J. Nat. Prod., 2009, 72, 2019–2023 CrossRef CAS.
- W. Lakornwong, K. Kanokmedhakul, S. Kanokmedhakul, P. Kongsaeree, S. Prabpai, P. Sibounnavong and K. Soytong, J. Nat. Prod., 2014, 77, 1545–1553 CrossRef CAS.
- M. A. Baghdadi, F. A. Al-Abbasi, A. M. El-Halawany and A. M. Al-Abd, Cancer Res., 2017, 77, 131 CrossRef.
- R. A. Algehani, R. Abou Khouzam, G. A. Hegazy, A. A. Alamoudi, A. M. El-Halawany, R. S. El Dine, G. A. Ajabnoor, F. A. Al-Abbasi, M. A. Baghdadi and I. Elsayed, Biomed. Pharmacother., 2021, 140, 111730 CrossRef CAS PubMed.
- S. B. Lin and T. H. Chen, Taiwan Pat., TWI462918B, 2013 Search PubMed.
- F. D. Prieto-Martínez, E. López-López, K. E. Juárez-Mercado and J. L. Medina-Franco, In Silico Drug Des., 2019, 19–44 Search PubMed.
- V. T. Sabe, T. Ntombela, L. A. Jhamba, G. E. Maguire, T. Govender, T. Naicker and H. G. Kruger, Eur. J. Med. Chem., 2021, 224, 113705 CrossRef CAS.
- A. A. Nasser, I. H. Eissa, M. R. Oun, M. A. El-Zahabi, M. S. Taghour, A. Belal, A. M. Saleh, A. B. M. Mehany, H. Luesch, A. E. Mostafa, W. M. Afifi, J. R. Rocca and H. A. Mahdy, Org. Biomol. Chem., 2020, 18, 7608–7634 RSC.
- D. T. Truong, K. Ho, H. T. Y. Nhi, V. H. Nguyen, T. T. Dang and M. T. Nguyen, Sci. Rep., 2024, 14, 12218 CrossRef CAS.
- S. Zare, L. Emami, Z. Faghih, F. Zargari, Z. Faghih and S. Khabnadideh, Sci. Rep., 2023, 13, 14461 CrossRef CAS PubMed.
- Y. Tu, C. Wang, S. Xu, Z. Lan, W. Li, J. Han, Y. Zhou, P. Zheng and W. Zhu, Bioorg. Med. Chem., 2017, 25, 3148–3157 CrossRef CAS PubMed.
- W. Hou, Y. Ren, Z. Zhang, H. Sun, Y. Ma and B. Yan, Bioorg. Med. Chem., 2018, 26, 1740–1750 CrossRef CAS.
- N. Sepay, R. Mondal, M. K. Al-Muhanna and D. Saha, New J. Chem., 2022, 46, 9735–9744 RSC.
- S. C. Yang, S. S. Chang, H. Y. Chen and C. Y. Chen, PLoS Comput. Biol., 2011, 7, e1002189 CrossRef CAS.
- P. Rangsinth, C. Sillapachaiyaporn, S. Nilkhet, T. Tencomnao, A. T. Ung and S. Chuchawankul, J. Tradit. Complement Med., 2021, 11, 158–172 CrossRef CAS PubMed.
- P. Jana and B. Bhardwaj, J. Comput. Biophys. Chem., 2021, 20, 251–266 CrossRef CAS.
- S. Maity, K. S. R. Pai and Y. Nayak, Pharmacol. Rep., 2020, 72, 799–813 CrossRef.
- G. P. Doss, B. Rajith, C. Chakraborty, N. NagaSundaram, S. K. Ali and H. Zhu, Sci. Rep., 2014, 4, 5868 CrossRef PubMed.
- C. H. Yun, K. E. Mengwasser, A. V. Toms, M. S. Woo, H. Greulich, K. K. Wong, M. Meyerson and M. J. Eck, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2070–2075 CrossRef CAS.
- K. Aertgeerts, R. Skene, J. Yano, B.-C. Sang, H. Zou, G. Snell, A. Jennings, K. Iwamoto, N. Habuka and A. Hirokawa, J. Biol. Chem., 2011, 286, 18756–18765 CrossRef CAS.
- A. Fiser, R. K. G. Do and A. Šali, Protein Sci., 2000, 9, 1753–1773 CrossRef CAS.
- B. Webb and A. Sali, Curr. Protoc. Bioinf., 2016, 54, 5.6.1–5.6.37 Search PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, Nucleic Acids Res., 2023, 51, D1373–D1380 CrossRef.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, 2016 Search PubMed.
- D. A. Case, H. M. Aktulga, K. Belfon, D. S. Cerutti, G. A. Cisneros, V. W. D. Cruzeiro, N. Forouzesh, T. J. Giese, A. W. Gotz, H. Gohlke, S. Izadi, K. Kasavajhala, M. C. Kaymak, E. King, T. Kurtzman, T. S. Lee, P. Li, J. Liu, T. Luchko, R. Luo, M. Manathunga, M. R. Machado, H. M. Nguyen, K. A. O'Hearn, A. V. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, A. Risheh, S. Schott-Verdugo, A. Shajan, J. Swails, J. Wang, H. Wei, X. Wu, Y. Wu, S. Zhang, S. Zhao, Q. Zhu, T. E. Cheatham, 3rd, D. R. Roe, A. Roitberg, C. Simmerling, D. M. York, M. C. Nagan and K. M. Merz, Jr., J. Chem. Inf. Model., 2023, 63, 6183–6191 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed.
- S. Pall, A. Zhmurov, P. Bauer, M. Abraham, M. Lundborg, A. Gray, B. Hess and E. Lindahl, J. Chem. Phys., 2020, 153, 134110 CrossRef CAS.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins, 2010, 78, 1950–1958 CrossRef CAS PubMed.
- P. Mark and L. Nilsson, J. Phys. Chem. A, 2001, 105, 9954–9960 CrossRef CAS.
- R. L. Davidchack, R. Handel and M. V. Tretyakov, J. Chem. Phys., 2009, 130, 234101 CrossRef PubMed.
- J. Åqvist, P. Wennerström, M. Nervall, S. Bjelic and B. O. Brandsdal, Chem. Phys. Lett., 2004, 384, 288–294 CrossRef.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- T. Darden, L. Perera, L. Li and L. Pedersen, Structure, 1999, 7, R55–R60 CrossRef CAS PubMed.
- J. Stourac, O. Vavra, P. Kokkonen, J. Filipovic, G. Pinto, J. Brezovsky, J. Damborsky and D. Bednar, Nucleic Acids Res., 2019, 47, W414–W422 CrossRef CAS.
- K. Ho, D. T. Truong and M. S. Li, J. Phys. Chem. B, 2020, 124, 5338–5349 CrossRef CAS.
- G. Hummer and A. Szabo, Biophys. J., 2003, 85, 5–15 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00817d |
| This journal is © the Owner Societies 2025 |