Modeling nitric oxide and its dimer: force field development and thermodynamics of dimerization†
Received
27th February 2025
, Accepted 8th June 2025
First published on 10th June 2025
Abstract
Nitric oxide, NO, is a free radical that forms dimers, (NO)2, at its vapor–liquid coexisting temperatures. In this work, we developed an all-atom force field for NO and (NO)2. To assess the performance of this force field, we computed the vapor–liquid equilibrium (VLE) properties of the reactive NO–(NO)2 system, as well as those of pure NO and pure (NO)2, using Continuous Fractional Component Monte Carlo (CFCMC) simulations. We then compared the results with the available experimental data and predictions from two previously developed force fields. For the reactive NO–(NO)2 system, we performed CFCMC simulations in the reactive Gibbs ensemble in which the formation of NO dimers, 2NO ⇌ (NO)2, is considered. The predicted coexistence vapor–liquid densities, dimer mole fractions in the liquid phase, saturated vapor pressures, and heats of vaporization using our force field in the temperature range 120 K to 170 K are in excellent agreement with experimental values. In addition, we conducted a systematic parameter study to analyze the sensitivity of the new force field parameters and the isolated molecule partition functions of (NO)2 on the VLE properties of the reactive NO–(NO)2 system. The results indicate that the VLE properties of the reactive NO–(NO)2 system are affected by both the force field parameters of the involved species as well as the isolated molecule partition functions of (NO)2.
1 Introduction
Electric gas discharge plasmas generated in O2 and N2 containing gases are sources of producing considerable amounts of nitric oxide (NO) radicals.4,5 The molecule NO serves as a precursor for production of other reactive nitrogen species such as nitrogen dioxide (NO2), dinitrogen trioxide (N2O3), dinitrogen tetraoxide (N2O4), nitrites (NO2−), nitrates (NO3−), peroxynitrite (ONOO−), peroxynitrous acid (ONOOH), and others, either directly in the gas phase plasma or upon absorption in aqueous solutions.6,7 NO and its byproducts play a key role in plasma processing of liquids and biological materials for applications in biomedicine8–10 and agriculture.11–13 Biomedical applications of plasmas are based on the selective delivery of reactive oxygen and nitrogen species to living tissues for the treatment of chronic wounds and skin infections, and in oncology.14,15 In plasma-agriculture, a specific mixture of reactive oxygen and nitrogen species, directly or dissolved in water, is used as fertilizer and/or pesticide for the growth of plants.16–18 For these applications, it is crucial to quantify the amount of plasma species that are absorbed by aqueous solutions from the gas phase plasmas. Macroscopic models of plasma–liquid systems are commonly used to predict the densities of species in the gas phase plasma and in the aqueous phase.19–23 In these models, the transfer of plasma species from the gas phase into aqueous solutions is incorporated using solubility coefficients of individual species. The solubility coefficients of individual plasma species are commonly used to interpret experimental findings.6,7 Experimental solubility coefficients, however, are only available for a few species and solutions, and with limited accuracy. This limits the predictive power of macroscopic models in determining the concentrations of plasma species in the aqueous phase.24 Molecular simulations are a natural choice to compute the solubility coefficients of plasma species. We aim to use force field-based molecular simulations to compute the solubility coefficients of NO species. It is important to note that the accuracy of force field-based molecular simulations depends on the quality of the force fields used to describe the solute species in the gas phase, the properties of aqueous solutions, and their interactions.25 To the best of our knowledge, there are no all-atom force field parameters for NO that can predict its vapor–liquid-equilibrium (VLE) properties in agreement with available experimental results. This motivated us to first develop a predictive all-atom force field for NO. In a future study, we plan to focus on interaction mechanisms of NO species with aqueous solutions and computing its solubility coefficients.
NO has an unpaired electron in the molecular orbital π* that is delocalized between the N and O atoms.26 NO tends to make dimers, trimers, and higher order oligomers with itself; the extend of which depends on temperature and pressure.27,28 Kohler29 reported that in the liquid phase, the existence of dimers is predominant, while the gas phase is mostly comprised of monomers. Marinakis et al.28 also reported that the liquid phase consists predominantly of dimers, but the presence of higher oligomers may also be possible. The mole fraction of dimers in the liquid phase decreases with increasing temperature as studied by Walsh et al.,30 Kohler,29 and Marinakis et al.28 According to Smith and Johnston,31 the mole fraction of dimers in the liquid phase in the temperature range of 110–120 K, is in the range of 0.95–0.91. High-level quantum mechanical calculations indicate that the planar cis-ONNO configuration is the most stable form of the NO dimer, with the non-planar trans-ONNO lying about 50 kJ mol−1 higher in energy.32,33 However, Marinakis et al.,28 suggested that non-planar NO dimers may still be present in the liquid phase. Therefore, the development of force field parameters for NO should account for dimer formation in the liquid phase.
Several force fields are available for NO.28,34–38 The formation of NO dimers was considered in the force fields developed by Kohler,34 Hirschfelder,35 and Lachet et al.36 using a single-site model for NO and a two-site model for (NO)2. The force field developed by Lachet et al.36 can predict the experimental thermodynamic and transport properties of the reactive NO–(NO)2 system better than the force fields developed by Kohler34 and Hirschfelder,35 as shown in the original paper by Lachet et al.36 As atomistic information is lacking in the force field developed by Lachet et al.,36 due to the treatment of NO as a single-site model, this force field is not suitable for applications where the atomistic information of the N and O atoms in NO molecules is needed.28 The force fields for NO developed by Marinakis et al.,28 Yang et al.,37 and Zhou et al.38 are of all-atom type. In the work by Marinakis et al.,28 the formation of NO dimer was implemented using additional attractive and repulsive Gaussian potential terms on top of van der Waals interactions, making it less compatible with widely used molecular simulation packages. Moreover, the transferability of the Gaussian potential to other systems, such as mixtures with water, is not well understood. For example, it remains unclear which mixing rules should be applied in such cases. The force field developed by Yang et al.37 for NO does not take dimer formation into account. Furthermore, this model consists of a 9-6 Lennard-Jones potential with a sixth-order combination law, which is not readily implemented in all popular simulation engines. The force field for NO developed by Zhou et al.38 only takes into account the non-bonded (electrostatic and van der Waals) interactions of the molecule. This force field was optimized to reproduce the experimental density and enthalpy of vaporization of liquid NO at its boiling point. However, it does not take into account the tendency of NO monomers to dimerize (i.e., no dimers are formed in the model). Therefore, this force field is incapable of reproducing the experimental (NO)2 mole fraction in the liquid phase.
In this work, we developed an all-atom force field for NO and its dimer, (NO)2, which include 12-6 Lennard-Jones and Coulombic potential energy functions. We assumed that NO exists only as monomers and dimers, and dimers exist in a planar cis-ONNO configuration.32 We computed the VLE properties of the reactive NO–(NO)2 system (e.g., coexistence densities, dimer mole fractions in liquid phase, saturated vapor pressures, and heats of vaporization) using continuous fractional component Monte Carlo (CFCMC) simulations in the reactive Gibbs ensemble.39–44 The VLE properties of the reactive NO–(NO)2 system computed using our force field parameters are in excellent agreement with the experimental values. The performance of our newly optimized force field is also compared with two existing force fields, Lachet et al.36 and Zhou et al.,38 that have been used extensively.45–48
The rest of the manuscript is organized as follows. The CFCMC simulations are described in Section 2. In Section 3, the details of the new force field parameters are provided. The isolated molecule partition functions for NO and (NO)2 are discussed in Section 4. The validation of the new force field with the experimental VLE properties and the comparison of the force field with existing NO force fields is investigated in Section 5. In Section 5 the sensitivity of the VLE properties on the isolated molecule partition functions of the dimer and the Lennard-Jones potential parameters is discussed. Finally, concluding remarks are provided in Section 6.
2 Methodology
CFCMC simulations49–51 using the open-source Brick-CFCMC software51–53 were used to assess the validity of our force field parameters for NO and (NO)2 compared to experimental findings. We performed a series of systematic CFCMC simulations in the reactive Gibbs ensemble (RGE)39–44 at constant temperature, number of atoms, and volume, to reproduce the experimental vapor–liquid coexistence densities, coexistence pressures, dimer mole fractions, and heats of vaporization of the reactive NO–(NO)2 system for the temperature range of 121.5 K to 165 K. From these, the critical temperature, density, and pressure of the system were calculated. RGE is a combination of the Gibbs ensemble (GE),54,55 which consists of two simulation boxes, one for each of the vapor and liquid phases to model the VLE, and the reaction ensemble,39,42,43 which in our case models the following equilibrium reactionWe also performed CFCMC simulations in the single-component Gibbs ensemble (GE)56,57 at constant temperature, number of molecules, and volume to compare the performance of our force field with the force fields developed by Lachet et al.36 and Zhou et al.38 To do this, we computed the coexistence densities and pressures of pure NO for the temperature range 120 K to 170 K and pure (NO)2 for the temperature range 120 K to 300 K.
In CFCMC simulations, fractional molecules (compared to normal or “whole” molecules) are introduced whose interactions with the rest of the molecules in the system are modulated by a continuous coupling parameter λ. The value of λ ranges from 0 to 1. λ = 0 means that the fractional molecules act as ideal gas molecules and do not have interactions with the rest of the molecules in the system. λ = 1 represents “whole” molecules that can fully interact with the other molecules. A fractional group is a collection of one or more fractional molecules. The fractional group must be charge neutral.51–53 To increase the efficiency of molecule transfers (insertions/deletions), the Wang–Landau algorithm58 is used to bias the coupling parameter λ with a weight function W(λ). This ensures a flat observed probability distribution of λ, therefore making sure that the sampling issues due to the energy barriers in λ space are not encountered. In our simulations, we used 100 bins to obtain a histogram for values of λ and their probability of occurrence p(λ). The Boltzmann average of an observable (A) is then computed using59
| | 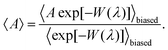 | (2) |
For the RGE, there are two sets of fractional groups;
59 (1) fractional group for individual components/molecules involved in the system for molecule transfers and (2) fractional group for reactions. The contributions of fractional components are not taken into account when computing the ensemble averages.
59
The fractional group for molecule transfers facilitates molecule transfers between the vapor and liquid simulation boxes. The reactive NO–(NO)2 system contains two such fractional groups, one each for NO and (NO)2. As mentioned earlier, the strength of the interactions of the molecules in these group are modulated by their corresponding λ values (λGibbs,NO and λGibbs,(NO)2). There are three trial moves associated with the fractional molecules for molecule transfers:59,60 (1) attempts to change the value of λGibbs; (2) attempts to transfer a fractional molecule from one simulation box to another; (3) attempts to convert a fractional molecule in a box to a “whole” molecule, and simultaneously, transform a “whole” molecule in the other box into a fractional molecule.
For the NO–(NO)2 system, the fractional group for reactions contains fractional molecules of either the reactants (2NO) or the reaction products (NO)2. The interactions of these molecules with the other molecules in the simulation is modulated by the parameter, λreaction. One fractional group for reactions was added each to the liquid and vapor simulation boxes. The trial moves associated with the fractional molecules in this group are:61 (1) attempts to change the value of λreaction; (2) attempts to convert the fractional molecules of reactant to fractional molecules of reaction product, or vice versa; (3) attempts to convert the fractional molecules of the reactant (or the reaction product) in a box to “whole” molecules of the reaction product (or the reactant), and simultaneously, transform “whole” molecules of the reaction product (or the reactant) to fractional molecules of reactant (or the reaction product).
The GE simulation of unreactive NO and (NO)2 systems contains the fractional group for molecule transfers and not the fractional group for reactions. In addition to the trial moves associated with the fractional molecules present in the RGE and the GE simulations, both the simulation setups have thermalization trial moves (molecule translations, rotations and volume changes). Since the molecules in the simulation are rigid, the sampling of the intramolecular degrees of freedom are not considered.
The simulation in the RGE is considered equilibrated when a combined phase equilibrium and chemical equilibrium is achieved. The two simulation boxes in this ensemble can exchange molecules and volume until the two phases reach phase equilibrium. Phase equilibrium is achieved when the liquid and vapor chemical potentials, μ, of individual species are equal (μNO,liquid = μNO,vapor and μ(NO)2,liquid = μ(NO)2,vapor). The chemical potential in the phase x (liquid or vapor) for the component i (NO or (NO)2) is given by59
| |  | (3) |
where
kB is the Boltzmann constant,
T is the temperature,
Vx is the volume of the simulation box that represents phase
x,
Λi is the thermal de Broglie wavelength of component
i,
Nx,i is the number of molecules of component
i in phase
x.
px,i(
λGibbs,i = 1) and
px,i(
λGibbs,i = 0) are the probabilities that
λGibbs,i (coupling parameter of fractional molecule for molecule transfers for component
i) are 1 and 0, respectively, in phase
x. Chemical equilibrium is attained when 2
μNO,liquid =
μ(NO)2,liquid and 2
μNO,vapor =
μ(NO)2,vapor.
62,63 For a system containing
S components in phase
x, where components 1 to
R are reactants and
R + 1 to
S are reaction products, the chemical potentials of the components follow the relations
42,52| |  | (4) |
| |  | (5) |
where,
qi is the isolated molecular partition function (excluding the translational part) of component
i,
νi is the stoichiometric coefficient of component
i in the reaction,
ρi,x is the number density of the species
i in phase
x.
pi(
λreaction,x = 1) and
pi(
λreaction,x = 0) are the probabilities that
λreaction,x (coupling parameter of fractional group for reactions for phase
x) approaches 1 and 0, respectively, for component
i.
For a two-component system such as NO–(NO)2, which follows the reaction in eqn (1), (4) and (5) simplify to
| |  | (6) |
where
i are the components NO and (NO)
2. From
eqn (4)–(6) it is clear that to perform CFCMC simulations in the RGE, partition functions of the isolated molecules involved in the reaction are required. More discussion about the partition functions of NO and (NO)
2 are given in Section 4. The simulation in the GE is equilibrated when there is equilibrium between the vapor and liquid phases (phase equilibrium).
The critical temperatures and densities were obtained by fitting the liquid and vapor phase densities to the law of rectilinear diameters64
| |  | (7) |
where
ρl and
ρg correspond to the density of liquid and vapor phases, respectively, along the VLE curve,
ρc is the critical density, and
Tc is the critical temperature.
A and
B are system dependent fitting parameters. For the RGE,
β is selected as the value that corresponds to the maximum of the coefficient of determination (
R2) for the linear regression fit of
 versus
versus temperature,
62 where
ρ0 is an arbitrary number density which is set to 1 Å
−3. In the single-component GE,
β is taken as 0.32.
65
To compute the coexistence pressures of the RGE and the GE systems, a series of NPT simulations for vapor phase were performed to match the vapor phase densities of the RGE and the GE. The P–T saturation points were fitted to a correlation of the form61
| |  | (9) |
where
A and
B are system-dependent fitting parameters. The critical saturated vapor pressure,
Pc, was obtained by extrapolating the above correlation to
Tc.
The molar heat of vaporization was calculated as the difference between the average molar enthalpy of the vapor and liquid simulation boxes of the RGE36
| | | Δhvap = hvapor − hliquid, | (10) |
where Δ
hvap is the molar heat of vaporization and
hliquid,
hvapor are the true molar enthalpies of the liquid and vapor simulation boxes, respectively. True molar enthalpy (
hx) is defined as
66| |  | (11) |
where
Hx is the total enthalpy in phase
x (liquid or vapor) and
ni,x is the number of moles of component
i (NO or (NO)
2) in phase
x. For the calculation of the molar heat of vaporization, the internal energies and volumes were obtained from RGE calculations and the saturated pressures were obtained from a series of NPT simulations. The total true molar enthalpy is taken as the sum of
hliquid and
hgas.
For all simulations in the RGE, 103 initialization cycles were performed followed by an equilibration of 8 × 106 cycles. This was followed by 5 independent production runs of 3 × 106 cycles, each. Note that one cycle refers to N number of trial moves, where N is the total number of molecules present in the system. The error bars are computed from the standard deviation of 5 independent production runs. The initial system size for the simulations in the RGE was a total of 400 molecules for temperatures below 140 K and 500 molecules for temperatures above 140 K. The trial moves were selected with the following probabilities: thermalization moves (which includes both translations and rotations) (69%), particle exchanges (15%), volume moves (1%) and reaction moves (15%). For the GE simulations, 103 initialization cycles were performed followed by an equilibration of 8 × 106 cycles and a production run of 8 × 106 cycles. The error bars for the GE simulations are computed from the standard deviation of 5 blocks into which the production run was divided. The trial moves were selected with the following probabilities: thermalization moves (84%), particle exchanges (15%) and volume moves (1%). Periodic boundary conditions were considered in all directions for both the RGE and the GE.
3 Force field
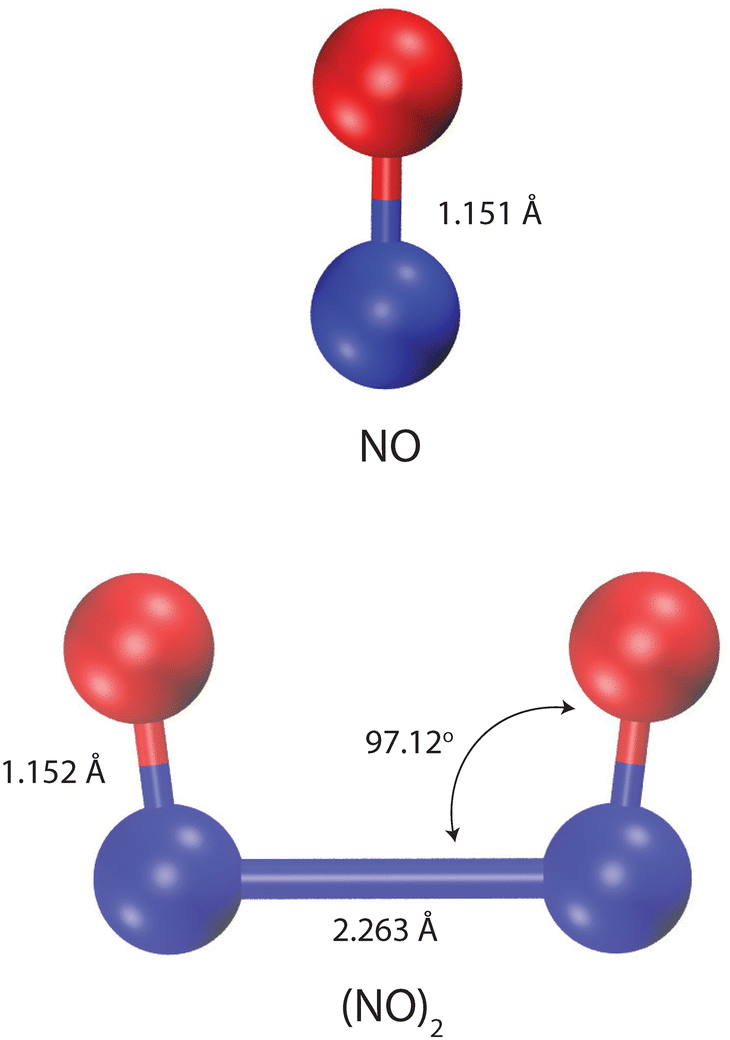
The structures of NO and (NO)2 were considered rigid according to the corresponding experimental geometries1,2 (see Fig. 1). We considered two terms in the total potential energy function (Etotal), electrostatic interactions (Eelectrostatic) and van der Waals interactions (EvdW),| | | Etotal = Eelectrostatic + EvdW. | (12) |
For the electrostatic interactions, the coulombic potential energy function is considered| |  | (13) |
where q represents the atomic partial charges, rij is the distance separating the charges, and ε0 is the permittivity of vacuum. The atomic partial charges of NO were obtained from the ratio between the experimental dipole moment67 and the experimental bond length of NO.1 The partial charges on the atoms of (NO)2 were taken to be the same as that of NO. For the van der Waals interactions, the 12-6 Lennard-Jones (L-J) potential energy function is considered| |  | (14) |
where σ represents the distance at which the particle–particle interaction energy is zero, and ε represents the depth of the potential well. The Lorentz–Berthelot68 mixing rules were used for the L-J parameters of two dissimilar non-bonded atoms, 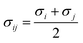 ,
,  . A cut-off radius of 10 Å was used for both the Lennard-Jones and Coulombic interactions. The Ewald summation method69,70 was used to treat long-range Coulombic interactions. The energies and pressures of the simulations were corrected with long-range tail corrections for the L-J interactions.65 After numerous trials of varying the σ and ε parameters (110 combinations were tested in total), we arrived at the values provided in Table 1. These parameters are referred to as the New FF in the rest of the manuscript. A detailed discussion on the sensitivity analysis of the New FF parameters is provided in Section 5.3.
. A cut-off radius of 10 Å was used for both the Lennard-Jones and Coulombic interactions. The Ewald summation method69,70 was used to treat long-range Coulombic interactions. The energies and pressures of the simulations were corrected with long-range tail corrections for the L-J interactions.65 After numerous trials of varying the σ and ε parameters (110 combinations were tested in total), we arrived at the values provided in Table 1. These parameters are referred to as the New FF in the rest of the manuscript. A detailed discussion on the sensitivity analysis of the New FF parameters is provided in Section 5.3.
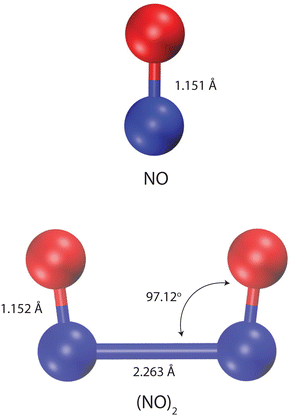 |
| | Fig. 1 Experimental values for bond distances and angles of NO and its dimer, (NO)2.1,2 The blue and red spheres represent N and O atoms, respectively. The visualization is created using the Visual Molecular Dynamics (VMD) software.3 | |
Table 1 Our new force field parameters for NO and (NO)2 molecules which are referred to as New FF in the text
| Atoms |
ε/kB/[K] |
σ/[Å] |
q/[e] |
|
|
|
NO |
|
| N |
48.50 |
3.09 |
0.0288 |
| O |
56.29 |
2.94 |
−0.0288 |
|
|
|
|
|
(NO)2 |
|
| N |
47.00 |
3.02 |
0.0288 |
| O |
54.52 |
2.87 |
−0.0288 |
Lachet et al.36 treat NO as a single L-J sphere with the 12-6 potential function. To model (NO)2, two NO L-J sites separated by a distance of 2.237 Å were used. The individual L-J parameters for each NO site in the dimer are considered to be the same as those for the monomer. The L-J parameters of the force field developed by Lachet et al.36 are listed in Table S9 of the ESI.† Lachet et al.36 neglect electrostatic interactions in their models of NO and (NO)2. The cut-off for the L-J potential was taken as half the length of the simulation box, and interactions beyond the cut-off distance were considered using the long-range tail correction.65 The force fields for NO and (NO)2 developed by Lachet et al.36 are referred to as Lachet FF in the rest of the manuscript.
In the work by Zhou et al.,38 NO is modeled as a two-site rigid body with partial charges and 12-6 L-J potential on both sites. In their model, the N and O atoms of NO are separated by a distance of 1.15 Å. The Lorentz–Berthelot mixing rules68 were used for the L-J parameters of two dissimilar nonbonded atoms. The L-J parameters and partial charges taken from the work by Zhou et al.38 are listed in Table S9 of the ESI,† and are referred to as Zhou FF in the rest of the manuscript. Zhou et al.38 do not parameterize a force field for (NO)2.
4 Isolated molecular partition functions
As explained in Section 2, to calculate the chemical potential of a component in a reaction, the isolated molecular partition function, qtotal(V, T), of that component is needed (see eqn (6)). We calculated the isolated molecular partition functions of NO and (NO)2 according to52| | | qtotal(V, T) = qtrans(V, T)qvib(T)qrot(T)qelec(T). | (15) |
The translational partition function, qtrans, is given by52| | 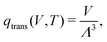 | (16) |
wherein Λ is the thermal de Broglie wavelength of the molecule,  . Here, h is the Planck constant, M is the sum of the masses of all atoms in the molecule, T is the temperature, and V is the volume. In Brick-CFCMC, a reference state of V = V0 = 1 Å3 is used for qtrans.52,71
. Here, h is the Planck constant, M is the sum of the masses of all atoms in the molecule, T is the temperature, and V is the volume. In Brick-CFCMC, a reference state of V = V0 = 1 Å3 is used for qtrans.52,71
The vibrational partition function, qvib, considering the vibrational ground state energy as the zero of the vibrational energy, is calculated using52
| |  | (17) |
in which
i runs from 1 to
α, where
α is the number of vibrational degrees of freedom. For a linear molecule,
α = 3
n − 5 and for a non-linear molecule,
α = 3
n − 6, where
n is the number of atoms in the molecule.

is the characteristic vibrational temperature corresponding to the
ith normal mode with vibrational frequency,
νi.
The energy levels (εJ) of a diatomic molecule such as NO, under the rigid-rotor approximation is given by52
| |  | (18) |
where
J is the rotational quantum number,
I is the principal moment of inertia. The energy level corresponding to
J = 0 is chosen as the zero of the rotational energy for the calculation of the rotational partition function,
qrot. The rotational constant of an axis,
X, is related to the moment of inertia of that axis,
I, by

. For a diatomic molecule such as NO,
qrot is given by
52| |  | (19) |
where

is the characteristic rotational temperature.
σrot is the rotational symmetry number of the molecule, which is the number of indistinguishable configurations the molecule can be brought into by rotation about its centre of mass.
72 For NO,
σrot = 1. The rotational partition function,
qrot, for (NO)
2, which is a non-linear polyatomic species, is calculated using
52| |  | (20) |
where,
σrot,(NO)2 is the rotational symmetry number of (NO)
2 and is equal to 1,
Θrot,A,
Θrot,B and
Θrot,C are the characteristic rotational temperatures corresponding to the principal moments of inertia,
IA,
IB and
IC, respectively. The rotational
73 and vibrational
2,74,75 partition functions of NO and (NO)
2 were calculated using experimental values of rotational and vibrational frequencies (see Tables S2 and S3 of the ESI
†).
Finally, the electronic partition function, qelec, is calculated using52
| |  | (21) |
where
ge1 is the degeneracy of the electronic ground state.
ge1,NO = 2
76 and
ge1,(NO)2 = 1.
77D0 is the atomization energy of the molecule which is defined as the difference between the enthalpy of formation of the molecule and the sum of the enthalpies of formation of the dissociated atoms which constituted the molecule.
52 The energy of the infinitely dissociated atoms at the ground electronic state is taken as the zero of the electronic energy.
The value of D0 for NO was calculated from the enthalpies of formation of NO, N and O, which were obtained from the JANAF tables;78D0,NO = 629.82 kJ mol−1. The atomization energy of (NO)2, D0,(NO)2, was not readily available in the JANAF tables,78 but can be calculated by
| | | D0,(NO)2 = 2D0,NO + BDE(NO)2, | (22) |
where BDE
(NO)2 is the bond dissociation energy of (NO)
2, which is the energy required to break the N–N bond of (NO)
2.
The values of BDE(NO)2 reported in literature, depending on the accuracy of the theoretical method or experimental technique used, are in the range of 7.64 to 13.8 kJ mol−1.2,33,39,79–83 Due to the large variations in the values of BDE(NO)2 reported in literature, an approach used by Turner et al.83 and Johnson et al.39 was to adjust the value of BDE(NO)2 to reproduce the experimental dimer mole fraction in the liquid phase. With this approach, Turner et al.83 obtained a value of BDE(NO)2 = 10.9 kJ mol−1 and Johnson et al.39 achieved a value of BDE(NO)2 = 13.6 kJ mol−1. In our work, we also varied the value of BDE(NO)2 between 7.64 and 13.8 kJ mol−1 to predict the mole fractions of dimer in the liquid phase as well as the VLE properties of the reactive NO–(NO)2 system in agreement the available data. After numerous iterations, we obtained a value of BDE(NO)2 = 13.06 kJ mol−1 which corresponds to a value of D0,(NO)2 = 1269 kJ mol−1.
The isolated molecular partition functions of NO and (NO)2 were calculated according to eqn (15) for temperatures ranging from 121.5 K to 165 K. A correlation was fitted to the respective isolated molecular partition functions, which is given by
| |  | (23) |
| |  | (24) |
where
q is the isolated molecule partition function excluding the translational contribution.
5 Results and discussion
The validation of the New FF with the experimental VLE properties of the reactive NO–(NO)2 system is discussed in Section 5.1. The performance of the New FF compared to the Lachet FF and Zhou FF in predicting the VLE properties of pure NO and pure (NO)2 are discussed in Section 5.2. Finally, the dependence of the VLE of the reactive NO–(NO)2 system on the D0,(NO)2 and L-J parameters (σ and ε) is investigated in Section 5.3.
5.1 Reactive NO–(NO)2 systems
5.1.1 Coexistence densities.
The coexistence densities of the liquid and vapor phases were computed at ten different temperatures ranging from 121.5 K to 165 K using the New FF and the Lachet FF. The results are presented in Fig. 2 along with the available experimental data.84 The raw data are available in Table S5 of the ESI.† The average absolute deviation between the liquid densities calculated using the New FF and the experimental liquid densities is 2.32 kg m−3 (0.2%). Our results using the Lachet FF are also in agreement with those presented in the original paper by Lachet et al.36 The Lachet FF overestimates the liquid densities at lower temperatures (120 K to 140 K) and underestimates the liquid densities at higher temperatures (>140 K). The average absolute deviation between the liquid densities calculated using the Lachet FF and the experimental liquid densities is 36.89 kg m−3 (3.3%) which is much larger than that calculated using the New FF.
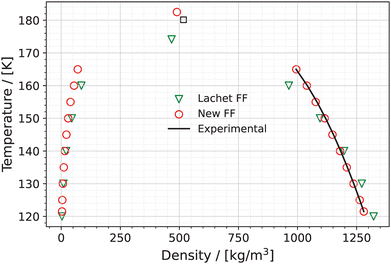 |
| | Fig. 2 Vapor–liquid coexistence densities of the NO–(NO)2 system predicted using the New FF as a function of temperature (red circles). Error bars for the New FF are estimated based on the standard deviation of 5 independent simulations and are much smaller than the symbols used in the figure. The orthobaric densities obtained using the Lachet FF36 (green triangles) along with the experimental liquid densities84 (black line) are added for comparison. The experimental critical point (black square) is also shown in the figure. | |
The predicted critical temperature (Tc), density (ρc), and pressure (Pc) using the New FF and Lachet FF are presented in Table 2 along with experimental data.84 The predicted critical density using the New FF is 489.59 kg m−3 which is within 27.76 kg m−3 (5.4%) of the experimental value. The critical temperature predicted using the New FF is 182.51 K which has a deviation of 1.3% from the experimental critical temperature. The critical density and temperature predicted by the Lachet FF deviate from the experimental values by 9.8% and 3.4%, respectively.
Table 2 Critical temperatures (Tc), densities (ρc), and pressures (Pc) calculated using the New FF and the Lachet FF.36 Errors (written as subscripts) are estimated using standard deviations of 5 independent simulations. Experimental values84 are added for comparison
|
|
T
c/[K] |
ρ
c/[kg m−3] |
P
c/[MPa] |
| New FF |
182.510.54 |
489.591.52 |
6.240.15 |
| Lachet FF36 |
174.03 |
466.60 |
6.37 |
| Experimental84 |
180.15 |
517.35 |
6.48 |
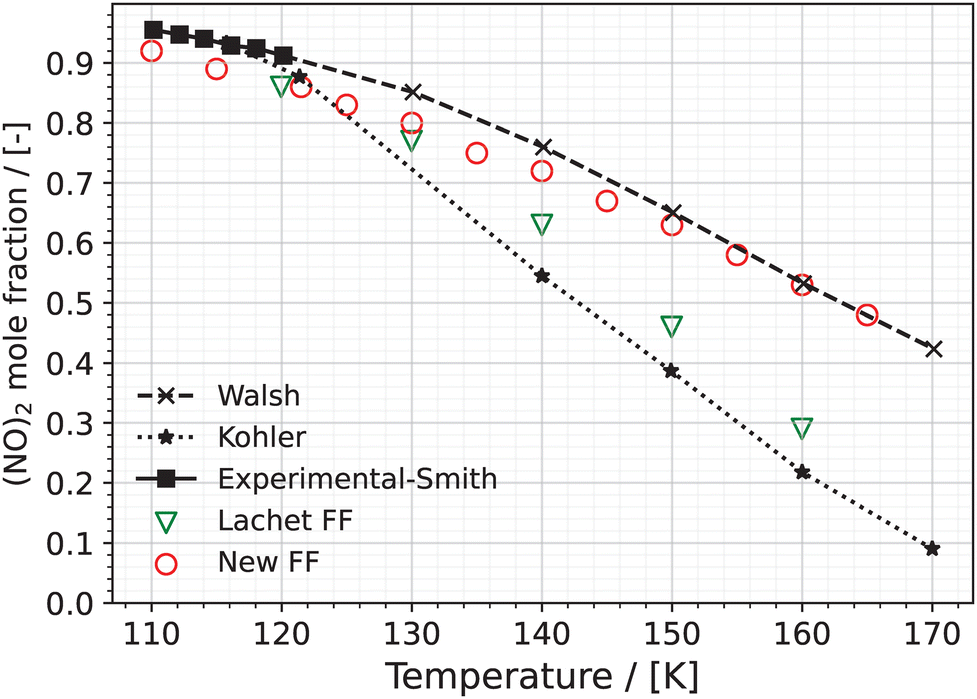
5.1.2 Dimer mole fractions.
The mole fraction of (NO)2 in the liquid phase of the NO–(NO)2 system obtained using the New FF and the Lachet FF is presented in Fig. 3. The raw data are presented in Table S6 of the ESI.† The experimental values, in the temperature range from 110 K to 120 K, are taken from the work of Smith and Johnston.31 Walsh et al.30 used a modified Wertheim's association theory and Kohler29 used the theory of corresponding states of mixture to theoretically estimate the dimer mole fractions in the liquid phase. The data from both works are presented in Fig. 3 for comparison.
 |
| | Fig. 3 Dimer mole fractions in the NO–(NO)2 liquid phase, which is at coexistence with the vapor phase, predicted using the New FF (red circles) as well as the Lachet FF36 (green triangles) as a function of temperature. Error bars for the New FF are estimated based on the standard deviation of 5 independent simulations and are much smaller than the symbols used in the figure. The experimental mole fractions (black squares) from 110 K to 120 K31 and extrapolated estimates of Walsh et al.30 and Kohler29 from 120 K to 170 K are added for comparison. | |
At 110 K, the predicted (NO)2 mole fraction using the New FF is 0.92, while the corresponding experimental value is 0.95. The mole fraction of dimers predicted using the New FF monotonically decreases with increasing temperature. (NO)2 mole fraction obtained using the New FF is in better agreement with the estimate values of Walsh et al.30 for the temperature range (121.5 K to 165 K). The dimer mole fractions predicted using the Lachet FF is in better agreement with the estimates by Kohler.29 We note that our calculations using the Lachet FF are in agreement with those presented in the original work by Lachet et al.36 In the vapor phase, monomers have a more dominant presence for the temperature range 121.5 K to 165 K. The presence of dimers increases as the temperature increases, although slightly (see Table S6 of the ESI†). At 121.5 K, the New FF predicts a dimer mole fraction of 6%, whereas the Lachet FF predicts a dimer mole fraction of 0.4%. One reason for the lower mole fraction of (NO)2 predicted in the work of Lachet et al.36 compared to our work using the New FF could be attributed to the differences between the standard chemical potential, μ0 (defined as  42,62) of (NO)2 and 2NO. The differences between the standard chemical potential of (NO)2 and 2NO used in the work of Lachet et al. favor more NO molecules in the system.36,85
42,62) of (NO)2 and 2NO. The differences between the standard chemical potential of (NO)2 and 2NO used in the work of Lachet et al. favor more NO molecules in the system.36,85
5.1.3 Coexistence pressures.
In Fig. 4, the inverse temperature dependence of the coexistence pressures of the NO–(NO)2 system predicted using the New FF and the Lachet FF are presented on a logarithmic scale. The experimental values are also added for comparison. The average absolute deviations between the computed vapor pressures using the New FF and the experimental vapor pressures is 15%. The corresponding value for Lachet FF is 17%. Higher values of vapor pressures predicted using the Lachet FF compared to the New FF could be traced back to the different isolated molecular partition functions used in Lachet et al.36 compared to our work; or alternatively the differences between the standard chemical potential  of (NO)2 and 2NO used in Lachet et al.36 compared to our work, which leads to differences in compositions. The isolated molecular partition functions in the work of Lachet et al.36 favor more NO molecules in both the vapor and liquid simulation boxes compared to our systems. This leads to higher vapor pressures for Lachet et al.36 The critical pressure is obtained by extrapolating the vapor pressure curve to the critical temperature using eqn (9). The critical pressure using the New FF is 6.24 MPa (3.9% deviation from the experimental critical pressure) and that predicted using the Lachet FF is 6.37 MPa (1.7% deviation from the experimental value). See Table 2.
of (NO)2 and 2NO used in Lachet et al.36 compared to our work, which leads to differences in compositions. The isolated molecular partition functions in the work of Lachet et al.36 favor more NO molecules in both the vapor and liquid simulation boxes compared to our systems. This leads to higher vapor pressures for Lachet et al.36 The critical pressure is obtained by extrapolating the vapor pressure curve to the critical temperature using eqn (9). The critical pressure using the New FF is 6.24 MPa (3.9% deviation from the experimental critical pressure) and that predicted using the Lachet FF is 6.37 MPa (1.7% deviation from the experimental value). See Table 2.
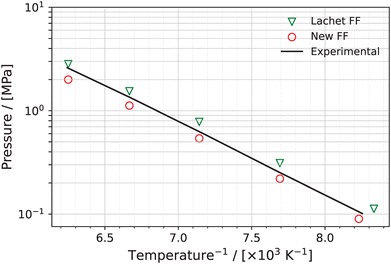 |
| | Fig. 4 Inverse temperature dependence of the coexistence pressures of the NO–(NO)2 system predicted using the New FF (red circles) and the Lachet FF36 (green triangles), in logarithmic scale. Error bars for the New FF are computed based on the standard deviation and are smaller than the symbols used in the figure. Experimental data84 for the coexistence pressures is represented by the black line. | |
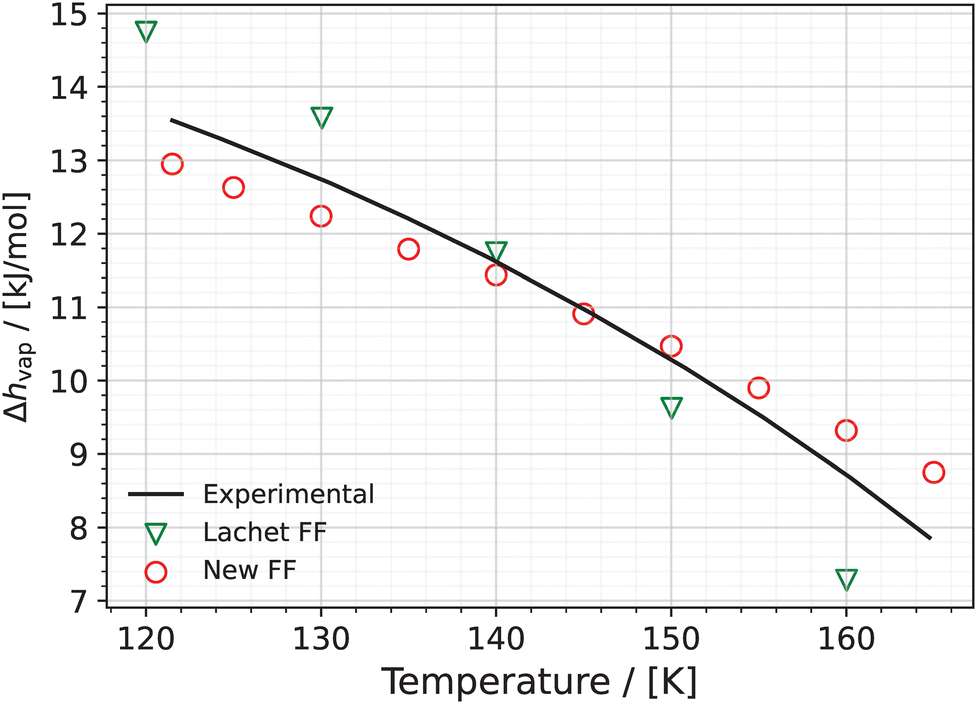
5.1.4 Heats of vaporization.
The temperature dependence of the heats of vaporization for the system containing NO and its dimer predicted using the New FF and the Lachet FF is shown in Fig. 5 (see eqn (10)). The experimental values are also shown for comparison. The results using the New FF are in good agreement with the experimental vaporization enthalpies. The average absolute deviation between the experimental enthalpies of vaporization and those predicted by the New FF is 0.46 kJ mol−1 (4.4%). This value for the Lachet FF is 0.82 kJ mol−1 (8%). The Lachet force field overestimates the heats of vaporization at lower temperatures (120 K and 130 K) and underestimates at higher temperatures (150 K and 160 K). At 140 K the Lachet FF is in good agreement with the experimental value.
 |
| | Fig. 5 Heat of vaporization versus temperature for the reactive NO–(NO)2 system obtained using the New FF (red circles) and the Lachet FF36 (green triangles). Error bars for the New FF are computed based on the standard deviation of 5 independent simulations and are much smaller than the symbols used in the figure. Experimental values84 (black line) are added for comparison. | |
5.2 Non-reactive pure NO and pure (NO)2 systems
5.2.1 Properties of pure NO.
The coexistence densities of pure (unreactive) NO computed using the New FF, the Lachet FF, and the force field developed by Zhou et al.38 (Zhou FF), as a function of temperature, are shown in Fig. 6. The saturated vapor pressures of pure NO for temperatures between 120 K and 160 K computed using these three force fields are shown in Fig. 7. The predicted critical densities, temperatures and pressures of pure NO are shown in Table S10 of the ESI.† The vapor phase densities of pure NO using the Lachet FF are in excellent agreement with the vapor phase densities computed using the New FF. The liquid densities of NO using the New FF are higher than those computed using the Lachet FF for the entire temperature range of 120 K to 160 K. The differences between the liquid densities of NO calculated using the Lachet FF and the New FF are reduced as the temperature reaches the critical temperature (168.20 K for Lachet FF and 167.34 K for the New FF). The vapor densities computed using the Zhou FF are much lower (on average 99%) than those calculated using the Lachet FF and the New FF, while the liquid densities predicted using the Zhou FF are much higher (on average 48%) than the densities of the other two force fields. This is because the Zhou FF was optimized to reproduce the experimental density of liquid NO–(NO)2 without taking the presence of dimer (NO)2 into account. The values of the L-J energy parameter ε of N and O in the Zhou FF are also higher (by 68%) than the New FF (see Table S8 of the ESI†) which leads to lower vapor phase densities, as higher ε contributes to higher intermolecular interactions in the liquid, making it difficult for molecules to move to the vapor phase. This is evident in Fig. 7, where the saturated vapor pressures (Psat) of the Zhou FF are lower, showing a difference of ca. 4 MPa at 160 K, compared to the New FF and the Lachet FF. The critical saturated vapor pressure (Pc) of the Zhou FF is larger than that of the New FF and Lachet FF as the critical temperature (Tc) of the Zhou FF is higher compared to the other two force fields. The New FF and the Lachet FF have comparable values of Psat as their vapor phase densities have similar values (see Fig. 6). The values of Pc of the New FF and the Lachet FF differ only by 0.18 MPa.
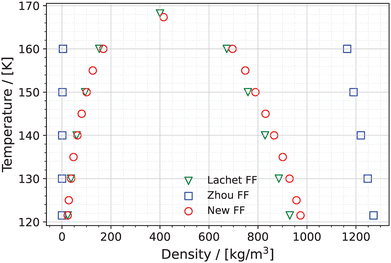 |
| | Fig. 6 Coexisting vapor and liquid densities of pure (unreactive) NO using the New FF (red circles), the Zhou FF38 (blue squares) and the Lachet FF36 (green triangles). Error bars are estimated based on the standard deviation of 5 blocks into which the production run was divided and are much smaller than the symbols used in the figure. The critical points of both the New FF and Lachet FF are indicated in the figure. The critical points of the Zhou FF is not shown in the figure, but it is listed in Table S9 (ESI†). | |
 |
| | Fig. 7 Inverse temperature dependence of the saturated vapor pressures of pure NO using the New FF (red circles), the Zhou FF38 (blue squares) and the Lachet FF36 (green triangles), on logarithmic scale. Error bars for the New FF are computed based on the standard deviation and are smaller than the symbols used in the figure. | |
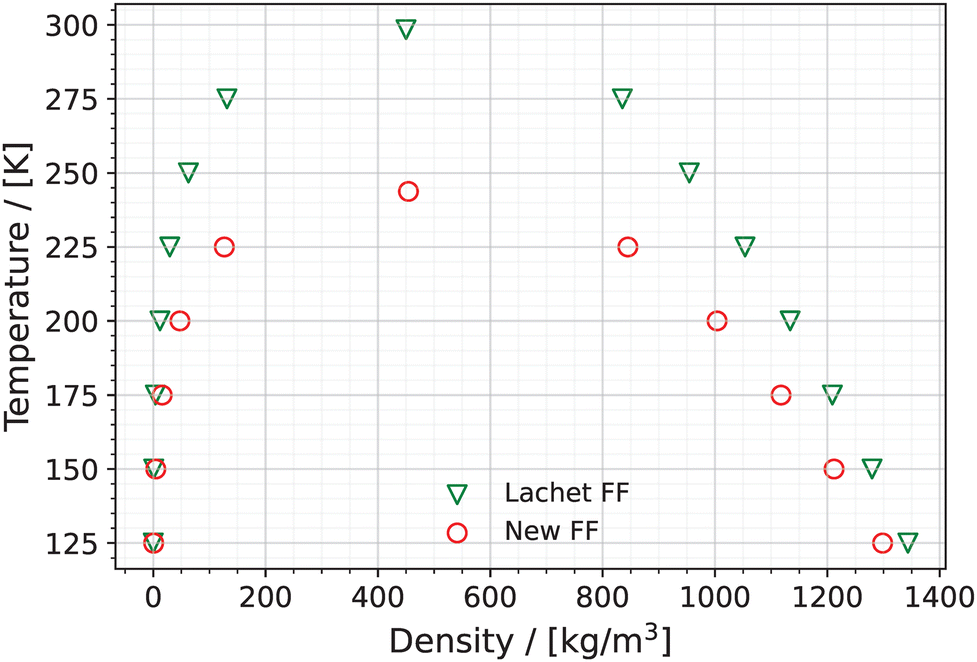
5.2.2 Properties of pure (NO)2.
Fig. 8 shows the vapor and liquid densities of pure (unreactive) (NO)2 as a function of temperature computed using the Lachet FF and the New FF. The saturated vapor pressures of pure (NO)2 as a function of the temperature computed using the New FF and Lachet FF are shown in Fig. 9. Note that the results for the Zhou FF is not included as there are no force field parameters for (NO)2 in the original work of Zhou et al.38
 |
| | Fig. 8 Coexisting vapor and liquid densities of pure (NO)2 using the New FF (red circles) and the Lachet FF36 (green triangles). Error bars are estimated based on the standard deviation of 5 blocks into which the production run was divided and are much smaller than the symbols used in the figure. The critical points are also shown in the figure. | |
 |
| | Fig. 9 Inverse temperature dependence of the saturated vapor pressures of pure (NO)2 using the New FF (red circles) and the Lachet FF36 (green triangles), on logarithmic scale. Error bars for the New FF are computed based on the standard deviation. | |
The predicted critical temperatures, densities, and pressures are listed in Table S9 of the ESI.† The predicted critical temperature, Tc, using the New FF is 243.78 K which is smaller than the predicted critical temperature using the Lachet FF (298.37 K). The critical densities (ρc) predicted by the two force fields are in agreement (454.28 kg m−3 for the New FF and 449.95 kg m−3 for the Lachet FF).
The saturated vapor pressures calculated using the Lachet FF and the New FF are shown in Fig. 9. Psat values computed using the Lachet FF are smaller (on average by 77.2%) than the Psat values computed using the New FF in the temperature range of 125 K to 225 K. However, the Pc computed using the Lachet FF is larger than the Pc calculated using the New FF by 0.8 MPa. This is attributed to the larger Tc predicted by the Lachet FF compared to the Tc predicted by the New FF (see Table S10 of the ESI†).
5.3 Sensitivity analysis
The VLE properties of the reactive NO–(NO)2 system are sensitive to the atomization energy of (NO)2 (D0,(NO)2) and the L-J parameters, σ and ε of NO and (NO)2. We performed a series of simulations to systematically analyze the dependence of the VLE properties of the reactive NO–(NO)2 system on D0,(NO)2 and L-J parameters, and hence reflect upon the uniqueness of our new force field parameters. These simulations and their main results are discussed below.
5.3.1 Effect of D0,(NO)2 on VLE properties of reactive NO–(NO)2 system.
In the first series of simulations, we considered three values for D0,(NO)2 which correspond to three values of BDE(NO)2 that are within the range of the reported experimental and theoretical values for BDE(NO)2 in the literature (7.64–13.8 kJ mol−1).2,32,75,79–82,86 These values are listed in Table S10 of the ESI.†D0,(NO)2 = 1269 kJ mol−1 is the value with which the VLE properties could be computed in agreement with experiments (see Section 5.1). In these simulations, the L-J parameters and the partial charges are fixed as in Table 1. These simulations are performed in the reactive Gibbs ensemble.
The vapor–liquid coexistence densities and (NO)2 mole fractions in the liquid phase of the reactive NO–(NO)2 system for these four values of D0,(NO)2 are shown in Fig. S2 and S3 of the ESI,† respectively. Even though the values of D0,(NO)2 are only different by 4 kJ mol−1 (difference between the highest and lowest value in the set), the differences in the corresponding dimer mole fractions in the liquid phase is significant (56% difference in the mole fractions at 160 K between the systems with the highest and lowest value of D0,(NO)2). Moreover, higher values of D0,(NO)2 tend to have more negative slopes for the liquid densities (see Fig. S2 of the ESI†). This could be attributed to the presence of more dimer species in the liquid phase. This results in a higher critical temperature compared to the systems with lower values of D0,(NO)2 (see Table S11 of the ESI†).
5.3.2 Effect of L-J parameters on VLE properties of reactive NO–(NO)2 system.
In a second series of simulations, the values of D0,(NO)2 = 1269 kJ mol−1 and partial charges (see Table 1) are fixed, while twelve different combinations of L-J parameters are considered. In each combination, the L-J parameters (σ or ε) of NO and/or (NO)2 are varied by ±10% compared to the respective parameters of the New FF. These parameters are labeled FF-a to FF-l: σ(NO)2 in FF-a (b), ε(NO)2 in FF-c (d), σNO in FF-e (f), εNO in FF-g (h), σNO and σ(NO)2 in FF-i (j), and εNO and ε(NO)2 in FF-k (l), are varied by −10% (10%). These parameters are shown in Table S12 of the ESI.† These simulations are also performed in the reactive Gibbs ensemble.
The coexistence densities, dimer mole fractions in the liquid phase, coexistence pressures, heats of vaporization, and total enthalpies of the reactive NO–(NO)2 system were computed using FF-a to FF-l and are shown in Fig. S4–S33 of the ESI.†
An increase in the L-J parameters of (NO)2, σ(NO)2 (FF-b) or ε(NO)2 (FF-d), or NO, σNO (FF-f) or εNO (FF-h), results in an increase in the mole fraction of that species, (NO)2 (see Fig. S5 and S10 of the ESI†) or NO (see Fig. S15 and S20 of the ESI†), in the liquid phase of the system. Increasing the values of the L-J parameters of a species makes it energetically favorable to increase the concentration of that species in the system. This can be seen by comparing two simple L-J systems that differ in the values of the L-J parameters, as shown in Fig. S34 of the ESI† (two systems with the same value of σ but different values of ε) and Fig. S35 of the ESI† (two systems with the same value of ε but different values of σ). With an increase in σ or ε, the area of the attractive potential L-J increases. Therefore, with the increase in the values of the L-J parameters (σ or ε) of NO or (NO)2, it becomes energetically favorable to increase the concentration of NO or (NO)2 in the NO–(NO)2 system.
The increase in the mole fraction of (NO)2 or NO affects the VLE properties of the reactive system, including the total enthalpies of the system. An increase in the dimer mole fraction lowers the total enthalpy of the system; see Fig. S8, S13, S18 and S23 of the ESI.† This is supported by the exothermic nature of dimer formation, which causes heat dissipation, and thereby, reduction of the total enthalpy of the system. However, an increase in monomer concentrations increases the total enthalpy of the system. This scenario may be supported by the endothermic nature of N–N bond dissociation between two NO entities in ((NO)2), which leads to an increase in the total enthalpy of the system. The dimer mole fractions in the liquid phase when the L-J parameters of NO and (NO)2 (σ in FF-i (j) and ε in FF-k (l)) are simultaneously varied by ±10% compared to the New FF, follow a similar trend as when only the L-J parameters of (NO)2 is varied (σ(NO)2 in FF-a (b) and ε(NO)2 in FF-c (d)); see Fig. S25 and S30 of the ESI.† This could be because the liquid phase is predominantly composed of dimers.
Although we demonstrated that the VLE properties of the reactive NO–(NO)2 system depends on both D0,(NO)2 (or subsequently BDE(NO)2) and the L-J parameters of NO and (NO)2 species, this may not be the case for all the reactive systems. An example is a reactive NO2–N2O4 system. The VLE properties of reactive NO2–N2O4 are almost exclusively affected by the values of BDEN2O4 and not by the values of L-J parameter as can be inferred by the work of Polat et al.62 As explained by Johnson et al.,39 the values of BDE determine the association strength of a fluid. A fluid is weakly (strongly) associating if the value of BDE is small (large).39 The concluded value of BDE for (NO)2 in our study is 13.06 kJ mol−1 (i.e., D0,(NO)2 = 1269 kJ mol−1), which is much smaller than value of BDE for N2O4 (∼50 kJ mol−1).61 Accordingly, NO2–N2O4 fluid is a more strongly associating fluid than NO–(NO)2 fluid. Therefore, the dependence of the VLE properties of a reactive fluid on the values of L-J parameters may be determined by the association strength of that fluid.
6 Conclusions
Our force field is capable of predicting the VLE properties of the reactive NO–(NO)2 system (i.e., coexistence vapor–liquid densities, dimer mole fractions in liquid phase, saturated vapor pressures, and heats of vaporization) in excellent agreement with experimental values for the temperature range 120 K to 170 K. For pure NO, the predicted VLE properties using our force field parameters agree well with those using the Lachet FF. However, there are large deviations between the predictions using the Zhou FF and predictions using our force field parameters or the Lachet FF. The Zhou FF was developed to reproduce the experimental density and heat of vaporization of NO at its boiling point without considering the formation of a dimer at this temperature. This may be the reason for the large deviations of the VLE properties of pure NO predicted by the Zhou FF from those predicted by the New FF and Lachet FF. For pure (NO)2, the liquid densities using the Lachet FF are larger (on average, by 11%) than the predictions using our force field parameters. The Lachet FF predicts smaller saturated vapor pressures (on average ca. 80%) compared to our force field. We also performed a systematic parameter study to analyze the impact of individual L-J parameters (σ and ε of NO and (NO)2) as well as the atomization energy of (NO)2, D0,(NO)2, or alternatively the bond dissociation energy of (NO)2, BDE(NO)2, on the VLE properties of the reactive NO–(NO)2 system. Our results show that the VLE properties of the reactive NO–(NO)2 system are affected by both the L-J parameters and the atomization energy of the NO dimer. This is in contrast to a previously studied reactive NO2–N2O4 system where the VLE properties are mainly affected by the atomization energy of NO2 dimer. This can be attributed to the association strength of reactive fluids. A fluid is weakly (strongly) associating if the value of BDE is small (large). The concluded value of the BDE for (NO)2 in our study is 13.06 kJ mol−1, which is much smaller than the BDE of N2O4 (∼50 kJ mol−1). Accordingly, the NO2–N2O4 fluid is a more strongly associating fluid than the NO–(NO)2 fluid. Therefore, the VLE properties of reactive NO–(NO)2 system are affected by both L-J parameters and the atomization energy of the dimer, while the VLE properties of reactive NO2–N2O4 are only affected by the atomization energy of the dimer.
Author contributions
All authors conceived the work. TS carried out the simulations and data analysis. All authors provided critical feedback on the interpretation of data analysis. BB supervised the project. TS and BB wrote the manuscript in collaboration with TV and SC.
Data availability
The data supporting this manuscript have been included as part of the ESI.† The input files to run Brick-CFCMC simulations as well as the output files are provided at https://doi.org/10.5281/zenodo.15524759.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
TS thanks the Institute for Complex Molecular Systems for financial support.
References
- E. F. Horn and F. P. Dickey, J. Chem. Phys., 1964, 41, 1614–1620 CrossRef CAS.
- A. R. McKellar, J. Watson and B. Howard, Mol. Phys., 1995, 86, 273–286 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- P. Lukes, E. Dolezalova, I. Sisrova and M. Clupek, Plasma Sources Sci. Technol., 2014, 23, 015019 CrossRef CAS.
- M. A. Malik, Plasma Chem. Plasma Process., 2016, 36, 737–766 CrossRef CAS.
- Z. Machala, B. Tarabová, D. Sersenová, M. Janda and K. Hensel, J. Phys. D: Appl. Phys., 2018, 52, 034002 CrossRef.
- K. Tachibana and T. Nakamura, J. Phys. D: Appl. Phys., 2019, 52, 385202 CrossRef CAS.
- D. B. Graves, J. Phys. D: Appl. Phys., 2012, 45, 263001 CrossRef.
- X. Lu, G. V. Naidis, M. Laroussi, S. Reuter, D. B. Graves and K. Ostrikov, Phys. Rep., 2016, 630, 1–84 CrossRef CAS.
- D. B. Graves, Plasma Processes Polym., 2014, 11, 1120–1127 CrossRef CAS.
- D. P. Park, K. Davis, S. Gilani, C.-A. Alonzo, D. Dobrynin, G. Friedman, A. Fridman, A. Rabinovich and G. Fridman, Curr. Appl. Phys., 2013, 13, S19–S29 CrossRef.
- N. Puač, M. Gherardi and M. Shiratani, Plasma Processes Polym., 2018, 15, 1700174 CrossRef.
- P. Ranieri, N. Sponsel, J. Kizer, M. Rojas-Pierce, R. Hernández, L. Gatiboni, A. Grunden and K. Stapelmann, Plasma Processes Polym., 2021, 18, 2000162 CrossRef CAS.
- C. V. Suschek and C. Opländer, Clin. Plasma Med., 2016, 4, 1–8 CrossRef.
- A. V. Butenko, A. B. Shekhter, A. V. Pekshev, A. B. Vagapov, A. L. Fayzullin, N. B. Serejnikova, N. A. Sharapov, V. A. Zaborova and V. N. Vasilets, Clin. Plasma Med., 2020, 19, 100112 CrossRef.
- A. Lindsay, B. Byrns, W. King, A. Andhvarapou, J. Fields, D. Knappe, W. Fonteno and S. Shannon, Plasma Chem. Plasma Process., 2014, 34, 1271–1290 CrossRef CAS.
- C. Bradu, K. Kutasi, M. Magureanu, N. Puač and S. Živković, J. Phys. D: Appl. Phys., 2020, 53, 223001 CrossRef CAS.
- D. Guo, H. Liu, L. Zhou, J. Xie and C. He, J. Sci. Food Agric., 2021, 101, 4891–4899 CrossRef CAS PubMed.
- W. Tian and M. J. Kushner, J. Phys. D: Appl. Phys., 2014, 47, 165201 CrossRef.
- A. M. Lietz and M. J. Kushner, J. Phys. D: Appl. Phys., 2016, 49, 425204 CrossRef.
- A. D. Lindsay, D. B. Graves and S. C. Shannon, J. Phys. D: Appl. Phys., 2016, 49, 235204 CrossRef.
- C. Verlackt, W. Van Boxem and A. Bogaerts, Phys. Chem. Chem. Phys., 2018, 20, 6845–6859 RSC.
- K. Ikuse and S. Hamaguchi, Jpn. J. Appl. Phys., 2022, 61, 076002 CrossRef CAS.
- P. J. Bruggeman, M. J. Kushner, B. R. Locke, J. G. Gardeniers, W. Graham, D. B. Graves, R. Hofman-Caris, D. Maric, J. P. Reid and E. Ceriani,
et al.
, Plasma Sources Sci. Technol., 2016, 25, 053002 CrossRef.
- T. H. G. Saji, J. M. Vicent-Luna, T. J. H. Vlugt, S. Calero and B. Bagheri, J. Mol. Liq., 2024, 401, 124530 CrossRef CAS.
- M. N. Hughes, Methods Enzymol., 2008, 436, 3–19 CAS.
- H. Hoshina, M. Slipchenko, K. Prozument, D. Verma, M. W. Schmidt, J. Ivanic and A. F. Vilesov, J. Phys. Chem. A, 2016, 120, 527–534 CrossRef CAS PubMed.
- S. Marinakis, C. Cockrell, K. Trachenko, T. F. Headen and A. K. Soper, J. Phys. Chem. B, 2022, 126, 9860–9870 CrossRef CAS PubMed.
- F. Kohler, H. J. Guedes, J. Revés and M. N. da Ponte, J. Mol. Liq., 1995, 67, 105–123 CrossRef CAS.
- J. M. Walsh, H. J. Guedes and K. E. Gubbins, J. Phys. Chem., 1992, 96, 10995–11004 CrossRef CAS.
- A. L. Smith and H. L. Johnston, J. Am. Chem. Soc., 1952, 74, 4696–4698 CrossRef CAS.
- J. K. Park and H. Sun, Chem. Phys., 2001, 263, 61–68 CrossRef CAS.
- R. Sayós, R. Valero, J. Anglada and M. González, J. Chem. Phys., 2000, 112, 6608–6624 CrossRef.
- F. Kohler, M. Bohn, J. Fischer and R. Zimmermann, Monatsh. Chem., 1987, 118, 169–182 CrossRef CAS.
-
J. O. Hirschfelder, C. F. Curtiss and R. B. Bird, The molecular theory of gases and liquids, John Wiley & Sons, 1964 Search PubMed.
- V. Lachet, B. Creton, T. de Bruin, E. Bourasseau, N. Desbiens, Ø. Wilhelmsen and M. Hammer, Fluid Phase Equilib., 2012, 322, 66–78 CrossRef.
- J. Yang, Y. Ren, A.-M. Tian and H. Sun, J. Phys. Chem. B, 2000, 104, 4951–4957 CrossRef CAS.
- Z. Zhou, B. Todd, K. P. Travis and R. J. Sadus, J. Chem. Phys., 2005, 123, 054505 CrossRef PubMed.
- J. K. Johnson, A. Z. Panagiotopoulos and K. E. Gubbins, Mol. Phys., 1994, 81, 717–733 CrossRef CAS.
- J. K. Johnson, Adv. Chem. Phys., 1999, 105, 461–481 CrossRef CAS.
- W. Smith and B. Triska, J. Chem. Phys., 1994, 100, 3019–3027 CrossRef CAS.
- A. Poursaeidesfahani, R. Hens, A. Rahbari, M. Ramdin, D. Dubbeldam and T. J. H. Vlugt, J. Chem. Theory Comput., 2017, 13, 4452–4466 CrossRef CAS PubMed.
- C. Heath Turner, J. K. Brennan, M. Lisal, W. R. Smith, J. Karl Johnson and K. E. Gubbins, Mol. Simul., 2008, 34, 119–146 CrossRef.
- S. C. Glotzer, D. Stauffer and N. Jan, Phys. Rev. Lett., 1994, 72, 4109 CrossRef CAS PubMed.
- J. Sterpenich, M.-C. Caumon, V. Lachet, B. Creton, M. El Jarmouni, A. Randi and P. Robert, J. Raman Spectrosc., 2022, 53, 645–653 CrossRef CAS.
- B. Creton, C. Nieto-Draghi, T. De Bruin, V. Lachet, E. El Ahmar, A. Valtz, C. Coquelet, S. Lasala, R. Privat and J.-N. Jaubert, Fluid Phase Equilib., 2018, 461, 84–100 CrossRef CAS.
- W. Sun, L.-C. Lin, X. Peng and B. Smit, AIChE J., 2014, 60, 2314–2323 CrossRef CAS.
- N. Qiu, X. Bai, J. Xu, N. Sun, J. S. Francisco, M. Yang, Q. Huang and S. Du, J. Phys. Chem. C, 2018, 123, 2691–2702 CrossRef.
- W. Shi and E. J. Maginn, J. Chem. Theory Comput., 2007, 3, 1451–1463 CrossRef CAS PubMed.
- W. Shi and E. J. Maginn, J. Comput. Chem., 2008, 29, 2520–2530 CrossRef CAS PubMed.
- A. Rahbari, R. Hens, M. Ramdin, O. Moultos, D. Dubbeldam and T. J. H. Vlugt, Mol. Simul., 2021, 47, 804–823 CrossRef CAS.
- R. Hens, A. Rahbari, S. Caro-Ortiz, N. Dawass, M. Erdös, A. Poursaeidesfahani, H. S. Salehi, A. T. Celebi, M. Ramdin and O. A. Moultos,
et al.
, J. Chem. Inf. Model., 2020, 60, 2678–2682 CrossRef CAS PubMed.
- H. M. Polat, H. S. Salehi, R. Hens, D. O. Wasik, A. Rahbari, F. de Meyer, C. Houriez, C. Coquelet, S. Calero, D. Dubbeldam, O. A. Moultos and T. J. H. Vlugt, J. Chem. Inf. Model., 2021, 61, 3752–3757 CrossRef CAS PubMed.
- A. Z. Panagiotopoulos, N. Quirke, M. Stapleton and D. Tildesley, Mol. Phys., 1988, 63, 527–545 CrossRef CAS.
- A. Poursaeidesfahani, A. Torres-Knoop, D. Dubbeldam and T. J. H. Vlugt, J. Chem. Theory Comput., 2016, 12, 1481–1490 CrossRef CAS PubMed.
- A. Z. Panagiotopoulos and M. R. Stapleton, Fluid Phase Equilib., 1989, 53, 133–141 CrossRef CAS.
- A. Panagiotopoulos, Fluid Phase Equilib., 1992, 76, 97–112 CrossRef CAS.
- F. Wang and D. P. Landau, Phys. Rev. Lett., 2001, 86, 2050–2053 CrossRef CAS PubMed.
- A. Poursaeidesfahani, A. Torres-Knoop, D. Dubbeldam and T. J. H. Vlugt, J. Chem. Theory Comput., 2016, 12, 1481–1490 CrossRef CAS PubMed.
- A. Rahbari, A. Poursaeidesfahani, A. Torres-Knoop, D. Dubbeldam and T. J. H. Vlugt, Mol. Simul., 2018, 44, 405–414 CrossRef CAS.
- S. Lasala, K. Samukov, H. M. Polat, V. Lachet, O. Herbinet, R. Privat, J.-N. Jaubert, O. A. Moultos, K. De Ras and T. J. H. Vlugt, Chem. Eng. J., 2024, 483, 148961 CrossRef CAS.
- H. M. Polat, S. Lasala, F. De Meyer, C. Houriez, O. A. Moultos and T. J. H. Vlugt, Fluid Phase Equilib., 2024, 582, 114084 CrossRef CAS.
- N. Ferrando, V. Lachet and A. Boutin, J. Phys. Chem. B, 2010, 114, 8680–8688 CrossRef CAS PubMed.
-
J. S. Rowlinson and B. Widom, Molecular theory of capillarity, Courier Corporation, 3rd edn, 2013 Search PubMed.
-
D. Frenkel and B. Smit, Understanding molecular simulation: from algorithms to applications, Elsevier, 3rd edn, 2023 Search PubMed.
- A. Tamir, C. Dragoescu, A. Apelblat and J. Wisniak, Fluid Phase Equilib., 1983, 10, 9–42 CrossRef CAS.
- C. Burrus and J. Graybeal, Phys. Rev., 1958, 109, 1553 CrossRef.
- H. A. Lorentz, Ann. Phys., 1881, 248, 127–136 CrossRef.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- H. M. Polat, F. de Meyer, C. Houriez, O. A. Moultos and T. J. H. Vlugt, J. Chem. Theory Comput., 2023, 19, 2616–2629 CrossRef CAS PubMed.
- F. Torrens, Toxicol. Environ. Chem., 1999, 73, 177–189 CrossRef CAS.
- C. M. Western, P. R. Langridge-Smith, B. J. Howard and S. E. Novick, Mol. Phys., 1981, 44, 145–160 CrossRef CAS.
- A. East, A. McKellar and J. Watson, J. Chem. Phys., 1998, 109, 4378–4383 CrossRef CAS.
- E. D. Glendening and A. M. Halpern, J. Chem. Phys., 2007, 127, 164307–164311 CrossRef PubMed.
- A. Malakhovskii and A. Gedanken, J. Chem. Soc., Faraday Trans., 1996, 92, 329–332 RSC.
-
H. C. Shi, Computational study of electronic excited states of nitric oxide monomer and nitric oxide dimer, Faculty of Graduate Studies and Research, University of Regina, 2007 Search PubMed.
-
M. W. Chase, NIST-JANAF thermochemical tables, American Chemical Society, Washington, DC, New York, 1998 Search PubMed.
- B. Howard and A. McKellar, Mol. Phys., 1993, 78, 55–72 CrossRef CAS.
- R. González-Luque, M. Merchán and B. O. Roos, Theor. Chim. Acta, 1994, 88, 425–435 CrossRef.
- A. Dkhissi, P. Soulard, A. Perrin and N. Lacome, J. Mol. Spectrosc., 1997, 183, 12–17 CrossRef CAS.
- M. Lsal, J. K. Brennan and W. R. Smith, J. Chem. Phys., 2006, 124, 064712–064725 CrossRef PubMed.
- C. H. Turner, J. K. Johnson and K. E. Gubbins, J. Chem. Phys., 2001, 114, 1851–1859 CrossRef CAS.
-
R. Rowley, W. Wilding, J. Oscarson, N. Zundel, T. Marshall, T. Daubert and R. Danner, DIPPR Data Compilation of Pure Chemical Properties, AIChE, 2010 Search PubMed.
-
J. Pedley, Thermochemical data and structures of organic compounds, CRC Press, 1994, vol. 1 Search PubMed.
- N. Taguchi, Y. Mochizuki, T. Ishikawa and K. Tanaka, Chem. Phys. Lett., 2008, 451, 31–36 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental values for bond distances and angles of NO and (NO)2 are presented in Table S1. The experimental rotational constants and vibrational frequencies of NO and its dimer are presented in Tables S2 and S3, respectively. The values of the isolated molecule partition functions for NO and (NO)2 for different temperatures are presented in Table S4. The VLE properties of the reactive NO–(NO)2 system predicted using the New FF as well as the respective experimental values are presented in Tables S5–S7. The force field parameters developed by Zhou et al.38 (Zhou FF) and Lachet et al.36 (Lachet FF) are listed in Table S9. The critical temperature, density, and pressure of pure NO and (NO)2 using the New FF, the Zhou FF, and the Lachet FF are listed in Table S10. The critical temperatures and densities of NO–(NO)2 systems for various values of D0,(NO)2 are listed in Table S11. The twelve different force field parameters used for our parameter study are listed in Table S12 with the respective predicted critical temperatures and densities in Table S13. Fig. S1 shows the vapor–liquid coexistence densities obtained with the the New FF along with the extrapolated densities at temperatures close to the critical temperature obtained by fitting the vapor and liquid phase densities to the law of rectilinear diameters. Fig. S2 and S3 show the changes in the orthobaric densities and dimer mole fractions using the different values of D0,(NO)2 listed in Table S10. Fig. S4–S33 show the variations in the coexistence densities, dimer mole fractions, saturated vapor pressures, heats of vaporization and total enthalpies of the reactive NO–(NO)2 system for the different force fields used for our parameter study. Fig. S34 and S35 show the difference in the area of the attractive L-J potential energy for systems with different values of ε and σ, respectively. See DOI: https://doi.org/10.1039/d5cp00784d |
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Thijs J. H.
Vlugt
ab,
Thijs J. H.
Vlugt

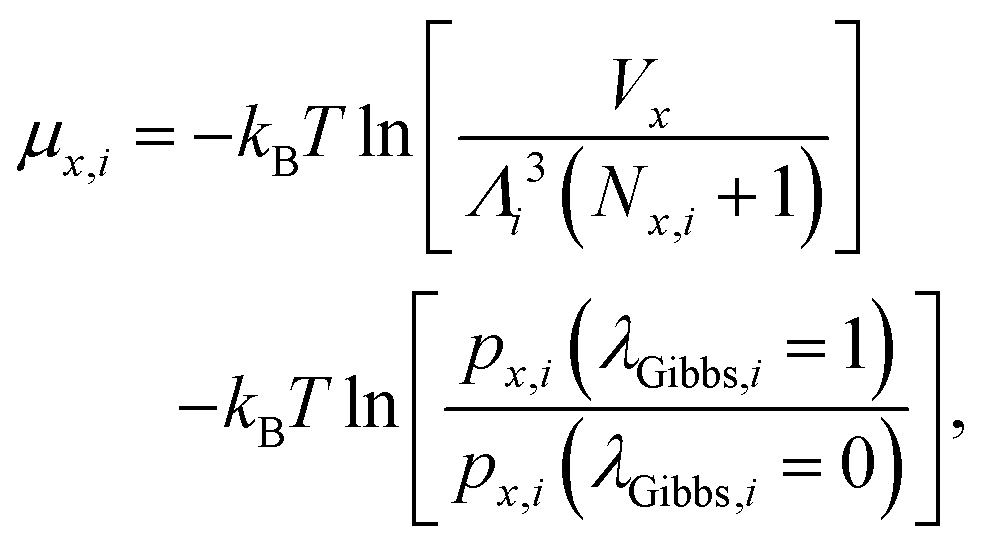




 versus temperature,62 where ρ0 is an arbitrary number density which is set to 1 Å−3. In the single-component GE, β is taken as 0.32.65
versus temperature,62 where ρ0 is an arbitrary number density which is set to 1 Å−3. In the single-component GE, β is taken as 0.32.65
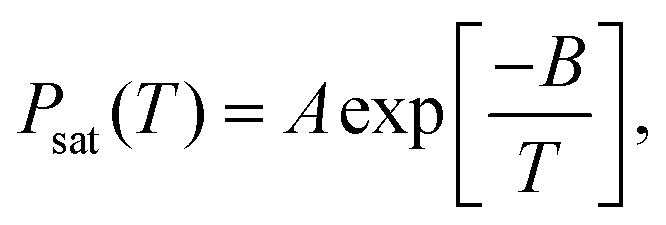



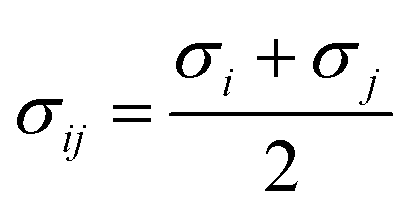 ,
,  . A cut-off radius of 10 Å was used for both the Lennard-Jones and Coulombic interactions. The Ewald summation method69,70 was used to treat long-range Coulombic interactions. The energies and pressures of the simulations were corrected with long-range tail corrections for the L-J interactions.65 After numerous trials of varying the σ and ε parameters (110 combinations were tested in total), we arrived at the values provided in Table 1. These parameters are referred to as the New FF in the rest of the manuscript. A detailed discussion on the sensitivity analysis of the New FF parameters is provided in Section 5.3.
. A cut-off radius of 10 Å was used for both the Lennard-Jones and Coulombic interactions. The Ewald summation method69,70 was used to treat long-range Coulombic interactions. The energies and pressures of the simulations were corrected with long-range tail corrections for the L-J interactions.65 After numerous trials of varying the σ and ε parameters (110 combinations were tested in total), we arrived at the values provided in Table 1. These parameters are referred to as the New FF in the rest of the manuscript. A detailed discussion on the sensitivity analysis of the New FF parameters is provided in Section 5.3.

 . Here, h is the Planck constant, M is the sum of the masses of all atoms in the molecule, T is the temperature, and V is the volume. In Brick-CFCMC, a reference state of V = V0 = 1 Å3 is used for qtrans.52,71
. Here, h is the Planck constant, M is the sum of the masses of all atoms in the molecule, T is the temperature, and V is the volume. In Brick-CFCMC, a reference state of V = V0 = 1 Å3 is used for qtrans.52,71

 is the characteristic vibrational temperature corresponding to the ith normal mode with vibrational frequency, νi.
is the characteristic vibrational temperature corresponding to the ith normal mode with vibrational frequency, νi.

 . For a diatomic molecule such as NO, qrot is given by52
. For a diatomic molecule such as NO, qrot is given by52
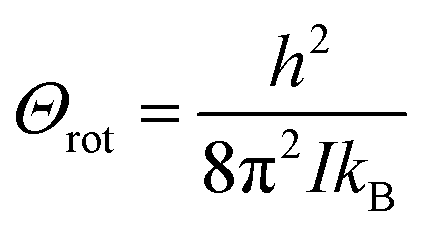 is the characteristic rotational temperature. σrot is the rotational symmetry number of the molecule, which is the number of indistinguishable configurations the molecule can be brought into by rotation about its centre of mass.72 For NO, σrot = 1. The rotational partition function, qrot, for (NO)2, which is a non-linear polyatomic species, is calculated using52
is the characteristic rotational temperature. σrot is the rotational symmetry number of the molecule, which is the number of indistinguishable configurations the molecule can be brought into by rotation about its centre of mass.72 For NO, σrot = 1. The rotational partition function, qrot, for (NO)2, which is a non-linear polyatomic species, is calculated using52


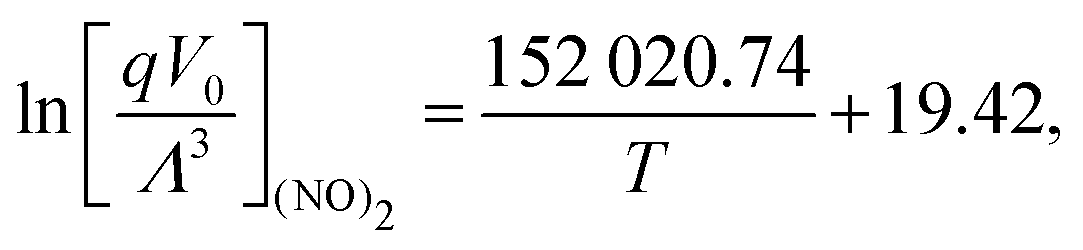

 42,62) of (NO)2 and 2NO. The differences between the standard chemical potential of (NO)2 and 2NO used in the work of Lachet et al. favor more NO molecules in the system.36,85
42,62) of (NO)2 and 2NO. The differences between the standard chemical potential of (NO)2 and 2NO used in the work of Lachet et al. favor more NO molecules in the system.36,85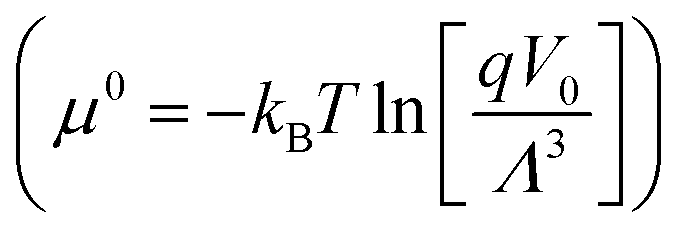 of (NO)2 and 2NO used in Lachet et al.36 compared to our work, which leads to differences in compositions. The isolated molecular partition functions in the work of Lachet et al.36 favor more NO molecules in both the vapor and liquid simulation boxes compared to our systems. This leads to higher vapor pressures for Lachet et al.36 The critical pressure is obtained by extrapolating the vapor pressure curve to the critical temperature using eqn (9). The critical pressure using the New FF is 6.24 MPa (3.9% deviation from the experimental critical pressure) and that predicted using the Lachet FF is 6.37 MPa (1.7% deviation from the experimental value). See Table 2.
of (NO)2 and 2NO used in Lachet et al.36 compared to our work, which leads to differences in compositions. The isolated molecular partition functions in the work of Lachet et al.36 favor more NO molecules in both the vapor and liquid simulation boxes compared to our systems. This leads to higher vapor pressures for Lachet et al.36 The critical pressure is obtained by extrapolating the vapor pressure curve to the critical temperature using eqn (9). The critical pressure using the New FF is 6.24 MPa (3.9% deviation from the experimental critical pressure) and that predicted using the Lachet FF is 6.37 MPa (1.7% deviation from the experimental value). See Table 2.